
IL SISTEMADI GESTIONE QUALITÀA ….pdf · ILSISTEMADI GESTIONE QUALITÀA...
67
IL SISTEMA DI GESTIONE QUALITÀ A SUPPORTO DELLA CERTIFICAZIONE DEI DISPOSITIVI MEDICI - LINEE ESSENZIALI SSFA / dic - 2016 - LINEE ESSENZIALI Marco Delbò – [email protected] - +393387210330 1
Transcript of IL SISTEMADI GESTIONE QUALITÀA ….pdf · ILSISTEMADI GESTIONE QUALITÀA...

IL SISTEMA DI GESTIONE QUALITÀ A
SUPPORTO DELLA CERTIFICAZIONE
DEI DISPOSITIVI MEDICI
- LINEE ESSENZIALI
SSFA / dic - 2016
- LINEE ESSENZIALI
Marco Delbò – [email protected] - +393387210330
1

a cura di: MARCO DELBÒ[email protected] / +39 338 7210330
Independent Medical Devices Professional
Microbiologist
Consultant: Regulatory Affairs and Quality Assurance; Certification of Medical Devices; Risk Analysis; Technical Files preparation; CE marking of MD; Quality Systems Certification, Canadian Regulations CMDCAS; FDA 510k; ISO 9001; ISO 13485; Internal Audit
Auditor Team Leader: ISO 9001; ISO 13485; CE marking; Canadian Regulations CMDCAS; Unannounced Audits
2
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: QUADRO LEGISLATIVO
DIRETTIVA 93/42/CEE DEL CONSIGLIO del 14 giugno 1993
concernente i dispositivi medici
M1 Direttiva 98/79/CE del Parlamento europeo e del Consiglio del 27
Modificata da:
Recepita DL46/97
3
Marco Delbò – [email protected] - +393387210330 +7
M1 Direttiva 98/79/CE del Parlamento europeo e del Consiglio del 27 ottobre 1998
M2 Direttiva 2000/70/CE del Parlamento europeo e del Consiglio del 16 novembre 2000
M3 Direttiva 2001/104/CE del Parlamento europeo e del Consiglio del 7 dicembre 2001
M4 Regolamento (CE) n. 1882/2003 del Parlamento europeo e del Consiglio del 29 settembre 2003
M5 Direttiva 2007/47/CE del Parlamento europeo e del Consiglio del 5 settembre 2007

MARCATURA CE: QUADRO LEGISLATIVO
CHANGES - UPCOMING NEW MD REGULATION
DIRETTIVA 93/42/CEE M5
NEW MDR (Medical Device Regulation)
Verrà sostituita da:
4
Marco Delbò – [email protected] - +393387210330 +3
NEW MDR (Medical Device Regulation)Proposal for a Regulation of the European Parliament and of the Council on medical devices, and amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009
Brussels, 27 June 2016

FAQCouncil of the European Union
Brussels, 27 June 2016 Proposal for a
REGULATION OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL
on medical devices, and amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No
Marcatura CE: quadro legislativoCHANGES - UPCOMING NEW MD REGULATION
Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009
This is based on the consolidated version of the Medical DevicesRegulation (MDR) that has been adopted by the EuropeanParliament and the Member States in June 2016. This text has beendescribed by one of the stakeholders as ‘Tentatively AgreedConsolidated Compromise Text’, which illustrates the delicatenature of this document. There is a small possibility that somechanges are made by the European Commission’s Legal Serviceswhen this compromise gets a final review for legal inconsistencies.In general it can be expected that most, if not all of therequirements in this version will be in the final version that will bepublished at the beginning of 2017.
5
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: QUADRO LEGISLATIVO
CHANGES - UPCOMING NEW MD REGULATION
NEW MDR (Medical Device Regulation)
modalità di transizione:
6
Marco Delbò – [email protected] - +393387210330 +2
After publication, there will be a three-year transition period. (early 2020 ??)
certificates issued prior to final implementation will have a maximum validity of five years. all CE Mark certifications issued before implementation of the new regulations will automatically expire four years after the new regulations come into force.

MARCATURA CE: PROCESSO
La marcatura CE di un Dispositivo Medico:
è un processo complesso
7
Marco Delbò – [email protected] - +393387210330 +4
con varie alternative
va correttamente pianificato

MARCATURA CE: PROCESSO: È UN MD?
1: è un dispositivo medico?
non è sempre scontato classificare
8
Marco Delbò – [email protected] - +393387210330 +2
scontato classificare un prodotto come MD

MARCATURA CE: PROCESSO: È UN MD?
1: è un dispositivo medico?«dispositivo medico»: qualunque strumento, apparecchio, impianto,software, sostanza o altro prodotto, utilizzato da solo o in
combinazione, compreso il software destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo, destinato dal fabbricante ad essere impiegato sull’uomo a fini di:
— diagnosi, prevenzione, controllo, terapia o attenuazione di una malattia;malattia;
— diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un handicap;
— studio, sostituzione o modifica dell'anatomia o di un processo fisiologico;
— intervento sul concepimento,la cui azione principale voluta nel o sul corpo umano non sia
conseguita con mezzi farmacologici né immunologici né mediante metabolismo, ma la cui funzione possa essere assistita da questi mezzi; 9
Marco Delbò – [email protected] - +393387210330 +1
MDD - COUNCIL DIRECTIVE 93/42/EEC M5

MARCATURA CE: PROCESSO: È UN MD?1: è un dispositivo medico?‘medical device’ means any instrument, apparatus, appliance, software, implant, reagent, material or other article, intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific medical purposes of: – diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease; – diagnosis, monitoring, treatment, alleviation of or compensation for an injury or disability, – investigation, replacement or modification of the anatomy or of a physiological or pathological process or state, physiological or pathological process or state, – providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations,
and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means. Products specifically intended for the cleaning, disinfection or sterilisation of medical devices and devices for the purpose of control or support of conception shall be considered medical devices
10
Marco Delbò – [email protected] - +393387210330 +1
Medical device regulation –negotiated text 27 June 2016 DRAFT

MARCATURA CE: PROCESSO: È UN MD?
1: è un dispositivo medico?
Poi ci sono le complicazioni:
accessorio
11
Marco Delbò – [email protected] - +393387210330
SI
NO FORSE
accessorio
+12
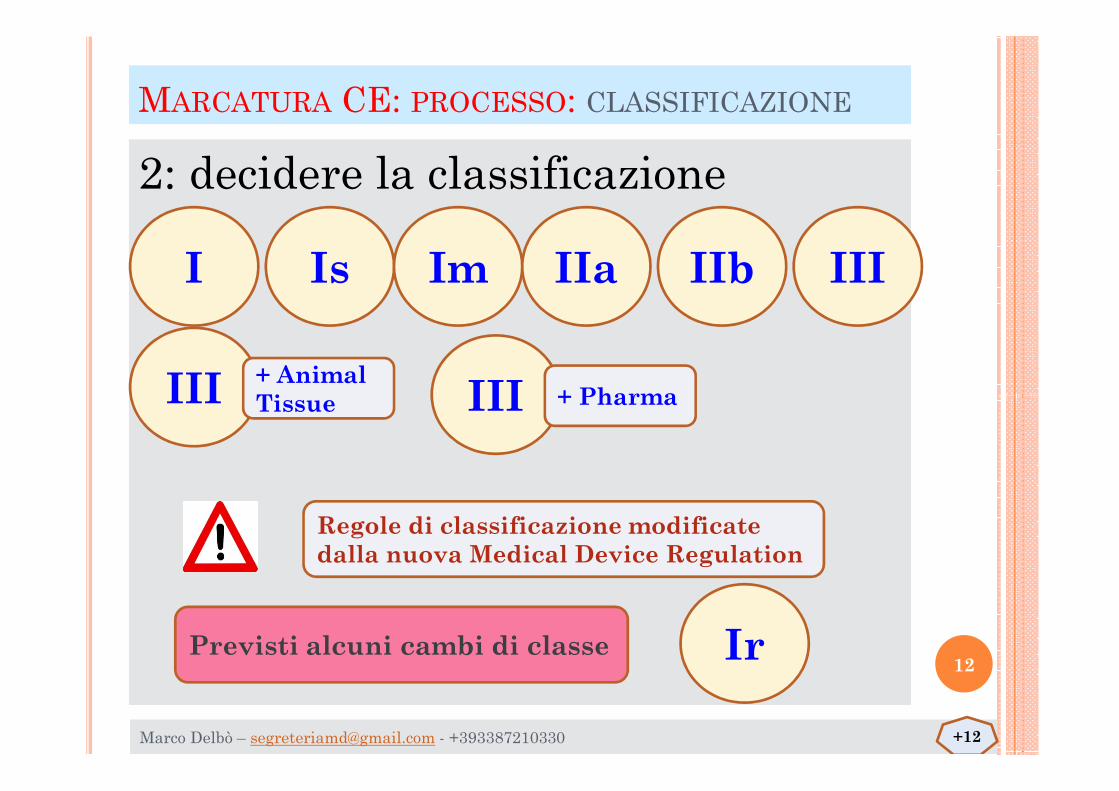
MARCATURA CE: PROCESSO: CLASSIFICAZIONE
2: decidere la classificazione
I Is Im IIa IIb III
III III+ AnimalTissue + Pharma
12
Marco Delbò – [email protected] - +393387210330 +12
Regole di classificazione modificate dalla nuova Medical Device Regulation
III III
Ir
Tissue + Pharma
Previsti alcuni cambi di classe

MARCATURA CE: PROCESSO: ITER DI CERTIFICAZIONE
Iter di Certificazione:
varie alternative per CE
All.
VIIAll.
VAll.
IIAll.
IIIAll.
IV
13
Marco Delbò – [email protected] - +393387210330 +10
nelle varie combinazioni
VII V II III IV
secondo la classe e le priorità aziendali
nuova Medical Device Regulation :cambieranno gli allegati di riferimento, ma il principio rimarrà lo stesso
All.
VI

MARCATURA CE: SGQ
Sistema Gestione Qualità
All.
V
All.
II All.
VIcompleto
14
Marco Delbò – [email protected] - +393387210330 +9
secondo la classe e le priorità aziendali
nuova Medical Device Regulation :cambieranno gli allegati di riferimento, ma il principio rimarrà lo stesso
VIcompletoescl. DD Escl. DD e Prod
Possono essere
obbligatori

MARCATURA CE: SGQ
SGQ: come supportarlo?
poco
EN ISO 9001 Serve?
SINO
Utile?
15
Marco Delbò – [email protected] - +393387210330 +12
GMP Serve?
NObuona base di partenza
SISI
Utile?

MARCATURA CE: SGQ
SGQ: come supportarlo?
è la norma di
EN ISO 13485 Serve?
SISINon obblig
Utile?
16
Marco Delbò – [email protected] - +393387210330 +7
norma di riferimento
SISISI
obbligatorio
Si presume la conformità ai requisiti per i sistemi di qualità che applicano le corrispondenti norme armonizzate

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN 46001:1997 +46002
(EN) ISO 13485:1996 +13488
(EN) ISO 13485:2003
17
Marco Delbò – [email protected] - +393387210330 +14
(EN) ISO 13485:2003/AC:2007
EN ISO 13485:2012 +allegato Z NORMA ARMONIZZATA
(EN) ISO 13485:2016 01/03/2016
three-year transition period > 28/02/2019
SARA’ NORMA ARMONIZZATA
No new ISO 13485:2003 certificates issued in final year of transition

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
Non ancora tradotta
21 set 2016
18
Marco Delbò – [email protected] - +393387210330 +3
In attesa di approvazione di nuovo CORRIGENDUM che modifica allegati Z della ISO13485:2016

MARCATURA CE: SGQ
EN ISO 13485:2016 �� EN ISO 9001:2015
Struttura NON più condivisa
19
Marco Delbò – [email protected] - +393387210330 +3
Struttura NON più condivisa
Le norme non saranno in contrasto, ma non più allineate

MARCATURA CE: SGQ
EN ISO 13485 �� GMP
≠ = Struttura NON condivisa
Management review
Produzione e controlli
calibrazioni
20
Marco Delbò – [email protected] - +393387210330 +10
vigilance
Scelte basate su RiskManagement, meno prescrittive
Ad es: classe cleanroom, validazioni ster. e non sterility test
Training, qualifica
ATTITUDINE
Le aziende GMP parlano la stessa lingua, hanno la giusta mentalità

MARCATURA CE: SGQ
EN ISO 13485 �� GMP
≠ general differences:
pharma is strictly regulated with clear indications, based on GMP guidelines and Pharmacopoeia
21
Marco Delbò – [email protected] - +393387210330
MD more flexible, based on risk management and applicable standards; more options based on company decisions

MARCATURA CE: SGQ
Estensione da GMP a EN ISO 13485
right attitude, correct approach
correct structure generally in place (i.e.: vigilance)
IMPLEMENTED PROCEDURES generally suitable: manufacturing, validations, controls,
22
Marco Delbò – [email protected] - +393387210330 +3
suitable: manufacturing, validations, controls, calibration, environmental controls, job descriptions and training, management of deviations

MARCATURA CE: SGQ
Estensione da GMP a EN ISO 13485
approach to apply the pharma rules for medical devices, resulting in overcomplicated and often misleading processes
main differences: TF management, risk
23
Marco Delbò – [email protected] - +393387210330 +3
main differences: TF management, risk management, vigilance, CAPA management, internal audit, D&D and certification process, management review

MARCATURA CE: SGQ – QUANDO CERTIFICARSI?
Finchè non ho il CE non vendo
Finchè non produco non posso chiedere il CE
FRETTA
Prima di audit di certificazione:
Marco Delbò – [email protected] - +393387210330 +7
24
Prima di audit di certificazione:
Almeno un paio di mesi di dati, di applicazione del SGQ

MARCATURA CE: SGQ – QUANDO CERTIFICARSI?
Almeno un paio di mesi di dati, di applicazione del SGQ
Almeno un esempio di applicazione dei processi, ad es:
Almeno un lot di produzione
anche pilota (prodotti rappresentativi)
Marco Delbò – [email protected] - +393387210330 +8
25
anche pilota (prodotti rappresentativi)
Se voglio certificare protesi sterili e suostrumentario non sterile
Ideale entrambi iprodotti

MARCATURA CE: SGQ – QUANDO CERTIFICARSI?
Almeno un paio di mesi di dati, di applicazione del SGQ
Almeno un esempio di applicazione dei processi, ad es:
Almeno un lot di produzione
Almeno un Riesame direzione
Marco Delbò – [email protected] - +393387210330 +5
26
Almeno un Riesame direzione
Almeno un ciclo V.I.I.
Almeno un piano per PMS
Almeno un progetto (se applicabile)

MARCATURA CE: SGQ – CHI SI DEVE AUDITARE?
Legal manufacturer
Fornitori critici, ad es:
sterilizzazione
filling
A meno che non siano certificati
ISO 13485 o
Tutte le location applicabili
Marco Delbò – [email protected] - +393387210330 +9
27
filling
Etichettatura / packaging
Materie prime critiche (es: ac. Ialuronico, principiattivi, tessuti d’origine animale, etc.)
Processi in outsourcing (es: DD, Vigilance, etc.)
ISO 13485 o GMP

MARCATURA CE: SGQ – CHI SI DEVE AUDITARE?
OBL
Virtual manufacturer:
Marco Delbò – [email protected] - +393387210330 +4
28
Virtual manufacturer:
Deve dimostrare competenza e avere i FT

MARCATURA CE: SGQ – COSA SI DEVE AUDITARE?
CERTIFICAZIONE
Fornitori critici / scopo / allegati
Implementati tutti i processi
Stage 1: si verifica che l’azienda sia pronta
Marco Delbò – [email protected] - +393387210330 +8
29
Stage 2: audit completo
SORVEGLIANZA
Almeno annuale
+ UAV (Verifiche NON annunciate)

MARCATURA CE: SGQ – COSA SI DEVE AUDITARE?
CERTIFICAZIONE
Se non già certificatiAnche processi in outsourcing
Tutti i processi, tutti i punti della ISO13485
i Fascicoli Tecnici
Marco Delbò – [email protected] - +393387210330 +8
30
i Fascicoli Tecnici
SORVEGLIANZA
processi, FT, punti della ISO13485, a campione
Sempre: Responsabilità Direzione, VII, NC, CAPA, Vigilance

MARCATURA CE: SGQ – DURATA CERTIFICATI
CERTIFICAZIONE ISO 13485
Max 3 ANNI
Non oltre transizione nuova edizione
Marco Delbò – [email protected] - +393387210330 +6
31
Marcatura CE (annex V, II, III)
Max 5 ANNI
Non oltre transizione nuovo regolamento

MARCATURA CE: SGQ – DURATA VISITA
Proporzionale alle dimensioni (numero dipendenti, FTE)
+ increasing factors
- Decreasing factors Processi ripetitivi
Marco Delbò – [email protected] - +393387210330 +4
32
Molte NC
Shift
ripetitivi
Processi non
applicabili
Molti processi
SGQ maturo
con poche NC
Processi comples
si
interprete

MARCATURA CE: SGQ – VITA VISSUTA
ERRORI PRINCIPALI – strategia errata
Scelta errata del NB (non è NB o non copre la famiglia di prodotto da certificare, o l’allegato)
Scelto Allegato errato (o non applicabile o inutile)
Marco Delbò – [email protected] - +393387210330 +5
33
Dispositivi border line: classificazione non accettata da NB (classe / non è Disp. Medico)

MARCATURA CE: SGQ – VITA VISSUTA
ERRORI PRINCIPALI – preparazione errata
Azienda non pronta (SGQ non ancora completato, nessuna produzione, nessun batch record)
Fornitori critici /location : elenco incompleto (se ne scoprono di nuovi durante audit)
Marco Delbò – [email protected] - +393387210330 +5
34
Visita ai fornitori: contratto non disponibile; nessuna produzione in corso ( o nessun prodotto simile da prendere ad esempio)
ne scoprono di nuovi durante audit)

MARCATURA CE: SGQ – VITA VISSUTA
ERRORI PRINCIPALI – preparazione errata
Risk management: non c’è collegamento tra SGQ e Analisi dei Rischi
VII non eseguite, o eseguite da personale non qualificato o non correttamente documentate
Marco Delbò – [email protected] - +393387210330 +7
35
Riesame Direzione: NON è una valutazione, non copre tutti i punti (es: nuovi requisiti regolatori)
qualificato o non correttamente documentate
Non è pianificata PMS
Fornitori critici non qualificati (anche consulenti)

MARCATURA CE: SGQ – VITA VISSUTA
ERRORI PRINCIPALI – attitudine errata
qual è il giusto approccio del fabbricante?
Marco Delbò – [email protected] - +393387210330 +4
36

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 – differenze principali della nuova edizione
general changes
Many additions
Some new requirements
37
Marco Delbò – [email protected] - +393387210330 +4
Some new requirements
Some expansion & clarification
Increased clarity of interrelationshipbetween clauses and requirements

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016
1 Scope
2 Normative references
3 Terms and definitions
changes
does not align with the new ISO 9001:2015 changes
standard’s numbering and clauses:
38
Marco Delbò – [email protected] - +393387210330 +4
standard’s numbering and clauses:- new clauses and sub-clauses have been added- In order to work with (MDSAP) NC-grading, (sub)clauses required formatting
facilitate global alignment (i.e. MDSAP)
allow organizations to be involved in different stages of the product life-cycle (extended scope)

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016
1 Scope
2 Normative references
3 Terms and definitions
changes
risk-based approach to control QMS processes
emphasis on understanding of, and adherence, to
39
Marco Delbò – [email protected] - +393387210330 +3
emphasis on understanding of, and adherence, to international regulatory requirements
a number of definitions that were added, i.e.: clinical evaluation, complaint, distributor, importer, lifecycle, manufacturer, medical device family, performance evaluation, postmarket surveillance, purchased product, risk, risk management and sterile barrier system.

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016
4 Quality management system4.1 General requirements
4.2 Documentation requirements
4.2.1 General
4.2.2 Quality manual
4.2.3 Medical device file4.2.3 Medical device file
4.2.4 Control of documents
4.2.5 Control of records
40
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016
4 Quality management system4.1 General requirements
4.2 Documentation requirements
4.2.1 General
4.2.2 Quality manual
4.2.3 Medical device file
changes
risk-based approach to control QMS processes (+ outsourced)
Roles to be documented to meet regulatory requirements (i.e., specifying the legal manufacturer, defining processes, and identifying records to be kept to meet regulatory requirements)
4.2.3 Medical device file
4.2.4 Control of documents
4.2.5 Control of records
41
Marco Delbò – [email protected] - +393387210330 +4
software used for the QMS to be validated prior to initial use and after any change
Medical Device File (similar to a technical file) for regulatory purposes, and the establishment of methods to protect confidential health information + Detailed list of items (a-f) to be included to meet regulatory requirements

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:20165 Management responsibility5.1 Management commitment5.2 Customer focus5.3 Quality policy5.4 Planning5.4.1 Quality objectives5.4.2 Quality management system planning5.4.2 Quality management system planning5.5 Responsibility, authority and communication5.5.1 Responsibility and authority5.5.2 Management representative5.5.3 Internal communication5.6 Management review5.6.1 General5.6.2 Review input5.6.3 Review output 42
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016
EN ISO 13485:20165 Management responsibility5.1 Management commitment5.2 Customer focus5.3 Quality policy5.4 Planning5.4.1 Quality objectives5.4.2 Quality management system planning
changes
Increased emphasis on regulatory requirements
Responsibilities and authority documented, including the 5.4.2 Quality management system planning5.5 Responsibility, authority and communication5.5.1 Responsibility and authority5.5.2 Management representative5.5.3 Internal communication5.6 Management review5.6.1 General5.6.2 Review input5.6.3 Review output
43
Marco Delbò – [email protected] - +393387210330 +3
Responsibilities and authority documented, including the interrelation of personnel affecting quality and liaison with external parties and regulatory authorities
Input and output requirements have been added to maintain the suitability and adequacy of the QMS

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:20166 Resource management6.1 Provision of resources6.2 Human resources6.3 Infrastructure6.4 Work environment and contamination control6.4 Work environment and contamination control6.4.1 Work environment6.4.2 Contamination control
44
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:20166 Resource management6.1 Provision of resources6.2 Human resources6.3 Infrastructure6.4 Work environment and contamination control
changes
Documented processes required to establish competence, provide training and ensure quality awareness throughout the organization
emphasis on defining, maintaining, and updating the 6.4 Work environment and contamination control6.4.1 Work environment6.4.2 Contamination control
45
Marco Delbò – [email protected] - +393387210330 +2
emphasis on defining, maintaining, and updating the competence of personnel involved with quality system compliance and regulatory requirements. required to evaluate effectiveness of any training commensurate with the risks associated with the work individuals perform

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:20166 Resource management6.1 Provision of resources6.2 Human resources6.3 Infrastructure6.4 Work environment and contamination control
changes
emphasis on maintenance-related activities of production, control, work env, monitor and measurement equipment - companies must have clearly documented
6.4 Work environment and contamination control6.4.1 Work environment6.4.2 Contamination control
46
Marco Delbò – [email protected] - +393387210330 +2
equipment - companies must have clearly documented procedures to specify how those activities are being performed.
new requirement to handle orders in a streamlined way to prevent mix-ups

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:20166 Resource management6.1 Provision of resources6.2 Human resources6.3 Infrastructure6.4 Work environment and contamination control
changes
control of work environment by specifying requirements for health, cleanliness, and clothing of personnel involved in production activities to prevent cross-contamination
adds a reference to ISO 14644 (air cleanliness classification 6.4 Work environment and contamination control6.4.1 Work environment6.4.2 Contamination control
47
Marco Delbò – [email protected] - +393387210330 +3
adds a reference to ISO 14644 (air cleanliness classification and monitoring) and ISO 14698 (cleanrooms and associated control environments). Manufacturers must consider conditions such as noise, temperature, humidity, lighting, and adequate infrastructure for manufacturing, inspection, storage, and distribution areas.
planned arrangements for controlling (potentially) contaminated product in order to prevent contamination of work environment, personnel, or product

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.1
7 Product realization7.1 Planning of product realization
changes
document how risk-management activities are being handled
48
Marco Delbò – [email protected] - +393387210330
Planned processes for verification, validation, monitoring, measurement, inspection, test activities, handling, storage, distribution, and traceability
Documented risk management throughout development-planning activities
+3

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.2
7 Product realization� 7.2 Customer-related processes� 7.2.1 Determination of requirements related to product
� 7.2.2 Review of requirements related to product
� 7.2.3 Communication� 7.2.3 Communication
49
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.2
7 Product realization� 7.2 Customer-related processes� 7.2.1 Determination of requirements related to product
� 7.2.2 Review of requirements related to product
� 7.2.3 Communication
changes
Manufacturers must determine whether there is a need for user training to ensure specified performance and safe use of the medical device + any user training identified is available or planned to be available…� 7.2.3 Communication
50
Marco Delbò – [email protected] - +393387210330
Manufacturers must ensure that applicable regulatory requirements are met as part of the product review requirements
documented arrangements must be in place for communicating with regulatory authorities ( product information, regulatory inquiries, complaints, and advisory notices)
+3
available or planned to be available…

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.3
7 Product realization� 7.3 Design and development� 7.3.1 General
� 7.3.2 Design and development planning
� 7.3.3 Design and development inputs� 7.3.3 Design and development inputs
� 7.3.4 Design and development outputs
� 7.3.5 Design and development review
� 7.3.6 Design and development verification
� 7.3.7 Design and development validation
� 7.3.8 Design and development transfer
� 7.3.9 Control of design and development changes
� 7.3.10 Design and development files 51
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.3
7 Product realization� 7.3 Design and development� 7.3.1 General
� 7.3.2 Design and development planning
� 7.3.3 Design and development inputs
changes
requirement to update documents with DD progress and to ensure traceability of outputs to inputs� 7.3.3 Design and development inputs
� 7.3.4 Design and development outputs
� 7.3.5 Design and development review
� 7.3.6 Design and development verification
� 7.3.7 Design and development validation
� 7.3.8 Design and development transfer
� 7.3.9 Control of design and development changes
� 7.3.10 Design and development files 52
Marco Delbò – [email protected] - +393387210330
competence of personnel in design and development activities must be defined and demonstrated, and a review by a specialist (independent reviewer) is now required
inputs must include usability - standards,- and should be verified and validated. Outputs must be in a form suitable for verification against inputs
+3

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.3
7 Product realization� 7.3 Design and development� 7.3.1 General
� 7.3.2 Design and development planning
� 7.3.3 Design and development inputs
changes
verification and validation plan must be documented, including methods, criteria for acceptance, failure, justification for sample sizes selected� 7.3.3 Design and development inputs
� 7.3.4 Design and development outputs
� 7.3.5 Design and development review
� 7.3.6 Design and development verification
� 7.3.7 Design and development validation
� 7.3.8 Design and development transfer
� 7.3.9 Control of design and development changes
� 7.3.10 Design and development files 53
Marco Delbò – [email protected] - +393387210330
verification and validation of connections and interfaces
Validation must be conducted on final production units (representative product) or documented equivalent devices
+4
verification + validation of user instructionsrequired, including review of failure modes

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.3
7 Product realization� 7.3 Design and development� 7.3.1 General
� 7.3.2 Design and development planning
� 7.3.3 Design and development inputs
changes
DD Transfer: new clause
requires a documented plan for design transfer to � 7.3.3 Design and development inputs
� 7.3.4 Design and development outputs
� 7.3.5 Design and development review
� 7.3.6 Design and development verification
� 7.3.7 Design and development validation
� 7.3.8 Design and development transfer
� 7.3.9 Control of design and development changes
� 7.3.10 Design and development files 54
Marco Delbò – [email protected] - +393387210330
requires a documented plan for design transfer to another facility or outsourcing partner. should include plans for supplier quality and capability, manufacturing personnel capability and training, manufacturing process and process validation, materials, manufacturing tools and methods, work environment, installation, and service
+2

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.3
7 Product realization� 7.3 Design and development� 7.3.1 General
� 7.3.2 Design and development planning
� 7.3.3 Design and development inputs
changesDD Changes and Files: new subclause, more detail
link to risk management and product realizationadded, + detail regarding determining significance of change� 7.3.3 Design and development inputs
� 7.3.4 Design and development outputs
� 7.3.5 Design and development review
� 7.3.6 Design and development verification
� 7.3.7 Design and development validation
� 7.3.8 Design and development transfer
� 7.3.9 Control of design and development changes
� 7.3.10 Design and development files 55
Marco Delbò – [email protected] - +393387210330
change
+4
requirements to control changes, documentation to be kept, approval requirements, reports and testing to justify changes, etc
DD files to be maintained for each medical device type /family. must include or reference records to demonstrate conformity to the requirements for D&D, and records to justify changes

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.4
7 Product realization
�7.4 Purchasing�7.4.1 Purchasing process�7.4.2 Purchasing information�7.4.2 Purchasing information�7.4.3 Verification of purchased product
56
Marco Delbò – [email protected] - +393387210330

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.4
7 Product realization
�7.4 Purchasing�7.4.1 Purchasing process�7.4.2 Purchasing information
changes
Criteria for evaluation and selection of suppliersincludes performance and risk
Supplier performance monitoring as part of re-evaluation process, additional record requirements�7.4.2 Purchasing information�7.4.3 Verification of purchased product
57
Marco Delbò – [email protected] - +393387210330
evaluation process, additional record requirements
+5
Purchasing information to include, as applicable, product specifications
Suppliers to agree to prior notification of changes
Verification of processes based on risks and results of evaluations and reevaluations; link to change control

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.5
7 Product realization
� 7.5 Production and service provision� 7.5.1 Control of production and service provision� 7.5.2 Cleanliness of product� 7.5.3 Installation activities� 7.5.4 Servicing activities� 7.5.5 Particular requirements for sterile medical devices� 7.5.5 Particular requirements for sterile medical devices� 7.5.6 Validation of processes for production and service provision� 7.5.7 Particular requirements for validation of processes for
sterilization and� sterile barrier systems� 7.5.8 Identification� 7.5.9 Traceability� 7.5.10 Customer property� 7.5.11 Preservation of product
Marco Delbò – [email protected] - +393387210330
58

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.5
7 Product realization
� 7.5 Production and service provision� 7.5.1 Control of production and service provision� 7.5.2 Cleanliness of product� 7.5.3 Installation activities� 7.5.4 Servicing activities� 7.5.5 Particular requirements for sterile medical devices
changes
Production / service to be planned, monitored, and controlled to ensure products conforms to specifications
Cleanliness: adds contamination control
Servicing: activity records to be analyzed to determine if should be considered and
� 7.5.5 Particular requirements for sterile medical devices� 7.5.6 Validation of processes for production and service provision� 7.5.7 Particular requirements for validation of processes for
sterilization and� sterile barrier systems� 7.5.8 Identification� 7.5.9 Traceability� 7.5.10 Customer property� 7.5.11 Preservation of product
Marco Delbò – [email protected] - +393387210330 +4
if events in the field should be considered and reported as complaints, or used to improve the product as part of design inputs
Validation: Processes must be validated where output cannot be, or is not, verified. + need for procedures. Statistical techniques applied and rationale for sample sizes must be documented, including approval of changes and validation of software after changes (risk based)
59

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.5
7 Product realization
� 7.5 Production and service provision� 7.5.1 Control of production and service provision� 7.5.2 Cleanliness of product� 7.5.3 Installation activities� 7.5.4 Servicing activities� 7.5.5 Particular requirements for sterile medical devices
changes
Validation of sterilization and sterile barriers
Validation required prior to implementation / Changes+ Document results, conclusions, actions� 7.5.5 Particular requirements for sterile medical devices� 7.5.6 Validation of processes for production and service provision� 7.5.7 Particular requirements for validation of processes for
sterilization and� sterile barrier systems� 7.5.8 Identification� 7.5.9 Traceability� 7.5.10 Customer property� 7.5.11 Preservation of product
Marco Delbò – [email protected] - +393387210330 +5
UDI (where required by regulations)
60
procedures for separation of returned products fromconforming product
shipping conditions and their impact on product and package integrity, including management of sterile device packaging, to be assessed

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §7.6
� 7.6 Control of monitoring and measuring equipment
changes
Requirements for the validation of the application of software used for monitoring and measurement of
Marco Delbò – [email protected] - +393387210330 +5
61
software used for monitoring and measurement of requirements. Risk based approach required.

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §8� 8 Measurement, analysis and improvement� 8.1 General� 8.2 Monitoring and measurement� 8.2.1 Feedback� 8.2.2 Complaint handling� 8.2.3 Reporting to regulatory authorities� 8.2.4 Internal audit� 8.2.5 Monitoring and measurement of processes� 8.2.5 Monitoring and measurement of processes� 8.2.6 Monitoring and measurement of product� 8.3 Control of nonconforming product� 8.3.1 General� 8.3.2 Actions in response to nonconforming product detected before delivery� 8.3.3 Actions in response to nonconforming product detected after delivery� 8.3.4 Rework� 8.4 Analysis of data� 8.5 Improvement� 8.5.1 General� 8.5.2 Corrective action� 8.5.3 Preventive action
Marco Delbò – [email protected] - +393387210330
62

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 §8� 8 Measurement, analysis and improvement� 8.1 General� 8.2 Monitoring and measurement� 8.2.1 Feedback� 8.2.2 Complaint handling� 8.2.3 Reporting to regulatory authorities� 8.2.4 Internal audit� 8.2.5 Monitoring and measurement of processes
changes
Feedback procedures --> input to risk management and improvement process. Clause strengthened
Complaint handling & Reporting to regulatoryauthorities (New Clauses)� 8.2.5 Monitoring and measurement of processes� 8.2.6 Monitoring and measurement of product� 8.3 Control of nonconforming product� 8.3.1 General� 8.3.2 Actions in response to nonconforming product detected before delivery� 8.3.3 Actions in response to nonconforming product detected after delivery� 8.3.4 Rework� 8.4 Analysis of data� 8.5 Improvement� 8.5.1 General� 8.5.2 Corrective action� 8.5.3 Preventive action
Marco Delbò – [email protected] - +393387210330 +5
63
authorities (New Clauses)
Monitoring and Measurement of Product: Records now must identify the test equipment used to perform measurement activities
NC products: + details in respect of controls, concessions, records (Clause restructured)
Verify that CAPA does not have an adverse effect; actions to be taken without undue delay

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 ANNEXESAnnex A (informative) Comparison of content
between ISO 13485:2003 and ISO 13485:2016
Annex B (informative) Correspondence between ISO 13485:2016 and ISO 9001:2015
Annex Z under revision (solo EN)European Annexes - ZA (AIMD), ZB (MDD) and ZC (IVD)Identifies relationship between the European
Standard (EN ISO 13485:2016) and Conformity Assessment Requirements of the respective EU Medical Device Directives via each conformity assessment route for each directive
Marco Delbò – [email protected] - +393387210330
64

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 transizione
Cosa fare?
Marco Delbò – [email protected] - +393387210330
65
+1

MARCATURA CE: SGQ - EN ISO 13485 OVERVIEW
EN ISO 13485:2016 transizioneCosa fare?
- Study the standard- gap analysis of current QMS Vs. new
requirements- Prepare transition plan, with timescales- Factor any additional resources & costs - Factor any additional resources & costs
into budgets- Review staff awareness / knowledge and
determine training required- Compile project / implementation plan- Look out for additional help, information
and resources
Marco Delbò – [email protected] - +393387210330
66

APPROFONDIMENTI:
Thank you for your attention
Any questions?Any questions?
67
Marco Delbò – [email protected] - +393387210330


















