
HER2/HER3 Signaling Regulates NK Cell-Mediated ... during cell transformation (4). Also, there is...
11
of May 25, 2018. This information is current as in Human Breast Cancer Cell Lines Chain-Related Molecule A and B Expression Cell-Mediated Cytotoxicity via MHC Class I HER2/HER3 Signaling Regulates NK Andreas Lundqvist, Kousaku Mimura and Rolf Kiessling Mao, Dhifaf Sarhan, Erik Wennerberg, Barbara Seliger, Riki Okita, Dimitrios Mougiakakos, Takashi Ando, Yumeng http://www.jimmunol.org/content/188/5/2136 doi: 10.4049/jimmunol.1102237 February 2012; 2012; 188:2136-2145; Prepublished online 1 J Immunol Material Supplementary 7.DC1 http://www.jimmunol.org/content/suppl/2012/02/01/jimmunol.110223 References http://www.jimmunol.org/content/188/5/2136.full#ref-list-1 , 24 of which you can access for free at: cites 51 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2012 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on May 25, 2018 http://www.jimmunol.org/ Downloaded from by guest on May 25, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of HER2/HER3 Signaling Regulates NK Cell-Mediated ... during cell transformation (4). Also, there is...

of May 25, 2018.This information is current as
in Human Breast Cancer Cell LinesChain-Related Molecule A and B ExpressionCell-Mediated Cytotoxicity via MHC Class I HER2/HER3 Signaling Regulates NK
Andreas Lundqvist, Kousaku Mimura and Rolf KiesslingMao, Dhifaf Sarhan, Erik Wennerberg, Barbara Seliger, Riki Okita, Dimitrios Mougiakakos, Takashi Ando, Yumeng
http://www.jimmunol.org/content/188/5/2136doi: 10.4049/jimmunol.1102237February 2012;
2012; 188:2136-2145; Prepublished online 1J Immunol
MaterialSupplementary
7.DC1http://www.jimmunol.org/content/suppl/2012/02/01/jimmunol.110223
Referenceshttp://www.jimmunol.org/content/188/5/2136.full#ref-list-1
, 24 of which you can access for free at: cites 51 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2012 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
HER2/HER3 Signaling Regulates NK Cell-MediatedCytotoxicity via MHC Class I Chain-Related Molecule A andB Expression in Human Breast Cancer Cell Lines
Riki Okita,* Dimitrios Mougiakakos,*,† Takashi Ando,‡ Yumeng Mao,* Dhifaf Sarhan,*
Erik Wennerberg,* Barbara Seliger,x Andreas Lundqvist,* Kousaku Mimura,{ and
Rolf Kiessling*
Overexpression of the receptor tyrosine kinases HER2 and HER3 is associated with a poor prognosis in several types of cancer.
Presently, HER2- as well as HER3-targeted therapies are in clinical practice or evaluated within clinical trials, including treatment
with mAbs mediating growth inhibition and/or activation of Ab-induced innate or adaptive cellular immunity. A better understand-
ing of how HER2/HER3 signaling in tumors influences cellular immune mechanisms is therefore warranted. In this study, we dem-
onstrate that HER2/HER3 signaling regulates the expression of MHC class I-related chain A and B (MICA and MICB) in breast
cancer cell lines. The MICA and MICB (MICA/B) molecules act as key ligands for the activating receptor NK group 2, member D
(NKG2D) and promote NK cell-mediated recognition and cytolysis. Genetic silencing of HER3 but not HER2 downregulated the
expression of MICA/B, and HER3 overexpression significantly enhanced MICA expression. Among the major pathways activated
by HER2/HER3 signaling, the PI3K/AKT pathway was shown to predominantly regulate MICA/B expression. Treatment with the
HER3-specific ligand neuregulin 1b promoted the expression in a process that was antagonized by pharmacological and genetic
interference with HER3 but not by the ataxia-telangiectasia–mutated (ATM) and ATM and Rad3-related protein kinases inhibitor
caffeine. These observations further emphasize that HER2/HER3 signaling directly, and not via genotoxic stress, regulates MICA/
B expression. As anticipated, stimulating HER2/HER3 enhanced the NKG2D-MICA/B–dependent NK cell-mediated cytotoxicity.
Taken together, we conclude that signaling via the HER2/HER3 pathway in breast carcinoma cell lines may lead to enhanced
NKG2D-MICA/B recognition by NK cells and T cells. The Journal of Immunology, 2012, 188: 2136–2145.
Natural killer cell activity is promoted via NK group 2,member D (NKG2D) on NK cells, CD8+ T cells, and gd
T cells (1, 2). The engagement between NKG2D and itsligands enhances cell-mediated cytotoxicity and cytokine pro-duction against transformed cells (3). An important NKG2D li-gand is the MHC class I chain A and B (MICA and MICB). BothMICA and MICB (MICA/B) are expressed by intestinal epithelial
cells and at low levels also by other nonmalignant cell types.Primary tumor cells and tumor cell lines frequently expressMICA/B molecules, but the mechanisms responsible for theinduction of MICA/B proteins and other NKG2D ligands(NKG2DLs) during oncogenesis are poorly understood. They aredescribed as stress-related proteins that can be induced by heatshock and that are regulated by heat shock transcription elementsactivated during cell transformation (4). Also, there is evidencethat the DNA damage pathway, induced by ionizing radiation andinhibitors of DNA replication, can induce NKG2DLs (5). A directcontrol of MICA by the BCR/ABL oncogene in chronic mye-logenous leukemia has also been described (6), but the regulationof MICA/B expression by oncogenes is incompletely understood.A better knowledge of this would seem important, because itis well established that NKG2DLs play an important role in therecognition of tumor cells by NK cells. Additionally, tumor cellsexpressing NKG2DLs can become susceptible to NK cell killingeven if they have normal MHC class I expression (1, 7). Thus, thebalance between NKG2DLs and MHC class I expression ontransformed cells is important for their survival under the immunesurveillance of their host (8).The epidermal growth factor (EGF) family consists of four
closely related transmembrane tyrosine kinase receptors (EGFR,also known as HER1, 2, 3, and 4). On binding small peptide ligandmolecules, HER receptors undergo homo- or heterodimerizationand autophosphorylate, which activates downstream signalingpathways resulting in enhanced cell proliferation, migration, in-vasion, and survival (9–11). The HER2/HER3 dimer is consideredto be the most active HER signaling dimer and is crucial for sig-naling in tumors containing amplification of HER2 (12–14). HER3
*Immune and Gene Therapy Laboratory, Department of Oncology and Pathology,Cancer Center Karolinska, Karolinska Institute, 171 76 Stockholm, Sweden;†Department of Internal Medicine 5, Hematology and Oncology, University of Erlan-gen–Nuremberg, D-91054 Erlangen, Germany; ‡Department of Orthopedic Surgery,University of Yamanashi, Yamanashi 409-3898, Japan; xInstitute of Medical Immu-nology, Martin Luther University Halle–Wittenberg, 06112 Halle, Germany; and{First Department of Surgery, University of Yamanashi, Yamanashi 409-3898, Japan
Received for publication August 2, 2011. Accepted for publication December 30,2011.
This work was supported by grants from the Cancer Society of Stockholm, theKarolinska Institutet, the Stockholm City Council, the Swedish Cancer Society,and the Swedish Research Council.
Address correspondence and reprint requests to Prof. Rolf Kiessling, Immune andGene Therapy Laboratory, Department of Oncology and Pathology, Cancer CenterKarolinska, R8:01, Karolinska Hospital, 171 76 Stockholm, Sweden. E-mail address:[email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: ATM, ataxia-telangiectasia–mutated proteinkinase; ATR, ataxia-telangiectasia–mutated and Rad3-related protein kinase; EGF,epidermal growth factor; MFI, mean fluorescence intensity; DMFI, increase in meanfluorescence intensity; MICA, MHC class I chain A; MICA/B, MHC class I chains Aand B; MICB, MHC class I chain B; NKG2D, NK group 2, member D; NKG2DL,NK group 2, member D ligand; NRG, neuregulin; rMFI, relative mean fluorescenceintensity; siRNA, small interfering RNA.
Copyright� 2012 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1102237
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

has several ligands, including the neuregulins (NRG) 1–4, but itlacks intrinsic tyrosine kinase activity. In contrast, HER2 has nodefined ligand but possesses an active tyrosine kinase domain (15,16). Ligand-induced heterodimerization of HER3 with HER2activates cell growth and cell proliferation via PI3K/AKT andRAS/MAPK signaling pathways (17, 18). HER2 overexpression isone of the poor prognostic factors in breast cancer (19, 20), andHER3 is often expressed together with HER2 in this disease (21–23). Both molecules are considered promising targets for cancertherapy, with HER2 targeting therapies already in clinical practice(24, 25), and HER3 targeting therapies currently in clinical trials(26, 27).Although HER2 and HER3 are considered potential targets for
cancer therapy, they have also been associated with an “immuneescape” phenotype. Thus, several groups reported that overex-pression of the HER2 oncogene can lead to tumor escape from thehost immune system via downregulation of MHC class I (28–30),and overexpression of HER2 severely impaired MHC class I-restricted recognition by CTLs (31, 32). These findings suggestthat activation of the HER2 signaling pathways leads to tumor es-cape from the adaptive immune system. However, because MHCclass I molecules activate the inhibitory signaling in NK cells (33),HER2-induced downregulation of MHC class I may instead makethem suitable targets for NK cell-mediated tumor rejection bya direct NK cell–tumor interaction or by NK cell-mediated Ab-dependent cellular cytotoxicity. Because NK cell-mediated cyto-toxicity is regulated by the balance between activating and inhibit-ing signals (34), it is of particular interest to understand whetherHER2/HER3 signaling also would affect the expression of activat-ing NK cell receptors. In this study, we demonstrate that HER2/HER3 signaling regulates the expression of MICA/B molecules inhuman breast carcinoma cell lines, resulting in enhanced sensitivityto NK cell-mediated recognition.
Materials and MethodsCell culture and reagents
Human breast cancer cell lines MDA-MB-231, MDA-MB-453, and T47Dwere obtained from the American Type Culture Collection. The genotypesof all cell lines were identified by short tandem repeat method using STRIdentifier (Applied Biosystems, Foster City, CA) and authenticated. Allcell lines were maintained in culture in RPMI 1640 medium with 2 mML-glutamine (Invitrogen, Carlsbad, CA) supplemented with 10% FBS(Invitrogen) and 50 U/ml penicillin and streptomycin (Sigma-Aldrich, St.Louis, MO) at 37˚C in a humidified atmosphere with 5% CO2. For cellculture work, LY294002 (Calbiochem, San Diego, CA), PD98059 (Cal-biochem), and lapatinib (GlaxoSmithKline, Brentford, U.K.) were dis-solved in DMSO (Sigma-Aldrich). NRG1b (R&D Systems, Minneapolis,MN), caffeine (Sigma-Aldrich), and doxorubicin hydrochloride (Sigma-Aldrich) were dissolved in PBS (2).
Analysis of cell surface molecules by flow cytometry
Tumor cells were washed in PBS (2), collected with EDTA (Sigma-Aldrich), and analyzed for cell surface expression of HER family pro-teins, MICA/B, MICA, MICB, and HLA class I by direct (for EGFR,HER2, HER3, HLA-A,B,C, MICA/B, MICA, and MICB) or indirect (forHER4) immunofluorescence using mAbs, including PE-labeled EGFR(EGFR.1; BD Biosciences, San Diego, CA), allophycocyanin-labeledHER2 (Neu 24.7; BD Biosciences), PE-labeled HER3 (1B4C3; Bio-Legend, San Diego, CA), FITC-labeled HLA-A,B,C (G46-2.6; BD Bio-sciences), PE/allophycocyanin-labeled MICA/B (6D4; BioLegend), PE-labeled MICA (159227; R&D Systems), allophycocyanin-labeled MICB(236511; R&D Systems), or purified HER4 (H4.77.16; Calbiochem) asfirst Ab, with PE-labeled anti-mouse Igs (RPE; affinity-isolated F(ab9)2;Dako, Humburg, Germany) as secondary Ab, and FITC-, PE- orallophycocyanin-labeled anti-mouse IgG1k (MOPC-21; BD Biosciences)or IgG2bk (27-35; BD Biosciences) as isotype controls. Stained cells wereacquired on a FACSCalibur cytometer (BD Biosciences) and analyzedusing FlowJo software 6.4.7 (Tree Star, Ashland, OR). The increase in
mean fluorescence intensity (DMFI) was calculated as: (MFI with specificmAb 2 MFI with isotype control)/MFI with isotype control. Relative MFI(rMFI) values were calculated to compare the differences between DMFIof a specific treatment and control as: 100 3 (DMFI of specific treatment/DMFI of control treatment).
Western blot analysis
Cells were washed in PBS (2), lysed during 15 min shaking in CelLyticlysis buffer (Sigma-Aldrich) with a protease inhibitor cocktail and a phos-phatase inhibitor cocktail (both from Sigma-Aldrich). Cell debris wascleared at 20,000 3 g for 15 min. The supernatants were collected whilecell lysates and the protein concentrations were analyzed using a BCAprotein assay (Thermo Scientific, Rockford, IL) according to the manu-facturer’s protocol. Equal amounts of protein were separated on 4–12%NuPage Bis-Tris acrylamide gels (Invitrogen) and transferred to polyvi-nylidene difluoride membranes (Immobilon-P; Millipore, Bedford, MA).Blots were blocked for 30 min in PBS (2) with 0.05% Tween 20 (Sigma-Aldrich) buffer (PBST) with 2% nonfat dry milk, followed by incubationovernight at 4˚C in PBSTwith 2% nonfat dry milk and primary Abs againstHER2 (Calbiochem), p-HER2 (Tyr1221/1222) (Cell Signaling Technology,Beverly, MA), HER3 (Upstate Biotechnology, Lake Placid, NY), p-HER3(Tyr1222), AKT, p-AKT (Ser473), p44/42 MAPK, p-p44/42 MAPK (Thr202/Try204) (Cell Signaling Technology), and b-actin (Sigma-Aldrich). Aftertwo washes in PBST, membranes were incubated with HRP-linked goatanti-rabbit or anti-mouse IgG Abs (Cell Signaling Technology) for 1 h atroom temperature. After blots were washed three times in PBST, enhancedchemiluminescence (GE Healthcare, Fairfield, CT) was performed, andimages were captured using an LAS-1000 camera system (Fujifilm, Tokyo,Japan). Membranes were stripped in strip buffer (PBS (2) with 2% SDS[Invitrogen] and 0.7% 2-ME [Sigma-Aldrich]) for 30 min at 50˚C andreprobed up to three times. Densitometric analyses for protein quantifica-tion of each blot were calculated using ImageJ 1.42q (http://rsb.info.nih.gov/ij). The experiments were repeated three times.
Cell proliferation assays
Cells were collected and stimulated with 2–250 ng/ml NRG1b for 15 min,then seeded in triplicate into 96-well flat-bottom plates at 5 3 103 cellsper 100 ml per well in RPMI 1640 with 10% FBS and 2 mM L-glutamineand incubated for 3 d. The relative percentage of metabolically activecells compared with that of untreated cells was determined on the basisof mitochondrial conversion of MTT (Molecular Probes, Eugene, OR)according the manufacturer’s protocol. In brief, after incubation, 10 mlMTT solution (5 mg/ml) was added to each well and incubated for 4 h.Thereafter, 50 ml DMSO was added to 25 ml cell supernatant and incu-bated for 10 min and plates were read at 540 nm using Versa Max (Mo-lecular Devices, Sunnyvale, CA).
Apoptotic assays
Cells were seeded into 24-well plates at 104 cells per well and treated with2–250 ng/ml NRG1b for 2 d. Both nonadherent and adherent cells werewashed in PBS (+), trypsinized, and collected. Cells were pelleted at4503 g, washed by PBS (–), repelleted, and labeled according to the man-ufacturer’s protocol. Briefly, each sample was stained with 50 ml mixtureregent containing annexin V binding buffer with 2.5 ml FITC-labeledannexin V (eBioscience) in a V-bottom 96-well plate at room tempera-ture for 15 min. The cells were resuspended in 300 ml annexin V bindingbuffer and transferred to FACS tubes for analysis. Cells were acquired ona FACSCalibur cytometer and analyzed using FlowJo software 6.4.7. Theexperiments were repeated three times.
DNA transfection
The vector pRK5/HER3 was a gift from Prof. Axel Ullrich (Max PlanckInstitute, Martinsried, Germany). Tumor cells were grown in 12-well platesand transfected upon reaching ∼70–90% confluence with pcDNA3/empty(Invitrogen) or pRK5/HER3. Cells were transfected in Opti-MEM I cellculture medium (Invitrogen) with 4.0 mg plasmid using 10 ml Lipofect-amine 2000 (Invitrogen) according to the manufacturer’s protocol. After24 or 48 h, transfected cells were collected for further experiments.
Small interfering RNA assay
Cell lines were grown in 12-well plates and transfected upon reaching 50%confluency with small interfering RNA (siRNA) targeting HER2 or HER3(SMARTpools; Dharmacon, Lafayette, CO) along with the control siRNA(Dharmacon). Cells were transfected in Opti-MEM I cell culture medium
The Journal of Immunology 2137
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

using Lipofectamine 2000 according the manufacturer’s protocol. After48 h, transfected cells were used for further experiments.
NK cell-mediated cytotoxicity assay
This work was approved by the Institutional Review Board of KarolinskaUniversity Hospital. PBMCs were collected from healthy donor buffy coatsthrough density centrifugation. PBMCs cultured with 100 IU/ml IL-2(Proleukin; Chiron, Emeryville, CA) for 3 d or negatively purified NKcells (NK cell isolation kit II; Miltenyi Biotec, Auburn, CA) rested overnightserved as the source of effector NK cells. The phenotype of the cells (CD32
CD56+) was confirmed by flow cytometric analysis using PE-labeled anti-CD3 (HIT3a) and allophycocyanin-labeled anti-CD56 (HCD56) Abs (bothfrom BioLegend). Treated or untreated target cells were tested for sensitivityto NK cell-mediated lysis in a standard chromium release assay (35). [51Cr]release in the supernatants was measured by a gamma counter (WallacSverige, Upplands Vasby, Sweden). The percentage of specific lysis wascalculated as follows: 1003 [(experimental release2 spontaneous release)/(maximum release 2 spontaneous release)]. To analyze the involvement ofNKG2D in the cytolytic activity of NK cells, effector cells were coincubatedwith 20 mg/ml anti-NKG2D blocking Ab (1D11; BioLegend) or an isotype-matched control Ab (11711; R&D Systems) at 4˚C for 30 min and washedbefore the cytolytic assay. To analyze the involvement of MICA/B, anti-MICA/B (6D4; BioLegend) or isotype-matched control Ab (MOPC-173;BioLegend) was added to the target cells at 4˚C for 30 min before the co-culture with NK cells to a final concentration of 10 mg/ ml.
Statistical analyses
Differences in means were evaluated with a Student t test or Fisher exacttest. All analyses were performed at a significance level of 5% (p , 0.05)using GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
ResultsHER3 influences expression of MICA/B in breast carcinomacell lines
The ability of HER2/HER3 signaling to influence MICA/B ex-pression levels was analyzed in breast cancer cell lines by flowcytometry. MDA-MB-231 (low HER2/HER3 levels; SupplementalFig. 1) and T47D and MDA-MB-453 (high HER2/HER3 levels;Supplemental Fig. 1) cell lines were chosen for these experiments.First, tumor cells were treated for 48 h with lapatinib, an EGFR
and HER2 tyrosine kinase inhibitor (36) that also can inhibit HER2/HER3 signaling (37). Expression of MICA/B was significantlydownregulated by lapatinib in the HER2/HER3 high-expressingT47D and MDA-MB-453 cell lines but was not affected in theHER2/HER3 low-expressing MDA-MB-231 cell line (Fig. 1A).These findings suggest that HER2/HER3 signaling is involved in
FIGURE 1. The expressions of MICA/B, MICA, and MICB are regulated by HER2/HER3 signaling in HER2/HER3 high-expressing breast carcinoma
lines. (A) Breast carcinoma cell lines were treated with 1 mM lapatinib (LAP) or as a control with 0.01% DMSO for 48 h. (i) The effects on the MICA/B,
MICA, and MICB expression were assessed by flow cytometry as shown in the representative histograms. (ii) The rMFI of MICA/B, MICA, and MICB were
calculated based on at least three independent experiments and evaluated with a Student t test. (B) T47D cells were transfected with siRNA targeting HER2
(siHER2), HER3 (siHER3), or control siRNA (siCtr). (i) Forty-eight hours after the transfection, the HER2 and HER3 levels were assessed by Western blot
analyses. (ii) The blots were semiquantified, and the columns represent the mean values of three independent experiments. The silencing effect was evaluated
with a Fisher exact test. (iii) Representative histograms demonstrating the MICA/B, MICA, and MICB expression in cells upon genetic silencing (as in-
dicated), and mean values of (iv) MICA/B and (v) MICB rMFI of three independent experiments were evaluated with a Fisher exact test. Bars indicate SEM.
*p , 0.05, ***p , 0.001. A.U., Arbitrary units; ISO, isotype control.
2138 HER2/HER3 SIGNALING REGULATES THE EXPRESSION OF MICA/B
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
the regulation of MICA/B expression in breast cancer cells, whenHER2/HER3 are expressed at high levels.Next, we silenced either HER2 or HER3 in the high-expressing
T47D breast carcinoma line using siRNA. Forty-eight hours aftersiRNA transfection, the expression of HER2 or HER3 in T47Dcells was found to be attenuated equally, as measured in Westernblot (Fig. 1Bi, 1Bii). Interestingly, the expression of MICA/B wassignificantly downregulated by siRNA of HER3 but not by siRNAof HER2 in T47D cells (Fig. 1Biii, 1Biv). MDA-MB-453 cellsalso showed similar results (Supplemental Fig. 2Ai).Next, MICA and MICB were assessed separately after lapatinib
or siRNA treatment. Whereas lapatinib had a strong effect onMICB and no effect on MICA in T47D cells, the opposite was truein MDA-MB-453 cells (Fig. 1Ai).Both HER2 and HER3 siRNA downregulated MICB expression
in T47D cells (Fig 1Biii, 1Bv). The effect, however, was signifi-cantly stronger for siRNA of HER3 as compared with siRNA ofHER2. Only siRNA of HER3, but not siRNA of HER2, was ableto downregulate MICA in MDA-MB-453 cells (Supplemental Fig.2A). These findings suggested that the expression level of HER3,but not of HER2, is important for the expression of MICA/Bmolecules, which is why for the subsequent experiments we fo-cused on the role of HER3 in regulating MICA/B expression.To investigate this further, the expressions of MICA and MICB
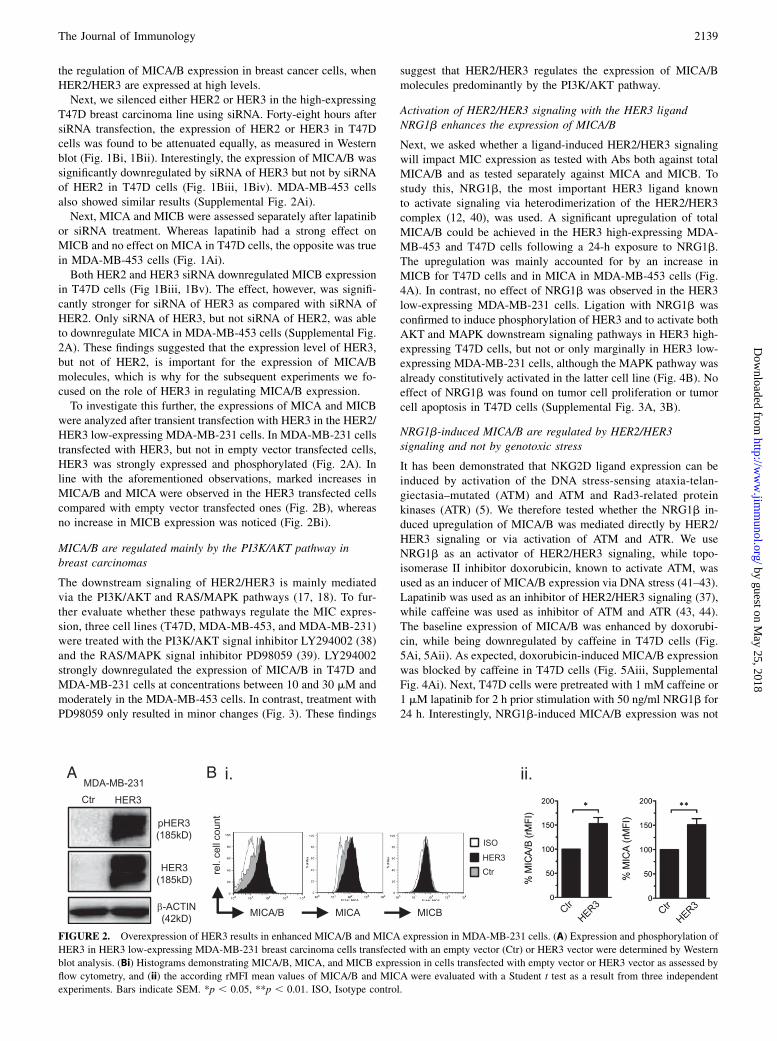
were analyzed after transient transfection with HER3 in the HER2/HER3 low-expressing MDA-MB-231 cells. In MDA-MB-231 cellstransfected with HER3, but not in empty vector transfected cells,HER3 was strongly expressed and phosphorylated (Fig. 2A). Inline with the aforementioned observations, marked increases inMICA/B and MICA were observed in the HER3 transfected cellscompared with empty vector transfected ones (Fig. 2B), whereasno increase in MICB expression was noticed (Fig. 2Bi).
MICA/B are regulated mainly by the PI3K/AKT pathway inbreast carcinomas
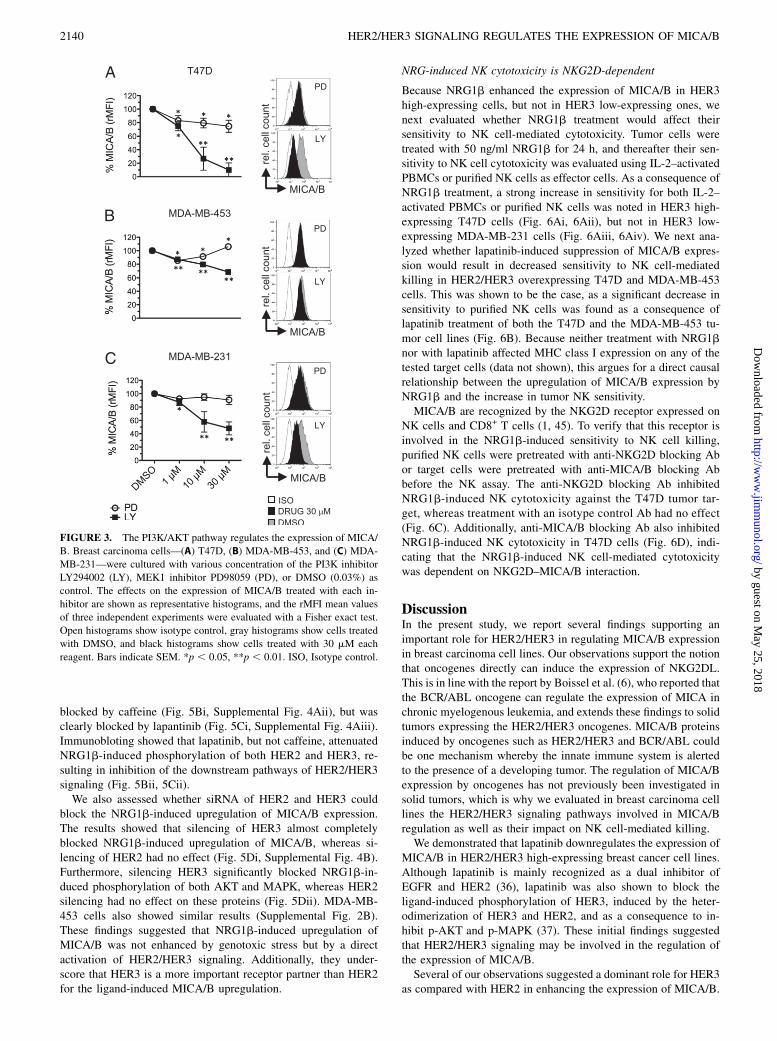
The downstream signaling of HER2/HER3 is mainly mediatedvia the PI3K/AKT and RAS/MAPK pathways (17, 18). To fur-ther evaluate whether these pathways regulate the MIC expres-sion, three cell lines (T47D, MDA-MB-453, and MDA-MB-231)were treated with the PI3K/AKT signal inhibitor LY294002 (38)and the RAS/MAPK signal inhibitor PD98059 (39). LY294002strongly downregulated the expression of MICA/B in T47D andMDA-MB-231 cells at concentrations between 10 and 30 mM andmoderately in the MDA-MB-453 cells. In contrast, treatment withPD98059 only resulted in minor changes (Fig. 3). These findings
suggest that HER2/HER3 regulates the expression of MICA/Bmolecules predominantly by the PI3K/AKT pathway.
Activation of HER2/HER3 signaling with the HER3 ligandNRG1b enhances the expression of MICA/B
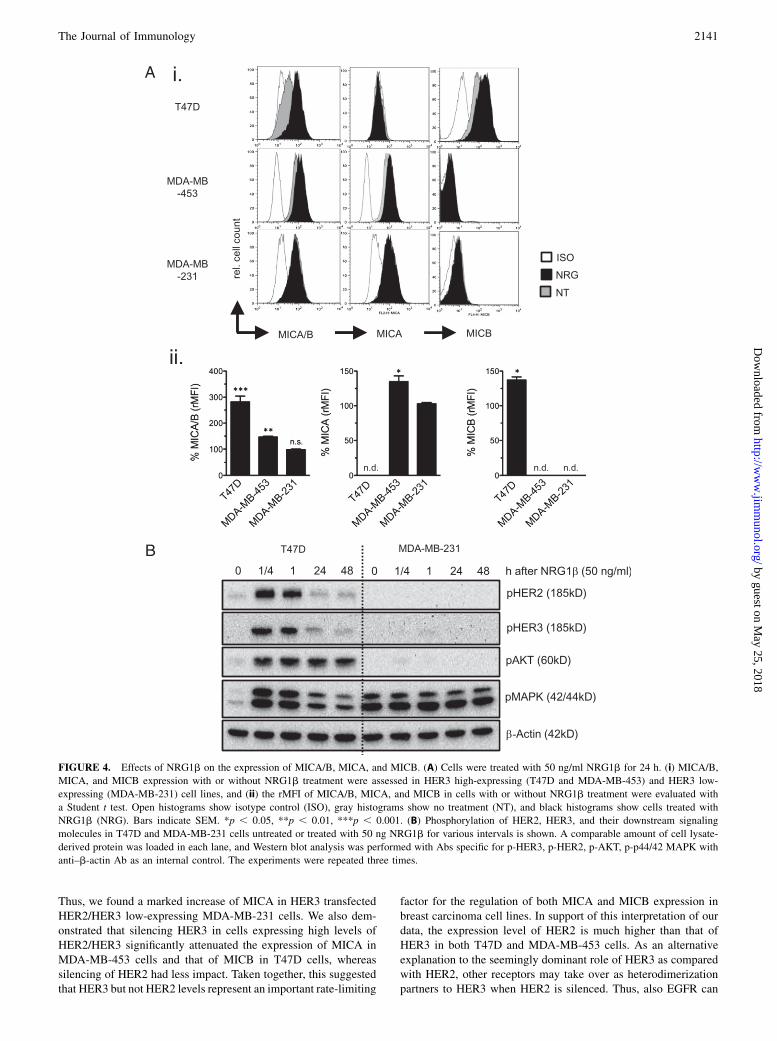
Next, we asked whether a ligand-induced HER2/HER3 signalingwill impact MIC expression as tested with Abs both against totalMICA/B and as tested separately against MICA and MICB. Tostudy this, NRG1b, the most important HER3 ligand knownto activate signaling via heterodimerization of the HER2/HER3complex (12, 40), was used. A significant upregulation of totalMICA/B could be achieved in the HER3 high-expressing MDA-MB-453 and T47D cells following a 24-h exposure to NRG1b.The upregulation was mainly accounted for by an increase inMICB for T47D cells and in MICA in MDA-MB-453 cells (Fig.4A). In contrast, no effect of NRG1b was observed in the HER3low-expressing MDA-MB-231 cells. Ligation with NRG1b wasconfirmed to induce phosphorylation of HER3 and to activate bothAKT and MAPK downstream signaling pathways in HER3 high-expressing T47D cells, but not or only marginally in HER3 low-expressing MDA-MB-231 cells, although the MAPK pathway wasalready constitutively activated in the latter cell line (Fig. 4B). Noeffect of NRG1b was found on tumor cell proliferation or tumorcell apoptosis in T47D cells (Supplemental Fig. 3A, 3B).
NRG1b-induced MICA/B are regulated by HER2/HER3signaling and not by genotoxic stress
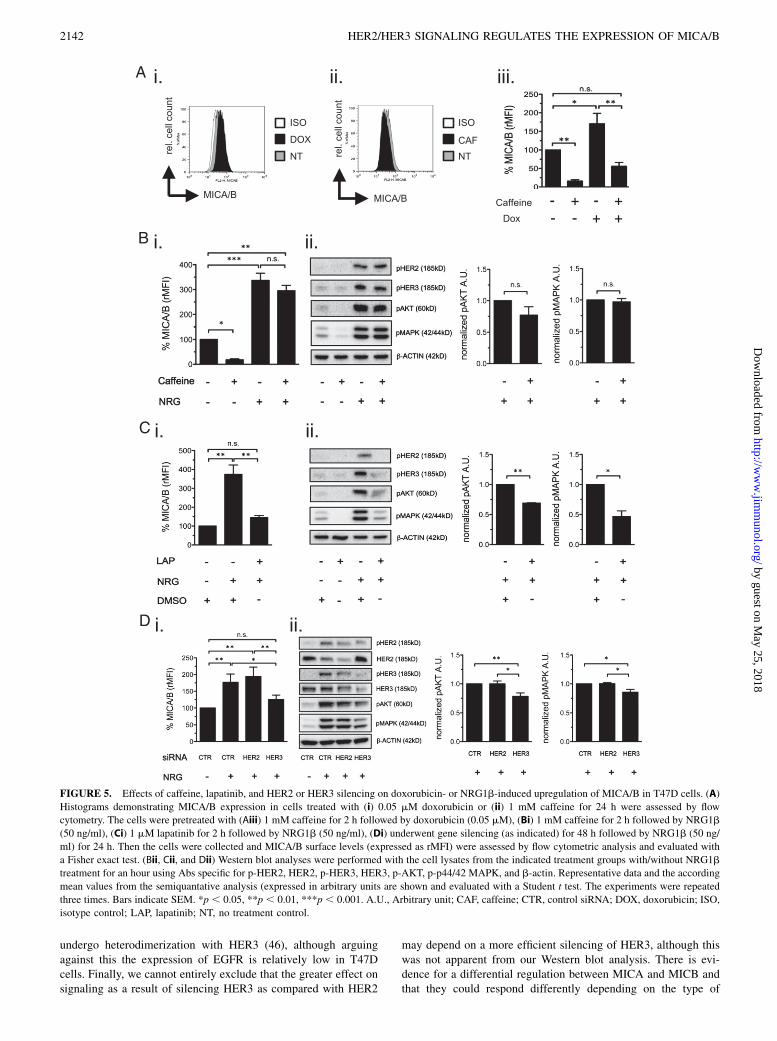
It has been demonstrated that NKG2D ligand expression can beinduced by activation of the DNA stress-sensing ataxia-telan-giectasia–mutated (ATM) and ATM and Rad3-related proteinkinases (ATR) (5). We therefore tested whether the NRG1b in-duced upregulation of MICA/B was mediated directly by HER2/HER3 signaling or via activation of ATM and ATR. We useNRG1b as an activator of HER2/HER3 signaling, while topo-isomerase II inhibitor doxorubicin, known to activate ATM, wasused as an inducer of MICA/B expression via DNA stress (41–43).Lapatinib was used as an inhibitor of HER2/HER3 signaling (37),while caffeine was used as inhibitor of ATM and ATR (43, 44).The baseline expression of MICA/B was enhanced by doxorubi-cin, while being downregulated by caffeine in T47D cells (Fig.5Ai, 5Aii). As expected, doxorubicin-induced MICA/B expressionwas blocked by caffeine in T47D cells (Fig. 5Aiii, SupplementalFig. 4Ai). Next, T47D cells were pretreated with 1 mM caffeine or1 mM lapatinib for 2 h prior stimulation with 50 ng/ml NRG1b for24 h. Interestingly, NRG1b-induced MICA/B expression was not
FIGURE 2. Overexpression of HER3 results in enhanced MICA/B and MICA expression in MDA-MB-231 cells. (A) Expression and phosphorylation of
HER3 in HER3 low-expressing MDA-MB-231 breast carcinoma cells transfected with an empty vector (Ctr) or HER3 vector were determined by Western
blot analysis. (Bi) Histograms demonstrating MICA/B, MICA, and MICB expression in cells transfected with empty vector or HER3 vector as assessed by
flow cytometry, and (ii) the according rMFI mean values of MICA/B and MICA were evaluated with a Student t test as a result from three independent
experiments. Bars indicate SEM. *p , 0.05, **p , 0.01. ISO, Isotype control.
The Journal of Immunology 2139
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
blocked by caffeine (Fig. 5Bi, Supplemental Fig. 4Aii), but wasclearly blocked by lapantinib (Fig. 5Ci, Supplemental Fig. 4Aiii).Immunobloting showed that lapatinib, but not caffeine, attenuatedNRG1b-induced phosphorylation of both HER2 and HER3, re-sulting in inhibition of the downstream pathways of HER2/HER3signaling (Fig. 5Bii, 5Cii).We also assessed whether siRNA of HER2 and HER3 could
block the NRG1b-induced upregulation of MICA/B expression.The results showed that silencing of HER3 almost completelyblocked NRG1b-induced upregulation of MICA/B, whereas si-lencing of HER2 had no effect (Fig. 5Di, Supplemental Fig. 4B).Furthermore, silencing HER3 significantly blocked NRG1b-in-duced phosphorylation of both AKT and MAPK, whereas HER2silencing had no effect on these proteins (Fig. 5Dii). MDA-MB-453 cells also showed similar results (Supplemental Fig. 2B).These findings suggested that NRG1b-induced upregulation ofMICA/B was not enhanced by genotoxic stress but by a directactivation of HER2/HER3 signaling. Additionally, they under-score that HER3 is a more important receptor partner than HER2for the ligand-induced MICA/B upregulation.
NRG-induced NK cytotoxicity is NKG2D-dependent
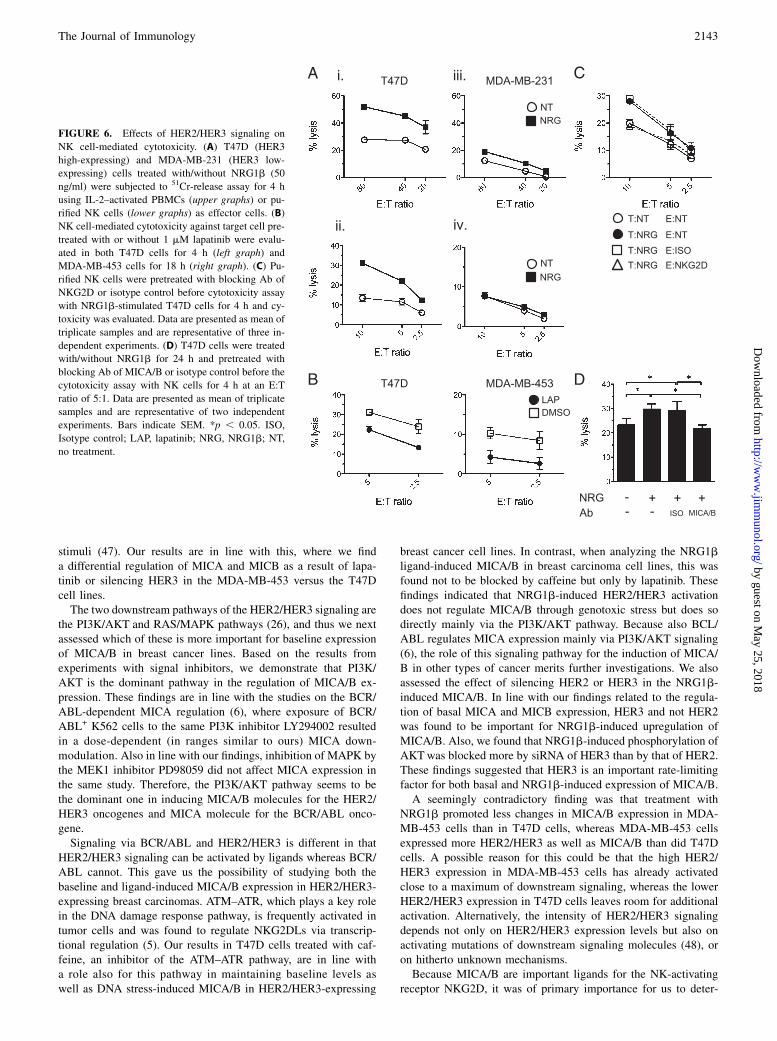
Because NRG1b enhanced the expression of MICA/B in HER3high-expressing cells, but not in HER3 low-expressing ones, wenext evaluated whether NRG1b treatment would affect theirsensitivity to NK cell-mediated cytotoxicity. Tumor cells weretreated with 50 ng/ml NRG1b for 24 h, and thereafter their sen-sitivity to NK cell cytotoxicity was evaluated using IL-2–activatedPBMCs or purified NK cells as effector cells. As a consequence ofNRG1b treatment, a strong increase in sensitivity for both IL-2–activated PBMCs or purified NK cells was noted in HER3 high-expressing T47D cells (Fig. 6Ai, 6Aii), but not in HER3 low-expressing MDA-MB-231 cells (Fig. 6Aiii, 6Aiv). We next ana-lyzed whether lapatinib-induced suppression of MICA/B expres-sion would result in decreased sensitivity to NK cell-mediatedkilling in HER2/HER3 overexpressing T47D and MDA-MB-453cells. This was shown to be the case, as a significant decrease insensitivity to purified NK cells was found as a consequence oflapatinib treatment of both the T47D and the MDA-MB-453 tu-mor cell lines (Fig. 6B). Because neither treatment with NRG1bnor with lapatinib affected MHC class I expression on any of thetested target cells (data not shown), this argues for a direct causalrelationship between the upregulation of MICA/B expression byNRG1b and the increase in tumor NK sensitivity.MICA/B are recognized by the NKG2D receptor expressed on
NK cells and CD8+ T cells (1, 45). To verify that this receptor isinvolved in the NRG1b-induced sensitivity to NK cell killing,purified NK cells were pretreated with anti-NKG2D blocking Abor target cells were pretreated with anti-MICA/B blocking Abbefore the NK assay. The anti-NKG2D blocking Ab inhibitedNRG1b-induced NK cytotoxicity against the T47D tumor tar-get, whereas treatment with an isotype control Ab had no effect(Fig. 6C). Additionally, anti-MICA/B blocking Ab also inhibitedNRG1b-induced NK cytotoxicity in T47D cells (Fig. 6D), indi-cating that the NRG1b-induced NK cell-mediated cytotoxicitywas dependent on NKG2D–MICA/B interaction.
DiscussionIn the present study, we report several findings supporting animportant role for HER2/HER3 in regulating MICA/B expressionin breast carcinoma cell lines. Our observations support the notionthat oncogenes directly can induce the expression of NKG2DL.This is in line with the report by Boissel et al. (6), who reported thatthe BCR/ABL oncogene can regulate the expression of MICA inchronic myelogenous leukemia, and extends these findings to solidtumors expressing the HER2/HER3 oncogenes. MICA/B proteinsinduced by oncogenes such as HER2/HER3 and BCR/ABL couldbe one mechanism whereby the innate immune system is alertedto the presence of a developing tumor. The regulation of MICA/Bexpression by oncogenes has not previously been investigated insolid tumors, which is why we evaluated in breast carcinoma celllines the HER2/HER3 signaling pathways involved in MICA/Bregulation as well as their impact on NK cell-mediated killing.We demonstrated that lapatinib downregulates the expression of
MICA/B in HER2/HER3 high-expressing breast cancer cell lines.Although lapatinib is mainly recognized as a dual inhibitor ofEGFR and HER2 (36), lapatinib was also shown to block theligand-induced phosphorylation of HER3, induced by the heter-odimerization of HER3 and HER2, and as a consequence to in-hibit p-AKT and p-MAPK (37). These initial findings suggestedthat HER2/HER3 signaling may be involved in the regulation ofthe expression of MICA/B.Several of our observations suggested a dominant role for HER3
as compared with HER2 in enhancing the expression of MICA/B.
FIGURE 3. The PI3K/AKT pathway regulates the expression of MICA/
B. Breast carcinoma cells—(A) T47D, (B) MDA-MB-453, and (C) MDA-
MB-231—were cultured with various concentration of the PI3K inhibitor
LY294002 (LY), MEK1 inhibitor PD98059 (PD), or DMSO (0.03%) as
control. The effects on the expression of MICA/B treated with each in-
hibitor are shown as representative histograms, and the rMFI mean values
of three independent experiments were evaluated with a Fisher exact test.
Open histograms show isotype control, gray histograms show cells treated
with DMSO, and black histograms show cells treated with 30 mM each
reagent. Bars indicate SEM. *p , 0.05, **p , 0.01. ISO, Isotype control.
2140 HER2/HER3 SIGNALING REGULATES THE EXPRESSION OF MICA/B
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Thus, we found a marked increase of MICA in HER3 transfectedHER2/HER3 low-expressing MDA-MB-231 cells. We also dem-onstrated that silencing HER3 in cells expressing high levels ofHER2/HER3 significantly attenuated the expression of MICA inMDA-MB-453 cells and that of MICB in T47D cells, whereassilencing of HER2 had less impact. Taken together, this suggestedthat HER3 but not HER2 levels represent an important rate-limiting
factor for the regulation of both MICA and MICB expression inbreast carcinoma cell lines. In support of this interpretation of ourdata, the expression level of HER2 is much higher than that ofHER3 in both T47D and MDA-MB-453 cells. As an alternativeexplanation to the seemingly dominant role of HER3 as comparedwith HER2, other receptors may take over as heterodimerizationpartners to HER3 when HER2 is silenced. Thus, also EGFR can
FIGURE 4. Effects of NRG1b on the expression of MICA/B, MICA, and MICB. (A) Cells were treated with 50 ng/ml NRG1b for 24 h. (i) MICA/B,
MICA, and MICB expression with or without NRG1b treatment were assessed in HER3 high-expressing (T47D and MDA-MB-453) and HER3 low-
expressing (MDA-MB-231) cell lines, and (ii) the rMFI of MICA/B, MICA, and MICB in cells with or without NRG1b treatment were evaluated with
a Student t test. Open histograms show isotype control (ISO), gray histograms show no treatment (NT), and black histograms show cells treated with
NRG1b (NRG). Bars indicate SEM. *p , 0.05, **p , 0.01, ***p , 0.001. (B) Phosphorylation of HER2, HER3, and their downstream signaling
molecules in T47D and MDA-MB-231 cells untreated or treated with 50 ng NRG1b for various intervals is shown. A comparable amount of cell lysate-
derived protein was loaded in each lane, and Western blot analysis was performed with Abs specific for p-HER3, p-HER2, p-AKT, p-p44/42 MAPK with
anti–b-actin Ab as an internal control. The experiments were repeated three times.
The Journal of Immunology 2141
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
undergo heterodimerization with HER3 (46), although arguingagainst this the expression of EGFR is relatively low in T47Dcells. Finally, we cannot entirely exclude that the greater effect onsignaling as a result of silencing HER3 as compared with HER2
may depend on a more efficient silencing of HER3, although thiswas not apparent from our Western blot analysis. There is evi-dence for a differential regulation between MICA and MICB andthat they could respond differently depending on the type of
FIGURE 5. Effects of caffeine, lapatinib, and HER2 or HER3 silencing on doxorubicin- or NRG1b-induced upregulation of MICA/B in T47D cells. (A)
Histograms demonstrating MICA/B expression in cells treated with (i) 0.05 mM doxorubicin or (ii) 1 mM caffeine for 24 h were assessed by flow
cytometry. The cells were pretreated with (Aiii) 1 mM caffeine for 2 h followed by doxorubicin (0.05 mM), (Bi) 1 mM caffeine for 2 h followed by NRG1b
(50 ng/ml), (Ci) 1 mM lapatinib for 2 h followed by NRG1b (50 ng/ml), (Di) underwent gene silencing (as indicated) for 48 h followed by NRG1b (50 ng/
ml) for 24 h. Then the cells were collected and MICA/B surface levels (expressed as rMFI) were assessed by flow cytometric analysis and evaluated with
a Fisher exact test. (Bii, Cii, and Dii) Western blot analyses were performed with the cell lysates from the indicated treatment groups with/without NRG1b
treatment for an hour using Abs specific for p-HER2, HER2, p-HER3, HER3, p-AKT, p-p44/42 MAPK, and b-actin. Representative data and the according
mean values from the semiquantative analysis (expressed in arbitrary units are shown and evaluated with a Student t test. The experiments were repeated
three times. Bars indicate SEM. *p , 0.05, **p , 0.01, ***p , 0.001. A.U., Arbitrary unit; CAF, caffeine; CTR, control siRNA; DOX, doxorubicin; ISO,
isotype control; LAP, lapatinib; NT, no treatment control.
2142 HER2/HER3 SIGNALING REGULATES THE EXPRESSION OF MICA/B
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
stimuli (47). Our results are in line with this, where we finda differential regulation of MICA and MICB as a result of lapa-tinib or silencing HER3 in the MDA-MB-453 versus the T47Dcell lines.The two downstream pathways of the HER2/HER3 signaling are
the PI3K/AKT and RAS/MAPK pathways (26), and thus we nextassessed which of these is more important for baseline expressionof MICA/B in breast cancer lines. Based on the results fromexperiments with signal inhibitors, we demonstrate that PI3K/AKT is the dominant pathway in the regulation of MICA/B ex-pression. These findings are in line with the studies on the BCR/ABL-dependent MICA regulation (6), where exposure of BCR/ABL+ K562 cells to the same PI3K inhibitor LY294002 resultedin a dose-dependent (in ranges similar to ours) MICA down-modulation. Also in line with our findings, inhibition of MAPK bythe MEK1 inhibitor PD98059 did not affect MICA expression inthe same study. Therefore, the PI3K/AKT pathway seems to bethe dominant one in inducing MICA/B molecules for the HER2/HER3 oncogenes and MICA molecule for the BCR/ABL onco-gene.Signaling via BCR/ABL and HER2/HER3 is different in that
HER2/HER3 signaling can be activated by ligands whereas BCR/ABL cannot. This gave us the possibility of studying both thebaseline and ligand-induced MICA/B expression in HER2/HER3-expressing breast carcinomas. ATM–ATR, which plays a key rolein the DNA damage response pathway, is frequently activated intumor cells and was found to regulate NKG2DLs via transcrip-tional regulation (5). Our results in T47D cells treated with caf-feine, an inhibitor of the ATM–ATR pathway, are in line witha role also for this pathway in maintaining baseline levels aswell as DNA stress-induced MICA/B in HER2/HER3-expressing
breast cancer cell lines. In contrast, when analyzing the NRG1bligand-induced MICA/B in breast carcinoma cell lines, this wasfound not to be blocked by caffeine but only by lapatinib. Thesefindings indicated that NRG1b-induced HER2/HER3 activationdoes not regulate MICA/B through genotoxic stress but does sodirectly mainly via the PI3K/AKT pathway. Because also BCL/ABL regulates MICA expression mainly via PI3K/AKT signaling(6), the role of this signaling pathway for the induction of MICA/B in other types of cancer merits further investigations. We alsoassessed the effect of silencing HER2 or HER3 in the NRG1b-induced MICA/B. In line with our findings related to the regula-tion of basal MICA and MICB expression, HER3 and not HER2was found to be important for NRG1b-induced upregulation ofMICA/B. Also, we found that NRG1b-induced phosphorylation ofAKTwas blocked more by siRNA of HER3 than by that of HER2.These findings suggested that HER3 is an important rate-limitingfactor for both basal and NRG1b-induced expression of MICA/B.A seemingly contradictory finding was that treatment with
NRG1b promoted less changes in MICA/B expression in MDA-MB-453 cells than in T47D cells, whereas MDA-MB-453 cellsexpressed more HER2/HER3 as well as MICA/B than did T47Dcells. A possible reason for this could be that the high HER2/HER3 expression in MDA-MB-453 cells has already activatedclose to a maximum of downstream signaling, whereas the lowerHER2/HER3 expression in T47D cells leaves room for additionalactivation. Alternatively, the intensity of HER2/HER3 signalingdepends not only on HER2/HER3 expression levels but also onactivating mutations of downstream signaling molecules (48), oron hitherto unknown mechanisms.Because MICA/B are important ligands for the NK-activating
receptor NKG2D, it was of primary importance for us to deter-
FIGURE 6. Effects of HER2/HER3 signaling on
NK cell-mediated cytotoxicity. (A) T47D (HER3
high-expressing) and MDA-MB-231 (HER3 low-
expressing) cells treated with/without NRG1b (50
ng/ml) were subjected to 51Cr-release assay for 4 h
using IL-2–activated PBMCs (upper graphs) or pu-
rified NK cells (lower graphs) as effector cells. (B)
NK cell-mediated cytotoxicity against target cell pre-
treated with or without 1 mM lapatinib were evalu-
ated in both T47D cells for 4 h (left graph) and
MDA-MB-453 cells for 18 h (right graph). (C) Pu-
rified NK cells were pretreated with blocking Ab of
NKG2D or isotype control before cytotoxicity assay
with NRG1b-stimulated T47D cells for 4 h and cy-
toxicity was evaluated. Data are presented as mean of
triplicate samples and are representative of three in-
dependent experiments. (D) T47D cells were treated
with/without NRG1b for 24 h and pretreated with
blocking Ab of MICA/B or isotype control before the
cytotoxicity assay with NK cells for 4 h at an E:T
ratio of 5:1. Data are presented as mean of triplicate
samples and are representative of two independent
experiments. Bars indicate SEM. *p , 0.05. ISO,
Isotype control; LAP, lapatinib; NRG, NRG1b; NT,
no treatment.
The Journal of Immunology 2143
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

mine the functional consequences of HER2/HER3-mediatedMICAand MICB regulations on tumor target cells. As anticipated, ac-tivation of HER2/HER3 signaling by NRG1b enhanced sensitivityto NK cell-mediated cytotoxicity in HER3 overexpressing breastcarcinoma cells. Conversely, inactivation of HER2/HER3 signal-ing by lapatinib attenuated the sensitivity to NK cell-mediatedcytotoxicity. These findings implicated a role for the NKG2D–MICA/B interaction in the modulation of the NK cell sensitivity inthese breast cancer lines. To more directly prove this, we dem-onstrated that NRG1b-enhanced NK cell-mediated cytotoxicitycould be blocked with an anti-NKG2D Ab. This result could notrule out that the interaction between NKG2D ligands other thanMICA/B could be of importance for the observed NRG1b-inducedenhancement of NK cell activity. Arguing for a predominant rolefor MICA and MICB as compared with other NKG2D ligands inthis regard, we were able to block the enhanced activity by an Abagainst MICA/B molecules. It is notable, however, that neitherblocking with anti-NKG2D Abs nor with anti-MICA/B Abs sig-nificantly decreased NK cell sensitivity of the untreated T47D line(data not shown). The reason for this could be either that NKG2Dis not involved in conferring “baseline” NK sensitivity of this cellline, or alternatively and more likely that these Abs are able toonly partially block NK-receptor/ligand interaction.NRGs are regarded as growth factors for cancer cells, as NRGs
activate cell growth and cell proliferation via activation of thedownstream signaling pathway of HER3 (17, 18). Indeed 1 nMNRG1b was shown to enhance the cell proliferation in 7 of 14ovarian cancer cell lines (49). We found that up to 250 ng/mlNRG1b could not affect tumor cell proliferation or apoptosis inthe T47D cell line. These findings support the conclusion thata high dose of NRG1b enhances breast carcinoma sensitivity forNK cell-mediated cytotoxicity without accelerating tumor growth.Conversely, the implication from our findings is that the HER2/HER3 signaling pathway targeting therapy may suppress tumorsensitivity to NK cells via downregulation of MICA/B, as wasshown by the attenuation of NK cell-mediated cytotoxicity bylapatinib. In theory, this indicates that therapies targeting HER2/HER3 signaling may be a double-edged sword as they block tu-mor cell-activating signaling while suppressing NKG2D-MICA/B–dependent immune surveillance by NK cells, CD8+ T cells, andgd T cells. However, because lapanitib pretreatment enhancedtrastuzumab-induced Ab-dependent cellular cytotoxicity by in-creasing the cell surface expression of HER2 through inhibition ofthe degradation of HER2 (50, 51), the clinical net effect of lapa-tinib treatment when it comes to NK cell-mediated tumor elimi-nation remains to be established.
AcknowledgmentsWe thank Juan Castro (Cancer Center Karolinska) for the cell line au-
thentication.
DisclosuresThe authors have no financial conflicts of interest.
References1. Bauer, S., V. Groh, J. Wu, A. Steinle, J. H. Phillips, L. L. Lanier, and T. Spies.
1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285: 727–729.
2. Wu, J., Y. Song, A. B. Bakker, S. Bauer, T. Spies, L. L. Lanier, and J. H. Phillips.1999. An activating immunoreceptor complex formed by NKG2D and DAP10.Science 285: 730–732.
3. Ogasawara, K., and L. L. Lanier. 2005. NKG2D in NK and T cell-mediatedimmunity. J. Clin. Immunol. 25: 534–540.
4. Groh, V., S. Bahram, S. Bauer, A. Herman, M. Beauchamp, and T. Spies. 1996.Cell stress-regulated human major histocompatibility complex class I gene
expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA 93: 12445–12450.
5. Gasser, S., S. Orsulic, E. J. Brown, and D. H. Raulet. 2005. The DNA damagepathway regulates innate immune system ligands of the NKG2D receptor. Nature436: 1186–1190.
6. Boissel, N., D. Rea, V. Tieng, N. Dulphy, M. Brun, J. M. Cayuela, P. Rousselot,R. Tamouza, P. Le Bouteiller, F. X. Mahon, et al. 2006. BCR/ABL oncogenedirectly controls MHC class I chain-related molecule A expression in chronicmyelogenous leukemia. J. Immunol. 176: 5108–5116.
7. Bryceson, Y. T., M. E. March, H. G. Ljunggren, and E. O. Long. 2006. Acti-vation, coactivation, and costimulation of resting human natural killer cells.Immunol. Rev. 214: 73–91.
8. Cerwenka, A., J. L. Baron, and L. L. Lanier. 2001. Ectopic expression of retinoicacid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejec-tion of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA 98:11521–11526.
9. Gschwind, A., O. M. Fischer, and A. Ullrich. 2004. The discovery of receptortyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 4: 361–370.
10. Guy, P. M., J. V. Platko, L. C. Cantley, R. A. Cerione, and K. L. Carraway, III.1994. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinaseactivity. Proc. Natl. Acad. Sci. USA 91: 8132–8136.
11. Sliwkowski, M. X., G. Schaefer, R. W. Akita, J. A. Lofgren, V. D. Fitzpatrick,A. Nuijens, B. M. Fendly, R. A. Cerione, R. L. Vandlen, and K. L. Carraway, III.1994. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinityreceptor for heregulin. J. Biol. Chem. 269: 14661–14665.
12. Pinkas-Kramarski, R., L. Soussan, H. Waterman, G. Levkowitz, I. Alroy,L. Klapper, S. Lavi, R. Seger, B. J. Ratzkin, M. Sela, and Y. Yarden. 1996.Diversification of Neu differentiation factor and epidermal growth factor sig-naling by combinatorial receptor interactions. EMBO J. 15: 2452–2467.
13. Tzahar, E., H. Waterman, X. Chen, G. Levkowitz, D. Karunagaran, S. Lavi,B. J. Ratzkin, and Y. Yarden. 1996. A hierarchical network of interreceptorinteractions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 16: 5276–5287.
14. Holbro, T., R. R. Beerli, F. Maurer, M. Koziczak, C. F. Barbas, III, andN. E. Hynes. 2003. The ErbB2/ErbB3 heterodimer functions as an oncogenicunit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl.Acad. Sci. USA 100: 8933–8938.
15. Burgess, A. W., H. S. Cho, C. Eigenbrot, K. M. Ferguson, T. P. Garrett,D. J. Leahy, M. A. Lemmon, M. X. Sliwkowski, C. W. Ward, and S. Yokoyama.2003. An open-and-shut case? Recent insights into the activation of EGF/ErbBreceptors. Mol. Cell 12: 541–552.
16. Hynes, N. E., and H. A. Lane. 2005. ERBB receptors and cancer: the complexityof targeted inhibitors. Nat. Rev. Cancer 5: 341–354.
17. Yarden, Y., and M. X. Sliwkowski. 2001. Untangling the ErbB signalling net-work. Nat. Rev. Mol. Cell Biol. 2: 127–137.
18. Sithanandam, G., L. W. Fornwald, J. Fields, and L. M. Anderson. 2005. Inac-tivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and in-vasiveness of human lung adenocarcinoma cell line A549. Oncogene 24: 1847–1859.
19. Tandon, A. K., G. M. Clark, G. C. Chamness, A. Ullrich, and W. L. McGuire.1989. HER-2/neu oncogene protein and prognosis in breast cancer. J. Clin.Oncol. 7: 1120–1128.
20. Witton, C. J., J. R. Reeves, J. J. Going, T. G. Cooke, and J. M. Bartlett. 2003.Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J.Pathol. 200: 290–297.
21. Bobrow, L. G., R. R. Millis, L. C. Happerfield, and W. J. Gullick. 1997. c-erbB-3protein expression in ductal carcinoma in situ of the breast. Eur. J. Cancer 33:1846–1850.
22. Travis, A., S. E. Pinder, J. F. Robertson, J. A. Bell, P. Wencyk, W. J. Gullick,R. I. Nicholson, D. N. Poller, R. W. Blamey, C. W. Elston, and I. O. Ellis. 1996.C-erbB-3 in human breast carcinoma: expression and relation to prognosis andestablished prognostic indicators. Br. J. Cancer 74: 229–233.
23. Quinn, C. M., J. L. Ostrowski, S. A. Lane, D. P. Loney, J. Teasdale, andF. A. Benson. 1994. c-erbB-3 protein expression in human breast cancer: com-parison with other tumour variables and survival. Histopathology 25: 247–252.
24. Slamon, D. J., B. Leyland-Jones, S. Shak, H. Fuchs, V. Paton, A. Bajamonde,T. Fleming, W. Eiermann, J. Wolter, M. Pegram, et al. 2001. Use of chemo-therapy plus a monoclonal antibody against HER2 for metastatic breast cancerthat overexpresses HER2. N. Engl. J. Med. 344: 783–792.
25. Gianni, L., U. Dafni, R. D. Gelber, E. Azambuja, S. Muehlbauer, A. Goldhirsch,M. Untch, I. Smith, J. Baselga, C. Jackisch, et al; Herceptin Adjuvant (HERA)Trial Study Team. 2011. Treatment with trastuzumab for 1 year after adjuvantchemotherapy in patients with HER2-positive early breast cancer: a 4-yearfollow-up of a randomised controlled trial. Lancet Oncol. 12: 236–244.
26. Baselga, J., and S. M. Swain. 2009. Novel anticancer targets: revisiting ERBB2and discovering ERBB3. Nat. Rev. Cancer 9: 463–475.
27. Schoeberl, B., A. C. Faber, D. Li, M. C. Liang, K. Crosby, M. Onsum,O. Burenkova, E. Pace, Z. Walton, L. Nie, et al. 2010. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 70:2485–2494.
28. Lollini, P. L., G. Nicoletti, L. Landuzzi, C. De Giovanni, I. Rossi, E. Di Carlo,P. Musiani, W. J. Muller, and P. Nanni. 1998. Down regulation of major histo-compatibility complex class I expression in mammary carcinoma of HER-2/neutransgenic mice. Int. J. Cancer 77: 937–941.
29. Herrmann, F., H. A. Lehr, I. Drexler, G. Sutter, J. Hengstler, U. Wollscheid, andB. Seliger. 2004. HER-2/neu-mediated regulation of components of the MHCclass I antigen-processing pathway. Cancer Res. 64: 215–220.
2144 HER2/HER3 SIGNALING REGULATES THE EXPRESSION OF MICA/B
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

30. Choudhury, A., J. Charo, S. K. Parapuram, R. C. Hunt, D. M. Hunt, B. Seliger,and R. Kiessling. 2004. Small interfering RNA (siRNA) inhibits the expressionof the Her2/neu gene, upregulates HLA class I and induces apoptosis of Her2/neu positive tumor cell lines. Int. J. Cancer 108: 71–77.
31. Vertuani, S., C. Triulzi, A. K. Roos, J. Charo, H. Norell, F. Lemonnier, P. Pisa,B. Seliger, and R. Kiessling. 2009. HER-2/neu mediated down-regulation ofMHC class I antigen processing prevents CTL-mediated tumor recognition uponDNA vaccination in HLA-A2 transgenic mice. Cancer Immunol. Immunother.58: 653–664.
32. Mimura, K., T. Ando, I. Poschke, D. Mougiakakos, C. C. Johansson, J. Ichikawa,R. Okita, M. I. Nishimura, D. Handke, N. Krug, et al. 2011. T cell recognition ofHLA-A2 restricted tumor antigens is impaired by the oncogene HER2. Int. J.Cancer 128: 390–401.
33. Karre, K., H. G. Ljunggren, G. Piontek, and R. Kiessling. 1986. Selective re-jection of H-2-deficient lymphoma variants suggests alternative immune defencestrategy. Nature 319: 675–678.
34. Orr, M. T., and L. L. Lanier. 2010. Natural killer cell education and tolerance.Cell 142: 847–856.
35. Torsteinsdottir, S., M. G. Masucci, B. Ehlin-Henriksson, C. Brautbar, H. BenBassat, G. Klein, and E. Klein. 1986. Differentiation-dependent sensitivity ofhuman B-cell-derived lines to major histocompatibility complex-restricted T-cellcytotoxicity. Proc. Natl. Acad. Sci. USA 83: 5620–5624.
36. Xia, W., R. J. Mullin, B. R. Keith, L. H. Liu, H. Ma, D. W. Rusnak, G. Owens,K. J. Alligood, and N. L. Spector. 2002. Anti-tumor activity of GW572016:a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 anddownstream Erk1/2 and AKT pathways. Oncogene 21: 6255–6263.
37. Gregory, C. W., Y. E. Whang, W. McCall, X. Fei, Y. Liu, L. A. Ponguta,F. S. French, E. M. Wilson, and H. S. Earp, III. 2005. Heregulin-inducedactivation of HER2 and HER3 increases androgen receptor transactivation andCWR-R1 human recurrent prostate cancer cell growth. Clin. Cancer Res. 11:1704–1712.
38. Vlahos, C. J., W. F. Matter, K. Y. Hui, and R. F. Brown. 1994. A specific inhibitorof phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269: 5241–5248.
39. Pang, L., T. Sawada, S. J. Decker, and A. R. Saltiel. 1995. Inhibition of MAPkinase kinase blocks the differentiation of PC-12 cells induced by nerve growthfactor. J. Biol. Chem. 270: 13585–13588.
40. Alimandi, M., A. Romano, M. C. Curia, R. Muraro, P. Fedi, S. A. Aaronson,P. P. Di Fiore, and M. H. Kraus. 1995. Cooperative signaling of ErbB3 andErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene10: 1813–1821.
41. Tewey, K. M., T. C. Rowe, L. Yang, B. D. Halligan, and L. F. Liu. 1984.Adriamycin-induced DNA damage mediated by mammalian DNA topoisomer-ase II. Science 226: 466–468.
42. Kurz, E. U., P. Douglas, and S. P. Lees-Miller. 2004. Doxorubicin activatesATM-dependent phosphorylation of multiple downstream targets in part throughthe generation of reactive oxygen species. J. Biol. Chem. 279: 53272–53281.
43. Soriani, A., A. Zingoni, C. Cerboni, M. L. Iannitto, M. R. Ricciardi, V. DiGialleonardo, M. Cippitelli, C. Fionda, M. T. Petrucci, A. Guarini, et al. 2009.ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on mul-tiple myeloma cells by therapeutic agents results in enhanced NK-cell suscep-tibility and is associated with a senescent phenotype. Blood 113: 3503–3511.
44. Sarkaria, J. N., E. C. Busby, R. S. Tibbetts, P. Roos, Y. Taya, L. M. Karnitz, andR. T. Abraham. 1999. Inhibition of ATM and ATR kinase activities by theradiosensitizing agent, caffeine. Cancer Res. 59: 4375–4382.
45. Groh, V., A. Steinle, S. Bauer, and T. Spies. 1998. Recognition of stress-inducedMHC molecules by intestinal epithelial gammadelta T cells. Science 279: 1737–1740.
46. Soltoff, S. P., K. L. Carraway, III, S. A. Prigent, W. G. Gullick, and L. C. Cantley.1994. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epi-dermal growth factor. Mol. Cell. Biol. 14: 3550–3558.
47. Venkataraman, G. M., D. Suciu, V. Groh, J. M. Boss, and T. Spies. 2007. Pro-moter region architecture and transcriptional regulation of the genes for theMHC class I-related chain A and B ligands of NKG2D. J. Immunol. 178: 961–969.
48. Berns, K., H. M. Horlings, B. T. Hennessy, M. Madiredjo, E. M. Hijmans,K. Beelen, S. C. Linn, A. M. Gonzalez-Angulo, K. Stemke-Hale, M. Hauptmann,et al. 2007. A functional genetic approach identifies the PI3K pathway as a majordeterminant of trastuzumab resistance in breast cancer. Cancer Cell 12: 395–402.
49. Gilmour, L. M., K. G. Macleod, A. McCaig, J. M. Sewell, W. J. Gullick,J. F. Smyth, and S. P. Langdon. 2002. Neuregulin expression, function, andsignaling in human ovarian cancer cells. Clin. Cancer Res. 8: 3933–3942.
50. Scaltriti, M., C. Verma, M. Guzman, J. Jimenez, J. L. Parra, K. Pedersen,D. J. Smith, S. Landolfi, S. Ramon y Cajal, J. Arribas, and J. Baselga. 2009.Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumu-lation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Onco-gene 28: 803–814.
51. Mimura, K., K. Kono, T. Maruyama, M. Watanabe, S. Izawa, S. Shiba,Y. Mizukami, Y. Kawaguchi, M. Inoue, T. Kono, et al. 2011. Lapatinib inhibitsreceptor phosphorylation and cell growth and enhances antibody-dependentcellular cytoxicity of EGFR- and HER2- overexpressing esophageal cancercell lines. Int. J. Cancer 129: 2408–2416.
The Journal of Immunology 2145
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from


















