Il dogma centrale della biologiadocente.unife.it/matteo.ramazzotti/bioinfo/01-Biology.pdf · 1....
26
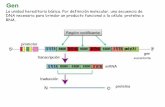
Trascrizione mRNA tRNA rRNA Aminoacil-tRNA TAA Sintesi proteica Struttura primaria Struttura secondaria Struttura terziaria Folding RBS ATG AAA TAC Il dogma centrale della biologia
Transcript of Il dogma centrale della biologiadocente.unife.it/matteo.ramazzotti/bioinfo/01-Biology.pdf · 1....

Trascrizione
mRNA
tRNA
rRNA
Aminoacil-tRNA
TAA
Sintesi proteica
Struttura primaria
Struttura secondaria
Struttura terziaria
Folding
RBS ATG AAA TAC
Il dogma centrale della biologia

Il dogma centrale della biologia (rivisto)
Il DNA, avvolto sugli istoni, deve essere reso accessibile alla polimerasi prima di essere trascritto.
L’mRNA deve essere correttamente modificato (polyA, CAP) perché sia poi possibile eseguire lo
Splicing, che in tessuti diversi o in momenti diversi può produrre diverse “varianti” dello steso gene.
La traduzione è un processo “assistito” da molte proteine che si occupano di proteggere le catene nascenti.
Solo proteine con la giusta strutturazione (folding + modificazioni post traduzionali) sono funzionanti e non vengono rimosse.
Ogni proteina è sottoposta ad un turnover mediato da apposite strutture cellulari.

Il gene procariotico
Nei procarioti non c’è il nucleo, quindi la trascrizione e la traduzione possono avvenire quasi contemporanamente : questa caratteristica influenza l’ organizzazione del gene procariotico.
Si possono riconoscere 3 regioni importanti:
1. Promotore : regioni a monte delle sequenza codificante su cui si legano fattori di trascrizione e di repressione e determinano l’arrivo della RNA polimerasi.
2. Regioni non codificanti (UTR): posti a monte (5’) e a valle (3’) del gene, lo delimitano e contengono i siti di attacco dei ribosomi (RBS) e i segnali per terminare la trascrizione.
3. Regione codificante , dove è contenuta l’informazione per la proteina.

Il gene eucariotico
Diversamente dai procarioti, l’organizzazione di un gene eucariotico è molto più complessa:
1. Esoni : porzioni del gene che contengono la sequenza codificante per la proteina, sono molto piccoli rispetto al gene intero.
2. Introni : porzioni del gene che interrompono gli esoni e che non contengono informazioni per la codifica. Costituiscono la maggior parte del gene.
3. TATA box : quattro basi canonioche a cui si lega la RNA polimerasi II e da cui inizia a svolgere il DNA per trascriverlo.
4. Promotori : regioni a monte delle sequenza codificante a cui si legano i fattori di trascrizione, che a loro volta chiamano la RNA polimerasi.
5. Enhancers : posti a monte e/o a valle del gene, potenziano/favoriscono l’azione dei promotori guidando dei particolari ripiegamenti nel DNA.

Trascritto primario e splicingLo splicing avviene dopo che un gene è stato trascritto. Il trascritto primario è molto più lungo di quanto serve perché contiene regioni non codificanti (introni) che vanno rimosse in modo da mettere insieme quelle codificanti (esoni). Il complesso detto spliceasoma si occupa di questo processo “taglia e cuci” riconoscendo sequenze specifiche sull’RNA (GT al 5’ e AG al 3’).

Strutture degli RNAL’RNA, essendo composto da 2 copie di basi complementari come il DNA, non si trova quasi mai nello stato di singolo filamento ma forma strutture secondarie con funzioni biologiche basilari.
l’RNA ribosomiale assume una struttura che guida l’assemblaggio del ribosoma, chiama a sé le proteine ribosomiali e costituisce il punto di aggancio per la formazione del complesso di inizio traduzione.
l’RNA transfer si struttura a trifoglio e in base alla struttura assunta viene riconosciuto da diverse aminoacil-tRNAsintetasi che guidano la formazione del legame ad alta energia tra Aminoacido e RNA, fondamentale per la sintesi proteica.

Si caratterizza come
- UNIVERSALE (…)
- DEGENERATO
- RIDONDANTE
E’ composto da 64 diversi codoni che codificano i 20 amino acidi.
La tebella accanto mostra il sistema di decodifica guidato dal dal ribosoma.
Sono evidenti le degenerazioni a carico delle basi 2 e 3 e la ridondanza complessiva.
Alanine Ala A GC[CATG]
Cysteine Cys C TG[CT]
Aspartic AciD Asp D GA[CT]
Glutamic Acid Glu E GA[AG]
Phenylalanine Phe F TT[CT]
Glycine Gly G GG[CATG]
Histidine His H CA[CT]
Isoleucine Ile I AT[CAT]
Lysine Lys K AA[AG]
Leucine Leu L CT[CATG], TT[AG]
Methionine Met M ATG
AsparagiNe Asn N AA[CT]
Proline Pro P CC[CATG]
Glutamine Gln Q CA[AG]
ARginine Arg R CG[CATG], AG[AG]
Serine Ser S TC[CTAG], AG[CT]
Threonine Thr T AC[CATG]
Valine Val V GT[CATG]
Tryptophan Trp W TGG
TYrosine Tyr Y TA[CT]
STOP - - TA[AG], TGA
Il codice genetico

Gli amino acidi e il legame peptidico
Agli amino acidi sono composti organici chirali che presentano almeno un gruppo carbossilico (-COOH) a funzione acida e un gruppo aminico (-NH2) a funzione basica.
Le proteine sono composte soltanto da α-amino acidi, legati tra loro mediante legami amidici detti PEPTIDICIche si instaurano tra il gruppo α-aminico e il gruppo α-carbossilico.
Ciò che diversifica i vari amino acidi è la catena laterale legata al carbonio α, che può conferire all’amino acido caratteristiche chimico-fisiche diverse.

Classi di amino acidi
Idrofobici
Aromatici
Acidi StrutturaliBasici
Polari, non carichi

I polimeri di α-amino acidi (le proteine) sono influenzati dalle caratteristiche chimico-fisiche delle catene laterali e, in base a principi di interazioni deboli di tipo idrofobico o elettrostatico, si possono ripiegare, fino a raggiungere la minor energia termodinamica.
ImportanteLa strutturazione delle proteine dipende principalmente dalla sequenza dei residui che la compongono, oltre che dall’ambiente in cui si strutturano
Questo processo, denominato FOLDING, è alla base del funzionamento delle proteine, visto che solo se sono correttamente strutturate esse assumeranno la loro forma e soprattutto la loro FUNZIONE definitiva.
Folding delle proteine

Il backbone delle proteine
Il legame peptidico ha delle caratteristiche di doppio legame e costringe i due atomi adiacenti C=O e N-H a giacere sullo stesso piano.
La rotazione della molecola può avvenire solo intorno al carbonio αααα, ma non tutti gli angoli di rotazione sono permessi a causa degli ingombri sterici delle diverse catene laterali e dello scheletro stesso.

Il legame peptidico genera una polarità negli scheletri proteici per cui si vengono a formare PONTI IDROGENO tra i gruppi amidici e i gruppi carbonilici di aminoacidi diversi. Queste interazioni deboli portano la struttura primaria della proteina (la sequenza dei suoi residui) a ripiegarsi in una STRUTTURA SECONDARIA in cui sono riconoscibili due formazioni
Alfa elica
struttura compatta e avvolta in cui i legami idrogeno sono disposti parallelamente allo scheletro, stabilizzando aminoacidi vicini tra loro.
Beta-strand
struttura rilassata in cui i ponti idrogeno si stabiliscono tra catene adiacenti che possono essere parallele o antiparallele, a formare i foglietti β
Struttura secondaria delle proteine

Il plot di Ramachandran
Nel 1963 GN Ramachandran, utilizzando un modello a sfere, gettò le basi che permisero di interpretare la stereochimica delle proteine.
Egli propose di descrivere gli aminoacidi di una proteina in termini di angoli torsionali del backbone: queste rotazioni dei vari angoli φ e ψnon potevano essere arbitrarie, ma dovevano seguire delle logiche di ingombro sterico delle catene laterali.
Dimostrò quindi che le strutture secondarie avevano tutte un inquadramento preciso in termini di φ e ψ.
Foglietto β: Φ tra - 80° e - 120°
Ψ tra 120° e 170°
Elica α: Φ tra - 40° e - 100°
Ψ tra -40° e -65°

Per ottenere un ripiegamento corretto le varie strutture secondarie si collegano tra loro mediante sequenze in cui non ci sono ponti idrogeno fissi e spesso nemmeno altri tipi di interazioni intra-molecolari, definiti “random coil”, o strutture disordinate.
Regioni ordinate e regioni non ordinate
In certe proteine però esistono adattamenti strutturali specifici che contribuiscono in modo decisivo alla strutturazione delle proteina e quindi alla sua funzione definitiva: sono i cosiddetti “loop” e sono stati classificati in base al tipo di struttura secondaria che congiungono in
alpha-alpha, alpha-beta, beta-alpha, beta-beta links e beta-hairpins.
http://www.bmm.icnet.uk/loop/index.html

β-turn-βup-down greca
super-barrel
jelly-roll α-turn-α
β-α-β
coiled-coil
four helixbundle
fold di Rossmann
Strutturazioni successiveCongiungendosi, le strutture secondarie formano motivi comuni e conservati che prendono il nome di strutture supersecondarie , dette anche MOTIVI. Tali strutture si organizzano poi a dare le vere strutture terziarie e quaste, assemblandosi, formano le strutture quaternarie.

Dominio singolo Dominio doppio
Dominio triplo Dominio quadruplo
Esempi di strutture terziarie

Strutture quaternarie e simmetrie

Targeting delle proteine

Denaturazione del DNA: la temperatura di melting
Tm°C = 2 × AT + 4 × GC
Tm°C = 69.3 + (0.41*GC%) – (650/N)
3’-tacgacactacgactacagacgaatactacgacatacagacgactag- 5’ 5’-atgctgtgatgctgatgtctgcttatgatgctgtatgtctgctgatc- 3’
c = 13g = 8a = 20t = 7tot = 48tot GC = 21 (44.75%)tot AT = 27 (56.25 %)Tm = 74.11
Se scaldo una soluzione contenente una molecola di DNA come quella scritta sopra fino a 75 °C, metà dei doppi filamenti saranno aperti. Usando il calore per svolgere il DNA simulo l’azione delle elicasi e delle topoisomerasi.
Alcune formule per calcolare la Tm

I primers - inneschi 3’-OH
3’-tacgacactacgactacagacgaatactacgacatacagacgactagt acgacatacagacgactag-5’ 5’-atgctgtgatgctgatgtctgcttatgatgctgtatgtctgctgatca tgctgtatgtctgctgatc-3’
Supponiamo di voler amplificare un tratto di DNA con questa sequenza
Devo creare due sequenze che si “fronteggiano” e che:
1. si leghino allo stampo (quindi devono essere complementari al DNA da copiare)2. forniscano un 3’-OH libero nella direzione di interesse.
3’-tacgacactacgactacagacgaatactacgacatacagacgactagt acgacatacagacgactag-5’ ||||||||||||||
5’-atgctgtgatgctg-3’ chiamiamolo primer diretto
chiamiamolo primer inverso 3’-tacagacgactag-5’|||||||||||||
5’-atgctgtgatgctgatgtctgcttatgatgctgtatgtctgctgatca tgctgtatgtctgctgatc-3’
Per “disegnare” dei primers bisogna conoscere la sequenza che si vuole amplificare, e per far questo si guardano le banche dati di DNA, cioè collezioni tratti di DNA ottenuti per esempio dai sequenziamenti genomici. Ottenuta la sequenza si procede alla produzione (oggi si acquistano) degli stessi.
Per ogni primer che si disegna bisogna calcolare la temperatura di melting (quindi sapere lunghezza e composizione in basi): primer diretto e inverso infatti devono avere Tm molto simili!

Enzimi di restrizione
Esistono tre tipi di enzimi di restrizione:
Tipo I. Riconoscono una sequenza specifica ma tagliano lontano da essa.
Tipo II . Riconoscono una sequenza specifica, spesso palindromica, e tagliano al suo interno.
Tipo III: Proprietà intermedie.
Gli enzimi di tipo II sono i più usati.
Ne sono stati caratteizzati circa 1000 e sono commercialmente disponibili.
La nomenclatura si basa su:
OrganismoCodice
aggiuntivo
Ordine di scoperta
EcoRI EcoRV HindIII

Il clonaggio di frammenti di DNA
Il clonaggio di un gene di interesse rappresenta oggi la tecnica di routine sia per il suo sequenziamento sia per lo studio delle sue funzioni.
Gli step da seguire sono:
0. Ottenimento del DNA
1. Taglio del DNA con ER.
2. Taglio di un plasmide con ER.
3. Incubazione dei due tagliati.
4. Aggiunta di ligasi
5. Inserimento in batteri
Se il sito di clonaggio si trova all’interno di una sequenza codificante (es. il gene LacZ), l’inserimento del gene provoca la perdita di quella funzione.
LacZ è in grado, se c’è, di reagire con un composto aggiunto nel terreno e dare colorazione blu.
=> Posso riconoscere i batteri con inserto.

Librerie di DNASono collezioni di frammenti di DNA ottenute per esempio dalla digestione con un enzima di restrizione di un intero genoma (librerie genomiche): se taglio anche il plasmide con lo stesso enzima, i frammenti e il plasmide si richiuderanno uno sull’altro. I plasmidi possono poi essere introdotti nei batteri e fatti moltiplicare.
Possono essere anche create librerie di cDNA: in questo modo nella libreria avrò i geni già maturati, quindi più piccoli e più gestibili.

Sequenziamento del DNALa possibilità di sequenziare il DNA è stata, una delle principali rivoluzioni per l’ingegneria genetica: fino ad allora si era solo in grado di lavorare sui caratteri fenotipici dei geni, non avendo modo di conoscere la loro seqerunza.
La tecnica oggi utilizzata, detta “ad interruzione di catena” fu introdotta nel 1975 da FrederickSanger e sfrutta la capacità della DNA polimerasi di polimer izzare nucleotidi su DNA stampo ma solo se presente un gruppo 3’OH libero . Fondamentali sono dei nucleotidi modificati detti di-deossi :
- nel ribosio i carboni 2 e 3 portano dei gruppi OH- nel desosiribosio il gruppo OH in posizione 2 è assente- del didesossiribosio anche il groppo 3-OH è rimosso
=> un dideossi-nucleotide inserito dalla polimerasi impedisce l’allungamento della catena
ribosio
desossiribosio
didesossiribosio

DNA MicroarrayE’ una tecnica relativamente nuova che permette il monitoraggio dell’espressione genica di tutti i geni di un genoma in un singolo esperimento.

Sintesi in situ di sondeLa fotolitografia permette la costruzione di array con alto contenuto di informazione permettendo di avere 500,000 probe in uno spazio di 1.28 cm2. Ogni singolo probe è costituito da milioni di oligonucleotidi identici, e un singolo array di 1.28 cm2 contiene probe set per circa 40,000 geni umani.