
Identificación y Caracterización de Un Homólogo a FixL en Brucella Abortus Tesis
67
Identificación y caracterización de un homólogo a FixL en Brucella abortus por Juan José Blesa Tesis de Licenciatura en Biotecnología Directora Dra. Marta A. Almirón Instituto de Investigaciones Biotecnológicas Escuela de Ciencia y Tecnología Universidad Nacional de San Martín Diciembre 2010
-
Upload
brucella2308 -
Category
Documents
-
view
6 -
download
0
description
Tesis de LicenciaturaMicrobiologíaBrucella abortus
Transcript of Identificación y Caracterización de Un Homólogo a FixL en Brucella Abortus Tesis

Identificación y
caracterización de un
homólogo a FixL en
Brucella abortus
por
Juan José Blesa
Tesis de Licenciatura en
Biotecnología
Directora
Dra. Marta A. Almirón
Instituto de Investigaciones Biotecnológicas
Escuela de Ciencia y Tecnología
Universidad Nacional de San Martín
Diciembre 2010

Identificación y
caracterización de un
homólogo a FixL en
Brucella abortus
Tesis de Licenciatura en Biotecnología
Dra. Marta A. Almirón
Directora
Juan José Blesa
Autor

3
Agradecimientos
A mis padres, por todo su apoyo y esfuerzo para que en estos días pueda estar
graduándome de la universidad. Sin su amor, y ayuda no hubiese sido
posible.
A todas esas grandes personas que tuve de compañeros en todos estos años,
siempre dando una mano desinteresadamente.
A Marta Almirón, por haberme dado la oportunidad de trabajar bajo su
tutela y compartir conmigo muchos de sus conocimientos y su tiempo.
A todos esos grandes profesores, que compartieron sus enseñanzas conmigo y
todos los alumnos de la universidad.
A todo el personal del Instituto de Investigaciones Biotecnológicas por su
colaboración y muy especialmente a la Universidad Nacional de General
San Martín por la posibilidad de estudiar la Licenciatura en
Biotecnología en un ambiente de excelencia y por el compromiso con la
enseñanza de todos los que allí trabajan.

4
Parte de los resultados obtenidos en este trabajo de Tesis han sido publicados en el siguiente congreso: LIV REUNION CIENTIFICA ANUAL de la Sociedad Argentina de Investigación Clínica (SAIC). Noviembre de 2009, Mar del plata, Buenos Aires, Argentina.
“SÍNTESIS DE UN POSIBLE SENSOR DE OXÍGENO EN BRUCELLA ABORTUS”. Almirón M.; Blesa J.; Ugalde R. Medicina, Buenos Aires, 69: 231, 2009.

Resumen

6
La capacidad de Brucella abortus para producir una enfermedad infecciosa
crónica como la brucelosis, se basa fundamentalmente en su habilidad para infectar y
replicar en macrófagos, diseminándose a través de ellos en el organismo infectado.
Además, esto le permite evadir los mecanismos de defensa naturales del huésped como
es la respuesta inmunológica. En las infecciones crónicas, estas bacterias residen en
granulomas. Estas son estructuras formadas principalmente por diferentes tipos
celulares del sistema inmunológico en donde coexisten áreas en condiciones de
microaerobiosis y de anaerobiosis. La expresión en Brucella de genes involucrados en
la adaptación a condiciones de menor tensión de oxígeno parecería ser esencial para su
supervivencia intracelular por largos períodos de tiempo. En organismos
filogenéticamente relacionados a Brucella, como Sinorhizobium meliloti y
Bradyrhizobium japonicum, se ha identificado un sistema de dos componentes llamado
FixL/FixJ, éstos constituyen un interruptor sensible al oxígeno. A través de su unión al
grupo hemo, regulan los genes responsables de la respiración microaeróbica entre otras
cosas. En el cromosoma II de la cepa salvaje de B. abortus 2308 existe una región que
codificaría para ambas proteínas. Esto nos llevó a plantear en una primera etapa la
identificación y la caracterización del posible homólogo a FixL en Brucella abortus.
Para cumplir con este objetivo nos propusimos, por un lado, expresar el DNA y
purificar la proteína para su posterior estudio. Por otro lado, obtener un mutante de B.
abortus en fixL y estudiar su fenotipo. La proteína recombinante expresada en
Escherichia coli y purificada por métodos cromatográficos resultó ser del tamaño
esperado. Estudios in vitro permitieron determinar que es capaz de unirse al grupo
hemo, una característica importante relacionada a la detección de oxígeno y en acuerdo
a lo descripto para FixL de otros microorganismos. El mutante obtenido por deleción
parcial de fixL pudo crecer al igual que la cepa salvaje en condiciones de
microaerobiosis, aunque resultados preliminares indicarían una diferencia en el patrón
de crecimiento entre ambas cepas.

7
Índice
Contenido
Resumen ...................................................................................................................... 5
Índice ............................................................................................................................. 7
Introducción .................................................................................................................. 9
Historia .................................................................................................................... 10
Características de Brucella ...................................................................................... 12
Patogenia y respuesta Inmune ................................................................................ 14
Sistema FixL/FixJ .................................................................................................... 16
Hipótesis y Objetivos .................................................................................................. 17
Hipótesis ................................................................................................................. 18
Objetivos ................................................................................................................. 18
Materiales y Métodos .................................................................................................. 19
1. Vectores de clonado, plásmidos y cepas utilizadas. ............................................ 20
Tabla 1. Cepas Bacterianas ........................................................................................ 20
Tabla 2. Plásmidos ..................................................................................................... 20
2. Medios y Condiciones de cultivo ......................................................................... 21
3. Estudio preliminar “in silico” ................................................................................. 22
4. Amplificación del gen por la Reacción en Cadena de la Polimerasa (PCR) ......... 23
5. Electroforesis en geles de agarosa...................................................................... 23
6. SDS-PAGE .......................................................................................................... 24
7. Ligaciones y Digestiones enzimáticas ................................................................. 24
8. Extracción de ADN a partir de un gel de Agarosa ................................................ 25
9. Transformación de células de E. coli competentes .............................................. 25
10. Extracción del ADN plasmídico “Miniprep” ......................................................... 25
11. Expresión y purificación de las proteínas recombinantes .................................. 26
11.a. Expresión en forma piloto ........................................................................... 26
11.b. Variación de los parámetros de incubación ................................................. 27
11.c. Expresión y purificación .............................................................................. 27
12. Ensayos de unión al Hemo ................................................................................ 29
13. Construcción del mutante en B. abortus ............................................................ 29
13.a. Preparación de células de B. abortus electrocompetentes .......................... 29

8
13.b. Generación de la cepa B. abortus 2308 fixL* ............................................ 29
14. Estudio del fenotipo del mutante ....................................................................... 30
14.a. Medición del crecimiento bacteriano ........................................................... 30
14.b. Determinación de la sobrevida bacteriana en microaerobiosis .................... 31
14.c. Test de la Catalasa ..................................................................................... 31
14.d. Crecimiento en tubos con medio tioglicolato ............................................... 31
14.e. Formación de biofilms ................................................................................. 32
Resultados y Discusión ............................................................................................... 33
1. Identificación del gen fixL en B. abortus y realización de predicciones
computacionales ..................................................................................................... 34
2. Amplificación de fixL* .......................................................................................... 36
3. Clonación de fixL* y construcción del plásmido pGEMfixL* ................................. 38
4. Mutación en fixL* ................................................................................................. 39
5. Comprobación de la mutación por secuenciación del ADN ................................. 40
6. Expresión y purificación de las proteínas recombinantes FixL* y FixL* ............. 41
6.a. Obtención de cepas de E. coli productoras de FixL* y FixL* ...................... 41
6.b. Pruebas piloto de expresión .......................................................................... 42
6.c. Influencia de los parámetros de incubación en la solubilidad de FixL* .......... 43
6.d. Expresión, purificación y renaturalización de las proteínas recombinantes ... 44
7. Capacidad de unión de FixL* al grupo hemo ....................................................... 46
8. Obtención del mutante en fixL de B. abortus ....................................................... 47
9. Fenotipo del mutante ........................................................................................... 50
10. Capacidad de formar biofilms por Brucella abortus ........................................... 52
Conclusión .................................................................................................................. 53
Perspectivas futuras ................................................................................................... 55
Bibliografía .................................................................................................................. 57
Apéndice ...................................................................................................................... 65

Introducción

10
Historia
La brucelosis, también conocida como enfermedad de Bang, fiebre ondulante o
fiebre de Malta, es una enfermedad infecciosa que afecta a muchas especies de
mamíferos y que se transmite al hombre, constituyendo una zoonosis. Esta es la
principal enfermedad transmitida por animales domésticos como cabras, cerdos,
vacunos y perros a los humanos y está distribuida en todo el mundo.
Los primeros reportes de esta enfermedad fueron a principios del siglo XIX en la
isla mediterránea de Malta, en donde algunos de sus habitantes fallecían como
consecuencia de un cuadro febril cuyas causas desconocían. Esta enfermedad llamó la
atención de los médicos del ejército inglés que ocupaban la isla ya que esta enfermedad
afectaba a parte de sus soldados. En 1886 el Coronel David Bruce viajó a la isla de
Malta para investigar qué aquejaba a los soldados ingleses. Luego de examinar
diferentes tejidos infectados de los soldados fallecidos observó la presencia de un gran
número de microorganismos cocoides Gram negativos. Estos fueron aislados y
cultivados luego de un año de trabajo (13).
Bruce, siguiendo los postulados de Robert Koch, realizó experimentos en monos
a los que inoculó con estos microorganismos cocoides, luego de dieciséis días
presentaban fiebre alta rozando los 41○
C y algunos de ellos morían. Sus pulmones se
encontraban colonizados por micrococos. Los animales que lograban sobrevivir
desarrollaban fiebre ondulante por dos o tres meses, como en los casos humanos. Estos
hallazgos fueron confirmados por Hughes en 1887. De esta forma Bruce logró
identificar el agente etiológico causal de la Fiebre de Malta, al que sugirió bautizar con
el nombre de Micrococcus melitensis (de Melitis, el término usado por los historiadores
latinos para nombrar la isla de Malta)(12).
En 1897, Wright desarrolló el test de seroaglutinación para el diagnóstico de la
enfermedad. En 1905 Zammit que era miembro de la comisión investigadora del
ejército inglés “Mediterranean Fever Comission” comprobó el papel epidemiológico
que desempeñaban las cabras al demostrar que el 40% de ellas sufría la enfermedad
(14).

11
Por otro lado en 1896, Bang con la colaboración de Stribolt lograron aislar el
agente epizoótico causante del aborto en bovinos, al que denominaron “abortus
bacillus”. Sin embargo, por muchos años creyeron que el bacilo de Bang no era
patógeno para los humanos. En 1918, la bacterióloga Alice Evans fue quien comprobó
la semejanza entre el bacilo de Bang y el Micrococcus melitensis de Bruce. En 1920
Meyer y Shaw proponen la creación de un nuevo género bacteriano al que denominaron
Brucella, en honor a Sir David Bruce (12, 14). Desde aquí en adelante varios autores
encuentran especies semejantes que pasan a formar parte del género Brucella.
En la Argentina, los primeros reportes de abortos epizoóticos fueron hechos por
el profesor Bernier en 1892. Villafañe, en 1913, se encargó de estudiar el tema en su
tesis de medicina veterinaria y en 1921, en el Instituto Biológico de la Sociedad Rural
Argentina, Rosenbusch declaró haber encontrado la naturaleza infecciosa de 3 abortos
vacunos, agregando que esta enfermedad causaba pérdidas de entre el 30 y el 50 % en
cada parición (15). En la década de 1920, el laboratorio de bacteriología del Ministerio
de Agricultura comenzó una investigación minuciosa de la enfermedad y se realizó un
examen de sangre a todos los animales importados al país. De esta investigación surgió
que en el país existían 12 focos de infección humana por brucelosis localizados en las
provincias de Mendoza, San Luis, Córdoba, Buenos Aires, Santa Fe, Santiago del
Estero, Tucumán, Catamarca, San Juan, Neuquén, Río Negro y la Capital Federal,
siendo los de mayor importancia los de la Capital Federal, Mendoza, Catamarca y San
Luis (16). Molinelli y Fernández Ithurrat demostraron, en 1932, que el 51.3% de las
vacas en establos de la ciudad de Buenos Aires estaban infectadas con Brucella (17).
Para 1933, la incidencia de la enfermedad en trabajadores de frigoríficos y mataderos en
Buenos Aires y La Plata era del 84.3% (18).
Por aquél entonces se definía como brucelosis del oeste a la vinculada con las
cabras, y brucelosis del este cuando la infección era causada por B. abortus o B. suis
(24). Un informe de la oficina sanitaria panamericana indica que entre los años 1945 y
1976 se lograron aislar 157 cepas de B. abortus, 20 cepas de B. suis y 22 de B.
melitensis a partir de 714 casos de brucelosis humana reportados en su mayoría por
trabajadores de granjas y mataderos (25). Entre los años 1993 y 1995 se reportaron 212
casos de brucelosis, 118 de éstos fueron en zonas rurales mientras que 18 de los casos se
reportaron en zonas urbanas asociados con el consumo de queso de cabra contaminado.

12
También se notificó sobre infecciones debidas a la auto inoculación con la cepa vacunal
B. abortus S19 (26) por trabajadores del campo.
Muchos avances y retrocesos se sucedieron en la historia respecto a la
brucelosis, en la actualidad según la World Organisation for Animal Health (OIE), esta
enfermedad está distribuida en todo el mundo pero es endémica en algunos países de
Sudamérica y del Mediterráneo, aunque también se está expandiendo en algunas zonas
de Asia. En África, Asia y Oceanía no existen muchos datos pero se conoce la
existencia de esta enfermedad. En Europa, no se presentan casos de brucelosis bovina
desde mediados del siglo pasado, o la incidencia es muy baja. La región más afectada de
este continente es la del mediterráneo, con una alta incidencia de brucelosis caprina. En
América, la mayor prevalencia de la enfermedad se encuentra en la parte central y sur
del continente.
En nuestro país, la brucelosis afecta enormemente la economía pecuaria
argentina ya que es uno de los principales países exportadores de carne del mundo (21).
Esta enfermedad es considerada endémica, con una prevalencia del 6 al 7% en bovinos
y aún mayor en cerdos y cabras. Se estima que produce pérdidas por 60 millones de
dólares anuales en la producción bovina y además es una enfermedad de lucha
obligatoria (SENASA, 22). Entre los animales domésticos afectados por la brucelosis
en nuestro país se encuentran los perros, el ganado porcino, caprino, ovino y bovino.
También se ha reportado la presencia de esta enfermedad en visones de criadero, zorro
pampeano y liebres (19, 20, 23).
En la actualidad B. abortus RB51 y B. abortus S19 se usan para inmunizar al
ganado vacuno, y B. melitensis REV.1 se usa para cabras y ovejas (30, 31, 32, 33), sin
embargo no existen vacunas para otros animales, ni tampoco para la brucelosis humana.
En Argentina se utiliza la cepa viva y atenuada B. abortus S19 para vacunar a las
terneras de entre 3-10 meses de edad, esta vacuna no es totalmente efectiva y retiene
cierta patogenicidad.
Características de Brucella
El agente etiológico causante de la brucelosis es una bacteria del género
Brucella, donde las especies conocidas y sus principales hospedadores se detallan a

13
continuación: B. melitensis, cabras y ovejas; B. suis, renos, liebres y cerdos; B. abortus,
ganado bovino; B. canis perros; B. neotomae, roedores del desierto del género Neotoma;
B. microti, algunos roedores; B. ceti y B. pinnipedialis mamíferos marinos.
Estas bacterias se encuentran en forma aislada o menos frecuentemente de a
pares o en pequeñas cadenas. Son bacterias Gram negativas, intracelulares, aeróbicas
(aunque algunas cepas pueden crecer mejor en microaerobiosis), no móviles, con
metabolismo de tipo respiratorio y un sistema de transporte de electrones que utiliza
oxígeno o nitrato como último aceptor. Muchas especies requieren un suplemento de
CO2 para crecer, especialmente en los aislamientos primarios. Las colonias en medios
sólidos translúcidos son transparentes, elevadas, convexas, con un borde entero, con una
superficie brillante y de un suave color miel a la luz transmitida. El rango de
temperatura para su crecimiento es entre 20°C y 40°C, siendo 37°C la óptima. El pH
ideal para su crecimiento es entre 6.6 y 7.4. Son catalasa y usualmente oxidasa
positivas. Son quimiorganotróficas y en general necesitan de medios complejos para
crecer. Son parásitos intracelulares. Su genoma típicamente está compuesto por dos
cromosomas circulares, a excepción de B. suis biovariedad 3 que posee un único
cromosoma. Pertenecen a la división α2-proteobacteria y al orden de los Rhizobiales
(34).
En cuanto a su morfología en medios ricos las células de Brucella son pequeños
bacilos o cocobacilos de extremos redondeados. Los aislamientos frescos suelen ser más
cocoides que las cepas adaptadas al cultivo en laboratorios. El lado exterior de la
membrana externa está compuesta por lipopolisacárido (LPS), diferentes proteínas de
membrana y es en donde se localizan la mayor parte de sus antígenos. Dependiendo del
tipo de LPS que poseen existen cepas lisas y rugosas. En su citoplasma existen
pequeñas vacuolas y gránulos de polisacáridos.
En su aislamiento primario las cepas de Brucella son de crecimiento lento y
difíciles de cultivar. No suelen ser visibles antes de las 48 hs. En medios semisólidos B.
abortus y B. suis, dependientes de CO2, forman un disco de crecimiento a unos pocos
milímetros por debajo de la superficie. Las especies no dependientes de CO2 crecen
formado una turbidez pareja desde la superficie hasta unos pocos milímetros por debajo
de ella. En ambos casos, en cultivos estáticos no hay crecimiento en la zona anaerobia
(1). En cuanto a su genoma, Brucella tiene dos cromosomas circulares y no posee

14
plásmidos, lo cual es una marcada diferencia con muchas otras bacterias que poseen un
cromosoma único (35, 36).
Brucella abortus tiene las mismas características morfológicas y bioquímicas
que las descriptas para el género. Su hospedador habitual es el ganado bovino, aunque
también infecta a ovinos, equinos, camélidos, a perros y al hombre.
Estas características de Brucella sumado a su potencial epidémico, la
inexistencia de una vacuna para humanos, la remanencia de patogenicidad de las
vacunas para el ganado y su eficiencia en infección mediante aerosoles hacen que este
patógeno esté clasificado bajo un nivel de bioseguridad 3 y sea considerado como un
potencial agente para el bioterrorismo (38).
Patogenia y respuesta Inmune
Las especies de Brucella son patógenas para un amplio rango de mamíferos,
incluyendo al hombre. Frecuentemente producen una infección generalizada, con una
bacteremia seguida de una localización en los órganos reproductivos y el sistema
mononuclear fagocítico. En hembras preñadas produce una infección placentaria y del
feto, habitualmente causando un aborto. Los microorganismos también pueden
Figura 1. Brucella abortus. Microscopia de fluorescencia.
Tinción con Rojo Congo.

15
colonizar las mamas de los animales infectados, produciendo la excreción de
microorganismos en la leche (1).
La brucelosis aguda en humanos se caracteriza por fiebre ondulante, cefalea,
dolor cervical y artritis. En los casos más graves puede causar endocarditis,
meningoencefalitis y neumonía. Para el hombre, las infecciones con B. suis y B.
melitensis suelen ser las más peligrosas, teniendo una mortalidad en pacientes no
tratados del 2% (39). Las otras especies no suelen provocar complicaciones graves, la
enfermedad suele auto limitarse dentro de un período que va desde unas pocas semanas
a más de seis meses. Por otra parte, la brucelosis no tiene un cuadro clínico
característico que permita la detección temprana de la infección lo que favorece la
evolución de la enfermedad hacia la cronicidad.
La infección en humanos se produce por un contacto directo o indirecto con
animales enfermos o por el consumo de productos derivados de éstos. Las infecciones
persona a persona son extremadamente raras. La entrada al organismo se produce por
vía respiratoria, por la mucosa gastrointestinal o por lesiones en la piel (37).
Los neutrófilos suelen ser las primeras células del huésped en entrar en contacto
con Brucella. Para que se produzca la muerte de las bacterias fagocitadas es necesario
la desgranulación de los neutrófilos, sin embargo se ha demostrado que Brucella posee
mecanismos que inhiben esta desgranulación evitando su eliminación (46). Si las
bacterias sobreviven y se multiplican dentro de las vacuolas de los neutrófilos y
macrófagos (27) llegarán a través del sistema linfático a los ganglios cercanos a los
que pueden colonizar y desde allí invadir el torrente sanguíneo.
Las infecciones crónicas pueden afectar a varios órganos, tales como el hígado,
el bazo o el cerebro en donde las bacterias residen en granulomas generados por el
sistema inmune. Los granulomas son estructuras formadas por diferentes tipos
celulares (macrófagos, células epitelioides, linfocitos, etc.) en donde coexisten áreas de
microaerobiosis y anaerobiosis (10).
La expresión de genes involucrados en la adaptación a condiciones de
oxigenación deficientes, parecería ser esencial para la sobrevida de Brucella por largos
períodos de tiempo (10). De hecho, las concentraciones de oxígeno en el interior del
fagosoma de los macrófagos activados serían menores que las del medio extracelular

16
(11). Además, es esencial para las bacterias tener un mecanismo que le permita detectar
los niveles de oxígeno externos e internos que le ayude a mantener el balance redox
intracelular (2).
Sistema FixL/FixJ
En muchas especies pertenecientes al orden de los Rhizobiales se produce un
cambio en su ciclo de vida, desde su estado de vida libre en condiciones aeróbicas a un
estado simbiótico en los nódulos de las plantas en condiciones de baja oxigenación. Es
importante para este tipo de bacterias poder detectar los niveles de oxígeno ambientales
para poder adaptarse a un nuevo nicho biológico e iniciar en muchos casos la fijación de
nitrógeno. Dado que Brucella pasa de un ciclo de vida libre en condiciones aeróbicas a
otro intracelular microaeróbico y pertenece al mismo orden, es posible plantear un
paralelismo en la adaptación entre estos microorganismos simbiontes fijadores de
nitrógeno y Brucella.
En Brucella todavía no se ha identificado ningún sistema que detecte oxígeno,
mientras que para organismos filogenéticamente relacionados como Bradyrhizobium
japonicum, Sinorhizobium meliloti o Azorhizobium caulinodans, se ha identificado un
sistema de dos componentes llamado FixL/FixJ (3, 7). En R. meliloti FixL es una
proteína de 505 aminoácidos, transductora de señales. Esta proteína puede ser separada
en tres dominios, posee una región de anclaje a la membrana, un dominio de unión al
grupo hemo y un dominio quinasa/fosfatasa (60, 61, 62, 65). En su N-terminal, FixL
tiene un dominio transmembrana que la localiza en el lado citoplasmático de la
membrana interna (3, 4, 63). La región central de la proteína posee un domino PAS
mediante el cual se acopla al grupo hemo, que le posibilita unirse reversiblemente al
oxígeno (62, 64, 77). En el dominio carboxilo terminal se encuentra el dominio
histidínquinasa. La histidina en la posición 285 es en donde se produce la
autofosforilación de la proteína (66). Esta proteína responde a bajas concentraciones de
oxígeno con un aumento de su actividad quinasa, autofosforilándose primero y luego
transfiriendo este fosfato a FixJ. FixJ fosforilado es activo, promoviendo la
transcripción de los genes involucrados entre otras cosas en la fijación de nitrógeno y en
la adaptación a condiciones de oxígeno limitado.

Hipótesis y
Objetivos

18
Hipótesis
En el cromosoma II de la cepa Brucella abortus 2308 existe una región que codificaría
un sistema bacteriano de dos componentes con similitud a FixL/J de Sinorhizobium
meliloti y de Bradyrhizobium japonicum. Esto nos llevó a plantear que en Brucella
abortus 2308 existiría un homólogo a FixL y que éste podría estar detectando los
niveles de oxígeno.
Objetivos
1. Establecer si el homólogo a FixL de Brucella abortus es funcional.
2. Expresar el DNA y purificar la proteína.
3. Obtener mutantes de B. abortus en fixL y estudiar su fenotipo.

Materiales y
Métodos

20
1. Vectores de clonado, plásmidos y cepas
utilizadas.
Las cepas bacterianas y los plásmidos utilizados en este trabajo de tesis se
detallan en las tablas 1 y 2 respectivamente.
Tabla 1. Cepas Bacterianas
Cepas Características Fuente
B. abortus 2308 Virulenta, aislada de campo, salvaje, lisa, NalR Stock del Laboratorio
B. abortus 2308 ∆fixL Mutante con deleción parcial en fixL Esta tesis
E. coli XL1- blue MRFA,b
(mcrA) 183 Delta(mcrB-hsdSMR-mrr)173 end A1 supE44 thi-1
recA1 gyrA96 relA1 lac[F' proAB lacIQZM15 Tn10(Tet
R)]C
(Jerpseth, 1992)
(88)
E. coli DH5α F'Iq
F' Φ80dlacZM15(lasZYA-argF)U169 deoR recA1 endA1
hsdR12(rk- mk
+)phoA supE44 λ- thi-1 gyrA96 relA1/F' proAB
+
lacIq ZM15 zzf::Tn5(Km
R)
Stock del
Laboratorio
E. coli Top 10
F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacΧ74 recA1
araD139 Δ(ara-leu) 7697 galU galK rpsL (StrR) endA1 nupG λ-
Invitrogen
Tabla 2. Plásmidos
Plásmido Características Fuente
pBlueKS2SacB/R sacB de B. subtilis, clonado en pBluescript-KSII, Amp
R
(R. Sieira et al., 2004)
(87)
pTrsHis C Vector de expresión, AmpR, N-Terminal 6xHis Invitrogen
pGEM-T easy Vector de clonado, AmpR Promega
pGEM fixL* Fragmento de fixL de B. abortus clonado en el vector
Esta tesis

21
pGEM-T easy. AmpR.
pGEMfixL*
Fragmento de fixL de B. abortus con una deleción de 231pb
con la enzima NruI clonado en el vector pGEM-T easy.
AmpR.
Esta tesis
pTrsHisfixL* Fragmento de fixL de B. abortus clonado en el vector
pTrsHis C. AmpR.
Esta tesis
pTrsHisfixL* Fragmento de fixL de B. abortus con una deleción de 231pb
con la enzima NruI clonado en el vector pTrsHis C. AmpR.
Esta tesis
pKS2 SacBfixL*
Fragmento de fixL de B. abortus con una deleción de 231pb
con la enzima NruI, clonado en el vector
pBlueKS2SacB/R. AmpR.
Esta tesis
2. Medios y Condiciones de cultivo
Las cepas de B. abortus se cultivaron en medio Luria Bertoni (LB) (89), brucella
broth (BB) (Disco laboratorios, Detroit, Mich.), SOB (89) o Medio Gerhardt
modificado1. Para Escherichia coli se utilizó medio LB. Para cultivos en placa se
adicionó 1.5% de agar al medio.
Las cepas fueron incubadas en estufas de cultivo a 37º C ó 28ºC, según se
indique, por 24 hs para E. coli y 48 hs o más para B. abortus. Según los requerimientos
de cada cepa los medios se suplementaron con: Ampicilina (Amp) 100 g/ml;
Carbenicilina (Cb) 100 µg/ml; Kanamicina (Km) 50 g/ml; Estreptomicina (Str)
10g/ml, 5-bromo-4-cloro-3-indolil-β-D-galactopiranósido (X-Gal) 20 mg/ml;
Isopropil-β-D-1-thiogalactopiranosido (IPTG) 100 mg/ml, o sacarosa al 10%.
Condiciones de microaerobiosis y anaerobiosis
Las condiciones de microaerobiosis se realizaron empleando tanto una jarra
(Oxoid) de incubación, en la que se introdujo un sobre generador de una atmosfera
1 Medio Gerhardt and Wilson modificado (MG*): ácido láctico anhidro 1ml, glicerol 6 ml,
glutamato monosodico 1 gr, NaCl 1.5 gr., K2PO4 2 gr., Na2S2O4:5H2O 0.02gr, extracto de levadura 0.1 gr.,
agua destilada suficiente para 200 ml. Llevar a pH 6.8 con NaOH.

22
microaeróbica (CampyGen, Oxoid), como una estufa de cultivo con regulación de la
presión parcial de CO2 fijada al 5%. Para las condiciones de anaerobiosis se utilizó la
misma jarra de incubación, pero esta vez con el sobre generador de una atmosfera
anaeróbica (CampyGen).
Ensayos en tioglicolato
Se realizaron en tubos de vidrio de 12 cm x 1.2 cm con tapa a rosca. Cada tubo
contenía 9 ml del medio Tioglicolato (Becton, Dickinson). Estos fueron calentados hasta
la ebullición por un minuto para eliminar el oxígeno disuelto y luego autoclavados. Los
tubos inoculados se incubaron por 28 días a 37○
C.
3. Estudio preliminar “in silico”
Con la secuencia de aminoácidos de FixL de Bradyrhizobium japonicum (Swiss-
Prot: P23222 ) se realizó una búsqueda con el programa tBLASTn (disponible en
internet a través de la página de “National Center for Biotechnology Information”
http://www.ncbi.nlm.nih.gov) en el genoma de B. abortus 2308 para corroborar la
identificación de una región homóloga.
El estudio de hidrofobicidad de la proteína que codificaría esta región del ADN
de B. abortus se realizó usando el programa TopPred (Disponible “on line” en:
http://mobyle.pasteur.fr ). Se utilizó el algoritmo de Kyte & Doolittle con los siguientes
parámetros: un valor de corte inferior de 1.25 y superior de 1.8, un “core” de 8 y un
“wedge” de 4.
Se realizó la búsqueda de patrones y dominios conservados dentro de la proteína
putativa FixL de B. abortus en la base de datos llamada PROSITE utilizando para ello
el programa ScanProsite (Disponible “on line” en:
http://www.expasy.org/tools/scanprosite/).
Los alineamientos múltiples entre secuencias proteicas se realizó con el
programa ClustalW2 (Disponible “on line” en: http://www.ebi.ac.uk/ ). Para los
mismos utilizó las secuencias de aminoácidos correspondientes a FixL de los siguientes
microorgenismos: B. abortus, Hyphomonas neptunium, R. leguminosarum, A.

23
caulinodans, Caulobacter crescentus, B. japonicum, S. meliloti, Oligotropha
carboxidovorans, R. etli, Mesorhizobium loti.
4. Amplificación del gen por la Reacción en
Cadena de la Polimerasa (PCR)
La secuencia codificante para la hipotética región citoplasmática de la proteína
en estudio (versión truncada del gen salvaje, desde ahora denominada: fixl*) fue
amplificada por PCR utilizando los oligonucleótidos RFIXL (5’-
GGACTAGTCGATCGAGGCAAACAGGTC-3’), y LFIXL (5’-
CGGGATCCAGATCGATGTGCGGGAG-3’). En azul se destacan los sitios
reconocidos por las enzimas de restricción SpeI y BamHI respectivamente. Su diseño se
realizó con ayuda del programa computacional OligoAnalyzer 3.0 (Disponible “on
line” en: http://www.idtdna.com/ ).
La muestra sobre la cual fue realizada la PCR fue obtenida a partir de colonias
de B. abortus 2308. La PCR fue llevada a cabo en un termociclador MiniCycler™
de MJ
RESEARCH. Se realizó un ciclo inicial de 2 minutos a 95º C para desnaturalizar el
ADN. Seguidamente se realizaron 30 ciclos de amplificación, cada uno con una primera
fase de desnaturalización a 95º C por 30 segundos, una segunda fase de alineamiento a
68º C por un minuto y finalmente la fase de extensión a 72º C por 3 minutos.
Terminados los ciclos se adicionó un último período de 4 minutos a 72º C.
Se probaron dos concentraciones de Mg2+
para evaluar en qué condiciones se
obtenía una mejor amplificación. Se utilizó agua miliQ estéril como control negativo y
5 µl de cada tubo de PCR se sometieron a una separación electroforética en un gel de
agarosa.
5. Electroforesis en geles de agarosa
Las separaciones electroforéticas en geles de agarosa fueron llevadas a cabo en
geles al 0.8% en buffer TBE (Tris-Borato-EDTA) conteniendo bromuro etidio (0.5
µg/ml), para luego visualizar los fragmentos de ADN en un transiluminador con luz
UV.

24
6. SDS-PAGE
Las corridas electroforéticas en geles de poliacrilamida de las proteínas en
estudio fueron realizadas en un Mini-PROTEAN®3 Cell de la empresa Bio-Rad,
siguiendo las instrucciones del manual. Las muestras fueron disueltas en Cracking
buffer (8M de Urea, 50mM de Tris/Cl pH:6.8, 2% SDS, 0.1% Azul de bromofenol, 5%
β-Mercaptoetanol, 10% glicerol, agua destilada c.s.), se calentaron a 100○
C por 4-5
minutos y se las sometió a electroforesis. Se utilizaron geles de poliacrilamida al 15%
de concentración. Cada gel fue teñido con una solución al 0.25% de Azul Brillante de
Coomassie R250 en 10% metanol y 10% ácido acético, para revelar las proteínas
incubándolos durante una noche. El exceso de colorante fue removido por sucesivos
lavados con una solución acuosa con 20% de metanol y 7% de ácido acético.
7. Ligaciones y Digestiones enzimáticas
Tanto las ligaciones como las digestiones enzimáticas con endonucleasas de
restricción fueron hechas siguiendo las instrucciones de cada fabricante, respetando los
tiempos, temperaturas de incubación y concentración de los reactivos.
Como ejemplo, podemos detallar una clásica reacción de digestión enzimática
con la enzima EcoRI de New ENGLAND BioLabs Inc. Esta fue llevada a cabo en un
volumen final de reacción de 10 µl, con 1µl de NEBuffer U 10X, 1 µl de enzima EcoRI,
1 µl del plásmido a ser digerido, 7 µl de agua miliQ. La mezcla de reacción se incubó 2
hs a 37○
C.
Como ejemplo de una típica reacción de ligación entre un vector y un fragmento
de ADN a ser clonado en él se detalla la siguiente: 1-3 µl del producto de PCR, 1µl del
vector, 1 µl de la enzima T4 DNA Ligasa (PROMEGA), 5 µl de “2X Rapid Ligation
Buffer, T4 DNA Ligase” (PROMEGA), agua miliQ c.s.p 10 µl. La mezcla se incubó una
hora a temperatura ambiente.

25
8. Extracción de ADN a partir de un gel de
Agarosa
La extracción de ADN a partir de una banda en un gel de agarosa se realizó
utilizando el kit comercial QUIAEX II (QUIGEN). Se siguió el protocolo que figura en
el manual del proveedor.
9. Transformación de células de E. coli
competentes
Las células utilizadas fueron E. coli Top 10 o DH5α según se indique. La
transformación se realizó descongelando en baño de hielo un tubo eppendorf con 200 l
de células competentes del stock del laboratorio. A cada tubo se le agregaron 5 µl de la
ligación con la que se deseaba transformar y se incubó en hielo por 30 minutos. A las
células que formaban el control negativo se las incubó con 5l de H2O miliQ.
Seguidamente los tubos se pusieron en un baño de agua a 42º C por 30 segundos y
después, otros dos minutos de incubación en baño de hielo. Luego las células se
cultivaron por 1h en tubos de ensayo con 1 ml de LB a 37º C con agitación (250 rpm).
Finalizada la incubación, los cultivos fueron centrifugados 5 minutos a 5000 rpm en una
microcentrífuga. Se descartó el sobrenadante, las células se resuspendieron en 100 l
de LB y se sembraron en placas con el medio adecuado. Se seleccionaron los
transformantes de acuerdo a la resistencia antibiótica de cada vector. A los resistentes se
les realizó una extracción del ADN plasmídico, que fue sometida a una digestión
enzimática para su verificación.
10. Extracción del ADN plasmídico “Miniprep”
El procedimiento para la extracción del ADN plasmídico consistió en inocular
con la cepa deseada un tubo de ensayo conteniendo 4 ml de LB suplementado con el
antibiótico correspondiente. Los tubos se incubaron toda la noche a 37º C con agitación
a 250 rpm. Seguidamente se centrifugaron 1.5 ml de cada cultivo a 6000 rpm por 5
minutos en una microcentrífuga, descartando el sobrenadante. Este procedimiento se
repitió una vez más en el mismo tubo, teniendo así en cada tubo el pellet

26
correspondiente a 3ml de cultivo. El pellet se resuspendió en 200 l del buffer P1 frío
(100g/ml de RNasa A; 50mM de Tris/Cl pH 8; 10 mM de EDTA, pH 8.0). Luego se
agregaron 200 l del buffer P2 (200 mM de NaOH; 1% de SDS) y se agitó por
inversión 5 veces dejándolo a temperatura ambiente por 5 minutos. Completada la
incubación se agregaron 200 l de acetato de potasio 3 M, pH 5.5, mezclando
suavemente, y se incubó por 10 minutos en hielo. Luego se centrifugó a 10.000 rpm en
una centrífuga refrigerada de mesada por 10 minutos a 4º C. Luego se tomó el
sobrenadante y se lo pasó a un tubo eppendorf con 0.7 volúmenes de isopropanol frío,
se agitó por inversión y se centrifugó a 15.000 rpm. por 30 minutos a 4º C. Finalmente
se descartó el sobrenadante y se lavó al ADN con 200 l de etanol al 70%, se centrifugó
5 minutos a 15.000 rpm, se descartó el sobrenadante, se dejó secar y finalmente se
resuspendió en 30 l de agua miliQ. El ADN obtenido se analizó por electroforesis en
geles de agarosa.
11. Expresión y purificación de las proteínas
recombinantes
11.a. Expresión en forma piloto
Como sistema heterólogo de expresión se utilizó E. coli DH5α y como vector de
expresión el pTrsHis C (Invitrogen). Este vector tiene un promotor trc (90) y una región
de anti terminación rrnB (91). Además el vector pTrsHis contiene una copia del gen
lacIq que codifica para la proteína represora del operon lac, necesaria para que no exista
expresión en ausencia del inductor. Este vector también tiene un gen potenciador de la
trascripción del bacteriófago T7.
A partir de cultivos en placa de las cepas E. coli / pTrsHisfixL* y E. coli /
pTrsHisfixL* se inocularon tubos Falcon de 15 ml conteniendo 2 ml del medio LB
suplementado con 50µg/ml de ampicilina. Estos se incubaron durante una noche en un
agitador rotatorio a 37○
C y a 250 rpm y se usaron como inóculo en erlenmeyers
conteniendo 20 ml del mismo medio de cultivo. Estos se incubaron por 3 hs a 37○
C y a
250 rpm, seguidamente se les agregó IPTG para alcanzar una concentración final de 1

27
mM. Cada cultivo se volvió a incubar a 37○
C y a 250 rpm, tomando muestras y
haciendo un recuento en placa, a los tiempos 0, 1.30 hs, 2.50 hs y 3.30 hs.
11.b. Variación de los parámetros de incubación
También se realizaron ensayos de expresión en diferentes condiciones para
intentar favorecer la producción de las proteínas recombinantes en forma soluble. Para
esto se probaron diferentes temperaturas de incubación, diferentes concentraciones del
inductor y el agregado de hemina al medio para intentar estabilizar las proteínas
recombinantes. Estos ensayos se llevaron a cabo sólo para la cepa de E. coli
transformada con el vector pTrsHisfixL*. Se usaron dos juegos de tres tubos Falcon de
15 ml conteniendo 2 ml de medio LB con 50µg/ml de ampicilina y suplementados con
1mM de IPTG, 0.05 mM de IPTG ó 1 mM de IPTG más 50 µg/ml de hemina.
Un juego de tubos se incubó a 37○
C y el otro a 28○
C, ambos con agitación a 250
rpm durante una noche. Finalizada la incubación 1 ml de cada cultivo se centrifugó a
10.000 rpm por 10 minutos a 4○
C en una centrífuga de mesa. A continuación se
descartó el sobrenadante, y el pellet se resuspendió en 100 µl de buffer de lisis (50mM
Tris/Cl pH: 8.8, 150mM ClNa, 1mM Fluoruro de Fenilmetilsulfonilo PMSF, hemina 50
µg/ml) luego, las muestras fueron sonicadas con 6 pulsos de 30 segundos cada uno a
una potencia de 6, con intervalos de un minuto entre cada pulso mientras permanecían
en hielo. A las muestras se les agregó Tritón-X al 1% y se las dejó en baño de hielo por
una hora con agitación cada 10 minutos. Seguidamente, las muestras fueron
centrifugadas a 14.000 rpm a 4○
C por 20 minutos. Finalmente se separó el sobrenadante
del pellet y ambos fueron sometidos a una corrida electroforética en geles de
poliacrilamida.
11.c. Expresión y purificación
A partir de cultivos en placa de las cepas E. coli / pTrsHisfixL* y E. coli /
pTrsHisfixL* se inocularon erlenmeyers conteniendo 10 ml del medio LB suplementado
con ampicilina. Estos se incubaron durante una noche a 37○
C en un agitador rotatorio a
250 rpm y se usaron como inóculo de erlenmeyers conteniendo 1l del mismo medio de
cultivo. Estos se incubaron por 3 hs a 37○
C y 250 rpm seguidamente se les agregó IPTG

28
para alcanzar una concentración final de 0.25 mM. Cada cultivo se volvió a incubar por
otras 3 hs a 37○
C y 250 rpm.
Finalizada la incubación, cada cultivo se centrifugó a 7.000 rpm por 15 minutos
a 4○
C, en una centrífuga Sorvall con un rotor GS-3. A continuación se separó al
sobrenadante del pellet y este último se resuspendió en 15 ml del buffer de lisis (20 mM
Tris/Cl pH 8.8, 150mM ClNa, 1 mM PMSF, 0.04 mg/ml de lisozima, 1 mM de EDTA,
10 mM β-Mercaptoetanol, una punta de espátula DNasa y hemina 50 µg/ml) luego las
muestras fueron sonicadas en un equipo BRANSON SONIFIER 450 usando 6 pulsos de
30 segundos cada uno a potencia 6, con intervalos de un minuto en hielo entre cada
pulso. A cada muestra se le agregó Tritón-X al 1% y se dejaron en baño de hielo por
una hora con agitación cada 15 minutos. Al finalizar, las muestras fueron centrifugadas
a 12.000 rpm a 4○
C por una hora en una centrífuga Sorvall con un rotor SS34.
Finalmente se separó el sobrenadante y el pellet se resuspendió en 5 ml del
buffer Tris/Cl 20 mM pH 8.8, éste se volvió a centrifugar a 18.000 rpm a 4○
C por 30
minutos. Nuevamente se guardó el segundo sobrenadante y el pellet se resuspendió en
2 ml del buffer Tris/Cl 20 mM pH 8.8. Tanto los sobrenadantes como los pellets se
almacenaron a -20○
C.
Los cuerpos de inclusión, presentes en los pellets, que contenían a las proteínas
recombinantes fueron desnaturalizados en un buffer con una concentración de urea 8 M,
a 50° C. Las muestras fueron filtradas y sembradas en una columna cromatográfica con
resina Ni-SepharoseTM
High Performance de Amersham Bioscience, siguiendo las
instrucciones del proveedor. La renaturalización de cada proteína fue realizada dentro
de la misma columna pasando en forma sucesiva buffers con concentraciones
decrecientes del agente desnaturalizante (urea 6, 4 y 3 M) (52). Finalmente, la elusión
fue hecha con concentraciones crecientes de imidazol (50, 100. 300 y 500 mM). Todas
las fracciones eluídas de la columna fueron colectadas, alícuotas de éstas fueron
sembradas en un gel al 15% de SDS-poliacrilamida y sometidos a electroforesis como
se describe con anterioridad en esta sección.
Las muestras fueron dializadas utilizando una membrana de celulosa con
exclusión para moléculas mayores de 12-14 KDa. La diálisis se realizó en 4 l del buffer
100 mM KCl, 50 mM Tris/Cl pH 8.2, a 4º C y con agitación. Luego de 4 hs se renovó el
buffer y se dejaron las muestras en las mismas condiciones toda la noche. Al día

29
siguiente se renovó el buffer por 600 ml del mismo conteniendo 20% de glicerol,
dejándolo 24 hs más a 4º C y con agitación. Las proteínas fueron concentradas
dispersando polivinilpirrolidona por encima de las membranas de diálisis.
Finalmente se calculó la concentración de las proteínas por el método de
Bradford utilizando un kit de BioRad y distintas concentraciones de BSA (seroalbumina
bovina) como patrón de referencia.
12. Ensayos de unión al Hemo
Para realizar este ensayo se incubó la proteína recombinante FixL* con hemina
(Sigma). Las reacciones se llevaron a cabo a temperatura ambiente empleando
concentraciones equimolares de la proteína purificada FixL* y la hemina. Tanto las
proteínas como la hemina estaban disueltas en buffer (10 mM de ClK, Tris/Cl 50 mM,
pH 8.2) y el volumen final de reacción fue de 600 µl. BSA y lisozima se emplearon
como controles positivo y negativo, respectivamente y en igual concentración que la
proteína en estudio. Las mezclas de reacción fueron analizadas en un espectrofotómetro
Beckman DU650, obteniéndose un espectro de absorción por barrido entre 300nm -
550nm para cada una.
13. Construcción del mutante en B. abortus
13.a. Preparación de células de B. abortus
electrocompetentes
A partir de un glicerol de B. abortus 2308 conservado a -70○
C se tomó una
muestra que se cultivó en placa. Se tomó una colonia para inocular 5 ml del caldo TSB.
Este se incubó con agitación a 37○
C y 250 rpm por 24 hs. Con este cultivo se inoculó
un erlenmeyer de 2 L conteniendo 500ml de medio TSB, este se incubó con agitación a
37○
C y 250 rpm hasta alcanzar la densidad óptica de 0.7 y se dejó en un baño de hielo
por 10 minutos. Luego se centrifugó la muestra por 20 minutos a 5500 rpm, el pellet se
resuspendió con 500 ml de agua bidestilada estéril a 4○
C y se volvió a centrifugar en las
mismas condiciones. Se repitió este procedimiento dos veces más reduciendo el
volumen de resuspensión a 250 ml y 125 ml respectivamente, seguidamente el pellet se

30
resuspendió en 50 ml de glicerol al 10% estéril a 4○
C y se centrifugó por 20 minutos a
5500 rpm. Finalmente las células se resuspendieron en 1 ml de glicerol al 10% estéril a
4○
C. Se fraccionaron en fracciones de 50 µl en tubos eppendorf que se guardaron a -
70○
C.
13.b. Generación de la cepa B. abortus 2308 fixL*
El plásmido pKS2SacBfixL* fue introducido en las células de B. abortus 2308
competentes por electroporación. Para esto, a 50 µl de células electrocompetentes se le
agregó 3 µl del plásmido pKS2SacBfixL* y la mezcla se depositó en una cuba de
electroporación de 2 mm. Se utilizó un equipo BTX ECM 600 con una capacitancia de
50 µF, una resistencia de 246 Ohm y un voltaje de 2.5 kV. Luego del pulso eléctrico, las
células se incubaron en 0.9 ml del medio SOB por 3 hs a 37○
C y los transformantes
fueron seleccionados en BB agar conteniendo Cb. Estas fueron repicadas en tubos con 6
ml de BB conteniendo sacarosa y se incubaron toda la noche en un agitador rotatorio a
37○ C y 250 rpm. Transcurridas 18 hs se sembraron 50 µl de cada cultivo en placas de
BB conteniendo sacarosa al 10%. Las colonias resistentes fueron replicadas en placas
con BB agar conteniendo Cb. Las colonias CbS que crecieron en presencia de sacarosa
fueron seleccionadas como posibles dobles recombinantes. El reemplazo de la versión
salvaje del gen fixL por la versión mutada ∆fixL* fue confirmado por análisis de PCR.
Este procedimiento generó la cepa B. abortus 2308 ∆fixL*.
El fragmento amplificado fue clonado en el vector pGEM-T easy para su
secuenciación. Estos fueron secuenciados en un secuenciador automático ABI 3130
(Applied Biosystem) empleando los oligonucleótidos T7 y SP6 (Promega). La
comparación de la secuencia obtenida con el gen salvaje, se realizó mediante un
alineamiento múltiple de las secuencias usando el programa computacional MegAlign
5.00 con el que también se realizó la representación gráfica de los alineamientos.
14. Estudio del fenotipo del mutante
14.a. Medición del crecimiento bacteriano
Se emplearon 6 tubos Falcon, cada uno con 3 ml de medio, tres con BB y otros
tres con MG*, estos fueron inoculados con 1 x 103 bacterias contenidas en 100 µl de
solución salina. Para cada cepa los tubos se incubaron a 37○
C en un agitador rotatorio a

31
250 rpm. Se realizaron recuentos de células viables por diluciones seriadas en placa para
cada cultivo a diferentes tiempos: tiempo cero, 4, 7, 22, 28, 48 horas y pasados los 6
días. (Ver Medios y Condiciones de Cultivo de Materiales y Métodos).
14.b. Determinación de la sobrevida bacteriana en
microaerobiosis
Se usaron 6 tubos Falcon, cada uno con 3 ml de medio, tres con BB y otros tres
con MG*, fueron inoculados con 1 x 103 bacterias contenidas en 100 µl de solución
salina. Los tubos se incubaron a 37○
C en un agitador rotatorio a 250 rpm hasta tener
cultivos en fase estacionaria. A partir de las 48 hs de incubación se cubrieron los
cultivos con 1 ml de aceite mineral estéril. Seguidamente se los incubó en forma
estacionaria a 37○C, realizando recuentos de células viables por diluciones seriadas en
placa para cada cultivo a las 24 y 48 hs. (Ver Medios y Condiciones de Cultivo de
Materiales y Métodos).
14.c. Test de la Catalasa
La actividad de la enzima catalasa se determinó al aplicar una gota de una
solución acuosa de peróxido de hidrogeno al 5% sobre placas con desarrollo fresco,
tanto a la cepa salvaje como a la del mutante de B. abortus. (Ver Medios y Condiciones
de Cultivo de Materiales y Métodos). Se cronometró el tiempo hasta la aparición de
burbujas.
14.d. Crecimiento en tubos con medio tioglicolato
Se estudio el patrón de crecimiento de la cepa salvaje y del mutante de B.
abortus en tubos con el medio tioglicolato. Para esto, una colonia de cada cepa fue
resuspendida en solución fisiológica y 100 µl de esta suspensión fue usada como
inóculo para cada tubo. Se registró periódicamente el patrón de crecimiento observado
de cada cepa (Ver Medios y Condiciones de Cultivo de Materiales y Métodos).
Diferentes muestras a partir del crecimiento de B. abortus en las paredes de los
tubos fueron tomadas para luego ser observadas en el microscopio electrónico de
barrido del museo de ciencia naturales “Bernardino Rivadavia”.

32
14.e. Formación de biofilms
Para evaluar la formación de biofilms por parte de B. abortus se realizaron
cultivos líquidos de esta cepa en el medio BB dentro de botellas de cultivo celular. Estas
fueron incubadas a 37○
C por 28 días. Terminada la incubación, se trató el cultivo con el
agregado de formaldehído, se desecho el caldo, y se lavó cinco veces la botella con agua
destilada estéril. Luego se le agregó una solución de cristal violeta al 0.35% por dos
minutos, seguidamente se descartó el colorante y se realizaron otros cinco lavados con
agua destilada.

Resultados
y Discusión

34
1. Identificación del gen fixL en B. abortus y
realización de predicciones computacionales
De la búsqueda en el genoma de B. abortus de un gen que codificara para una
proteína homóloga a FixL de B. japonicum se obtuvo en el cromosoma II de B. abortus
2308 un gen (Locus: BAB2_0040) con un 33% de identidad y una similitud del 53%.
Corriente abajo de este gen se encontró una secuencia (Locus: BAB2_0041) que
codificaría para una proteína reguladora perteneciente a la familia de LuxR con
homología a FixJ de B. japonicum.
La proteína putativa FixL de B. abortus, de 356 aminoácidos, es identificada en
dicha base de datos como una proteína sensora en su amino terminal, con un dominio
de unión a ATP y de histidínquinasa en su extremo carboxilo. Estos mismos dominios
son los que encontramos en FixL de microorganismos relacionados tales como,
Bradyrhizobium japonicum, Rhizobium meliloti, Mesorhizobium loti, Caulobacter
crescentus y Rhizobium leguminosarum (60, 61, 62, 65). La zona más conservada es en
la región que codificaría para el dominio quinasa, mientras que el dominio sensor es el
menos conservado. Esto está de acuerdo a lo observado en otras especies en las que el
dominio sensor de la proteína es el más divergente.
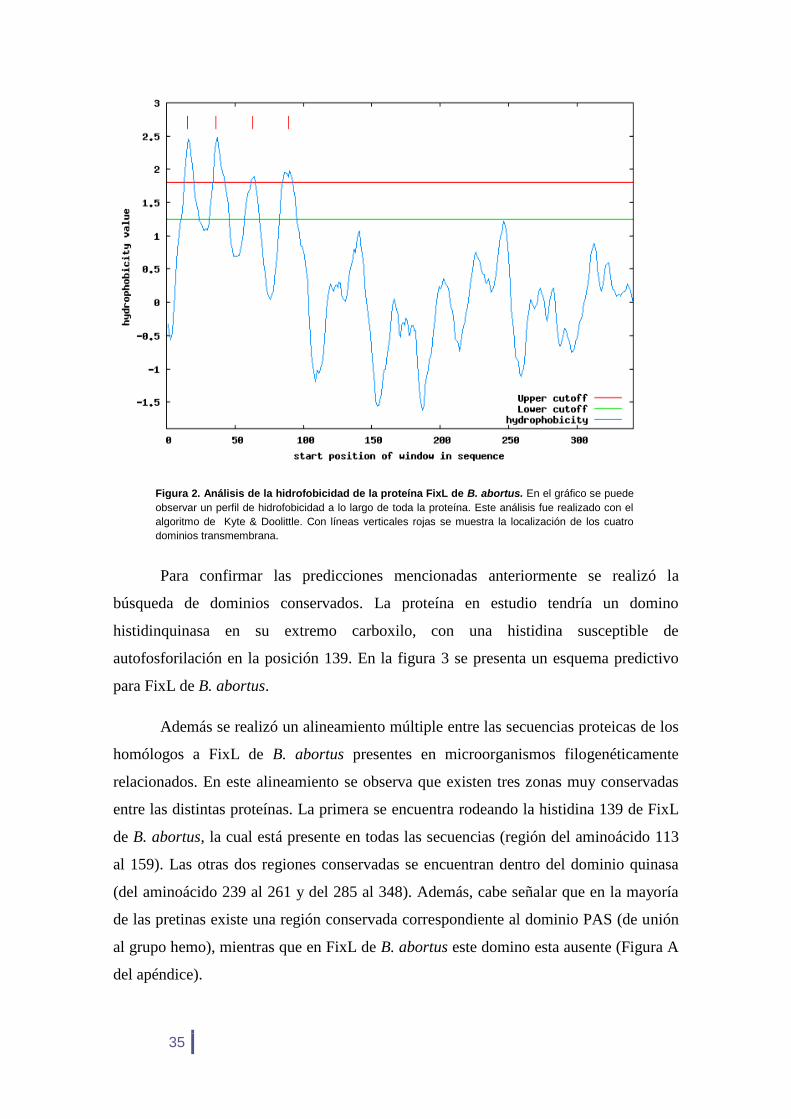
El estudio de su hidrofobicidad nos indica que se trata de una proteína de
membrana que tendría cuatro dominios transmembrana (Figura 2). Esta arquitectura
dejaría a la proteína situada del lado citoplasmático de la membrana. Estos resultados
predictivos coinciden con aquellos hechos con otros algoritmos, todos ellos predicen la
existencia de 3 ó 4 dominios transmembrana, dependiendo de los parámetros
ingresados.
35
Para confirmar las predicciones mencionadas anteriormente se realizó la
búsqueda de dominios conservados. La proteína en estudio tendría un domino
histidinquinasa en su extremo carboxilo, con una histidina susceptible de
autofosforilación en la posición 139. En la figura 3 se presenta un esquema predictivo
para FixL de B. abortus.
Además se realizó un alineamiento múltiple entre las secuencias proteicas de los
homólogos a FixL de B. abortus presentes en microorganismos filogenéticamente
relacionados. En este alineamiento se observa que existen tres zonas muy conservadas
entre las distintas proteínas. La primera se encuentra rodeando la histidina 139 de FixL
de B. abortus, la cual está presente en todas las secuencias (región del aminoácido 113
al 159). Las otras dos regiones conservadas se encuentran dentro del dominio quinasa
(del aminoácido 239 al 261 y del 285 al 348). Además, cabe señalar que en la mayoría
de las pretinas existe una región conservada correspondiente al dominio PAS (de unión
al grupo hemo), mientras que en FixL de B. abortus este domino esta ausente (Figura A
del apéndice).
Figura 2. Análisis de la hidrofobicidad de la proteína FixL de B. abortus. En el gráfico se puede
observar un perfil de hidrofobicidad a lo largo de toda la proteína. Este análisis fue realizado con el
algoritmo de Kyte & Doolittle. Con líneas verticales rojas se muestra la localización de los cuatro dominios transmembrana.

36
2. Amplificación de fixL*
Los cebadores RFIXL y LFIXL fueron diseñados para amplificar sólo la
secuencia que codificaría para la región soluble de la proteína, de 830pb (fixL*),
excluyendo la región del gen que codificaría para los dominios transmembrana. Esta
estrategia de amplificar sólo la región soluble de la proteína fue para optimizar la
purificación de la misma debido a que la actividad catalítica estaría en esta región. Sin
embargo en algunos trabajos se demuestra que estos dominios transmembrana son
necesarios para una total funcionalidad de la proteína (85). A continuación se muestra
un esquema en donde se puede observa la región del gen que fue amplificada.
Figura 3. Esquema de FixL de B. abortus. Este es un esquema de la estructura de
FixL de B. abortus. Fue realizado teniendo en cuenta las predicciones de los
dominios transmembrana de la proteína hechos con el algoritmo Kyte & Doolittle y
las búsquedas de dominios conservados hechas con el programa ScanProsite. En el
esquema se observan los cuatro dominios transmembrana, y el dominio histidin
quinasa, con una hitidina en la posición 139 susceptible de ser fosforilada. Los
números indican la posición en forma aproximada de cada aminoácido.
37
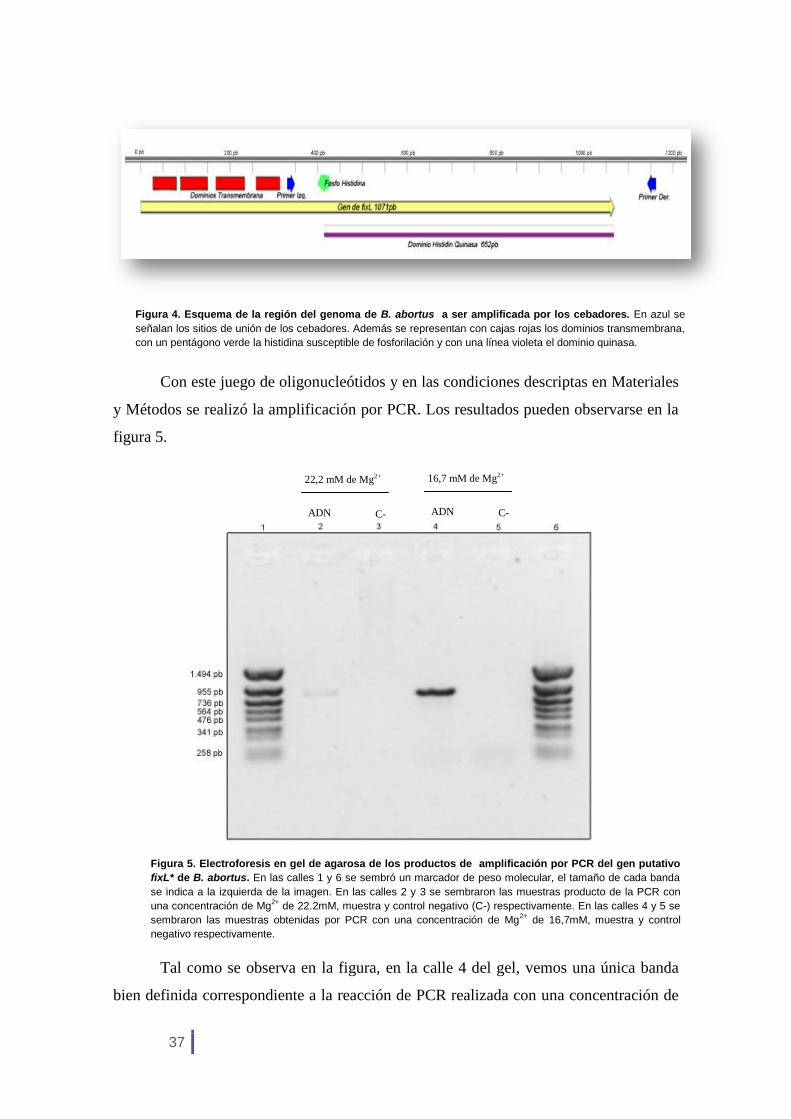
Con este juego de oligonucleótidos y en las condiciones descriptas en Materiales
y Métodos se realizó la amplificación por PCR. Los resultados pueden observarse en la
figura 5.
Tal como se observa en la figura, en la calle 4 del gel, vemos una única banda
bien definida correspondiente a la reacción de PCR realizada con una concentración de
Figura 5. Electroforesis en gel de agarosa de los productos de amplificación por PCR del gen putativo
fixL* de B. abortus. En las calles 1 y 6 se sembró un marcador de peso molecular, el tamaño de cada banda
se indica a la izquierda de la imagen. En las calles 2 y 3 se sembraron las muestras producto de la PCR con
una concentración de Mg2+
de 22.2mM, muestra y control negativo (C-) respectivamente. En las calles 4 y 5 se
sembraron las muestras obtenidas por PCR con una concentración de Mg2+
de 16,7mM, muestra y control
negativo respectivamente.
Figura 4. Esquema de la región del genoma de B. abortus a ser amplificada por los cebadores. En azul se
señalan los sitios de unión de los cebadores. Además se representan con cajas rojas los dominios transmembrana,
con un pentágono verde la histidina susceptible de fosforilación y con una línea violeta el dominio quinasa.
22,2 mM de Mg2+
ADN C-
16,7 mM de Mg2+
ADN C-

38
16.7 mM de Mg2+
. En la calle 2, en donde se sembró la muestra amplificada con 22.2
mM de Mg2+
, se observa una banda del mismo tamaño que la anterior pero muy tenue.
En ambos casos se logró amplificar un fragmento con el tamaño esperado de 830pb. En
ninguno de los controles negativos hubo amplificación (calles 3 y 5). Para las
subsiguientes reacciones de PCR hechas en este trabajo se utilizó una concentración
16.7 mM de Mg2+
, ya que con esta se obtuvo una mayor amplificación.
3. Clonación de fixL* y construcción del plásmido
pGEMfixL*
El producto de la amplificación fue clonado en el vector pGEM-T easy y con
esta construcción (pGEMfixL*) se transformaron las células de E. coli Top 10
competentes, tal como se detalla en Materiales y Métodos. Se seleccionaron las colonias
resistentes a ampicilina (AmpR) y blancas (LacZ
-) esperando que contengan el plásmido
recombinante y a cada una se le realizó una extracción del ADN plasmídico. Para
verificar la construcción el plásmido pGEMfixL* fue digerido con la enzima EcoRI, que
libera el inserto, y sometido a una separación electroforética en un gel de agarosa cuya
fotografía se observa en la figura 6.
En la segunda calle del gel, donde se ha sembrado la digestión, se observan dos
bandas. La primera de aproximadamente 3.000 pb corresponde al vector sin el inserto A B
Figura 6. A) Electroforesis en gel de agarosa de la digestión del vector pGEMfixL* con
la enzima EcoRI. En la calle 1 se sembró el marcador de peso molecular λ /Hind III, el tamaño
de cada banda se indica a la izquierda de la imagen. En la calle 2 se sembró la digestión del
vector pGEMfixL* con la enzima EcoRI. B) Esquema del vector pGEMfixL* .
A) B)

39
(3.015 pb). La segunda, más abajo, de aproximadamente 800 pb, corresponde al inserto
fixL* (830 pb). De esta forma se pudo chequear la incorporación del fragmento fixL* en
el vector y a partir de esto seguir con la construcción de los vectores necesarios para la
expresión de la proteína recombinante y la obtención del mutante en B. abortus.
4. Mutación en fixL*
Con fixL* clonado en el vector pGEM-T easy (pGEMfixL*) realizamos una
deleción parcial del gen. Para ello se lo digirió con la enzima de restricción NruI que
reconoce dos sitios de corte dentro del gen quitando un fragmento de 231pb, tal como se
muestra en la figura 7. La deleción se realizó dentro de la región que codificaría para el
dominio quinasa, eliminando de la proteína la región conservada que va desde el
aminoácido 239 al 261(Figura B del apéndice).
Los productos de digestión se sometieron a una separación electroforética.
Seguidamente se purificó el ADN de la banda correspondiente al vector con parte del
inserto y se lo religó obteniéndose de esta forma el plásmido pGEM∆fixL*.
Seguidamente el fragmento ∆fixL* fue purificado. Para esto dicho fragmento fue
escindido del plásmido pGEM∆fixL*, digiriéndolo con las enzimas de restricción
BamHI y SpeI (incorporadas en cada cebador), y purificado a partir de la banda
correspondiente en un gel de agarosa luego de su separación electroforética. El tamaño
del fragmento ∆fixL* es de 600 pb.
Deleción de231pb
Figura 7. A) Esquema de la deleción del fragmento fixL* con la enzima NruI. Arriba se observa un esquema
general de fixL* presente en el vector pGEMfixL*. Se observan los sitios reconocidos por la enzima NruI en las
posiciones 632 y 863. Abajo se muestra el esquema del plásmido resultante pGEM∆fixL*. B) Electroforesis en gel
de agarosa del fragmento ∆fixL*. En la calle 1 se sembró el marcador de peso molecular λ /Hind III; el tamaño de
cada banda se indica a la izquierda de la imagen. En la calle 2 se sembró el fragmento ∆fixl* purificado a partir de
la digestión del vector pGEM∆fixL* con las enzimas BamHI y Spe I que reconocen los sitios presentes en los
cebadores utilizados en la amplificación del fragmento originalmente.
pGEM∆fixL*
A) B)
pGEMfixL*
NruI (863) NruI (632)

40
5. Comprobación de la mutación por
secuenciación del ADN
La presencia de la mutación fue comprobada por secuenciación. Para esto, el
fragmento ∆fixL* fue clonado en el vector pGEM-T easy para su análisis en un
secuenciador automático ABI3130 (Applied Biosystem). La secuencia consenso del gen
mutado (∆fixL*) se muestra a continuación:
AGATCGATGTGCGGGAGTCGCGCGAACGGCTTTTGCGTCTGGCGCGAATGACCACGCTCGGT
CAGTTGACAAGCTCCATCGCTCACGAAGTGAGCCAGCCGCTTGTCGCCATTGAGATCAGCGC
AAGCGCCGGCAGGCGCTGGCTTGCGCAATCGCCCCCCAATAATGAGCGCGCCGCGGCAGCG
ATCGAGCGCATCTCGGCGGATTCGCATCGGGCGATTGAAATTCTCGACCGGGTGCGGCGGCT
GTCTCGAGGCGAAAAGCCCCATGTTGCGCCCTTCGATATCAACGAGGTGGTTCGCGATAGTG
GCGGCGGCATAGCGCCCGCAGATCAGGAACGGCTGTTCGACGCATTCTGGACCACGAAGAA
GGGGGGCTTCGGCCTCGGACTCACCATCAGCCGCACCATCGTCGAAAGCAGTGGCGGCACC
ATTGCCCTGGGGTCCACGACGCCCCATGGAACGACAATCCTGTTTGAAGTGCCAACGACACA
GGAGACGGAGATATGAACTGCGCATATGAACGCGATCCTGTCGTCTTTATCGTCGACGACGA
TCATTCCGTCCGCCTTGGTCTGGTGGACCTGTTTGCCTCGATCG
La deleción mantiene el marco de lectura del gen por lo cual la secuencia de
ADN codificaría para una proteína de menor tamaño que preserva su carboxilo terminal.
La secuencia obtenida luego fue examinada y comparada contra la secuencia salvaje del
gen mediante alineamientos múltiples.
Figura 8: Secuenciación de la versión mutante del gen ∆fixL*: En verde se destacan las secuencias de los
cebadores utilizados. En azul se presenta la primer parte de ∆fixL* hasta el lugar de la deleción, y en negro desde
el lugar de la deleción al final del mismo.
Secuencia mutante ∆fixL*
Secu
en
cia
salv
aje
de
fix
L
Figura 9. Representación gráfica de
las regiones homólogas entre la
secuencia salvaje de fixL y la versión
mutante ∆fixL*: Las líneas oblicuas
indican las zonas con una homología del
100%. De manera horizontal se
representa la secuencia mutante ∆fixL* y
de manera vertical la secuencia salvaje
de fixL.

41
Al comparar la versión mutada del gen ∆fixL* con la secuencia salvaje del
mismo, en un gráfico tipo Dotplot (Figura 9), se observa que ambas secuencias difieren
al comienzo y en la región central. La primera diferencia se debe a que la secuencia
mutante no incluye la región del gen codificante para los dominios transmembrana. La
segunda diferencia, en la parte central, es debida a la ausencia de los 231 pb en ∆fixL*
producto de la deleción realizada.
6. Expresión y purificación de las proteínas
recombinantes FixL* y FixL*
6.a. Obtención de cepas de E. coli productoras de
FixL* y FixL*
Para los experimentos de expresión se clonaron las secuencias fixL* y fixL* en
el vector pTrsHis C. Cada inserto de ADN se posicionó corriente abajo y en el mismo
marco de lectura de un iniciador de traducción (codón ATG) y de una secuencia que
codifica para un péptido de fusión en su amino terminal. Este péptido que consta de seis
residuos de histidina actúa como dominio de unión a iones metálicos. Esta característica
fue elegida para facilitar la purificación de las proteínas recombinantes en una
cromatografía de afinidad iónica.
En un primer paso se digirieron los vectores pGEMfixL* y pGEMfixL* con las
enzimas de restricción BamHI y EcoRI en forma secuencial para liberar los insertos que
luego se purificaron. Cada uno de estos fragmentos luego fue ligado al vector pTrsHis
C, previamente digerido con las mismas enzimas y purificado, dando lugar a la creación
de los plásmidos pTrsHisfixL* y pTrsHisfixL*.
Con estos plásmidos se transformaron células de E. coli DH5α competentes,
seleccionando los transformantes por la resistencia antibiótica conferida por el vector.
Las características de este no permitían distinguir entre las cepas que incorporaron el
vector y las que incorporaron el vector con el inserto, por lo que la confirmación final
de los transformantes se realizó por Colony – PCR utilizando los cebadores RFIXL y
LFIXL teniendo en cuenta que E. coli no posee este gen fixL. Los resultados se
muestran en la figura 10.

42
En la imagen A, calle 4, observamos la presencia de una banda que corre entre
las bandas de 955pb y 736pb del marcador. Esto está de acuerdo con lo esperado ya que
el fragmento fixL* tiene un tamaño de 820pb, confirmando la incorporación del
plásmido pTrsHisfixL* en el transformante. Por otro lado, en la imagen B calle 2,
observamos la presencia de una banda que corre entre las bandas de 736pb y 586pb del
marcador, ya que el fragmento fixL* tiene un tamaño de 600pb. De esta forma se
confirma la incorporación del plásmido pTrsHisfixL* en dicha cepa. Ambos
transformantes fueron conservados a -70○
C para su posterior uso.
6.b. Pruebas piloto de expresión
Los primeros ensayos de expresión fueron hechos en forma piloto, para evaluar
si realmente era posible expresar las proteínas recombinantes FixL* y FixL* en E. coli
y en qué momento se presentaba el pico máximo de expresión. Para esto, se tomaron
muestras de los cultivos inducidos a distintos tiempos y se sometieron a electroforesis
en geles de poliacrilamida. En la figura 11 se observa una fotografía del gel en el cual se
analizaron las muestras de proteínas totales. La cantidad de proteínas sembradas
correspondió a igual número de unidades formadoras de colonia por mililitro del
cultivo.
A partir de estos resultados podemos concluir que las proteínas de interés están
siendo expresadas en E. coli a partir de su inducción. Para el pTrsHisfixL* se observa
B) A)
Figura 10. Confirmación de los plásmidos pTrsHisfixL* (A) y pTrsHisfixL* (B) por Colony PCR. A)
En las calles 1 y 2 se observa marcadores de peso molecular donde el tamaño de cada banda se indica a
los costados de la imagen. En la calle 3 se muestra el control negativo y en la calle 4 la amplificación a
partir de un transformante. B). En la calle 1 se observa un marcador de peso molecular. En la calle 2 se
observa la amplificación a partir de un transformante y en la calle 3 se muestra el control negativo.
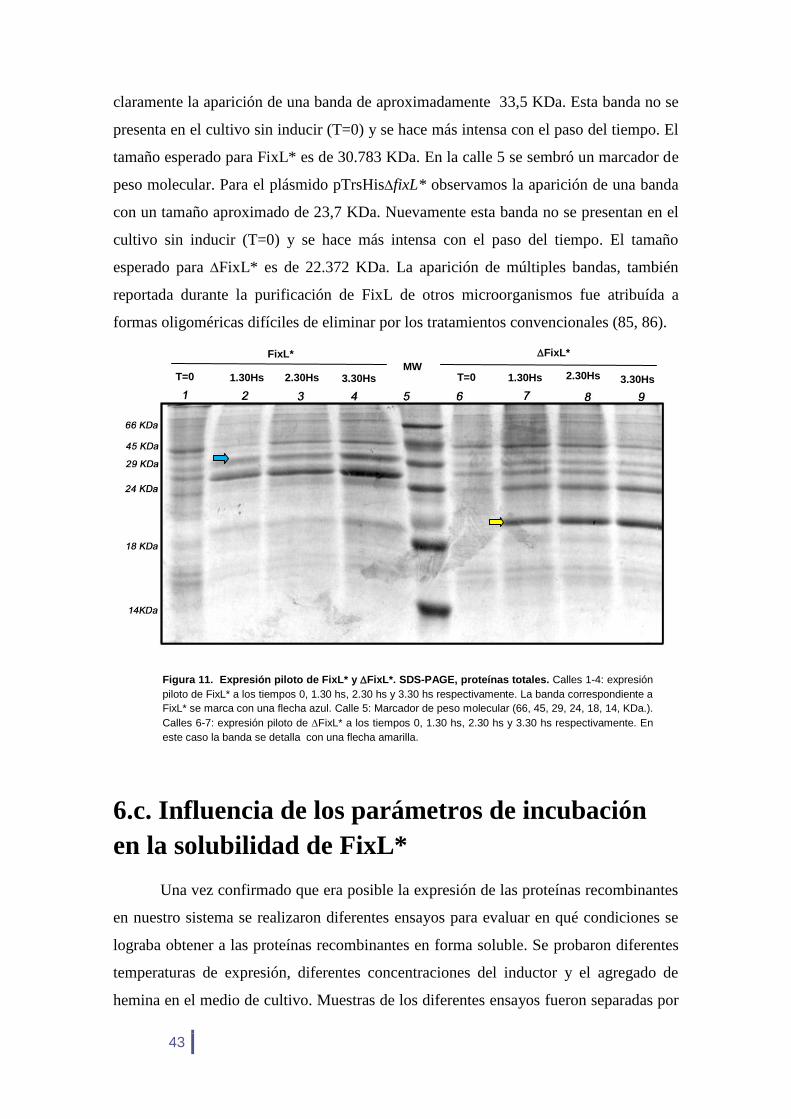
43
claramente la aparición de una banda de aproximadamente 33,5 KDa. Esta banda no se
presenta en el cultivo sin inducir (T=0) y se hace más intensa con el paso del tiempo. El
tamaño esperado para FixL* es de 30.783 KDa. En la calle 5 se sembró un marcador de
peso molecular. Para el plásmido pTrsHisfixL* observamos la aparición de una banda
con un tamaño aproximado de 23,7 KDa. Nuevamente esta banda no se presentan en el
cultivo sin inducir (T=0) y se hace más intensa con el paso del tiempo. El tamaño
esperado para FixL* es de 22.372 KDa. La aparición de múltiples bandas, también
reportada durante la purificación de FixL de otros microorganismos fue atribuída a
formas oligoméricas difíciles de eliminar por los tratamientos convencionales (85, 86).
6.c. Influencia de los parámetros de incubación
en la solubilidad de FixL*
Una vez confirmado que era posible la expresión de las proteínas recombinantes
en nuestro sistema se realizaron diferentes ensayos para evaluar en qué condiciones se
lograba obtener a las proteínas recombinantes en forma soluble. Se probaron diferentes
temperaturas de expresión, diferentes concentraciones del inductor y el agregado de
hemina en el medio de cultivo. Muestras de los diferentes ensayos fueron separadas por
Figura 11. Expresión piloto de FixL* y FixL*. SDS-PAGE, proteínas totales. Calles 1-4: expresión
piloto de FixL* a los tiempos 0, 1.30 hs, 2.30 hs y 3.30 hs respectivamente. La banda correspondiente a
FixL* se marca con una flecha azul. Calle 5: Marcador de peso molecular (66, 45, 29, 24, 18, 14, KDa.).
Calles 6-7: expresión piloto de FixL* a los tiempos 0, 1.30 hs, 2.30 hs y 3.30 hs respectivamente. En
este caso la banda se detalla con una flecha amarilla.
MW
T=0
1.30Hs
2.30Hs
3.30Hs
T=0
1.30Hs
2.30Hs
3.30Hs
FixL* FixL*
44
electroforesis en geles desnaturalizantes de poliacrilamida, a continuación presenta una
fotografía de un gel.
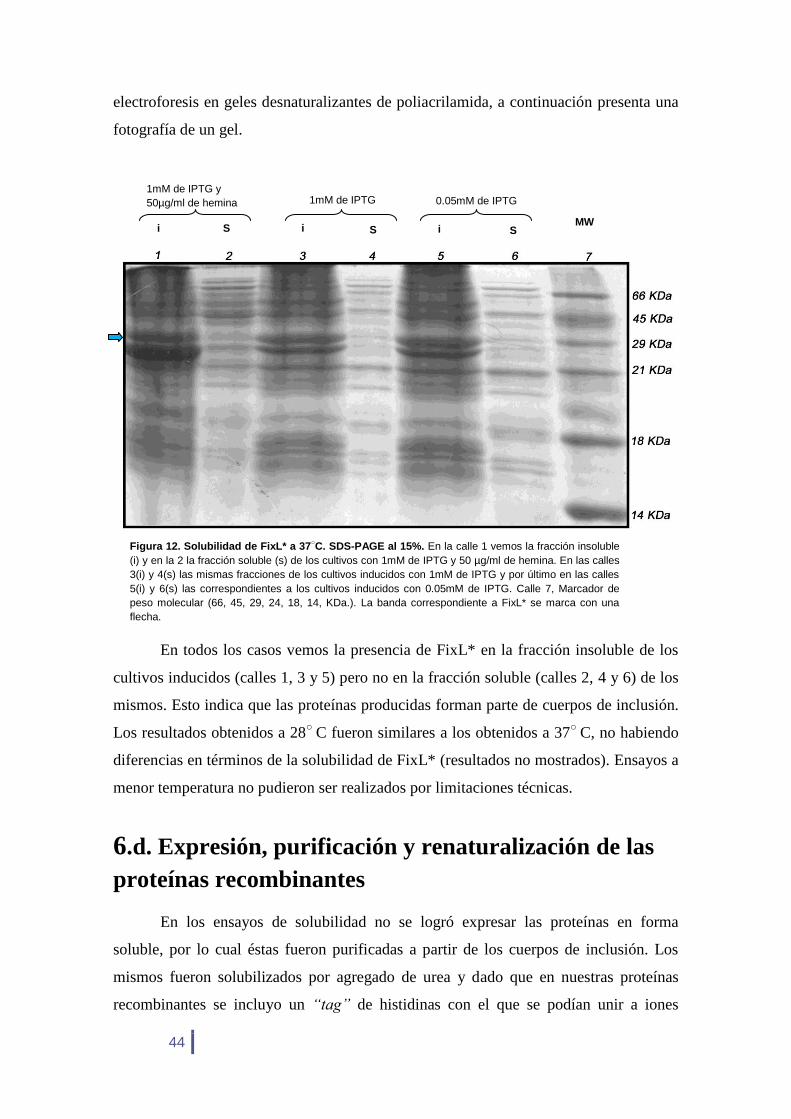
En todos los casos vemos la presencia de FixL* en la fracción insoluble de los
cultivos inducidos (calles 1, 3 y 5) pero no en la fracción soluble (calles 2, 4 y 6) de los
mismos. Esto indica que las proteínas producidas forman parte de cuerpos de inclusión.
Los resultados obtenidos a 28○
C fueron similares a los obtenidos a 37○
C, no habiendo
diferencias en términos de la solubilidad de FixL* (resultados no mostrados). Ensayos a
menor temperatura no pudieron ser realizados por limitaciones técnicas.
6.d. Expresión, purificación y renaturalización de las
proteínas recombinantes
En los ensayos de solubilidad no se logró expresar las proteínas en forma
soluble, por lo cual éstas fueron purificadas a partir de los cuerpos de inclusión. Los
mismos fueron solubilizados por agregado de urea y dado que en nuestras proteínas
recombinantes se incluyo un “tag” de histidinas con el que se podían unir a iones
Figura 12. Solubilidad de FixL* a 37○C. SDS-PAGE al 15%. En la calle 1 vemos la fracción insoluble
(i) y en la 2 la fracción soluble (s) de los cultivos con 1mM de IPTG y 50 µg/ml de hemina. En las calles
3(i) y 4(s) las mismas fracciones de los cultivos inducidos con 1mM de IPTG y por último en las calles
5(i) y 6(s) las correspondientes a los cultivos inducidos con 0.05mM de IPTG. Calle 7, Marcador de
peso molecular (66, 45, 29, 24, 18, 14, KDa.). La banda correspondiente a FixL* se marca con una
flecha.
i
i
i
S
S
S
MW
1mM de IPTG y
50µg/ml de hemina 1mM de IPTG 0.05mM de IPTG
45
metálicos, éste se utilizó para la purificación en una columna de afinidad iónica. La
renaturalización se llevó a cabo mientras la proteína recombinante se encontraba
inmovilizada en la columna mediante el pasaje de buffers con concentraciones
decrecientes del agente desnaturalizante. Diferentes muestras fueron tomadas durante el
proceso de expresión y posterior purificación de las proteínas recombinantes,
procesadas y luego fueron separadas por electroforesis en geles desnaturalizantes de
poliacrilamida. En las figuras 13 y 14 se muestran los resultados obtenidos, en donde
podemos observar la expresión y posterior purificación de las proteínas FixL* y FixL*
respectivamente.
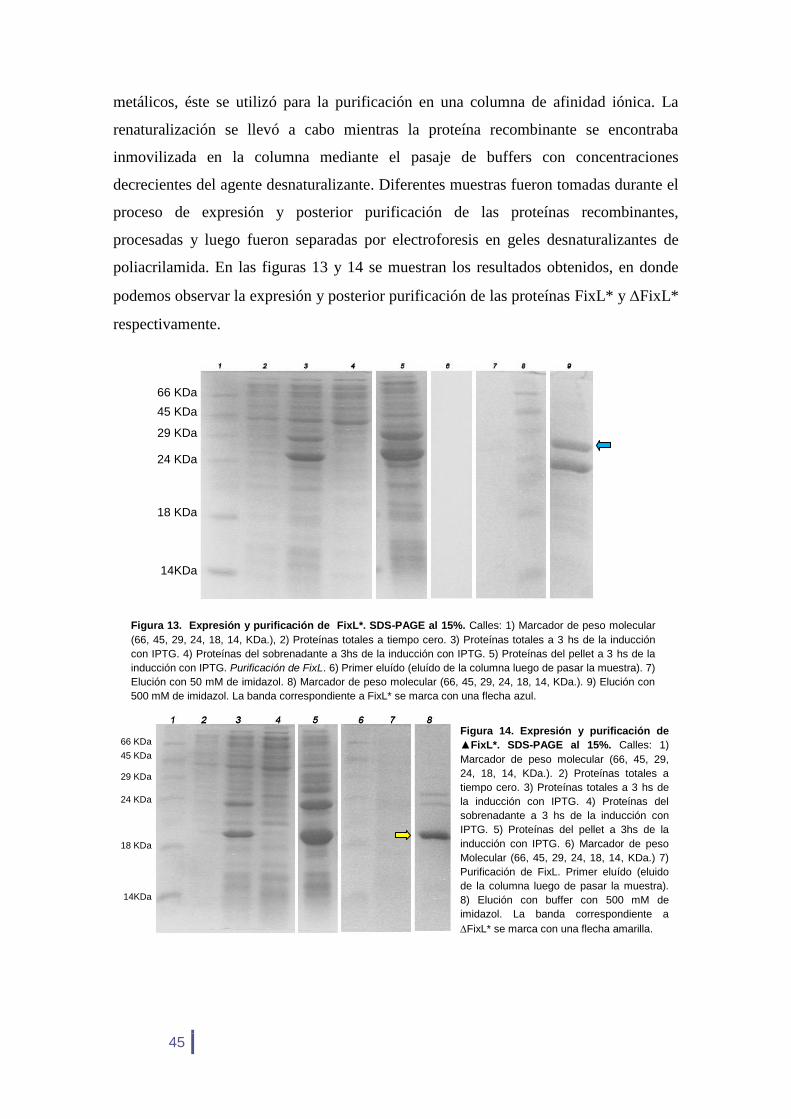
Figura 13. Expresión y purificación de FixL*. SDS-PAGE al 15%. Calles: 1) Marcador de peso molecular
(66, 45, 29, 24, 18, 14, KDa.), 2) Proteínas totales a tiempo cero. 3) Proteínas totales a 3 hs de la inducción
con IPTG. 4) Proteínas del sobrenadante a 3hs de la inducción con IPTG. 5) Proteínas del pellet a 3 hs de la
inducción con IPTG. Purificación de FixL. 6) Primer eluído (eluído de la columna luego de pasar la muestra). 7)
Elución con 50 mM de imidazol. 8) Marcador de peso molecular (66, 45, 29, 24, 18, 14, KDa.). 9) Elución con
500 mM de imidazol. La banda correspondiente a FixL* se marca con una flecha azul.
Figura 14. Expresión y purificación de
▲FixL*. SDS-PAGE al 15%. Calles: 1)
Marcador de peso molecular (66, 45, 29,
24, 18, 14, KDa.). 2) Proteínas totales a
tiempo cero. 3) Proteínas totales a 3 hs de
la inducción con IPTG. 4) Proteínas del
sobrenadante a 3 hs de la inducción con
IPTG. 5) Proteínas del pellet a 3hs de la
inducción con IPTG. 6) Marcador de peso
Molecular (66, 45, 29, 24, 18, 14, KDa.) 7)
Purificación de FixL. Primer eluído (eluido
de la columna luego de pasar la muestra).
8) Elución con buffer con 500 mM de
imidazol. La banda correspondiente a
FixL* se marca con una flecha amarilla.
66 KDa
45 KDa
29 KDa
24 KDa
18 KDa
14KDa
66 KDa
45 KDa
29 KDa
24 KDa
18 KDa
14KDa

46
Los tamaños esperados son de 30.781 Da para FixL* y de 22.371 Da para
FixL*. Tal como se muestra en las figuras 13 y 14, en la calle 5, las proteínas
recombinantes quedaron en la fracción insoluble luego de la expresión. A partir de los
procedimientos de desnaturalización y renaturalización de la muestras en la columna de
afinidad iónica, se logró la purificación de las proteínas expresadas. Las proteínas
fueron luego concentradas mediante una diálisis y cuantificadas por el método de
Bradford. La concentración final para FixL fue de 822.32 µg/ml y para FixL 539.86
µg/ml.
7. Capacidad de unión de FixL* al grupo hemo
Con la proteína FixL* purificada se decidió estudiar su capacidad de unión al
grupo hemo. Esta característica es importante ya que en otras especies la región sensora
se encuentra compuesta por un dominio PAS, de unión al grupo hemo, con el cual la
proteína se une transitoriamente al oxígeno. En Brucella, en un principio se descartó la
presencia de FixL debido a que éste no posee el dominio PAS, sin embargo, en este
trabajo de tesis y en otros trabajos (84) se demostró que ciertas proteínas de Brucella
pueden unirse al grupo hemo aún en ausencia de un dominio PAS.
En la figura 15 observamos que FixL* provocó un corrimiento del pico de
absorción de la hemina libre de 385 nm a 412 nm. El corrimiento de la banda de Soret
se evidencia mejor cuando al espectro del complejo se le resta el de la hemina libre, por
este motivo se grafican las absorbancias corregidas. También podemos apreciar los
distintos espectros de barrido de absorción evidenciando la semejanza del espectro del
complejo hemina-FixL* con el del control positivo (BSA). El espectro del control
negativo (Lisozima), cuando se le resta el de la hemina libre no presenta absorbancia,
indicando que no se une al hemo.
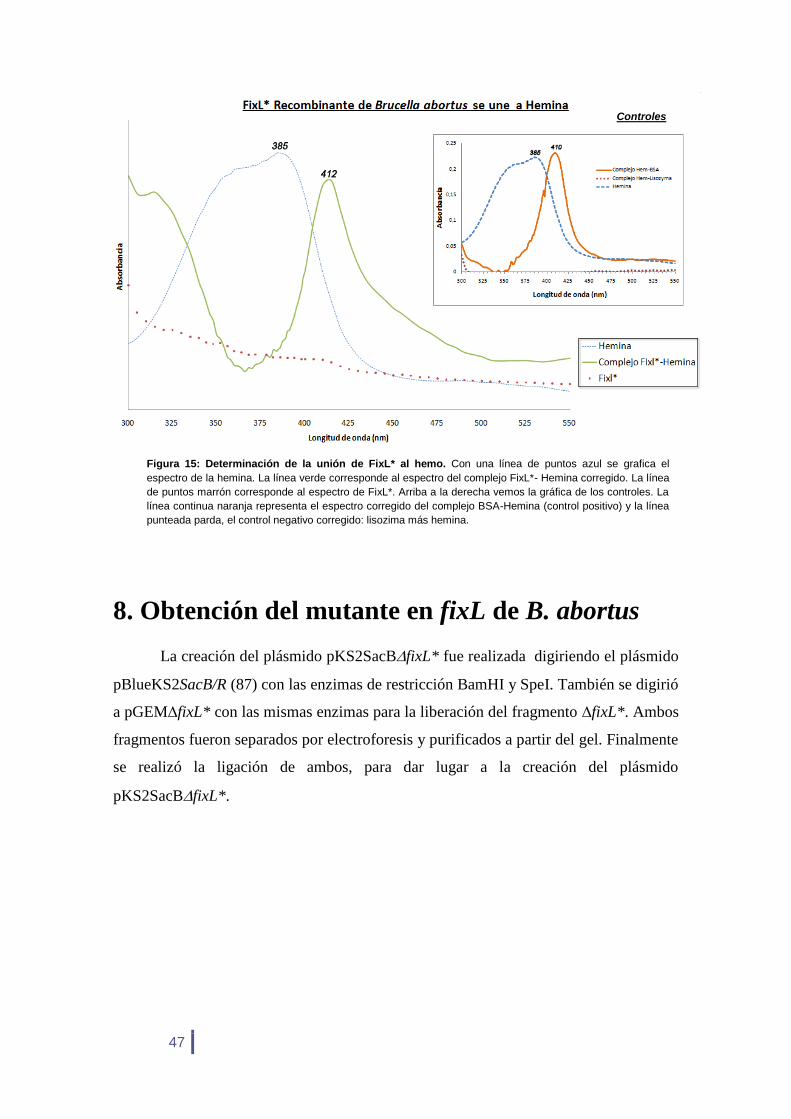
47
8. Obtención del mutante en fixL de B. abortus
La creación del plásmido pKS2SacBfixL* fue realizada digiriendo el plásmido
pBlueKS2SacB/R (87) con las enzimas de restricción BamHI y SpeI. También se digirió
a pGEM∆fixL* con las mismas enzimas para la liberación del fragmento ∆fixL*. Ambos
fragmentos fueron separados por electroforesis y purificados a partir del gel. Finalmente
se realizó la ligación de ambos, para dar lugar a la creación del plásmido
pKS2SacBfixL*.
Figura 15: Determinación de la unión de FixL* al hemo. Con una línea de puntos azul se grafica el
espectro de la hemina. La línea verde corresponde al espectro del complejo FixL*- Hemina corregido. La línea
de puntos marrón corresponde al espectro de FixL*. Arriba a la derecha vemos la gráfica de los controles. La
línea continua naranja representa el espectro corregido del complejo BSA-Hemina (control positivo) y la línea
punteada parda, el control negativo corregido: lisozima más hemina.
Controles
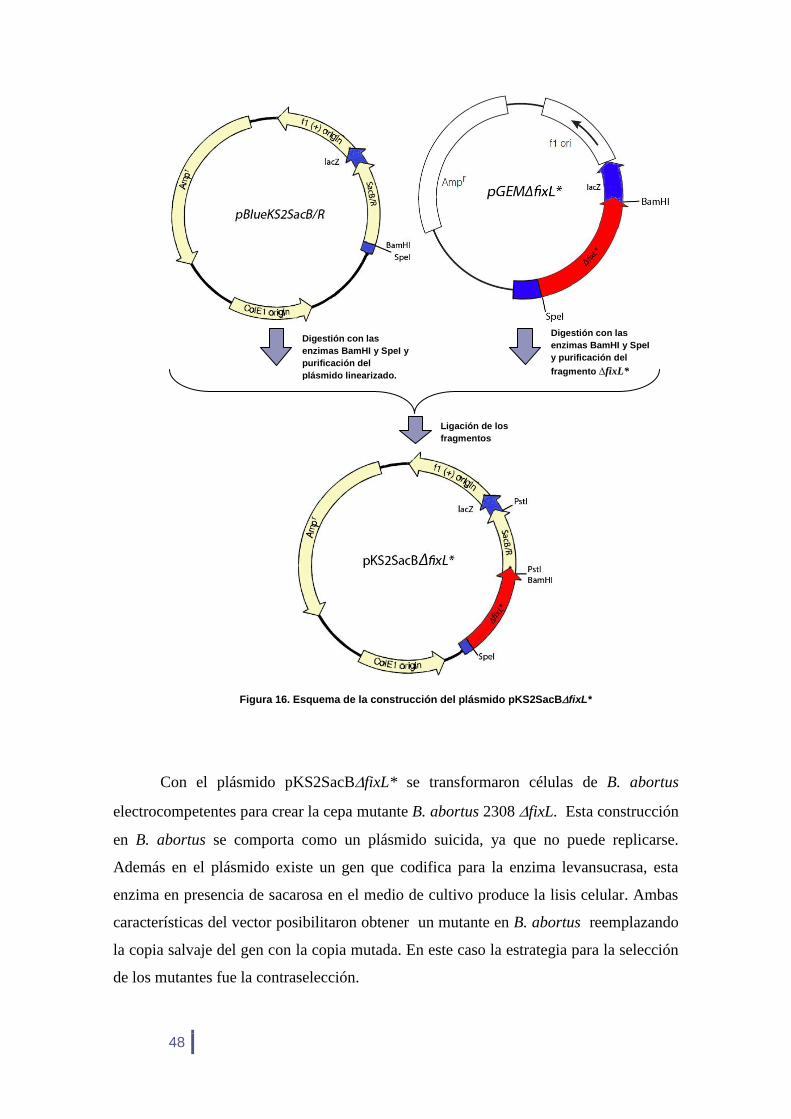
48
Con el plásmido pKS2SacBfixL* se transformaron células de B. abortus
electrocompetentes para crear la cepa mutante B. abortus 2308 fixL. Esta construcción
en B. abortus se comporta como un plásmido suicida, ya que no puede replicarse.
Además en el plásmido existe un gen que codifica para la enzima levansucrasa, esta
enzima en presencia de sacarosa en el medio de cultivo produce la lisis celular. Ambas
características del vector posibilitaron obtener un mutante en B. abortus reemplazando
la copia salvaje del gen con la copia mutada. En este caso la estrategia para la selección
de los mutantes fue la contraselección.
Digestión con las
enzimas BamHI y SpeI y
purificación del
plásmido linearizado.
Digestión con las
enzimas BamHI y SpeI
y purificación del
fragmento ∆fixL*
Ligación de los
fragmentos
Figura 16. Esquema de la construcción del plásmido pKS2SacBfixL*
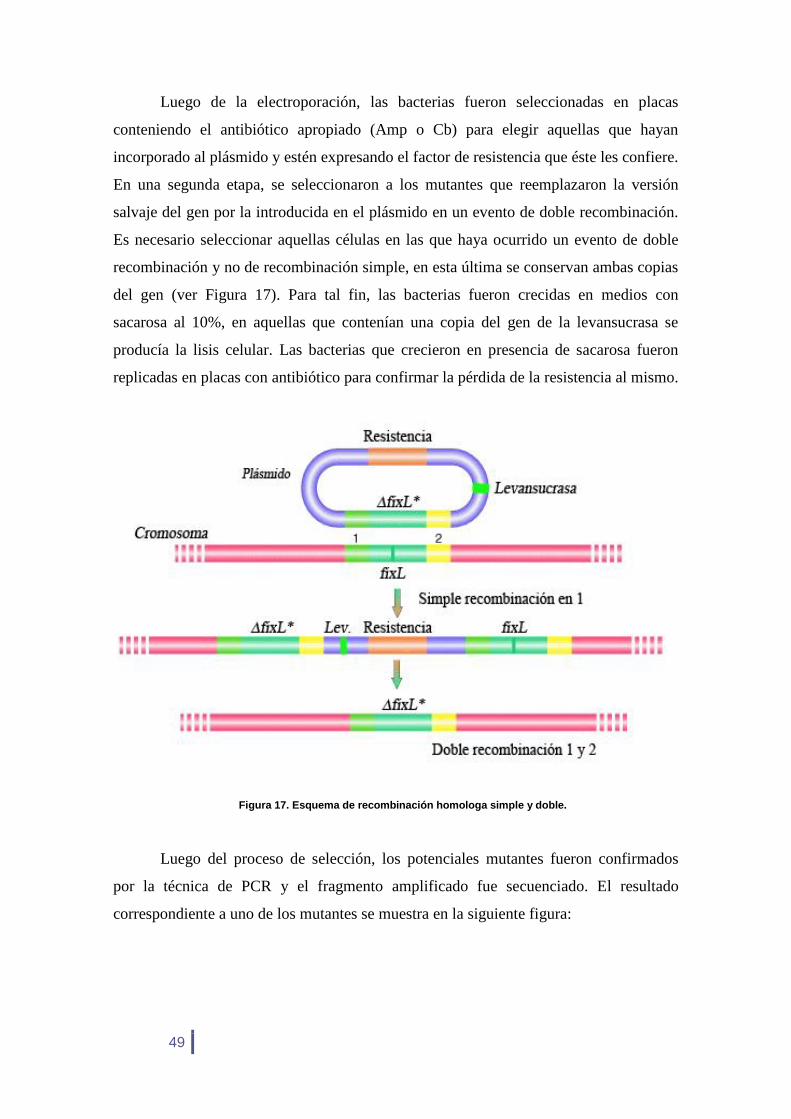
49
Luego de la electroporación, las bacterias fueron seleccionadas en placas
conteniendo el antibiótico apropiado (Amp o Cb) para elegir aquellas que hayan
incorporado al plásmido y estén expresando el factor de resistencia que éste les confiere.
En una segunda etapa, se seleccionaron a los mutantes que reemplazaron la versión
salvaje del gen por la introducida en el plásmido en un evento de doble recombinación.
Es necesario seleccionar aquellas células en las que haya ocurrido un evento de doble
recombinación y no de recombinación simple, en esta última se conservan ambas copias
del gen (ver Figura 17). Para tal fin, las bacterias fueron crecidas en medios con
sacarosa al 10%, en aquellas que contenían una copia del gen de la levansucrasa se
producía la lisis celular. Las bacterias que crecieron en presencia de sacarosa fueron
replicadas en placas con antibiótico para confirmar la pérdida de la resistencia al mismo.
Luego del proceso de selección, los potenciales mutantes fueron confirmados
por la técnica de PCR y el fragmento amplificado fue secuenciado. El resultado
correspondiente a uno de los mutantes se muestra en la siguiente figura:
Figura 17. Esquema de recombinación homologa simple y doble.

50
En la calle dos, observamos la amplificación del control positivo,
correspondiente a la cepa salvaje. En esta calle observamos una banda de
aproximadamente 800pb correspondiente al fragmento fixL*. En la calle 4, se observa
una única banda que corre entre la tercer y cuarta banda del marcador, la misma
coincide con el peso esperado para el fragmento ∆fixL*, de aproximadamente 600 pb.
De esta forma se confirmó la obtención del mutante B. abortus 2308 ∆fixL.
9. Fenotipo del mutante
Una vez obtenido el mutante se intentó caracterizar su fenotipo. Los resultados
aquí mencionados son de carácter preliminar.
Se analizaron la capacidad de crecimiento tanto en medio sólido como líquido en
condiciones de oxigenación, microaerobiosis y anaerobiosis. No hubo diferencias
significativas entre el mutante y la cepa salvaje para ninguno de los ensayos realizados.
Es decir, ambas cepas presentaron curvas de crecimiento semejantes cuando fueron
incubadas con agitación en medio rico o mínimo y ambas sobrevivieron de igual modo
a las condiciones de microaerobiosis expuestas. En anaerobiosis ninguna de las cepas
fue capaz de desarrollar. Tampoco hubo diferencias entre ambas cepas en la actividad
de la enzima catalasa. Curiosamente, durante el estudio del crecimiento en tubos con
medio tioglicolato pudo observarse diferencias en el patrón de crecimiento entre el
mutante y la cepa salvaje (Figura 19). En la cepa salvaje el desarrollo se concentró en la
zona microaerobia (parte superior del tubo) con desarrollo por paredes del tubo y sin
desarrollo en la zona anaerobia (parte inferior del tubo). Esto está de acuerdo a lo
descripto en la bibliografía para B. abortus (1). Por otro lado, el mutante se desarrolló
desde la zona microaerobia con un intenso desarrollo por el centro y paredes del tubo
Figura 18. Comprobación por PCR del
mutante de B. abortus 2308 ∆fixL. En la calles
1 se sembró un marcador de peso molecular, el
tamaño de cada banda se indica a la izquierda
de la imagen. En la calle 2, se sembró una
muestra correspondiente al control positivo (B.
abortus 2308 wt) y en la calle 3 se sembró una
muestra del control negativo de la PCR. En la
calle 4 la muestra de un posible mutante
B.abortus 2308 fixL.

51
hacia la zona de menor tensión de oxígeno en las profundidades del tubo, colonizando la
mayor parte de éste.
Este resultado podría significar que la expresión de fixL restringe el crecimiento
de B. abortus a la zona de microaerobiosis y su ausencia o no funcionalidad serían
necesarias para el crecimiento a menores tensiones de oxígeno. Esto sería diferente a lo
planteado en una primer instancia, pero también es cierto que todavía no se han hecho
estudios para saber en qué momento el gen es expresado en B. abortus. Una posibilidad
por la que no hayamos observado más características fenotípicas diferenciales es quizás,
debido a que la actividad de esta proteína se presente sólo en una fase del desarrollo en
condiciones especiales de crecimiento que no hemos podido reproducir en el
laboratorio.
Figura 19. Crecimiento en Tioglicolato. Desarrollo de
la cepa salvaje B. abortus 2308 (izquierda) y del
mutante B. abortus 2308 ∆fixL* (derecha) en tubos con
medio tioglicolato. Fotografía tomada a los 5 días de
incubación.

52
10. Capacidad de formar biofilms por Brucella
abortus
Durante la realización de los experimentos mencionados en el punto anterior
pudo observarse que B. abortus crecía formando películas delgadas denominadas
biofilms, este tipo de crecimiento nunca había sido descripto para esta especie
bacteriana. Estas observaciones fueron confirmadas al realizar cultivos estáticos de B.
abortus en botellas con medios líquidos, que luego de ser lavadas se tiñeron con
colorantes que evidenciaban la presencia de films biológicos. Se tomaron muestras de
esta particular forma de crecimiento de B. abortus pare ser observadas en un
microscopio electrónico de barrido (Figura 20).
Es importante destacar que este descubrimiento abre nuevos caminos en la
investigación de Brucella. Los biofilms son estructuras de resistencia que protegen a las
bacterias de los antibióticos y del sistema inmune.
Figura 20. Desarrollo de B. abortus 2308 en forma de biofilm. A la izquierda se muestra una fotografía del
cultivo de B. abortus en un medio líquido sin agitación. Luego de eliminar el caldo y lavar la botella, se realizó
una coloración con cristal violeta para evidenciar su presencia. A la derecha se muestra, una imagen obtenida
en un microscopio electrónico de barrido, en la cual se observan las células unidas por una matriz extracelular
(barra: 2 µm).

Conclusión

54
Para concluir, podemos decir que en este trabajo de tesis se han realizados
avances claves para la identificación y caracterización de FixL en Brucella abortus.
Pudimos establecer que la región en estudio, con homología a FixL, es funcional en
Brucella abortus y codifica para una proteína que logramos expresar en forma
recombinante en E. coli. Una vez expresada, fue posible purificarla y determinar que es
capaz de unirse al grupo hemo. También se logró obtener un mutante de B. abortus en
fixL que presentó un patrón de crecimiento diferencial en cuanto a sus requerimientos de
oxigenación.

Perspectivas
futuras

56
Partiendo de que se ha logrado expresar y purificar la proteína recombinante, se
puede profundizar el estudió “in vitro” de la misma. Todavía queda trabajo por hacer en
este terreno, ya sea estudiando la capacidad de la proteína de unirse al oxígeno o su
actividad quinasa y los mecanismos de autofosforilación.
Para un completo estudio del fenotipo del mutante deben realizarse más ensayos,
cuidando sobre todo las condiciones de oxigenación. Por este motivo para continuar con
el estudió de FixL es deseable investigar en qué momento de su ciclo de vida se expresa
y para ello el estudio del promotor usando un gen reportero sería de gran ayuda.
También sería deseable estudiar el fenotipo de los mutantes “in vivo”, ya sea en
infecciones en ratones o macrófagos. Como hemos mencionado anteriormente, en los
Rhizobiales, FixL juega un rol fundamental durante la simbiosis en los nódulos de las
plantas. Si hacemos un paralelismo entre lo que ocurre en estos microorganismos y
Brucella podemos especular que FixL en este último juegue un rol importante durante la
infección, detectando los niveles de oxígeno tanto en el exterior de su célula huésped
como una vez fagocitado en el interior del fagosoma.

Bibliografía

58
1. Don J. Brenner, Noel R. Krieg , James T. Staley, George M. Garrity. 2005.
Bergey's Manual of Systematic Bacteriology, Volume Two. United States of
America. Springer.
2. Jeffrey Green, Mark S. Paget. Bacterial redox sensors. 2004. Nature Reviews
Microbiology.2, 954-966.
3. Rodgers K. R., Lukat-Rodgers, G. S. 2005. “Insights into Heme-based O2
Sensing from Structure Function Relationships in the FixL Proteins” J. Inorg.
Biochem. 99, 963-977.
4. Lois, A. F., G. S. Ditta, and D. R. Helinski. 1993. The oxygen sensor FixL of
Rhizobium meliloti is a membrane protein containing four possible
transmembrane segments. J. Bacteriol. 175, 1103-1109.
5. Crosson, S., P. T. McGrath, C. Stephens, H. H. McAdams, and L. Shapiro. 2005.
Conserved modular design of an oxygen sensory/signaling network with
species-specific output. Proc. Natl. Acad. Sci. 102, 8018-8023.
6. Pellequer J.-L.; Brudler R.; Getzoff E.D. 1999. Biological sensors: More than
one way to sense oxygen. Curr. Biol. 9, 416-418.
7. Rodgers KR. Heme-based sensors in biological systems. 1999. Curr. Opin.
Chem. Biol. 3, 158-157.
8. Tuckerman, J. R., Gonzalez, G., Dioum, E. M., and Gilles Gonzalez, M. A.
2002. Ligand and Oxidation-State Specific Regulation of the Heme-Based
Oxygen Sensor FixL from Sinorhizobium meliloti. Biochemistry. 41, 6170–
6177.
9. Séverine Loisel Meyer, Maria Pilar Jiménez de Bagués, Stephan Kohler, Jean-
Pierre Liautard, and Véronique Jubier-Maurin. 2005. Differential Use of the
Two High Oxygen-Affinity Terminal Oxidases of Brucella suis for In Vitro and
Intramacrophagic Multiplication. Infect Immun.73, 7768–7771.
10. Jiménez de Bagüés MP, Loisel Meyer S, Liautard JP, Jubier Maurin V. 2007.
Different roles of the two high-oxygen-affinity terminal oxidases of Brucella
suis: Cytochrome c oxidase, but not ubiquinol oxidase, is required for
persistence in mice. Infect Immun.75, 531-5.
11. James, P. E., O. Y. Grinberg, G. Michaels, and H. M. Swartz. 1995.
Intraphagosomal oxygen in stimulated macrophages. J. Cell Physiol. 163, 241-
247.
12. Enrique Laval R. 2006. Contribución al estudio histórico de la brucelosis en
Chile. Rev. Chil. Infect. 23, 362-366.
13. Correa, W. M.; Correa, C. N. 1992. Enfermidades Infecciosas dos Mamíferos
Domésticos. 2 ed. Rio de Janeiro. Medsi.
14. José Jerónimo Estévez. 2000. Anales de la Real Academia Ciencias Veterinarias
de Andalucía Oriental. Granada. 13, 33-70.

59
15. Quiroga, S. A. 1933. Día Médico.
(hist.library.paho.org/Spanish/BOL/v13n7p630.pdf).
16. Apud. 1934. La Semana Médica.
(hist.library.paho.org/Spanish/BOL/v13n7p630.pdf).
17. Molinelli, E. A., Fernandez Ithurrat, E. M. 1934. La Semana Médica.
(hist.library.paho.org/Spanish/BOL/v13n7p630.pdf).
18. Molinelli, E. A. 1934. La Semana Médica.
(hist.library.paho.org/Spanish/BOL/v13n7p630.pdf).
19. Hozbor, F.A.; Fiorentino, M.A., Malena, R.C., Terzolo H. y F.A. Paolicchi.
Reunión Anual de la AAVLD (Asociación Argentina de Veterinarios de
Laboratorios de Diagnóstico) XIVa. Noviembre de 2002, Villa Gral. Belgrano,
Sierras de Córdoba. Bruselosis en visones (Mustela vison) de criadero.
20. Lumila Fuchs, Valeria Baldone, Marcelo Fort, María del Carmen Rojas, Luis
Samartino, Hugo Giménez. 2009. Brucelosis en el zorro gris pampeano
(Pseudalopex gymnocercus) en la provincia de La Pampa (Argentina). Acta
Bioquím. Clín. Latinoam. 43, 227-31.
21. Instituto Nacional de Tecnología Agropecuaria (INTA). 2000. Documento del
Programa de Sanidad Animal, Mimeo, 85.
22. Servicio Nacional de Sanidad y Calidad Agroalimentaria (SENASA). 1999. Plan
nacional para la erradicación de Brucelosis y Tuberculosis Bovina. Resolución
115/99. Mimeo, 62.
23. Acha, F., Szyfres, B. 1986. Brucellosis. Zoonosis y enfermedades trasmisibles
comunes al hombre y a los animales. Pan-American Health Organization
(PAHO).
24. Luis E. Samartino. 2002. Brucellosis in Argentina. Veterinary microbiology. 90,
71-80.
25. Carrillo, C. G. 1990. Animal and human brucelosis in the Americas. Office
Internacional des Epizooties, 4-30.
26. Wallach, J.C.; Lucero, N.; Efron, A.; Casimir, L.; Baldi, P. Congreso Argentino
y Latinoamericano de Zoonosis, Buenos Aires. 1995. Estudio
seroepidemiológico y microbiológico en infecciones humanas por Brucella. I.
27. Pizarro-Cerda J, Meresse S, Parton RG, van der Goot G, Sola-Landa A, Lopez-
Goni I, et al. 1998. Brucella abortus transits through the autophagic pathway
and replicates in the endoplasmic reticulum of nonprofessional phagocytes.
Infect Immun. 66, 5711-5724.

60
28. Pontow S, Kery V, Stahl D. 1992. Mannose receptor. Int Rev Cytol. 137, 221-
241.
29. Canning P. C, Roth J. A, Deyoe B. L. 1986 Release of 5'-guanosine
monophosphate and adenine by Brucella abortus and their role in the
intracellular survival of the bacteria. J Infect Dis.154, 464-470.
30. Blasco, J. M. 1997. A review of the use of B. melitensis Rev 1 vaccine in adult
sheep and goats. Prev. Vet. Med. 31, 275–283.
31. Bosseray, N. 1991. Brucella melitensis Rev. 1 living attenuated vaccine:
stability of markers, residual virulence and immunogenicity in mice. Biologicals
19, 355–363.
32. Corbel, M. J. 1997. Brucellosis: an overview. Emerg. Infect. Dis. 3, 213–221.
33. Jimenez de Bagues, M. P., C. M. Marin, M. Barberan, and J. M. Blasco. 1989.
Responses of ewes to B. melitensis Rev1 vaccine administered by subcutaneous
or conjunctival routes at different stages of pregnancy. Ann. Rech. Vet. 20, 205–
213.
34. Moreno, E., E. Stackebrandt, M. Dorsch, J. Wolters, M. Busch, and H. Mayer.
1990. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship
with members of the alpha-2 subdivision of the class Proteobacteria. J.
Bacteriol. 172, 3569–3576.
35. Michaux, S., J. Paillisson, M. J. Carles-Nurit, G. Bourg, A. Allardet-Servent,
and M. Ramuz. 1993. Presence of two independent chromosomes in the
Brucella melitensis 16M genome. J. Bacteriol. 175, 701–705.
36. Michaux-Charachon, S., G. Bourg, E. Jumas-Bilak, P. Guigue-Talet, A.
Allardet-Servent, D. O’Callaghan, and M. Ramuz. 1997. Genome structure and
phylogeny in the genus Brucella. J. Bacteriol. 179, 3244–3249.
37. Brinley Morgan, W. J., and M. J. Corbel. 1990. Brucella infections in man and
animals: contagious equine metritis. 547–570.
38. Kaufmann, A. F., M. I. Meltzer, and G. P. Schmid. 1997. The economic impact
of a bioterrorist attack: are prevention and postattack intervention programs
justifiable? Emerg. Infect. Dis. 3:83–94.
39. Madkour M. M. 2001. Brucellosis: overview. Brucellosis, 2nd edn. Berlin,
Springer-Verlag.
40. Canning, P. C., J. A. Roth, and B. L. Deyoe. 1986. Release of 5_-guanosine
monophosphate and adenine by Brucella abortus and their role in the
intracellular survival of the bacteria. J. Infect. Dis. 154, 464–470.
41. Jones, S. M., and A. J. Winter. 1992. Survival of virulent and attenuated strains
of Brucella abortus in normal and gamma interferon-activated murine peritoneal
macrophages. Infect. Immun. 60, 3011–3014.

61
42. Robertson, M. 1998. Innate immunity: Curr. Biol. 8, R595–R597.
43. Fernandez-Prada, C. M., M. Nikolich, R. Vemulapalli, N. Sriranganathan, S. M.
Boyle, G. G. Schurig, T. L. Hadfield, and D. L. Hoover. 2001. Deletion of wboA
enhances activation of the lectin pathway of complement in Brucella abortus
and Brucella melitensis. Infect. Immun. 69, 4407–4416.
44. Ko J., Splitter G. A. 2003. Molecular host-pathogen interaction in brucellosis:
Current understanding and future approaches to vaccine development for mice
and humans. Clin Microbiol Rev. 1, 65-78.
45. Young, E. J., M. Borchert, F. L. Kretzer, and D. M. Musher. 1985. Phagocytosis
and killing of Brucella by human polymorphonuclear leukocytes. J. Infect. Dis.
151, 682–690.
46. Teixeira Gomes A., Cloeckaert A., Zygmunt M. S. 2000. Characterization of
heat, oxidative, and acid stress responses in Brucella melitensis. Infect Immun.
68, 2954-2961.
47. Castro Hugo Abel, González Sofía Raquel, Prat María Inés. 2005 Brucelosis:
una revisión práctica. Acta bioquím. clín. Latinoam. 39, 203-216.
48. Zhan Y, Cheers C. 1995. Endogenus Interleukin-12 is involved in resistance to
Brucella abortus infection. Infect Immun. 63, 387-389.
49. Ahmed K, Al-Matrouk KA, Martínez G, Oishi K, Rotimi VO, Nagatake T.
1999. Increased serum levels of Interferon-γ and Interleukin-12 during human
brucellosis. Am J Trop Med Hyg. 61, 425-427.
50. Winter, A. J., J. R. Duncan, C. G. Santisteban, J. T. Douglas, and L. G. Adams.
1989. Capacity of passively administered antibody to prevent establishment of
Brucella abortus infection in mice. Infect. Immun. 57, 3438–3444.
51. Montaraz, J. A., and A. J. Winter. 1986. Comparison of living and nonliving
vaccines for Brucella abortus in BALB/c mice. Infect. Immun. 53, 245–251.
52. Wang C., Wang L., Geng X. 2009. Optimization of refolding with simultaneous
purification of recombinant human granulocyte clolony-stimulating factor from
Escherichia coli by immobilized metal ion affinity chromatography.
Biochchemical engineering journal 43, 197-202.
53. David, M., M.-L. Daveran, J. Batut, A. Dedieu, 0. Domergue, J. Ghai, C. Hertig,
P. Boistard, and D. Kahn. 1988. Cascade regulation of nif gene expression in
Rhizobium meliloti. Cell. 54, 671-683.
54. David, M., 0. Domergue, P. Pognonec, and D. Kahn. 1987. Transcription
patterns of Rhizobium meliloti symbiotic plasmid pSym; identification of nifA-
independent fix genes. J. Bacteriol. 169, 2239-2244.
55. Hans-Martin Fischer. 1994. Genetic Regulation of Nitrogen Fixation in
Rhizobia. Microbiological Reviews. 58, 352-386

62
56. Anthamatten, D., and H. Hennecke. 1991. The regulatory status of the fixL-like
and fixJ-like genes in Bradyrhizobium japonicum may be different from that in
Rhizobium meliloti. Mol. Gen.Genet. 225, 38-48.
57. Batut, J., B. Terzaghi, M. Gherardi, M. Huguet, E. Terzaghi, A. M. Garnerone,
P. Boistard, and T. Huguet. 1985. Localization of a symbiotic fix region on
Rhizobium meliloti pSym megaplasmid more than 200 kilobases from the nod-
nif region. Mol. Gen. Genet. 199, 232-239.
58. Kaminski, P. A., and C. Elmerich. 1991. Involvement of fixLJ in the regulation
of nitrogen fixation in Azorhizobium caulinodans. Mol. Microbiol. 5, 665-673.
59. Virts, E. L., S. W. Stanfield, D. R. Helinski, and G. S. Ditta. 1988. Common
regulatory elements control symbiotic and microaerobic induction of nifA in
Rhizobium meliloti. Proc. Natl. Acad. Sci 85, 3062-3065.
60. Gilles Gonzalez M.A., Ditta G.S., Helinski D.R. 1991. A haemoprotein with
kinase activity encoded by the oxygen sensor of Rhizobium meliloti. Nature.
350, 170-172.
61. A F Lois, G S Ditta, and D R Helinski. 1993.The oxygen sensor FixL of
Rhizobium meliloti is a membrane protein containing four possible
transmembrane segments. J Bacteriol. 175, 1103–1109.
62. E K Monson, M Weinstein, G S Ditta, and D R Helinski. 1992. The FixL protein
of Rhizobium meliloti can be separated into a heme-binding oxygen-sensing
domain and a functional C-terminal kinase domain. Proc. Nat. Acad. Sci. 89,
4280-4284.
63. Lois, A. F., G. S. Ditta, and D. R Helinski. 1993. The oxygen sensor FixL of
Rhizobium meliloti is a membrane protein containing four possible
transmembrane segments. J. Bacteriol. 175, 1103-1109.
64. Perutz M.F., Paoli M., Lesk A.M. 1999. Fix L, a haemoglobin that acts as an
oxygen sensor: signalling mechanism and structural basis of its homology with
PAS domains. Chem Biol. 6, R291-297.
65. Lois AF, Weinstein M, Ditta GS, Helinski DR. 1993. Autophosphorylation and
phosphatase activities of the oxygen-sensing protein FixL of Rhizobium meliloti
are coordinately regulated by oxygen. 268, 4370-4375.
66. Ellen K. Monson, Gary S. Ditta and Donald R. Helinski. 1995. The Oxygen
Sensor Protein, FixL, of Rhizobium meliloti. Role of histidine residues in heme
binding, phosphorylation, and signal transduccitions. Journal of biological
chemistry. 270, 5243-5250.
67. Parkinson, J. S., and E. C. Kofoid. 1992. Communication modules in bacterial
signaling proteins. Annu. Rev. Genet. 26, 71-112.
68. Kahn, D., and G. Ditta. 1991. Modular structure of FixJ: homology of the
transcriptional activator domain with the -35 binding domain of sigma factors.
Mol. Microbiol. 5, 987-997.

63
69. Kurashima-Ito K, Kasai Y, Hosono K, Tamura K, Oue S, Isogai M, Ito Y,
Nakamura H, Shiro Y. 2005. Solution structure of the C-terminal transcriptional
activator domain of FixJ from Sinorhizobium meliloti and its recognition of the
fixK promoter. Biochemistry. 44, 14835-14844.
70. Batut, J., M.-L. Daveran-Mingot, M. David, J. Jacobs, A. M. Garnerone, and D.
Kahn. 1989. fixK, a gene homologous with fnr and crp from Escherichia coli,
regulates nitrogen fixation genes both positively and negatively in Rhizobium
meliloti. EMBO J. 8, 1279-1286.
71. Mandon, K., H. Hillebrand, C. Mougel, N. Desnoues, B. Dreyfus, P. A.
Kaminski, and C. Elmerich. 1993. Characterization of fixK-regulated
Azorhizobium caulinodans genes, p. 478. In R. Palacios, J. Mora, and W. E.
Newton (ed.), New horizons in nitrogen fixation. Kluwer Academic Publishers,
Dordrecht, The Netherlands.
72. Mandon, K., P. A. Kaminski, and C. Elmerich. 1994. Functional analysis of the
fixNOQP region of Azorhizobium caulinodans. J. Bacteriol. 176, 2560-2568.
73. Reyrat, J. M., M. David, P. de Philip, A. M. Garnerone, J. Batut, and P.
Boistard. 1993. In vivo and in vitro oxygen control of nitrogen fixation gene
expression in Rhizobium meliloti, p. 399- 403. In R. Palacios, J. Mora, and W. E.
Newton (ed.), New horizons in nitrogen fixation. Kluwer Academic Publishers,
Dordrecht, The Netherlands.
74. Garnerone A. M., Cabanes D., Foussard M., Boistard P., Batut J. 1999.
Inhibition of the FixL sensor kinase by the FixT protein in Sinorhizobium
meliloti. J Biol Chem. 274, 32500-32506.
75. O. Preisig, Rachel Zufferey and H. Hennecke. 1996. The Bradyrhizobium
japonicum fixGHIS genes are required for the formation of the high-affinity
cbb3-type cytochrome oxidase. Arch. Microbiol. 165, 297-305
76. Soupene, E., Foussard, M., Boistard, P., Truchet, G. & Batut, J. 1995. Oxygen as
a key developmental regulator of Rhizobium meliloti N2 fixation gene expression
within the alfalfa root nodule. Proc. Natl Acad. Sci. 92, 3759-3763.
77. Jason R. Tuckerman, Gonzalo Gonzalez and Marie-Alda Gilles Gonzalez. 2001.
Complexation Precedes Phosphorylation for Two-component Regulatory
System FixL/FixJ of Sinorhizobium meliloti. J. Mol. Biol. 308, 449-455.
78. Gilles Gonzalez, M. A., Gonzalez, G. & Perutz, M. F. 1995. Kinase activity of
oxygen sensor FixL depends on the spin state of its heme iron. Biochemistry. 34,
232-236.
79. Gilles-Gonzalez, M.A., and Gonzalez, G. 1993. Regulation of the kinase activity
of heme protein FixL from the two component system FixL/FixJ of Rhizobium
meliloti. J Biol Chem. 268, 16293–16297.
80. Ken Saito, Eiichi Ito, Kaito Hosono, Kayako Nakamura, Kiyohiro Imai,
Tetsutaro Iizuka, Yoshitsugu Shir and Hiro Nakamura. 2003. The uncoupling of

64
oxygen sensing, phosphorylation signalling and transcriptional activation in
oxygen sensor FixL and FixJ mutants. Molecular Microbiology. 48, 373–383.
81. Pascale de Philip, Jacques Baut, and Pierre Boistard. 1990. Rhizobium meliloti
FixL Is an Oxygen Sensor and Regulates R. meliloti nifA and fixK Genes
Differently in Escherichia coli. Journal of Bacteriology. 172, 4255-4262.
82. Da Re, S., Schumacher, J., Rousseau, P., Fourment, J., Ebel, C. & Kahn, D.
1999. Phosphorylationinduced dimerization of the FixJ receiver domain. Mol.
Microbiol. 34, 504-511.
83. Sean Crosson, Patrick T. McGrath, Craig Stephens, Harley H. McAdams, and
Lucy Shapiro. 2005. Conserved modular design of an oxygen sensory_signaling
network with species-specific output. PNAS. 102, 8018–8023.
84. Marta Almirón, Marcela Martínez, Norberto Sanjuan, and Rodolfo A. Ugalde.
2001. Ferrochelatase Is Present in Brucella abortus and Is Critical for Its
Intracellular Survival and Virulence. Infect Immun. 69, 6225–6230.
85. I. D’hooghe, J. Michiels, J. Vanderleyden. 1998. The Rhizobium etli FixL
protein differs in structure from other known FixL proteins. Mol Gel Genet.
257, 576-580.
86. E. K. Monson, M. Weinstein, G. S. Ditta, and D. R. Helinski. 1992. The FixL
protein of Rhizobium meliloti can be separated into a heme-binding oxygen-
sensing domain and a functional C-terminal kinase domain. Proc. Natl. Acad.
Sci. 89, 4280–4284.
87. Rodrigo Sieira, Diego J. Comerci, Lía I. Pietrasanta and Rodolfo A. Ugalde.
2004. Integration host factor is involved in transcriptional regulation of the
Brucella abortus virB operon. Molecular Microbiology. 54, 808–822.
88. Jerpseth, B., Greener A., Short, J.M., Viola, J., and kretz, P.L. 1992. Strategies
5, 81-83.
89. Sanbrook, J., Fritsch, E. F. and Maniatis, T. 1989. Molecular cloning: a
laboratory manual, 2nd
ed. Cold Spring Harbor laboratory Press. Cold Spring
Harbor, N.Y.
90. Egon, A., Brosius, J., and Ptashne, M. 1983. Vectors Bearing a Hybrid trp-lac
Promoter Useful for Regulated Expression of Cloned Genes in Escherichia coli.
Gene 25, 167-178.
91. Li, S. C., Squires, C. L., and Squires, C. (1984). Antitermination of E. coli
rRNA Transcription is Caused by a control Region Segment Containing Lambda
nut-like Sequences. Cell 38, 851-860.

65
Apéndice
Fig
ura
A
. A
lin
eam
ien
tos
m
últ
iple
s
de
las d
isti
nta
s s
ecu
en
cia
s F
ixL
. E
n
la
fig
ura
se
observ
a
tres
zonas
conserv
adas.
Cada
una
de
ella
s
se
colo
rea
de
dos
form
as
altern
ativas,
arr
iba
según
el
porc
enta
je
de
identid
ad
de
los
am
inoácid
os.
Abajo
según
las
cara
cte
rísticas
fisic
oquím
icas d
e c
ada a
min
oácid
o.
La
secuencia
de F
ixL de B
. abort
us se
desta
ca
entr
o
dos
líneas
para
lela
s
roja
s.
Bru
ce
lla a
bo
rtu
s
Hy
ph
om
on
as
ne
ptu
niu
m
Rh
izo
biu
m le
gu
min
osa
rum
Azo
rhiz
ob
ium
ca
ulin
od
an
s
Ca
ulo
ba
cte
r c
resc
en
tus
Bra
dy
rhiz
ob
ium
ja
po
nic
um
Sin
orh
izo
biu
m m
elilo
ti
Olig
otr
op
ha
ca
rbo
xid
ov
ora
ns
Rh
izo
biu
m e
tli
Me
sorh
izo
biu
m lo
ti
Bru
ce
lla a
bo
rtu
s
Hy
ph
om
on
as
ne
ptu
niu
m
Rh
izo
biu
m le
gu
min
osa
rum
Azo
rhiz
ob
ium
ca
ulin
od
an
s
Ca
ulo
ba
cte
r c
resc
en
tus
Bra
dy
rhiz
ob
ium
ja
po
nic
um
Sin
orh
izo
biu
m m
elilo
ti
Olig
otr
op
ha
ca
rbo
xid
ov
ora
ns
Rh
izo
biu
m e
tli
Me
sorh
izo
biu
m lo
ti
Bru
ce
lla a
bo
rtu
s
Hy
ph
om
on
as
ne
ptu
niu
m
Rh
izo
biu
m le
gu
min
osa
rum
Azo
rhiz
ob
ium
ca
ulin
od
an
s
Ca
ulo
ba
cte
r c
resc
en
tus
Bra
dy
rhiz
ob
ium
ja
po
nic
um
Sin
orh
izo
biu
m m
elilo
ti
Olig
otr
op
ha
ca
rbo
xid
ov
ora
ns
Rh
izo
biu
m e
tli
Me
sorh
izo
biu
m lo
ti
Bru
ce
lla a
bo
rtu
s
Hy
ph
om
on
as
ne
ptu
niu
m
Rh
izo
biu
m le
gu
min
osa
rum
Azo
rhiz
ob
ium
ca
ulin
od
an
s
Ca
ulo
ba
cte
r c
resc
en
tus
Bra
dy
rhiz
ob
ium
ja
po
nic
um
Sin
orh
izo
biu
m m
elilo
ti
Olig
otr
op
ha
ca
rbo
xid
ov
ora
ns
Rh
izo
biu
m e
tli
Me
sorh
izo
biu
m lo
ti

66
Fig
ura
B.
Esq
uem
a d
e l
a r
eg
ión
de
l g
en
om
a d
e B
. ab
ort
us
en
la c
uá
l se r
ealizó
la d
ele
ció
n.
En a
zul se s
eñala
n lo
s s
itio
s d
e u
nió
n d
e lo
s c
ebadore
s.
Adem
ás s
e r
epre
senta
n c
on
caja
s r
oja
s l
os d
om
inio
s t
ransm
em
bra
na,
con u
n p
entá
gono v
erd
e l
a h
istid
ina s
usceptib
le d
e f
osfo
rila
ció
n y
con u
na l
íne
a v
iole
ta e
l dom
inio
quin
asa.
La
regió
n q
ue f
ue d
ele
cio
nada s
e
mu
estr
a c
on u
na lín
ea a
zul.

67


















