
Industry experience with Change Management/ … · Glossary: RRF: Risk ranking and filtering;...
39
Industry experience with Change Management/ Comparability Protocols: a biologics perspective Dr. Markus Goese EMA Liaison Pharma Technical Regulatory Policy F. Hoffmann-La Roche Ltd. CH-4070 Basel, Switzerland 26th Annual EuroMeeting 25-27 March 2014 ACV, Vienna Austria
Transcript of Industry experience with Change Management/ … · Glossary: RRF: Risk ranking and filtering;...

Industry experience withChange Management/Comparability Protocols:a biologics perspective
Dr. Markus GoeseEMA LiaisonPharma Technical Regulatory PolicyF. Hoffmann-La Roche Ltd.CH-4070 Basel, Switzerland
26th AnnualEuroMeeting
25-27 March 2014ACV, Vienna
Austria

DisclaimerThe views and opinions expressed in the following PowerPoint slides are those of the individual presenter and should not be attributed to Drug Information Association, Inc. (“DIA”), its directors, officers, employees, volunteers, members, chapters, councils, Special Interest Area Communities or affiliates, or any organization with which the presenter is employed or affiliated.
These PowerPoint slides are the intellectual property of the individual presenter and are protected under the copyright laws of the United States of America and other countries. Used by permission. All rights reserved. Drug Information Association, DIA and DIA logo are registered trademarks or trademarks of Drug Information Association Inc. All other trademarks are the property of their respective owners.
2

Presentation Outline
• Typical CMC biologics changes & current challenges
• Regulatory tools to facilitate post-approval changes:– EU: The Change Management Protocol (CMP)– US: The (expanded) Comparability Protocol (eCP)– US/ EU: The Post-Approval Lifecycle Management
(PALM)-Plan
• Summary/ Outlook
3

Post-approval CMC Changes/ VariationsPost-approval CMC changes/ variations for biologics are typically occurring in the following areas:
Maintenance of Global Supply– Manufacturing site transfer/ addition (including scale-up) – Process improvements to increase product titer to meet forecasted
demand: Process Improvements
– New manufacturing technology• e.g., Media treatment technology to prevent Drug Substance
Contamination, isolator filling technology to prevent drug product contamination
– Improved controls• e.g., Improvements in microbiological controls, new methods to
detect raw material adulteration– Improved testing methodology
• e.g., Mass Spectrometry
4

Our approach towards a proactive Lifecycle Management Based on Modern Risk-based Concepts (ICH Q 5E, 8, 9, 10, 11, Draft WHO Guidelines)
• Initial BLA/ NDA contains:
– Overall control strategy proposal including a design space proposal
– A Post Approval Lifecyle Management (PALM) Plan with commitments to monitor product quality, process performance and verify changes within the design space
• Changes within the design space do not require pre-approval
• In some regions (US, EU), the PALM is supplemented with Comparability/ Change Management Protocols to facilitate stream-lined regulatory management of planned changes outside the design space:
– Site & scale changes
– Raw material changes
– Working Cell Bank Replacements
– Planned process improvements
5

Example (EU): Biologics DS manufacturing site transfer -Benefit of CMP Approach vs. „Traditional“ Approach*
3-5 months faster approval of the site change using a CMP
*Note: approval timelines for type II variation in this scheme include positive CHMP opinion and Commission Decision
6

The US-”counterpart” of the EU-CMP:the Change (Comparability) Protocol
– FDA published post-approval change regulations which contained section on comparability protocols (CPs) already in 2004 (see 21 CFR 314.70 / 601.12(e))
– Prospectively defines the change and acceptance criteria
– First step submitted as Prior Approval Supplement (PAS, 4 month approval timeline)
– Prospective definition of change requirements allow for reduction in submission category second step upon change implementation (e.g., PAS CBE-30)
7

Example (US): Manufacturing Site Transfer of Product A from Site X to Site Y Leveraging ‘traditional’ CP
Product A
Execute transfer per defined
requirements in CP
Drug Substance Receiving Site Y
D
CBE-30 Supplement with data
demonstrating acceptance
criteria met
Drug Substance Donor Site X
A B C
Submit CP describing site
transfer acceptance criteria for
product-site combination
Defined business need
A
Time savings for US-CP-approach comparable to EU-CMP, eg. up to 5 months for site-transfer (product release to market)
8

Short “excursion”: What is Quality by Design?Definition
QbD is a systematic approach to development that
• Leverages knowledge of structure-function relationship to define product attributes that are important
• Uses science-based and risk-based approaches to define the commercial manufacturing process
• Aims at developing deeper product & process understanding throughout the lifecycle of a product
Control system tailored to product requirements
Process robustness enhanced
Deviation and change assessments facilitated
9

“CP + QbD Concepts = Expanded Change Protocol (eCP)”• The term eCP was first coined at the US July CASSS CMC Forum (2009) on QbD as
concept for how to manage lifecycle changes based on risk
• Leverages existing regulations for change protocols: move from one protocol for one discreet change to an expanded protocol that can be applied to groups of similar changes
• A range of QbD concepts (ICH Q8, Q9, Q10, Q11) can be applied:
– Leverages historical knowledge, Systematic risk assessment and change requirements
Genentech’s drug substance manufacturing site transfer eCP proposal was accepted in FDA’s QbD pilot program in 2009
Primary objective of the site transfer eCP: support the mobility of approved products to licensed drug substance facilities within Roche/Genentech’smanufacturing network (“trans-BLA approach”: multiple products/ multiple sites)
Effectively leverages Quality Risk Management (ICH Q9) and builds upon Genentech’s experience with site transfers across products
Reduces the number of regulatory submissions of similar content and drives consistency
10

Concept of eCP to Support Multi-Product, Multi-Site TransfersA network of Drug Substance
Donor Sites
B C
D
A
Execute transfer per defined
requirements in eCP
B
C
D
Site Y or Z
Site X or Z
Site X or Z
Site X or Y
Site X
Site Z
SiteY
A network of Drug Substance Receiving Sites
Submit eCP describing acceptance criteria
broadly for both Site and Product
CBE-30 Supplement with data
demonstrating acceptance criteria
met
Potential Future Network
Requirements
A
11

Design of Site Transfer eCP Risk Assessment Tools
Important questions to be asked:
→ What should be assessed in considering GMP or inspectional risk?
→ What should be tested to confirm comparability?
→ What are the impacts of ‘facility fit’ changes?
→ What products and facilities should be in scope?
Site Transfer Risk Assessment/ Quality Risk Management
Scope and Limitations
Comparability & Validation
Risk-Based Approach to process/facility
Facility modifications
Product and Process Evaluation
GMP/ComplianceSite
Inspection
eCPCBE-30
12
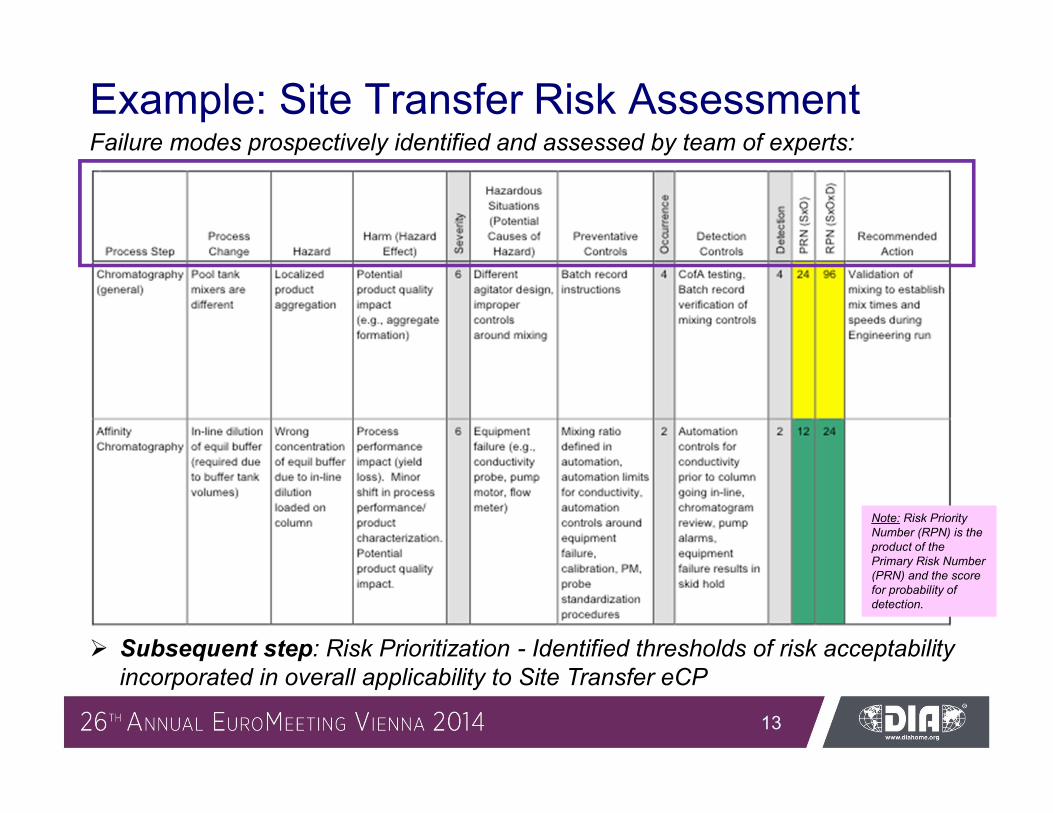
Example: Site Transfer Risk AssessmentFailure modes prospectively identified and assessed by team of experts:
Subsequent step: Risk Prioritization - Identified thresholds of risk acceptability incorporated in overall applicability to Site Transfer eCP
Note: Risk Priority Number (RPN) is the product of the Primary Risk Number (PRN) and the score for probability of detection.
13

Multi-Site, Multi-Product eCP Approved in FDA QbD Pilot Program for Biotech ProductsGoal of eCP filing:
Justify that historical experience with Genentech & Roche’s network of production facilities and product characterization was sufficient to support product transfer with a CBE-30 approval (subsequent to approval of eCP PAS)
Accomplished: Agreed upon scope: products and facilities (scope excludes opportunistic process
changes including changes to increase yield; included within scope: facility driven process modifications, eg. scale changes, disposables, raw material sourcing)
Justified that historical knowledge of product characterization, stability & process performance is sufficient to support comparability with post-approval real-time stability commitment (successful demonstration of comparability using stress stability studies)
Only licensed facilities in “a current state of GMP compliance” are allowed, so that Pre-Approval Inspection (PAI) may be waived
Agreed future products and facilities could cross-reference the eCP if pre-defined criteria were approved
Overall: ability to quantify risk related to facility and process change:eCP PAS approved in 2011 ✔ , Subsequent drug substance transfers approved under eCP criteria with CBE-30 ✔
14

“e”-CP concepts (eg. site transfer): applicable to EU?eCP Strategic Component Change Initiation
(EU Change Management Protocol – Type II
Variation)
Change Implementation
(EU Type IB Variation)
Scope and Limitations Defined scope and limitations Demonstrate requirements of scope are met, including process
changes associated with transfer
Quality Risk Management (QRM) Description of QRM program and approach to site
transfer risk assessment
Documented risk control strategy and executed risk management
report
Comparability and Stability Product-specific comparability plan and real-time
stability commitments and acceptance criteria.
Data demonstrating that acceptance criteria are met
Process Validation/Evaluation Overview of validation program Summary of facility/equipment differences and applicable validation;
Validation summary data support the process, facility/equipment,
and method transfer activities
Questions:
How can EU inspectional/ GMP aspects be addressed appropriately?
Is «worksharing procedure» a way forward in the EU to involve all (co-)rapporteurs?
15

Our approach towards a proactive Lifecycle Management Based on Modern Risk-based Concepts (ICH Q 5E, 8, 9, 10, 11, Draft WHO Guidelines)
• Initial BLA/ NDA contains:
– Overall control strategy proposal including a design space proposal
– A Post Approval Lifecyle Management (PALM) Plan with commitments to monitor product quality, process performance and verify changes within the design space
• Changes within the design space do not require pre-approval
• In some regions (US, EU), the PALM is supplemented with Comparability/ Change Management Protocols to facilitate stream-lined regulatory management of planned changes outside the design space:
– Site & scale changes (see previous slides)
– Raw material changes
– Working Cell Bank Replacements
– Planned process improvements
• New products may be added to exisiting approved expanded change protocols
16
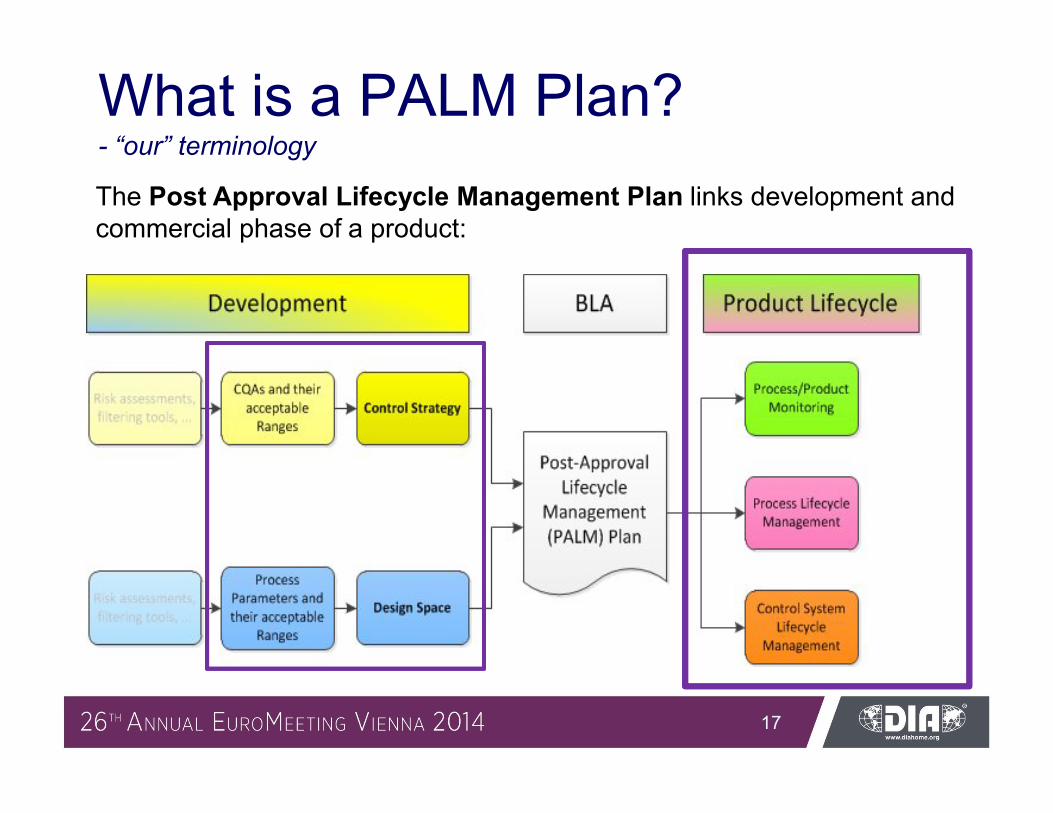
What is a PALM Plan?- “our” terminology
The Post Approval Lifecycle Management Plan links development and commercial phase of a product:
17

What is a Critical Quality Attribute (CQA)? “A physical, chemical, biological or microbiological property or characteristic
that should be within an appropriate limit, range, or distribution to ensure the desired product quality” (ICH Q8).
Example: Monoclonal Antibody (MAb):
Sequence Variants
Glycosylation Variants
Glycation
DeamidationAspartic Acid Isomerization
Cysteine Forms
N-terminal Variants
C-terminal Lysine
Proline Amidation
Oxidation Variants
Host Cell Protein
Host Cell DNA
LeachedProtein A
Adventitious Agents
Composition andStrength
Raw Materials
18
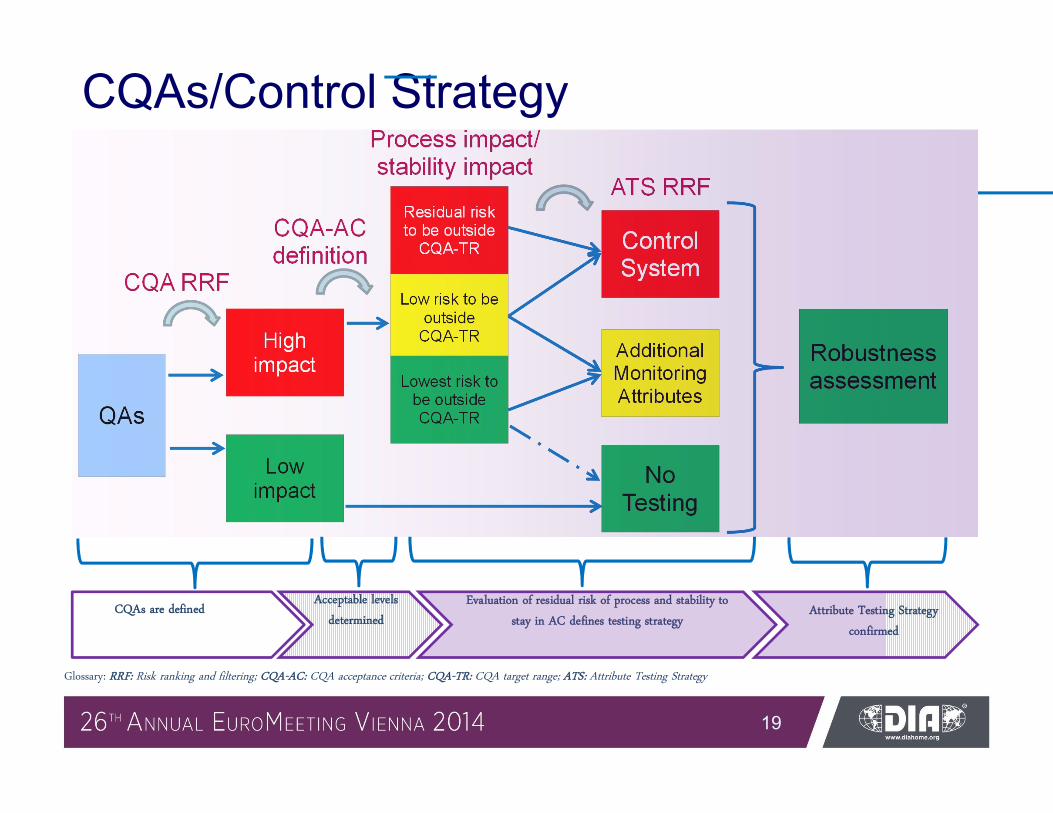
CQAs/Control Strategy
QAs
High impact
Low impact
Low risk to be outside
CQA-TR
Process impact/stability impact
CQA RRF
Lowest risk to be outside CQA-TR
ATS RRF
ControlSystem
Additional Monitoring Attributes
No Testing
Robustness assessment
Residual risk to be outside
CQA-TR CQA-AC definition
CQAs are definedAcceptable levels
determined
Evaluation of residual risk of process and stability to
stay in AC defines testing strategyAttribute Testing Strategy
confirmed
Glossary: RRF: Risk ranking and filtering; CQA-AC: CQA acceptance criteria; CQA-TR: CQA target range; ATS: Attribute Testing Strategy
19
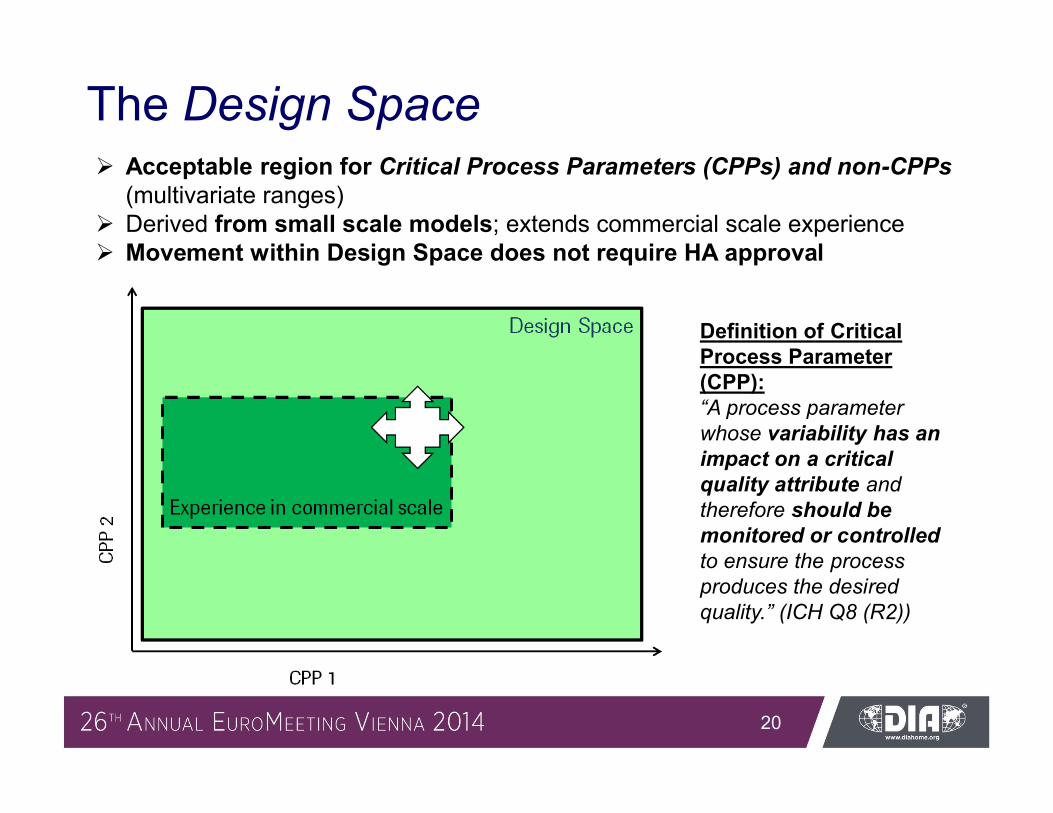
The Design Space Acceptable region for Critical Process Parameters (CPPs) and non-CPPs
(multivariate ranges) Derived from small scale models; extends commercial scale experience Movement within Design Space does not require HA approval
Definition of Critical Process Parameter (CPP): “A process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality.” (ICH Q8 (R2))
20

Typical elements/ definition of a PALM plan
The PALM Plan is a regulatory agreement between the MAH and the Health Authorities that specifies how the MAH will:
• Monitor the MAb process and product quality attributes to ensure that both remain within a controlled state post-approval
• Manage changes to process parameters within the design space
• Update the control system as necessary based on further process and product knowledge
• Elements of the PALM plan are described in the Quality System
21

Changes to (non) CPPs within Design Space:Examples for Required Assessments - PALM plan
Risk Examples for Process Change Assessment Requirements
Low
(e.g., change of non-CPPs or one
Low-Impact CPP)
• Control system testing done at manufacturing scale.
• No influence on testing frequency of monitoring attributes.
Medium
(e.g., change of multiple
Low-Impact CPPs or one
High-Impact CPP)
• Testing of control system and appropriate monitoring attributes done at manufacturing scale under
validation protocol to verify model prediction (minimum of one batch).
• No influence on testing frequency of monitoring attributes.
High
(e.g., change of multiple
High-Impact CPPs)
• Testing of control system and appropriate monitoring attributes done at manufacturing scale under
validation protocol to verify model prediction (minimum of three batches).
• Increased testing frequency of relevant monitoring attributes,
as appropriate.
22

PALM Plan - Process Parameter Change Control- our approach
• Change management includes assessment against ranges in risk assessments, PC/ PV or development studies; impact on quality and regulatory relevance is assessed.
23

PALM Plan - CQA Change Control- our approach (2)
24

Summary/ Outlook
• EU-Change Management Plans, US-(expanded) Change/ Comparability Plans, as well as the Post-Approval Lifecycle Management Plan (related to the Design Space) have proven to be very useful instruments for our industry since they provide:
– A more predictable framework and timeline for managing changes– A science and risk-based approach based on principles set forth in ICH Q 8-11– Harmonization of requirements, and increased regulatory flexibility post-approval – Minimized administrative burden on industry and health authorities– Support in rapid and timely introduction of continuous quality improvements and technological
advances, ensuring availability of high quality products for patients– Tools to classify criticality that will also help to group changes into classes with higher or lower risk
can also help to drive wider, global adoption (ICH?) of instruments like comparability/ change protocols that allow for pre-defined changes of a defined risk class to be implemented in a more streamlined manner if predefined criteria are met
• We also believe CPs/CMPs will play an important role in implementing new, important regulatory initiatives aiming at providing life-saving new products to patients in a highly accelerated manner(US breakthrough initiative; EU adaptive licensing/ new R&D models)
25

Back-ups
26

Variation categories (example EU)Most variations for biologics are major (type II) changes –incl. manufacturing site transfers
Category Impact on quality, safety or efficacy? Notification and implementation Review time
Type IA None / minimal impact
Notification: within 12 months after implementation 30 days
Type IAINNone / minimal impact
Notification: immediate following implementation 30 days
Type IB
Minor potential impact;Classified by default, if change does not fall into another category
Notification: before implementation, but no formal approvalImplementation: ‘tell, wait and do’ – MAH implements changes unless negative opinion received within 30 days
30 days
Type II Major potential impact
Notification: EMA forwards positive CHMP opinion to EU Commission, and they amend the MA within one yearImplementation: following CHMP opinion, without waiting for Commission approval
60 days normally 90 days for indication
changes 30 day for urgent safety
issues
27

«New» tool in the EU: The ChangeManagement Protocol (CMP)• Since 2010, the EU offers the possibility of a post-approval
change managment protocol (CMP or PACMP) • CMP describes specific changes that the MAH would like to
implement following marketing authorization and how these would be prepared and verified
• CMP applies to all types of products and incorporates a science and risk-based approach to evaluate impact of change on product quality in a proactive manner
• CMPs may be included in an original marketing authorization application (MAA) or be submitted as a stand-alone type II-variation
Company will have obtained agreement from Competent Authority about the proposed strategy before implementation of the change; implementation itself is done via a downgraded variation (default: type IB variation for biologics)
Requires pre-work, but is an option for a faster and more predictable implementation of changes post-approval, more flexibility for MAH
28

The principle of the Change ManagementProtocol: a 2-step approach
Source: EMA CMP Questions & Answers: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/04/WC500125400.pdf
29

The Contents of a Post-Approval CMPAccording to EMA CMP Q&A: Justification that there is a recognized future need for the specific change
within a reasonable timeframe.
A detailed description of the proposed change.
Risk assessment of the impact of the change on product quality.
Discussion on the appropriateness of the approved control strategy to identify and manage these risks.
Description of the studies to be performed, the test methods and acceptance criteria that will be used to fully assess the effect of the proposed change on product quality.
For biologics, the approach to be used to demonstrate the comparability of the pre- and post- change product.
A plan for stability studies should be included, if appropriate.
In case that the protocol describes several changes, a justification showing that the changes are related, and that a simultaneous review under a single protocol is meaningful.
30
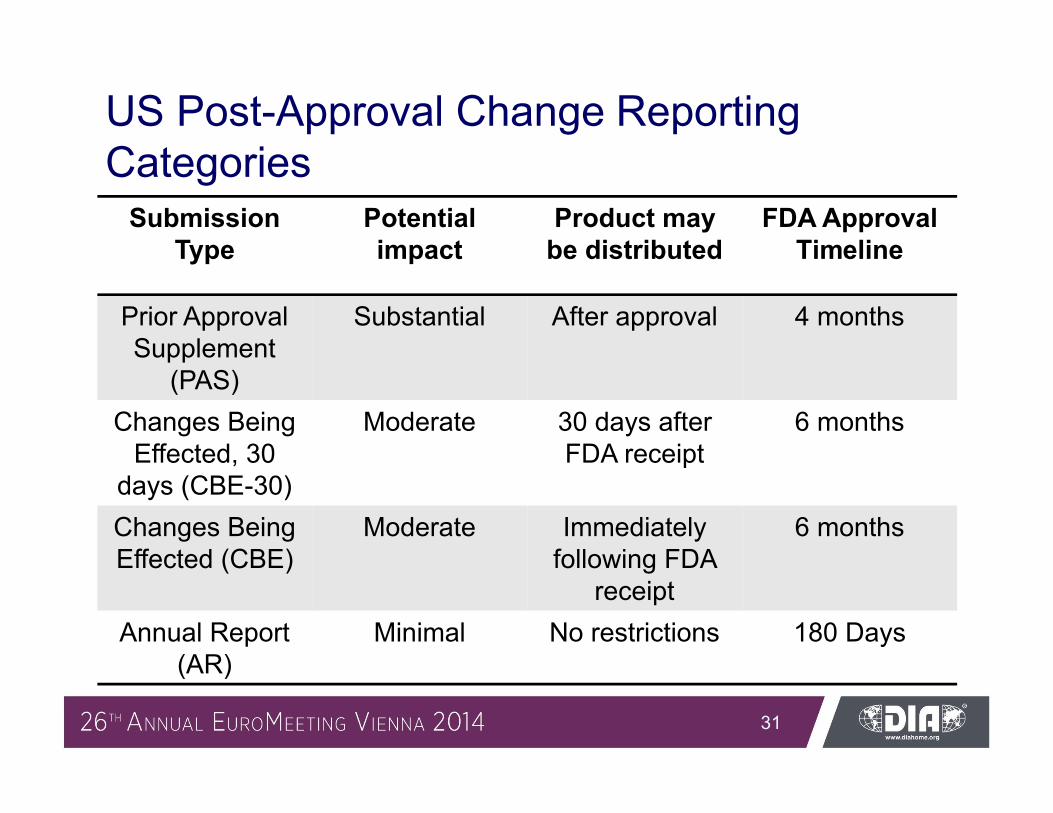
US Post-Approval Change Reporting Categories
Submission Type
Potential impact
Product may be distributed
FDA Approval Timeline
Prior Approval Supplement
(PAS)
Substantial After approval 4 months
Changes Being Effected, 30
days (CBE-30)
Moderate 30 days after FDA receipt
6 months
Changes Being Effected (CBE)
Moderate Immediately following FDA
receipt
6 months
Annual Report (AR)
Minimal No restrictions 180 Days
31
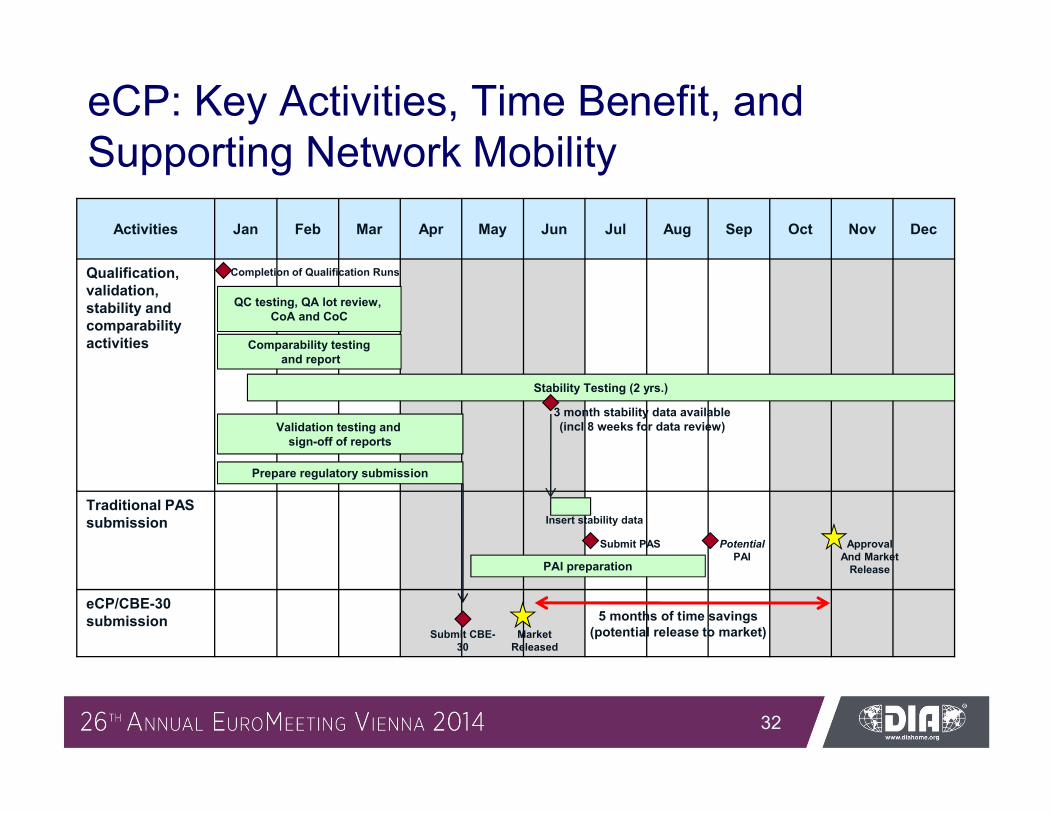
eCP: Key Activities, Time Benefit, and Supporting Network Mobility
Activities Jan Feb Mar Apr May Jun Jul Aug Sep Oct Nov Dec
Qualification, validation, stability and comparability activities
Traditional PAS submission
eCP/CBE-30 submission
3 month stability data available (incl 8 weeks for data review)
Stability Testing (2 yrs.)
Submit PAS ApprovalAnd Market
Release
QC testing, QA lot review, CoA and CoC
Comparability testingand report
Validation testing and sign-off of reports
Prepare regulatory submission
Completion of Qualification Runs
Insert stability data
Potential PAI
Submit CBE-30
MarketReleased
5 months of time savings (potential release to market)
PAI preparation
32

Genentech’s Path to eCPs
Data and information supporting comparability
Data and requirements submitted for defined product and change
Comparability Protocols (CPs) with pre-defined requirements and acceptance criteria
Site Transfers submitted as Prior Approval Supplements (PAS)
Formalized approach to risk management and adoption of ICH Q9 principles and practices
eCP supporting product introductions for multiple products and sites and addressed cross-contamination Extension of both
comparability and Quality Risk Management concepts to Site Transfers
Site Transfer eCP accepted to FDA QbD Pilot Program
Site Transfer eCP approved by FDA (first QbD Pilot Program approval)
Routine execution of eCP requirements to facilitate post-approval changes
The Site Transfer eCP was FDA’s 1st QbD Approval in the QbD Pilot Program
33

Quality Risk Management Plan
PHAHACCP FMEA
Approve/Implement Risk Control Strategy
Develop Quality Risk Management Report
Update and review on periodic basis or when significant
events/changes occur
Initiate Quality Risk Management Process
Ris
k C
om
mu
nic
atio
n
Risk M
anag
emen
t too
ls
Risk Assessment
Hazard Identification
Risk Analysis
Risk Evaluation
Risk Control
Risk Reduction
Risk Acceptance
Output / Report and Implementation
Risk Review
Risk Event
unacceptable
Communicate Plan, high level
risks, and Risk Control
Strategy to appropriate management
team
Quality Risk Management ICH Q9
34

Site Transfer Risk AssessmentConsiderations• Performed for each site transfer
• Identifies, scores, and documents the potential hazard and harm associated with each unit operation and process change, as well as the prevention and detection controls
• Accounts for known elements of the process
– Robustness
– Existing controls
– Potential impact to product quality
• Considers the subjectivity of risk assessment
– Team
– Facilitator
– Training
– Consistency and calibration of risks
– Severity, Occurrence, Detection scoring definitions
35

Comparability Approach
Category Components
A The basic package:
• QC lot release data, including potency
• extended physico-chemical characterization methods
• accelerated degradation, with real-time stability and commitments
• process-related impurity levels (host cell proteins, DNA, Protein A)
A+B Biological characterization:
• Fcg receptor assays, FcRn, ADCC
• Biacore or other binding assays
A+B+C Animal PK or PK/PD studies:
• rodent PK may suffice
• may need primates for PD
A+B+C+D Clinical PK (comparability bridging study):
• direct comparison to licensed process material in human subjects
A+B+C+E Clinical experience or efficacy
• may need to confirm efficacy, lack of AEs, lack of immunogenicity
• might be a “clinical experience” study, or head-to-head vs. licensed process
Transfer to a new site with facility fit changes, with no expected change to product characteristics
36

The importance of stressed conditions stability testing: Comparison of Product A Drug Substance IEC Profiles at T14 days, 40°C/75% RH
Similar to the real-time figures, an increase in acidic variants (30% to 44%) and a decrease in main peak (66% to 49%) are observed at the stressed conditions (40°C/75% RH), to a higher degree
37

Product A Drug Product IEC Profile Timepoints T0 and T 730 Days, 5°C
Comparing T0 to end of shelf-life under real-time conditions (5°C), the predominant changes are an increase in acidic variants (26% to 28%) and a decrease in main peak (69% to 66%)
38

ICH Q8(R2) Pharmaceutical Development: Definition of Design Space
39


















