
Hidróxi-Selenetose Hidróxi-Te'luretos Quirais...ASL CALB CALA CCD CG CRL DIBAL-H DMF DMSO E e.e....
210
1_" _,?;_?a_'.5_-g_1 * t INSTITUTO DE QUi MICA UNIVERSIDADE DE SÃO PAULO UNIVERSIDADE DE SÃO PAULO INSTITUTO DE QUíMICA Biotransformações na Obtenção de Hidróxi-Selenetos e Hidróxi- Te'luretos Quirais Carlos Eduardo da Costa f Tese de Doutoramento ProL Dr. João V. Comasseto Orientador São Paulo 2006
Transcript of Hidróxi-Selenetose Hidróxi-Te'luretos Quirais...ASL CALB CALA CCD CG CRL DIBAL-H DMF DMSO E e.e....

~ 1_"_,?;_?a_'.5_-g_1 *tINSTITUTO DE QUi MICAUNIVERSIDADE DE SÃO PAULO
UNIVERSIDADE DE SÃO PAULO
INSTITUTO DE QUíMICA
Biotransformações na Obtenção de
Hidróxi-Selenetos e Hidróxi-Te'luretos
Quirais
Carlos Eduardo da Costaf
Tese de Doutoramento
ProL Dr. João V. Comasseto
Orientador
São Paulo2006

<.: L,. : .... !i;' :


---- ~
DEDALUS • Acervo· CQ
IIIIIIIIII~ IIIIII~IIIIIIIIIIIIIIIIIIIIII11I1I ~IIIIII~ 11111111
30100012254

Aos meus queridos pais, José Cesarioe Isaura (in memorian), e a minha
querida irmã-mãe, Marilene,pelo seu amor e apoio sem restrições.

A minha amada noiva, Mariana,pelo seu amor e sua
compreensão.


Agradecimentos...


À Deus e ao meu anjo da guarda, pela constante proteção e por sempre
iluminarem meu caminho...
Ao Professor Comasseto pela sua orientação, pelas ótimas condições de
trabalho e pela liberdade de desenvolver projetos em seu laboratório. Por todo
apoio e encorajamento dado durante todo o tempo e principalmente por ensinar
muito mais que química aos seus alunos...
Aos professores, Helena Ferraz, Liliana Marzorati e Hélio Stefani, pela ajuda e
convivência durante esses anos. Também aos seus alunos por toda a ajuda,
pelos favores e pela amizade. Em especial ao Eduardo da Costa Ramos pelo
compartilhamento de experiências e sobre tudo pela amizade incondicional.
As professoras Maria da Graça e Sandra Zanotto, pela breve orientação, pelos
trabalhos em colaboração e principalmente pelo apoio e a amizade.
Aos amigos que estão ou estiveram no laboratório durante todo este período:
Dennis, Priscila, Rodrigo, Takeo, Maurício, Mirian, F. Kauer, Nathalia, Cristiano,
Solange, Artur, Martin, Fabiano, Ivani, Silas, Zinc, João, Bruno, Alcindo, Zé
Fogo, Felipe, Nilson, Mulla, Gabriel e Vinícios, pelo bom ambiente de trabalho
no laboratório. Ao Lauri por ter me apresentado a "Biocatálise". Em especial ao
Giuliano por ter sempre compartilhado suas experiências em síntese e
biocatálise e pelos trabalhos em colaboração.
A amiga Sabrina pela amizade e pelas boas e divertidas conversas durante
nossos almoços.
Aos colegas e amigos do bloco zero: Pedro, Luís, Leonardo, Roberto, Roberta,
Piovan, Thaís, Adriana, Alexandre e Edna, por toda a ajuda e favores. Em
especial aos professores Leandro e André pela amizade, pelo apoio e pelos
trabalhos em colaboração.
xi

Aos meus alunos de Iniciação Científica: Guilherme, Henrique e Renan por
toda a ajuda e ensinamentos. A participação de vocês foi muito importante
neste trabalho e principalmente na minha formação.
Ao Alexandre Sardelli e a Dona Rosa pela boa e agradável convivência ao
longo desses anos.
A toda a equipe da Central Analítica, especialmente Luzia, Adriana, Mirian e
Alessandra pela pronta ajuda na execução das análises.
Ao pessoal da Secretaria de Pós-Graduação, por sempre terem sido muito
prestativos.
A FAPESP, pelas bolsas de Iniciação Científica e Doutorado Direto.
Aos meus sogros, Leila e João, pelo apoio, pela amizade e pela cumplicidade
em todos os momentos em que compartilhamos. Em especial aos meus
cunhados, Vinicius e Mayra que da maneira deles, sei que torcem muito por
mim...
Aos meus queridos pais, José Cesario e Isaura (in memorian), a minha irmã
mãe, Marilene, ao meu irmão, José Teodoro, e aos meus sobrinhos, Thiago e
Guilherme, pelo amor, pelo carinho, pela compreensão, pelo total e irrestrito
apoio e pelo compartilhamento de todas a minhas alegrias e frustrações. Muito
obrigado!
A minha querida noiva, Mariana Simplício Touzarim, pelo seu amor, carinho e
compreensão, que foram e são muito importantes para mim, como aluno de
doutorado e como pessoa. Obrigado por tudo! Você é muito importante para
mim...
Xll

Lista de Abreviaturas


ASL
CALB
CALA
CCD
CG
CRL
DIBAL-H
DMF
DMSO
E
e.e.
E.M.
I.V.
J
LDA
MSL
m/z
PPL
PSLPLERMN 1H
RMN 13C
RMN 77SeRMN 125Te
t.a.
THF
TLL
- lipase de Ascaligenes sp.
- lipase de Candida antartica fração B
- lipase de Candida antartica fração A
- cromatografia em camada delgada
- cromatografia a gás
- lipase de Candida rugosa
- hidreto de di-isobutilaluminio.
- N,N-dimetilformamida
- dimetil sulfóxido
- razão enantiomérica
- excesso enantiomérico
- espectro de massas I espectrometria de massas
- infravermelho
- constante de acoplamento em Hz
- diisopropilamideto de lítio
- íon molecular
- lipase de Mucor javanicus
- relação massa-carga
- lipase de pâncreas de porco
- lipase de Pseudomonas sp.
- esterease de fígado de porco
- ressonância magnética nuclear de hidrogênio
- ressonância magnética nuclear de carbono
- ressonância magnética nuclear de selênio
- ressonância magnética nuclear de telúrio
- temperatura ambiente
- tetraidrofurano
- lipase de Thermonyces /anuginosa
xv


Lista de Estruturas

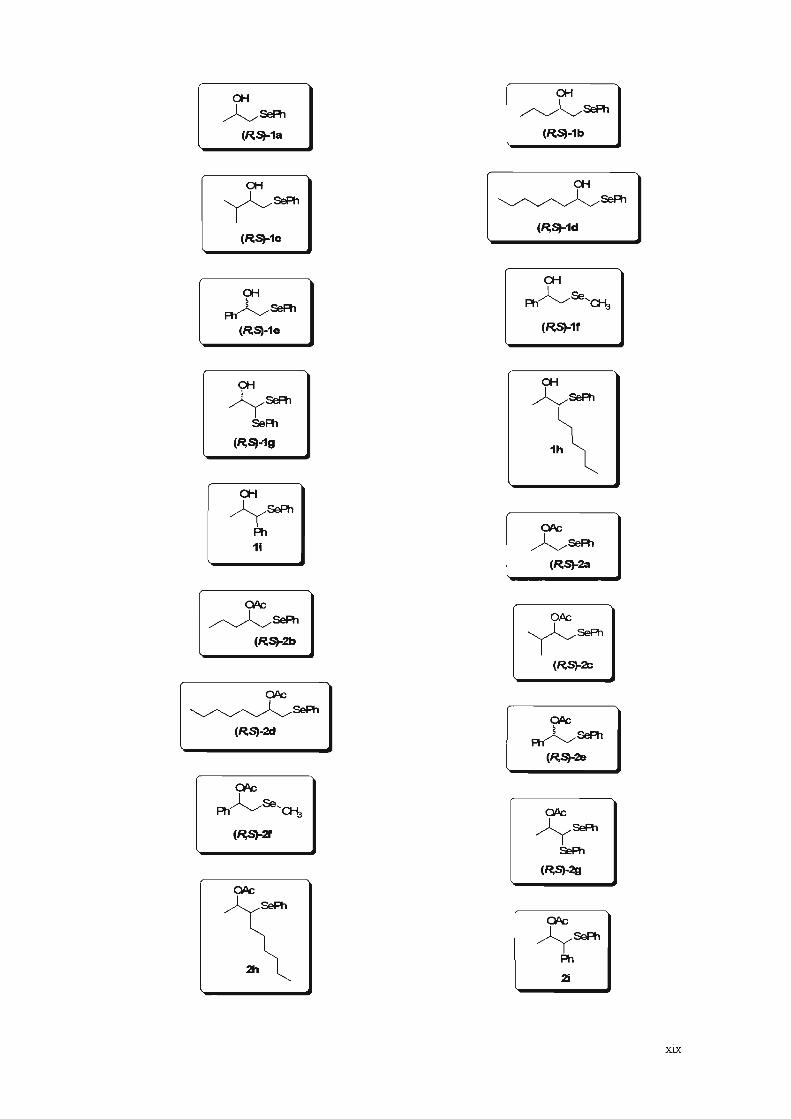
I
a-f
~5ePh(~S)-1a
(~S)-1c
a-f
~5ePhR1(~Sl-1e
a-f
~5ePh
(~S)-1b
I
a-f
Ar5ePh
SeR!
I (~S)-1gI
5ePh
1h
OPC
~5ePh(~~2c
2hI
xix
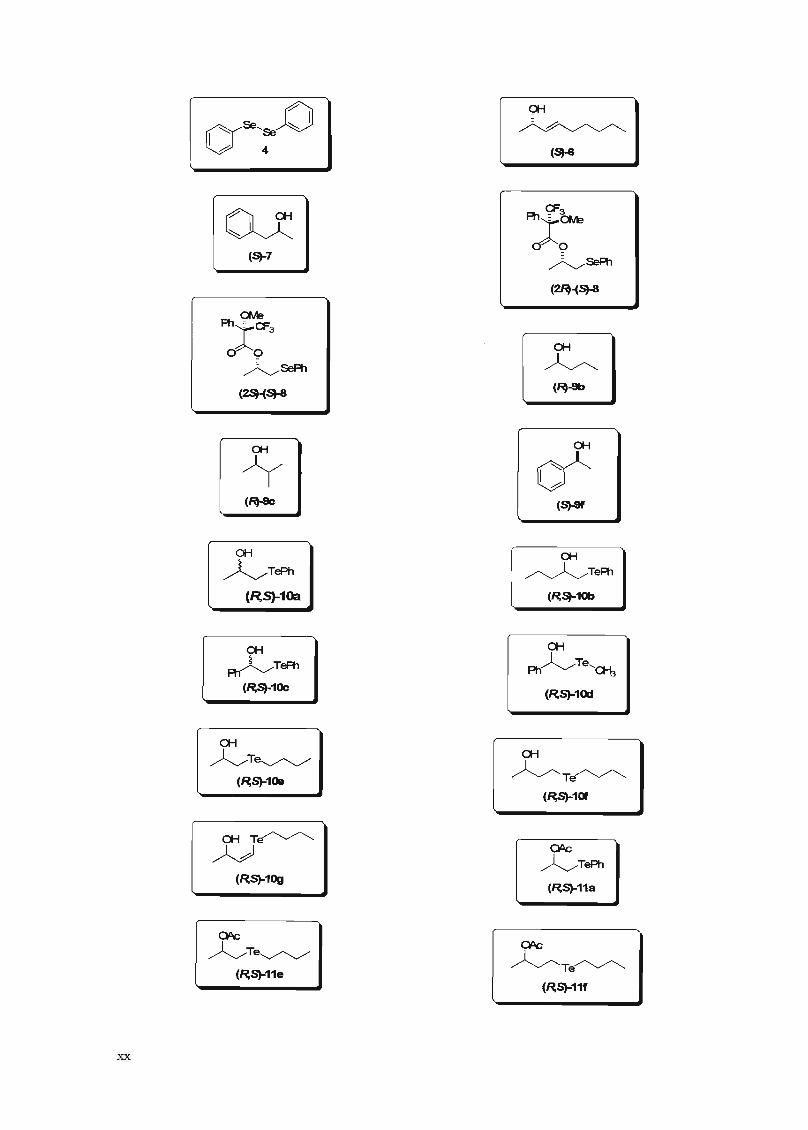
À) OH
~ry5e'5e ~I b 4 (5)-6
CF3Q;: '):~(5)-7
~5ePh
(~-(~
<:Me
')::3 OH
~~5ePh
(~-Sb(2SH~
WOH
if(S)-8f
OH OH
~TePh ~TeA1
(R,S)-1Cla (~5)-1Ob
OHOH
Ph~Te,~~TePhPh
(~5)-1Oc(~S)-1Od
xx
OH
~T~(~S)-1Oe
a;Te~
~(~S)-10g
QOc
~T~(~S)-11e
QOc
~TePh
(~S)-11a
QOc
~Te~(~S)-11f
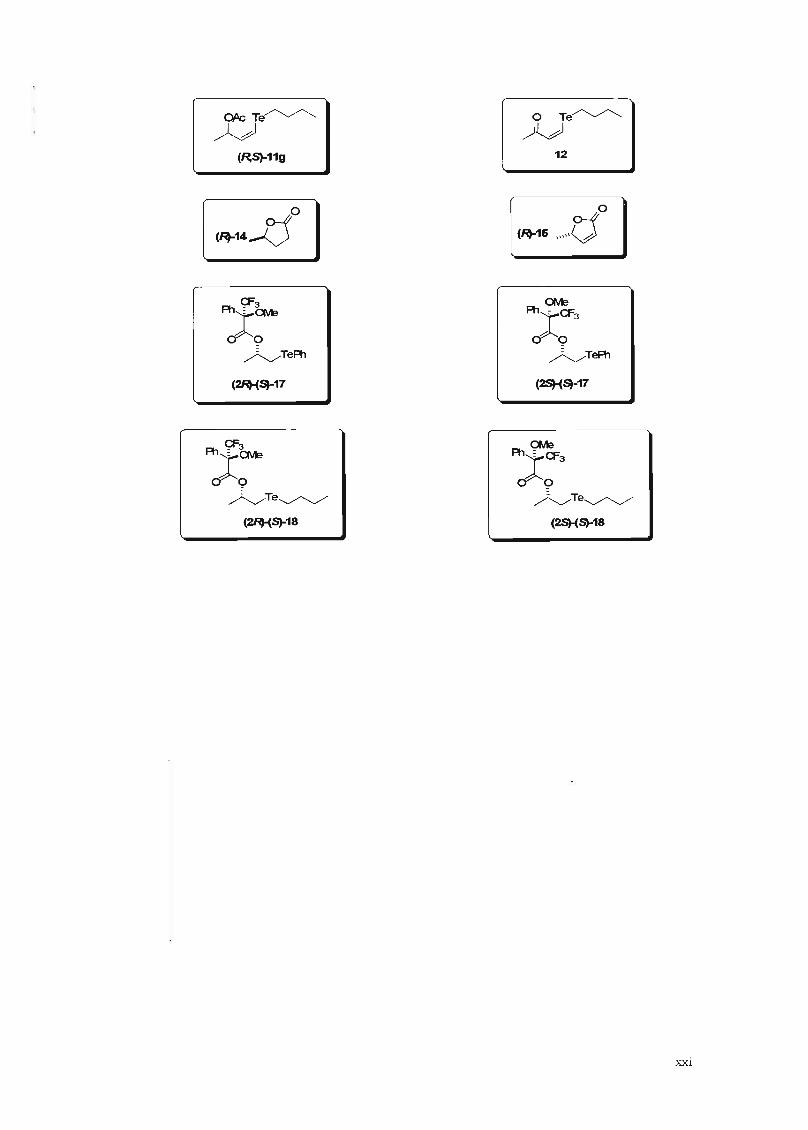
OkTe~
~(R,S)-11g
o Te~~
12
o
(~14Y:5 (~16
(2It'(~-17
(2It'(S)-18
(2So}(S)-17
(2So}(Sl-18
xxi


índice

".

1Iiil" Lo# ~ I' __ • - -~-
INSTITUTO DE QUíMICAUniversidade de São Paulo
Lista de Abreviaturas
Lista de Estruturas
Índice
Resumo
Abstract
Objetivos
1. Introdução
1.1. Quiralidade e Atividade Biológica
1.2. Biotransformações
1.2. 1. Enzimas
1.2.1.1. Aspectos Mecanísticos
1.2.1.2. Aplicação em Síntese Orgânica
1.2.1.3. Hidrolases
1.2.1.4. Técnicas Especiais em Síntese Orgânica
1.3. Selênio e Telúrio
2. Resultados e Discussão
21.11idróx~Seknetos
2.1.1. Síntese Racêmica
2.1.2. Biotransformações
2.1.2.1. Resolução em Meio Orgânico
2.1.2.2. Resolução em Meio Aquoso
2.1.2.3. Biotransformações Utilizando Células Inteiras de Fungos
2.1.3. Aplicação Sintética
2.1.4. Determinação da Pureza ÓpticalEstereoquímica
2.1.4.1. Determinação de Excesso Enantiomérico
2.1.4.2. Determinação da Configuração Absoluta
2.2. llidróxi-Teluretos
2.2.1. Síntese Racêmica
2.2.2. Biotransformações
2.2.2.1. Resolução em Meio Orgânico
2.2.3. Aplicação Sintética
2.2.4. Determinação da Pureza ÓpticalEstereoquímica
2.2.4.1. Determinação de Excesso Enantiomérico
2.2.4.2. Determinação da Configuração Absoluta
Xl11
XVIl
XXI II
XXVIX
XXXl11
XXXVIl
1
9
13
14
17
19
23
27
31
33
35
38
38
45
49
52
53
53
54
57
59
62
62
67
68
68
69
xxv

3.Conclusões 71
4. Parte Experimental 75
4.1. Materiais e Métodos 77
4.2. Procedimentos Gerais 83
4.2.1. Preparação do Disse1eneto de Difenila 85
4.2.2. Preparação dos Hidróxi-selenetos (R,8)-la e (R,8)-le 85
4.2.3. Preparação do Bis(fenilsse1eno)metano 87
4.2.4. Preparação dos Hidróxi-selenetos (R,8)-lb-d 87
4.2.5. Preparação do Hidróxi-seleneto (R,8)-lf 89
4.2.6. Preparação do Diisopropilamideto de lítio (LDA) 90
4.2.7. Preparação do Hidróxi-seleneto (R,8)-lg 90
4.2.8. Preparação dos Fenilsselenoacetais 91
4.2.9. Preparação dos Hidróxi-selenetos (±)-lh-i 91
4.2.10. Preparação do Ditelureto de Difenila 93
4.2.11. Preparação dos Hidróxi-teluretos (R,8)-lOa e (R,.S)-lOc 94
4.2.12. Preparação do Diazometano 95
4.2.13. Preparação do Bis((fenilteluro)metano 96
4.2.14. Preparação do Hidróxi-telureto (R,8)-lOb 96
4.2.15. Preparação dos Hidróxi-teluretos (R,8)-lOd e (R,8)-lOe 97
4.2.16. Preparação do Hidróxi-te1ureto (R,8)-lOf 98
4.2.17. Preparação da (Z)-butilteluro-enona 99
4.2.18. Preparação do Hidróxi-telureto (R,8)-lOg 100
4.2.19. Preparação dos Acetatos dos Hidróxi-calcogenetos 101
4.2.20. Resolução Enzimática em Meio Orgânico 106
4.2.21. Imobilização Enzimática 111
4.2.21. 1. Gel de Agar 111
4.2.21.2. Filme de Caseinato de Sódio 111
4.2.21.3. Sílica ou Montmorilonita K-10 (Silicato de Alumínio) 111
4.2.21.4. Poli(oxietileno) - PEO 111
4.2.22. Triagem Enzimática 112
4.2.23. Hidrólise Enzimática 113
4.2.24. Biotransformação Empregando Células Inteiras de Fungos 113
4.2.24.1. Crescimento dos Fungos em Meio Sólido 114
4.2.24.2. Crescimento dos Fungos em Meio Líquido 114
xxvi

4.2.24.3. Obtenção das Células Úmidas de Fungos 114
4.2.24.4. As Biotransformações 114
4.2.25. Preparação do Álcool Alílico - Reação de Eliminação de Se1enóxido 115
4.2.26. Preparação das lactonas 115
4.2.27. Determinação dos Excessos Enantioméricos 116
4.2.27.1. Hidrólise Química 117
4.2.27.2. Desselenização Redutiva 117
4.2.27.3. Derivatização Utilizando Anidrido Trif1uoracético 118
4.2.28. Determinação da Configuração Absoluta 118
4.2.28.1. Preparação do Cloreto de Ácido de Mosher 118
4.2.28.2. Preparação dos Derivados de Mosher 118
4.2.28.3. Desselenização Redutiva 121
4.2.28.4. Eliminação de Selenóxido 122
4.2.28.5. Reação de Lactonização 123
4.3. Espectros Selecionados 125
5. Bibliografia 167
6. Curriculum Vitae 173
xxvii


Resumo


Neste trabalho foi estudado o comportamento de hidróxi-calcogenetos
(Se e Te) frente a biotransformações, empregando enzimas isoladas em meio
orgânico ou aquoso e empregando microorganismos (fungos). Estudos
comparativos sobre a influência de diversas variáveis, como solvente,
temperatura, imobilização enzimática e estrutura do hidróxi-calcogeneto, foram
realizados.
Inicialmente os compostos foram sintetizados utilizando métodos
descritos na literatura, em seguida foi estudada a resolução de hidróxi
selenetos em meio orgânico empregando lipases isoladas (Esquema 1),
incluindo um estudo de imobilização da PSL em diversos suportes, além do
estudo da influência da variação do solvente, da temperatura, da lipase, etc.
a, R1 =A1, R2=H, R'=~b, R1 =A1, R2=H, R'=(~~C, R1 =A1, R2=H, R'=(~~d, R1 = A1, R2 = H, R' = a-t(~>:ze, R1 = A1, R2 = H, R' = A1
f, R1=~ R2=H, R3=A190 R1 =A1, R2=SePh, R'=~h, R1 = A1, R2 = (~013, R' =~i, R1 =A1, R2=A1, R'=013
o)l
Esquema 1. Resolução enzimática de hidróxi-selenetos em meio orgânico.
Na resolução em meio aquoso empregando enzimas isoladas,
primeiramente os hidróxi-selenetos foram acetilados quimicamente e depois
realizado uma triagem (com dez enzimas de diferentes fontes) empregando
indicador de pH colorimétrico. Posteriormente os acetatos dos hidróxi-selenetos
(Esquema 2) foram submetidos à resolução enzimática em meio aquoso
empregando as enzimas que foram selecionadas na triagem enzimática.
AcO I" AcOT SePh Ipase. '''''rSePhR Tampão \ Acetona R
Esquema 2. Hidrólise enzimática.
HO~SePh+ I +
R
o
)lOH
As biotransformações utilizando fungos foram realizadas empregando
células inteiras de algumas linhagens de Aspergillus terreus.
xxxi
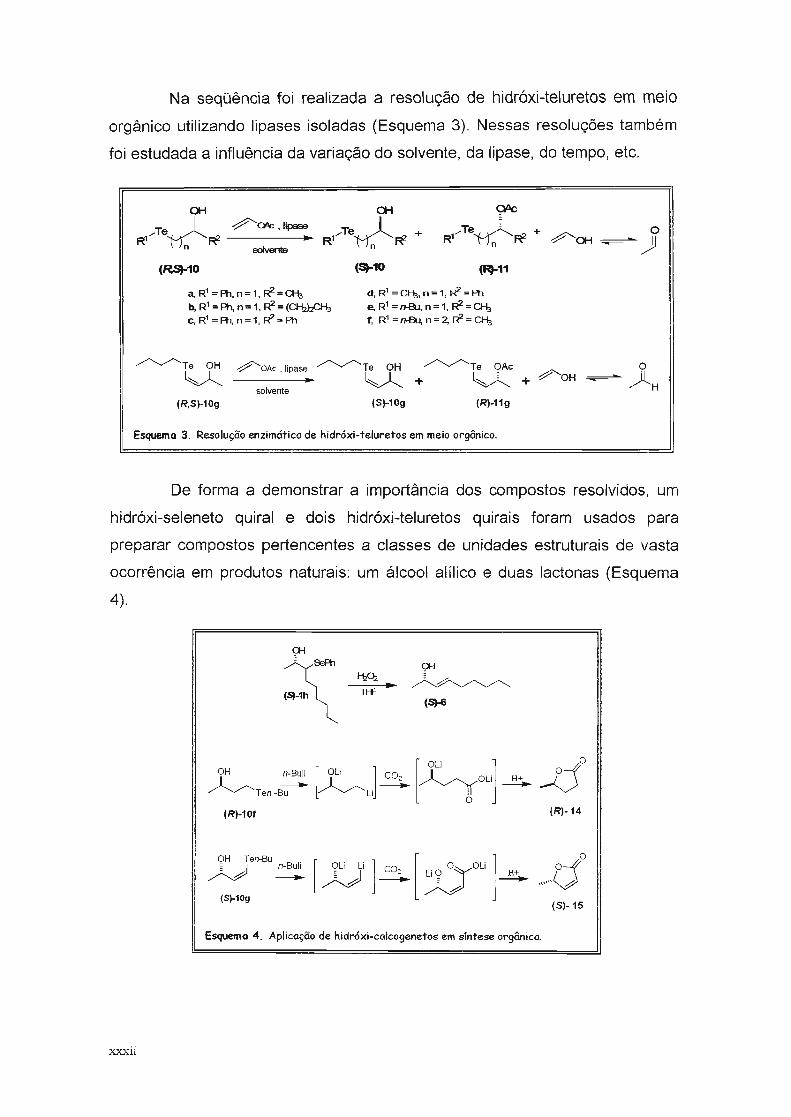
Na seqüência foi realizada a resolução de hidróxi-teluretos em meio
orgânico utilizando lipases isoladas (Esquema 3). Nessas resoluções também
foi estudada a influência da variação do solvente, da lipase, do tempo, etc.
a-t a-t Qllc
/Te0 -::?'a>c .lipase
/Te0 / Te0 + o+
R1 R1 ~ R1n R1 R1
n R1 -::::?'a-t~ )Jn soIvenIe
(~S)-10 (5)-10 (R)-11
a, R1 =Ph.n=1.~=%
b, R1 =Ph, n=1.~=(~c, R1 =Ph, n=1, ~=Ph
d,R1 =%.n=1, ~=Ph
e. R1 =n-8.J. n=1.~=%f, R1 =1H3u, n=2, R2 =%
~Te OH
~
(R.S)-10g
~OAc.lipase ~Te OH ~Te OAc O
------ ~ + ~ + Ç'OH ~~Hsolvente
(S)-10g (R)-11g
Esquema 3. Resolução enzimático de hidróxi-teluretos em meio orgânico.
De forma a demonstrar a importância dos compostos resolvidos, um
hidróxi-seleneto quiral e dois hidróxi-teluretos quirais foram usados para
preparar compostos pertencentes a classes de unidades estruturais de vasta
ocorrência em produtos naturais: um álcool alílico e duas lactonas (Esquema
4).
Ç»-I
~
(5)-6
[ali ]OH n-Buli [ali ] CO ~
~ ~: OLi H+- - -Ten -Bu Li
O
(R)-10f
JjO(R)-14
OH Ten-Bu
~ ~(S)-10g
[?Li Li] CO: [X)iº° OLi] H+ rj0~-, -"""h-.&
(S)-15
Esquema 4. Aplicação de hidróxi-calcogenetos em síntese orgânica,
xxxii

Abstract


In this work, the behavior of hydroxy chalcogenides (Se and Te)
towards the biotransformations using isolated enzymes in organic media or
aqueous media and using microorganisms (fungi) was studied. A comparative
study of the effect of temperature, solvent, enzyme immobilization and structure
of the substrates on the resolution was performed.
Initially, the compounds had been synthesized using described
methods in the literature, after, the resolution of hydroxy selenides in organic
media using isolated lipases was carried out (Scheme 1), including a study on
the immobilization of PPL on some supports, as well studies on the influence of
the variation of the solvent, the temperature, lipase, etc.
a, R1 =RI, R2=H, R3=0i3b, R1 =RI, R'2=H,R3=(~c, R1 =RI, R2 = H, R3 =(CH$%d, R1 =RI, R2 =H, R3 =Oi0i.3he, R1 =RI, R2=H, R3=R1
f, R1 =0i3, R2=H, R3=A1g, R1 =RI, R2 =SePh, R3 =0i.3h, R1 =RI, R2 =(0i:2>s0i.3, R3 =0i3i, R1 =A1, R2=R1, R3=%
o~
Scheme 1. Enzymatic resolution of hydroxy selenides in organic media.
In the resolution in aqueous media using isolated lipases, initially the
hydroxy selenides were transformed into their acetates by convertional
chemical methods, and then, a screening with ten enzymes from different
sources was carried out using pH indicator. In the following, the enzymatic
resolution of the selanyl acetates in aqueous media using the enzymes selected
in the screening step was performed (Scheme 2).
AcO - AcO HO oY"'sePh II
pase. ""'rSePh + YSePh + .,)l
R Tampão I Acetona R R OH
Scheme 2. Enzyma Suffe, I acetona selanyl acetates in aqueous media.
The biotransformations by fungi were performed using whole cells of
some Aspergillus terreus strains.
xxxv

In the sequence, the resolution of hydroxy tellurides in organic media
using isolated lipases was carried out (Scheme 3). In these resolutions, the
influence of the variation of the solvent, lipase and the reaction time was also
studied.
a-i a-i Q6c
/ Te0~01'c , lipase ~/Te0R1 + / Te0 + o
R1 R1 • R1 n R1 -::?'a-i~ ,Jn solverte R n
(RS)-10 (5)-10 (R)-11
a, R1 =Ph, n=1. R2=Q-f.3b, R1 =Ph, n=1,R2=(~c. R1 =Ph, n=1. R2=Rl
d R1 =CH.3 n=1 R2=Rl~ R1=~ n=1·. R2=%f. R1 =n-B.J, n=2, R2 =%
~Te OH
~(R,S)-10g
Ç'OAc . Iipase ~Te OH ~Te OAc O
----~. ~ + ~ + ~OH ~ )lHsolvente
(S)-10g (R)-11g
Scheme 3. Enzymatic resolution of hydroxy tellurides in organic media.
In order to demonstrate the potencial of the resolved compounds, one
chiral hydroxy selenide and one chiral hydroxy telluride were used to prepare
compounds belonging to classes of building blocks of wide occurrence in
natural products: an allylic alcohol and a lactone (Scheme 4).
ÇlH
OH n-Buli [ OLi ] CO [AnoLi
] -es0
~ __ I ~ OLi~
Ten-Bu ~LiO
(R)-10f (R)-14
OH Ten-Bu
~ ~(51-109
[?Li Li] COz [X)'i'?° OLi] H+ [j0~ -, -",," ~
.ó
(S)-15
Scheme 4. Application of hydroxy chalcogenides in organic synthesis.
xxxvi

Objetivos


o objetivo do presente trabalho foi avaliar o comportamento de hidróxi
selenetos e hidróxi-teluretos frente a reações de biotransformações,
empregando principalmente enzimas isoladas em meio orgânico, de modo a
obter compostos orgânicos opticamente puros ou enriquecidos contendo
selênio e telúrio, os quais podem ser precursores sintéticos na preparação de
compostos bio-ativos. Em adição a estes estudos, foi desenvolvida uma
metodologia de preparação de hidróxi-selenetos quirais em meio aquoso,
contribuindo significativamente no desenvolvimento de técnicas de obtenção de
bloco quirais que agridem cada vez menos o nosso meio ambiente. Dessa
forma, daremos continuidade à união dessas ferramentas sintéticas
(biotransformações e a química de organo-selenetos e organo-teluretos),
aumentando a versatilidade dos compostos de selênio e telúrio em síntese
orgânica e o lêque de opções para o químico orgânico sintético na síntese de
fármacos, fragrâncias, pesticidas, feromônios, etc.
xxxix


1. :Introdução


1.1. Quiralidade e Atividade BiológÍca
1.1. Quiralidade e Atividade Biológica


Figu-a 1. 1.1. Imagem Especular da mão.
Figu-O 1.1.2. Centro estereogênico.
Centro CHEstereogênico, .-L 3 ,...
~H C02H
1.1. Quiralidade e Atividade Biológica
A quiralidade é um conceito que ultrapassa o domínio da química. Ela
se refere a uma determinada característica geométrica: a sobreposição (ou
não) da imagem especular de um objeto sobre ele mesmo. Assim,
denominamos um objeto como quiral quando a sua imagem especular não
pode ser sobreposta ao objeto original. Existem diversos objetos quirais ao
nosso redor, como por exemplo, as conchas marinhas, as nossas mãos (Figura
1.1.1), etc. 1
Essa propriedade também é
observada em moléculas orgânicas, as
quais podem ser quirais ou aquirais
(imagem especular pode ser superposta à
molécula original). A idéia de molécula
quiral remonta ao século XIX quando Louis
Pasteur separou os enantiômeros do ácido
tartárico. 1 Geralmente, a quiralidade em moléculas orgânicas é resultado da
presença de um centro quiral localizado em um átomo que possui um conjunto
de ligantes (ou átomos) num arranjo espacial que não permite a superposição
da sua imagem especular. Nesse sentido, em compostos orgânicos, o centro
de quiralidade (também chamado de centro assimétrico ou centro
estereogênico) geralmente se encontra sobre um átomo de carbono de
hibridização Sp3 que está ligado a quatro átomos ou grupos diferentes (Figura
1.1.2). 1
Designamos por estereoisômero os
isômeros (do grego isoméres = partes iguais)
que apresentam diferenças entre si na
orientação espacial de seus Iígantes.
Denominamos de diastereoisômeros os
estereoisômeros que não possuem uma relação objeto-imagem especular,
resultante, por exemplo, da presença de mais de um centro estereogênico na
estrutura da molécula orgânica. Por outro lado, denominamos de enantiômeros
(do grego enantio =opostos) os estereoisômeros que são imagens especulares
um do outro (Figura 1.1.3). Uma mistura em partes iguais de dois
enantiômeros é denominada mistura racêmica (ou racemato). 1
5

Figt.l'o 1. 1.3. Enantiômeros.
Figt.l'Q 1.1.4. Exemplos de enantiômeros com diferentespropriedades.
(5)-Talidorrida
figlrO 1.1.5. Fármaco com atividadebiológica distinta para cada enantiômero.
1. Introdução
. Os diastereoisômeros possuem
propriedades físicas e químicas distintas,
enquanto que, os enantiômeros apresentam
propriedades físicas e químicas idênticas,
exceto quando submetidos à luz plano
polarizada, onde um dos enantiômeros desvia a luz polarizada em um sentido e
o outro em sentido contrário. Devido à interação entre as soluções dos
enantiômeros e a luz polarizada, os enantiômeros são também designados por
isômeros ópticos. 1
Enantiômeros podem apresentar atividades biológicas completamente
distintas, como diferenciação de odores, sabores, etc. 2,3 Dos vinte
aminoácidos existentes nos organismos, dezenove são encontrados apenas na
forma de um único enantiômero (a forma L), sendo que apenas um, a glicina,
não é um composto quiral, o que demonstra a relevância da quiralidade em
organismos vivos. 4
Como exemplo das diferenças de atividades biológicas de
enantiômeros, podemos citar a (-)-carvona e a (+)-carvona, responsáveis,
respectivamente, pelos
aromas da menta e do
cominho, duas plantas
utilizadas na culinária, e o (+)
limoneno e o (-)-limoneno,
responsáveis pelos aromas
da laranja e do limão,
respectivamente (Figura 1.1.4). 5
Um outro exemplo das diferenças de atividade biológica entre
enantiômeros, com conseqüências bem mais sérias do que apenas diferenças
de aromas, é o da talidomida (Figura 1.1.5),
um fármaco muito utilizado, entre os anos
de 1957 e 1962 em todo o mundo, no
tratamento de enjôos e náuseas, muito
comum no período inicial da gravidez
humana. 1
6

1.1. Quiralidade e Atividade Biológica
Quando a talidomida foi lançada no mercado sua utilização por
grávidas era considerada segura, sendo administrada a mistura racêmica.
Entretanto, na época não havia estudos sobre a interação de cada enantiômero
com o organismo. Mais tarde foi descoberto que o enantiômero "R' apresenta a
atividade sedativa, mas o enantiômero "s' apresenta ação teratogênica (do
grego terás = monstro; gene = origem), levando à má formação congênita,
afetando principalmente no desenvolvimento dos braços e das pernas do feto.
O uso indiscriminado deste fármaco levou ao nascimento de milhares de bebês
com gravíssimos defeitos físicos (Figura 1.1.6). 1
Esse lamentável
acontecimento despertou a
atenção da comunidade
científica e das autoridades
farmacêuticas sobre a
importância da pureza óptica
Figura 1.1.6. Anomalias causadas pela talidomida. de fármacos na atividade
biológica dos mesmos, visto
que a maioria dos fármacos é constituída por substâncias quirais. 1 Nesse
sentido, várias providências foram tomadas a fim de evitar que uma tragédia
como esta se repita. Um exemplo das mudanças ocorridas pode ser observado
nos Estados Unidos da América, onde o FDA (United States Food and Drug
Administration - órgão que regulamenta a comercialização de fármacos
naquele país) exige uma experimentação completa de cada enantiômero e da
mistura racêmica para que seja aprovada a comercialização de um fármaco.
No Brasil, apesar de diversos fármacos serem comercializados como
racematos, os fármacos quirais já são uma realidade na indústria farmacêutica.
Em 2000, esse ramo da indústria faturou mais de 130 bilhões de dólares nas
vendas de fármacos quirais (em todo o mundo), num mercado que cresce
aproximadamente 13% ao ano. 6 Neste conexto, a indústria farmacêutica tem
um grande interesse em preparar drogas enantiopuras. 6
Nesse sentido, a indústria tem ajustado seus processos para o
controle da estereoquímica de novas drogas através de sínteses assimétricas.
Deste modo, nenhum aspecto da síntese orgânica recebeu nas últimas
7

1. Introdução
figura 1.1.7. Ganhadores do Premio Nobelde química em 2001.
K.BarrySharplessNasceu: 1941PhD: 1968(StanfordUniversity)
RVoji NovoriNasceu: 1938PhD: 1967(KyotoUniversity)
WilliamS.KnowlesNasceu: 1917PhD: 1942(ColumbiaUniversity)
décadas tanta atenção quanto à preparação de compostos
enantiomericamente puros ou enriquecidos. Em 2001, os químicos Willian S.
Knowles (Monsanto Company, St. Louis, Missouri/USA), Ryoji Noyori (Nagoya
University, Chikusa, NagoyalJapan) e K. Barry Sharpless (the Scripps
Research Institute, La Jolla, California/USA) foram agraciados pela Royal
Swedish Academy of Sciences com o prêmio Nobel de Química pelos seus
trabalhos em síntese catalítica assimétrica (Figura 1.1.7). 7
As descobertas destes três
químicos tiveram um grande impacto, tanto
na pesquisa acadêmica como na indústria
farmacêutica, pois possibilitaram o
desenvolvimento de novas drogas e outras
substâncias biologicamente ativas
(fragrâncias, pesticidas, feromônios, etc.)
com alta pureza enantiomérica.
8

1.2. Biotransformações
1.2. Biotransformaçães


figura 1. 2.1. Serina.
1.2. Biotransforrnações
Nos últimos anos, o desenvolvimento de produtos e/ou processos~
químicos que reduzem ou eliminem o uso e a geração de substâncias
perigosas vem ganhando muita atenção dentro da comunidade científica e fora
dela. Isso devido à conscientização da necessidade da preservação do meio
ambiente. Nesse sentido a Química Verde vem trabalhando para reduzir e,
preferencialmente, eliminar a produção e o uso de substâncias prejudiciais ao
meio ambiente e ao homem. 8
A biotransformação, ou seja, a utilização de microorganismos ou
enzimas isoladas (ou na forma de extrato enzimático) em transformações
químicas possibilita a redução e/ou a eliminação da utilização de substâncias
perigosas em reações químicas. Por exemplo, podem catalisar reações de
oxidação que geralmente são realizadas em solvente orgânico (prejudicial ao
meio ambiente) e empregando oxidantes que contém metais pesados (ex.:
PCC - Clorocromato de piridínio). Além disso, os microorganismos, as enzimas
e os resíduos gerados durante uma biotransformação são geralmente
biodegradáveis, o que não compromete o meio ambiente. 9 As
biotransformações são geralmente realizadas utilizando água como solvente,
ao invés de solventes orgânicos, o que contribui para o uso das mesmas em
reações químicas como forma de preservação do meio ambiente. 9
Somando-se a estas vantagens ambientais, as biotransformações
possibilitam a obtenção de compostos enantiomericamente puros ou
enriquecidos, ou a transformação de um grupo funcional de maneira seletiva,
ou ainda, permitir distinguir um mesmo grupo funcional em ambientes
diferentes num mesmo substrato, etc, 9 diferentemente de reações realizadas
usando reagentes químicos convencionais, quando geralmente não se observa
seletividade.
Por exemplo, ao tentarmos sintetizar a serina (aminoácido encontrado
em organismos vivos - Figura 1.2.1) sob condições convencionais (sem a
utilização de reagentes ou catalisadores quirais), será
obtida uma mistura de dois compostos, a (S)-serina e a
(R)-serina (mistura racêmica).Para obtermos somente
um dos enantiômeros, precisamos optar por uma
síntese assimétrica, que é capaz de produzir, ao
11

1. Introdução
I Prochiml ~Ihsrmrc.s I
c.: 7)ik çheriç~J
ElUllI>.)mericaJly pure compOllll<lS
'''T I ~;~~.c...wu· ~I
,.ym~~is
kinelic l"f}'l<1a1lizalÍOll cllrolf1aJ.O"rJpllv/ \ < •
Neste contexto, as
biotransformações vêm se
destacando rapidamente,
devido a suas características
menos, um excesso de uma das formas, ou, a partir da mistura racêmica,
aplicar uma técnica de resolução, como por exemplo, a cristalização, que
permita a separação dos enantiômeros, ou utilizar um reagente opticamente
puro (Figura 1.2.2). 10
de compostosFigura 1. Z. Z Rotas para obtençãoenantioméricamente enriquecidos.
ambientamente adequadas e
as suas propriedades
catalíticas e seletivas. Fato esse notado pelo volume crescente de publicações
nos últimos anos sobre o assunto,11 pelo número de indústrias envolvidas
direta ou indiretamente em pesquisa e aprimoramento de tecnologias de
modificações genéticas, bem como pela aplicação direta de biotransformações,
em substituição a processos químicos clássicos.
A utilização de biocatalizadores em transformações sintéticas pela
indústria tem crescido rapidamente nas últimas décadas, e a previsão é que a
presença de biocatalizadores na indústria cresça muito mais. 12
OXidoreductases
Figura 1. Z. 3. Tipo de enzimas usadas na indústria.
'\Reducing I \cells ' 1
Isomerases . /1 j\~ //Hydrolases
Lyases •~..-...-.
Em um estudo sobre processos industriais (amostra de 134
processos), as hidrolases estão presentes em quase metade dos processos,
enquanto que as oxi-redutases representam cerca de 25% (Figura 1.2.3).
Nesse estudo, observou-se que geralmente em reações redox são empregadas
células inteiras, enquanto que enzimas isoladas (livres ou imobilizadas) são
mais comuns em reações que empregam hidrolases. 13
A chave de todo o processo biocatalítico são as enzimas, 9 que são
produzidas pelas células e possuem
a habilidade de atuar como
catai isadores altamente estéreo-
específicos. Esses catalisadores
possibilitam a obtenção de
compostos enantiomericamente
puros ou enriquecidos em condições
brandas, gerando muito poucos
12

1.2. Biotransformações
resíduos, os quais são geralmente biodegradáveis, conforme salientado
anteriormente, tornando os processos biocatalíticos muito atraentes do ponto
de vista estereoquímico e ambiental. Também podemos citar como vantagens
do emprego da biocatálise em síntese orgânica, a alta eficiência catalítica das
enzimas, que podem aumentar a velocidade de reação na ordem de 108-101°, e
a tolerância a substratos não naturais. 9
1.2.1. Enzimas
As enzimas governam as reações químicas nos processos biológicos
vitais, sendo responsáveis por uma ampla variedade de transformações
metabólicas, catalisando quase todos os tipos de reações organrcas
conhecidas. Desta forma, uma grande variedade de processos catalisados por
enzimas têm uma reação orgânica sintética equivalente. 9
As enzimas são proteínas formadas por subunidades de aminoácidos
e possuem em suas estruturas grupos polares como -COOH, -OH, -NH2 , -SH e
-CONH2 (Figura 1.2.1.1), que atuam como catalisadores. Uma vez elaboradas
por uma célula, as enzimas podem atuar independentemente da mesma.
IsoLeucine Pb.eo,\'Jalanine
COfT+ i
H.N C Hi.CHsOR
Sel;ne
coo+ I
H·,N-C-H, I .
H
Glycine
coo+ I
H.N-O-HI
H-C-·~OH
ICH.
Threonine
(',()()
+ IR3N-C-H
ieH.
AJanine
COO-+ 1
H~-C-H
ICH.
H
Cy.steine
cool/H
+/C......H.,N CH.
- I I·H sC--CH2
Pl'()line
(',()()-
Hil-C-H
CH;lI .
OH/' """:'
OH;, OH.
000+ I
H~-C-H
IeH
/"CHs CHsValin"
+ yOO-HaN-C-H
IH-Y--cH.CH
~
coo-• I
H3N-C-HIGHoI .000-
Aspartote
coo+ i
IIaN- y-IIeH.
6coo-
H:i~ C H
tH.I?H2
too-Glul{uuole
cooH.N c li
TYJ'OSine
COO-o I
H,N-C-HiCH.
kH~'mI
UTtyptopluHI
cooHiN-C-H
:..1, .:?H;;,
...0H2N'~'~O
Aapa....gine O1utamiue L:raine
000
H,N-C-H, I
6H.j
CRsIqH.1NHI +C=NH2jNR. .
A11,rinine
000-
H.N-C-H
(-.H.I
] -t-l..HbH11
Ü-N
Hist.idine
COO.. I
H-N-C-Ho I
T~CR.ISICR.
Methionine
Figura 1.2.1.1. Aminoácidos naturais.
13

1. Introdução
Todas as enzimas possuem estruturas complexas de cadeias
polipeptídicas que se mantêm estáveis em meio aquoso, seu ambiente natural.
Entretanto, poucas enzimas possuem sua estrutura tridimensional determinada
ou seu mecanismo de ação descrito. 14
As enzimas são classificadas pela "lnternational Union of
Biochemistry" em seis classes, de acordo com o tipo de reação que catalizam
(Tabela 1.2.1.1). 9
Tabela 1.2.1.1. Classificação das Enzimas.
Classificação da Enzima Tipo de Reação
1. Oxi-redutases
2. Transferases
3. Hidrolases
4. Liases
5. Isomerases
6. Ligases
Oxidação-redução: oxidação de C-H, C-C, C=C,remoção ou adição de hidrogênios.
Transferência de grupos: Aldeídicos, cetônicos, acila,açucares, etc.
Formação e hidrólise de ésteres, amidas, lactonas,lactamas, epóxidos, nitrilas, anidridos, etc.
Adição-eliminação de pequenas moléculas em C=C,CN, C=O.
Isomerização, como racemização e epimerização.
Formação e c1ivagem de ligações do tipo C-O, C-S,C-N, C-C, com concomitante c1ivagem de trifosfato.
1.2.1.1. Aspectos Mecanísticos
1.2.1.1.1 ).
Slbstrate
Dentre as teorias que vem sendo desenvolvidas para entender a
catálise enzimática, o modelo mais ilustrativo continua sendo o clássico chave
fechadura, que foi desenvolvido por Emil Fischer em 1894. O mesmo propõe
que o substrato deve ser complementar ao sítio catalítico da enzima, levando a
formação do complexo enzima
substrato, caso contrário a catálise não
ocorreria. 15 Entretanto, este modelo
não explica porque algumas enzimas
aceitam substratos não naturais (Figura
fnlym.
Figura 1.2. 1. 1. 1. Mecanismo chave-fechadura. Um modelo proposto por
Kosland no final da década de sessenta
tenta explicar melhor o mecanismo das enzimas, assumindo que as mesmas
14

1.2. Biotransformações
não são estruturas rígidas. 16 De acordo com essa proposta, conhecida como
encaixe induzido, durante a aproximação entre a enzima e o substrato, várias
interações ocorrem, resultando em mudanças conformacionais no sítio ativo da
enzima, facilitando assim a formação do complexo "enzima-substrato" (Figura
1.2.1.1.2).
r--"'"i /-.\\ i\. • b ( )
'''-----../
A
Como já destacado, as reações
enzimáticas são muito mais rápidas que
as reações químicas convencionais ..""....+ ~
análogas. Tentando explicar essas ~\ ,{.......\
observações, Dewar postulou uma \ 'í: ,)
teoria conhecida como teoria da .~.,Eo..,....
dessolvatação, 17 na qual as reações figlro 1.2.1.1.2. Mecanismo encaixe induzido.
enzimáticas são comparadas àquelas
que ocorrem em fase gasosa. De acordo com essa teoria, quando o substrato
entra no sítio ativo da enzima, expulsa todas as moléculas de água presentes.
Num ambiente anidro as reações ocorrem mais facilmente, levando a um
aumento na velocidade da reação. Esta teoria levou à outra, conhecida como
teoria da "solvatação-substituição". 18 A mesma está baseada na suposição de
que a enzima não poderia simplesmente expulsar todas as moléculas de água,
pois este processo é energeticamente desfavorável. Provavelmente, as
moléculas de água são acomodadas em outras partes da enzima, liberando o
sítio hidrofóbico da enzima, o que favorece a formação do complexo "enzima
substrato" .
A regra dos três pontos, utilizada por Ongston em 1948, 19 tenta
explicar de forma simplificada a
enantiosseletividade enzimática.
Partindo-se da premissa que a
quiralidade é uma característica
espacial, para que a
enantiosseletividade seja máxima, o
substrato necessita estar firmemente
posicionado em três dimensões no sítio figlrO 1.2.1.1.3. Regra dos três pontos.
enzimático. Para tal, é necessário que
15

1. Introdução
existam pelo menos três pontos de ligação entre o substrato e o sítio ativo da
enzima. Como pode ser observado na Figura 1.2.1.1.3, um dos enantiômeros
se encaixa bem nos três pontos do sítio ativo da enzima, enquanto que o outro
enantiômero se encaixa em apenas dois pontos do sítio ativo, não se
encaixando no terceiro ponto.
Figtra 1.2.1.1.4. Diagrama de energia de reaçãocatalisada VS. não catalisada.
p
Coordenada de reação
S •. -
p
Em termos físico-químicos, as enzimas, assim como os demais
catalisadores, aceleram a velocidade das reações, diminuindo a barreira
energética (energia de ativação - Ea) entre reagentes e produtos, devido à
estabilidade do complexo "enzima-substrato" formado no estado de transição
(Figura 1.2.1.1.4). 9,11
De forma análoga, a .'>G
estereosseletividade das enzimas
também se origina da diferença de
energia entre os estados de
transição dos complexos formados
entre os enantiômeros de um
substrato quiral {[EA]#, [EBt}, ou
das duas formas pró-quirais de um
substrato, envolvendo seus grupos ou faces enantiotópicas com o sítio ativo da
enzima.
Os complexos diastereoisoméricos "enzima-substrato" formados
apresentam diferentes valores de energia livre para seus respectivos estados
de transição, conseqüentemente, um enantiômero se formará
preferencialmente com relação ao outro. A diferença de energia livre de
ativação (~~G#) caracteriza a seletividade da enzima na reação, sendo
diretamente proporcional à razão entre as velocidades de formação de cada
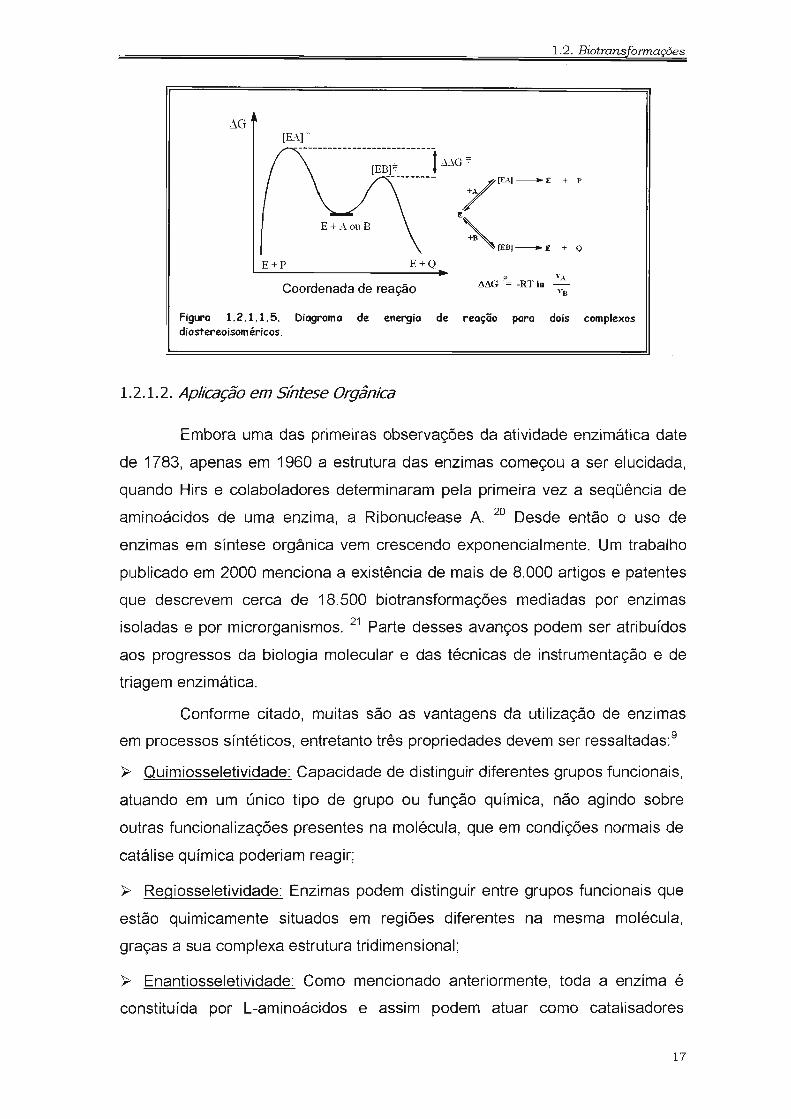
um dos enantiômeros (VANs), de acordo com a Figura 1.2.1.1.5. Portanto,
quanto maior a diferença de energia livre de ativação entre os dois complexos
"enzima-substrato" formados (~~G#), maior será a enantiosseletividade
enzimática e maior será o excesso enantiomérico obtido. 9,11
16
,
[EA.(---------------------------1 ,',..\0 ;=
+A/[EAI-E
;/E
~IEBI_E
1.2. Biotransforrnações
+ p
+ Q
E+P E+Q
Coordenada de reação- '"A
ôôG ~= -RT In -'-"B
FiglrO 1.2.1.1.5. Diagrama de energia de reação para dois complexosdiastereoisoméricos.
1.2.1.2. Aplícação em Síntese Orgâníca
Embora uma das primeiras observações da atividade enzimática date
de 1783, apenas em 1960 a estrutura das enzimas começou a ser elucidada,
quando Hirs e colaboladores determinaram pela primeira vez a seqüência de
aminoácidos de uma enzima, a Ribonuclease A. 20 Desde então o uso de
enzimas em síntese orgânica vem crescendo exponencialmente. Um trabalho
publicado em 2000 menciona a existência de mais de 8.000 artigos e patentes
que descrevem cerca de 18.500 biotransformações mediadas por enzimas
isoladas e por microrganismos. 21 Parte desses avanços podem ser atribuídos
aos progressos da biologia molecular e das técnicas de instrumentação e de
triagem enzimática.
Conforme citado, muitas são as vantagens da utilização de enzimas
em processos síntéticos, entretanto três propriedades devem ser ressaltadas: 9
~ Quimiosseletividade: Capacidade de distinguir diferentes grupos funcionais,
atuando em um único tipo de grupo ou função química, não agindo sobre
outras funcionalizações presentes na molécula, que em condições normais de
catálise química poderiam reagir;
~ Regiosseletividade: Enzimas podem distinguir entre grupos funcionais que
estão quimicamente situados em regiões diferentes na mesma molécula,
graças a sua complexa estrutura tridimensional;
~ Enantiosseletividade: Como mencionado anteriormente, toda a enzima é
constituída por L-aminoácidos e assim podem atuar como catalisadores
17

1. Introdução
quirais. Como conseqüência, algum tipo de quiralidade presente na molécula é
reconhecido e um substrato pró-quiral pode ser transformado em um produto
opticamente ativo ou ambos enantiômeros de um composto podem reagir em
diferentes velocidades, implicando em uma resolução cinética.
Em algumas biotransformações utilizando enzimas isoladas existe a
necessidade de utilizar cofatores, os quais são muitos caros. 9 Em vista disso,
microorganismos vêm sendo empregados em reações de oxidação-redução e
de epoxidação, evitando utilização de cofatores e/ou coenzimas, visto que os
mesmos possuem os cofatores e/ou coenzimas necessários. 22,23
A escolha entre enzimas isoladas ou microorganismos, livres ou
imobilizados, em meio reacional orgânico ou aquoso, depende de vários
fatores, tais como a escala em que será realizada a reação, a necessidade da
reciclagem de cofatores, e a disponibilidade de infra-estrutura para a
manipulação de microorganismos, entre outras. Assim é necessário avaliar as
vantagens e as desvantagens de cada processo em cada caso.
As biotransformações em síntese orgânica podem ser realizadas
basicamente em três sistemas de solventes distintos: sistema água e co
solvente orgânico miscível; sistema água e solvente orgânico imiscível (sistema
bifásico) e sistema solvente orgânico anidro. A influência do solvente orgânico
utilizado pode ser relacionada diretamente com sua constante dielétrica: quanto
menor for a constante dielétrica, maiores serão as interações eletrostáticas
entre as cargas dos resíduos de aminoácidos que compõem a estrutura
protéica da enzima. Esse efeito pode levar a uma redução da flexibilidade e
conseqüentemente à redução da atividade catalítica. 24
A água (seu meio natural) ativa a enzima porque possibilita uma maior
flexibilidade. Sua presença é imprescindível nas reações catalisadas por
enzimas. Estudos mostram que existe um mínimo de moléculas de água para
cada molécula de enzima. Essa quantidade de água corresponde a uma mono
camada de solvatação, proporcionando que a enzima não seja inativada.
Mesmo nos sistemas de solventes anidros, existe uma pequena quantidade de
água presente no solvente. 25
O pH é outro fator muito importante em reações biocatalizadas, com a
variação do pH ocorrem mudanças na conformação protéica da enzima, devido
18

1.2. Biotransformaçães
à ionização dos resíduos de aminoácidos que compõem a estrutura enzimática.
O fator pH é específico para cada enzima e a conformação obtida com a
variação do pH pode ou não corresponder à conformação ativa da enzima, o
que pode se tornar um fator crítico em certas reações. 26
1.2.1.3. Hidrolases
R'-OH
HJ.s ... Int~nuedL~lno
~ T si f ~\Cil-Euzima
I ~ n 0--....# •oAO- H N N-H~ \
~v~ 7 R1
Nu
Esquema 1.2.1.3.1. Mecanismo de serina-hidrolase.
Etapa li
Etapa I
arranjo espacial destes
grupos favorece o
aumento da nucleofilicidade do grupo hidróxi da serina, que pode então atacar
um grupo carboxila de um substrato do tipo R1-CO-OR2 (Esquema 1.2.1.3.1
Etapa I). Dessa forma o intermediário acil-enzima é formado liberando o grupo
de saída (R2-OH). Em seguida um nucleófilo (em geral a água) ataca o
intermediário acil-enzima, regenerando a enzima e formando um ácido
carboxílico do tipo R1-CO-OH (Esquema 1.2.1.3.1 - Etapa 11). 9
As enzimas hidrolíticas são muito uilizadas em síntese orgânica. Nesta
classe estão incluídas as amidases, proteases, esterases, nitrilases, fosfatases
e epoxido hidrolases, sendo de particular e grande interesse as lipases. 9
O mecanismo de hidrólise enzimática de amidas e ésteres é muito
similar ao observado para a hidrólise química convencional utilizando base. Um
grupo nucleofílico do sítio ativo da hidrolase ataca o grupo carboxílico do éster
ou amida do substrato. Este nucleófilo pode ser o grupo hidróxi da serina ou o
grupo carboxi do ácido aspártico ou ainda o grupo tiol da cisteína.
O mecanismo elucidado em detalhes é o da serina. Dois aminoácidos
localizados próximos a
serina (geralmente uma
aspartina e uma
histidina) auxiliam na
catálise e juntos formam
a tríade catalítica. O
Em baixas concentrações de água, outros nucleófilos podem competir
com a .água no ataque ao intermediário acil-enzima, levando a formação de
outros compostos, aumentando a aplicação dehidrolases em síntese orgânica
(Esquema 1.2.1.3.2). Quando enzimas hidrolíticas são empregadas na hidrólise
19

1. Introducão,
de racematos, podemos
observar a discriminação dos
enantiômeros. Isto ocorre
Esquema 1.2.1.3.2. formação de outros compostos.
devido à quiralidade existente
no sítio ativo da enzima, com
o qual um enantiômero
interage com mais facilidade
que o outro, e assim um dos enantiômeros é convertido mais rapidamente,
resultando em uma resolução cinética do racemato, conforme destacado
anteriormente na figura 1.2.1.1.5.
K m= constante d~ 1fich.~dis·1knt~r
Em casos ideais, onde a enzima apresenta alta enantiosseletividade, a
diferença de velocidade é tão grande que apenas um enantiômero é convertido
em produto (excesso enantiomérico >99%) e o outro praticamente não reage.
Entretanto, há casos em que a enzima não demonstra alta enantiosseletividade
e ambos os enantiômeros são convertidos a produto.
O parâmetro E (razão enantiomérica), desenvolvido por C. J. Sih em
1982, 9 traduz em termos numéricos a seletividade enzimática, descrevendo a
relação entre a conversão (c) e os excessos enantioméricos do substrato (e.e. s )
e do produto (e.e. p) (Figura 1.2.1.3.1). Assim, esse parâmetro é uma
ferramenta muito útil no estudo de resoluções enzimáticas.
As reações de hidrólise em meio
aquoso são consideradas irreversíveis, pois a
concentração de água é alta (55,5 MoI/L).
Nestas condições, o parâmetro E pode ser
determinado facilmente a partir dos valores de
conversão e dos valores de excessos
Figura 1.2.1.3.1. Determinação de E enantioméricos do substrato (e.e. s ) e do
produto (e.e. p) utilizando a equação da Figura
1.2.1.3.2. Esta equação é derivada do estudo cinético enzimático empregando
a equação de Michaelis-Menten e partindo-se do princípio que não existe
inibição enzimática. 9
20

1.2. Biotransformações
FigtrG 1.2.1.3.2. Dependência da seletividade e daconversõo nas resoluções.
c = conversão: e.e.p = excesso enalltiomérico do produto:
e.e.s = excesso eUaIltiomérico do substrato:
E = raziio en3ntiomérica
Para o substrato:
E = _ll1-=[:....l_-e....:.(_1+_e_.t--'>.s:;:..:.)~]
ln[l-e(1+e.e·s)]
Para o produto:
ln[l-c(1+e.e.p)]E = ---''-----'---''-'-~
111[l-c(l-e .e.p)]
Entretanto, na prática,
a determinação exata da
conversão em resoluções, é
prejudicada por erros oriundos
da manipulação da amostra.
Nestes casos, o emprego da
equação da Figura 1.2.1.3.3 é
sugerido, visto que nesta
equação apenas os valores de excesso enantiomérico do substrato (e.e.s ) e do
produto (e.e. p) são necessários. 9
(e.e.p +e.e.8)
(e.e.p +e.e'8)
E=-------[e.e.p(1 +e.e.8)]
In-----
[e.e.p(1-e.e·s)]111-----
e.t.?p = exc~so enantionlirico do pn.xiuto:tIJ!.s = excC$so en31\b.oluérico do substrato:E = razão enantiomáica
FiQ'rO 1.2.1.3.3. Determinação de E
De todas as hidrolases, as lipases são
as mais utilizadas em biotransformações. Elas
são muito empregadas devido à sua
versatilidade catalítica, disponibilidade
comercial e baixo custo. Nos organismos elas
hidrolisam triglicerídeos em ácidos graxos e
glicerol. Apesar de catalisarem reações
semelhantes das proteases e esterases o seu
mecanismo molecular é diferente.
Figuro 1.2.1.3.4. Cinética de esterases vs. Cinética de Iipases.
Lipase
inSOIÜVel~
fsolúvel
CMCconcentração do substrato [5]
saturação
Esrerase
concentração do substrato [5]
otividode
A mais importante diferença entre as lipases e as esterases é a
interação físico-química com o substrato. Enquanto as esterases apresentam
dependência direta entre a atividade enzimática e a concentração do substrato
(normal Michaelis-Menten), as Iipases apresentam baixa atividade enzimática
enquanto o substrato
permanecer solúvel no meio
(Figura 1.2.1.3.4). Porém,
quando a concentração do
substrato é elevada
gradualmente, após o limite
de solubilidade do mesmo
no meio (formando uma
segunda fase), a atividade
enzimática aumenta
21

1. Introdução
gradualmente. Em outras palavras, Iipases não hidrofisam substratos abaixo da
concentração micelar crítica (CMC - concentração micelar crítica - Figura
1.2.1.3.4).9
Esse comportamento das lipases pode ser explicado por um processo de
rearranjo que ocorre com a enzima na interface do sistema bifásico (Figura
2.1.3.5). A lipase dissolvida sem a presença de um sistema bifásico está na
forma inativa {[Enz]}, pois existe uma parte da estrutura da enzima (uma
pequena a-hélice - a tampa) que cobre o sítio ativo, bloqueando a passagem
do substrato (baixa atividade enzimática). Quando a enzima entra em contato
com a interface do sistema bifásico, uma reacomodação estrutural dobra a
pequena a-hélice liberando a entrada do sítio ativo da lipase, ativando a
enzima {[ENZ]*}, possibilitando assim o aumento da atividade enzimática com o
aumento da concentração do substrato. 9
s~nica
/ ~ P ~ I
" [E~][EnzSP~ "I II I
~/~/ ~ 1'"
Enz fase aquosa
interface + SEnz~ [Enz]Jf~ [EnzS]Jf _ [Enz]Jf + p
Figtro 1.2.1.3.5. Comportamento de lipases em sistemas bifásicos.
Em solventes orgânicos, as lipases catalisam a transferência de
grupos acila de compostos doadores para uma ampla faixa de compostos
aceptores diferentes da água. As reações catalisadas por lipases incluem
esterificação, transesterificação, 27 amidação, 28 síntese de peptídeos 29 e
formação de lactonas. 30 Dentre estas reações, a transesterificação
enantiosseletiva é a de grande interesse aos químicos sintéticos, pois
possibilita a fácil preparação de álcoois e ácidos opticamente ativos, os quais
são largamente utilizados na síntese de produtos biologicamente ativos.
A resolução de álcoois secundários empregando lipases em meio
-: .., orgânico é uma prática muito utilizada, pois, além da grande importância em
~" síntese orgânica de álcoois enantiomericamene puros ou enriquecidos, as
lipases apresentam maior enantiosseletividade para esses compostos
22

1.2. Biotransformações
• M = Substituinte médio; L = Substituinte grande.
Figtra 1. 2. 1. 3.6. Regra de Kazlauskas.
a teoria estereo-eletrônica. 34
HO H
~-cD
(comparando com a resolução de álcoois primários e terciários). 10 Númerosos
exemplos de resolução enzimática de álcoois secundários empregando lipases
livres e imobilizadas, com razão enantiomérica superior a cem (E > 100),
podem ser encontrados na literatura, 9, 10 inclusive na resolução de alguns
compostos orgometál icos.23
A estereoseletividade das lipases comumente utilizadas nas
resoluções enzimáticas (exemplo: lipase de Candida antartica, lipase de
Pseudomonas sp. , lipase de pâncreas de porco, etc.) segue um modelo
empírico conhecido como a 'regra de Kazlauskas': 31 lipases preferem o
enantiômero com a apresentada na Figura 1.2.1.3.6. 32
Esta regra empírica está baseada
na observação de que muitas lipases
preferem catalisar a conversão de um
mesmo enantiômero na reação de síntese ou
na reação de hidrólise de ésteres. Esta
estereosseletividade é explicada pelo arranjo
espacial dos resíduos catalíticos da lipase,
baseado em estudos de raios-X 33 e via
Entretanto, esta regra não prediz o grau de enantiosseletividade da lipase na
resolução dos álcoois.
Em geral, altos valores de excesso enantiomérico são observados
quando os substituintes ligados ao carbono carbinólico do álcool secundário
possuem tamanhos diferentes. 9 Em um estudo sobre resoluções de álcoois
usando CALB, foi demonstrado que álcoois secundários que possuem um
substituinte de tamanho médio maior que um grupo etila ou um substituinte de
tamanho grande menor que um grupo n-propila apresentam baixos valores na
razão enantiomérica (conseqüentemente baixos valores de excesso
enantiomérico), salientando a necessidade da diferença de tamanhos entre os
substituintes do álcool secundário para uma resolução eficiente. 35
1.2.1.4. Técnicas Especiais em Síntese Orgânica
Em reações de transesterificação empregando lipases em meio
orgânico a concentração do nucleófilo é sempre limitada, diferentemente das
23

1. Introdução
reações de hidrólise, onde o nucleófiló está sempre em excesso (a água - 55
Moi/L). Como resultado, as transesterificações em meio orgânico utilizando
ésteres como doadores de acila, são geralmente reversíveis, chegando ao
equilíbrio e levando a rendimentos menores que os observados em reações
pseudo-irreversíveis (como as hidrólises). A reversibilidade das
transesterificações é causada pelas nucleofilicidades relativas do nucleófilo
(Nu1) e do grupo de saída do doador de acila (Nu2
), já que ambos competem
pelo intermediário "acil-enzima" (Esquema 1.2.1.4.1). 9
processo, e poderia
influenciar
negativamente na
atividade enzimática,
Grupodt!+ Saído
Nu2
('H
~DoalÚ'r tk ilpas<
+ --RI Rl Adla--solv~nte
orgânlc-l"\
Esquema 1.2.1.4.1. Acilação enzimática em meio orgânico.
Para contornar este problema, podemos utilizar um excesso do doador
de acila, o que
elevaria os custos do
tornando essa alternativa pouco atraente.
Uma alternativa interessante é a utilização de doadores de acila
especiais, os quais podem transformar as reações em meio orgânico em
reações irreversíveis. Neste contexto, a utilização de enol ésteres em
resoluções empregando lipases apresenta bons resultados, pois, doadores de
acila como ésteres de vinila ou de isopropenila liberam enóis instáveis como
grupo de saída, os quais tautomerizam para formar aldeídos ou cetonas
estáveis (Esquema 1.2.1.4.2). Dessa maneira, a resolução se torna irreversível,
I fi Jl ' + R-0HR/"-..O R- .. O
RI = n-a1quiL ari!. aril-alquil. haloalquil Rl = H CH3
Esquema 1.2.1.4.2. Acilação enzimática em meio orgânico.
contribuindo para o aumento do rendimento e da seletividade enzimática. 9
Uma outra técnica muito utilizada em biotransformações é a
imobilização s;le enzimas. Tal técnica é uma estratégia empregada para
solucionar prqblemas causados pela utilização de solventes orgânicos e/ou por
24

1.2. Biotransformações
mudanças bruscas de pH em biotransformações, além de facilitar a
recuperação da enzima após a reação, possibilitando a sua reutilização e
diminuindo os custos. 9
Muitas vezes as enzimas não são estáveis em meio orgânico,
ocorrendo sua desnaturação pelo solvente. Em outros casos, mesmo em meio
aquoso, pode ocorrer uma auto-oxidação ou uma inativação pela variação do
pH. 36 A imobilização permite a estabilização da enzima, possibilitando a
realização de biotransformações em condições adversas às normalmente
empregadas com enzimas. Essa técnica consiste na ligação da enzima em um
suporte sólido insolúvel ou em ligações cruzadas com reagentes bifuncionais
(ou multifuncionais), ou ainda, no confinamento da enzima em uma matriz
sólida (gelou polímero) onde ela permanece cataliticamente ativa (Figura
1.2.1 .4.1 ).
@enzymeCo-Cxou:-UnkingCroee.L.Ir\king
Entropmem
Coupllng In_ ......----- ~ by-
.~- ------ ------ - / ~ / "B: "'te ~~~ "~- ~~ ~ [~;;IC-® cB ~~/l\~ ;@l; ~
-® B E~ Enz Enz C Gel Cf PoIymer ~ HolIow ='"- (MEEe-Tedlnlque)
AdsorplivelIonIc
figuro 1.2.1.4.1. Princípio da técnica de imobilização enzimática.
25

-.

1.3. Selênio e Telúrio
1.3. Selênio e TelúTÍo


i.·~I!,I\JIV V'- "",~".,, -,
'03 T 'cid? ele 90 Pall!º 1.3. Selênio e Telúno
A versatilidade dos compostos orgânicos de selênio e telúrio em
síntese orgânica pode ser notada pela reatividade peculiar apresentada por
cada um desses organo-calcogenetos. Essa reatividade está ligada à
habilidade desses heteroátomos (Se e Te) em estabilizar cargas positivas ou
negativas adjacentes, à labilidade das ligações C-Se e C-Te e à facilidade de
oxidação de selenetos à selenóxidos associada à eliminação de forma "syn".
Por exemplo, a estabilização de cargas negativas pelo átomo de selênio
permite a reação de a-desprotonação dos selenetos vinílicos com bases fortes
não nucleofílicas como o LDA, sendo o carbânion vinílico resultante capturado
com diferentes eletrófilos. 37 Também podemos destacar a reação de oxidação
de selenetos à selenóxidos seguida de eliminação syn, a qual representa um
método versátil na preparação de álcoois alílicos. 38
É interessante ressaltar que a química de compostos orgânicos de
telúrio não é uma extensão da química de compostos orgânicos de selênio,
sendo observadas diversas diferenças na reatividade destas classes de
compostos. Dependendo das condições reacionais, reagentes alquil-Iítio
podem efetuar adições do tipo Michael em selenetos vinílicos, clivar a ligação
C-Se e promover a a-desprotonação, levando a mistura de compostos. 37 Por
outro lado, quando te/uretos orgânicos são tratados com reagentes alquil-Iítio,
não se observam produtos de adição do tipo Michael ou decorrentes da
a-desprotonação, sendo apenas observados produtos oriundos da clivagem da
ligação C-Te. Dessa forma, teluretos orgânicos podem ser considerados
precursores de reagentes organometálicos sinteticamente valiosos. 39
Um aspecto interessante da metodologia de preparação dos teluretos
vinílicos a partir de uma olefina ou um alcino (hidroteluração), é a alta
estereosseletividade apresentada para produtos com a configuração Z,40
diferentemente de outras hidrometalações que levam a produtos de
configuração E ou misturas de estereoisômeros. Além disso, a geometria da
ligação dupla (Z) dos teluretos vinílicos é mantida em reações de troca
metalóide-metal, o que torna essa estratégia muito interessante para a síntese
de moléculas insaturadas com essa geometria.
Síntese de sistemas deste tipo já foram alvos de estudos, como a
preparação da (-)-Macrolactina A, 41 um agente antiviral e citotóxico, e o
29

1. Introdução
Siphonodio/, um metabólito bioativo originado de organismos marinhos (Figura
1.3.1), 42 demonstrando a grande potencialidade dos compostos orgânicos de
telúrio em síntese orgânica.
OH
~ ~OH
O O ,,,,,,OH
H li;
~li;
HO""" ~
l\facrolactina ASiphonodiol
* Em vermelho estão indicados as unidades estruturais provinientes de teluretos vinílicos.
Figura 1. 3.1. Emprego de teluretos na síntese de compostos bioativos.
Em vista do potencial sintético dos compostos orgânicos de selênio e
telúrio, aliado à importância das sínteses enantiosseletivas (seção 1.1.) é de
grande interesse que se desenvolvem métodos de preparação de compostos
desses elementos na sua forma enantiomericamente pura. Os mesmos
constituiriam importantes blocos de construção quirais, ao se explorar a
química orgânica do selênio43 e do telúrio44, hoje em dia já bem estabelecidas.
30

2. Resultados e Discussão


2 .. 1.. Hidróxi-Selenetos
2.1 Hidróxi-Selenetos


2.1 Hidróxi-Selenetos
2.1.1. Síntese Racêmica
Os hidróxi-selenetos utilizados nas biotransformações foram
preparados empregando metodologias descritas na literatura (Figura 2.1.1.1).
OH OH OH OH
~SéPh ~SéPh ~SéPh ~SéPhl:t lb le ld
OH OH OH OH OH
~SéPh ~Sé ~SéPh SéPh 6'"Ph Ph 'CHlle 1f SéPh
19 Ih O li
Figura 2. 1. 1. 1. Hidróxi-selenetos empregados nas biotransformações.
Os hidróxi-selenetos 1a e 1e foram sintetizados, respectivamente,
empregando a reação de abertura do óxido de propileno e óxido de estireno
com fenil-selenolato de sódio, preparado "in situ" pela redução de disseleneto
de difenila com NaBH4 (Esquema 2.1.1.1). 38
O composto 1f foi preparado de maneira análoga, porém utilizando
metil-selenolato de lítio, o qual foi preparado "in situ" pela adição de metil-Iítio
sobre selênio elementar em THF (Esquema 2.1.1.1). De maneira geral, a
abertura de epóxidos mono-substituídos com nucleófilos de selênio são regio
específicas, originando apenas o álcool secundário, isto é, a abertura ocorre no
carbono menos substituído
(Esquema 2.1.1.1). 38
Entretanto, a
abertura do óxido de estireno
pode originar uma mistura de
..... SéPh NaBH4PhSé EtOH
SeO M.:LiTHF
o EtOHou OH
R'S8+/\ ~ ~. é '-----'o""", SéR'Ret.38 R R
(R,S)-lin R=CH3 éR'=Ph (Rend.=89%)
le R = Ph é R' = Ph (Rend. = 55%)1f R = Ph é R' = CH3 (Rend. = 77%)
dois regio-isômeros (álcool Esquema 2.1.1.1. Preparação dos compostos la, le e lf.
primário e o álcool
secundário). 45 A presença dos dois regio-isômeros é explicada pela abertura
do óxido de estireno na posição benzílica, gerando o álcool primário (Esquema
2.1.1.2).
35

Esquema 2.1.1.2. Mistura de régio-isômeros.
2. Resultados e Discussão
CoRSee+~ ~
PhReJ.45
(iH
I SeRPh~
I
(IH
+ yseR
Ph
Os hidróxi-selenetos
1b-d foram sintetizados por
reação de adição de
fenilseleno-metil-Iítio ao aldeído
correspondente, levando a
formação do hidróxi-seleneto de interesse com elevada pureza. 46 O
fenilseleno-metil-Iítio foi gerado in situ pela reação de troca metalóide-metal
entre o bis(fenilseleno)metano e n-SuLi.· 46 O bis(fenilseleno)metano foi
preparado pela reação do fenilselenolato de sódio com dibromometano
(Esquema 2.1.1.3). 38
2 RS e CH B EtOH I7-BuLi ~Ph' ~,e + 2 f1 - PhSe"-./SePh~L se"-./LJ+PhSe~
(Rend. = 88"/..) ReJ.44 1()
~~ ~ ~~
Ib R = tCHê)êCH3 (Rend. = 72%)le R = CHtCH3h (RemI. = 78%)Id R = (CHêhCH\ (Rend. = 83%)
OH
R~sePhI
Esquema 2.1.1.3. Modo de preparação do bis(fenilseleno)metono e seu emprego napreparação dos compostos lb-d.
o composto 19 foi sintetizado a partir da desprotonação do
bis(feni!séleno)metano empregando o LDA, gerando um carbânion, o qual foi
capturado com acetaldeído, produzindo assim o hidróxi-seleneto (R,S)-1 9 em
bom rendimento (Esquema 2.1.1.4). 46,47
Devido à presença
compostos (t)-1 h e (t)-1 i, a
síntese dos mesmos via a
estereogênicos[ ]
o OH
LDA PhSysePh ~ ~PhSe~SePh ------=--- _ SePh
TIIF Li TIIFReJ.47 Ret. 47 Ig SePh
(Rend. = 73%)
Esquema 2.1.1.4. Rota sintética para obtenção de 19.
de dois centros
nos
reação de troca metalóide
metal entre selenoacetal substituído e n-SuLi seguida da adição de
acetaldeído, gerou os compostos (t)-1 h-i como uma mistura de quatro
estereoisômeros (dois pares de enatiômeros - Esquema 2.1.1.5). 46
36

2.1 Hídróxí-Selenetos
OH
~S~PhR
LhS~yL] l~+ lHF
PhS~~ Ret.46
Ih R = (CH~)sCH3 (Rend. = 86%)li R = Ph (Rend. = 88%)
Esquema 2.1.1.5. Síntese dos compostos lh e li.
aComo
separação dos PhSYS~Ph ~
diastereoisômeros por R THFRef.46
cromatografica em
coluna não foi possível,
as biotransformações
foram realizadas utilizando-se essa mistura estereoisomérica.
A síntese dos selenoacetais substituídos empregados nessas reações
foi a etapa mais delicada na preparação desses hidróxi-selenetos [(t)-1 h e (t)
1i]. Os selenoacetais foram sintetizados por selenoacetalização do aldeído
apropriado com fenil-selenol, utilizando o H2S04. 48 O cuidado nesta etapa de
síntese foi com o H2S04 utilizado, pois o mesmo é um agente oxidante. Para a
obtenção do selenoacetal em bons rendimentos foi necessário utilizar o H2S04
13M, evitando assim a oxidação do fenil-selenol a disseleneto de difenila
(Esquema 2.1.1.6). 48
Esquema 2.1.1.6. Preparação dos selenocetais.
Os
racêmicos
selenetos
dos
1a-i
acetatos
hidróxi
foram
o2 PhSeH + Il
R Ret.48
sintetizados quimicamente
utilizando anidrido acético em piridina seca. 49 Essas reações não
apresentaram maiores problemas, sendo os produtos facilmente purificados e
caracterizados (Esquema 2.1.1.7).
o o
)loA..~
pindUl<l
Ret.492
(l.-\"
~sePh2a
O.-\c
~sePh2b
OAc
........ ~ /SePh
T 2C--
O.-\c
~S.Ph
2d
0.-\"
~SoPhSePh
2g
O.-\.c
2h
SePh ~sePh
©2iFigtra 2. 1. 1.7. Acetatos dos hidróxi-selenetos sintetizados.
37

2. Resultados e Discussão
2.1.2. Biotransformações
2.1.2.1. Resolução em Meio Orgânico
Esquema 2.1.2.1.1. Resolução enzimática do (R.S)-10 em meioorgânico.
o)
(~S>-1a
OH
R1Se~
biocatalizadores: a PPL
(Iipase de pâncreas de
porco - livre), a PSL
(AMANO PS - lipase de
Pseudomonas sp. -livre)
e a CALB (NOVOZYM 435® - lipase de Candida antartica - Fração B
imobilizada). Na tabela 2.1.2.1.1 estão resumidos os resultados observados na
resolução do composto (R,S)-1a com essas enzimas em hexano seco e em
diferentes temperaturas. Os resultados demonstram que a atividade enzimática
depende da temperatura empregada. A temperatura apresentou um
considerável efeito na enantiosseletividade da reação, mas não apresentou
influência sobre o enantiômero preferencialmente acetilado (enantiômero-(R)].
A enantio-preferência observada está de acordo com a regra de Kazlauskas
(Sessão 1.2.1.3). 31
A resolução enzimática de hidróxi-selenetos foi estudada variando-se o
tipo de lipase, o suporte da enzima (quando imobilizada), o solvente, a
temperatura e a estrutura do hidróxi-seleneto, a fim de avaliarmos a influência
desses fatores sobre as resoluções enzimáticas desses compostos em meio
orgânico. Dessa forma o (R, S)-hidróxi-seleneto 1a foi utilizado como padrão
neste estudo (Esquema 2.1.2.1.1).
Três lipases
foram escolhidas como
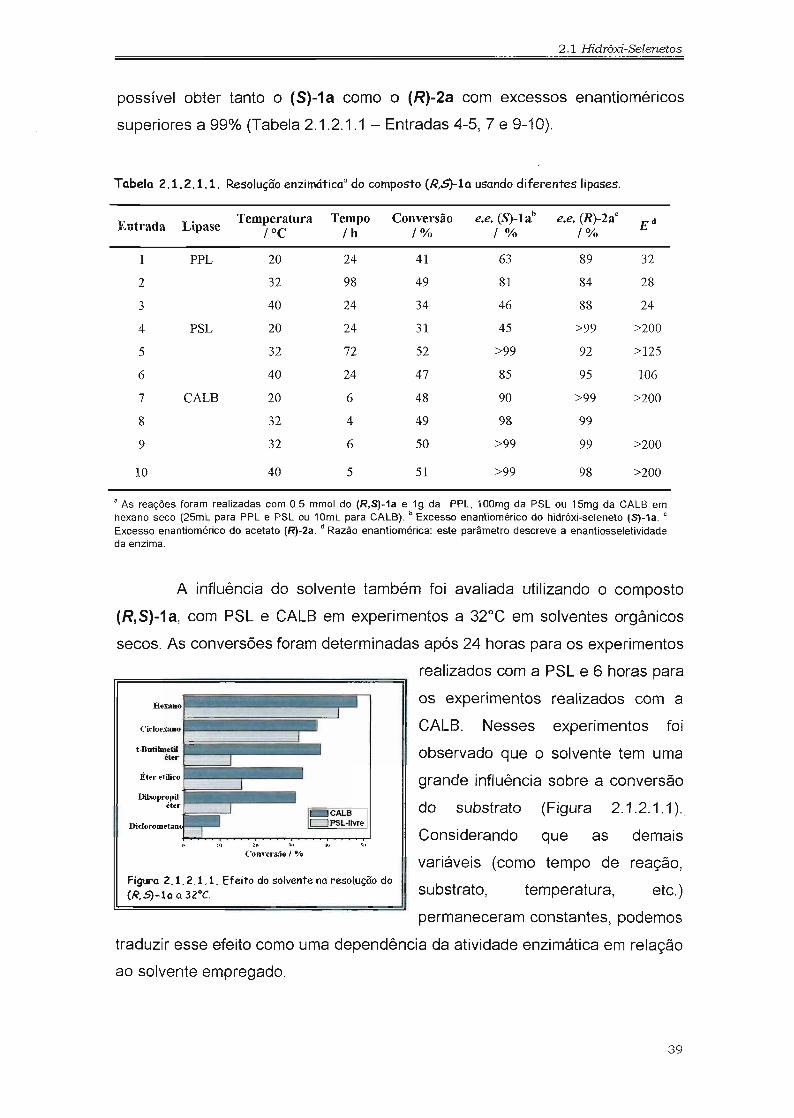
As melhores enantiosseletividades foram obtidos utilizando PSL e
CALB entre 20 e 40°C e a melhor relação entre excesso enantiomérico e
conversão foi observado a 32°C. Na reação com a PSL, o (S)-1a e o (R)-2a
foram obtidos com excessos enantioméricos superiores a 92% a 32°C (Tabela
2.1.2.1.1 - entrada 5), enquanto que a melhor enantiosseletividade foi
observada com a CALB. Na reação com a CALB o (S)-1a e o (R)-2a foram
obtidos com excessos enantioméricos superiores a 98% a 32°C (Tabela
2.1.2.1.1 - entradas 8 e 9). Controlando o tempo de reação e a temperatura foi
38
2.1 Hidróxí-Selenetos
possível obter tanto o (S)-1a como o (R)-2a com excessos enantioméricos
superiores a 99% (Tabela 2.1.2.1.1 - Entradas 4-5,7 e 9-10).
Tabela 2.1.2.1.1. Resolução enzimáticaO do composto (R,S)-la usando diferentes lipases.
Entrada LipaseTemperatura Tempo Conversão e.e. (S)-lab e.e. (R)-2aC
E d
fOC fh f% f% f%
1 PPL 20 24 41 63 89 32
2 32 98 49 81 84 28
3 40 24 34 46 88 24
4 PSL 20 24 31 45 >99 >200
5 32 72 52 >99 92 >125
6 40 24 47 85 95 106
7 CALB 20 6 48 90 >99 >200
8 32 4 49 98 99
9 32 6 50 >99 99 >200
10 40 5 51 >99 98 >200
• As reações foram realizadas com 0.5 mmol do (R,S)-1a e 19 da PPL, 100mg da PSL ou 15mg da CALB emhexano seco (25mL para PPL e PSL ou 10mL para CALB). b Excesso enantiomérico do hidróxi-seleneto (S)-1a. c
Excesso enantiomérico do acetato (R)-2a. d Razão enantiomérica: este parãmetro descreve a enantiosseletividadeda enzima.
CALS. Nesses experimentos foi
observado que o solvente tem uma
grande influência sobre a conversão
do substrato (Figura 2.1.2.1.1 )"
Considerando que as demais
variáveis (como tempo de reação,
substrato, temperatura, etc.)
oI
I
I
I
'1 IlêS~ALB~ c:::::J PSL-Ilvre
1 J
He~n
CidoexaDO
Dü....prOI'i1eter
Éte.. etilico
10 ~ ~ ~ ~
Cooyers..;o I e/.
t-Butilmeliléter
Didorometan
Figtro 2.1.2.1.1. Efeito do solvente no resolução do(R,S)-10 a 32°C.
A influência do solvente também foi avaliada utilizando o composto
(R,S)-1a, com PSL e CALS em experimentos a 32°C em solventes orgânicos
secos. As conversões foram determinadas após 24 horas para os experimentos
realizados com a PSL e 6 horas para
os experimentos realizados com a
permaneceram constantes, podemos
traduzir esse efeito como uma dependência da atividade enzimática em relação
ao solvente empregado.
39

2. Resultados e Discussão
Apesar da dependência da atividade enzimática com o solvente, não
foi observada mudança na enantiosseletividade e nem da enantiopreferência
da enzima, sendo sempre acetilado preferencialmente o enantiômero-(R)-1a.
Em todos os solventes o (R)-2a foi obtido com excesso enantiomérico
superior a 97% usando a PSL e superior a 99% usando a CALB. As maiores
atividades enzimáticas (isto é, maiores conversões) foram observadas em
solventes não polares, como hexano e cilcloexano, enquanto que em solventes
polares, como diclorometano, a atividade foi menor (Figura 2.1.2.1.1 ).
Como salientado, a imobilização enzimática é uma ferramenta muito
útil em biotransformações (Sessão 1.2.1.4). Nesses sentido, a PSL foi
imobilizada em alguns suportes: gel de agar, sílica, filme de caseinato,
Montmorilonita K-10 e óxido de poli(etileno) (PEO). Os experimentos utilizando
a PSL imobilizada foram conduzidos nas mesmas condições empregadas para
a enzima livre. Todos os experimentos de imobilização foram realizados com a
supervisão da Profa. Ora. Maria da Graça Nascimento do Departamento de
Química da Universidade Federal de Santa Catarina.
'. 40 ~
.. So
oPSL -PEO
PSL-IivrePSL-Silica #S
PSL - Caseinato S\S~
Figtro 2.1.2.1.2. PSL imobilizada versus tempode resolução do {R,S)-lo em hexano a 35°C.
Os resultados evidenciaram a dependência da atividade enzimática
com o tipo de suporte utilizado (Figura 2.1.2.1.2). Nenhum produto acetilado foi
detectado com a PSL imobilizada em gel de ágar ou em Montmorilonita K-10,
enquanto que em sílica e em filme de caseinato apresentaram um efeito
negativo sobre a atividade enzimática, sendo observadas conversões inferiores
às observadas com a lipase livre. Por outro lado, o óxido de poli(etileno)
apresentou bons resultados, aumentando sensivelmente a atividade enzimática
da PSL (Figura 2.1.2.1.2). Uma
racionalização para os resultados
observados com a imobilização da
PSL em diversos suportes pode ser
baseada nos processos de difusão de
reagentes e produtos, do suporte para
o meio reacional, e também da
manutenção da integridade da
estrutura ativa da Iipase. 50
A estereoquímica preferencial
40

2.1 Hidróxi-Selenetos
para o enantiômero-(R) na acetilação foi mantida, independente do suporte
utilizado. Após 24 horas de reação a 35°C em hexano, a resolução utilizando a
PSL livre apresentou uma conversão de 45% e o substrato (S)-1a. foi
recuperado com excesso enantiomérico de 80%, enquanto que o produto (R)
2a foi obtido com excesso enantiomérico de 97%. Entretanto, a mesma
resolução empregando a PSL imobilizada em PEG apresentou uma conversão
de 52% e o substrato (S)-1 a foi recuperado com excesso enantiomérico de
>99%, enquanto que o produto (R)-2a foi obtido com excesso enantiomérico de
92%.
Analisando os resultados obtidos, verificamos que apesar da melhora
do desempenho da PSL com a imobilização em PED, a CALS (Iipase adquirida
imobilizada) apresentou os melhores resultados na resolução do hidróxi
seleneto (R,S)-1a. Assim, o sistema CALS em hexano a 32°C foi escolhido
para avaliar o comportamento da estrutura do hidróxi-seleneto nas
biotransformações. Nesse sentido, as resoluções dos hidróxi-selenetos (R,S)
1b-e foram realizadas utilizando a CALS como biocatalizador em hexano seco
(Esquema 2.1.2.1.2).
Esquema 2.1.2.1.2. Resolução enzimático de hidróxi-selenetos em meioorgânico.
o)J
(R}-2
crcR"Se~w + ~G1
(5)-1
e, R'=Al, W=Phf, R' = Cf1,. W= Ph
b,R'=Al, W=(~C, R' =Al, W-(~d, R' = Al, W= Cl-(a-13>2
(~S)-1
Inicialmente, a resolução do (R,S)-1b foi realizada a 32°C, porém a
esta temperatura a reação enzimática foi muito lenta, necessitando mais de
quatro dias para uma conversão de 50% (Tabela 2.1.2.1.2 - entradas 1-3),
apesar do aumento da quantidade de lipase utilizada (de 15mg passamos a
utilizar 300mg por 0,5 mmol de substrato). Como a CALS apresentou
conversões maiores a 40°C (Tabela 2.1.2.1.1 - entrada 10), optamos por
realizar essas resoluções nesta temperatura.
Apesar do
aumento da
temperatura tenha
influenciado
negativamente na
enantiosseletividade
enzimática na
resolução do (R,S)-1b, os resultados da Tabela 2.1.2.1.2, mostram que o
tempo de reação e a enantiosseletividade enzimática são profundamente
41
2. Resultados e Discussão
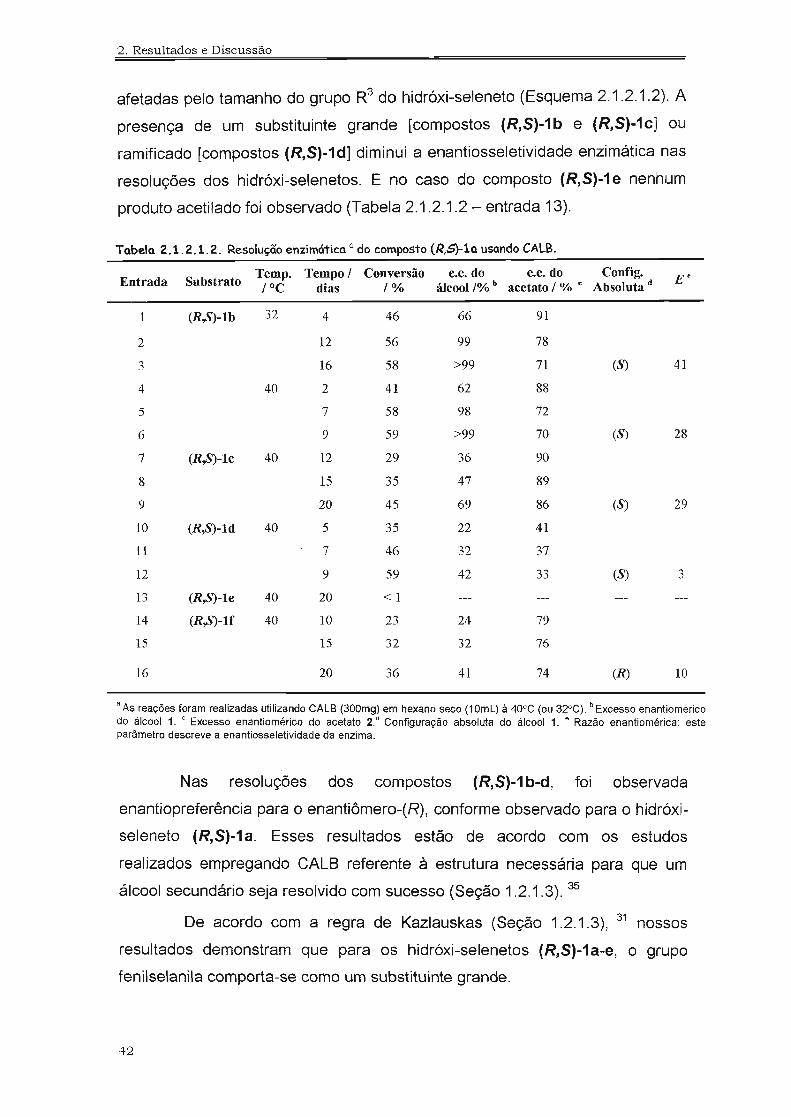
afetadas pelo tamanho do grupo R3 do hidróxi-seleneto (Esquema 2.1.2.1.2). A
presença de um substituinte grande [compostos (R,S)-1b e (R,S)-1c] ou
ramificado [compostos (R,S)-1d] diminui a enantiosseletividade enzimática nas
resoluções dos hidróxi-selenetos. E no caso do composto (R,S)-1e nenhum
produto acetilado foi observado (Tabela 2.1.2.1.2 - entrada 13).
Tabela 2.1.2.1.2. Resolução enzimática a do composto (R,S)-la usando CALB.
Entrada SubstratoTemp. Tempo 1 Conversão e.e. do e.e.do Config. E e
1°C dias 1% álcool 1% b acetato 1% c Absoluta d
1 (R,8)-lb 32 4 46 66 91
2 12 56 99 78.., 16 58 >99 71 (8) 41.)
4 40 2 41 62 88
5 7 58 98 72
6 9 59 >99 70 (8) 28
7 (R,8)-lc 40 12 29 36 90
8 15 35 47 89
9 20 45 69 86 (8) 29
10 (R,8)-ld 40 5 35 22 41
11 7 46 32 37
12 9 59 42 33 (8) 3
13 (R,8)-le 40 20 < 1
14 (R,8)-lf 40 10 23 24 79
15 15 32 32 76
16 20 36 41 74 (R) 10
a As reações foram realizadas utilizando CALS (300mg) em hexano seco (10mL) à 40°C (ou 32"C). b Excesso enantioméricodo álcool 1. c Excesso enantiomérico do acetato 2. d Configuração absoluta do álcool 1. e Razão enantiomérica: esteparâmetro descreve a enantiosseletividade da enzima.
Nas resoluções dos compostos (R,S)-1 b-d, foi observada
enantiopreferência para o enantiômero-(R), conforme observado para o hidróxi
seleneto (R,S)-1a. Esses resultados estão de acordo com os estudos
realizados empregando CALB referente à estrutura necessária para que um
álcool secundário seja resolvido com sucesso (Seção 1.2.1.3). 35
De acordo com a regra de Kazlauskas (Seção 1.2.1.3), 31 nossos
resultados demonstram que para os hidróxi-selenetos (R,S)-1a-e, o grupo
fenilselanila comporta-se como um substituinte grande.
42

2.1 Hidróxi-Selenetos
Para avaliar o efeito do tamanho do grupo selanila na resolução dos
hidróxi-selenetos, o composto (R,S)-1f foi sintetizado. Este composto possui
um grupo fenila (como o grupo R3- grupo grande) e um grupo metilselanila
(como grupo R1- Esquema 2.1.2.1.2). A resolução enzimática do (R,S)-1f
apresentou a estereoquímica preferencial para o enantiômero-(S),
diferentemente dos demais compostos [compostos (R,S)-1a-e / Tabela
2.1.2.1.2 - entradas 1-12] e com baixa enantiosseletividade. De acordo com a
regra de Kazlauskas (Seção 1.2.1.3), 31 o grupo metilselenila comporta-se
como um substituinte médio, porém maior que um grupo etila, levando a baixa
enantiosseletividade (E = 10) observada para a resolução utilizando CALS
(Tabela 2.1.2.1.2 - entradas 14-16).
Considerando os resultados obtidos com a resolução dos hidróxi
selenetos (R,S)-1a-f empregando a CALS como biocatalisador e a regra de
Kaslauskas (Seção 1.2.1.3), 31 concluímos que para um 13-hidróxi-seleneto ser
eficientemente resolvido pela CALS em meio orgânico é necessário que o
substituinte de tamanho médio seja menor que o grupo etila, visto que o grupo
selanila se comporta melhor como substituinte grande, como ocorre no
composto (R,S)-1a.
Assim, as resoluções dos 13-hidróxi-selenetos (R,S)-1g e (t)-1h-i (os
quais possuem o grupo metila como substituinte de tamanho médio - Esquema
2.1.2.1.3) foram realizadas nas mesmas condições empregadas na
biotransformação do composto (R,S)-1 a, uma maneira de comprovarmos os
resultados e as conclusões obtidas nesse estudo.
+ ~OH
(~S)-1g, Fi'=SEA1(:I:}-1h, Fi' = {o-t,),;CH,(:I:}-11. Fi'=A1 o
)JEsquema 2.1.2. 1.3. Resolução enzimático de hidróxi-selenetos
(R,S)-lg e (±)-lh-i em meio orgânico.
resolvido pela CALS (E
> 200) e o produto
acetilado foi obtido
com excessos enantioméricos superiores a 99% (Tabela 2.1.2.1.3), de maneira
análoga ao observado com o hidróxi-seleneto (R,S)-1a.
Nessas
condições o composto
(R,S)-1g foi
eficientemente
43

2. Resultados e Discussão
Na resolução dos compostos (t)-1 h-i, foi empregada a mistura dos
quatro estereoisômeros (dois pares de enantiômeros - Esquema 2.1.2.1.3).
Nessas resoluções não foi observada nenhuma diastereopreferência.
Como a discriminação de cada um dos quatro enantiômeros via CG
Quiral não foi possível, o grupo fenilselanila foi removido da estutura dos
álcoois (t)-1h-i, desfazendo-se assim, um dos centros estereogênicos. Foram
utilizadas reações que não modificam a configuração do carbono carbinólico
(vide seção 2.1.4.1), possibilitando a determinação da enantiosseletividade
enzimática relativa ao centro estereogênico localizado no carbono carbinólico.
Assim, após as reações de eliminação do grupo fenilselanila, foi
observada uma alta enantiosseletividade para o centro carbinólico dos
compostos (t)-1 h-i, observando-se excessos enantioméricos superiores a 99%
para os produtos acetilados (sem o grupo fenilselanila - Tabela 2.1.2.1.3),
também de maneira análoga ao observado com o hidróxi-seleneto (R,S)-1 a .
Tabela 2.1.2.1.3. Resolução enzimáticaQ dos compostos 19-i usando CALB.
Entrada SubstratoTempo Conversão e.e. do substrato e.e. do produto E"Ih 1% 1% 1%
1 (R,s)-lg 25 50 99° >99d >200
2 (±)-lb 50 48 93e >99f >200
3 (±)-li 72 48 92g >99h >200
a As reações foram realizadas utilizando CALS (15mg) em hexano (1 OmL) à 32°C e utilizando 0,5 mmol do substrato.b Razão enantiomérica: este parâmetro descreve a enantiosseletividade da enzima c Excesso enantiomérico do álcool19. d Excesso enantiomérico do acetato 2g. e Excesso enantiomérico do álcool após a reação de eliminação deselenóxido (vide seção 2.1.4.1) . f Excesso enantiomérico do acetato após a reação de eliminação de selenóxido (videseção 2.1.4.1). 9 Excesso enantiomérico do álcool após a remoção do grupo fenilseleno (vide seção 2.1.4.1). h Excessoenantiomérico do acetato após a remoção do grupo fenilseleno(vide seção 2.1.4.1).
Nas resoluções dos compostos (R,S)-1 9 e (t)-1 h-i, foi observado que
a CALB apresenta preferência em acetilar o álcool de configuração (R)
(carbono carbinólico), em concordância com os resultados observados
anteriormente.
Todos os experimentos realizados neste estudo demonstraram que
l3-hidróxi-selenetos que possuem substituinte ligado ao carbono carbinólico
menor que um grupo etila, podem ser obtidos opticamente puros empregando
se lipases como biocatalisadores em meio orgânico.
44

2.1.2.2. Resolução em Meio Aquoso
2.1 Hidróxi-Selenetos
o emprego de água como solvente em reações orgânicas sempre
representa uma contribuição valiosa para a preservação do meio ambiente. Em
biotransformações, as vantagens podem ir além do ganho ambiental, pois o
meio natural da enzima é o meio aquoso, proporcionando uma melhor
conformação e conseqüentemente uma maior atividade enzimática, o que pode
contrubuir para um aumento na eficiência na seletividade da enzima.
Assim, com o intuito de encontrarmos um processo de resolução
enzimática de hidróxi-selenetos menos agressivo ao meio ambiente e mais
eficiente, foram realizados estudos de hidrólise enzimática dos respectivos
acetatos dos hidróxi-selenetos (R,S)-1 b-f (Esquema 2.1.2.2.1).
AcO~SePh Iipase AcO" /' .I ---.~ "~'I SePhR Tampllo \ Acetona R
HO~SePh+ I +
R
o.)lOH
AcoiSePh AcoisePh AcOXSePh
(RS)-2a ((RS)-2b (RS)-2c
ACOrsePh
g(RS)-2e
ACOrSe
, CH3
[QJ (RS)-2f
J=ACO
SePh
(RS)-2d
SePh
ACo0SePh
(RS)-2g
Esquema 2.1.2.2.1. Resolução enzimática em meio aquoso.
Nesse sentido, realizamos primeiramente uma triagem com dez
enzimas comerciais (não imobilizadas) de fontes naturais diferentes, que
geralmente apresentam boa atividade em meio aquoso à temperatura ambiente
e a pH neutro: [CALA (Iipase de Candída antarctíca Tipo A - Roche Co.),
CALB (Iipase de Candída antarctíca Tipo 8 - Roche Co.), CRL (Iipase de
Candída rugosa - Roche Co.), PSL (Iipase de Pseudomonas sp. - Roche Co.),
PLE (esterase de fígado de porco - Roche Co.), PPL (Iipase de pâncreas de
porco - Roche Co.), TLLa (Iipase de Thermomyces lanugínosa - Roche Co.),
ASL (Iipase Alcalígenes sp. - Roche Co.), MJL (Iipase de Mucor Javanícus
Sigma Co.) e TLLb (L1POZYME Tl 1DDl® - Iipase de Thermomyces fanugínosa
- Novozymes Co.)].
45

2. Resultados e Discussão
Para agilizarmos o processo de triagem com as enzimas e os
substratos de interesse, optamos por realizar a triagem em placas de "Elisa"
(com 96 poços - Figura 2.1.2.2.1) utilizando um indicador de pH colorimétrico,
visto que na reação de hidrólise ocorre a produção de ácido acético (Esquema
2.1.2.2.1), permitindo assim avaliar a conversão no processo de hidrólise pela
mudança de cor do indicador. 51 Dessa maneira foi possível realizar várias
micro-reações ao mesmo tempo e verificar rapidamente quais lipases
promoviam a hidrólise dos compostos (R,S)-2a-g.
Bra :Substrato· Ind,c.d
Br:anco:E,wma + indIcador
Subiualn
lipas'>
1 2 3 4 5 6 7 8 9 10 11 12A X I-
B O>< >< ><CDE I-FG Õ?" ?' ?'
'X 'X 'XH .....
I IBranco:
Substrato· ln,hcad r
Placa de Elisa
figlrO 2. 1.2.2.1. Disposição das soluções na placa de Elisa.
Nesta triagem utilizamos como indicador de pH azul de bromo timol
(0,005%; azul = meio básico \ verde = meio neutro \ amarelo = meio ácido),
tampão fosfato (pH=7,2 /20.10-3 Moi L-1) e duas placa de "Elisa" com 96 poços
cada. Todas as micro-reações foram realizadas em duplicatas e sempre
acompanhadas com os brancos: enzima + indicador e substrato + indicador, na
mesma placa. A disposição das soluções (brancos e reações) na placa seguiu
o padrão monstrado na Figura 2.1.2.2.1 .
Seis enzimas (CALA, CALS, CRL, PSL, PLE, ASL e MJL)
apresentaram atividade hidrolítica para alguns acetatos utilizados na triagem
(Figura 2.1.2.2.2). Entretanto, nenhuma enzima apresentou atividade hidrolítica
frente aos substratos possuidores de dois ligantes volumosos [ligados ao
carbono carbinólico \ (R,S)-2c-e - Figura 2.1.2.2.2], provavelmente devido à
dificuldade na formação do complexo enzima-substrato no sítio catalítico da
enzima, causada pelo volume dos ligantes, conforme observado na resolução
em meio orgânico (Seção 2.1.2.1).
46
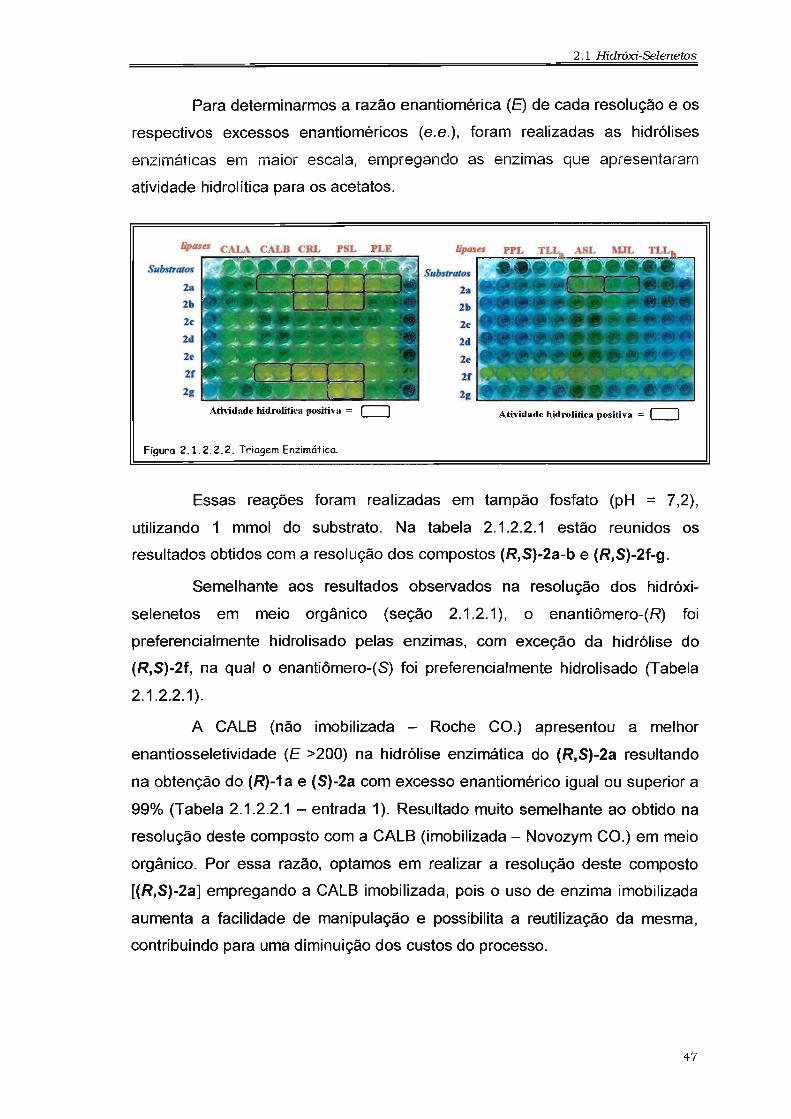
2. 1 Hidróxi-Selenetos
Para determinarmos a razão enantiomérica (E) de cada resolução e os
respectivos excessos enantioméricos (e.e.), foram realizadas as hidrólises
enzimáticas em maior escala, empregando as enzimas que apresentaram
atividade hidrolítica para os acetatos.
2e
2f
2g
2d
2f
2g
2e
liptun PSL PLE
Substratos t(~etll~tí~jiiifJ Substratos
2a 2a
2b 2b
2c 2c
2d
Athidade hidI·olítica posiô"a = c=J Atividade hidrolitica positiva = c=J
Figura 2.1.2.2.2. Triagem Enzimática.
Essas reações foram realizadas em tampão fosfato (pH = 7,2),
utilizando 1 mmol do substrato. Na tabela 2.1.2.2.1 estão reunidos os
resultados obtidos com a resolução dos compostos (R,S)-2a-b e (R,S)-2f-g.
Semelhante aos resultados observados na resolução dos hidróxi
selenetos em meio orgânico (seção 2.1.2.1), o enantiômero-(R) foi
preferencialmente hidrolisado pelas enzimas, com exceção da hidrólise do
(R,S)-2f, na qual o enantiômero-(S) foi preferencialmente hidrolisado (Tabela
2.1.2.2.1 ).
A CALS (não imobilizada - Rache CO.) apresentou a melhor
enantiosseletividade (E >200) na hidrólise enzimática do (R,S)-2a resultando
na obtenção do (R)-1a e (S)-2a com excesso enantiomérico igualou superior a
99% (Tabela 2.1.2.2.1 - entrada 1). Resultado muito semelhante ao obtido na
resolução deste composto com a CALS (imobilizada - Novozym CO.) em meio
orgânico. Por essa razão, optamos em realizar a resolução deste composto
[(R,S)-2a] empregando a CALS imobilizada, pois o uso de enzima imobilizada
aumenta a facilidade de manipulação e possibilita a reutilização da mesma,
contribuindo para uma diminuição dos custos do processo.
47
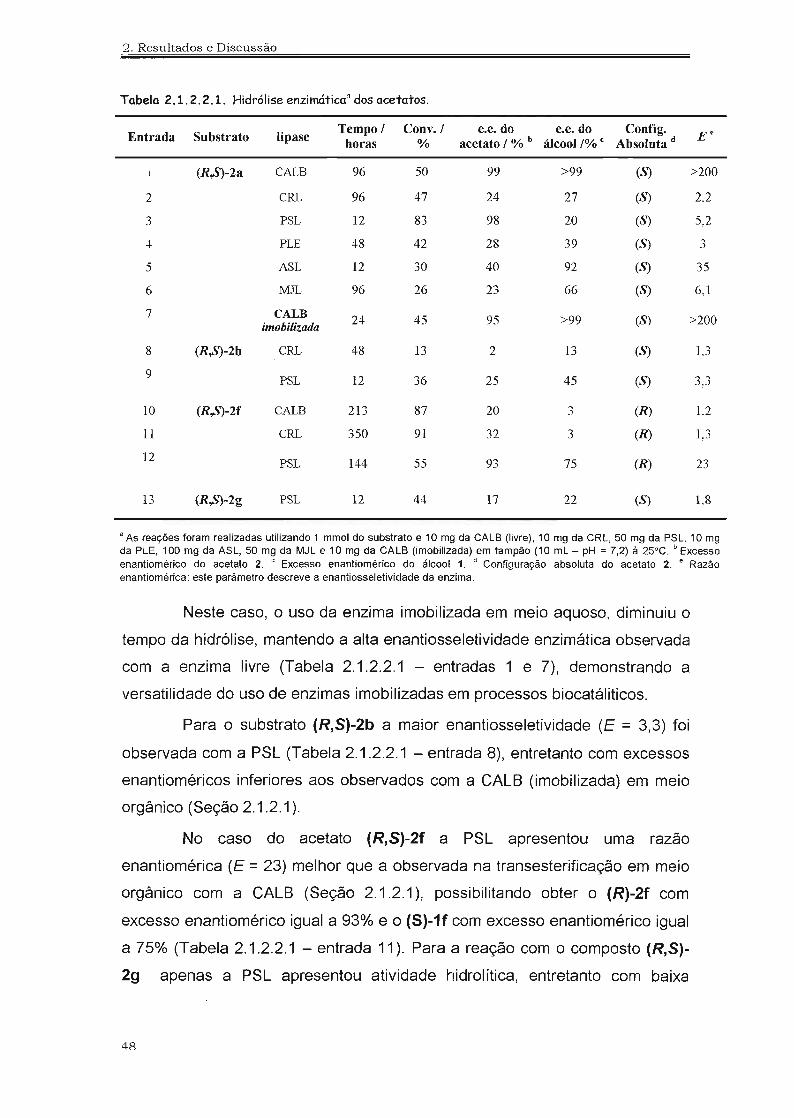
2. Resultados e Discussão
Tabela 2.1.2.2.1. Hidrólise enzimáticaO dos acetatos.
Entrada Substrato lipaseTempo! Conv.! e.e. do e.e. do Config. E e
horas % acetato! % b álcool!% c Absoluta d
1 (R,s)-2a CALB 96 50 99 >99 (S) >200
2 CRL 96 47 24 27 (S) 2,2-. PSL 12 83 98 20 (S) 5,2.)
4 PLE 48 42 28 39 (S) 3
5 ASL 12 30 40 92 (S) 35
6 MJL 96 26 23 66 (S) 6,1
7 CALB 24 45 95 >99 (S) >200imobilizada
8 (R,s)-2b CRL 48 13 2 13 (S) 1,3
9 PSL 12 36 25 45 (S) 3,3
10 (R,s)-2f CALB 213 87 20 3 (R) 1,2
11 CRL 350 91 32 3 (R) 1,3
12 PSL 144 55 93 75 (R) 23
13 (R,s)-2g PSL 12 44 17 22 (S) 1,8
a As reações foram realizadas utilizando 1 mmol do substrato e 10 mg da CALB (livre), 10 mg da CRL, 50 mg da PSL, 10 mgda PLE, 100 mg da ASL, 50 mg da MJL e 10 mg da CALB (imobilizada) em tampão (10 mL - pH =7,2) à 25°C. b Excessoenantiomérico do acetato 2. c Excesso enantiomérico do álcool 1. d Configuração absoluta do acetato 2. e Razãoenantiomérica: este parâmetro descreve a enantiosseletividade da enzima.
Neste caso, o uso da enzima imobilizada em meio aquoso, diminuiu o
tempo da hidrólise, mantendo a alta enantiosseletividade enzimática observada
com a enzima livre (Tabela 2.1.2.2.1 - entradas 1 e 7), demonstrando a
versatilidade do uso de enzimas imobilizadas em processos biocatáliticos.
Para o substrato (R,S)-2b a maior enantiosseletividade (E = 3,3) foi
observada com a PSL (Tabela 2.1.2.2.1 - entrada 8), entretanto com excessos
enantioméricos inferiores aos observados com a CALB (imobilizada) em meio
orgânico (Seção 2.1.2.1).
No caso do acetato (R,S)-2f a PSL apresentou uma razão
enantiomérica (E = 23) melhor que a observada na transesterificação em meio
orgânico com a CALB (Seção 2.1.2.1), possibilitando obter o (R)-2f com
excesso enantiomérico igual a 93% e o (S)-1f com excesso enantiomérico igual
a 75% (Tabela 2.1.2.2.1 - entrada 11). Para a reação com o composto (R,S)
29 apenas a PSL apresentou atividade hidrolítica, entretanto com baixa
48

2.1 Hídróxi-Selenetos
enantiosseletividade (E = 1,8), gerando compostos com baixos excessos
enantioméricos (Tabela 2.1.2.2.1 - entrada 12).
Analisando os resultados obtidos na hidrólise dos seleno-acetatos,
podemos observar a semelhança na resolução dos respectivos hidróxi
selenetos em meio orgânico, principalmente com relação ao volume dos
substituintes e a enantiosseletividade enzimática. Esse resultado evidênciou
que a estrutura do seleno-acetato foi um fator importante para uma boa
resolução do mesmo empregando enzimas hidrolíticas.
2.1.2.3. Biotransformações UtlYizando Células Inteiras de Fungos
R=NOz. F,a
Esquema 2.1.2.3.1. Desracemização utilizandocélula inteira de Aspergil/us terreus.
Em experimentos anteriormente realizados em nosso laboratório
utilizando células inteiras de fungos (Aspergi//us terreus) , foi observado que as
mesmas são capazes de catalisar a desracemização de alguns álcoois
secundários (Esquema 2.1.2.3.1). 52
Esses resultados incentivaram a
realização de experimentos com os
hidróxi-selenetos, pois na resolução o
rendimento máximo é de 50%,
enquanto que na desracemização (ou
resolução dinâmica) o rendimento pode chegar a 100%. Além disso, o uso de
fungos poderia permitir a desracemização de alguns compostos que não
haviam sido produzidos em excessos enantioméricos satisfatórios quando
utilizamos enzimas isoladas.
Foram empregadas duas
linhagens de fungos AspergiJIus
terreus (URM 3571 e CCT 3320)
na desracemização dos hidróxi-Esquema 2.1.2.3.2. Desracemizaçãoutilizondo Fungos.
Uma proposta para o processo de desracemização (ou resolução
dinâmica) empregando fungos consiste em duas reações redox catalisadas por
enzimas, sendo a primeira reação uma oxidação reversível seguida de uma
redução irreversível, gerando apenas um dos enantiômeros (Esquema
2.1.2.3.2).
49

2. Resultados e Discussão
selenetos (R,S)-1a-c e (R,S)-1e (Figura 2.1.2.3.1).
OH OH OH
~sePh ~sePh "- ~ /SePh
(RS)-la (R,S)-lb T(R;lC
OHI SePh
Ph~(RS)-1e
Nos primeiros
testes, foram observados
excessos enantioméricos
figu-a 2.1.2.3.1. Hidróxi-selenetosutilizados. maiores que 99% na
biotransformação do
hidróxi-seleneto (R,S)-1a utilizando cepas de Aspergillus terreus URM 3571. Os
hidróxi-selenetos (R,S)-1b-c também foram obtidos com bons excessos
enantioméricos (e.e. > 95%). Um resultado interessante foi a biotransformação
do composto (R,S)-1e, pois este não havia sido biotransformado em reações
empregando enzimas isoladas (seção 2.1.2.1 e 2.1.2.2).
Entretanto, analisando com mais cuidado os resultados das
biotransformações empregando fungos, foi observada a presença de um novo
composto: o seleneto de fenil-metila (3) (Tabela 2.1.2.3.1 e Esquema
2.1.2.3.3).
Realizando a biotransformação do hidróxi-seleneto (R,S)-1a em escala
maior, foi obtido o hidróxi-seleneto quiral em 50% de rendimento (e.e.>99%) e
o seleneto de fenil-metila (3) em 40% de rendimento (Tabela 2.1.2.3.1
entrada 3). Ao empregarmos uma quantidade maior de células (ou um maior
tempo de reação) a quantidade de seleneto de fenil-metila aumentou (Tabela
2.1.2.3.1 ).
A terreus
Com base nos resultados observados, verificamos que a
biotransformação de hidróxi-selenetos empregando fungos não consistia em
uma reação de desracemização, como imaginado inicialmente, e sim em uma
oxidação enantiosseletiva, seguida
de uma biometilação, gerando o
seleneto de fenil-metila.
Esquema 2.1.2.3.3. Hidráxi-selenetos utilizados.
3
oIIcrse'OH
I~5
Para avaliar a capacidade
das cepas de Aspergillus terreus em
biometilar organoselenetos,
realizamos a biotransformação de
outros organoselenetos: difenil disseleneto (4) e o ácido selenínico (5)
(Esquema 2.1.2.3.3). Novamente, em ambas as reações, o seleneto de fenil-
50

2.1 Hidróxi-Selenetos
metila (3) foi observado como produto principal da biotransformação,
comprovando a capacidade das cepas de Aspergillus terreus em biometilar
organosselenetos.,.'
Tabela 2.1.2.3.1. Biotransformações dos hidróxi-selenetos com fungoso•
,.
Tempo Massa de A. terreus URM 3571 A. terreus CCT 3320 RendimentoEntrada Substrato 1 dias células 1 g
e.e.1/% 3/% e.e.11 % 3/% 1/% 3/%
(R,s)-la 5 >99 2 31 2
2 5 3 O 100 98 51
" (R,s)-la# 6 5 >99 50 40-'
4 (R,s)-lb 14 30 16 98 18
5 14 3 95 13 O 100
6 (R,s)-lc 5 1 1 10 12 20
7 14 5 1 18 95 14
8 (R,s)-le 7 2 24 20
9 142 O 100
a As reações foram realizadas utilizando 20IJL do substrato em tampão (pH =7,2) à 25°C.
# Foi utilizado 1OOIJL do substrato.
Uma peculiaridade da química de compostos orgânicos de selênio é a
fácil oxidação de selenetos a selenóxidos (Seção 1.3). Estes, devido à baixa
estabilidade sofrem reação de eliminação, gerando alcenos
espontaneamente.38 Assim, o mecanismo deste processo de biometilação
provavelmente envolve a oxidação do seleneto a selenóxido, seguida de uma
eliminação. 53 A biometilação pode ocorrer no produto de eliminação (ácido
selenenico) ou ocorrer uma nova oxidação (ácido selenínico) e depois a
""- A. te"eus. ~bi()-metilaçãO
(R)-1a
+
(5)-1 a
r
0--- HII~OH
rArse Se-OHA[;rreus. g U - ©r +
selenóxldo acldo selenenlco
H20 eloulA. terreus
O
11 .. ©rse,()Yse-OH____ '~ Aterreus
-
-
Rendimento = 50%e.e. >99%
acldo setenínlco fenllofl1etll seleneto(Rendimento = 40%)
Esquema 2.1. 2. 3. 4. Proposta para Q resolução do hidroxi-seleneto com Aspergillus terreus.
51

2. Resultados e Discussão
biometilação (Esquema 2.1.3.3.4). No caso dos ~-hidróxi-selenetos, ocorre uma
oxidação enantiosseletiva, sendo que um dos enantiômeros é oxidado
preferencialmente e em seguida é biometilado, gerando o seleneto de fenil
metila, enquanto que o outro enantiômero é preservado (Esquema 2.1.2.3.4).
Este é o primeiro relato de biometilação envolvendo organo-selenetos
e células inteiras de fungos.
2.1.3. Aplicação Sintética
Apesar do trabalho desenvolvido ser de cunho metodológico,
consideramos relevante sua aplicação sintética. Nesse sentido e em conjunto
com os objetivos iniciais do trabalho (aliar a química de selênio com a
biocatálise), escolhemos a reação de eliminação de selenóxidos na preparação
de álcoois alílicos para exemplificar o potencial de nossa metodologia.
Para isso foi utilizado o hidróxi-seleneto 1h obtido pela resolução da
sua mistura racêmica empregando CALB em hexano seco (Seção 2.1.2.1
Esquema 2.1.2.1.3).
A reação de eliminação de selenóxidos em hidróxi-selenetos é estéreo
seletiva, levando a formação apenas da olefina de configuração "E".38 Essa
reação também não interfere no centro estereogênico, mantendo-se a
configuração original.
52
Çl-i
~
(5)-6 e.e.=92%
Esquema 2.1.3. 1. Preparação do álcool alílico quiral poreliminação oxidativa.
Assim, a
preparação do álcool
alílico a partir do hidróxi
seleneto 1h utilizando esta
reação gerou apenas o
álcool alílico de
configuração E, (5)-6, com
total integridade do centro
estereogênico (Esquema
2.1.3.1 ).

2.1 Hidróxi-Selenetos
~. ,2.1.4. Determinação da Pureza Optica/Estereoquímica
Em estudos relacionados a enantiômeros, a espectroscopia aliada a
outras técnicas análiticas são fundamentais para a determinação da
especificidade dos procedimentos utilizados e de suas eficiências. Assim, as
técnicas analíticas para determinação de excessos enantioméricos e
determinação de configuração absoluta foram essenciais para a condução do
trabalho realizado.
2.1.4.1. Determinação de Excesso Enantiomérico
Figu-a 2.1.4.1. Exemplo de comparação de cromatogramasobtidos em CG-Quiral.
(Rf-2.
+
.-\cO. ,/"--~J'I SePh
R
(S)-la
(R,.S)-l
Ac'Ü"'f'sePhR
e.e.>~"Ó
Os excessos enantioméricos (e.e.) dos álcoois 1a-i e dos ésteres 2a-i
foram calculados a partir de cromatogramas obtidos em cromatografo a gás
equipado com coluna com fase estacionária quiral (CG-Quiral). As análises
foram conduzidas comparando os cromatogramas obtidos com cromatogramas
de amostras racêmicas (Figura 2.1.4.1 ).
Para os compostos
19-i e 2c-i, análises diretas
em CG-Quiral não foram
possíveis, pois não foi obtida
a perfeita discriminação dos
enantiômeros destes com-
postos via CG-Quiral. Nesse
sentido, o composto 19 foi
transformado no hidróxi-
selento 1a através da reação de deselenização redutiva, 54 a qual não interfere
no centro assimétrico, permitindo desta forma a determinação indireta do
excesso enantiomérico do composto 19 (Esquema 2.1.4.1.1).
O hidróxi-seleneto 1h foi submetido à reação de eliminação de
selenóxido gerando o álcool alílico 6, possibilitando assim a determinação do
excesso enaniomérico relativo ao carbono carbinólico via CG-Quiral, pois tal
reação não modifica a configuração do carbono carbinólico (Seção 2.1.3
Esquema 2.1.4.1.1). 38
53

2. Resultados e Discussão
De forma similar, o hidróxi-seleneto 1i foi transformado no álcool
correspondente 7, pela deselenização redutiva com n-Bu3SnH (Esquema
2.1.4.1.1). 54 Os enantiômeros do composto 7 foram facilmente discriminados
utilizando CG-Quiral.
Os ésteres 2c-2i foram hidrolisados, gerando seus respectivos hidroxi
selenetos e em seguida submetidos aos procedimentos realizados para
determinar os excessos enantioméricos dos respectivos álcoois (Esquema
2.1.4.1.1 ).
Q-l
~0Sl!Ph 19
1h
1eq. ~Srj-j.CCN, ToIuero
Ret.531a
ll-F6
Ret.43
c, R = Ph, R = H, R" =Q-I(~d. R = Ph, R = H. R" = (0iVs0'3e. R = Ph, R = H. R" = Ph
Q-l
-"C: 1HJl.bsnH Q-l. ~A1CCN, ToIuero
Rpf !'l::l 7
Ql\c Q-l
~se0~K:zCOa R/se0~.
HP/MeCHR R2c.f Ret.49 1c.f
f, R=a-\s, R=H, R"=Ph9, R = Ph, R = sePh, R" =~h. R=Ph, R=(~.R"=~
i. R= Ph, R= Ph, R"=~
Esquemo 2.1.4.1.1. Transformações para a determinação de excesso enantiomérico.
2.1.4.2. Determinação de Configuração Absoluta
A configuração absoluta do hidróxi-seleneto 1a foi determinada por
espectroscopia de ressonância magnética nuclear (RMN). O enantiômero do
composto 1a (e.e. > 99%) não acetilado pela lipase foi derivatizado
separadamente com os dois enantiômeros do cloreto de a-metoxi-a
trifluormetil-fenilacetila (MTPA-CI).
O espectro de 1H-RMN dos derivados diastereoisômericos formados
apresentam deslocamentos químicos diferentes. De acordo com o modelo de
Mosher, 55 as diferenças de deslocamento químico nos espectros de 1H-RMN
dos substituintes ligado ao carbono carbinólico dos diastereoisômeros são
originadas pelo efeito anisotrópico do anel aromático em uma conformação
preferencial. Este efeito permite determinar a posição dos substituintes ligados
ao carbono carbinólico, pois o cone de proteção do anel benzênico (resultado
54

2.1 Hidróxi-Selen.etos
do campo magnético gerado pelo movimento dos elétrons da nuvem TI do anel
benzênico) blinda diferentes ligantes em cada diastereoisâmero.
Assim, imaginado que o hidróxi-seleneto 1a não acetilado pela lipase
tenha a configuração (S), a derivatização gerou os estereoisâmeros de
configuração (R,5)-8 e (5,5)-8. No estereoisâmero (R,5)-8 o cone de proteção
irá blindar o substituinte -CH2SePh, enquanto que no estereoisâmero (5,5)-8
o cone de proteção irá blindar o substituinte -CH3 (Figura 2.1.4.2.1). Como
resultado, os deslocamentos químicos no espectro de 1H-RMN do substituinte
-CH2SePh no estereoisâmero (R,5)-8 deverá cair em campo mais alto que no
espectro do estereoisâmero (5,5)-8. Por outro lado, o deslocamento químico
no espectro de 1H-RMN do substituinte -CH3 no estereoisâmero (R,5)-8 deverá
cair em campo mais baixo que no estereoisâmero (5,5)-8.
o (S) MeVista Me ~ ~CH2SePh- Ph-::T-(· 'H
f ,C (R) O
Ph (S) MeVista ~~CH2sePh- MeD_i li H
f,C (S) O
(RS)-8
llH,ü>pm) 3.U
IlHb(ppm) 2.93
IlH...,(ppm) = 1.44
1ls.(ppm) = 269.99
Ilc.s,(ppm) = 32.20
IlC•M,(ppm) = 19.70
(SS)-8
3.17
3.02
1.35
271.12
32.40
19.60
FiQ\ro 2.1.4.2.1. Modelo de Mosher oara o composto la.
Analizando O espectro de 1H-RMN dos estereoisâmeros obtidos,
podemos observar exatamente o padrão de deslocamento químico descrito
anteriormente, (Figura 2.1.4.2.1), confirmando a configuração absoluta do
nosso hidroxi-seleneto 1a não acetilado pela lipase como sendo (S).
Os espectros de 13C-RMN e 77Se-RMN apresentaram a mesma
tendência em seus deslocamentos químicos, confirmando o efeito anisotrópico
55

2. Resultados e Discussão
A configuração
absoluta do composto 1dsfoi
determinada diretamente
pela comparação da sua
rotação óptica com a
publicada na literatura para
9
CCN, ToIuene
b,R1 =RI, R2=H,R3=(~c, R1 = RI, R2=H, R3=a-(~f, R1 = 0-0, R2 = H, R3 = RI
ESQUema 2.1.4.2.2. Reacõescom os compostos lb. ld e lf.
do anel benzênico sobre os substituintes ligados ao carbono carbinólico e a
configuração absoluta determinada (Figura 2.1.4.2.1 ).
A configuração absoluta dos hidróxi-selenetos 1b, 1d e 1f foi
determinada de maneira indireta, comparando-se as rotações ópticas {[ajo} dos
seus derivados sem o grupo selanila, com os valores da literatura para os
mesmos compostos. 56, 57 Para isso, os compostos 1b, 1d e 1f foram
derivatizados via reação de deselenização redutiva usando n-Bu3SnH
(Esquema 2.1.4.2.2).
o enantiômero-(R). 58
Para a determinação da configuração absoluta do composto 19 foi
necessário transformá-lo no composto 1a, via reação de deselenização
redutiva (Esquema 2.1.4.1.1). Assim sua configuração absoluta foi determinada
pela comparação das rotações ópticas dos compostos.
A determinação da configuração absoluta dos dois centros
estereogênicos dos compostos 1h e 1i não foi realizada, sendo determinada
apenas a configuração do centro estereogênico localizado no carbono
carbinólico. Assim os compostos 1h e 1i foram transformados em seus
respectivos álcoois sem o grupo selanila, conforme realizado na determinação
dos excessos enantioméricos (Seção 2.1.4.1 - Esquema 2.1.4.1), e a rotação
óptica dos mesmos foram comparadas com os valores descritos na literatura.58, 59
56

2.2. Hidróxi-Teluretos
2.2. Hidróxi-Teluretos


2.2 Hidróxi-Teluretos
2.2.1. Síntese Racêmica
Partindo dos resultados obtidos com as biotransformações dos hidróxi
selenetos (Seção 2.1.2), foram sintetizados alguns hidróxi-teluretos com
estruturas particulamente selecionadas para o estudo das biotransformações
destes compostos (Figura 2.2.1.1).
OH
~TePh
(R.5)-lO:t
OH
~TePh
(R,s)-lOb
OH
I TePhPh~
(R,S)-lOc (R,5)-lOd
OH OH
~Te~~Te~OH Te~
N(R,5)-lOe (R's)-lor (R,5)-lOg
Figura 2.2.1.1. Hidróxi-teluretos sintetizados.
De maneira análoga à reação do disseleneto de difenila com NaBH4
(Seção 2.1.1), a reação de ditelureto de difenila com NaBH4 gera o
feniltelurolato in situ, 60 que reage prontamente com óxido de propileno
produzindo o composto (R,S)-10a (Esquema 2.2.1.1). 54 Diferentemente da
reação do fenilselenolato com óxido de estireno, que gera dois régio-isômeros
(Seção 2.1.1.), a reação do feniltelurolato com óxido de estireno produz apenas
um régio-isômero: o hidróxi-telureto (R,S)-10c (Esquema 2.2.1.1). 54
o EtOHo\l OH
[RITee] +~R2 THF .. R2~TeRl
(R,5')-lO
Ref.60
Teo RlLiTHF
Ret. &1U)-.t RI = CH3" R2
= Ph (Rendm~o)
lOe R1 =PheR2 = Ph (Rend.=7l%)
lOd RI = Ph e R2 = CH3 (Rend. = 60%)
lOe RI = n-Bu e R2 = CH3 (Rend. = 80%)
Os hidróxi-teluretos (R,S)-10d e (R,S)-10e foram preparados pela
reação do alquil telurolato
de lítio apropriado, obtido ;'TePh NaBH~PhT" EtOH
via reação de alquil lítio
com telúrio elementar em
THF seco, 61 com óxido de
estireno e óxido de
Esquema 2.2.1.1. Preparação dos compostos 10a, lOc, lOd e lOe.propileno respectivamente
(Esquema 2.2.1.1). 54
O Telureto (R,S)-10b foi preparado pela reação de troca telúrio/lítio do
bis(fenilteluro)metano com n-Buli, gerando o fenilteluro-metil lítio in situ, o qual
59

2. Resultados e Discussão
se adiciona à carbonila do butanal (Esquema 2.2.1.2).62 O
bis(fenilteluro)metano foi preparado a partir da adição de diazometano (CH2N2)
sobre solução de ditelureto de difenila em éter etílico (Esquema 2.2.1.2). 63
O hidróxi-
(R,S)-10ftelureto
foi preparado
através da reação
de adição 1,4 do n
Butil-telurol à metil
OH
~TePh(R,s)-10b
Ret.62 (I !~H (Rend.=61%)
Ret.62
Esquema 2.2.1.2. Síntese do composto lObovinil cetona,
seguida de redução
da carbonila com NaBH4 . 64 Alternativamente, o telureto (R,S)-10 foi obtido em
70% de rendimento isolado através de reações seqüênciais one-pot (adição 1,4
seguida de redução - Esquema 2.2.1.3). 65
<)
1)/I-BuLi ~2)HoO I. J OTeO - .. ~-BllTeH .. 1ITHF THF ~TeBu
(Rend. = 70%)
Esquema 2.2.1.3. Obtenção do hidróxi-telureto (R.S)-lOf.
NaBH~ ..HO
~TeBu(RS}lOf
Ret. 63 e 64
Ret.66
Esquema 2.2. 1.4. Hidroteluracao de álcoois propargílicos ea1quinonas.
~"TeBu
o TeBu
~R1EtOH
n-BuTeH
_I n-BuTeH IH rBU
R"" ~ EtOH" ~R1 +
R1 Ref.65
A hidroteluração direta de álcoois propargílicos produz uma mistura de
dois teluretos vinílicos (Esquema 2.2.1.4), 66 os quais possuem uma polaridade
muito próxima, o que dificulta sua separação e conseqüentemente prejudica os
rendimentos, inviabilizando a obtenção de álcoois alílicos y-butiltelurados, como
o composto (R,S)-10g. Por outro lado, a hidroteluração de alquinonas produz
apenas um regioisômero
(Esquema 2.2.1.4). 67 Assim,
o telureto vinílico (R,S)-10g
foi sintetizado por redução
da (Z)-~-butilteluro-enona
(12) com NaBH4 em 93% de
rendimento (Esquema
2.2.1.5).
60

2.2 Hidróxi-Teluretos
Esquema 2.2.1.5. Redução da (Z)-~-butilteluro
enona (12) com NABH4 •
o TeBu Na8H4 OH TeBu
)V MeOHo N. (R,s)-10g
(Rend. = 93%)12
Apesar de encontrarmos relatos na literatura. de que a redução de
compostos carbonílicos a, ~-insaturados utilizando NaBH4 leva a uma mistura
do álcool alílico e do álcool
saturado,68 foi observado que (Z)-~
butilteluro-enonas são reduzidas
seletivamente e em bons rendimentos
aos álcoois alílicos correspondentes,
utilizando NaBH4 ou DIBAL-H como
agente redutor, provavelmente devido à presença do grupo n-butilteluro na
posição ~ das enonas. 69
o emprego de um procedimento seqüencial em larga escala (0,1 moi),
utilizando um organozinco como intermediário na preparação da alquinona,
possibilitou a obtenção da (Z)-~-butilteluro-enona (12) em bom rendimento
(Esquema 2.2.1.6).
H---'---H a, b~ GI---'=--Z"CJ C.[/l] d •~"B"
(a) n-BuLi I THF. ooc(b)ZnCI2
o
(c) .)lei(d) [BuTeLij I H20
(RemI. = 62%)
12
Esquema 2.2.1.6. Síntese da (Z)-~-butilteluro-enona(12).
Os respectivos acetatos racêmicos dos hidróxi-teluretos 10a-g foram
sintetizados quimicamente utilizando anidrido acético em piridina seca, 49
utilizando procedimento análogo ao empregado com os hidróxi-selenetos
(Seção 2.1.1). Novamente, essas reações não apresentaram maiores
problemas, sendo os produtos facilmente purificados e caracterizados
(Esquema 2.2.1.7).
61

2. Resultados e Discussão
o oOH )l. 11 '\cC!
~TeRl 0/"0...... '~T R'2 .,.., eR R-
n pindina n
(R,S)-IO Ret.49 (R'sl-ll
.'\c<)
~TePh
(R,S)-lla (R,S)-lle (R,S)-llf
OH Te~
N pindma
AcO Te~
N(R'sl-llg (R,S}-llg
Figura 2.2.1.7. Acetato dos hidl'Óxi-teluretos sintetizados.
2.2.2. Biotransformações
2.2.2.1. Resolução em Meío Orgâníco
o)J
(Rt-11a
?""A1Te~ + "'"
-17 -o;
II
o;
A1T~+o;
A1Te~
Esquema 2.2.2.1.1. Resolução enzimática do (R,S)-10a.
A avaliação da enantiosseletividade enzimática nas transesterificações
.dos hidroxi-teluretos em meio orgânico foi realizada inicialmente utilizando o
composto (R,5)-10a como substrato e acetato de vinila como doador do grupo
acila (Esquema 2.2.2.1.1 ). Quatro lipases foram escolhidas como
biocatalisadores: a PPL (Iipase de pâncreas de porco - livre), a PSL (AMANO
PS lipase de
Pseudomonas sp.
livre), CRL (Iipase de
Candída rugosa - livre)
e a CALB (NOVOZYM
43S® lipase de
Candída antarlíca - Fração B - imobilizada).
As maiores enantiosseletividades foram observadas com a PSL e a
CALB (Tabela 2.2.2.1.1). Na reação com PSL foi obtido o hidráxi-telureto (5)
10a com excesso enantiomérico igual a 94% (Tabela 2.2.2.1.1 - entrada 4) e o
produto (RF11a com excesso enantiomérico igual a 98% (Tabela 2.2.2.1.1
entrada 3). A melhor enantiosseletividade observada foi na resolução
62

2.2 Hidráxi-Teluretos
empregando a CALS como biocatalisador, onde tanto o produto [(R)-11a] como
o substrato [(S)-10a], foram obtidos com excessos enantioméricos superiores a
98% (Tabela 2.2.2.1.1 - entrada 7).
Tabela 2.2.2.1.2. Resolução enzimáticaQ do composto (R,S)-10a usando diferentes lipases.
Entrada LipaseTempo! Conversão e.e. do (S)- e.e. do (R)- Config. E'
horas !% lOa!%b lla! % c Absoluta d
1 PPL 6 32 44 93
2 24 41 63 91 (R) 40
3 PSl 6 36 56 98
4 24 49 94 96 (R) 174
5 CRL 1 21 13 48
6 3 46 33 41 (S) 3,3
7 CALB 2 50 >99 98 (R) >200
a As reações foram realizadas com 0.5 mmol do (R,S)-10a e 19 da PPL, 300 mg da PSL, 75 mg da CRL ou 30 mg daCALS em hexano seco (10mL). b Excesso enantiomérico do álcool 10a. c Excesso enantiomérico do acetato 11a.d Configuração absoluta do acetato 11a.· Razão enantiomérica: este parãmetro descreve a enantiosseletividade daenzima.
A enantiopreferência para o enantiômero-(R) foi observada nas
resoluções utilizando a PPL, PSL e CALS. Entretanto, a CRL apresentou
enantiopreferência para o enantiômero-(S) na resolução do hidróxi-telureto
(R,S)-10a (Tabela 2.2.2.1.1).
A partir dos resultados observados, a influência do solvente na
resolução do hidróxi-telureto (R,S)-10a foi estudada empregando a CALS (a
maior enantiosseletidade observada em hexano - Tabela 2.2.2.1.2) e a CRL
(enantiopreferência inversa das demais lipases em hexano - Tabela 2.2.2.1.2).
Os experimentos foram realizados a 30°C, usando alguns solventes secos
comumente empregados em estudos de resolução cinética. As conversões
foram determinadas após 3 horas para os experimentos realizados com a CRL
e 1 hora para os experimentos realizados com a CALS.
Em comparação a variação do solvente na resolução dos hidróxi
selenetos em meio orgânico (Seção 2.1.2.1), a variação do solvente na
resolução dos hidróxi-teluretos apresentou uma grande influência sobre a
conversão do substrato (Figura 2.2.2.1.1), em outras palavras, sobre a
atividade enzimática considerando que as demais variáveis (como tempo de
63
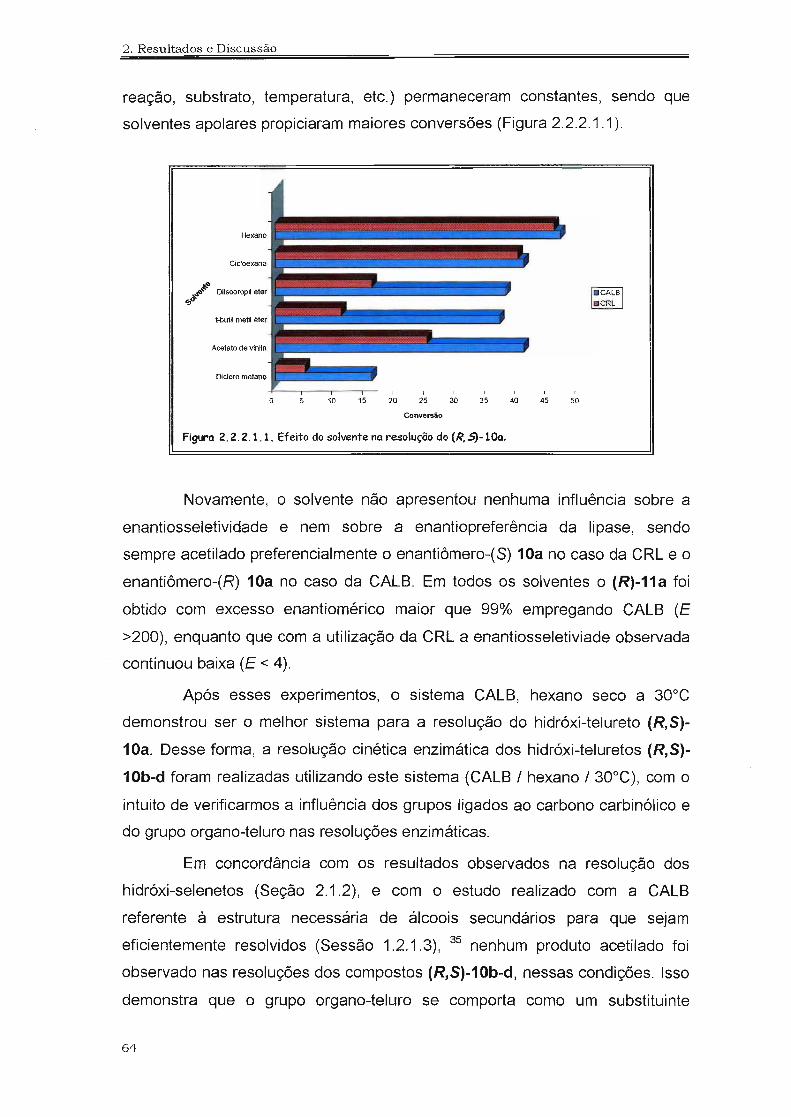
2. Resultados e Discussão
reação, substrato, temperatura, etc.) permaneceram constantes, sendo que
solventes apoiares propiciaram maiores conversões (Figura 2.2.2.1.1).
Hexano
Cicloexano
-s!'# Diisopropil éter
<á'
t-bulil metil éter
Acetato de vinHa
Oielora metano
BCALBIICRL
o 5 10 15 20 25 30 35 40 45 50
Conversão
FigtrQ 2.2.2. 1. 1. Efeito do solvente na resolução do (R. 5)-100.
Novamente, o solvente não apresentou nenhuma influência sobre a
enantiosseletividade e nem sobre a enantiopreferência da lipase, sendo
sempre acetilado preferencialmente o enantiômero-(S) 10a no caso da CRL e o
enantiômero-(R) 10a no caso da CALB. Em todos os solventes o (R)-11a foi
obtido com excesso enantiomérico maior que 99% empregando CALB (E
>200), enquanto que com a utilização da CRL a enantiosseletiviade observada
continuou baixa (E < 4).
Após esses experimentos, o sistema CALB, hexano seco a 30°C
demonstrou ser o melhor sistema para a resolução do hidróxi-telureto (R,S)
10a. Desse forma, a resolução cinética enzimática dos hidróxi-teluretos (R,S)
10b-d foram realizadas utilizando este sistema (CALB I hexano 130°C), com o
intuito de verificarmos a influência dos grupos ligados ao carbono carbinólico e
do grupo organo-teluro nas resoluções enzimáticas.
Em concordância com os resultados observados na resolução dos
hidróxi-selenetos (Seção 2.1.2), e com o estudo realizado com a CALB
referente à estrutura necessária de álcoois secundários para que sejam
eficientemente resolvidos (Sessão 1.2.1.3), 35 nenhum produto acetilado foi
observado nas resoluções dos compostos (R,S)-10b-d, nessas condições. Isso
demonstra que o grupo organo-teluro se comporta como um substituinte
64

2.2 Hidróxi-Teluretos
grande na resolução dos hidróxi-teluretos. Assim, para que um hidróxi-telureto
seja eficientemente resolvido pela CALS em meio orgânico é necessário que o
substituinte de tamanho médio seja menor que o grupo etila.
Assim, as resoluções enzimáticas dos hidróxi-telureto (R,S)-10e-g
foram conduzidas empregando a CALS em hexano seco a 30°C (Esquema
2.2.2.1.2). Nessas condições esses compostos foram resolvidos, sendo
observada a acetilação preferencial do enantiômero-(R) dos compostos (R,S)
10e-f, em concordância com os resultados observados e com a regra de
Kazlauskas. 31
Oi Oi Q!lc
~Te~ ~QAc ,C'\LB ~Te~+~Te~+~Oin solvente n n
(RS)-1Oe-f (S)-1Oe-f (~-11e-f
e, n=1f, n=2
o~ ,.ÂH
~Te ai
~
(RS)-10g
~Qllc CALB ~Te ai ~Te QOc ~---'-__ ~ + ~ + -:?' ai
heKaro
(S)-10g (~11g
o,.ÂH
Esquema 2.2.2.1.2. Resolução enzimática de hidróxi-teluretos (R,S)-8e-g em meio orgânico
empregando CALB.
Na resolução do composto (R,S)-10e, o produto (R)-11e foi obtido com
excesso enantiomérico maior que 99% e o substrato (S)-1 Oe com excesso
enantiomérico igual a 97%, demonstrando a alta enantiosseletividade
enzimática (E> 200) da CALS para este composto (Tabela 2.2.2.1.3).
No caso da resolução do composto (R,S)-10f, também foi observada
uma alta enantiosseletivídade enzimática (E = 120) da CALS, entretanto o
rendimento isolado do produto enriquecido enantiomericamente foi menor que
5%. Esse baixo rendimento está relacionado com a baixa estabilidade de
teluretos alquílicos em hexano, evienciado pela formação de um pó branco
(provavelmente teluróxido) no decorrer das reações (utilizando hexano
deaerado). Por esse motivo, a resolução do hidróxi-telureto (R,S)-1Of foi
efetuada em THF, pois observamos que teluretos orgânicos apresentam maior
estabilidade neste solvente. Entretanto, a enantiosseletividade enzimática em
solventes polares é prejudicada, conforme resultados observados
65
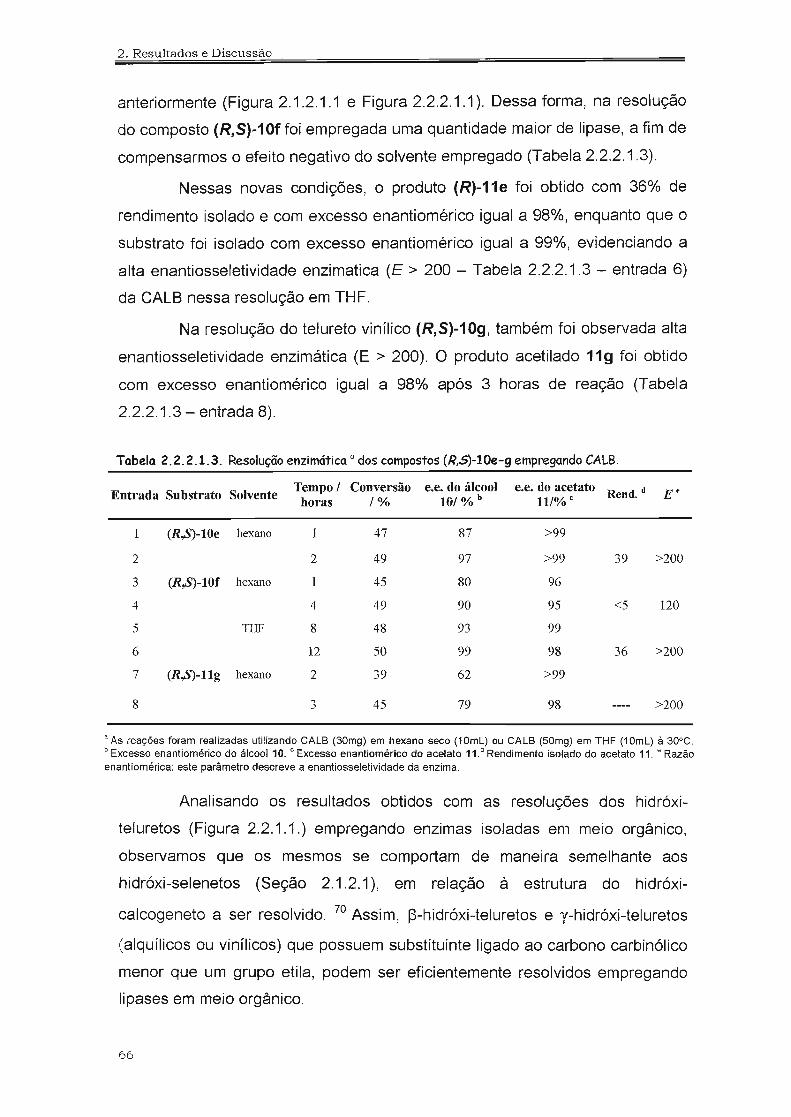
2. Resultados e Discussão
antériormente (Figura 2.1.2.1.1 e Figura 2.2.2.1.1). Dessa forma, na resolução
do composto (R,S)-1 Of foi empregada uma quantidade maior de lipase, a fim de
compensarmos o efeito negativo do solvente empregado (Tabela 2.2.2.1.3).
Nessas novas condições, o produto (R)-11e foi obtido com 36% de
rendimento isolado e com excesso enantiomérico igual a 98%, enquanto que o
substrato foi isolado com excesso enantiomérico igual a 99%, evidenciando a
alta enantiosseletividade enzimatica (E > 200 - Tabela 2.2.2.1.3 - entrada 6)
da CALB nessa resolução em THF.
Na resolução do telureto vinílico (R,S)-10g, também foi observada alta
enantiosseletividade enzimática (E > 200). O produto acetilado 11g foi obtido
com excesso enantiomérico igual a 98% após 3 horas de reação (Tabela
2.2.2.1.3 - entrada 8).
Tabela 2.2.2.1.3. Resolução enzimática a dos compostos (R,S)-10e-g empregando CALB.
Entrada Substrato SolventeTempo / Conversão e.e. do álcool
horas / % 10/ % b
e.e. do acetato Rend. d
11/% cE e
1 (R,s)-lOe hexano 1 47 87 >99
2 2 49 97 >99..,
(R,s)-lOf hexano 1 45 80 96.)
4 4 49 90 95
5 TIIF 8 48 93 99
6 12 50 99 98
7 (R,s)-l1g hexano 2 39 62 >99
8 3 45 79 98
39 >200
<5 120
36 >200
>200
a As reações foram realizadas utilizando CALB (30mg) em hexano seco (10mL) ou CALB (SOmg) em THF (10mL) à 30°C.b Excesso enantiomérico do álcool 10. c Excesso enantiomérico do acetato 11 d Rendimento isolado do acetato 11. e Razãoenantiomérica: este parãmetro descreve a enantiosseletividade da enzima.
Analisando os resultados obtidos com as resoluções dos hidróxi
teluretos (Figura 2.2.1.1.) empregando enzimas isoladas em meio orgânico,
observamos que os mesmos se comportam de maneira semelhante aos
hidróxi-selenetos (Seção 2.1.2.1), em relação à estrutura do hidróxi-
calcogeneto a ser resolvido. 70 Assim, f3-hidróxi-teluretos e y-hidróxi-teluretos
(alquílicos ou vinílicos) que possuem substituinte ligado ao carbono carbinólico
menor que um grupo etila, podem ser eficientemente resolvidos empregando
lipases em meio orgânico.
66

2.2 Hidróxi-Teluretos
2.2.3. Aplicação Sintética
A fim de demonstrarmos o potencial sintético da nossa metologia de
obtenção de hidráxi-teluretos enantiomericamente puros, dois teluretos quirais
foram empregados na preparação de lactonas. O produto acetilado (R)-11f
(e.e.>99%) obtido na resolução do composto (R,5)-10f empregando CALS em
THF seco (Seção 2.2.2), foi utilizado na preparação da (R)-y-valerolactona,
enquanto que o hidráxi-telureto (5)-10g (e.e. = 98%) não acetilado na
resolução do composto (R,5)-10g empregando CALS em hexano seco, foi
empregado na preparação da (S)-~-angelicalactona (Esquema 2.2.3.1 ).
Inicialmente, foi realizada a hidrálise química do produto acetilado (R)
11f, gerando o hidráxi-telureto (R)-10f. Na seqüência, a adição de dois
equivalentes de n-SuLi a -78DC, levou a formação do di-ânion, o qual foi
capturado com CO2 formando o (R)-hidráxi-ácido 13. A adição de ácido (HCI -
50%) levou a ciclização, resultando na (R)-y-valerolactona [(R)-14] com
excesso enantiomerico igual a 98%. De forma análoga, a (S)-angelicalactona
[(5)-15] foi obtida com excesso enantiomérico igual a 98% (Esquema 2.2.3.1),
demonstrando uma aplicação para os hidráxi-teluretos quirais obtidos
empregando biotransformações.
o
)lÇ> l<:zOO.l <?"I- .. =~Ten-a. IIIleCH-H,O ~Ten-a.
(~-11f (~1Of
ÇlH Ten-BJ n-8Ui [ Qj U] [ o Qj]~ ~I ~ .I co.z ~·o H+-
~-~-~ -1HF ~
(S}-1Og.7f!JC
Esquema 2.2.3.1. Síntese do (Rh-valerolactona e da (s)-Il-angelicalactona.
o
M e.e. = 98"k'\I\I\V(R)-15
67

2. Resultados e Discussão
, .. : .. ~....
2.2.4. Determinação da Pureza Óptica/Estereoquímica
Como salientado anteriormente no trabalho com hidróxi-selenetos, as
técnicas analíticas para determinação de excessos enantioméricos e
determinação de configuração absoluta foram essenciais para a condução do
trabalho realizado com os hídróxi-teluretos.
2.2.4.1. Determinação de Excesso Enantiomérico
Os excessos enantioméricos dos hidróxi-teluretos 10a, 10e-g e dos
seus respectivos ésteres 11 a, 11e-g foram calculados a partir de
cromatogramas obtidos em cromatografo a gás equipado com coluna com fase
estacionária quiral (CG-Quiral). Conforme realizado no estudo das
biotransformações dos hidróxi-selenetos, os cromatogramas obtidos foram
sempre comparados com cromatogramas de amostras racêmicas (Seção
2.1.4.1 ).
discriminados via CG-quiral,
possibilitando a
determinação indireta dos
excessos enantioméricos do
hidróxi-telureto 10f.
o o o
F~O~F oJl t FF F (""'~~F
n-F Te16
o oQ-i ..Jl.oÁ.. O"c
~TeA -Pl-.ridi-na-· ~TeA10e Ref.49 11e
Q-i
~Te~10f
Esquema 2.2.4.1. Derivatizações para determinação dos e.e.
Para os compostos 10e e 10f análises diretas em CG-quiral não foram
possíveis, pois não houve discriminação dos enantiômeros em nenhuma
coluna disponível no laboratório. Assim, o hidróxi-telureto 10e foi transformado
quimicamente em seu respectivo acetato (11e) e depois submetido à análise
em CG-Quiral. O hidróxi-telureto 10f foi convertido no seu respectivo trifluor
acetato 16, utilizando método convencional (Esquema 2.1.4.1). 49 Os
enantiômeros do composto
16 foram facilmente
68

INSTITUTO DE OUIMIQAOntversidade de São F'aulo
2.2.4.2. Determinação da Configuração Absoluta
2.2 Hidróxí-Teluretos
De maneira análoga à utilizada na determinação da configuração
absoluta do hidróxi-seleneto 1a (Seção 2.1.4.2.), as configurações absolutas
dos hidróxi-teluretos 10a e 10e foram determinadas por espectroscopia de
ressonância magnética nuclear (RMN). Os enantiômeros dos compostos 8a
(e.e. > 99%) e 8e (e.e. > 99%) não acetilados pela lipase foram derivatizados
com os dois enantiômeros do cloreto de a-metóxi-a-trifluormetil-fenilacetila
(MTPA-CI).
Os espectros de 1H-RMN dos derivados diastereoisoméricos formados
apresentam deslocamentos químicos diferentes e, de acordo com o modelo de
Mosher, 55 as diferenças de deslocamento químico são originadas pelo efeito
anisotrópico do anel aromático em uma conformação preferencial. Este efeito
permite determinar a posição dos substituintes ligados ao carbono carbinólico,
pois o cone de proteção do anel benzênico (resultado do campo magnético
gerado pelo movimento dos elétrons da nuvem TT do anel benzênico) blinda
diferentes ligantes em cada diastereoisômero.
Assim, imaginado que os hidróxi-teluretos 10a e 10e não acetilados
pela lipase tenham a configuração (S), a derivatização gerou os
estereoisômeros (R,5)-17, (R,5)-18, (5,5)-17 e (5,5)-18. No caso dos
estereoisômeros (5,5)-17 e (5,S)-18 o substituinte -CH3 ligado ao carbono
carbinólico será blindado pelo cone de proteção, enquanto que nos
estereoisômeros (R,S)-17 e (R,S)-18 o cone de proteção irá blindar o
substituinte -CH2TeR (Figura 2.2.4.2.1). Assim, os deslocamentos químicos
no espectro de 1H-RMN do substituinte -CH3 ligado ao carbono carbinólico dos
estereisômeros (R,S)-17 e (R,S)-18 aparecerá em campo mais baixo quando
comparado os deslocamentos químicos do mesmo substituinte nos
estereoisômeros (5,5)-17 e (5,5)-18. O efeito inverso deverá ser observado
para o substituinte -CH2TeR (Figura 2.2.4.2.1).
Analizando o espectro de 1H-RMN dos estereoisômeros, podemos
observar exatamente o padrão de deslocamento químico descrito
anteriormente (Figura 2.2.4.2.1), confirmando a configuração absoluta dos
hidróxi-teluretos 8a e 8e não acetilados pela lipase como sendo (S).
69

2. Resultados e Discussão
Como observado na determinação da configuração absoluta do
hidróxi-seleneto 1a, os espectros de 13C-RMN e 125Te-RMN para os
éstéreoisômeros apresentaram o mesmo efeito em seus deslocamentos
químicos, confirmando o efeito anisotrópico do anel benzênico sobre os
substituintes ligados ao carbono carbinólico e a configuração absoluta
determinada (Figura 2.2.4.2.1).
°(S) Me
Me , a--.-.::.CH TeR~ Ph'j-(· 'H 2
f,C (R) °Ph (S),.Me
Vista .:............-O~CH2TeR
- Meo.í II 'Hf,C (S) O
~.(ppm) =
ÕHb(ppm) =
~.M.(ppm)=
(~.(ppm) =
()C.T.(ppm) =
()C'M.(ppm)=
{R,S)-17 {R,S)-183.14' 2,70
2.98 i 2.86
lA5.' 1,48
440.9 l 204.9
13,35' 7.23
20.86 ' 20.70
17 R = Ph18 R =n-Bu
(S.S}-17 I (S.S)-183,17' 2.79
3.06 2.91
1.38 1.40
441.9 206.3
13.40 i 7.30
20.71 20,56
Figuro 2.2.4.2.1. Modelo de Mosher para os derivados dos compostos 100 e 10e.
A configuração dos hidróxi-teluretos 10f e 109 foi determinada de
maneira indireta. Foi com parado a rotação óptica {[aJD} da y-valerolactona 14 e
da S-angelicalactona 15 preparadas, respectivamente, a partir do hidróxi
telureto 10f e do 109 enantiomericamente enriquecidos (Seção 2.2.3
Esquema 2.2.3.1) com o valor descrito na literatura. 71,72 Dessa forma, foram
determinadas as configurações absolutas dos hidróxi-teluretos 10f e 109, não
acetilados preferencialmente pela lipase, como sendo (S).
70

3. Conclusões


A presença do átomo de Se ou de Te na estrutura dos hidróxi
calcogenetos não ocasionou a inativação das enzimas, possibilitando a
aplicação de biotransformações na resolução cinética dos mesmos, levando à
hidróxi-selenetos e hidróxi-teluretos em altos excessos enantioméricos. Alguns
destes foram usados na preparação de blocos de construção quiral
encontrados em várias classes de produtos naturais, o que demonstra o
potencial sintético da metodologia.
O controle de variáveis, tais como o solvente, a lipase, o suporte, a
estrutura do álcool, a temperatura e o tempo de reação nas biotransformações
de hidróxi-calcogenetos (Se e Te), foi fundamental para uma boa resolução.
Além de permitir a reciclagem das enzimas, o uso de suportes
aumentou sensivelmente a atividade e a estabilidade das mesmas, diminuindo
o tempo de reação e a quantidade de enzima utilizada nas biotransformações.
O emprego de água como solvente na resolução de selenetos
possibilitou a obtenção dos mesmos enriquecidos enantiomericamente sem a
necesidade do emprego de solventes orgânicos, valorizando o processo
biocatálitico pela redução na utilização de substâncias nocivas ao meio
ambiente. Além disso, o uso concomitante de enzimas imobilizadas possibilitou
uma diminuição nos tempos de hidrólise, tornando o processo mais atraente.
A triagem enzimática empregando indicador de pH permitiu realizar
uma análise qualitativa prévia sobre a atividade hidrolítica de dez enzimas
frente a vários compostos concomitantemente, possibilitando escolher as
enzimas mais adequadas para a resolução dos selenetos em meio aquoso.
O emprego de cepas de Aspergillus terreus nas biotransformações de
hidróxi-selenetos levou à oxidação enantiosseletiva, seguida de biometilação
do produto de eliminação de selenóxido.
A Ressonância Magnética Nuclear como ferramenta para a
determinação da configuração absoluta de alguns hidróxi-calcogenetos,
permitiu observarmos o efeito anisotrópico do anel aromático sobre os átomos
de Se e Te, em comparação ao efeito observado sobre os átomos de H e C
nesses compostos.
73


4. Parte Experimental


· "
4.1. Materiais e Métodos
4.1. Materiais e Métodos


4.1. Materiais e Métodos
Os solventes foram purificados e secos antes do uso, empregando
procedimentos usuais. 73 Os reagentes comerciais foram convenientemente
purificados. O THF e o éter etilico foram destilados sob sódio e benzofenona
imediatamente antes do uso. Para evaporar as soluções orgânicas foi utilizado
um evaporador rotativo Büchi, operando a pressão reduzida.
As análises por cromatografia a gás (CG) foram efetuadas nos
cromatógrafos SHIMAOZU GC-2010 e HEWLETT PACKARO - 5730,
equipados com um detector de ionização de chama e acoplado a um
integrador. O gás de arraste foi o nitrogênio ou hidrogênio (colunas quirais) e
as colunas empregadas foram de sílica fundida revestida com goma de poli
SPB-35 (35% difenil /65% dimetilsilicone), a-ciclodextrina de 30m x 0,2mm x
O,22~m (ALFA OEX™120), [3-ciclodextrina de 30m x O,2mm x O,221lm (BETA
DEX™120), y-c1clodextrina de 30m x 0,2mm x 0,221lm (GAMA DEX™120),
todas da SUPELCO® e a [3-ciclodextrina de 25m x O,25mm x O,251lm
(CRHOMOPACK / CHIRASIL-DEX CB) da Varian®.
Nas cromatografias em coluna (CC) foi utilizada sílica gel (60-30mesh)
60A da Aldrich Chemical Company, Inc. A sílica Kieselgel 60 PF254 da Merck foi
utilizada na preparação de placas preparativas. As análises cromatográficas
em camada delgada (CCO) foram efetuadas empregando-se as placas
comerciais da Merck (E. Merck, tipo 5544, 0,2mm), as quais foram reveladas
sob luz ultravioleta (À=254nm) e/ou com solução de vanilina em uma mistura de
ácido sulfúrico-etanol (6%vanilina m/v, 4% ácido sulfúrico e 10% água, v/v, em
etanol).
Os espectros no infravermelho foram registrados em um
espectrofotômetro Bomen MB-100.
Os espectros de massas de baixa resolução (LRMS) foram obtidos no
espectrômetro EM da Hewlett-Packard modelo 5988A acoplado a um
cromatógrafo Hewlett-Packard modelo 5890 e as análises elementares foram
efetuadas em um equipamento Perkin Elmer Series 11, CHNS/O 2400.
Os espectros de ressonância magnética nuclear de 1H (RMN-1H),
ressonância magnética nuclear de 13C (RMN-13C), ressonância magnética
nuclear de 77Se (RMN-77Se) e ressonância magnética nuclear de 125Te (RMN
125Te) foram registrados em um espectrômetro Varian-Inova (300MHz) ou
79

4. Parte Experimental
espectrometro da Sruker (200MHz ou 500 MHz) utilizando padrão interno
tetrametilsilano (TMS), como solvente COCb e nos casos de RMN-77Se e RMN
125Te foi utilizado um capilar selado com solução 1M de difenil dicalcageneto
(Se ou Te, correspondentemente) em COCb como padrão interno. Os
deslocamentos químicos estão expressos em ppm (8) e as constantes de
acoplamento (J) em Hertz (Hz). Para indicar a multiplicidade dos sinais foram
adotadas as seguintes abreviações: s (singleto), d (dubleto), dd (duplo dubleto),
t (tripleto), q (quarteto), quint (quinteto), sext (sexteto) e m (multipleto).
Os desvios ópticos foram determinados em um polarimetro JASCO
OIP 3170, sendo que a concentração (c) foi estabelecida em g/100mL.
As enzimas empregadas pertencem ao grupo dos triglicerol acil
hidrolases, sendo que a PPL (Iipase de pâncreas de porco - livre, 42 units/mg
prot.), a CRL (Iipase de Candida rugosa - livre, 1.440 units/mg prot.) e a MJL
(Iipase de Mucor Javanicus - livre, 536 units/mg) foram adquiridas da Sigma
Comp. A CALS (NOVOZYM 435® - lipase de Candida antartica - Fração S
imobilizada, 10.000 U/g) e a TLLb (L1POZYME TL 1OOL® - lipase de
Thermomyces lanuginosa - livre, >100 KU/g) foram doadas pela NOVOZYMES
CO. e a PSL (AMANO PS - lipase de Pseudomonas sp. - livre, 30.000 u/g) foi
doada pela Amano S/A. As demais enzimas [CALA (Iipase de Candida
antarctica Tipo A - livre - >30 U/mg), CALS (Iipase de Candida antarctica Tipo
S - livre - >120 U/mg), CRL (Iipase de Candida rugosa - livre - >250 U/mg),
PSL (Iipase de Pseudomonas sp. - livre - 400 U/mg), PLE (esterase de fígado
de porco - livre - 40 U/mg), PPL (Iipase de pâncreas de porco - livre - 20
U/mg), TLLa (Iipase de Thermomyces lanuginosa - livre - >800 U/mg) e ASL
(Iipase Alcaligenes sp. -livre - >20 U/mg)] foram adquiridas da Roche CO.
As manipulações dos fungos foram efetuadas numa capela de fluxo
laminar utilizando os métodos de assepsia adequados para evitar
contaminações das colônias. Os meios de cultura foram preparados utilizando
água destilada e esterilizados a 120°C por vinte minutos. Todo o material
utilizado na manipulação de microorganismo (erlenmayers, pipetas, tubos de
ensaio, etc.) foi autoclavado e lavado, exceto materiais descartáveis, os quais
foram acondicionados em sacos de lixo especiais (tipo hospitalar).
80

4.1. Materiais e Métodos
o descarte dos resíduos foi realizado no laboratório seguindo-se as
normas estabelecidas pelo Instituto de Química-USP. Os resíduos orgânicos,
incluindo os solventes orgânicos utilizados nos experimentos foram todos
descartados em frascos próprios para depois serem encaminhados pelo
Instituto de Química para a incineração. O etanol, utilizado para lavagem da
vidraria, foi reciclado por destilação em nosso laboratório. Ácidos e bases
foram neutralizados antes de serem descartados. Soluções aquosas de sais
(NaCI, NH4CI) provenientes de lavagem de fase orgânica, após diluição, foram
descartadas.
As vidrarias com resíduos de teluretos foram lavadas primeiramente
com solução 5% de hipoclorito de sódio, para que ocorresse a oxidação dos
teluretos a teluróxidos, compostos pouco solúveis em água, que foram
descartados com os resíduos orgânicos. A sílica utilizada nas colunas
cromatográficas foi armazenada em tambores apropriados que foram
encaminhados pelo Instituto de Química para serem descartados em locais
designados para tal fim.
81


4.2. Procedimentos GeralS
4.2. Procedimentos Gerais


4.2. Procedimentos Gerais
4.2.1. Preparação do Disseleneto de Difenila 74
Em um balão de três bocas equipado com um condensador de refluxo,
agitador mecânico e um funil de adição, contendo raspas de Mg (24,31 g; 1,05
moi) e alguns cristais de iodo (b) em éter etílico seco (100 mL), foi adicionado
uma solução de bromobenzeno (3,925g - 25 mmol) em éter etílico seco (70mL).
A reação começou rapidamente (observada pela descoloração do iodo) e
violentamente, necessitando um banho de água e gelo para controlar a reação.
Em seguida foi adicionada mais solução de bromobenzeno (85 mmol) em éter
etílico seco (930 mL). Após toda a adição da solução de bromobenzeno, a
mistura reacional foi agitada mecanicamente e mantida sobre refluxo até todo o
consumo do Mgo. Na seqüência foi adicionado Seo em pó (1 moi) em pequenas
porções, de modo a manter o refluxo suave, e deixou-se reagir por 30min. A
solução foi então transferida para um kitassato de 500mL munido de uma
entrada de ar e uma saída para vácuo contendo KOH e etano!. A solução foi
agitada por 12 horas. Em seguida filtrou-se e evaporou-se o solvente. O
produto foi recristalizado em etanol 95% obtendo-se disseleneto de difenila
com pureza analítica. O rendimento total da reação foi de 225g (72%).
4.2.2. Preparação dos Hidróxi-selenetos (R,S)-la e (R,S)-le 38
Em um balão de duas bocas, equipado com agitação magnética sob
nitrogênio, o disseleneto de difenila (1,56g - 5mmol) foi dissolvido em álcool
etílico seco (30mL). Em seguida foi adicionado lentamente NaBH4 em
pequenas porções, até a solução permanecer incolor. Após a formação do
selenolato, foi adicionado lentamente sobre a mistura reacional o epóxido
(óxido de propileno ou estireno - 11 mmol). A agitação foi mantida durante uma
hora. Ao final desse tempo, a mistura reacional foi lavada três vezes com
solução 10% de NaC03 , extraída com acetato de etila e seca com MgS04
85

4. Parte Experimental
anidro. O solvente foi rotoevaporado e a purificação foi realizada em coluna de
sílica utilizando como eluente uma mistura de hexano/acetato de etila (85: 15 ou
9~ 1).
(R,S)-1-fenilsseleno-2-propanol: Óleo; rendimento = 1,9139 (89%); N° CAS:
25570-56-3; RMN 1H(300MHz, CDCb), 8(ppm): 1,27 (d; J =6,19Hz; 3H), 2,46
(s; 1H), 2,88 (dd; J = 12,71Hz; J = 8,31Hz; 1H), 3,10 (dd; J = 12,71Hz; J =
3,95Hz; 1H), 3,84-3,88 (m; 1H), 7,23-7,28 (m; 3H), 7,50-7,56 (m; 2H); RMN 13C
(75MHz, CDCb), 8(ppm): 22,4; 38,5 (f13c-77se = 64 Hz); 66,1; 127,3; 129,3;
129,2; 133,1; RMN 77Se(95MHz, CDCb), 8(ppm): 240,8; LV. (filme - KBr) Banda
de absorção (cm-1): 691, 736,1371,1478,1578,2927,2971,3016,3056,3070,
3390; E.M., m/z (%reL): 216 (60),215 (M+, 4), 172 (47), 158 (57), 157 (50),91
(100), 77 (72), 78 (89), 59 (68), 51 (63), 45 (53); Microanálise (C9H120Se)
Calculado: C = 50,24% e H = 5,62% e observado: C = 49,86% e H = 5,63%.
(R,S)-1-fenil-2-fenilsseleno-etanol: Óleo; rendimento = 1,5239 (55%); N°
CAS: 51558-95-3; RMN 1H (300MHz, CDCI3), 8(ppm): 2,83 (d; J =2,78Hz; 1H),
3,13 (dd; J = 12,75Hz; J = 9,31 Hz; 1H), 3,29 (dd; J = 12,75Hz; J = 3,77Hz; 1H),
4,71-4,76 (m; 1H), 7,24-7,33 (m; 8H), 7,52-7,55 (m; 2H); RMN 13C (75MHz,
CDCI3 ), 8(ppm): 38,5; 72,3; 125,8; 127,4; 127,9; 128,5; 129,3; 133,1; 133,2;
142,5; RMN 77Se (95MHz, CDCb), 8(ppm): 251.9; LV. (filme - KBr) Banda de
absorção (cm-\ 693, 736, 1085, 1437, 1453, 1477, 1578, 2880, 2932, 2983,
3000,3029,3058,3907; E.M., m/z (%rel.): 278 (25), 277 (M+, 3),172 (100), 157
(18), 121 (4), 107 (60),91 (54),79 (83),77 (83); 51 (43),43 (29); Microanálise
(C14H140Se) - Calculado: C = 60,66% e H = 5,09% e observado: C = 60,51 % e
H =5,34%.
86

4.2. Procedimentos Gerais
4.2.3. Preparação do Bis(fenilsseleno)metano 38
Em um balão de duas bocas equipado com septo e sob nitrogênio
disseleneto de difenila (3,433g - 11 mmol) foi dissolvido em etanol seco (50
mL). Em seguida, NaBH4 foi adicionado em pequenas porções até a solução
permanecer incolor. Na seqüência foi adicionado CH2Br2 (0,77mL - 1,9039
11 mmol) em THF (4mL) com uma seringa e a mistura reacional foi agitada por
4h a temperatura ambiente. Ao final das 4h a mistura reacional foi transferida
para um funil de extração contendo solução saturada de NH4CI (100mL) e
extraída com acetato de etila. A fase orgânica foi lavada com solução saturada
de NaCl, seca com MgS04e o solvente evaporado. O produto foi purificado por
cromatografia utilizando hexano como eluente. O composto foi obtido puro.
Rendimento da reação foi de 88% (3,157g).
4.2.4. Preparação dos Hidróxi-selenetos (R,S)-lb-d 46
Em um balão de duas bocas de 50mL equipado com septo, sob
agitação magnética e sob nitrogênio o bi(fenilsseleno)metano (0,652g - 2mmol)
foi dissolvido em THF seco (8mL). A mistura foi resfriada com um banho de
acetona e gelo seco (-78°C) e foi adicionado uma solução de n-butil lítio
(1,43mL - 1,4MoIL-1 em hexano - 2mmol) em hexano. A mistura reacional foi
mantida sobre agitação durante 30 minutos, em seguida foi adicionado o
aldeído correspondente (2mmol). A agitação foi mantida por 10 minutos a
-78°C e por 30 minutos a temperatura ambiente. Ao final desse período à
mistura reacional foi transferida para um funil de extração (100 mL) contendo
solução saturada de NH4CI (30mL) e extraída com acetato de etila. A fase
orgânica foi lavada com solução saturada de NaCI, seca com MgS04 e o
solvente evaporado. O produto obtido foi pré-purificado numa coluna
cromatográfica utilizando primeiramente apenas hexano e depois apenas
acetato de etila como eluentes. A fase de acetato de etila foi então evaporada e
87

4. Parte Experimental
purificada por cromatografia usando uma solução hexano/acetato (9: 1) como
eluente.
(R,S)-1-fenilsseleno-2-pentanol: Óleo; rendimento =0,350g (72%); RMN 1H
(300MHz, COCb), o(ppm): 0,89 (t; J =7,20Hz; 3H), 1,33-1,37 (m; 1H); 1,44
1,54 (m; 3H); 2,18 (s; 1H), 2,88 (dd; J =12,73Hz; J =8,61 Hz; 1H), 3,14 (dd; J =12,73Hz; J =3,51 Hz; 1H), 3,66-3,71 (m; 1H), 7,24-7,27 (m; 3H), 7,52-7,53 (m,
2H); RMN 13C (75MHz, COCb), o(ppm): 14,2; 19,2; 37,5 (f13C-77se = 64 Hz);
39,0; 69,91; 127,5; 129,4; 129,7; 133,3; RMN 77Se (95MHz, COCb), o(ppm):
234,2; I.V. (filme - KBr) Banda de absorção (cm-\ 691, 737,1436,1459,1474,
1578,2870,2928,2958,3063,3401; E.M., m/z (%rel.): 244 (60),243 (M+, 4),
172 (100),158 (63),157 (61),91 (86),78 (81),77 (74), 69 (44),55 (85),51 (51),
45 (90), 41 (89); Microanálise (C11 H160Se) - Calculado: C = 54,32% e H =
6,63% e observado: C = 54,08% e H = 6,85%.
(~~1c
(R,S)-1-fenilsseleno-3-metil-2-butanol: Óleo; rendimento = 0,379g (78%);
CAS NR: 68395-98-2; NMR 1H (500MHz): o 0,89-0,93 (m; 6H), 1,74-1,80 (m;
1H), 2,89 (dd; J = 9,5 Hz; J = 12,7Hz; 1H), 3,17 (dd; J = 3,0 Hz; J = 12,7Hz;
1H), 3,40-3,44 (m; 1H), 7,23-7,28 (m; 3H), 7,50-7,54 (m; 2H); RMN 13C
(125MHz, COCb), o(ppm): 17,7; 18,7; 33,3; 34,9; 74,5; 127,2; 129,2; 129,3;
133,0; RMN 77Se (95MHz, COCb), o(ppm): 240,8; I.V. (filme - KBr) Banda de
absorção (cm-1): 691,737, 1472, 1578, 1801, 1874,2874,2960,3429; E.M.,
m/z (% rel.): 244 (57), 183 (4), 172 (71), 157 (72), 91 (74), 77 (76), 69 (100), 51
(60),45 (44); Microanálise (C11 H160Se) - Calculado: C =54,32% e H =6,63% e
observado: C = 54,75% e H = 7,05%.
88

4.2. Procedimentos Gerais
(~5}-1d
(R,S)-1-fenilsseleno-2-octanol: Óleo; rendimento = O,473g (83%); N° CAS:
52954-45-7; RMN 1H (500MHz, CDCb), o(ppm): 0,87 (t; J = 7,1 Hz; 3H), 1,25
1,31 (m; 8H), 1,51-1,54 (m; 2H), 2,14 (s; 1H), 2,87 (dd; J = 8,6Hz; J = 12,7Hz;
1H), 3,14 (dd; J= 3,5Hz; J= 12,7Hz; 1H), 3,63-3,68 (m; 1H), 7,24-7,27 (m; 3H),
7,51-7,54 (m; 2H); RMN 13C (125MHz, CDCb), o(ppm): 14,0; 22,5; 29,23; 31,7;
36,6; 37,2; 69,8; 127,2; 129,2; 133,0; RMN 77Se (95MHz, CDCI3), o(ppm):
237,0; I.V. (filme - KBr) Banda de absorção (cm-1): 691, 736, 1024, 1070, 1478,
1578,1712,1799,1873,1946,2855,2928,3056, 3070, 3399cm-1; E.M. m/z(%
rel.): 286 (68), 172 (96), 158 (71), 129 (23), 91 (78), 77 (70), 69 (96), 55 (100),
51 (48), 45 (42); Microanálise (C14H220Se) - Calculado: C = 58,94% e H =
7,77% e observado: C = 59,31 % e H = 7,97%.
4.2.5. Preparação do Hidróxi-seleneto (R,S)-l f 45
Em um balão de duas bocas equipado com septo, agitação magnética,
sob nitrogênio e contendo Seo em pó (0,395g - 5 mmol) em THF seco (25mL)
foi adicionada lentamente uma solução de MeLi (1,3MoIL-1 em hexano) até o
ponto de viragem (solução incolor). Em seguida foi adicionado óxido de
estireno (O,57mL - 0,600g - 5 mmol) e foi mantida a agitação sob refluxo
durante 24 horas. Ao final deste período a mistura reacional foi transferida para
um funil de extração contendo solução saturada de NH4CI e extraída com
acetato de etila. A fase orgânica foi lavada com solução saturada de NaCI,
seca com MgS04 e o solvente rotaevaporado. O produto obtido foi pré
purificado numa coluna cromatográfica utilizando primeiramente apenas
hexano e depois apenas acetato de etila como eluentes. A fase de acetato de
etila foi então evaporada e purificada por cromatografia usando uma solução
hexano/acetato (9: 1) como eluente.
89

4. Parte Experimental
(R,S)-1-metilsseleno-2-fenil-etanol: Óleo; rendimento = 0,828g (77%); N°
CAS: 56051-09-3; RMN 1H (200MHz, COCb), õ(ppm): 1,95 (s; 3H), 2,79 (dd; J
= 8,8Hz; J = 12,7Hz; 1H), 2,92 (dd; J = 3,9Hz; J = 12,7Hz; 1H), 4,77 (dd; J =3,9Hz; J =8,8Hz; 1H), 7,24-7,39 (m; 5H); RMN 13C (50MHz, COCI3), o(ppm):
4,7; 35,8; 72,1; 125,7; 127,7; 128,4; RMN 77Se (95MHz, COCb), o(ppm): 30,6;
E.M. m/z (% rel.): 216 (53),121(65),107(94),91(80),77(100),51(66).
4.2.6. Preparação da Diisopropilamideto de lítio (LDA)
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, a diisopropilamina (destilada recentemente
0,202g - 2mmol) foi dissolvida em THF seco (4mL). A solução foi resfriada a
78°C e lentamente foi adicionada a mesma uma solução de n-BuLi (1,43mL
1,4MoIL-1 em hexano - 2mmol). Após a adição a mistura reacional foi mantida
sobre agitação a temperatura ambiente por 30 minutos.
4.2.7. Preparação do Hidróxi-seleneto (R,$)-lg 47
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio o bis(fenilsseleno)metano (0,652g - 2mmol) foi
dissolvido em THF seco (8mL). A solução foi resfriada a -78°C e lentamente foi
adicionada à solução de LOA (2mmol). A agitação foi mantida por mais uma
hora e em seguida foi adicionado acetaldeído (0,14mL - 0,11 Og - 2,5 mmol).
Após a adição, a agitação foi mantida por 10 minutos a -78°C e 30 minutos a
temperatura ambiente. Ao final deste período a mistura reacional foi transferida
para um funil de extração conten"do solução saturada de NH4CI e extraída com
acetato de etila. A fase orgânica foi lavada com solução saturada de NaCI,
seca com MgS04 e o solvente rotaevaporado. O produto obtido foi pré-
90

4.2. Procedimentos Gerais
purificado numa coluna cromatográfica utilizando primeiramente apenas
hexano e depois apenas acetato de etila como eluentes. A fase de acetato de
etila foi então evaporada e purificada por cromatografia usando uma solução
hexano/acetato (9: 1) como eluente.
(R,S)-1,1-difenilsseleno-2-propanol: Óleo; rendimento = 0,540g (73%); N°
CAS: 77461-31-5; RMN 1H (200MHz, CDCb), õ(ppm): 1,38 (d; J =6,1Hz; 3H),
2,46 (s; 1H), 3,89-3,99 (m; 1H), 4,49 (d; J = 3,1 Hz; 1H), 7,24-7,30 (m; 6H),
7,53-7,62 (m; 4H); RMN 13C (50MHz, CDCI3), õ(ppm): 20,8; 55,3 (i13C-77S~ =86.8 Hz); 69,2; 128,1; 128,2; 129,2; 129,3; 129,9; 130,3; 134,3; 135,5; RMN
77Se (95MHz, CDCI3), õ(ppm): 355,0; 357,9; E.M. mlz (% rel.): 372 (6), 314
(13), 215 (31), 157 (62), 117 (22), 91 (77), 77 (93), 51 (72), 43 (100).
4.2.8. Preparação dos Fenilselenoacetais 48
AlSey seR1
R
R=(~A1
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio contendo PhSeH (1,570g - 10 mmol) e o alde.ído
correspondente (10mmol) a O°C, foi adicionada solução de H2S04 (13M
12mmol). A agitação foi mantida por 20 minutos e em seguida foi adicionado
acetato de etila. A mistura foi lavada com solução 10% de NaHC03 e seca com
MgS04. O resíduo foi purificado por coluna cromatografica utilizando hexano
como eluente.
4.2.9. Preparação do Hidróxi-seleneto (R,S)-1h-i 46
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, o selenocetal (2 mmol) de interesse foi dissolvido
em THF seco (8ml). Em seguida, foi adicionada lentamente uma solução de
n-BuLi (1 ,43mL - 1,4MoIL-1 em hexano - 2mmol) a -78°C e mantida a agitação
por 30 minutos. Na seqüência, acetaldeído foi adicionado (0,14mL - 0,11 Og
2,5 mmol) e a agitação mantida por 20 minutos a -78°C e 1 hora a temperatura
ambiente. Ao final deste período a mistura reacional foi transferida para um
91

4. Parte Experimental
funil de extração contendo solução saturada de NH4CI e extraída com acetato
de etila. A fase orgânica foi lavada com solução saturada de NaCI, seca com
MgS04 e o solvente rotaevaporado. O produto obtido foi pré-purificado numa
coluna cromatográfica utilizando primeiramente apenas hexano e depois
apenas acetato de etila como eluentes. A fase de acetato de etila foi então
evaporada e purificada por cromatografia usando uma solução hexano/acetato
(85: 15) como eluente.
OH
(+/-)-1-hexil-1-(fenilsseleno)-2-propanol: Óleo; mistura diatereoisomérica;
rendimento = O,514g (86%); RMN 1H (300MHz, CDCb), 8(ppm): O,85-0,93(m;
6H), 1,18-1,76 (m; 26H), 2,08 (s; 1H), 2,12 (s; 1H), 2,97-3,03 (m; 1H), 3,25-3,76
(m; 1H), 3,65-3,76 (m; 1H), 3,80-3,88 (m; 1H), 7,20-7,30 (m; 6H), 7,56-7,60 (m;
4H); RMN 13C (75MHz, CDCb), 8(ppm): 14,0; 19,8; 20,5; 22,6; 28,1; 28,4; 29,0;
29,1; 31,1; 31,6; 31,7; 31,9; 57,8; 58,0; 68,9; 69,3; 127,5; 127,6; 129,0; 129,1;
129,6; 134,4; 134,9; E.M. m/z (% rel.): 300 (30),255 (6), 172 (5), 158 (59), 157
(24),91 (16),78 (49),77 (40), 69 (100),55 (96),51 (20),45 (63); Microanálise
(C15H240Se) - Calculado: C =60,19% e H =8,08% e observado: C =60,34% e
H =8,37%.
~SeFhA1
(+1-)-1 i
(+/-)-1-fenil-1-(fenilsseleno)-2-propanol: Óleo; mistura diatereoisomérica;
rendimento = O,512g (88%); N° CAS: 623575-61-1; RMN 1H (200MHz, CDCI3),
8(ppm): 1,15 (d; J = 5,7Hz; 3H); 8 1,24 (d; J = 5,7Hz; 3H); 1,95 (s; 2H); 4,07
4,26 (m; 4H); 7,02-7,37 (m; 20H); RMN 13C (75MHz, CDCb), 8(ppm): 20,5;
20,6; 57,3; 60,1; 69,2; 69,6; 127,1; 127,4; 128,3; 128,4; 128,9; 129,0; 135,1;
135,5; E.M. m/z (% rel.): 292 (11), 247 (4),158 (30),157 (15),135 (86), 117
(51), 91 (62), 78 (32), 77 (39), 57 (73), 51 (25), 43 (100).
92

Universidade de São Paul
4.2.10. Preparação do Ditelureto de Difenila 75
4.2. Procedimentos Gerais
Em um balão de três bocas, equipado com um funil de adição de
sólidos, funil de adição, agitador mecânico, contendo raspas de MgO (6,07 g
0,25 moi) e alguns cristais de iodo (12) em THF (25 ml), foi adicionada
lentamente uma solução de bromobenzeno (0,08 moi) em THF (20 mL). A
reação começou rapidament.e (observada pela descoloração do iodo) e
violentamente, necessitando um banho de água e gelo para controlar a reação.
Em seguida foi adicionada mais solução de bromobenzeno (0,20 moi) em THF
seco (150 mL). Após toda a adição da solução de bromobenzeno, a mistura
reacional foi agitada mecanicamente e mantida sobre refluxo até todo o MgO
ser consumido. Após a formação do reagente de Grignard, adicionou-se (sob
agitação) 33 g (0,25 moi) de telúrio em pó sobre a solução de Grignard. A
reação começa em poucos minutos. Logo que algum ditelureto tenha sido
formado (caracterizado pela coloração vermelha intensa) resfriamos a solução
a zero grau e trocamos o funil de adição por uma tampa de borracha com um
tubo de vidro provido de uma torneira ligada a um balão contendo 2,5 litros de
oxigênio (02). A cada dez minutos a torneira era aberta para o oxigênio fluir
para dentro do balão. Depois que todo o oxigênio foi consumido
(aproximadamente 1h) elevamos a temperatura até a temperatura ambiente e
agitamos por mais 1,5h. Ao final de 1,5h despejamos a mistura em um Becker
grande e deixamos durante toda à noite em contato com o ar atmosférico. No
dia seguinte tratamos a mistura com solução saturada de NH4CI, extraímos
com éter dietílico, rotaevaporamos o solvente e obtivemos cristais laranja
avermelhados de ditelureto de difenila. Os cristais foram recristalizados em
álcool 95% obtendo-se ditelureto de difenila com pureza analítica. O
rendimento total da reação foi de 59,36 g (58%).
93

4. Parte Experimental
4.2.11. Preparação dos Hidróxi-teluretos (R..s)-10a e (R. .s)-lOc 54
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, o ditelureto de difenila (2,252 g - 5,5 moi) foi
dissolvido em etanol seco (30 mL). Em seguida foi adicionado lentamente
NaBH4 em pequenas porções, até a solução permanecer incolor. Após a
formação do telurolato, foi adicionado lentamente sobre a mistura reacional o
epóxido (óxido de propileno ou estireno - 11 mmol). A agitação foi mantida
durante uma hora. Ao final desse período, a mistura reacional foi lavada três
vezes com solução 10% de NaC03, extraída com acetato de etila e secada com
MgS04 anidro. O solvente foi rotaevaporado e a purificação foi feita em coluna
de sílica utilizando como eluente uma mistura de hexano/acetato de etila (85: 15
ou 9:1).
OH
~TePh
(~S)-1Oa
(R,S)-1-fenilteluro-2-propanol: Óleo; rendimento = 2,350g (81 %); N° CAS:
133617-20-6; RMN 1H (300MHz, COCI3), o(ppm): 1,30 (d; J =6,12Hz; 3H), 2,16
(s; 1H), 2,96 (dd; J = 12,25Hz; J = 7,59Hz; 1H), 3,14 (dd; J = 12,25Hz; J =
4,50Hz; 1H), 3,89-3,95 (m; 1H), 7,17-7,28 (m; 3H), 7,72-7,76 (m; 2H); RMN 13C
(75MHz, COCb), õ(ppm): 21,5(f13c-125Te = 163 Hz); 23,7; 67,4; 111,2; 127,7;
129,2; 138,4; RMN 125Te (157MHz, 300K, COCI3), o(ppm): 367.1; I.V. (filme
KBr) Banda de absorção (cm-1): 692, 732, 1058, 1320, 1405, 1433, 1451, 1472,
1573,2873,2891,2924,2968,3061,3371; E.M., m/z (%rel.): 264 (M+, 21), 222
(19),207 (18),130 (10),91 (26),77 (100), 59 (96),51 (60),45(10); Microanálise
(C9H120Te) - Calculado: C =40,98% e H =4,59% e observado: C =40,63% e
H =4,46%.
OH
~TeA1(~S)-1OC
(R,S)-1-fenil-2-fenilteluro-etanol: Óleo; rendimento = 2,510g (70%); N° CAS:
55136-89-5; RMN 1H (300MHz, COCb), õ(ppm): 2,59 (d; J =3,51 Hz; 1H), 3,26
(dd; J =12,13Hz; J =8,11Hz; 1H), 3,32 (dd; J= 12,13Hz; J =4,99Hz; 1H), 4,84-
94

4.2. Procedimentos Gerais
4,90 (m; 1H), 7,15-7,35 (m; 8H), 7,69-7,72 (m; 2H); RMN 13C (75MHz, CDCb),
õ(ppm): 20,6; 73,6; 111,5; 125,6; 127,7; 127,8; 128,4; 129,2; 138,3; 143,4; I.V.
(filme - KBr) Banda de absorção (cm-1) 697, 734, 1047, 1434, 1452, 1475,
1493, 1574, 2874, 2930, 2989, 3029,3059, 3407; E.M., m/z (%rel.): 326 (14),
325 (M+, 2), 220 (16),205 (15), 121 (18), 103 (33),91 (50),77 (100),51 (58);
Microanálise (C14H140Te) - Calculado: C =51,60% e H =4,33% e observado:
C =51,43% e H =4,25%.
4.2.12. Preparação do Diazometano 76
Utilizando um aparato de destilação Sem juntas esmerilhadas, as
diversas partes do conjunto (condensador de tubo reto longo, balão, funil de
adição, frascos coletores) foram conectadas com rolhas. O condensadõr foi
conectado a uma alonga e esta a dois frascos coletores conectados em série, o
primeiro com 500 mL de capacidade e o segundo com 100 mL; ambos os
frascos foram resfriados com uma mistura de gelo picado e sal. No segundo
frasco foi adicionado éter etílico (40 mL) e um tubo de vidro foi conectadõ dê
modo que a ponta deste permanecesse abaixo do éter (isto servirá para que
qualquer traço de diazometano que escape do primeiro frasco seja capturado
neste segundo frasco coletor). Esta preparação foi executada numa capela
com exaustão potente em virtude da toxicidade do diazometano. Assim, em Um
balão de destilação (250 mL) foi depositada uma solução contendo hidróxido
de potássio (10 g; 180 mmol), água (15 mL) e etanol (96% - 50 mL). A solução
no balão de destilação foi aquecida com banho de água (60-65°C). Em
seguida, uma solução de N-metil-N-nitrosotolueno-p-sulfonamida (43 g; 200
mmol) em éter etílico (250 mL) foi adicionada lentamente, de forma que a
velocidade de adição fosse igual a velocidade de destilação. Após a adição,
novas porções de éter etílico foram adicionadas até que o destilado não ficasse
mais amarelado (indicando a presença de diazometano). Após a destilação, as
soluções de éter dos frascos coletores continham cerca de 5,9 a 6,1 g de
diazometano (rendimento = 70-73 %).
95

4. Parte Experimental
4.2.13. Preparação do Bis(fenilteluro)metano 63
Em um kitassato de 250 mL sob nitrogênio, equipado com agitação
mecânica e com um funil de extração, ditelureto de difenila (2,047 g - 5 mmol)
dissolvido em éter dietílico foi tratado gota a gota com solução 'de diazometano
(em éter dietílico) a O°C até o desaparecimento da cor vermelha do ditelureto
de difenila. O produto foi obtido por evaporação do solvente e purificado em
coluna cromatográfica utilizando hexano como eluente.
4.2.14. Preparação do Hidróxi-telureto (R.S)-10b 62
OH
~TeA1
(RS)-1Ob
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, o bis(fenilteluro)metano (0,847 g - 2 mmol) foi
dissolvido em THF seco (8 mL). A mistura foi resfriada a -78°C e foi adicionado
n-BuLi (1,43 mL - 1,4MoIL-1 em hexano - 2 mmol). A mistura reacional foi
mantida sobre agitação durante 30 minutos, em seguida, foi adicionado o
butanal (0,18 mL - 0,144g - 2 mmol) e foi mantida a agitação por mais 10
minutos a -78°C e 30 minutos a temperatura ambiente. Ao final desse período a
mistura reacional foi transferida para um funil de extração (100 mL) contendo
solução saturada de NH4CI (30 mL) e extraída com acetato de etila. A fase
orgânica foi lavada com solução saturada de NaCl, seca com MgS04 e o
solvente evaporado. O produto obtido foi pré-purificado numa coluna
cromatográfica utilizando primeiramente apenas hexano e depois apenas
acetato de etila. A fase de acetato de etila foi então evaporada e purificada por
cromatografia usando uma solução hexano/acetato (9: 1) como eluente.
(R,S)-1-fenilteluro-2-pentanol: Óleo; rendimento = 0,355g (61 %); RMN 1H
(500MHz, CDCb), o(ppm): 0,89 (t; J = 7,32Hz; 3H), 1,32-1,37 (m; 1H), 1,40
1,47 (m; 1H), 1,50-1,61 (m; 2H), 2,14 (s; 1H), 2,98 (dd; J = 12,26Hz; J =
96

4.2. Procedimentos Gerais
7,94Hz; 1H), 3,16 (dd; J = 12,26Hz, J = 4,03Hz; 1H), 3,69-3,74 (m; 1H), 7,17
7,29 (m; 3H), 7,73-7,76 (m, 2H); RMN 13C (125MHz, COCb), õ(ppm): 13,9; 19,2;
20,4; 40,1; 70,8; 111,2; 127,8; 129,3; 138,5; E.M., m/z (%rel.): 292 (40), 222
(40), 207 (45), 130 (18), 91 (43), 77 (93), 69 (63), 45 (100), 43 (64);
Microanálise (C11 H160Te) - Calculado: C = 45,27% e H = 5,53% e Observado:
C= 45,62% e H= 5,66%.
4.2.15. Preparação dos Hidróxi-teluretos (R,S)-10d e (R,S)-10e 54
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, telúrio elementar (0,638 g - 5 mmol) foi suspenso
em THF seco (20 mL), adicionando-se a seguir lentamente MeU ou n-BuLi até
a formação de uma solução límpida amarela. Em seguida, foi adicionado o
epóxido correspondente (óxido de estireno ou óxido de propileno - 5 mmol). A
agitação foi mantida por 4 horas. Ao final deste período a mistura reacional foi
transferida para um funil de extração contendo solução saturada de NH4CI e
extraída com acetato de etila. A fase orgânica foi lavada com solução saturada
de NaCl, seca com MgS04 e o solvente rotaevaporado. O produto. obtido foi
pré-purificado numa coluna cromatográfica utilizando primeiramente apenas
hexano e depois apenas acetato de etila como eluentes. A fase de acetato de
etila foi então evaporada e purificada por cromatografia em coluna usando uma
solução hexano/acetato (85: 15) como eluente.
(R,S)-1-metilteluro-2-fenil-etanol: Óleo; rendimento = 0,791g (60%); RMN 1H
(200MHz, COCb), õ(ppm): 1,77 (s; 3H); 2,98-3,03 (m; 2H); 4,78-4,84 (m; 1H);
7,27-7,33 (m; 5H); RMN 13C (75MHz, COCb), õ(ppm): 17,3; 73,8; 125,7; 127,8;
128,4; 143,8; RMN 125Te (157MHz, 298K, COCb), õ(ppm): 25,1; E.M. m/z (%
rel.): 226 (32); 160 (61); 140 (9), 121 (71), 103 (73),91 (44),77 (100), 65 (14),
43 (80); Microanálise (C9H120Te) - Calculado: C = 40,98% e H = 4,59% e
observado: C = 40,71 % e H = 4,36%.
97

4. Parte Experimental
a-I
~Te~
(~S)-1oe
(R,S)-1-(n-butilteluro)-2-propanol: Óleo; rendimento = 0,794 (80%); N° CAS:
414902-88-8; RMN 1H (200MHz, COCb), õ(ppm): 0,92 (t; J = 7,5Hz; 3H), 1,29
(d; J = 5,7Hz; 3H), 1,32-1,47 (m; 2H), 1,72 (quint.; J = 7,5Hz; 2H), 2,54-2,91
(m; 4H), 3,86 (sext.; J = 5,7Hz; 1H), RMN 13C (50MHz, COCb), õ(ppm): 3,16;
13,2; 15,6; 23,5; 24,79 (J1C-Te = 45,9 Hz); 34,07; 67,4; RMN 125Te (157MHz,
298K, COCI3), õ(ppm): 118,5; E.M. mlz (% rel.): 246 (13); 204 (5); 186 (2), 168
(10), 145 (3), 126 (2), 57 (55), 55 (29) e 41 (100); Microanálise (C7H160Te)
Calculado C =34,49% e H =6,61 % e observado: C =34,33% e H =6,32%.
4.2.16. Preparação do Hidróxi-telureto (R.S)-lOf 63.64
a-I
~Te~(~1or
Em um balão de duas bocas equipado com septo, agitação magnética e sob
nitrogênio, telúrio elementar (0,638 g - 5 mmol) foi suspenso em THF seco (10
mL), adicionando-se a seguir lentamente n-BuLi (3,57 mL - 1,4MoIL-1 em
hexano - 5 mmol) até a formação de uma solução límpida amarela, a qual foi
adicionada água desoxigenada (10mmol - 0,18 mL). A mistura foi mantida
sobre agitação por 5 minutos e foi adicionada de uma só vez a metil vinil
cetona (MVK - 5 mmol; 0,35 g; 0,4 mL). A mistura vermelha resultante foi
mantida sobre agitação por 30 minutos e utilizando um funil de adição de
líquidos, foi adicionada uma solução de NaBH4 em etanol (6 mmol, 0,22 g; 6
mL de uma solução 1 moIL-1). A reação foi acompanhada por TLC seguida de
diluição em água (20 mL) e extraida com acetato de etila:hexano 1:1 (50 mL).
As fases foram separadas e a fase aquosa lavada com acetato de etila:hexano
(1: 1 - 2x 50 mL). Na seqüência as fases orgânicas foram combinadas e secas
com MgS04 anidro. O solvente foi rotaevaporado e a purificação foi realizada
por cromatografia em coluna empregando uma solução hexano!acetato (15:1)
como eluente.
98

4.2. Procedimentos Gerais
(R,S)-4-(n-butilteluro)-2-butanol: Óleo; rendimento = 0,901 g (70%); N° CAS:
861399-10-2; N° CAS: 861399-10-2; RMN 1H (200 MHz, COCb), o(ppm): 0,85
(t; J = 7,2 Hz; 3H); 1,13 (d; J = 6,3 Hz; 3H); 1,31 (sext; J = 7,2 Hz; 2H); 1,65
(quint; J =7,2 Hz; 2H); 1,76-1,85 (m; 2H); 2,53-2,69 (m; 4H); 3,70-3,81 (m; 1H);
RMN 13C (50 MHz, COCb), o(ppm): -2,3; 2,7; 13,4; 23,2; 25,0; 34,2; 41,1; 69,1;
RMN 125Te (157MHz, 298K, COCb), o(ppm): 251.43; MS m/z (reI. int.): 260 (13);
258 (13); 256(7); 255 (3); 254 (2); 215 (3); 186 (8); 72 (5); 57 (73); 55(100); 45
(44).
4.2.17. Preparação da (Z)-butilteluro-enona 66,68
o Te~~
12
Em um balão de duas bocas equipado com um septo, agitação
magnética e sob nitrogênio, foi adicionado THF seco (200 mL). O sistema foi
resfriado a O°C e saturado com acetileno, através do borbulhamento de
acetileno sobre a solução. Em seguida, n-BuLi (38,46 mL - 1,3MoIL-1 em
hexano - 50 mmol) foi adicionado lentamente por um período de 20 minutos,
através de um agulha mergulhada na solução, a fim de evitar a formação de
carbeto de lítio, o qual provoca o entupimento da agulha. Na seqüência o
sistema foi novamente saturado com acetileno e foi adicionada solução de
ZnCb (6,815g - 50 mmol) em THF (10mL) gota-a-gota via seringa. Após a
adição, a solução resultante foi aquecida à temperatura ambiente, agitada por
30 minutos e resfriada a O oCo Em seguida, foi adicionada lentamente uma
solução de cloreto de acetila (3,925 g - 50 mmol) em THF (10 mL), mantendo
se a agitação por 30 minutos a O °C e na seqüência o sistema foi aquecido a
temperatura ambiente.
Em um segundo balão de 1L contendo telúrio elementar (6,6352 g - 52
mmol) em THF seco (150 mL) a O °C, foi adicionado n-BuLi (40 mL - 1,3 MoIL-1
em hexano - 52 mmol) até a formação de uma solução límpida amarelo claro.
Em seguida foi adicionada água desoxigenada (10 mL), mantendo-se a
agitação por 10 minutos a temperatura ambiente. A alquinona preparada no
99

4. Parte Experimental
primeiro balão foi transferida via cânula para o segundo balão, o qual contem o
sistema hidrotelurante. O progresso da reação foi monitorado por TLC. Depois
de 5 minutos a mistura reacional foi diluída com hexano (200 mL), lavada com
solução saturada de NaCI (3x 100 mL) e seca com MgS04. O resíduo foi
purificado por cromatografia em coluna empregando uma solução
hexano/acetato (20: 1) como eluente.
(Z)-4-(butilteluro)but-3-en-2-ona: Óleo; rendimento =7,868g (62%); 1H-RMN
(CDCb, 200 MHz) o (ppm): 0,92 (t; J =7,4 Hz; 3 H), 1,40 (sext.; J =7,4 Hz; 2
H), 1,80 (quint.; J =7,4 Hz; 2 H), 2,24 (s; 3 H), 2,51 (t; J =7,4 Hz; 2H), 7,53 (d;
J =9,2 Hz; 1H), 8,78 (d; J =9,2 Hz; 1 H), 13C-RMN (CDCb, 50 MHz) o (ppm):
10,5; 13,3; 24,9; 29,9; 33,7; 129,5; 139,8; 197,1; RMN 125Te (157MHz, 300K,
CDCb), o(ppm): 606,7; E.M. m/z (% rel.): 256 (M+ +3, 8); 254 (M+ +1, 8); 252
(5); 251 (2); 200 (2); 199 (46); 196 (45); 195 (25); 193 (10); 192 (7); 190 (3); 57
(10); 55 (11); 43 (100); I.v. (filme KBr) Banda de absorção (cm-\ 2957; 2922;
2730; 2376; 2064; 1852; 1750; 1644; 1506; 1360; 1334; 1189; 973; 709; 669;
Microanálise (CSH140Te) - Calculado C =37,86% e H =5,38% e observado: C
= 38,03% e H = 5,38%.
4.2.18. Preparação do Hidróxi-telureto (R,S)-10g 68
a;Te~
~(~S)-10g
Em um balão de duas bocas equipado com um septo, agitação
magnética e sob nitrogênio, a (Z)-butilteluro-enona (0,253g - 1 mmol) foi
dissolvida em metanol (5 mL) a O°C. Para a solução resultante, uma solução de
NaBH4 em etanol (1 mmol - 3- mL etanol) foi adicionada gota-a-gota. A reação
foi monitorada por TLC. Depois do consumo de todo o material de partida, a
mistura reacional foi diluída com hexano:éter etílico (1:1 - 15 mL), lavada com
água (3x10 mL) e solução saturada de NaCI (2x 10 mL). O solvente foi
evaporado e o resíduo foi purificado por cromatografia em coluna empregando
uma solução de hexano:acetato de etila (5: 1) como eluente.
100

4.2. Procedimentos Gerais
(R,S)-(Z)-4-(butilteluro)but-3-en-2-ol: Óleo; rendimento = O,205g (80%); N°
CAS: 835594-73-5; 1H-RMN (COC/3, 300 MHz) o (ppm): 0,92 (t; J = 7,2 Hz; 3
H), 1,27 (d; J =6,3 Hz; 3 H), 1,39 (sext.; J =7,2 Hz; 2 H), 1,78 (quint.; J =7,2
Hz; 2 H), 1,94 (s; 1 H), 2,65 (t; J =7,2 Hz; 2 H), 4,34 (quint.; J =6,6 Hz; 1 H),
6,28 (dd; J =6,6Hz; 9,9 Hz; 1H), 6,70 (dd; J = 1,2Hz; 9,9 Hz; 1H); 13C-RMN
(COCb, 75 MHz) o (ppm): 7,2; 13,3; 22,4; 24,9; 34,0; 70,5; 103,4; 141,8; RMN
125Te (157MHz, 295K, COCI3), o(ppm): 280,9; E.M. m/z (% rel.): 258 (13); 256
(12); 201 (15); 199 (15); 71 (100); 57 (60); 55 (40); 53 (35); 45 (22); 43 (81); 41
(55).
4.2.19. Preparação dos Acetatos dos Hidróxi-calcogenetos 49
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, o hidróxi-calcogeneto (3,6 mmol) foi dissolvido em
piridina seca (5 mL). A solução foi resfriada a zero grau (O°C) e em seguida foi
adicionado lentamente anidrido acético (1,99 mL - 2,144 g - 21 mmol). Ao final
da adição a temperatura foi elevada para a temperatura ambiente. A reação foi
acompanhada por CCO. Ao final da reação a mistura reacional foi lavada três
vezes com solução 10% de HCI, três vezes com solução saturada de CUS04 e
extraída com acetato de etila. A fase orgânica foi lavada com solução saturada
de NaCl, seca com MgS04 e o solvente rotaevaporado. A purificação foi
realizada por coluna cromatogratográfica usando uma solução hexano/acetato
(95:5) como eluente.
(R,S)-O-acetil-1-fenilsseleno-2-propanol: Óleo; rendimento =O,842g (91 %);
RMN 1H (300MHz, COCI3), o(ppm): 1,33 (d; J =6,30Hz; 3H), 1,94 (s; 3H), 2,99
(dd; J =12,80Hz e J =6,23Hz; 1H), 3,10 (dd; J =12,80Hz e J =6,13Hz; 1H),
5,04-5,10 (m; 1H), 7,21-7,30 (m; 3H), 7,50-7,54 (m; 2H); RMN 13C (75MHz,
COCI3), o(ppm): 19,8; 21,1; 33,0; 70,3; 127,1; 129,1; 129,9; 132,8; 170,4; RMN
77Se (95MHz, COCI3), o(ppm): 264,3; I.V. (filme - KBr) Banda de absorção(cm
\ 691, 738, 1242, 1371, 1438, 1453, 1478, 1578, 1737,2932,2980,3057,
101
t.

4. Parte Experimental
3071; E.M., m/z (%rel.): 257 (M+; 1),215 (1), 198 (12), 181 (4), 157(5), 101
(13), 78 (7), 77 (11), 43 (100); Microanálise (C11 H1402Se) - Calculado: C =51,37% e H =5,49% e observado: C =51,50% e H =5,54%.
(R,S)-O-acetil-1-fenilsseleno-2-pentanol: Óleo; rendimento = 0,9449 (92%);
RMN 1H (500MHz, CDCI3), õ(ppm): 0,89 (t; J =7,37Hz; 3H), 1,24-1,36 (m; 2H),
1,63-1,68 (m; 2H), 1.94 (s; 3H), 3,07 (d; J =6,OOHz; 2H), 5.01-5.06 (m; 1H),
7.21-7.33 (m; 3H), 7.51-7.54 (m; 2H); RMN 13C (125MHz, CDCI3), õ(ppm): 13,8;
18,5; 21,0; 31,6; 35,9; 73,2; 127,1; 129,1; 130,1; 132,8; 170,6; RMN 77Se
(95MHz, CDCh), õ(ppm): 260,3; I.V. (filme - KBr) Banda de absorção (cm-1):
692,738, 1237, 1372, 1435, 1459, 1474, 1578, 1739,2872,2960,3057; E.M.,
m/z (%rel.): 286 (27),285 (M+, 3), 226 (51),157 (35),129 (41),116(49),91
(52), 78 (45), 77 (51), 69 (80), 51 (44), 43 (100); Microanálise (C13H1802Se)
Calculado: C =54,74% e H =6,36% e observado: C =54,60% e H =6,36%.
(R,S)-O-acetil-1-fenilsseleno-3-metil-2-butanol: Óleo; rendimento = 0,9859
(96%;) RMN 1H (200MHz, CDCI3), õ(ppm): 0,77-0,98 (m; 7H), 1,95 (s; 3H),
3,05-3,09 (m; 2H), 4,85-4,97 (m; 1H), 7,23-7,32 (m; 3H), 7,50-7,55 (m; 2H);
RMN 13C (125MHz, CDCh), õ(ppm): 17,3; 18,4; 18,7; 20,8; 47,9; 77,4; 127,0;
129,0; 129,1; 132,9; 170,7; NMR 77Se (95MHz, CDCh), õ(ppm): 265,4; I.V.
(filme - KBr) Banda de absorção (cm-\ 694, 739, 1021, 1301, 1474, 1578,
1744,2874,2934,2965,3032,3058; E.M. m/z (% rel.): 286 (20),226 (44), 171
(10), 157 (24), 91 (45), 77 (42), 69 (96), 43 (100); Microanálise (C13H1802Se)
Calculado: C =54,74% e H =6,36% e observado: C =54,91% e H =6,31%.
102

4.2. Procedimentos Gerais
(R,S)-O-acetil-1-fenilsseleno-2-octanol: Óleo; rendimento = 0,965g (82%); N°
CAS: 67007-28-7; RMN 1H (500MHz, CDCb), õ(ppm): 0,86 (t; J = 7,2Hz; 3H),
1,23-1,29 (m; 8H), 1,62-1,70 (m; 2H), 1,93 (s; 3H), 3,06 (d; J = 6,OHz; 2H),
4,99-5,04 (m; 1H), 7,21-7,28 (m; 3H), 7,51-7,54 (m; 2H); RMN 13C (125MHz,
CDCb), õ(ppm): 14,0; 21,0; 22,5; 25,2; 29,1; 31,6; 33,8; 73,4; 127,6; 129,9;
132,8; 140,9; 170,6; RMN 77Se (95MHz, CDCb), õ(ppm): 264,0; I.V. (filme
KBr) Banda de absorção (cm-\ 691, 736, 1022, 1239, 1579, 1738, 2858, 2959,
2955, 3016, 3058; E.M. m/z (% rel.): 328 (15), 268 (44), 171 (25), 158 (6), 91
(46), 77 (38), 69 (89), 43 (100); Microanálise (C16H2402Se) - Calculado: C =
58,71 % e H = 7,39% e observado: C = 59,12% e H = 7,67%.
(R,S)- O-acetil-1-fenil-2-fenilsseleno-etanol: Óleo; rendimento = O,838g
(73%); RMN 1H (300MHz, CDCb), õ(ppm): 2,01 (s; 3H), 3,22 (dd; J = 12,8Hz e
J = 5,7Hz; 1H), 3,37 (dd; J = 12,8Hz; J = 7,9Hz; 1H), 5,93 (dd; J = 7,9Hz; J =
5,7Hz; 1H), 7,23-7,55 (m; 10H); RMN 13C (75MHz, CDCb), õ(ppm): 21,0; 33,4;
75,2; 126,6; 127,2; 128,3; 128,5; 129,1; 133,1; 140,4; 170,0; RMN 77Se
(95MHz, CDCb), õ(ppm): 275,6; Microanálise (C16H1602Se) - Calculado: C =
60,19% e H = 5,05% e observado: C = 59,97% e H = 5,39%
(R,S)-O-acetil-1-metilsseleno-2-fenil-etanol: Óleo; rendimento = 0,851 9
(92%); N° CAS: 56268-17-8; RMN 1H (200MHz, CDCb), õ(ppm): 1,91 (s; 3H),
2,09 (s; 3H), 2,87 (dd; J = 6,6Hz; J =12,9Hz; 1H), 3,00 (dd; J = 7,46; J =12,9Hz;
1H), 5,86-5,93 (m; 1H), 7,26-7,38 (m; 5H); RMN 13C (50MHz, CDCI3), õ(ppm):
5,0; 21,1; 30,7; 75,4; 126,7; 128,3; 128,5; 139,6; 170,0; RMN 77Se (95MHz,
103

4. Parte Experimental
COCb), 6(ppm): 64,3; E.M. m/z (% rel.): 258 (14), 198 (48), 181 (10), 163 (42),
149 (6), 104 (50), 91 (37), 77(48), 43(100).
(R,S)-O-acetil-1,1-difenilsseleno-2-propanol: Óleo; rendimento = 1,424g
(96%); RMN 1H (200MHz, COCb), 6(ppm): 1,45 (d; J = 6,6Hz; 3H), 1,88 (s; 3H),
4,52 (d; J = 3,5Hz; 1H), 5,14-5,25 (m; 1H), 7,22-7,33 (m; 6H), 7,52-7,55 (m;
4H); RMN 13C (50MHz, COCb), 6(ppm): 18,2; 20,8; 48,8; 72,7; 128,1; 128,2;
129,1; 129,2; 130,2; 130,5; 134,2; 134,9; 170,2; NMR 77Se (95MHz, COCI3),
6(ppm): 374,7; 382,4; E.M. m/z (% rel.): 414 (7),257 (46), 215 (58), 197 (52),
157 (46), 116 (34), 91 (31), 77 (64), 51 (52), 43 (100); Microanálise
(C17H1802Se2) - Calculado: C =49,53% e H =4,40 e observado: C =49,41 e H
=4,68.
(+f-)-2h
(R, S)-O-aceti1-1-hexi1-1-(fenilsseleno)-2-propanol: Mistura diastereomérica;
óleo; rendimento =1,154g (94%); RMN 1H (300MHz, COCI3), 6(ppm): 0,84-0,90
(m; 6H), 1,24-1,29 (m; 16H), 1,31 (d; J = 6,3Hz; 3H), 1,36 (d; J = 6,3Hz; 3H),
1,50-1,80 (m; 4H), 1,81 (s; 3H), 1,99 (s; 3H), 3,17-3,23 (m; 1H), 3,27-3,34 (m;
1H), 5,01-5,16 (m; 2H), 7,24 - 7,27 (m; 6H), 7,55-7,59 (m; 4H); RMN 13C
(75MHz, COCb), 6(ppm): 14,0; 17,2; 17,2; 20,9; 21,1; 22,5; 28,0; 29,0; 31,4;
31,6; 31,7; 50,5; 51,7; 72,6; 73,0; 127,2; 127,4; 128,9; 129,9; 130,0; 134,0;
134,7; 170,4; E.M. m/z (% rel.): 342 (2), 282 (4), 157 (20), 91 (4), 77 (30), 69
(100),55 (94),51 (18); Microanálise (C17H2602Se) - Calculado: C = 59,82% e H
=7,68% e observado: C =60,4% e H =7,82%.
104

4.2. Procedimentos Gerais
ooc/yseAl
Al(of{-)-2i
(R,S)-O-acetil-1-fenil-1-(fenilsseleno)-2-propanol: Mistura diastereomérica;
óleo; rendimento =1,1039 (92%); RMN 1H (200MHz, COCb), õ(ppm): 1,20 (d; J
=6,1 Hz; 3H), 1,30 (d; J =6,1 Hz; 3H), 1,90 (s; 3H), 1,98 (s; 3H), 4,27-4,37 (m;
2H), 5,33-5,49 (m; 2H), 7,11-7,28 (m; 16H), 7,35-7,43 (m; 4H); RMN 13C
(125MHz, COCb), õ(ppm): 18,9; 20,9; 21,1; 53,4; 53,7; 72,4; 73,4; 127,3; 127,7;
127,9; 128,2; 128,4; 128,7; 128,9; 129,1; 129,5; 135,0; 135,4; 170,2; 170,4;
E.M. m/z (% rel.): 334 (11), 274 (8),177 (52),158 (11),157 (19),135 (73),117
(61), 91 (41), 78 (39), 77 (44), 57 (38), 51 (35), 43 (100); Microanálise
(C17H1802Se) - Calculado: C =61,26% e H =5,44% e observado: C =61,42%
e H =5,56%.
(R,S)-O-acetil-1-fenilteluro-2-propanol: Óleo; rendimento = 0,9259 (84%);
RMN 1H (300MHz, COCI3), õ(ppm): 1,34 (d; J = 6,24Hz; 3H), 1,93 (s; 3H), 3,08
(d; J =6,19Hz; 2H), 5,03-5,13 (m; 1H), 7,17-7,28 (m; 3H), 7,73-7,76 (m; 2H);
RMN 13C (75MHz, COCb), õ(ppm): 14,8(f13c-125Te = 173 Hz); 21,1; 21,2; 71,4;
111,5; 127,8; 128,2; 138,5; 170,3; RMN 125Te (157MHz, 300K, COCb), õ(ppm):
440,5; I.V. (filme - KBr) Banda de absorção (cm-\ 692, 733,1241,1372,1434,
1574,1735,2870,2932,2979,3059; E.M. m/z (%rel.): 306 (M+, 5), 249 (2),222
(1),207 (5),101 (36),77 (26),43 (100); Microanálise (C11H1402Te) - Calculado:
C =43,20% e H =4,61 % e observado: C =43,10% e H =4,61 %.
Ok
~T~(~S)-11e
(R,S)-O-acetil-1-(n-butilteluro)-2-propanol: Óleo; rendimento = 0,8759 (85%);
RMN 1H (500MHz, COCI3), õ(ppm): 0,92 (t; J = 7,4Hz; 3H), 1,35 (d; J = 6,2Hz;
3H), 1,36-1,42 (m; 2H), 1,73 (sext.; J = 7,4Hz 2H), 2,04 (s; 3H), 2,69 (t; J =
7,4Hz; 2H), 2,77 (dd; J =7,2; J =12,2Hz; 1H), 2,83 (dd; J =5,6; J =12,2Hz;
105

4, Parte Experimental
1H), S,OO (sext.; J = 6,2Hz 1H); RMN 13C (125MHz, COCb), o(ppm): 3,7 (f13C
125Te = 147 Hz); 8,7 (J113c-125Te = 173 Hz); 13,4; 20,9; 21,3; 2S,O; 34,2; 72,0;
170,4; 125Te NMR (1S7MHz, 300K, CDCb), o(ppm): 189,0; E.M. m/z (% rel.):
288 (6), 258(2), 187 (3), 172 (8), 101 (29), S7 (18), 43 (100); Microanalise
(C9H180 2Te) - Calculado: C =37,82% e H =6,3S% e observado: C =37,80 e H
=6,38.
Ql)c
~Te~(~S)-11f
(R,S)-O-acetil-4-(n-butilteluro)-2-butanol: Óleo; rendimento = O,993g (92%);
RMN 1H (300MHz, CDCb), o(ppm): 0,92 (t; J =7,SHz; 3H), 1,23 (d; J =6,3Hz;
3H), 1,38 (sext.; J =7,SHz; 2H), 1,72 (quint.; J =7,SHz; 2H), 2,04 (s; 3H), 1,87
2,11 (m; 2H), 2,49-2,67 (m; 4H); RMN 13C (7SMHz, CDCI3), o(ppm): -3,6; 2,8;
13,4; 19,5; 21,3; 2S,O; 34,2; 38,8; 72,2; 170,6; 125Te NMR (157MHz, 298K,
CDCI3 ), o(ppm):.270,1; Microanalise (C1QH200 2Te) - Calculado: C =40,OS% e H
=6,72% e observado: C =40,02 e H =6;5~.
Ql)cTe~
~(~S)-11g
(R;S)-(Z)-O-acetil-4-(butiltêluró)but-3-en-2-ol: Óleo; rendimento = 1,008g
(94%); RMN 1H (300MHz; CDCb), o(ppm): 0,92 (t; J = 7,SHz; 3H), 1,30 (d; J =6,3Hz; 3H), 1,39 (sext.; J = 7,SHz; 2H), 1,78 (quint.; J =7,SHz; 2H), 2,06 (s;
3H), 2,67 (t; J =7,SHz; 2H),S,2S-S,37 (m, 1H); 6,2S (dd; J =7,2Hz e J =9,9Hz;
1H), 6,83 (d; J = 9,9; 1H); RMN 13C (7SMHz, CDCI3), o(ppm): 7,2; 13,4; 19,6;
21,26; 24,9; 34,0; 72,9; 10S,6; 138,0; 170,1; 125Te NMR (1S7MHz, 298K,
CDCI3), o(ppm): 293,6 Microanalise (C1QH180 2Te) - Calculado: C =40,32% e H
= 6,09% e observado: C = 40,48 e H = S,92.
4.2.20. Resolução Enzimática em Meio Orgânico
Em um Erlenmayer de SO mL provido de rolha de silicone ou septo, o
(±)-hidróxi-calcogeneto (O,S mmol) foi dissolvido em solvente orgânico seco (10
106

4.2. Procedimentos Gerais
mL - 25 mL). Em seguida, foi adicionada a lipase e acetato de vinila (3 mmol).
A mistura resultante foi mantida sob agitação contínua (160 r.p.m.) e
temperatura constante (1 O-40°C - Figura 3.2.20.1). A reação foi acompanhada
por CG-quiral. Ao final da reação (aproximadamente 50% de conversão) a
enzima foi removida por filtração simples ou por centrifugação, e o solvente
rotaevaporado. A separação dos produtos foi realizada por coluna
cromatogratográfica usando uma solução hexano/acetato (9: 1) como eluente.
EnzimaT
f f CG-QWaI
Substrato
Solnnte
Doador de ~Adia
Agitador Rotativo
Figtra 3.2.20.1. Procedimento experimental nas resoluções enzimáticas em meioorgânico.
ÇlH
~SeR1
(S)-1a
(S)-1-fenilsseleno-2-propanol: Óleo; >99% e.e.; rendimento =O,049g (46%);
[o.]D22 =+56 (c 1,0; CH2Cb).
(R)-O-acetil-1-fenilsseleno-2-propanol: Óleo; >99% e.e.; rendimento =0,054g (42%); [o.]D22 =-7 (c 1,0; CH2Cb).
107

4. Parte Experimental
a-t~SePh
(~1b
(S)-1-fenilsseleno-2-pentanol: Óleo; '>99% e.e.; rendimento = O,036g (30%);
[a]o23 = +41 (c 1,0; CH2CI2).
(R)-O-acetil-1-fenilsseleno-2-pentanol: Óleo; 94% e.e.; rendimento = O,050g
(35%); [a]o28 =+1 (c 1,0; CH2Cb).
(st-1c
(S)-1-fenilsseleno-3-metil-2-butanol: Óleo; 69% e.e.; [a]o20 = +47 (c 1,0;
CH2Cb).
(R)-O-acetil-1-fenilsseleno-3-metil-2-butanol: Óleo; 86% e.e.; [a]o20 = -12 (c
1,0; CH2CI2).
(S)-1-fenilsseleno-2-octanol: Óleo; 70% e.e.; [a]o22 = +12 (c 1,0; CH2CI2); na
literatura para o enantiomero (R): 93% e.e.; [alo19 = - 35,5 (c 0,986; CHCb).58
(R)-O-acetil-1-fenilsseleno-2-octanol: Óleo; 33% e.e.; [a]o22 = +2 (c 1,0;
CH2CI2).
108

4.2. Procedimentos Gerais
(R)-1-metilsseleno-2-fenil-etanol: Óleo; 41% e.e.; [a]o22 = -55 (c 1,0; CH2Cb).
(S)-O-acetil-1-metilsseleno-2-fenil-etanol: Óleo; 74% e.e.; [a]o22 =+12 (c 1,0;
CH2Cb).
~: SeRl
~SeRl
(5)-19
(S)-1,1-difenilsseleno-2-propanol: Óleo; >99% e.e.; [a]o21 = -43 (c 1,0;
CH2Cb).
(R)-O-acetil-1,1-difenilsseleno-2-propanol: Óleo; 99% e.e.; [a]o21 = +1 (c 1,0;
CH2Cb).
OH~TePh
(5)-108 .
(S)-1-fenilteluro-2-propanol: Óleo; >99% e.e.; [a]o28 =+5 (c 1,0; CH2Cb).
QOc
~TePh
(~11a
(R)-O-acetil-1-fenilteluro-2-propanol: Óleo; >99% e.e.; [a]o28 = -6 (c 1,0;
CH2Cb).
109
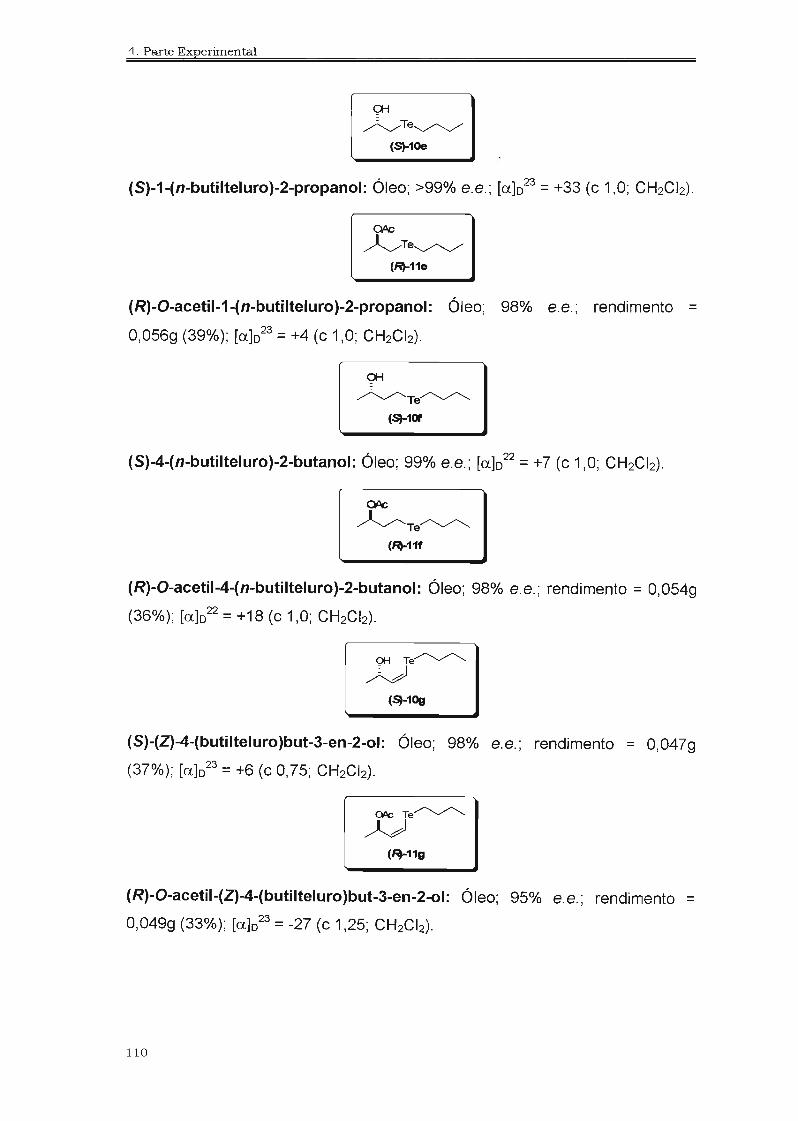
4. Parte Experimental
Q-I
~Te~
(8)-108 -
(S)-1-(n-butilteluro)-2-propanol: Óleo; >99% e.e.; [a]o23 =+33 (c 1,0; CH2CI2).
Q6c
~Te~(R)-11e
(R)-O-acetil-1-(n-butilteluro)-2-propanol: Óleo; 98% e.e.; rendimento =0,0569 (39%); [a]o23 = +4 (c 1,0; CH2CI2).
ÇH
~Te~
(S)-1or
(S)-4-(n-butilteluro)-2-butanol: Óleo; 99% e.e.; [a]o22 = +7 (c 1,0; CH2Cb).
QOc
~Te~(~11f
(R)-O-acetil-4-(n-butilteluro)-2-butanol: Óleo; 98% e.e.; rendimento =0,0549
(36%); [a]o22 = +18 (c 1,0; CH2CI2).
OH Te~
~(S)-10g
(S)-(Z)-4-(butilteluro)but-3-en-2-ol: Óleo; 98% e.e.; rendimento = 0, 0479
(37%); [a]o23 =+6 (c 0,75; CH2Cb).
QI)c Te~
~(~11g
(R)-O-acetil-(Z)-4-(butilteluro)but-3-en-2-o1: Óleo; 95% e.e.; rendimento =
0,0499 (33%); [a]o23 = -27 (c 1,25; CH2CI2).
110

4.2. Procedimentos Gerais
4.2.21. Imobilização Enzimática
o procedimento para imobilização de lipases depende do suporte a ser
empregado:
4.2.21.1. Gel de Agar
Em um tubo de ensaio foi colocado Agar (0,4g) e a seguir foi
adicionada água destilada (9mL). Esta mistura foi levada a um aquecimento de
aproximadamente 100oe, (temperatura no qual o Agar se dissolve). Após o
Agar ter sido dissolvido, a mistura foi resfriada à temperatura de 400e(naturalmente). Em seguida foi adicionada uma solução aquosa (ou suspensão
em H20) da enzima (100mg), esperando-se a formação do gel. O gel foi
cortado em cubos que foram mantidos em solvente orgânico.
4.2.21.2. Filme de caseinato de Sódio
Em um pequeno béquer (25mL) foram colocados caseinato de sódio
(1,50g), glicerol (O,50g - plastificante), água destilada (15mL) e a enzima
(100mg). Em seguida, essa solução foi colocada em uma placa de petri de
poliestireno (d ~ 8,7cm), para a evaporação do solvente à temperatura
ámbiente e formação do filme. Após a evaporação, o filme foi retirado, cortado
em pequenas porções e mantido em solvente orgânico.
4.2.21.3. SJ7ica ou Montmorílonita K-l0 (Silicato deAluiriínlo)
A imobilização foi realizada misturando-se uma solução (ou
suspensão) da enzima (100mg) em água destilada (5mL) com uma suspensão
de sílica ou Montmorilonita K-10 (500mg) em água destilada (10mL) a
temperatura ambiente. A mistura resultante foi agitada mecanicamente por 5
horas. Ao final, essa mistura foi colocada em uma placa de Petri (d ~ 8,7cm)
para a evaporação do solvente à temperatura ambiente.
4.2.21.4. Poli(oxietíleno) - PEO
O filme de PEO foi obtido misturando-se uma solução (ou suspensão)
da enzima (100mg) em água destilada (5mL) com uma solução de PEO
(500mg) em água destilada (20mL), previamente agitada por 12 horas a
111

4. Parte Experimental
temperatura ambiente. A mistura resultante foi agitada mecanicamente por 5
horas. Ao final, essa mistura foi colocada em uma placa de petri (d :::::: 8,lcm)
para a evaporação do solvente à temperatura ambiente.
4.2.22. Triagem Enzimática
Inicialmente foram preparadas soluções dos acetatos (30mg) em
DMSO (dimetilsulfóxido - 1mL) e das enzimas (15mg) em tampão fosfato (1 mL
- pH = 1,2). O tampão foi preparado pela adição de uma solução de NaH2P04
(0,002 MoIL-1) sobre uma solução de Na2HP04 (0,002 MoIL-1
) ate atingir
pH=1,2, controlada por pHmetro. Também foi preparada a solução do indicador
colorimétrico (0,005% - azul de bromotimol) em tampão. Em seguida, foram
adicionadas as soluções nos pocinhos da placa de Elisa obedecendo ao
seguinte padrão (Figura 3.2.22.1): No pocinho A1 apenas solução tampão
(180IJL), nos demais pocinhos da linha A foram adicionadas as soluções
enzimaticas (40 IJL \ branco - uma enzima para cada pocinho) e a solução do
indicador (lOIJL), fazendo-se sempre em duplicata.
Indicador- Branco:
Enzima
Branco:Substrato - Indicador
1 2 3 4 5 6 7 8 9 1011 12A J(j(J()( )( )(
B r;)C )( ?~ )~ d C)( )~ rC b< x OOx .:J<X x QO<x ';.:D~~~~~~~*~~04E - Subsh-ato
F "--/.~ '---' "--/ '-" >-./'---'.~'---' "--/ '-'O'--A
G ~õocl~P)CXooCr<
H ~POOO~=SOOL~I I I
lipa~'eBranco:
Substrato Indicador
Figura 3.2.22.1. Disposição das soluções na placa de Elisa.
Na primeira coluna (exceto o pocinho A1) foram adicionados os
brancos do substrato (40 IJL) mais a solução do indicador (lOIJL). Os demais
pocinhos foram completados com soluções da enzima e do substrato,
respectivos a coluna e linha que ele se encontra, acrescido da solução do
112

4.2. Procedimentos Gerais
indicador conforme Figura 3.2.22.1. Em alguns casos a enzima apresentou
acidez, acarretando mudança de cor no momento da adição (mudança de pH).
Esta mudança de pH foi corrigida pela adição da solução de Na2HP04 ate pH
neutro (cor verde). Ao final das adições o volume foi ajustado com tampão
(180~L) e a placa incubada a 3rC.
4.2.23. Hidrolise Enzimática
Em um Erlenmayer de 125mL provido de rolha de silicone ou septo, o
(±)-hidróxi-calcogêneto (0,5mmol) foi dissolvido em acetona (3mL). Em
seguida, foi adicionado tampão fosfato (pH = 7,2 - 10mL) e a enzima (10
100mg). A mistura resultante foi mantida sobre agitação continua (160 r.p.m.) e
incubada a 32°C. A reação foi acompanhada por CG-quiral. Ao final da reação
(aproximadamente 50% de conversão) a enzima foi removida por filtração
simples ou por centrifugação, e os produtos extraídos com acetato de etila. A
fase orgânica foi seca com MgS04 e o solvente rotoevaporado. A separação
dos produtos foi realizada por coluna cromatogratográfica usando uma solução
hexano/acetato (9: 1) como eluente.
4.2.24. Biotransformação Empregando Células Inteiras de Fungos
Substrato
J
Figura 3.2.24.1. Procedimento nas biotransformações empregando células inteiras de fungos.
Inicialmente, os fungos foram crescidos em meio sólido e
posteriormente em meio líquido, com o objetivo de aumentar a massa celular e
113

4. Parte Experimental
conseqüentemente à quantidade de enzimas. Após O crescimento em meio
líquido, o meio reacional foi filtrado, separando as células do meio de cultura
(Figura 3.2.24.1). Em seguida as células foram ressuspensas em tampão,
adicionando-se o substrato e mantendo a agitação a 30°C. Alíquotas de 3mL
foram retiradas periodicamente para o acompanhamento da reação via CG
quiral.
4.2.24.1. Crescimento dos Fungos em Meio Sólido
Os fungos Aspergillus terreus foram adquiridos da Coleção de Culturas
Tropicais da Fundação André Tosello. 77 Posteriormente foram feitos repiques
em tubos inclinados contendo um meio de cultura constituído por extrato de
malte (2g), agar (2g) e água destilada (1 OOmL). Os fungos foram colocados
para crescer em estufa a 32°C.
4.2.24.2. Crescimento dos Fungos em Meio Líquido
Os fungos foram transferidos dos tubos inclinados para frascos
Erlenmeyer contendo meio de cultura líquido composto por extrato de malte
(2g) e água destilada (100 mL). Em alguns casos foram utilizados Erlenmeyers
de 1-3L. O crescimento dos fungos foi realizado em agitador rotativo a 170 rpm
e 32°C por72-96 horas.
4.2.24.3. Obtenção das Células Úmidas de Fungos
Após o crescimento dos fungos, as células foram filtradas em funil
Buchner. A quantidade de células empregadas de fungos foi de 1-5g de células
úmidas.
4.2.24.4. As Bíotransformaçães
As células úmidas de fungos foram resuspensas em solução tampão
fosfato S6rensen (Na2HP04 e KHP04 0,1 MoIL-1) em frascos Erlenmeyer de 250
mL (50mL de tampão), 125 (30mL de tampão) ou de 50 mL (20mL de tampão).
Em seguida foi adicionado o substrato (20I-'L) e mantido em agitador rotativo a
170 rpm e 32°C (Figura 3.2.24.1). A reação foi acompanhada por CG-Quiral,
retirando-se alíquotas (3mL) periodicamente em tubos de centrifuga,
adicionando-se 0,5mL de acetato de etila, agitando e centrifugando (6000 rpm,
5 minutos).
114

4.2. Procedimentos Gerais
,4.2.25. Preparação do Alcool Alílico - Reação de Eliminação de
Selen6xido 38
Em um balão, o (S)-1-hexil-1-(fenilsseleno)-2-propanol com 92% e.e.
[(S)-1h] (0,057g - 0,2 mmol) foi dissolvido em THF (5 mL). Em seguida H20 2
(30% sol. - 0,6 mmol) foi adicionada lentamente à temperatura ambiente. A
mistura reacional foi mantida sob agitação durante 4 horas, diluída com água e
extraída com várias porções de éter etílico. A fase orgânica foi lavada com
solução concentrada de Na2C03 e NaCI e seca com MgS04. O resíduo foi
purificado por cromatografia utilizando hexano:acetato de etilei (9: 1).
(S)-3-nonen-2-ol: Óleo; rendimento = 0,027g (95%); N° CAS: 173144-01-9;
92% e.e.; [a]21 D = -8 (c 1.5, CHCb); RMN 1H (200MHz, CDCb), õ(ppm): 0,89 (t;
J = 4,50Hz; 3H), 1,25-1,39 (m; 8H), 1,97-2,04 (m; 3H), 4,22-4,31 (m; 1H), 5,50
(dd; J = 4,35Hz; J =1 0,45Hz; 1H), 5,67 (dt; J = 4,40Hz; J = 10,45Hz; 1H); E.M.
m/z (% rel.): 142 (1), 124 (25),84 (23),71 (100),68 (72),58 (30),54 (55),43
(12).
4.2.26. Preparação das Lactonas
Em um balão de duas bocas equipado com um septo, agitação
magnética e sob nitrogênio, o hidróxi-telureto enantiomericamente enriquecido
(1 mmol), foi dissolvido em THF seco (5mL). Em seguida, a solução foi
resfriada a -78°C e n-SuLi (1,69mL - 1,3MoIL-1 em hexano - 2,2mmol) foi
adicionado lentamente. Após 20 minutos, CO2 seco foi borbulhado na solução
por 30 mino Na seqüência, a temperatura foi elevada gradualmente à
temperatura ambiente. A mistura reacional foi lavada com solução de HCI
(50%) e extraída com éter etílico. A fase orgânica foi seca com MgS04 ,
sõlvente destilado e o resíduo purificado por cromatografia em coluna utilizando
pentano:éter etíico (1 :1).
115

4. Parte Experimental
o
(~14J:5
(R)-y-valerolactona: Óleo; rendimento =53%; N° CAS: 58917-25-2; 98% e.e.;
(a]o23 =+30 (c 1,0; CHCI3); RMN 1H (300 MHz, CDCI3), õ(ppm): 1,43 (d; J =6,3
Hz; 3H), 1,78-1,91 (m; 1H), 2,54-2,59 (m; 2H), 4,6-4,71 (m;1 H), RMN 13C (75
MHz, CDCb), õ(ppm): 21,0; 29,1; 29,8; 77,4; 177,6; I.V (filme KBr) banda de
absorção (cm-1): 2998, 1777, 1432, 1130; E.M. m/z (% rel.): 41 (47), 43 (34), 56
(100),85 (45),100 (8).
o
(~16 ",,,(j
(S)-J3-angelica: Óleo; rendimento = 51 %; N° CAS: 92694-51-4; 98% e.e.; [a]o23
=+100 (c 0,25; CHCb); RMN 1H (200 MHz, CDCb), õ(ppm): 1,46 (d; J =6,6 Hz;
3H), 5,09-5,21 (m; 1H), 6,11 (dd; J =5,7 Hz e J =2,2 Hz; 1H), 7,47 (dd; J =5,7
Hz e J =1,7 Hz; 1H); RMN 13C (50 MHz, CDCb), õ(ppm): 18,7; 79,6; 121,2;
157,3; E.M. m/z (% rel.): 43 (58),55 (100),69 (12) 83 (40),98 (34).
4.2.27. Determinação dos Excessos Enantioméricos
As condições das análises em CG-Quiral para cada composto foram
as seguintes: (R,S)-1a: coluna GAMA DEX™; pressão =100 Kpa; isoterma a
135°C; tR1 = 24,7 mino e tR2 = 25,2 min.; (R,S)-1b: coluna GAMA DEX™;
pressão = 80 Kpa; isoterma a 140°C; tR1 = 59,5 mino e tR2 = 61,2 min.; (R,S)
1c: coluna GAMA DEX™; pressão = 100 Kpa; isoterma a 135°C; tR1 = 42,0
mino e tR2 = 43,0 min.; (R,S)-1d: coluna GAMA DEX™; pressão = 100 Kpa;
isoterma a 150°C; tR1 = 106,2 mino e tR2 = 107,9 min.; (R,S)-1f: coluna GAMA
DEX™; pressão = 80 Kpa; isoterma a 125°C; tR1 = 75,6 mino e tR2 = 77,6 min.;
(R,S)-2a: coluna BETA DEX™; pressão = 115 Kpa; isoterma a 125°C; tR1 =38,0 mino e tR2 = 38,8 min.; (R,S)-2b: coluna BETA DEX™; pressão =90 Kpa;
isoterma 120°C; tR1 =54,6 mino e tR2 =55,1 min.; (R,S)-6: coluna ALFA DEX™;
pressão = 70 Kpa;50°C-60 min, 50-190°C, 1°C/min; tR1 = 171,8 mino e tR2 =173,7 min.; (R,S)-7: coluna CHIRASIL-DEX CB; pressão = 85 Kpa; 60°C-40
min, 60-120°C, O,5°C/min, 120°C-15 min; tR1 =92,0 mino e tR2 =95,2 min.;
116

4.2. Procedimentos Gerais
(R,S)-10a: coluna GAMA DEX™; pressão = 70 Kpa; 120°C-100 min, 120
170°C, 0.5°C/min, 170°C-30 min; tR1 = 119,1 mino e tR2 = 121,5 min.; (R,S)
10e: coluna GAMA DEX™; pressão = 90 Kpa; 65°C-150 min, 65-98°C,
O,5°C/min; tR1 = 198,4 mino e tR2 = 200,2 min.; (R,S)-10g: coluna CHIRASIL
DEX CB; pressão = 120 Kpa; 80°C-120 min, 80-160°C, 2°C/min, 160°C-30 min;
tR1 = 144,1 mino e tR2 = 145,2 min.; (R,S)-11a: coluna CHIRASIL-DEX CB;
prêssão =90 Kpa isoterma a 120°C; tR1 =35,3 mino e tR2 =39,6 min.; (R,S)
11e: coluna CHIRASIL-DEX CB; pressão = 125 Kpa 80°C-120 min., 80-110°C,
2°C/min, 11 0°C-1 O min; tR1 =102,2 mino e tR2 = 125,1 mino ;(R,S)-11f: coluna
CHIRASIL-DEX CB; pressão = 80 Kpa; 80°C-90 min, 80-140°C, 1°C/min,
140°C-30 min; tR1 = 126,8 mino e tR2 = 134,3 min.; (R,S)-11g: coluna
CHIRASIL-DEX CB; pressão = 120 Kpa; 80°C-120 min, 80-160°C, 2°C/min,
160°C-30 min; tR1 = 142,5 mino e tR2 = 143,3 min.; (R,S)-14: coluna CHIRASIL
DEX CB; pressão =100 Kpa; 60°C-3 min, 60-190°C, 5°C/min, 190°C-10 min;
tR1 =10,0 mino e tR2 =10,3 min; (R,S)-15: coluna CHIRASIL-DEX CB; pressão
=100 Kpa; 60°C-10 min, 60-90°C, 2°C/min, 90°C-30 min; tR1 =15,6 mino é tR2
= 16,0 min.; (R,S)-16: coluna CHIRASIL-DEX CB; pressão = 100 Kpa; 80°C-90
min, 80-175°C, 1°C/min, 175°C-30 min; tR1 =78,6 mino e tR2 =83,1 min;
Os valores de E (razão enantiomérica) foram calculados a partir dos
excessos enantioméricos dos produtos e substratos, de acordo com a equação
dê Sirh, Sharpless e Fajans {E = In [1-c (1 +e.e. p)]/In [1-c (1-e.e. p)), onde c =e.e'$/ (e.e'$ + e.e. p)}.9. 78
4.2.27.1. Hidrólise Química
Em um balão o acetato (1 mmol) foi dissolvido em metanol (5mL) em
seguida foi adicionando H20 (2 mL) e K2C03 (0,2 mmol). A mistura foi agitada
durante toda a noite à temperatura ambiente. Depois deste período, a mistura
foi extraída com acetato de etila e a fase orgânica foi lavada com solução
saturada de NaCI e seca com MgS04. O produto obtido foi purificado por
cromatografia usando uma mistura de hexano acetato (85: 15).
4.2.27.2. Desselenização Redutiva S4
Em um balão de duas bocas equipado com septo, com agitação
f:i'iªQnética e sob nitrogênio, uma quantidade catalítica de CCN [1,1'-
117

4. Parte ExperlinenUli
azobis(cicloexanacarbonitrila)] e o 13-hidróxi-seleneto (0,2 mmol) foram
dissolvidos em tolueno seco. Em seguida a solução foi pré-aquecida e o
n-BuSnH foi adicionado (0,2 mmol). A mistura reacional foi agitada sob refluxo
durante 4 horas. Após este período, o sistema foi resfriado a temperatura
ambiente hexano foi adicionado e a mistura foi extraída com acetonitrilá. A,
fase acetonitrila foi evaporada e o resíduo purificado por cromatografia
utilizando hexano:acetato (9: 1).
4.2.27.3. Derívatização Utilizando Anidrido Tri-fluoracético
Em um viaI de 2mL, o hidróxi-telureto 10f (O,05mmol) foi dissolvido em
THF (1 mL) em seguida foi adicionado anidrido de tri-fluoracético (duas gotas).
A mistura reacional foi agitada durante 5 rYíinut~$.
4.2.28. Determinação da Configuração Absoluta
4.2.28.1. Preparação do Cloreto de Ácido de Mosher 55, 79
Em um balão de duas bocas equipado com septo, com agitação
mã§A~tica e sob nitrogênio, o ácido (MTPA; O,5mmol) foi dissolvida o em 5ml
de hexano seco. Em seguida, adicionou-se Cloreto de oxalila (5mmol) e DMF
(1 gota) a temperatura ambiente. Após dois dias, o solvente foi rotaevaporado.
4.2.28.2. Preparação dos Derivados de Mosher 79
Em um balão de duas bocas equipado com septo, com agitação
magnética e sob nitrogênio, o cloreto de ácido (MTPA-CI; 0,5 mmol) foi
dissolvido em CH2CI2 seco (5mL). Em seguida, foi adicionada uma solução do
hidróxi-calcogeneto (0,1 mmol), Et3N (1,2 mmol) e DMPA (0,1 mmol) em
CH2Cb seco (10 mL). A agitação foi mantida por 30 minutos a temperatura
ambiente. Depois deste período a solução resultante foi lavada com água,
extraída com acetato de etila, lavada com solução saturada de NaCl, seca com
MgS04 e o solvente rotaevaporado. O resíduo foi purificado em placa
preparativa utilizando uma mistura hexano:acetato de etila (85: 15) como
eluente. Entretanto, para o derivado do hidróxi-telureto Se, devido à
118

4.2. Procedimentos Gerais
instabilidade do éster formado, não foi possível realizar a purificação. Assim,
após os 30 minutos de agitação o solvente foi evaporado e o resíduo filtrado
rapidamente em coluna de sílica utilizando uma mistura hexano:acetato de etila
(85: 15) como eluente.
(2R)-(5)-3,3,3-Trifluoro-2-metóxi-2-fenil-p(oPilnoato de 1-fenilseleno-2
propila: Óleo; rendimento =0,029g (67%); RMN 1H (500MHz, CDCb), 6(ppm):
1,44 (d; J =6.29Hz; 3H), 2,93 (dd; J =12.82Hz e J =6.76Hz; 1H), 3,14 (dd; J =
12,82Hz e J =6,30Hz; 1H), 3,55 (s; 3H), 5,25-5,31 (m; 1H), 7,23-7,55 (m; 10H);
RMN 13C (125MHz, CDCb), 6(ppm): 19,7; 32,2; 55,5; 73,1; 165,9; RMN 77Se
(95MHz, CDCI3), 6(ppm): 269,9; E.M., m/z (%rel.): 431 (M+,19), ·275 (43)", 217
(11),198 (68),189 (100),157 (64),105(49),91 (37),77 (78), 69 (17),51 (32).
AlÇlMe;::3~5eR1
(2S)-(~
(25)-(5)-3,3,3-Trifluoro-2-rtIefóxi-2-fenil-propanoato de 1-fenilseleno-2
propila: Óleo; rendimento = 0,030g (69%); RMN 1H (500MHz, CDCb), 6(ppm):
1,35 (d; J =6,26Hz; 3H), 3,02 (dd; J = 12,88Hz; J =6,07Hz; 1H), 3,17 (dd; J =
12,88Hz e J =6.99Hz; 1H), 3,58 (s; 3H), 5,24-5,30 (m; 1H), 7,25-7,57 (m; 10H);
RMN 13C (125MHz, CDCI3), 6(ppm): 20,0; 32,8; 56,1; 73,5; 166,4; RMN 77Se
(95MHz, CDCb), 6(ppm): 271,1; E.M., m/z (%rel.): 431 (M+, 13), 275 (36), 217
(11), 198 (65), 189 (100), 157 (68), 105 (54), 91 (42), 77 (88), 69 (20), 51 (39).
119

4. Parte Experimental
(~S)-17
(2R)-( 5)-3,3,3-Trifluoro-2-metóxi-2-fenil-propanoato de 1-fenilteluro-2
propila: Óleo; rendimento =0,0259 (52%); RMN 1H (500MHz, CDCb), ú(ppm):
1,45 (d; J =6,23Hz; 3H), 2,98 (dd; J =12,32Hz e J =7,32Hz; 1H); 3,14 (dd; J =12,32Hz e J =5,84Hz; 1H), 3,56 (5; 3H), 5,30-5,36 (m; 1H), 7,27-7,64 (m; 10H);
RMN 13C (125MHz, CDCI3), ú(ppm): 13,35; 20,86; 55,50; 74,80; 111,38; 127,34;
128,41; 129,39; 129,60; 130,02; 138,50; 165,84; RMN 125Te (157MHz, 300K,
CDCb), ú(ppm): 440,9.
(25)-(5.)-17
(25)-(5)-3,3,3-Trifluoro-2-metóxi-2-fenil-propanoato de 1-fenilteluro-2
propila: Óleo; rendimento =0,0249 (50%); RMN 1H (500MHz, CDCb), ú(ppm):
1,38 (d; J =6,21 Hz; 3H), 3,06 (dd; J =12,35Hz e J =7,05Hz; 1H), 3,17 (dd; J =
12,35Hz e J =6,25Hz; 1H), 3,56 (5; 3H), 5,29-5,34 (m; 1H), 7,26-7,59 (m; 10H);
RMN 13C (125MHz, CDCb), ú(ppm): 13,39; 20,72; 55,63; 74,92; 111,32; 127,71;
128,41; 129,43; 129,60; 130,03; 138,55; 165,94; RMN 125Te (157MHz, 300K,
CDCb), ú(ppm): 441,9.
(~S)-18
(2R)-(5)-3,3,3-Trifluoro-2-metóxi-2-fenil-propanoato de 1-(n-butilteluro)-2
propila: Óleo; RMN 1H (500MHz, CDCb), ú(ppm): 0,90 (t; J = 7,3Hz; 3H), 1,31
1,38 (m; 2H), 1,48 (d; J = 6,2Hz; 3H), 1,65-1,71 (m; 2H), 2,57-2,67 (m; 2H),
2,70 (dd; J =12,3Hz e J =7,9Hz; 1H), 2,86 (dd; J =12,3Hz e J =5,3Hz; 1H),
120

4.2. Procedimentos Gerais
3,57 (s;3H), 5,23-5,30 (m; 1H), 7,38-7,43 (m; 3H), 7,53-7,55 (m; 2H); RMN 13C
(125MHz, CDCI3), õ(ppm): 4,1; 7,2; 13,4; 20,7; 24,9; 34,1; 55,5; 75,4; 84,5;
127,3; 128,4; 129,5; 132,4; 165,9; RMN 125Te (157MHz, 300K, CDCI3), õ(ppm):
204,9.
A1Çltv1e;::3~Te~
(2Sl-(~18
(25)-(5)-3,3,3-Tritluoro-2-metóxi-2-tenil-propanoato de 1-(n-butilteluro)-2
propila: Óleo; RMN 1H (500MHz, CDCb), õ(ppm): 0,91 (t; J = 7,3Hz; 3H), 1,32
1,39 (m; 2H), 1,40 (d; J =6,2Hz; 3H), 1,68-1 ,74(m; 2H), 2,67-2,70 (m; 2H), 2,79
(dd; J = 12,3Hz e J = 7,7Hz; 1H), 2,91 (dd; J = 12,3Hz e J = 5,7Hz; 1H), 3,57 (s;
3H), 5,23-5,28 (m; 1H), 7,39-7,45 (m; 3H), 7,53-7,56 (m; 2H); RMN 13C
(125MHz, CDCb), õ(ppm): 4,2; 7,3; 13,4; 20,6; 25,0; 34,1; 55,5; 75,6; 84,5;
127,5; 128,4; 129,6; 132,3; 166,0; RMN 12sre (157MHz, 300K, CDCb), õ(ppm):
206,3.
4.2.28.3. Desselenização Redutiva 54
Eªsas reações foram realizadas seguindo o procedimento descrito na
~~9~€l 3;~.~7.2, utilizando os hidróxi-selénetos correspondentes não acetilados
preferencialmente pela liji)~~~;
(R)-2-pentanol [obtido do (S)-1-fenilsseleno-2-pentanol (S)-1b]: >99% e.e.;
[a]o19 =-12 (c 1,0; CHCb); na literatura para o enantiômero (S): 79% e.e.; [alo =+10,25 (c 0,986; CHCb). 56
121

4. Parte Experimental
(R)-3-metil-2-butanol [obtido do (S)-1-fenilsseleno-3-meti1-2-butanol (5)-1 c]:
69% e.e.; [a]o20 =-2 (c 2,0; CHCb); na literatura para o enantiômero (R): 86%
e.e.; [alo = -2,74 (c 0,986; CHCb). 56
(8)«
(5)-1-feniletanol [obtido do (R)-1-metilsseleno-2-fenil-etanol (R)-1f]: 41 % e.e.;
[a]o20 =-24 (c 2,0; CHCb); na literatura para o enantiômero (S): >99% e.e.; [alo
= -55,1 (c 1,63; CHCb). 57
(5)-1-fenil-2-propanol [obtido da mistura diastereoisomérica dos (1 R,2S)-1
fenil-1-(fenilsseleno)-2-propan~1 e (1 S,2S)-1-fenil-1-(fenilsseleno)-2-propanol]:
92% e.e.; [a]o21 = +37 (c 1,0; CHCI3); na literatura para o enantiômero (S):
>99% e.e.; [alo =+41,7 (c 1,19; CHCb). 57
4.2.28.4. Eliminação de 5elenóxida 38
Essa reação foi realizada seguindo o procedimento descrito na seção
4.2.25, utilizando os hidróxi-seleneto 1h não acetilado preferencialmente pela
lipase.
[~l(5)-3-nonen-2-ol [obtido da mistura diastereoisomérica dos (1 R,2S)-1-hexil-1
(fenilsseleno)-2-propanol e (1 S,2S)-1-hexil-1-(fenilsseleno)-2-propanol]: 92%
122

4.2. Procedimentos Gerais
e.e.; [a]21 o =-8 (c 1,5, CHCI3); na literatura para o enantiômero (R): 97% e.e.;
[a] 19 = +1068 (c 1 03' CHCI ) 58o , " 3 .
4.2.28.5. Reação de Lactonízação
Essa reação foi realizada seguindo o procedimento descrito na seção
4.2.26, utilizando o telureto (R)-11f (e.e. = 98%) acetilado preferencialmente
pela lipase e o hidróxi-telureto (S)-109 (e.e. = 98%), não acetilado
preferencialmente na resolução.
Assim, inicialmente foi realizada a hidrólise química do telureto (R)
11f, gerando o hidróxi-telureto (R)-10f e em seguida a reação de lactonização
foi realizada gerando as lactonas de interesse.
o(~14 Jj
(R)-y-valerolactona [obtido do (R)-4-(n-butilteluro)-2-butanol (R)-10f]: 98%
e.e.; [a]o23 =+30 (c 1,0; CHCb); na literatura para o enantiômero (S): 74% e.e.;
[alo =-26,7 (c 1,0; CHCb). 71
o
(~16 "",(j
(S)-J3 -angelicalactona [obtido do (S)-(Z)-4-(butilteluro)-but-3-en-2-ol (S)-1 09]:
98% e.e.; [a]o23 =+100 (c 0,25; CHCb); na literatura para o enantiômero (S):
>99% e.e.; [a]o21 = + 117 (c 3,60; CHCI3). 72
123


4.3. Espectros Selecionados
4.3. Espectros Selecionados

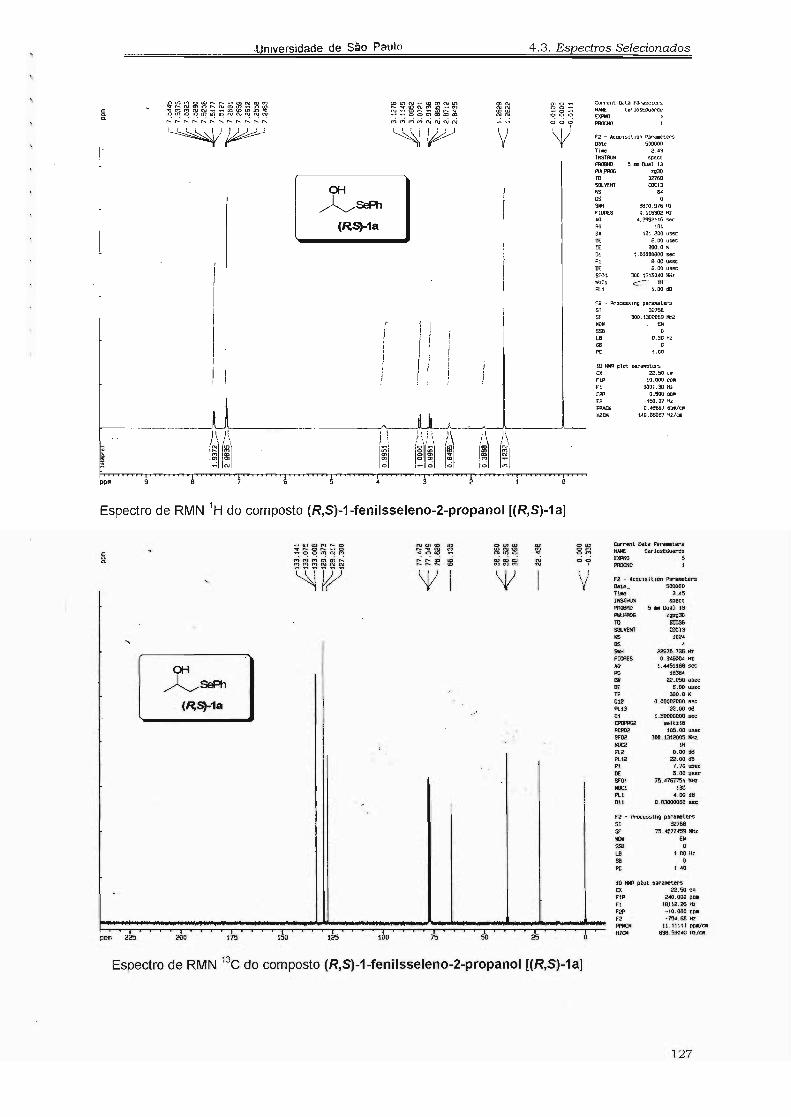
Universidade de são Paulo 4.3. Espectros Selecionados
f2 - kouisjtlan Paril1lelel"'5
Currenl DaLa ParilfttersNME Cal'lcsElkhlNloEXPNJ .4
PROCNO I
5Oll00ll2.43
SGe.ct5 .. Dulll t3,,30
327..com...
o38S0.976 ff7O.JleJ02 Hz
".2992H5 sec
."t3i.2OO ustC6.00 usee
300'.0 KLOOOOOOQO sec
8.00 usec6.00 U~
300. t3t~O Mi2.:""~~-"-lti·
1.00 dO
"'''Ti..INST1U<......,F'l.V'llOO'0so.Y<H1NSoss.,FImESIollA6OWDETEo.P,DESfOt·OUC.PlI
~~~
\Vv~~gj~~~;:~- ..... OOClaJ(Dm
t"if'ÍP'if'Ínfni~N
~~~~~~ruiíi~~:R~
~~:cp~~t'1~~~~~~~,....,.....,...,...r-.,...,.... ..... " ..... ,...."
rI
Ji
.I
I !i [11, ,
F2 - ProctS$íng gar••tIf'5
SI 32768Sf 300.1300069 ItilWOIf _. EMSS8 oLa 0.30 Hz.. oPC LOO
tU ~ plot uera.!lersCll 22.50 ,aFtP 10.000 Jip.FI 3001.30 Hl-F2P -O.!OQ pp.f2 -150.07 HlPA«Jl O.046667~ctI
IilCM 140.06067 tt2/CIl
00.
Espectro de RMN 1H do composto (R,S)-1-fenilsseleno-2-propanol [(R,S)-1a]
Currenl Data ParaftttrsNME Car lost:CSuarooOI'NJ 5
"""'"' .-olCllOM,...O
~~~ ~ g~~ "'"'If ,... C),... ..... o '"g _OOC"1NPl V o '" 0."'0 vMMMcnair--: ~~~ :8 ~~~ '"
~1P'"
\V I \V
o'"o'"oM
00
; J
Y Oatl!_Ti.INSfIQIPRIIlIIlPIl.PAOG10so..VENTllSOSSII<FrMfSAORG!lim:TE012RI3O,CPIJ'I'G2PCPII2Sf02NU:2R2PU"P!DESfO'1llC'RI011
5000003."'5
spect5. DJal t3
zgpg3065536CPCl3
t02-4,22675.736 Hz
0.34600-4 H!1.-4-451188 sec
"...22.050 usec
6.00 usec300.0 K
0.00002000 !!JI!C
22.00 lJ81.50000000 sec
waltzj6tOS.DO usec
300 . .13t2005 ~IH
D.OO dO22.00 da7.70 U$ltC6.00 usec
75..t76nSt NH2.30
4.00 da0.03000000 sec
F2 - Protl!sslllQ par&l!llltersSI 32768Sf 75 . .4677459 ...,z""" EMS58 olO LDO H.zGIl oPC 1."0
oom 225 200 125 100 75 50
to ft4P plGt oiJN~lers
ex 22.50 "C.F1P 240.000 DIlIIf1 18112.26 Hzf2P -10.000 111111F.2 -154.68 HZPflMCN 11.11111 opll/clllHZCM 838.53040 1l1/cll
Espectro de RMN 13C do composto (R,S)-1-fenilsseleno-2-propanol [(R,S)-1a]
127
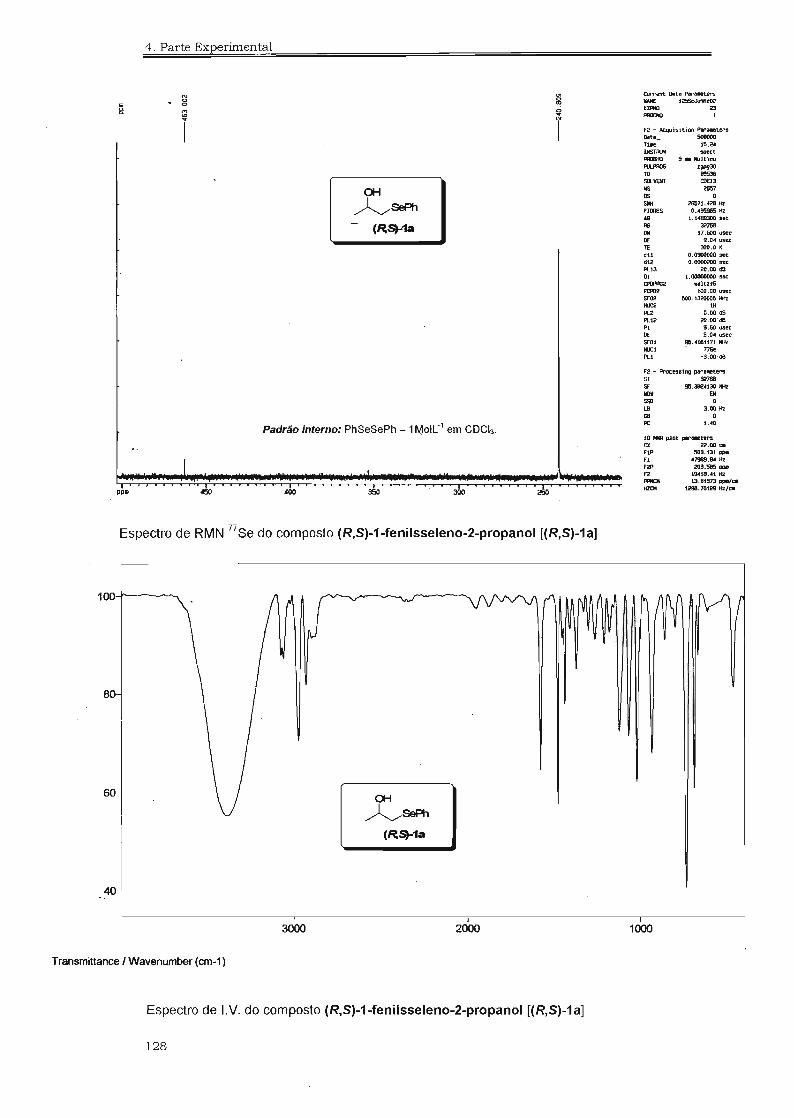
Padrão interno: PhSeSePh - 1MoIL-' em CDCh.
4. Parte Experimental
no• g
i
3SO 300
'"ãl
iQ.lrrent. o.t. Plr_ttNlrWE 125Sc.)JMo02.-, 23PIlllClIO I
F2 - Jcqutsttian ~UrllDlte_ 500000TiR 15.2~
lMSTRM sPeCtPIIIHl 5 _ MJltlpu
PlJJlfQi Igpg30T1l _
StLYENT mel]1IS 2l!l1ll5 oSItf 28571.42& HlFIIJIES 0.435965 ...AQ 1. t4693DCI secA6 327118DI 17.500 usecCE g.Q~ U$tC
TE 300.(1 K
ou 0.0300000 seed12 O.()()()0200 secflt.13 20.00 d8Dl t •oooooooo secCPlJ1RG2 ..lut&PQ3D2 100.00 usecSF02 !5OO.132OOQ5 MHz!IlC2 tl<...2 0.00 dePLt2 2O.00'dBPt e.tiO usec[E .9.,,º4 uneSFOt 9!5 ••16117J NHzUi ~ 17SII:PU -3.00 d8
F2 - PrOCtsstno par..tet'SSI 327118SF 95.382.130 Mil:lIlIf DISSB OLB 3.00 H2... OPC 1.«1
10 _ pJot or_ursCJ 22.00 e-FtP !1Q3.t3t c.-F! ·47989.84 Hz:F3' 203.585~f2 19418.41 ttIllfW]I 13.61573 ppa!C8Hl(]It 129&.70129 ttz/ca
Espectro de RMN 77Se do composto (R,S)-1-fenilsseleno-2-propanol [(R,S)-1a]
80
6
Transmittance I Wavenumber (cm-1)
3000 2000 1000
Espectro de I.V. do composto (R,S)-1-fenilsseleno-2-propanol [(R,S)-1a]
128
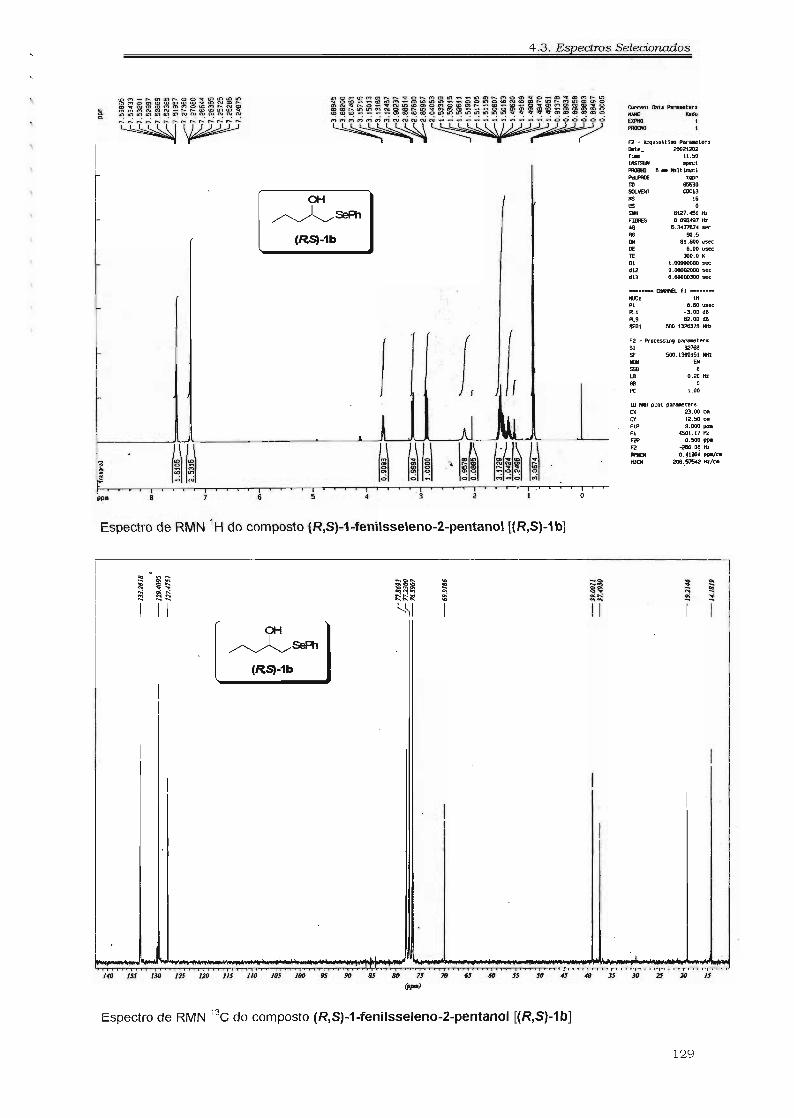
4.3. Espectros Selecíonados
Curf'elt Dat., Pv..,tef'S
"""' ......E""'" I........ t
23.00 c.12.50 CIl9.000 ~
4501.t7 Hl-o.MO ppe.;!50_~ tU
o.~l»4 ""'aI206 ',57'542 til/c.
exCY
'........F2-..HZOI
F2 - Pracl!5Si.n9 ~tersSI ,.,..Sf 500.1300151 M1Z.... EMSS8 •18 0.20 H2... oPC. l.OO
_ ..... DWte.. fi _ •••-
fU:l IHPI e.60 usecPLt. -3.00 dBPl.9 62.00 d8SOl 5OO.1326378..-n:
F2 - lcqu1s1tton Pal"HeteM:OItl!_ 20021202n.. 11.50lNSfFUI spectPfUII) 5 _ ...lt LrMK1
PI1J'AlG .....m l!i536Sll.VDfT a:JC13lOS I.os o
. s.t 6121.61 H1:FIlIES 0_ Dfi497 ttr:AO 5.3411874 secAS 90.5til 81.600 ~ec
tE 6.00 I.IUCTE lOC).O KDl LOOlIOGOOO secdl2 Q.OCIOá2OOO RC1113 a.oeoGOlOO 5H
-Oi
~5eRl
(~S)-1b
I f I r rr ~ AI Ul·I
Espectro de RMN 1H do composto {R,S)-1-fenilsseleno-2-pentanol [(R,S)-1b]
.. .'" -
~ ~~ "'8-- '" -co ~ ~...... S~~ ~ 8~~ ~~
;;;::~~ ~ ~~ ~ ~
I I , \~I I II I IOi
~5eRl
(~S)-1b
I
I I
I I
II
I
m m m m m m m _ m ~ ~ ~ • n ~ ~ ~ » ~ C ~ D ~ D ~ li
p)
Espectro de RMN 13C do composto (R,S)-1-fenilsseleno-2-pentanol [(R,S)-1b]
129
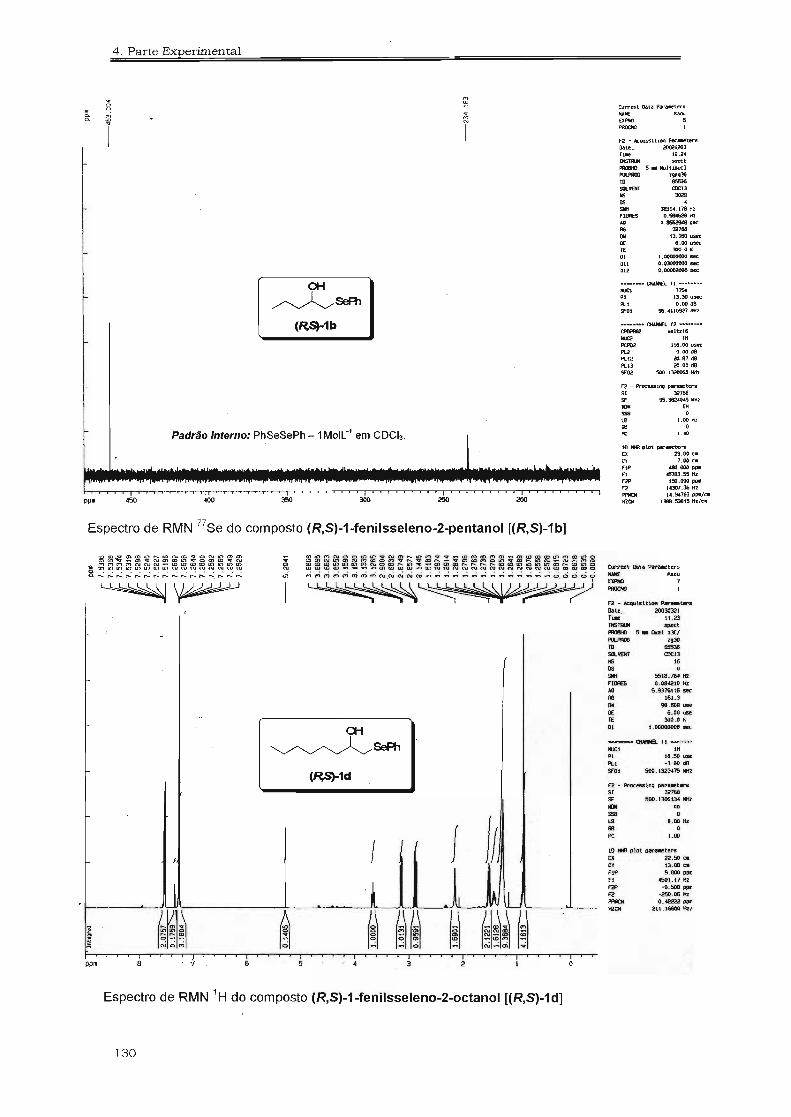
4. Parte Experimental
~
oo...
i
OH
~seRl
(~1b
Padrão Interno: PhSeSePh - 1Moll" em CDCh.
iQrTenl nau: Par~lers
IUoNE l{&)u
EX""" 5""""'" ,F2 - AcquislÜQfl Pw..tersDlte_ 2OOlt203Ti.. 10.2:4tNS1lUO _.
PRlJIIK) s. NuJtinuclPllPRlG :lgpgJOm lI55lllSIIL\OT aJe)]
'" 302!lllS •SItI )8'u4. 176 H2FUJIES O.!511M628 H2AO O.~.ac:
AO 327llllQN n.05O UMe
DE fi.OO usecTE 300.0 K01 1.OOOOOOOO 15ec1111 O.OllXlOOOO Meali! 0.000Q2000 Sk
·_·..·QtAJII€lu ..·_···IU:I 7?SePI 13.30 u..c.PlI 0.00 d8SfOl 9I5 .•UII)927 MHt
_ .... Oio\NIEl f2 .-........
CJI(Il9G2 .. lt216lU:2 'H~ lt6.00UMCPU 0.00 d81IL12 20.8708PlIJ 20.00 d8SF02 500.1320005 JI12
F2 - PracUSIl"J praetet".SI 32168SF 915.]824049 MtU.... ..... .LB 1.00 Hz
'" .PC t.40
PP' 400 300 250
•tO tHI plot PIJ'.n:eMex 23.00 acy 7.DOe.FtP •.000 Pe-Ft fi183.!55 "2F2fJ 15O.00G PPIIF2 1007.36 H1?fI'IiOl 1".34783 DPlllCItliZDl 1]111.53015 ttt/c.
Espectro de RMN 77Se do composto (R,S)-1-fenilsseleno-2-pentanol [(R,S)-1b]
IOH
t~SePh
(~~1d
I I J fI
I .,li ~UJ
I !~~I~ ~ !~\ !~H~\ I~I ~I~\~\ !~\pp. 8 '7 • 4
~ffi';~~~~~~:R::g~~~ffi
~~~~~~~~~~~~~~~~~Q~~~~~~~~--~~~~--
l l l L '>~ ~",J.J_-LJ
m~~NOO~~.Nm_~m~v_~~m~m_m~mm~mm~owmN~m~m~o~~_.m __ vm~~o~vm_~N_N_~Om~~~~~MNom_~~_mmm ww~~~~m_~~o
mw~w--__ m~~~_~NNNNNNNNNNNNN~m~mo
~~~~~~~~~N~NN~~~~~~~~~~~~~~OOOOO
LLlk~~~~LLG~dJ~
eur~llt Dlh Pr_U.rs_ ..ouElPlCl 7
'"""'" I
F2 - ACQuisi.tion ParaRtel"SDate 20030321fiale 11.23IHS11UI spectPAOIH) 5 _ Dual IX/J:It.LFlRJG zg30TO ...,.sa..V9fT ClC13
N5 "OS osatl 5518.764 HiFIOAES 0.084210 HZAO S.!U76116 ~AS 161.3I* 9Q. 6GO use~ 6.DO useTE 300.0 KOI 1.OOOOOQOQ sec
--- 0WfrEL fi~••UI IHPI ID.50 usePLl -3.00 d8SfOt 500.1323475 Jttl
f2 - Pl"'DC~ing Claf',*tel"'$SI 32768SF" 500.1300134 !til101 ••SSO •18 0.00 H:zli! •PC l.OO
10 fIlfI plot p..~tersex 22.50 c.CY 13.00 c.FlP 9.000 P9IFi 4501.17 tU:F2P -O.soa ppt'
FZ -250.06 Hl
PPMC:M O~42222 ~
I12CM Zlt.16E1OOHzJ
Espectro de RMN 1H do composto (R,S)-1-fenilsseleno-2-octanol [(R,S)-1d]
130
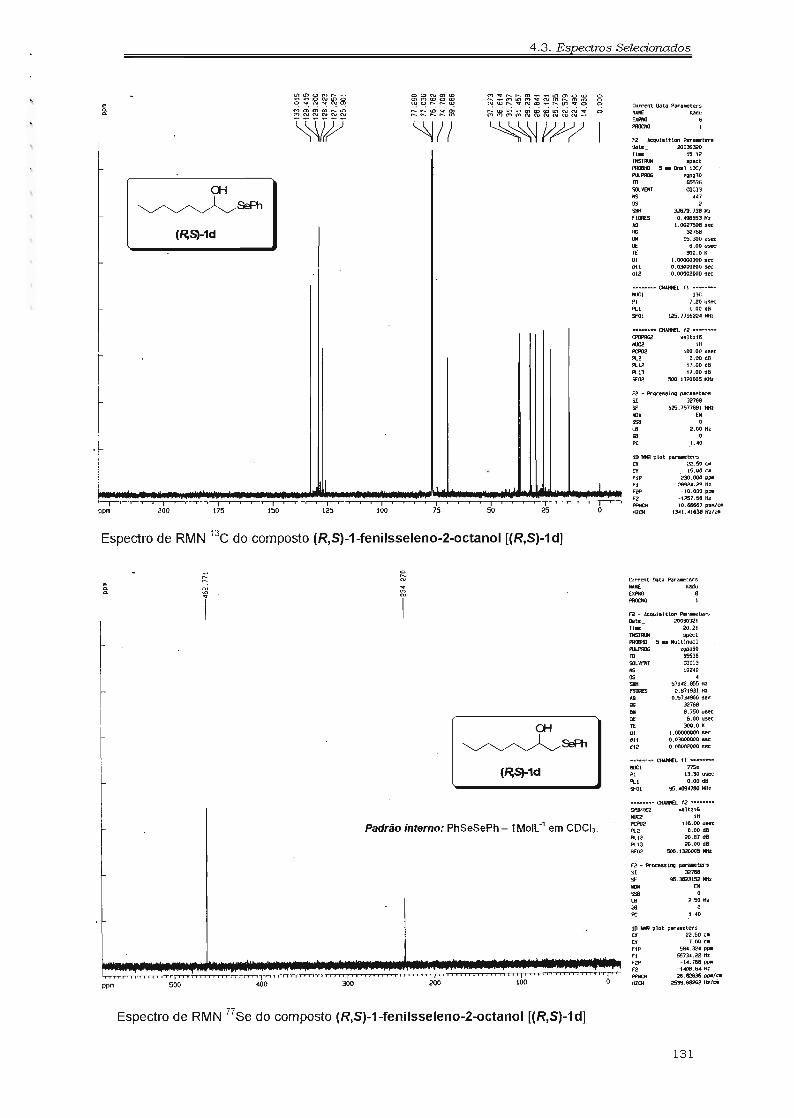
4.3. Espectros Selecionados
a'llnO(O")r--_
ô;~~~~. . . . . .
~tJJ~~~Rii..... ,... (Q ... O),...,... ...... ,... tO
~;/
('I"J ..... ,....,...m .......... ltlClOtO <:),...._(rIIt'JPl~{\ItO..... mLO o~~":~~~-:":~""':~ o..... ta- ..... cnCD<OlnN('\l..,(O") ('I"J ("'1.('1') (\IC\I C\lC\I CVN_
l::\IrTent Diltll Par._t~.l'WIE Kadu
"""" ."""'"' I
f2 - q,ilsition Ptr..-tersDIltl!!_ 200J0320n. IS.t2IftST$IIf S1JKt~ 5 _ Dual sr.!PUI..PMG 19P1llOlD ....,.
SOlVen aJClJMS 441os 2SWH 32679.138 Hz_FlDAE5 0.<l9B653 Ht10 t . OO275Oe sec11;; 32768IJI 15.300 uwcDE 6.00 usecTE 300.0 K -01 t.OOOOOooo 8ft:di! 0.03000000 .s«dt2 0.00002000 5«
...._ ••• CHAItEl f t ...._ ..-.
PlJCI IJCPI 7.20 U5«P\.l loOO d8SFOt 125.7716224 NH:l
......... CfAJtlEL t2 ....... _ ..QlCFRõZ Ifaltll6llJC2 IHPCP02 100.00 US«
R.i? 0.00 d8Pl.12 17.00 c2BPl13 17.00dB$fOZ 500.1320005 Nttz
F2 - ~in; ~8I11!t~
SI 32768$f 125.7577881 Mth:.... EHSS8 oLB 2.00 H1GIl oPC .1.40
pp. 200 175 150 125 100 75 50 25
I
o
tD _ plot ~ttnOI 22.50 c.Cf • 15.00 ".FJP 230.000 p~FI 2&9i!04.29 ttzF2P -10.000 ~F2 -1251.58 HlPPHOl 10 .lS6667 ~c.ttZOI 1341.4t638 Hllca
Espectro de RMN 13C do composto (R,S)-1-fenilsseleno-2-octanol [(R,S)-1 d]
i
~~I(~S)-1d .
Padrão Interno: PhSeSePh - 1MoIL-1 em COCh.
CUrrent DlIta Para-etl!l'5fWiIE. Kadu
"""" .""""" I
F2 - AequisiUon Par.etersOIte_ 20030321Ti. 2O.2JDtSflUl lIpectPAIBII 5 _ Nult tnuc]f'l.I.PfIlG 19pQ30lD 65530SOLYEHT coei)MS 102.40os •SIIl 57J.42.85S HzFItJlES 0.87t931 HJAO 0.5134900 5"R6 327680If 8.750uHCCE 6.00 UfJec:
TE 300.0 I( ~
Dl t .OOOOOOOO secd1t O.OlOOOOÓQ sec:dt2 0.000020OO sec
...__.. CHAffEl ti ....._.
UI 77SeP1 n.JO ~c
A.I 0.00 dBSFOI 95.409.t780 ~
•.--........ 0i.UfrEL f2 _ .....--
~ ..ltzl6"fi1JC2 IHPCP02 1t6. 00 U:HC
1l..2 0.00 d8Pl.!2 2O.S7 dePt.t3 20.00 111':;02 500.1320005 __
F2 - PrDCn'S b19 para.t:ersSI 32168SI' 95.3823l!i2 NH:i:_ EM
SS8. oL8 2.50 HzGIl oPC 1.40
pp, 500 400TI"""I""I"" i "1""""""'1 " "'''''''''''''''1''
~ m ~ o
10 "" plat par..tarsex 22.50 c-o 1.DOc.FtP '584.324 ~FI 5!5n4.22 HzF2P -14.7eS PP'IF2 -t408.64 HzPPNCH 26.$2635 IJt*/c.I'\ZOC 2519.6&262 Hz/c.
Espectro de RMN 77Se do composto (R,S)-1-fenilsseleno-2-octanol [(R,S)-1d]
131
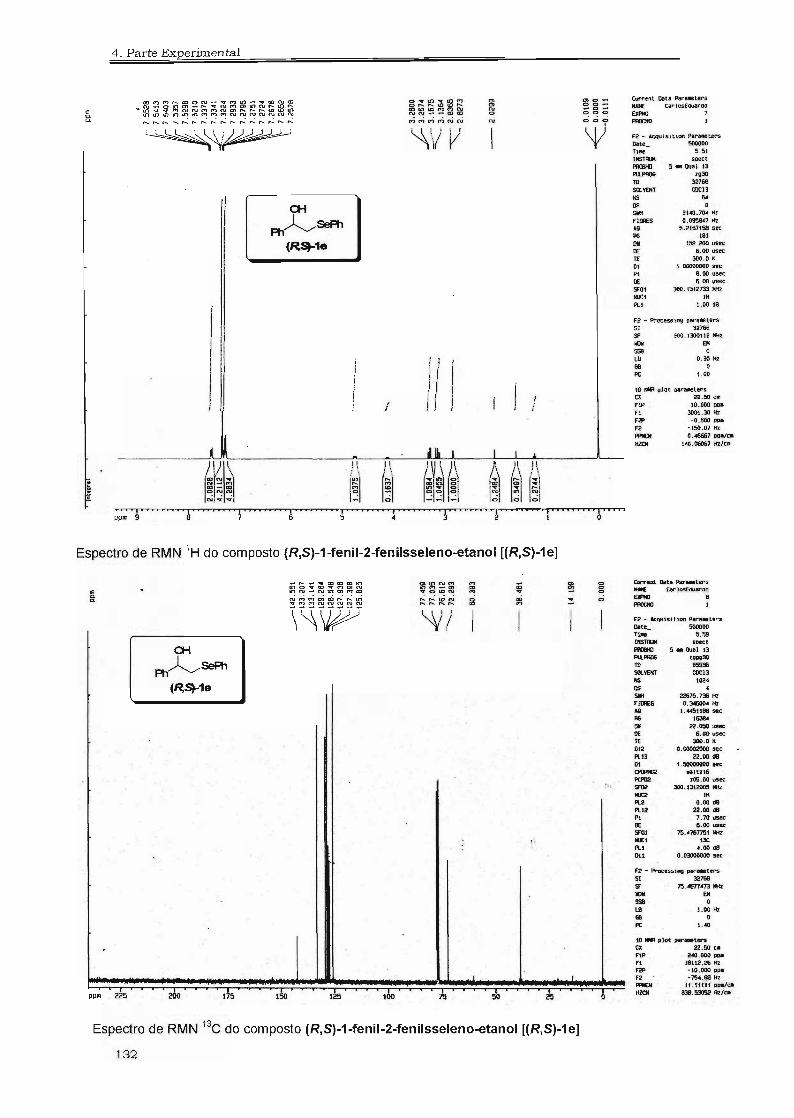
4. Parte Experimental
5000005.5.
SDect5 _ l:kJal 13
zg3032768alel3
6AO
3,..0.70.t HlO.O~7 Hl
5.2167158 sec18•
159.2'00 usec6.00 usec
lOO.O KS. OOOOOOOO sec
8.00 usec6.00 Utiec
300.1312133 NHz
'H1.00 er3
F2 - AtQUiSttHM'I ~aralll!urs
F2 - Process mg DaretltlersSI 32768SF 300. i3OO112 NH;z:
10< E.MSS8 OLS 0.30 lt2
.. OPC 1.00
1D MAR p)ol oaretleltrsO 22.50 ceHP JO.OOO gp•
n 3001.30 Hz
f2P -0.500 ppa,f2 ~t50.07 tilPPIOI 0.46667 ooalc.l1lCM 1tD.06067 ltt/CII
Ct.rr'enl Data ParalllelersfWE CarlClsEduarOo
0ACl 7l'RIOlO •
OIt-e_h ..IhSTlU<AQlI(J
Al.PRJGm5a.VENTNSOSSItlrHJESAlI..llIIllI'TEDIP,llI'srOt!l.C1PlI
'" <><> <>_ <>-~~~
Wre<>
'"
O<q'U'1"C1'lI1MO""''''''~UJ'''''CDtOU)rlt-lNNN--~CO
rririr'irrinini
'\Vv
!\
I~I 1\ )\
(~1 (~ (~l f~)I:I , ..... ,. i 1, •••• 1. (" fi ••••••• ", •••5 4 3 2 I oI
6
re~a~~2~~~;:I~Vi~)e~:e·~~~~NN~~Nm~~~~~~
1t1U'lIllIl1.ntnMMMNNNC'UiNNNro.:.,...:,...:,...:,...:,..:,...:,...:,...:,...:,..:,...:"":"':''''':''':
IIr! r
! I !lI I
. ~'l. __"'___'_'----=--_..JI....II.LJ1__---L_L-....1 ........JI__
~~~".'."""" 1"'" i IIIP~ 9 B 7
Eg
Espectro de RMN 1H do composto (R,S)-1-fenil-2-fenilsseleno-etanol [(R,S)-1e]
_ .... _ .. aJU)Q)\CJ
~~~s: g:~
<> CUrTem. Dacte Per..let'5lflO~tD""'~OJN '" <> lIME Car-1D5EdWroo
ã ,"('U-NU1m~m .... o ta N .., v <>
" ~~~gjre~~Ki ""''''''ION 2 li> :!:0ACl 8
..... ,... ..... ,.... ..,PRlCHO I
\~~ ~/ I F2 - AcQUisitJon PII"'.lt1"SDate_ 500000Ti_ 5.59IflST1UI
._,Oi -.o 5 _ 3Jal 13
Ph~5eR1PlLPAOG zgpg30T1l 65536Sll.VENT alel3OS 1024
(~~1e OS •SII1 22615.736 H1FI1lRES 0.346004 HzAlI 1.445U.88 sec.. 183Il'I
"" 22.050 usecDE 6.00 usecTI' 300.0 kO•• Q.OOOO2OOO se:cPU3 õ'2.ood8O. f.50000000 $teCPIFIlG2 Wiltzl6P<Pll2 1OS.00 ~ec
~-. : SF02 300.1312005 tti2
lU:2 IH
l't2 0.00 d8pt.2 22.oocIBP. 7.70 USI!C
llI' 15..00 U5U
STOI 75.061151 Mil'~ .~ IU:' 'JC
pt, ".00 da011 0.03000000 sec
F2 - Processing P....ter-sSI 32768ST 15.JI617473 ~101 EIIS58 OUI 1.00 Hl... OPC l.40
10 ~ olot parllletersO 22.50 c.FlP 240.000 pplI
ri 18112.26 Hz
FCP -10.000 pge,F2 -7S4.~ Hz....,. . JLtltlt D~C.
PP" 225 200 175 150 125 100 75I H1Q4 838·,53052 Hl/C.
50 25 O
Espectro de RMN 13C do composto (R,S)-1-fenil-2-fenilsseleno-etanol [(R,S)-1e]
132
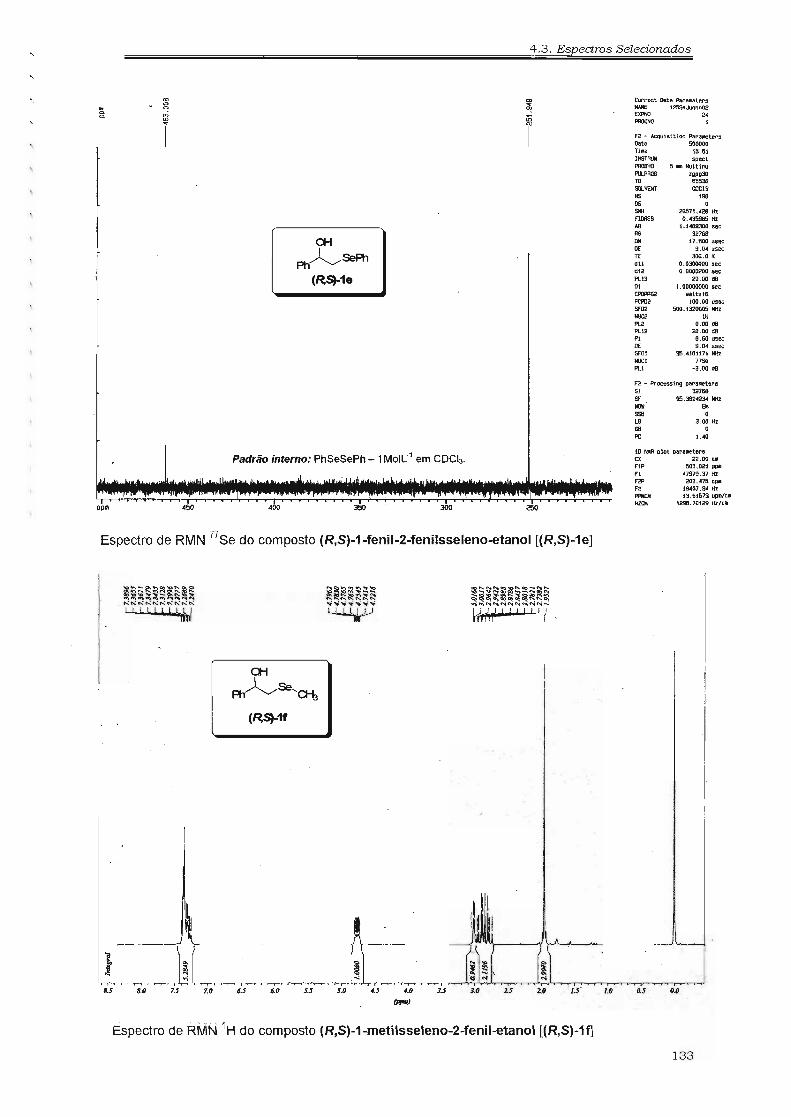
4.3. Espectros Selecionados
f2 - AcQuisitian Par.-eten
Cuf'rent OBta PerallelersNAIE 12SS1tJuohoOêEXPND 24PIlOCNIJ I
g"'
i i Date_Ti.tIlSTRl.I<PR08HIlPlJLPAQG
lUSlLVENTNSosS"FlORESAORG01111'TEaudl2PU,DIa>IlPllG2PO'Il2SFD2MJC21'1.21'1.12PIDESFOIIlUCI1'1.1
ooסס50
15.51specl
5 _ Hultinu
IQJIg3065536coeI'
196a
28571."28 HzO.ofJ5!l65 til
i .J«i93OQ ,ec32768
11.500 usec9.04 usec
300.0 K0.0300000 SK0.0000200 sec
20.00 dBt 0oooooס0• sec
lAltz16100.00 une
500.1320005 liIfZIH
0.00 dB20.00 dBB.60 usec9.0-4 usec
95 .•U6H71 liIiz7750
-3.00 Cle
Padrão Interno: PhSeSePh - 1MoIL'1 em COCh.
DOm 400 350 300 250
F2 - Protessiog pM'8l1ttel"lISI 32768SF 95.382..... IttzlOt EMSSB aLB 3.00 Hz68 aPC 1.-40
10 _ plot per8t1etersa ~.OO ~
F1P 503.021 ppaFI <17979.37 tfZF2P 203."75 opaf2 19401.84 Hz'PPIDt 13.61573 PPl'/c.HZ04 1298.701.29 Hz/t.•
Espectro de RMN 77Se do composto (R,S)-1-fenil-2-fenilsseleno-etanol [(R,S)-1ej
~~:::~::~~~~~ ~;;:~::~~~ ~i~~!!~i~~~~~~~:::~~t'oI~ ......~t:::~~~~"t-,;"""""t'"" ..,;...;. ...... ~ ...... "";-it'i~o:ooi~f'>i~"'f>i-:
I I I I I!! I.,f I ~ h! 11
J! J ! I J (
11 RlMI ' ..
Q-I IPh~Se'013
(~~1f
I·
,
I
I II
~ - Wlli A
} ~ ~ij1
~ fl" ~ \ ..
.; ..; oi oi
11.5 &0 7.0 6.5 6.0 S.O 4.5 U
m-)li 3.0 2S 1.0 1.S /.() O.S
Espectro de RMN 1H do composto (R,S)-1-metilsseleno-2-fenil-etanol [(R,S)-1f]
133
4. Parte Experimental
;:: .... .., .... "'r- .....-~ ......~ ~~ ....~~ ~ ~
~'O-
~~ ~~;s
.. ~:g~ i!i:~~~ ::! ~"""'" "i.......... 'Ó
I ~II ~I I I~ I I
.
I a-l
I Ph~Se,~I
(~sr1f
III
II i
I I
II
,I! II
II/6(J UO IM 119 /lI) /tIO 7f) M 111 lO O
Espectro de RMN 13C do composto (R,S)-1-metilsseleno-2-fenil-etanol [(R,S)-1f]
ppa 500
v• g
i
400
Padrão Interno: PhSeSePh - 1MoIL-1 em COCh.
300 100
F2 - M:.QUlslUon PlrllRterso.te_ ~.407
n. 10.012IJISTRIt SPKtPRIBIJ 5 _ MuJtinucllUPRII& ....,.
TU 655J6stl.YEltT CDCI]
'" 389llS •SIH 57142.855 lt2FIMES 0.811931 HzAO O.!5n.900 sec:FI; 32768tIf 8.750 usec.I:E 6.00 usec.TE m.o Kai 1.oooooooo seccru O.OlOOOOOO secdiZ G.O()(JQ2()OO sec
-..__.. 0UIIEl. t t _ ....--
IIJCt 77SePt 12.00 usecPU 0.00 d8SFOI 95."0632J6 HHz
--- 010UI€L f2 ---..0I'I:FAG2 ..azi6llJC2 IHPCAJ2 IOS.OO US«
PU -3.00 d8PLt2 19.00 d8PL13 19.00 d8SF02 . 5OD.13õ!0005 MHz
F2 - Procnsjl19 " .._ters51 32768SF 95 .1823625 lI1Z... EMSS8 olB 5.00 HzIil oPC 1.40
to tllAplotpr_tef"Sex 23.oo"CIICY 10.00 aFtP 550.956 ~
~ ,~:.F2 -45!II.-41 H:!FqDI 26.~J!50 --'aiH20f ~.'72H 11l/c.
Espectro de RMN 77Se do composto (R,S)-1-metilsseleno-2-fenil-etanol [(R,S)-1f]
134
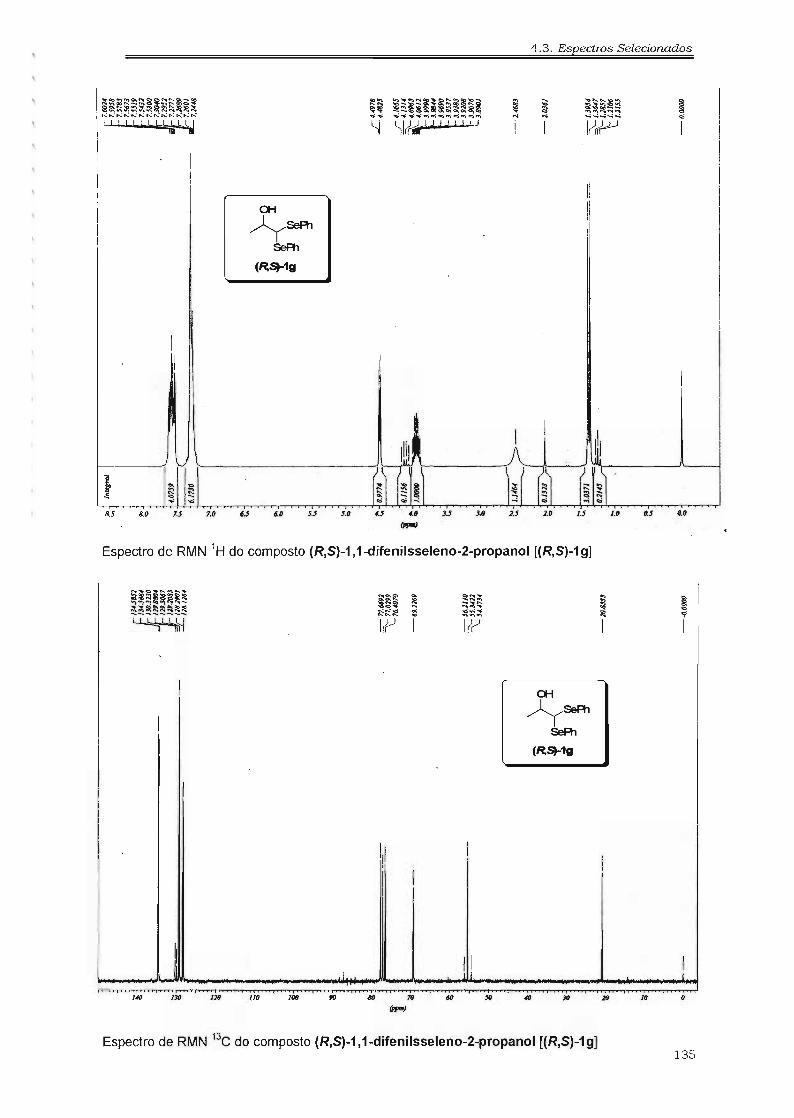
4.3. Espectros Selecionados
OH
/y5eR15eR1
(~1g
I I
I
r~1a5 au h5 hU ~5 ~D ~5 ~u ~5 ~D ~ LO ~5 ~O L5 LO a5 ~O
f-'
Espectro de RMN 1H do composto (R,S)-1,1-difenilsseleno-2-propanol [(R,S)-1g]
~i~i~~~~ i~~ ~..... ~ l:l
~-... ..,-~~ ~
~=:~~~~~~.....~
~~~ ~ ~~~ lO! <?LLb4L 4~ I~ I I(~ I I
.
I OH
I/y5eR1
5eR1(~~1g
I
I., III
I
~ .1II !11.
/-HI /Jf) 120 //U /Ot/ 50 Jf) 10 10 o
Espectro de RMN 13C do composto (R,S)-1,1-difenilsseleno-2-propanol [(R,S)-1 g]135
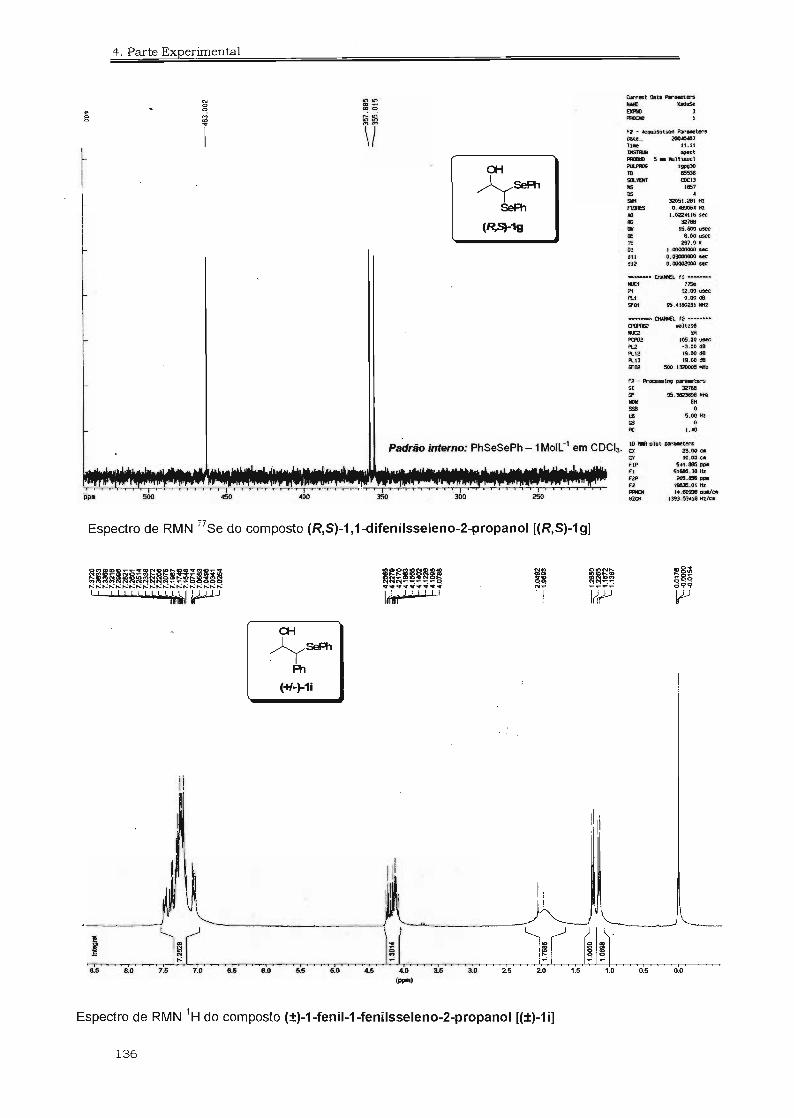
Padrão interno: PhSeSePh - 1Moll'l em COCh.
4. Parte Experimental
'"o~..,
i
500 J50 300
",l.ll. J.
250
.......
t.rrent 1lItl Prueun_ UlIUSc
...... 3'-"" I
F2 - Aequisition Pr_t.ersiJlte_ 2(lO.<<)401Ti. 11.11DlS1RIl ....tPAI8IJ 5 _ Mui t\nucl
PlI..F'fOõ lllP9JOm .....SIL't'BIT mel]JIIS S157DS •5liIt 32051. 2!U HtfIlIES O.4l!!i064 tft10 L0Z24U6 SKli; 327680lI: 15.600 u-=DE 6.00 USKTE 297.01(OI 0oooooס1.0 ucCSI1 O.OlOOOOOO Me
cU 2' O. 000020OO SIC
__lo OUJIIEL fi ....
lU:1 7)Se
Pt 12.00 usecPU 0.00 dBSF'Ot 95 .• 180231 MH1'
~-~'2"·_·-"C1UWi2 waltll6ou:> IHPCAJ2 '05 .00 URCPU -].00 dEIPL12 Ig.oo d8Pl13 19.00 d8r:FQ2 500. tl2OOO!! tttz
f2 - ~tll9 O....trssr 32768g: 95 .l82Jfi98 NHI
- E.ssa oLB 5.00 Hz... oPC 1..0
tO_plot. or_unex 23.00 c.Cf 10.00 c.FtP 541._".FI 5,_.)0 HzF2P . 20!5.!156 p~f2 liI35.01 tUPADI 14.6D998 pl»Jeataoc 1193.53418 Ht/c.
Espectro de RMN 77Se do composto (R,S)-1,1-difenilsseleno-2-propanol [(R,S)-1 g]
~~~~J~~~~~~~~~~~i!~i ~~~~l~~~~ il ~~§~ ~i~~~~~~~~~~~~~~~~~~~~~ ....................... ..- .:...:..:..: ~q9111111'1!!lll~~~ IJ I J " 11 I 11 I(~ ~nu .Ii
a-i
/y~Ph
(+f-)-1i
, .
II
II11
III
II I
'! )( )t( JLllj'Ij 1
'8:0' 8'.5'.,
5:5i ú i as ' f
0:5i
8.5 7.5 7.0 8.0 8.0 4.0 3.0 2.5 2.0 1.5 1.0 0.0(ppn)
Espectro de RMN iH do composto (±)-1-fenil-1-fenilsseleno-2-propanol [(±)-1 i]
136
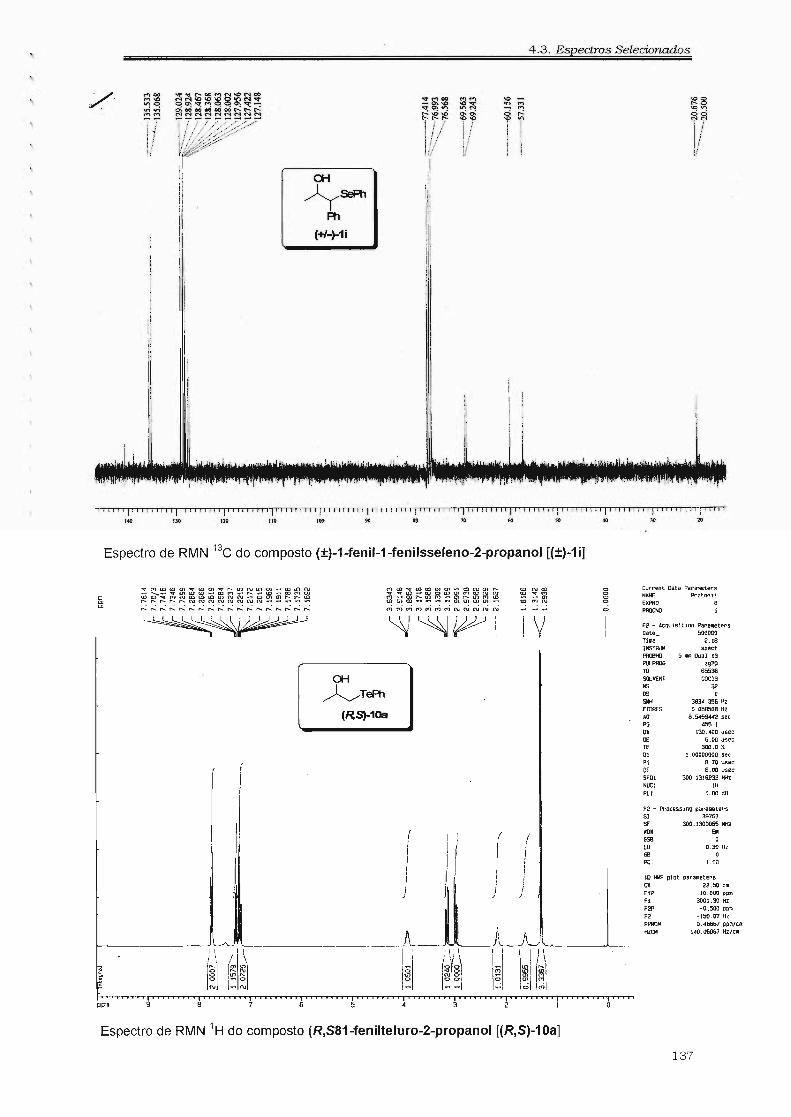
4.3. Espectros Selecionados
(]-i
~5eRlA1
(+I+1i
..,..,~~$$I !! !,/
V
~..,..,
i TI I
It'ili
I!
li
1 I I j I I II I I I I I j I ! I i I I I I i I I I 11 1 I I i I , i I I i , I I f , 11 I II I I I i , I I I I t I i j I i I I ! ! I I I I I I I 1 I I FI I I I I I fi _I I I i , I I i li ( r I I I " I i I I I i i I I 1 I (I I I j I i I I I J i I I I " I
140 l30 120 no 100 90 80 70 60 50 40 30 20
Espectro de RMN 13C do composto (±)-1-tenil-1-tenilsseleno-2-propanol [(±)-1i]
F2 - .a.t:Quisitlon Parelleters
Current Data P~"alRetef'S
NoUE Proton11EXPNO BPRllCI<D t
5Ollooo2.18
sneet5 ... Qual t3
zg3l165536COCl'
'"D3834.356 H20.058508 m
8.5459.4.q ,ec456.\
t3O.400 usec6.00 usec
300.0 K1.00000000 sec
8.70 usec6.00 usec
300.13.16232 NHIIH
1.00 d8
Date_T,..INSTFUlPRlIllIJPW'!106TO5OC.VENTHSos5,*,FIlIESA<l
"GO'llETEDlPlDEsrDlHOC!PU
ogoo
IV
t"')co'q"rocomm ..... CDrumr--..'<:r "<I"tn_WOI(1OlC"laJCUNrrl-m"Inf"1_ml'tnC"11DOl(1)CD-- ..... -OImCJltJ1-. . . . . . . . . . .
a;
~TePh
(RS)-1oa
rI
~~mwm~wm~~IDrumm-mIDru
~~;~~~~~ffi~N~~~~~~m~~~~~NNNNruNNru-- _
~~~~~~~~~~~~~~~~~~
j(I
Ii
)
F2 - Pt'oceSSlng pBra.tersSI 32768SF 300. 1300066 ttiZWDW EMSSB OLB 0.30 H2GIl OPC 1.00
tO NWl plDt Ollr"lIetersex 22.50 c.F1P 10.000 PPIlFi 3001.30 HzF2P -0.500 DDJIF2 -150.01 ltZPPMCN 0.46667 pptVCIJHZCM t40.06067 Hzlc,.,
ppm 9 7 6 5 3 2
Espectro de RMN 1H do composto (R,S81-tenilteluro-2-propanol [(R,S)-1Da]
137
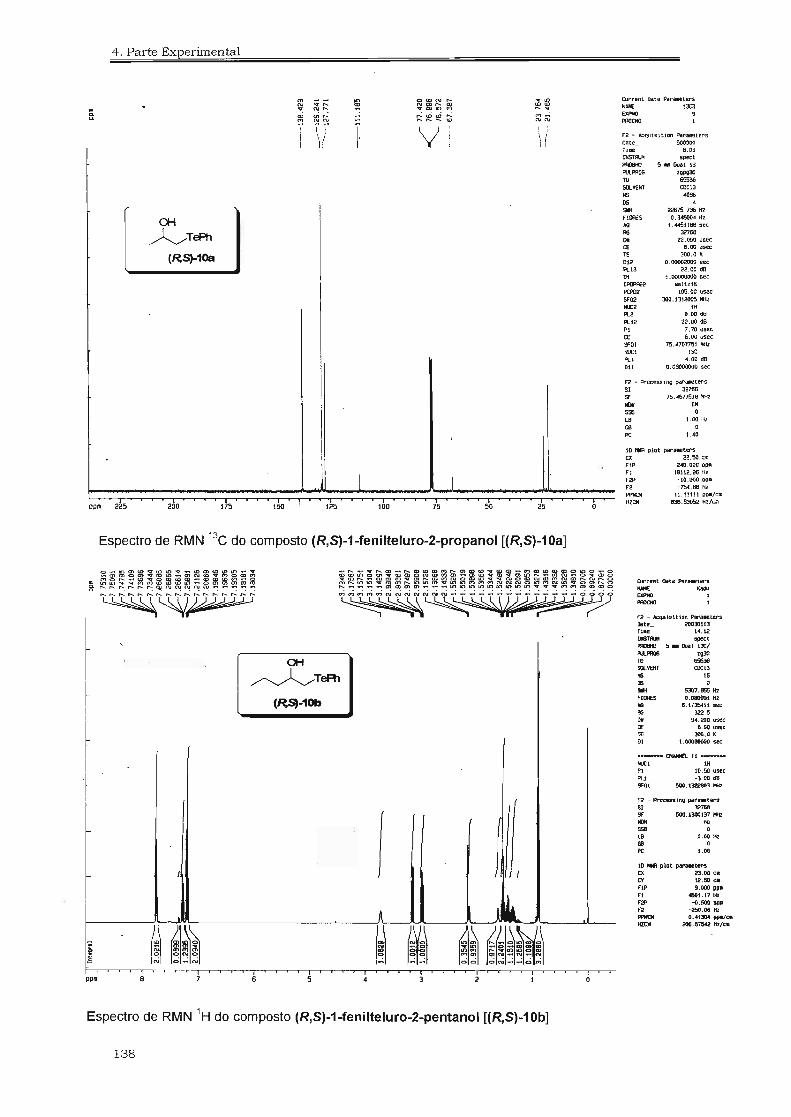
F2 - ÃC.QUlISltion Par'lMler's
Corrent Oat~ Para.elet"SIWE 'X3ElCPOCl 9
"""""" 1
4. Parte Experimental
a;
~TePh
(R~1oa
ao ... ~ on~ Si~ m .on
tl' "'~ ~ ~~
~ ao ...~~~~ .... -
~~NN
i \/ j. \V I \I _o_h ..IHST.....PU.PROGTUSCl.YENTloSosSIIffllRSAQ
R6001CETI'012RI3D1
"""""'2PCP02Sf02lU:2R2F\. 12PlCESfOl
lIlC'PLl011
5000006.01
soect5 _ OUil 13
'gjl93065536coe]34096
•22675 .736 Hz
0,3450(1.4 Hz1.4451188 51!C
3276822.050 uaec
6.00 usec~.O k
o .00002000 sec22.00 de
1.OOOOOOOO lH!Cul1216
105.00 usec300.1312005 Itil
IH0.00 da
22.00 dB7.70 usec6.00 usec
75 ..41677'Jl MHz'3e
".00 dB0.030000OO sec
200 175 150 125
I100 75 25
F2 - Pr1Xessinç para-elersSI 32768SI' 15.-4617518 N!1llOI eN
"'" oL8 1.00 HlG8 oPC 1..40
10 ~ plot Dara-lersa 22.00~
F1P 2040.tlQO Ps-Ft 18112.26 HzF2P ·tO.OOO opa
F2 -75.4.68 HlPPNCN 11.U1U ~c.
HZCH 838.53052 Hz/cm
Espectro de RMN 13C do composto (R,S)-1-fenilteluro-2-propanol [(R,S)-10a]
Qrrent Data ParHei.er$- ....,EllPl«l 1PIIlCNO 1
OlIt!_n..lIISTIUIPRIIHlPIl.l'IlOSTUSCLVE!lTNSIIS....f1lI8..I!GrJICETE01
2003011314.12ooect
5 _ Ml 13C/
'030655J6coel3
16
•5307.855 Hz0.080991 Hz
6.1735411 sec322.5
9<4.200 USf!!C
6.00 """300.0 I(
t .00000000 5CC
•••_- OUHNB. fi --
lIlCI '"Pt ID.S) usec~ ~.OO~
SOl 500. t322803 Jtt1
F2 - Pr"ocessituj par.-te:rs$[ 12168~ 500.1300137 MillIllI noSSIl •L8 .0.00 Hzfil . •PC -1.00
10 JMI plOt ~teM.ex 23.00 c.CY 12.50 c.FlP 9.000 ppFI 4601.17 Hz
Fa' -1.500 ....F2 -250.06 HzPPM:M 0.41J04 ppe/c.H'lDI 206.57542 tU/c.
Ppq 5 2 o
Espectro de RMN 1H do composto (R,S)-1-fenilteluro-2-pentanol [(R,S)-10b]
138
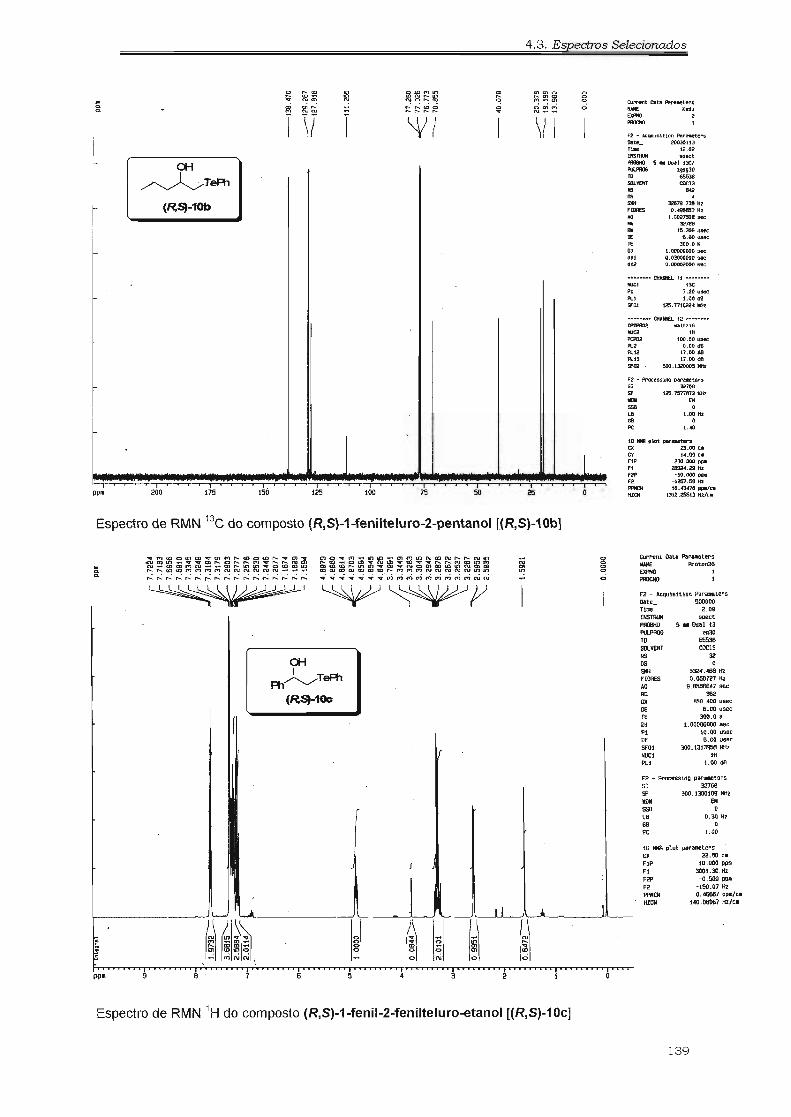
4.3. Espectros Selecionados
OH
~TeA1
(~S)-1Ob
I
e.,.,
pp. 200 175 150
o ~.,
~ .,-.,. ~~
., '" ~
I \/
125 100
'VI
75 50
\/1
go
• O
eu.....ent Data Pa,...ter.fiWE KaduEXPIlO ,PIlOCItO I
F2 - AcquisU\on Pw'Meter-sDate_ 2OOlO1t3TIa 12.02lHSlllJf ~«t.
PR(B«J 5. Dual IX!Pl.lPflIJG 2gp9lOm 1\55305OL.VEltT CDC13... ...OS •5»t 32fS1'9.738H2Fro:aES 0.498853 HtIJIJ 1.0027!D& sec.. ",768[IN 15.300 UHCtE 6.00 usecTE :300.0 I(
OJ 1.00000oo0 St!Cdi I 0.03000000 aecd12 O. tIOOQlOOO uc
·.._--CttAIftEl fl-....••••NJCI IlCPI 7.20 usecPLI 1.00 d851'01 125.7716224 MHl
--_.... 0IAtfEL fi! -_...-c::PIIWl2 ..Uzt6MJC2 IHPCJlD2 loo.oa u:secPL2 0.00 daPlJ2 17.00dBR.n 17.00 di!~ !SOO.llõ!OOO5Mttz
fi! - fhJcess}ng 1Ja"_ten:51 3i!7HSf U5.~tItz
tIllI E.SS8 OlB 1.00 Hz
'" OPC 1.40
iO fIfI Illat 1Ia"..et_OI 23.00 c.CY U.OO c.FtP 230.000~FI 28924.29 HZF2P -10.000 p.-F2 -1251.58 tI:1:PPMCN 10."~78 pp&lClIltlOI 1312 .25&13 tU/c.
Espectro de RMN 13C do composto (R,S)-1-fenilteluro-2-pentanol [(R,S}-10b]
~m~~~~;~g~~g~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
~~~~~~~i;re~~~~~re~~~~~~~~~~~~~~~~~~~~~ •• vV~~~M~M~MM~~ruN
Ct.Il"r~nt Data Paralll81ersNANE Prot.0n36El<I'MJ IPROCIll I
F2 - Acquisition Par-aIIIters
F2 - PrlXtSSing Qer_tersSI 3276851' 300.1300109 1117IlllW EWSSIl OLB 0.30 11268 OPC 1.00
10~ plot parnelersex 22.50 r::.F1P 10.000 pp.FI 3001.30 HlF2P -0.500 !XJ1lF2 -t5O.07 H2PPMCN 0."'6667 PPII/c.HlDl 140.06067 Hz/c.
5000002.09
...ct5. OUal t3
1.3065536CDC13
32O
3JôM.468 Hz0.050727 tu
9. 85866.47 sec3112
t50.AQO uSt!c6.00 usee
300.0 K1.00000000 .sec
10.00 U9~C
6.00 USt!c300. tJt~ .MHl
IH1.00 d9
Date_Ti.INST"'"PROIIIIlPlU'ROGTOSll.YENTMS05SIIlfImESAORGDIf11"TEDIPIDE51'01PiUCjPlI
-
OH
Ph~TeA1(~S)-1Oc
r
J
.,10 il ,\, I'--'
i !~~~~, o
!~ I!I I~Ig ~o o- N
9 8 6 5 4 3 2
Espectro de RMN 1H do composto (R,S)-1-fenil-2-fenilteluro-etanol [(R,S)-10c]
139
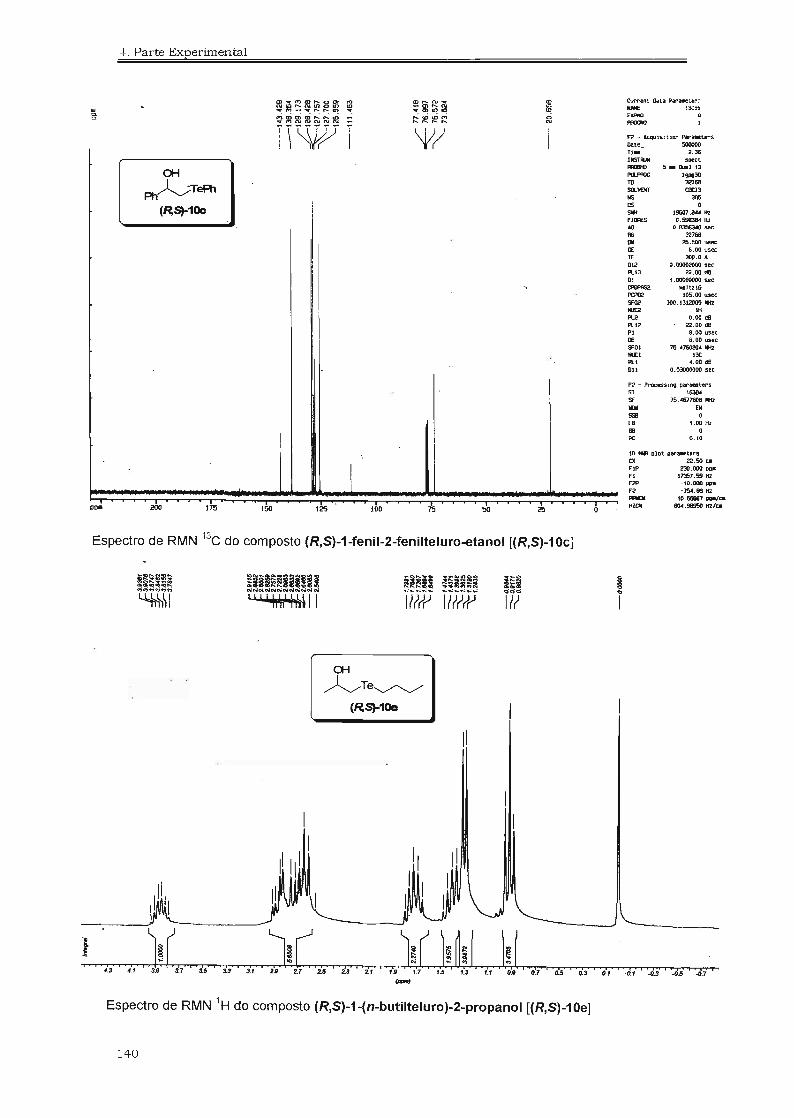
-+. Parte Experimental
.'"'"
~iil~~i!!~!! !i~~Ri~~~~ ;::
I\\VP íCD .... C'\t ...-CJ)"C\I-.CDVlID
~~~~
ClrriRt DIta ~_t.rt
IWE 13::1t.,."., 8
PRJDIl 1
F2 - kquisitian Pr_lar'SOIte_ 5DOOOOTlae 2.36IJCST1I..It SOIctRUIf) 5 _ ria) 13
Pll.flRJG llJPglD
TO 32166stLVDfT ax:l3llS 385CS •SIitf 19607.S4A taFlIHS C.59l!38of Itl.. O.B3563otO secAS 32166[JII 25.500 usecI::E 6.00 usecTE 300.0 KD12 O.000020QQ ateRt3 ~.OO~
01 1..OOOOOOOO $ecCPrPAG2 ..I tt 16PCPD2 105.00 IS5eCSF02 300.1312005 tIUlIC! IHPl2 0.00 C19A..12 22.00 CIBPt 8.00 usecCE &.00 usecg:-Of 1'5, ..76Q2O.oI)ffZ
tlLl 13CRi •. 00 d8Ou 0.0300000O sec
F2' - Processing par_tersSI l63O-iSI' 15.47760tl IttzIOf EM!SI •LB t.ooHz... .PC 0.10
200 175 150
I125 100 75 50
ID -' plot ...._t..,ex 22.50 c-FIP 23).000 PQlIFi 17351.59 Klf2fJ -10.ODD PPIaF2 -754•• tilFAOI 10.66667 PDllceHlDI BQ.4. 98950 tU/c.
Espectro de RMN 1:;C do composto (R,S)-1-fenil-2-fenilteluro-etanol [(R,S)-10c]
Iô
I
. ~~ ( )~ n! I \! I. ......................................-.-':....8~.............(i...7 ..........-5............:-3.........4...·'......' ...,..i.....,..,.'~~...'3........• ,..,..i......3"""-..,.'.7....,.....'-
IIIII I
i ,!L. JL, . . ...,:, 15 . 'u f.7Ú i3 i4.3 '.1 3.7 U 3., 2.,
Espectro de RMN 1H do composto (R,S)-1-(n-butilteluro)-2-propanol [(R,S)-10e]
140
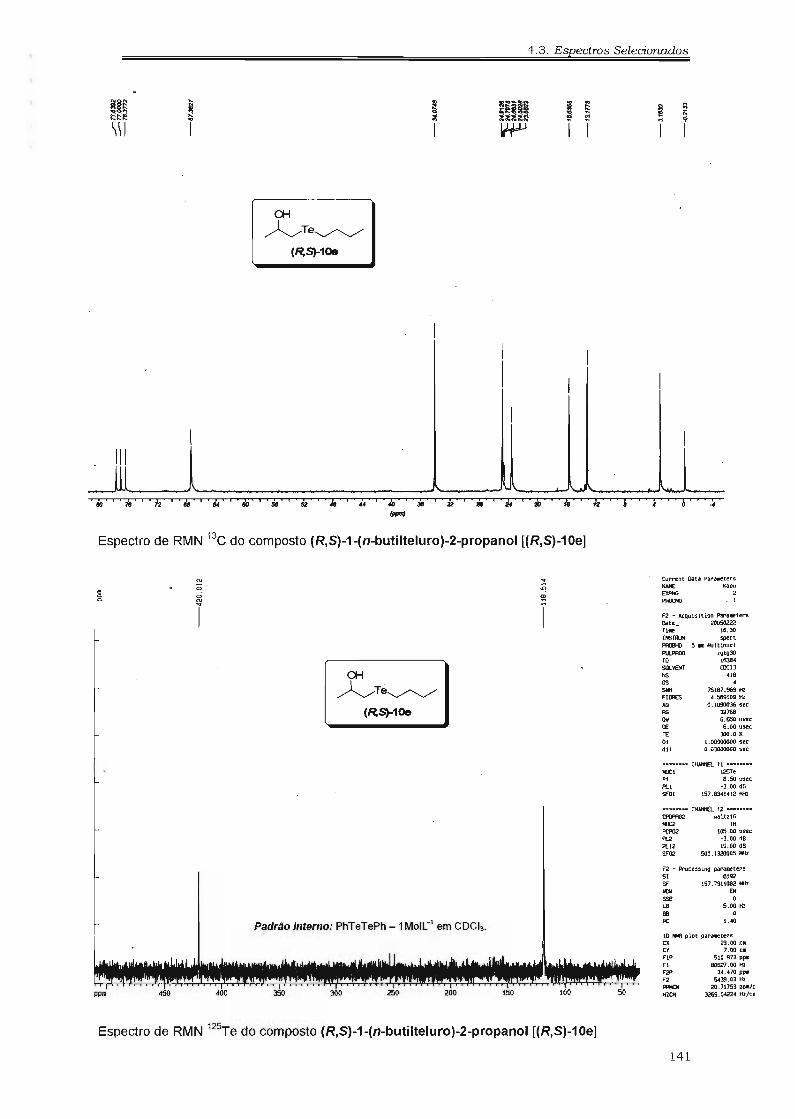
!I
Q-i
~Te~
(~S)-1Oe
4.3. Espectros Selecionados
ªriI
ill'lo ia fz • I $.' i
~ 5z 18 4f i 38 i.. ao «J 3Z-- iI-
Espectro de RMN 13C do composto (R,S)-1-(n-butilteluro)-2-propanol [(R,S)-10e]
'";;;'"
iQ-i
~Te~
(~S)-1Oe
íCt.orN:nt o.u Pa,.a.e[e"~_ lCallu
EXPIlO 2PRlDtJ ,
F2 - At:QulS1tion PJrawtersOate_ 2OOfJ0222n~ 16.30IH5TlUt spectPRlB() 5 .. "'lt1nuelPtLPROG Z91930TO 16384so..>e<T aJel3MS 418os •SNi 75187.969 tilFtCflES 4.589109 I-!ZAO O. lQ9OO36 StCAG 32768ON 6.650 usectE 6.00 uneTE 300.0 "01 1.OOOOOOOO sec411 O.03DCJ(lOQQ Sl!!C
•••••••• C1WfEl fi .~~•••Ui 125ftPt 8.50 usecPlI ').00 dSSF'Ot 157.8342412 Miz
••••_ •• CH.WEL (2 ••••••••
CPl:FfG2 -.all2t6lU2 IHPCP02 105.00 usecPl.2 -3.00 dePLt2 19.00 daSF02 500.1320005 Iffz
Padrão Interno: PhTeTePh -1MoIL" em COCho
ppm ~50 400 300 250 200 150 \00 50
F2 - PNIcuslng paNIle:tersSI 8192SF 157.]gll082 liIUNIlII EMSSll oLB 5.00 H2GIl o~ 1.40
tO ~ plot p.,...tet'sex 23.00 .c.CV 7.00 c.FiP 510.9]3 ppaFi 8062J .00 tizF2P 34.470 ppaF2 5439.03 tt2~ 20.71753 D"IVCHICH 32'69.04224 til/CI
Espectro de RMN 125Te do composto (R,S)-1-(n-butilteluro)-2-propanol [(R,S)-10e]
141
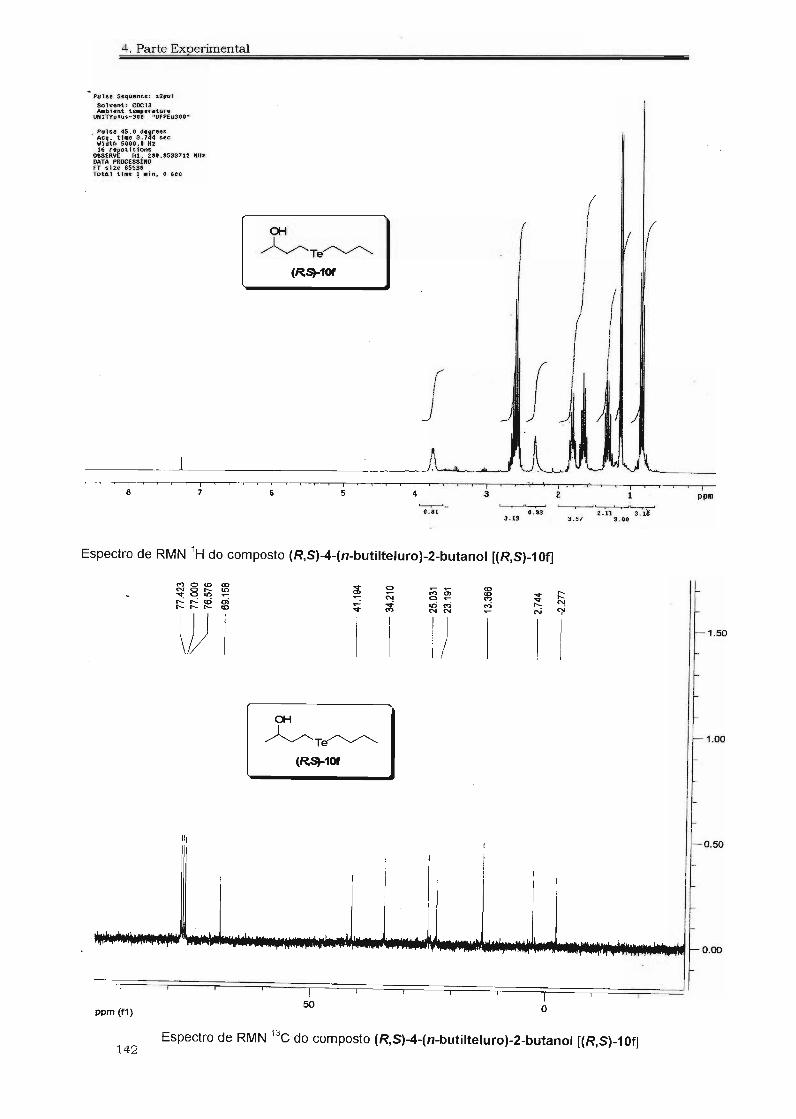
4. Parte Experimental
Pu15e Sequence: 52pUl
Solvent: COCt3-.bi ent te~er.ture
UNITYplus-300 "UfPEuSOO"
Pulse 4S.0 degrau" Acq. tf.e 3.144 uc
Wt dth 5800. O Hz
O~fR~fetf~~~~".'S39712 ""z:DATA PRDCESSIIIGfT 51 ze 65536Total tt •• ~ .in, O seç
a-f
~Te~(~S)-101'
(
Ippll
I . I .,' • i I ' •
Z 1L..-.r--'~ '---T--'~~
0.t3 2.n 3.183.13 3.57 3.00
3
Jfi
I4
~-0.11
I5
I6
I7
I8
-----..-l--- --'~___"'___J
Espectro de RMN 1H do composto (R,S)-4-(n-butilteluro)-2-butanol [(R,S)-10f]
1.50
1.00
0.50
0.00
Io
I50
"'g<O co (J; o ;;;0; <O ....e-> .... U>
N <O •- • lO o ~ (') .... .........,:r--:có ai~ !riM ,.; .... N............ U> ;: N N ~ N ~
\V I I I I) I I II-
a-f
~Te~ f-
(~S)-101'
-
I1/,
I I~I I I
II ,
I I
.L • 11
~I I I I I i I I , I
ppm (f1)
142Espectro de RMN 13C do composto (R,S)-4-(n-butilteluro)-2-butanol [(R,S)-10f]
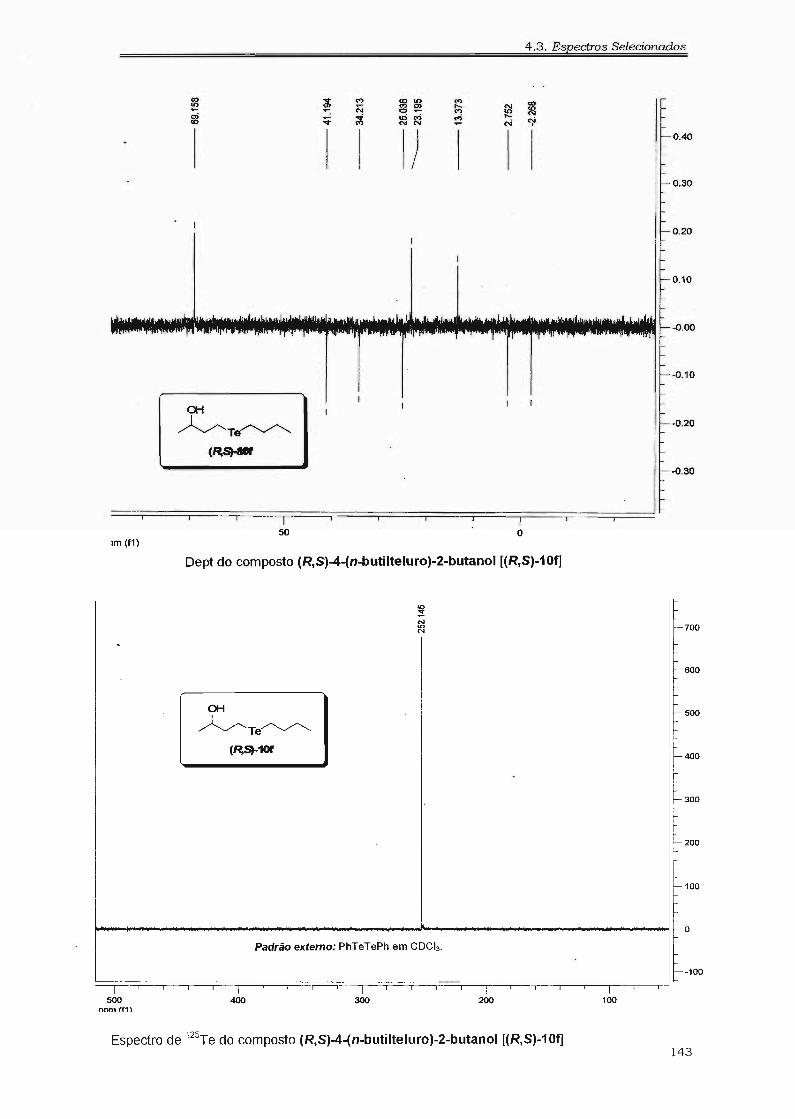
4.3. Espectros Selecionados
Q) et ,.,~U)
,.,~ N .... N
Q)
.,; O~ ..., lO lEl~ ~ 1ÓC"i M ....
~<O N N N
1 I I)1 1 1
0.40
0.30
0.20
0.10
-0.00
-0.10
lm (f1)
I50
IO
-0.20
-0.30
Dept do composto (R,S)-4-(n-butilteluro)-2-butanol [(R,S)-10f]
Padrão externo: PhTeTePh em CDCI3.
ISOO
nnm If1)
OH
~Te~(~S)-1or
I400
I300
I200
-700
t-600
f-SOO
f-400
f-300
-200
-100
o
--100
I100
Espectro de 125Te do composto (R,S)-4-(n-butilteluro)-2-butanol [(R,S)-10f)143
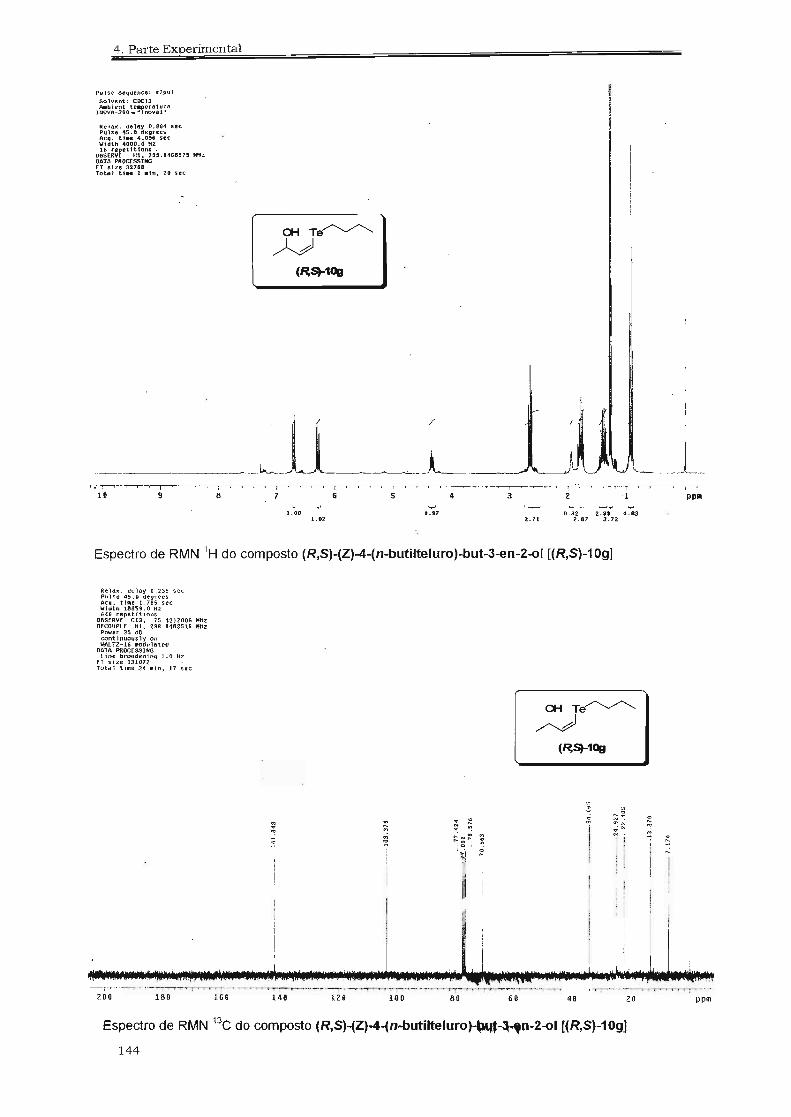
4. Parte Experimental
PulSe: S.quenC8: s2pul
Solvent: CDC13Mbient t~eratur8
I NOVA-300 ... , nova 1"
Relax. delay 0.904 secPulse 45. O degree$ACIlI. t t.. ".0'6 sec\Jidth 4010.0 Hz1$ rapettttons -
OBSERVE Hl't 2U.'468515 ,,"zDATA PROCESSlflfOrT 't>1n 32768Total t.i.. 1 _in, 29 cec
a-I Te~
~(~sr10g
'. I
10iII
i6
... y1.00
1.02
,5
I.. I'.
I I~
3 2 ·1 pp.'---.---' y ...... '--,-' ...
0.82 2." 4.132.71 2.8" 3.12
Espectro de RMN 1H do composto (R,S)-(Z)-4-(n-butilteluro)-but-3-en-2-o1 [(R,S)-10g]
Re!14X. delay 0.235 secPulse 45.0 degreeSAcq. tl.e 1.765 secWidth 18859. O Hz6~O repetftiom.
06SERY[ CU. 75.4217006 "HzDECOUPlf Hl, 299.5483516 "HZ
POWflr 3S d8<;ont I nuously OnIiMLTZ-16 .adulated
DATA PROCESSIHGUne br~denf",;l 1.0 Hz
fT s1ze 131072Total t1.e 34 IIln, J7 $8C
a-I Te~
~(~sr10g
200 180 160 140 120 100 80 60 40 20 pprn
Espectro de RMN 13C do composto (R,SHZ)-4-(n-butilteluro)~'-~'1o·2-o1 [(R,S)-10g]
144
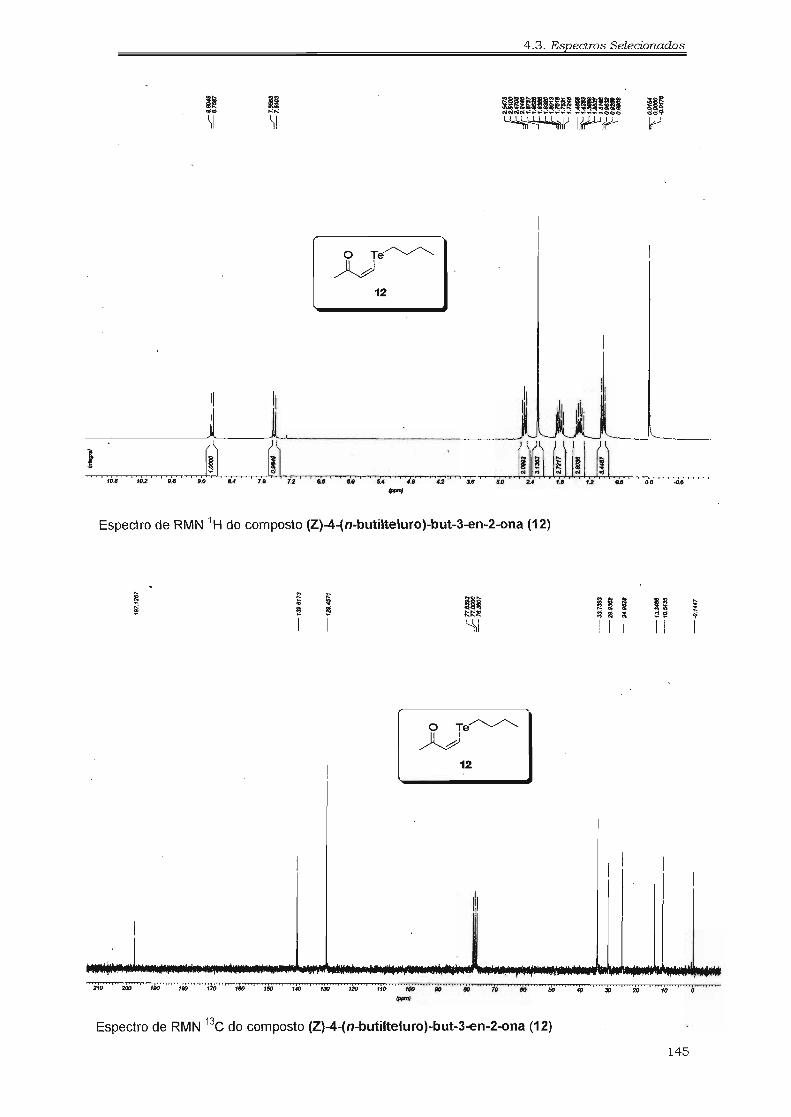
~,
4.3. Espectros Selecionados
o Te~~
12
" ,i1i'I
u' ' ... ' ' ... ' , .. ' ' ... ' , U: '... ' 'u'~ ... ' .... ' .... , ,-Espectro de RMN 1H do composto (Z)-4-(n-butilteluro)-but-3-en-2-ona (12)
o Te~~
12
I
I II
I II
1II
i210 • 2bo I 100 i 180 ' áo ' 1" I 150 • f~ i· fio • do i 110 100' Qo I ia to i tio I tio 40 3D 20 1'0_J
Espectro de RMN 13C do composto (Z)-4-(n-butilteluro)-but-3-en-2-ona (12)
145
eo.o.
4. Parte Experimental
'"...,oo
i
o Te~~
12
Current Dala Para_tersMAtE: KlCluEXPIlO 9PAllCNlJ l
Fi:! - ÃCQUisitlon ~_UrsDate 2OQ5022'3n~ 9.41lHSTlUI 'PeetPAID'IJ 5 _ '*' lt tnuc 1Pl1.PAJG Ig1g30m 163SASll.VEHT ax:13NS 256OS •Slti 75187.969 HzFrHS ".589109 112~ 0.1051)036 setRG 32768ON 6.650 usecDE 6.00~TE :JlO.O K01 1.0000000o secdll 0.03000000 sec
--....-- CHAfofEL ri·····..·tt..Ct 125ft!Pl 8.50 USKPLl -3.00 d8SFOl 157.8609665 Mil
••--- CHANIEL '2 .CPlPRi2 valU16
"-"2 '"PCP02 105.00 U$t!CPl2 -3.00 (l8
Pl12 19.00 d8SF02: 500.1320005 ...u.
Padrão interno: PhTeTePh -1 MoIL-1 em COCh.F'2 - Pf'Ocessl.n9 parHlttersSI 81112$F 157. 79U321 Mil.... ElOSS8 OLB 5.00 HzG8 OPC 1.40
ppm 600 550 500 • 450 400 350 300 250
tO l'Ifl pIot plt'aatersex 23.00 aiCY 10.00 caFtP 670.000 ~Ft 1~.~~
F2P 210.000 ~f'2 33136.t4 H2PAOf 20.00000 rx-/c.HZCM 3155.82275 H2/c.
F2 - AcQUiSHiao Parneters
tu"f"ent DiJliI Pal"MetarsNãNE 141.152ElIPIIIl IPIlOCNO I
Espectro de RMN 12~e do composto (Z)-4-(n-butilteluro)-but-3-en-2-ona (12)
_N~_~NoNm_~~~owm
~~lillll(91 ... C\tmU).,ocn ... ;:o; ; ... '" '" :igrD:grom~o~voMmNm_~~~~ "mteC'Uol'tn_cn "'''''''g ~~~~~mre~~re~~~~~~
m,.... U) 'f') ocu_om_om,....~ ao '" no o o o C'l
C!~'!~ =---0000'101 ao ~~C"! 00<::10. . . . . . . . . . . . . . .rtitfl~tflt'iM~niN,...,...,...,...,...,...,...,...",....,.... ..... ,...,...,...." lllUl1tJ1tl 0000
I I
w+S~~~~ ~ I~p I IV ~ ( Oite_Ti..INSTlUOPllPllOCmSlI.YOO'NSOSSII<FIlR:SAI)
A6llIItETEDlPltEsrOllU'PU
sooooo1·4.22spect
5 _ tlJlt1nu
19306!i536alCl3
32o
55:92.641 Hz0.085340 Hz
!L 9589883 5ec90.5
89.400 UNe
ti.DO usec300.0 K
1.OOOOOOOO $Cc
9.60 usec6.00 U$ec
500.1322456 ~IH
-3.00 dO
j
'd
' .
!~ ~I~I~.~
pp. 8 6
f
5
11
3
f
.1 ~
2
lu 1.1
F2 - Pl'"ocusJng par_tiros51 32768Sf 500.13001043 MillIIlll EM5SIl OLB 0.30 HlGIl OPC 1.00
10 ... p101 per_ttMiex 22.00 c;.aFtP 9.000 QptI
Ft ..501.11 tilF2P -0.500 ~F2 -25CUJ7 HlPADI O,CIB2 ..-!C11HZOt 215.915523 11l/c.
Espectro de RMN 1H do composto (R,S)-O-acetil-1-fenilsse'leno-2-propanol [(R,S)-2a]
146
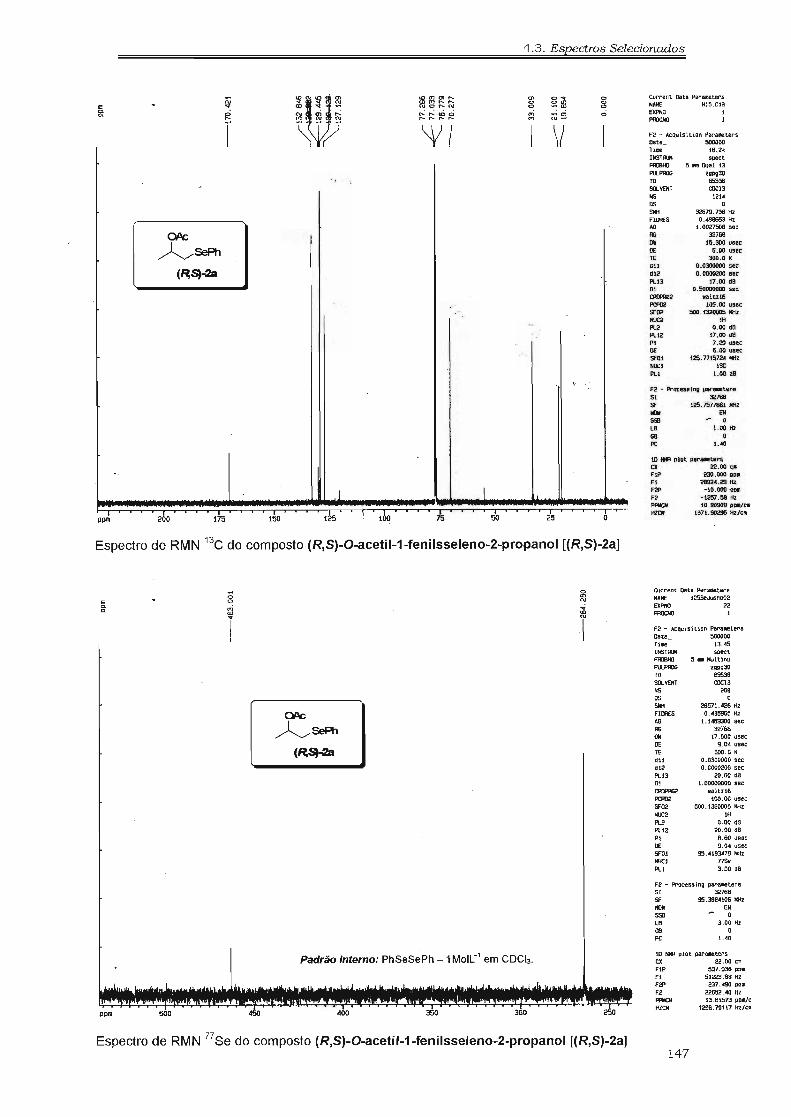
4.3. Espectros Selecionados
N
·~r:§3trJOI,... :?l o .... o CUrNlnt Data PirMeters
...... no .......... O", 8 NAME MiS.CJ3li ": co ... - NO ..... C\l o -coo. o ~ &} ~ ~;::~~
.., -cn .,; EXf'>IO l
I.., no- PIlOCHO I
~-p ~I \I F2 - lLQuisition Para.ter'sDate_ 500000TiIIe 18.2.IIlSTIUl soectPRlIlIIl 5 ... [)Jal 13Pll.PROG zgpg30TO 655385O..Vl:rI! coel3MS 121..llS oSlOt 32679. 73B HzFIlIES 0.-498653 HzAO 1.0027508 see
Qllc RG 32768
~5eA1O. 15.300 usecIJE 6.00 usecTE 300.0 K
O" 0.0300000 see(~S}-.2a 0.2 0.000020O SlC
""3 17.00 d8O. 0.500000OO sec~2 waltz16
"""'" 105.00 useeSFQ2 500. t320005 IrI1.zN!,C2 'HR2 0.00 dEIFU2 n.oocIBF. 7.20 usecIJE 6.00 usecSFO' 125.771572< Itl,1IlC' lXFU 1.00 de
\.F2 - PnJtes&ing ,.--ttrsSI 32768SF 125.1577IEJ NHz:- aoSS8 oL8 1.00 HzGIl oPC 1.<10
10 .- plot oere.t.,.,ex 22.00 e..-FiP 2'30.000 QPIt
I I IF' 28924.29 HzF2P -10.000 0CtfIF2 -1257.58 Hz.-.. tO.90909 ppa/c•
200 175 150 125 100 75 50 25 o HZOI 1371.904!96 Hz/clIopm
Espectro de RMN 13C do composto (R,S)-O-acetil-1-fenilsseleno-2-propanol [(R,S)-2a]
CUl"rent Dala Parueters"AlE 12SSe..umo02EXPMJ 22FIlllCIOO I
oo
I Oate_filieINSTIUlPRIIlHOIUPROGTOS(LvENTNSllSSIItFIIHSAOAli0lIIJETE
O"012
""3OICI'ff>AG2PCI'll2SF02NUC2R21'1.12PIIJESFO'NUCl...,
500000\3.45._,
5 _ NulUnu
zggg3Q55536CIlCI3
269O
28571.<28 til0.~35$5 til
1.1469300 see32768
17.500 U58Cg.04 usee
300.0 K0.0300000 see0.000020Q sec
20.00081. OOOOOOOD see
~ltlt6
100.00 uleC'500. 1320005 Itlz
IH0.00 dS
20.00 aBB.60 usac9.04 usec
95.0419311B teil:ns"
-3.00 de
Padrão interno: PhSeS~Ph - 1MoIL-1 em COCh.
350 300 250
F2 - F'rocusing pW'll_tersSI 32768SF 95.3824106 NHl1IlIf EMSS8 oLB 3.00 lUGIl OPC t .40
to ~ p)ot p.r-.tusex 22.00 c.F1P 531.036 PP.fl 51223.83 Hz
F2P m.490 PPIlFZ 22652.40 HzAItDt 13.6J573 ppa/cHZCIt t29B.7Ott7 Hl'/CJ
Espectro de RMN 77Se do composto (R,S)-O-acetil-1-fenilsseleno-2-propanol [(R,S)-2a]147
4. Parte ExperÚTIenau
8
lL-----r-1----.--,------,,--3000 2000 1()()()
Transmittance I Wavenumber (cm-1 )
Espectro de I.V. do composto (R,S)-O-acetil-1-fenilsseleno-2-propanol [(R,S)-2a]
l l l hH~ ~d.J ..J.-J
(\/ Lfl (9'1 (U toOCDtOU't"'l(lU1(l"J ('1')(91 (\J
~~~~~iOlOl,OCOLn
~~~:~~~~~~~~~~~~~~ó~~~~g~~~~~~~~~~~~~~~~~~~ro~~~g~mN~~~~~~~~~~~~~~~~~OOOO
~llLLL*~~;
r
Current Det. Para-!te~-- .....0l'lIl 1l'IlOOCl 1
F2 - AcQUiSltion Pra-eter.sOate_ 20021.209fia 15.46DfSTJUII SDKlPROa«J 5. IilJUinlJClP.lLPf1J6 tlJlOm 6li536sa.\EHT coeI3HS lOOS O51ft 56&8.9301 HZfIlIES O.08S501 HzMI 5. 78Q325.4 se(
RG 128til 88.200 usec(E 6.00 usecTE 3OO.D KOI 1. OOOOOOOO uc
----- l:Ji.WEl fi ••••--.UI tHPt 7.50 u.scc1'\.1 -~.OO '"Sf'OI 500. 1323417 ~
ff
)
t. :
I l À ...
~ !~~I~ . I~\ !~\ ~ I~I \~II~I~
pp. 8 5 '4 2 o
Espectro de RMN 1H do composto (R,S)-O-acetil-1-fenilsseleno-2-pentanol [(R,S)-2b]
148
F2 - ProceS5iAV par..tersS[ 32768SF 500.13001"1 IR
- no95ll Ol8 0.00 HzGIl OPC 1.00
10 NIfl p lat Plf"lIl!trl'tex 23.00 c.CY 12.00 c.FIP· 9.000 PpIlFI 4501.17 tilF2P -o.!500 PPIlF2 -250.011 H.z.-.. 6.413114pt1O(aHZCN 206.51542 Hz/c.
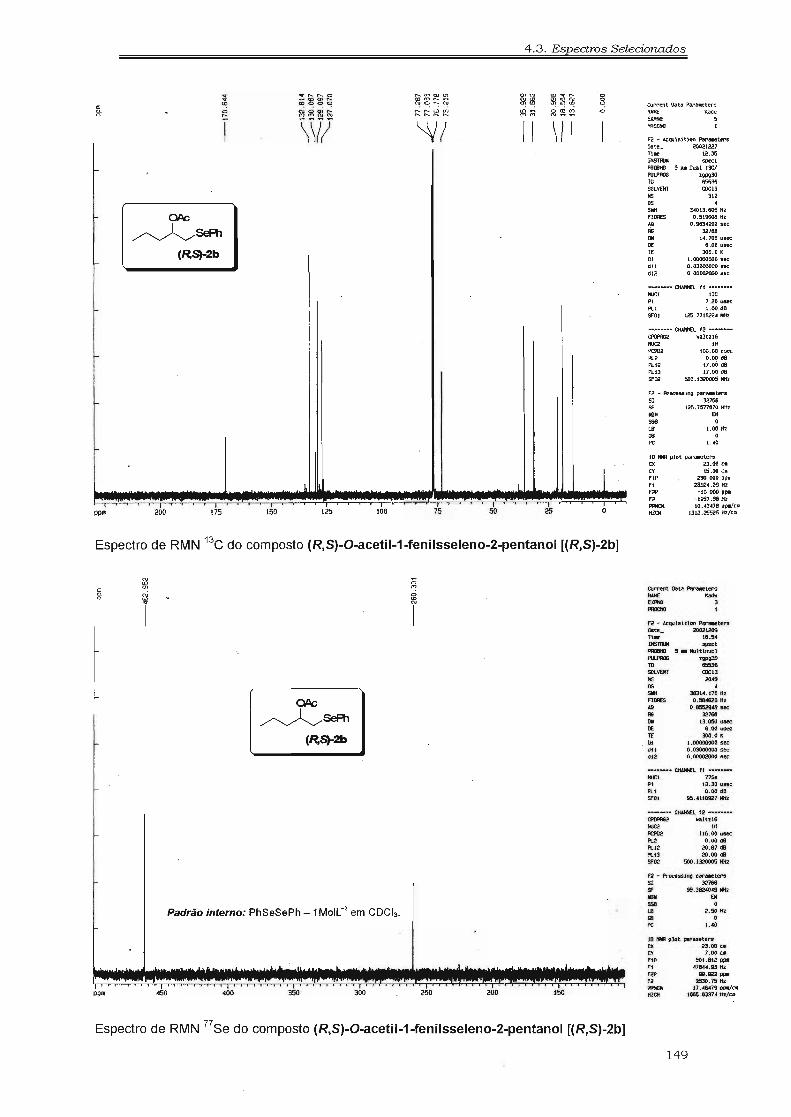
4.3. Espectros Selecionados
aoc~SeR1
(R,S)-2b
~ I I
..CoCo
pp. 200 175 150
'"lI'''''''' o_ to m,.....~~~~(\.lom,...,~~~~
\\11
125 100
r-- ~ CP CTlQ) t't1 ..........C'\,l 0.,....<\1
~;:::::e~
75 50
II \1I
25
goo
o
OJffent Date ParaEteraR.QE Kaclu
ExPIlO •PIlOCIIl ,
F2 - Acquisition ParMMltersOlIte_ 20021227n. 12.35IHSTJUt lijIl:ctPfKDI) 5 _ Dual sr.!PUt.PROO lQPq30lU ...36SOtvOO me}]MS 312OS ,SWt 304013.605 HzFIlIES 0.519006 HzAO O.96342512 secAG 32768111 ,<C. 700 UH<:IE 6.00 USKTE 300.0 K01 1.0000000o 5ltC'O 11 0.0300000o se<;
fU2 0.000020OO sec
........_ 0iAIfrtEt ti .
MJCj tJCPl 7.20 uSKP\.I 1.00 dO5F01 125.771622" ttftl
..---. CHAJtEL f2 _-.....-CPtflREi2 Naltzl6
PlJC2 'HPCf'D2 100.00 URtPl2 0,00 d8F\.t2 11.00 elePU] 17.00 aB9='02 500 _1320005 *tz
F2 - Pracusi.ng p.._tet'5SI 32758g 125.m7870 Mttl1Il" ..558 oL8 1.00 H1G8 oPC 1.40
10 fIII plot ~.eteNlex 23.00 c.CY lS.OO caFIP, ~30.000 PP8FI 2892-4.29 H:zF2P -10.000 PPIIF2 -1251.58 H:zllPMCM 10 •.43478 ~c.HZCH 1312.25525 Hz/C.
Espectro de RMN 13C do composto (R,S)-O-acetil-1-fenilsseleno-2-pentanol [(R,S)-2b]
ru
:5lru
i
Padrão interno: PhSeSePh - 1MoIL-1 em COCho
iDJrrent Data Parll_ters_ .-ExPIlO Jl'IlllCI«l ,
f2 - Acquisl.tion Paraaetl'r"5llate_ 20021209n.. 16.54lHSfR.14 spec:tPRlJH) 5 .....ltimel~ zQPD30lU ll55J6nvau aJeJJNS 2048OS ,SIH 38314.176 HzFIIHS O.5&f628 HZ.t.O 0.8!il52948 secAG 32768lJI 13.050 U5KDE 6.00 usec:Jf 300.0 K0' i .000000lXl SI!CdU 0.0300000O SI!CIU2 0.000020OO see
......... CHAJfrEl fl .
NJCt 77SePS 13.30 Uft(Pl.l 0.00 dBSfOl 95.4110927 MHz
••_ CH.UfEl. f2 ....
~ -alttt6
PlJC2 'HPCPQ2 Jt6.00 usecPL2 0.00 dBPl.12 2O.8J daPLt3 20.00 dBSf02 500. t 320005 tItz
F2 - Processing Dar..t ....ase 32768SF 95.382.4049 NHz.... E",.., oLB 2.50 HzG8 oPC 1."0
~50 ~oo 350 300 250 200 150
tO _ p)at pr..etet'Sex 23.00 c.c:Y 1.00uFtP 5OL612 ~FI 47844.93 HzF2P 99.922 11.-F2 9530.75 taPPMCM f7 .46479 D_c.tiZCM 1685.8331" mie-
Espectro de RMN 77Se do composto (R,S)-O-acetil-1-fenilsseleno-2-pentanol [(R,S)-2b]
149
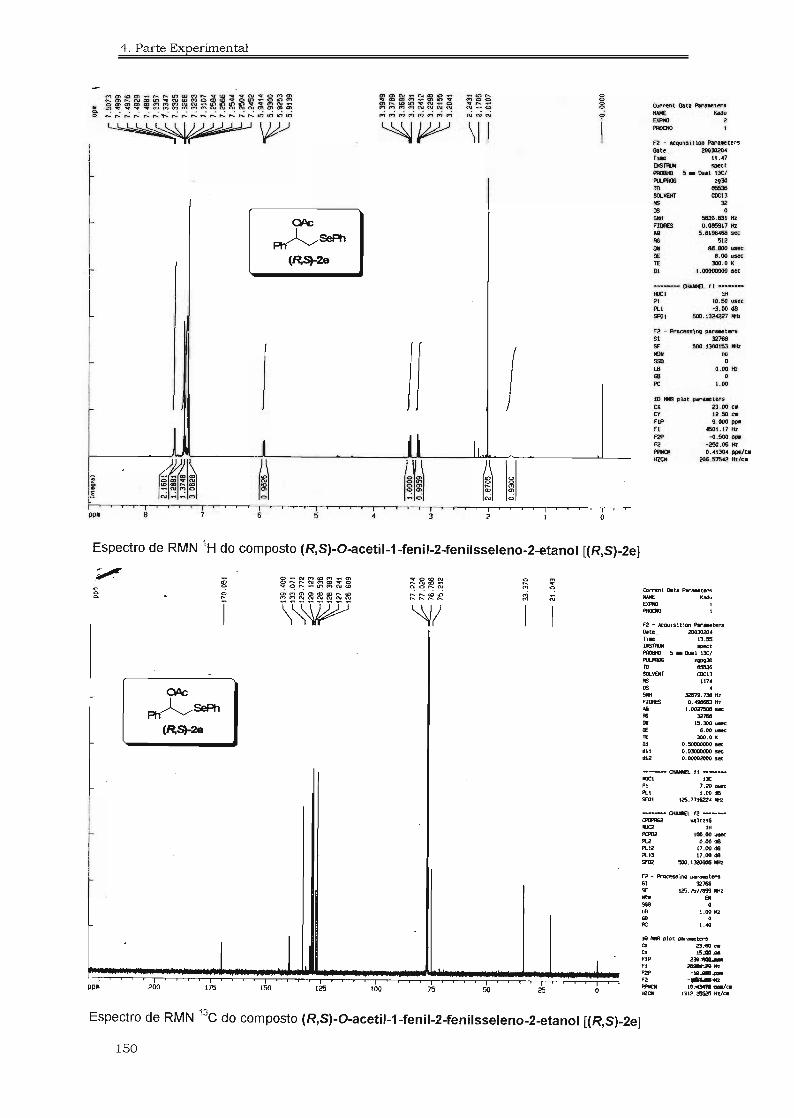
4. Parte Experimental
CJ\OlC\t_C\lCDIO_~Q)cn~ ... mLn.(n1'.1DU'1'V C\I_OtrItr1MMNNC\lCU
rriPiMMM~P;rri
~~ \11
go Current Data Plre.tcrs
- "<luEXI'ICl 2PAllO«l I
F2 ~ Aequ151tlon PlI'uetersOIte_r...lNSl1U<......,PU.I'AIllimS(L'IEIfT..OSSItlFIllRESAllRGllIfOETEDI
2003Q2O.<11 .41spoc.
5 _ tual IX/
'930ll5636CllCl3
32O
5630.631 ttl0.085917 Hl
5.8196«18 Sei:512
88.800 usec8.00 use(
300.0 I(
1.OOOOOOOO 5ec
I fi J1 t U I,
~i (~I~ !~\ !!l~\ ~ ~II i
PIlO e 5 o
•••--- OfIVIEl fi ._......fU:1 IHPt 10.50 usecPU ~3.00 deSFO 1 500. t324227 Jttz
F2 - ProCI!5sill9 ~rueters
SI 32768SF 500.1300153 R
""" noSSll OLB 0.00 H2GIl OPC 1.00
lD .. plot p.r..tersex 23.00 c.CY 12.50 c.FlP 9.000 ~Fi 4501.17 H2F2P -0.500 OPaF2 -250.06 Hz -PPNCM 0.41304 PP&/C.HZCJt 206.S7'542 HJle.
Espectro de RMN 1H do composto (R,S)-O-acetil-1-fenil-2-fenilsseleno-2-etanol [(R,S)-2e]
.~ O_t'lMlOtrI_cn~~~~ o ~
'" O,..,,..,Ntrlcn ... O ~ .o v o,... ... "'(7)C'U<D~~"':~ ~ oe
o ~:;igjm~re~re '"d
~~~:e ;;;d
I~
\\\W;V ~!
Cbl'1"'ett Data Pv_t.....
"""' ."'""""'" I....... I
Fi! - Acquisitian Pr.etel"SDltl!!_ 2OOJ0204n. 13.55lIISTlUf _,
PAGMJ 5 _ OuIl t]C/........ ZQ09lOm l5!53li5m...YOIT meultS 117..05 •9Ii 32679.738 HlFnJIES 0 . .191153 ttlMil I .0G2750I Me.
.. 32"'"DI 15.JOO U:IK:
DE 8.00~TE 300.0 lCDi 0.500000OO Me:
dU O.OlODOOOO .cOJ2 0.000020OO Me
-- otAIIEL '1 ---llJC1 1XPl 1.20 u&eC
PU 1.00 dBSFQ1 125.77t622.c -.z
-.._.~f2--CJl(JIRG2 ,.lUU5JUC2 IHPCflU2 100.00 ueK
PU! 0.00 d8PLt2 11.00 diíllt3 11.00 di~ 500.ll2OOO5 Mz
f2 - Pr'octssing ~ttrsSI 32JI8SF 125 •J517II9 'ItJ:
- EMSS8 OlB 1.00 tf1'111 •PC 1 . .tO
PCO 200
J
175 150 125 100 75 50 25
Io
!D ... plGt .....tensex 23:10 c:eCY IS.GO.mflP 2lt~
FI ~tt1
F2P -11...-...F2 -~
FflliDt Ilt.~-'ra
H20t 1112.2525 "r/e.
Espectro de RMN 13C do composto (R,S)-O-acetil-1-fenil-2-fenilsseleno-2-etanol [(R,S)-2e]
150
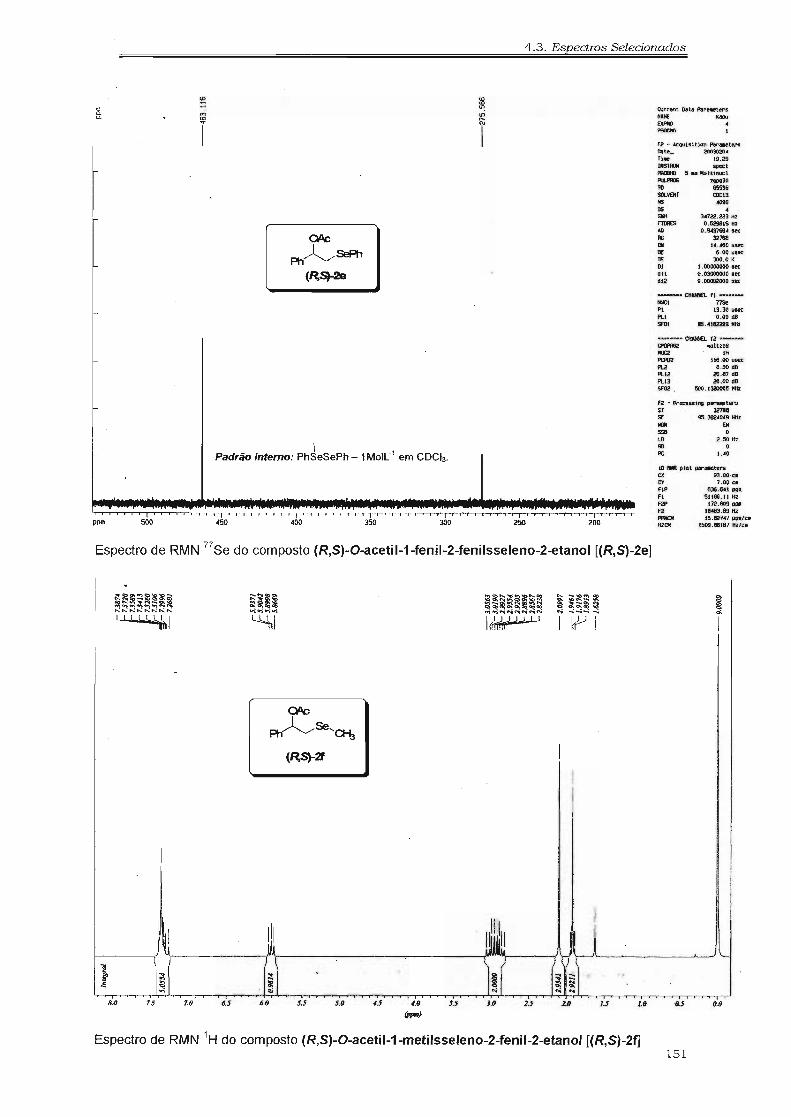
....
i
\Padrão Interno: PhSeSePh - 1MoIL'1 em COCho
4.3. Espectros Selecionados
CUN'ent Data Para.tet"'sIWE .....EXPIlO •PIlIDIll I
f2 - Ai::qutstUon Pra.etef"sDate_ 2003020.Ti. 19.29OOTU soect:f'AmIt) 5 .. Mult,nuclAJl.I'lD; """,30l1l ll5536Stt..va.T COC13NS <096llS •SIIt 34122.223 "2FIlIES O.5a8t9 H2AO O.9.Q768oi iSllCRG !2168'* 1....«)0 usecDE 6.00 U'SK
TE 300.0 K01 t . oooaoooo secdU O.OlOClOOOO se«:a12 O.~uc
----CttAtflEl fi _ .......""'"IIJCt 7&Pt tJ.30 usecPLt 0.00 dBG;Ot t5. "'162219 NH7
----- CHANE:l 12 _ ...~ -a'Ut6lIJC2 IHll'CflO2 H6.00 uneA..2 0.00 dBPLJ2 20.81 dOPlt3 ao.oo d8SF"0i? • 5OO.1S!0005 MHl
F2 - A-oceMi09 p....ter5SI 32768SF fti.382.fOoC9- MKzIlO1O EMSS8 oLfI 2.50 Hz
'" oPC 1."0
ppm, i '500
• I i
450I I I
~oo
i I'350
I I I
300I I i
250i I i
200
10 ,. plot par_tersex 23.oo-c.o 7.00 ~FtP 536.&U PPlfi SUes,SI HzF2P 112.609 NaF2 11463.89 ttzPPMCN 15.827"7 pp./CIIKZa. 1509.•l81 Hl/c.
Espectro de RMN 77Se do composto (R,S)-O-acetil-1-fenil-2-fenilsseleno-2-etanol [(R,S)-2e]
.~§~~~~~~ ~il~ :a!~~$ll~~ i .... 'O~ ao
~~ 19 ~c 0..0. Q)Ql) !~i5r--:r-.:r-:.t-.:t-.:....:r...:to..: 1o"i'riW\Y'i ....;"'i""~ ..... "''''''f'i ..; ...;....;...;....; <S
III Ll~l L1.!n1 l~fflTJ ' J I J I ler I Ili
.
Ofi'c
~Se,%
(~~2r II
I,
~! ~ , i'I!(
\......
} ~ ~.
~ ::::s li: ;;: ~..; <S ..; ..;
8.11 7.5 7.11 6.5 6.0 5.5 5.0 4.0
r-J3.11 1.5 1./1 1.5 1.11 11.0
Espectro de RMN 1H do composto (R,S)-O-acetil-1-metilsseleno-2-fenil-2-etanol [(R,S)-2f]151
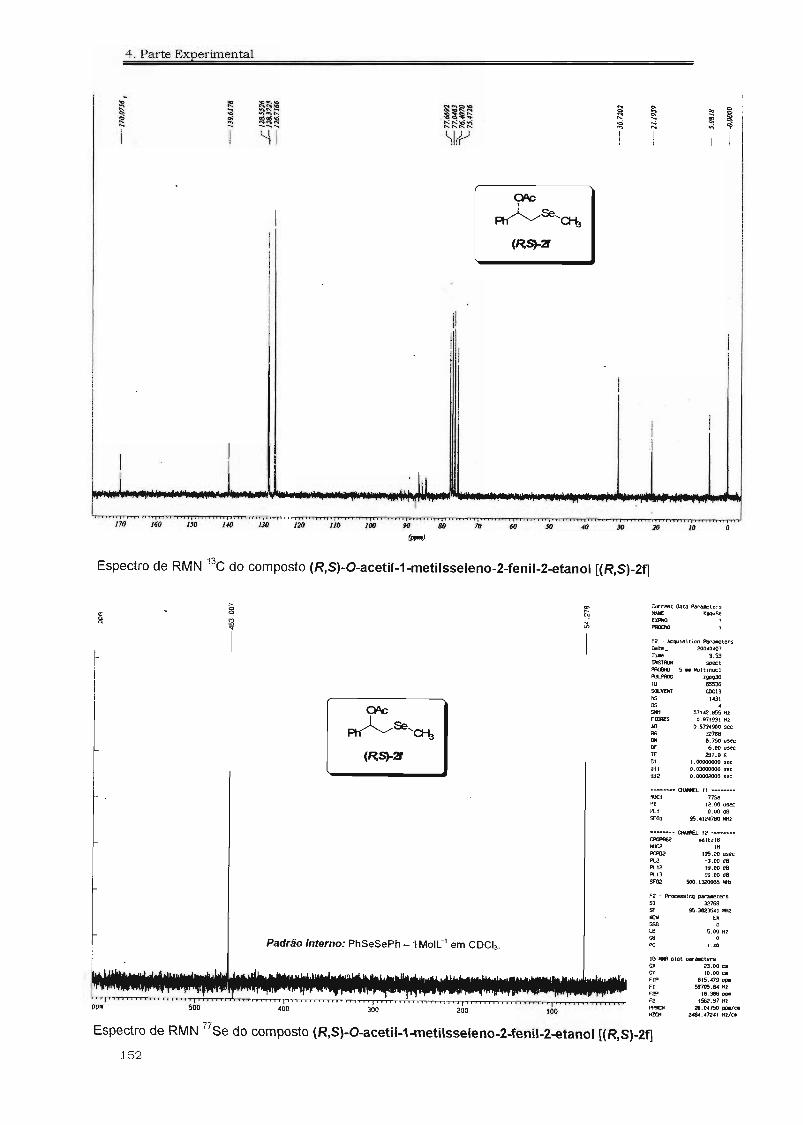
4. Parte Experimental
~ ~ ~~~;;; ~~~
~o< ~~'Õ
~ ~~~
~I
~~~~~ '" ~ §~ .....~" ~"' ........ ~ '""':t-.:'Owi Q ...;
'" 9r-.. r-.."" .., ...\Irf-J I I I
o1010BO111(}1/0110IJ()UlJ/SIl160170
I (~S)-2I'
I IIII
IIII
I,
i II
I I
I i I.. 111jI I1"'''11'' ""I'I",,,,,,ljilll,II"ji
Espectro de RMN 13C do composto (R,S)-O-acetil-1-metilsseleno-2-fenil-2-etanol [(R,S)-2f]
'"....N
~
'"
~elt OIIta PaI"..tU5- ............, 1
""""" 1
r
F2 - AcQUlsltlon PraeeteNDatl!_ 200<40407Ti. 9.53DISTO soe:ctPRCIJC) '5 _ ...Jt iouc 1
PlI...JIIO; 19P9lOro ll5036sa.VDlT alC13k5 leios ,S»l 57142.855 ttzFrmES 0.911931 HzAO O.5~'ee
AO ..768DI 8.7'50 lIset
DE 6.00 uscc:TE 291.01(01 t 0oooooס0. sec\J 11 O. 0300000O secd12 O.lJOOO2OO0 Se(
• __........ OiANEl f t ....--.
NJCI ns.1'1 12.00 lt5~
PL.l O.DadSSFOt 95.4IZ.780 M1l
...-- ...... 0iAIIEL '2 ......--..G't'PA&2 .... ltli!0llC2 IHPCPD2 tas.DO une.PL2 -1.00 dBPU2 19.00 dePll3 19.00 dBSf02 !iOO.I320005 JItt1
Padrão Interno: PhSeSePh - 1Moll-' em COCho
f2 - Pracess iJ\Q Pan.! tI!"51 ..768Sf 95.382)5.(1 fI1llIllI ElISS8 oLO 5.00 HzGB oPC 1.40
tO,.. p)ot __teM
o 2].00 C'a
CY 10.00 caF1P 615.479 p.-fi 58105.8.. IflF2P 16.l!l6 PPIIf2 t56ê!.97 ttlPADI 26.04750 ~ct1tflOt 2......72.' "l/C.
Espectro de RMN 77Se do composto (R,S)-O-acetil-1 ~etilsseleno-2-fenil-2-etanol[(R,S)-2f]
152
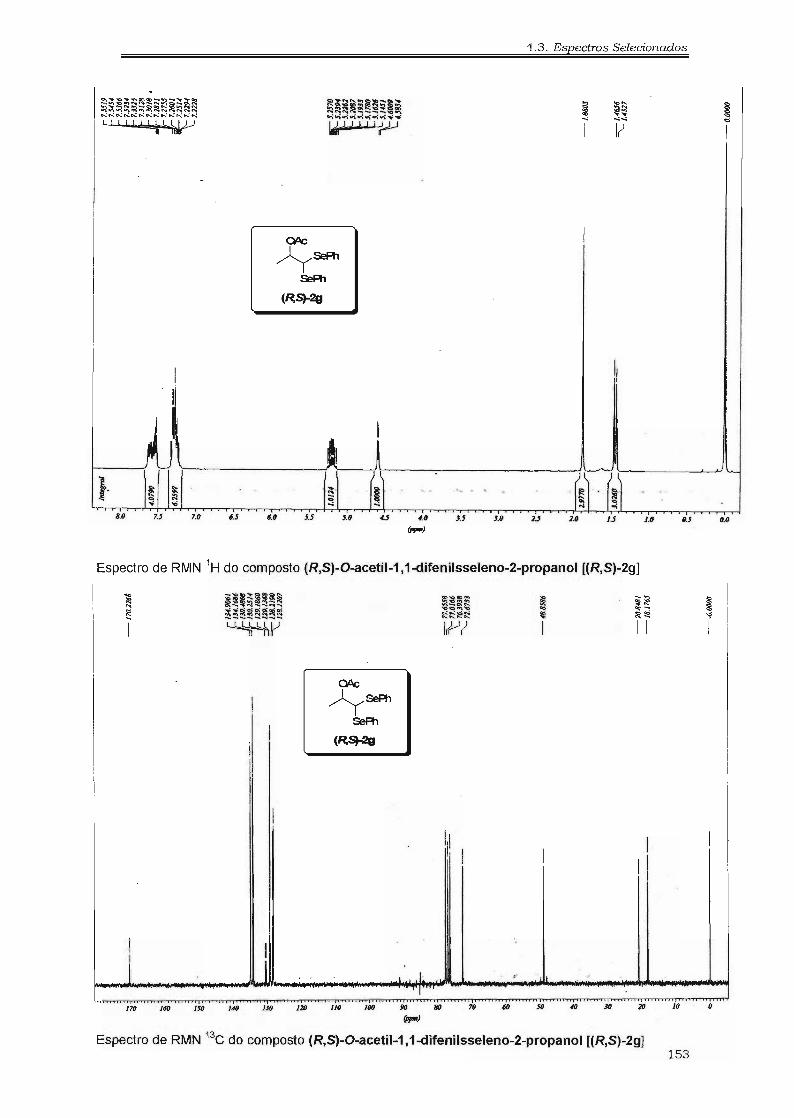
4.3. Espectros Selecionados
I(
Espectro de RMN 1H do composto (R,S)-O-acetil-1,1-difenilsseleno-2-propanol [(R,S)-2g]
I i ('
,CGc
I ~5eR1!seRl
I(~
I1.1 I I,. I I III I
I i'.
.. .1. . , .,
I
m ~ = ~ ~ m ~ m ~ ~ ro ~ ~ ~ ~ M ro(ípt)
Espectro de RMN BC do composto (R,S)-O-acetil-1,1-difenilsseleno-2-propanol [(R,S)-2g]153
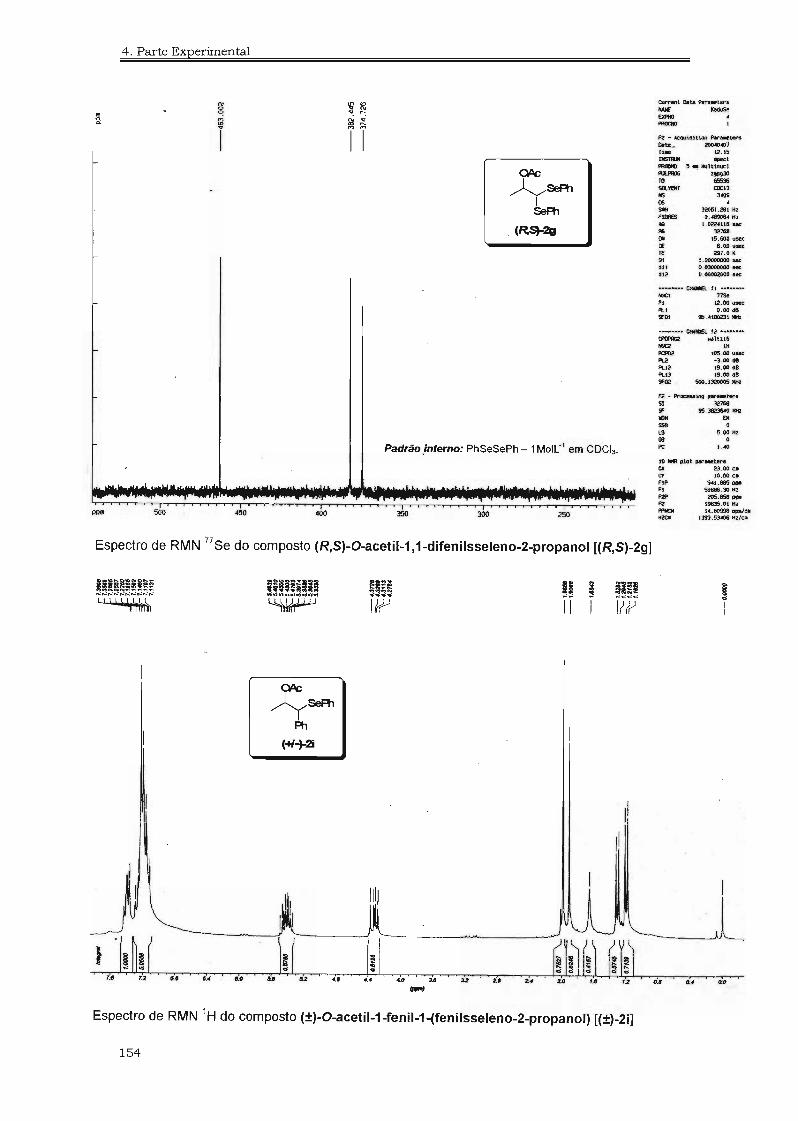
4. Parte Experimental
'" ... '"g " '"" ~ r;g ....
~ ~
i 11
Padrão .Interno: PhSeSePh - 1MoIL-' em CDCh.
eurrent OIt.. P.,...etu5lWI' KaduS<EJl1'IIll ,
"""'"' I
F2 - ACQ.,üslUon p.,._t..-sOlIu_ 20040107n. 12.J5IJISTRM __I
PRlIIG 5 lIl!I ...,lt inuC IPW'IIlli .-m .....$Cl.YBtT aJe13
'" .409os •SIIIt 32051.281 H2flORES O•.c8906" H:rAO 1.0224118 sec... '''!7ll8<11 15.600 u-ec:DE 6.00~TE 297.0kDI 1.OOOOOOOO secdi ! 0.030000OO S«d12 O,OOOQ2OOO SlI(:
.._ ...~ fi ...-.._.
IU:t 17S.e~I 12.00 usecPU 0.00 dBSfOI 95.·4180231 NHl:
••••_ •• CHMIEL. fê ..CPIJlA:i2 ~ltzll5
llJC2 IHPCPD2 105.00 lIMe
Pl.2 -3.00 diPL12 19.00 08PL13 19.00 08SF02 • 500.1.320005 Nftz
F'2 - ProaainO paraeeters51 ...,..SI'" 95.l82364O Nttt- ..SS8 oL8 5.00 HzG8 oPC 1.40
500 450 400 350 JOO 250
tD .... plat or_t.enex 23.00 C8Cf 10.00 c.ftP 541.885 p.-FI 51.:t.30"1F2P 205.855 '*F'2 19635.01 HzPPIltM 14.60998 P4)a/c.HZOf I]g]. 53406 ":r/c:.
Espectro de RMN 77Se do composto (R,S)-O-acetil-1,1-difenilsseleno-2-propanol [(R,S)-2g]
II
,I
II
I~I
~~l, , : , : W·5.2 .... ... .:0 ie 12 ... ... 2.0 1.6 1.2 0.. ti. rio-
Espectro de RMN 1H do composto (±)-O-acetil-1-fenil-1-(fenilsseleno-2-propanol) [(±)-2i]
154

4.3. Espectros Selecionados
~~ .i!~i!!~ii~i!~; il§n !!l: ~~~~ !ih~!!~~J~~~~~~s~ ~:::~~~ aa ...: ..~ ~~!\l~
.. ~I(~ Ir ~ I I
1~ 1~·. ~ • ~ ~ ~ ~ ••• ~ 1~ ~ • ~ • ~ • ~ • ~ ••• ~ ~ • ~ •• ~0>J00!I
Espectro de RMN BC do composto (±)-O-acetil-1-fenil-1-(fenilsseleno-2-propanol) [(±)-2i]
~U'100omruo~m<n~-~w~mmm
~~~~fflM~~~~~N~Õ~~~ffi""~~~""""NrurururuNNN _
~~~~~~~~~~~~~~~~~~
I
I
I:,I1I/1
"'lf"" o P'JCT)C'UC'U .....-0l1'U'1~~~~1010 lO Ln
QOc
~TePh
(R&}-11a
rI
i
vo l.Cl ..... tO mlCl'V tDOlaJ"II;f f'l C) V C\latOl UJI'TlC'l.....
VIV
Ii i
CUrrent Data PiIr.UlEtersNAME CarlosEOUarooEXPMl IPROCNO I
F2 - ACQuisitlon P"raee:tersIJate_ 500000Tll1e 6.49y,.STJIJM' ~ect
PROOtt) !i .. Qus) 13PU..PPOG 11;130TO 32768stl.VENT aJeJJNS 32OS OSIIH 3834.356 H1FIMES 0.111015 H.zAO ".27299741 seeRG \81DN J3O.400 usecDE 6.00 usetTE 300.0 KDl 1.000000Q0 ~cPt 8.00 usecDE 6.00 u5ecSfQI 3GQ. '31-4166 MH7MJCJ IH"'-, 1.00 oB
F2 - Processlng parallll:l8NiSI '32768
. Sf 9OO.13DOO66 HHzllllII EIISSIl OLB 0.30 HzG8 OPC 1.00
10 MI=l ,,10t (ull"'l!lllletersex 22.50 CIIF1P 10.000 OpllFi 3001.30 tilF2P -0.500 OPIIf2 -150.07 HZPPICH (1.116667 DOlO/C"HltM 140.06067 Hz/c"
ppm 9 B 6 5 3 2
Espectro de RMN 1H do composto (R,S)-O-acetil-1-fenilteluro-2-propanol [(R,S)-11 a]155
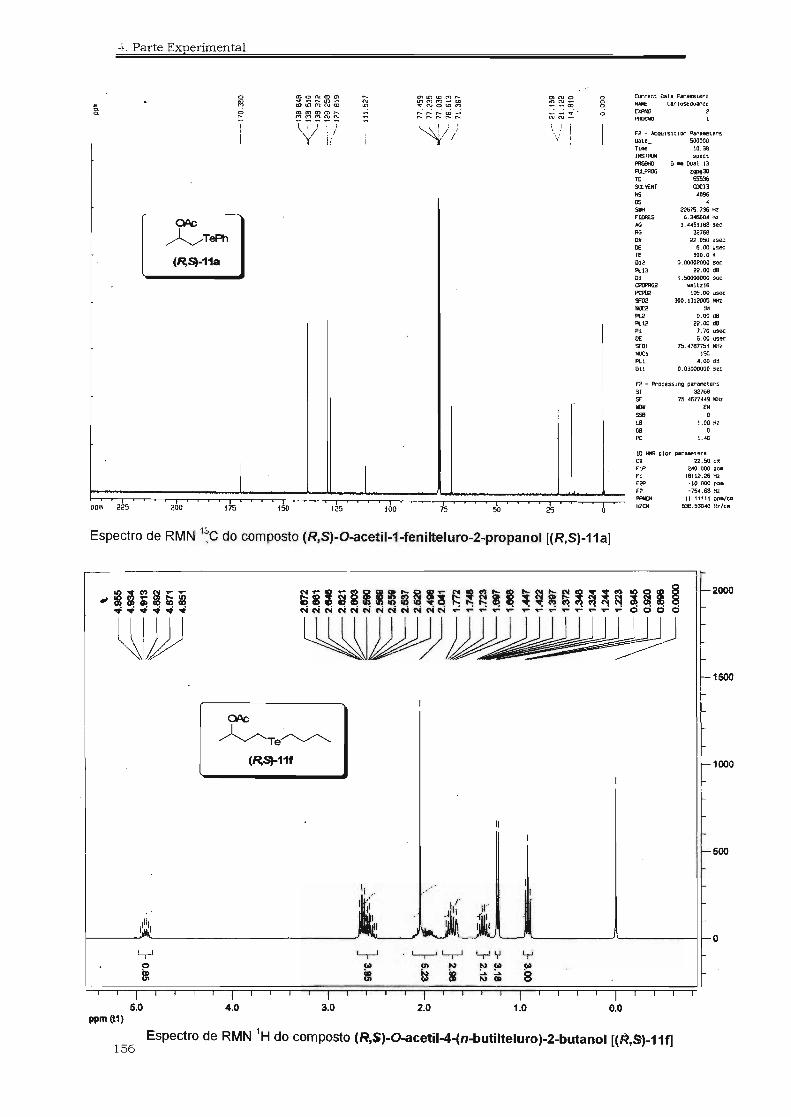
.+. Parte Experimental
QOc
~TeR1
(~S)-11a
I
I Ippm 225 200 175
<>
"',.,<>~
I
150 125
í
100
~')
! /i
75 50
~g:~- - <D....: ...; ~ ~~C\!C\I ...
\11
25
oooo
Current Data ParMeter!iNIrIIE CII"JostdlJarCOEl<f'l«l 2l'IlllOlll t
F2 - AcQ,dsluan ParaeeUrsDate_ 500000T1me 10.38IfISTIUl saectPfIB() 5 ., Dua1 t 3~G z!PJ30TU 65536Sll.V9tT coe13NS 4096os 4
Swt m15.736 Hlr1CJE5 o.346QO.4 ftlAO J ••~ttsa se:cRG 32168Ofli 22 •050 usecDE 6.00 usecTE 300.0 K012 0.000020OO sec~13 n.oo~
01 1.500000OO sec:CA:PAG2 we Itz 16PCRJ2 105.00 lJSeCSF02 JOO. J312005 fIt1Z
lllX2 '"Pl.2 0.00 cIB~t2 ~.OO~
PI 1.70 usecm:: 6.00 usecsr01 75.47'61751 NHl10:1 13C.PU 4.00 d9ou 0.03000000 sec
F2 - Pr'acessing parametersSI 32768SF 75...677....9 tI"l2.... ENSSB oLa 1.00 H1GIl oPC 1.-'0
lD Nfl pJOt p.ra.etersex 22.5O-cillF"1P Z"O.OOO DOaF! 18112.26 Hlf2P ·10.000 P:MF2 -]5.4.68 l'tZPfIM:N H 1H ti DD~C.
HlOl 838.530"0 H,z/cfIl
Espectro de RMN 1~C do composto (R,S)-O-acetil-1-fenilteluro-2-propanol [(R,S)-11a]
-2000
-1500
QOc
~Te~(~~11f -1000
-500
'-------.J'--~----...H-O
I
liI
110,o' I..... I
11": . i- U'.'
,I
~ ,I{ I\Ik' I I'" ~ '------'
y L,-J L...,-J L...,J yy yp ~ !" N N CIO ~g: S ~ li :... :... 8N Clt
I I I I I I I I I I5.0 •.0 3.0 2.0 1.0 0.0
ppm (t1)
156 Espectro de RMN 1H do composto (R,$)-O-acetil-4-(n~utilteluro)-2-butanol [(~,SJ-11f]
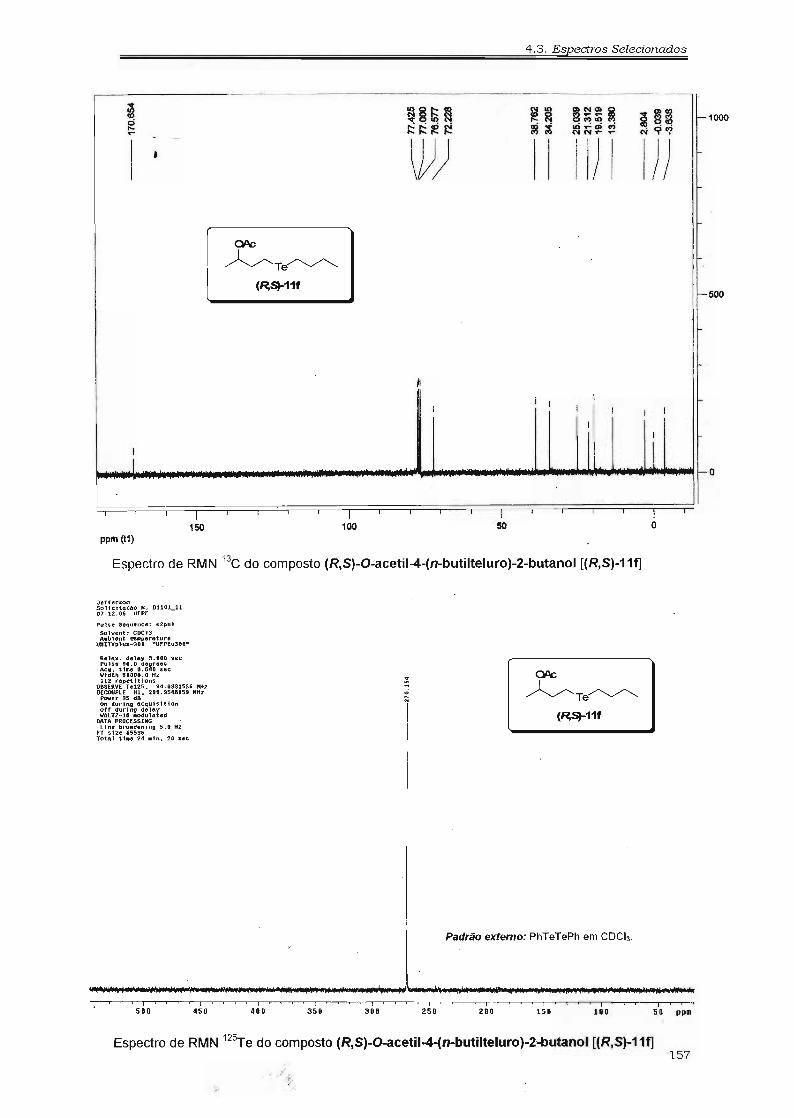
4.3. Espectros Selecionados
o
500
1000
IO
I50
I100
I150
~ ~§I::~ ~~ ~~~ ~ 2 ~~<O t-~
.. O/) N 0<')0/) ocoJ::J::Ié~ ~~ &ri~c:D ... N q c?..- - NN"- ..-
I-
\1)) 11) I I))•
Qllc
~Te~(~S)-11'
t-
~
I II
I I I I I
I
ilI
I f-
I I I I I I I I I I I I I I
ppm (11)
Espectro de RMN 13C do composto (R,S)-O-acetil-4-(n-butilteluro)-2-butanol [(R,S)-11f]
JeffersonSol1cit4cao N. D1101_1101.12.05 UfPE
Pulse Sequcnce: s2pul
So'vent: CDC13AnIb1.nt ~perature
U"1TYplul-300 "UfP[u3eo"
Rel«x. delay S.OOO $SePulse '0.0 dear••sACCl. t1ln. 0.640 ucWtdth $0000.0 Hz112 repetfttons
otSERV[ Te12S. 94.6331555 ,,"2OfCOUPU "1, 2". !l54865' ""z
Power 35 d8on clurtng aCllIuistttono1't durlng dela.yWAl TZ-lI .odulated
DATA PROCfSSINGltne broadenlng 5.0 Hz
fT s1ze 65536Total ti.. 24 .tn~ 20 uc
Qllc
~Te~(~S)-11'
Padrão externo: PhTeTePh em COCh.
• I •510
I I i
450I i
400I '
350I •
391i I I
250I '
200I I
150I
100I
50 ppm
Espectro de RMN 12Sr'e do composto (R,S)-O-acetil-4,n-butilteluro)-2-butanol [(R,S)-11f]'157
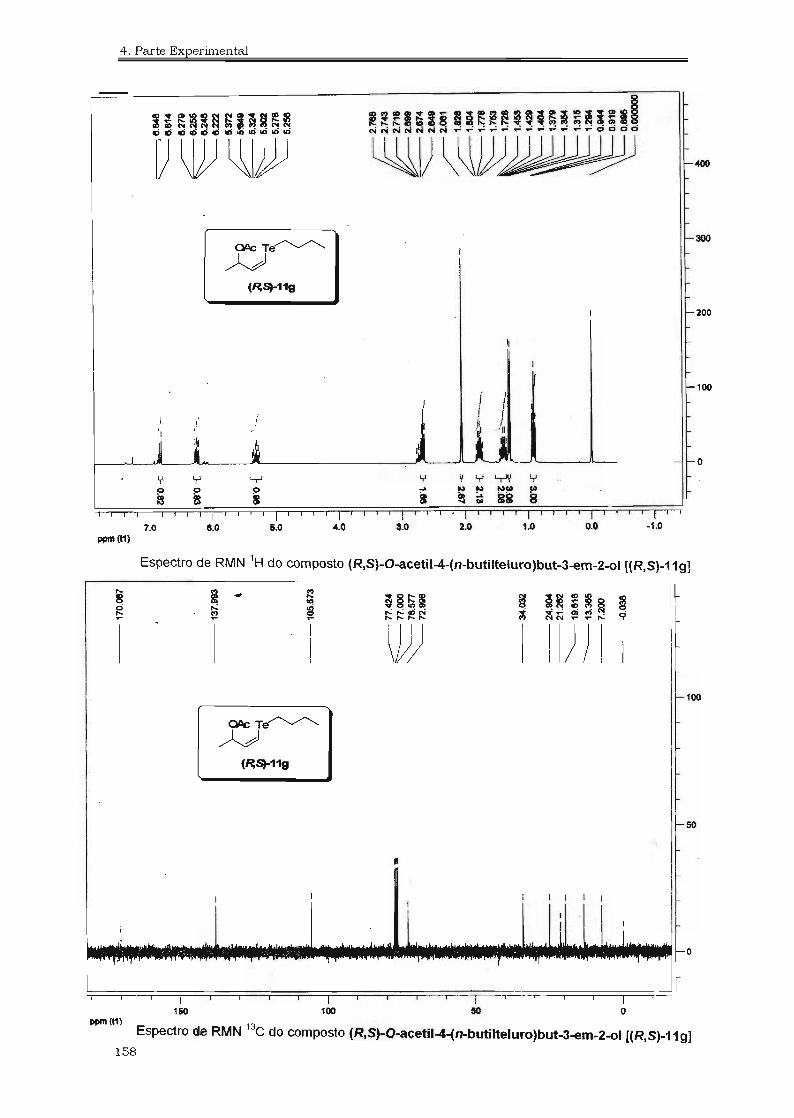
4. Parte Experimental
o
200
100
300
-1.00.01.02.03.0I
".0I
5.0I
e.oI
7.0
!;~~!a~I~~~~ ~~~1~18~i~~~~~~~~~~J~.1ci ci G cici ci IÓ IÓ lO lO 10 lO
UlJJ)~~w»@JI) \lV l\WJ -,
.
f-
QllcTe~ I
~(~S}-11g
I f-
~
I
f-
i Ir j II ,I /
i ,In
.J,
J_11 j S-l jY y y y y y yy yo ; !:> i ~ ~ NflO !A
~:.. ii 8i.J! I (o>
i I I I i i I i I I I i i i i I I I I I I i I I i I I i I I I I II I I I I i i I I I
ppm (t1)
Espectro de RMN 1H do composto (R,S)-O-acetil-4-(n-butilteluro)but-3-em-2-ol [(R,S)-11g]
o
100
50
oI
50I
100I
150
~I .. ~ ~§~I li ~~ ~ ~ ~ ~~ ~ J:::::::lé~ ,; ........ ai C') • <:;>N N or- 'r ,....
I.
I I \})) I 11) ) II
I~
QllcTe~
~(~S}-11g
-
•I I I I I I I
I
.lI
1I
IIII ..,
f-, 1 r ",I
I I I I I I I I I I I I I i I I i
ppm (\1)
Espectro de RMN 13C do composto (R,S)-o-acetil-4-(n-butilteluro)but-3-em-2-o1 [(R,S)-11 g]158
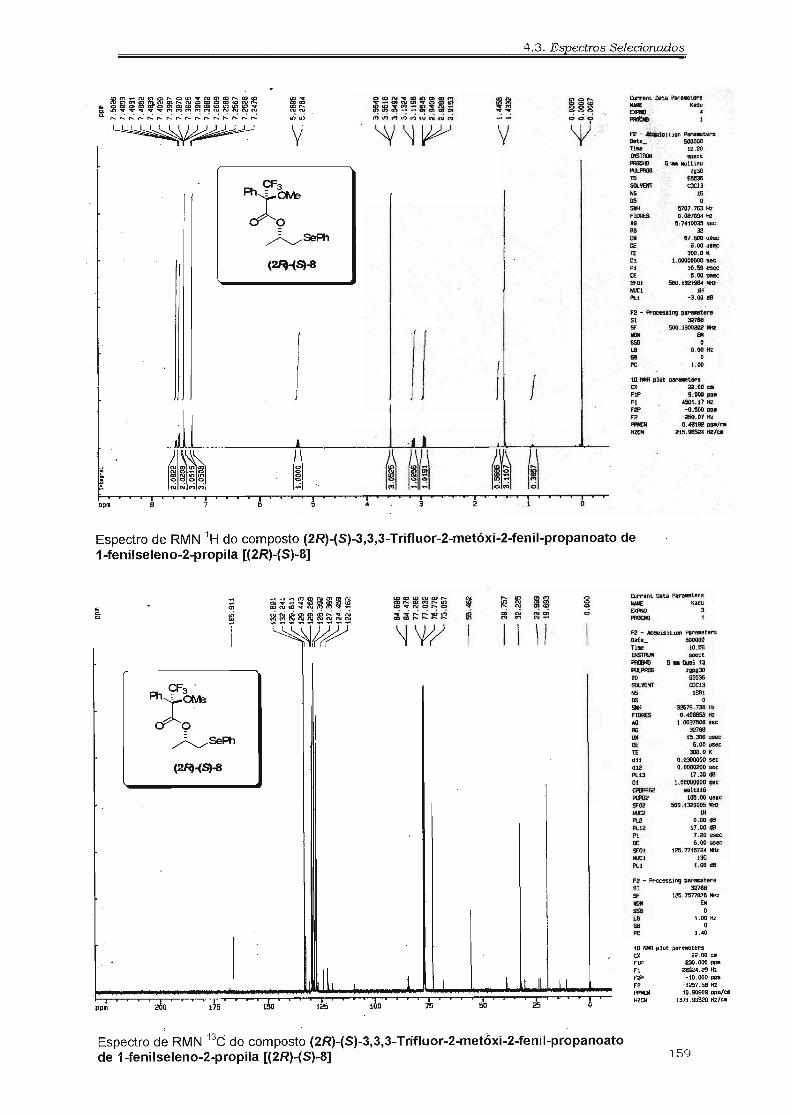
4.3. Espectros Selecionados
~li1 Sim-~~ i~ã 8fB~~ ~ ~~g~~~~~~~m~_~NNNNN
~~~~~~~~~~~~~~~~~
LLLL~~))..J...J y~~~~i!O~CI):ri~:g~~::~~~õ:"';~rriC"icYicVNnini
y~ent Data Pí1rHeters.. K_EXAIJ-.' ..
PRIÍ:lI!' I
F'2 - '.'.~lian PaNI_tersDlte_ ' 500000n. 12.20II'SllUt sglllctPFIIH] -5~. NvlUnuPll.PA06 z;30m 65536stLYEKI ctlC13NS 16OS OSIIf 5707.763 H7F1IJES O.087Q9l1 H2AQ 5.7"10035 secR6 32DIII 81.600 USlt:tE 6.0(1 use,:TE 300.0 KDl 1.DOOOOOOO secP! 10.50 usec:m: 6.00 usêcSFDt 5OO.I32t98A JItttM.Cl 1H'A,.I -3.00 d8
2 .1
1 ))
tilI 1 .i •
~ ~I~~) I!\ I!I !~\!5\fSppm 8 ~ ~ 5
f
F2 - PrOCt:SSing ~ttt'1l
SI !l:!7IBSF 500. 1300202 lI1ZllOII EMSSIl OLB 0.00 Hz6ll OPC 1.00
1.0 PIlR plot ~tersO ~.OO~
FtP 9.000 POlIFI <I!JOI.11 '"f2P ·O.~_
F2 -250.01 HZPPICM 0 ..(3192 ptla/CIl
HlCM 21..5.9852" Hzlca
Espectro de RMN 1H do composto (2R)-(S)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-fenilseleno-2-propila [(2R)-(S)-8]
':17~SeRl
(~-(~
IIIr I I I 111 11 IIpp. 200 175 ISo 125 100 75 50 25 ~
10 NHA plot per_te,.,
eurrent Data parall8lersMAME KaCltlEmIJ 3PRIClIO ,
22.00 c.230.000 PSJII
2ll924.29 '"-10.000 PPII
-1257.5& HZ10.90909 DPlIIca
t371.9032O Hz/c.
exHPfIf2Pf2
-="'CN
F2 - PrOCe$Sing plIl"_tersSI 3216851 125.1517876 NHzIOf EMs!ia o18 t.DO Hz68 OPC 1...0
F2 - AtQUisiLion Par_lersDail!_ 500000Tiae 10.56IHS1fUl spe1:tPfQK) 5 • tlHIl 13AJ..PAOG liKJg30m 65536stLYENT ctlCl3NS 199!OS O9I'f 32679. 738 HlFItlES O.~ HzAO 1.0027508 secA6 32768DW 15.300 usecDE 6.00 LtSIlCTE :lOO.O ktlU 0.030000O sectl12 O.OOOQ200 :recpua 17.00 dO"OI 1.00000000 SecCPtFRG2 WllU16PCPD2 105.00 uneSF02 500.1320005 tIqlIJC2 IHPl2 0.00 48PL12 17.00 lJ1Pl 7.20 lpt(:oc 6.00 as.cSfOJ t25.17t572.4 JIiltu:! 131:PU 1,00 4B
g~C>
~~Na>N-
I I \I·m~reã;::~~~::::~:e~
,"C"1mNa'ltnC\l_,-lOcnCOItlU)lDVN~tTl ... _
RillÍgj!\i:;i~~
~WIII
íeli
Espectro de RMN 13C do composto (2R)-(S)-3,3,3-Trifluor-2-metoxi-2-fenil-propanoatode 1-fenilseleno-2-propila [(2R)-(S)-8] 159
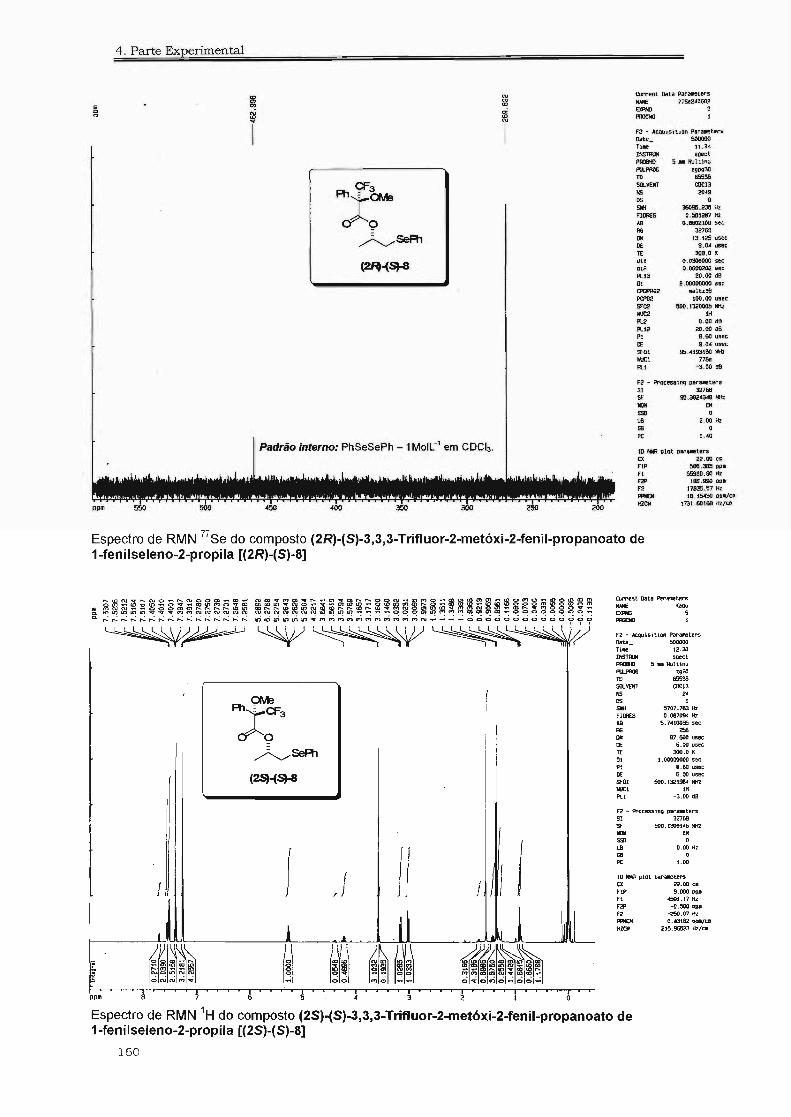
4. Parte Experimental
F2 - AtQUi$ltlQn p.t'a.eters
Currel'lt Data PitaHlersIWE 77Se2.40602OPNO 2P!IlC!Il I
i i 0..._
Ti.II<STIUIPlOIlfJPlt.PIlQ(;
mSll.YEN'NSOSSItfFIIJlESAORGDII!lOTEOildi.PlI'OICPIJ'R62PCI'Il2SFD2lllC2Pl2PU.P!!lOSFOI111:11'1.1
5ODOOO11.34SDect
S.ltsltinuZQDg3065536aleI'200l
aJ80g5.238 Hz0.581287 HZ
0.8602100 sec32766
13.125 tJsec9.().oI usec
300.0 K0.0300000 se:c0.000020O sec
2O.00dS2. oooooooo sec
lMIU16100.00 usec
500. 1320005 M1ZIH
0.00 "820.00 da8.60 usec9.04 usec
g:j ...U93180 \11Hz7750
-•.aa d9
Padrão interno: PhSeSePh - 1MoIL-' em COCb.
FZ - Processing par-••tet'sSI 327689" 95.382"348 lrIi2lIJlI E>lSS8 aLB 2.00 HzGIl aPC 1.010
10 'ItR plOl P....t~sex 22.00 eaFtP 586.385 pp.Fi 55930 .80 HzF2P 186.990 PIS_F2 t78J5.57 HzPADI t6.1SC30 opa/c.H2a. 1731.60168 Hz/Clll
Espectro de RMN 77Se do composto (2R)-(5)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-fenilseleno-2-propila [(2R)-(5)-8]
Al~
)::3~5eAl
(2SH~
tuN"enl Data Plf'a.ters- --OPNO 5l'IIEIIl I
5QQQQQ12.30spect
fi _ lLlltinu
'0""65536ale13
2'o
5101.763 H70.Q8709.4 ~
5.IJtl0035 .sec2!i6
87.600 UNe6.00 UR(;
300.0 K1.OOOOOOOO 9te
8.60~6.00 usee
500. t321!B( lIizIH
-3.00 1I8
Oite_n..D<STIUO-.,l'Il.PAQGm5ll.lEJfTOSOS5IIHFlIJlESAO..lIII!lOTEaI1'1!lOSFOIIOJClPlI
m~~~~~~~~~~~~§~~m~~g~~~~~~i~g~~m~g~~~NNNNNNN~~~~ ooog~~~~~~~~_OOOOOQOO_
~~~\\P~~h~Óóoó0!p
~~~~!;:~~õ!;~~~~õ~ã)!~m~~~~~~~~~~~~re~
,..,:,...:,..:,..:.,..:",..:,...:,..:,..:,..:,..,:,..:,..:,..:,..:
f f
6
11
F2 - PNK:tsSlhg nr_te,.,SI 32768SF 500.13001046 ltfZ_ E>l
9511 oli 0.00 Hz... oPC 1.00
lD JNl, plot O""U:r$ex 22.00 (11
FiP 9.000 PPIFi 601.17 HlF2P -0.500 0IMlF2 -250.07 ItlPPICM Q•.0182 ~e.KlQI 215.~ ltZ/C11
Espectro de RMN 1H do composto (25)-(5)-3,3,3-Trifluor-2-rnetóxi-2-fenil-propanoato de1-fenilseleno-2-propila [(25)-(5)-8]
160
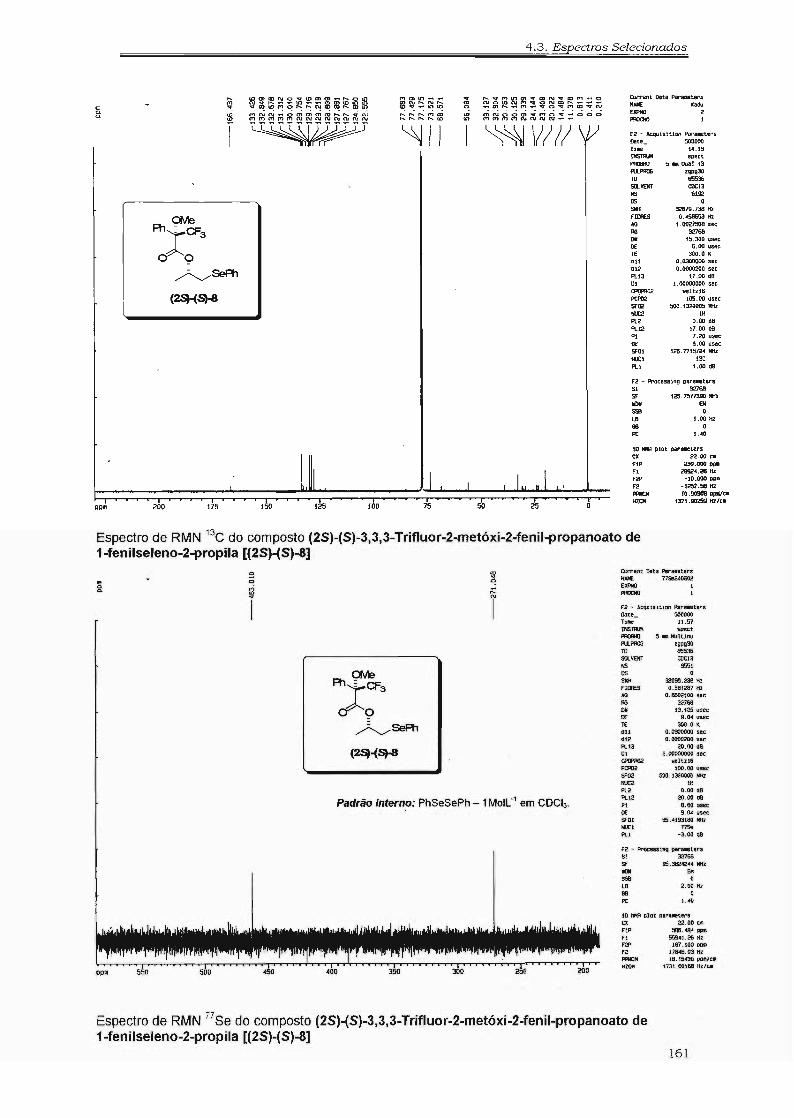
4.3. Espectros Selecionados
Ctrrent Dita fW'..tIf'S-- .....EllI'IIl 2FIlllCNO •
....~!~~~~~~2iieiiiB MlClen:;:: ;J. ~i~~~~~~ii;;~;~g ~ ~~~~~ ~
'" Pinini";óaiaioiai"':"':":N ,... ,.... ,... .., cu '" m~~~re~rJ~~::ÓÓÓ'"~~
'" ,.... ,..., ,.... U) '"I ~ I I I ~~I VI/!I
R1~;r:3~SeRl
(2S)-(S)-8
,,",0Ti_IICS1RJIPRlIHlPW'AIlGlUSlL'IEIITlOSOSSlttFIlHSAOR6DO~
TEou0121'1..3OIlPlPRG2PCi>o29'02lU21'1.21'1..2
P'~
9'0'lllC'PU
sooooo14.19_ct
5 _llIal 13z_65!536aJC)]
8.92O
32679.738 Hz0.-498&53 Hz
1.00Z1508 sec32768
15.300 useI;6.00 usec
300.0 K0.030000O srt:Q.OOOO2OCl seç
t7.00dQ0oooooס1.0 soe
",ltU6105.00 o,e:e
500 _1320005 Mil:
'H0.00 dB
H.CO d87.20 'USK6.00 USIC
125. nt5724 ttQIX .
1.00 d8
F2 - Pr>octwln!l per..tel"tiiSI l2768Sf 125.7!17139O .-ulOI ao9SIl O
18 1.00 '"GIl OPC 1.-40
10 ... plot ~ters
ppll 200 175 ISO 125 100 75 SO 25' o
exF.PFIF2PF2-..H20I
22.00c:a2JO~OOO ..
i?8!I24.28 ftl-10...oao~
-S2!)1t~ KzIO.~,,,,,co
1371.9D2S!!J Hz/CII
Espectro de RMN 13C do composto (25)-(5)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-fenilseleno-2-propila [(25)-(5)-8]
~M
i r
(2St-(S)-8
Padrão intern,o: PhSeSePh - 1MoIL'1 em CDCh,
CL-rre1lt Oata P...... tIb"SlrWE 77Se2~
E1<PICl ,PRlOIO ,
F2 - Ac.nuisit..lOtl Par_UrsDate_ 500000
lute 11.'57IMSTAAC suectPRJHJ S _ ~ltJ"lJ
FU.PROG zgplI30TO 65536sa..veNT coeI3lOS 9&.l'OS OSlIH 38C95. 23e HzfImES 0.581287 HzAO 0.8602100 :fICFI(; ~ê768
Oi' 13.i25 ~eC'
a: 9.041 usecTE 300.0 K011 0.0300000 SKd12 O.ססoo200 secPl13 20.00 d8Dl 0oooooס2.0 secc::PlFR6C welU16PCPD2 100.00 ~ec
Sf02 500. t 3?0005 NHzMJ:2 lHPl2 0.00 dePLt2 20.00 dOFI 8.60 usectE 9.DA useeSFQI 55 ... t93t80 teil
lU' ns.PU -3.00 @
FZ - Proces!1ng par..elers51 327689' !fi. 3e242"" jIf{z
.... EMSSll OlB 2.00 H1GIl OPC 1.-40
1D JH:l plot par,.,tersex 22.00 uFtP 586.-49-4 apaFI 55941.26 HzF2P 187.100 CDIlFZ 17346.03 HZPPMCN 18. '5.00 poa/c.H2CM 173.1.60168 l-h/c.
Espectro de RMN f7Se do composto (2S)-(S)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-fenilseleno-2-propila [(25)-(5)-8]
161
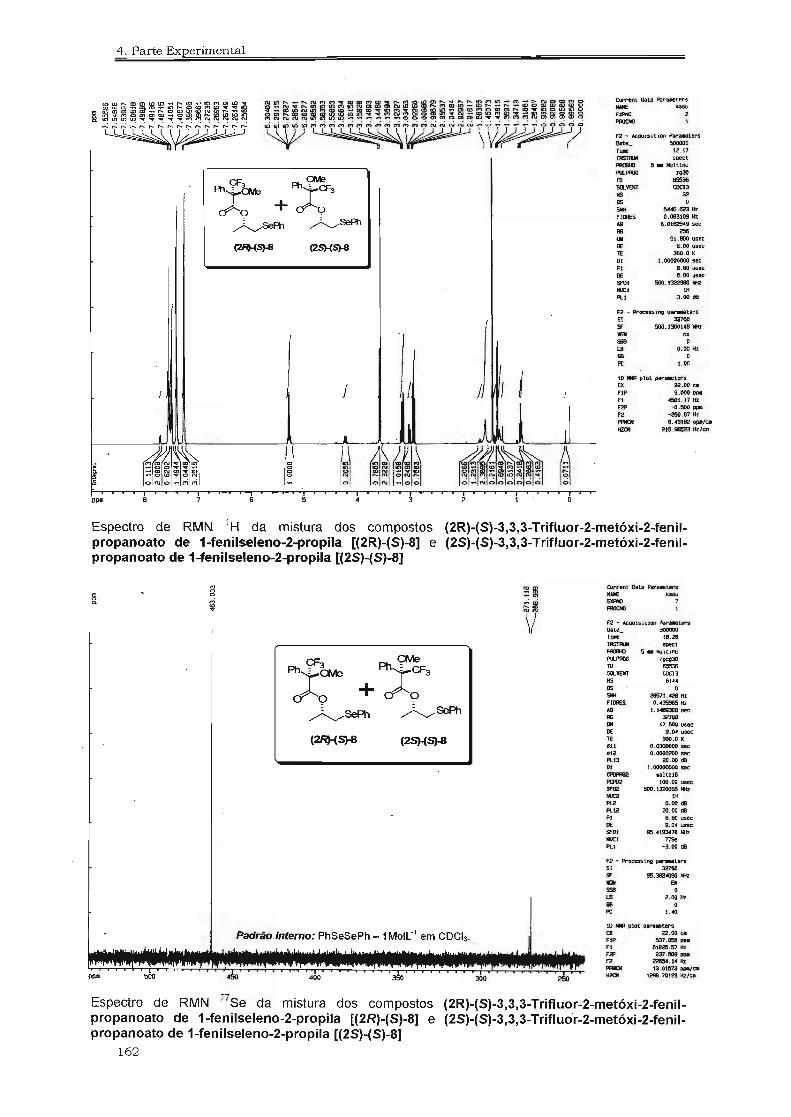
4. Parte Experimental
10 ... p}ot prawtet':;ex 22.00 c.FtP 9.000 ppefi 4501.S1HIF2P -0.500 ~F2 ·250.07 HzFfIN:H o.43182 ~cetilCM 215.96523 tn/CIl
CurTenl Dala PilrueterslIME ......-, ,"""""' 1
F2 - Proc.easl!1g p.,._tINlSI 3V5Ilg: SOO.t300t.e Mil..,. noSSIl oLB 0.00 Hz... oPC 1.00
F2 - Acauisltlon P;rMllttll"'SDlte_ 5000CXlTi_ 12.17J1CSllU( soec: tPRmtt:I 5 _ ..,i ti nuPU.PAOG lQ30ro 6!i'S36so..YENT alC13MS ...os o~ 54«i.623 HzFImES 0.083109 Hz:AO 6.0t62Sf9 sec
"" 25ti.. 91.800 lJfil!CIE 6.00 useeTE 300.0 KDI 1.OOOOOOOO secFI 8.60 UIlI!CCE 6.00 USf(SFQI 500. t322980 ~MlCI 111RI -3.00 dB
2567B
Ph ÇF3 o.1e
r ;;r~ + )::3~8ePh ~8ePh
~S)-8 (2SHS)-8
"I IIr ! ili f
,
I ! 1
j I . ,I 1 ~ ,Ili (~~)) r~1 r~1 ~~~~I~ ~'I~~ ~c
I
Espectro de RMN 1H da mistura dos compostos (2R)-(S)-3,3,3-Trifluor-2-metóxi-2-fenilpropanoato de 1-fenilseleno-2-propila [(2R)-(S)-8] e (2S)-(S)-3,3,3-Trifluor-2-metóxi-2-fenilpropanoato de 1-fenilseleno-2-propila [(2S)-(S)-8]
(2S)-(S)-8
\iCUrf'ent Data ParaaetersN_ Kaou..- ,F!lOCl<O I
F2 - ~is}tion Par.etft"SDlt~_ 50000Ch. 18.26IHSTFUt soectPFIJH) 5 • tlJll111\JFUPROG zgpg30ro 6!i536stLlEJIfT a:J:13MS 6tA.lIos oSJIi Z8S71.Q8 HZFüJES 0.0415965 H2AO J. 1469300 5!C
"" 3V5IlOI 17.500 usecDE 9.0.. userTE 300.0 KoU 0.0300000 s«d1.2 6.000020O SKfll13 20.00 dBDj t . OOOOOOOC serlJUlAIli2 waltUePCPD2 J00.00 ust!cSF02 500. t 320005 !fizM.C2 111PL2 0.00 daPL12 20.00 teFI B.60 USf:C:DE 9.0.c USft:srOf 95.4t93A7B )fiz
OU:I mePL1 -3.00 dB
F2 - ~lng pre-elltf'SSJ 327tiIlSF 95.3824080 ltf2101 EJlSSIl OLB 2.00 Hz... OPC 1.40
ppo
Padrão Interno: PhSeSePh - 1MoIL-1 em COCho
350 300 250
10 _ plot o""'_tersex 22.DO c.Ftp 531.055 PPfIfi 51225.57 taF2P 237.!509 ~F2 2285C.).oi KZPfIIEN 13.61573 ~OIHZDIl t2!:B.70129 ttz/CII
Espectro de RMN i7Se da mistura dos compostos (2R)-(S)-3,3,3-Tritluor-2-metóxi-2-fenilpropanoato de 1-fenilseleno-2-propila [(2R)-(S)-8] e (2S)-(S)-3,3,3-Trifluor-2-metóxi-2-fenilpropanoato de 1-fenilseleno-2-propila [(2S)-(S)-8]
162
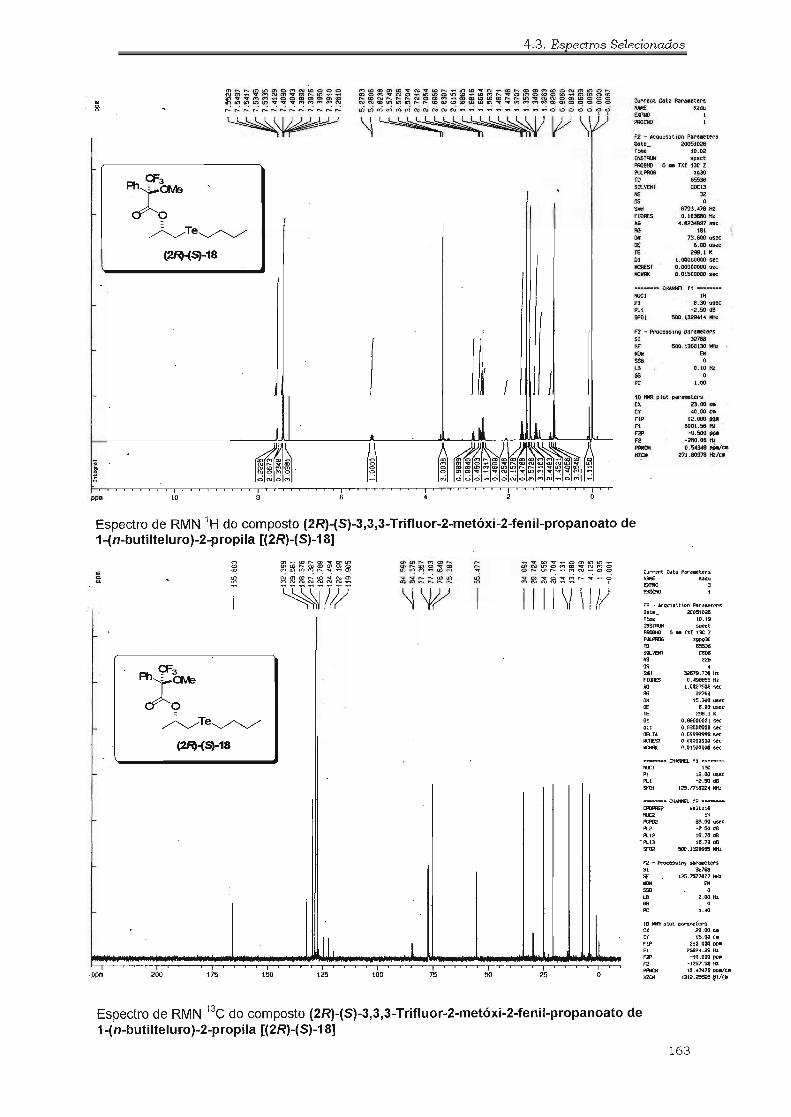
4.3. Espectros Selecionados
"~~
m""''''''Ull!lOlO(Y}(\;IWOOO('\,lQ) ..... 'q"MNcn~m,....It'l .... _
~~~~~~~~~~~~~,....,.... .... ,....,....,...,,....,....,....,... ...... ,...,....
(2R)-(~18
~~~~reó~~ffi~~~~~~~~~~~~~~~~~g~~~~~~~~~mm~mm~~~~~~~~~gm~ggg~~~~~~NNNNN~~~~~~~~~~OOOOO~?
~ LLLb~H),~~ V \Curre.nt Data ParaaetersNAIifE KaduEXPfll) ,
l'!IOCNl I
F'2 - ~Quisition Para.t.er,OlIIte 20051026
'filie tO.02OOHUt Spl!ctPROBKl 5. TU 13C ZFUPAOG 2930m 65536sa....VE'NT coeI3HS 32OS aS)tf 6193.478 HzFlORES 0.103660 HzAQ ".8234991 secFIG 181DN 73. 600 usectE' 6.00 usecTE 298.1 K01 1.00000000 secNCRESr 0.00000000 SE:C
telllR<. 0.01500000 sec
I II
_ ..__••• 0iAI'IfEL '1 •..,........lU' iHPl 8.30 useeP\.1 -2.50 d8SFOl 500.1329414 Itil
F2 - Pracesslflg pariJaetersSI 32768SF 500.1300130 ~.... EMssa olJ!, 0.10 HzG8 aPC 1.00
\0 fMl: plot paraw.ters
pp, 10 O
exCfFlPFIF2PF2......H2CM
23.00 c.AO.OO c.
12.000 PQII6001.56 Hl .
-Q.5OO pp8
-250.06 tU0.543048 .ppa/Cla
211.80978 Hz/ClfI
Espectro de RMN 1H do composto (2R)-{S)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-(n-butilteluro)-2-propila [(2R)-(S)-18]
,
RI ÇF3
):~~Te~
(2R)-(~-18
11. I .1, , , I , ,
23.00 c•IS.OO c.
230.000 PPlI28924.29 H2.-to.ooa ppa
-l2S7.58 HI:lQ.4).478 o04llc.
1312.25525 ttt/~
F2 - AeQUisltlon Para_te~
Date 2005\026fllll! 10.19IttSTRJil s~tMlIHI S _ TU 13C l
N..PfIIG z9P93010 .....so..VBn C606
"" 228as ,SWt 126'XJ.738 H1flmES 0.49tli53 tU.lO 1.0021'508ucAS 327680If 15.300 usec0If 6.00 usecn: 298.1 IC.
DI 0.80000001 5~
cUS 0.03000000 se(
OELTA O.69999999 secIDEsr 0.00000000 SKlDlfI( 0.CJI500000 se(
excrFIOFIF>P'2,-..H204
F'2 - f'rocn.slr'l9 Dal"_t..ssr 32768SF t25.7517en ltt1.... EMSSII OLS 2.00 HZ.. OPC 1.40
..--QtAftEl.fS··_....•fU:J 13CPl 12.00 UUCPU -2.50 d8SfQI t2!f.nl&22A NiZ
Ctlt'roent Data P4t'a.tl!M..... ,...."""'" ,- ,
••.......·Oi.tJt'El.f2 ..--..r:JlDfIRi2 W61n16
><JC2 'HPCP02 83.00 usec:PL2 -2.50 d8PLI2 16.10 «11
# PLI3 IS.70 d69112 500 .1320005 ttI2
O25
..... q-W"l;" ..... omU1U1_mNIt'JOP')Q)VNP10o,...m,..... MC\J OO- .
111\ \/ \ li!
5075
mm ....... {"'ICJ),.....m ...... anO'O;fCDIl"l p') C""J .... <D C'l
"Q' q' ,... ,.... CD lí'IQ) (I) r-- ,.... ,.... r-
100
cn_m,...cn"OJ"CTI/l'J
~:fi!~~~~~~NC)c;gr-..W""""'(f)
~JP
125150175200ppm
Espectro de RMN 13C do composto (2R)-(S)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-(n-butilteluro)-2-propila [(2R)-(S)-18]
163
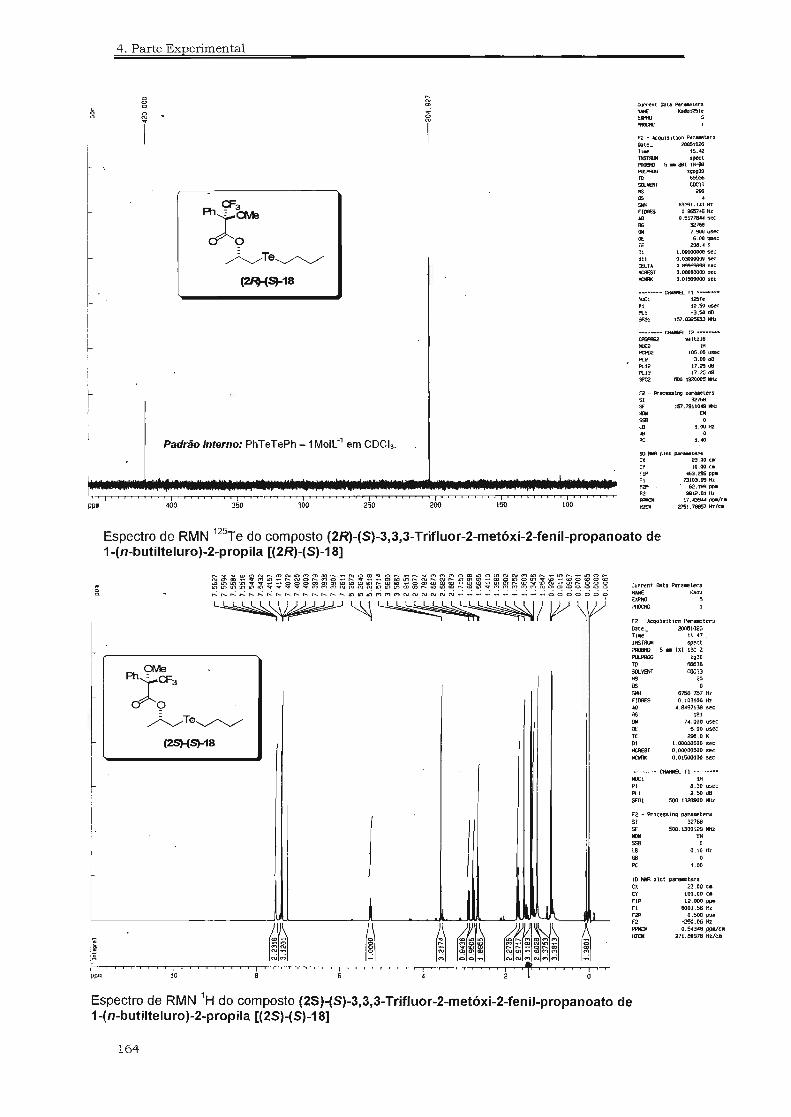
eo.o.
4. Parte Experimental
oooo
r
(~~18
Padrão interno: PhTeTePh - 1MoIL-1 em CDCI3 _
i~t Data flV"lCf'S/IWE k.dJ125TeEJPI«J ,.....,., .Fê - AcQU1$~Uon PM'..l~Glll!_ 200~H026
Tl. 15. .&2-IJCSlIUf apectf'fIlIIH) 5. BlliI ttHIB~ zgpglOm .....SIl.'IelT CDC13
'" ...as ,SMH Ii329L 141 HtF~ 0.9&5146 HzfoIJ Q.5177844 set11> 32768(11 7.900 UU(
CE 6.00 usecTE 298.4 fCDS 1.OOOOOOOO serlUtO.030000OO S«
lELTA Q.~HC
lCt2ST O.CIOOOOOoo $eCMOA( 0.01500000 CK
•__aQilll'fllEl..fl __
IU:t 125ft!Pt 12.50 U$«PLt -3.50 d8!IIFOI 157.8325&33 lfiZ
l]I[fIR;2 Wlllt6
lU:2 '"PCP02 105 _00 USKR,2 -3.GO CJ8PL12 17.2'5 dBPt.13 17.25 CJ8SF02 500. 1·32ll005 lI'It
F"2-Pr"ocns1.....tl!t'SSI 32768SF 157.7911049 »tl..... El<
"" ,LB 3.00 H25il ,
PC 1.40
pp. 400 350 300 250 200 150 100
to~ plot pr_tet'Sex 23,00 c..Cf 10.00 caftp <463.296 ps*
F'J 73.10].95 H:zF2P 62.t89 poli
f2 9812.81 HJ:fIA0 17. 0!:U4 ppa/caH2Dl ml.7f!JI!I51 Hz/ca
Espectro de RMN 125Te do composto (2R)-(S)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-(n-butilteluro)-2-propila [(2R)-(S)-18]
~~~~~N~~NQ~m~~_No~~O~_~~~M~om~O~NN~~~_~~_~O~Nmm_VM~_~NO~~o_~v_~mW~~N~N~~~~_mo~o~~~_wo~oww~~~~~__ ooommmw~w~~w~_ommmw_m~_mm~WV~N_m~oco~~~~~~vvvvvm~~NNNN~~~~m~www~w~vmmmmmNmmmooco
~~~~~~~~~~~~~~~~~~M~~NNNNNN~~~~~~~~~~ooooóo9
LLLL\~Wdd.J~~~ LLL~H--;Y+\~/~ \Current Data Paralleters"WE KacuEXPf«l 5PROCKJ I
F2 - AcQui,.it ion PlIrurtrrs
(25)-(5)-18
Date_fi..IltST1UlPRlIlIIlPI.U'ROGmSQ.YeiT
'"OS
""rtlRS.......11'TED'ICREST..:-
2005102611.47.....,1
5 _ TXt IX Z,,30
65536COC13
20O
6756.757 H20.103100 H:z
4.8497138 sec18•
704.000 usee6.00 usee
298.0 te1.00000000 ueO•00(100000 see0.01500000 ~c
II j U 11
I ~r~ !~I ~\ ~~I~ (~~~ ~-pp. 10 e 6 4 2 r o
••••_. 0Uit'EL f t _ ........
lU' IHPI 8.30 usecPU -2.50 dOsrOI 500.1328900 Mil
F.2 - Processing PI1'HetrrsSI 32768Sf 500 .1300129 Mil_ ElO
SS8 OLa 0.10 Hl.. ,PC 1.00
10 ~ p lat paraRter,ex 23.00 c.Cf 100.00 c.FtP 12.000 ,..FI 6001.56 HzF2P -o .500 OP.F2 ·250.06 HzPPtDI 0.54348 ~e.Hlt:N 271.80978 Hzlc.
Espectro de RMN 1H do composto (2S)-(S)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-(n-butilteluro)-2-propila [(2S)-( 5)-18]
164
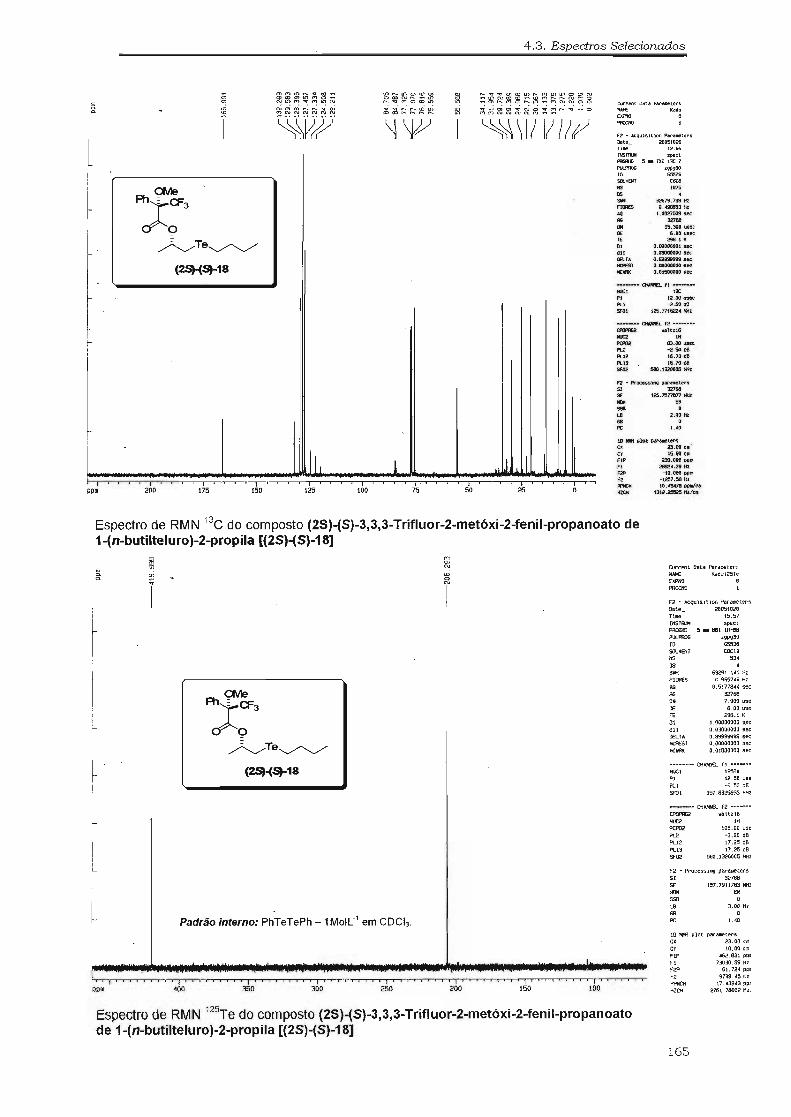
4.3. Espectros Selecionados
'"ln(O')lnr-.'Q'W_
~~~~~~ fg ""<l' ...... allXlll)r--MmlC1a>WC\l(7)(D(J)LOMO- ~1Ot\l(IJCC_<Ot"'J"r--.C'\l[""JO
E '" "!""!~~~"':~ -:O!"":"'!~'"':""!~!'"!~~~~
~~ (\I cn a> ,.... ,.... '" C\I ................ ,.... ID 10 ~ ... _mcn""'C\lO...,.,.,,.... ........ O
i ~VPCDCDr-.",... ...... ~ {O')(rJC\lC\lC\lC\lf\J ........
~ \Y~ ~\I/ I/ /1//,
aII1e -
:X:3
~Te~
(2S)-(S)-18
~!
I I" J lI, lippo 200 175 150 125 100 75 50 25 o
Cl.rretlt Data P.tt'uetersHNE luulu
""'" ,.....,., I
F2 - AtQU15U.lol'l ~rMllerso.te_ 2005IG26H~ 12.06[NS1lUl spectPIQH) 5 _ U[ 13C l
Pl.I.JIR:lG z9J)930TIl .....so..vElU C606MS J075os •SIfI ~79.na H2nftS O.<C986SlHl.AO (.0027508 secRei 32768011 15.300 usecDE: 6.00 usecTE 298.1 j(
OI O.aoooooo1 seeali 0.030000OO secIEUA 0.69999999 UI:MUEST 0.00tI00000 _tc)IJR{ 0.01500000 sec
........-.. CHAIftL ti ......_ •••Ut t3CPI 12.00 usecflLl -2.50 deSRli 125.77t6224 tcHz
~_ O4.UIEL f2 a __"_
CflIFfIi2 IIIltzl6
fU2 '"!'CPOé! 83.00 usecPI..2 -2.50 e1BflLl2 16.70 dBflL13 16.7(Jd8SflJi2 5OO.ll2OOO5 Niz
f"2 • PrOCKslflll par_ters51 32768Sf 125.75778:17 !ltiZ
- ElO... .La 2.00 KlG8 oPC 1.40
1!IMlfiDl'otlWf'Mllet'Sex 23.00 c.'CY 15.00 (11,
flP 230.000 PP.fi 2&924.29 HI.f2P ~IO.OOO pp.1'2 -\257.58 tuPfIOt IO.43C8~c.H20C 1312.25525 HI/t.
Espectro de RMN 13C do composto {2S)-(5)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoato de1-{n-butilteluro)-2-propila ({25)-(5)-18]
Il~i
(2S)-(Sl-18
Padrão interno: PhTeTePh -1MoIL-1 em CDCh.
Mm
'"
iCUl"l"Mlt OlJt3 Paraoetl!l"s/WlIE K<Wu1251e
ExPNO •PIlOCl«l I
F2 ~ AeQlJ1SJl1Q(l Parllltetfl'S
Date_ 2C051026n_ 15.51IttSfAJM speclPRC&I) 5 .. sal tH-8BPll.PRJG 201)9:30TO 65536SOLVENT alC13l'fS 534
os •SMH 63291.1041 HzFICIES 0.96571116 Hl.MI 0.517780114 secRei 32768OH 7.900 1llIl!DE 6.00 lIserE 290.1K~i 1.00000000 see(lU 0.03000000 seeDELTA 0.89999998 Sl"<:JC:AEST 0.00000000 secMCNAl< 0.01500000 sec
__...... 01MfEL fi ......_:11.fU:l 125ft!Pl 12.50 uS-t
PU -3.50 e18SFOl 151.8325633 HH2
--_ cttAlf'El 12 .CPOPRi2 IfUtztij
NUC2 '"PO'D2 105.00 U$ePL2 -3.00 (\8PL12 t7.25dBJ:Il.l3 17.25 (19SfD2 ~.1320005 Iolt1l
.2 - Processtl'lg paraJletet"sSI 127W5F 151.7911]83 Hffi.... EMSS8 oLa 3.00 til:GIl oPC 1.40
ppmI i i
400i I i
350I I I
300I I i
250• I I
200i I i
150• I i
100
10 fNl plot Di!II"aJll!tel"$ex 23.00 c.CY 10.00 til
F1F 462.831 pplfi 73030.59 HtF2P 61.724 pplF2 9739 • .c5 HzPPM04 17.43943 pplHlCH 2751. 78882 HL
Espectro de RMN 125Te do composto (2S)-(5)-3,3,3-Trifluor-2-metóxi-2-fenil-propanoatode 1-(n-butilteluro)-2-propila [(25)-(5)-18]
165


S. Bibliograf'ia


(1) Coelho, F. A S. Cadernos Temáticos de Química Nova na Escola 2001, 3,23.
(2) Tumlinson, J. H.; Klein, M. G.; Doolitle, R. E.; Ladd, T L.; Proveaux, A TScience 1977, 197, 789.
(3) Mori, K. Chirality 1998, 10, 578.
(4) a) Nilson, D.L.; Cox, M.M. Lehninger Principies of Biochemistry, 3th ed.;W.H. Freeman: New York, 2000. b)http://quark.qmc.ufsc.br/qmcweb/artigos/drogas_quirais.htm/.
(5) http://www.spq.ptlboletim/docs/boletimSPQ_096_072_11.pdf.(6) Stinson, S. C. Chem. & Eng. 2001, 40, 79.
(7) Borman, S. C&EN 2001, 79, 5.
(8) http://academic.scranton.edu/faculty/CANNM1/introport.html.
(9) Faber, K. Biotransformations in Organic Chemistry, 4th ed.; Springer-Verlag: Berlin, 2000.
(10) Ghanem, A; Hassan, Y. A Tetrahedran:Asymmetry 2004, 15, 3331.
(11) Liese, A; Filho, M. V. Chemical Biotechno/1999, 10,595.(12) Roberts, S. M. J. Chem. Soe. Perkin Trans 2000, 1,611.
(13) Straathof, A J. J.; Schmid, A; Panke, S. Curr Opin Biotechno/2002, 13,548.
(14) Fulkul, T; Soda, K. Molecular Aspects of Enzyme Catalysis; Kadansha:Toquio, 1994.
(15) Fischer, E. Ber. Dtsch. Chem. Ges. 1894, 27,2985.
(16) Kosland, D. E.; Neet, K. E. Ann. Rev. Biochem. 1968,37,359.(17) Dewar, M. S. J. Enzyme 1986,36,8.(18) Warshel, A; Aqvist, J.; Creighton, S. Prac. Nat!. Acad. Sei. 1989, 86,
5820.(19) Ongston, A G. Nature 1948, 162,963.(20) Hirs, C. H. W.; Moore, S.; Stein, W. H. J. Biol. Chem. 1960, 235, 633.
(21) Carrea, G.; Riva, S. Angew. Chem. Int. Ed. 2000, 39, 2226.
(22) Holland, H. L. Nat. Prado Rep. 2001, 18, 171.
(23) Roberts, S. M. (ed.); Biocatalysts for Fine Chemicals Synthesis John Wiley& Sons: Chichester, 1999.
(24) Ching-Shih, C.; Cahrles, J. S. Angew. Chem. Int. Engl. 1989, 28,695.
(25) Koskinem, A M.; Klibanov, A M. Enzymatic Reactions in Organic Media,1st ed.; Chapman & Hall: New York, 1995.
(26) Wong, C.; Whitesides, G. M. Enzymes in synhetic organic chemistry;Pergamon Press: London, 1995.
(27) Cambou, B.; Klibanov, A M. Biotechnol. and Bioeng. 1987, 26, 1449.
(28) Djeghaba, Z.; Deleuze, H.; Dejeso, B.; Messadi, D.; Maillard, B.Terahedran 1991, 32,761.
(29) Margolin, A L.; Klibanov, A M. J. Am. Chem. Soe. 1987, 109, 3802.
(30) Uno, Y.; Tanaka, A.; Yamashita, K. Agric. Biolog. Chem. 1972, 36, 2505.
(31) Kazlauskas, R. J.; Weissfloch, A N. E.; Rappaport, A T; Cuccia, A J.Org. Chem. 1991, 56, 2656.
169

5. Bibliografia
(32) Hanefeld, U. Org. Biomo/. Chem. 2003, 1,2405.
(33) Cygler, M.; Grochulski, P.; Kazlauskas, R J.; Schrag, J. O.; 8uothillier, F.;Rubin, 8.; Serreqi, A N.; Gupta, A K J. Am.Chem. Soe. 1994, 116,3180.
(34) Ema, T; Kobayashi, J.; Maeno, S.; Sakai, T; Utaka, M.; Buli. Chem. Soe.Jpn. 1998, 71, 443.
(35) Rotticci, O.; Ha3ffner, F.; Orrenius, C.; Norin, T; Hult, K; J. Mo/. CataI.B:Enzym. 1998, 5, 267.
(36) Mojovic, L.; Knezevic, Z.; Popadic, R; Jovanovic, S. App/. Mie. Biotech1998, 50, 676.
(37) Comasseto, J. V. J. Organomet. Chem. 1983, 253, 131.
(38) 8ack, T (ed); Organose/enium Chemistry Oxford University Press: Oxford,1999.
(39) Comasseto, J. V. Rev. Heteroatom. Chem. 1993, 9,61.
(40) Comasseto, J. V.; 8arrientos-Astigarraga, R E. A/drichimica Acta 2QOO,33,66.
(41) Marino, J. P.; McClure, M. S.; Holub, O. P.; Comasseto, J. V.; Tucci, F.C. J. Am. Chem. Soe. 2002, 8, 1664.
(42) Ellensohn, R M.; Tese de doutorqdo, Universidqde de São Paulo: SãoPaulo, 20Q9. .,
(43) a) Coma$~eto, JV. J. Org~nomet. Ch~m'1~83, 253, ~31; b) Comasseto,J. V.; Lo, W. L.; Petragnam, N.; Stefanl, ,H. A. Synthes/s 1997,373.
(44) Petragnani, N.; Stefani, H. A Tetrahedron 2q05, 61,1613-1679.
(45) Liotta, O. Organose/enium Chemistry; John Wiley: New York, 1987.
(46) (a) Sebach, O.; Peleties, N. Angew. Chem. Int. Ed. Eng/. 1969, 8, 450-451;(b) Oumont, W.; 8ayet,P; Krief, A Angew. Chem. Int. Ed. Engl. 1974, 13,804.
(47) Reich, H.J.; Willis, W. W. Jr.; Clark, M. C. J. Org. Chem. 1981, 46, 2775.
(48) Clarembeau, M.; Cravador, A; Oumont, W.; Hevesi, L.; Krief, A; Lucchetti,J.; Vanende, O. Tetrahedron 1985,41,4793.
(49) (a) Weber, H.; Khorana, H. G.; J. Mal. Biol. 1972, 72,219; (b) Zhdanov, R1.; Zhenodarova, S. M. Synthesis 1975, 222.
(50) Queiroz, N.; Nascimento, M. G. Tetrahedron Letters 2002, 43,5225-5227.
(51) Móris-Varas, F.; Shah, A; Aikens, J.; Nadkarni, N.P.; Rozzell, J.O.;Oemirjian, O.C. Bioorg. Med. Chem. 1999, 7,2183.
(52) Andrade, L. H.; Porto, A L. M.; Comasseto, J. V.; Omori, AT; Assis, L.F.J. Mal. CataI. B: Enzym. 2004, 29, 55.
(53) Chasteen, T G., 8entley, R Chf;m. Rev. ~003, 1,103.
(54) Clive, O.L.; Chittattu, G.J.; Farin~, V.; Kiel, W.A.; Menchen, S.M.; Russel,C.G.; Singh, A; Wong, C. K.; C~rtis, N. J. J. Am. Chem. Soe. 1980, 102,4438. \
(55) Oale, J.A; Mosher, H.S. J. Am. Chem. Soe. 1973,95:2,512.
(56) Keinan, E.; Hafeli, E. K.; Se'h, KK; Lamed, R J. Am. Chem. Soe. 1986,108, 162.
(57) Nakamura, K; Matsuda, T J. Org. Chem. 1998, 63,8957.
170

(58) Toshimitso, A; Nakano, K.; Mukai, T; Tamao, K. J. Am. Chem. Soe. 1996,118,2756.
(59) Ohkuma, T; Koizumi, M.; Doucet, H.; Pham, T; Kozawa, M.; Murata, K.;Katayama, E; Yokozawa, T.; Ikariya, T; Noyori, R J. Am. Chem. Soe.1998, 120, 13529.
(60) Comasseto, J. V.; Ling, L. W.; Petragnani, N.; Stefani, H. A Synthesis1997, 4,373.
(61) Zeni, G.; Formiga, H. B.; Comasseto, J. V. Tetrahedron Lett. 2000, 41,1311.
(62) Seebach, D.; Beck, A K. Chem. Ber. 1975, 108,314.
(63) Petragnani, N.; Schill, Chem. Ber., 1970, 103,2271.
(64) Zinn, F. K.; Righi, V. E; Luque, S. C.; Formiga, H. B.; Comasseto, J. V.Tetrahedron Lett. 2002, 43, 1625.
(65) Princival, J. L.; Barros, S. M. G., Comasseto, J. V.; Dos Santos, A A .Tetrahedron Lett. 2005,46, 4423.
(66) (a) Barrietos-Astigarra, R E; Castelani, P.; Comasseto, J. V.; Formiga, H.B.; Silva, N. C.; Sumida, C. Y.; Vieira, M. L. J. Organomet. Chem.2001,43,623; (b) Raminelli, C.; Silva, N. C.; Dos Santos, A A; Porto, A L.M.;Andrade, L. H.; Comasseto. J. V. Tetrahedron 2005, 61,409.
(67) Vieira, M. L.; Zinn, F. K.; Comasseto, J. V. J. Braz. Chem. Soe. 2001, 12,586.
(68) (a) Wheeler, J. W.; Chungo, RH. J. Org. Chem. 1969, 34, 1149; (b)Johnson, M. R; Rickborn, B. J. Org. Chem. 1970, 35,1041.
(69) Dos Santos, AA; Castelani, P.; Bassora, B.K.; Junior, J.C.F.; Costa, C.E;Comasseto, J.V. Tetrahedron, 2005, 61,9173.
(70) Costa, C.E; Clososki, G.C.; Barchesi, H. B.; Zanotto, S. P.; Nascimento,M.G.; Comasseto, J. V. Tetrahedron:Asymmetry 2004, 15,3945.
(71) Clososki, G.C.; Costa, C.E; Missio, L.J.; Cass, a.B.; Comasseto, J.V.Synthetie Comm. 2004, 34, 817.
(72) Tsuboi, S.; Sakamoto, J.; Kawano, T Utaka, Takeda, A J. Org. Chem.1991,56,7177.
(73) Perrin, D. D.; Amarego, W.L.F. Purifieation of Laboratory Chemiea/s;Pergamon Press: London, 1980.
(74) Sharpless, K. B.; Lauer, R F. J. Am. Chem. Soe. 1973, 95,2697.
(75) Haller, W. S.; Irgolic, K. J. J. Organomet. Chem. 1972, 38,97.
(76) Tatchell, A R. Et a/In Vogers Textbook of Praetieal Organie Chemistry, 5th
ed., Wiley: New York, 1989,432.
(77) Rua Latino Coelho, 1301, Parque Taquaral, CEP 13087-010 - Campinas SP - Brasil.
(78) Sir, C. J.; Wu, S. H. Topies Stereoehem 1989, 19, 63.
(79) Abad, J. L.; Fabriás, G.; Camps, F.; J. Org. Chem. 2000,65,8582.
171


















