
Hematological & systemic disorders (1)
77
Good Morning Dr.Ram
-
Upload
drram-thiramdas -
Category
Health & Medicine
-
view
61 -
download
4
Transcript of Hematological & systemic disorders (1)

Dr.Ram
Good Morning

Dr.Ram
HEMATOLOGICAL & SYSTEMIC DISORDERS

Dr.Ram
Study of blood and blood forming tissues is called as hematology.
Components of Blood:-
Plasma 55%
Blood Cells 45% Three types
Erythrocytes/RBCs Leukocytes/WBCs Thrombocytes/Platelets

Dr.Ram
RBC OR ERYTHROCYTES
Composed of hemoglobin Erythropoiesis
= RBC production Stimulated by hypoxia Controlled by erythropoietin
Hormone synthesized in kidney
Hemolysis = destruction of RBCs Releases bilirubin into blood stream Normal lifespan of RBC = 120 days

Dr.Ram
DISEASES OF RBCANEMIA:- Anemia is defined as abnormal reduction in the number of circulating
RBC,the quantity of hemoglobin & the volume of packed red cells in a given unit of blood.
THALASEMIA:- Thalasemia is genetically determined disorder of hemoglobin synthesis
with decreased production of either alpha or beta chains of hemoglobin.
POLYCYTHEMIA:- Abnormal increase in the number of RBC in the peripheral blood, with
an increased hemoglobin content

Dr.Ram
CLASSIFICATION OFANEMIA PATHOPHYSIOLOGIC CLASSIFICATION
1)Anemias due to increased blood loss.
Acute post hemorrhagic anemia
Chronic blood loss
2)Due to impaired cell function
a) cytoplasmic maturation defects:
Iron deficiency anemia
Thalassaemic syndromes
b) Nuclear maturation defects:
Vit B12 deficiency-Pernicious anemia
Megaloblastic anemia
c) Defects in stem cell proliferation: Aplastic anemia
Pure red cell aplasia

Dr.Ram
d)Anemia of chronic disorders
e)Bone marrow infiltration
f)Congenital anemia
3)Hemolytic anemias:Extracorpuscular red cell abnormalities
Intracorpuscular red cell abnormalities
MORPHOLOGIC CLASSIFICATION
1)Microcytic hypochromic anemia
2)Normocytic normochromic anemia
3)Macrocytic normochromic anemia

Dr.Ram
PERNICOUS ANEMIA It is common chronic hematologic disorder. Associated with deficiency of vitamin B12,due to lack of
production of intrinsic factor.
C/F:- Seen in individuals above 30 yr. F>M Triad of symptoms:
Generalized weakness
Sore & painful neck
Numbness or tingling of extremities Pt with severe anemia yellowish tinge of skin & sclera is
seen. Degeneration of peripheral nerves is seen.
Dr.Ram
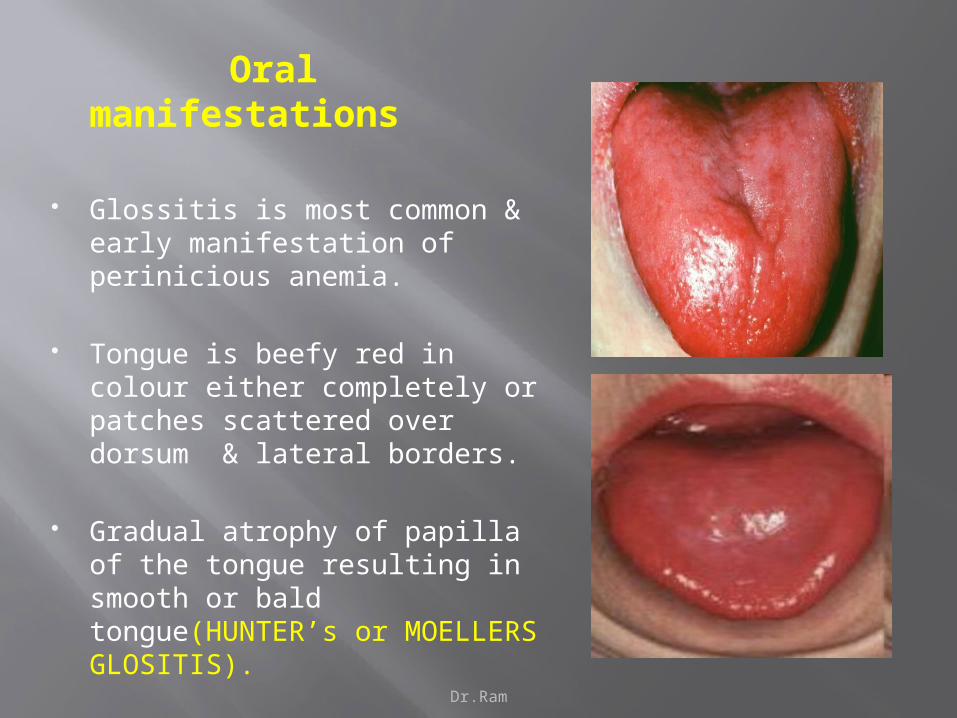
Oral manifestations
Glossitis is most common & early manifestation of perinicious anemia.
Tongue is beefy red in colour either completely or patches scattered over dorsum & lateral borders.
Gradual atrophy of papilla of the tongue resulting in smooth or bald tongue(HUNTER’s or MOELLERS GLOSITIS).
Dr.Ram
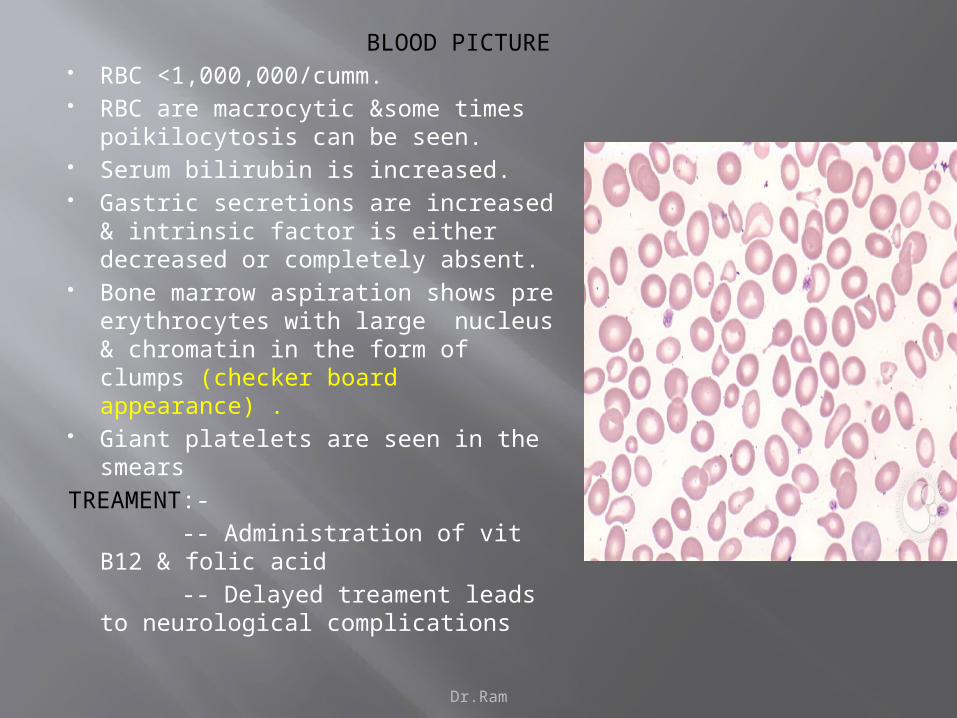
BLOOD PICTURE RBC <1,000,000/cumm. RBC are macrocytic &some times
poikilocytosis can be seen. Serum bilirubin is increased. Gastric secretions are increased &
intrinsic factor is either decreased or completely absent.
Bone marrow aspiration shows pre erythrocytes with large nucleus & chromatin in the form of clumps (checker board appearance) .
Giant platelets are seen in the smears
TREAMENT:-
-- Administration of vit B12 & folic acid
-- Delayed treament leads to neurological complications

Dr.Ram
IRON DEFICIENCY ANEMIACommon type of anemia in females.
Etiology:-
Ch blood loss
Inadequate dietary intake
Faulty iron absorption o Plummer–Vinson syndrome presents as a triad of dysphagia , esophageal
webs, and iron deficiency anemia.o It most usually occurs in postmenopausal women.
C/F:-o Can occur at any age.o F>Mo Angular chelitiso Dysphagia to solid foodo Lemon tint pallor of skino Koilonychiao Splenomegaly
Dr.Ram
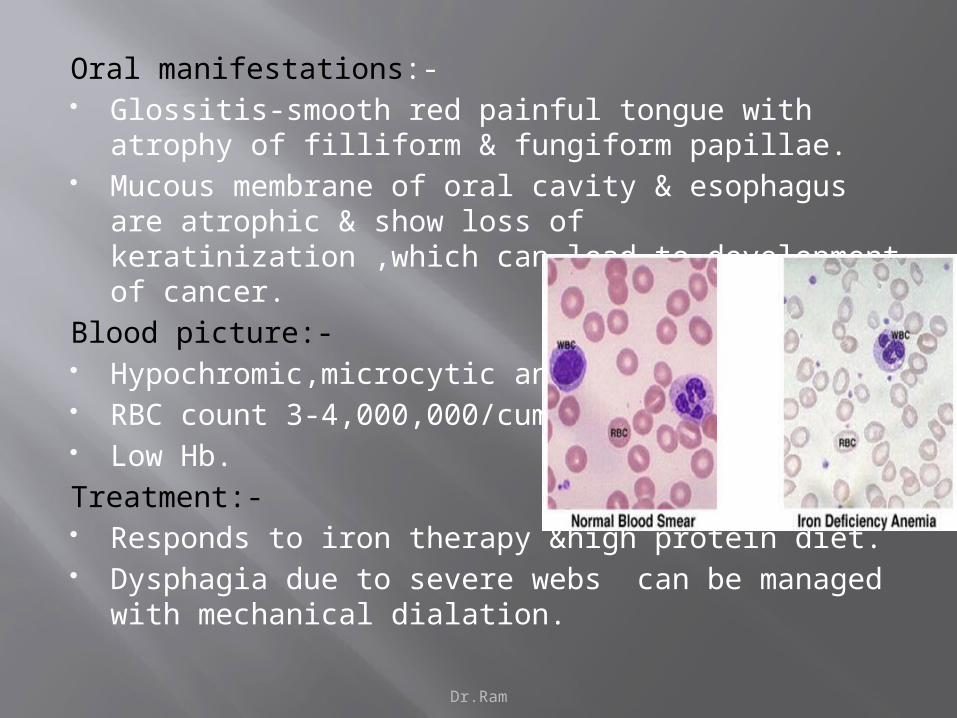
Oral manifestations:- Glossitis-smooth red painful tongue with atrophy of filliform
& fungiform papillae. Mucous membrane of oral cavity & esophagus are atrophic
& show loss of keratinization ,which can lead to development of cancer.
Blood picture:- Hypochromic,microcytic anemia. RBC count 3-4,000,000/cumm. Low Hb.
Treatment:- Responds to iron therapy &high protein diet. Dysphagia due to severe webs can be managed with
mechanical dialation.

Dr.Ram
SICKLE CELL ANEMIA
Sickle cell anemia is autosomal dominant chronic hemolytic anemia.
The name is given due to microscopic appearance of sickle or crescent shaped RBC in the circulating blood.
In this adult hemoglobulin( HbA) is altered by the substitution of glutamine with valine at 6th position of beta globulin chain.
This produces sickle hemoglobin (Hbs) which gives sickle shape to RBC .
Life cycle of sickle-shaped RBC is very short-10-20 days.

Dr.Ram
C/F:-
Common in females Age-below 30yrs Impaired growth Increased susceptibility to infections due to impaired
function of spleen Weak Short of breath Pain in joints limbs & abdomen Nausea& vomiting Systloic murmur & cardiomegaly
Dr.Ram
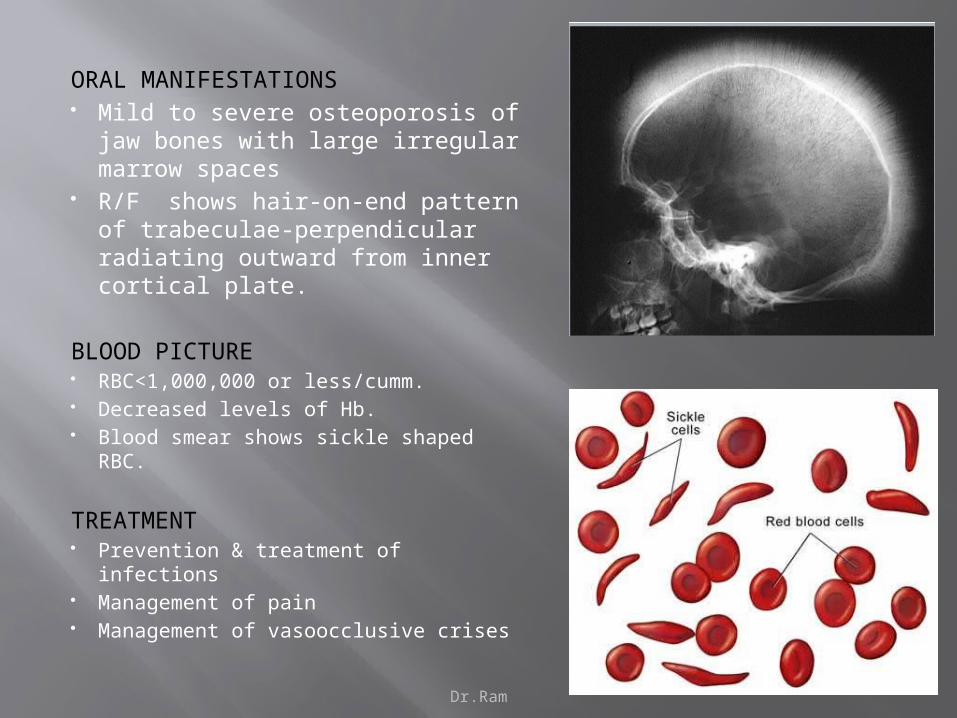
ORAL MANIFESTATIONS Mild to severe osteoporosis of jaw
bones with large irregular marrow spaces
R/F shows hair-on-end pattern of trabeculae-perpendicular radiating outward from inner cortical plate.
BLOOD PICTURE RBC<1,000,000 or less/cumm. Decreased levels of Hb. Blood smear shows sickle shaped RBC.
TREATMENT Prevention & treatment of infections Management of pain Management of vasoocclusive crises

Dr.Ram
APLASTIC ANEMIA Aplastic anemia is a bone marrow failure syndrome
characterized by peripheral pancytopenia & general lack of bone marrow activity.
Two types:-
Primary aplastic anemia
Secondary aplastic anemia
Primary aplastic anemia is of unknown etiology. Effects young adults, develops rapidly & terminates fatally.
FANCONI’S SYNDROME:- Familial or congenital aplastic anemia Bone abnormalities Microcephaly Generalized olive-brown pigmentation of the skin

Dr.Ram
Secondary aplastic anemia:- Known etiology Occurs at any age
Etiology:- Drugs or chemicals X-rays, radium or radio active isotopes Infections
C/F:- Severe weakness even with slight physical exertion Pallor of the skin Numbness & tingling of extremities Patechiae of skin & mucous membrane Decreased resistance to infections

Dr.Ram
Oral manifestations:- Petechiae & hematomas of oral mucosa Spontaneous gingival bleeding Ulcerative lesions of oral mucosa & pharynx
Blood picture:- RBC<1,000,000 or less/cumm Reduced Hb Thrombocytopenia Pancytopenia –hypoplasia of all marrow elements Severe-hypocellular bone marrow with fatty replacement.
Treatment:- Management of infections Bone marrow transplantation Immunosuppressive therapy

Dr.Ram
THALASSEMIA(cooley’s or erythroblastic anemia)
Thalasemia is genetically determined disorder of hemoglobin synthesis with decreased production of either alpha or beta chains of hemoglobin.
HbA=Heme +Globin(2 alpha & 2 beta chains)
Thalasemia -decreased synthesis of either alpha or beta chains.
Alpha thalasemia –Alpha chain is deficient
Beta thalasemia- Beta chain is deficient ,excess alpha chains unstable hemoglobin.

Dr.Ram
Thalasemia minor:-mild ,effects both alpha & beta chains
Thalasemia major:-
Production of beta chains is decreased.
decreased synthesis of hemoglobin(hypochromic)
Due to excess alpha chains inclusion bodies are formed with in
erythocytes (Fessas bodies).
Leads to destruction of primitive red cell & severe ineffective
hemopoisis.
70-80% of normoblasts are destroyed.
Hemoglobin H disease-mild –deficiency of alpha chains.
Hemoglobin bart’ diseases- still born infants
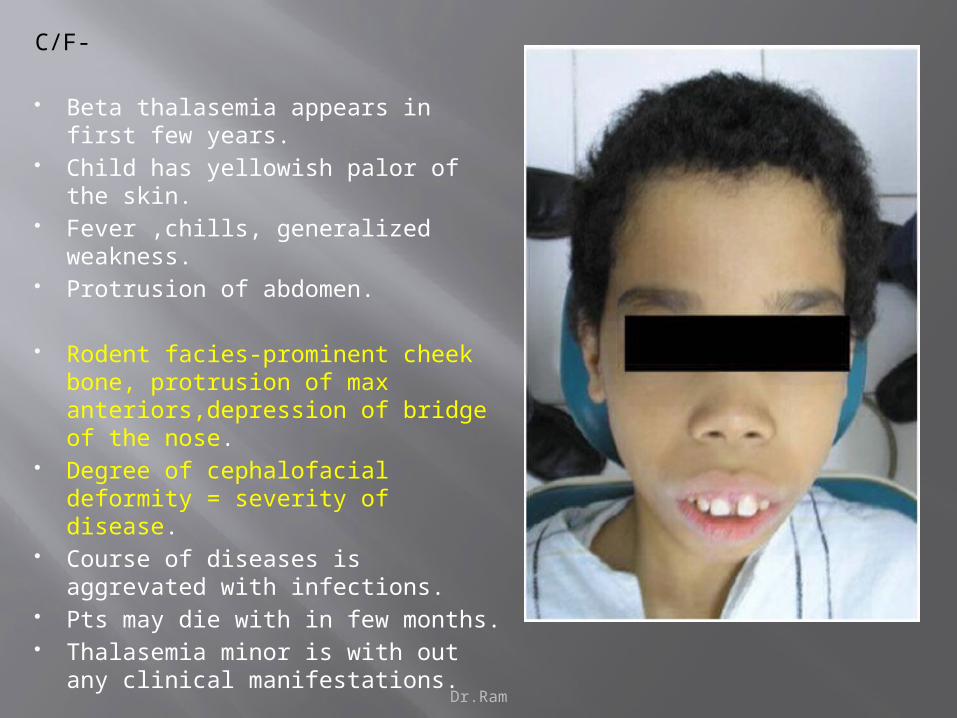
Dr.Ram
C/F-
Beta thalasemia appears in first few years.
Child has yellowish palor of the skin. Fever ,chills, generalized weakness. Protrusion of abdomen.
Rodent facies-prominent cheek bone, protrusion of max anteriors,depression of bridge of the nose.
Degree of cephalofacial deformity = severity of disease.
Course of diseases is aggrevated with infections.
Pts may die with in few months. Thalasemia minor is with out any
clinical manifestations.
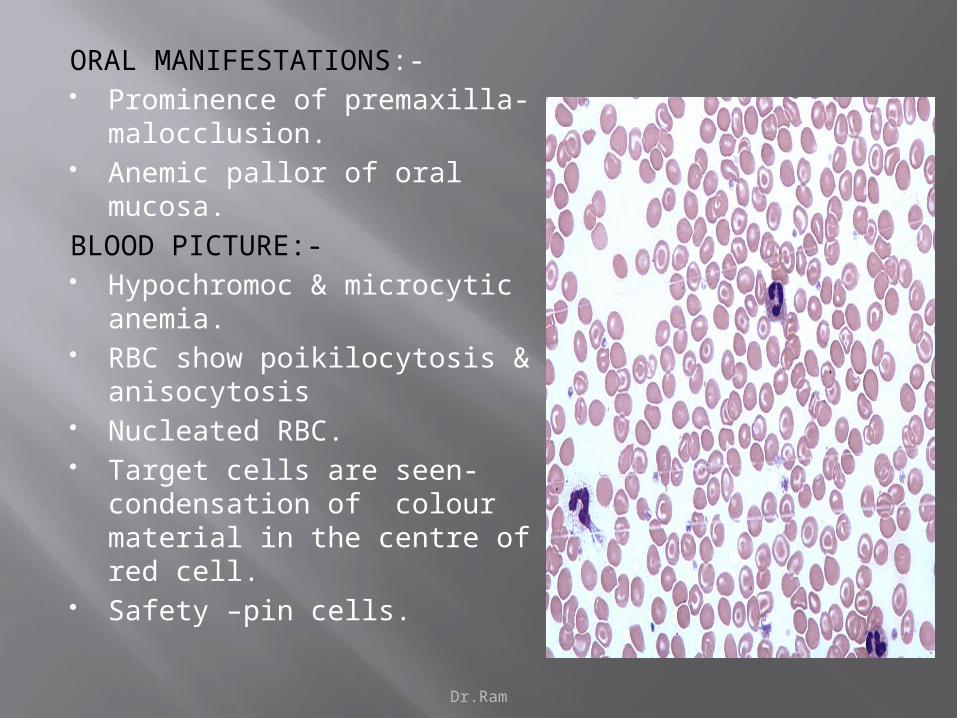
Dr.Ram
ORAL MANIFESTATIONS:- Prominence of premaxilla-
malocclusion. Anemic pallor of oral mucosa.
BLOOD PICTURE:- Hypochromoc & microcytic
anemia. RBC show poikilocytosis &
anisocytosis Nucleated RBC. Target cells are seen-
condensation of colour material in the centre of red cell.
Safety –pin cells.
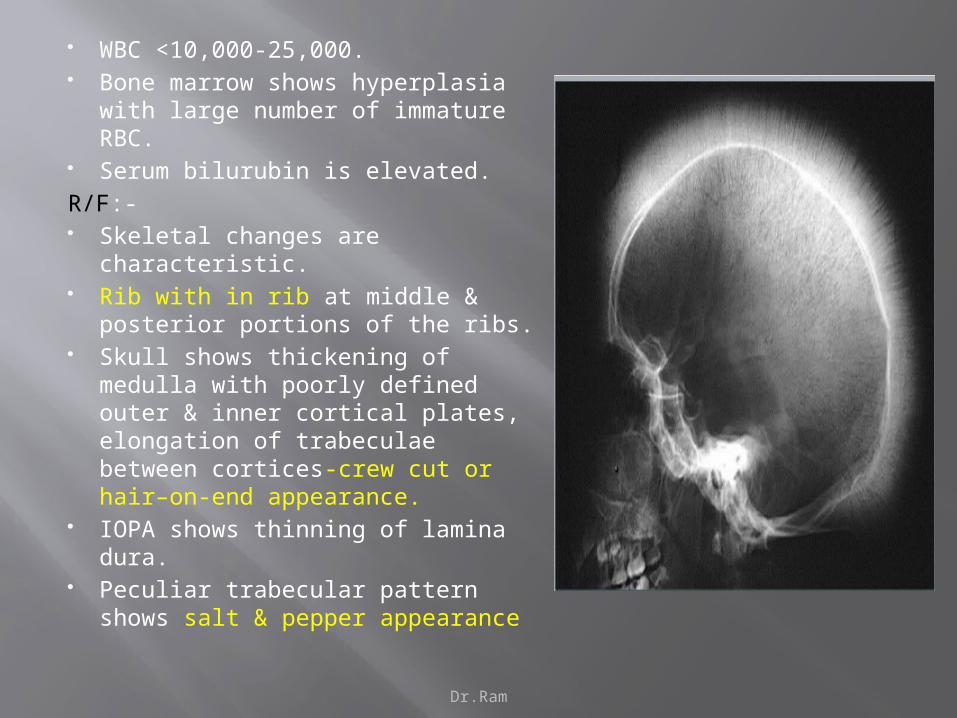
Dr.Ram
WBC <10,000-25,000. Bone marrow shows hyperplasia with
large number of immature RBC. Serum bilurubin is elevated.
R/F:- Skeletal changes are characteristic. Rib with in rib at middle & posterior
portions of the ribs. Skull shows thickening of medulla
with poorly defined outer & inner cortical plates, elongation of trabeculae between cortices-crew cut or hair–on-end appearance.
IOPA shows thinning of lamina dura. Peculiar trabecular pattern shows salt
& pepper appearance

Dr.Ram
.
TRETMENT:-
No treatment. Blood transfusions give temporary remission. Bone marrow trasplantation Disease is fatal & death can be due to infections, cardiac
damage because of anoxia or liver failure.

Dr.Ram
DISEASES OF WBC

Dr.Ram
White blood cells are cells of the immune system involved in defending the body against both infectious disease and foreign materials.
Two types –
Granulocytes:Neutrophills
Basophills
Eosinophills
Agranulocytes:Lymphocytes
Monocytes

Dr.Ram
Diseases of WBC
Leukemia
Agranulocytosis
Neutropenia
Infectious mononucleosis

Dr.Ram
leukemia Leukemia is characterized by progressive production of
WBC,which circulate in blood in immature form. It is a true malignant which is often fatal . Any of WBC can be involved.
CLASSIFICATION
Based on course of disease:
Acute leukemia
sub acute leukemia
Chronic leukemia
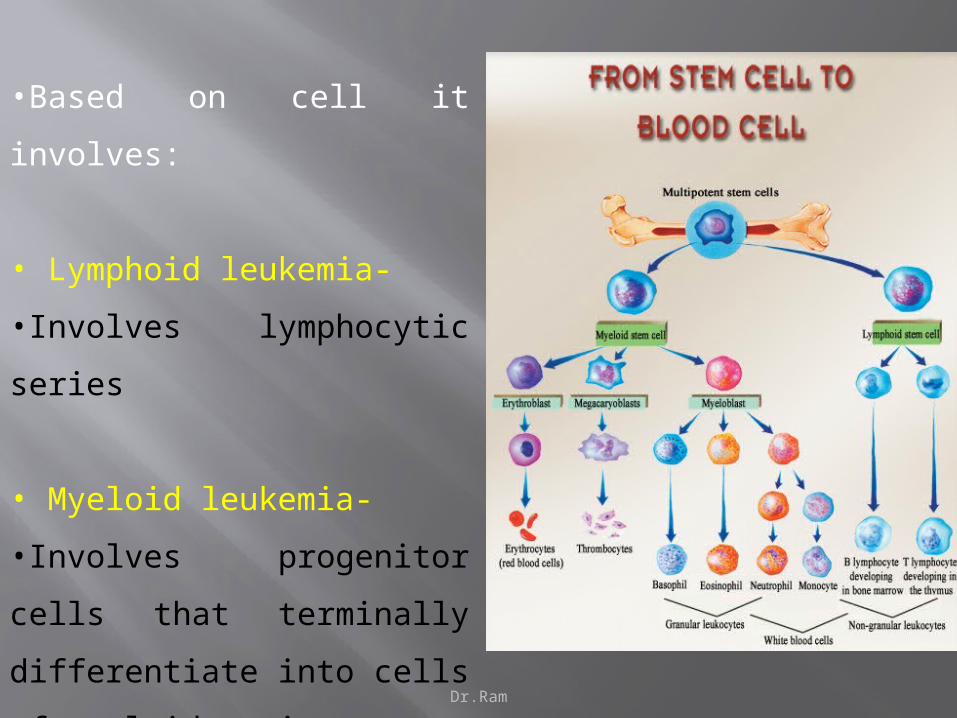
Dr.Ram
•Based on cell it involves:
• Lymphoid leukemia-
•Involves lymphocytic series
• Myeloid leukemia-
•Involves progenitor cells that
terminally differentiate into cells of
myeloid series

Dr.Ram
ETIOLOGY:- Etiology of leukemia is unknown. Can be infectious origin-Ebstein-Bar virus
Human T-cell leukamia virus-1 Ionizing radiation Ch exposure to chemicals like benzol,aniline dyes.
Chromosomal abnormalities- philadelphia chromosome Seen in 85-95% of ch myeloid leukemia Translocation of chromosomal material from chromosome
22 to 9. Downs syndrome is due to trisomy of chromosome 21

Dr.Ram
C/F:- Acute leukemia-children & young adults Ch leukemia-middle age or older age M>F
Acute leukemia:-Acute lymphoblastic leukemia(ALL)
Acute myeloblastic leukemia(AML)
Leukemic cells develop from immature blast cells Weakness,fever, head ache. Swelling of lymph nodes-first sign. Petechial & echymotic hemorrhages in skin & oral mucosa. Splenomegaly& hepatomegaly due to leukemic infiltration. Hemorrhages due to decrease in platelets. Infections due to crowding myeloid tissue.

Dr.Ram
Chronic leukemia:-2 types:CML-20%
CLL
Cells involved are mature cell. Slow before symptomatic. Found on routine hematological examination. Anemic pallor & emaciation. Lymphadenopathy-ch lymphoblastic leukemia. Sphenomegaly & hepatomegaly. Enlargement of salivary glands & tonsils-xerostomia Skin lesions-papules, pustule, bullae, areas of pigmentation,
nodular lesions having leukemic cells. Destructive lesions of bone –ch leukemia-pathologic fracture
& osteomyelitis.

Dr.Ram
ORAL MANIFESTATIONS Common in acute monocytic leukemia. Absent in young & edentulous individuals. Least in acute lymphocytic leukemia. Gingivitis. Gingival hyperplasia-gingiva is edematous, boggy,& deep
red.
Gingiva bleeds easily
In severe teeth are completely hidden. Gingival hemorrhage due to ulceration& necrosis of
underlying tissue. Moblity of teeth due to necrosis of PDL & destruction of Al
bone.
Dr.Ram
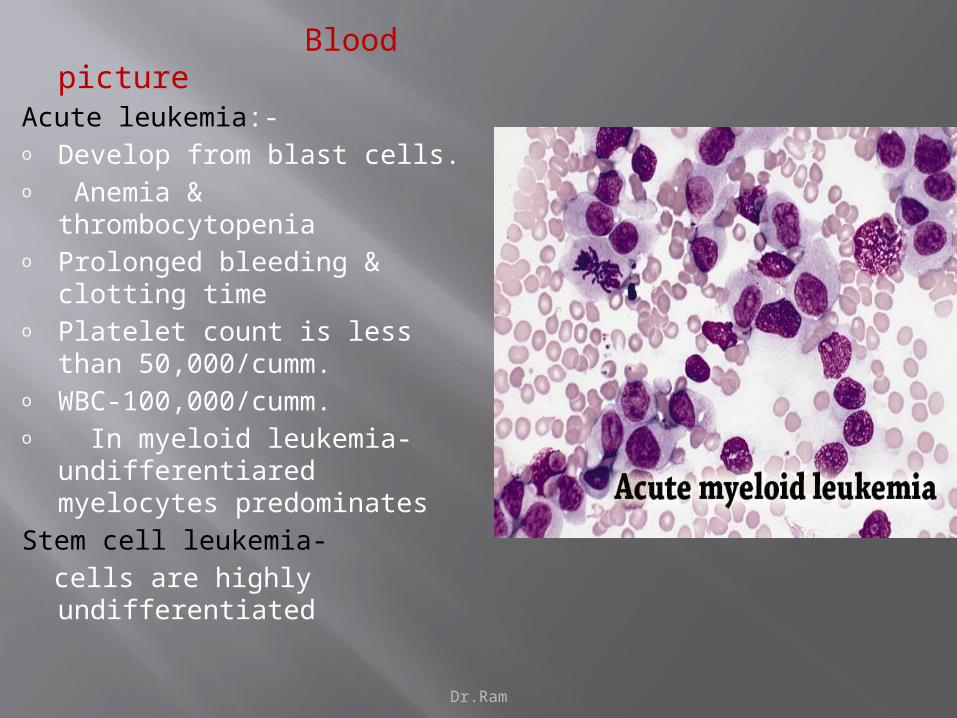
Blood pictureAcute leukemia:-o Develop from blast cells.o Anemia & thrombocytopeniao Prolonged bleeding & clotting
timeo Platelet count is less than
50,000/cumm.o WBC-100,000/cumm.o In myeloid leukemia-
undifferentiared myelocytes predominates
Stem cell leukemia-
cells are highly undifferentiated
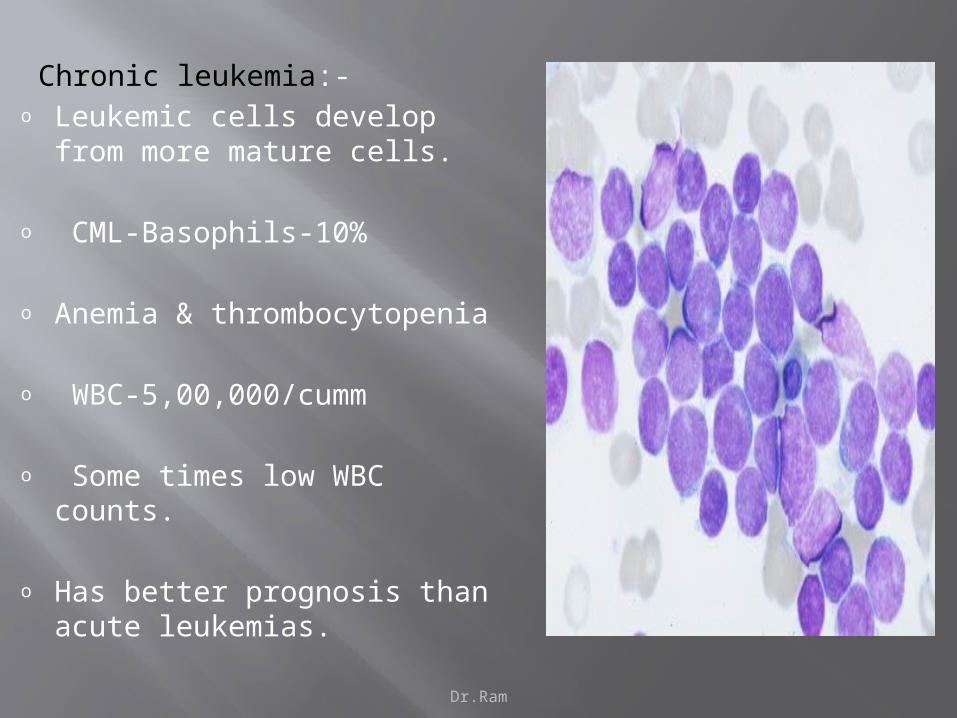
Dr.Ram
Chronic leukemia:-o Leukemic cells develop from
more mature cells.
o CML-Basophils-10%
o Anemia & thrombocytopenia
o WBC-5,00,000/cumm
o Some times low WBC counts.
o Has better prognosis than acute leukemias.

Dr.Ram
TREATMENT
Chemotherapy
Radiotherapy
corticosteroids

Dr.Ram
INFECTIOUS MONONUCLEOSIS Also known as glandular fever or kissings disease. It is an infectious, widespread viral disease caused by the
Epstein–Barr virus(EBV). Incubation period-4-7weeks. It is a clinical syndrome characterized by fever,pharyngitis
and adenopathy.
C/F:- Age-15-20yrs. Fever ,chills ,head ache. Sore throat, cough, nausea & vomoting. Bilateral & symmetrical lymphadenopathy. Sphenomegaly & hepatits. Pharyngitis & laryngitis.

Dr.Ram
ORAL MANIFESTATIONS
Acute gingivitis & stomatitis.
Palatal patechiae at the junction of hard & soft palate is the
early manifestation.
Lesions persist for 3-11 days & fade gradually.
Edema of soft palate & uvula.
Gingival & oropharyngeal bleeding is seen.

Dr.Ram
LABORATORY FINDINGS:-
Atypical lymphocytes , resembling monocytes.
Antibodies to EB virus.
Positive paul- Bunnel test- A test for the presence of heterophile antibodies in the serum produced in
infectious mononucleosis; agglutination of sheep red cells is a positive test.
ESR is high.
Thrombocytopenia.
TREATMENT:-
Bed rest & adequate diet.
Steroid therapy can be given.
Disease persist for 2-4 weeks .

Dr.Ram
AGRANULOCYTOSIS Agranulocytosis is the disease involving WBC which is
characterized by decreased number of circulating granulocytes.
Two types ;- Primary agranulocytosis- un known etiology. Secondary granulocytosis
Etiology:- Infections Hemopoitic disorders Chemical agents Physical agents It mainly in individuals who manifest allergic reactions .

Dr.Ram
Mechanism:
In drug induced agranulocytosis the drug may act as antigen & induce
antibody formation.
These antibodies destroy granulocytes or form immunocomplexes
which bind to neutrophils & destroy them.
KOSTMANN SYNDROME: Inherited autosomal recessive pattern in
which severe congenital neutropenia is seen.
C/F:-
Can occur at any age.
F>M
Mainly affects health workers .

Dr.Ram
Disease commences with high fever, chills & soar throat.
Skin pale & anemic some times yellow tinged.
Characteristic feature is presence of infections mainly in oral cavity , GIT, GUT,RT,& skin.
Regional lymphadenitis .
If not treated in time infection can become generalized sepsis& may be fatal.
Clinical symptoms develop rapidly with a few days & death may occur with in a week.

Dr.Ram
O/M:-
Necrotizing ulcerations of oral mucosa mainly palate &
gingiva, tonsils ,pharynx.
Lesions appear as ragged necrotic ulcers covered by gray or
black membrane.
Necrosis of gingiva begins adjacent to sulcus -free gingiva -
PDL -Al bone.
Excessive salivation
Oral surgical procedures are contraindicated.

Dr.Ram
Laboratory findings:- WBC below 2000/cumm. Complete absence of granulocytes. Bone marrow shows myeloblasts, premyelocyte,
metamyelocytes & myelocyte.
Treatment:- Recognition & with drawel of causative agent.
Administration of antibiotics for the control of infections.
Agranulocytosis secondary to viral diseases is self limiting & has good prognosis.

Dr.Ram
CYCLIC NEUTROPENIA Cyclic neutropenia is an unusual form of agranulocytosis
characterized by periodic diminution in circulating PNL.
Rare congenital neutropenia. Etiology is unknown. Can be an autosomal dominant condition due to
germline mutations in GRANULOCYTE –COLONY STIMULATING FACTOR. (G-CSF).
C/F:- Can occur at any age ,infants & young adults. Fever, sore throat, stomatitis,& regional lymphadenopathy. Head ache, arthritis,cutaneous infection, conjunctivitis can be
seen. The cycle will be ussually for 3 weeks, so there won’t be
significant bacterial infection.

Dr.Ram
O/M:-
Patients exhibit severe gingivitis,somatitis with ulceration, during the period of neutropenia.
With neutrophil count normal the oral mucosa assumes normal clinical appearance.
In children repeated infection leads to loss of supporting bone of the teeth.
Isolated painful ulcers occur which persist for 10-14 days & heal with scarring.

Dr.Ram
R/F:-
IOPA shows severe loss of superficial alveolar bone, due to repeated
cyclic gingivitis ,advancing to periodontitis.
Loss of bone around teeth in children is termed as prepubertal
periodontitis.
LABORATORY FINDINGS:-
Pt exhibits normal blood count for a period of 4-5 days followed by
decrease in granulocytes for a period of 3weeks or for several months.
At height of disease neutrophils may completely disappear for 1 or
2days.

Dr.Ram
Treatment & prognosis:- No specific treatment is necessary,
Prognosis is better than typical agranulocytosis.

Dr.Ram
DISEASES OF PLATELETS

Dr.Ram
PLATELETS
o Platelets, or thrombocytes are small, irregularly shaped cells, 2–3 µm in diameter.
o Platelets are derived from megakaryocytes.
o Normal count is 1,50000-4,00000
o The average lifespan of a platelet is 5 to 9 days.
o Platelets provide lipoprotein surface for the conversion of prothrombin to thrombin.

Dr.Ram
Thrombin helps in conversion of fibrinogen to fibrin which causes the aggregation of platelets.
Platelet disorders are the most common cause of bleeding.
The disorder could be decrease in the number
(thrombocytopenia) or defective function.

Dr.Ram
Diseases of platelets are:-
Thrombocytopenic purpura
Thrombocytopathic purpura
Thrombocytosis

Dr.Ram
THROMBOCYTOPENIC PURPURA
Thrombocytopenia is a disease which is characterized by abnormal reduction in number of circulating platelets.
Purpura is defined as purplish discolouration of skin or mucous membrane due to spontaneous extravassation of blood.
Purpura can be due to reduction in platelets or increased capillary fragility.

Dr.Ram
Based on the etiology purpura is of 2 types:-
- Non thrombocytopenic purpura –due to increased capillary fragility.
- Thrombocytopenic purpura
Thrombocytopenic purpura is of 2 types:-
Primary thrombocytopenic purpura –unknown etiology
Secondary thrombocytopenic purpura

Dr.Ram
Etiology:-
.1. Failure of platelets production most common cause, Megakaryocytes are in the bone marrow e.g. Ionizing radiation Drugs & chemicals Aplastic anemia Infections 2. rate of removal of platelets from the circulation
Drugs
Infections
Hemolytic anemia
3.Diceases causing excessive utilization of platelets
Splenomegaly
Platelet sequestration
Intravascular coagulation

Dr.Ram
Primary thrombocytopenic purpura :
It is thought to be an autoimmune disorder in which a person develops anitibodies to his own platelets.
It can also be due to absence of platelet stimulating or megakaryocyte –ripening factor.
Acute form occurs in children following some viral infections.
Chronic form occurs mostly in adult females.
Dr.Ram
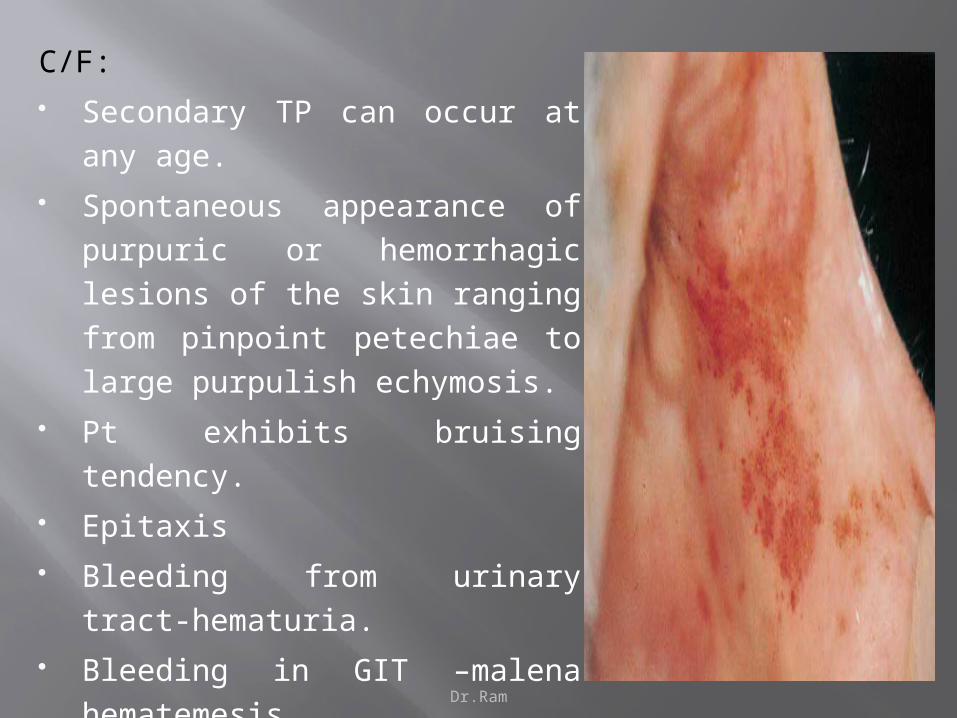
C/F:
Secondary TP can occur at any age.
Spontaneous appearance of purpuric or
hemorrhagic lesions of the skin ranging
from pinpoint petechiae to large
purpulish echymosis.
Pt exhibits bruising tendency.
Epitaxis
Bleeding from urinary tract-hematuria.
Bleeding in GIT –malena hematemesis.
Intracranial hemorrhage - hemiplegia.
Dr.Ram
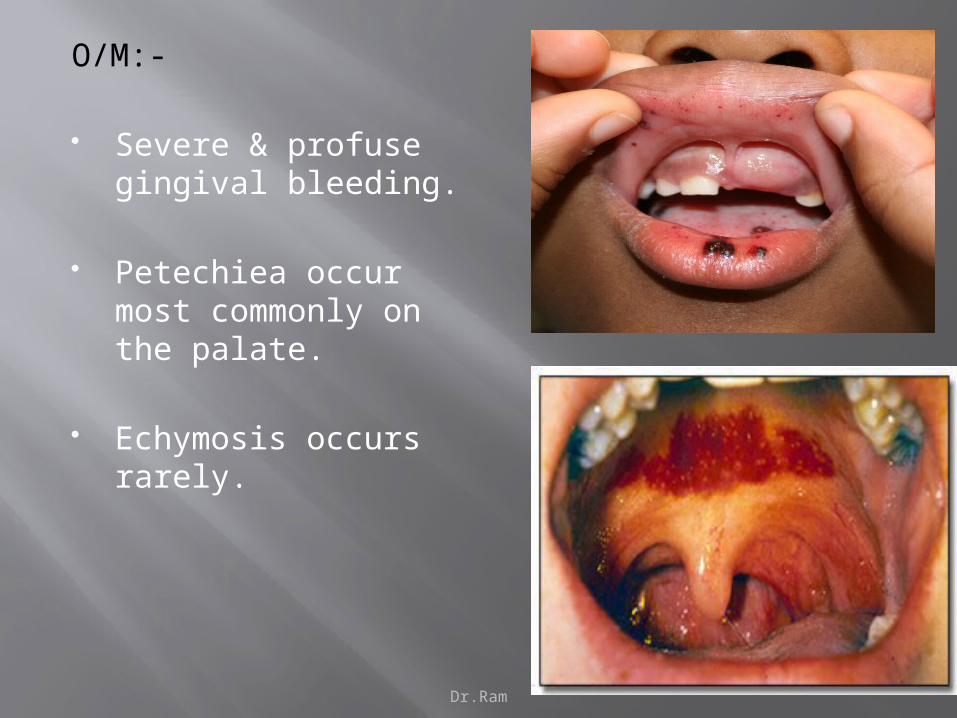
O/M:-
Severe & profuse gingival bleeding.
Petechiea occur most commonly on the palate.
Echymosis occurs rarely.

Dr.Ram
Laboratory findings
Platelets >60,000/cumm
BT-more than one hr.
CT is normal but clot shows failure of retraction.
Capillary fragility is increased
Giant platelets in peripheral smears-congenital
thrombocytopenia

Dr.Ram
Treatment & prognosis:-
No specific treatment
Splenectomy can be done.
Corticosteroids
Pt shows fairly good prognosis with intervals of remission &
exacerbation
Death may be due sudden ,severe hemorrhage.
Oral surgical procedures are contra indicated until deficiency
is cleared

Dr.Ram
Diseases of blood factors

Dr.Ram
HEMOPHILIA-(bleeders disease or disease of kings)
Hemophilia is characterized by prolonged coagulation time & hemorrhagic tendencies.
It is a heriditary disease carried by X- chromosome .
It is transmitted as a gender- linked mandolin recessive trait.
Females act as carriers ,rarely affected by diseases.

Dr.Ram
ETIOLOGY:-
Mainly 3 types of hemophilia.
Hemophilia A-factor VIII or Anti hemophilic globulin deficiency(AHG).
Hemophilia B-IX plasma thromboplastin component (PTC).christmas disease.
Hemophilia C-XI plasma thromboplastin antecedent (PTA)

Dr.Ram
Genes for AHG & PTC are located on long arm of X chromosome in q28 & q27.
Mutations in these genes can lead to hemophilia.
Gene for PTA is located on chromosome 4.
Hemophilia A is the most common type & is also called as classic hemophilia.

Dr.Ram
AHG is a glycoprotein which has 3 components.
1)clot promoting factor-
corrects the coagulation defects in pt with classic hemophilia.
2) factor VIII antigen-
present in pt with classic hemophilia & absent in pt with Von Willebrand’s disease.
3) Von Willebrand factor-
synthesized by endothelial cells which corrects platelet adhesion defects in Von Willebrand’s disease.

Dr.Ram
Based on the severity of disease divided into 3 types-
Mild hemophilia-hemorrhage is due to major trauma or surgery.
Moderate hemophilia-mild to moderate trauma.
Severe hemophilia-spontaneous & soft tissue bleeding ,hemarthrosis.
Dr.Ram
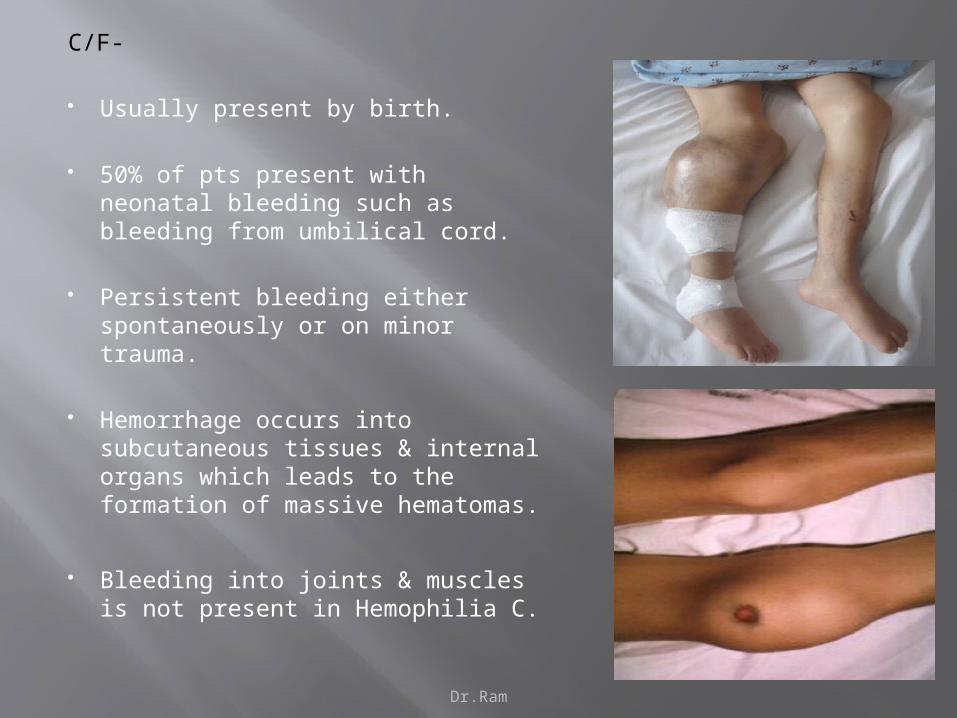
C/F-
Usually present by birth.
50% of pts present with neonatal bleeding such as bleeding from umbilical cord.
Persistent bleeding either spontaneously or on minor trauma.
Hemorrhage occurs into subcutaneous tissues & internal organs which leads to the formation of massive hematomas.
Bleeding into joints & muscles is not present in Hemophilia C.

Dr.Ram
O/M:-
Gingival bleeding is massive & severe.
Even physiologic process of tooth erruption & exfoliation can lead to severe & prolonged bleeding.
Due to subperiosteal bleeding & reactive new bone formation there will be formation of tumour like expansion of mandible- pseudotumour.

Dr.Ram
Surgical procedures should be carried out with proper premedication.
Tooth extractions can be carried out by means of rubber bands.
The rubber band is placed around cervical portion of the tooth & allowed to migrate apically, causing exfoliation of tooth through pressure necrosis of periodontal ligament.

Dr.Ram
LABORATORY FINDINGS:- C .T –increased B.T ,prothrombin time –normal Plasma of hemophilic pts show deficiency of clot –
promoting factor.
Treatment :-
Pts should be protected from traumatic injuries.
Preoperative transfusion of whole blood & administration of antihemophilic factor concentrate is recommended.
Prognosis is variable ,& pts may die in childhood.

Dr.Ram
VON WILLEBRAND’S DISEASE Also known as pseudohemophilia or vascular hemophilia.
It is characterized by excessive bleeding in pts with normal platelet count, normal CT,normal serum fibrinogen & normal prothrombin time.
BT is prolonged.
It is hereditary disease inherited as an autosomal dominant trait.
Transmitted & manifested by both males & females ,but females are most commonly effected.
Caused by abnormalities of Von Willebrand factor(VWF).

Dr.Ram
vWF is a large glycoprotein that acts as a carrier protein for factor VIII.
It helps in platelet aggregation (primary hemostasis) & prevents the degradation of factor VIII & delivers it to the site of injury.(secondary hemostasis).
vWF gene is present on chromosome 12.

Dr.Ram
vWD is classified into 3 types:-
Type 1- partial decrease in vWF & factor VIII.
Type 2- qualitative defects in vWF.
Type 3- Severe & rarest form.
Deficiency 0f both vWF & factor VIII in the plasma .
vWF is absent in both platelets & endothelial cells.
Severe clinical bleeding, inherited as an autosomal recessive trait

Dr.Ram
C/F:-
Acquired form is seen in willim’s tumour & systamic lupus erythmatosis.
Most of the children are asymptomatic .
Diagnosed on routine hematological examination.
Excessive bleeding either spontaneously or on minor trauma.
Bleeding from nose ,gingiva & skin.
Bleeding into GIT common.
Hemarthrosis is rare .
Bleeding tendency may be spontaneous or cyclic.

Dr.Ram
O/M:-
Bleeding of gingiva may be spontaneous or followed by minor trauma.
Disease may be discovered after extraction due to prolonged & excessive bleeding following procedure.
Laboratory findings:-
BT is prolonged- min - hrs.
Prothrombin time & CT are normal.
Capillary fragility is increased & positive tourniquet test.
Poor platelet adherence.

Dr.Ram
Treatment:-
Transfusion of plasma/ AHF.
Pts may become refractory after repeated
tranfussions ,can develop antibodies to AHF.

Dr.Ram
THANK YOU


















