Glycosaminoglycans and Glycoproteins UNIT II: Intermediary Metabolism.
54
Glycosaminoglycans and Glycoproteins UNIT II: Intermediary Metabolism
-
date post
19-Dec-2015 -
Category
Documents
-
view
264 -
download
1
Transcript of Glycosaminoglycans and Glycoproteins UNIT II: Intermediary Metabolism.

Glycosaminoglycans and Glycoproteins
UNIT II:
Intermediary Metabolism

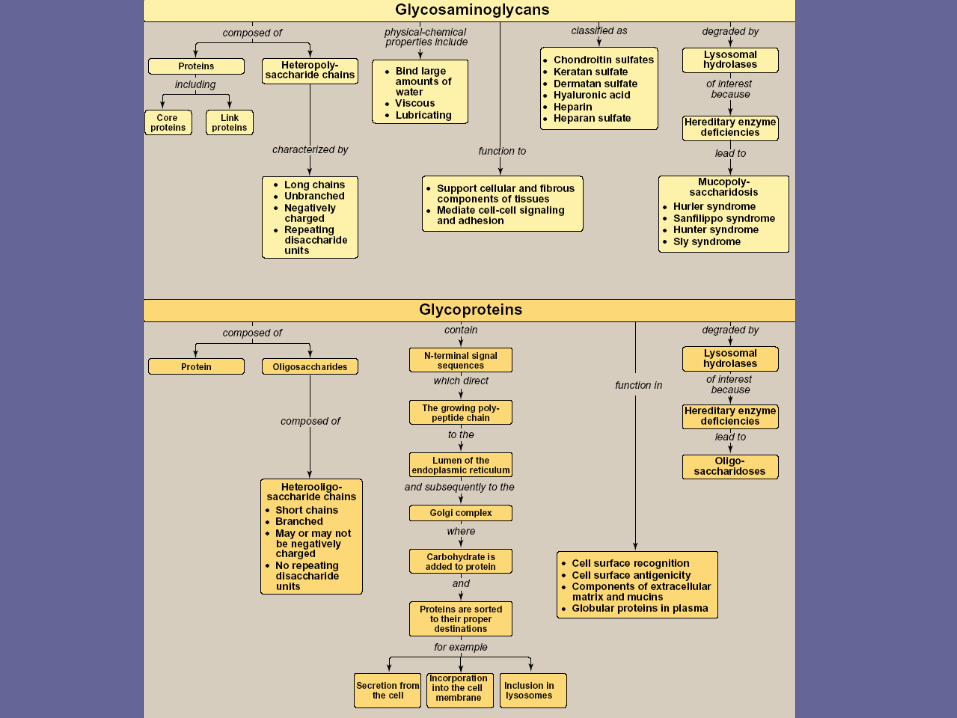
Overview of glycosaminoglycans
• Glycosaminoglycan (GAGs) are large complexes of negatively charged heteropolysaccharide chains. They are generally associated with a small amount of protein, forming proteoglycans, which typically consist of > 95% CHO.
Note: this is in comparison to the glycoproteins, which consist primarily of protein with a small amount of CHO
• Glycosaminoglycans have the special ability to bind large amounts of water, thereby producing the gel-like matrix that forms the basis of the body’s ground substance
• The viscous, lubricating properties of mucous secretions also result from the presence of GAGs, which led to the original naming of these cpds as mucopolysaccharides

II. Structure of GAGs
• GAGs are long, unbranched, heteropolysaccharide chains generally composed of a repeating disacch unit [acidic sugar-amino sugar]n
• The amino sugar is either D-glucosamine or D-galactosamine, in which the amino group is usually acetylated, thus eliminating its +ve charge. The amino sugar may also be sulfated on C-4 or 6 or on a non-acetylated nitrogen
• The acidic sugar is either D-glucuronic acid or its C-5 epimer, L-iduronic acid
Note: a single exception is keratan sulfate, in which galactose rather than an acidic sugar is present
• These acidic sugars contain carboxyl groups that are negatively charged at physiologic pH &, together with the sulfate groups, give GAGs their strongly –ve nature
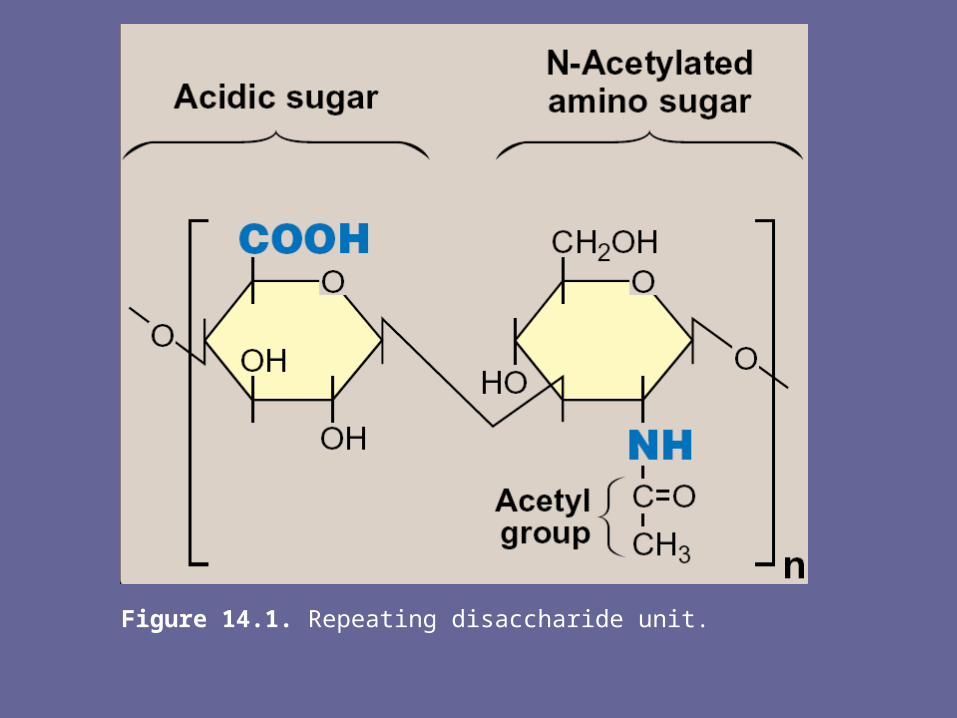
Figure 14.1. Repeating disaccharide unit.
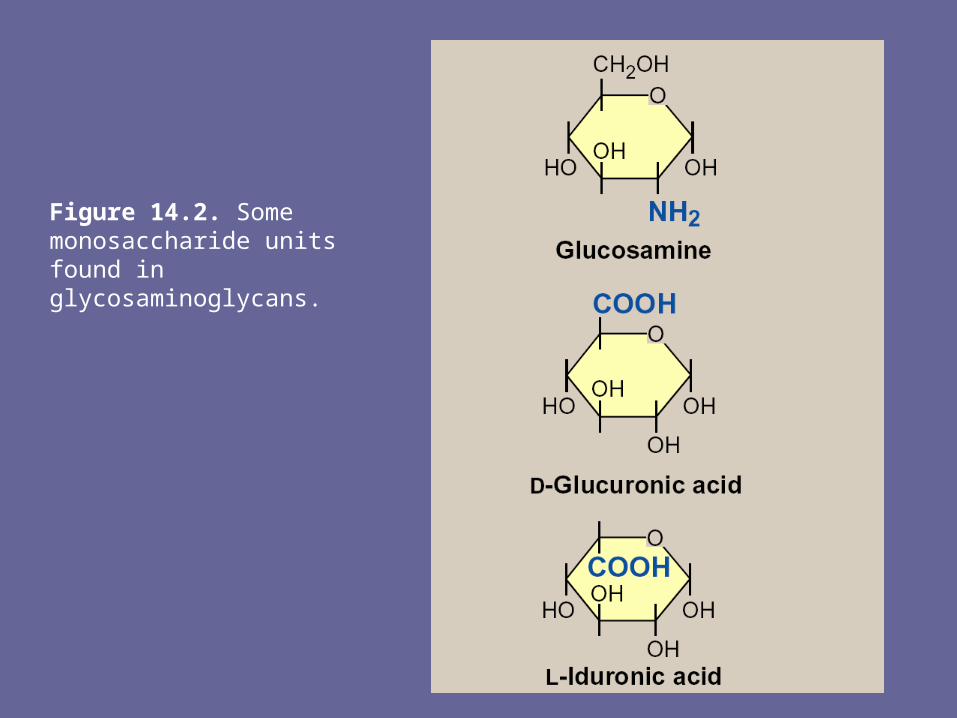
Figure 14.2. Some monosaccharide units found in glycosaminoglycans.

A. Relationship between GAG structure and function- Because of their large # of –ve charges, these
heteropolysacch chains tend to be extended in solutions. They repel each other and are surrounded by a shell of water molecules.
- When brought together, they “slip” past each other, much as two magnets with the same polarity seem to slip past each other. This produces “slippery” consistency of mucous secretions & synovial fluid
- When a soln of GAGs is compressed, the water is “squeezed out” & the GAGs are forced to occupy a smaller volume. When the compression is released, the GAGs spring back to their original, hydrated volume because of repulsion of their –ve charges. This property contributes to the resilience of synovial fluid & the vitreous humor of the eye.
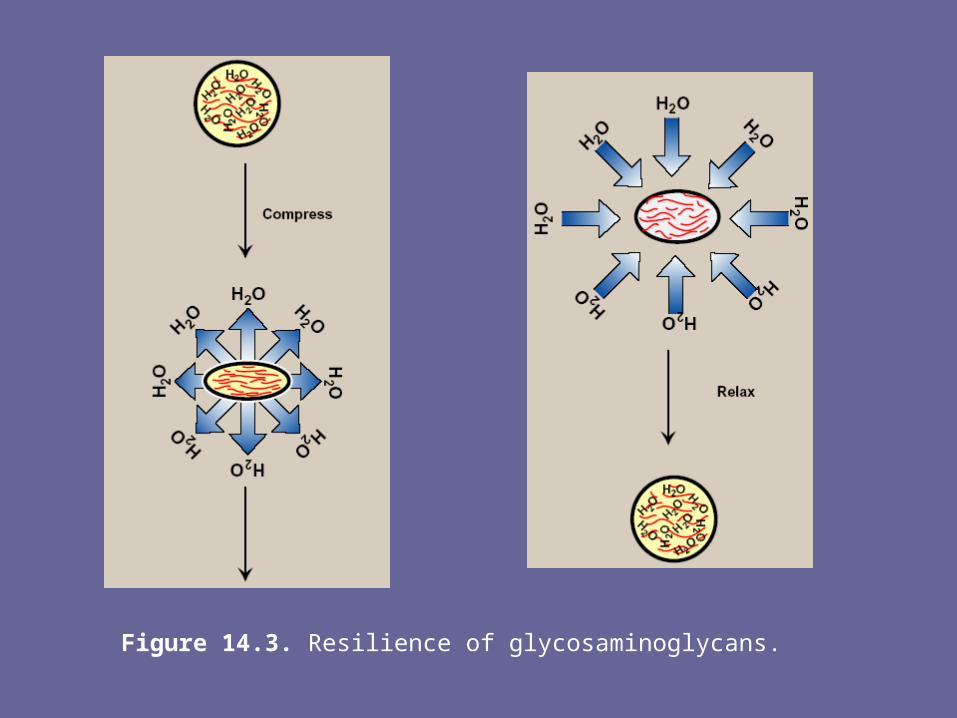
Figure 14.3. Resilience of glycosaminoglycans.

B. Classification of GAGs
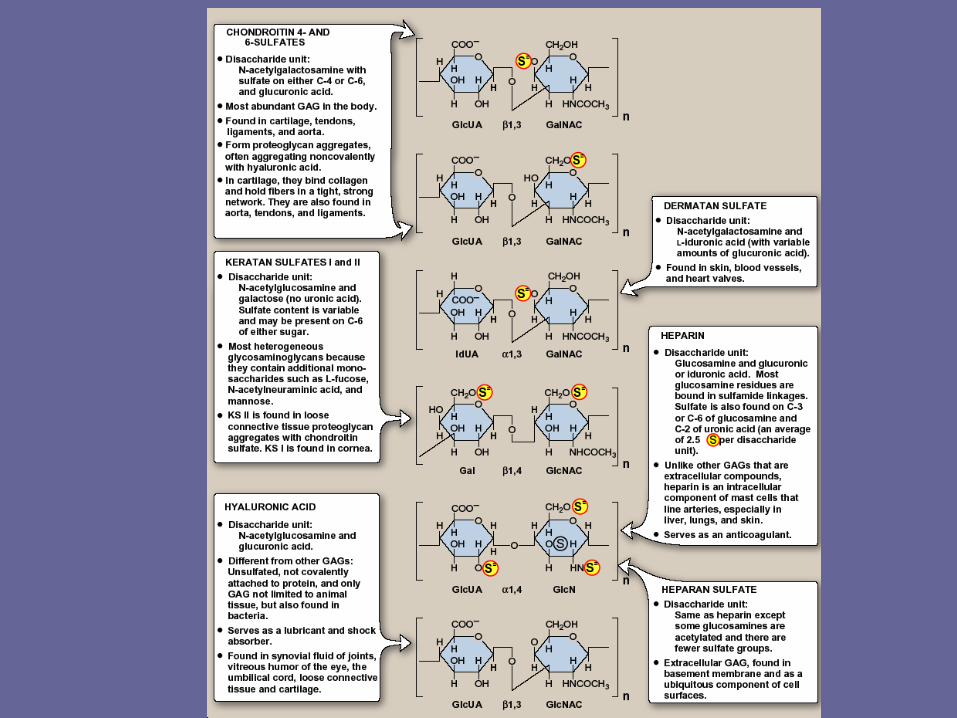
- The six major classes of GAGs are divided according to monomeric composition, type of glycosidic linkages, & degree & location of sulfate units.
- The structure of the GAGs & their distribution in the body illustrated in Fig. 4.

C. Structure of proteoglycans- All of the GAGs, except hyaluronic cid, are found covalently
attached to proteins, forming proteoglycan monomers.1. Structure of proteoglycan monomer: - A proteoglycan monomer found in cartilage consists of a core protein
to which the linear GAG chains are covalently attached- These chains which may each be composed of >100 monosacch,
extend out from the core protein & remain separated from each other because of charge repulsion. The resulting structure resembles a “bottle brush”
- In cartilage proteoglycan, the species of GAGs include chondroitin sulfate & keratan sulfate
Note: a number of proteoglycans have been characterized & named based on their structure & functional location. E.g., syndecan is an integral memb proteoglycan, versican & aggrecan are the predominant extracellular proteoglycans, & neurocan & cerebrocan are found primarily in the NS.
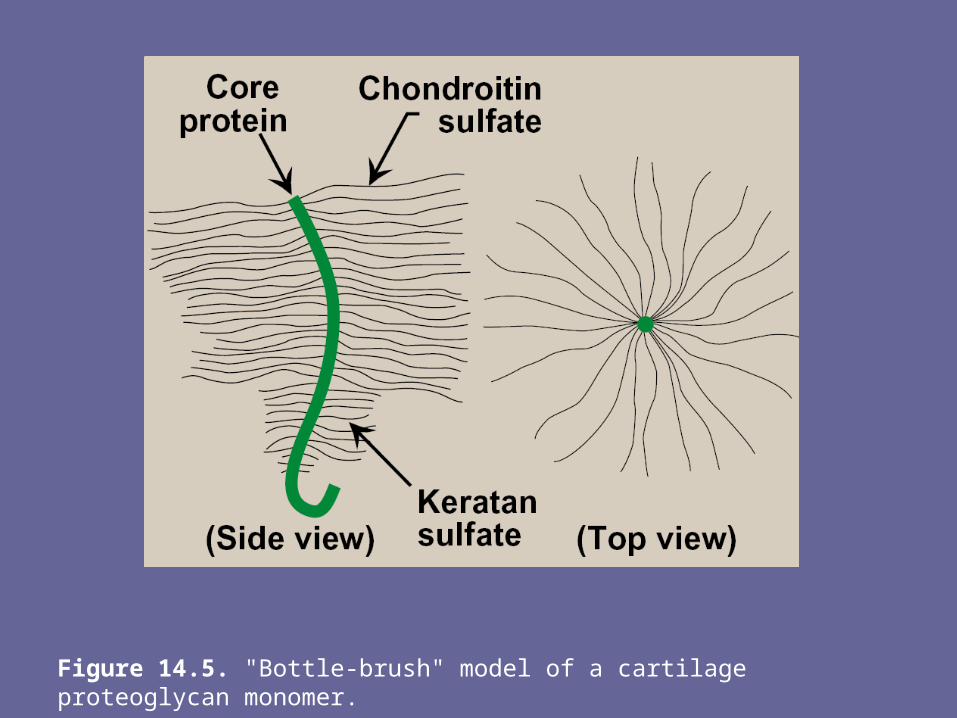
Figure 14.5. "Bottle-brush" model of a cartilage proteoglycan monomer.
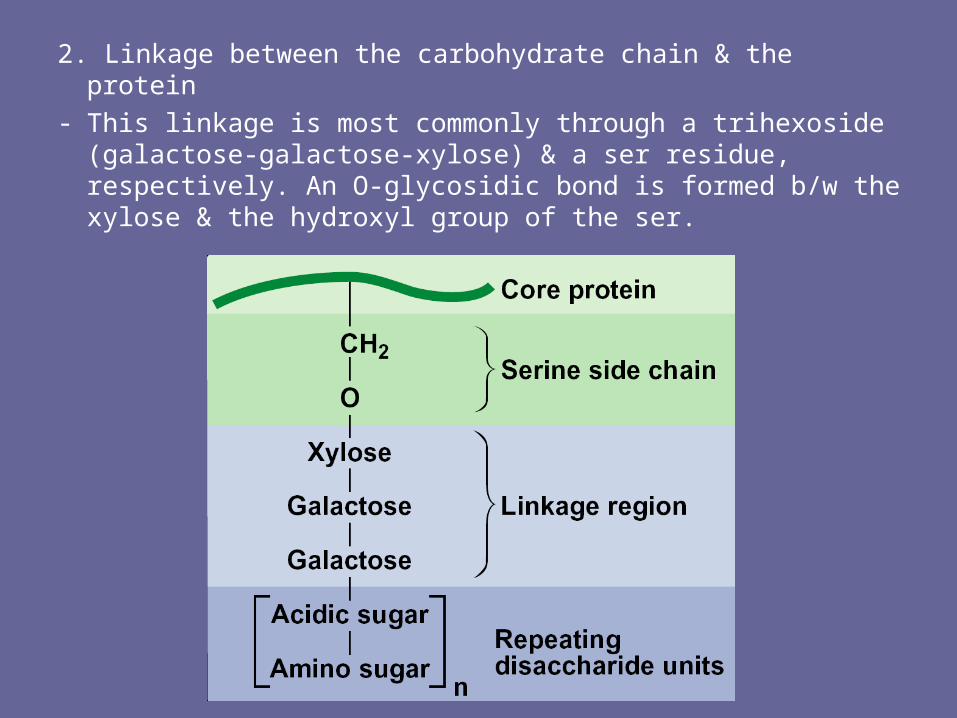
2. Linkage between the carbohydrate chain & the protein- This linkage is most commonly through a trihexoside
(galactose-galactose-xylose) & a ser residue, respectively. An O-glycosidic bond is formed b/w the xylose & the hydroxyl group of the ser.

3. Proteoglycan aggregates
- The proteoglycan monomers associate with a molecule of hyaluronic acid to form proteoglycan aggregates. The association is not covalent, but occurs primarily through ionic interactions b/w core protein & the hyaluronic acid
- The association is stabilized by additional small proteins called link proteins
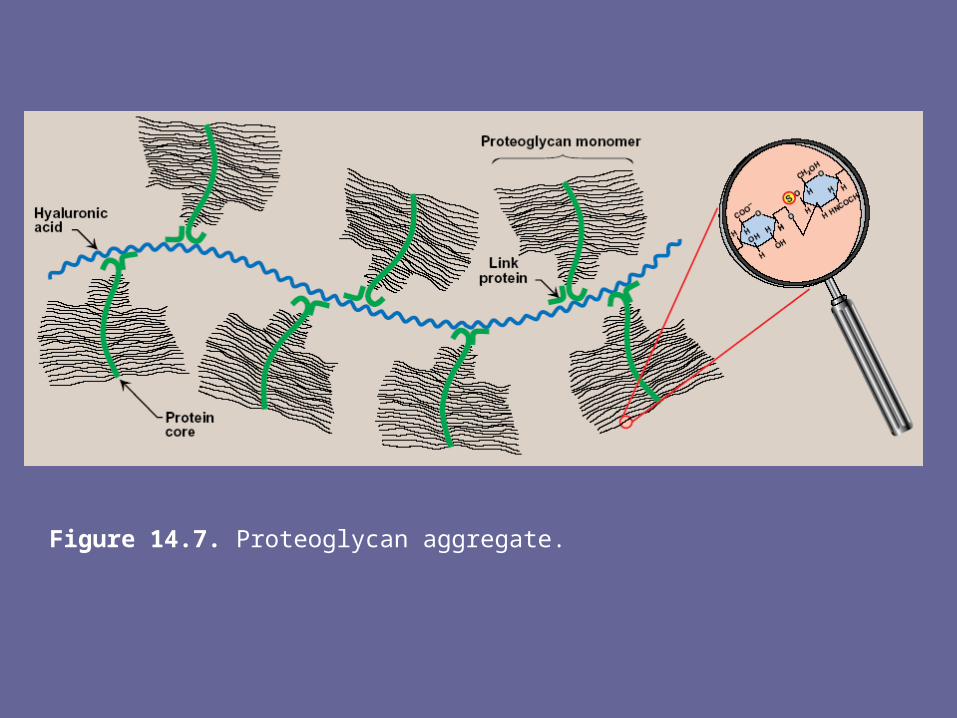
Figure 14.7. Proteoglycan aggregate.

III. Synthesis of glycosaminoglycans- The polysacch chains are elongated by the
sequential addition of alternating acidic & amino sugars, donated by their UDP-derivatives
- The reactions are catalyzed by specific transferases
- The synthesis of the GAGs is analogous to that of glycogen except that the GAGs are produced for export from the cell. The synthesis occurs, therefore, in the ER & the Golgi, rather than in the cytosol.

A. Synthesis of amino sugars- Amino sugars are essential components of GAGs, glycoproteins,
glycolipids, & certain oligosaccharides, & are also found in some antibiotics. The synthetic pathway of amino sugars is very active in connective tissues, where as much as 20% of gluc flows through the pathway.
1. N-acetylglucosamine (gluNAc) & N-acetylgalactosamine (galNAc):- The monosacch F-6-P is the precursor of gluNAc, galNAc, & the sialic
acids, including N-acetylneuraminic acid (NANA, a nine-carbon, acidic monosacch).
- In each of these sugars, a hydroxyl group of the precursor is replaced by an amino group donated by the aa, glutamine
Note: the amino groups are almost always acetylated- The UDP-derivatives of gluNAc & galNAc are synthesized by
reactions analogous to those described for UDP-glucose synthesis. These are activated forms of the monosaccharides that can be used to elongate the CHO chain

2. N-Acetylneuraminic acid:- NANA is a member of the family of sialic acids, each of
which is acylated at a different site. These cpds are usually found as terminal CHO residues of oligosaccharide side chains of glycoproteins, glycolipids, or, less frequently, of GAGs
- The carbons & nitrogens in NANA come from N-acetylmannosamine & PEP
Note: before NANA can be added to a growing oligosacch, it must be converted into its active form by reacting with cytidine triphosphate (CTP). The enz N-acetylneuraminate-CMP-pyrophosphorylase removes pyrophosphate from CTP & attaches the remaining CMP to the NANA. This is the only nucleotide sugar in human metabolism in which the carrier nucleotide is a monophosphate
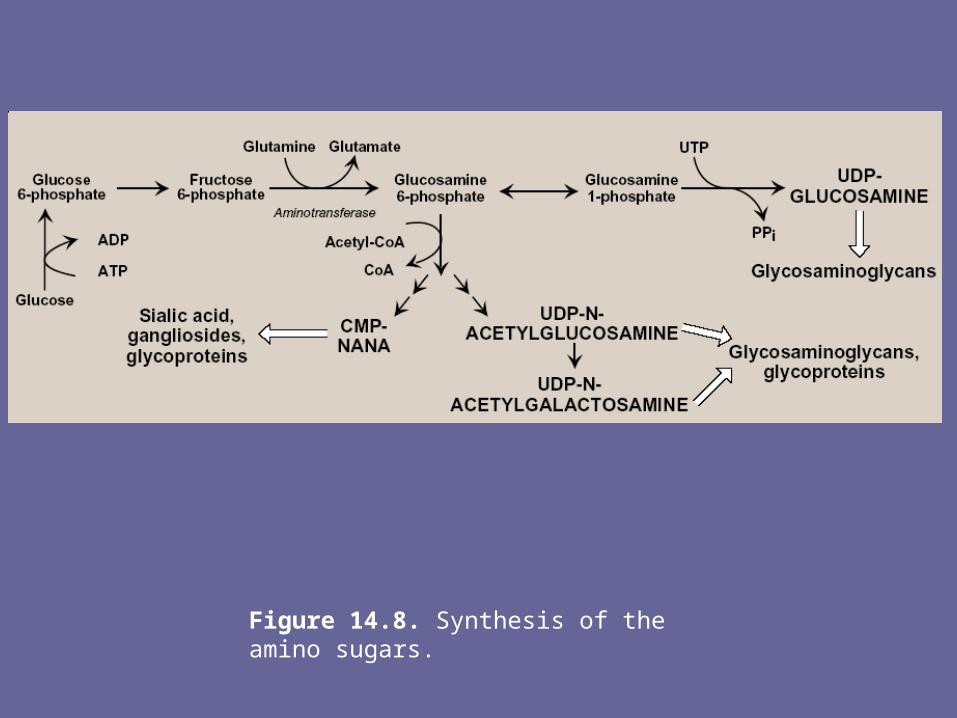
Figure 14.8. Synthesis of the amino sugars.

B. Synthesis of acidic sugars- D-glucuronic acid, whose structure is that of gluc with an
oxidized C-6 (-CH2OH → -COOH), & its C-5 epimer, L-iduronic acid, are essential components of GAGs.
- Glucuronic acid is also required in detoxification reactions of a number of insoluble cpds, e.g., bilirubin, steroids, & several drugs.
- In plants & mammals (other than guinea pigs & primates, including man), glucuronic acid serves as a precursor of ascorbic acid (vitamin C).
- The uronic acid pathway also provides a mechanism by which dietary D-xylulose can enter the central metabolic pathways

1. Glucuronic acid:- Glucuronic acid can be obtained in small amounts from diet.
It can also be obtained from the intracellular lysosomal degradation of GAGs, or via the uronic acid pathway.
- The end-product of glucuronic acid metabolism in humans is D-xylulose 5-P, which can enter the hexose monophosphate pathway & produce the glycolytic intermediates GA-3P & F-6-P
- The active form of glucuronic acid that donates the sugar in GAG synthesis & other glucuronylating reactions is UDP-glucuronic acid, which is produced by oxidation of UDP-glucose
2. L-iduronic acid synthesis:- Synthesis of L-iduronic acid residues occurs after D-
glucuronic acid has been incorporated into the CHO chain.- Uronosyl 5-epimerase causes epimerization of the D- to L-
sugar
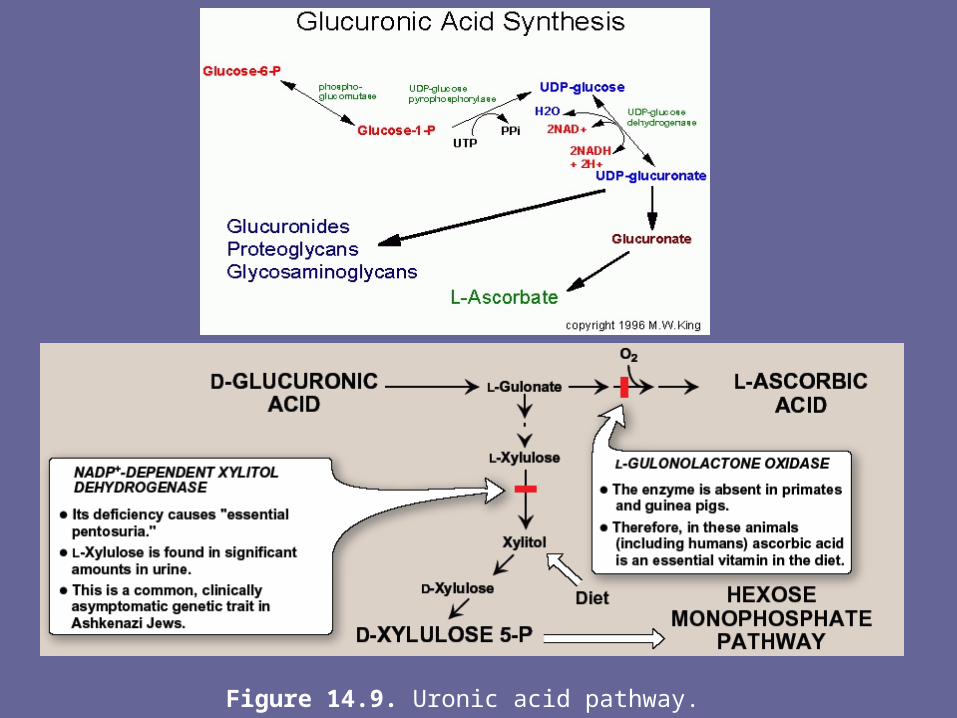
Figure 14.9. Uronic acid pathway.
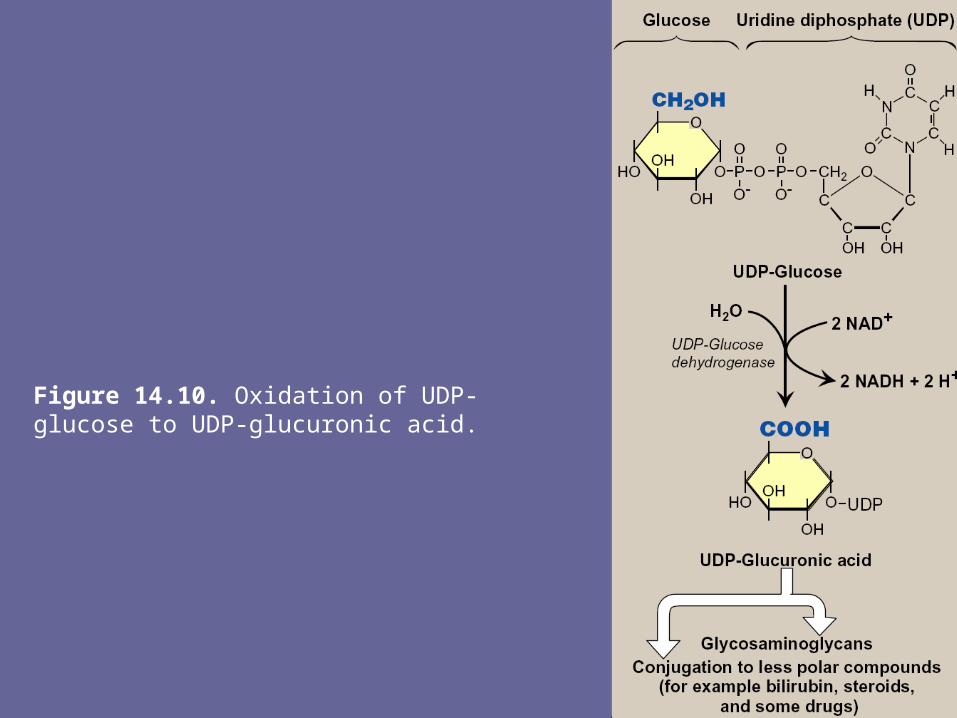
Figure 14.10. Oxidation of UDP-glucose to UDP-glucuronic acid.

C. Synthesis of the core protein- The core protein is synthesized on & enters rER. The
protein is then glycosylated by memb-bound transferases as it moves through ER.
D. Synthesis of the carbohydrate chain- CHO chain formation begins by synthesis of a short
linkage region on the core protein on which CHO chain synthesis will be initiated.
- The most common linkage region is formed by the transfer of xylose from UDP-xylose to the hydroxyl group of a ser (or thr) catalyzed by xylosyltransferase.
- Two galactose molecules are then added, completing the trihexoside.
- This is followed by sequential addition of alternating acidic & amino sugars, & conversion of some D-glucuronyl to L-iduronyl residues
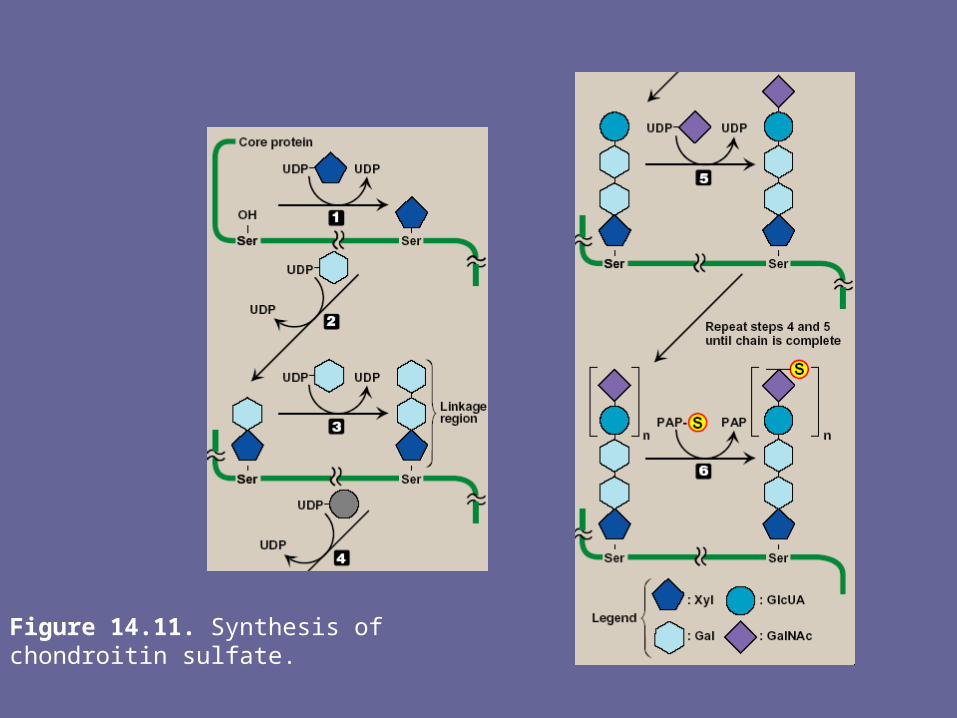
Figure 14.11. Synthesis of chondroitin sulfate.

E. Addition of sulfate groups- Sulfation of CHO chain occurs after the
monosacch to be sulfated has been incorporated into the growing CHO chain
- The source of the sulfate is 3`-phosphoadenosyl-5`-phosphosulfate (PAPS, a molecule of AMP with a sulfate group attached to the 5`-phosphate).
- Sulfotransferases cause the sulfation of the CHO chain at specific sites.
Note: a defect in the sulfation process results in one of several autosomal recessive disorders that affect the proper development & maintenance of the skeletal system. This illustrates the importance of the sulfation step

IV. Degradation of glycosaminoglycans- GAGs are degraded in lysosomes, which contain
hydrolytic enz’s that are most active at a pH of ~ 5 [Note: therefore, as a group, these are called acid hydrolases]
- The low pH optimum is a protective mechanism that prevents the enz’s from destroying the cell should leakage occur into cytosol where pH is neutral.
- With exception of keratan sulfate, which has a half-life of > 120 days, the GAGs have a relatively short half-life, ranging from ~ 3 days for hyaluronic acid to 10 days for chondroitin & dermatan sulfate

A. Phagocytosis of extracellular glycosaminoglycans
- Because GAGs are extracellular or cell-surface cpds, they must be engulfed by an invagination of the CM (phagocytosis), forming a vesicle inside of which the GAGs are to be degraded.
- The vesicle then fuses with a lysosome, forming a single digestive vesicle in which GAGs are efficiently degraded
B. Lysosomal degradation of GAGs- The lysosomal degradation of GAGs requires a large # of
acid hydrolases for complete digestion.- 1st , the polysacch chains are cleaved by
endoglycosidases, producing oligosaccharides. Further degradation of the oligosacch’s occurs sequentially from the non-reducing end of each chain, the last group (sulfate or sugar) added during synthesis being the 1st to be removed. Examples of some of these enz’s & the bonds they hydrolyze Fig 12.

V. Mucopolysaccharidosis- Mucopolysaccharidoses are hereditary disorders that are
clinically progressive. They are characterized by accumulation of GAGs in various tissues, causing varied symptoms, such as skeletal & extracellular matrix deformities, & mental retardation.
- Mucopolysaccharidoses are caused by a deficiency of one of the lysosomal hydrolases normally involved in degradation of heparan sulfate and/or dermatan sulfate.
- This results in the presence of oligosacch’s in urine, because of incomplete lysosomal degradation of GAGs. These fragments can be used to diagnose the specific mucopolysaccharidosis, namely by identifying the structure present on the non-reducing end of oligosacch. That residue would have been the substrate for the missing enz.

- Diagnosis can be confirmed by measuring the patients cellular level of lysosomal hydrolases. Children who are homozygous for one of these diseases are apparently normal at birth, then gradually deteriorate. In severe cases, death occurs in childhood.
- All of the deficiencies are autosomal & recessively inherited except Hunter syndrome, which is X-linked
- Bone marrow transplants are currently being used successively to treat Hunter syndrome; the transplanted macrophages produce the sulfatase needed to degrade GAGs in the extracellular space.
Note: some of lysosomal enzymes required for degradation of GAGs also participate in degradation of glycolipids & glycoproteins. Therefore, an individual suffering from a specific mucopolysaccharidosis may also have a lipidosis or glycoprotein-oligosaccharidosis
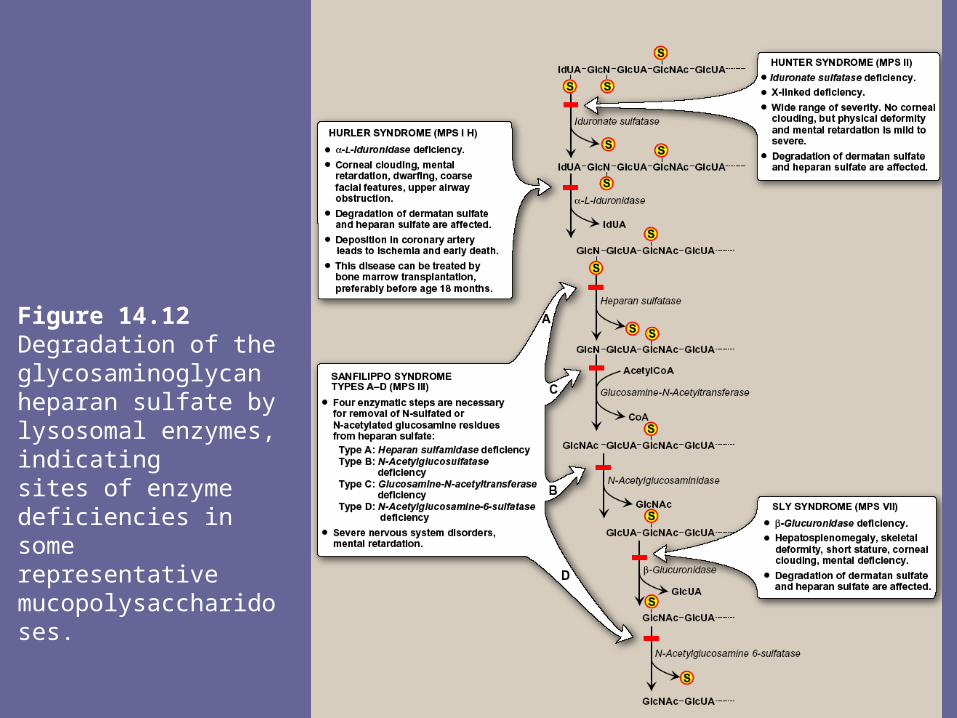
Figure 14.12Degradation of the glycosaminoglycan heparan sulfate by lysosomal enzymes, indicatingsites of enzyme deficiencies in some representative mucopolysaccharidoses.
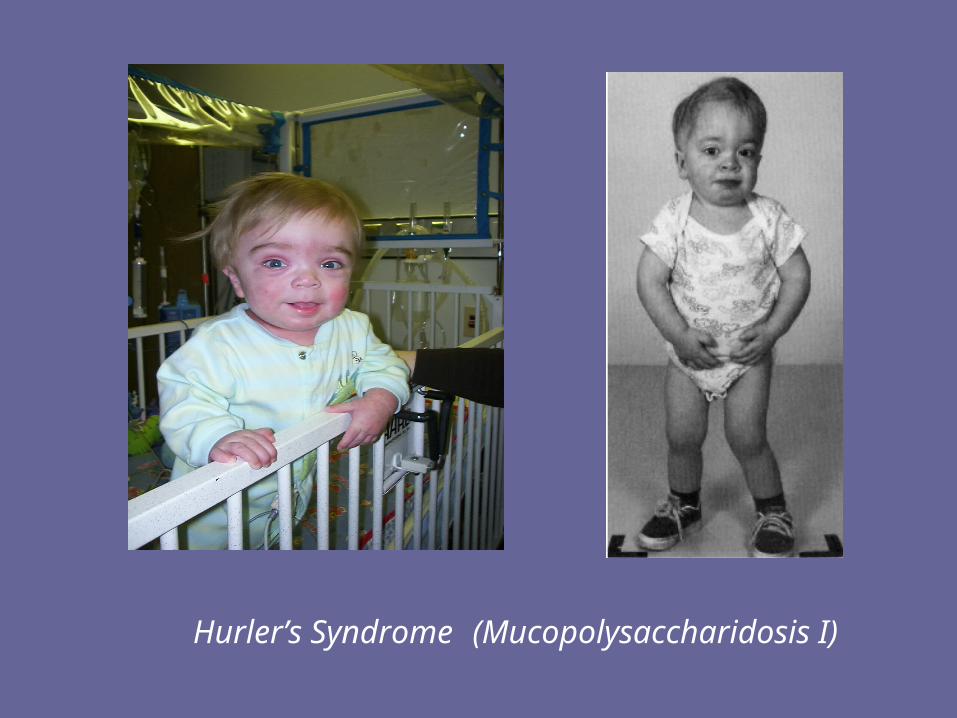
Hurler’s Syndrome (Mucopolysaccharidosis I)

Facies of a male with the mucopolysaccharidosis, Hunter syndrome

VI. Overview of glycoproteins- Glycoproteins are proteins to which oligosacch’s are covalently attached.
They differ from proteoglycans (which might be considered a special case of glycoproteins) in that length of glycoproteins’ CHO chain is relatively short (usually 2-10 sugar residues in length, although they can be longer), whereas it can be very long in the GAGs.
- In addition, whereas GAGs have diglucosyl repeat units, the CHO’s of glycoproteins do not have serial repeats.
- The glycoprotein CHO chains are often branched instead of linear, & may or may not be negatively charged.
- Glycoproteins contain highly variable amounts of CHO. e.g., the immunoglobulin IgG, contains < 4% of its mass as CHO, whereas human gastric glycoprotein (mucin) contains > 80% CHO
- Membrane-bound glycoproteins participate in a broad range of cellular phenomena, including cell surface recognition (by other cells, hormones, viruses), cell surface antigenicity (such as the blood group antigens), and as components of the extracellular matrix & of the mucins of the gastrointestinal & urogenital tracts, where they act as protective biologic lubricants.
- In addition, almost all of the globular proteins present in human plasma are glycoproteins
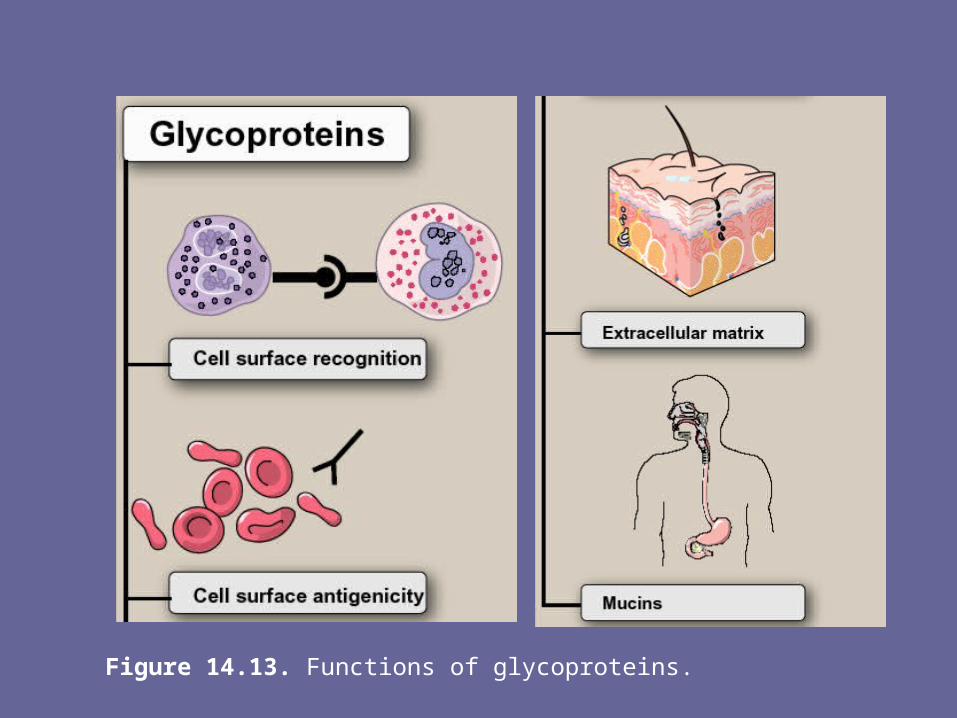
Figure 14.13. Functions of glycoproteins.

VII. Structure of glycoprotein oligosaccharides- The oligosaccharide components of glycoproteins are
generally branched heteropolymers composed primarily of D-hexoses, with the addition in some cases of neuraminic acid, & L-fucose, a 6-deoxyhexose
A. Structure of the linkage between carbohydrate and protein
- The oligosacch may be attached to the protein through an N- or O-glycosidic link.
- In the former case, sugar chain is attached to the amide group of an asparagine side chain, & in the latter case, to a hydroxyl group of either ser or thr R-group.
Note: in case of collagen, there is an O-glycosidic linkage b/w galactose or gluc & hydroxyl group of hydroxylysine

B. N- and O-linked oligosaccharides- A glycoprotein may contain only one type of
glycosidic linkage (N- or O-linked), or may have both O- and N-linked oligosacchs within same molecule
1. O-linked oligosaccharides:- The O-linked oligosacch’s may have one or
more of a wide variety of sugars arranged in either a linear or branched pattern.
- Many O-linked oligosacch’s are found as memb glycoprotein components or in extracellular glycoproteins. E.g., O-linked oligosacch’s help provide the ABO blood group determinants

2. N-linked oligosaccharides:
- The N-linked oligosacch’s fall into 2 broad classes: complex oligosacch’s & high-mannose oligosacch’s. both contain same core pentasaccharide (Fig 14), but the complex oligosacch’s contain a diverse group of additional sugars, e.g., N-acetylglucosamine (GlcNAc), L-fucose (Fuc), N-acetylneuraminic acid (NANA), whereas the high-mannose oligosacch’s contain primarily mannose (Man)
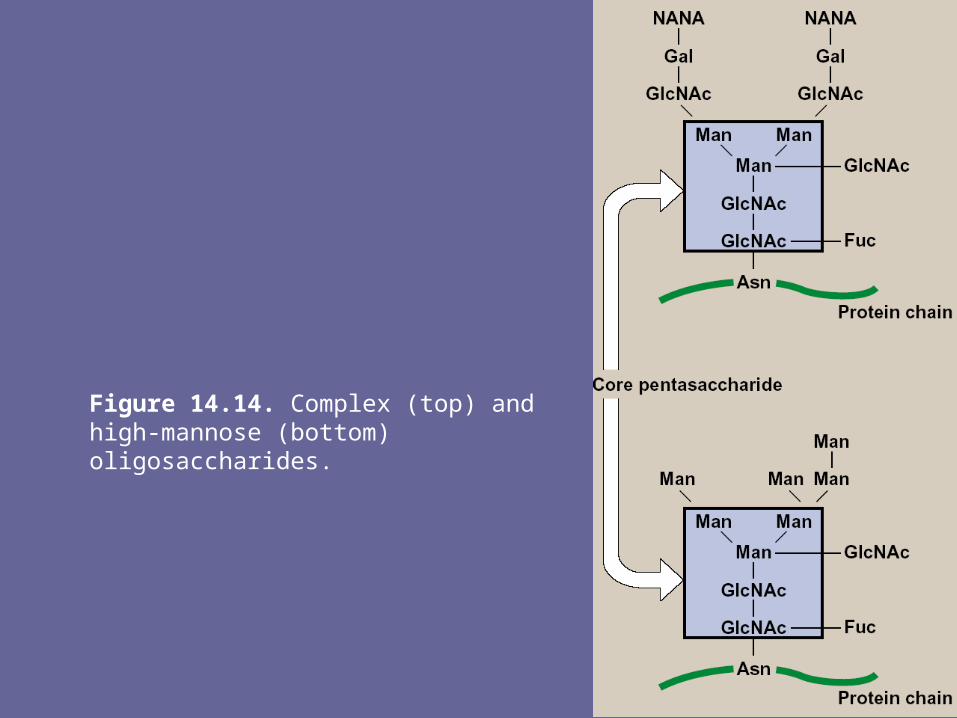
Figure 14.14. Complex (top) and high-mannose (bottom) oligosaccharides.

VIII. Synthesis of glycoproteins- Most proteins are destined for the cytoplasm & are synthesized on
free ribosomes in cytosol. However, proteins, including many glycoproteins, that are destined for cellular memb’s, lysosome, or to be exported from cell, are synthesized on ribosomes attached to rER.
- These proteins contain specific signal sequences at their N-terminal end that act as molecular “address labels” which direct the proteins to their proper destinations
- These signal sequences allow growing polyp to be extruded into lumen of rER. The proteins are then transported via secretory vesicles to Golgi complex, which acts as a sorting center.
- In Golgi those glycoproteins that are to be secreted from cell (or are targeted for lysosomes) remain free in lumen, whereas those that are to become components of the CM become integrated into Golgi memb, their CHO portions oriented toward lumen.
- Vesicles bud off from Golgi & fuse with the CM, either releasing the free glycoproteins, or adding the memb-bound proteins of the vesicle to the CM. The memb glycoproteins are thus oriented with the CHO portion on the outside of the cell.
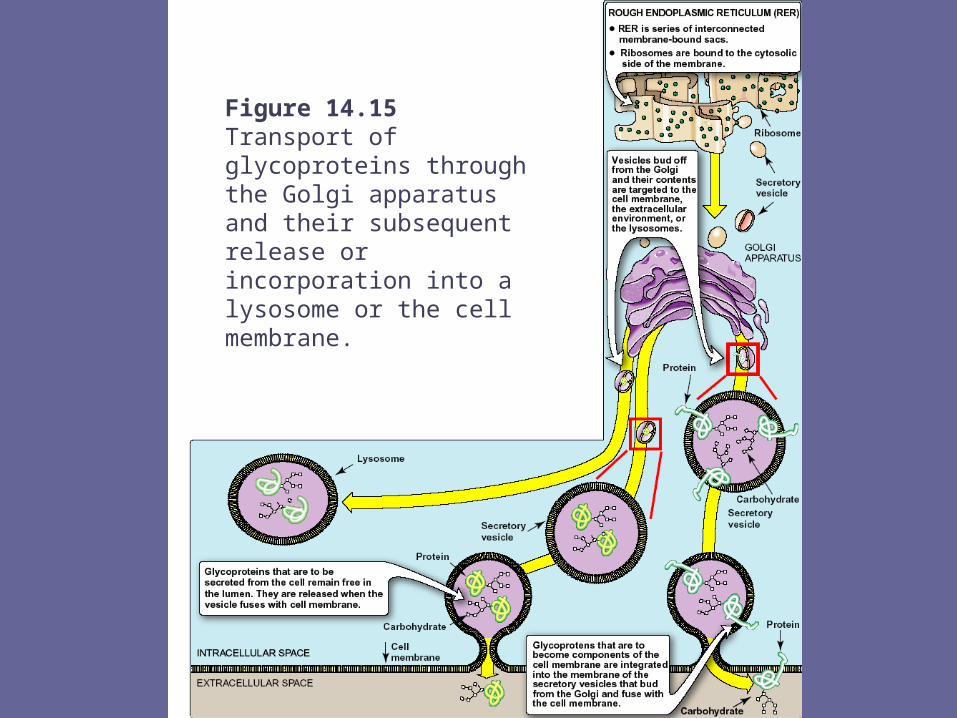
Figure 14.15Transport of glycoproteins through the Golgi apparatus and their subsequent release orincorporation into a lysosome or the cell membrane.

A. Carbohydrate components of glycoproteins• The precursors of the CHO components of glycoproteins
are sugar nucleotides, which include UDP-glucose, UDP-galactose, UDP-N-acetylglucosamine & UDP-N-acetylgalactosamine. In addition, GDP-mannose, GDP-L-fucose (which is synthesized from GDP-mannose), & CMP-N-acetylneuraminic acid may donate sugars to the growing chain.
Note: when NANA is present, the oligosacch has a –ve charge at physiologic pH.
• The oligosacch’s are covalently attached to specific aa R-groups of the protein, where the 3-D structure of the protein determines whether or not a specific aa R-group is glycosylated

B. Synthesis of O-linked glycosides- Synthesis of O-linked glycosides is very similar to that of the GAGs.
1st the protein to which the oligosacch’s are to be attached is synthesized on the rER, & extruded into its lumen.
- Glycosylation begins immediately, with the transfer of an N-acetylgalactosamine (from UDP-N-acetylgalactosamin) onto a specific seryl or threonyl R-group.
1. Role of glycosyltransferases- The glycosyltransferases responsible for the stepwise synthesis of
the oligosacch’s are bound to the memb’s of the ER or the Golgi apparatus.
- They act in a specific order, without using a template as is required for DNA, RNA, & protein synthesis, but rather by recognizing the actual structure of the growing oligosacch as the appropriate substrate

C. Synthesis of the N-linked glycosides• The synthesis of N-linked glycosides also occurs in the
lumen of the ER & in Golgi. However, these structures undergo additional processing steps, & require the participation of a lipid (dolichol) & its phosphorylated derivative, dolichol pyrophosphate
1. Synthesis of dolichol-linked oligosaccharide• 1st, as with O-linked glycosides, protein is synthesized on
rER & enters its lumen. The protein itself does not become glycosylated with individual sugars at this stage of glycoprotein synthesis, but rather a lipid-linked oligosacch is 1st constructed
• This consists of dolichol (an ER memb lipid 80-100 carbons long) attached through a pyrophosphate linkage to an oligosacch containing N-acetylglucosamine, mannose, & glucose.

• The sugars to be added to the dolichol by the memb-bound glycosyltransferases are first N-acetylglucosamine, followed by mannose & gluc.
• The oligosacch is transferred from the dolichol to an asparagine side group of the protein by a protein-oligosaccharide transferase present in the ER
2. Final processing of N-linked oligosaccharides:• After incorporation into the protein, the N-linked oligosacch is
processed by removal of specific mannosyl & glucosyl residues as the glycoprotein moves through the ER
• Finally, the oligosacch chains are completed in Golgi by addition of a variety of sugars (e.g., N-acetylglucosamine, N-acetylgalactosamine, and additional mannoses, and then fucose or NANA as terminal groups) to produce a complex glycoprotein, or they are not processed further, leaving branched, mannose-containing chains in a high-mannose glycoprotein
• The ultimate fate of N-linked glycoproteins is the same as that of the O-linked, e.g., they can be released by cell, become part of a CM, or alternatively, N-linked glycoproteins can be translocated to the lysosomes
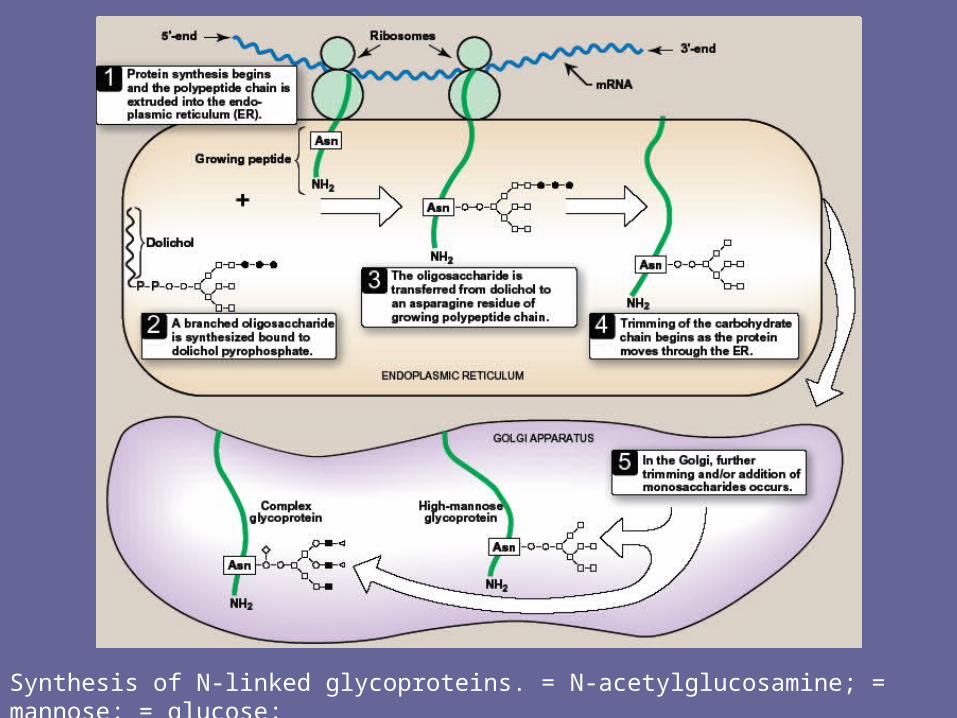
Synthesis of N-linked glycoproteins. = N-acetylglucosamine; = mannose; = glucose;= N-acetylgalactosamine; or for example, fucose or N-acetylneuraminic acid.

3. Enzymes destined for lysosomes:• N-linked glycoproteins being processed through Golgi
can be phosphorylated at one or or more specific mannosyl residues.
• Mannose 6-P receptors, located in Golgi, bind the mannose 6-P residues of these targeted enz’s, resulting in their translocation to the lysosomes
• I-cell disease is a rare syndrome in which the acid hydrolase enz’s normally found in lysosomes are absent, resulting in an accumulation of substrates normally degraded by lysosomal enz’s within these vesicles
Note: I-cell disease is so-named because of the large inclusion bodies seen in cells of patients with this disease
• In addition, high amounts of lysosomal enz’s are found in patient’s plasma, suggesting that targeting process to lysosomes (rather than the synthetic pathway of these enzymes) is deficient.

• It has been determined that individuals with I-cell disease are lacking the enzymic ability to phosphorylate the mannose residues of potential lysosomal enz’s, causing an incorrect targeting of these proteins to extracellular sites, rather than lysosomal vesicles
• I-cell disease is characterized by skeletal abnormalities, restricted joint movement, coarse facial features, & severe psychomotor impairment. Death usually occurs by age 8 yrs.
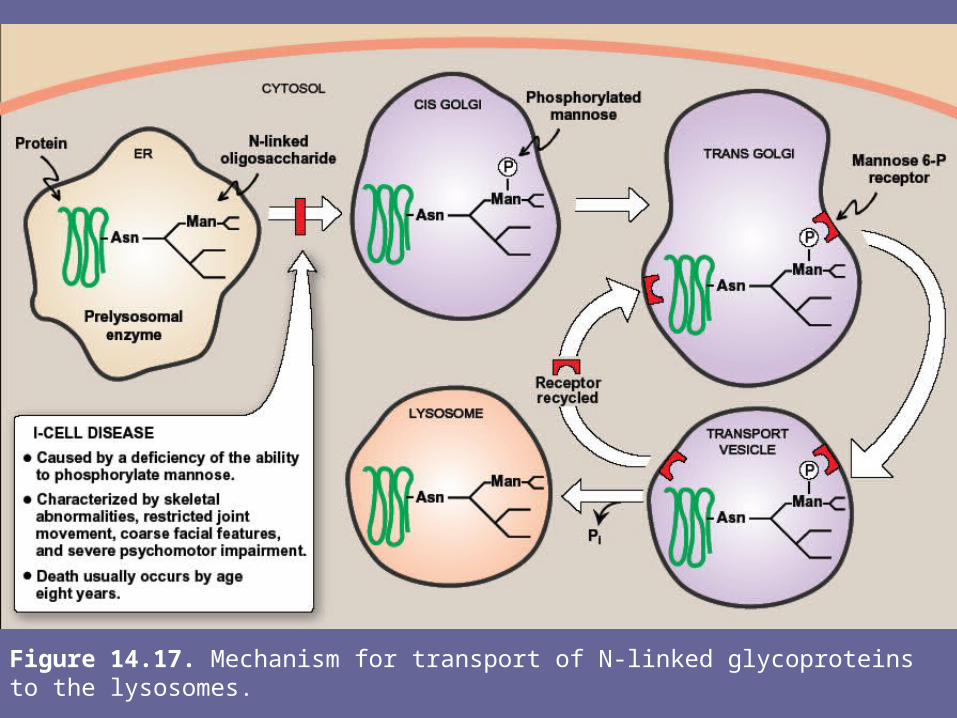
Figure 14.17. Mechanism for transport of N-linked glycoproteins to the lysosomes.

IX. Lysosomal degradation of glycoproteins• Degradation of glycoproteins is similar to that of GAGs. • The lysosomal hydrolytic enz’s are each generally specific
for the removal of one component of the glycoprotein.• They are primarily exoenzymes that remove their
respective groups in sequence in the reverse order of their incorporation (“last on, first off”)
• If any one degradative enz is missing, degradation by the other exoenzymes can’t continue.
• A group of genetic diseases called the glycoprotein storage diseases (oligosaccharidoses), caused by a deficiency of one of the degradative enz’s, results in accumulation of partially degraded structures in lysosomes.
• After cell death, the oligosacch fragments appear in the urine
Note: these disorders are very often directly associated with the same enzyme deficiencies involved in mucopolysaccharidoses & the inability to degrade glycolipids

Summary
• GAGs are long, negatively charged, unbranched heteropolysacch chains generally composed of a repeating disaccharide unit [acidic sugar-amino sugar]n
• The amino sugar is either D-glucoamine or D-galactosamine in which the amino group is usually acetylated, thus eliminating its +ve charge
• The amino sugar may also be sulfated on C-4 or 6 or on a non-acetylated nitrogen.
• The acidic sugar is either D-glucuronic acid or its C-5 epimer, L-iduronic acid. These cpds bind large amounts of water, thereby producing the gel-like matrix that forms the basis of the body’s ground substance
• The viscous, lubricating properties of mucous secretions are also caused by the presence of GAGs which led to the original naming of these cpds as mucopolysaccharides
• As essential components of cell surfaces, GAGs play an important role in mediating cell-cell signaling & adhesion.

• There are 6 major classes of GAGs, including chondroitin 4- & 6-sulfates, keratan sulfate, dermatan sulfate, heparin, heparan sulfate & hyaluronic acid.
• All of the GAGs, except hyaluronic acid, are found covalently attached to protein, forming proteoglycan monomers, which consist of a core protein to which the linear GAG chains are covalently attached.
• The proteoglycan monomers associate with a molecule of hyaluronic acid to form proteoglycan aggregates
• GAGs are synthesized in the ER & Golgi. The polysacch chains are elongated by sequential addition of alternating acidic & amino sugars, donated by their UDP-derivatives. The last step in synthesis is the sulfation of some of the amino sugars. The source of the sulfate is 3’-phosphoadenosyl-5`-phosphosulfate.

• GAGs are degraded by lysosomal hydrolases. They are 1st broken down to oligosacch’s, which are degraded sequentially from the non-reducing end of each chain.
• A deficiency of one of the hydrolases results in a mucopolysaccharidoses. These are hereditary disorders in which GAGs accumulate in tissues, causing symptoms such as skeletal & extracellular matrix deformities, & mental retardation. Examples of these genetic diseases include Hunter & Hurler syndromes.
• Glycoproteins are proteins to which oligosacch’s are covalently attached. They differ from proteoglycans in that length of glycoprotein’s CHO chain is relatively short (usually 2-10 sugar residues long, although they can be longer). The CHO’s of glycoproteins do not have serial repeats as do GAGs.
• Memb-bound glycoproteins participate in a broad range of cellular phenomena, including cell surface recognition (by other cells, hormones, viruses), cell surface antigenicity (e.g., blood group antigens), & components of the extracellular matrix & of the mucins of the GI & urogenital tracts, where they act as protective biologic lubricants.

• In addition, almost all of the globular proteins present in human plasma are glycoproteins
• Glycoproteins are synthesized in the ER & Golgi. The precursors of the CHO components of glycoproteins are sugar nucleotides. O-linked glycoproteins are synthesized by the sequential transfer of sugars from their nucleotide carriers to the protein.
• N-linked glycoproteins contain varying amounts of mannose. They are synthesized by the transfer of a pre-formed oligosacch from its membrane lipid carrier, dolichol, to the protein.
• They also require dolichol, an intermediate carrier of the growing oligosacch chain.
• A deficiency in the phosphorylation of mannose residues in N-linked glycoprotein pre-enzymes destined for the lysosomes results in I-cell disease.
• Glycoproteins are degraded in lysosomes by acid hydrolases. A deficiency of one of these enz’s results in a glycoprotein storage disease (oligosaccharidosis), resulting in accumulation of partially degraded structures in the lysosome.