
HD-Wegleitung Zulassung Humanarzneimittel nach Art. 13 HMG · PMDA Pharmaceuticals Medical Devices...
17
HD-Wegleitung Zulassung Humanarzneimittel nach Art. 13 HMG QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 1 / 17 Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12 Inhaltsverzeichnis 1 Definitionen, Begriffe und Abkürzungen .........................................................................2 1.1 Definitionen und Begriffe .....................................................................................................2 1.1.1 Länder mit vergleichbarer A rzneimittelkontrolle ..................................................................2 1.1.2 Referenzbehörde .................................................................................................................2 1.2 Abkürzungen .......................................................................................................................2 2 Einleitung und Zielsetzung ...............................................................................................3 3 Geltungsbereich ................................................................................................................4 4 Rechtsgrundlagen .............................................................................................................4 5 Mitgeltende Dokumente ....................................................................................................4 6 Anforderung an die Dokumentation (Art. 5a VAM) ..........................................................5 6.1 Bei der Referenzbehörde eingereichte Unterlagen ..............................................................5 6.2 Datum der Zulassung oder der letzten Überarbeitung der Dokumentation ...........................6 6.3 Ergebnisse der Begutachtung und Entscheide der Referenzbehörde ..................................6 6.4 Spezifische Anmerkungen zum Schweizerischen Modul 1 ..................................................7 6.5 Arzneimittelinformation ........................................................................................................7 6.6 Anforderungen an Sprache und Übersetzung der Dokumentation .......................................8 6.7 Abweichungen gegenüber dem von der Referenzbehörde zugelassenen Präparat .............8 6.8 Einhaltung spezifischer schweizerischer Vorgaben .............................................................8 6.9 Weitere behördenspezifische Unterlagen ............................................................................8 6.10 Information und Dokumentation nach der Zulassung durch Swissmedic .............................8 7 Arzneimittel mit bekannten Wirkstoffen (Art. 5b VAM) ...................................................9 7.1 BWS mit Zulassung einer ausländischen Behörde jedoch ohne zentrale europäische oder FDA-Zulassung (Art. 5b Abs. 1 VAM) ..................................................................................9 7.2 BWS mit zentraler europäischer Zulassung oder FDA-Zulassung (Art. 5b Abs. 2 VAM) .....9 7.3 Transparenz betreffend wesentlicher Bedenken ................................................................ 10 8 NAS oder deren Indikationserweiterung (Art. 5c VAM) ................................................. 10 8.1 Einschränkung der Begutachtung auf Gesuch hin ............................................................. 10 8.2 Einschränkung der Begutachtung von Amtes wegen ......................................................... 10 9 Parallele Verfahren in der Schweiz und im Ausland (Art. 5d VAM) .............................. 10 10 Prozess bei Swissmedic ................................................................................................. 11 10.1 Gesuchsbearbeitung ......................................................................................................... 11 10.2 Dauer und Kosten des Verfahrens ..................................................................................... 11 10.3 Öffentlichkeit und Transparenz .......................................................................................... 11 11 Anhang ............................................................................................................................. 12 11.1 Unterlagen, die für Gesuche aller Referenzbehörden eingereicht werden müssen ............ 12 11.2 Zulassung bezogen auf EU Centralised Procedure (CP) ................................................... 13 11.3 Zulassung bezogen auf EU Mutual Recognition Procedure MRP und Decentralised Procedure DCP ................................................................................................................. 13
Transcript of HD-Wegleitung Zulassung Humanarzneimittel nach Art. 13 HMG · PMDA Pharmaceuticals Medical Devices...

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 1 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Inhaltsverzeichnis
1 Definitionen, Begriffe und Abkürzungen ......................................................................... 2
1.1 Definitionen und Begriffe ..................................................................................................... 2
1.1.1 Länder mit vergleichbarer A rzneimittelkontrolle .................................................................. 2
1.1.2 Referenzbehörde ................................................................................................................. 2
1.2 Abkürzungen ....................................................................................................................... 2
2 Einleitung und Zielsetzung ............................................................................................... 3
3 Geltungsbereich ................................................................................................................ 4
4 Rechtsgrundlagen ............................................................................................................. 4
5 Mitgeltende Dokumente .................................................................................................... 4
6 Anforderung an die Dokumentation (Art. 5a VAM) .......................................................... 5
6.1 Bei der Referenzbehörde eingereichte Unterlagen .............................................................. 5
6.2 Datum der Zulassung oder der letzten Überarbeitung der Dokumentation ........................... 6
6.3 Ergebnisse der Begutachtung und Entscheide der Referenzbehörde .................................. 6
6.4 Spezifische Anmerkungen zum Schweizerischen Modul 1 .................................................. 7
6.5 Arzneimittelinformation ........................................................................................................ 7
6.6 Anforderungen an Sprache und Übersetzung der Dokumentation ....................................... 8
6.7 Abweichungen gegenüber dem von der Referenzbehörde zugelassenen Präparat ............. 8
6.8 Einhaltung spezifischer schweizerischer Vorgaben ............................................................. 8
6.9 Weitere behördenspezifische Unterlagen ............................................................................ 8
6.10 Information und Dokumentation nach der Zulassung durch Swissmedic ............................. 8
7 Arzneimittel mit bekannten Wirkstoffen (Art. 5b VAM) ................................................... 9
7.1 BWS mit Zulassung einer ausländischen Behörde jedoch ohne zentrale europäische oder
FDA-Zulassung (Art. 5b Abs. 1 VAM) .................................................................................. 9
7.2 BWS mit zentraler europäischer Zulassung oder FDA-Zulassung (Art. 5b Abs. 2 VAM) ..... 9
7.3 Transparenz betreffend wesentlicher Bedenken ................................................................ 10
8 NAS oder deren Indikationserweiterung (Art. 5c VAM) ................................................. 10
8.1 Einschränkung der Begutachtung auf Gesuch hin ............................................................. 10
8.2 Einschränkung der Begutachtung von Amtes wegen ......................................................... 10
9 Parallele Verfahren in der Schweiz und im Ausland (Art. 5d VAM) .............................. 10
10 Prozess bei Swissmedic ................................................................................................. 11
10.1 Gesuchsbearbeitung ......................................................................................................... 11
10.2 Dauer und Kosten des Verfahrens ..................................................................................... 11
10.3 Öffentlichkeit und Transparenz .......................................................................................... 11
11 Anhang ............................................................................................................................. 12
11.1 Unterlagen, die für Gesuche aller Referenzbehörden eingereicht werden müssen ............ 12
11.2 Zulassung bezogen auf EU Centralised Procedure (CP) ................................................... 13
11.3 Zulassung bezogen auf EU Mutual Recognition Procedure MRP und Decentralised
Procedure DCP ................................................................................................................. 13

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 2 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
11.4 Zulassung bezogen auf EU und EFTA-Staaten: Nationale Zulassungen ........................... 13
11.5 Zulassung bezogen auf Zulassung USA / FDA .................................................................. 13
11.6 Zulassung bezogen auf Zulassung Japan.......................................................................... 13
11.7 Zulassung bezogen auf Zulassung Kanada ....................................................................... 13
11.8 Zulassung bezogen auf Zulassung Australien ................................................................... 14
11.9 Zulassung bezogen auf Zulassung Singapur ..................................................................... 14
11.10 Zulassung bezogen auf Zulassung Neuseeland ................................................................ 14
11.11 Flowcharts der Gesuchsabläufe ........................................................................................ 14
Änderungshistorie
Mindestens letzte Versionsänderung sichtbar, wobei nicht zwingend bei 01 beginnend.
Version Gültig und
verbindlich
ab
Ohne
Versions-
änderung
angepasst
Beschreibung, Bemerkung (durch Autor/in erstellt) Visum
(Kürzel)
Autor/in
01 30.05.17 Formale Anpassung an Vorgabenmanagement der
Swissmedic: Umwandlung Verwaltungsverordnung in
Wegleitung; keine inhaltlichen Veränderungen vorgenommen
fua
1 Definitionen, Begriffe und Abkürzungen
1.1 Definitionen und Begriffe
1.1.1 Länder mit vergleichbarer Arzneimittelkontrolle
Als Länder mit vergleichbarer Arzneimittelkontrolle anerkennt Swissmedic gestützt auf Art. 5a Abs. 4
VAM die folgenden Staaten (Stand Januar 2013):
Australien
EU und EFTA-Länder
Japan
Kanada
Neuseeland
Singapur
USA
Die Arzneimittelbehörden dieser Länder werden in dieser Wegleitung auch summarisch als
ausländische Behörden bezeichnet.
1.1.2 Referenzbehörde
Mit dem Begriff Referenzbehörde ist diejenige ausländische Behörde gemeint, welche das
betreffende Arzneimittel bereits zugelassen hat und auf deren Prüfungsergebnisse die
Gesuchstellerin ihr Zulassungsgesuch in der Schweiz abstützt.
1.2 Abkürzungen
AMZV Verordnung des Schweizerischen Heilmittelinstituts über die Anforderungen an die
Zulassung von Arzneimitteln (Arzneimittel-Zulassungsverordnung) (SR 812.212.22)
ASMF Active Substance Master File
BWS Arzneimittel mit bekannten Wirkstoffen
CHMP Committee for Medicinal Products for Human Use der EMA
COMP Committee for Orhan Medicinal Products
CP Centralised Procedure

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 3 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
CxMP Committee for Medicinal Products der EMA
DCP Decentralised Procedure der EU
DMF Drug Master File
eCTD Elektronische Einreichung als Common Technical Document
EFTA European Free Trade Association
EMA European Medicines Agency
ERA Environmental Risk Assessment
EU Europäische Union
EU-SmPC Summary of Product Characteristics (EU)
FDA Food and Drug Administration (USA)
FI Fachinformation
GCP Good Clinical Practice
GLP Good Laboratory Practice
GMP Good Manufacturing Practice
GxP Good x Practices
HGebV Verordnung über die Gebühren des Schweizerischen Heilmittelinstituts (SR 812.214.5)
HMG Heilmittelgesetz vom 15. Dezember 2000 (SR 812.21)
HMPC Committee on Herbal Medicinal Products
ICH E2E International Conference of Harmonisation Guideline Pharmacovigilance Planning
IE Neue Indikation
KPA Komplementär- und Phytoarzneimittel
LoQ List of Questions
LoOI List of Outstanding Issues
MRP Mutual Recognition Procedure der EU
NAS Arzneimittel mit neuen aktiven Substanzen
NOC Notice of Compliance
NtA Notice to Applicant
Ph. Eur. Pharmacopoea Europaea
Ph. Helv. Pharmacopoea Helvetica
PI Patienteninformation
PMDA Pharmaceuticals Medical Devices Agency (Japan)
RiskMAP Risk Minimization Action Plan
RMP Risk Management Plan
RMS Reference Member State der EU
SBA Summary Basis of Approval
VAM Verordnung über die Arzneimittel vom 17 Oktober 2001 (SR 2812.212.21)
VAZV Verordnung des Schweizerischen Heilmittelinstituts vom 22. Juni 2006 über die
vereinfachte Zulassung von Arzneimitteln und die Zulassung von Arzneimitteln im
Meldeverfahren (VAZV SR 812.212.23)
WL Wegleitung
2 Einleitung und Zielsetzung
Beantragt eine Gesuchstellerin die Zulassung für ein Arzneimittel, für welches die Zulassung in einem
Land mit vergleichbarer Arzneimittelkontrolle bereits erteilt worden ist, so berücksichtigt das
Schweizerische Heilmittelinstitut (in der Folge Swissmedic oder das Institut) die Ergebnisse der von
der ausländischen Arzneimittelbehörde durchgeführten Prüfungen, sofern bestimmte Anforderungen
erfüllt sind. Diese Anleitung präzisiert diese Anforderungen und beschreibt den institutsinternen
Prozess, mit dem ihre Erfüllung überprüft wird.
Es handelt sich bei dieser Wegleitung um eine Anleitung, die sich an die Verwaltungsorgane richtet
und somit nicht unmittelbar Rechte und Pflichten von Privaten festlegt. Diese Wegleitung dient
Swissmedic in erster Linie als ein Hilfsmittel, um die gesetzlichen Bestimmungen einheitlich und
rechtsgleich anzuwenden. Durch die Publikation der Anleitung soll transparent gemacht werden,
welche Anforderungen gemäss Praxis der Swissmedic zu erfüllen sind, damit entsprechende
Gesuche möglichst effizient behandelt und abgeschlossen werden können.

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 4 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Bei der Zulassung im Ausland bereits zugelassener Arzneimittel soll unter den nachfolgend
beschriebenen Voraussetzungen eine Beschleunigung des Zulassungsverfahrens vor dem
Schweizerischen Heilmittelinstitut erzielt werden.
Die Berücksichtigung der Ergebnisse ausländischer Zulassungsverfahren soll dazu beitragen die
Zulassung von Arzneimitteln in der Schweiz so abzuwickeln, dass im Ausland bereits zugelassene
Arzneimittel den Patientinnen und Patienten in der Schweiz möglichst rasch zur Verfügung stehen
und die Ressourcen des Instituts gezielt und risikobasiert eingesetzt werden (Art. 1 Abs. 3 Bst.a
HMG).
3 Geltungsbereich
Die Anleitung gilt für den Bereich Zulassung von Swissmedic und ist anwendbar bei:
Erstzulassungen inkl. wesentlicher Änderungen von Humanarzneimitteln
genehmigungspflichtigen Änderungen bereits nach Art. 13 HMG zugelassener Arzneimittel,
sofern ein Assessment Report zur jeweiligen Änderung vorliegt
genehmigungspflichtigen Änderungen von Präparaten, die ohne Bezugnahme auf Art. 13 HMG
zugelassen worden sind, sofern eine Bestätigung betreffend gleicher Stand des Dossiers und ein
Assessment Report zur jeweiligen Änderung vorliegen.
Diese Wegleitung gilt:
für Zulassungsgesuche, die sich auf Art. 5a - 5c VAM abstützen und sich auf folgende von einer
ausländischen Behörde (gemäss Kapitel 1.1.1) zugelassenen Arzneimittel beziehen:
Arzneimittel mit bekannten Wirkstoffen (BWS)
Arzneimittel mit neuen aktiven Substanzen (NAS) und deren Indikationserweiterung, sofern
sie die in Kapitel 8 aufgeführten Kriterien erfüllen
Arzneimittel, die gestützt auf Art. 12 Abs. 4 VAZV nicht vereinfacht zugelassen werden
können, sofern sie die in Kapitel 8 aufgeführten Kriterien erfüllen
für parallele Verfahren in der Schweiz und bei der EMA gemäss Art. 5d VAM
sinngemäss für die Zulassung von Verfahren nach Art. 9 Abs. 3 HMG.
Die Wegleitung ist nicht anwendbar bei:
genehmigungspflichtigen Änderungen für die kein Assessment Report zur jeweiligen Änderung
vorliegt
meldepflichtigen Änderungen
Anerkennungen von Chargenfreigaben.
4 Rechtsgrundlagen
Das Verfahren für die Berücksichtigung der Prüfungsergebnisse, die im Rahmen von ausländischen
Zulassungsverfahren ergangen sind, richtet sich insbesondere nach folgenden Rechtsgrundlagen
(Gesetzes- und Verordnungsbestimmungen):
Bundesgesetz vom 15. Dezember 2000 über Arzneimittel und Medizinprodukte (Heilmittelgesetz,
HMG1):
Art. 13 im Ausland zugelassene Arzneimittel und Verfahren
Verordnung vom 17. Oktober 2001 über die Arzneimittel (Arzneimittelverordnung, VAM2):
Art. 5a - 5d im Ausland zugelassene Arzneimittel und Verfahren (Art. 13 HMG)
5 Mitgeltende Dokumente
Dokumentenidentifikation
ZL000_00_012d_CL Formale Kontrolle Zulassungsgesuch Humanarzneimittel Art.13 HMG
1 SR 812.21 2 SR 812.212.21

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 5 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
ZL000_00_003d_FO Gesuch Zulassung / Änderung für Humanarzneimittel
ZL000_00_007d_FO Status Zulassungsgesuche im Ausland
ZL302_00_001d_FO Genehmigungspflichtige Änderung Qualität
ZL101_00_003d_FO Informationen zur Qualität bei Antrag Art. 13 HMG
ZL000_00_005d_WL Zulassung Humanarzneimittel mit bekanntem Wirkstoff
ZL101_00_003d_WL Zulassung Biosimilar
ZL000_00_009d_WL Zulassung Allergenpräparat
ZL000_00_003d_WL Zulassung Radiopharmazeutikum
Anleitung zum Einreichen von Zulassungsgesuchen für pflanzliche Arzneimittel der Humanmedizin (Phytoanleitung)
ZL000_00_008d_WL Zulassung Antidot
ZL000_00_007d_WL Zulassung Medizinalgas
ZL000_00_006d_WL Fristen Zulassungsgesuche
ZA000_00_001d_VZ Liste aller Länder mit vergleichbarer Arzneimittelkontrolle
ZL000_00_001d_WL Formale Anforderungen
ZL000_00_002d_VZ Tabelle einzureichende Unterlagen
Guidance Paragraph 13 TPA v1.3.
6 Anforderung an die Dokumentation (Art. 5a VAM)
Wurde einem Arzneimittel die Zulassung in einem Land mit vergleichbarer Arzneimittelkontrolle nach
Kapitel 1.1.1 bereits erteilt, werden im Rahmen des Zulassungsverfahrens vor dem Institut die
Prüfungsergebnisse der Referenzbehörde berücksichtigt, sofern die Gesuchstellerin dies im Formular
Gesuch Zulassung / Änderung für Humanarzneimittel explizit beantragt. In diesem Fall prüft das
Institut zugleich, ob auch alle notwendigen Unterlagen für dieses Verfahren vollständig eingereicht
wurden.
6.1 Bei der Referenzbehörde eingereichte Unterlagen
Übereinstimmung zwischen ausländischer und schweizerischer Dokumentation
Grundsätzlich muss die bei Swissmedic eingereichte Dokumentation mit derjenigen identisch sein,
auf Grund derer die Referenzbehörde die Zulassung des Arzneimittels resp. dessen Änderung
genehmigt hat3 (finale Version).
Erfolgte die Zulassung in mehr als einem Land mit vergleichbarer Arzneimittelkontrolle, so muss die
Zulassungsdokumentation nur einmal vollständig, so wie sie der Referenzbehörde vorgelegt wurde,
eingereicht werden. Für nachfolgende Änderungsgesuche muss die einmal gewählte
Referenzbehörde beibehalten werden.
Die vollständige Dokumentation ist im CTD-Format4, mit dem von der Referenzbehörde begutachteten
länderspezifischen sowie dem schweizerischen Modul 1 bei Swissmedic einzureichen. Falls die
Dokumentation im NtA Format (Teil I - IV) von der Referenzbehörde zugelassen wurde, kann sie auch
bei Swissmedic in diesem Format eingereicht werden.
Dokumentation bei Änderungsgesuchen
Für die Anwendung von Art. 13 HMG für Änderungsgesuche von Präparaten muss ein Assessment
Report der Referenzbehörde vorliegen. Für Änderungsgesuche von Präparaten, die primär ohne
Bezugnahme auf Art. 13 HMG von Swissmedic zugelassen wurden, ist zudem eine Bestätigung einer
unterschriftsberechtigen Person oder des Regulatory Affairs-Verantwortlichen notwendig, dass die
Dokumentation der Referenzbehörde (vor Genehmigung der Änderung) und in der Schweiz identisch
sind. Bei Gesuchen für wesentliche Änderungen ohne vorherige Bezugnahme auf Art. 13 HMG
3 Zu den Ausnahmen: vgl. Kapitel 6.7 4 Common Technical Document gemäss Leitlinie M2 (eCTD) und M4 (CTD) der ICH (International Conference on
Harmonization). Besteht aus den folgenden Modulen: 1 (Country specific), 2 (Summary), 3 (Quality), 4 (Preclinical), 5 (Clinical).

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 6 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
gemäss Kapitel 7 sind die Dokumentation und entsprechenden Unterlagen für die Erstzulassung
ebenfalls einzureichen, wenn mehrheitlich darauf verwiesen wird. Wird ein Gesuch für eine
wesentliche Änderung neu im eCTD eingereicht, ist es nicht erforderlich für die früher eingereichte
Papier-Dokumentation ein elektronisches Baseline-File zu erstellen.
Änderungen und / oder Ergänzungen seit dem ausländischen Entscheid
Gleichzeitig mit dem Gesuch müssen auch die seit dem Zulassungsentscheid der Referenzbehörde
genehmigten Änderungen und / oder Ergänzungen Swissmedic vorliegen. Diese zusätzlichen oder
ausgetauschten Dokumentationen können entweder in die Dokumentation resp. das betroffene Modul
integriert sein oder separat eingereicht werden. Die Änderungen sind im Begleitschreiben zu
erwähnen und eine Gegenüberstellung der Änderungen (alt / neu) muss mit dem dazugehörigen
finalen Assessment Report beigelegt werden.
Informationen betreffend Sicherheitssignalen Nur in Zusammenhang mit aktuellen, national und international erfassten Sicherheitssignalen sind
auch alle relevanten Informationen und die einschlägige Korrespondenz mit der Referenzbehörde,
wie Eröffnungsbriefe eines Verfahrens, LoQ-Schreiben, Experten Berichte, Interimsergebnisse
(Milestones) und Abschlussberichte, einzureichen. Gegebenenfalls sind entsprechende
Aktualisierungen während des Zulassungsverfahrens nachzureichen. Für Sicherheitssignale, die seit
der Zulassung im Ausland und der Einreichung bei Swissmedic erfolgten und die abgeschlossen sind,
ist nur der Schlussbericht und die allenfalls angepasste Arzneimittelinformation einzureichen.
GLP / GMP / GCP
Die GLP- / GMP- / GCP-Konformität ist zu bestätigen. Laufende Untersuchungen (z.B. Behebung von
Mängeln, erforderliche Nachinspektionen) sind im Begleitschreiben anzugeben.
Risk Management Plan nach ICH E2E
Der Risk Management Plan ist für NAS und / oder deren Indikationserweiterung nach den Vorgaben
der ICH E2E einzureichen.
Drug Master File (DMF / ASMF)
Wurde für das betreffende Gesuch ein DMF / ASMF bei der Referenzbehörde eingereicht, muss der
DMF / ASMF Holder Swissmedic eine identische Kopie des Restricted Part inklusive Letter of Access,
Assessment Report des Restricted Part, LoQ und Antworten der Firma zum Restricted Part
einreichen. Wurde das DMF / ASMF in der Zwischenzeit geändert, sind die genehmigten Änderungen
mit dem dazugehörigen Assessment Report separat einzureichen und im Begleitschreiben mit einer
Gegenüberstellung der Änderungen (alt / neu) aufzuführen.
6.2 Datum der Zulassung oder der letzten Überarbeitung der Dokumentation
Die Zulassung oder die letzte von der Referenzbehörde genehmigte aufdatierte Version der
gesamten Dokumentation, insbesondere die Module 2.3, 2.4 und 2.5 (Quality, Non-Clinical Overview,
Clinical Overview) resp. Teil IC2, IC3 und IC4, darf zum Zeitpunkt der Einreichung des Gesuchs bei
Swissmedic nicht älter als 5 Jahre5 sein. So kann sichergestellt werden, dass die Begutachtung durch
die Referenzbehörde nach dem aktuellen Stand von Wissenschaft und Technik erfolgt ist.
Unterschiede zu aktuell gültigen Richtlinien (Guidelines), die zum Zeitpunkt der Zulassung im Ausland
noch nicht in Kraft waren, sind möglich, wenn sie kritisch bewertet werden und im Begleitschreiben
erwähnt sind.
6.3 Ergebnisse der Begutachtung und Entscheide der Referenzbehörde
Dem Institut müssen die Ergebnisse der Begutachtung vorliegen, die es ihm erlauben, die Entscheid-
findung der Referenzbehörde nachzuvollziehen. Die dafür erforderlichen Dokumente sind im Anhang
aufgeführt (siehe Kapitel 11).
5 Datum der Verfügung der Zulassung resp. der Genehmigung

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 7 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Wurde ein Arzneimittel in mehreren Ländern mit vergleichbarer Arzneimittelkontrolle zugelassen, sind
dem Institut nur der Zulassungsentscheid (Verfügung) und die Ergebnisse der Begutachtung (gemäss
Anhang) der von der Gesuchstellerin herangezogenen Referenzbehörde einzureichen.
Beantragt die Gesuchstellerin jedoch die Zulassung oder die Änderung einer Zulassung für ein
Arzneimittel, für welches sowohl ein Entscheid der EMA als auch ein Entscheid der FDA vorliegen, so
sind dem Institut bei abweichenden Entscheiden und / oder bei einem Gesuchrückzug die
Begutachtungsergebnisse beider Behörden einzureichen.
Im Formular Status Zulassungsgesuche im Ausland muss zum angemeldeten Präparat auch ein
negativer Zulassungsentscheid, ein Rückzug durch die Gesuchstellerin, ein laufendes
Überprüfungsverfahren oder eine Sistierung für alle ausländischen Behörden nach Kapitel 1.1.1
aufgelistet werden. Im Begleitschreiben müssen solche abweichenden Zulassungsentscheide anderer
Behörden (Abweisung Mitteilungen, die zum Rückzug des Gesuches führten Abweichungen
betreffend Indikationen, Dosierung, Lagerungshinweis, Laufzeit resp. weitere Einschränkungen und
ähnliches) transparent dargestellt werden.
6.4 Spezifische Anmerkungen zum Schweizerischen Modul 1
Das Institut benötigt zusätzlich zu den bei der Referenzbehörde vorgelegten Unterlagen die
administrativen Daten des schweizerischen Modul 1 gemäss Wegleitung Formale Anforderungen und
das dazugehörigen Verzeichnis Tabelle Einzureichende Unterlagen.
Auf dem Formular Gesuch Zulassung / Änderung für Humanarzneimittel muss die Gesuchstellerin die
Berücksichtigung der Prüfungen ausländischer Behörden nach Art. 13 HMG / Art. 5a - 5d VAM
beantragen.
Alle erforderlichen Beilagen und Formulare des Modul 1 (resp. für KPA: Teile 1A und 1B NtA) sind in
der Checkliste Formale Kontrolle Zulassungsgesuch Humanarzneimittel Art. 13 HMG, die ebenfalls
eingereicht werden muss, aufgeführt. Zusätzliche, in der Checkliste nicht aufgeführte Unterlagen, sind
im Begleitbrief zu erwähnen.
Belege für die Einhaltung der aktuellen Anforderungen der Ph. Eur. / Ph. Helv. können im Modul 3
CTD (für NtA: Part II) integriert oder separat beigelegt sein und in der Checkliste Formale Kontrolle
Zulassungsgesuch Humanarzneimittel Art. 13 HMG zu bestätigen. Werden statt der entsprechenden
Methoden der Ph. Eur. / Ph. Helv. andere Methoden verwendet, so ist deren Äquivalenz zu den
Methoden der Ph. Eur. / Ph. Helv. zu belegen.
Das ERA (Modul 1) muss nur für Arzneimittel eingereicht werden, die in einem Land mit
vergleichbarer Arzneimittelkontrolle zugelassen worden sind, welches nicht Mitglied der EU ist.
Falls spezifische Anforderungen an die Umsetzung der Spontanerfassung vermuteter unerwünschter
Arzneimittelwirkungen in der Schweiz vorgesehen sind, (z.B. spezielle Fragebogen im Rahmen von
„enhanced Pharmacovigilance“), ist dies bei der Einreichung des Gesuchs im Begleitschreiben
speziell zu vermerken.
6.5 Arzneimittelinformation
Swissmedic muss für die Schweiz spezifische Punkte, wie die Einhaltung der Vorgaben für die
Arzneimittelinformation (z.B. Schwangerschaftshinweis, Lagerungshinweise) oder die Kongruenz mit
dem Wortlaut der Arzneimittelinformation von Arzneimitteln mit vergleichbarer Datenlage,
sicherstellen. Eine wörtliche Übernahme der von der Referenzbehörde genehmigten Arzneimittel-
information ohne Überprüfung durch Swissmedic ist somit in der Regel nicht möglich.
Liegt hingegen eine im zentralisierten Verfahren erteilte Zulassung (Scientific Decision EMA) oder
eine Zulassung in einem Mitgliedstaat der EU oder der EFTA vor, so kann als Arzneimittelinformation
die gültige Arzneimittelinformation, die EU-SmPC, genehmigt werden (Art. 5a Abs. 3 VAM). Bestehen
inhaltliche Unterschiede zu schweizerischen Vorgaben, so muss Swissmedic diese für die Schweiz
oben beschriebenen spezifischen Punkte prüfen. In jedem Fall eingehalten werden müssen die
sprachlichen Vorgaben und die Deklarationspflicht für gentechnisch veränderte Organismen.
Die schweizerischen Anforderungen an die Angaben und Texte auf Behälter und Packungs-
materialien sind einzuhalten (Art. 12 und Anhänge AMZV6).
6 SR 812.212.22

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 8 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
6.6 Anforderungen an Sprache und Übersetzung der Dokumentation
Die Dokumentation (Module 2 bis 5 sowie die länderspezifischen Module 1 resp. Teil I - IV) und die
geforderten Unterlagen gemäss Anhang (siehe Kapitel 11) müssen dem Institut in einer
schweizerischen Amtssprache oder in Englisch vorliegen. Akzeptiert werden auch Übersetzungen in
diese Sprachen, sofern die Zulassungsinhaberin die Korrektheit der Übersetzung schriftlich bestätigt.
Das spezifische schweizerische Modul 1 (bzw. für KPA Teile 1A und 1B) sowie die
Arzneimittelinformation und die Packungselemente müssen in einer schweizerischen Amtssprache (FI
/ PI in Deutsch oder Französisch) eingereicht werden.
6.7 Abweichungen gegenüber dem von der Referenzbehörde zugelassenen Präparat
Grundsätzlich muss das im Ausland zugelassene Präparat mit dem in der Schweiz angemeldeten
identisch sein. Abweichungen sind in folgenden Fällen möglich:
Abweichungen betreffend den Herstellungsort des Fertigproduktes
Abweichungen betreffend die Chargenfreigabe
Abweichungen betreffend die Qualitätskontrolle(n)
Abweichungen betreffend die Primärverpackung oder den Primärverpacker
Abweichungen betreffend die Sekundärverpackung oder den Sekundärverpacker
Abweichungen betreffend die Packungsgrösse, wenn diese nicht im Widerspruch zur Anwendung
stehen
Abweichung von der im Ausland zugelassenen Präparatebezeichnung
Bei Abweichungen zwischen der Zulassung im Ausland und dem Gesuch an Swissmedic ist primär
die der Referenzbehörde zur Zulassung vorgelegte Dokumentation einzureichen. Die Abweichungen
sind im Begleitschreiben darzulegen. Abweichungen werden wie eine Änderung durch Swissmedic
begutachtet, führen aber zu keiner längeren Bearbeitungszeit.
6.8 Einhaltung spezifischer schweizerischer Vorgaben
Die je nach Gesuchstyp spezifischen Anforderungen folgender Anleitungen des Schweizerischen
Heilmittelinstituts sind zwingend einzuhalten und die damit verbundene Dokumentation dem Institut
zusammen mit dem Zulassungsgesuch einzureichen:
Wegleitung für die Zulassung Humanarzneimittel mit bekanntem Wirkstoff
Wegleitung für die Zulassung von Biosimilar
Wegleitung für die Zulassung von Allergenpräparat
Anleitung zum Einreichen von Zulassungsgesuchen für pflanzliche Arzneimittel der
Humanmedizin
Wegleitung für die Zulassung von Antidot
Wegleitung für die Zulassung von Medizinalgas
Wegleitung für die Zulassung von Radiopharmazeutikum.
6.9 Weitere behördenspezifische Unterlagen
Die einzureichenden Dokumente der Referenzbehörde sind im Anhang (siehe Kapitel 11) aufgeführt.
Swissmedic bezieht sich bei ihrer Beurteilung ausschliesslich auf Unterlagen, welche durch die
Gesuchstellerin eingereicht werden. Eine direkte Übermittlung der Unterlagen der Begutachtung im
Ausland durch die dort zuständige Behörde ist nicht möglich.
6.10 Information und Dokumentation nach der Zulassung durch Swissmedic
Mit dem Erlass der Verfügung Gutheissung oder Abweis durch Swissmedic ist das Zulassungs-
verfahren nach Art. 5a - 5d VAM abgeschlossen.
Durch die Referenzbehörde verfügte Auflagen, die zum Zeitpunkt des Zulassungsentscheids des
Instituts noch nicht erfüllt worden sind, werden in der Regel von Swissmedic ebenfalls verfügt.
Nach der Zulassungserteilung in der Schweiz ergehende Entscheide der Referenzbehörde betreffend
Erfüllung der Auflagen sind dem Institut innert angemessener Frist nachzureichen.

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 9 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
7 Arzneimittel mit bekannten Wirkstoffen (Art. 5b VAM)
Die nachfolgenden Ausführungen gelten für Gesuche um Zulassung eines BWS, d.h. von
Arzneimitteln, die einen Wirkstoff enthalten, der bereits in einem anderen vom Institut zugelassenen
Arzneimittel enthalten ist oder war7.
Die Berücksichtigung der Prüfungsergebnisse ausländischer Behörden im Rahmen eines Gesuchs
um Zulassung einer neuen galenischen Form, einer neuen Dosisstärke, eines neuen
Verabreichungsweges und / oder einer neuen Dosierungsempfehlung für ein Originalpräparat im
Sinne von Art. 12 HMG richtet sich nach den Vorgaben von Kapitel 7.1 resp. 7.2.
Die Berücksichtigung der Prüfungsergebnisse ausländischer Behörden im Rahmen eines Gesuchs
um Zulassung einer weiteren galenischen Form, Dosisstärke, eines weiteren Verabreichungsweges,
einer weiteren Indikation und / oder Dosierungsempfehlung für ein Arzneimittel mit bekanntem
Wirkstoff richtet sich ebenfalls nach den Vorgaben von Kapitel 7.1 resp. 7.2.
Wenn sich die für die Begutachtung des Präparates geltenden schweizerischen Vorschriften von
denen der Referenzbehörde wesentlich unterscheiden (z.B. Komplementär- und Phytoarzneimittel
Festlegung der Abgabekategorie), so behält sich Swissmedic vor, eine eigene Begutachtung
durchzuführen. Die Ergebnisse der ausländischen Behörde werden dabei, soweit als möglich,
mitberücksichtigt.
7.1 BWS mit Zulassung einer ausländischen Behörde jedoch ohne zentrale europäische oder FDA-Zulassung (Art. 5b Abs. 1 VAM)
Bei Gesuchen um Zulassung eines Arzneimittels mit bekannten Wirkstoffen (siehe Flowchart I im
Anhang) beschränkt sich das Institut in der Regel auf eine Prüfung der Ergebnisse der Begutachtung
der Referenzbehörde, sofern die Anforderungen nach Art. 5a VAM eingehalten wurden (vgl. Kapitel
6).
Anhand des Assessment Reports der Referenzbehörde wird die Qualität, Wirksamkeit und Sicherheit
des Arzneimittels begutachtet und beurteilt, ob die Prüfungsergebnisse der Referenzbehörde für den
Zulassungsentscheid durch Swissmedic übernommen werden können. Dabei können sich
begründete Anhaltspunkte für ein möglicherweise ungünstiges Nutzen-Risiko Verhältnis ergeben, die
anhand einer gezielten Einsicht in die Grunddokumentation vertiefter beurteilt werden.
Zudem überprüft Swissmedic in einer Analyse der Vorgeschichte und des Umfeldes, ob Gründe für
eine eigene wissenschaftliche Begutachtung bestehen. Zu diesen Gründen gehören ein früherer
Abweis resp. Rückzug eines Gesuchs für dieses Arzneimittel oder für eines aus der gleichen
Substanzklasse in der Schweiz oder in einem anderen Land mit vergleichbarer Arzneimittelkontrolle
im Sinne von Kapitel 1.1.1 sowie das Vorliegen neuer wissenschaftlicher Erkenntnisse seit der
Zulassungserteilung im Ausland.
7.2 BWS mit zentraler europäischer Zulassung oder FDA-Zulassung (Art. 5b Abs. 2 VAM)
Bei Gesuchen für BWS mit Zulassung der EMA und / oder der FDA führt das Institut in der Regel
keine Prüfung des Assessment Reports durch, sofern die Anforderungen nach Art. 5a VAM
eingehalten wurden (vgl. Kapitel 6).
Swissmedic analysiert bei diesen Gesuchen aufgrund der Vorgeschichte, des Umfeldes und der
Fachinformation, ob Sicherheitssignale vorliegen, welche speziell berücksichtigt werden müssen.
Ergibt diese Analyse wesentliche Bedenken hinsichtlich Qualität, Sicherheit und / oder Wirksamkeit
aufgrund einer eigenen früheren Begutachtung eines Präparates mit gleichem Wirkstoff oder der
gleichen Substanzklasse oder aufgrund materieller Unterschiede zwischen den
Zulassungsentscheiden der EMA und der FDA (Zulassung der einen und Ablehnung oder
Teilabweisung der andern Behörde, unterschiedliche Indikationen und / oder Therapieschemata oder
ähnliche Unterschiede) erfolgt eine Prüfung der Assessment Reports und bei Unklarheiten eine
gezielte Einsicht in die Grunddokumentation.
7 Vgl. Art. 12 Abs. 1 der Verordnung des Schweizerischen Heilmittelinstituts vom 22. Juni 2006 über die vereinfachte
Zulassung von Arzneimitteln und die Zulassung von Arzneimitteln im Meldeverfahren (VAZV; SR 812.212.23)

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 10 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Können die gegenüber dem Zulassungsentscheid der Referenzbehörde bestehenden Bedenken
durch eine Prüfung der Assessment Reports nicht ausgeräumt werden, führt das Institut eine auf
diese Punkte bezogene wissenschaftliche Begutachtung unter Einbezug weiterer Unterlagen durch.
7.3 Transparenz betreffend wesentlicher Bedenken
Die Gründe für Bedenken, welche gegebenenfalls eine eigenständige Begutachtung durch
Swissmedic zur Folge haben können, werden der Gesuchstellerin im Schreiben „Mitteilung Termin
LoQ“ dargelegt.
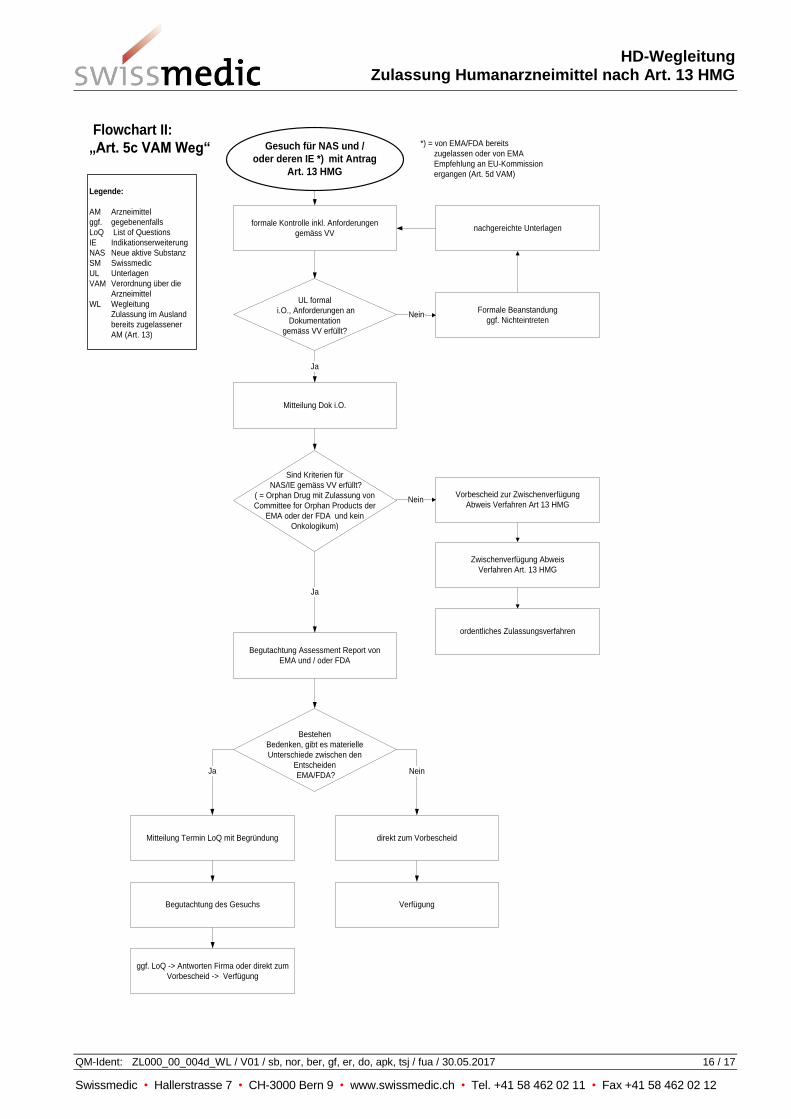
8 NAS oder deren Indikationserweiterung (Art. 5c VAM)
Gesuche um Zulassung eines Arzneimittels mit einer neuen aktiven Substanz (New Active
Substance) oder deren Indikationserweiterung werden vom Institut in der Regel eigenständig und
umfassend anhand aller vorgelegten Unterlagen begutachtet, ebenso Gesuche für die Zulassung von
Präparaten nach Art. 12 Abs. 4 VAZV.
Swissmedic kann die Begutachtung in begründeten Fällen auf Gesuch hin oder von Amtes wegen,
gestützt auf entsprechende ausländische Prüfungsergebnisse, angemessen reduzieren. (siehe
Flowchart II im Anhang).
8.1 Einschränkung der Begutachtung auf Gesuch hin
Eine reduzierte Begutachtung unter Anwendung von Art. 13 HMG für NAS oder deren
Indikationserweiterungen resp. nach Art. 12 Abs. 4 VAZV zuzulassende Präparate ist nur bei
Arzneimitteln, die durch das Committee for Orphan Medicinal Products (COMP) der EMA oder durch
den Orphan Drug Act der FDA als „Orphan Drug“ eingestuft und zugelassen sind, möglich. Davon
ausgenommen sind onkologische Arzneimittel diese werden immer eigenständig von Swissmedic
begutachtet.
Für ein Gesuch um Berücksichtigung der Prüfungsergebnisse der ausländischen Behörde(n) eines
„Orphan Drugs“ ist die entsprechende Status-Anerkennung und Zulassung durch die EMA und / oder
FDA vorzulegen.
Die reduzierte Begutachtung beschränkt sich in der Regel auf eine Prüfung der vorgelegten
Zulassungsentscheide und Assessment Reports von EMA und / oder FDA. Das Institut geht hierbei
wie in Kapitel 7.1 dargelegt, vor.
Werden die oben aufgeführten Kriterien (Orphan Drug und kein Onkologikum) nicht erfüllt, wird eine
Zwischenverfügung (Abweisung Anwendung von Art. 13 HMG) erlassen und das Gesuch unterliegt
dann dem normalen Zulassungsverfahren.
8.2 Einschränkung der Begutachtung von Amtes wegen
Das Institut kann in begründeten Fällen auch von Amtes wegen eine eingeschränkte eigene
Begutachtung beschliessen namentlich dann, wenn es im Interesse der öffentlichen Gesundheit
erforderlich ist, das Verfahren zu beschleunigen (z.B. Zulassung eines Impfstoffs im Pandemiefall).
Das Institut wird ein diesbezügliches Vorgehen mit der Antragstellerin fallweise regeln.
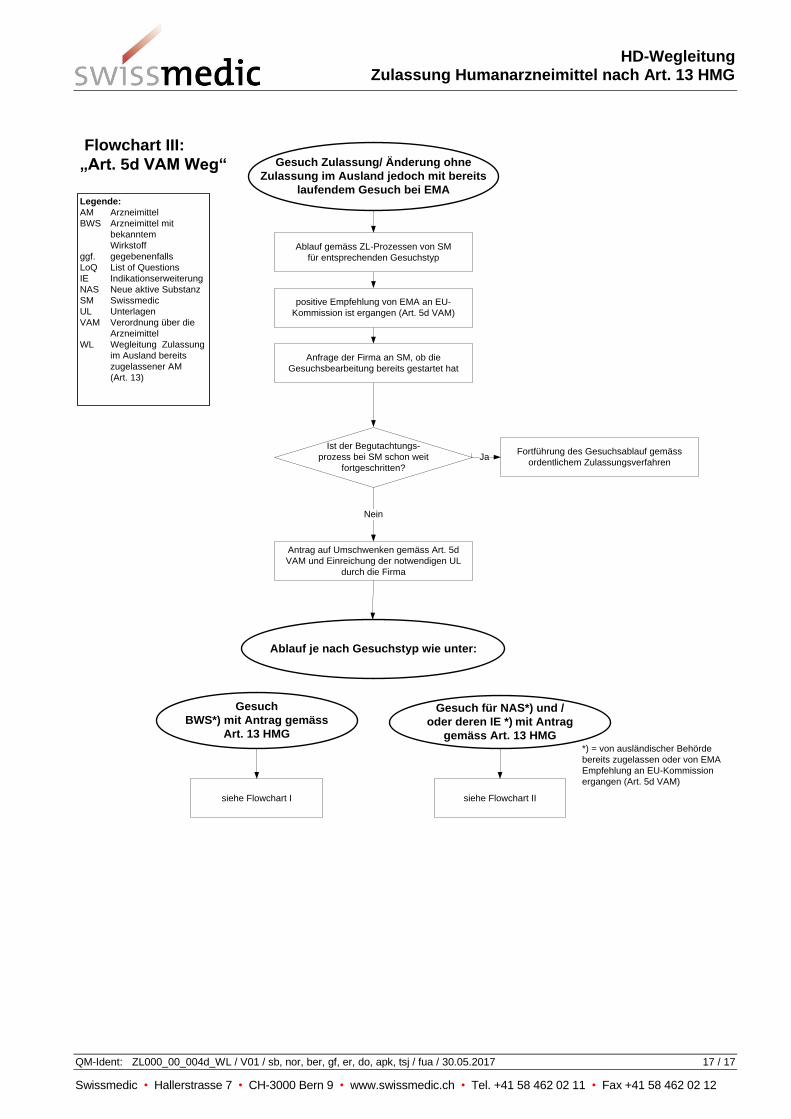
9 Parallele Verfahren in der Schweiz und im Ausland (Art. 5d VAM)
Bei der Einreichung eines Gesuchs im normalen Zulassungsverfahren wird im Formular Status
Zulassungsgesuche im Ausland angegeben, ob eine Zulassung für dasselbe Arzneimittel bei der
EMA bereits beantragt wurde.
Sobald eine Empfehlung der EMA an die EU-Kommission ergangen ist, wendet das Institut auf
entsprechendes Gesuch der Firma hin die Artikel 5a - 5c VAM analog an, sofern auf Grund der
bereits erfolgten eigenen Begutachtung keine wesentlichen Bedenken bestehen und es absehbar ist,
dass dieses Vorgehen (Ablauf nach Art. 13 HMG) zu einem rascheren Entscheid führen wird (siehe
Flowchart III im Anhang).

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 11 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
10 Prozess bei Swissmedic
10.1 Gesuchsbearbeitung
Bei der formalen Kontrolle wird geprüft, ob die Antragstellerin die Übereinstimmung der
Dokumentation mit der im Ausland zugelassenen Dokumentation bestätigt und die geforderten
Unterlagen vollständig vorliegen. Die Checkliste Formale Kontrolle Zulassungsgesuch
Humanarzneimittel Art. 13 HMG bildet einen integrierten Bestandteil dieser Prüfung. Ebenfalls wird
geprüft, ob die Anforderungen an die Dokumentation nach Art. 5a VAM entsprechend eingehalten
wurden. Die Abteilung Submission informiert die Gesuchstellerin über das Ergebnis der formalen
Kontrolle.
Alle BWS-Gesuche bzw. alle Änderungsgesuche gemäss Kapitel 7, welche die formalen
Anforderungen erfüllen, werden immer unter Anwendung von Art. 13 HMG begutachtet.
Bei Gesuchen für NAS oder deren IE resp. bei Präparaten nach Art. 12 Abs. 4 VAZV wird geprüft, ob
die Kriterien gemäss Kapitel 8 erfüllt sind und das Gesuch folglich für den Prozessablauf nach Art. 5a
- 5d VAM qualifiziert ist.
Werden keine Fragen identifiziert, welche eine LoQ notwendig machen, wird der Gesuchstellerin
direkt der Vorbescheid zugestellt.
10.2 Dauer und Kosten des Verfahrens
Stellt die Zulassungsinhaberin ein Gesuch um Berücksichtigung der Prüfungen ausländischer
Behörden nach Art.13 HMG / Art.5a - 5d VAM, erfüllt sie die in dieser Anleitung beschriebenen
Voraussetzungen und kann der Zulassungsentscheid des Instituts in der Folge auf die Ergebnisse der
Prüfungen der Referenzbehörde abgestützt werden, so reduzieren sich die im Einzelfall anwendbaren
Pauschalgebühren gemäss gültiger Gebührenverordnung (Bst. B Ziff. 8 des Anhangs 2 zur
Verordnung über die Gebühren des Schweizerischen Heilmittelinstituts [HGebV SR 812.214.5]). Die
Bearbeitungszeit wird verkürzt, wenn sich keine Fragen (LoQ-Schreiben) ergeben.
10.3 Öffentlichkeit und Transparenz
Über die Anzahl Gesuche mit Nachvollzug einer ausländischen Zulassung führt Swissmedic eine
interne Liste mit Angabe der betroffenen Arzneimittel. Diese kann für statistische Zwecke verwendet
und summarisch publiziert werden.

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 12 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
11 Anhang
Folgende Unterlagen (Entscheide und zusätzliche Dokumentation) müssen im Einzelnen ergänzend
eingereicht werden:
11.1 Unterlagen, die für Gesuche aller Referenzbehörden eingereicht werden müssen
Modul 1 bis 5 resp. Teil I bis IV (NtA) wie bei der ausländischen Behörde eingereicht. (Bei
Gesuchen für wesentliche Änderungen ohne vorherige Bezugnahme auf Art. 13 HMG gemäss
Kapitel 7 sind die Dokumente für die Erstzulassung ebenfalls einzureichen, wenn mehrheitlich auf
diese verwiesen wird.)
Sofern zutreffend: bei DMF / ASMF ist eine identische Kopie des Restricted Part inklusive Letter
of Access, Assessment Report des Restricted Part, LoQ und Antworten der Firma zum Restricted
Part vom Holder einzureichen.
Schweizerisches Modul 1 (gemäss Wegleitung Formale Anforderungen und das dazugehörigen
Verzeichnis Tabelle Einzureichende Unterlagen) inkl. CL Formale Kontrolle Zulassungsgesuch
Humanarzneimittel Art. 13 HMG
Begleitschreiben (gemäss Wegleitung Formale Anforderungen und das dazugehörigen
Verzeichnis Tabelle Einzureichende Unterlagen). Falls zutreffend sind in folgenden Situationen
Bestätigungen, Erklärungen, kritische Beurteilungen oder zusätzliche Dokumentationen
einzureichen:
bei abweichenden Zulassungsentscheiden z.B. Abweichungen betreffend Indikationen,
Dosierung, Lagerungshinweis, Laufzeit resp. weitere Einschränkungen und ähnlichem, bei
Rückzug, Abweisung, Sistierung resp. laufendem Überprüfungsverfahren
bei Abweichungen oder Ergänzungen der Dokumentation und / oder des DMF / ASMF
(Applicant‘s Part und Restricted Part) seit dem Zulassungsentscheid: eine Gegenüberstellung
(alt / neu) inkl. kritischer Beurteilung und Assessment Report.
werden statt der entsprechenden Methoden der Ph. Eur. / Ph. Helv. andere Methoden
verwendet, so ist deren Äquivalenz zu den Methoden der Ph. Eur. / Ph. Helv. zu belegen.
bei Änderungsgesuchen ohne frühere Bezugnahme auf Art. 13 HMG, eine Bestätigung durch
eine unterschriftsberechtige Person oder des Regulatory Affairs-Verantwortlichen, dass die
schweizerische Dokumentation und die der Referenzbehörde identisch sind.
bei laufenden GxP Untersuchungen (z.B. Behebung von Mängeln, erforderliche
Nachinspektionen)
bei spezifischen Anforderungen an die Umsetzung der Spontanerfassung vermuteter
unerwünschter Arzneimittelwirkungen in der Schweiz (z.B. spezielle Fragebogen im Rahmen
von „enhanced Pharmacovigilance“)
bei Unterschieden zu aktuell gültigen Richtlinien (Guidelines), die zum Zeitpunkt der
Zulassung im Ausland noch nicht in Kraft waren
bei Übersetzungen Bestätigung der Korrektheit der Übersetzung
bei erforderlichen Informationen betreffend Sicherheitssignalen
Zulassungsentscheid inkl. zusätzliche Dokumentation (Prüfungsergebnisse) der Referenzbehörde
gemäss Kapitel 11.2 – 11.10. (Bei abweichendem Entscheid zwischen FDA und EMA siehe
Kapitel 6.3)

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 13 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
11.2 Zulassung bezogen auf EU Centralised Procedure (CP)
Basis ausländischer Entscheid: CxMP / HMPC Opinion
Sobald der Entscheid (Decision) der EU-Kommission vorliegt, ist
dieser nachzureichen.
Zusätzliche Dokumentation: Day 80 Assessment Report
Day 120 LoQ
Day 180 LoOI
Answers to Day 120 LoQ
Answers to Day 180 LoOI
Day 210 Assessment Report
Risk Management Plan RMP (für NAS und deren IE)
Pediatric Investigation Plan and amendments (sofern vorhanden)
11.3 Zulassung bezogen auf EU Mutual Recognition Procedure MRP und Decentralised Procedure DCP
Basis ausländischer Entscheid: Marketing Authorisation in RMS (Letter of approval oder Letter
end of procedure)
Zusätzliche Dokumentation: LoQ
Answers to LoQ
Day 90 RMS Assessment Report (für MRP)
Day 70 Preliminary Assessment Report (für DCP)
Final Assessment Report (MRP = Day 90 DCP ≥ Day 105)
Bei Arbitration to CHMP (EMA) sofern zutreffend ist die Opinion
der EMA einzureichen
11.4 Zulassung bezogen auf EU und EFTA-Staaten: Nationale Zulassungen
Basis ausländischer Entscheid: Marketing Authorisation (Letter of approval oder Letter end of
procedure)
Zusätzliche Dokumentation: LoQ
Answers to LoQ
Assessment Report resp. Evaluationsbericht
11.5 Zulassung bezogen auf Zulassung USA / FDA
Basis ausländischer Entscheid: Approval Letter (keine Rolling Submission)
Zusätzliche Dokumentation: LoQ
Answers to LoQ
Assessment Report: Standard oder Priority Review
Sofern vorhanden: Summary Basis of Approval SBA
Sofern gefordert: Risk Minimization Action Plan (RiskMAP)
ERA
11.6 Zulassung bezogen auf Zulassung Japan
Basis ausländischer Entscheid: Marketing Authorisation
Zusätzliche Dokumentation: LoQ (übersetzt d, f oder e)
Answers to LoQ (übersetzt d, f oder e)
Review Reports of New Drug Applications PMDA (übersetzt d, f
oder e)
Review Summaries und Overall Summary Basis of Decision
(übersetzt d, f oder e)
ERA
11.7 Zulassung bezogen auf Zulassung Kanada
Basis ausländischer Entscheid: Notice of Compliance (NOC)

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 14 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Zusätzliche Dokumentation: LoQ
Answers to LoQ
Assessment Report
Sofern vorhanden: Summary Basis of Decision
ERA
11.8 Zulassung bezogen auf Zulassung Australien
Basis ausländischer Entscheid: Marketing Authorisation
Zusätzliche Dokumentation: LoQ
Answers to LoQ
Assessment Report
ERA
11.9 Zulassung bezogen auf Zulassung Singapur
Basis ausländischer Entscheid: Marketing Authorisation
Zusätzliche Dokumentation: LoQ (übersetzt d, f oder e)
Answers to LoQ (übersetzt d, f oder e)
Assessment Report (übersetzt d, f oder e)
ERA
11.10 Zulassung bezogen auf Zulassung Neuseeland
Basis ausländischer Entscheid: Marketing Authorisation
Zusätzliche Dokumentation: LoQ
Answers to LoQ
Assessment Report
ERA
11.11 Flowcharts der Gesuchsabläufe
Flowchart I: Gesuch BWS und Änderungen mit Antrag Art. 5a - 5d VAM
Flowchart II: Gesuch NAS und deren IE mit Antrag Art. 5a - 5d VAM
Flowchart III: Gesuch Zulassung / Änderung ohne Zulassung im Ausland jedoch mit laufendem
Gesuch bei EMA

HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 15 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
nachgereichte Unterlagen
Verfügung
Verfügung
Gesuch BWS und Änderungen *)
mit Antrag Art. 13 HMG
direkt zum
Vorbescheid
Begutachtung
Assessment Report
EMA/FDA bezüglich
identifizierter
Bedenken
Begutachtung des
Gesuchs bezügl.
Bedenken
Mitteilung „Dok i.O.“
Ja
Nein
direkt zum
Vorbescheid
Ja
Nein
ggf. LoQ -> Antworten
Firma oder direkt zum
Vorbescheid ->
Verfügung
*) von ausländischer Behörde bereits
zugelassen oder von EMA Empfehlung
an EU-Kommission ergangen (Art. 5d
VAM)
Verfügung
Begutachtung des
Gesuchs bzgl.
Bedenken
Legende:
AM Arzneimittel
AR Assessment Report
BWS Arzneimittel mit
bekanntem Wirkstoff
ggf. gegebenenfalls
LoQ List of Questions
SM Swissmedic
UL Unterlagen
VA Verordnung über die
Arzneimittel
WL Wegleitung
Zulassung im
Ausland bereits
zugelassener AM
(Art. 13)
Flowchart I:
„Art. 5b VAM Weg“
Nein
Begutachtung
Assessment Report
der Referenzbehörde
formale Kontrolle inkl. Anforderungen
gemäss WL und Art. 5a-5d VAM
Formale Beanstandung ggf.
Nichteintreten
Bestehenmaterielle Widersprüche
zwischen EMA/FDA Zulassung oder Bedenken
aufgrund früherer Ergebnisse?
Bestehen noch
Bedenken ?
formal i.O.,
Anforderungen
an Dokumentation
erfüllt?
Welche
Referenzbehörde ?
direkt zum
Vorbescheid
Nein
Mitteilung Termin LoQ
mit Begründung
Mitteilung Termin LoQ
mit Begründung
Ja
ggf. LoQ -> Antworten
Firma oder direkt zum
Vorbescheid ->
Verfügung
JaBestehen noch
Bedenken ?
Zulassung
EMA u/o FDA
andere Behörden
gemäss WL
HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 16 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Gesuch für NAS und /
oder deren IE *) mit Antrag
Art. 13 HMG
*) = von EMA/FDA bereits
zugelassen oder von EMA
Empfehlung an EU-Kommission
ergangen (Art. 5d VAM)
Verfügung
Mitteilung Termin LoQ mit Begründung direkt zum Vorbescheid
Nein
Begutachtung des Gesuchs
ggf. LoQ -> Antworten Firma oder direkt zum
Vorbescheid -> Verfügung
Mitteilung Dok i.O.
ordentliches Zulassungsverfahren
Legende:
AM Arzneimittel
ggf. gegebenenfalls
LoQ List of Questions
IE Indikationserweiterung
NAS Neue aktive Substanz
SM Swissmedic
UL Unterlagen
VAM Verordnung über die
Arzneimittel
WL Wegleitung
Zulassung im Ausland
bereits zugelassener
AM (Art. 13)
nachgereichte Unterlagen
Begutachtung Assessment Report von
EMA und / oder FDA
Ja
Flowchart II:
„Art. 5c VAM Weg“
formale Kontrolle inkl. Anforderungen
gemäss VV
Formale Beanstandung
ggf. Nichteintreten
Bestehen
Bedenken, gibt es materielle
Unterschiede zwischen den
Entscheiden
EMA/FDA?Ja
Sind Kriterien für
NAS/IE gemäss VV erfüllt?
( = Orphan Drug mit Zulassung von
Committee for Orphan Products der
EMA oder der FDA und kein
Onkologikum)
Zwischenverfügung Abweis
Verfahren Art. 13 HMG
Ja
Vorbescheid zur Zwischenverfügung
Abweis Verfahren Art 13 HMG
UL formal
i.O., Anforderungen an
Dokumentation
gemäss VV erfüllt?
Nein
Nein
HD-Wegleitung
Zulassung Humanarzneimittel nach Art. 13 HMG
QM-Ident: ZL000_00_004d_WL / V01 / sb, nor, ber, gf, er, do, apk, tsj / fua / 30.05.2017 17 / 17
Swissmedic • Hallerstrasse 7 • CH-3000 Bern 9 • www.swissmedic.ch • Tel. +41 58 462 02 11 • Fax +41 58 462 02 12
Gesuch Zulassung/ Änderung ohne
Zulassung im Ausland jedoch mit bereits
laufendem Gesuch bei EMA
Fortführung des Gesuchsablauf gemäss
ordentlichem Zulassungsverfahren
Legende:
AM Arzneimittel
BWS Arzneimittel mit
bekanntem
Wirkstoff
ggf. gegebenenfalls
LoQ List of Questions
IE Indikationserweiterung
NAS Neue aktive Substanz
SM Swissmedic
UL Unterlagen
VAM Verordnung über die
Arzneimittel
WL Wegleitung Zulassung
im Ausland bereits
zugelassener AM
(Art. 13)
Ablauf je nach Gesuchstyp wie unter:
Ja
Nein
Gesuch
BWS*) mit Antrag gemäss
Art. 13 HMG
Gesuch für NAS*) und /
oder deren IE *) mit Antrag
gemäss Art. 13 HMG *) = von ausländischer Behörde
bereits zugelassen oder von EMA
Empfehlung an EU-Kommission
ergangen (Art. 5d VAM)
Flowchart III:
„Art. 5d VAM Weg“
Antrag auf Umschwenken gemäss Art. 5d
VAM und Einreichung der notwendigen UL
durch die Firma
siehe Flowchart I siehe Flowchart II
Ablauf gemäss ZL-Prozessen von SM
für entsprechenden Gesuchstyp
positive Empfehlung von EMA an EU-
Kommission ist ergangen (Art. 5d VAM)
Anfrage der Firma an SM, ob die
Gesuchsbearbeitung bereits gestartet hat
Ist der Begutachtungs-
prozess bei SM schon weit
fortgeschritten?


















