
Hemolytic anemia ( immun/ nonimmun) and Hemorrhagic disease of the newborn Dr Ebru Tuğrul...
66
Hemolytic anemia ( immun/ nonimmun) and Hemorrhagic disease of the newborn Dr Ebru Tuğrul Sarıbeyoğlu
-
Upload
melina-fleming -
Category
Documents
-
view
224 -
download
1
Transcript of Hemolytic anemia ( immun/ nonimmun) and Hemorrhagic disease of the newborn Dr Ebru Tuğrul...
Hemolytic anemia
( immun/ nonimmun) and
Hemorrhagic disease of the
newborn
Dr Ebru Tuğrul Sarıbeyoğlu

Goals:
• Define hemolysis• Understand causes of hemolysis• Clinical presentation of hemolysis• Immune and non immune
mechanisms of hemolysis• Diagnostic tests for immun hemolysis• pathogenesis, clinical course and
treatment of the hemorragic disease of the newborn

Hemolysis
• Premature!!! (normal RBC survival time:110-120 days) destruction of red blood cells (RBCs)
• Anemia results when the rate of destruction exceeds the capacity of the marrow to produce RBCs

Evidence of RBCs break down
• Serum bilirubin level↑• Serum haptoglobulin level ↓• Plasma hemoglobin level ↓• Urinary uroblinogen ↑• Hemoglobinuria• Blood smear: red cell fragments
(schistocytes, spherocytes, target cells)

Evidence of increased erythropoeisis
• Reticulocytosis
• Increased MCV ( as a result of normoblasts and reticulocyts)
• Erythroid hyperplasia of the bone marrow
• Chronic: expansion of marrow space

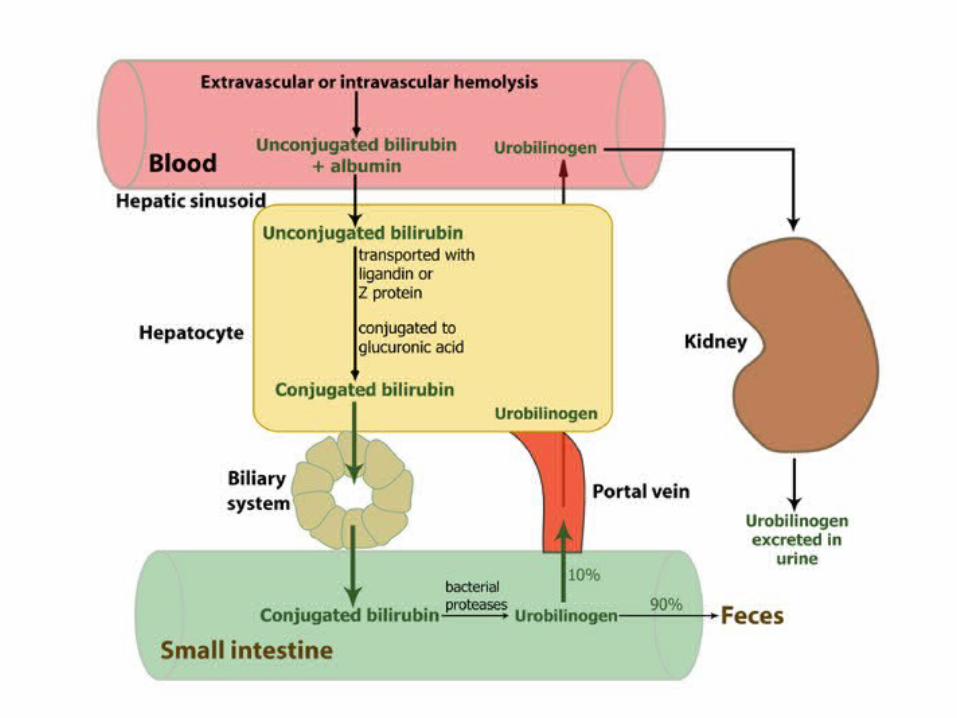
Intra/Extravascular hemolysis
• The site of hemolysis may be intravascular→ the erythrocyte is destroyed in the circulation,
• or extravascular →the red cell destruction occurs within macrophages in the spleen, liver, or bone marrow


Intravascular hemolysis is typically severe and results from mechanical damage to the red cell due to prosthetic valves, the presence of fibrin within the vasculature (microangiopathic hemolytic anemia), or thermal injury to the erythrocytes from serious burns; infections or toxins, such as Clostridium perfringens bacteremia, severe falciparum malaria, or certain snake venoms; or complement-mediated damage to red cells, as with paroxysmal nocturnal hemoglobinuria, ABO-incompatible blood transfusions, and cold agglutinins.

Laboratory Observations
Laboratory abnormalities seen with hemolysis:
•increased RBC destruction,
•increased erythropoiesis by the bone marrow,
•disease-specific findings.
The unconjugated bilirubin is elevated, accounting for more than 80% of the total bilirubin, and is not excreted in the urine.
Unlike in liver disease, in patients with hemolysis pruritus is usually absent.

Intravascular hemolysis liberates hemoglobin into the bloodstream, where it binds to haptoglobin.
The haptoglobin/hemoglobin complex is then removed by the liver. A reduced serum haptoglobin level is one of many findings in intravascular hemolysis, but it also occurs in extravascular hemolysis.
Haptoglobin is an acute-phase reactant and levels increase in response to inflammation, infection, and malignancy.
One needs to be aware of nonhemolytic conditions that can result in low haptoglobin levels (liver disease, hereditary haptoglobin deficiency after red cell transfusions), and normalized haptoglobin levels despite hemolysis (acute phase surges) during work-up of such patients.

When the amount of free hemoglobin in the circulation exceeds the binding capacity of haptoglobin, it makes the plasma pink and is filtered through the kidneys.
The urine may become red, and urine iron levels increase. The urine proves positive for blood upon dipstick testing in the absence of erythrocytes on urine microscopy (not hematuria!!!!).
Other than hemolysis, only in hemochromatosis and nephrotic syndrome can one detect increased urinary iron levels.
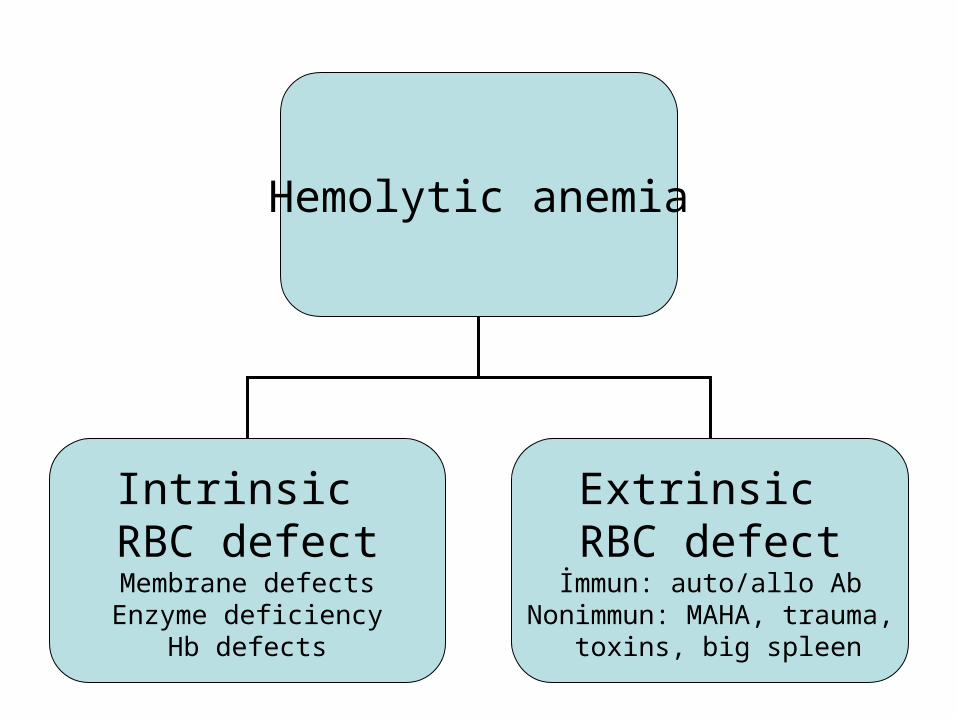
Hemolytic anemia
Intrinsic RBC defectMembrane defectsEnzyme deficiency
Hb defects
Extrinsic RBC defectİmmun: auto/allo Ab
Nonimmun: MAHA, trauma, toxins, big spleen


RBC Membrane Disorders
• Hereditary Spherocytosis• Hereditary Elliptocytosis• Acanthocytosis usually seen with chronic liver disease, and is the
result of cholesterol accumulation in the red cell membrane.
• RBC Metabolic Disorders: Pyruvate Kinase Deficiency, G6PD Deficiency
• Wilson’s Diseasecopper overload. • Congenital Hemoglobinopathies: Sickle Cell
Anemia, Hemoglobin C Disease, Hemoglobin SC Disease, Hemoglobin E Disease, Thalassemias

Another classification of hemolytic anemias distinguishes between disorders intrinsic to the red cell, generally hereditary, and those extrinsic to the red cell, generally acquired. The intrinsic disorders: abnormal hemoglobins( HbS or HbC), enzyme defects( deficiencies in G6PD), membrane abnormalities (hereditary spherocytosis or elliptocytosis). The extrinsic abnormalities: immunologic—alloantibodies (ABO incompatibility), autoantibodies (warm (IgG) or cold (IgM) antibody hemolytic anemias), drug-induced antibodies, mechanical factors ( trauma from prosthetic valves or fibrin deposition in small vessels, as in microangiopathic hemolytic anemias), infections and toxins( falciparum malaria or certain snake venoms),

Most hemolytic disorders are extravascular. The causes of extravascular hemolysis include
•infections, •drugs, •immunologic processes •red cell membrane defects, such as hereditary spherocytosis;• erythrocyte metabolic defects, such as deficiencies in pyruvate kinase or G6PD; •hemoglobin structural defects, such as sickle cell anemia or hemoglobin C.

Most of the disorders that lead to hemolysis are not specific to any race.
It can occur in persons of any age.
Most cases are not sex specific.
Autoimmune hemolytic anemia is slightly more likely to occur in females than in males.
G6PD deficiency is an X-linked recessive disorder. Males are usually affected, and females are carriers.Hereditary disorders are usually evident early in life.

Chronic Congenital Hemolytic Anemias
Even though there are numerous congenital hemolytic disorders, their clinical features are very similar.
Chronic congenital hemolytic anemias are usually characterized by anemia, jaundice, periodic crises, splenomegaly, and black pigment gallstones ( family history!!)
Other than during a crisis, symptoms are usually mild to moderate because of compensation by the bone marrow. Chronic symptoms may become severe at times of crisis. (Aplastic crisis with human parvovirus type B19 ,splenic crisis).

Acquired Hemolytic Anemias
If hemolytic anemia develops acutely, as in hemolytic transfusion reaction or G6PD deficiency, the symptoms may suggest an acute febrile illness with skeletal pains, headache, malaise, fever, and chills.
Symptoms of shock, renal failure, jaundice, and anemia may be evident in severe cases.


G6PD catalyzes the oxidation of glucose-6-phosphate to 6-phosphogluconate while concomitantly reducing the oxidized form of nicotinamide adenine dinucleotide phosphate (NADP+) to nicotinamide adenine dinucleotide phosphate (NADPH). NADPH, a required cofactor in many biosynthetic reactions, maintains glutathione in its reduced form. Reduced glutathione acts as a scavenger for dangerous oxidative metabolites in the cell. With the help of the enzyme glutathione peroxidase, reduced glutathione also converts harmful hydrogen peroxide to water. Red blood cells rely heavily upon G6PD activity because it is the only source of NADPH that protects the cells against oxidative stresses; therefore, people deficient in G6PD are not prescribed oxidative drugs, because their red blood cells undergo rapid hemolysis under this stress

Erythrocytes manufacture ATP through glycolysis. A deficiency in pyruvate kinase,which potentiates the last step of glycolysis (phosphoenolpyruvate converted to pyruvate), results in RBCs with decreased energy.The events leading to hemolysis are still not well understood, but it seems that the lack of ATP impairs the Na+/K+-ATPase and other ATP dependent processes leading to a cellular loss of K+ and water. This causes rigidity of the RBC and eventual splenic hemolysis

Clinical presentation depends on whether the onset of hemolysis is gradual or abrupt and on the severity of erythrocyte destruction. A patient with mild hemolysis may be asymptomatic. In more serious cases, the anemia can be life threatening, The clinical presentation also reflects the underlying cause for hemolysis.

A patient with mild hemolysis may have normal hemoglobin levels if increased production matches the rate of erythrocyte destruction.
Skull and skeletal deformities can occur with a marked increase in hematopoiesis, expansion of bone in infancy, and early childhood disorders such as sickle cell anemia or thalassemia.

Blood smear is the single most valuable test in defining the underlying disorder causing hemolysis.
Spherocytes are the hallmark of hereditary spherocytosis, sickle cells of sickle cell anemia, target cells of thalassemia, schistocytes of RBC fragmentation, erythrophagocytosis of red cell surface damage by complement-fixing antibodies and infections, auto agglutination of cold agglutinin disease, and elliptocytes of hereditary elliptocytosis.
The bone marrow usually shows erythroid hyperplasia.

IMMUNE HEMOLYSİS
• Autoimmune hemolytic anemia (AIHA) is caused by autoantibodies that react with RBCs at temperatures ≥ 37° C (warm antibody hemolytic anemia) or < 37° C (cold agglutinin disease).
• Hemolysis is usually extravascular.

AIHA is diagnosed by detection of autoantibodies with the direct antiglobulin (direct Coombs') test.
Antiglobulin serum is added to washed RBCs from the patient; agglutination indicates the presence of immunoglobulin or complement bound to the RBCs.
Generally IgG is present in warm antibody hemolytic anemia, and C3 (C3b and C3d) in cold antibody disease.
The test is ≤ 98% sensitive for AIHA; false-negative results can occur if antibody density is very low or if the autoantibodies are IgA or IgM.
In general, the intensity of the direct antiglobulin test correlates with the number of molecules of IgG or C3 bound to the RBC and, roughly, with the rate of hemolysis.

Coombs’ test The Coombs' test looks for antibodies that may bind to your red blood cells and cause hemolysis.
There are two forms of the Coombs' test: direct and indirect.

The direct Coombs' test is used to detect antibodies that are already bound to the surface of red blood cells. Many diseases and drugs (quinidine, methyldopa, and procainamide) can lead to production of these antibodies. These antibodies sometimes destroy red blood cells and cause anemia. The direct antiglobulin test detects the presence of antibodies and complement on erythrocytes by using a reagent that contains antibodies directed against human immunoglobulin and complement components (primarily C3). This test is nearly always positive in warm AHA, but when IgG is present in very small quantities, other diagnostic techniques may be necessary to detect them.


A positive direct Coombs' test means you have antibodies that act against your red blood cells. This may be due to:Autoimmune hemolytic anemia without another cause Chronic lymphocytic leukemia or other lymphoproliferative disorder Drug-induced hemolytic anemia (many drugs have been associated with this complication) Erythroblastosis fetalis (hemolytic disease of the newborn) Infections: infectious mononucleosis, Mycoplasmal infection, Syphilis Systemic lupus erythematosus or another rheumatologic condition Transfusion reaction, such as one due to improperly matched units of blood

Autoantibodies unattached to erythrocytes may be present in the serum and are detectable by incubating the patient’s serum or plasma with normal red cells, to which the antibodies then attach. These erythrocytes are then tested for the presence of autoantibodies with the Coombs reagent. This is the indirect antiglobulin or Coombs test.
The indirect Coombs' test looks for unbound circulating antibodies against a series of standardized red blood cells.
The indirect Coombs' test is used to determine whether a person might have a reaction to a blood transfusion.
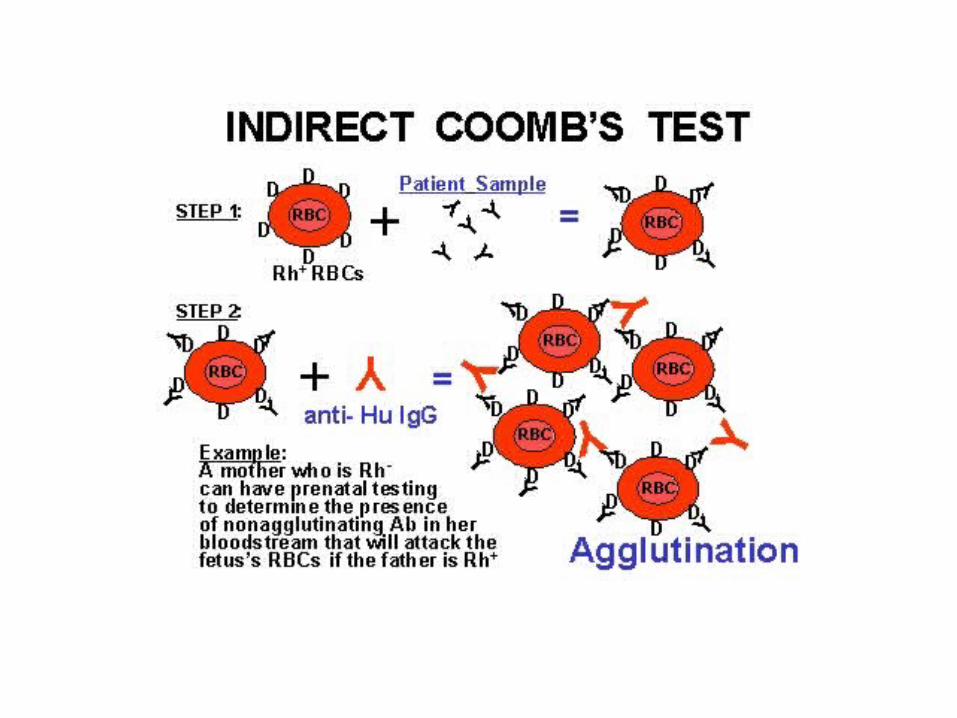

A positive indirect Coombs' test means you have antibodies that will act against red blood cells your body views as foreign. This may suggest:
Autoimmune or drug-induced hemolytic anemia
Erythroblastosis fetalis hemolytic disease
Incompatible blood match (when used in blood banks)

Warm antibody hemolytic anemia: Warm antibody hemolytic anemia is the most common form of AIHA; it is more common among women.
Autoantibodies in warm antibody hemolytic anemia (AHA), generally react at temperatures ≥ 37° C.
In AHA, RBC life span decreases because autoantibodies (usually polyclonal IgG) are optimally active against erythrocytes at body temperature.
These warm antibodies account for about 80% to 90% of acquired AHA, and only about 10% are caused by antibodies maximally active at lower temperatures (cold-reactive autoantibodies).

Immune HemolysisWarm-Antibody Acquired AHA
In warm antibody AHA, hemolysis occurs primarily in the spleen. It is often severe and can be fatal. Most of the autoantibodies in warm antibody hemolytic anemia are IgG.
In about one-half of cases, an underlying disorder is present—most commonly a lymphoproliferative disease such as chronic lymphocytic leukemia or lymphoma, but also systemic lupus erythematosus, other inflammatory conditions, some infections (cytomegalovirus), and nonlymphoid malignancies. Certain drugs can also play a role in the development of these warm antibodies and hemolysis: levodopa (Dopar, Larodopa) and penicillin can cause IgG, and quinidine can cause IgM-type antibodies and AHA.

In warm AHA, IgG coats many red cells with or without complement.
Macrophages in the spleen and Kupffer cells in the liver trap these erythrocytes, sometimes ingesting them whole.
The reticulocyte count is usually increased, as are the serum indirect bilirubin and LDH levels.

Cold Agglutinin DiseaseCold agglutinins are IgM antibodies that bind red cells at cold temperatures. Nearly all healthy people have low titers of clinically insignificant cold agglutinins. Cold AHA can be idiopathic or secondary (infection, lymphoma).
EBV and Mycoplasma pneumoniae are the two most common causes; Hemolysis is usually intravascular and in the liver. Agglutination is seen when blood is cooled. In certain infections, transient, high titers of cold agglutinins appear, causing the abrupt onset of an anemia that is short-lived, but occasionally severe.
Cold agglutinin disease is mediated by a complement-fixing monoclonal IgM antibody either in an acute (mycoplasma or Epstein-Barr virus infection) or chronic setting (lymphoproliferative disorders).

Cold Agglutinin DiseaseIn cold agglutinin disease, RBCs clump on the peripheral smear, and İn addition to anemia, the automated blood count often reveals an increased mean corpuscular hemoglobin concentration (MCV), reflecting the presence of the spherocytes. and spuriously low Hb due to such clumping; hand warming of the tube and recounting result in values significantly closer to normal.
Warm antibody hemolytic anemia can often be differentiated from cold agglutinin disease by the temperature at which the direct antiglobulin test is positive; a test that is positive at temperatures ≥ 37° C indicates warm antibody hemolytic anemia, whereas a test that is positive at lower temperatures indicates cold agglutinin disease.


Hemorrhagic Disease of Newborn
• Vitamin K is an essential cofactor for γ -glutamyl carboxylase enzymatic activity that catalyses the γ -carboxylation of specific glutamic acid residues in a subclass of proteins.3 These vitamin K–dependent proteins are known as Gla-proteins. Media file 1 outlines the vitamin K cycle.

Hemorrhagic Disease of Newborn
• Coagulation factors II, VII, IX, and X and other Gla-proteins (eg, protein C, protein S, protein Z) also depend on the presence of vitamin K for their activity.
• In vitamin K deficiency, des-carboxylated proteins are formed that are functionally defective because they can not bind calcium and phospholipid. These abnormal coagulation factors are called protein-induced by vitamin K absence (PIVKA). PIVKA-II is des-carboxylated prothrombin.

Currently, the following 3 forms of vitamin K are known:
K1: Phylloquinone is predominantly found in green leafy vegetables, vegetable oils, and dairy products. Vitamin K given to neonates as a prophylactic agent is an aqueous, colloidal solution of vitamin K1.
K2: Menaquinone is synthesized by gut flora.
K3: Menadione is a synthetic, water soluble form that is no longer used medically because of its ability to produce hemolytic anemia.

Vitamin K supplementation given after the birth for early onset vitamin K deficiency bleeding may be too late to prevent this disease, especially if vitamin K supplementation was not provided during pregnancy. Numerous maternal medications and/or exposure to toxins during pregnancy are associated with vitamin K deficiency bleeding in neonates (eg, anticonvulsants [eg, phenytoin, barbiturates, carbamazepine], antitubercular drugs [eg, rifampin, isoniazid], vitamin K antagonists [eg, warfarin, phenprocoumon]).

Hemorrhagic Disease of Newborn• Newborn infants are at risk of developing vitamin K
deficiency, and this coagulation abnormality leads to serious bleeding.
• Placental transfer of vitamin K is very limited, and phylloquinone (vitamin K1) levels in umbilical cord blood is very low.
• the storage of vitamin K in neonatal liver is also limited.
• This makes the newborn infant uniquely vulnerable to hemorrhagic disorders unless exogenous vitamin K is given for prevention of bleeding immediately after birth.Once the infantile gut is colonized with bacterial flora, the microbial production of vitamin K results in a lower risk of infantile vitamin K deficiency bleeding.
• A gut-related microbial source of vitamin K is particularly important if dietary phylloquinone is restricted.

Hemorrhagic Disease of Newborn
• The newborn infant’s intestinal tract is relatively sterile and takes some time to colonize with bacteria, which may have a role in synthesizing vitamin K2 (menaquinones).
• Because Bacteroides species are among the most common bacteria that inhabit the human intestinal tract, and because strains such as Bacteroides fragilis synthesize vitamin K, Bacteroides species are more significant in producing human vitamin K in the intestine than Escherichia coli.

Hemorrhagic Disease of Newborn
• Breast milk is a poor source of vitamin K (breast milk levels are 1-4 μ g/L). The recommended dietary intake of vitamin K is 1 μ g/kg/d.
• Breastfed infants have intestinal colonization with lactobacilli that do not synthesize vitamin K; thus, reduced production of menaquinones increases the neonatal risk of developing a hemorrhagic disorder if not supplemented with vitamin K.
• Formula-fed infants have higher fecal concentrations of vitamin K1 because of dietary intake and significant quantities of fecal menaquinones, reflecting the gut’s microflora.
Preterm infants who are receiving total parenteral nutrition (TPN) are not at risk because they are receiving vitamin K via the multivitamin additive to the TPN.

Hemorrhagic Disease of Newborn
vitamin K deficiency bleeding is usually classified by 3 distinct time periods after birth,
• Early-onset vitamin K deficiency bleeding in the newborn
• Classic vitamin K deficiency bleeding in the newborn
• Late-onset vitamin K deficiency bleeding in the newborn

Early-onset vitamin K deficiency bleeding usually occurs during first 24 hours after birth.
It is seen in infants born to mothers taking anticonvulsant or antituberculosis medication.
Serious hemorrhagic complications can occur in this type of hemorrhage.
The mechanisms by which anticonvulsant and antituberculosis medications cause vitamin K deficiency bleeding in neonates is not clearly understood, but limited studies suggest that vitamin K deficiency bleeding is a result of vitamin K deficiency and can be prevented by administration of vitamin K to the mother during the last 2-4 weeks of pregnancy.

Classic vitamin K deficiency bleeding usually occurs after 24 hours and as late as the first week
of life.
Classic VKDB is observed in infants who have not received prophylactic vitamin K at birth.
The incidence of classic vitamin K deficiency bleeding ranges from 0.25-1.7 cases per 100 births.
Usually the disease occurs from the second day of life to the end of the first week; however, it can occur during first month and sometimes overlaps with late-onset vitamin K deficiency bleeding.

Infants who have classic vitamin K deficiency bleeding are often ill, have delayed feeding, or both.
Bleeding commonly occurs in the umbilicus, GI tract (ie, melena), skin, nose, surgical sites (ie, circumcision), and, uncommonly, in the brain.

Late-onset VKDB This usually occurs between age 2-12 weeks; however, late-onset vitamin K deficiency bleeding can be seen as long as 6 months after birth.
This disease is most common in breastfed infants who did not receive vitamin K prophylaxis at birth.
Industrial contaminants in breast milk have been implicated in promoting vitamin K deficiency bleeding.
More than half of these infants present with acute intracranial hemorrhages.

Hemorrhagic Disease of Newborn
• Frequency• United States• In the United States, routine intramuscular
administration of vitamin K immediately after birth has made vitamin K deficiency bleeding an uncommon occurrence. The frequency of vitamin K deficiency bleeding varies from 0.25-1.7% in the first week of life in infants not receiving vitamin K prophylaxis. Late vitamin K deficiency bleeding appears to be reduced or prevented with I.M. administration of vitamin K at birth.
• Late vitamin K deficiency bleeding has fallen from 4.4-7.2 cases per 100,000 births to 1.4-6.4 cases per 100,000 births in reports from Asia and Europe after regimens for prophylaxis were instituted.

Hemorrhagic Disease of Newborn
• The most common sites of hemorrhage or bleeding are the umbilicus, mucus membrane, the GI tract, circumcision, and venipuncture sites.
• Hematomas frequently occur at the sites of trauma (large cephalohematomas, scalp bruising related to instrumentation used at delivery, and, rarely, intracranial hemorrhage).

Hemorrhagic Disease of Newborn
• Intracranial hemorrhage is uncommon in classic vitamin K deficiency bleeding but can be observed in more than 50% of infants with late-onset vitamin K deficiency bleeding. Intracranial hemorrhage is responsible for nearly all mortality and long-term sequelae due to vitamin K deficiency bleeding.
• No racial predilection is noted, but breastfeeding practices can result in apparent racial disparities.
• No predilection to vitamin K deficiency bleeding based on gender is apparent.
• Neonatal mortality and long-term neurologic morbidity are severe consequences of vitamin K deficiency bleeding.

Hemorrhagic Disease of Newborn
• History• The maternal history is very important
when assessing vitamin K deficiency bleeding (VKDB), especially the medications used during pregnancy, the presence of medical conditions such as short gut syndrome, and unusual dietary intakes.
• Better surveillance during pregnancy and careful medical evaluation of neonate after delivery are essential.

Hemorrhagic Disease of Newborn
• Physical• Most newborn infants are healthy upon examination,
even if early onset bleeding is present; however, intracranial hemorrhage can occur during the birthing process and can lead to severe complications.
• Signs of intracranial hemorrhage include apnea with or without seizures and a shocklike syndrome.
• Internal hemorrhage of organs other than the brain may be difficult to detect; however, if they are suspected, careful physical monitoring and serial imaging after birth are indicated.
• Soft tissue hemorrhage is easier to recognize, but sequential measurements of the bleeding into soft tissues or muscle are mandatory.

Hemorrhagic Disease of Newborn
• Causes• Vitamin K deficiency in the newborn can be present for
various reasons• Maternal medications that interfere with vitamin K stores or
function (eg, carbamazepine, phenytoin, barbiturates, some cephalosporins, rifampin, isoniazid, warfarin or warfarinlike drugs) can result in vitamin K deficiency bleeding in the infant.
• In addition to breastfeeding, clinical states that are risk factors for late-onset vitamin K deficiency bleeding include: – Diarrhea – Hepatitis – Cystic fibrosis – Celiac disease – Alpha1-antitrypin deficiency – Short bowel syndrome – Intestinal bacterial overgrowth – Chronic exposure to broad spectrum antimicrobials

Hemorrhagic Disease of Newborn• Laboratory Studies• A prothrombin time (PT), activated partial thromboplastin
time (aPTT), fibrinogen levels, and a platelet count should be included in the initial workup for vitamin K deficiency bleeding (VKDB) in a newborn. – A prolonged PT is usually the first laboratory test result to
be abnormal in VKDB however, no laboratory test result can confirm the diagnosis of vitamin K deficiency bleeding.
– A direct blood measurement of vitamin K is not useful because levels normally are low in newborns.
– levels of protein induced by vitamin K antagonism (PIVKA II) are increased in vitamin K deficiency bleeding, but this test is generally not available outside of research laboratories.
– Infants with vitamin K deficiency bleeding typically have a prolonged PT with platelet counts and fibrinogen levels within the normal range for newborns. Thrombocytopenia or a prolonged aPTT should prompt workup for other causes of bleeding during the neonatal period.
• The diagnosis of vitamin K deficiency bleeding is confirmed if administration of vitamin K halts the bleeding and reduces the PT value.

Hemorrhagic Disease of Newborn
• Medical Care• Prevention of VKDB with intramuscular vitamin K is of
primary importance in the medical care of neonates. • A single dose of intramuscular vitamin K after birth
effectively prevents classic vitamin K deficiency bleeding. • Conversely, oral vitamin K prophylaxis improves
coagulation test results at 1-7 days, but vitamin K administered by this route has not been tested in randomized trials for its efficacy in preventing either classic or late vitamin K deficiency bleeding.
The American Academy of Pediatrics in their policy statements has endorsed the universal supplementation of vitamin K using the intramuscular injection (IM) because no vitamin K preparation is licensed for oral use in the United States.

Hemorrhagic Disease of Newborn
• Immediately administer vitamin K subcutaneously (hold pressure on the site) for any infant in whom vitamin K deficiency bleeding is suspected or who has serious, unexplained neonatal bleeding.
• IM administration can result in a hematoma because of the coagulopathy.
• Intravenous (IV) administration of vitamin K has been associated with anaphylactoidlike reactions.
• Fresh frozen plasma may be considered for moderate-to-severe bleeding.
• Life-threatening bleeding may also be treated with prothrombin complex concentrates (PCC).

Hemorrhagic Disease of Newborn
• Because the bleeding in classic vitamin K deficiency bleeding usually is not life threatening, a single dose of parenteral vitamin K is sufficient to stop the bleeding and return prothrombin time (PT) values to the reference range.
• In the early 1990s, an association between parenteral vitamin K and the later occurrence of childhood cancer was reported; however, a large cohort study and a large retrospective analysis of a database in the United States could not confirm this association. Because this association is weak at best, routine vitamin K prophylaxis is recommended and supported by the American Academy of Pediatrics.
• Oral vitamin K has been studied as an alternative and can improve clotting studies and vitamin K levels, but it has not been studied in large randomized controlled trials to determine if this strategy effectively prevents early and late vitamin K deficiency bleeding.

Take home message
•Administer to all new borns routinly 1 mg
intramuscular vitamin K immediately after birth

Further reading• Schwartz RS. Autoimmune and intravascular
hemolytic anemias. In: Goldman L, Ausiello D, eds. Cecil Medicine. 23rd ed. Philadelphia, Pa: Saunders Elsevier; 2007: chap 164.
• Powers A, Silberstein LE. Autoimmune hemolytic anemia. In: Hoffman R, Benz EJ, Shattil SS, et al, eds. Hematology: Basic Principles and Practice. 5th ed. Philadelphia, Pa: Elsevier Churchill Livingstone;2008:chap 47.
• Schrier SL, Price EA. Extrinsic nonimmune hemolytic anemias. In: Hoffman R, Benz EJ, Shattil SS, et al, eds. Hematology: Basic Principles and Practice. 5th ed. Philadelphia, Pa: Elsevier Churchill Livingstone;2008:chap 48.
• Clarke P, Shearer MJ. Vitamin K deficiency bleeding: the readiness is all. Arch Dis Child. Sep 2007;92(9):741-3.


















