![Adrenal Imaging - University of Floridaxray.ufl.edu/files/2010/02/Adrenal-Imaging.pdfadrenal glands [3], and a metastasis might ... CT, adrenal imaging, adrenal lymphoma imaging, adrenal](https://static.fdocuments.net/doc/165x107/5b26814c7f8b9a8c0f8b4820/adrenal-imaging-university-of-glands-3-and-a-metastasis-might-ct-adrenal.jpg)
INSUFICIÊNCIA ADRENAL
27
INSUFICIÊNCIA ADRENAL
-
Upload
melyssa-patterson -
Category
Documents
-
view
89 -
download
0
description
INSUFICIÊNCIA ADRENAL. FISIOLOGIA. Os hormônios produzidos pela adrenal são chamados de esteróides . Todos derivam de um precursor comum, o colesterol. Todos são constituídos por diversos anéis aromáticos e contêm 19 ou 21 átomos de carbono. - PowerPoint PPT Presentation
Transcript of INSUFICIÊNCIA ADRENAL

INSUFICIÊNCIAADRENAL


FISIOLOGIA• Os hormônios produzidos pela adrenal são chamados de esteróides.
Todos derivam de um precursor comum, o colesterol.• Todos são constituídos por diversos anéis aromáticos e contêm 19 ou
21 átomos de carbono.• Os C19-esteróides são representados pelos androgênios e como
possuem um grupamento cetona no carbono 17, são também chamados de 17-cetosteróides.
• Os C21-esteróides podem atuar como glico ou mineralocorticóides. Ao receberem um radical hidroxila no carbono 17, ganham o nome de 17-hidroxicorticosteroides, sendo classificados como glicocorticoides. Na ausência da 17-hidroxilação, são classificados como mineralocorticóides.
• Os estrógenos contém 18 carbonos e não produzidos pelas adrenais, tendo origem nos ovários ou na conversão periférica dos androgênios em estrogênios.
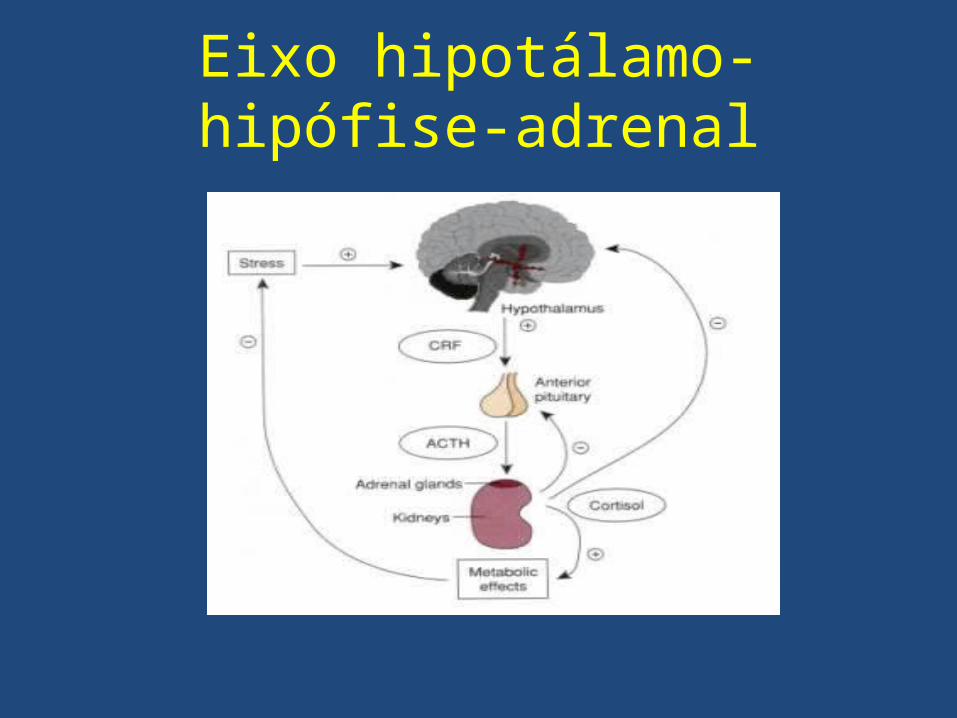
Eixo hipotálamo-hipófise-adrenal

Eixo hipotálamo-hipófise-adrenal• O cortisol é regulado pelo ACTH;• Pró-opiomelanocortina (POMC): precursor do
ACTH. A POMC também origina em menor escala, endorfinas e MSH (hormônio melanotrófico).
• ACTH estimula as zonas fasciculada e reticular, promovendo a secreção de cortisol e androgênio.
• O ACTH possui um efeito trófico sobre as adrenais.

Rítmo circadiano do cortisol

Ações do cortisol
• Hiperglicemiantes• Cutâneo-musculares• Ósseas• Antiinflamatórias• Ações sobre o sistema nervoso central• Controle da pressão arterial

Insuficiência adrenal• Deficiência dos hormônios do córtex adrenale suas consequentes manifestações clínicas.• IA 1ª: destruição de 90% ou mais do córtex
adrenal, mas pode advir de condições que reduzam a síntese de esteróides adrenais, levando a produção subnormal de cortisol, aldosterona e androgênios.
• IA 2ª: decorre da deficiência do ACTH que pode ser resultado ou de um comprometimento hipofisário ou da deficiente liberação de CRH pelo hipotálamo.

Causas de IA primária

Etiologia: doença de Addison
• Causas mais comuns:- Adrenalite auto-imune (isolada ou associada aSindrome Poliglandular Autoimune);- Infecção por tuberculose, histoplasmose e paracoccidioidomicose;- Adrenoleucodistrofia;- HIV (SIDA, drogas e infecções oportunistas);- Hemorragia adrenal bilateral.

Síndrome poliglandular auto-imune tipo 1
• Rara. Transmissão autossômica recessiva (gene AIRE).• Manifesta-se em geral, na infância.• Forma esporádica ou familiar.• Leve predomínio em mulheres (0,8 a 2,4).• Tríade: hipoparatireoidismo + doença de Addison + candidíase
muco-cutânea crônica (CMC).• Pode ocorrer também: ceratoconjuntivite, distrofia ungueal, e
formação defeituosa do esmalte dentário.• CMC: manifestação inicial e envolve a mucosa oral e unhas e
menos comumente, a pele e o esôfago.• Doença de Addison é vista em 60-100% dos casos e
geralmente ocorre após a CMC e o hipopara. A idade do surgimento é variável.

Síndrome poliglandular auto-imune tipo 2 (Síndrome de Schmidt)
• Autossômica dominante;• Predomina no sexo feminino e em adultos;• Tríade: DA (100%) + DAT (75-85%) + DM1 (28-50%);• Se apresentam em uma seqüência específica: DM1 se
desenvolve antes da DA. DAT pode se desenvolver antes, concomitantemente ou após a DA.
• Incompleta e potencial: quando o paciente apresenta uma doença auto-imune clínica característica da síndrome com um ou mais marcadores imunológicos de outra doença fundamental, mas com função normal dos órgãos-alvo.
• Incompleta e subclínica: na presença de alterações subclínicas da função dos órgãos-alvo.

SPA tipo 3 e 4
• SPA tipo 3:Dç. tireoidiana auto-imune (TH, mixedema idiopático, tireoidite silenciosa e DG) + uma ou mais doenças auto-imunes, descartando-se a DA e o hipoparatireoidismo.• SPA tipo 4:Duas ou mais doenças auto-imunes órgão-específicas (exceto hipopara, candidíase muco-cutânea crônica, DAT ou DM1).

Tuberculose• O acometimento ocorre por disseminação
hematogênica e uma doença extra adrenal geralmente é evidente.
• Destruição do córtex adrenal e substituição por extensos granulomas epitelioides e caseificação, o que ocasiona o aumento da glândula. Posteriormente, surge a fibrose e a adrenal se mostra de tamanho normal ou reduzido, com calcificação evidente em 50% dos casos.
• Na doença infecciosa há também comprometimento da medula adrenal.

Aids
• Adrenais são as glândulas endócrinas mais frequentemente afetadas.
• O acometimento pode ser secundário a infecções oportunistas (tuberculose, citomegalovírus, micobactérias atípicas, micoses, etc), fármacos (rifampicina, cetoconazol, etc) ou a lesões metastáticas (sarcoma de Kaposi, linfomas).

Fármacos
• Inibem a esteroidogênese adrenal: cetoconazol, fluconazol, itraconazol, etomidato, mitotano, metirapona, trilostano, aminoglutetimida e etc).
• Aumentam a depuração metabólica dos esteróides adrenais: rifampicina, fenitoína, fenobarbital, troglitazona e etc).
• Causam hemorragia adrenal: heparina e dicumarol.

Adrenoleucodistrofia
• Doença recessiva ligada ao X;• Produção de uma proteína transportadora
anormal dentro dos peroxissomos que impede a oxidação dos ácidos graxos de cadeia muito longa, provocando o seu acúmulo no cérebro, córtex adrenal, testículos, fígado e plasma.
• Surge desmielinização do SNC e IA primária.

Hiperplasia adrenal congênita
• É a causa mais usual de IA no período neonatal.
• Deficiência de enzimas envolvidas na biossíntese do cortisol.
• A forma mais comum é a deficiência de 21-hidroxilase.
• Os níveis baixos de cortisol promovem um aumento na síntese e liberação do ACTH e este estimula a adrenal levando a hiperplasia e acúmulo de metabólitos intermediários.

Etiologia: IA secundária
• Congênita- Deficiência congênita de ACTH- Pan-hipopituitarismo congênito
• Adquirida- Uso crônico de glicocorticóide exógeno (por ex. prednisona >7,5mg/dia por >15 dias).- Após TCE, necrose hipofisária pós-parto, apoplexia hipofisária, cirurgia, radioterapia ou qualquer outra patologia que envolva a hipófise e o hipotálamo.

Insuficiência adrenal: quadro clínicoSinais / sintomas dependentes da deficiência de:Glicocorticoide:- astenia, fraqueza, indisposição- anorexia, emagrecimento- hipotensão- hipoglicemiaMineralocorticóide:- hiponatremia- hipovolemia / desidratação- hipercalemia, acidose metabólicaAndrogênios adrenais- perda dos pelos axilares e pubianos e redução da libido ( ).♀

Apresentação clínica:IA primária X IA secundária
• IA primária: ACTH alto = hiperpigmentaçãoIA secundária: deficiência de ACTH / CRH =sem hiperpigmentação!
• IA secundária: camada glomerulosapreservada (dependente de aldosterona) =sem hipovolemia, sem hipercalemia!

Hiperpigmentação

Hiperpigmentação

Insuficiência adrenal: diagnóstico
• Dosagem do cortisol as 8h:- Valores ≤ 3µg/dl confirmam- Valores ≥ 18µg/dl excluem- Entre 3 e 18mg/dl = teste confirmatório• Testes confirmatórios:- Estimulo com cortrosina (ACTH sintetico)- Hipoglicemia insulinica

Insuficiência adrenal: testes confirmatórios
• Estimulo com cortrosina (ACTH sintetico)( 250mcg de cortrosina EV; coleta cortisoltempos 0, 30 e 60 minutos)Valores de pico < 18- 20 mcg/dl: confirmam o diagnóstico de insuficiência adrenal. Não discrimina se o problema está nas adrenais ou se é hipotálamo-hipofisário.→Fazer a dosagem do ACTH plasmático (alto na IA 1ª e baixo ou no LIN na IA 2ª).Não descarta os casos de deficiência leve de ACTH!!!

Insuficiência adrenal: testes confirmatórios
• Teste de tolerância a insulina:- Indicado para os pacientes com suspeita de IA
2ªfranca ou parcial.- 0,05 UI/kg de insulina. Dosagem do cortisol
nos tempos 0, 30 e 60 minutos.- Resposta normal: incremento > 8µg/dl e um
pico >18 a 20 µg/dl.- Está contra-indicado em idosos e pacientes
com doença cardiovascular e cerebrovascular.

Insuficiência adrenal crônica: tratamento
• Reposição glicocorticóide:Prednisona 2,5-5mg/dia OUHidrocortisona 15-20mg manhã e 5-10mg a tarde.
• Reposição mineralocorticóide:Fludrocortisona 0,1 (0,05 a 0,2) mg/dLivre ingestao salinaMonitorar: hipotensão postural, hiperpotassemia.Na IA 2ª raramente é necessária a reposição de mineralocorticóide.