
Hemostasis
44
Hemostasis Cynthia J. Rutherford, M.D. Hematology-Oncology Division January 28, 2015
-
Upload
katejohnpunag -
Category
Health & Medicine
-
view
384 -
download
2
Transcript of Hemostasis

Hemostasis
Cynthia J. Rutherford, M.D.
Hematology-Oncology Division
January 28, 2015

Phases of hemostasis
• Primary hemostasis
– platelet adhesion and aggregation
• platelet number and function
• von Willebrand factor
• Secondary hemostasis
– activation of the coagulation system
• extrinsic
• intrinsic
• common pathway

The bleeding patientClues from the history
• Life long history of bleeding
• Family history of bleeding
• History of liver disease
• History of hematologic disorder – ITP, leukemia
• Medication history – ASA, NSAIDS . . .

The bleeding patientClues from the physical exam
• Pattern of skin and mucosal bleeding seen in defects of primary hemostasis - suggest thrombocytopenia, platelet defect or vWD
• Pattern of muscle/deep bleeding suggesting defect in secondary hemostasis - coagulopathy
• Stigmata of liver disease – arterial nevi, splenomegaly, palmar erythema
• Pattern and clinical scenario of DIC
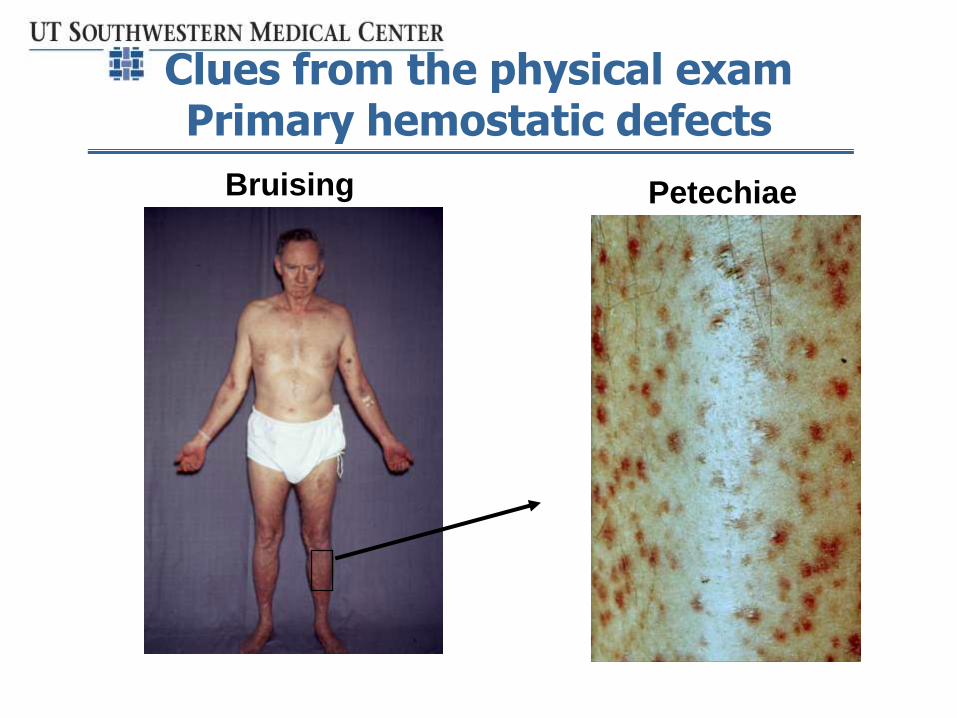
Clues from the physical exam Primary hemostatic defects
PetechiaeBruising

Clues from the physical examSevere primary hemostatic defects
Mucosal bleeding = “wet” purpura
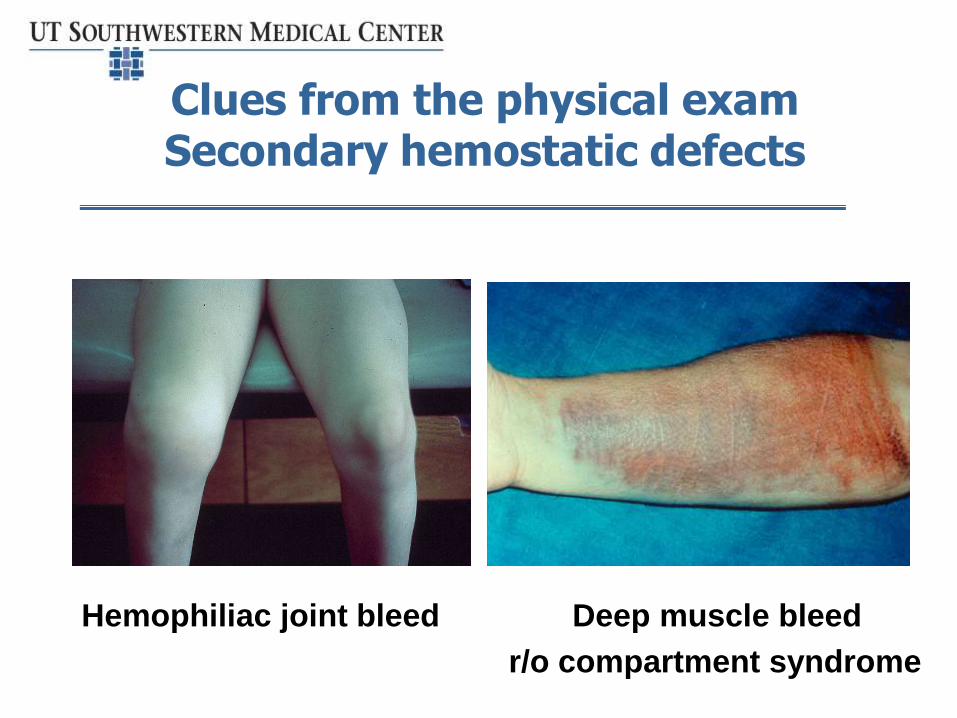
Clues from the physical examSecondary hemostatic defects
Hemophiliac joint bleed Deep muscle bleed
r/o compartment syndrome
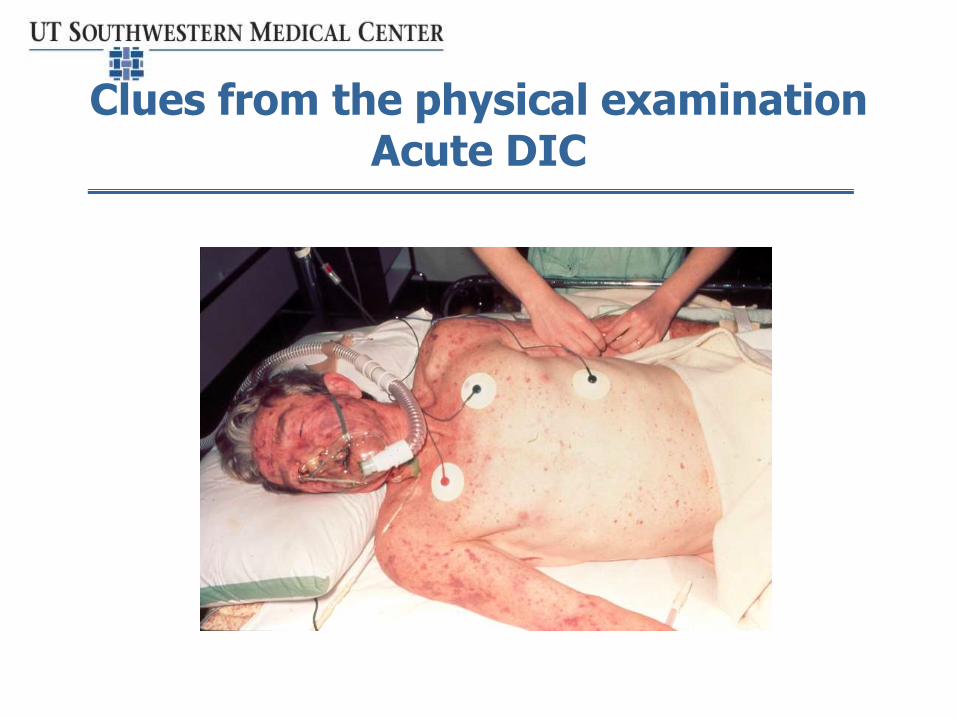
Clues from the physical examinationAcute DIC
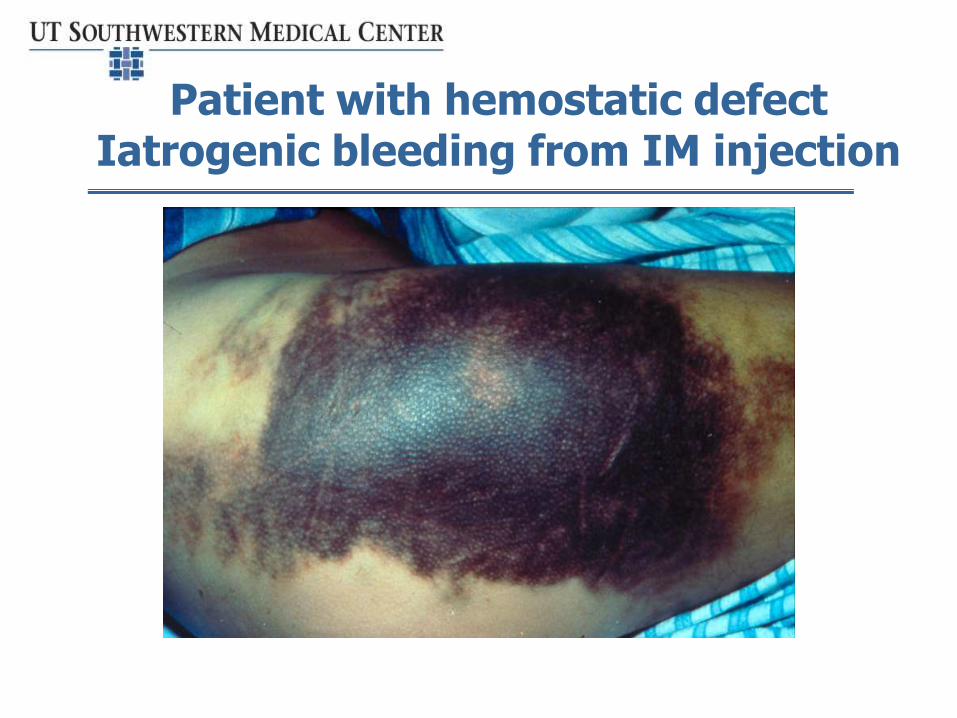
Patient with hemostatic defectIatrogenic bleeding from IM injection

Laboratory evaluation of the bleeding patient
• Hemoglobin/hematocrit
– to assess effect of bleeding
• Platelet count
– bruising below 50K
– bleeding below 10-20K
• Platelet function – PFA-100
– Replaces “bleeding time”
• Basic tests of coagulation – PT, PTT
– Use these to guide specific factor testing

PFA-100 testing
Platelet Function Analyzer
Tests 1º hemostasis with collagen-ADP and collagen-epinephrine agonists
Very sensitive (90%) for - vWD diagnosis - platelet dysfunction
Useful in monitoring DDAVP therapy
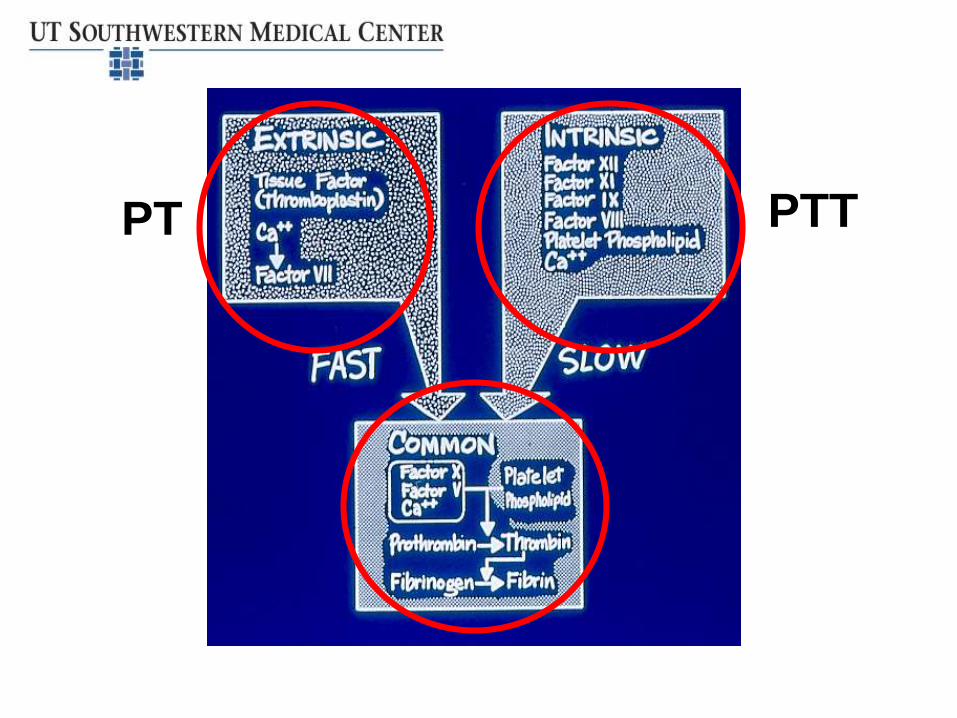
PT PTT

Common causes of coagulopathy• Advanced liver disease – especially low factor VII
– liver makes all coagulation factors, except factor VIII
• Anticoagulants
– Warfarin inhibits factor II, VII, IX and X
– “new” oral anticoagulants – anti-Xa and anti thrombin inhibitors
• DIC
• Von Willebrand disease
• Hemophilia
• Other inherited factor deficiencies – afibrinogenemia, factor VII deficiency, etc.
• Coagulation inhibitors

Treating the common causes of coagulopathy
• Advanced liver disease – especially low factor VII Treat only with bleeding
• Disseminated intravascular coagulation (DIC) – Treat cause– Maintain circulation– Transfuse platelets if platelets <50K and patient is
bleeding, aiming to maintain platelets 50-75K– Transfuse cryoprecipitate to keep fibrinogen
>50mg/dL
Note: Plasma infusion not often needed as other coagulation factors not as depleted as fibrinogen
•

Cryoprecipitate
• Contains only
– Fibrinogen
– VIII and vWD factor
– Factor XIII
• Cryoprecipitate packs pooled from 5 donors
• Short shelf life after thawing – 4 hours

Von Willebrand disease
• Autosomal dominant inherited condition
• Reduced or dysfunctional von Willebrand factor affects:
– Platelet adhesion and aggregation
– Initiation of secondary hemostasis
– Transport of factor VIII
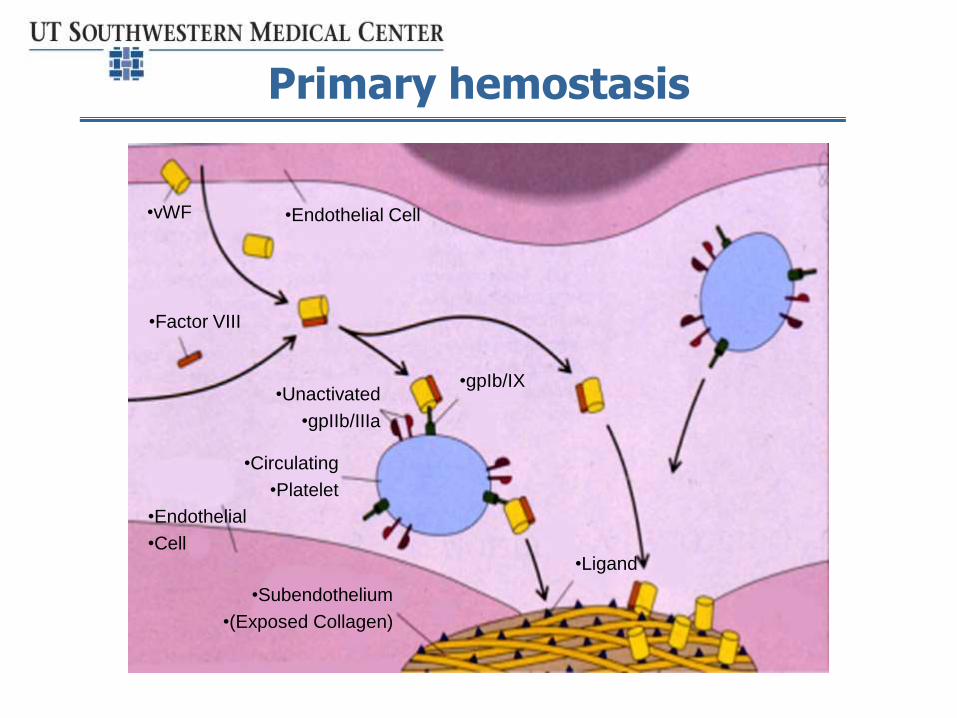
Primary hemostasis
•vWF
•Factor VIII
•Endothelial Cell
•Endothelial
•Cell
•Unactivated
•gpIIb/IIIa
•Circulating
•Platelet
•Subendothelium
•(Exposed Collagen)
•gpIb/IX
•Ligand

Primary hemostasisAggregation of activated platelets
•Fibrinogen
•Activated
Platelet
•Activated
gpIIb/IIIa

Von Willebrand’s original family

Classification of von Willebrand’s Disease
Type 1 Quantitative deficiency of vWF 75-80%
Autosomal dominant
Type 2 Qualitative deficiency of vWF 15-20%
Autosomal dominant
Type 3 Absence of vWF 1:1,000,000Autosomal recessive

Laboratory Diagnosis of von Willebrand Disease
• General screening tests
– PFA – 100 (replaces bleeding time)
– Activated partial thromboplastin time
• Tests specific to vWD
– Factor VIII coagulant function
– vW factor antigen
– vW factor (Ristocetin co-factor) assay
– Ristocetin-induced platelet aggregation
– vW factor Multimer analysis

Type 2 vWD:Qualitative Abnormalities in vWF
Type 2A Impaired secretion or increased proteolysis
Absence of large and intermediate multimers
Type 2B Increased affinity for gpIb
Absence of large multimers
Type 2M Decreased platelet-dependent function
Normal multimer pattern
Type 2N Decreased affinity for factor VIII
Normal multimers

Patterns of multimers in von Willebrand disease subtypes
•Largest Multimers
•Intermediate
•Multimers
•Small Multimers
•Normal •Type I •Type IIA •Type IIB •Type III

Treatment of von Willebrand Disease
• DDAVP to release vWF from endothelial cells and platelets
• Replacement of vWF with plasma-derived products - Humate-P, Alphanate or Wilate
• Prevention of fibrinolysis – mucous membrane bleeding only – e-amino caproic acid (Amicar®)
• General measures – estrogens, topical agents

DDAVP in von Willebrand’s Disease
Releases stored DDAVP from endothelial cells
Increases vWF 2-3 fold – need therapeutic trial
Route Dose Time to Peak
Intravenous 0.3 µg/Kg 30 minutes
Intranasal 300 µg 60 minutes
• Side effects: flushing, headache, hyponatremia, ? myocardial infarction
• Modest tachyphylaxis

Hemophilia
Inherited deficiency or dysfunction of factor:
VIII Hemophilia A 85% (Classic hemophilia)
IX Hemophilia B 15%(Christmas disease)

Types of Hemophilia
# in # inTYPE FACTOR INCIDENCE USA TEXAS
Hemophilia A VIII 1:7000 17,500 1100
(Classical
hemophilia)
Hemophilia B IX 1:30,000 4000 250
(Christmas
disease)

Signs and Symptoms of Hemophilia
• Hemarthroses – joint bleeding
• Muscle hemorrhage
• Soft tissue hematomas
• CNS bleeding
• Uncommon: epistaxis, other mucosal bleeding, prolonged bleeding from cuts, petechiae

Hemophilia Genetics
• X-linked recessive inheritance
• Disease almost exclusively in males
• High rate of spontaneous mutation
– one third new cases have negative
family history
• Carrier testing & pre-natal diagnosis available
(RFLP)

Degrees of Severity of Hemophilia
Factor level Frequency and Severity Type
(Normal 50-150%) of bleeding of bleeding
Severe <1% 30-50 times per year Spontaneous
Moderate 1-5% 3-10 times per year Occ spontaneous,
usually post trauma
Mild 5-30% <1 per year Post-trauma or
surgery only

Signs and Symptoms of Hemophilia
• Soft tissue hematomas
• Hemarthroses
• Muscle hemorrhage
• CNS bleeding
• Uncommon: epistaxis, other mucosal bleeding, prolonged bleeding from cuts, petechiae

Spontaneous Head “Bump”Severe Hemophilia

Hemophilic Arthropathy
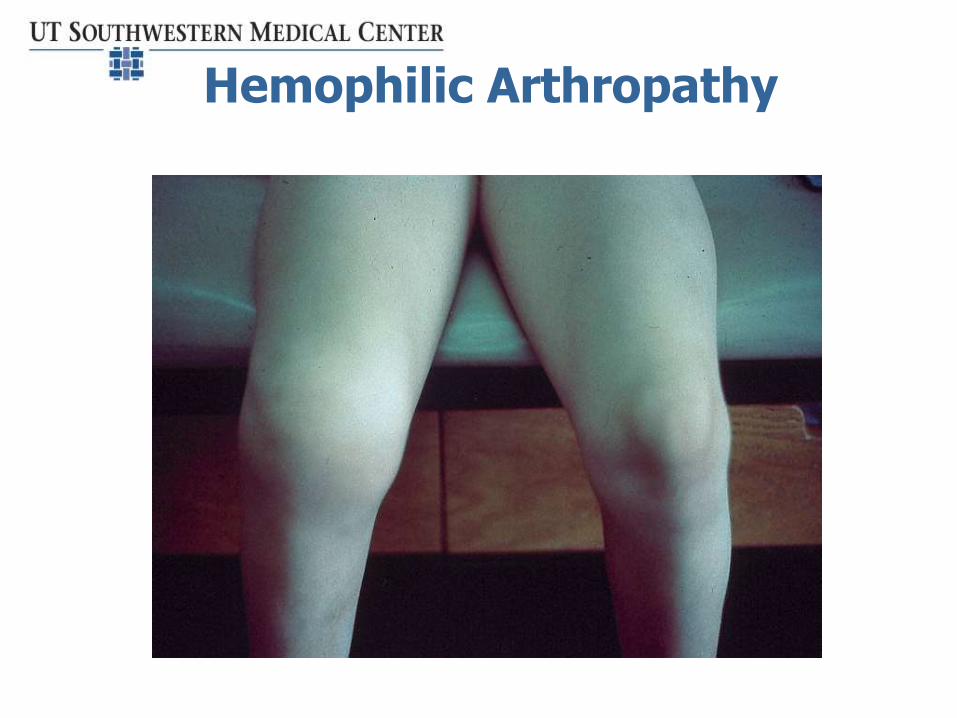
Hemophilic Arthropathy
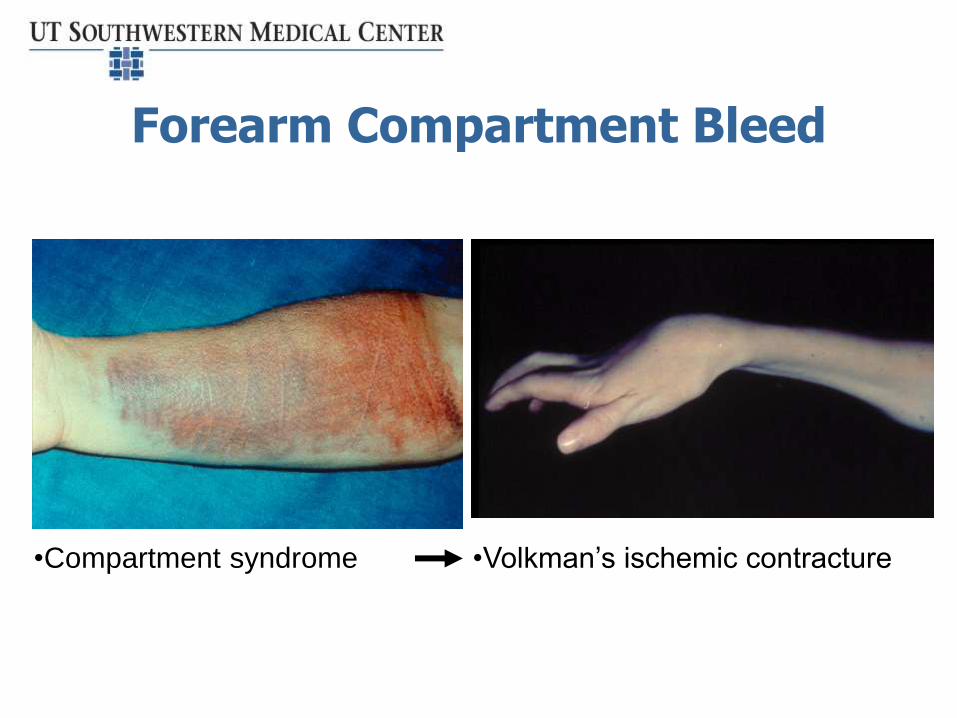
Forearm Compartment Bleed
•Volkman’s ischemic contracture•Compartment syndrome
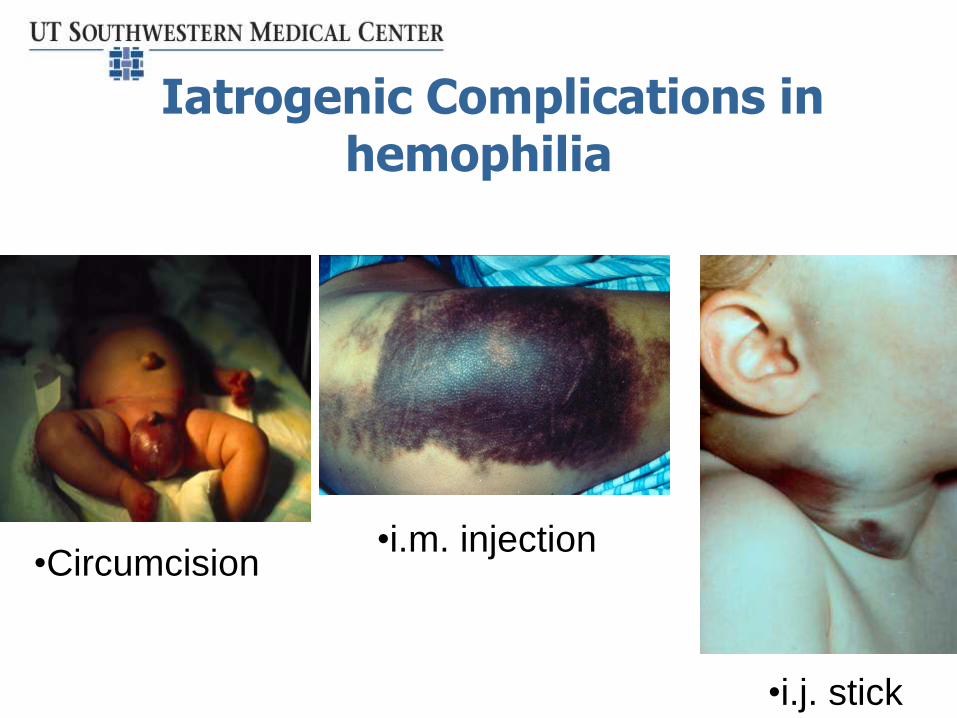
Iatrogenic Complications in hemophilia
•Circumcision•i.m. injection
•i.j. stick

Ilio-Psoas Hemorrhage

Laboratory Diagnosis of Hemophilia
• aPTT Prolonged • Prothrombin time - INR Normal • (Bleeding time) Normal• PFA-100 Normal • Platelet count Normal • von Willebrand factor Normal • Factor VIII One or
the other • Factor IX reduced •}

Treatment in hemophilia
• For mild hemophilia A – always consider DDAVP – will raise factor level 2-3X
• Factor replacement in doses related to the severity of the bleed or procedure

HemophiliaFactor VIII and IX Concentrates
• Heat or solvent-detergent treated– “intermediate” purity factors, e.g. Humate-P
• Purified with– Monoclonal antibodies
– Chromatography
• Recombinant factors
• In 2014 – factors with longer T1/2

Major Emergencies in Hemophilia
• Intra-cranial hemorrhage
• Iliopsoas hemorrhage
• Bleeding around airway
• Uncontrolled external bleeding

Management “pearls” for hemophiliacs in the ER
• Most patients are very knowledgeable about their
disease, its treatment with concentrate and which
veins are best for infusion
• Laboratory testing (PT/PTT, factor assays, CBC, etc.)
or x-rays (e.g, of joints) not routinely necessary
• Following trauma or when any serious illness is
suspected, treat with factor concentrates first and
then do laboratory tests and x-rays

The bleeding patient
• Don’t panic!
• Try to figure the cause from history and physical examination
• Order laboratory tests before blood or other products given
• Ask for help – there’s a lot of it around!

Questions?


















