
INCORPORATION OF PELLETS AND MICROGRANULES INTO …
59
KAUNAS UNIVERSITY OF MEDICINE FACULTY OF PHARMACY DEPARTAMENT OF PHARMACEUTICAL TECHNOLOGY AND SOCIAL ORGANIZATION Edita Eidukaityt MASTER THESIS INCORPORATION OF PELLETS AND MICROGRANULES INTO PRESSED AND LYOPHILIZED TABLETS, RESPECTIVELY Master thesis is accomplished at a Departament of Pharmaceutical Technology, Medical University of Gdansk Head of thesis: prof. hab. dr. Malgorzata Sznitowska- Departament of Pharmaceutical Technology, Medical University of Gdansk, prof. Dr. Vitalis Briedis- Departament of Pharmaceutical Technology and social orgnization, Kaunas University of Medicine KAUNAS 2007
Transcript of INCORPORATION OF PELLETS AND MICROGRANULES INTO …

KAUNAS UNIVERSITY OF MEDICINE FACULTY OF PHARMACY
DEPARTAMENT OF PHARMACEUTICAL TECHNOLOGY AND SOCIAL ORGANIZATION
Edita Eidukaityt�
MASTER THESIS
INCORPORATION OF PELLETS AND MICROGRANULES INTO PRESSED AND
LYOPHILIZED TABLETS, RESPECTIVELY
Master thesis is accomplished at a Departament of Pharmaceutical Technology, Medical University of Gdansk
Head of thesis: prof. hab. dr. Małgorzata Sznitowska- Departament of Pharmaceutical Technology,
Medical University of Gdansk, prof. Dr. Vitalis Briedis-
Departament of Pharmaceutical Technology and social orgnization, Kaunas University of
Medicine
KAUNAS
2007

2
Content
1. Introduction………………………………………………………..……4 1.1.The importance of multiparticulate dosage forms……………………...…………4
1.2.The aim of the study……………………………………………………………....4
1.3.The tasks of the study…………………………………………………….……….5
2. Literature review………………………………………………….……..6 2.1. Pellets and microparticles – characteristics and methods of preparation………..6
2.1.1. Pellets........................................................................................................6
2.1.2. Microcapsules............................................................................................8
2.1.3. Microspheres...........................................................................................10
2.1.4. Lipospheres..............................................................................................11
2.1.5. Microgranules..........................................................................................13
2. 2. The influence of pellet core composition and coating film type on the release of
active substance .........................................................................................................14
2. 3. Lyophilized tablets.......................... ...................................................................18
2.3.1. Lyophilization process.............................................................................19
3. Experimental part……….........................................................................21
3.1. Incorporation of microgranules with selegiline hydrochloride into
lyophilized tablets………………..…………………………………………21 3.1.1. Reagents, materials and equipment...................................................................21
3.1.2. Methods ............................................................................................................23
3.1.2.1. Microgranules formation......................................................................23
3.1.2.1.1. Preparation of microgranules type XPS1.................................23
3.1.2.1.2. Preparation of microgranules type XPS3.................................23
3.1.2.1.3. Coating of the microgranules (type XPS3P preparation).........23
3.12.2. Visual inspection of microgranules ......................................................24
3.12.3. Evaluation of selegiline hydrochloride content in the cross-linked
microgranules ...................................................................................................24
3.1.2.4. The release of selegiline hydrochloride from cross-linked
microgranules....................................................................................................24
3.1.2.5. Preparation of lyophilized tablets containing microgranules with
selegiline hydrochloride....................................................................................24
3.1.2.6. Visual inspection of lyophilized tablets ..............................................25

3
3.1.2.7. Release test of selegiline hydrochloride from lyophilized tablets........25
3.1.2.8. Quantitative analysis of selegiline hydrochloride by HPLC method...26
3.2. Incorporation of floating pellets into pressed tablets –
tableting of floating pellets……………………………………...28
3.2.1. Reagents, materials and equipment...................................................................28
3.2.2. Preparation of floating pellet cores with verapamil hydrochloride by extrusion-
spheronization method................................................................................................30
3.2.3. Coating of pellet cores with Eudragit NE 40 D................................................31
3.2.4. Tableting of floating pellets with verapamil hydrochloride by using impact
tableting machine........................................................................................................32
3.2.5. Analysis of pellets and tablets...........................................................................34
3.2.5.1. Measurement of film thickness............................................................34
3.2.5.2. In vitro drug release test from pellets...................................................34
3.2.5.3. Tablets appearance and size.................................................................34
3.2.5.4. Content of VH in tablets.......................................................................34
3.2.5.5. Determination of single tablet mass uniformity...................................35
3.2.5.6. Evaluation of tablets resistance to crushing (hardness of tablets)........35
3.2.5.7. Evaluation of tablets friability..............................................................35
3.2.5.8. In vitro drug release test from tablets and flotation start time of
pellets………………………………………………………………………….35
4. Results and discussion………...................................................................36
5. Conclusions................................................................................................54
6. Acknowledgments………………………………………………………..55
7. References..................................................................................................56

4
1. Introduction
1.1. The importance of multiparticulate dosage forms
Multiparticulate dosage form contains actives divided into many individual units, so-called
subunits, each exhibiting some desired characteristics. These subunits usually are microparts
such as microcapsules, microspheres, lipospheres, microgranules or larger particles - pellets.
Multiparticulate dosage forms are more expensive to manufacture and develop, but despite of it
are widely used in pharmaceutical formulations. They are more reliable in their
biopharmaceutical behaviour and considered to provide pharmacokinetic advantages compared
with monolithic dosage forms [27].
When compared with single-unit dosage forms, oral multiparticulate drug-delivery systems
offer biopharmaceutical advantages:
� Microparticles can be used to prepare pharmaceutical formulations composed of
incompatible drugs or to obtain delivery systems with different release profiles [16].
� Can be divided into desired doses without formulation and process changes.
� Possibility to produce modified release dosage forms as very significant means of drug
delivery nowadays [16, 27].
� More even and predictable distribution and transportation in the gastrointestinal tract,
which is fairly independent of the nutritional state [26]. When taken orally
multiparticulates generally disperse freely in the gastrointestinal tract, thus maximize
drug absorption, reduce peak plasma fluctuation, minimize side effects and reduce inter-
and intrapatient variability [15].
� Are less susceptible to dose dumping than the reservoir-type, single unit formulations [4].
These dosage forms have several disadvantages, like the risk of tampering with capsules or
the rupturing of the coating during compression resulting in a loss of the modified drug-release
properties [27].
1.2. The aim of the study
With regard to the final dosage form, the multiparticulates can be filled into hard gelatin
capsules or be incorporated into tablets. The compression of multiparticulates into tablets is
becoming more popular. The advantages of tableting multiparticulates include a reduced risk of
tampering and fewer difficulties in oesophageal transport when compared with capsules. Large

5
volume tablets generally have a higher patient compliance than capsules; higher dose strength
could be administered with tablets. Tablets with pellets can be prepared at lower cost when
compared to pellet-filled capsules because of higher production rate of tablet presses. The
expensive control of capsule integrity after filling is also eliminated. In addition, tablets allow a
more flexible dosing regimen [3, 39].
The aim of the study was to obtain multiparticulate prolonged release drug delivery
systems by incorporating microgranules with selegiline hydrochloride (SCh) and pellets with
verapamil hydrochloride (VH) into lyophilized and pressed tablets, respectively. Also it was
assumed to preserve primary dosage form properties unchanged in final drug form. Controlled
SCh release of microgranules was achieved by cross - linking pectin with 20% ZnSO4 solution,
whereas controlled VH release from pellets was gained by coating it with methacrilic acid
copolymer – Eudragit NE 40 D in a fluidized-bed coater.
Preparing lyophilized buccoadhesive tablets with microgranules and compressed oral
tablets with floating pellets and maintaining the same drug release profile for the resulting tablets
as for the microgranules or pellets, respectively, was the goal of the study.
1.3. The tasks of the study
The tasks of the study were as following:
1. To obtain microgranules with SCh by two different methods of preparation;
2. To incorporate these microgranules into lyophilized tablets;
3. To test and compare release profiles of microgranules before and after incorporation;
4. To prepare floating pellets with VH by extrusion-spheronization method;
5. To coat pellets by fluidized-bed method with Eudragit NE 40;
6. To tablet these floating pellets with various tableting excipients by means of single
stroke machine with 12 kN and 18 kN compression force;
7. To test and compare release profiles of floating pellets before and after tableting.

6
2. Literature review
The main types of multiparticulate dosage forms (pellets, microcapsules, microspheres,
lipospheres, and microgranules) are discussed in this chapter.
2.1. Pellets and microparticles – characteristics and methods of preparation
2.1.1. Pellets
Pellets are uniformly sized spherical granules, which range in size from 0.5 – 1.5 mm and
in some applications may be as large as 3.0 mm. This drug form can contain 10 – 90% of active
pharmaceutical ingredient (API) and usually is obtained by coating the cores, wet granulation,
hot extrusion or extrusion-spheronization method. The last mentioned method is applied mostly
[17] (Fig. 1).
Fig. 1. Pellets, prepared by extrusion-spheronization method (interspace on the scale=1
mm)
Preparation of pellets by extrusion and spheronization offers numerous advantages over
other methods:
� Ease of operation;
� High throughput with low wastage;
� Narrower particle size distribution;
� Production of pellets with low friability;
� Production of pellets that are suitable for film coating;

7
� More sustained and better controlled drug-release profile when compared with other
techniques [15].
The major advantage is the ability to incorporate high levels of active ingredients without
producing exclusively large particles.
The main steps of the process are [33]:
1. Dry mixing of ingredients to achieve homogenous powder dispersion;
2. Granulation (wet massing) to produce a sufficiently plastic wet mass;
3. Extrusion to form rod-shaped particles of uniform diameter;
4. Spheronization to round off these rods into spherical particles;
5. Drying to achieve the desired final moisture content;
6. Screening (optional) to achieve the desired narrow size distribution.
Different steps, parameters and equipment used in the process are summarized in Fig. 2
[15].
Fig. 2. Flow diagram showing different steps, process parameters and equipment involved in extrusion and
spheronization to produce spherical modified release pellets [15]
Pellets are usually used to prepare multiparticulate drug forms both with immediate and
modified drug release [47]. However nowadays, when in certain circumstances it is needed a
constant drug concentration in the blood, it is preferred to produce medications with not only
prolonged, but also controlled drug release. It is well known, that using these drug forms, a drug
plasma level is more constant, which is associated with less adverse side-effects, a more constant
and prolonged therapeutic effect and a better compliancy [19]. Controlled release dosage forms
are especially applied for drugs with very low therapeutic index, the median effective dose being
close to the median lethal dose. Moreover, the main advantage is the possibility of once-daily
Powder dry mixing
Granulating liquid
Mixer
Wet mixing
� Granulator type
� Granulation liquid
� Mixing time
Extruder
Extrusion
Spheronizer
Spheronization
Dryer
Drying
Coater
Coating
� Extruder type � Extrusion speed � Screen opening
size � Extrusion
temperature
� Spheronizer type � Plate type � Plate speed � Spheronization
time � Spheronizer load
� Dryer type � Drying
temperature
Coating solution

8
dosing. This simplification in the therapy improves compliance and leads to a decrease in overall
costs. Controlled release dosage forms have essentially been implemented in the treatment of
hypertension, inflammatory processes, obstructive pulmonary, Parkinson’s diseases [12].
2.1.2. Microcapsules
Microcapsules are kind of capsules, which size varies from 5 to 1000 �m, commonly they
have a diameter between 100 – 500 �m (fig. 3).
Fig. 3. SEM photograph of microcapsules (bar=5 �m) [20]
They consist of an active agent or core material which is surrounded by coating or shell.
The core can contain solid, liquid (solution, suspension, emulsion) or gaseous substance. The
mass of the core usually works out 30 – 90% of the all microcapsule mass. Depending on the
core composition microcapsules have spherical or irregular, close to core substance, shape. A
wide range of core materials have been encapsulated, including pharmaceuticals, adhesives,
agrochemicals, live cells, active enzymes, flavors, fragrances and inks. Most microcapsule shell
materials are synthetic, but natural ones are also used. Gelatin, arabic gum, shellac, colophony,
ethyl cellulose, carboxymethyl cellulose, methylcellulose, cellulose nitrate, cellulose acetate,
polyvinyl alcohol, polyamides, polyoxyethyl glycol, polypropylene, polyvynilpirolidone are
applied mostly. The mixtures of polymers for example ethylcellulose and methylcellulose are
also used frequently. Depending on the kind of core, the substance passes through the film faster
or slower. The films may be stiff, elastic or porous [23, 45].
Methods of preparation
I Chemical methods
Coacervation. There are two types of coacervation: simple and complex. Only one
colloid, e. g., gelatin in water, it is used in the simple coacervation and the cores – solid or liquid

9
substances – are suspended or emulsified in the water solution of shell material. The associated
water from around the dispersed colloid is removed by adding agents with a greater affinity for
water, such as alcohols, salts or by changing environmental parameters: temperature, pH,
concentration. The dehydrated molecules of polymer tend to aggregate with surrounding
molecules to form coacervate (it is coacervation in aqueous media). Soluble and insoluble in
water substances can be encapsulated using coacervation in nonaqueous media. The substances
can not be soluble in the organic solvent, in which the process is accomplished. Shell materials
also must be insoluble in the organic solvent, for this reason usually cellulose derivates (cellulose
nitrate, cellulose acetate, ethylcellulose) are applied. The coacervation comes when reciprocal
organic solvent, miscible with polymer solution, is added [9, 23].
Complex coacervation involves the use of more than one colloid. This process occurs with
reciprocal neutralization of two oppositely charged polymers. Method is based on the ability of
cationic charged gelatin and negatively charged polymer (for example arabic gum or acacia) to
interact in water and form a liquid, polymer rich phase called a coacervate. This method is rather
used to encapsulate water-immiscible liquids [9, 45].
Polymer – polymer incompatibility. Polymer – polymer incompatibility occurs because
two chemically different polymers dissolved in a common solvent are incompatible and do not
mix in solution (e.g.ethyl cellulose and polyethylene in hot (80oC) cyclohexane). These polymers
form two separate liquid phases: one phase is rich in polymer which acts as the capsule shell (in
this case ethyl cellulose), the other is rich in the second, incompatible polymer. This polymer
causes formation of two phases and it is not designed to be part of the final capsule shell. Then
the core material (small particles) is dispersed in two-phase system. Since the ethyl cellulose is
more polar than polyethylene, it adsorbs on core material and forms a thin coating. When the
system is cooled to room temperature the ethylcellulose precipitates forming solid microcapsules
[45].
Interfacial polymerization (IFP). During this process the capsule shell is formed at or on
the surface of encapsulated particle by polymerization of reactive monomers. IFP allows
encapsulate a wide range of core materials, including aqueous solutions, water-immiscible
liquids, and solids.
Vinyl monomers that polymerize by free radical reactions generally are used to encapsulate
solids. The solid is dispersed in aqueous media along with a dispersing agent. Vinyl monomer is
added to the system and free radical polymerization is initiated by redox initiation system [45].

10
In situ polymerization. In situ polymerization is closely related to IFP. The difference is
that with in situ encapsulation processes, no reactive agents are added to the core material.
Polymerization occurs on the continuous-phase side of the interface formed by the dispersed core
material and continuous phase. Polymerization of reagents located there produces relatively low
molecular weight prepolymer. As this prepolymer grows in size, it deposits onto the surface of
the dispersed core material being encapsulated where polymerization with cross-linking
continues to occur thereby generating a solid capsule shell [45].
II Physical methods
Spray drying. Microencapsulation by spray drying is based on two steps: emusification or
dispersion of the core substance in the polymer solution and removal of the solvent by a hot
stream of air. The core material is generally water-immiscible. The shell material normally is
water soluble polymer like gum arabic or a modified starch [45, 28].
Fluidized bed method. Fluidized bed coaters suspend solid particles in a moving gas
stream, usually air. A liquid coating formulation is sprayed onto the individual particles. Freshly
coated particles are moved into a zone where the coating formulation is dried either by solvent
evaporation or cooling. This coating and drying sequence is repeated until a desired coating
thickness is achieved [45].
Rotational suspension separation. During this process, core material dispersed in a liquid
shell formulation is fed onto a rotating disk. Individual core particles coated with a film of shell
formulation are flung off the edge of the rotating disk along with droplets of pure coating
material. The shell mass is solidified, e.g., by cooling, and discrete microcapsules are produced.
The excess of pure shell material also solidify, but it is collect in a discrete zone away from the
capsules [23, 45].
�
2.1.3. Microspheres
Microspheres are monolithic, porous spheres, composed of various polymers, in which
active pharmaceutical substances are dispersed or dissolved. The size of microspheres varies
from 1 to 500 �m (fig. 4). Although the size and the shape sometimes are very close to
microcapsules, essential difference is composition of microspheres. Microcapsules possess active
pharmaceutical ingredient in liquid or solid form, enclosed in polymeric membrane. Meanwhile
in the microspheres API is incorporated in polymeric matrix. Therefore, microspheres might be
called micrometric matrix systems.

11
Fig. 4. SEM picture of microspheres (bar=10 µm) [14]
The most suitable polymeric materials for producing microspheres are synthetic
hydroxyacids polyesters: polylactic acid (PLA) and polylacticglycolic acid (PLGA). These
polymers undergo biodegradation to natural products of metabolism - lactic and glycolic acids.
The main method of preparation of these drug delivery systems is solvent evaporation.
This technique is based on removing the hydrophobic polymer solvent by evaporation [29].
Polymeric material is dissolved in a volatile organic solvent. The API is then dispersed or
dissolved in the organic solution. In the following step, a dispersing phase, consisting of
nonsolvent of the polymer and immiscible with the organic solvent also containing appropriate
tensioactive substance, is added by continuous mechanical stirring. The solvent evaporates after
diffusing through the continuous phase and the result is creation of solid microspheres.
Microspheres also can be produced by polymer-phase separation, spray-drying, milling
methods or methods using fluids under supercritical conditions [2].
�
2.1.4. Lipospheres
Lipospheres are a new type of lipid based drug delivery system developed for parenteral or
topical action of bioactive compounds. They are composed of solid hydrophobic fat core
(triglycerides) stabilized by one monolayer of phospholipid molecules embedded in their surface.
Particle size ranges between 0.2 to 100 �m in diameter (fig. 5).

12
Fig. 5. Stereomicrograph of lipospheres (bar=100 �m) [46]
The internal core contains the bioactive compound dissolved or dispersed in the solid-fat
matrix. The lipospheres system has been used for the controlled delivery of various types of
drugs including anti-inflammatory compounds, local anaesthetics, antibiotics and anticancer
agents. They have also been used successfully as carriers of vaccines and adjuvants.
Lipospheres have several advantages over the other drug delivery systems: better physical
stability, low cost ingredients, ease of preparation and scale-up, high dispersability in aqueous
medium, high entrapment of hydrophobic drugs, controlled particle size, extended release of
incorporated drug after a single injection from a few hours to several days.
The main hydrophobic core constituents are tricaprin, trilaurin, tristearin, stearic acid, ethyl
stearate, hydrogenated vegetable oil. The phospholipids used to form the surrounding layer of
lipospheres are pure-egg phosphatidylcholine, soybean phosphatidylcholine, dimyristoyl
phosphatidylglycerol and phosphatidylethenolamine.
Liposphere formulations are prepared by a solvent or melt process. In the melt method, the
API is dissolved or dispersed in the melted solid carrier and then a hot buffer solution is added at
once with the phospholipid powder. The hot mixture is homogenized for about 2-5 min using
homogenizer or ultrasound. The milky formulation is rapidly cooled by immersing the flask with
mixture in an acetone-dry ice bath while homogenization is continued to yield a uniform
dispersion of lipospheres.
In the solvent technique, the active agent, the solid carrier and phospholipid are dissolved
in an organic solvent (e.g.acetone, ethyl acetate, ethanol or dichlormethane). The solvent is
evaporated; the resulting solid is mixed with warm buffer solution. Mixing is continued until a
homogenous dispersion of lipospheres is obtained [11].
�

13
2.1.5. Microgranules
Microgranules are compacted powder particles with a size range between 200 and 500 �m
[50] (fig. 6). In order to obtain required release profile microgranules can be coated. It is difficult
to prepare microgranules of a proper size and density using conventional dry/wet granulation or
the other methods. Usually microgranules, prepared by these methods, have bigger size and
lower density than the required ones, e.g. for coating or incorporation into lyophilized tablets.
Fig. 6. Microgranules (bar=100 �m)
During the process in a fluid-bed apparatus, the granulation powder is kept in suspension
by an appropriate air flow, while the granulation fluid is simultaneously sprayed. The resulting
product has an even shape, but is very porous and has a low density. Granules are therefore
unsuitable for subsequent coating, as they are inclined to break. Furthermore, the material to be
subjected to a fluid-bed coating process must be made up of particles of sufficient density to
avoid the agglomeration phenomenon. Otherwise, the particles tend to occupy the upper section
of the apparatus, are not subject to the normal movement inside the apparatus, and thus do not
receive an appropriate gradual coating.
A conventional mixer-granulator consists of a vessel, which may be of varying shape,
equipped with an agitator that keeps the powder moving while the granulation fluid is being
added. The motion is slow and the resulting granules, even though suitable for making
conventional dosage forms such as tablets or capsules, does not possess the density, shape and
particle-size distribution suitable for subsequent coating.
Unlike conventional mixer-granulators, extruder-spheronizers can produce spherical
particles of homogeneous sizes and even shapes and surfaces. The limitation that prevents their
application to producing the microgranules is the average product size, which is rarely smaller
than 1-2 mm and in any case never smaller than 500 �m.

14
A high-shear mixer-granulator is made up of a vessel in which the mixture to be granulated
is introduced that is equipped with a mixer and a mill that rotate with a normal mixer motion.
Since the mixer and the mill have variable and adjustable speeds, they ensure densification and
preparation of the granulate in shorter times as compared to conventional granulators.
It has now been found that, using high-shear mixer-granulators and operating within
specific critical ranges of the parameters that control the granulation process, it is possible to
obtain a microgranulate of a size smaller than 500 �m. It is an object of the present invention to
provide a size distribution, density, surface and shape of the particles produced that makes them
particularly suitable for preparing the final drug forms, e.g. coating and for suspension in low
density fluids [35].
The other method, which can be used for preparation of microgranules, is modified wet
granulation. Wet granulation generally involves the wetting of a mixture of dry primary powder
particles using a granulating fluid. The fluid contains a solvent which must be volatile so that it
can be removed by drying, and be non-toxic. Typical liquids include water, ethanol and
isopropanol, either alone or in combination [50]. In a modified granulation method, applied for
preparing microgranules with selegiline, as granulating liquid aqueous zinc sulphate solution was
used. Bivalent ions such as zinc, calcium etc. cross-link the polymer (in this case – pectin). This
modification served as a good mean for achieving prolonged drug release.
2.2. The influence of pellet core composition and coating film type on the
release of active substance
In order to obtain a medication with modified drug release quality, it is important to choose
appropriate constituents of the core and the coating formulation. As the film is commonly
responsible for controlled release action, in most studies the main attention is paid exactly for the
influence of coating type on releasing of active substances. Dissolution tests are performed under
in vitro conditions, but to optimize a dosage form the in vivo investigations are also necessary.
During the study of in vitro and in vivo dissolution of theophylline from pellets, coated
with Eudragit L, the in vivo liberation of theophylline was studied in rabbits. Finally, it was
concluded, that there was no great difference in the maximum values (all the pellets gave cmax >
10 µg/ml) between the uncoated and the Eudragit L-coated pellets, but a significant shift in tmax
was found for both Eudragit-coated pellets. The difference was 4h as compared with the
uncoated preparation (fig. 7). This result exhibits a relationship between the in vitro dissolutions
(3 h), which confirms the reliability of the in vitro dissolution method. The slower decreases in

15
theophilline concentration in the cases of the Eudragit L (64 µm) and Eudragit L (85 µm) films
demonstrate the effectiveness of the enteric coating process [30].
Fig. 7. Plasma level of theophylline after administration in rabbits [30].
In the study of Hu et al. [21], the influence of surface modification by talc, the effects of
Eudragit types and ratios, as well as the correlation between in vitro release and in vivo
absorption were investigated in detail in metformin hydrochloride (MH) sustained release
pellets. Three pellets formulations were prepared: formulation 1 (F-1): coated with Eudragit
NE30D, resulting in 10% coat loading; formulation 2 (F-2): coated with Eudragit L30D-55:
Eudragit NE30D (1:20), resulting in 7% coat loading; formulation 3 (F-3): coated with Eudragit
L30D-55: Eudragit NE30D (1:20), resulting in 10% coat loading. The relative bioavailability of
the sustained release pellets was studied in 12 healthy volunteers after oral administration in a
fast state using a commercially available immediate release (IR) tablet (Glucophage) as a
reference. The results suggest that talc modification effectively controls drug release and avoids
drug dumping. The in vivo bioavailability showed varying sustained-release characteristics for
the coated pellets when compared with IR MH tablets (fig. 8).

16
�
Fig. 8. Mean plasma metformin concentration-time profiles of three sustained MH pellets
and IR MH tablets [21]
When coated with a blend of Eudragit L30D-55 and Eudragit NE30D (1:20) to a loading
weight of 7% or 10%, pellets exhibited excellent sustained-release effects and high relative
bioavailability. Restricted delivery of metformin hydrochloride to the small intestine from
differently coated pellets resulted in increased relative bioavailability and a sustained release
effect. The adoption of several different pH dissolution media (0.1 M HCl, distilled water and
phosphate buffer (pH 6.8) established a better relationship between the in vitro release and in
vivo absorption of the sustained-release pellets.
Zhou et al. [51] tested the bioavailibilty of ibuprofen from pellets based on
microcrystalline wax (Lunacera P® and Lunacera M®) and starch derivatives. This matrix
system provides a flexible drug delivery system, whereby the drug release rate depends on the
type and the concentration of the hydrophobic and the hydrophilic component. During the
studies three pellets formulations were prepared, which had the following composition: F-1:
ibuprofen–waxy maltodextrin (WMD)–Lunacera P® and Lunacera M® mixture (ratio 3/7)
60/15/25 (w/w/w); F-2: ibuprofen-waxy maltodextrin-Lunacera P 60/15/25 (w/w/w). Both
formulations were filled into hard gelatine capsules. Formulation F-3 (ibuprofen–drum dried
corn starch (DDCS) –Lunacera P® 30:40:30 (w:w:w)) pellets were compressed into tablets.
Pellets (F-1) formulated with the wax having the highest melting range gave the slowest drug
release rate while F-3 pellets failed to form a sustained release matrix system as 90% of the dose
was released within 45 min of the dissolution test. In vivo evaluation was performed with

17
healthy human volunteers. The plasma profiles also indicated that the absorption of ibuprofen
depended on the composition of the matrix pellet formulation. From these in vivo studies it can
be concluded that the bioavailability of pellet formulations based on the combination of
microcrystalline waxes and starch derivatives can be adjusted by means of varying the type and
the content of both the waxes and the starch derivatives. Pellets with a sustained as well as an
immediate drug release could be formulated using the wax–starch delivery system.

18
2.3. Lyophilized tablets
The main method for obtaining this drug form is freeze-drying or lyophilization. It is a
process in which water is sublimated from the product after freezing [10]. The main advantage
being that pharmaceutical substances can be processed at non-elevated temperatures that enables
drying labile substances, excluding the action of enzymes and microorganisms [13]. Final
product can be stored in a dry state with relatively few shelf life stability problems. Another
reason for applying this method is that freeze-dried forms offer more rapid dissolution than other
available solid products. The lyophilization process imparts a glassy amorphous structure to the
bulking agents and, sometimes, to the drug, thereby enhancing the dissolution characteristics of
the formulation. After placing it in the mouth, these dosage forms immediately disperse/dissolve
in the saliva and are then swallowed in the normal way without the need for water. This is very
important especially in the paediatric and geriatric patients, who have difficulty swallowing
tablets and hard gelatine capsules. Also lyophilized tablets offer a convenience during travels.
The bioavailability of a drug from fast dispersing formulations may be even grater than observed
for standard dosage forms. Furthermore, side-effects may be reduced, if they are caused by first-
pass metabolites [43]
The use of lyophilization however is strongly limited by the long time and handling
required for processing. Freeze-drying is also energy- intensive process, having limited amount
of materials processed for each batch. Other major disadvantages of the final dosage forms
include the lack of physical resistance and limited ability to accommodate adequate
concentrations of active [10].
Lyophilized tablets are prepared by sublimation of solutions after dosing it to blisters.
Polymers and sugars that give for lyophilized tablet appropriate structure are applied in this
process as excipients (table 1).

19
Table 1. The main excipients applied for preparation of lyophilized tablets [43]
Kind of substance Sample Properties
Binders Gelatine, alginates, gums (xanthan, arabic), cellulose derivatives, povidone
� Determine the structure of tablets � Have influence on it hardness � Ensure amorphous structure � Make easier suspending of active substance particles
Fillers Mannitol, sorbitol, saccharose, lactose,
glucose, maltodextrins, erytritol, lactitol
� Commonly forms the main tablets mass � Fill the structure created by polymers � Have sweet flavour
Sweeteners Aspartame, sodium saccharine � Mask bitter taste of API
Aroma Mostly mint, cherry, orange or vanilla � Make drug application more attractive
2.3.1. Lyophilization process Generally the complete freeze-drying process comprises three stages: freezing, primary
drying, and secondary drying [44].
Freezing
Freezing is an efficient desiccation step where most of the solvent, typically water, is
separated from the solutes to form ice. As freezing progresses, the solute phase becomes highly
concentrated and is termed the “freeze concentrate.” By the end of freezing, the freeze
concentrate usually contains only about 20% of water (w/w), or less than 1% of total water in the
solution before ice formation. The freezing stage typically takes several hours to finish. In this
step, it is important to freeze the material at a temperature below the eutectic point of the
material. Since the eutectic point occurs at the lowest temperature where the solid and liquid
phase of the material can coexist, freezing the material at a temperature below this point ensures
that sublimation rather than melting will occur in the following steps.
Primary drying
Primary drying, or ice sublimation, begins whenever the chamber pressure is reduced and
the shelf temperature is raised to supply the heat removed by ice sublimation. During primary
drying, the chamber pressure is well below the vapour pressure of ice, and ice is transferred from
the product to the condenser by sublimation and crystallization onto the cold coils/plates
(<−50°C) in the condenser. Typically, the primary drying stage is the longest stage of freeze
drying and optimization of this stage has a large impact on process economics.
Secondary drying
The secondary drying phase aims to sublimate the water molecules that are adsorbed
during the freezing process, since the mobile water molecules were sublimated in the primary
drying phase. This part of the freeze-drying process is governed by the material’s adsorption

20
isotherms. In this phase, the temperature is raised even higher than in the primary drying phase
to break any physico-chemical interactions that have formed between the water molecules and
the frozen material. Usually the pressure is also lowered in this stage to encourage sublimation.
After the freeze drying process is complete, the vacuum is usually broken with an inert gas,
such as nitrogen, before the material is sealed.
Tablets obtained by lyophilization can also be applied as buccal drug delivery systems.
These formulations may prove to be an alternative to the conventional oral medications as they
can be readily attached to the buccal cavity retained for a longer period of time and removed at
any time. In order to achieve buccal adhesive drug delivery systems quite a wide variety of
bioadhesive substances can be used including such polymers as pectin, gelatine, sodium CMC,
HPMC, sodium alginate, xanthum gum, acacia. Bioadhesive formulations use polymers as the
adhesive component. These formulations are water soluble and when in a dry form attract water
from the biological surface and this water transfer leads to a strong interaction with mucous
membrane. In order to retain drug delivery, polymer can be transformed into insoluble form
those prolonging the release of active substance [42]. This may be performed using bivalent ions,
e.g. Zn, Ca etc, which cross-link the polymer and makes it insoluble [48].

21
3. Experimental part
3.1. Incorporation of microgranules with selegiline
hydrochloride into lyophilized tablets
3.1.1. Reagents, materials and equipment
Reagents
� Gelatin powder pure, POCh, Gliwice, Poland
� Pectin classic (type CU 701), Herbstreith & Fox, Neuenburg, Germany
� Selegiline hydrochloride (series 03/1-6), Dipharma, Milan, Italy
� Sodium citrate, POCh, Gliwice, Poland
� Sodium carboxymethylcellulose (type 7HXF), Hercules, Wilmington, USA
� Zinc sulfate × 7H2O, POCh, Gliwice, Poland
� Water purified through ion change and reversed osmosis (system Elix 3), Millipore,
Bedford, USA
Materials
� PCV blisters – gift from Polpharma, Starogard Gdanski, Poland
Equipment
� Freeze-dryer – Alpha 2-4, Christ, Osterode am Harz, Germany
� Vacuum-pump – type RZ 2, Vaccumbrand, Wertheim, Germany
� Magnetic stirrer – type MR 3001 K, Heidolph, Kelhaim, Germany
� Magnetic stirrer – type MS 11 HS, Wigo, Piastow, Poland
� Analytical balance – type WAX 62 RPT022146, Radwag, Radom, Poland
� Sieves 45, 100 and 200 µm, Rotsch, Germany
� Electronic balance – type WPS 600/C RPT 9553, Radwag, Radom, Poland
� pH-meter Orion, model 350, Orion Research, Boston, USA

22
- Microscope – type B1 223A (Motic, Welzlar, Germany), equipped with digital
camera – type GP-KR 222, Panasonic, Osaka, Japan
� HPLC set (Merck Hitachi, Darmstad, Germany):
- integrator – model D-2500A
- detector – UV-Vis – L-4250
- pump – type L-6200A
� Vibration mill – type KM1, Heinz – Janetzki, Germany
� Coffee grinder (type 651), Z. S. P. Niewiadow, Poland

23
3.1.2. Methods 3.1.2.1. Microgranules formation In order to obtain prolonged-release microgranules containing selegiline hydrochloride
(SCh) two different methods were applied. In the first method SCh was introduced in powdered
form (granulate type XPS1), while in the second one SCh was added to pectin in form of 20%
aqueous solution (granulate type XPS3). The exact composition of these granules is listed in
table 2.
Table 2. Ingredients utilized for preparing cross-linked microgranules containing SCh
Amount [g] Ingredient
XPS1 XPS3
Pectin 4.0 4.0
Purified water 13.3 3.3
Selegiline hydrochloride 2.0 (powder form) 10.0 (20% solution)
20% ZnSO4 solution 13.3 13.3
3.1.2.1.1.Preparation of microgranules type XPS1
The weighed amount of pectin was transferred to a mortar and 13.3 g of water was added
by continuous mixing. The mixture was left for 30 min for swelling of pectin. Then 2.0 g SCh
was added and mixed using a pestle. The pectin was cross-linked by dripping 20% ZnSO4
solution to the mixture of powders. During the cross-linking process, large granules were
formed, which were transferred to a dryer (35 ºC) for 12 h.
In order to obtain microgranules, the dried large granules were milled in a vibration mill
and sieved with 3 sieves set (200, 100 and 45 �m).
3.1.2.1.2.Preparation of microgranules type XPS3
Four grams of pectin was transferred to mortar and 3.3 g of water was added. The mixture
was left for 10 min. Next 10.0 g of 20% SCh solution were dripped to the wetted mass, and the
mass was left for additional 30 min. For cross-linking of pectin 13.3 g of 20% ZnSO4 solution
was added. Next steps were the same as described for XPS1 granules (the milling process was
performed by means of a coffee-grinder).
3.1.2.1.3. Coating of the microgranules (type XPS3P preparation)
In order to evaluate, how the additional coating influences the properties of microgranules,
the microgranules (100 – 200 µm) were coated in the mortar by adding dropwise 2% pectin

24
solution, while microgranules were stirred. For 1.0 g of microgranules 3.3 g of 2% pectin
solution were added. The coated microgranules were dried as described above.
3.1.2.2. Visual inspection of microgranules The shape, surface and colour of the microgranules was observed using optic microscope
equipped with digital camera. The images of microgranules were analyzed by means of Multi
Scan (version 12.07) program.
3.1.2.3. Evaluation of selegiline hydrochloride content in the cross-linked
microgranules Three samples (about 50 mg) of each microgranules batch were dissolved in 10 ml of 2%
sodium citrate solution using magnetic stirrer. The content of SCh in the solution was determined
by HPLC method. Before testing the solution was diluted 100-fold with HPLC mobile phase.
Composition of the phase and the analysis parameters are listed in section 3.1.2.8.
3.1.2.4. The release of selegiline hydrochloride from cross-linked
microgranules Three samples, approximately 50 mg of the cross-linked microgranules (corresponding to
about 10 mg of selegiline hydrochloride), were analyzed. The test was accomplished in water
bath shaker, using following conditions:
� medium: 20 ml of purified water
� temperature: 37ºC ± 1ºC;
� shaking amplitude: 10 (apparatus scale)
� shaking speed: 70 c.p.m.
After 1, 2, 4, 6, and 24 h a 0.5 ml sample of the medium was taken. After diluting the
samples 10-fold with HPLC mobile phase, the concentration of SCh was measured by HPLC
(the parameters are described in section3.1.2.8.).
3.1.2.5. Preparation of lyophilized tablets containing microgranules with
selegiline hydrochloride
For preparation of the freeze-dried tablets two different formulations of polymers as matrix
forming agents were used. Microgranules of type XPS1 were incorporated in the pectin matrix,
while microgranules of type XPS3 and XPS3P - in the Orabase®. Both solutions - 2% solution of

25
pectin and Orabase (2.0 g pectin, 2.0 g sodium caramellose and 2.0 g of gelatin in 94.0 g of
water) were prepared by dissolving polymers in warm (60 ºC) water and mixing with a magnetic
stirrer.
Lyophilization was performed after dosing of the solutions and microgranules to blisters by
two methods:
a) the blisters were filled with 0.35 g polymer solution, then microgranules were placed
on the polymer solution layer and the blisters were filled up with polymer solution to 0.70 g;
b) microgranules were suspended in the polymer solution and then the blisters were
filled up with 0.70 g of this suspension.
The composition of the mixture, transferred into blister cavity, was set that each
lyophilized tablet contained 10 mg of SCh.
The filled blisters were transferred to a freeze-dryer and the lyophilization process was
accomplished as shown in the table 3.
Table 3. Freeze-drying parameters
Stage Shelf temperature [ºC] Duration time [h] Chamber pressure [mbar]
Freezing - 45 2.5 -
Primary drying
- 20
- 5
+ 5
+ 20
2.5
14
3
2
0.08
0.08
0.08
0.08
Secondary drying + 30 1 - 2 0.08
3.1.2.6. Visual inspection of lyophilized tablets The surface, edges, porosity and mechanical properties of the tablets were evaluated. The
diameter was measured using a slide calliper. Photos of freeze-dried tablets were also made by a
digital camera.
3.1.2.7. Release test of selegiline hydrochloride from lyophilized tablets
The experiment was performed for three tablets from each batch (every tablet contains the
amount of granules, corresponding to about 10 mg of SCh). Each tablet was placed separately
into flasks containing 20.0 ml of water. The next steps were accomplished according to 2.4.
section.
26
3.1.2.8. Quantitative analysis of selegiline hydrochloride by HPLC
method The quantitative analysis of SCh was performed by reverse phase HPLC method using
mobile phase consisting of buffer and acetonitril 80:20 (v/v). The composition of the buffer was
as follows: 0.1 mol/l ammonium dihydrogenphosphate and 0.08 % triethylamine (pH of the
buffer was adjusted to 3.1 with phosphoric acid).
HPLC parameters:
� volume injected: 20 µl;
� flow rate: 1 ml/min;
� column: Lichrospher RP-18 (250 mm, 5 �m)
� analytical wavelength: 215 nm,
� retention time: about 10 min;
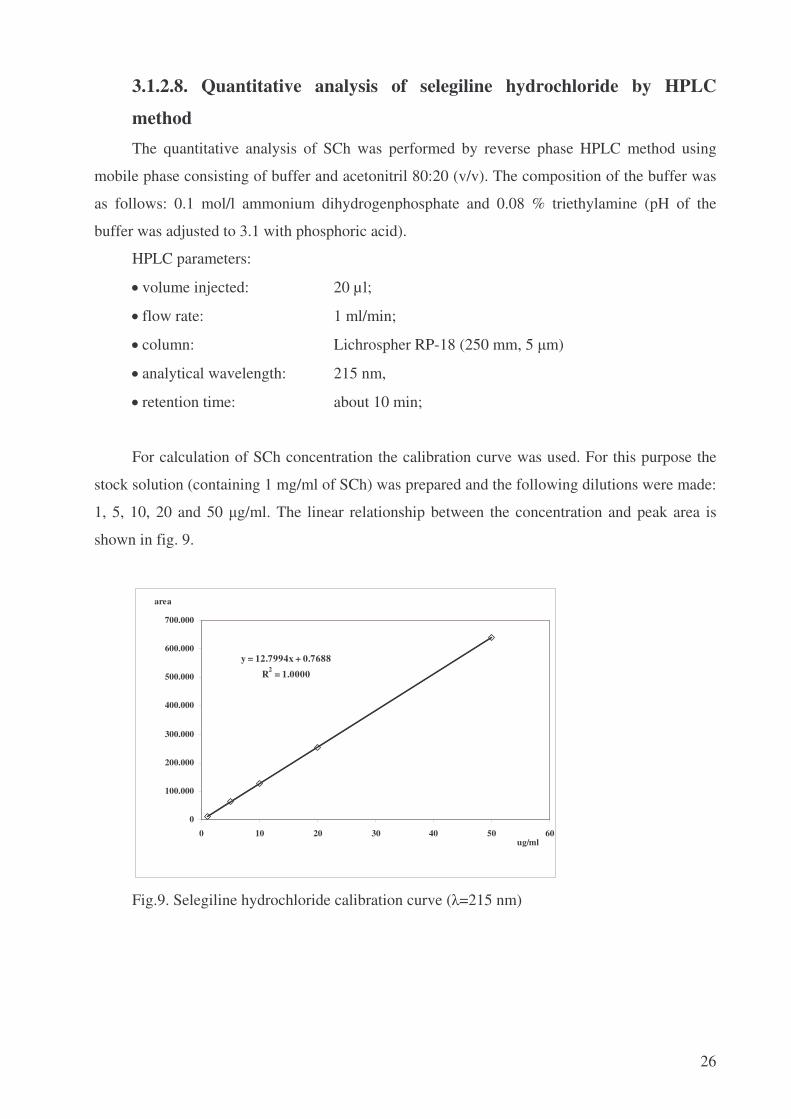
For calculation of SCh concentration the calibration curve was used. For this purpose the
stock solution (containing 1 mg/ml of SCh) was prepared and the following dilutions were made:
1, 5, 10, 20 and 50 �g/ml. The linear relationship between the concentration and peak area is
shown in fig. 9.
y = 12.7994x + 0.7688R2 = 1.0000
0
100.000
200.000
300.000
400.000
500.000
600.000
700.000
0 10 20 30 40 50 60ug/ml
area
Fig.9. Selegiline hydrochloride calibration curve (�=215 nm)

27
The concentration of SCh in the analyzed solutions C (µg/ml) was calculated using the
following equation:
C = (A – 0.7688) / 12.7994,
where:
A – peak area (in thousands).
The sample chromatogram obtained for a standard solution (50 µg/ml) is presented in fig.
10.
Fig. 10. Chromatogram of a standard selegiline hydrochloride solution (50 µg/ml).

28
3.2. Incorporation of floating pellets into pressed tablets –
tableting of floating pellets
3.2.1. Reagents, materials and equipment
Reagents
� Aqueous dispersion of methacrilic acid copolymer – Eudragit NE 40 D, Röhm, Pharma
Polymers - Darmstadt, Germany
� Calcium hydrophosphate · 2H2O, Merck – Darmstadt, Germany
� Calcium hydrophosphate anhydrous, Merck, Darmstadt, Germany
� Cross-linked polyvidon – Kollidon CL, BASF – Ludwigshafen, Germany
� Hydrochloric acid – 0.1 mol/l solution, P.O.Ch. – Gliwice, Poland
� Lactose and polyvidon (96.5:3.5) for direct tableting – Ludipress LCE, BASF –
Ludwigshafen, Germany
� Lactose for direct tableting – Tablettose 80, Meggle – Walsenburg, Germany
� Macrogol 6000 S, Fluka Chemie AG – Buchs, Switzerland
� Magnesium stearate, Riedel-de Hean, Seelze, Germany
� Mannitol, P.O.Ch. – Gliwice, Poland
� Microcrystalline cellulose and guar gum – Avicel CE-15, FMC Europe, Bruccels,
Belgium
� Microcrystalline cellulose, particle size 100 µm – Avicel PH 102, FMC Europe, Brussels,
Belgium
� Microcrystalline cellulose, particle size 50 µm – Avicel PH 101, FMC Europe, Brussels,
Belgium
� Modified starch for direct tableting – Starch 1500, Colorcon – Dartford, UK
� Monohydrated lactose and corn starch (85:15) for direct tableting – StarLac, BG
Excipients &Tech. – Walsenburg, Germany
� Povidone K-30, ICN Biomedicals – Aurora, Ohio, USA
� Powdered cellulose, particle size 200 µm – Viva Pur 200, Rettenmeier & Söhne –
Rosenberg, Germany
� Powdered cellulose, particle size 70 µm – Arbocel 290, Rettenmeier & Söhne –
Rosenberg, Germany
� Sodium hydrocarbonate, Farm Impex – Gliwice, Poland

29
� Sorbitol, BASF - Ludwigshafen, Germany
� Talc, Ph. Eur.
� Verapamil hydrochloride, Recordati – Mediolan, Italy
� Water purified through ion change and reversed osmosis
Equipment
� Analytical electronic balance WAX 62 RPT 0246, Radwag – Radom, Poland
� Apparatus for granulation and coating Uni-Glatt, Dresden, Switzerland � Apparatus for water purification and ion exchange, system Elix 3, Millipore, Bedford,
USA
� Automatic hardness tester TBH 20, Erweka, Hensenstamm, Germany
� Electronic balance WPS 600/C RPT 9553, Radwag – Radom, Poland
� Extruder model 25, Caleva – Dorset. UK
� Friabiliator TAP, Erweka – Frankfurt, Germany
� High shear mixer Cucina, Philips – Budapest, Hungary � Microscope – type B1 223A (Motic, Wetzlar, Germany), equipped with digital camera –
type GP-KR 222, Panasonic, Osaka, Japan
� Ph. Eur. paddle apparatus, Erweka DT 800, Frankfurt, Germany � Sieves 80, 100µm, 1.0 and 1.25 mm, Retsch, Germany
� Single - stroke tablet press machine EK0, Korsch – Berlin, Germany, integrated with
compression force measurement system UCT 5882/S, Spais – Gdansk, Poland
� Spectrophotometer UV VIS V 530, Jasco – Tokyo, Japan
� Spheronizator 120 MP, Caleva – Dorset. UK
� Tachometer, Caleva – Dorset, UK

30
3.2.2. Preparation of floating pellet cores with verapamil hydrochloride
by extrusion-spheronization method
Pellets were prepared by extrusion-spheronization method. The composition of pellet cores
is showed in table 4.
Table 4. The composition of pellet cores.
Substance Composition of granulated mass (g) Floating pellet cores
(dry mass)
Verapamil hydrochloride 20.5 20.0
Sodium hydrocarbonate 20.5 20.0
Avicel PH 101 46.4 45.3
Lactose 12.6 12.2
Povidone K-30 - 2.5
VH (verapamil hydrochloride) and excipients were dry mixed in a high shear mixer
(Philips H 7720/06, Budapest, Hungary) for 2-3 min. The mixture was granulated using a
granulation fluid (~50 g of 5% Polividon K-30 solution for 100 g of granulation mass) to achieve
the appropriate level of moisture content for extrusion and spheronization. The wet granulation
mixture was extruded through the sieve of 1.2 mm using the extruder model 25 (Caleva – Dorset,
UK). The rotation speed of extruder carried out 30 rpm.
About 20 g portions of the wet extrudate were immediately introduced into the
spheronizator’s chamber (Caleva - Dorset, UK). Spheronization process parameters were as
follows:
� the pressure of incoming air 2.0 bar;
� the speed of spheronizator disk 1500-1550 rpm;
� spheronization time 1.5 min.
Obtained pellets were dried in the dryer at 40ºC for 24 h. In order to achieve desired
narrow size distribution (1.00 – 1.25 mm) the dried pellets were then separated using 1.00 mm
and 1.25 mm sieves and stored in screw-capped, high-density polyethylene bottles.

31
3.2.3. Coating of pellet cores with Eudragit NE 40 D
Different methods and coating agents can be combined to achieve a specific release profile.
In this case pellets cores were coated with coating mixture based on Eudragit® NE 40 D. The
composition of coating mixture is showed in the table 5.
Coating mixture was prepared according to the following steps: the appropriate amount of
Eudragit NE 40 D was introduced to a beaker with a magnetic stirrer. Next, approximately 2 g
portions of talc were added during stirring process (before this procedure talc was sieved through
a sieve size 80 �m). At the same time Macrogol 6000 S was dissolved parallel in the water
earlier and added to the beaker. Eventually all mixture was stirred for 2 h. At last coating mixture
was perfused through the sieve size 200 �m.
Table 5. The composition of coating mixture.
Substance Content (%)
Eudragit NE 40 D 43.2
Talc (ø 80 �m) 6.9
Macrogol 6000 S 2.6
Purified water 48.6
Core coating was prepared by fluidized-bed/bottom-spray technique using Uni-Glatt
apparatus (Glatt Systemtechnik, Dresden, Germany). The process parameters were as follows:
� incoming air temperature 40 °C;
� outgoing air temperature 30 °C;
� incoming air pressure 6 bar,
� air pressure in spray nozzle 2 bar;
� peristaltic pump feeding rate 3 ml/min.
At once 200 g of pellet cores were given in for coating and also about 130 ml of coating
mixture were used. Coated pellets were dried in a blow-dryer at 40 °C for 24 h. It was prepared
four batches of floated pellets with VH, which differs from each other in film thickness, table 13.

32
3.2.4. Tableting of floating pellets with verapamil hydrochloride by using
impact tableting machine The composition of tableting masses, which were used for compression using the
laboratory single stroke Korsch tablet press (Korsch EK0, Berlin, Germany), is shown in tables 6
– 9. The masses were prepared in such way, that one 550 mg tablet contained 40 mg of VH. For
tableting 12 mm diameter round punches were used. The matrix was filled up manually. The
tableting mass was prepared for about 60 tablets in each batch.
Table 6. Tableting mass composition [%]; formulations I – IV.
Formulation Substance
I II III IV
Floating pellets with VH 38.2 38.2 38.2 38.2
Avicel PH 101 13.5 - - 51.3
Avicel PH 102 - 13.5 51.3 -
Mannitol 37.8 37.8 - -
Kollidon CL 9.5 9.5 9.5 9.5
Magnesium stearate 1.0 1.0 1.0 1.0
Table 7. Tableting mass composition [%]; formulations V – VIII.
Formulation Substance
V VI VII VIII
Floating pellets with VH 38.2 38.2 38.2 38.2
Avicel PH 102 13.5 13.5 - 13.5
Tablettose 80 37.8 - - -
Ludipress LCE - 37.8 - -
Arbocel 290 - - 51.3 -
D-Sorbitol - - - 37.8
Kollidon CL 9.5 9.5 9.5 9.5
Magnesium stearate 1.0 1.0 1.0 1.0

33
Table 8. Tableting mass composition [%]; formulations IX – XII.
Formulation Substance
IX X XI XII
Floating pellets with VH 38.2 38.2 38.2 38.2
Avicel PH 102 13.5 13.5 13.5 13.5
Polividon K-30 37.8 - - -
Star Lac - 37.8 - -
Starch 1500 - - 47.3 -
Calcium hydrophosphate
�2H2O
- - - 37.8
Kollidon CL 9.5 9.5 9.5 9.5
Magnesium stearate 1.0 1.0 1.0 1.0
Table 9. Tableting mass composition [%]; formulations XIII – XVI.
Formulation Substance
XIII XIV XV XVI
Floating pellets with VH 38.2 38.2 38.2 38.2
Avicel PH 102 13.5 13.5 13.5 13.5
Calcium hydrophosphate
anhydrous
37.8 - - -
Vivapur 200 - 37.8 - -
Avicel CE-15 - - 37.8 -
Macrogol 6000 S - - - 37.8
Kollidon CL 9.5 9.5 9.5 9.5
Magnesium stearate 1.0 1.0 1.0 1.0

34
3.2.5. Analysis of pellets and tablets
3.2.5.1. Measurement of film thickness
In order to determine the film thickness twenty randomly selected floating pellets were
coss-sectioned. Film thickness was measured using microscope (Motic, Wetzlar, Germany)
equipped with digital camera (Panasonic, Osaka, Japan), connected to PC. For survey of film
thickness software image analysis Multi Scan program v. 12.07 (Computer scanning systems,
Warsaw, Poland) was applied.
3.2.5.2. In vitro drug release test from pellets
Dissolution studies of VH (verapamil hydrochloride) pellets (equivalent to 40 mg VH)
were conducted using the Ph. Eur. paddle apparatus, Erweka DT 800 (Erweka, Frankfurt,
Germany). Test specifications were as follows: USP I paddle rotating at 75 rpm with 750 ml of
hydrochloric acid (0.1 mol/l) maintained at temperature of 37 ± 0.5 °C as dissolution medium
and accurate amount floating pellets corresponding to 120 mg VH. The 5 ml samples were
withdrawn at 1 h time intervals during a 6 h time period, and the volume was immediately
replaced with a fresh medium. The concentration of VH in the samples, after 5-fold dilution, was
analyzed spectrophotometrically (Jasco V-530, Jasco Corporation, Tokyo, Japan) at a
wavelength 278 nm.
3.2.5.3. Tablets appearance and size
Tablets were estimated visually especially paying attention on the surface and edge.
Tablets thickness and diameter was measured using a slide calliper with 0.1 mm precision.
3.2.5.4. Content of VH in tablets
10 randomly selected one series tablets were pulverized in mortar. By means of analytical
balances four accurate samples of 550 mg (what correspond tablet mass) were weighted. Every
sample was dissolved in 100 ml of 0.1 mol/l hydrochloric acid in volumetric flask. The flask
content was shaken throughout 6 h at 37°C ± 0.5°C and then incubated in the same temperature
during 18 h. The 5 ml samples were withdrawn using a pipette, which ends with a glass filter.
The concentration of VH in the samples, after 5-fold dilution, was analyzed
spectrophotometrically at a wavelength 278 nm.
The content of VH in each tablet should be comprised between 85 and 115% of average
content.

35
3.2.5.5. Determination of single tablet mass uniformity
Single tablet mass uniformity determination was performed according to Eur. Ph. 5.
3.2.5.6. Evaluation of tablets resistance to crushing (hardness of tablets)
Tablets resistance to crushing was determined by using automatic hardness tester type
TBH 20 (Erweka, Hensenstamm, Germany). Resistance to crushing was evaluated for 10
randomly selected tablets.
3.2.5.7. Evaluation of tablets friability
The tablets’ friability was performed according to Eur. Ph. 5.
3.2.5.8. In vitro drug release test from tablets and flotation start time of pellets
The test was performed according to p. 4.2. Six samples were examined; each of them
contained three randomly selected tablets. During in vitro test, the process of tablet disintegration
in appropriate beakers was observed. Consequently, by means of stop-watch, the time of pellets
flotation start was measured. As pellet flotation start time was considered the moment, in which
almost all pellet agglomerates undergo disintegration into individual floating pellets.

36
4. Results and discussion
Incorporation of microgranules with selegiline hydrochloride into
lyophilized tablets
Parkinson's disease is a degenerative disease of the nervous system which affects mainly
elderly people. The common manifestations involve trembling of the arms and legs, stiffness and
rigidity of the muscles and slowness of movement (bradykinesia). The disease is caused by the
progressive loss of brain cells (neurones) in a part of the brain - substantia nigra, which produces
the chemical dopamine. As the cells die, less dopamine is produced and transported to the
striatum, the area of the brain that co-ordinates movement [40].
Dopamine is the chemical in the brain known to be absent or reduced in Parkinson's
disease. Dopamine is broken down by a chemical called monoamine oxidase. Selegiline is a
selective irreversible monoamine oxidase (MAO) inhibitor, used alone or with levodopa in the
treatment of Parkinson's disease. Selegiline prevents monoamine oxidase from breaking down
dopamine, which results in an increased amount of active dopamine in the brain. In this way the
symptoms of mild Parkinson's disease can be reduced [32].
On the Polish pharmaceutical market only conventional tablets, containing 5 and 10 mg
selegiline, are present: Apo-Selin, Jumex, Segan, Selerin, Selgin, Parhinil and Selgres [34].
Considering Lithuania as a smaller country it also has enough preparations with selegiline to
fulfil its demands. These preparations comprise coated tablets Cognitive also conventional
tablets Jumex, Segan and Selegiline Alpharma Tablets [6].
Since selegiline has an extensive presystemic metabolism resulting in inadequate oral
absorption (absolute bioavailability is only about 10%), there is a need for developing a new
drug delivery systems, which provide higher drug concentration in the blood [18].
Pharmaceutical scientists throughout the world are trying to explore transdermal (e.g. selegiline
based transdermal system in USA) and transmucosal routes as an alternative to conventional oral
forms and injections. Among the various transmucosal sites available, mucosa of the buccal
cavity was found to be the most convenient and easily accessible site for the delivery of
therapeutic agents for both local and systemic delivery as retentive dosage forms, because it has
expanse of smooth muscle which is relatively immobile, abundant vascularization, rapid
recovery time after exposure to stress and the nearly absence of the Langerhans cells. Direct
access to the systemic circulation through the internal jugular vein bypasses drugs from the

37
hepatic first pass metabolism leading to high bioavailability. Further, these dosage forms are
self-administrable, cheap and have superior patient compliance [42].
In a year 1996 on pharmaceutical market an oral freeze-dried form of SCh (Selegiline
Zydis) was introduced. This tablet after placing in oral cavity rapidly disintegrates and do not
require water to aid swallowing. During the studies of this new formulation of selegiline, it was
ensured, that absorption of selegiline from “Zydis “, was more efficient and less variable than
from conventional selegiline tablets. The concentrations of selegiline in plasma after Zydis
Selegiline (1.25 mg) administration were similar to those following conventional selegiline
tablets 10 mg. Selegiline was shown to be rapidly and extensively absorbed pre-gastrically from
this form [7]. Because until this time there are no commercial buccal prolonged drug delivery
systems containing selegiline, the goal of the study was to investigate such drug form.
In order to realize the aim of the study, multiparticulate lyophilized tablets, a new buccal
delivery system based on cross-linked pectin was developed displaying a prolonged drug release
and mucoadhesive properties. In the study all the batches of microgranules (XPS1, XPS3 and
XPS3P) were prepared by cross-linking the pectin with Zn2+ ions. During this process the active
pharmaceutical ingredient (SCh) was incorporated in the cross-linked granules (SCh to pectin
ratio was 1:2). In this case namely cross-linking the pectin with Zn ions has the main influence in
gaining drug prolonged release properties. In order to evaluate, how the coating can change
microgranule properties, they were additionally coated with 2% pectin solution (XPS3P).
For achieving good drug distribution in microgranules, two methods of preparation were
applied: SCh was introduced to pectin in a powder form or as a solution. For achieving narrow
size microparticles, XPS1 microgranules were grinded using a vibration-mill. The process
endured 24 h. On purpose to shorten the procedure, other milling methods were searched.
Milling granules by means of a coffee grinder was tried and accepted as suitable. Microgranules
between 100 and 200 �m in size were used in the following studies. This middle fraction
comprised approximately 50 % of all grinded microgranules.
Despite of the two separate methods applied for microgranules XPS1 and XPS3, no
difference between the shapes of microparticles after milling was noted. Both types of
microgranules had rough surface, also were transparent and irregular in shape. They were
insoluble in water (fig. 11).

38
XPS1 XPS3 XPS3P
Fig. 11. The shape and size of three different types of microgranules with selegiline (type
XPS1, XPS3 and XPS3P) (bar=100 �m)
Microscopic analysis showed that microgranules coated with an excess of pectin were
smaller than XPS1 and XPS3. The reduction in particle size was caused likely by repeated
attrition of microgranules during the coating.
For determination of SCh content, microgranules were dissolved in 2% sodium citrate
solution because citrate anion binds Zn2+ and causes complete disintegration of the
microgranules. The determined SCh content for three analyzed samples is presented in table 10.
Table 10. Content of SCh in microgranules type: XPS1, XPS3, XPS3P (n=3)
Type [%] ± SD [mg] ± SD
XPS1 25.2 ± 0.4 12.7 ± 0.2
XPS3 24.8 ± 0.4 12.5 ± 0.1
XPS3P 22.5 ± 0.5 11.2 ± 0.3
Water was chosen as dissolution medium for drug release studies. After 1, 2, 4, 6 and 24 h,
0.5 ml samples of the dissolution medium were taken.
The results obtained for drug release studies from microgranules are presented in table 11
and figure 12.

39
Table 11. Released amount [%] of SCh from microgranules type: XPS1, XPS3 and XPS3P
(� ± SD, n=3)
Time [h] XPS1 XPS3 XPS3P
1 33.5 ± 6.4 36.5 ± 4.7 31.7 ± 6.5
2 65.7 ± 27.3 64.7 ± 7.7 48.1 ± 4.6
4 81.9 ± 4.5 98.1 ±0.7 80.2 ± 4.1
6 88.8 ± 8.0 97.2 ± 1.8 96.5 ± 3.8
24 - 93.7 ± 2.4 100.4 ± 1.4
The microgranules released SCh in an extended manner. During the first hour releasing of
SCh from all types of microgranulate was almost the same (about 30%). After 2 h a difference in
the release profile was observed: from XPS1 and XPS3 65% of the drug was released while from
XPS3P the release was slower (50%). Complete liberation of the drug occurred after 6 – 7 h
from all types of microgranules. As the aim of study was to develop dosage form for selegiline,
which enables prolonged drug release at least for 4 h, optimally for 4 – 6 h, the achieved release
rate is regarded as satisfactory.
0
20
40
60
80
100
120
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of S
Ch
[%]
XPS1
XPS3
XPS3P
Fig. 12. The release of SCh from microgranules type: XPS1, XPS3 and XPS3P.

40
Coating of microgranules with pectin solution had a favourable influence on the release of
active pharmaceutical ingredient during 4 h – the release was slower when comparing with
uncoated microgranules. However after 5 h the release from the coated microgranules was
almost the same comparing with microgranules type XPS3 and XPS1, respectively.
It was necessary to use polymers with proven mucoadhesive properties as a matrix for
lyophilized tablets. Within this study the following polymers were tested: pectin, sodium
caramellose, carbomer and Orabase (mixture of pectin, sodium CMC and gelatin). During
suspending the microparticles in the solutions of these polymers, an interaction between
microgranules and polymers was observed. Visible changes in viscosity of polymer solution
occurred probably because of the excess of zinc ions used for cross-linking process and
remaining in the microgranules. The main changes were observed in carbomer and sodium
CMC. In the both cases the polymer solutions liquefied and it was impossible to incorporate
microgranules. For this reason in further studies only pectin and Orabase solutions were used,
where this phenomenon was not observed.
Two methods of incorporation of microparticles in polymer solutions were proposed, as
described in the section 2.5. When in the first method microgranules were introduced between
two layers of the polymer solution, the obtained lyophilized tablets did not have smooth surface
at the edges and were even inclinable to lamination when being removed from the blister (fig.
13).
Fig. 13. Lyophilized tablets based on Orabase prepared by introducing microgranules as a
central layer.
Lyophilized tablets prepared by the second method (the microgranules were carefully
suspended in the solution of the matrix polymer) had uniform mass throughout the tablet surface
and also were more mechanically resistant – when being removed from blister tablets kept their

41
first appearance (fig. 14). Therefore the second method is more suitable for preparation of
lyophilized tablets with microgranules.
A) B)
Fig. 14. Lyophilized tablets containing cross-linked pectin microgranules based on (A)
Orabase, or (B) pectin solution.
The results of release studies from the lyophilized tablets containing SCh microgranules
are showed in table 12 and fig. 15.
Table 12. Released amount [%] of SCh from lyophilized tablets
Time [h] LT XPS1 LT XPS3 LT XPS3P
1 73.5 ± 4.2 63.2 ± 11.9 67.09 ± 2.1
2 92.8 ± 5.1 92.8 ± 1.0 89.66 ± 2.1
4 95.2 ± 2.1 101.2 ± 0.1 107.81 ± 10.6
6 90.5 ± 8.2 101.6 ± 2.4 111.92 ± 12.1
24 93.0 ± 3.5 104.2 ± 2.2 105.47 ± 7.4

42
0
20
40
60
80
100
120
140
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d a
mo
un
t o
f S
Ch
[%
]
LT XPS1
LT XPS3
LT XPS3P
Fig. 15. Release of SCh from lyophilized tablets containing SCh microgranules (type
XPS1, XPS3 and XPS3P)
The release test of SCh from lyophilized tablets showed unexpected results. It was
considered that lyophilized tablets would release the active ingredient with a slower rate than the
microgranules themselves, because microgranules were incorporated into a jellifying matrix.
However after 1 h of the test more than 60% of SCh was released. Almost complete drug
liberation appeared after 2 h. The faster release of SCh from lyophilized tablets than from
microgranules can be interpreted, that microgranules had a direct contact with a matrix medium
during preparation of the lyophilized tablets or during freezing of tablets the structure of
microgranulates changes. Therefore bulk of SCh is probably released into the matrix of tablets,
causing easier liberation of the drug into a dissolution medium.

43
Incorporation of floating pellets into pressed tablets – tableting of
floating pellets
Verapamil hydrochloride (VH) is a calcium ion influx inhibitor (slow channel blocker or
calcium ion antagonist), which reduces the intracellular concentration of free calcium ions in the
arterial smooth muscle as well as in conductive and contractile myocardial cells [31].
Consequently, excitation-contraction coupling and cardiac work is depressed. Also it causes
peripheral vasodilatation, which further reduces cardiac work load. Therefore this drug mainly is
used in the treatment of angina pectoris, cardiac dysrhythmias and hypertension [4].
On the Polish pharmaceutical market VH is available for oral administration in film-coated
tablets containing 40 mg, 80 mg or 120 mg of active pharmaceutical ingredient (API) (Isoptin,
Lekoptin, Novo-Veramil, Staveran); also there are sustained release tablets in doses of 120 and
240 mg of VH (Isoptin SR, Isoptin SR-E240, Lekoptin Retard, Staveran Prolongatum) and
injection solution, containing 5 mg of VH in 2 ml (Isoptin, Lekoptin) [34]. As for Lithuania, it
also has the same drug forms, just some commercial names differ, e.g. despite of above
mentioned names, Finoptin, Verapamil-Ratiopharm as film-coated tablets are present. Besides
Isoptin, there is Verogalid ER 240 mg as sustained release form. Additionally Verapamil
Alpharma in suppositories and conventional tablets steps out [6].
There is no VH in a multiple-unit dosage form – pellets in a tablet form. Namely
compression of pellets into tablets is much more ideal than enclosing them to hard gelatine
capsules [37]. Firstly, a higher dose of API could be administered with tablets. Secondly, this
drug form enables division of the tablet into appropriate parts. Further large volume tablets
generally have a higher patient compliance than capsules because they offer less difficulties in
oesophageal transport and more even distribution in the gastrointestinal tract, which is fairly
independent of nutritional state [15]. Also these drug delivery systems are less susceptible to
dose dumping than single unit formulations [17]. Tablets from pellets can be obtained by a
simpler method, at lower cost and with a higher production rate comparing to pellet-filled
capsules. Eventually, there is a reduced risk of tampering the original product [3].
Approximately 90% of the orally administered dose of verapamil hydrochloride is
absorbed. Because of rapid biotransformation of verapamil during its first pass through the portal
circulation, bioavailability ranges from 10% to 20% [25]. Improvement of VH bioavailability
comparing to conventional tablet Staveran 40 mg was achieved in Department of Pharmaceutical
Technology, Medical University of Gdansk: the multi-unit dosage form (capsules filled with
floating pellets) having modified drug release in stomach was prepared [38]. It was found that
VH is more than 6-fold higher soluble in 0.1 mol/l hydrochloric acid than in water. It is the key

44
argument for better VH absorption in the stomach. The better solubility of VH in acidic
environment of the stomach may result in a larger amount of the drug absorbed and its higher
concentration in plasma [36].
The reason of preparing floating pellets was to retain them in the stomach for a longer time
and thus improve drug’s bioavailability. This phenomenon was achieved by adding sodium
hydrocarbonate to pellets core. The product of sodium hydrocarbonate and hydrochloride acid
reaction is carbon dioxide, which results in bubble, adsorbing on the spherical core surface and
causing their flotation both in the dissolution medium in vitro and gastric juices in vivo [5].
First of all in realizing the aim of the study, pellet cores with VH, as constituents of multi-
unit dosage form, were prepared by extrusion-spheronization technique. Pellet cores were based
on microcrystalline cellulose (MCC) as a very widely used pharmaceutical excipient in oral solid
drug forms. The structure of MCC is essentially fibrous and highly porous and is able to absorb
large quantities of water. When under compression some of this moisture can be liberated and
become available to plasticise the wet mass and permit it to flow through the die. On the die exit
the pressure is released and the free solvent re-absorbed, returning the extrudate to its earlier
“stiffer” state [24]. Therefore MCC is suitable for preparation of wet mass with required
properties (not too wet and not too dry). The second significant factor in wet mass formulation is
the proper amount of granulation liquid. The most commonly used granulating liquid is water,
although in some cases use of alcohol or a water-alcohol mixture has also been reported [49].
Different amounts of water as a granulating liquid affects the hardness and particle size
distribution of final pellets [15]. Increased amount of an aqueous granulating liquid in pellets
formulation B comparing with formulation A, caused reduction in a pellet core size [41].
Therefore within this study formulation A as distributing a higher amount of proper pellet cores
(size 1.00 – 1.25 mm) was prepared.
Pellet cores were coated in fluidized-bed apparatus type Uni-Glatt. The main coating
mixture constituent was aqueous dispersion of Eudragit NE 40 D (table 5). It is film-forming
compound, often applied in the solid modified release drug forms This derivative of methacrilic
acid (polymethyl methacrylate) is water-insoluble over the entire pH range, but swells in
digestive fluids independently of pH. In the swollen state Eudragit NE is permeable to water and
dissolved actives. The Eudragit NE film is flexible and the plasticiser is not required.
Furthermore it is pharmacologically indifferent to the body tissues and fluids also highly stable
to environmental influences. All these properties make Eudragit NE suitable for controlled
release dosage forms [1]. Macrogol 6000 S served as porophor – the substance which after
dissolving gradually increases film permeability. Addition of talc helped to avoid agglomeration

45
of pellets while coating or drying. Individual series of pellets differed in film thickness (table
13), what had an influence in VH release rate (table 14, 15).
Table 13. Thickness of the coating film in pellets (� ± SD, n=50), measured with
microscope
Series Thickness [�m])
I 38.6 ± 3.52
II 35.6 ± 3.84
III 39.9 ± 2.83
IV 41.2 ± 1.84
Mixture I –IV 38.8 ± 2.39
Table 14. Release [%] of verapamil hydrochloride from floating pellets individual batches
(� ± SD, n=6)
Series Time [h]
I II III IV
1 11.27 ± 2.47 15.16 ± 1.48 3.07 ± 0.36 0.17 ± 0.06
2 33.89 ± 2.08 59.82 ± 1.37 17.11 ± 1.34 3.46 ± 0.39
3 58.26 ± 1.41 86.11 ± 1.10 44.27 ± 2.87 12.72 ± 1.63
4 75.27 ± 0.92 91.63 ± 0.60 70.10 ± 4.42 21.24 ± 1.40
5 82.80 ± 1.35 92.19 ± 0.77 83.90 ± 3.91 36.01 ± 2.36
6 85.98 ± 2.06 85.23 ± 8.48 50.38 ± 1.80
Table 15. Release [%] of verapamil hydrochloride from floated pellet mixtures A and B
(� ± SD, n=6)
Mixture Time [h]
A B
1 20.73 ± 1.93 21.92 ± 1.72
2 32.27 ± 4.00 29.02 ± 2.79
3 54.22 ± 2.59 44.00 ± 3.60
4 70.71 ± 2.32 61.88 ± 3.71
5 80.62 ± 1.91 75.24 ± 2.48
6 85.95 ± 2.43 81.69 ± 2.11

46
For compression of tablets by means of single-stroke Korsch tablet press machine, sixteen
tableting masses, based on various filling substances, were prepared (tables 6 – 9). As filling
substances Avicel PH 101, Avicel PH 102, mannitol, Tablettose 80, Ludipress LCE, Arbocel P
290, D-sorbitol, PVP K-30, Star Lac, Starch 1500, calcium hydrophosphate � 2H2O, calcium
hydrophosphate anhydrous, Vivapur 200, Avicel CE-15 and Macrogol 6000 S were applied.
Magnesium stearate was used as lubricant. Kollidon CL possessing swelling properties,
accelerated tablets disintegration into floating pellets. Formulation XI was prepared without
Kollidon CL because in this case used filling substance Starch 1500 also has swelling properties.
Individual formulations differed in tableting mass properties subject to applied filling
substances. Formulations I, II, VIII, IX, X, XV, XVI had characterized uniformity. In these
mixtures separation of pellets from powders was not observed. Pellets were apparently and
equally surrounded by powders. Whereas residual tableting masses had obvious separation of
pellets on the bottom of container.
For tableting, mixture of coated and uncoated pellets was used, that is obviously seen in
achieved tablets cross-sections, fig. 16.
Fig. 16. Cross – section of tablet: uncoated and coated pellets (bar =100 µm)
In order to examine the influence of different compression force on tablets properties, all
sixteen formulations were pressed with 12 and 18 kN compression force.
All the tablets had distinctive white colour with visible yellowish pellets both on the
surface and in cross-sections, fig. 17.
uncoated pellet
polymer film coated pellet

47
A) B)
Fig. 17. Tablets with pellets: A) general view; B) cross-section (bar =100 µm)
The main physical properties of tablets are presented in tables 16 and 17. Tablets’
thickness was approximately 5.0 mm and diameter 12 mm – as used punches. Every tablet
constituted approximately 40 mg of VH as intended before this study. The uniformity and
friability of tablets fulfilled the requirements according to Eur. Ph. 5.

48
Table 16. Physical properties of tablets with verapamil hydrochloride floating pellets,
prepared by using impact tableting machine with the compression force of 12 kN.
Formulation The content of VH
[mg] ± SD; (n=10)
Tablet mass [mg] ±
SD; (n=20)
Hardness [N] ± SD;
(n=10)
Tablet thickness
[mm] ± SD; (n=20)
I 40.6 ± 1.2 551.7 ± 16.9 91.4 ± 3.9 5.1 ± 0.12
II 39.7 ± 1.5 553.8 ± 14.5 78.9 ± 5.4 5.1 ± 0.11
III 44.0 ± 0.7 546.6 ± 17.5 248.4 ± 12.8 5.0 ± 0.13
IV 44.5 ± 1.8 541.3 ± 12.5 229 ± 18.0 4.9 ± 0.14
V 41.2 ± 1.9 554.8 ± 10.1 72.1 ± 9.7 5.1 ± 0.10
VI 40.6 ± 4.3 557.9 ± 15.3 111.9 ± 13.8 5.1 ± 0.09
VII 43.4 ± 3.7 552.1 ± 14.6 155.9 ± 25.5 5.1 ± 0.09
VIII 43.3 ± 0.5 545.3 ± 13.4 259.9 ± 15.2 5.0 ± 0.1
IX 38.5 ± 1.7 555.6 ± 12.1 66.9 ± 9.6 5.5 ± 0.09
X 39.5 ± 1.5 554.6 ± 9.4 91.8 ± 6.8 5.1 ± 0.06
XI 40.7 ± 1.3 550.5 ± 14.6 35.7 ± 3.9 5.1 ± 0.08
XII 39.7 ± 2.4 550.4 ± 13.4 68.9 ± 6.8 4.7 ± 0.07
XIII 38.2 ± 1.5 552.9 ± 7.8 78.9 ± 8.0 4.5 ± 0.07
XIV 39.5 ± 2.3 558.1 ± 11.9 159.7 ± 8.0 5.1 ± 0.09
XV 37.2 ± 2.5 553.1 ± 12.7 105.8 ± 9.7 5.1 ± 0.09
XVI 38.7 ± 1.9 543.8 ± 17.1 182.0 ± 5.3 5.1 ± 0.12

49
Table 17. Physical properties of tablets with VH floating pellets, prepared by using impact
tableting machine with the compression force of 18 kN.
Formulation The content of VH
[mg] ± SD; (n=4)
Tablet mass [mg] ±
SD; (n=20)
Hardness [N] ±
SD; (n=10)
Tablet thickness
[mm] ± SD;
(n=20)
I 35.0 ± 1.6 552.2 ± 15.4 80.1 ± 1.7 4.8 ± 0.08
II 34.6 ± 3.8 549.0 ± 13.1 86.75 ± 5.3 4.8 ± 0.04
III 39.7 ± 4.3 547.8 ± 8.3 272.9 ± 18.8 4.8 ± 0.07
IV 39.5 ± 1.5 552.7 ± 13.2 244.7 ± 7.1 4.7 ± 0.05
V 36.6 ± 2.1 552.5 ± 6.3 77.1 ± 3.4 4.9 ± 0.05
VI 38.6 ± 2.3 546.8 ± 10.3 131 ± 9.8 4.8 ± 0.06
VII 38.9 ± 1.2 551.2 ± 15.3 144.6 ± 7.8 4.9 ± 0.05
VIII 39.8 ± 3.2 549.0 ± 10.0 282.1 ± 3.7 4.8 ± 0.03
IX 37.5 ± 4.8 552.8 ± 11.9 52.1 ±1.8 5.3 ± 0.02
X 41.4 ± 2.1 558.0 ± 10.7 120.1 ± 7.5 4.9 ± 0.02
XI 44.0 ± 1.5 553.9 ± 11.3 42.4 ± 5.9 5.0 ± 0.02
XII 42.9 ± 1.9 555.1 ± 12.4 89.1 ± 6.9 4.6 ± 0.07
XIII 44.8 ± 1.7 550.6 ± 17.3 107.0 ± 5.2 4.4 ± 0.04
XIV 38.8 ± 3.7 552.6 ± 10.8 264.3 ± 6.6 4.8 ± 0.04
XV 40.2 ± 2.0 546.1 ± 13.8 141.0 ± 6.9 4.9 ± 0.05
XVI 41.4 ± 2.1 552.4 ± 9.5 194.6 ± 8.7 4.9 ± 0.05
It was noticed that bigger part of formulations (I, II, VIII, IX, X, XI, XII, XIII), pressed
with 18 kN strength, had lower friability comparing to tablets, compressed with 12 kN strength
(table 18).

50
Table 18. Tablets friability [%]
Tableting compression force Formulation
12 kN 18 kN
I 0.38 0.29
II 0.41 0.3
III 0.11 0.00
IV 0.04 0.09
V 0.31 0.34
VI 0.13 0.15
VII 0.15 0.24
VIII 0.18 0.14
IX 0.58 0.43
X 0.24 0.23
XI 1.57 0.11
XII 0.42 0.27
XIII 0.38 0.34
XIV 0.09 0.06
XV 0.18 0.24
XVI 0.23 0.27
For comparison of physical tablet properties, their resistance to crushing was tested, (tables
15, 16). Formulation VIII, prepared on the base of D-sorbitol compressed both with 12 and 18
kN strength, was the hardest (259.9 N and 282.1 N respectively). Formulation III and
formulation IV, based on Avicel PH 102 and Avicel PH 101, respectively (compression force 12
kN) as well as III and XIV tablets formulations, based on Avicel PH 102 and Vivapur 200,
respectively (compression force 18 kN) also possessed relatively high hardness. Pressing of VII
formulation required markedly higher pressure strength. Formulation XI, prepared on the base of
Starch 1500 and compressed both with 12 and 18 kN strength, possessed the lowest hardness
(35.7 N and 42.4 N respectively). Tablets of this formulation had insufficient mechanical
resistance because during preservation a part of them underwent crumbling. It is evident, that
tablets, pressed with higher strength were more mechanically resistant to crushing .
The main criterion for evaluation individual tablets formulations was release test. Results
are presented in fig. 18, 19.

51
0102030405060708090
100
0 1 2 3 4 5 6 7Time [h]
Rel
ease
d a
mou
nt o
f VH
[%]
I
II
III
Pellets
0102030405060708090
100
0 1 2 3 4 5 6 7Time [h]
Rel
ease
d am
ount
of V
H [%
]
IV
VVI
Pellets
01020304050
60708090
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
oun
t of V
H [%
]
VII
VIII
IXPellets
0102030405060708090
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]X
XI
XII
Pellets
0102030405060708090
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]
XIII
XIV
Pellets
0102030405060708090
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]
XV
XVI
Pellets
Fig. 18. In vitro verapamil hydrochloride release from uncompressed pellets with
comparison to tablets formulations I – XVI. Compression force 12 kN (Pellets=uncompressed
pellets)
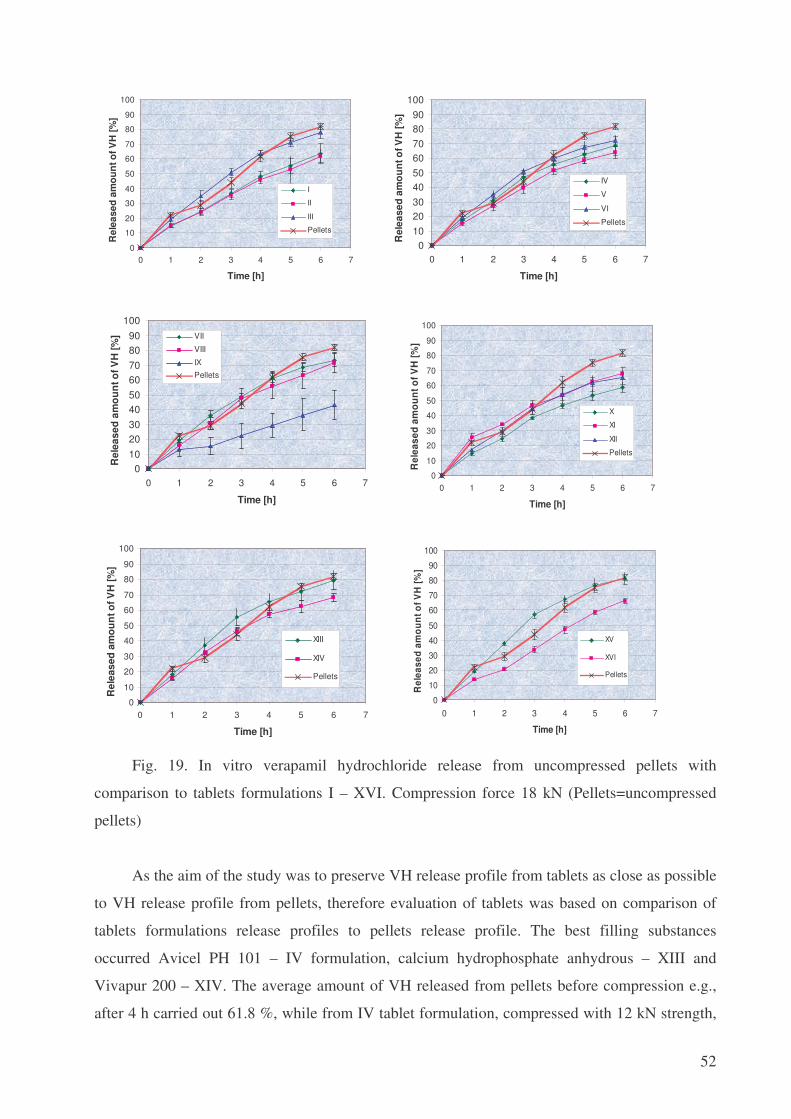
52
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d a
mou
nt o
f VH
[%]
I
II
III
Pellets
0102030405060708090
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]
IV
V
VI
Pellets
0102030405060708090
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
] VII
VIII
IXPellets
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]X
XI
XII
Pellets
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]
XIII
XIV
Pellets
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5 6 7
Time [h]
Rel
ease
d am
ount
of V
H [%
]
XV
XVI
Pellets
Fig. 19. In vitro verapamil hydrochloride release from uncompressed pellets with
comparison to tablets formulations I – XVI. Compression force 18 kN (Pellets=uncompressed
pellets)
As the aim of the study was to preserve VH release profile from tablets as close as possible
to VH release profile from pellets, therefore evaluation of tablets was based on comparison of
tablets formulations release profiles to pellets release profile. The best filling substances
occurred Avicel PH 101 – IV formulation, calcium hydrophosphate anhydrous – XIII and
Vivapur 200 – XIV. The average amount of VH released from pellets before compression e.g.,
after 4 h carried out 61.8 %, while from IV tablet formulation, compressed with 12 kN strength,

53
59.4 %. After compression of this formulation with 18 kN strength, release of VH was slower –
55.8 %. Worthy to say, that release of VH after compression the other formulations with 18 kN
strength, increased; just from I, II, IV and IX formulation the release of VH was slower.
Formulation XIII and XIV, compressed with 12 kN strength, also had close release profiles to
pellets – 56.5 % and 55.5 % respectively. After compression of these tableting masses with 18
kN strength, release profile was even more close to the goal than after compression of them with
a lesser strength (65.5 % and 57.3 %).
The biggest difference in release of VH towards pellets had IX formulation, based on
Povidone K-30. This formulation released the active substance the slowest manner: after 4 h
released amount of VH carried out 30.09 % (compression force of 12 kN) and 29.1 %
(compression force 18 kN). Also tablets of IX formulation possessed the longest disintegration
and floatation start time (about 30 min.) while the other formulations disintegrated and started to
float within a few minutes (1 – 9 min.). Such results can be due to good binding properties of
Povidone K-30, that can cause retention in VH release.
Among applied filling substances, there were modified pharmaceutical excipients,
definitely designed for direct compression, e.g. in V, VI, X, XI formulations. However release
profiles from these formulations were unsatisfactory, fig.18 and 19.
In previous studies on tableting the pellets [41] was stated that pressing the tablets with
even with small compression force (6 kN) exclusively by upper punch, followed to pellet core
deformation or even disruption what caused increased VH release rate. This study results showed
that it is possible to obtain tablets with very close release profile to pellets not only by means of
rotary tableting machine, but also with single stroke tablet press machine, applying higher
compression strength (12 kN and 18 kN). Retention in the release time could be interpreted by
stronger compaction of the film to pellet core. For this reason diffusion time of acceptory liquid
elongates, therefore decreases VH release rate.
Coating film elasticity and resistance properties could also influence VH dosing rate from
compressed pellets. Diffusion rate through film depends on the kind of polymer and plasticizer
amount [8].
In the studies of tableting of pellets with propranolol hydrochloride (PH) coated with
ethylocellulose with adding 25 % triethylcitrate (TC) as plasticizer (Dashevsky et al. [8]), was
stated, that compression even with 5 kN strength causes film deformation and increase of PH
release rate. While coating the same pellets with Kollicoat SR 30 D with 2.5-fold lower amount
of TC, ensured gaining very close release profile to pellets before compression, what proves, that
the polymer film was not damaged.

54
5. Conclusions
1. Cross-linking the mixture of pectin and selegiline hydrochloride with zinc ions allowed
preparation of microgranules displaying prolonged release profile of the drug.
2. Additional coating of microgranules with pectin causes slower release of SCh.
3. Carbomer and sodium CMC solutions were unsuitable for preparation of lyophilized tablets
with SCh microgranules because an interaction between these polymers and uncoated
microgranules occurred.
4. When coated SCh microgranules were incorporated in the matrix of lyophilized tablets based
on pectin or Orabase, the release of the drug was much faster comparing with the free
microgranules. For understanding this phenomenon, further studies are required.
5. Extrusion- spheronization is a suitable method for preparing floating pellets with verapamil
hydrochloride.
6. Coating of pellet cores with Eudragit NE 40 D prolonged the release of active substance.
7. Tableting of floating pellets with verapamil hydrochloride by means of single-stroke tablet
press machine with 12 kN and 18 kN compression force allowed to achieve release profiles
similar to those from pellets before compression. The closest release profiles were noted
from IV, XIII and XIV formulations.
8. Some tablet formulations possessed slower release profiles comparing to pellets and this was
due to prolonged disintegration of tablets into individual pellets (e.g. formulation IX, based
on Povidone K-30).
9. Together with a growth of compression force from 12 to 18 kN increase of tablet hardness
and decrease of friability was observed.

55
6. Acknowledgments
I am very grateful Mr. Marcin Płaczek and Mr. Rafał Łunio at Department of
Pharmaceutical Technology, Medical University of Gda�sk for their indispensable and excellent
technical advice, valuable suggestions, attention and support during the experiments. Also prof.
hab. dr. Małgorzata Sznitowska and Dr. Wiesław Sawicki (Department of Pharmaceutical
Technology, Medical University of Gda�sk) are gratefully acknowledged for critical reading of
manuscript.

56
7. References
1. Bajdik J., Pintye– Modi K., Regdon Jr. G., Fezkaz P., Szavo – Revesz P.: The effect of
storage on the behaviour of Eudragit NE free film, J. Therm. Anal. Calor., 2003;73:607–
613
2. Benoit et al., Biodegradable microspheres in: Benita S.: Microencapsulation, Methods
and industrial applications, Hon Kong; 1996.p.36-53
3. Bodmeier R.: Tableting of coated pellets, Eur. J. Biopharm., 1997;43:1-8
4. Bowman W. C., Rand M. J.: Textbook of pharmacology, Oxford; 1980.p.22.70
5. Choi B.Y., Park H. J., Hwang S. J., Park J. B.: Preparation of alginate beads for floating
drug delivery: effects of CO2 gas formation, Int. J. Pharm. 2002;239:81
6. �iburien� E., Griškevi�ius L., Gulbinovi� J. et al.: Vaist� žinynas, Vaist� žinios, Vilnius,
2006;105 (SCh) and 153, 154 (VH)
7. Clarke A., Brewer F., Johnson E. S., Mallard N., Hartig F., Taylor S. and Corn T. H.: A
new formulation of selegiline: improved bioavailability and selectivity for MAO-B
inhibition, J. Neural Transm., 2003;110:1241–1255
8. Dashevsky A., Kolter K., Bodmeier R.: Compression of pellets coated with various
aqueous polymer dispersions, Int. J. Pharm., 2004; 79:19–26
9. Deasy P. B.: Microencapsulation and related drug processes”, printed in the USA;
1984.p.64-69
10. Dobetti L.: Fast-melting tablets: developments and technologies, Pharm. Techn. Eur.,
2000;12:32-42
11. Domb et al.: Lipospheres for controlled delivery of substances in: Benita S.:
Microencapsulation, Methods and industrial applications, Hon Kong; 1996.p.378-379
12. Dominguez-Gil, A.: Contribution of biopharmaceutics and pharmacokinetics to improve
drug therapy, Eur. J. Drug Metab. Pharmac., 1993;18:1-5
13. Fiebig A., Wójcik P.: Suszenie In: Janicki S., Fiebig A., Sznitowska M. (red.): Farmacja
Stosowana, podrecznik dla studentów farmacji, Wydawnictwo Lekarskie PZWL, wyd.
IV, Warszawa; 2002.p.56 – 58
14. Fu Y. J., Shyu S. S., Su F. H., Yu P. C.: Development of biodegradable co-poly
microspheres for the controlled release of 5-FU by the spray drying method, Coll. Surf.,
2002;25:269-279
15. Gandhi R., Kaul C.L.: Panchagnula R.: Extrusion and spheronization in the development
oral controlled-release dosage forms, PSTT, 1999;4:160-170

57
16. Ghebre – Sellassie I.: Multiparticulate oral drug delivery, Marcel Dekker Inc., New York;
1994.p.1-4
17. Ghebre – Sellassie I.: Pharmaceutical Pelletization Technology, Marcel Dekker Inc., New
York; 1989.p.1–13
18. Harbele D., Szoko E., Magyar K.: The influence of metabolism on the MAO-B inhibitory
potency of selegiline, Curr. Med. Chem., 2002;9:47-51
19. Heilman, K.: Therapeutic systems, Georg Thieme, Shtuttgart; 1983.p.1-33
20. Hong K., Park S.: Melamine resin microcapsules containing fragrant oil: synthesis and
characterization, Mater. Chem. Phys., 1999;58:130
21. Hu L. D., Liu Y., Tang X., Zhang Q.: Preparation and in vitro/in vivo evaluation of
sustained-release metformin hydrochloride pellets, Eur. J. Pharm. Biopharm.,
2006;64:185–192
22. Janicki S., Granulaty w: JanickS. i, Fiebig A., Sznitowska M. (red.): Farmacja
Stosowana, podrecznik dla studentów farmacji, Wydawnictwo Lekarskie PZWL, wyd.
IV, Warszawa; 2002.p.165–171
23. Janicki S., Kapsułki in: Janicki S., Fiebig A., Sznitowska M. (red.): Farmacja
Stosowana, podrecznik dla studentów farmacji, Wydawnictwo Lekarskie PZWL, wyd.
IV, Warszawa; 2002.p.222–225
24. Kibbe A. H.: Handbook of Pharmaceutical Excipients, London; 2000.p.102
25. Kirsten R., Nelson K., Kirsten D., Heintz B.: Clinical pharmacokinetics of vasodilatators,
Clin. Pharmacokinet. 1998;34:457
26. Kraömer, J., Blume H.: Biopharmaceutical aspects of multiparticulates, In: Ghebre-
Selassie, I. (Ed.), Multiparticulate Oral Drug Delivery. Marcel Dekker, New York;
1994.p.307–332
27. Lehmann K. (ed.): Practical course in film coating of pharmaceutical dosage forms with
Eudragit, Dremstadt; 1999.p.125 - 128
28. Magdassi, Vinetsky: Microencapsulation of oil in water emulsions in: Benita S. :
Microencapsulation, Methods and industrial applications, Hon Kong; 1996.p.22-25
29. McGinity JW., O’Donnell PB.: Preparation of microspheres by the solvent evaporation
technique, Adv. Drug Deliv. Rev., 1997;1:25-42
30. Muskó Zs., Pintye-Hódi K., Gáspár R., Pintye J., Szabó-Révész, Ers I., Falkay G.: Study
of in vitro and vivo dissolution of theophylline from film coated pellets, Eur. J. Pharm.
Biopharm., 2001;51:143-146
31. Mutschler E., Derendorf H.: Drug actions, scientific publishers, Stuttgart; 1995.p.374

58
32. Mycek M. J., Harvey R. A., Champe P. C.: Lippincott’s illustrated reviews:
Pharmacology, Lippincitt Williams & Willkins, 2nd edition; 2000.p.81-88
33. Nurnberg E., Wunderlich J.: Manufacturing pellets by extrusion-spheronization, Pharm.
Technol. Eur., 1998;11:41 - 47
34. Piwowarczyk K. et al.: Pharmindex. Kompendium leków, CMP Medica Poland;
2005.p.41, 680 (SCh) and 24, 538, 539 (VH)
35. Santus G., Bottoni G., Golzi R.: Process for the preparation of microgranules suitable for
suspension in fluids, United States; 1995, patent Nr.: 5460828
36. Sawicki W., Janicki S., Pietkiewicz P.: A method of obtaining floating tablets with VH,
Farm. Pol. 1997;53:698
37. Sawicki W., Łunio R., Walentynowicz O., Kubasik-Juraniec J.: Influence of the type of
cellulose on properties of multi-unit target releasing in stomach dosage form with
verapamil hydrochloride, Acta Pol. Pharm. – drug research; 2007;64:81–88
38. Sawicki W.: Pharmacokinetics of verapamil and norverapamil from controlled release
floating pellets in humans, Eur. J. Pharm. Biopharm., 2002;59:29–35
39. Schmidt Ch., Bodmeier R.: A multiparticulate drug-delivery system based on pellets
incorporated into congealable polyethylene glycol carrier materials, Int. J. Pharm.,
2001;216: 9-16
40. Shiel W.C. (reviewing ed.).: Parkinson’s disease, available at
http://www.medicinenet.com/parkinsons_disease/article.htm
41. Skoczen P.: Evaluation of tableting of floating pellets coated with Eudragit NE, master
work, MUG, Gdansk; 2004
42. Sudhakar Y., Kuotsu K., Bandyopadhyay A.K.: Buccal bioadhesive drug delivery – a
promising option for orally less efficient drugs, J. Contr. Release , 2006;114:15–40
43. Sznitowska M., Płaczek M.: Lyophilized and other fast melting oral tablets, Ordynator
Leków, 2004;4:33-38
44. Tang X. C., Pikal M. J.: Design of freeze-drying process for pharmaceuticals: practical
advice, Pharm. Research, 2004;21:191-200
45. Thies C.: A survey of microencapsulaton processes in: Benita S.: Microencapsulation,
Methods and industrial applications, Hon Kong; 1996.p.1-17
46. Tosi A., Mazzitelli S., Bozzuto N., Bertini B., Luca G., Nastruzzi C.: Tripalmitin based
cationic lipospheres: preparation, characterization and in lab-on-a-chip applications,
Ninth European Symposium on controlled drug delivery, 2006:165 - 168

59
47. Tunon, E. Borjesson, G. Frenning, Alderborn G.: Drug release from reservoir pellets
compacted with some excipients of different physical properties, Eur. J. Pharm. Sci.,
2003;20:469–479
48. Vandamme Th. F., Lenourry A., Charrueau C., Chaumeil J-C.: The use of
polysaccharides to target drugs to the colon, Carbohydr. Polym., 2002;48: 219-231
49. Vervaet C., Baert L., Remon J. P.: Extrusion-Spheronisation, A literature review,
1995;116:131-146
50. Wørts O.: Wet granulation – fluidized bed and high shear techniques compared, Pharm.
Tech. Europe, 2000;10:27 - 30
51. Zhou F., Vervaet C., Schelkens M., Lefebvre R., Remon J.P.: Bioavailibility of ibuprofen
from matrix pellets based on the combination of waxes and starch derivatives, Int. J.
Pharm., 1998;168:79-84


















