
Guía de desarrollos preclínicos - icono.fecyt.es · Fuente: Predictive ADME and Toxicology...
87
Guía de desarrollos preclínicos
Transcript of Guía de desarrollos preclínicos - icono.fecyt.es · Fuente: Predictive ADME and Toxicology...

Guía de desarrollos preclínicos

Guía de desarrollos preclínicos
© Fundación para el Desarrollo de la Investigación
en Genómica y Proteómica (Genoma España)
Coordinación y redacción:
Emilia Gómez (Genoma España)
Eduardo Cunchillos (Innoqua)
Autores:
Mercedes de Miguel (Genoma España)
Cristina Gracia (Genoma España)
Luis Martín (Genoma España)
Mercedes Escribano (Genoma España)
Colaboradores:
Cristina González (Genoma España)
Edición:
Cheo Machín
Fecha:
Febrero de 2012

Guía de desarrollos preclínicos 3
Índice
1. JUSTIFICACIÓN DE LA GUÍA....................................................................................................5
2. EL DESARROLLO DE MEDICAMENTOS: INTRODUCCIÓN .....................................................6
A. FASE DE DESCUBRIMIENTO ............................................................................................... 7
B. FASE PRECLÍNICA.............................................................................................................. 10
C. FASE CLÍNICA.................................................................................................................... 13
D. FASE DE APROBACIÓN Y REGISTRO................................................................................ 14
E. FASE DE DESARROLLO QUÍMICO FARMACÉUTICO......................................................... 16
3. ¿QUÉ ES LA FASE PRE‐CLÍNICA? ...........................................................................................19
A. ENTORNO REGULATORIO................................................................................................ 21
A.1 ORGANISMOS REGULADORES .................................................................................. 21
ICH: INTERNATIONAL CONFERENCE OF HARMONISATION .......................................... 21
EMA: EUROPEAN MEDICINES AGENCY......................................................................... 23
AEMPS: AGENCIA ESPAÑOLA DEL MEDICAMENTO Y PRODUCTOS SANITARIOS........ 24
OCDE: ORGANIZACIÓN PARA LA COOPERACIÓN Y EL DESARROLLO ECONÓMICO .... 25
FDA: FOOD AND DRUG ADMINISTRATION ................................................................... 26
A.2. BASE LEGISLATIVA: COMUNITARIA Y ESPAÑOLA.................................................. 26
A.3. ASESORÍAS CIENTÍFICAS.......................................................................................... 29
A.4. REDUCCIONES DE TASAS.......................................................................................... 33
B. CLASIFICACIÓN DE MEDICAMENTOS ............................................................................. 33
B.1. CLASIFICACIÓN DE LOS MEDICAMENTOS POR SU COMPOSICIÓN ....................... 34
MEDICAMENTOS QUÍMICOS (SMALL MOLECULES) ........................................................ 34
MEDICAMENTOS BIOLÓGICOS ........................................................................................ 35
ASOCIACIONES DE PRINCIPIOS ACTIVOS ........................................................................ 40

Guía de desarrollos preclínicos 4
B.2. CLASIFICACIÓN DE LOS MEDICAMENTOS POR SU INDICACIÓN TERAPÉUTICA .. 41
ANTITUMORALES ............................................................................................................ 41
MEDICAMENTOS HUÉRFANOS........................................................................................ 42
C. EFICACIA VERSUS SEGURIDAD ....................................................................................... 43
C.1. EFICACIA .................................................................................................................... 43
¿POR QUÉ SON NECESARIOS LOS ESTUDIOS?................................................................. 43
¿QUÉ ENSAYOS ES NECESARIO CONSIDERAR?............................................................... 44
C.2. SEGURIDAD................................................................................................................ 45
¿POR QUÉ SON NECESARIOS LOS ESTUDIOS?................................................................. 45
¿QUE ENSAYOS ES NECESARIO CONSIDERAR?............................................................... 45
D. ¿QUÉ TIPO DE MODELOS DEBO INCLUIR EN MIS ENSAYOS? ¿“IN VITRO”, “IN VIVO”,
“IN SILICO”?........................................................................................................................... 52
E. ¿CUÁNDO USAR GLP, GCP, GMP?................................................................................ 58
F. PREFORMULACIÓN Y PREPARACIÓN DE LOTES. ¿QUÉ DEBO TENER EN CUENTA?...... 60
4. CASOS PRÁCTICOS ................................................................................................................61
A. QUÍMICO (SMALL MOLECULE) ....................................................................................... 61
B. BIOTECNOLÓGICO: PROTEÍNA RECOMBINANTE ........................................................... 66
C. ANTITUMORAL ................................................................................................................. 71
D. DESARROLLO BIOLÓGICO ............................................................................................... 74
5. PROVEEDORES .......................................................................................................................77
A. ¿POR QUÉ CONTRATAR UN PROVEEDOR ESPECIALIZADO DE SERVICIOS?................. 77
B. ¿POR QUÉ LA NECESIDAD DE ELABORAR UN PLAN DE DESARROLLO PRECLINICO? .. 78
6. GLOSARIO ..............................................................................................................................80

Guía de desarrollos preclínicos 5
1. JUSTIFICACIÓN DE LA GUÍA
La Fundación Genoma España pretende, con esta iniciativa, facilitar
una guía de referencia para todos aquellos que, disponiendo de un
potencial agente terapéutico, quieran abordar su desarrollo como
medicamento. Dentro del desarrollo global de un nuevo producto
definimos el ámbito y la necesidad del desarrollo preclínico, así como
su ubicación en todo su recorrido desde el laboratorio al paciente.
Los estudios preclínicos constituyen una etapa crucial en el
desarrollo de nuevas moléculas y terapias, y actualmente constituyen
un cuello de botella en el desarrollo farmacéutico, concretamente para el paso de moléculas a
fase clínicas.
En el entorno académico, fundamentalmente debido al elevado coste, y muchas veces al
desconocimiento de los requisitos mínimos para llevar a cabo este tipo de ensayos, se
produce un retraso importante en el desarrollo de terapias susceptibles de pasar a fase
clínicas. Se hace necesario por tanto el fomento de este tipo de estudios, dirigidos a
profesionalizar estas actividades e impulsar el paso de moléculas y terapias a fases clínicas.
El objetivo principal de esta iniciativa es añadir valor a los resultados de investigación
científica fomentado la realización de actividades de investigación preclínica con moléculas o
terapias de alto potencial farmacológico. Se pretende con ello favorecer la transferencia de
resultados de investigación al mercado, reforzando la competitividad y capacidad de la
I+D+i de la Biotecnología en España y de las empresas relacionadas con el sector.
Así pues los dos objetivos de la guía son:
Proporcionar una hoja de ruta para emprender un desarrollo preclínico regulatorio,
es decir un documento guía que no sólo describa las diferentes etapas a seguir, sino
que también sirva de ayuda en la toma de decisiones en entornos públicos
(promoción de programas y políticas de fomento de estudios preclínicos) y privados
(diseño de estrategias de desarrollo de productos y servicios).

Guía de desarrollos preclínicos 6
Identificar a los actores implicados en desarrollos pre-clínicos, proporcionando un
directorio en el que encontrar los principales centros que ofrecen servicios
relacionados con estudios preclínicos, como por ejemplo CROs (Contract Research
Organizations), CMOs (Contract Manufacturing Organizations), consultoras, empresas
biotecnológicas, laboratorios y centros de investigación, etc..
2. EL DESARROLLO DE MEDICAMENTOS: INTRODUCCIÓN
El desarrollo de un nuevo medicamento es un proceso largo y costoso cuyo objeto es
demostrar en distintas fases, descubrimiento, preclínica y clínica, que el nuevo fármaco
reúne los requisitos de eficacia, seguridad y calidad exigidos para su comercialización y
administración al ser humano.
Las fases de este proceso son:
A. Fase de descubrimiento
B. Fase preclínica
C. Fase clínica (humanos)
D. Fase de aprobación y registro
E. Fase de desarrollo químico farmacéutico

Guía de desarrollos preclínicos 7
Confluyen en este proceso numerosas actividades y disciplinas, todas ellas dirigidas a
demostrar seguridad, eficacia, y una relación coste-eficacia óptima lo que se traduce en un
considerable esfuerzo intelectual y en un enorme costo económico.
A. FASE DE DESCUBRIMIENTO
A lo largo de la historia muchos medicamentos fueron descubiertos por mediación del azar.
Así, sin conocer el origen de las dolencias o el efecto sobre el organismo de los remedios
tradicionales ya se disponía de tratamientos eficaces para diversas patologías. Muchos de
estos remedios tradicionales se basaban en productos naturales que, por el método del
ensayo y error, habían logrado remediar un mal o enfermedad.
Hoy en día sin embargo poco se deja al azar, y el descubrimiento de fármacos, así como su
posterior desarrollo, se ha convertido en un proceso largo y complejo, muy estructurado y
que además requiere un concienzudo diseño previo.
El primer paso siempre es definir el objetivo; así basándose en intereses científicos y
estratégicos se decide en qué patología trabajar. Una vez definido esto se buscarán moléculas
que presenten actividad biológica relacionada con esta patología.
Aunque habitualmente, y para simplificar, nos referiremos a estas sustancias como fármacos
conviene, al menos inicialmente, definir unas diferencias que ilustren además la sucesión de
etapas que implica el desarrollo de fármacos.
Figura 1. Sustancias con actividad biológica
Fuente: elaboración propia

Guía de desarrollos preclínicos 8
Esta búsqueda de posibles fármacos para una determinada enfermedad puede hacerse a
través de un abordaje biológico o a través de un abordaje químico:
Abordaje biológico: consiste en definir el objetivo terapéutico, es decir, el
componente del organismo implicado en el desarrollo de la patología sobre el cual se
desea actuar, y buscar posibles fármacos que alteren su función.
Abordaje químico: consiste en probar una batería de compuestos previamente
disponibles en un sistema modelo de la enfermedad hasta observar cuáles de ellos
tienen un efecto sobre el desarrollo de la misma.
Figura 2. Abordajes biológico y químico en el descubrimiento de fármacos.
Fuente: Predictive ADME and Toxicology Strategies. Dr. Nelesh Patel. 2006. Business Insights Ltd
Estos ensayos de cribado de compuestos o posibles fármacos resultan complejos debido al
elevado número de moléculas disponibles y/o generadas que se prueban así como a la
reducida calidad y disponibilidad (cantidad) de las mismas.
Por eso se utilizan técnicas asociadas al High Throughput Screening (HTS), o cribado de alta
capacidad, que permiten seleccionar rápidamente las nuevas moléculas capaces de reconocer
una determinada diana. Estas moléculas son conocidas en el sector como Hits.

Guía de desarrollos preclínicos 9
Estos hits son de gran interés porque informan sobre los requerimientos estructurales
necesarios para interacciones con el receptor, sin embargo a menudo su actividad es débil, su
estructura químicamente inestable, y resultan tóxicos o poco selectivos.
Para mejorar sus propiedades se emprende un proceso de optimización de los hits que
incluye ensayos para confirmar su afinidad por la diana, modificaciones para aumentar su
afinidad y finalmente la selección de aquellos que, por tener mejores propiedades que los
demás, serán los potenciales candidatos al desarrollo. Estos candidatos a fármacos se
conocen como Leads.
Tal y como se ilustra en la siguiente imagen el proceso comúnmente conocido como “From
hit to lead” no es lineal. En cada ensayo realizado, así como en la documentación consultada,
se va obteniendo nueva información que ayuda a definir las relaciones estructura-actividad
que permite identificar los grupos funcionales, los fragmentos, o las propiedades
moleculares que favorecen el que algunos compuestos resulten más o menos activos en los
tests. La continua incorporación de este conocimiento hace que este proceso de optimización
sea cíclico.
Figura 3. Etapas de la fase del descubrimiento de fármacos
Fuente: El fármaco y su desarrollo: diez preguntas y una consideración. An. Quim. 2006, 102 (3), 13‐22 (www.rseq.org)

Guía de desarrollos preclínicos 10
B. FASE PRECLÍNICA
Una vez seleccionados los “leads” se diseñará el programa de la fase preclínica para
caracterizar principalmente el perfil de seguridad del candidato a fin de reducir y anticipar
en lo posible el riesgo existente para humanos antes de comenzar el ensayo clínico.
Tal y como se indica en la figura el paso de la fase de “Drug Discovery” a la preclínica lo
determina la identificación de “leads” definidos, según criterios seleccionados
individualmente, por el promotor del desarrollo (empresa, grupo de investigación, etc.). Sin
embargo, el paso de la fase preclínica a la clínica está sujeto a la obtención de la aprobación
por parte de las Autoridades Regulatorias pertinentes (FDA - Food and Drug Administration,
EMA - European Medicines Agency, etc.).
Para enmarcar mejor el desarrollo de fármacos en la siguiente figura se pueden apreciar los
tres pasos básicos: descubrimiento, preclínica y clínica
*ADME: Absorción, Distribución, Metabolismo y Excreción Figura 4. Etapas del desarrollo de un fármaco Fuente: elaboración propia

Guía de desarrollos preclínicos 11
En términos generales se puede decir que la preclínica regulatoria, realizada exclusivamente
en laboratorio (ya sea en animales o en cultivos de células), sirve sobre todo para demostrar
la falta de efectos adversos, mientras que es en la fase clínica (que ya se realiza en humanos)
cuando se enfatiza en probar la eficacia terapéutica. Esto se debe a que una de las causas más
importantes de la interrupción del desarrollo de un lead es la aparición de efectos tóxicos,
mientras que la falta de eficacia terapéutica contribuye en una proporción más reducida.
Figura 5. Principales factores que provocan la interrupción del desarrollo de un fármaco.
Fuente: Predictive ADME and Toxicology Strategies. Dr. Nelesh Patel. 2006. Business Insights Ltd.
A lo largo de la historia ha habido casos catastróficos de toxicidad para humanos (ejemplo,
caso de la talidomida) y por eso las autoridades sanitarias incrementaron sus medidas de
seguridad, exigiendo un gran número de pruebas toxicológicas antes de dar su aprobación a
la administración a humanos de los posibles fármacos. Todas estas exigencias están
recogidas en numerosas guías publicadas por las agencias responsables de la aprobación de
fármacos.
Aunque es acertado afirmar que dicha regulación exige que un número importante de
pruebas de toxicología se realicen antes del primer ensayo en humanos, algunas pruebas no
se exigen hasta avanzada la fase clínica o incluso el registro. Por eso, tal y como se indica en
la figura 4, se engloba bajo el concepto de “regulatorio preclínico” todas las pruebas de
toxicología realizadas en laboratorio (“in vitro” y/o en animales “in vivo”, pero no en
humanos) exigidas por las autoridades regulatorias, ya deban realizarse antes del primer
ensayo en humanos o antes de las restantes fases clínicas del desarrollo del fármaco.

Guía de desarrollos preclínicos 12
El tipo de pruebas requeridas para aprobar el desarrollo de un fármaco dependerá de
diferentes factores:
Tipo de medicamento: hay regulaciones diferentes para medicamentos de origen
químico, biológico, biotecnológico o terapia avanzada.
Tipo de pacientes: hay grupos poblacionales para los cuales se exigen requisitos
especiales, por ejemplo mujeres embarazadas, niños, ancianos…
Tipo de patologías: existen patologías, como por ejemplo las oncológicas para las
cuales se exigen requisitos especiales.
Distribución geográfica: existen diferentes agencias regulatorias para diferentes
territorios.
Hasta el momento de iniciar el primer ensayo en humanos, se habrían evaluado parámetros
de toxicidad y eficacia tan sólo en cultivos “in vitro” y en animales “in vivo”. Estas opciones,
aunque apropiadas, representan una variabilidad biológica limitada en comparación con la
inmensa variabilidad que supone la población humana mundial. Por otro lado, no sería
adecuado pensar que las reacciones al tratamiento en los animales se pueden extrapolar
directamente a los humanos en todos los casos, ya que existen diferencias de respuesta entre
especies. Además existen ciertas reacciones de difícil determinación en animales como, por
ejemplo, las cefaleas o las depresiones. De ahí que el salto a humanos siempre constituye un
riesgo y para poder optar a la solicitud de un ensayo clínico tiene que existir un balance
beneficio-riesgo adecuado.
Una vez realizadas las pruebas exigidas en la preclínica, se integran los resultados en un
único informe que se presenta a la autoridad regulatoria para que se autorice el comienzo de
la fase clínica, es decir, la realización de pruebas en humanos.
A medida que avanza el desarrollo clínico, debe exponerse a un mayor número de pacientes
al medicamento, con una duración de tratamiento más prolongada y grupos de población
más extensos, pudiéndose incluir mujeres en edad fértil con posibilidad de quedar
embarazadas durante el tratamiento, ancianos, niños, etc. Cada una de estas circunstancias

Guía de desarrollos preclínicos 13
debe haberse evaluado previamente en estudios de seguridad en modelos animales
adecuados para que puedan ser, a su vez, autorizadas.
C. FASE CLÍNICA
Es necesario que antes de comercializar un fármaco éste sea probado en humanos. Esto se
hace, con la finalidad de asegurar la bondad del nuevo fármaco, en tres fases consecutivas en
diferentes grupos poblacionales.
Para comenzar cada una de estas fases se debe solicitar la autorización a las Autoridades
Regulatorias correspondientes presentando los resultados de la fase anterior.
Figura 6. Del descubrimiento a la comercialización. Fuente: Pharmaceutical Research and Manufactures of America
Fase I: es aquella en la que se administra por primera vez el fármaco a humanos y se
hace a un grupo reducido (menos de 100) de voluntarios sanos, generalmente adultos
jóvenes de género masculino. El principal objetivo es detectar signos de toxicidad.
Esta fase puede durar aproximadamente entre 6 meses y un año.

Guía de desarrollos preclínicos 14
Fase II: es aquella en la que, por primera vez, se administra el fármaco a pacientes. Se
administra a un grupo relativamente homogéneo de entre 100 y 200 individuos que
se dividen en dos grupos cuyos resultados posteriormente serán comparados. A un
grupo se le suministra el fármaco y al otro (grupo control) se le administra o bien el
mejor medicamento del mercado contra la patología o un placebo
El principal objetivo es verificar la eficacia del fármaco. Esta fase es más larga que la
anterior, suele durar aproximadamente entre 2 y 3 años.
Fase III: los ensayos de esta fase se caracterizan por ser multicéntricos y en
numerosos pacientes (cientos o miles) con características heterogéneas, así se
consigue una gran diversidad biológica que ayuda a concretar los perfiles de
seguridad y eficacia del fármaco. En esta fase además se pueden detectar
manifestaciones de toxicidad que no se habían sospechado previamente. La duración
de estos ensayos suele ser de entre 3 y 5 años, lo cual permite evaluar efectos de
seguridad y toxicidad a largo plazo.
Fase IV: tiene lugar una vez ya ha sido autorizado el fármaco y consiste en realizar
un seguimiento del mismo después de su comercialización por eso también se
conocen como estudios de farmacovigilancia. Básicamente se buscan efectos adversos
raros (frecuencia menor a 1/1000) o a muy largo plazo derivados, por ejemplo, de una
exposición continuada al fármaco. Así mismo, también puede servir para identificar
posibles efectos terapéuticos no detectados anteriormente.
El desarrollo de un nuevo medicamento es, por tanto, un proceso largo, complejo y costoso
cuyo objeto es demostrar en distintas fases que el nuevo fármaco reúne los requisitos de
eficacia y seguridad (relación beneficio-riesgo aceptable), y de calidad exigidos para su
comercialización y administración al ser humano.
D. FASE DE APROBACIÓN Y REGISTRO
El resultado de los estudios de la Fase Clínica III proporciona la base para la aprobación.

Guía de desarrollos preclínicos 15
Una vez que el registro ha sido presentado en la Administración correspondiente, la revisión
y aprobación, en su caso, supone unos dos o tres años.
En la Unión Europea existen varias modalidades para la aprobación (o autorización de
comercialización) de un medicamento. Ya que cada procedimiento es complejo en sí mismo,
aquí nos limitaremos a indicar que los tipos de registro pueden ser por procedimiento
centralizado, por reconocimiento mutuo y por procedimiento descentralizado. Estos
mecanismos permiten autorizar de forma más breve y eficiente el medicamento, en todos o
muchos de los países de la Unión Europea, sin necesidad de realizar solicitudes
independientes en cada país. Para más detalle se recomienda consultar la web de la EMA
(http://www.ema.europa.eu) y la legislación correspondiente (Reglamento Nº 726/2004 del
Parlamento Europeo y del Consejo). Dependiendo del procedimiento elegido los plazos para
la aprobación varían desde 9 meses a dos o tres años. Hay que destacar que algunos tipos de
medicamentos, entre los que se encuentran los medicamentos de origen biotecnológico,
necesitan ser autorizados mediante procedimiento centralizado.
La agencia reguladora evalúa los estudios presentados por el promotor, y sus resultados
relativos a la seguridad y eficacia del medicamento para el uso propuesto. Ningún
medicamento es completamente seguro ya que todos los medicamentos tienen efectos
secundarios. “Seguro” hace referencia a que los beneficios del medicamento aparentan ser
mayores que los riesgos. Es por ello que si los beneficios de un medicamento compensan sus
riesgos, el medicamento recibe la aprobación.
El producto aprobado, se convierte en medicamento innovador, y el promotor realiza el
lanzamiento de los medicamentos derivados. Pero la responsabilidad con el producto
generado continúa, ya que, con el lanzamiento al mercado comienza la Fase Clínica IV, que
implica el seguimiento de los efectos del producto o farmacovigilancia. La razón de este
seguimiento está en el hecho de que, aunque el producto se haya estudiado en sus fases
clínicas previas sobre grupos de pacientes relativamente grandes, cuando sale al mercado su
uso se expande a muchos miles de pacientes en todo el mundo, con una diversidad genética

Guía de desarrollos preclínicos 16
y una variedad de condiciones y de posibles interacciones como no se ha dado en ninguno
de los grupos utilizados en su fase de desarrollo, por muy compleja que esta haya sido.
Por ello, el promotor realiza un seguimiento detallado de las incidencias que el uso del
producto pudiese generar, bien para modificar la información técnica relativa al producto,
bien para modificar las recomendaciones de uso, o bien para retirar el producto del mercado,
en caso de que apareciesen efectos secundarios adversos.
E. FASE DE DESARROLLO QUÍMICO FARMACÉUTICO
Durante el desarrollo de un medicamento se deben desarrollar y validar una serie de
procesos relacionados con la correcta fabricación del principio activo y del medicamento
como producto acabado, así como realizar numerosos estudios que tienen en conjunto como
finalidad caracterizar el medicamento y establecer los controles necesarios para garantizar la
calidad de éste. Al conjunto de estudios y procesos encaminados a caracterizar la calidad del
medicamento se le llama desarrollo químico-farmacéutico. También se utilizan a menudo las
siglas en inglés CMC que significan “Chemistry, Manufacturing and Controls”.
Muy brevemente, podemos definir los procesos involucrados en la caracterización del
principio activo y del medicamento.
Caracterización del principio activo
El principio activo (o “drug substance”, en inglés) debe estar muy bien definido químicamente
desde los inicios del desarrollo de un medicamento. La composición exacta, tanto si es una
sustancia activa como si es una combinación, la estructura química y la pureza deben estar
definidas. Debemos pensar que cualquier pequeña modificación en la composición de un
principio activo o de sus propiedades físico-químicas (cristalización, tamaño de partícula,
estructura tridimensional, estereiosomería, etc.) podría dar lugar a diferencias importantes
en actividad farmacológica, seguridad y eficacia. Por lo tanto se debe poder garantizar la
homogeneidad de la sustancia a lo largo de todo el desarrollo en los diferentes lotes.

Guía de desarrollos preclínicos 17
En el caso de los principios activos de origen biológico o biotecnológico es a veces muy difícil
definir una composición o una estructura química exacta o incluso una estructura
tridimensional (estructura terciaria). Por eso, el esfuerzo que se debe hacer en los inicios con
este tipo de productos para definir la estructura específica responsable de la actividad
farmacológica, es quizá mayor. Muchas veces, estas sustancias complejas no se pueden
definir bien únicamente desde un punto de vista químico y se debe recurrir también a
ensayos biológicos para medir su potencia o actividad biológica.
Síntesis química: Es necesario establecer los pasos de la ruta de síntesis del principio activo,
a fin de que el proceso sea lo más corto posible en el tiempo, más rentable, utilice reactivos lo
menos contaminantes posible y dé lugar a un menor contenido de impurezas residuales en la
sustancia final. La ruta de síntesis se va a ir optimizando conforme avanza el desarrollo del
medicamento y en paralelo, se va a ir dimensionando progresivamente para la obtención de
lotes de cada vez mayor tamaño. A este último proceso se le llama escalado, para referirse a
la evolución gradual de la síntesis desde escala piloto o laboratorio, hasta escala industrial.
Desarrollo de métodos analíticos: tan pronto como sea posible, se deberán desarrollar
métodos analíticos que permitan garantizar la identidad del principio activo, así como
establecer la pureza del lote acabado y detectar las posibles impurezas y cuantificarlas en
proporción al principio activo. Una vez los métodos analíticos están desarrollados y
optimizados, se deberán validar.
Estabilidad: A lo largo del desarrollo del medicamento se deberá averiguar la estabilidad del
principio activo en diferentes condiciones estándar de temperatura y humedad. Con ello se
podrá, no solo conocer la vida útil de la sustancia, sino estudiar los productos de
degradación y la evolución de éstos, así como programar las necesidades de nuevas síntesis y
su periodicidad.
Especificaciones de sustancia: Cuando ya se tiene suficiente conocimiento del
comportamiento físico-químico del principio activo, y siempre antes del primer ensayo
clínico, deberán definirse las especificaciones de la sustancia. Las especificaciones establecen
los márgenes dentro de los cuales un lote puede darse por aceptable desde un punto de vista

Guía de desarrollos preclínicos 18
de calidad. Principalmente definen los límites aceptables en cuanto a pureza, contenido de
humedad, de impurezas individuales y totales, etc. Además, se deben establecer las
especificaciones tanto para la liberación del lote como para el final de la vida útil de la
sustancia o fecha de caducidad.
Caracterización del medicamento
El medicamento (o “drug product” en inglés) es la forma farmacéutica acabada, tal como
deberá ser administrada a los pacientes. Contiene el/los principio/s activo/s y una serie de
sustancias no activas que ayudan de diferentes maneras a que el producto final sea
adecuado, ya sea ayudando en su conservación, como contribuyendo a su presentación
farmacéutica final (cápsulas, comprimidos, soluciones inyectables, etc.), a su solubilidad,
dispersión, liberación modificada, etc. Las sustancias no activas se llaman excipientes.
Selección de la formulación clínica: Los excipientes se seleccionan por su función
determinada, pero deben ser ingredientes que estén previamente aprobados para su uso en
medicamentos, so pena de tener que realizar numerosos estudios para garantizar la
seguridad de cada nuevo excipiente. Además se deben estudiar las posibles interacciones o
incompatibilidades entre excipientes, a fin de evitar la aparición o el aumento de productos
de degradación en el medicamento. Por otro lado, los excipientes deben de estar justificados,
no debiendo añadirse más excipientes de los necesarios ni en cantidad mayor.
Debemos tener en cuenta que, por lo general, los estudios preclínicos por vía oral en
animales no necesitan ser realizados con la formulación propuesta para clínica, pero si la vía
propuesta en humanos es distinta a la oral (inyectable, dérmica, ocular, inhalatoria, etc.) es
muy importante evaluar la fórmula clínica, ya que algunos aspectos como la tolerancia local,
la capacidad de penetración o la solubilidad, dependerán en buena parte de los excipientes
que acompañan al principio activo. Cambios en la formulación final a lo largo del desarrollo
pueden suponer tener que repetir algunos estudios preclínicos.
Desarrollo analítico: También en el caso del medicamento se deben desarrollar métodos
analíticos que permitan analizar correctamente el contenido del principio activo y que

Guía de desarrollos preclínicos 19
aseguren que la presencia de otras sustancias como los excipientes no interfiere en su
cuantificación. De forma similar ocurre con las impurezas, ya que los métodos analíticos
deben garantizar la correcta cuantificación de éstas en el producto acabado.
Fabricación: Al igual que para el principio activo, se deben definir y validar los pasos para la
fabricación del producto acabado, desde la escala piloto a la escala industrial. El estándar de
cumplimiento GMP en la fabricación del lote de producto se exige para ensayos clínicos,
pero no para ensayos preclínicos.
Envase: Se debe definir el envase en el que se dispensará el medicamento tan pronto como
sea posible, ya que se debe estudiar la compatibilidad del medicamento con los materiales
con los que va a estar en contacto. Estos materiales pueden adsorber el principio activo o
determinado excipiente, o pueden liberar extractos o dar lugar a interacciones químicas en el
sistema cerrado. La estabilidad del producto acabado se debe de realizar dentro del envase
final, ya que en numerosas ocasiones las impurezas y productos de degradación dependen
de los materiales en contacto con el medicamento.
Estabilidad: Al igual que con el principio activo, se debe establecer la estabilidad del
producto acabado en diferentes condiciones de temperatura y humedad, para poder
establecer la vida útil del producto y la fecha de caducidad.
Especificaciones: Al igual que para el principio activo, se deben fijar especificaciones para el
medicamento, que establecerán los límites de aceptación de los diferentes parámetros que
componen la calidad final del producto. Estas especificaciones deberán establecerse antes del
primer ensayo clínico y deberán cumplirse en cualquier lote que se vaya a administrar a
humanos, pero no necesariamente en los lotes destinados a estudios preclínicos.
3. ¿QUÉ ES LA FASE PRE‐CLÍNICA?
El desarrollo preclínico de un medicamento hace referencia al conjunto de
estudios de eficacia y seguridad del principio activo que se deben realizar
en sistemas biológicos diferentes al ser humano. Debido a que un

Guía de desarrollos preclínicos 20
determinado grupo de estudios debe de realizarse antes de comenzar las primeras pruebas
en humanos (ensayo clínico) y otros antes de comenzar las restantes fases clínicas, es por lo
que a esta parte del desarrollo se le llama “preclínico”. El objeto de la fase preclínica es
caracterizar la eficacia y seguridad del medicamento en animales o sistemas “in vitro”. Es
requisito indispensable para obtener la autorización por parte de las entidades reguladoras a
proceder al ensayo en humanos, presentar en un dossier toda la información preclínica
obtenida.
El desarrollo preclínico comprende una fase inicial de selección de las nuevas moléculas
candidatas, seguida de la investigación de su potencial acción farmacológica, y finalmente de
la evaluación de su seguridad. La clave del éxito es que las moléculas candidatas posean
ciertas propiedades favorables: actividad biológica y solubilidad adecuada, capacidad para
atravesar barreras críticas, razonable estabilidad metabólica y seguridad en su
administración al hombre.
En la fase preclínica hay que considerar que la investigación ha de ir unida a una simultánea
toma de decisiones, decisiones que abarcan qué hacer, cuándo hacerlo y cómo y con quién
hacerlo.
Cada fármaco particular necesita su propio desarrollo preclínico debiendo tenerse en cuenta
diversos factores: tipo de compuesto, mecanismo de acción e indicaciones clínicas. Es
especialmente importante conocer el ámbito regulatorio y las guías que aplican. En este
sentido es fundamental contemplar dónde se va a realizar el desarrollo clínico, ya que en
ocasiones los requisitos regulatorios de EEUU (“Food and Drug Administration”, FDA) y
Europa (“European Medicines Agency”, EMA) pueden diferir, etc. Igualmente importante para
definir el desarrollo preclínico más adecuado es tener en cuenta el diseño del ensayo clínico,
así como duración del tratamiento, vía y pauta de administración, tipo de población sujeta a
estudio, etc.

Guía de desarrollos preclínicos 21
A. ENTORNO REGULATORIO
Los ensayos preclínicos están ampliamente regulados ya que suponen la llave de entrada
para un ensayo clínico en humanos.
Son varios los organismos reguladores, directrices, legislaciones y guías de diferente
aplicación según el país.
La FDA tiene el control exclusivo sobre las decisiones ejecutivas en relación con la
aprobación de fármacos en los Estados Unidos. Sin embargo, en Europa, es posible tener un
fármaco aprobado por vías diferentes. Esto se debe a que en la Unión Europea (UE) se puede
solicitar la aprobación europea a través de la EMA (“European Medicines Agency”) o a través
de las agencias nacionales (en el caso de España, la AEMPS - Agencia Española de
Medicamentos y Productos Sanitarios). No obstante, desde noviembre de 2005, todos los
nuevos fármacos destinados al tratamiento de determinadas enfermedades (SIDA, cáncer,
diabetes, enfermedades neurodegenerativas) así como los fármacos desarrollados por medio
de procesos biotecnológicos y los que constituyen terapias avanzadas (génica y celular),
deben ser aprobados por la EMA.
Con la globalización de la industria farmacéutica, la Conferencia Internacional sobre
Armonización (ICH) implantó una normalización en el contenido de las solicitudes de
autorización de fármacos. Japón, Estados Unidos y la UE deben cumplir con los requisitos de
calidad, seguridad y eficacia de nuevos fármacos.
A.1 ORGANISMOS REGULADORES
ICH: International Conference of Harmonisation
http://www.ich.org/ La Conferencia Internacional para la Armonización de los Requisitos Técnicos para el Registro de
Productos Farmacéuticos para Uso Humano es el organismo, por excelencia a nivel mundial, que
reúne a las autoridades reguladoras y a la industria farmacéutica de Europa, Japón y Estados
Unidos.

Guía de desarrollos preclínicos 22
Su misión es lograr una armonización en las directrices técnicas y los requisitos para el
registro de productos farmacéuticos, para asegurar la inocuidad y eficacia de los
medicamentos.
La armonización se logra con el consenso entre científicos, expertos reguladores y la
industria de las tres regiones de la ICH. Fruto de ello se elaboran guías aceptadas por las
autoridades reguladoras de estas regiones:
EMA (European Medicines Agency): en Europa
FDA (Food and Drug Administration): en Estados Unidos
MHLW (Ministry of Health, Labour and Welfare): en Japón
La armonización en la reglamentación ofrece muchos beneficios directos a los organismos
reguladores de cada región. Entre estos beneficios cabe destacar:
Evitar la duplicación de los ensayos clínicos en seres humanos y reducir al mínimo
los ensayos con animales sin comprometer la seguridad y la eficacia.
Racionalizar el proceso de evaluación legal de las solicitudes de nuevos
medicamentos.
Reducir los tiempos de desarrollo y recursos para el desarrollo de medicamentos.
La ICH emite guías de interés estructuradas en cuatro ámbitos:
Guías en el área de calidad (Nomenclatura: código Q- seguido de un número): relativas
a la realización de estudios de estabilidad, la definición de límites relevantes para
pruebas de impurezas y un enfoque a la calidad de productos farmacéuticos basados
en GMP (Good Manufacturing Practices).
Guías en el área de seguridad (Nomenclatura: código S- seguido de un número):
directrices para riesgos potenciales como la carcinogenicidad, genotoxicidad y
toxicidad para la reproducción.

Guía de desarrollos preclínicos 23
Guías en el área de eficacia (Nomenclatura: código E- seguido de un número): para el
diseño, desarrollo, seguridad y comunicación de ensayos clínicos. También hay
directrices para medicamentos derivados de procesos biotecnológicos, uso de
farmacogenética y técnicas genómicas para producción de medicamentos más
específicas.
Guías en áreas multidisciplinarias (Nomenclatura: código M- seguido de un número):
para temas transversales que no se ajustan únicamente a una de las categorías
anteriores (calidad, seguridad y eficacia).
EMA: European Medicines Agency
http://www.ema.europa.eu Es un organismo descentralizado de la Unión Europea responsable de la evaluación
científica de los medicamentos desarrollados por las compañías farmacéuticas para su uso en
la Unión Europea. La EMA se responsabiliza de:
Evaluación científica de solicitudes de autorización para comercialización en Europa
de medicamentos humanos y veterinarios mediante un procedimiento centralizado.
Aprobación de todos los medicamentos para usos humanos y animales, generados
biotecnológicamente o mediante otros procesos, por el procedimiento centralizado.
Así mismo para los medicamentos de terapia avanzada de uso humano, para el
tratamiento del VIH / SIDA, cáncer, diabetes, enfermedades neurodegenerativas,
trastornos autoinmunes, enfermedades virales, así como todos los medicamentos
huérfanos para tratamiento de enfermedades raras.
Supervisión constante de la seguridad de los medicamentos a través de una red de
farmacovigilancia.
Estimulación de la innovación y la investigación en el sector farmacéutico. La
Agencia ofrece asesoramiento científico y ayuda a las empresas para el desarrollo de
nuevos medicamentos. Emite directrices de calidad, seguridad y eficacia.

Guía de desarrollos preclínicos 24
La EMA está estructurada en seis Comités Científicos:
Comité de Medicamentos de Uso Humano (CHMP)
Comité de Medicamentos de Uso Veterinario (CVMP)
Comité de Medicamentos Huérfanos (COMP)
Comité de Medicamentos a Base de Plantas (HMPC)
Comité Pediátrico (PDCO)
Comité de Terapias Avanzadas (CAT)
Dentro de cada uno de estos comités científicos (Scientific Committees) la Agencia tiene una
serie de grupos de trabajo (Working Parties) que están formados por expertos. Realizan
evaluación científica de las solicitudes de autorización de comercialización así como
redacción y revisión de las guías que emite la EMA.
En el contexto del desarrollo preclínico, la EMA emite guías en el ámbito de farmacología,
farmacocinética, toxicología, plantas medicinales, además de otra serie de guías generales.
AEMPS: Agencia Española del Medicamento y Productos Sanitarios
http://www.aemps.gob.es/
Es una Agencia estatal por la que España participa, a través de la Red de Agencias Europeas
de Medicamentos, en un modelo en el que las evaluaciones, inspecciones y demás
actuaciones técnicas o científicas se realizan por los equipos de las agencias nacionales, en un
marco de cooperación gestionado por la Agencia Europea del Medicamento (EMA),
organismo de la UE que aúna los recursos de todas las agencias nacionales europeas.
La AEMPS actúa con arreglo a la normativa y directrices técnicas de la Unión Europea. Su
objeto es garantizar que tanto los medicamentos de uso humano como los de uso veterinario
y los productos sanitarios, cosméticos y productos de higiene personal cumplan con estrictos
criterios de calidad, seguridad, eficacia y correcta información con arreglo a la normativa
vigente sobre dichas materias en el ámbito estatal y de la Unión Europea.

Guía de desarrollos preclínicos 25
La AEMPS tiene asignadas muchas competencias entre las que destacan evaluar, autorizar,
modificar, renovar, restringir, suspender o revocar la autorización de comercialización de los
medicamentos de uso humano y veterinario elaborados industrialmente. También es de su
competencia realizar la inscripción de autorizaciones y mantenimiento en el Registro de
Medicamentos, etc.
Como órganos de asesoramiento y coordinación, la Agencia y su Consejo rector, cuentan con
una serie de Comités Técnicos adscritos a la misma. Entre estos, destaca el Comité de
Medicamentos de Uso Humano (CMH) que representa los intereses de la sociedad y vela por
la transparencia, objetividad y rigor científico de las decisiones de la Agencia, en materia de
comercialización de medicamentos.
OCDE: Organización para la Cooperación y el Desarrollo Económico
http://www.oecd.org/
Su misión es la de promover políticas que mejoren el bienestar económico y social de las
personas alrededor del mundo.
La OCDE emite guías muy específicas acerca de cómo llevar a cabo los estudios preclínicos
de seguridad. Estas guías están enfocadas a la evaluación de sustancias químicas en general
para muy diferentes aplicaciones, sin embargo en la mayoría de casos, las recomendaciones
que establecen son aplicables también a medicamentos, por lo que habitualmente a la hora
de diseñar un estudio preclínico siempre se tiene en cuenta o se hace referencia a estas guías
de la OCDE.
Estas directrices de la OCDE para la evaluación de productos químicos son una compilación
de los ensayos más relevantes acordados internacionalmente y utilizados, tanto por los
laboratorios del gobierno como por la industria, para evaluar la seguridad de los productos
químicos.
http://www.oecd-ilibrary.org/environment/oecd-guidelines-for-the-testing-of-chemicals-
section-4-health-effects_20745788

Guía de desarrollos preclínicos 26
Asimismo la OCDE es el organismo que ha emitido una serie de guías o directrices acerca de
las Buenas Prácticas de Laboratorio (BPLs-GLPs). Estas guías se consideran de referencia
para la implementación de las GLPs en los laboratorios preclínicos, así como el seguimiento
y la ejecución de los ensayos preclínicos de seguridad.
http://www.oecd-ilibrary.org/environment/oecd-series-on-principles-of-good-laboratory-
practice-and-compliance-monitoring_2077785x
FDA: Food and Drug Administration
http://www.fda.gov/
La FDA estadounidense también emite directrices para la realización de ensayos preclínicos
dirigidos a soportar ensayos clínicos y para la autorización de comercialización de
medicamentos en Estados Unidos.
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm065014.htm
A.2. BASE LEGISLATIVA: COMUNITARIA Y ESPAÑOLA
Legislación comunitaria (publicada en el Diario Oficial de la Unión Europea)
Las principales directivas que aplican son:
La Directiva sobre medicamentos (Directiva 2001/83/EC) en la que se establecen los
requisitos que deben cumplir todos los medicamentos de uso humano fabricados
industrialmente que pretenden ser comercializados en un Estado Miembro europeo.
En ella se especifican las normas y protocolos analíticos, fármaco-toxicológicos y
clínicos relativos a la realización de pruebas con medicamentos, así como los
requisitos en cuanto al contenido del expediente de solicitud de autorización de
comercialización.
Reglamento Nº 726/2004 del Parlamento Europeo y del Consejo, en el que se
establecen los procedimientos comunitarios para la autorización de comercialización

Guía de desarrollos preclínicos 27
de los medicamentos de uso humano y veterinario y su supervisión, una vez éstos ya
han sido autorizados (farmacovigilancia). Asimismo, se definen las tareas,
responsabilidades y funcionamiento de la EMA.
En lo que concierne a la solicitud y realización de ensayos clínicos aplican las siguientes
directivas:
La Directiva sobre ensayos clínicos (Directiva 2001/20/CE del Parlamento Europeo y
del Consejo) entró en vigor en abril de 2001, armonizando las disposiciones legales,
reglamentarias y administrativas de los Estados miembros, en relación a la aplicación
de las buenas prácticas clínicas en la realización de ensayos clínicos de medicamentos
para uso humano.
La Directiva 2010/C 82/01 especifica unas Directrices detalladas sobre la presentación
a las autoridades competentes de la solicitud de autorización de un ensayo clínico de
un medicamento para uso humano, la notificación de modificaciones relevantes y la
comunicación de finalización del ensayo. Explicita en qué consiste el IMPD
(“Investigational Medicinal Product Dossier”), Dossier de un Medicamento en
Investigación, y la base para la aprobación de los ensayos clínicos por las autoridades
competentes en la UE y como deben de presentarse, entre otros, los datos
farmacológicos y toxicológicos no clínicos siguiendo la estructura del Documento
Técnico Común (CTD – Common Technical Document) establecido por la ICH y que es
obligatorio.
ICH M4 CTD The Common Technical Document. ICH M8 eCTD Electronic Common Technical Document.
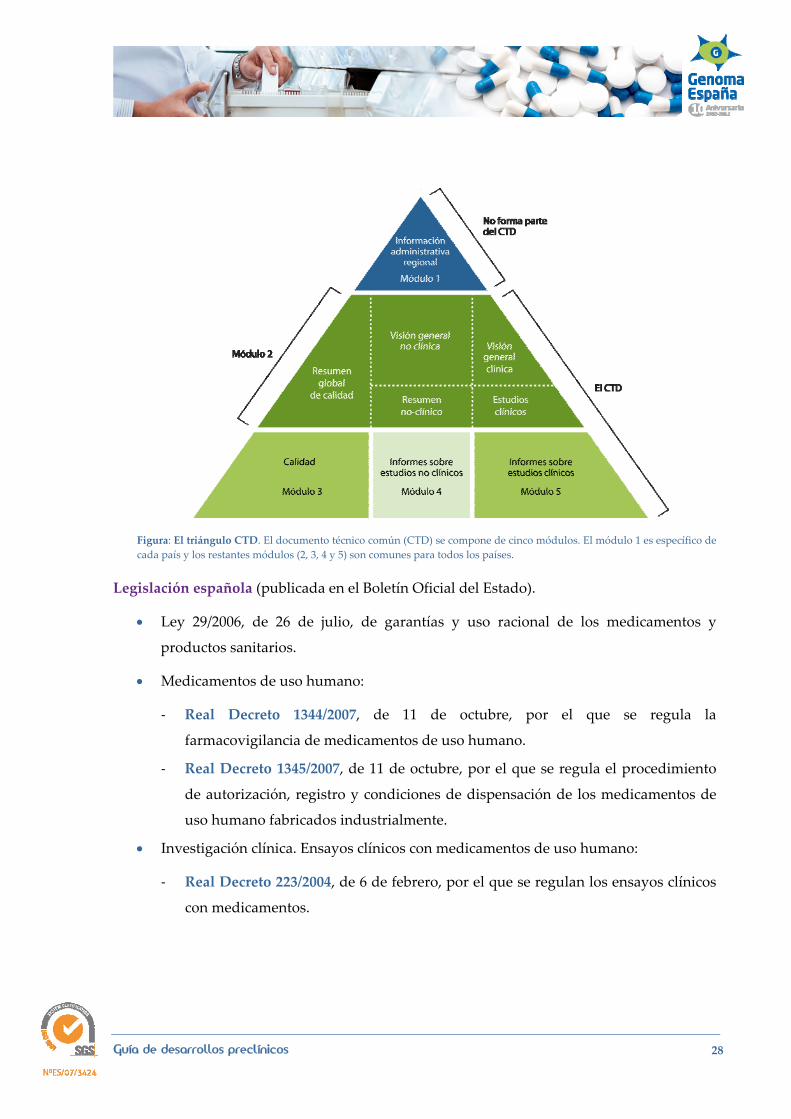
Guía de desarrollos preclínicos 28
Figura: El triángulo CTD. El documento técnico común (CTD) se compone de cinco módulos. El módulo 1 es específico de cada país y los restantes módulos (2, 3, 4 y 5) son comunes para todos los países.
Legislación española (publicada en el Boletín Oficial del Estado).
Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y
productos sanitarios.
Medicamentos de uso humano:
‐ Real Decreto 1344/2007, de 11 de octubre, por el que se regula la
farmacovigilancia de medicamentos de uso humano.
‐ Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento
de autorización, registro y condiciones de dispensación de los medicamentos de
uso humano fabricados industrialmente.
Investigación clínica. Ensayos clínicos con medicamentos de uso humano:
‐ Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos
con medicamentos.

Guía de desarrollos preclínicos 29
A.3. ASESORÍAS CIENTÍFICAS
La complejidad del proceso de desarrollo de un medicamento ha llevado a la necesidad de
regular el mecanismo de solicitud de asesoramiento a las agencias implicadas, tanto a nivel
Nacional como Comunitario. Es importante llevarlas a cabo porque permiten consensuar con
las agencias las estrategias a seguir en el desarrollo del producto. El asesoramiento científico
no debe considerarse como una pre-evaluación de un producto. El objetivo es solicitar la
opinión de la Agencia sobre cuestiones específicas acerca de lo que podría hacerse con
respecto a una situación o problema en particular, en relación con tres áreas principales de
desarrollo de productos:
Clínica
Preclínica - Toxicología
Calidad
Farmacovigilancia
Cuestiones regulatorias
Concretamente, y a nivel Nacional, la AEMPS ha emitido una Circular (12/2002) con
“Instrucciones para la solicitud de asesoramiento científico sobre la calidad, seguridad y
eficacia de un medicamento de uso humano o de uso veterinario, durante las etapas de
investigación y desarrollo del mismo, para iniciar un procedimiento de reconocimiento
mutuo y otro tipo de asesoramientos”.
Hay tres tipos diferentes de procedimientos de asesoramiento:
Asesoramiento científico inicial
Seguimiento de asesoramiento científico
Asesoramiento previo a la presentación de reuniones de solicitud de autorización

Guía de desarrollos preclínicos 30
Dependiendo del tipo de asesoramiento solicitado (según las preguntas planteadas), la
AEMPS decide sobre la conveniencia de un informe escrito con su opinión o una reunión de
asesoramiento científico, siendo esta última la opción más frecuente.
Las solicitudes se presentarán en el impreso de solicitud de asesoramiento científico
facilitado por la AEMPS, donde se explicita la documentación que será presentada en papel
y soporte electrónico, siempre que éste último sea posible.
A nivel comunitario y previo al asesoramiento de la EMA, los promotores pueden solicitar
reuniones con el “Innovation Task Force” (ITF, un grupo multidisciplinar con competencias
científicas, regulatorias y legales) que proporciona un foro para el diálogo inicial con los
solicitantes. Está pensado para dar un asesoramiento inicial antes de iniciar otros
procedimientos en la EMA. El ITF puede orientar en etapas tempranas del desarrollo con el
máximo de efectividad.
El asesoramiento científico de la EMA es de carácter prospectivo. No es una pre-evaluación
de los datos para respaldar una solicitud de autorización de comercialización, sino que se
centra en las estrategias de desarrollo. Por tanto, el asesoramiento científico recibido por
parte de la Agencia no es jurídicamente vinculante con respecto a cualquier futura
autorización de comercialización.
La EMA presta dos tipos de asesoramiento en el desarrollo de medicamentos:
Asesoramiento científico (“Scientific Advice”): asesoramiento sobre los ensayos y
estudios adecuados para el desarrollo de un medicamento para facilitar el desarrollo
y la disponibilidad de medicamentos de elevada calidad, eficacia y seguridad.
Protocolos de asistencia (“Protocol Assistance”): forma específica de asesoramiento
científico para desarrollos de medicamentos huérfanos para tratamiento de
enfermedades raras.
Para solicitar asesoramiento la EMA tiene establecido un procedimiento, detallado en su
Web, y para ello dispone de modelos específicos de documentos a presentar.

Guía de desarrollos preclínicos 31
Las empresas pueden solicitar asesoramiento científico de la Agencia Europea de
Medicamentos en cualquier etapa del desarrollo de un medicamento.
El Grupo de Trabajo (“Scientific Advice Working Party” - SAWP) y el Comité de
Medicamentos de Uso Humano (“Committee for Medicinal Products for Human Use” - CHMP)
proporcionan asesoramiento científico al responder a las preguntas planteadas por las
empresas.
Se pueden realizar solicitudes de asesoramiento científico en relación a:
Calidad
No clínica (toxicología y farmacología)
Clínica (seguridad y eficacia)
También existe la posibilidad de realizar consultas científicas y regulatorias a la “Food and
Drug Administration” (FDA) de EE.UU. En lugar de llamarse “asesoramiento científico”
(“Scientific Advice”) las consultas a la FDA se llaman “meetings”. El procedimiento de
solicitud está establecido por guías propias de la FDA, pero en definitiva, al margen de los
aspectos administrativos que difieren bastante de los establecidos por la EMA, la estructura
del procedimiento es similar. Se debe de enviar una carta de solicitud de reunión, preparar
un documento sumarial de las actividades realizadas, del momento del desarrollo en el que
se encuentra el medicamento y de los planes futuros de desarrollo tanto en el área de
calidad, preclínica como clínica, y presentar las preguntas con las que se desea compartir la
opinión de la FDA.
Las reuniones pueden tener lugar en cualquier momento del desarrollo. Sin embargo, hay
unas ocasiones específicas en las que las reuniones son más convenientes y frecuentes. Por
ejemplo, las reuniones antes de solicitar un estudio clínico de Fase I o II (pre-IND—
Investigational New Drug—meetings), la correspondiente al final de Fase II, con vistas a
consensuar con la FDA como se va a plantear la Fase III (“end‐of‐phase” II meeting, EOP2) y
otra antes de enviar el dossier para la aprobación y registro (pre-NDA—New Drug
Application—meeting).

Guía de desarrollos preclínicos 32
Es importante tener en cuenta que no es imprescindible realizar un estudio clínico en EEUU
para poder tener una reunión con la FDA. Por otro lado, actualmente es más frecuente que
las reuniones se realicen vía telefónica. Las reuniones presenciales son posibles, aunque
suelen estar más restringidas a casos que requieren discusión científica o regulatoria más
intensa.
A diferencia de lo que ocurre con la EMA o las agencias nacionales europeas, la FDA envía
las respuestas a las preguntas antes de que tenga lugar la reunión. Esto permite que la
reunión, que es de tiempo limitado, pueda centrarse en los aspectos que no quedan claros o
bien aquellos en los que se necesita recabar más detalles sobre los puntos de vista de la FDA,
consiguiéndose que la reunión sea más productiva. Transcurridos unos días después de la
reunión, la FDA envía un acta de la reunión. La posición de la FDA se suele considerar
bastante vinculante, en especial en las reuniones que tiene lugar en fases avanzadas del
desarrollo como la end‐of‐phase II meeting. Ello es porque los mismos evaluadores que se
implican en la reunión normalmente estarán también implicados en la evaluación del dossier
para su aprobación, y por tanto aplicarán criterios uniformes.
La Agencia Europea del Medicamento (EMA) y la FDA de EE.UU. tienen un programa para
proporcionar asesoramiento científico en paralelo. El objetivo del programa es proporcionar
un mecanismo para que los evaluadores de la EMA y la FDA y los sponsors intercambien sus
puntos de vista sobre temas científicos durante la fase de desarrollo de nuevos
medicamentos. Estas interacciones permiten un mayor diálogo entre los dos organismos y
los sponsors desde el inicio del ciclo de vida de un nuevo producto y la oportunidad de
optimizar el desarrollo de productos y evitar la duplicidad innecesaria de pruebas.
El procedimiento paralelo de asesoramiento científico se centra principalmente en
medicamentos innovadores o problemas de seguridad importantes en las siguientes áreas
que han sido identificadas como grupos de intereses entre las agencias: oncología, vacunas,
medicamentos huérfanos, fármacos para la población pediátrica, nanotecnologías, terapias
avanzadas, farmacogenómica y productos sanguíneos.

Guía de desarrollos preclínicos 33
Como en todos los casos de Scientific Advice el procedimiento de asesoramiento científico en
paralelo no es garantía para que se autorice la solicitud del sponsor.
A.4. REDUCCIONES DE TASAS
Concepto Pequeñas y medianas
empresas (SMEs)
Medicamentos
huérfanos
Medicamentos para
terapias avanzadas
Asesoramiento científico 90% reducción 100% reducción 65% reducción
90% para SMEs
Certificación 90% reducción - -
Nueva solicitud dossier
completo
Aplazamiento o exención
condicional
50% reducción
100% para SMEs 50% reducción
Dosis o forma farmacéutica
adicional
Aplazamiento o exención
condicional
50% reducción
100% para SMEs 50% reducción
Formato adicional Aplazamiento o exención
condicional
50% reducción
100% para SMEs 50% reducción
Inspección 90% reducción - -
Según reglamento (CE) Nº 2049/2005
Hasta que se notifique la decisión final sobre la autorización de comercialización o hasta que se retire la solicitud.
Exención condicional: sólo deberá abonarse si se concede la autorización de comercialización
Figura 7.
Fuente: Industria Farmacéutica, Vol. 156 Pág. 48‐56.
B. CLASIFICACIÓN DE MEDICAMENTOS
Antes de iniciar la clasificación de medicamentos, sería conveniente aclarar las diferencias
existentes entre principio activo, medicamento y producto terapéutico, ya que su uso puede
inducir a errores y en algunos casos se utilizan como sinónimos cuando en realidad no lo
son.
Principio activo; cuando se habla de principio activo se está haciendo referencia a la
sustancia que produce un efecto clínico determinado, ya esté relacionado con la
prevención, el diagnóstico, tratamiento o cura de una patología concreta.
Medicamento; en este caso se trata de un conjunto de sustancias compuesto por uno
o más principios activos y los excipientes correspondientes, formulado de tal manera

Guía de desarrollos preclínicos 34
que quede listo para su uso directo o indirecto (en el caso de que se tenga que
reconstituir con algún tipo de solución acuosa).
Producto terapéutico; en esta categoría se englobarían los medicamentos y
complementos médicos como suplementos de dieta, además de productos sanitarios,
ayudas a la audición, a la visión y otros.
Para simplificar la terminología de esta guía, en el contexto de la misma vamos a referirnos a
medicamentos para hablar no sólo de los más clásicos sino también a los de origen biológico,
aunque en algunos casos se considera que éstos se encuentran en la categoría más amplia de
producto terapéutico.
A la hora de realizar una clasificación de medicamentos se pueden utilizar infinidad de
criterios: composición química, origen, forma de administración, indicación terapéutica, tipo
de efecto… En esta guía vamos a abordar la clasificación desde dos perspectivas que son las
que tienen más implicaciones desde el punto de vista regulatorio y, por tanto, mayor
relevancia en el ámbito de la preclínica y los ensayos clínicos. En concreto, nos centraremos
en una clasificación de medicamentos por composición y por indicación terapéutica, que
lejos de ser exhaustiva, pone énfasis en las principales circunstancias especiales que hacen
que el desarrollo preclínico deba seguir un camino u otro.
B.1. CLASIFICACIÓN DE LOS MEDICAMENTOS POR SU COMPOSICIÓN
Desde este punto de vista, los medicamentos se pueden clasificar en dos grandes grupos que
a su vez pueden estar divididos en subgrupos de mayor o menor complejidad. Estos grupos
son el de las Moléculas Químicas de bajo peso molecular o “small molecules” y el de
medicamentos Biológicos. También se podría considerar como un grupo el de los
medicamentos formados por una combinación de principios activos.
Medicamentos químicos (small molecules)
En lo que se refiere a desarrollo de medicamentos, una “small molecule” se considera una
molécula que generalmente tiene un peso molecular inferior a 1000 Da y no es polimérica,

Guía de desarrollos preclínicos 35
aunque algunos oligómeros se pueden considerar como tales. Pueden ser de origen natural o
resultado de un proceso de selección química de una familia de compuestos a partir de una
sustancia natural, pero en general, una vez que se localiza un compuesto químico con una
actividad biológica deseable para la clínica se desarrolla el proceso para poder obtenerlo por
síntesis química; de esta manera se evita depender de su fuente natural, que puede acarrear
complicaciones logísticas y se puede contar con una sustancia de gran pureza y calidad
homogénea entre distintos lotes.
El mecanismo de acción suele estar bien caracterizado y fundamentado en principios de
farmacología clásica, siendo el más frecuente la unión a moléculas biológicas como
receptores específicos, enzimas, etc., lo que produce un efecto determinado, normalmente de
inhibición o activación, en dicha molécula.
Debido a su pequeño tamaño, este tipo de moléculas difunde con gran facilidad a través de
sus tejidos diana. Esta característica les confiere claras ventajas desde el punto de vista
funcional, ya que llegan a sus sitios de actuación prácticamente sin sufrir alteraciones en su
estructura. No obstante, sí que pueden sufrir alteraciones debido a la acción del metabolismo
en su proceso de excreción, lo que tiene importantes implicaciones desde el punto de vista
regulatorio, ya que se debe estudiar la seguridad del principio activo y de todos los
compuestos intermedios resultantes del metabolismo que tiene lugar antes de su excreción,
como veremos en otros capítulos de esta guía.
La mayoría de los medicamentos clásicos que se han desarrollado hasta la fecha son small
molecules, como son los analgésicos, antihistamínicos, antibióticos, inhibidores de canales de
iones, agonistas o antagonistas de receptores de membrana, etc.
Medicamentos biológicos
En este tipo de medicamentos, el principio activo es de origen biológico.
Una sustancia biológica es aquella que se produce o se extrae a partir de una fuente biológica
y que necesita, para su caracterización y determinación de su calidad, una combinación de
ensayos físico-químicos y biológicos junto con el proceso de producción y su control.

Guía de desarrollos preclínicos 36
Debido a la variabilidad y heterogeneidad en el origen de los materiales de partida y los
procesos de producción, los medicamentos biológicos se consideran medicamentos
especiales y requieren un tratamiento particular a efectos de demostrar su calidad, seguridad
y eficacia. En el caso concreto de la seguridad, las ventajas de los biológicos frente a los
medicamentos basados en moléculas pequeñas están bastante claras. La toxicidad de los
primeros es prácticamente inexistente ya que el material de partida es muy compatible con
los organismos receptores. No obstante, sí que puede haber reacciones adversas, a veces muy
remarcables, debidas a farmacología exagerada o de tipo inmunológico si la(s) molécula(s)
que se utiliza(n) tiene un alto comportamiento inmunogénico.
En principio, se consideraban medicamentos biológicos sólo los medicamentos
inmunológicos y los medicamentos derivados de la sangre o el plasma humanos
(hemoderivados). Con los avances tecnológicos que se han producido a partir del último
cuarto del siglo XX, han surgido nuevas categorías de medicamentos biológicos con
características especiales como son los biotecnológicos y aquellos empleados para las
denominadas terapias avanzadas. Estas dos categorías presentan condiciones tan
particulares que algunos autores las consideran aparte de los biológicos. No obstante, debido
a que en ambos casos los productos son claramente de origen biológico, consideramos más
adecuado tratar estos medicamentos como casos especiales entre los de este tipo.
Medicamentos inmunológicos
Son todos aquellos medicamentos cuya acción está directamente ligada a la activación del
sistema inmunitario del paciente, como son las vacunas, toxinas, sueros y alérgenos.
Las Vacunas son preparaciones que contienen antígenos de microorganismos patógenos
como principio activo. Los antígenos son sustancias capaces de inducir inmunidad activa
específica frente a agentes infecciosos completos o partes de éstos, así como frente a
moléculas que producen estos microorganismos.
Las vacunas pueden contener:
Organismos que han sido inactivados por medios químicos o físicos pero mantienen
sus propiedades inmunogénicas.

Guía de desarrollos preclínicos 37
Organismos vivos que de forma natural carecen de potencial virulento o que han sido
tratados para atenuar su virulencia conservando propiedades inmunogénicas.
Antígenos extraídos de organismos, secretados por éstos o bien producidos mediante
tecnología recombinante de ADN.
Las vacunas representan una clase de agentes muy heterogénea, por lo que el programa de
desarrollo preclínico de una vacuna se deberá adaptar al producto en cuestión y estar
adecuadamente justificado en un informe de experto fármaco-toxicológico.
Los anticuerpos monoclonales son también agentes inmunógenos, así como las vacunas a
base de proteínas recombinantes. Pero debe de tenerse en cuenta que, debido a su modo de
producción, deben ser considerados asimismo como productos biotecnológicos.
Las Toxinas y sueros, en general, son agentes utilizados para diagnosticar el estado de
inmunidad (por ejemplo, tuberculina) y agentes utilizados para provocar una inmunidad
pasiva, como la antitoxina diftérica o antitetánica.
Los Alérgenos son cualquier medicamento destinado a detectar o provocar una alteración
adquirida y específica en la respuesta inmunológica a un agente alergizante. Su finalidad es
el diagnóstico “in vivo” o el tratamiento de enfermedades alérgicas. Los productos pueden
contener uno solo o una mezcla definida de alérgenos.
Medicamentos hemoderivados
Son medicamentos a base de constituyentes sanguíneos preparados industrialmente por
establecimientos públicos o privados; dichos medicamentos comprenden, en particular,
albúmina, factores de coagulación e inmunoglobulinas de origen humano.
Medicamentos biotecnológicos
Un medicamento biotecnológico es aquél cuyo(s) principio(s) activo(s) se ha(n) obtenido por
técnicas de biotecnología (expresión en sistemas celulares, tecnología del ADN
recombinante...).

Guía de desarrollos preclínicos 38
A diferencia de las small molecules suelen ser moléculas biológicas de alto peso molecular, con
un tamaño de hasta 1000 veces el de las moléculas de síntesis química.
La mayoría de las moléculas activas obtenidas por estos métodos son proteínas, péptidos o
sus derivados, aunque podrían ser de otra naturaleza química siempre que sean obtenidas
mediante biotecnología. En el caso concreto de las proteínas, su actividad se verá
condicionada principalmente por su estructura tridimensional y su patrón de modificaciones
post-traduccionales el cual es muy dependiente de la especie en la que se genera la proteína,
lo que tiene importantes implicaciones a la hora de determinar el sistema de producción de
la misma.
Los fármacos biotecnológicos se obtienen a partir de procesos de producción que pueden
durar meses y que comprenden varias etapas complejas. La complejidad de este proceso
convierte a la molécula final en un producto totalmente dependiente de cada una de las
etapas del proceso de fabricación, de manera que pequeños cambios en el mismo podrían
comportar alteraciones clínicamente significativas en términos de seguridad y eficacia del
producto final. De hecho, existe la afirmación de que, en el caso de productos
biotecnológicos, “el proceso es el producto” para recalcar la importancia de éste en la
producción de este tipo de medicamentos.
Una de las características diferenciales fundamentales entre las moléculas de síntesis química
y aquellas obtenidas por biotecnología es el riesgo de inmunogenicidad inherente a estas
últimas, esto es, la capacidad para activar en el organismo respuesta inmune frente a la
propia sustancia, por tratarse de moléculas biológicamente activas derivadas de células
vivas.
En el caso de medicamentos biotecnológicos, se habla de actividad biológica, que sería el
equivalente a farmacodinamia en small molecules. Este concepto es importante tanto para el
desarrollo clínico como preclínico ya que la actividad biológica es muy específica de especie.
De modo que un anticuerpo monoclonal, que puede tener actividad biológica en humanos,
puede que no tenga ninguna en rata.

Guía de desarrollos preclínicos 39
Otro concepto interesante es el de compuesto biosimilar frente a bioequivalente. En el caso
de los medicamentos de tipo small molecule se dice que los genéricos deben ser
bioequivalentes, es decir deben tener los mismos principios activos y en la misma
concentración que los medicamentos originales; se supone que en estas condiciones deben
tener la misma actividad pero en realidad se permite un margen de tolerancia de hasta el
20% con respecto al original. En el caso de biotecnológicos se habla de biosimilares, ya que el
principio activo nunca puede ser el mismo al original debido a la fuerte dependencia del
proceso de producción al que están sujetos, como hemos explicado antes. Esto tiene
consecuencias regulatorias, ya que para demostrar la seguridad y eficacia, a un biosimilar se
le exigen nuevos ensayos que cubran estos aspectos, mientras que al bioequivalente no.
La importancia de los medicamentos biotecnológicos es cada vez mayor, de hecho se calcula
que alrededor del 50% de los medicamentos en fase clínica es de este tipo.
Ejemplos de medicamentos biotecnológicos son citoquinas, activadores del plasminógeno,
factores de crecimiento, proteínas de fusión, enzimas, receptores, hormonas y anticuerpos
monoclonales.
Terapias avanzadas
Por medicamento de terapia avanzada se entiende cualquiera de los siguientes
medicamentos para uso humano:
Un medicamento de terapia génica: producto obtenido mediante un conjunto de
procesos de fabricación destinados a transferir, “in vivo” o “ex vivo”, un gen
profiláctico, de diagnóstico o terapéutico, tal como un fragmento de ácido nucleico, a
células humanas/animales y su posterior expresión “in vivo”.
Un medicamento de terapia celular somática: se entiende la utilización en seres
humanos de células somáticas vivas tanto autólogas (procedentes del mismo
paciente) como alogénicas (procedentes de otro ser humano) cuyas características
biológicas han sido modificadas sustancialmente como resultado de su manipulación
para obtener un efecto terapéutico o preventivo basado en su actividad metabólica,
farmacológica e inmunológica.

Guía de desarrollos preclínicos 40
Un medicamento de ingeniería tisular: se entiende como tal aquél que contiene o
está formado por células o tejidos manipulados por ingeniería, y del que se alega que
tiene propiedades, se emplea o se administra a las personas para regenerar, restaurar
o reemplazar un tejido humano.
Un producto de ingeniería tisular podrá contener células o tejidos de origen humano,
animal, o ambos. Las células o tejidos podrán ser viables o no. Podrá también
contener otras sustancias, como productos celulares, biomoléculas, biomateriales,
sustancias químicas, soportes o matrices.
La evaluación de los medicamentos de terapia avanzada suele requerir conocimientos y
experiencia muy específicos, que van más allá del ámbito farmacéutico tradicional y abarcan
zonas limítrofes de otros sectores, como los de la biotecnología y los productos sanitarios.
Los medicamentos de terapia avanzada están sometidos a los mismos principios reguladores
que otros tipos de medicamentos obtenidos por biotecnología. Sin embargo, los requisitos
técnicos, en particular el tipo y la cantidad de datos sobre calidad, así como de datos
preclínicos y clínicos necesarios para demostrar la calidad, seguridad y eficacia del producto,
pueden ser muy específicos.
Asociaciones de principios activos
Son medicamentos que asocian dos o más principios activos, ya sean químicos o biológicos,
en proporciones fijas. Los principales objetivos buscados con las asociaciones terapéuticas y
medicamentos de combinación fija a menudo consisten en que las interacciones
farmacológicas o farmacocinéticas den como resultado una mejor eficacia o perfil de
seguridad. También se combinan sustancias con el único propósito de facilitar la dosificación
o la comodidad del paciente.
Desde un punto de vista regulatorio y en especial, teniendo en cuenta las implicaciones en el
desarrollo preclínico, las combinaciones pueden plantear las siguientes situaciones:
Combinación fija de compuestos ya aprobados anteriormente para ser utilizados en
asociación libre.

Guía de desarrollos preclínicos 41
Combinación fija de compuestos aprobados anteriormente pero que no han sido
aprobados para ser utilizados en asociación.
Combinación fija en la que una o más sustancias son Nueva Entidad Química o
biológica. A su vez, este grupo puede consistir en la combinación de una nueva
entidad química o biológica con una o más sustancias previamente aprobadas y bien
conocidas, o bien, una combinación de dos o más nuevas entidades químicas o
biológicas.
Cada una de estas situaciones requerirá desarrollos preclínicos diferentes.
B.2. CLASIFICACIÓN DE LOS MEDICAMENTOS POR SU INDICACIÓN
TERAPÉUTICA
La clasificación de medicamentos por indicación terapéutica es muy extensa, ya que hay una
categoría por cada sistema orgánico y a su vez por cada tipo de patología. De hecho, los
medicamentos se pueden clasificar como destinados a sistema gastrointestinal,
cardiovasculares, sistema nervioso, contra infecciones, anti-alérgicos, etc. En general, desde
el punto de vista regulatorio todos tienen que seguir un esquema parecido, teniendo en
cuenta que el desarrollo preclínico puede necesitar aproximaciones a la medida en casos
particulares.
En el marco de esta guía no vamos a hablar de todos las posibles categorías de medicamentos
por indicación terapéutica, sino que nos centraremos en aquellas que presentan diferencias
más claras a la hora de establecer su camino regulatorio. Estos casos son el de los
medicamentos para uso compasivo, en concreto antitumorales, y medicamentos para
enfermedades de baja prevalencia, denominadas raras, que se conocen habitualmente como
medicamentos huérfanos.
Antitumorales
A efectos de considerar requerimientos propios al grupo terapéutico en los programas de
desarrollo preclínico, antitumorales son medicamentos tanto de origen químico como

Guía de desarrollos preclínicos 42
biotecnológico, citotóxicos o no, dirigidos a tratar el cáncer en estado avanzado que amenaza
la vida del paciente.
Se benefician de este estatus regulatorio los medicamentos en etapas tempranas de desarrollo
clínico (fase I y II) cuyos ensayos clínicos se van a llevar a cabo en pacientes terminales o con
una esperanza de vida corta y con alternativas terapéuticas limitadas.
Quedan excluidos de esta categoría regulatoria los medicamentos que se van a probar en
población sana o con esperanza de vida prolongada, así como aquellos cuya indicación sea la
prevención del cáncer o el tratamiento de los síntomas o efectos adversos de los agentes
quimioterápicos.
Medicamentos huérfanos
Medicamentos huérfanos son aquellos dirigidos a prevenir, diagnosticar o tratar
enfermedades huérfanas. Enfermedades huérfanas son aquellas cuya prevalencia es muy
baja y no existen alternativas terapéuticas adecuadas para cubrir las necesidades.
Los requerimientos en cuanto a prevalencia son diferentes entre regiones y en función de la
gravedad de las patologías, pero en general por ejemplo en Europa, la prevalencia debe de
ser menor a 5 o 10 pacientes por cada 10.000 habitantes, para que un medicamento pueda
obtener la designación de medicamento huérfano.
Esta categoría de medicamentos goza de una serie de beneficios en cuanto a costes
administrativos en los procedimientos regulatorios y derechos de exclusividad en las ventas
que tiene por objetivo incentivar el desarrollo de este tipo de medicamentos, ya que de otro
modo podría no quedar compensada la inversión realizada en el desarrollo con los beneficios
esperados en su comercialización. Asimismo, el hecho de que la prevalencia de la
enfermedad sea muy baja lleva a que el número de pacientes y número de estudios clínicos
necesarios para someter a registro dichos medicamentos sea más reducido. Todo ello en
conjunto comporta que el desarrollo de un medicamento huérfano implique costes globales
más reducidos y ayudas gratuitas por parte de la EMA (European Medicines Agency) o FDA
(Food and Drug Administration) en el asesoramiento científico y en la elaboración de los

Guía de desarrollos preclínicos 43
protocolos clínicos. Así pues, se trata de medicamentos que se benefician de ventajas
estratégicas, económicas y regulatorias.
Sin embargo se debe de tener muy en cuenta que, al contrario que en el desarrollo clínico, el
desarrollo preclínico no se ve reducido ni simplificado por el hecho de que un medicamento
tenga la designación de huérfano. Por lo tanto ésta es una condición que no se traduce en
programas de desarrollo preclínico especiales.
Es importante hacer hincapié en que hay compuestos que pueden estar englobados en dos o más categorías diferentes y por tanto a la hora de elaborar el plan de desarrollo preclínico se tendrá en cuenta los requisitos regulatorios que aplicarían a ambas categorías. Ejemplos: - Un compuesto puede estar clasificado como biotecnológico, y que su indicación
sea oncológica con lo cual también sería un compuesto antitumoral. - Otro ejemplo sería una vacuna de DNA para la que aplicarían tanto los
principios y requisitos regulatorios de biotecnológicos, como los de biológicos
por ser vacuna.
C. EFICACIA VERSUS SEGURIDAD
C.1. EFICACIA
¿Por qué son necesarios los estudios?
En las fases iniciales del desarrollo de un medicamento las
agencias regulatorias enfocan su interés en los estudios de
seguridad, por lo que suelen ser comparativamente menos
exigentes en cuanto a la caracterización de la eficacia del
compuesto, permitiendo aportar todavía información
reducida acerca del mecanismo de acción del mismo con
objeto de justificar la exposición de los pacientes a esa
nueva terapia.

Guía de desarrollos preclínicos 44
En consecuencia, existe muy poca regulación -apenas hay guías y documentos regulatorios-
en torno a los estudios necesarios para probar la eficacia de los compuestos en esta fase
temprana del desarrollo de un medicamento.
Por otro lado, el promotor del compuesto es el primer interesado en conocer su eficacia, y en
que esta sea máxima, antes de llegar al desarrollo clínico, donde el incremento de los costes
es muy significativo.
Los estudios de eficacia tienen como objetivo:
Evaluar el mecanismo de acción.
Verificar la actuación frente a la diana terapéutica seleccionada.
Establecer la dosis mínima eficaz y la curva dosis-respuesta que posteriormente, en
futuros ensayos clínicos, ayudarán a estimar una dosis terapéutica.
Establecer la pauta y vía de administración más adecuadas en función de la
farmacodinamia del compuesto y su perfil farmacocinético.
¿Qué ensayos es necesario considerar?
Estudios preclínicos relativos a eficacia. La Farmacodinamia (PD) examina más
específicamente como el compuesto ejerce su acción farmacológica, como interactúa y el tipo
de reacciones que tienen lugar a nivel celular, tisular y de órgano, así como la curva de dosis-
respuesta. La PD es crucial para el diseño de los estudios de toxicología así como para la
evaluación y extrapolación de los datos toxicológicos a humanos.
Los estudios de eficacia se realizan tanto “in vitro” como “in vivo”. Los aspectos que hay que
considerar a la hora de su diseño son la relación entre la estructura y la actividad, la correcta
elección de los sistemas experimentales tanto “in vitro”, líneas y cultivos celulares, como la
elección de los modelos animales con respecto a la patología humana a tratar, modelos
regulatoriamente aceptados, modelos con disfunción inducida (farmacológica, patología,
etc.).

Guía de desarrollos preclínicos 45
C.2. SEGURIDAD
¿Por qué son necesarios los estudios?
El objetivo de estos estudios del desarrollo preclínico es salvaguardar, ya en esta fase tan
temprana del desarrollo de un medicamento, la seguridad del paciente, es decir, detectar y
evitar todo riesgo o posible daño para el ser humano.
Estos estudios deben dar respuesta a las siguientes cuestiones:
Caracterización de los efectos tóxicos y reversibilidad de los mismos.
Identificación de los órganos diana.
Determinación de la relación dosis-respuesta: qué dosis ejercen qué efectos.
Establecimiento del NOAEL (“Non‐observed Adverse Effects Level”).
Así mismo, se deben correlacionar los efectos tóxicos con la exposición sistémica al
compuesto mediante la toxicocinética e identificar biomarcadores para la estimación de una
dosis inicial segura y un diseño adecuado en los futuros ensayos clínicos en humanos.
Así pues, estos estudios están sujetos a estrictas regulaciones internacionales existiendo una
gran cantidad de guías de las diferentes agencias acerca de cómo han de llevarse a cabo.
Es por todo esto que los estudios preclínicos de seguridad se consideran estudios
regulatorios y se han de llevar a cabo siguiendo unos requisitos determinados de acuerdo
con las guías en vigor y las Buenas Prácticas de Laboratorio (BPLs-GLPs).
¿Que ensayos es necesario considerar?
Farmacocinética/toxicocinética y metabolismo (ADME-Tox)
La farmacocinética (PK) describe lo que le sucede al fármaco por acción del organismo. La
farmacocinética depende de la dosis administrada, de la vía de administración y del estado
fisiológico del organismo así como de las propiedades físico-químicas del compuesto
(solubilidad, estabilidad,…). Un estudio PK típico supone la administración de una cantidad

Guía de desarrollos preclínicos 46
fija del compuesto (dosis) al sujeto y la recogida de muestras de sangre a diferentes tiempos
después de la administración para la determinación de las concentraciones del compuesto y
de sus metabolitos. Esto implica la necesidad de disponer de métodos analíticos validados,
sensibles y reproducibles que permitan la detección del fármaco y de sus metabolitos así
como, en determinados tipos de ensayos, la necesidad de marcar los compuestos
radiactivamente. En esta fase se estudia la Absorción (biodisponibilidad dependiente de la
forma de administración y de las propiedades físico-químicas), la Distribución del
compuesto (unión a proteínas plasmáticas y tisulares, distribución en tejidos, distribución en
hembras gestantes, etc.), su Metabolismo (estabilidad metabólica en microsomas y
hepatocitos, citocromos empleados, comparación ínter-especies, identificación de
metabolitos, etc.) y la Excreción (incorporación y excreción en suspensión de hepatocitos,
balance de masas, etc.)
El estudio permite identificar, si los hubiera, efectos tóxicos del compuesto así como la forma
de administración más apropiada y la dosis a utilizar. Tradicionalmente los estudios de
ADME (Absorción, Distribución, Metabolismo y Excreción) se realizan en dos especies de
mamíferos, una de ellas de roedor, utilizando diferentes dosis y aplicadas a hembras y
machos.
Pruebas ADME
Parámetro Experimento
Absorción
‐ Células Caco-2
‐ Células MDCK
‐ Transporte PGP
‐ Perfil farmacocinético "in vivo"
Distribución ‐ Unión a proteínas plasmáticas "in vitro"
‐ Distribución en tejidos "in vivo"
Metabolismo
‐ Estabilidad metabólica (microsomas, fracciones subcelulares, hepatocitos)
‐ Estudios de inhibición de P450 (microsomas)
‐ Estudios de inducción de P450 (chips de ADN, dosis múltiple)
Eliminación ‐ Cuantificación del compuesto y sus metabolitos en fluidos biológicos
Figura 8. Experimentos tipo para evaluar las propiedades ADME de los candidatos a medicamentos.

Guía de desarrollos preclínicos 47
Mientras que los modelos animales se siguen utilizando para los estudios tóxico-cinéticos, en
la última década se ha desarrollado una importante variedad de ensayos “in vitro” que
ayudan a predecir las propiedades ADME-Tox. Entre estos destacan los cultivos celulares
(Caco-2; HT 29, T-84, etc.) y modelos fisiológicos (Tecnomouse, Log P, etc.).
Un solo ensayo no puede sustituir la complejidad del sistema experimental “in vivo”, por lo
que sigue la estrategia de intentar validar métodos que puedan sustituir algunos aspectos o
etapas de los estudios principales en el animal. Así pues, toda la aproximación “in vitro” a la
predicción de la toxicidad, debe de considerar una batería de ensayos diferentes tendientes a
cubrir el mayor número de respuestas posibles y permitir la confirmación de los resultados.
El uso de estos ensayos permite obviar en parte las limitaciones de los “in vivo”, alto costo,
variaciones ínter-especies y problemas éticos. La combinación de estudios “in vitro” junto
con los datos toxicocinéticos “in vivo” obtenidos en los estudios de toxicología han permitido
relegar otros análisis “in vivo” más costosos para fases posteriores del desarrollo (antes de
fase III) y/o siempre que este justificada su realización. La incorporación de otros modelos
animales alternativos a los mencionados, tales como invertebrados o pequeños vertebrados
(Zebrafish) a la preclínica temprana, refuerza la tendencia a posponer los ensayos “in vivo” de
validez regulatoria con mamíferos a la fase clínica. Si bien estos últimos ensayos, hoy por
hoy, no tienen validez regulatoria, se consideran una herramienta muy útil de “screening”
para discriminar productos con distintos niveles de seguridad, permitiendo descartar
aquellos no seguros.
ICH S3A Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies
ICH S3B Pharmacokinetics: Guidance for Repeated Dose Tissue Distribution Studies.
Farmacología de seguridad
Los estudios de farmacología de seguridad están concebidos para caracterizar las acciones
farmacológicas no deseadas del medicamento sobre los diferentes sistemas funcionales del
organismo. En primera instancia, se evaluarán los efectos del fármaco sobre órganos o

Guía de desarrollos preclínicos 48
funciones vitales, lo que incluye el sistema nervioso central (SNC) (actividad motora
espontánea, Irwin, etc.), sistema cardiovascular (SCV) (hERG – human Ether‐à‐go‐go‐Related
Gene-, evaluación de la prolongación del intervalo QT, función cardiaca en animal
anestesiado, etc.) y sistema respiratorio (SR) (Pletismografía de todo el cuerpo, presión
arterial pulmonar, etc.). Puede requerirse también la evaluación de los efectos sobre otros
sistemas como renal, gastrointestinal o nervioso autónomo si existen indicios o alertas
específicas para el compuesto o para la clase farmacológica a la que pertenece.
ICH S7A Safety Pharmacology Studies for Human Pharmaceuticals. ICH S7B Nonclinical evaluation of QT Interval prolongation.
Toxicidad
Con estos estudios se pretende identificar la potencial toxicidad del candidato tan temprano
como sea posible. Con el fin de armonizar los requerimientos, en cuanto al tipo y duración de
estudios necesarios para cada fase del desarrollo de un medicamento, se han establecido
numerosas guías. Las dos siguientes son las de jerarquía superior.
ICH M 3(R2) Non clinical safety studies for the conduct of human Clinical trials and marketing authorization for Pharmaceuticals.
ICH S6 Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals.
La definición de los ensayos toxicológicos a realizar es altamente dependiente del producto y
de su aplicación así como de la fase clínica a la que tienen que dar cobertura.
Es decir, hay baterías de ensayos que son de estricto cumplimiento para la solicitud del
ensayo clínico y conforme se va avanzando en la fase clínica se van incorporando nuevos
datos al expediente. Es por esto que es muy importante definir exactamente que tenemos que
hacer y que no, caso a caso, así como el momento y las condiciones que aplican para no
desperdiciar recursos y tiempo. (Figura 9)
Para una completa caracterización del perfil de toxicidad de un compuesto se deben realizar
estudios de toxicidad sistémica general, tolerancia local, genotoxicidad, efectos

Guía de desarrollos preclínicos 49
inmunológicos adversos como la sensibilización, y, en algunos casos, estudios sobre la
función reproductora y sobre la descendencia. Las pruebas preclínicas de toxicidad a dosis
única y en dosis repetidas brindan una valiosa información sobre la seguridad del producto,
al incluir el estudio macroscópico e histopatológico de todos los órganos, así como
evaluaciones de las vías de administración y el establecimiento de la dosis segura.
Los estudios toxicológicos se clasifican en:
Toxicología general: su objetivo es demostrar cualquier tipo de efecto tóxico que
afecte morfológica o funcionalmente a los distintos órganos o sistemas del individuo.
ICH S4 Duration of chronic toxicity testing in animals (rodent and non rodent toxicity testing).
EMA Guidance on repeated dose toxicity.
- Toxicidad a dosis única. Evaluación de los efectos adversos resultado de una sola
exposición a una sustancia. Este tipo de estudios fueron un requerimiento
importante en el pasado. Sin embargo en la actualidad solamente se recomiendan
en casos muy particulares.
- Toxicidad por dosis repetida. Se pretende evaluar el efecto tóxico de la
administración reiterada durante un periodo de tiempo más largo (desde dos
semanas hasta varios meses). Puede revelar efectos acumulativos e identificar los
órganos diana. Para que sea lo más predictiva posible de los efectos para el ser
humano, se replica en la medida de lo posible la pauta de administración
propuesta en humanos, maximizando la exposición mediante períodos más
prolongados y/o múltiplos elevados de la dosis.
Genotoxicidad. El objetivo es evaluar los efectos del medicamento sobre el material
genético (genes y cromosomas). No existe un ensayo único que permita determinar el
potencial genotóxico de los fármacos en desarrollo. Se determina mediante una
batería de estudios acotada y que incluye ensayos “in vitro” e “in vivo”. Como la Fase
clínica I se limita a un número pequeño de voluntarios solamente se requieren
resultados de los test “in vitro” de mutagénesis bacteriana y estudios en líneas

Guía de desarrollos preclínicos 50
celulares de mamífero en cultivo. Para fases mas avanzadas se requieren datos más
exhaustivos “in vivo”.
ICH S2(R1) Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use.
Toxicología de la reproducción y desarrollo. Sin embargo, hay circunstancias
especiales en las que es necesario acometer ensayos “in vivo” para la autorización de
la fase clínica. Tales son el caso de ensayos en los que hay que reclutar mujeres
embarazadas, en edad fértil, o en niños. Consiste en el estudio de los efectos del
medicamento sobre la capacidad reproductiva de los progenitores y sobre la
descendencia a lo largo de los tres segmentos de la reproducción: fertilidad y
desarrollo embrionario temprano, desarrollo embriofetal (o período de
organogénesis) y desarrollo peri y posnatal.
ICH S5(R2) Detection of toxicity to reproduction for medicinal products & toxicity to male fertility.
Potencial carcinogénico. Consiste en evaluar la capacidad de un medicamento en
inducir o aumentar la probabilidad de que aparezcan tumores cuando es
administrado a lo largo de toda la vida del animal. Son estudios de dos años de
duración que se realizan en roedores. Deben realizarse para dar soporte a la fase final
de registro en la mayoría de casos en los que el medicamento vaya a administrarse de
forma crónica en el ser humano.
ICH S1A Guideline on the need for carcinogenicity studies of pharmaceuticals
ICH S1B Testing for carcinogenicity of pharmaceuticals. ICH S1C(R2) Dose selection for carcinogenicity studies of pharmaceuticals
Tolerancia local. El objetivo es evaluar el efecto tóxico en el sitio de administración y
la regresión de las lesiones en caso de producirse. La prueba de tolerancia local tiene
como objetivo evaluar el daño local provocado por la administración de un
medicamento, diferenciando entre el daño tisular causado por efectos puramente

Guía de desarrollos preclínicos 51
físicos (temperatura, volumen de muestra, método de administración) o químicos
(pH, corrosividad) y el originado por los efectos fármaco-toxicológicos. Asimismo,
existen ensayos específicos para poner de manifiesto el potencial de sensibilización
(alergia de contacto) y la fototoxicidad.
Toxicología especial: son aquellos estudios que se clasifican en función del efecto que
se quiere investigar ya sea por el órgano (Ej. hepatotoxicidad) o sistema (Ej.
neurotoxicidad) o para entender mecanismos de toxicidad.
- Inmunotoxicología. Consiste en la evaluación de la capacidad del medicamento
para inducir efectos tóxicos sobre el sistema inmunitario. No existe un tipo de
ensayo único, ya que dependiendo de si se observan o no determinados efectos
sobre el sistema inmunitario en los estudios de dosis repetida, se deberán hacer
estudios adicionales que van a variar según el tipo de efecto encontrado. Un caso
particular que a menudo se debe investigar en medicamentos de origen
biotecnológico es la inducción de inmunogenicidad, es decir, la capacidad del
compuesto para inducir la formación de anticuerpos anti-fármaco.
ICH S8 Immunotoxicity Studies for Human Pharmaceuticals.
Tipo de estudio En soporte a
Farmacodinamia Fase I
Farmacología de seguridad (“core battery”)
- SNC
- SCV
- S. Respiratorio
Fase I
Toxicocinética Fase I
Toxicidad repetida (duración ajustada a ensayo clínico) Fase I
Genotoxicidad “in vitro” Fase I
Tolerancia local Fase I
Metabolismo “in vitro” Fase I o Fase II
Toxicidad repetida (duración ajustada a ensayo clínico) Fase II
Genotoxicidad “in vivo” Fase II
ADME in vivo Fase III
Toxicidad a dosis repetida crónica (excepciones) Fase III

Guía de desarrollos preclínicos 52
Tipo de estudio En soporte a
Fertilidad desarrollo embrionario Fase III
Desarrollo peri-postnatal Fase III
Caso especiales
Tipo de estudio En soporte a
Cancerogénesis Registro
Desarrollo embrio-fetal Según reclutamiento de mujeres embarazadas
Toxicidad aguda No se requiere (salvo en casos particulares)
Figura 9. Fases de la Clínica en las que son necesarias las distintas pruebas toxicológicas.
Fuente: Innoqua Toxicology Consultants, S.L.
D. ¿QUÉ TIPO DE MODELOS DEBO INCLUIR EN MIS ENSAYOS? ¿“IN
VITRO”, “IN VIVO”, “IN SILICO”?
La sociedad demanda medicamentos cada vez más eficaces y seguros a la vez que demanda
una utilización racional de animales de ensayo.
Directiva 86/609/EEC. Siempre que haya una alternativa “in vitro” viable y validada que
sustituya a un ensayo con animales, no se deberán utilizar animales
El valor predictivo de los ensayos, “in vivo” e “in vitro”, en la evaluación del riesgo de
toxicidad para el hombre de nuevas moléculas depende de un diseño y elaboración correcto
de la batería de ensayos así como de un análisis muy cuidadoso de la información obtenida
primeramente “in vitro” y secundariamente “in vivo” y de su, también, correcta
interpretación.
Esta tendencia ha propiciado de forma extraordinaria el desarrollo de modelos “in vitro”
alternativos a la experimentación animal para estudios de actividad farmacológica y
toxicológica en las fases iniciales de desarrollo, lo que permite de manera eficaz reducir el
numero de posibles candidatos a fármacos.
Por su propia naturaleza los modelos “in vitro” son una simplificación de una realidad
mucho mas compleja, el ser vivo, por eso en muchas ocasiones la información que son

Guía de desarrollos preclínicos 53
capaces de proporcionar es limitada y, a menudo, tienen una mala correlación con los
resultados obtenidos “in vivo”. Aun así, no cabe ninguna duda de que ofrecen ventajas
intrínsecas muy destacables para evaluar características esenciales de los futuros
medicamentos, dada su simplicidad, disponibilidad, bajo coste, fácil control de las variables
experimentales, necesidad de cantidades muy pequeñas de la molécula en estudio y la
posibilidad de realizar estudios en etapas muy tempranas de desarrollo.
Ensayos de genotoxicidad “in vitro” más empleados
Objetivo de la
demostración Ensayos
Mutaciones génicas o
puntuales
- Ensayo de mutaciones reversas en Salmonella typhimurium
- Ensayo de mutaciones reversas en Escherichia coli
- Ensayo de mutaciones génicas en Saccharomyces cerevisiae
- Ensayo de mutaciones génicas en células de mamífero en cultivo: CHO,
L5178Y TK+/- (este último es actualmente uno de los más recomendados)
- Ensayos de transformación en células de mamífero
- Ensayo de mutaciones génicas en Aspergillus nidulans
- Ensayo de mutaciones génicas en Neurospora crassa
Aberraciones
cromosómicas - Ensayo citogenético en células de mamífero: CHO, CHL o linfocitos
Otros tipos de efectos sobre
los cromosomas
- Ensayo de intercambio de cromátidas hermanas.
- Ensayo de recombinación mitótica en Saccharomyces cerevisiae.
- Lesión y reparación de ADN en células de mamífero.
Figura 10. Ensayos de genotoxicidad “in vitro” más empleados, según el objetivo de la demostración.
La disponibilidad de cultivos de células humanas de diferentes órganos y tejidos así como de
diferentes patologías, permite una mayor aproximación al ser humano. Constituyen un
alternativa capaz de reemplazar a los animales para determinados estudios del desarrollo
preclínico, o de complementarlos. Sin embargo, los tejidos, sólo proporcionan una condición
“ex vivo” aislada, que no es del todo representativa de la respuesta “in vivo” debido a que la
acción del fármaco a menudo implica al metabolismo y la interacción entre diferentes tejidos.
Por ejemplo, los efectos de un fármaco sobre el músculo pueden involucrar al intestino en su
absorción y al hígado para metabolizarlo. En este contexto, los modelos celulares hepáticos
de origen humano, constituyen una herramienta idónea para abordar estudios encaminados
a predecir en humanos la toxicidad y el metabolismo hepático de nuevas moléculas. En

Guía de desarrollos preclínicos 54
cualquier caso son de gran ayuda para la toma de decisiones hasta el punto de poder detener
el desarrollo del medicamento, a la vista de los resultados con el consiguiente ahorro
económico y de tiempo.
Sin embargo el uso de animales en las pruebas preclínicas es obligado, ya que datos
procedentes de los distintos modelos animales validados forman parte de la información
contenida en el Dossier remitido a las autoridades regulatorias para la autorización del
ensayo clínico. Una parte de los estudios encaminados a evaluar el riesgo de toxicidad del
compuesto se debe de llevar a cabo en animales de experimentación, que aun siendo de gran
valor para la evaluación del riego tóxico, proporcionan resultados que no siempre han
podido ser extrapolados al hombre. De hecho, hay compuestos que fallan en etapas tardías
del desarrollo lo que demuestra que los animales modelo no son suficientemente predictivos
de la situación real en humanos.
Conocer de antemano cual es el modelo animal idóneo es la manera mas eficaz de reducir el
uso innecesario de animales, y una de las maneras de contribuir a una investigación mas
racional y sólida desde el punto de vista científico y más humana desde el punto de vista
ético.
En la Figura 11 se indica el porcentaje de utilización de los distintos modelos en los estudios
de ADME/toxicocinética.
Figura 11. Porcentaje de utilización de los distintos modelos en los estudios de ADME‐Tox.

Guía de desarrollos preclínicos 55
Además de los modelos tradicionalmente usados, roedores, cerdos, perros y primates no
humanos, algunos de los cuales deben de ser necesariamente incluidos en las pruebas
toxicológicas de validez regulatoria, existen otros modelos, vertebrados e invertebrados,
cuya inclusión en el desarrollo no clínico debe de ser considerada.
Los modelos de organismos invertebrados más típicos son Drosophila melanogaster y
Caenorhabditis elegans. El uso de estos modelos se basa en la existencia de rutas moleculares
muy conservadas entre estos invertebrados y los humanos que combinándolo con la
genética, la biología celular y las herramientas disponibles en biología molecular, hacen que
estos sistemas modelo sean adecuados para investigación.
Drosophila melanogaster: Se han desarrollado modelos para muchas patologías
como por ejemplo, metástasis, Alzheimer, enfermedad de Huntington.
Caenorhabditis elegans: Su estructura es sencilla y es transparente lo que permite la
observación de fenotipos celulares. El amplio estudio de la apoptosis en C. elegans ha
demostrado una regulación muy similar a la de mamíferos. Por lo tanto, este
nematodo resulta ser un buen modelo para el estudio de genes implicados en
apoptosis y para la detección de compuestos que modulan este proceso, lo cual tiene
importantes aplicaciones en el desarrollo de tratamientos contra el cáncer. Además, el
sistema nervioso de C. elegans es muy simple lo que lo convierte en un buen modelo
para el estudio de enfermedades neurodegenerativas.
Danio rerio (pez cebra): es uno de los nuevos modelos de vertebrados más utilizados
en los últimos años. Su genoma está casi finalizado y la disponibilidad de las
secuencias génicas ha facilitado el aislamiento y la manipulación de su genoma.
Los embriones de pez cebra son transparentes lo que permite la evaluación directa e “in
vivo” del efecto del fármaco sobre los órganos y tejidos. El pez cebra es fácil de criar y
mantener, y su fecundidad es alta (cada hembra puede producir 100 a 200 huevos por
apareamiento, ofreciendo un gran número de animales para ensayos).

Guía de desarrollos preclínicos 56
Dado que el pez cebra tiene órganos similares a los de mamíferos, resulta ser un
modelo mucho más útil que D. melanogaster y C. elegans para el estudio de la función
de los genes y el efecto de los fármacos en seres humanos.
Como conclusiones más importantes cabe destacar que:
Los modelos “in vivo” de invertebrados son éticamente más aceptables y pueden
complementar los ensayos con vertebrados.
Aportan considerables ventajas: amplia disponibilidad, genomas secuenciados, pocos
requerimientos para su cría y mantenimiento, alta fecundidad, capacidad de genética
de modificación y trazabilidad experimental
Los nuevos modelos de Danio rerio (pez cebra) permiten realizar pruebas preclínicas
“in vivo” más rápidas, precisas y rentables.
Los modelos de invertebrados de Drosophila melanogaster, Caenorhabditis elegans, y
nuevos insectos como Locusta migratoria, son fáciles de reproducir y baratos.
En relación a nuevos modelos con mayor valor predictivo o alternativas en las tecnologías
“in silico”, existe actualmente una gran demanda. Se han hecho avances significativos en el
desarrollo de estos modelos para predecir las propiedades ADME-Tox en fármacos
candidatos. Por ejemplo, la convergencia de aspectos estructurales de compuestos
potenciales, con datos de investigación “in vivo” e “in vitro”, permite el modelado
computacional y matemático del comportamiento de los compuestos, lo que puede a su vez
ser utilizado para predecir las respuestas humanas “in vivo”. Además, las tecnologías “in
silico” están avanzando en su capacidad para trazar respuestas en relación con una
enfermedad.
El modelado molecular y las simulaciones fisiológicas son aspectos importantes del
descubrimiento de fármacos porque se pueden predecir las principales propiedades
fisicoquímicas de los compuestos y su comportamiento biológico.

Guía de desarrollos preclínicos 57
Las aproximaciones del modelado molecular están basadas en evaluaciones computacionales
de las relaciones cuantitativas estructura-actividad (Structure Activity Relationships ‐ SAR) o
sistemas basados en conocimiento, como la quimioinformática.
Los modelos “in silico” son programas de software de gran alcance que integran datos de, por
ejemplo, transcripción, regulación, traducción, redes intracelulares de la señal transducción,
activación transcripcional, genómica, proteómica, división celular, por nombrar sólo algunos.
En general, los modelos de predicción “in silico” se han desarrollado para hacer frente al
cuello de botella asociado al descubrimiento y desarrollo de fármacos “in vitro” e “in vivo”
que resulta ser muy costoso.
La base metodológica de modelos “in silico” implica la manipulación de datos de estudios
empíricos. Los modelos establecidos se están complementando con aproximaciones de
inteligencia artificial (AI) entre las que se incluyen las redes neuronales artificiales y la
programación genética.
Conclusiones más importantes de los ensayos “in silico”:
Las plataformas “in silico” proporcionan información predictiva de ADME-Tox,
rápida y rentable, reduciendo los amplios requisitos que son necesarios en estudios
“in vivo”.
Se están desarrollando potentes modelos “in silico” para integrar, analizar, visualizar,
interpretar y manipular datos de diferente naturaleza.
Las tendencias actuales en los modelos computacionales incluyen aplicaciones
virtuales de estructuras moleculares en 3D, relaciones estructura-actividad, perfiles
metabólicos, creación de pacientes virtuales, desarrollo de programas traslacionales
basados en genómica y desarrollo de software de nueva generación para
secuenciación genómica.
Las capacidades de las plataformas “in silico” acelerarán y apoyarán la toma de
decisiones en las pruebas ADME-Tox en preclínica.

Guía de desarrollos preclínicos 58
E. ¿CUÁNDO USAR GLP, GCP, GMP?
Existe cierta confusión entre los promotores de ensayos clínicos acerca de cuando usar
condiciones GLP (Good Laboratory Practice) y/o GMP (Good Manufacturing Practice). En
términos generales condiciones GLP no son necesarias hasta que no se hagan los estudios de
seguridad preclínicos que den soporte a la aprobación de los ensayos clínicos.
Así pues se requieren condiciones GLP solo para los estudios de seguridad, incluyendo
toxicidad general, genotoxicidad, toxicocinética y farmacología de seguridad y para aquellos
que dan soporte a los estudios de seguridad, como por ejemplo las validaciones de métodos
analíticos y bioanalíticos. No se requiere para los estudios de eficacia, tanto los “in vitro”
como los “in vivo”. No se requiere para la batería de ensayos ADME (Absorción,
Distribución, Metabolismo y Excreción) “in vitro” o “in vivo”.
Las Buenas Prácticas de Laboratorio, BPL o GLP, es un sistema de calidad relacionado con el
proceso de organización y las condiciones en las que los estudios preclínicos de seguridad
(relacionados tanto con la salud como con el medio ambiente) son planificados, realizados,
controlados, registrados, archivados e informados.
Los principios de las GLP se desarrollaron en su momento para promover la calidad y
validez de los datos de los ensayos que se llevaban a cabo para determinar la seguridad de
sustancias y productos químicos. Dichos principios deben ser seguidos por las instalaciones
que realizan este tipo de estudios de seguridad.
La calidad de los datos obtenidos en estos estudios preclínicos tiene una importante
dimensión internacional, ya que si las autoridades reguladoras en los diferentes países
pueden confiar en los datos de ensayos de seguridad que se llevan a cabo en el exterior, la
duplicación de ensayos se pueden evitar y se ahorran costes tanto para el gobierno como la
industria. Por otra parte, estos principios comunes de BPL facilitan el intercambio de
información al tiempo que contribuye a la protección de la salud y el medio ambiente.

Guía de desarrollos preclínicos 59
Es importante destacar que en Europa existe un sistema de acreditación y verificación del
cumplimiento de GLPs. Sin dicha certificación, una instalación no puede declarar que realiza
estudios bajo GLPs.
Los principios de las buenas prácticas de laboratorio los establece la OECD (Organización
para la Cooperación y el Desarrollo Económico), la cual ha emitido una serie de guías en la
que se desarrollan las diferentes facetas de las BPL:
http://www.oecd-ilibrary.org/environment/oecd-series-on-principles-of-good-laboratory-
practice-and-compliance-monitoring_2077785x
Las Buenas Prácticas Clínicas (“Good Clinical Practices” - GCP) requeridas en la fase clínica
son equivalentes a las GLP esto es, conjunto de requisitos éticos y científicos de calidad
reconocidos a escala internacional, que deben cumplirse en la planificación, la realización, el
registro y la comunicación de los ensayos clínicos en que participen seres humanos. Su
cumplimiento garantiza la protección de los derechos humanos, le seguridad y el bienestar
de los sujetos del ensayo, así como la fiabilidad de los resultados del ensayo clínico.
Las Buenas Prácticas de Fabricación (“Good Manufacturing Practice“ - GMP) es un sistema de
autorización de fabricación que asegura que todos los productos autorizados en la unión
europea se fabrican / o son importados por fabricantes autorizados y siguiendo unas normas
de calidad y cuyas actividades son inspeccionadas regularmente por las autoridades
competentes.
Las GMPs son obligatorias para todos los laboratorios farmacéuticos fabricantes de la Unión
Europea tanto si los productos se venden dentro o fuera de la Unión.
La comisión europea aprobó dos directivas que establecen principios y directrices de buenas
prácticas de fabricación (GMP), la Directiva 2003/94/CE se aplica a medicamentos de uso
humano y la Directiva 91/412/CEE a los de uso veterinario.
Asimismo la comisión ha emitido guías más detalladas en las que se abordan los principios
de las GMPs en el ámbito de la Guía de Buenas Prácticas de Fabricación.

Guía de desarrollos preclínicos 60
Es importante añadir que así como para los ensayos clínicos se considera obligatorio utilizar
lotes fabricados bajo GMPs, para los estudios preclínicos esto no es un requisito, en éste
último caso es aconsejable utilizar lotes bien caracterizados y lo más similares posibles a los
que se usarán en clínica pero no han de estar fabricados necesariamente bajo GMPs.
El desarrollo químico y la producción de los fármacos no químicos, tal y como figura en el
siguiente esquema precisa de condiciones GMP:
F. PREFORMULACIÓN Y PREPARACIÓN DE LOTES. ¿QUÉ DEBO TENER EN
CUENTA?
En el momento de comenzar los estudios preclínicos con finalidad regulatoria, es decir, los
estudios orientados a dar soporte para la autorización de ensayos clínicos, el compuesto debe
de estar bien caracterizado químicamente, se debe conocer su pureza, contenido de
impurezas, demostrar que es estable a lo largo del estudio y haber desarrollado un método
analítico que permita emitir un certificado de análisis BPL (Buenas Prácticas de Laboratorio)
asociado al lote que se va a utilizar. Además, los lotes a utilizar en el desarrollo preclínico
deben ser representativos de los que después se utilizarán en los ensayos clínicos. Sin
embargo, a diferencia de los lotes para ensayos clínicos, no es necesario que cumplan con
requisitos de producción de Buenas Prácticas de Fabricación (o GMP, -Good Manufacturing
Practices- por sus siglas en inglés).

Guía de desarrollos preclínicos 61
Conforme avanza el desarrollo de un medicamento la escala de producción de los lotes pasa
de ser de escala piloto (muy reducida) a escala industrial, de tamaño cada vez mayor. Es
recomendable que los estudios preclínicos principales para el soporte de un ensayo clínico
utilicen lotes de compuesto producidos a escala similar a la que se va a utilizar en el lote
clínico. De esta manera se garantiza mejor la representatividad entre lotes preclínicos y
clínicos.
4. CASOS PRÁCTICOS
A continuación se facilitan una serie de ejemplos de programas de desarrollo preclínico de
diferentes tipos de compuestos para dar cobertura al primer ensayo en humanos, de acuerdo
con el marco regulatorio que aplica en cada caso. El objetivo de esta sección no es suministrar
la estrategia a seguir para cada tipo de medicamento, ya que es imposible y excede el alcance
de esta Guía, sino orientar al usuario en los elementos a tener en cuenta e ilustrar la
complejidad y variabilidad que puede existir en el desarrollo, dependiendo de cada caso
particular. La elección adecuada de los estudios necesarios y sus diseños tiene amplias
repercusiones en calendarios, presupuestos y probabilidades de éxito regulatorio, por lo que
la responsabilidad última en la selección de un programa es del promotor ayudado de su
equipo asesor.
A. QUÍMICO (SMALL MOLECULE)
A la hora de diseñar un programa de desarrollo preclínico es importante
contar con la información suficiente acerca del diseño clínico, indicación,
pauta y duración del ensayo clínico.
El presente ejemplo de programa preclínico corresponde a una small
molecule que además es nueva entidad química (NCE). Se puede considerar programa
estándar para soportar el primer ensayo clínico o una prueba de concepto en humanos.
Marco regulatorio
ICH Topic M3 (R2): Non‐Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals

Guía de desarrollos preclínicos 62
ICH Topic S2 (R1): Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use
ICH Topic S7A: Safety Pharmacology studies for human pharmaceuticals
ICH Topic S7B: Nonclinical Evaluation of QT Interval Prolongation
ICH S3A: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies
ICH S8: Immunotoxicity studies for human Pharmaceuticals
FDA: Guidance for Industry: Bioanalytical Method Validation
EMA: Guideline on Bioanalytical Method Validation
EMA: Note for Guidance on Repeated Dose Toxicity
EMA: Guideline on the evaluation of control samples in nonclinical safety studies: checking for contamination with the test substance
Propuesta de programa preclínico
Farmacodinamia
La información de estos estudios debe estar disponible antes de la administración en humanos.
Las agencias requieren datos de eficacia del producto en modelos animales o “in vitro” así
como una idea del mecanismo de acción del compuesto para justificar la exposición de
pacientes a dicho compuesto en desarrollo.
Unión a receptores
Es un estudio en el que se somete el compuesto y sus metabolitos principales a una batería
conocida de receptores celulares, para identificar sobre cuál de ellos presenta actividad
farmacológica, ya sea desde un punto de vista agonista o antagonista.

Guía de desarrollos preclínicos 63
Estudios farmacodinámicos en animales
Son estudios en los que se pretende valorar la actividad farmacológica del producto sobre
modelos animales. Dependiendo de la indicación terapéutica, los modelos experimentales
pueden ser muy diversos.
Farmacocinética y metabolismo
Farmacocinética
Son estudios complementarios pero recomendables en una etapa inicial del desarrollo que
permiten definir el perfil farmacocinético del producto en las especies que se usarán en
toxicología.
Metabolismo “in vitro”
Estudio “in vitro” comparativo utilizando microsomas o hepatocitos con la finalidad de
establecer el perfil metabólico del producto desde un punto de vista cualitativo, así como
poder identificar posibles metabolitos y posibles diferencias entre especies. Este estudio es
predictivo del metabolismo “in vivo”.
Toxicología
En general, los estudios de toxicología deberán diseñarse de forma que se pueda caracterizar
la naturaleza de los efectos tóxicos del compuesto, órganos diana y capacidad para la
reversibilidad de los efectos. Además, es necesario que los estudios permitan establecer la
NOAEL (dosis máxima sin efectos adversos), la cual permitirá establecer márgenes de
seguridad y estimar las dosis seguras para administrar en humanos. Estos estudios deberán
ser realizados bajo BPL (Buenas Prácticas de Laboratorio).
Rango de dosis en roedor
Este estudio de corta duración se realizará con la finalidad de obtener una información
preliminar sobre la toxicidad del producto, establecer la MDT (Máxima Dosis Tolerada) y la
MDL (Mínima Dosis Letal). Esta información permitirá escalar las dosis en el estudio
principal en roedor. Las especies de roedor preferentes para toxicología son rata y ratón.

Guía de desarrollos preclínicos 64
Rango de dosis en no‐roedor
Se realizará un estudio de dosis ascendentes, con la finalidad de obtener un perfil
toxicológico preliminar en una segunda especie y obtener información para escalar las dosis
del estudio principal en dicha especie. Las especies de no-roedor preferentes para toxicología
son perro, mono y minipig.
Estudio de administración repetida en roedor
Se realizará un estudio por una duración igual o superior a la exposición en humanos en el
ensayo clínico. Se evaluarán signos clínicos, peso corporal y consumo de comida. Se
realizarán análisis bioquímico-hematológicos, oftalmoscopia, determinaciones de los niveles
del compuesto en plasma para su valoración toxicocinética, observación macroscópica de
órganos e histopatología de los principales tejidos. Se evaluará asimismo la reversibilidad de
los posibles signos toxicológicos que se hayan presentado durante el periodo de exposición.
Estudio de administración repetida en no‐roedor
Se realizará un estudio por una duración igual o superior a la exposición en humanos en el
ensayo clínico. Se evaluarán prácticamente los mismos parámetros descritos para roedor y
además, se obtendrán electrocardiogramas de los animales, con la finalidad de identificar
posibles efectos del fármaco sobre el sistema cardiaco.
Estudios de Genotoxicidad
Test de Ames: Es un test de referencia en el que se establece el posible potencial mutagénico
de un compuesto utilizando como modelo diferentes cepas de Salmonella typhimurium y E.
coli, expuestas al compuesto, con y sin activación metabólica (S9).
Estudio de genotoxicidad “in vitro” en células de mamífero: En esta fase del desarrollo
preclínico, las guías establecen la necesidad de evaluar la posible genotoxicidad del fármaco
cómo mínimo en un estudio que no utilice un modelo microbiológico. Se realizará in test “in
vitro” en células de mamífero para identificar efectos genotóxicos sobre los cromosomas.

Guía de desarrollos preclínicos 65
Estudios de Inmunotoxicidad
Si durante la valoración de los estudios estándares de toxicología, se observan alteraciones
que puedan ser asociadas a efectos sobre el sistema inmunitario, será necesaria la realización
de estudios específicos para tipificar los efectos sobre el sistema inmune.
Estudios de toxicología de la reproducción y el desarrollo
Aunque a menudo estos estudios no son necesarios, habrá que valorar en cada caso
particular la necesidad de realizarlos, adecuando siempre el programa preclínico a las
necesidades del ensayo clínico que se desea soportar.
Toxicocinética
Validación de métodos analíticos: Los métodos bioanalíticos utilizados en la determinación
de un fármaco en muestras de plasma han de ser validados siguiendo unos parámetros
determinados que se establecen en las guías correspondientes de la EMA (European Medicines
Agency) y FDA (Food and Drug Administration), con la finalidad de demostrar su idoneidad en
el análisis de las muestras biológicas obtenidas en los estudios.
Evaluación toxicocinética: En los estudios toxicológicos “in vivo” definidos anteriormente, se
realizará la determinación bioanalítica del fármaco en las muestras de plasma obtenidas a
diferentes tiempos después de la administración con el fin de verificar la exposición
sistémica al compuesto en cuestión durante los estudios. Se debe valorar caso por caso la
necesidad de evaluar también la exposición a metabolitos principales del fármaco.
Farmacología de seguridad
Antes de que un fármaco sea probado en un estudio clínico con humanos, se deben realizar
estudios con la finalidad de evaluar efectos farmacológicos sobre órganos o funciones vitales:
Estudios sobre el sistema nervioso central
Estudios sobre el sistema cardiovascular
Estudios sobre el sistema respiratorio

Guía de desarrollos preclínicos 66
Dependiendo de los resultados de los estudios de farmacología de seguridad estándar o de la
clase farmacológica a la que pertenezca el compuesto, es posible que se deban realizar
estudios complementarios para evaluar efectos sobre los sistemas indicados o sobre otros
sistemas.
Cronograma
Se presenta a continuación una simulación de la duración y secuencia de los estudios
mencionados, asumiendo una progresión estándar. Plazos más cortos son posibles si se
solapan más algunas actividades, a la vez que se asumen más riesgos. La duración
representada en cada estudio corresponde al tiempo total aproximado desde el inicio hasta la
finalización del informe.
Cronograma
Estudio 1M 2M 3M 4M 5M 6M 7M 8M 9M 10M 11M 12M
Estudio farmacodinamia en animales e “in vitro”
Unión a receptores Metabolismo “in vitro” Validación del método bioanalítico Rango de dosis en roedor Toxicidad a dosis repetidas en roedor
Rango de dosis en no-roedor Toxicidad a dosis repetidas en roedor
Test de Ames Genotoxicidad en células de mamífero
Farmacología de seguridad Inmunotoxicidad (si se requiere) Farmacocinética complementaria en 2 especies
B. BIOTECNOLÓGICO: PROTEÍNA RECOMBINANTE
En el presente ejemplo se propone el plan preclínico necesario para
soportar un ensayo clínico de fase I en el que se llevará a cabo una
administración única con un medicamento biotecnológico, una
proteína recombinante.

Guía de desarrollos preclínicos 67
El siguiente plan de desarrollo se ha diseñado siguiendo principalmente los criterios de la
guía ICH S6 Preclinical Safety Evaluation of Biotechnology‐Derived Pharmaceuticals.
Marco regulatorio
ICH Topic M3 (R2): Non‐Clinical Safety Studies for the Conduct of Human Clinical Trials
and Marketing Authorization for Pharmaceuticals
ICH Topic S6: Preclinical Safety Evaluation of Biotechnology‐Derived Pharmaceuticals
Addendum to ICH S6: Preclinical safety evaluation of biotechnology‐derived
pharmaceuticals S6 (R1)
ICH S3A: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in
Toxicity Studies
EMA: Note for Guidance on Repeated Dose Toxicity
ICH topic S7 A: Safety Pharmacology Studies for Human Pharmaceuticals
FDA: Guidance for Industry: Bioanalytical Method Validation
EMA: Guideline on Bioanalytical Method Validation
EMA: Guideline on strategies to identify and mitigate risks for first‐in‐human clinical trials
with investigational medicinal products
Propuesta de programa preclínico
Farmacodinamia
La principal peculiaridad de los medicamentos biotecnológicos es que la actividad biológica,
o lo que sería equivalente a la farmacodinamia, es muy específica de especie. De modo que
una proteína recombinante que puede tener actividad biológica en humanos puede que no
tenga ninguna actividad en rata, por lo que no sería una especie relevante para llevar a cabo
los estudios necesarios para evaluar la seguridad del medicamento biotecnológico.

Guía de desarrollos preclínicos 68
Selección de la especie relevante
Las primeras fases de un programa de desarrollo preclínico de un medicamento
biotecnológico se centrarán en la selección de la especie relevante para los estudios tanto de
eficacia “in vivo” como los estudios regulatorios de seguridad.
Dado que cada medicamento biotecnológico tiene su propia actividad biológica y un
mecanismo de acción muy especifico, los estudios que se llevarán a cabo encaminados a
seleccionar la especie relevante podrán ser muy diversos y adaptados a las particularidades
de cada compuesto.
La selección del abordaje más adecuado y de los ensayos “in vitro” e “in vivo” necesarios para
seleccionar la especie relevante requiere en muchos casos de la evaluación por parte de
expertos en inmunología y biología molecular.
Estudios en las potenciales especies relevantes
Tras hacer una preselección de las potenciales especies relevantes, se llevarán a cabo los
siguientes estudios para realizar una selección definitiva.
Estudios farmacodinámicos “in vivo”: el objetivo de estos estudios será demostrar el perfil
farmacodinámico o actividad biológica similar entre las especies seleccionadas usando
biomarcadores de la actividad biológica.
Reactividad cruzada en tejidos: antes de la exposición a humanos ha de llevarse a cabo un
estudio de reactividad cruzada de la proteína recombinante con una batería completa de los
diferentes tejidos de humanos.
Toxicología
De acuerdo con guía ICH S6, los estudios regulatorios de toxicidad han de llevarse a cabo en
2 especies (una de roedor y otra de no roedor).
En función de los resultados de los estudios de selección de la especie relevante se decidirá
en qué especies se llevarán a cabo los estudios de seguridad. En el caso de que no se pueda

Guía de desarrollos preclínicos 69
encontrar más de una especie relevante o incluso ninguna, mediante la adecuada
justificación se realizarán los estudios de seguridad en sólo una especie.
En general, los estudios de toxicología deberán diseñarse de forma que se pueda caracterizar
la naturaleza de los efectos biológicos adversos y/o tóxicos del compuesto, órganos diana y
capacidad para la reversibilidad de los efectos. Además, es necesario que los estudios
permitan establecer la NOAEL (dosis máxima sin efectos adversos) o en los casos de
medicamentos que den lugar a efectos en cascada y multiorgánicos, la MABEL (dosis
mínima con efecto biológico). Estos índices permitirán establecer márgenes de seguridad y
estimar las dosis seguras para administrar en humanos. Estos estudios deberán ser
realizados bajo BPL (Buenas Prácticas de Laboratorio).
Rango de dosis en roedor y no roedor
Este estudio de hasta 1 semana de duración se realizará en cada una de las especies
seleccionadas con la finalidad de obtener una información preliminar sobre la toxicidad del
producto, anticipando la MTD (Maximum Tolerated Dose) para permitir el escalado de
estudios de mayor duración.
Estudios de dosis repetida
Dado que en este ejemplo se pretende dar una sola administración del medicamento en
humanos en un ensayo clínico, los estudios principales de toxicología serán de 2 semanas de
duración (duración mínima aceptada) en las 2 especies seleccionadas como relevantes. Las
dosis se seleccionarán en los estudios de rango de dosis. El compuesto se administrará
diariamente, o a intervalos pertinentes, durante 14 días.
Se evaluarán signos clínicos, peso corporal y consumo de comida. Se realizarán análisis
bioquímico-hematológicos, oftalmoscopia, determinaciones de los niveles del compuesto en
plasma para su valoración toxicocinética, observación macroscópica de órganos e
histopatología de los principales tejidos. En los estudios en especie no-roedor además se
obtendrán electrocardiogramas de los animales. Se evaluará asimismo la reversibilidad de
los posibles signos toxicológicos que se hayan presentado durante el periodo de exposición.

Guía de desarrollos preclínicos 70
Estudios de Genotoxicidad
Por regla general, no será necesario realizar estudios específicos de genotoxicidad con
compuestos de origen biotecnológico, pero se debe valorar caso por caso.
Toxicocinética
Validación de métodos analíticos: Los métodos bioanalíticos utilizados en la determinación
de un fármaco en muestras de plasma han de ser validados siguiendo unos parámetros
determinados que se establecen en las guías correspondientes de la EMA (European Medicines
Agency) y FDA (Food and Drug Administration), con la finalidad de demostrar su idoneidad en
el análisis de las muestras biológicas obtenidas en los estudios.
Evaluación toxicocinética: En los estudios toxicológicos “in vivo” definidos anteriormente, se
realizará la determinación bioanalítica del fármaco en las muestras de plasma obtenidas a
diferentes tiempos después de la administración con el fin de verificar la exposición
sistémica al compuesto en cuestión durante los estudios.
Farmacología de seguridad
Dado que los medicamentos biotecnológicos suelen ejercer una acción muy específica como
resultado de su unión a un receptor o epítopo, de acuerdo con la guía en vigor, no se
requerirán estudios específicos de farmacología de seguridad, sino que se incluirán los
parámetros como parte de los estudios de dosis repetidas. Sin embrago, en caso de que
hubiera algún efecto de clase, sí que habría que llevar a cabo estudios específicos de
farmacología de seguridad.
Metabolismo
En el ejemplo propuesto, no será necesario llevar a cabo estudios clásicos de metabolismo, ya
que al tratarse de una proteína recombinante, ésta se hidrolizará en péptidos y aminoácidos
que no se espera posean actividad biológica.
Cronograma
Se presenta a continuación una simulación de la duración y secuencia de los estudios
mencionados, asumiendo una progresión estándar. Plazos más cortos son posibles si se

Guía de desarrollos preclínicos 71
solapan más algunas actividades, a la vez que se asumen más riesgos. La duración
representada en cada estudio corresponde al tiempo total aproximado desde inicio hasta
finalización de informe.
Cronograma
Estudio 1M 2M 3M 4M 5M 6M 7M 8M 9M 10M 11M 12M
Selección de la especie relevante
Estudio farmacodinamia en animales
Reactividad cruzada en tejidos
Validación del método bioanalítico
Rango de dosis en roedor
Toxicidad a dosis repetidas en roedor
Rango de dosis en no-roedor
Toxicidad a dosis repetidas en roedor
C. ANTITUMORAL
En el caso de aquellos compuestos antitumorales cuyo ensayo clínico
de fase I o IIa se va a llevar a cabo en pacientes terminales o con una
esperanza de vida corta, los requerimientos preclínicos son algo
menores que para un desarrollo estándar, según la guía en vigor (ICH
S9). En ella se definen los estudios de seguridad necesarios para soportar el inicio del
desarrollo clínico de un fármaco antitumoral cuando la población diana son pacientes
terminales.
Marco regulatorio
ICH Topic S9: Nonclinical Evaluation for Anticancer Pharmaceuticals
ICH Topic M3 (R2): Non‐Clinical Safety Studies for the Conduct of Human Clinical Trials
and Marketing Authorization for Pharmaceuticals
ICH S3A: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in
Toxicity Studies
FDA: Guidance for Industry: Bioanalytical Method Validation
EMA: Guideline on Bioanalytical Method Validation

Guía de desarrollos preclínicos 72
EMA: Note for Guidance on Repeated Dose Toxicity
EMA: Guideline on the evaluation of control samples in nonclinical safety studies: checking
for contamination with the test substance
Programa de desarrollo preclínico
Farmacodinamia
Al inicio del desarrollo de un fármaco antitumoral es necesario demostrar eficacia ante uno o
varios tumores, realizándose inicialmente un estudio “in vitro” en líneas celulares y
posteriormente uno o más estudios “in vivo” con ratones, también con varias líneas celulares
tumorales.
Eficacia “in vitro”
Son estudios en los que se expone el fármaco a una batería establecida de líneas celulares
tumorales humanas, identificándose aquellas en las que presenta una mayor incidencia sobre
la inhibición del crecimiento celular.
Estudios de Xenograft
Se realizan en ratones inmunodeprimidos a los que se les induce la expresión de uno o varios
tumores. Después de una fase de crecimiento tumoral se administran los animales a
diferentes dosis del compuesto y se evalúa la reducción del tamaño de los nódulos y/o el
aumento de la supervivencia de los animales.
Estos estudios ayudarán a establecer la dosis mínima eficaz o dosis terapéutica en la cual nos
basaremos a su vez para definir las dosis a administrar en los estudios de toxicología que se
mencionan a continuación.
Toxicología
En general, los estudios de toxicología deberán diseñarse de forma que se pueda caracterizar
la naturaleza de los efectos tóxicos del compuesto, órganos diana y capacidad para la
reversibilidad de los efectos. Además, es necesario que los estudios permitan establecer la
máxima dosis no severamente tóxica, la cual permitirá establecer márgenes de seguridad y

Guía de desarrollos preclínicos 73
estimar las dosis seguras para administrar en humanos. Estos estudios deberán ser
realizados bajo BPL (Buenas Prácticas de Laboratorio).
Rango de dosis en roedor
Este estudio de corta duración se realizará con la finalidad de obtener una información
preliminar sobre la toxicidad del producto, establecer la MDT (Máxima Dosis Tolerada) y la
MDL (Mínima Dosis Letal). Esta información permitirá escalar las dosis en el estudio
principal en roedor. Las especies de roedor preferentes para toxicología son rata y ratón.
Rango de dosis en no‐roedor
Se realizará un estudio de dosis ascendentes, con la finalidad de obtener un perfil
toxicológico preliminar en una segunda especie y obtener información para escalar las dosis
del estudio principal en dicha especie. Las especies de no-roedor preferentes para toxicología
son perro, mono y minipig.
Estudio de 4 semanas en roedor
En el caso de los antitumorales, la duración del estudio no necesita igualar o exceder la
duración del tratamiento en los pacientes. Por lo general, estudios de toxicidad de duración
hasta 28 días permiten administrar el compuesto en pacientes con cáncer durante períodos
más prolongados. Se evaluarán signos clínicos, peso corporal y consumo de comida. Se
realizarán análisis bioquímico-hematológicos, oftalmoscopia, determinaciones de los niveles
del compuesto en plasma para su valoración toxicocinética, observación macroscópica de
órganos e histopatología de los principales tejidos. Además, se podrán incluir parámetros de
farmacología de seguridad y en determinados casos, también de inmunotoxicidad. Se
evaluará asimismo la reversibilidad de los posibles signos toxicológicos que se hayan
presentado durante el periodo de exposición.
Estudio de 4 semanas en no‐roedor
Este estudio tendrá asimismo una duración de 28 días, y se evaluarán prácticamente los mismos
parámetros descritos arriba para roedor y además, se obtendrán electrocardiogramas de los
animales, con la finalidad de identificar posibles efectos del fármaco sobre el sistema cardiaco.

Guía de desarrollos preclínicos 74
Toxicocinética
Validación de métodos analíticos: Los métodos bioanalíticos utilizados en la determinación de un
fármaco en muestras de plasma han de ser validados siguiendo unos parámetros determinados
que se establecen en las guías correspondientes de la EMA y FDA, con la finalidad de demostrar su
idoneidad en el análisis de las muestras biológicas obtenidas en los estudios.
Evaluación toxicocinética: En los estudios toxicológicos “in vivo” definidos anteriormente, se
realizará la determinación bioanalítica del fármaco en las muestras de plasma obtenidas a
diferentes tiempos después de la administración con el fin de verificar la exposición
sistémica al compuesto en cuestión durante los estudios.
Cronograma
Se presenta a continuación una simulación de la duración y secuencia de los estudios
mencionados, asumiendo una progresión estándar. Plazos más cortos son posibles si se solapan
más algunas actividades, a la vez que se asumen más riesgos. La duración representada en cada
estudio corresponde al tiempo total aproximado desde inicio hasta finalización de informe.
Cronograma Estudio 1M 2M 3M 4M 5M 6M 7M 8M 9M 10M 11M 12M Estudio eficacia “in vitro” Estudios de xenograft Validación del método bioanalítico
Rango de dosis en roedor Toxicidad a dosis repetidas en roedor
Rango de dosis en no-roedor Toxicidad a dosis repetidas en roedor
D. DESARROLLO BIOLÓGICO
A continuación se citan muy brevemente a modo de ejemplo para un
desarrollo de producto biológico, los ensayos preclínicos que se
deberán realizar con una vacuna en desarrollo sin adyuvante o que
contenga un adyuvante autorizado, para poder soportar el primer
ensayo de Fase I en humanos.

Guía de desarrollos preclínicos 75
Los estudios a que se hace referencia se deberán hacer por la vía de administración por la
que se administrará la vacuna en humanos.
Marco regulatorio
Note for Guidance on preclinical pharmacological and toxicological testing of vaccines
(CPMP/SWP/465/95)
Guideline on adjuvants in vaccines for human use (EMEA/CHMP/VEG/134716/2004)
Propuesta de programa preclínico
Farmacodinamia
Caracterización de la respuesta inmune
Son estudios dirigidos a determinar la producción de anticuerpos, caracterización de la clase
de anticuerpos, respuesta humoral y celular, y duración de la respuesta inmune. Estos
estudios, además de tener la aplicación directa de demostrar eficacia en modelos animales,
servirían para justificar con criterios objetivos la selección de la especie adecuada para
realizar los estudios de toxicología.
Estudio confirmatorio de la idoneidad de la formulación
Es un estudio en el que se debe verificar y justificar que los componentes de la vacuna son
los idóneos y se encuentran en las proporciones óptimas para producir la máxima respuesta
inmune y los mínimos efectos adversos. Este es un estudio que está a caballo entre eficacia y
seguridad y del que depende la selección de fórmula final, por lo tanto, tiene también
implicaciones galénicas.
Toxicología
Es muy importante realizar los estudios con la combinación de antígeno y adyuvante
idéntica a la que se vayan a exponer los humanos.

Guía de desarrollos preclínicos 76
Toxicidad aguda en una especie de roedor
El estudio tiene como objetivo evaluar los efectos adversos de una administración única a
dosis superiores a las que se van a dar en humanos. Los efectos locales y sistémicos se
registrarán durante un periodo de observación de 14 días.
Toxicidad a dosis repetida en una especie de roedor
La administración deberá realizarse con un intervalo entre dosis suficientemente largo para
que se produzca reacción inmune y con una duración total del tratamiento dependiente del
que se va a hacer en humanos. Como pauta general, se administra en animales el mismo
número de dosis +1 (n+1) que el que se va a administrar en humanos, seguida de un período
de recuperación. Los ensayos se realizan en 1 especie en la que se haya demostrado
capacidad para generar respuesta inmune.
Se evaluarán signos clínicos, pirogenicidad, peso corporal y consumo de comida. Se
realizarán análisis bioquímico-hematológicos, oftalmoscopia, observación macroscópica de
órganos e histopatología de los principales tejidos. Se evaluarán asimismo los efectos
producidos a largo plazo o la recuperación de los mismos.
Tolerancia local en una especie
En este estudio se evalúa el efecto local de la vacuna por la misma vía de administración que
se va a utilizar en humanos. Este estudio podría formar parte del estudio de dosis repetida,
como parámetro adicional a evaluar.
Farmacología de seguridad
Los estudios de farmacología de seguridad deben incluir la evaluación de los efectos sobre
sistema nervioso central y autónomo, cardiovascular y respiratorio. Al igual que se ha
comentado para otros tipos de medicamentos, esta evaluación también podría formar parte
del estudio de dosis repetida incluyendo determinaciones adicionales.

Guía de desarrollos preclínicos 77
Cronograma
Se presenta a continuación una simulación de la duración y secuencia de los estudios
mencionados, asumiendo una progresión estándar. Plazos más cortos son posibles si se
solapan más algunas actividades, a la vez que se asumen más riesgos. La duración
representada en cada estudio corresponde al tiempo total aproximado desde inicio hasta
finalización de informe.
Cronograma
Estudio 1M 2M 3M 4M 5M 6M 7M 8M 9M 10M 11M 12M
Caracterización de la respuesta inmune
Idoneidad de componentes de la formulación
Validación del método de marcadores de respuesta
Toxicidad aguda en roedor
Toxicidad a dosis repetidas en roedor
Farmacología de seguridad
5. PROVEEDORES
A. ¿POR QUÉ CONTRATAR UN PROVEEDOR ESPECIALIZADO DE
SERVICIOS?
Tanto si se trata de estudios de seguridad como
de eficacia o ADME (Absorción, Distribución,
Metabolismo y Excreción), estos estudios por lo
general son de gran complejidad, requieren
equipos sofisticados y especializados, y personal
con gran experiencia y conocimiento de los
sistemas y técnicas experimentales. En el caso de

Guía de desarrollos preclínicos 78
los estudios de seguridad, además es necesario que el laboratorio disponga de la certificación
de BPLs (Buenas Prácticas de Laboratorio). Por ello, aunque las compañías que desarrollen
un medicamento pueden realizar directamente algunos estudios en el caso de que dispongan
de personal, instalaciones y equipos cualificados, la tendencia habitual es que los estudios
preclínicos se subcontraten a empresas especializadas denominadas CROs preclínicas las
cuales, tal y como requieren las agencias reguladoras, tienen certificación BPL.
A la hora de seleccionar el laboratorio (propio o subcontratado), en donde se realizarán los
estudios preclínicos, hay que tener en cuenta la experiencia del mismo en estudios similares.
Al tratarse de estudios tan complejos, no todos los laboratorios tienen la experiencia
necesaria o pueden hacer cualquier tipo de ensayo. En el caso de las CROs, algunas optan
por adquirir experiencia y elevado grado de especialización en determinados tipos de
estudios, vía de administración e incluso en ensayos con determinadas especies.
La subcontratación necesita de un seguimiento intenso y una continua toma decisiones de
tipo técnico por parte del promotor del desarrollo del medicamento. El responsable de esta
misión deberá además conocer la globalidad del proyecto, el entorno regulatorio que aplica,
las necesidades estratégicas de la empresa y además poder interactuar con las áreas
regulatoria, de desarrollo farmacéutico y clínico (ya sean parte del mismo laboratorio o estén
a su vez subcontratadas). Solo la alta profesionalización de cada uno de los implicados en el
desarrollo de un medicamento podrá asegurar el éxito con plazos de tiempo y costes
óptimos.
B. ¿POR QUÉ LA NECESIDAD DE ELABORAR UN PLAN DE DESARROLLO
PRECLINICO?
El objetivo de la elaboración de una Hoja de ruta o Plan de desarrollo preclínico es identificar de
forma prospectiva los estudios preclínicos a realizar necesarios para soportar la autorización
de un ensayo clínico o el registro de un medicamento, así como su duración y secuencia
temporal, con el fin de anticipar y designar recursos humanos, materiales y económicos de
forma racional y ajustados a las necesidades.

Guía de desarrollos preclínicos 79
En el plan de desarrollo se especifican los estudios necesarios para llevar a cabo la evaluación
de la eficacia y toxicidad del fármaco en cuestión.
La hoja de ruta es específica para cada medicamento en investigación y no puede ser
adaptada a otros desarrollos. Los desarrollos similares sólo pueden servir como orientación,
de lo que sería necesario hacer en nuestro caso particular, pero no hay una única estrategia
efectiva aplicable a cada tipo de fármaco.
El plan incluye una serie de pasos no siempre secuenciales. Una vez diseñada la primera hoja
de ruta con la batería de ensayos propuestos, se deben realizar a posteriori, y a la vista de
resultados, revisiones sistemáticas acerca de los distintos estudios que se van realizando.
Esto justifica las sucesivas reuniones de trabajo que deberán realizarse, es decir, el
seguimiento de los resultados de los ensayos no puede considerarse como una simple
subcontratación, ya que el éxito es fruto de un trabajo en equipo.
La hoja de ruta debe ser diseñada con el objetivo de asegurar que los resultados obtenidos
sean, además de científicamente válidos, relevantes en la toma de decisiones.
La combinación de los ensayos no clínicos a desarrollar, depende de las características del
fármaco, sus indicaciones y la población humana que será tratada entre otras variables. El
Plan ayudará a establecer la ruta general y los pasos posteriores deben estar justificados
sobre la base de resultados previos y ofrecer información útil para evaluar el fármaco.
La batería de estudios consta de estudios farmacodinámicos, farmacocinéticos y
toxicológicos. La ausencia de ciertos estudios también debe ser justificada fundamentando
que los estudios previos son suficientes para permitir la valoración del fármaco.
El plan de desarrollo preclínico debe tener en cuenta la normativa a aplicar en cada caso, ya
que los estudios deben ajustarse a las normativas aceptadas que se encuentran debidamente
establecidas en las guías de la Organización Mundial de la Salud (OMS), la Conferencia
Internacional para la Armonización (ICH), la Agencia Europea de Medicamentos (EMA), la

Guía de desarrollos preclínicos 80
Organización para la Cooperación y el Desarrollo Económico (OCDE) y la Food and Drug
Administration de Estados Unidos (FDA).
Se especificarán los ensayos en los que es obligatoria su realización siguiendo las Buenas
Prácticas de Laboratorio (BPL) según las Agencias Reguladoras de Referencia.
La hoja de ruta justifica los estudios que deben realizarse “in vivo” y aquellos que deben
llevarse a cabo “in vitro”. También puede orientar en la selección de las especies animales en
las que realizar los ensayos.
La importancia de un buen diseño de un plan de desarrollo preclínico, es vital para el éxito
del proyecto en su conjunto, y justificará la correcta documentación a presentar a las
autoridades competentes (el Documento Técnico Común o CTD).
Por todo lo indicado anteriormente, el diseño del plan de desarrollo preclínico debe estar a
cargo de un equipo de profesionales, ya que requiere personal con gran experiencia y
conocimientos muy amplios y específicos tanto técnicos como regulatorios. Existen servicios
de asesoría y consultoría altamente cualificados que pueden desempeñar este cometido
aportando valor al proyecto
6. GLOSARIO
Asesoramiento Científico (Scientific Advice): asesoramiento prestado por la EMA,
FDA o agencias sanitarias nacionales sobre los ensayos y estudios adecuados para el
desarrollo de un medicamento para facilitar el desarrollo y la disponibilidad de
medicamentos de alta calidad, eficaces y seguros.
Buenas Prácticas Clínicas (Good Clinical Practice - GCP): conjunto de requisitos éticos
y científicos de calidad reconocidos a escala internacional, que deben cumplirse en la
planificación, la realización, el registro y la comunicación de los ensayos clínicos en
que participen seres humanos. Su cumplimiento garantiza la protección de los
derechos humanos, le seguridad y el bienestar de los sujetos del ensayo, así como la
fiabilidad de los resultados del ensayo clínico.

Guía de desarrollos preclínicos 81
Buenas Prácticas de Fabricación (Good Manufacturing Practice – GMP): Se encuentran
incluidas dentro del concepto de Garantía de Calidad, constituyen el factor que
asegura que los productos se fabriquen en forma uniforme y controlada, de acuerdo
con las normas de calidad adecuadas al uso que se pretende dar a los productos y
conforme a las condiciones exigidas para su comercialización. Las reglamentaciones
que rigen las buenas prácticas de fabricación tienen por objeto principal disminuir los
riesgos inherentes a toda producción farmacéutica. Las GMPs son aplicables a las
operaciones de fabricación de medicamentos, cosméticos, productos sanitarios,
alimentos y medicamentos, en sus formas definitivas de venta al público incluyendo
los procesos a gran escala en hospitales y la preparación de suministros para el uso de
ensayos clínicos para el caso de medicamentos.
Buenas Prácticas de Laboratorio –BLP– (Good Laboratory Practice – GLP): conjunto de
reglas, procedimientos operacionales y prácticas establecidas y promulgadas por
determinados organismos como la Organization for Economic Cooperation and
Development (OCDE) o la Food and Drug Administration (FDA), etc., que se consideran
de obligado cumplimiento para asegurar la calidad e integridad de los datos
producidos en determinados tipos de investigaciones o estudios.
Common Technical Document (CTD): es el formato obligatorio para presentar todas
las solicitudes de autorización de medicamentos en el que se recoge toda la
información de calidad, seguridad y eficacia.
Contract Manufacturing Organization (CMO): es una organización que presta
servicios a la industria farmacéutica y proporciona a sus clientes servicios integrales
de fabricación en el desarrollo de fármacos. Entre los servicios que ofrecen las CMOs
se puede incluir, aunque no de forma limitativa: pre-formulación, formulación,
estudios de estabilidad, el desarrollo de métodos, escalado, registro de lotes y
producción comercial.
Contract Research Organization o Clinical Research Organization (CRO): es una
organización (comercial, académica o de otro tipo) que presta servicios contratados
por un promotor (industria farmacéutica, biotecnológica…), en forma de servicios

Guía de desarrollos preclínicos 82
externalizados, tanto para medicamentos como para dispositivos médicos. Los
servicios que ofrecen las CROs son: investigación en fases de tempranas (discovery o
screening), ensayos preclínicos, ensayos clínicos y asuntos regulatorios. Muchas CROs
están especializadas en cada una de estas áreas, mientras que algunas son globales
cubriendo todos los aspectos de la I+D de un medicamento.
Contract Service Organization (CSO): es una organización que presta servicios a la
industria. Las CSOs pueden estar especializadas en uno de los muchos aspectos de la
empresa, incluidos servicios de fabricación, legales, o de otro tipo.
La subcontratación de tareas por parte de la industria cada vez es más frecuente ya
que permite a las empresas centrarse en sus competencias básicas, mejorando la
rentabilidad.
Dossier de Medicamento en Investigación (IMPD): es la base para la aprobación de
los ensayos clínicos por las autoridades competentes en la UE. Contiene la
información relacionada con calidad, fabricación, control de medicamentos en
investigación, datos de los estudios preclínicos y su uso clínico.
Ensayo clínico: toda investigación efectuada en seres humanos, con el fin de
determinar o confirmar los efectos clínicos, farmacológicos y/o los demás efectos
farmacodinámicos de uno o varios medicamentos en investigación, y/o de detectar las
reacciones adversas a uno o varios medicamentos en investigación, y/o de estudiar la
absorción, la distribución, el metabolismo y la eliminación de uno o varios
medicamentos en investigación con el fin de determinar su inocuidad y/o su eficacia.
Fármaco: sustancia (principio activo) que produce un efecto clínico determinado, ya
esté relacionado con la prevención, el diagnóstico, tratamiento o cura de una
patología concreta.
Farmacocinética (PK): rama de la farmacología que estudia los procesos a los que un
fármaco es sometido a través de su paso por el organismo. Trata de dilucidar qué
sucede con un fármaco desde el momento en el que es administrado hasta su total
eliminación del cuerpo. El estudio detallado de los sucesivos pasos que atraviesa el

Guía de desarrollos preclínicos 83
fármaco en el organismo, se agrupan en el ADME (Absorción, Distribución,
Metabolismo, Eliminación).
Farmacodinamia (PD): es el estudio de los efectos bioquímicos y fisiológicos de los
fármacos y de sus mecanismos de acción y la relación entre la concentración del
fármaco y el efecto de éste sobre un organismo.
Innovation Task Force (ITF): grupo multidisciplinar (con competencias científicas,
regulatorias y legales) que proporciona un asesoramiento inicial, antes de iniciar otros
procedimientos en la EMA, orientando en etapas tempranas del desarrollo.
Investigational New Drug (IND): documento que se presenta en la FDA para
conseguir el status de medicamento en desarrollo y conseguir la autorización de un
ensayo clínico.
Medicamento: conjunto de sustancias compuesto por uno o más principios activos y
los excipientes correspondientes, formulado de tal manera que quede listo para su
uso directo o indirecto (en el caso de que se tenga que reconstituir con algún tipo de
solvente).
Medicamento en Investigación (MI): forma farmacéutica de un principio activo o
placebo que se utiliza como referencia en un ensayo clínico, incluidos los productos
con autorización de comercialización cuando se utilicen o combinen de una forma
diferente a la autorizada, o cuando se utilicen para tratar una indicación no
autorizada, o para obtener más información sobre un uso autorizado.
Medicamento huérfano: es aquel dirigido a prevenir, diagnosticar o tratar una
enfermedad huérfana, que es aquella cuya prevalencia es baja y no existe alternativa
terapéutica adecuada.
La Organización Mundial de la Salud (OMS) define enfermedad huérfana, rara o de
baja prevalencia como “toda condición patológica que afecte de 650 a 1.000 personas por
millón de habitantes”. Sin embargo, la legislación de cada país establece ese límite de
una forma variable.

Guía de desarrollos preclínicos 84
Medicamentos para Terapias Avanzadas (ATMP): medicamento de terapia génica,
de terapia celular somática o de ingeniería tisular.
Pletismografía: Es un examen utilizado para medir cambios en volumen en
diferentes partes del cuerpo. Estos cambios en el volumen ayudan a verificar la
circulación en muchas partes del cuerpo. Este examen se puede hacer para
inspeccionar si hay coágulos sanguíneos en los brazos y las piernas o para medir
cuánto aire puede usted contener en sus pulmones.
Producto terapéutico: incluye medicamentos y complementos médicos como
suplementos de dieta, productos sanitarios, ayudas a la audición y visión y otros.
Promotor o Sponsor: individuo, empresa, institución u organización responsable del
inicio, gestión y/o financiación de un ensayo preclínico, clínico y de calidad.
El Promotor sólo podrá iniciar el ensayo clínico una vez que el Comité Ético haya
emitido un dictamen favorable, y siempre y cuando las autoridades competentes del
estado miembro de la UE interesado no le hayan comunicado objeciones motivadas.
"Sponsor" se refiere a:
(a) el sponsor de un Investigational New Drug (IND) o Medicamento en
Investigación (MI) en los EE.UU.
(b) el solicitante, que presenta una solicitud de nuevo fármaco (New Drug
Application ‐ NDA) o Biologics License Application (BLA) en los EE.UU.
(c) un posible solicitante en proceso de autorización de comercialización en la
Unión Europea.
Protocolos de asistencia (Protocol Assistance): forma específica de asesoramiento
científico prestado por la EMA para el desarrollo de medicamentos huérfanos.
Solicitud de Autorización de Ensayo (Clinical Trial Application - CTA):
procedimiento, en la Unión Europea, por el que una compañía farmacéutica u otro
grupo de investigación solicita el permiso para llevar a cabo ensayos clínicos en

Guía de desarrollos preclínicos 85
humanos. La normativa europea exige la obtención de una autorización de ensayo
clínico expedida por las autoridades sanitarias competentes.
Working Parties: grupos de trabajo del ICH que emiten guías especializados en cada
una de las áreas de interés.
GLOSARIO DE ACRÓNIMOS
AARR: Autoridades Reguladoras
ADME: Absorción, Distribución, Metabolismo y Excreción
ADME‐Tox: Absorción, Distribución, Metabolismo y Excreción y toxicología
ADN: Ácido Desoxirribonucleico
AEMPS: Agencia Española de Medicamentos y Productos Sanitarios
AI: Artificial Intelligence - Inteligencia Artificial
BPL: Buenas Prácticas de Laboratorio
CAT: Comité de Terapias Avanzadas (EMA)
CHMP: Comité de Medicamentos de Uso Humano (EMA)
CMH: Comité de Medicamentos de Uso Humano (AEMPS)
CMOs: Contract Manufacturing Organizations
COMP: Comité de Medicamentos Huérfanos (EMA)
CROs: Contract (o Clinical) Research Organizations
CTD: Common Technical Document - Documento Técnico Común
CVMP: Comité de Medicamentos de Uso Veterinario (EMA)
EMA: European Medicines Agency
FDA: Food and Drug Administration
GCP: Good Clinical Practice
GLP: Good Laboratory Practice

Guía de desarrollos preclínicos 86
GLOSARIO DE ACRÓNIMOS
GMP: Good Manufacturing Practice
hERG: Human Ether‐à‐go‐go‐Related Rene
HMPC: Comité de Medicamentos a Base de Plantas (EMA)
HTS: High‐throughput Screening
ICH: International Conference of Harmonisation
IMPD: Investigational Medicinal Product Dossier - Dossier de Medicamento en Investigación
IND: Investigational New Drug - nuevo medicamento en investigación
ITF: Innovation Task Force – grupo de trabajo en innovación
MABEL: Dosis Mínima con Efecto Biológico
MDL: Mínima Dosis Letal
MDT: Máxima Dosis Tolerada
MHLW: Ministry of Health, Labour and Welfare
MTD: Maximum Tolerated Dose
NCE: New Chemical Entity - Nueva Entidad Química
NDA: New Drug Application – solicitud de nuevo medicamento
NOAEL: Non‐observed Adverse Effects Level – Dosis Máxima sin Efectos Adversos
OCDE: Organización para la Cooperación y el Desarrollo Económico
OMS: Organización Mundial de la Salud
PD: Pharmacodynamics – Farmacodinamia
PDCO: Comité Pediátrico (EMA)
PK: Pharmacokinetics – Farmacocinética
SAR: Structure Activity Relationships – Relación entre Estructura y Función

Guía de desarrollos preclínicos 87
GLOSARIO DE ACRÓNIMOS
SAWP: Scientific Advice Working Party (EMA)
SIDA: Síndrome de Inmunodeficiencia Adquirida
UE: Unión Europea
VIH: Virus de la Inmunodeficiencia Humana


















