
Introduction to the GCG Wisconsin Package
64
Introduction to the GCG Wisconsin Package The Center for Bioinformatics UNC at Chapel Hill Jianping (JP) Jin Ph.D. Bioinformatics Scientist Phone: (919)843-6105 E-mail: jjin@email. unc . edu Fax: (919)843-3103
-
Upload
leonard-hopper -
Category
Documents
-
view
144 -
download
23
description
Introduction to the GCG Wisconsin Package. The Center for Bioinformatics UNC at Chapel Hill Jianping (JP) Jin Ph.D. Bioinformatics Scientist Phone: (919)843-6105 E-mail: [email protected] Fax: (919)843-3103. What is GCG. - PowerPoint PPT Presentation
Transcript of Introduction to the GCG Wisconsin Package

Introduction to the GCG Wisconsin Package
The Center for BioinformaticsUNC at Chapel Hill
Jianping (JP) Jin Ph.D.
Bioinformatics ScientistPhone: (919)843-6105E-mail: [email protected]: (919)843-3103

What is GCG An integrated package of over 130
programs (the GCG Wisconsin Package).
For extensive analyses of nucleic acid and protein sequences.
Associated with most major public nucleic acid and protein databases.
Works on UNIX OS.

Why use GCG Removes the need for the constant collection of
new software by end users. Removes the need to learn new interface as new
software is released. Provides a flow of analyses within a single
interface. Unix environment allows users to automate
complex, repetitive tasks. Allows users to use multiple processors to
accelerate their jobs. Supports almost all public databases that can be
updated daily. Fast local search.

Flexibility or Automation
1. MEME: upstream regulatory motifs;2. MotifSearch: genes sharing these
potential regulatory motifs;3. PileUp: multiple sequence alignment;4. Distances: extract pairwise distances
from the alignment;5. GrowTree: a phylogenetics tree.

InterfacesCommand Line: Running programs from
UNIX system prompt. SeqLab: Graphic User’s Interface,
requiring an X windows display.SeqWeb: to a core set of sequence
analysis program.

Limitations with GCG
The GUI interface does not give the users the full access to the power of the command line, nor to the complete set of programs.
Many programs place a limit of the maximum size of the sequences that they can handle (350 Kb). This limitation will be removed in version 11.

Databases GCG Supports
Nucleic acid databases GenBank EMBL (abridged)
Protein databases NRL_3D UniProt (SWISS-PROT, PIR, TrEMBL) PROSITE, Pfam,
Restriction Enzymes (REBASE)

Database Update Services DataServe: Automatically updates nucleic
acid on a daily basis via FTP. DataExtended: the most compete set of
nucleic acid and protein data. The timing of the release is coordinated with the major GenBank release, 2-3 months.
DataBasic: Similar to DataExtended, but excludes EST and GSS data from GenBank and EMBL.

File Importing and ExportingReformatFromEMBLFromGenBankFromPIR ToPIRFromStaden ToStadenFromIG ToIGFromFastA ToFastA

File Formats with GCGSingle sequence files (in GCG format)List (a list of files)MSF (multiple sequence format)RSF (rich sequence format)

Typical program

Result from MAP analysis

X-Windows server must be running

SeqLab Main Window (List Mode)
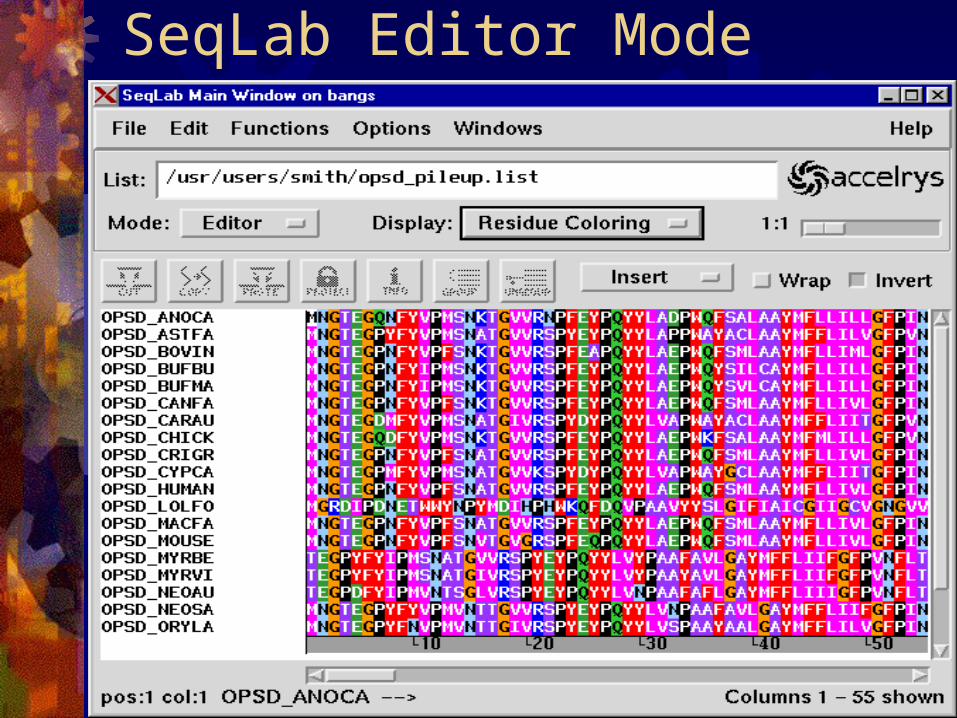
SeqLab Editor Mode

Display by Features

SeqLab Editor Mode (cont.)

SeqLab Output Manager

GCG Programs 1. Comparison 2. Database Searching and Retrieval 3. DNA/RNA Secondary Structure 4. Editing and Publication 5. Evolution 6. Fragment Assembly 7. Importing and exporting 8. Mapping 9. Primer Selection 10. Protein Analysis 11. Translation

Create your own sequence

PlasmidMap

FindPatterns

HmmerPfam Analysis

Gene Finding (FRAME)
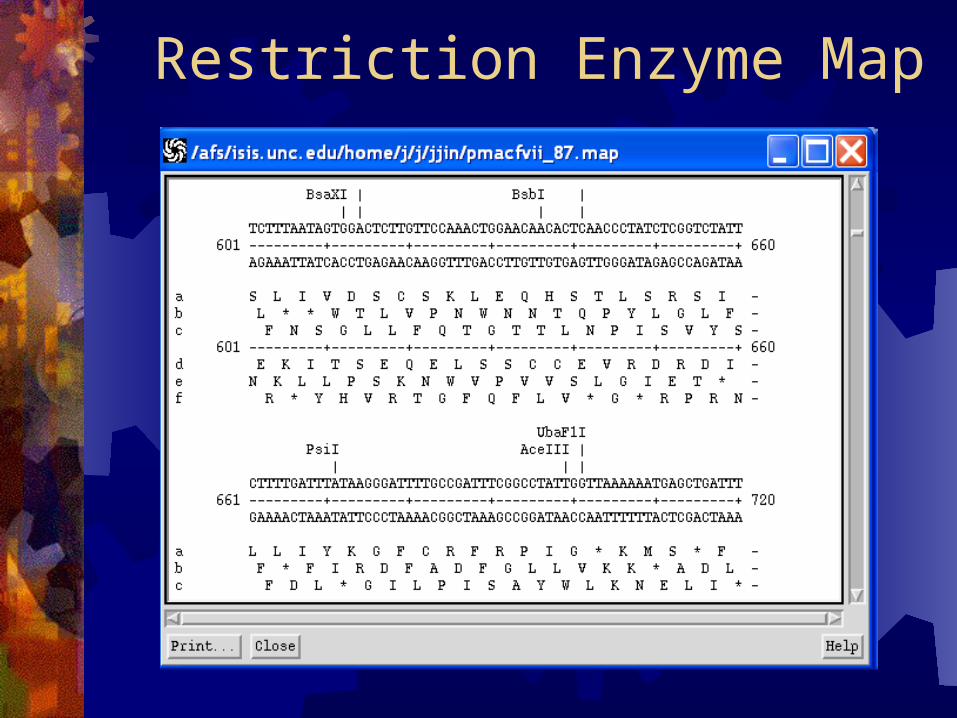
Restriction Enzyme Map

Consensus Sequence

Phylogenetic Tree (Cladogram)

Peptide Structure

Peptide Structure (2)

Isoelectric Analysis

Transmemberane Domains

Neucleic Acid 2nd Structure

Pairwise Comparison (Gap)
Neelman & Wunsch algorithm.A global alignment covering the whole
length of both sequences and the resulting sequences are of the same length with inserted gaps.
Good when two sequences are closely related.

Pairwise Comparison (BestFit)Algorithm of Smith and Waterman. Local homology alignment that finds the
best segment of similarity b/w two sequences.
The most sensitive sequence comparison method available.

Comparison of two sequences

GapShow

Multiple Comparison (PileUp)The method of Feng and Doolittle
similar to Higgins & Sharp.A series of progressive pairwise
alignments (up to 500 seq.) generate a final alignment.
An extension of Gap, not ideal for finding the best local region of similarity, such as a shared motif.

Multiple Comparison by Pileup

Multiple Comparison by Pileup

Dendrogram by Pileup

Database Search Nearly always employ local alignment
algorithms.Often use “heuristic” methods (for a
screen), FASTA and BLAST.Assures the seq.are given correct local
similarity score, but no guarantee that all seq. with high Smith-Waterman scores pass through the screen.

BLAST Accepts a number of sequences as input and
specify any number of DBs. $Blast –INfile2=PIR,SWPLUS; -INfile=hsp70.msf{*}.
Support 5 BLAST programs, but no gap alignment available for TBLASTX.
For non-coding nucleotide homology search, considering either reducing the word size from 11 to 6/7, or using the FASTA.
The number of scoring matrices is limited, BLOSUM62/45/80 and PAM70 available for –MATRix parameter.

Database Search (SSearch)A rigorous Smith-Waterman search for
similarity between a query sequence and a group of sequences of the same type.
The most sensitive method available for similarity search.
Very slow.

HmmerSearchUse a profile HMM as a query to search
a sequence database. Profile HMM: a position specific scoring
table, a statistical model of the consensus of a multiple sequence alignment.
Output can be used for any GCG program that accepts list file.

Profile Hidden Markov Model

HmmerSearch

HmmerSearch (cont.)

HmmerSearch (cont.)

HS (cont.Histogram of scores)

HS (cont. resulting alignment)

NetBLASTSends your query sequences over the
internet to a server at NCBI, Bethesda.Some limitations on NetBLAST, e.g.
prohibiting TBLASTX search vs. the nr database, only Alu, EST, GSS, STS.
Not support as many options as are available with BLAST.

NetBLAST

PSIBLASTSimilar to BLAST, except using position-
specific scoring matrices during the search.
Use protein sequence(s) to iteratively search protein database(s).

MEME and MotifSearch Multiple EM Motif Elicitation, a tool for
discovering motifs in a group of DNA or protein sequences.
Motif: a sequence pattern that occurs repeatedly in a group of related sequences.
Use a set of MEME profiles to search a database for new sequences similar to the original family.

MEME PROFILE

MEME (cont.)

GrowTree (Cladogram)

Access to GCG on Campus
1. Onyen and password plus sign up to BioSci service at http://onyen.unc.edu;
2. Computer connected to the Campus network;
3. Postscript printer connected to the campus network;
4. SSH Secure Client; 5. X-Windows Server (optional).

Sign up BioScience

Log onto GCG

Log onto GCG (cont.)

GCG Welcome Page

How to get seqlab to run Open X-Windows; Logon to the GCG server, nun.isis.unc.edu,
through SSH Secure Shell Client; At the prompt ($) enter the command “export
DISPLAY=yourMachineIP:0.0; Enter the command “xterm &” to activate the
xterm window; On the GCG main window enter the
command “seqlab &” to activate the SeqLab GUI.

How to get SeqLab to run (cont.)


















