
Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons
11
Journal of Physiology Abdominal pain is a major cause of morbidity for patients who suffer from intestinal disorders, such as inflammatory bowel disease (IBD) and irritable bowel syndrome. Antecedent infection or inflammation has been implicated in the visceral hyperalgesia associated with the irritable bowel syndrome (IBS). The cell bodies of sensory neurons which initiate these sensations originate in the dorsal root ganglia (DRG) (Mayer & Gebhart, 1994). Sub-populations of DRG neurons respond to innocuous and noxious mechanical stimulation, as well as chemical and thermal stimulation, and thus are thought to be polymodal sensory neurons (Mayer & Gebhart, 1994). These polymodal neurons play a central role in responding to conditions that are potentially injurious to tissues. Inflammatory mediators cause a reduction in the threshold and an increase in the gain of the transduction process of these neurons in a process referred to as ‘peripheral sensitization’. Also, under certain pathological conditions, there is evidence that an additional population of normally mechanically insensitive nociceptive fibres can be recruited (Gebhart, 2000). Following inflammatory stimulation these ‘silent afferent’ fibres become active at rest and begin respond to mechanical stimulation. Together, these afferent pathways have been suggested to contribute to disproportionate pain states in response to injury and may persist even after the inflammatory state has resolved (Al Chaer et al. 2000; Collins et al. 2001). In studies in the somatic nervous system, peripheral sensitization appears to involve both acute and chronic mechanisms (Woolf & Costigan, 1999). Acute mechanisms of increased nociceptive stimulation include both direct depolarization of nerve terminals by neuroactive agents and alterations in ionic currents in the membrane terminals (Rang et al. 1991). Also, later and longer lasting transcription-dependent changes occur and appear to be evoked by either signalling molecules, such as nerve growth factor and/or activity-dependent second messenger cascades (McCleskey & Gold, 1999). These transcription-dependent events can result in altered expression of voltage-gated ion channels, such as increased expression of TTX-resistant sodium channels, and increased TTX-resistant currents (TTX-R I Na ) (Khasar et Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons Timothy Stewart*, Michael J. Beyak* and Stephen Vanner Gastrointestinal Diseases Research Unit, Queen’s University, Kingston, Ontario, Canada Intestinal inflammation induces hyperexcitability of dorsal root ganglia sensory neurons, which has been implicated in increased pain sensation. This study examined whether alteration of sodium (Na + ) and/ or potassium (K + ) currents underlies this hyperexcitability. Ileitis was induced in guinea pig ileum with trinitrobenzene sulphonic acid (TBNS) and dorsal root ganglion neurons innervating the site of inflammation were identified by Fast Blue or DiI fluorescence labelling. Whole cell recordings were made from acutely dissociated small-sized neurons at 7–10 days. Neurons exhibited transient A-type and sustained outward rectifier K + currents. Compared to control, both A-type and sustained K + current densities were significantly reduced (42 and 34 %, respectively; P < 0.05) in labelled neurons from the inflamed intestine but not in non-labelled neurons. A-type current voltage dependence of inactivation was negatively shifted in labelled inflamed intestine neurons. Neurons also exhibited tetrodotoxin-sensitive and resistant Na + currents. Tetrodotoxin-resistant sodium currents were increased by 37 % in labelled neurons from the inflamed intestine compared to control (P < 0.01), whereas unlabelled neurons were unaffected. The activation and inactivation curves of these currents were unchanged by inflammation. These data suggest ileitis increases excitability of intestinal sensory neurons by modulating multiple ionic channels. The lack of effect in non-labelled neurons suggests signalling originated at the nerve terminal rather than through circulating mediators and, given that Na + currents are enhanced whereas K + currents are suppressed, one or more signalling pathways may be involved. (Received 7 May 2003; accepted after revision 12 August 2003; first published online 15 August 2003) Corresponding author S. Vanner: 166 Brock Street, Hotel Dieu Hospital, Kingston, Ontario, Canada K7L 5G2. Email: [email protected] J Physiol (2003), 552.3, pp. 797–807 DOI: 10.1113/jphysiol.2003.046409 © The Physiological Society 2003 www.jphysiol.org * Timothy Stewart and Michael J. Beyak contributed equally to this work.
-
Upload
timothy-stewart -
Category
Documents
-
view
212 -
download
0
Transcript of Ileitis modulates potassium and sodium currents in guinea pig dorsal root ganglia sensory neurons

Jou
rnal
of P
hysi
olog
y
Abdominal pain is a major cause of morbidity for patients
who suffer from intestinal disorders, such as inflammatory
bowel disease (IBD) and irritable bowel syndrome.
Antecedent infection or inflammation has been implicated
in the visceral hyperalgesia associated with the irritable
bowel syndrome (IBS). The cell bodies of sensory neurons
which initiate these sensations originate in the dorsal root
ganglia (DRG) (Mayer & Gebhart, 1994). Sub-populations
of DRG neurons respond to innocuous and noxious
mechanical stimulation, as well as chemical and thermal
stimulation, and thus are thought to be polymodal sensory
neurons (Mayer & Gebhart, 1994). These polymodal
neurons play a central role in responding to conditions
that are potentially injurious to tissues. Inflammatory
mediators cause a reduction in the threshold and an
increase in the gain of the transduction process of these
neurons in a process referred to as ‘peripheral
sensitization’. Also, under certain pathological conditions,
there is evidence that an additional population of normally
mechanically insensitive nociceptive fibres can be
recruited (Gebhart, 2000). Following inflammatory
stimulation these ‘silent afferent’ fibres become active at
rest and begin respond to mechanical stimulation.
Together, these afferent pathways have been suggested to
contribute to disproportionate pain states in response to
injury and may persist even after the inflammatory state
has resolved (Al Chaer et al. 2000; Collins et al. 2001).
In studies in the somatic nervous system, peripheral
sensitization appears to involve both acute and chronic
mechanisms (Woolf & Costigan, 1999). Acute
mechanisms of increased nociceptive stimulation include
both direct depolarization of nerve terminals by
neuroactive agents and alterations in ionic currents in the
membrane terminals (Rang et al. 1991). Also, later and
longer lasting transcription-dependent changes occur and
appear to be evoked by either signalling molecules, such as
nerve growth factor and/or activity-dependent second
messenger cascades (McCleskey & Gold, 1999). These
transcription-dependent events can result in altered
expression of voltage-gated ion channels, such as increased
expression of TTX-resistant sodium channels, and
increased TTX-resistant currents (TTX-R INa) (Khasar et
Ileitis modulates potassium and sodium currents in guineapig dorsal root ganglia sensory neuronsTimothy Stewart*, Michael J. Beyak* and Stephen Vanner
Gastrointestinal Diseases Research Unit, Queen’s University, Kingston, Ontario, Canada
Intestinal inflammation induces hyperexcitability of dorsal root ganglia sensory neurons, which has
been implicated in increased pain sensation. This study examined whether alteration of sodium
(Na+) and/ or potassium (K+) currents underlies this hyperexcitability. Ileitis was induced in guinea
pig ileum with trinitrobenzene sulphonic acid (TBNS) and dorsal root ganglion neurons
innervating the site of inflammation were identified by Fast Blue or DiI fluorescence labelling.
Whole cell recordings were made from acutely dissociated small-sized neurons at 7–10 days.
Neurons exhibited transient A-type and sustained outward rectifier K+ currents. Compared to
control, both A-type and sustained K+ current densities were significantly reduced (42 and 34 %,
respectively; P < 0.05) in labelled neurons from the inflamed intestine but not in non-labelled
neurons. A-type current voltage dependence of inactivation was negatively shifted in labelled
inflamed intestine neurons. Neurons also exhibited tetrodotoxin-sensitive and resistant Na+
currents. Tetrodotoxin-resistant sodium currents were increased by 37 % in labelled neurons from
the inflamed intestine compared to control (P < 0.01), whereas unlabelled neurons were unaffected.
The activation and inactivation curves of these currents were unchanged by inflammation. These
data suggest ileitis increases excitability of intestinal sensory neurons by modulating multiple ionic
channels. The lack of effect in non-labelled neurons suggests signalling originated at the nerve
terminal rather than through circulating mediators and, given that Na+ currents are enhanced
whereas K+ currents are suppressed, one or more signalling pathways may be involved.
(Received 7 May 2003; accepted after revision 12 August 2003; first published online 15 August 2003)
Corresponding author S. Vanner: 166 Brock Street, Hotel Dieu Hospital, Kingston, Ontario, Canada K7L 5G2. Email: [email protected]
J Physiol (2003), 552.3, pp. 797–807 DOI: 10.1113/jphysiol.2003.046409
© The Physiological Society 2003 www.jphysiol.org
* Timothy Stewart and Michael J. Beyak contributed equally to this work.

Jou
rnal
of P
hysi
olog
y
al. 1998; Tanaka et al. 1998) or decreased availability of
voltage-gated K+ channels (Yoshimura & de Groat, 1999).
Compared to the somatic nervous system, there is
considerably less known about the ionic mechanisms
underlying inflammation-induced changes in sensory
neurons innervating the gastrointestinal tract. Recent
studies in the rat stomach have shown that experimentally
induced gastric ulcers increase TTX-R INa in vagal and
DRG neurons innervating the stomach (Bielefeldt et al.2002a)and to a lesser degree in mild gastritis (Bielefeldt etal. 2002b). This latter study, however, found that outward
K+ currents were unchanged. In contrast, studies in viscera
outside the gastrointestinal (GI) tract suggest that
inflammation may also modulate K+ currents (Yoshimura
& de Groat, 1999) Taken together, it is likely that changes
in ionic mechanisms underlying inflammation-induced
plasticity depend on the organ involved and the
inflammatory repertoire which follows the initiation of
inflammation.
We have recently used trinitrobenzene sulphonic acid
(TNBS) ileitis in the guinea pig, as a model of
inflammatory bowel disease (Moore et al. 2002), to
examine the effects of intestinal inflammation on DRG
neurons. We found hyperexcitability of dissociated DRG
neurons innervating the intestine, manifested by
decreased threshold and repetitive firing, properties
consistent with hyperalgesia and allodynia. This study
employs whole cell voltage clamp techniques in this model
to determine if changes in Na+ and/ or K+ currents underlie
this hyperexcitability.
METHODS Animal model and neuron isolationGuinea pigs (140–225 g) of either sex were obtained from CharlesRiver Laboratories (Montreal, Quebec, Canada). Experimentswere performed according to the guidelines of the CanadianCouncil of Animal Care and approved by the Queen’s Universityanimal care committee.
Animals were anaesthetized using a combination of Hypnorm(0.315 mg ml_1 fentanyl citrate and 10 mg ml_1 fluanisone) andmidazolam (5 mg ml_1) (0.0025 ml g_1 each, I.P.), and surgery wasperformed under aseptic conditions to exteriorize the terminalileum as described previously (Moore et al. 2002). Ileal-projectingneurons were labelled by injecting the retrograde tracer Fast Blue(dissolved in distilled water) or DiI (dissolved in EtOH) into theileal wall using a Hamilton syringe fitted with a 30 Ga needle. Atotal injection volume of 15–20 ml was given via 10–15 injectionsites. DiI was used in later experiments since Fast Blue becameunavailable. Ileitis was induced by injection of 0.5 ml of TNBS(25 mg ml_1 in 25 % EtOH; TNBS kindly provided by Dr G.Morris, Department of Biology, Queen’s University, Kingston,Ontario, Canada K7L 5G2) into the ileal lumen using a 30 Ganeedle. The intestine was replaced in the abdominal cavity and thewound sutured with 4–0 silk. Buprenex (Buprenophine0.0225 mg g_1
I.P., Reckitt and Colman) was given to all animals tocontrol post-operative pain. Animals recovered from the
anaesthetic in a quiet environment, on an electric thermal blanketto maintain normothermia. The post-operative condition of theanimals was monitored at least twice daily by trained animal carestaff. Animals that were failing to thrive or showed behavioursuggestive of persistent pain were killed. After 7–10 days, animalswere anaesthetized by inhalation of isofluorane and killed bycervical dislocation and exsanguination.
The terminal ileum was removed from each animal to establishthe degree of TNBS induced ileitis. The ileum was opened alongthe mesenteric border and pinned flat with the mucosal surfaceuppermost. Inspection of the mucosa revealed mucosalhemorrhage, ulceration and thickening of the tissue, as describedin our previous studies (Moore et al. 2002). In the current study,inspection of the mucosa revealed similar damage followinginflammation.
DRG neurons were isolated from thoracic vertebra Th10_13 asdescribed previously (Moore et al. 2002). Briefly, isolated DRGwere dissected free of adherent connective tissue and thenincubated at 37 °C in HBSS with 0.2 mg ml_1 papain and0.4 mg ml_1 cysteine for 10 min. This was followed by washing inL-15 medium (GIBCO-BRL) with 10 % fetal bovine serum. TheDRGs were then incubated in HBSS containing 1 mg ml_1 Type 1collagenase (Worthington) and 4 mg ml_1 dispase II (BoeringerManneheim). The ganglia were then titurated with a fire-polishedPasteur pipette. Neurons were maintained in MEM culturemedium with Earle’s salts and HCO3 (GIBCO-BRL) containing1 % penicillin–streptomycin, 2 mM glutamine and 0.2 % (w/v)glucose. The cell suspension was plated onto rat tail collagen-coated glass coverslips and stored in a humidified incubator at37 °C under 95 % air and 5 % CO2 until they were retrieved for usein electrophysiological experiments 4–24 h later.
SolutionsFor current clamp experiments, the control solution used tosuperfuse the cells contained (mM): NaCl, 140; KCl, 5; MgSO4, 1;CaCl2, 1; Hepes, 5; pH adjusted to 7.4 using NaOH. Identicalcontrol solutions were used to superfuse the cells in K+ currentvoltage clamp experiments, except for the equimolar replacementof NaCl with N-methyl-D-glucamine. Potassium channelrecording solutions were adjusted to pH 7.4 using HCl. Thecontrol filling solution contained (mM): KCl, 140; Hepes, 5;MgSO4, 1; EGTA, 1; pH adjusted to 7.2 using KOH. For therecording of sodium currents, solutions of the followingcomposition were used: extracellular solution (mM): NaCl, 100;NMDG, 50; Hepes, 10; MgCl2, 10; D-Glucose, 10; pH adjusted to7.4 with HCl. Pipette solution (mM): Cs, 115; NaCl, 25; Hepes, 10;MgCl2, 3; EGTA, 11; pH adjusted to 7.2 with CsOH.
Electrophysiological proceduresCoverslips supporting adherent DRG neurons were placed in aRC-26 recording chamber (Warner Instrument Corporation) andmounted on an inverted microscope (IX-70, Olympus, Japan)fitted for both fluorescence and bright field microscopy. Labelledneurons were identified by their fluorescence under brief (< 15 s)exposure to ultraviolet light, using a U-MWIG2 filter for Fast Blueor a U-MWU2 (Olympus, Japan) for DiI, after which cells wereviewed under bright field illumination. Whole cell recordingswere made using variations of the patch clamp technique (Hamillet al. 1981) and an Axopatch-200B amplifier (Axon Instruments).Patch electrodes were fabricated using thin-walled borosilicateglass (Kimble Products) and a PP-830 electrode puller (NarishigeInstruments) or P-97 (Sutter Instruments). After fire polishing,pipettes used for whole cell voltage clamp experiments had DC
T. Stewart, M. J. Beyak and S. Vanner798 J Physiol 552.3

Jou
rnal
of P
hysi
olog
y
resistances of 1.5–3.0 MV when filled with the control fillingsolution. Electronic compensation of total series resistance wasused in all experiments. Capacitive transients were correctedusing analog circuitry. Drugs were added to the bath solution, ordelivered by a fast-flow solution switching system (VC-6, WarnerInstrument Corporation). Membrane currents were filtered at2 kHz and sampled at 5 kHz and stored on a microcomputer forlater analysis using pCLAMP 8.2 software (Axon Instruments).Cells were included for analysis if in current clamp mode theresting membrane potential was more negative than _40 mV andthe cells displayed overshooting action potentials or in voltageclamp mode the series resistance error (after compensation) wasless than 5 mV and seal and access resistances remained stable.The mean number of cells studied per animal was 3 (range 1–10).
Drugs and chemicalsAll chemicals and drugs were from Sigma Chemical Co.4-Aminopyridine (4-AP) was dissolved in distilled water to a stockconcentration of 100 mM and the pH was adjusted to 7.4 using 2 N
HCl. Tetrodotoxin (TTX) was dissolved in water at a stockconcentration of 1 mM. Drugs were diluted to their finalconcentration in the bath solution. Drugs were administeredeither by direct addition to the bath solution, or directly to the cellunder study using a fast-flow solution switching system (WarnerInstruments).
Statistical analysisStudent’s t test or ANOVA with Student-Newman-Keuls post hoctest were used where appropriate. P values < 0.05 were consideredsignificant.
RESULTSGeneral propertiesSuccessful recordings were obtained from 149 DRG
neurons. Three groups of neurons were examined in this
study; Fast Blue- or DiI-labelled neurons from control
animals (Control) or TNBS-treated animals (TNBS) and
neurons from TNBS-treated animals which were not
labelled (non-labelled TNBS). Approximately 3–5 % of
the dissociated cells were labelled with Fast Blue or DiI.
The mean resting membrane potential in current clamp
recordings from labelled control neurons was
–58.1 ± 1.1 mV (range _48 to _64 mV; n = 13). Our
previous studies of TNBS ileitis using intracellular
recording techniques demonstrated that small cell size
correlated very closely with the following properties: TTX-
resistant action potentials with inflections on the
repolarizing phase and capsaicin sensitivity (Moore et al.2002). These properties have been shown to be present in
small-diameter somatic and visceral unmyelinated
afferents, a proportion of which are nociceptive afferents
(Sengupta & Gebhart, 1994; Blackshaw & Gebhart, 2002).
Using whole cell current clamp recordings, we found that
small neurons had similar properties, i.e. TTX-resistant
action potentials with a prominent inflection of the
repolarizing phase (5/5). Measurements of cell capacitance
were used to monitor size, and only cells with capacitance
< 40 pF were examined, as these cells consistently
expressed these properties. Larger cells (mean capacitance
81 ± 7.4 pF) had narrow, TTX-sensitive action potentials
(4/4). In our previous work using the same model we
demonstrated inflammation-induced changes in
excitability of this sub-population of small neurons. In
these studies, intracellular recordings were obtained from
neurons from control (n = 12) and animals with ileitis
(n = 17) and we found that TNBS ileitis caused significant
reductions in rheobase, and increased the number of
action potentials evoked at 2 w rheobase. Furthermore
TNBS ileitis resulted in increased action potential
upstroke velocity and increased input resistance. (Moore
et al. 2002). In the present study, using whole cell
recording techniques, we demonstrated a similar effect of
TNBS ileitis on rheobase (> 65 % decrease compared to
control and non-labelled cells) and number of action
potentials elicited at twice rheobase (> 2.7 times compared
to control and non-labelled cells) (Fig. 1).
Characterization of voltage-gated K+ currentsPrevious studies of unidentified DRG neurons have
identified two kinetically distinct voltage-dependent K+
currents, a transient ‘A-type’ current (IA) and ‘sustained
delayed rectifier type’ (IK) (McFarlane & Cooper, 1991;
Akins & McCleskey, 1993; Gold et al. 1996; Everill et al.1998). In the present study, using a voltage clamp protocol
with 5 mV steps (400 ms) from a holding potential of
–100 mV, both the IA and sustained IK were evident
(Fig. 2A). The reversal potential of these currents was
_75.5 ± 2.2 mV, near the predicted reversal potential for
potassium (_84 mV). Inactivating IA currents were
adequately fitted by a monoexponential decay with a mean
time constant of 171 ± 16 ms (range 133–210 ms).
IA and IK currents were isolated based on their contrasting
biophysical and pharmacological properties. In
preliminary experiments, we established that neither
current was significantly inactivated when the membrane
potential was held at –100 mV (Fig. 2). However, the IA was
selectively inactivated when the membrane potential was
held at –60 mV, whereas inactivation of IK at this holding
potential was minimal (Fig. 2). Thus, the sustained current
was isolated by holding the membrane potential at –60 mV,
with the sustained IK measured isochronally, 400 ms after
the onset of the pulse, at which time IA was largely
inactivated, minimizing contamination by this current
(Fig. 2). The IA was isolated by subtracting the sustained IK
from the total K+ current recorded from a holding potential
of –100 mV. Peak IA was measured as the peak of the
transient component of this subtracted current (Fig. 2).
IA was also isolated pharmacologically using the K+
channel blocker 4-aminopyridine (4-AP) (Fig. 2B).
Preliminary experiments demonstrated that 100 mM 4-AP
(n = 6), and 600 mM 4-AP (n = 3) caused partial
suppression of the IA currents whereas 2 mM (n = 3)
completely suppressed IA and caused significant
suppression of the IK currents. 4-AP most selectively
Ileitis modulates sensory nerve K+ and Na+ currentsJ Physiol 552.3 799

Jou
rnal
of P
hysi
olog
y
blocked the IA at a concentration of 1 mM. Therefore the IA
was isolated by subtracting the current in the presence of
1 mM 4-AP from the total control current, yielding the
4-AP-sensitive IA (Fig. 2B). Experiments using both of
these approaches in the same control neuron revealed no
significant difference in the IA current density obtained by
either method (80.3 ± 19.7 vs. 99.1 ± 10.0 pA pF_1, n = 5)
IA and IK current density following TNBS-inducedinflammationThe density of IA and IK elicited by a depolarizing pulse to
+50 mV was compared in labelled neurons from control
animals and both labelled and unlabelled neurons from
TNBS animals (Fig. 2C and D). Current density was
T. Stewart, M. J. Beyak and S. Vanner800 J Physiol 552.3
Figure 1. Effects of TNBS ileitis on intestinal sensory neuron excitabilityA, representative current clamp traces of action potentials elicited at rheobase and two times rheobase incontrol labelled, TNBS labelled and TNBS non-labelled neurons. TNBS results in a reduction of therheobase, while increasing the number of spikes evoked by a 2 w rheobase current injection. Inflections onthe falling phase of the action potential are not obvious due to the time scale. B, mean rheobase in controllabelled (n = 13), TNBS labelled (n = 4) and TNBS non-labelled (n = 5) neurons (* P < 0.05). C, meannumber of spikes elicited by a 2 w rheobase current injection in these same groups of neurons (* P < 0.05).

Jou
rnal
of P
hysi
olog
yIleitis modulates sensory nerve K+ and Na+ currentsJ Physiol 552.3 801
Figure 2. Voltage-gated potassium currents and the effect of TNBS ileitis on current densityRepresentative traces from one cell showing currents separated biophysically by manipulating the holdingpotential (A) and pharmacologically by 4-AP (B). Currents were elicited in response voltage steps from_90 mV to +50 mV in 5 mV increments. A, left: at holding potentials of _100 mV, two currents wereapparent, a transient, inactivating ‘A’ type current, and a non-inactivating sustained IK type current. A,middle: resultant current when membrane potential is held at _60 mV. Note disappearance of the transientcomponent. Only the sustained component remains, and was measured at 400 ms. A, right: subtraction ofthe sustained from the total current yields IA. IA amplitude was measured as the peak of the transientcomponent. B, left: in the pharmacological experiments, the holding potential was _100 mV. B, middle:when the voltage steps were repeated in the presence of 1 mM 4-AP the transient component was significantlyinhibited. B, right: results of subtracting current obtained in the presence of 4-AP from control, revealing the4-AP-sensitive IA current. C, effect of TNBS ileitis on mean peak IA density obtained using either biophysical(left) or pharmacological separation (right). TNBS ileitis results in a significant reduction in IA densitycompared to control, or non-labelled TNBS neurons (* P < 0.05). D, effect of TNBS ileitis on mean peak IK
density. TNBS ileitis results in a significant reduction in peak IK density, compared to control and non-labelled cells (* P < 0.05).

Jou
rnal
of P
hysi
olog
y
determined by normalizing the current amplitude by the
cell’s capacitance. The IK amplitude was measured 400 ms
following the onset of a depolarizing pulse from a holding
potential of –60 mV. The IA amplitude was measured as
the peak current amplitude of the subtracted current as
described above (Fig. 2A and B). Figure 2C–E illustrates
the reduction of IA and IK in labelled neurons from TNBS
animals. When IA was isolated biophysically, the IA density
was 42 % less (P < 0.05) in labelled neurons from TNBS
animals (n = 22) than in labelled neurons from control
animals (n = 14) and 65 % less than in unlabelled neurons
from TNBS animals (n = 19) (Fig. 2C). IA was also isolated
pharmacologically using 4-AP (1 mM). The 4-AP-sensitive
IA in labelled neurons was reduced by 33 % in labelled
TNBS neurons (n = 6, P < 0.05) compared to the currents
in labelled neurons from control animals (n = 4) and was
43 % less than in non-labelled TNBS neurons (n = 7,
P < 0.01, Fig. 2D). The current density of IK from labelled
TNBS neurons (n = 20) was 34 % less (P < 0.05) than in
labelled neurons from control animals (n = 15) and 31 %
less (P < 0.05) than in unlabelled neurons from TNBS
animals (n = 20) (Fig. 2E).
We also attempted to isolate IK pharmacologically, using
TEA. However, we found, as have others (Everill et al.1998; Gold et al. 1996) that TEA in doses sufficient to block
IK, also cause significant blockade of the IA.
Voltage dependence of activation and inactivationVoltage dependencies of activation and inactivation were
compared in labelled control cells and labelled TNBS cells
to determine if changes in TNBS animals could contribute
to the reduced current density.
To determine IA K+ conductance and IK K+ conductance,
peak IA amplitudes and isochronally measured end-pulse
current amplitudes were measured, respectively.
Conductance was then determined using the relation
G = I/(Vm – EK), where G is the conductance, I is the
measured membrane current, Vm is the voltage step, and
EK is the equilibrium K+ potential, which was calculated to
be –84 mV in control solutions. Average K+ conductance
was plotted against membrane potential (Fig. 4) and the
continuous line is an average of individual fits to a
Boltzmann function of the form:
G/Gmax = 1/(1 + exp[VÎ _ Vm/k]),
where G is the conductance, Gmax is the fitted maximal
conductance, VÎ is the membrane potential for half-
activation, Vm is the command potential, and k is the slope
factor. No differences in the voltage dependencies of
activation for IA or IK were found between control and
TNBS neurons (Fig. 3A and B).
To examine the voltage dependencies of inactivation for IA
and IK, we employed two-pulse voltage protocols as
described by others (Philipson et al. 1991; Yoshimura & de
Groat, 1999). Residual IA currents were measured
following short conditioning pulses (1 s duration) which
allowed inactivation of only the rapidly inactivating IA
currents. Longer conditioning pulses (8 s duration)
allowed inactivation of both IA and IK currents. Therefore
residual currents were isochronally measured at the end of
a 1 s test pulse to minimize the contribution of IA currents
to the measured residual current. The residual current
amplitude was plotted against conditioning pulse
T. Stewart, M. J. Beyak and S. Vanner802 J Physiol 552.3
Figure 3. Activation and inactivationcurves of voltage-gated K+ currentsActivation curves were generated by voltage pulsesin 5 mV steps from -80 to +50 mV. Each curverepresents the mean of curves fitted to theBoltzmann equation (n = 8 cells each). A,activation curves for IA in control and TNBSneurons. TNBS did not affect the activationproperties of IA.B, inactivation curves for IA.
Inactivation curves were constructed using a two-pulse protocol, a 1 s prepulse varying between_120 and 0 mV, followed by a 400 ms test pulse of+50 mV. The peak of the transient component wasmeasured. TNBS treatment resulted in a slightleftward shift in the inactivation curve of IA.Activation (C) and inactivation (D) curves for IK.Inactivation curves for IK were generated using atwo-pulse protocol, with an 8 s prepulse varyingbetween _80 and 0 mV, followed by a 1 s test pulseof +50 mV. End-pulse IK amplitude was measured.TNBS had no effect on voltage dependence ofeither activation or inactivation of IK.
Jou
rnal
of P
hysi
olog
y
potential and the continuous line is an average of fits to a
negative Boltzmann function:
I/Imax = 1/(1 + exp[VÎ _ Vm/k]),
where I is the current, Imax is the maximal current, VÎ is the
membrane potential for half-activation, Vm is the
command potential, and k is the slope factor (Philipson etal. 1991). Significant differences in IA were found between
control and TNBS neurons with VÎ (control
_65.4 ± 2.0 mV, TNBS 85.2 ± 2.4 mV, P < 0.05) and
slope factors (control 6.9 ± 0.9, TNBS 14.3 ± 1.1,
P < 0.05) (Fig. 3C). However, the voltage dependence and
voltage sensitivity of inactivation for IK were not different
between control and TNBS neurons (VÎ: control
_47.6 ± 2.8 mV, TNBS _50.2 ± 3.0 mV; slope: control
7.5 ± 1.5, TNBS 9.4 ± 1.2, P > 0.05; Fig. 3D).
We also employed a pharmacological approach using
4-AP (1 mM) to isolate IA and examine its voltage
dependence. Using the above two-pulse protocol, in the
absence and presence of 4-AP, the currents obtained in the
presence of 4-AP were subtracted from those obtained in
its absence (n = 10). These experiments indicated that
sensitivity to 4-AP varied significantly with the prepulse
voltage thus making analysis of the voltage dependence of
inactivation of the subtracted 4-AP sensitive current
impractical. This finding has been reported by others
(Thompson, 1982; Rasmusson et al. 1995).
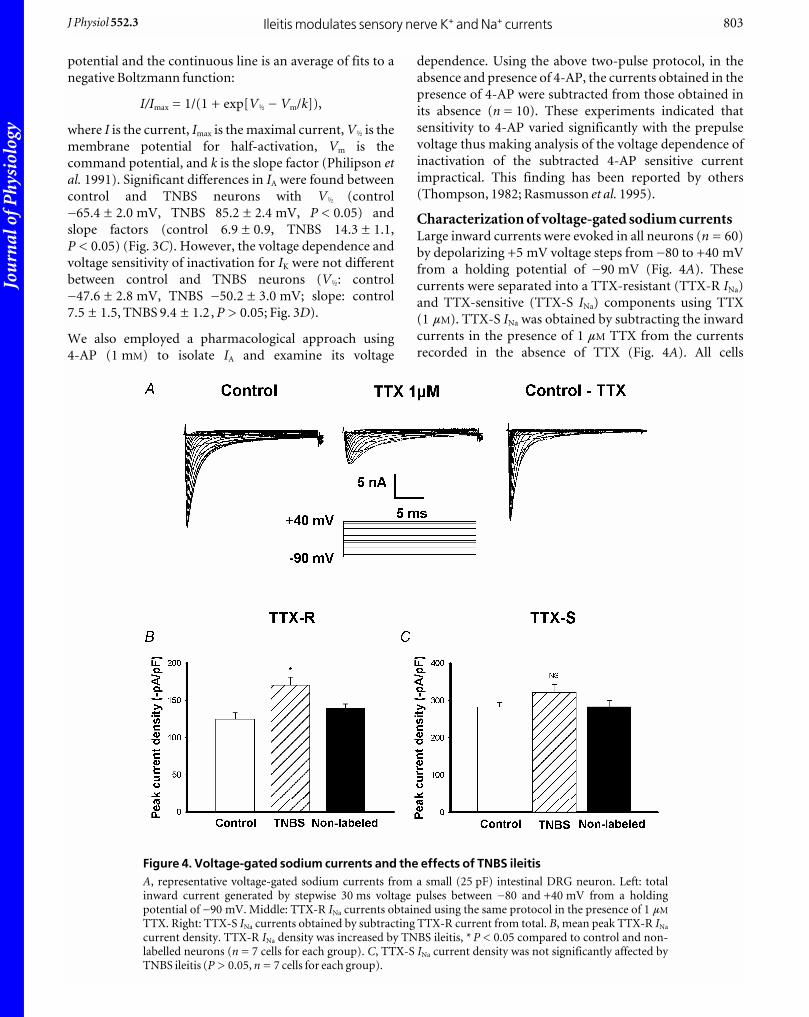
Characterization of voltage-gated sodium currentsLarge inward currents were evoked in all neurons (n = 60)
by depolarizing +5 mV voltage steps from _80 to +40 mV
from a holding potential of _90 mV (Fig. 4A). These
currents were separated into a TTX-resistant (TTX-R INa)
and TTX-sensitive (TTX-S INa) components using TTX
(1 mM). TTX-S INa was obtained by subtracting the inward
currents in the presence of 1 mM TTX from the currents
recorded in the absence of TTX (Fig. 4A). All cells
Ileitis modulates sensory nerve K+ and Na+ currentsJ Physiol 552.3 803
Figure 4. Voltage-gated sodium currents and the effects of TNBS ileitisA, representative voltage-gated sodium currents from a small (25 pF) intestinal DRG neuron. Left: totalinward current generated by stepwise 30 ms voltage pulses between _80 and +40 mV from a holdingpotential of _90 mV. Middle: TTX-R INa currents obtained using the same protocol in the presence of 1 mM
TTX. Right: TTX-S INa currents obtained by subtracting TTX-R current from total. B, mean peak TTX-R INa
current density. TTX-R INa density was increased by TNBS ileitis, * P < 0.05 compared to control and non-labelled neurons (n = 7 cells for each group). C, TTX-S INa current density was not significantly affected byTNBS ileitis (P > 0.05, n = 7 cells for each group).
Jou
rnal
of P
hysi
olog
y
examined exhibited both the TTX-R INa and TTX-S INa
currents.
Effects of TNBS ileitis on Na+ current densityTTX-R INa and TTX-S INa peak current densities were
obtained by normalizing the current amplitude by the
individual cell’s capacitance. TTX-R INa was increased
significantly in the labelled TNBS group (n = 10) compared
to labelled control neurons (n = 10) (37 %, P < 0.01) or
non-labelled control neurons (n = 10) (32 %, P < 0.05)
(Fig. 4B). TTX-S INa was not significantly different between
inflamed labelled neurons (282.0 ± 12.7 pA pF_1, n = 10),
labelled control neurons (312.2 ± 22.4 pA pF_1, n = 10,
P > 0.05) or TNBS non-labelled neurons (282.3 ±
17.0 pA pF_1, n = 10, P > 0.05) (Fig. 4C).
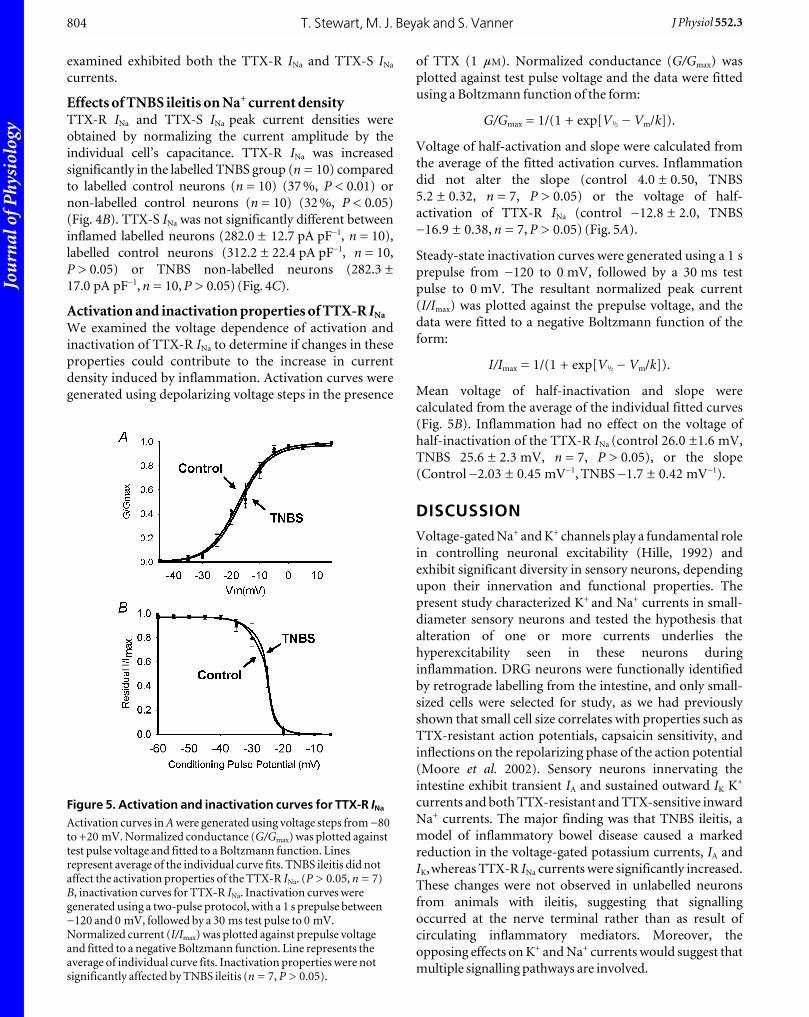
Activation and inactivation properties of TTX-R INa
We examined the voltage dependence of activation and
inactivation of TTX-R INa to determine if changes in these
properties could contribute to the increase in current
density induced by inflammation. Activation curves were
generated using depolarizing voltage steps in the presence
of TTX (1 mM). Normalized conductance (G/Gmax) was
plotted against test pulse voltage and the data were fitted
using a Boltzmann function of the form:
G/Gmax = 1/(1 + exp[VÎ _ Vm/k]).
Voltage of half-activation and slope were calculated from
the average of the fitted activation curves. Inflammation
did not alter the slope (control 4.0 ± 0.50, TNBS
5.2 ± 0.32, n = 7, P > 0.05) or the voltage of half-
activation of TTX-R INa (control _12.8 ± 2.0, TNBS
_16.9 ± 0.38, n = 7, P > 0.05) (Fig. 5A).
Steady-state inactivation curves were generated using a 1 s
prepulse from _120 to 0 mV, followed by a 30 ms test
pulse to 0 mV. The resultant normalized peak current
(I/Imax) was plotted against the prepulse voltage, and the
data were fitted to a negative Boltzmann function of the
form:
I/Imax = 1/(1 + exp[VÎ _ Vm/k]).
Mean voltage of half-inactivation and slope were
calculated from the average of the individual fitted curves
(Fig. 5B). Inflammation had no effect on the voltage of
half-inactivation of the TTX-R INa (control 26.0 ±1.6 mV,
TNBS 25.6 ± 2.3 mV, n = 7, P > 0.05), or the slope
(Control _2.03 ± 0.45 mV_1, TNBS _1.7 ± 0.42 mV_1).
DISCUSSIONVoltage-gated Na+ and K+ channels play a fundamental role
in controlling neuronal excitability (Hille, 1992) and
exhibit significant diversity in sensory neurons, depending
upon their innervation and functional properties. The
present study characterized K+ and Na+ currents in small-
diameter sensory neurons and tested the hypothesis that
alteration of one or more currents underlies the
hyperexcitability seen in these neurons during
inflammation. DRG neurons were functionally identified
by retrograde labelling from the intestine, and only small-
sized cells were selected for study, as we had previously
shown that small cell size correlates with properties such as
TTX-resistant action potentials, capsaicin sensitivity, and
inflections on the repolarizing phase of the action potential
(Moore et al. 2002). Sensory neurons innervating the
intestine exhibit transient IA and sustained outward IK K+
currents and both TTX-resistant and TTX-sensitive inward
Na+ currents. The major finding was that TNBS ileitis, a
model of inflammatory bowel disease caused a marked
reduction in the voltage-gated potassium currents, IA and
IK,whereas TTX-R INa currents were significantly increased.
These changes were not observed in unlabelled neurons
from animals with ileitis, suggesting that signalling
occurred at the nerve terminal rather than as result of
circulating inflammatory mediators. Moreover, the
opposing effects on K+ and Na+ currents would suggest that
multiple signalling pathways are involved.
T. Stewart, M. J. Beyak and S. Vanner804 J Physiol 552.3
Figure 5. Activation and inactivation curves for TTX-R INa
Activation curves in A were generated using voltage steps from _80to +20 mV. Normalized conductance (G/Gmax) was plotted againsttest pulse voltage and fitted to a Boltzmann function. Linesrepresent average of the individual curve fits. TNBS ileitis did notaffect the activation properties of the TTX-R INa. (P > 0.05, n = 7)B, inactivation curves for TTX-R INa. Inactivation curves weregenerated using a two-pulse protocol, with a 1 s prepulse between_120 and 0 mV, followed by a 30 ms test pulse to 0 mV.Normalized current (I/Imax) was plotted against prepulse voltageand fitted to a negative Boltzmann function. Line represents theaverage of individual curve fits. Inactivation properties were notsignificantly affected by TNBS ileitis (n = 7, P > 0.05).

Jou
rnal
of P
hysi
olog
y
Voltage-gated K+ currents in neurons innervatingcontrol and inflamed ileumThe IA current plays an important role in setting high
thresholds for spike activation and suppression of
repetitive firing (Rudy, 1988; Hille, 1992; Yoshimura & de
Groat, 1999). Several types of IA current have been
described in DRG sensory neurons (Gold et al. 1996;
Yoshimura & de Groat, 1999) and can be separated into
those with relatively fast and slow inactivation rates based
on their biophysical and pharmacological properties
(Gold et al. 1996; Yoshimura & de Groat, 1999). Several
studies have shown that the fast inactivating IA currents are
confined to the large-sized neurons with TTX-sensitive
spikes (Yoshimura et al. 1996; Gold et al. 1996). The slowly
inactivating IA current has an inactivation time constant of
between 150 and 300 ms and the voltage of half-maximal
inactivation is displaced more positively than the fast
inactivating IA (Akins & McCleskey, 1993; Fjell et al. 1999).
In the present study the transient IA inactivation was fitted
by a single monoexponential function with a time constant
of 171 ms, typical of that described for the slowly
inactivating IA. These slowly inactivating IA currents, in
contrast to the fast inactivating currents, have been found
to be selectively expressed in the small-sized neurons
exhibiting TTX-resistant action potentials (Yoshimura etal. 1996). Taken together, the data suggest that slowly
inactivating IA current is the dominant form of IA in
sensory afferent DRG neurons innervating the small
intestine.
In addition to the transient IA current, studies of DRG
sensory neurons have identified a sustained delayed
rectifier current in all cells (Akins & McCleskey, 1993;
Gold et al. 1996). In the present study, these currents could
be separated from the IA currents by prepulse inactivation
or by 1 mM 4-AP which blocked the IA currents but had
little effect on the sustained current, as reported by others
(Gold et al. 1996). Some studies have suggested that this
sustained current may represent several different currents,
based largely on their steady-state inactivation properties
(Akins & McCleskey, 1993; Gold et al. 1996). These
sustained currents, however, often could not be separated
by voltage protocols or pharmacological agents (Gold et al.1996) making the study of changes in individual sustained
currents impractical.
TNBS ileitis caused a significant reduction in both IA and
sustained IK currents identified in this study and we have
previously shown that pharmacological suppression of K+
currents such as IA can significantly increase the
excitability of sensory neurons innervating the intestine
(Moore et al. 2002). It is unclear whether these changes in
K+ currents occur elsewhere in the GI tract because there
has been relatively little work done in this area and in
studies conducted in a gastritis model of inflammation
similar changes were not observed (Bielefeldt et al. 2002b).
It is possible that this difference can be explained by
variations in the degree of inflammation or experimental
protocols. Our finding that visceral inflammation
suppresses IA and correspondingly increases excitability of
sensory neurons is similar to previous studies examining
the inflamed urinary bladder (Yoshimura & de Groat,
1999). This reduction in peak current density was
associated with a hyperpolarizing shift in the inactivation
curve, as described in the current study, suggesting there
was an associated change in the biophysical properties of
the channels. The shift of the inactivation curve in a
hyperpolarizing direction would make fewer IA channels
available, at or near resting membrane potentials, and lead
to a further increase in excitability of the cell, leading to
repetitive firing. In contrast to the study in the rat urinary
bladder, we also observed a marked reduction in the
current density of IK. Whether these differences also reflect
degrees of inflammation in these organs or are unique to
the sensory neurons and the organs they innervate remains
to be established. Given that the role of these voltage-gated
potassium currents is to raise the action potential
threshold and limit firing (Hille, 1992) suppression of
these currents would be expected to increase excitability.
Voltage-gated Na+ currents in neurons innervatingcontrol and inflamed ileumThree types of sodium channels predominate in DRG
neurons; NaV1.7 channels, which are responsible for the
fast, rapidly inactivating TTX-S INa current, NaV1.8
channels, which result in a more slowly inactivating TTX-R
INa and are relatively selectively expressed in nociceptors,
and the recently described NaV1.9 which is responsible for a
TTX-R persistent current that is ultra-slowly inactivating
(Dib-Hajj et al. 2002).The activation and inactivation
properties of our TTX-R INa currents fit the known
properties of the NaV1.8 channel, although a detailed
molecular or immunocytochemical characterization of
guinea pig DRG sodium channels has yet to be performed.
We observed a significant increase in TTX-R INa in sensory
neurons innervating the inflamed ileum, as has been
implicated in other viscera (Yoshimura et al. 2001;
Bielefeldt et al. 2002a,b) and the somatic nervous system.
In the somatic nervous system (Khasar et al. 1998; Tanaka
et al. 1998) these changes were also accompanied by
neuronal hyperexcitability and molecular evidence of
increased expression of sodium channels (Gould et al.1998, 1999), specifically the TTX-R channel subtype,
NaV1.8 (Tanaka et al. 1998). Moreover, mice deficient in
Nav1.8 exhibit decreased neuronal excitability
(Renganathan et al. 2001) and decreased visceral pain
(Laird et al. 2002). Antisense NaV1.8 oligodeoxynucleotide
can also prevent inflammation-induced mechanical
hyperalgesia (Khasar et al. 1998) and cyclophosphamide
cystitis-induced bladder hyperreflexia (Yoshimura et al.2001). Taken together, these studies suggest that
Ileitis modulates sensory nerve K+ and Na+ currentsJ Physiol 552.3 805

Jou
rnal
of P
hysi
olog
y
inflammation-induced increased expression of TTX-R INa
in nociceptive neurons is common to inflammation in
both the somatic and visceral organs, including the GI
tract, and is an important mechanism underlying
neuronal hyperexcitability and visceral hyperalgesia.
Potential mechanisms mediating inflammation-induced changes in ion channelsThe inflammatory milieu surrounding the nerve terminal
contains numerous inflammatory mediators, such as
PGE2, and 5-HT, which have been shown to augment
TTX-R Na+ currents in vitro, in both visceral (Gold et al.2002) and somatic afferents (Cardenas et al. 2001; Gold etal. 2002). PGE2 has also been shown to decrease IK
potassium currents through the same cyclic AMP–protein
kinase C-dependent mechanism which alters Na+ currents,
in unidentified DRG neurons (Nicol et al. 1997; Evans etal. 1999). These changes occur within minutes and are
dependent on the activation of intracellular second
messenger systems, and probably subsequent
phosphorylation of ion channels or other regulatory cell
signalling proteins. These actions cannot account for the
findings in the present study because recordings were
obtained from cell bodies of DRG neurons located in
ganglia outside the inflammatory milieu of the intestine
and recordings were made at least 6–8 h after removal of
the neurons from the animal at a time when unlabelled
neurons were unaffected. Consequently, changes in ionic
currents appear to result from longer term alterations such
as transcriptional events altering the number of channels,
their subunits or other biophysical properties of the
membrane channels themselves.
Both activity-dependent and growth factor signalling
pathways have been proposed to activate transcription
factors which can alter ion channel expression. In
particular there is considerable evidence that the
neurotrophin nerve growth factor (NGF) can increase
expression of TTX-R INa (Fjell et al. 1999; Bielefeldt et al.2003). In addition NGF and other neuotrophins have been
shown to modulate various potassium channels, via
activation of tyrosine kinases (Yang et al. 2001) or via
activation of ceramide (Zhang et al. 2002). Thus NGF
appears to be an important candidate for modulating ion
channel activity in visceral inflammation. However,
experiments specifically designed to test this hypothesis
are needed. It is also unknown whether the diverging
effects on these currents are due to signalling through a
single pathway or whether multiple pathways are involved.
ConclusionsThis study demonstrates that an animal model of
inflammatory bowel disease is associated with a
suppression of transient and sustained K+ currents and
augmentation of TTX-R INa currents in intestinal sensory
neurons. The implications are that these mechanisms are
important in the increased neuronal excitability seen in
these neurons as well as in the increased nociceptive
trafficking which occurs during intestinal inflammation,
such as in Crohn’s disease. Furthermore, there is
increasing evidence that visceral hyperalgesia underlies
many functional bowel disorders, such as post-infectious
irritable bowel syndrome. Whether the findings observed
in the current study are present following the resolution of
macroscopic inflammation, or whether low levels of
microscopic inflammation, which are suggested to persist
in these conditions (Collins et al. 2001; Chadwick et al.2002; Tornblom et al. 2002), are sufficient to sustain these
changes remains to be established.
REFERENCESAkins PT & McCleskey EW (1993). Characterization of potassium
currents in adult rat sensory neurons and modulation by opioids
and cyclic AMP. Neuroscience 56, 759–769.
Al Chaer ED, Kawasaki M & Pasricha PJ (2000). A new model of
chronic visceral hypersensitivity in adult rats induced by colon
irritation during postnatal development. Gastroenterology 119,
1276–1285.
Bielefeldt K, Ozaki N & Gebhart GF (2002a). Experimental ulcers
alter voltage-sensitive sodium currents in rat gastric sensory
neurons. Gastroenterology 122, 394–405.
Bielefeldt K, Ozaki N & Gebhart GF (2002b). Mild gastritis alters
voltage-sensitive sodium currents in gastric sensory neurons in
rats. Gastroenterology 122, 752–761.
Bielefeldt K, Ozaki N & Gebhart GF (2003). Role of nerve growth
factor in modulation of gastric afferent neurons in the rat. Am JPhysiol Gastrointest Liver Physiol 284, G499–507.
Blackshaw LA & Gebhart GF (2002). The pharmacology of
gastrointestinal nociceptive pathways. Curr Opin Pharmacol 2,
642–649.
Cardenas LM, Cardenas CG & Scroggs RS (2001). 5HT increases
excitability of nociceptor-like rat dorsal root ganglion neurons via
cAMP-coupled TTX-resistant Na(+) channels. J Neurophysiol 86,
241–248.
Chadwick VS, Chen W, Shu D, Paulus B, Bethwaite P, Tie A &
Wilson I (2002). Activation of the mucosal immune system in
irritable bowel syndrome. Gastroenterology 122, 1778–1783.
Collins SM, Piche T & Rampal P (2001). The putative role of
inflammation in the irritable bowel syndrome. Gut 49, 743–745.
Dib-Hajj S, Black JA, Cummins TR & Waxman SG (2002).
NaN/Nav1.9: a sodium channel with unique properties. TrendsNeurosci 25, 253–259.
Evans AR, Vasko MR & Nicol GD (1999). The cAMP transduction
cascade mediates the PGE2-induced inhibition of potassium
currents in rat sensory neurones. J Physiol 516, 163–178.
Everill B, Rizzo MA & Kocsis JD (1998). Morphologically identified
cutaneous afferent DRG neurons express three different
potassium currents in varying proportions. J Neurophysiol 79,
1814–1824.
Fjell J, Cummins TR, Davis BM, Albers KM, Fried K, Waxman SG &
Black JA (1999). Sodium channel expression in NGF-
overexpressing transgenic mice. J Neurosci Res 57, 39–47.
Gebhart GF (2000). Pathobiology of visceral pain: molecular
mechanisms and therapeutic implications IV. Visceral afferent
contributions to the pathobiology of visceral pain. Am J PhysiolGastrointest Liver Physiol 278, G834–838.
T. Stewart, M. J. Beyak and S. Vanner806 J Physiol 552.3

Jou
rnal
of P
hysi
olog
y
Gold MS, Shuster MJ & Levine JD (1996). Characterization of six
voltage-gated K+ currents in adult rat sensory neurons.
J Neurophysiol 75, 2629–2646.
Gold MS, Zhang L, Wrigley DL & Traub RJ (2002). Prostaglandin
E(2) modulates TTX-R I(Na). in rat colonic sensory neurons.
J Neurophysiol 88, 1512–1522.
Gould HJ III, England JD, Liu ZP & Levinson SR (1998). Rapid
sodium channel augmentation in response to inflammation
induced by complete Freund’s adjuvant. Brain Res 802, 69–74.
Gould HJ III, Gould TN, Paul D, England JD, Liu ZP, Reeb SC &
Levinson SR (1999). Development of inflammatory
hypersensitivity and augmentation of sodium channels in rat
dorsal root ganglia. Brain Res 824, 296–299.
Hamill OP, Marty A, Neher E, Sakmann B & Sigworth FJ (1981).
Improved patch-clamp techniques for high-resolution current
recording from cells and cell-free membrane patches. Pflugers Arch391, 85–100.
Hille B (1992). In Ion Channels of Excitable Membranes. Sinauer
Associates, Sunderland, MA, USA.
Khasar SG, Gold MS & Levine JD (1998). A tetrodotoxin-resistant
sodium current mediates inflammatory pain in the rat. NeurosciLett 256, 17–20.
Laird JM, Souslova V, Wood JN & Cervero F (2002). Deficits in
visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null
mice. J Neurosci 22, 8352–8356.
McCleskey EW & Gold MS (1999). Ion channels of nociception.
Annu Rev Physiol 61, 835–856.
McFarlane S & Cooper E (1991). Kinetics and voltage dependence of
A-type currents on neonatal rat sensory neurons. J Neurophysiol66, 1380–1391.
Mayer EA & Gebhart GF (1994). Basic and clinical aspects of visceral
hyperalgesia. Gastroenterology 107, 271–293.
Moore BA, Stewart TM, Hill C & Vanner SJ (2002). TNBS ileitis
evokes hyperexcitability and changes in ionic membrane
properties of nociceptive DRG neurons. Am J Physiol GastrointestLiver Physiol 282, G1045–1051.
Nicol GD, Vasko MR & Evans AR (1997). Prostaglandins suppress an
outward potassium current in embryonic rat sensory neurons. JNeurophysiol 77, 167–176.
Philipson LH, Hice RE, Schaefer K, Lamendola J, Bell GI, Nelson DJ
& Steiner DF (1991). Sequence and functional expression in
Xenopus oocytes of a human insulinoma and islet potassium
channel. Proc Natl Acad Sci U S A 88, 53–57.
Rang HP, Bevan S & Dray A (1991). Chemical activation of
nociceptive peripheral neurones. Br Med Bull 47, 534–548.
Rasmusson RL, Zhang Y, Campbell DL, Comer MB, Castellino RC,
Liu S & Strauss HC (1995). Bi-stable block by 4-aminopyridine of
a transient K+ channel (Kv1.4). cloned from ferret ventricle and
expressed in Xenopus oocytes. J Physiol 485, 59–71.
Renganathan M, Cummins TR & Waxman SG (2001). Contribution
of Na(v)1.8 sodium channels to action potential electrogenesis in
DRG neurons. J Neurophysiol 86, 629–640.
Rudy B (1988). Diversity and ubiquity of K channels. Neuroscience25, 729–749.
Sengupta JN & Gebhart GF (1994). Characterization of
mechanosensitive pelvic nerve afferent fibers innervating the
colon of the rat. J Neurophysiol 71, 2046–2060.
Tanaka M, Cummins TR, Ishikawa K, Dib-Hajj SD, Black JA &
Waxman SG (1998). SNS Na+ channel expression increases in
dorsal root ganglion neurons in the carrageenan inflammatory
pain model. Neuroreport 9, 967–972.
Thompson S (1982). Aminopyridine block of transient potassium
current. J Gen Physiol 80, 1–18.
Tornblom H, Lindberg G, Nyberg B & Veress B (2002). Full-
thickness biopsy of the jejunum reveals inflammation and enteric
neuropathy in irritable bowel syndrome. Gastroenterology 123,
1972–1979.
Woolf CJ & Costigan M (1999). Transcriptional and
posttranslational plasticity and the generation of inflammatory
pain. Proc Natl Acad Sci U S A 96, 7723–7730.
Yang F, Feng L, Zheng F, Johnson SW, Du J, Shen L, Wu CP & Lu B
(2001). GDNF acutely modulates excitability and A-type K(+).
channels in midbrain dopaminergic neurons. Nat Neurosci 4,
1071–1078.
Yoshimura N & de Groat WC (1999). Increased excitability of
afferent neurons innervating rat urinary bladder after chronic
bladder inflammation. J Neurosci 19, 4644–4653.
Yoshimura N, Seki S, Novakovic SD, Tzoumaka E, Erickson VL,
Erickson KA, Chancellor MB & De Groat WC (2001). The
involvement of the tetrodotoxin-resistant sodium channel
Na(v)1.8 (PN3/SNS). in a rat model of visceral pain. J Neurosci 21,
8690–8696.
Yoshimura N, White G, Weight FF & de Groat WC (1996). Different
types of Na+ and A-type K+ currents in dorsal root ganglion
neurones innervating the rat urinary bladder. J Physiol 494, 1–16.
Zhang YH, Vasko MR & Nicol GD (2002). Ceramide, a putative
second messenger for nerve growth factor, modulates the TTX-
resistant Na(+) current and delayed rectifier K(+) current in rat
sensory neurons. J Physiol 544, 385–402.
Acknowledgements This work was supported by a grant from The Crohn’s and ColitisFoundation of Canada. M. J. Beyak was supported by a CIHR/CAG/Astra-Zeneca Research Initiative Award. The authors wishto thank Margaret O’Reilly and Iva Kosatka for their experttechnical assistance in the performance of these experiments.
Ileitis modulates sensory nerve K+ and Na+ currentsJ Physiol 552.3 807


















