
Introduction to EMBOSS and JEMBOSS
55
Introduction to EMBOSS and JEMBOSS Lecture Note for Computational Biology 1 (LSM 5191) Jiren Wang http://www.bii.a-star.edu.sg/~jiren BioInformatics Institute Singapore
Transcript of Introduction to EMBOSS and JEMBOSS

Introduction to EMBOSS and JEMBOSSLecture Note for Computational Biology 1 (LSM 5191)
Jiren Wanghttp://www.bii.a-star.edu.sg/~jiren
BioInformatics InstituteSingapore

What is EMBOSS?
EMBOSS – European Molecular Biology Open Software Suite
A free open source software package specially developed for the needs of the molecular biology user community.Public domain (GNU public license).Written by HGMP / Sanger / EBI / NorwayHGMP – Human Genome Mapping Project Resource Centre

The History of EMBOSS
Winconsin package, GCG – Genetics Computer GroupWidely used, source available for inspection
1988 – EGCG – academic add-on startedGCG commercial – source not freely available1999 – EGCG split from GCG to become EMBOSS

EMBOSS Suite
Provides a comprehensive set of sequence analysis programs.Provides a set of core software libraries.Integrates other public available packages.Encourages the use of EMBOSS in sequence analysis training.Encourages developers elsewhere to use the EMBOSS libraries.Supports all common Unix platforms including Linux, Digital Unix, Irix and Solaris.

EMBOSS Suite (cont’d)
They share a common look and feel.Easy to run from the command line.Easy to call from other programs (e.g. Perl).Easy to set up behind GUIs and Web interfaces.

Scope of Applications
Sequence alignmentNucleotide sequence pattern analysisSimple and species-specific repeat identificationCodon usage analysis for small genomesRapid identification of sequence patterns in large scale sequence setsPresentation tools for publicationEST analysis

Running EMBOSS Programs
EMBOSS programs are run by typing them at the Unix prompt, or by using an interface.The EMBOSS command syntax follows normal Unix command conventions.programname –help
to get some help on the options.
programname –opt to make the program prompt you for common options.
tfm programname to get the full help on a program.

Global Qualifier Description
Program will also prompt for optional qualifiers-options
When used with -help also gives the associated qualifiers and the general qualifiers
-verbose
Will give usage information of this program-help
Writes by default to stdout, but still prompts the user-stdout
Reads from stdin and writes to stdout and implies -auto-filter
Turns on debugging with ajDebug calls-debug
Turns off any prompting of the user-auto
Outputs a nicely formatted ACD file-acdpretty
Turns on logging of ACD file processing-acdlog
DescriptionQualifier definition

ACD Files
Every EMBOSS program will be accompanied by a so called ACD (Ajax Command Definitions) file, which describes the parameters that the program needs. It contains information about its input and output files and other parameters the program may need. It will indicate if any of the parameters are mandatory or that certain parameters are within certain limits.It can also indicate whether one parameter's value is dependent on the value or the presence of another.

Input and Output of EMBOSS Programs
InputA number of input file(s) and some parameters that are important to the function of the program.
OutputIn the form of files, plots, web pages, or simple text output.

What is Jemboss?
Jemboss is the Graphical User Interface (GUI) to EMBOSS.It is being developed at the Human Genome Mapping Project Resource Centre.The software is free and is part of the EMBOSS distribution.It is written in Java, enabling the interface to be used in both Unix and Windows environments.

Jemboss Server at BII
Jemboss Server is running on machine mammoth.bii.a-star.edu.sg, i.e. 203.116.36.122, at port 5060.Make sure you can access the page athttp://mammoth.bii.a-star.edu.sg:5060/index.html.

Setting Up Jemboss Client
StepsInstall Java 2 Platform, Standard EditionDownload, Unzip, and / or Untar the file JEMBOSS.tar.gz / JEMBOSS.zip.Set the Java ClasspathStart Jemboss client

Install Java 2 platform
Install Java 2 Platform, Standard Edition, that can be downloaded from http://java.sun.com/j2se/1.3/download.html.

Download, Unzip and/or Untar the File
For Linux / UnixDownload, Unzip, and Untar the File JEMBOSS.tar.gz at http://mammoth.bii.a-star.edu.sg/JEMBOSS/
gunzip JEMBOSS.tar.gz tar -xvf JEMBOSS.tar
For WindowsDownload, Unzip the File JEMBOSS.zip at http://mammoth.bii.a-star.edu.sg/JEMBOSS/. Unzip the file JEMBOSS.zip by WinZip at http://www.winzip.com/ddca.htm

Set the Classpath
For Linux / UnixSTORE=/location_to_put_downloaded_jar_file (e.g. STORE=/tmp/test)(These files are soap.jar, xerces.jar, mail.jar, activation.jar, jakarta-regexp-1.2.jar,jalview.jar, jsse.jar, jcert.jar, jnet.jar, and Jemboss.jar, that are included in JEMBOSS.tar.) export STORE CLASSPATH=$STORE/soap.jar:$STORE/xerces.jar:$STORE/mail.jar:$STORE/activation.jar:$STORE/jakarta-regexp-1.2.jar:$STORE/jalview.jar:$STORE/jsse.jar:$STORE/jcert.jar:$STORE/jnet.jar: $STORE/Jemboss.jar:. export CLASSPATH
For Windows(Suppose unzipped jar files soap.jar, xerces.jar, mail.jar, activation.jar, jakarta-regexp-1.2.jar, jalview.jar, jsse.jar, jcert.jar, jnet.jar, and Jemboss.jar are moved and stored in the directory c:\JEMBOSS) set CLASSPATH=c:\JEMBOSS\soap.jar;c:\JEMBOSS\xerces.jar;c:\JEMBOSS\mail.jar; c:\JEMBOSS\activation.jar;c:\JEMBOSS\jakarta-regexp-1.2.jar; c:\JEMBOSS\jalview.jar;c:\JEMBOSS\jsse.jar;c:\JEMBOSS\jcert.jar; c:\JEMBOSS\jnet.jar; c:\JEMBOSS\Jemboss.jar;.

Start Jemboss Client
Run the following command and the Jemboss client application will be started.
java org/emboss/jemboss/Jemboss &

wossname
FunctionFinds programs by keywords in their one-line documentation.
DescriptionAllows a user to search for keywords or parts of words in the brief documentation.Output is the program name and the brief description. If no words to search for are specified, then details of all the EMBOSS programs are output.

wossname example$ wossnameFinds programs by keywords in their one-line documentationKeyword to search for, or blank to list all programs: proteinSEARCH FOR 'PROTEIN'antigenic Finds antigenic sites in proteinsbacktranseq Back translate a protein sequencecharge Protein charge plotchecktrans Reports STOP codons and ORF statistics of a protein sequencedigest Protein proteolytic enzyme or reagent cleavage digestemowse Protein identification by mass spectrometryfuzzpro Protein pattern searchfuzztran Protein pattern search after translationgarnier Predicts protein secondary structureiep Calculates the isoelectric point of a proteinoctanol Displays protein hydropathyoddcomp Finds protein sequence regions with a biased compositionpatmatdb Search a protein sequence with a motifpatmatmotifs Search a PROSITE motif database with a protein sequencepepnet Displays proteins as a helical netpepstats Protein statisticspepwheel Shows protein sequences as helicespepwindow Displays protein hydropathypepwindowall Displays protein hydropathy of a set of sequencespreg Regular expression search of a protein sequencepscan Scans proteins using PRINTSsigcleave Reports protein signal cleavage sitessiggen Generates a sparse protein signaturesigscan Scans a sparse protein signature against swissprottranalign Align nucleic coding regions given the aligned proteins

seqretFunction
Reads and writes sequence.Description
seqret can read a sequence or many sequences from databases, files, files of sequence names, the command-line or the output of other programs and then can write them to files, the screen or pass them to other programs.seqret can read sequences in any of a wide range of standard sequence formats.seqret can read in the reverse complement of a nucleic acid sequence.seqret can read in a sequence whose begin and end positions you have specified and write out that fragment.seqret can change the case of the sequence being read in to upper or to lower case.

seqret Example seqret test.fasta –outseq embl::test.embl
>gi|1708198|sp|P80487|HHP_THICU HETEROTROPH-SPECIFIC PROTEINAADDVTVVIGSAAPMSGPQ>gi|13878750|sp|Q9CDN0|RS18_LACLA 30S ribosomal protein S18MAQQRRGGFKRRKKVDFIAANKIEVVDYKDTELLKRFISERGKILPRRVTGTSAKNQRKVVAIKRARVMALLPFVAEDQN
ID HHP_THICU STANDARD; PRT; 19 AA.AC P80487;DE HETEROTROPH-SPECIFIC PROTEINSQ SEQUENCE 19 AA; 1786 MW; ED8E6EAF CRC32;
AADDVTVVIG SAAPMSGPQ//ID RS18_LACLA STANDARD; PRT; 81 AA.AC Q9CDN0;DE 30S ribosomal protein S18SQ SEQUENCE 81 AA; 9371 MW; 3BD4FDD1 CRC32;
MAQQRRGGFK RRKKVDFIAA NKIEVVDYKD TELLKRFISE RGKILPRRVT GTSAKNQRKVVNAIKRARVM ALLPFVAEDQ N
//

infoseq
FunctionDisplays some simple information about sequences
DescriptionThis is a small utility to list the sequences' USA, name, accession number, type (nucleic or protein), length, percentage C+G, and/or description.USA - Uniform Sequence Address is a standard sequence naming used by all EMBOSS applications.The USA syntax is basically one of following:
Format::fileFormat::file:entrydbname:entry@listfile

infoseq Examplecat testone.fasta>gi|1708198|sp|P80487|HHP_THICU HETEROTROPH-SPECIFIC PROTEINAADDVTVVIGSAAPMSGPQ>gi|13878750|sp|Q9CDN0|RS18_LACLA 30S ribosomal protein S18MAQQRRGGFKRRKKVDFIAANKIEVVDYKDTELLKRFISERGKILPRRVTGTSAKNQRKVVNAIKRARVMALLPFVAEDQN
infoseq testone.fastaDisplays some simple information about sequences# USA Name Accession Type Length Descriptionncbi::testone.fasta:HHP_THICU HHP_THICU P80487 P 19 HETEROTROPH-SPECIFIC PROTEINncbi::testone.fasta:RS18_LACLA RS18_LACLA Q9CDN0 P 81 30S ribosomal protein S18
cat myseqone.gcg!!AA_SEQUENCE 1.0
HETEROTROPH-SPECIFIC PROTEIN
HHP_THICU Length: 19 Type: P Check: 4589 ..
1 AADDVTVVIG SAAPMSGPQ
!!AA_SEQUENCE 1.0
30S ribosomal protein S18
RS18_LACLA Length: 81 Type: P Check: 9484 ..
1 MAQQRRGGFK RRKKVDFIAA NKIEVVDYKD TELLKRFISE RGKILPRRVT
51 GTSAKNQRKV VNAIKRARVM ALLPFVAEDQ N
infoseq myseqone.gcg Displays some simple information about sequences# USA Name Accession Type Length Descriptiongcg::myseqone.gcg:HHP_THICU HHP_THICU - P 19gcg::myseqone.gcg:RS18_LACLA RS18_LACLA - P 81

Pairwise Sequence Alignment
An alignment is an arrangement of two sequences which shows where the two sequences are similar, and where they differ.There is no unique, precise, or universally applicable notion of similarity.

Three Categories of Methods
Segment methods compare all overlapping segments of a predetermined length from one sequence to all segments from the other.Optimal global alignment methods allow the best overall score for the comparison of the two sequences to be obtained, including a consideration of gaps.Optimal local alignment methods seek to identify the best local similarities between two sequences but include explicit consideration of gaps.

Global Alignment
A global alignment is one that compares the two sequences over their entire lengths, and is appropriate for comparing sequences that are expected to share similarity over the whole length.The alignment maximizes regions of similarity and minimizes gaps using the scoring matrices and gap parameters provided to the program.

needle
FunctionNeedleman-Wunsch global alignment.
DescriptionThis program uses the Needleman-Wunsch global alignment algorithm to find the optimum alignment (including gaps) of two sequences when considering their entire length.The computation is rigorous.It can be time consuming to run if the sequences are long.

needle example needle -sequencea test.fa -seqall db.fa -gapopen 10.0 -gapextend 0.5 -outfile result.txt
>730305 MATHHTLWMGLALLGVLGDLQAAPEAQVSVQPNFQQDKFLGRWFSAGLASNSSWLREKKAALSMCKSVVAPATDGGLNLTSTFLRKNQCETRTMLLQPAGSLGSYSYRSPHWGSTYSVSVVETDYDQYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDTIVFLPQTDKCMTEQ>404390APEAQVSVQPNFQPDKFLGRWFSAGLASNSSWLQEKKAALSMCKSVVAPAADGGFNLTSTFLRKNQCETRTMLLQPGDSLGSYSYRSPHWGSTYSVSVVETDYDHYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGF TEDSIVFLPQTDKCMTEQ
Needleman-Wunsch Global Alignment ######################################## # Program: needle # Rundate: Wed Jul 31 14:58:56 2002 # Report_file: /tmp/o40075 ######################################## #======================================= ## Aligned_sequences: 2 # 1: 730305 # 2: 404390 # Matrix: EBLOSUM62 # Gap_penalty: 10.0 # Extend_penalty: 0.5 # # Length: 190 # Identity: 160/190 (84.2%)# Similarity: 162/190 (85.3%)# Gaps: 22/190 (11.6%) # Score: 843.0###=======================================

needle example (cont’d)
730305 1 MATHHTLWMGLALLGVLGDLQAAPEAQVSVQPNFQQDKFLGRWFSAGLAS 50 |||||||||||||.||||||||||||||
404390 1 APEAQVSVQPNFQPDKFLGRWFSAGLAS 28730305 51 NSSWLREKKAALSMCKSVVAPATDGGLNLTSTFLRKNQCETRTMLLQPAG 100
|||||:||||||||||||||||.|||.|||||||||||||||||||||..404390 29 NSSWLQEKKAALSMCKSVVAPAADGGFNLTSTFLRKNQCETRTMLLQPGD 78730305 101 SLGSYSYRSPHWGSTYSVSVVETDYDQYALLYSQGSKGPGEDFRMATLYS 150
||||||||||||||||||||||||||.|||||||||||||||||||||||404390 79 SLGSYSYRSPHWGSTYSVSVVETDYDHYALLYSQGSKGPGEDFRMATLYS 128730305 151 RTQTPRAELKEKFTAFCKAQGFTEDTIVFLPQTDKCMTEQ 190
|||||||||||||||||||||||||:||||||||||||||404390 129 RTQTPRAELKEKFTAFCKAQGFTEDSIVFLPQTDKCMTEQ 168

Local Alignment
Local alignment searches for regions of local similarity and need not include the entire length of the sequences.Local alignment methods are very useful for scanning databases or other circumstances when you wish to find matches between small regions of sequences, for example, between protein domains.

water
FunctionSmith-Waterman local alignment.
Descriptionwater uses the Smith-Waterman algorithm (modified for speed enhancements) to calculate the local alignment.

Input Sequences for water
Sequence 1
MATHHTLWMGLALLGVLGDLQAAPEAQVSVQPNFQQDKFLGRWFSAGLASNSSWLREKKAALSMCKSVVAPATDGGLNLTSTFLRKNQCETRTMLLQPAGSLGSYSYRSPHWGSTYSVSVVETDYDQYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDTIVFLPQTDKCMTEQ
Sequence 2
APEAQVSVQPNFQPDKFLGRWFSAGLASNSSWLQEKKAALSMCKSVVAPAADGGFNLTSTFLRKNQCETRTMLLQPGDSLGSYSYRSPHWGSTYSVSVVETDYDHYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDSIVFLPQTDKCMTEQ
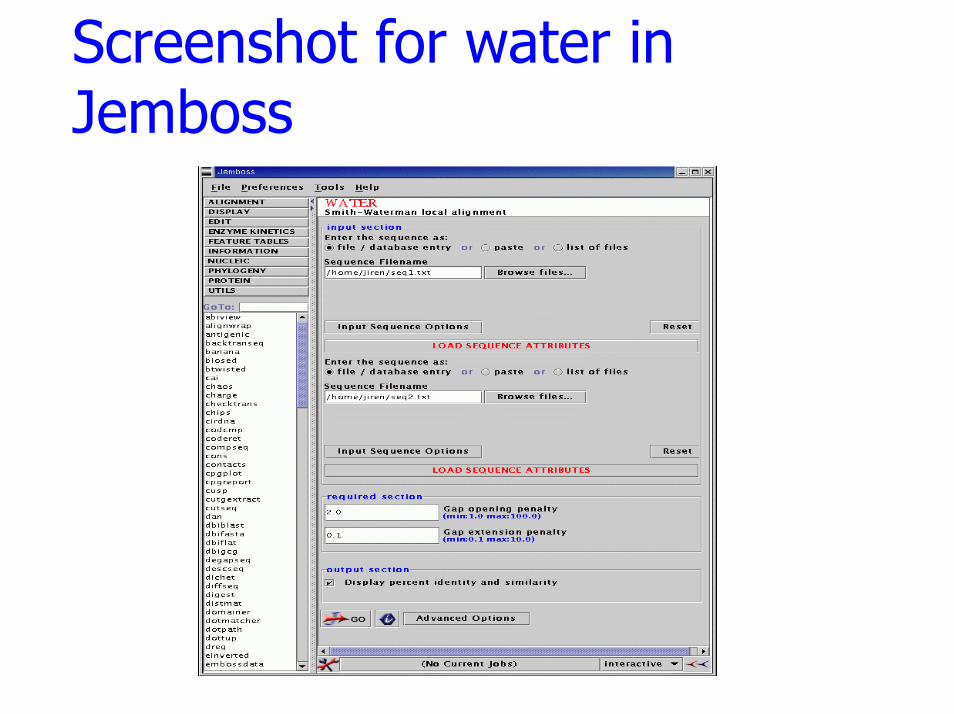
Screenshot for water in Jemboss

Screenshot of the Result in water

getorf
FunctionFinds and extracts open reading frames (ORFs).
DescriptionThe program finds and outputs the sequences of open reading frames (ORFs).The START and STOP codons are defined in the Genetic Code tables.The ORFs can be defined as regions of a specified minimum size between STOP codons or between START and STOP codons.

getorf Examplegetorf -minsize 500 bigdna.seq
>HSA1280_1 [210 - 707]TGSHVFSWEGCYLSRGCRLEIFKEVEIGNWLCVSVVFGEGDYKEGLTPSPYSLRLDHRDEVVRYKDKGGMGNTMMWYFSFLFFLFDNIYPWLRGGRSSAAEIKRAVAGANRLPALSLTCGGRTCLWLKPSPGSDSHVCTGGAETWTGVHQPHTGCSGCSLLFLLLF >HSA1280_2 [1211 - 1726]GCIVVVSSSSPSSSSPSSSPFCSCSPSSSSSFFSLFFLFTLLSSLLFPFPLLSLSSGHSGVNQLGGVFVNGRPLPDSTRQKIVELAHSGARPCDISRILQVILPAPPHSPPPRPSTLNALSSFLTVNDANYGPPHPHPPQSPVPPPTPLPSHFPLSFHHPSLSLSTWVSYIR >HSA1280_3 [7968 - 7468]TPGAGALANFRGQNFEATMSADNKRILLRRAAGHIVLQEAQSTDFSVKCVNPSALLSLITVRRAGASVLFRALGWGDGGWMGGPGPSMGGGRESGRGPGLGSYIEEMEGRQDGNQRWGWKQGGRIGGPGLGERQRAMEGKRGRWRGKEGSKNIEARKVFLKKDRKEC >HSA1280_4 [5579 - 4998]QSPPKNLTCGWGGGDCQLTQNFLATRRVKVKSPWTSSTSGPNHHRRTLADLTLTSRPQTAGDENPPESGATSGGSCCPRLLPNSKKRKGGVSCAPSSWEPTHRGEPGVEALGSALPGLGHRALSPSPRLLGLSGPDSCQGRDHQDLRLLGAEGWTLVSAKFGCHLGVSSVYKWESYGFKS QAGMGLPTLGGVNR >HSA1280_5 [4652 - 4053]AITERQDCSHNKFLRVTEASFVPDISHTVTDFPFPRPTPWRRRASGAGPRAHGCAGSLLAVPRAAPGPEPLGSVVLLGVRRHVPLSRILSPCHEDNKYRLIFGVNCFLFLTKLPRGVEGSGGEPTDPPLRPTPPIPRSSSSSPSHRTWGVSGPISPPRPERLPASRRPAHSIFTSPLPHS
RHPQRVRKWDPEGRTGEGQG

transeq
FunctionTranslate nucleic acid sequences
DescriptionThis translate nucleic acid sequences to the corresponding protein sequence.It can translate in any of the 3 forward or 3 reverse frames, or in all three forward or reverse frames, or in all six frames.It can translate specified regions corresponding to the coding regions of your sequences.

transeq Example
> testgatccggagcgacttccgcctatttccagaaattaagctcaaacttgacgtgcagctagttttattttaaagacaaatgtcagagaggctcatcatattttcccccctcttctatatttggagcttatttattgctaagaagctcaggctcctggcgtcaatttatcagtaggctccaaggagaagagaggagaggagaggagagctgaacagggagccacgtcttttcctgggagggct
transeq testdna.txt -frame=6 stdoutTranslate nucleic acid sequences>test_1DPERLPPISRN*AQT*RAASFILKTNVREAHHIFPPLLYLELIYC*EAQAPGVNLSVGSKEKRGEERRAEQGATSFPGRA>test_2IRSDFRLFPEIKLKLDVQLVLF*RQMSERLIIFSPLFYIWSLFIAKKLRLLASIYQ*APRRREERRGELNREPRLFLGG>test_3SGATSAYFQKLSSNLTCS*FYFKDKCQRGSSYFPPSSIFGAYLLLRSSGSWRQFISRLQGEERRGEES*TGSHVFSWEG>test_4SPPRKRRGSLFSSPLLSSLLLGAY**IDARSLSFLAINKLQI*KRGENMMSLSDICL*NKTSCTSSLSLISGNRRKSLRI>test_5PSQEKTWLPVQLSSPLLSSPWSLLIN*RQEPELLSNK*APNIEEGGKYDEPL*HLSLK*N*LHVKFELNFWK*AEVAPDX>test_6ALPGKDVAPCSALLSSPLFSLEPTDKLTPGA*AS*Q*ISSKYRRGGKI**ASLTFVFKIKLAARQV*A*FLEIGGSRSGS

Multiple Sequence Analysis
Multiple sequence alignments are used To find patterns to characterize protein families.To detect or demonstrate homology between new sequence and existing families of sequences.To help predict the secondary and tertiary structures of the new sequences.As an essential prelude to molecular evolutionary analysis.

emma
FunctionMultiple alignment program – interface to ClustalW program.
Descriptionemma calculates the multiple alignment of nucleic acid or protein sequences according to the method of Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994).

The Main Reference for ClustalW
Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice." Nucleic Acids Research, 22:4673-4680.

emma Example mulseq.txt>gi|730305|MATHHTLWMGLALLGVLGDLQAAPEAQVSVQPNFQQDKFLGRWFSAGLASNSSWLREKKAALSMCKSVVAPATDGGLNLTSTFLRKNQCETRTMLLQPAGSLGSYSYRSPHWGSTYSVSVVETDYDQYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDTIVFLPQTDKCMTEQ>gi|404390|APEAQVSVQPNFQPDKFLGRWFSAGLASNSSWLQEKKAALSMCKSVVAPAADGGFNLTSTFLRKNQCETRTMLLQPGDSLGSYSYRSPHWGSTYSVSVVETDYDHYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDSIVFLPQTDKCMTEQ>gi|895868MAALRMLWMGLVLLGLLGFPQTPAQGHDTVQPNFQQDKFLGRWYSAGLASNSSWFREKKAVLYMCKTVVAPSTEGGLNLTSTFLRKNQCETKIMVLQPAGAPGHYTYSSPHSGSIHSVSVVEANYDEYALLFSRGTKGPGQDFRMATLYSRTQTLKDELKEKFTTFSKAQGLTEEDIVFLPQPDKCIQE

emma Example (cont’d)cat outseq.txt>730305MATHHTLWMGLALLGVLGDLQAAPEAQVSVQPNFQQDKFLGRWFSAGLASNSSWLREKKAALSMCKSVVAPATDGGLNLTSTFLRKNQCETRTMLLQPAGSLGSYSYRSPHWGSTYSVSVVETDYDQYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDTIVFLPQTDKCMTEQ>404390----------------------APEAQVSVQPNFQPDKFLGRWFSAGLASNSSWLQEKKAALSMCKSVVAPAADGGFNLTSTFLRKNQCETRTMLLQPGDSLGSYSYRSPHWGSTYSVSVVETDYDHYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDSIVFLPQTDKCMTEQ>895868MAALRMLWMGLVLLGLLGFPQTPAQGHDTVQPNFQQDKFLGRWYSAGLASNSSWFREKKAVLYMCKTVVAPSTEGGLNLTSTFLRKNQCETKIMVLQPAGAPGHYTYSSPHSGSIHSVSVVEANYDEYALLFSRGTKGPGQDFRMATLYSRTQTLKDELKEKFTTFSKAQGLTEEDIVFLPQPDKCIQE-

cusp
FunctionCreate a codon usage table
DescriptionReads one or more coding sequences (CDS sequence only) and calculates a codonfrequency table.

cusp Examplecusp bigdna.seq# CUSP codon usage file# Codon Amino acid Fract /1000 NumberGCA A 0.342 16.042 119GCC A 0.230 10.785 80GCG A 0.098 4.583 34GCT A 0.330 15.503 115TGC C 0.365 14.020 104TGT C 0.635 24.400 181…TAC Y 0.342 9.302 69TAT Y 0.658 17.929 133TAA * 0.334 15.368 114TAG * 0.264 12.133 90TGA * 0.402 18.469 137
The 'Fract' column gives the faction of all amino acids coded for by this codon triplet. The /1000 column represents the number of codons, given the input sequence(s), there are per 1000 bases. This will be an extrapolation if the sequence is shorter than 1000 bases. If multiple sequences are input then the statistics are given for all of the sequences together, not individually.

preg
FunctionRegular expression search of a protein.
DescriptionA regular expression is a way of specifying an ambiguous pattern to search for. Regular expressions are commonly used in some computer programming languages.

preg Example> test1MATHHTLWMGLALLGVLGDLQAAPEAQVSVQPNFQQDKFLGRWFSAGLASNSSWLREKKAALSMCKSVVAPATDGGLNLTSTFLRKNQCETRTMLLQPAGSLGSYSYRSPHWGSTYSVSVVETDYDQYALLYSQGSKGPGEDFRMATLYSRTQTPRAELKEKFTAFCKAQGFTEDTIVFLPQTDKCMTEQ
preg -sequence test1.txt -pattern S[A-Z] stdoutRegular expression search of a protein sequencepreg search of test1.txt with pattern S[A-Z]Matches in test1
test1 29 SVtest1 45 SAtest1 50 SNtest1 52 SStest1 63 SMtest1 67 SVtest1 81 STtest1 101 SLtest1 104 SYtest1 106 SYtest1 109 SPtest1 114 STtest1 117 SVtest1 119 SVtest1 133 SQtest1 136 SKtest1 150 SR

dreg
FunctionRegular expression search of a nucleotide sequence
DescriptionThis searches for matches of a regular expression to a nucleic acid sequence.

splitter
FunctionSplit a sequence into (overlapping) smaller sequences.
DescriptionThis simple editing program allows you to split a long sequence into smaller, optionally overlapping, subsequences.

splitter Example>HSA1280 gatccggagcgacttccgcctatttccagaaattaagctcaaacttgacgtgcagctagttttattttaaagacaaatgtcagagaggctcatcatattttcccccctcttctatatttg gagcttatttattgctaagaagctcaggctcctggcgtcaatttatcagtaggctccaag gagaagagaggagaggagaggagagctgaacagggagccacgtcttttcctgggagggct Gctatctaagtcggggctgcaggttggagatttttaaggaagtggaaattggcaattggc
splitter mbigdna.seq many_small.seq -size=50 -over=10 >HSA1280_1-60 gatccggagcgacttccgcctatttccagaaattaagctcaaacttgacgtgcagctagt >HSA1280_51-110 tgcagctagttttattttaaagacaaatgtcagagaggctcatcatattttcccccctct >HSA1280_101-160 tcccccctcttctatatttggagcttatttattgctaagaagctcaggctcctggcgtca >HSA1280_151-210 cctggcgtcaatttatcagtaggctccaaggagaagagaggagaggagaggagagctgaa >HSA1280_201-260 gagagctgaacagggagccacgtcttttcctgggagggctgctatctaagtcggggctgc >HSA1280_251-300 tcggggctgcaggttggagatttttaaggaagtggaaattggcaattggc

showfeat
FunctionShow features of a sequence.
Descriptionshowfeat reads a protein or nucleic sequence and its feature table, and writes a text representation of the features to standard output.

showfeat Example LOCUS GMGL01 3400 bp DNA linear PLN 01-OCT-1996DEFINITION Glycine max leghemoglobin gene or pseudogene (no mRNA detected).ACCESSION V00451 L00005 L00006VERSION V00451.1 GI:18592KEYWORDS leghemoglobin; pseudogene.SOURCE soybean.ORGANISM Glycine max
Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta;Spermatophyta; Magnoliophyta; eudicotyledons; core eudicots;Rosidae; eurosids I; Fabales; Fabaceae; Papilionoideae; Phaseoleae;Glycine.
REFERENCE 1 (bases 1 to 3400)AUTHORS Wiborg,O., Hyldig-Nielsen,J.J., Jensen,E.O., Paludan,K. and
Marcker,K.A.TITLE the structure of an unusual leghemoglobin gene from soybeanJOURNAL EMBO J. 2, 449-452 (1983)
COMMENT On Jan 11, 2002 this sequence version replaced gi:170001 gi:170002.

showfeat Example (cont’d)
FEATURES Location/Qualifierssource 1..3400
/organism="Glycine max"/db_xref="taxon:3847"
CDS join(363..460,555..663,2182..2286,3065..3208)/codon_start=1/product="leghemoglobin"/protein_id="CAA23729.1"/db_xref="GI:313502"/db_xref="SPTREMBL:Q42801"/translation="MGAFTEKQEALVNSSFEAFKANLPHHSVVFFNSILEKAPAAKNMFSFLGDAVDPKNPKLAGHAEKLFGLVRDSAVQLQTKGLVVADATLGPIHTQKGVTDLQFAVVKEALLKTIKEAVGDKWSEELSNPWEVAYDEIAAAIKKAMAIGSLV"
exon <363..460/number=1
intron 461..554/number=1
exon 555..663/number=2
intron 664..2181/number=2
exon 2182..2286/number=3
intron 2287..3064/number=3
exon 3065..>3208/number=4
BASE COUNT 931 a 310 c 334 g 975 t 850 others

showfeat Example (cont’d)ORIGIN
1 ttttactcaa atcaatgata tatattttgg taactttttt tcttttactt ataattttgt61 ttacgttaaa agtcaaaaaa gaatacatta aaaaattaaa aattcaccga acaacttaaa
121 ttatttattt actttgacta agtgaaaaat tacttgatta agtttttgaa aaggtcgttg181 tgtcttcata atgccgattg atacgctcca cattcaataa gccaagagag acatattcaa241 taacaatcgc aacaaatttt ttttcagtct ccaaaccatc tatataaaca agtattggat301 gtgaacttat aactggattg aaaatagaaa ttaaataaca gaaaattaca aaagatcgaa361 atatgggtgc ttttacagag aagcaagagg ctttggtgaa tagctcgttt gaagcattca421 aggcaaacct tcctcaccac agcgttgtat tcttcaattc gtaatttttc tctctcaccc481 tatgtttccc ttgagttgaa aagaggtagt gtacataata gtgtctttgg tttgattaaa541 aaacaaaata ataggatatt ggagaaagca ccagcagcaa agaacatgtt ctcattttta601 ggtgatgcag tagatccgaa aaatcctaag ctcgcgggcc atgctgaaaa gctttttgga661 ttggtaagtg ttagtcaact aaaattatag ttattttatg tgattttagg gatgtatact
……3181 ggcaatggct ataggatcat tagtataaag tctagtagta ataaataaat tttgtttcac3241 taaaatttgt tattaacttc ttgatataaa tgtcggttac attaggtaaa atacagtact3301 tgtctttgaa taaacaatat taaattattt gcctcagggt ttatgtttat gaatcacaat3361 cgatacttta tacatgtttt aaaattattt taataagctt
//

showfeat Example (cont’d)showfeat showfeat.txt stdoutShow features of a sequence.GMGL01Glycine max leghemoglobin gene or pseudogene (no mRNA detected).|==========================================================| 3700|-----------------------------------------------------> source
|-> CDS|-> exon
|> intron|> CDS|> exon|------------------------> intron
|-> CDS|-> exon
|-----------> intron|--> CDS|--> exon


















