
GLUCONACETOBACTER HANSENII CELLULOSE SYNTHESIS
184
The Pennsylvania State University The Graduate School Department of Biochemistry and Molecular Biology BIOCHEMICAL CHARACTERIZATION OF THE PROTEINS INVOLVED IN GLUCONACETOBACTER HANSENII CELLULOSE SYNTHESIS A Dissertation in Integrative Biosciences by Radhakrishnan Iyer Prashanti 2012 Radhakrishnan Iyer Prashanti Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2012
Transcript of GLUCONACETOBACTER HANSENII CELLULOSE SYNTHESIS

The Pennsylvania State University
The Graduate School
Department of Biochemistry and Molecular Biology
BIOCHEMICAL CHARACTERIZATION OF THE PROTEINS INVOLVED IN
GLUCONACETOBACTER HANSENII CELLULOSE SYNTHESIS
A Dissertation in
Integrative Biosciences
by
Radhakrishnan Iyer Prashanti
2012 Radhakrishnan Iyer Prashanti
Submitted in Partial Fulfillment
of the Requirements
for the Degree of
Doctor of Philosophy
August 2012

ii
The dissertation of Radhakrishnan Iyer Prashanti was reviewed and approved*
by the following:
Ming Tien
Professor of Biochemistry and Molecular Biology
Dissertation Advisor
Chair of Committee
B. Tracy Nixon
Professor of Biochemistry and Molecular Biology
Nicole R. Brown
Associate Professor of Wood Chemistry
Charles T. Anderson
Assistant Professor of Biology
Peter J Hudson
Program Chair, Integrative Biosciences
Director, The Huck Institutes of the Life Sciences
* Signatures are on file in the Graduate School

ABSTRACT
Gluconacetobacter hansenii is a Gram-negative bacterium, considered as the
model organism for studying the process of cellulose biogenesis. This is due to its unique
ability to synthesize and secrete copious amounts of cellulose as an extracellular
polysaccharide, in its growth medium. G. hansenii is therefore an ideal bacterium to
study cellulose as a material as well as cellulose synthesis as a biological process. We
have therefore employed this bacterium as our subject for our inquiry into the
biochemistry of the process of cellulose synthesis.
In this work, the main area of focus is towards understanding the bacterial cellulose
synthesis and secretion complex, in terms of its component proteins, their structure,
organization and their interactions. Two parallel approaches were used to understand and
gain insights into the cellulose synthesis complex. One was to heterologously express and
purify the proteins that are known to be involved in cellulose synthesis for structural
studies or for generation of antibodies. Another method was to isolate the cellulose
synthase complex from the G. hansenii cells and dissect its component proteins to reveal
as-yet-unknown constituents of this complex.
The cellulose synthase operon encodes for three proteins, AcsAB, AcsC and
AcsD. AcsD protein was heterologously expressed and purified. The pure protein was
employed for structural characterization as well as for antibody generation. Using the
specific anti-AcsD antibody, the subcellular localization of this protein was identified to
be the periplasmic space. Studies of AcsD using gel filtration, analytical
ultracentrifugation and dynamic light scattering revealed that it exists as an octamer in
solution. Structural characterization of AcsD using small angle-X-ray scattering reveals

iv
that, in accordance with its crystal-structure, the protein forms a complex of a tetramer of
dimers that assumes a cylindrical conformation with a central pore.
The predicted non-membrane regions of the cytoplasmic membrane-bound cellulose
synthase (AcsAB) protein were heterologously overexpressed and purified using affinity
methods. Specific antibodies generated against these regions revealed that the protein,
though encoded by a single gene, is actually processed into three polypeptides AcsA (45
kDa), AcsB1 (34 kDa) and AcsB2 (95 kDa). Western blot of the fractions from a sucrose
density centrifugation combined with sequence-based analysis revealed that the AcsB2
protein is localized in the periplasmic region of the bacterial cell.
The genome of G. hansenii 23769 was sequenced to provide a database for mass-
spectrometry based-proteomic studies of cellulose synthesis. The completed genome is
now a public database in NCBI. These studies involved Multidimensional Protein
Identification Tool (MudPIT) analysis of the total membrane (TM), outer membrane
(OM) and the cytoplasmic membrane (CM) fractions of the cells for the comparison of
the proteomic profile of the three compartments. This revealed that the AcsB, AcsC and
the AcsD were largely concentrated in the OM whereas the CM compartment contains
lower abundance of AcsAB protein.
Using blue native polyacrylamide gel electrophoresis (BN-PAGE), the protein
complexes in solubilized G. hansenii TM were isolated and the complex containing the
proteins involved in cellulose was located using the specific antibodies against AcsD and
AcsA. As revealed by LC-MS analysis of the antibody cross-reacting gel-band, this
complex also contains phosphoglucomutase, glucose-6-phosphate isomerase and UDP-
glucose pyrophosphorylase. These proteins were known to be involved in cellulose

v
biosynthesis pathway, but this work presents evidence for the existence of these proteins
in association with the proteins involved in the cellulose synthesis and secretion complex.
In addition to using gel-based methods for identifying the components of the cellulose
synthesis complex, zymography was used to demonstrate in-vitro cellulose synthesis
using detergent solubilized membranes. This study directs towards a greater efficiency of
the detergent dodecyl maltoside compared to Triton-X 100, in solubilization of the TM,
while retaining the enzyme-activity.
In summary, we have used a combination of traditional and modern biochemical
approaches to study the protein components of the cellulose synthesis machinery. Our
work has resulted in a sequenced genome, structural analysis and localization of AcsD,
and identification of processing of the AcsAB. We have also presented evidence that the
proteins involved in the cellulose biosynthetic process, indeed exist as a complex and
have identified other proteins relevant to the process of cellulose synthesis, to be
components of this complex. Based on our findings, we have proposed our model for the
bacterial cellulose synthesis and extrusion complex.

TABLE OF CONTENTS
LIST OF ABBREVIATIONS ............................................................................. x
LIST OF FIGURES…………………………..................................................... xi
LIST OF TABLES ............................................................................................. xiii
ACKNOWLEDGEMENTS ............................................................................... xiv
CHAPTER I BIOCHEMISTRY OF CELLULOSE SYNTHESIS ...................... 1
Cellulose and its impact on our lives .................................................................... 1
Chemical structure of cellulose ........................................................................... 4
Physical properties of cellulose ........................................................................... 6
G. HANSENII AS THE MODEL ORGANISM FOR CELLULOSE
SYNTHESIS ………………… ...... .................................................................... 9
Bacterial cellulose: properties and uses ……...................................................... 11
Visualization of cellulose-synthesizing complexes ............................................ 13
Uridine diphosphate glucose (UDP-glucose) ..................................................... 15
Cyclic diguanylate (c-di-GMP) .......................................................................... 16
In vitro cellulose synthesis in presence of the regulator ….…........................... 19
Identification of the genes involved in cellulose synthesis ............................... 20
BACTERIAL CELLULOSE SYNTHASE OPERON……………................... 22
acsAB ……….................................................................................................... 23
AcsC .................................................................................................................. 25
AcsD................................................................................................................... 26
acs operon is flanked by genes that modulate cellulose synthesis .................... 27
dgc and pdeA genes ......................................................................................... 28
Cellulose biosynthetic pathway and mechanism of cellulose synthesis ..….… 30
STATEMENT OF THE PROBLEM ................................................................ 33
SUMMARY ...................................................................................................... 34
CHAPTER II WHOLE GENOME SEQUENCING OF
GLUCONACETOBACTER HANSENII 23769.................................................. 38
INTRODUCTION ............................................................................................. 38
STEPS IN GENOME SEQUENCING……..…................................................. 40
General outline of sequencing protocol ............................................................. 40

vii
reparation of single stranded DNA library ..................................................... 41
Amplification of the library by emulsion PCR .............................................. 41
Sequencing by synthesis (Pyrosequencing) ......................................................41
Paired-end library generation ......................................................................... 43
SOLid sequencing ........................................................................................... 45
ANALYSIS OF THE SEQUENCE USING SOFTWARE TOOLS ............... 46
Assembly ......................................................................................................... 46
Generation of contigs ...................................................................................... 46
Scaffolds .......................................................................................................... 47
Gene prediction and annotation ....................................................................... 48
MATERIALS AND METHODS ……..……….…......................................... 49
Isolation of genomic DNA ..…......................................................................... 49
Processing and analysis of the sequenced data using software tools …........... 51
RESULTS ……….…....................................................................................... 55
Assembly metrics ............................................................................................. 55
Finishing, annotation and databank entry ........................................................ 55
Genome features ……….……......................................................................... 56
Features relevant to cellulose synthesis …....................................................... 56
Proteomic analysis of the membrane compartments ……............................... 57
DISCUSSION ….…......................................................................................... 60
CONCLUSIONS …………………………………………………..……….... 62
CHAPTER III LOCALIZATION OF THE ACSD PROTEIN IN THE
PERPIPLASM OF G. HANSENII CELLS …….…........................................... 63
INTRODUCTION ...............................................................................................63
MATERIALS AND METHODS ………........................................…...….……65
Bacterial strains and culture conditions ….....................................................…66
AcsD cloning, expression and protein purification ……...................................66
Protein expression and purification ….…….…............................................... 68
Antibody preparation …...…......................................................................….. 69
Preparation of membrane fractions .................................................................. 69
Preparation of periplasmic and cytoplasmic fractions ..................................... 70

viii
Detection of AcsD using Western blot ….…................................................... 72
RESULTS ……................................................................................................ 72
Purification of AcsD ........................................................................................ 73
Specificity of anti-AcsD antibody …......................................................…..... 73
Subcellular fractionation ................................................................................. 74
Detection of AcsD in the periplasmic fraction ............................................... 75
DISCUSSION.................................................................................................. 77
CONCLUSION ……...................................................................................... 79
CHAPTER IV DETERMINATION OF THE SOLUTION-STRUCTURE
OF ASCD ………….......................................................................................... 81
INTRODUCTION...............................................................................................81
Principles behind structural analysis by SAXS…............................................. 84
MATERIALS AND METHODS.…. ................................... ........................... 85
AcsD overexpression and purification ….....................................................…. 85
Sample preparation for AUC .. ................................... .......................................86
DLS experiment …………................................................................................ 86
Analytical ultracentrifugation ………......................................................…..... 86
Gel filtration ……………................................................................................. 88
Sample preparation for SAXS ......................................................................... 88
Data acquisition ………..….............................................................................. 89
SAXS data analysis ….........................................................................................90
RESULTS ………............................................................................................. 92
DLS experiment …..............................................................................………. 92
Gel filtration ……………..…............................................................................ 94
Sedimentation velocity experiments ……...................................................….. 95
Determination of the AcsD structure using SAXS…................................……..99
DISCUSSION ……….……..……….…………….….................................... 107
CONCLUSION ……….……..……….……............................................…. . 109
CHAPTER V BIOCHEMICAL CHARACTERIZATION OF
ACSAB, THE CELLULOSE SYNTHASE PROTEIN ....................................110
INTRODUCTION …........................................................................................110

ix
MATERIALS AND METHODS ..................................................................... 110
Cloning and heterologous expression of the acsAB gene regions …..….......…113
Expression of the AcsAB soluble regions ........................................................ 114
Protein purification using denaturing method ...................................................115
Antibody generation, purification and Western blot ........................................ 117
Sucrose density gradient centrifugation …...................................................… 118
RESULTS ……................................................................................................. 118
Predicting the soluble regions of the AcsAB protein …................................... 118
Western blot using anti-AcsAB1 and anti-AcsAB2 antibodies ....................... 119
Western blot using anti-peptide antibodies ...................................................... ...119
Sucrose density gradient centrifugation …...................................................… 122
DISCUSSION ................................................................................................... 124
CONCLUSIONS ............................................................................................... 128
CHAPTER VI ISOLATION OF THE CELLULOSE SYNTHASE COMPLEX
USING ELECTROPHORETIC TECHNIQUES ...................................... ........ 129
INTRODUCTION ................................................................................... ........ 129
MATERIALS AND METHODS …................................................................. 131
Solubilization of the TM proteins …………...................................................... 131
Native gel electrophoresis of the solubilized TM proteins ……........................ 132
Zymography .…................................................................................................... 133
Blue native polyacrylamide gel electrophoresis (BN-PAGE) ……….….…...... 133
RESULTS ……................................................................................................... 139
Zymography for in vitro cellulose synthase activity ….………. ....................... 139
Selection of an efficient detergent for BN-PAGE ……….……........................ 139
BN-PAGE of DDM-solubilized TM …….…….........,...................................... 140
DISCUSSION ……............................................................................................ 144
CONCLUSIONS …........................................................................................... 147
CHAPTER VII SUMMARY AND FUTURE DIRECTIONS ................….…. 148
REFERENCES .................................................................................................. 153

x
ABBREVIATIONS
Acs: Acetobacter cellulose synthase
AUC: Analytical ultracentrifugation
BN-PAGE: Blue native polyacrylamide gel electrophoresis
Brij 58: Polyethylene glycol hexadecyl ether
c-di-GMP: Cyclic di-guanosine monophosphate
CM: Cytoplasmic membrane
DDM: Dodecylmaltoside
EDTA: Ethylene diamine tetraacetate
DLS: Dynamic Light Scattering
IPTG: Isopropyl thiogalactopyranoside
Ni-NTA: Nickel nitriloacetate
OM: Outer membrane
MudPIT: Multidimensional Protein Identification Tool
PCR: Polymerase chain reaction
SDS-PAGE: Sodium dodecyl sulphate electrophoresis
SAXS: Small angle X-ray scattering
SDS: Sodium dodecyl sulphate
TM: Total membrane
UDP-glucose: Uridine diphosphate glucose
WC: Whole cells

xi
LIST OF FIGURES
Figure 1.1 Chemical structure of cellulose …………...................................... 5
Figure 1.2 Turnover of c-di-GMP in bacterial cells ....................................... 18
Figure 1.3 Structure of cellulose synthase operon in related strains
of Acetobacter ………………………………………………………………. 22
Figure 2.1 Steps involved in a genome sequencing project............................ 39
Figure 2.2 Generation of a DNA library by paired-end method ………..... ... 44
Figure 2.3. Agarose gel electrophoresis of genomic DNA …………............. 50
Figure 2.4 Subsystem catalogue of the genes ………..................................... 59
Figure 3.1 Cloning of the acsD gene …........................................................... 66
Figure 3.2 Overexpression and purification of recombinant AcsD ….…...... 69
Figure 3.3 Determining the specificity of the anti-AcsD antibody ……..….. 73
Figure 3.4 Subcellular fractionation and detection of AcsD ……………....... 76
Figure 3.5 The amino acid sequence of AcsD …………………………..….. 77
Figure 4.1 SAXS experiment ………………………………….………….…. 83
Figure 4.2 Anion exchange chromatography of AcsD …………………….... 87
Figure 4.3 DLS analysis of AcsD .............................................................. 92, 93
Figure 4.4 Gel filtration of AcsD …................................................................. 94
Figure 4.5a Sedimentation coefficient distributions: An overlay showing
normalized distribution plots for AcsD…………..……............... 96
Figure 4.5b,c Continuous sedimentation coefficient distribution c(s) ………..98
Figure 4.6a Experimental scattering profiles ..…………….….…….…….... 99
Figure 4.6b Buffer subtracted scattering curves for AcsD ………………..... 100

xii
Figure 4.6c Guinier plot ..…………………………………………………......... 101
Figure 4.7 Plot for pair distribution function derived using GNOM ………….. 102
Figure 4.8 Representative bead models of AcsD generated by GASBOR.........104,105
Figure 4.9: Comparison of the contours of the solution structure and
the crystal structure of AcsD ………………………………………….………….…106
Figure 5.1 Depiction of the heterologously expressed
regions of AcsAB protein ... ........................................................................................113
Figure 5.2 Agarose gel of amplified products of acsAB gene regions ……………..116
Figure 5.3 SDS-PAGE of heterologously-expressed AcsAB polypeptides ……….. 121
Figure 5.4 Western blot using specific polypeptide and peptide antibodies ………. 121
Figure 5.5 Graph for Molecular weight determination of processed
AcsA polypeptide …………………………………………………….... 123
Figure 5.6 Western blot of fractions from sucrose density gradient of TM …. ……125
Figure 5.7a Analysis of AcsB2 sequence using Signal P (Gram negative) ..............125
Figure 5.7b Lipo P prediction of a signal sequence in the AcsB polypeptide …….. 126
Figure 6.1. Zymogram of detergent-solubilized G. hansenii TM ……………….... 138
Figure 6.2 Comparison of the second dimension gel profiles ……………………... 141
Figure 6.3 BN PAGE of G. hansenii TM…..………................................................. 142
Figure 7.1 Working model for cellulose synthesis complex ………………………. 152

xiii
LIST OF TABLES
Table 2.1 Proteins relevant to cellulose biosynthesis identified by MudPIT. …. ….. 59
Table 3.1 Marker enzyme assays for sub-cellular fractions ….….............................. 74
Table 3.2 Protein identification by LC-MS of trypsin-digested 17kDa gel band…... 77
Table 6.1a Composition of polyacrylamide gradient BN-gel …………………….. 135
Table 6.1b Buffers for BN-PAGE ………………………………………………..... 136
Table 6.2 Proteins detected after LC-MS of the BN-gel band ………………. ........ 143

xiv
ACKNOWLEDGEMENTS
When I think about my life as a graduate student, I feel like I have been looking at
the world of research, standing on the shoulders of giants. I am fortunate to have had
mentorship from outstanding scientists, within and outside Penn State. I am thankful to
my advisor Dr. Ming Tien whose academic experience and able guidance have been
invaluable for me. He has always motivated me in order to bring out the best in me. I
would like to extend my sincere thanks to Dr. Tracy Nixon, Dr. Nicole Brown and Dr.
Charles Anderson, for being part of my thesis committee. I am extremely thankful to Dr.
Nicole Brown for being very supportive and for introducing me to the world of cellulose
synthesis. I am very grateful to Dr. Charles Anderson for accepting to be a part of my
committee at the very last minute. This kind gesture by him, will always be remembered.
I have had the good fortune of working with Dr. Tracy Nixon, whose easy grasp
of structural analysis of proteins, have helped me through my learning curve on this
subject. His patience and dedication while teaching me and training me during the SAXS
experiments, are something I will remember forever and would like to imbibe in my own
teaching methods. It is a dream-come-true, to be guided by the experts in the field of
plant cell wall themselves, Dr. Candace Haigler and Dr. Daniel Cosgrove. Through the
platform of the CLSF, they have given me the opportunity to present my work in front of
varied audiences in numerous conference presentations. Their kindness, encouragement
and faith in me, has helped me more than they know.
I would like to acknowledge Dr. Teh-Hui Kao and Dr. Jeffrey Catchmark for
having fruitful discussions with me over the years. I also want to thank Dr. Yara
Yingling, Dr. James Kubicki, Dr. Alan Esker and Dr. Linghao Zhang for lifting-up my

xv
spirits before all my presentations and treating me like their own student. A heartfelt
thanks goes to Laura Ullrich and Liza, who have been such a good friends. Laura took
care of all the arrangements for my presentations and even made room reservations for
my thesis defense.
I am really thankful to my colleagues who have enriched my research through
their inputs. My initial training in the Tien lab was by Dr. Shin Sato, who trained me in
the area of fungal biochemistry, while I was a rotation student in this lab. He trained me
in techniques that I now routinely use in my work, and am extremely indebted to him for
initiating me to lab work. I am very grateful to Dr. Scott Geib who was instrumental in
teaching me the ropes of bioinformatic techniques required for genome sequencing. This
work would not have been possible without his guidance. My dear friends and senior
graduate students Camille Stephens and Tatyana Sysoeva have always been a good
support throughout my graduate life. I am greatly thankful to them for teaching me
crucial biochemistry skills. I would also like to thank everyone in Tein lab for the being a
very supportive group of friends.
The road to my graduate career was a winding one. But I would have not taken
this path without the love for learning which was instilled in me, very early in my life.
My family and my teachers had envisioned my life in academics, much before I knew
about myself. My teachers have been instrumental in motivating me to perform better
than I thought I am capable of. I thank my teachers, Mrs. Kumar, Mrs. Sophy Verghese,
Mrs. Sreeja Prakash, Mrs. Aparna Kulkarni, Mrs. Vimala Srinivasan, Mrs. P. Katre, Mrs
D. Majumdar, Mrs.Kulkarni, Dr. DN Mishra, Dr. A. Maniyar, Dr. T. Sheikh, Dr. Rama

xvi
Kannan, Mrs. Aparna Rajagopalan and Mrs Radhika. You saw in me what I did not know
about myself.
In my life I have had the privilege of some of the greatest teachers. My first
teachers were my parents, uncles, aunts and grandparents. Through their innumerable
story-telling sessions, reading time and family discussions, I was initiated into my
informal education. My parents are extremely ambitious for me and have provided me
with the best of education at the expense of their own comfort. I am grateful to my dear
sister Priya for teaching me through her example, to be dedicated to a cause and to
achieve it against all odds. My parents chose an equally loving and caring family for me
to get married into. The affection and blessings showered on me by my mother- and
father-in-law, is the greatest treasure in my life. Without the encouragement and
motivation from my in-laws (parents-, uncles-, aunts-, sisters- and brothers-) and my
husband, this Ph.D. would have been just a dream. I have learnt the art of enjoying
research from my husband, who has taught me, that enthusiasm and passion are the
biggest skill-sets of a good scientist. My conversations with my family members were my
source of rejuvenation and strength throughout my graduate life. It is their goodness of
heart that has translated into my good fortune. To this wonderful family of amazing
people, I dedicate this thesis.

CHAPTER I
BIOCHEMISTRY OF CELLULOSE SYNTHESIS
Cellulose and its impact on civilization
Cellulose is a homopolysaccharide that is the most abundant naturally-occurring
macromolecular polymer on earth, being produced at the rate of 180 billion tons per year
(Brown and Montezinos 1976). Cellulose is the major component of the plant cell wall
where it is embedded in a matrix of lignin, hemicelluloses and pectin. On average,
cellulose comprises around 45% of the plant lignocellulosic biomass but the content of
this polymer varies with the plant type (Matrone, Ellis et al. 1946; Meier 1964;
Toyoshima, Onda et al. 1990). In woody plants, cellulose constitutes almost half the
weight of the biomass (Meier 1964), whereas in grasses the content of cellulose is
roughly 20-30% (Matrone, Ellis et al. 1946; Toyoshima, Onda et al. 1990). The
secondary cell walls of cotton, with their cellulose content of almost 100% are the purest
form of cellulose in nature (Itoh 1990; Haigler 2007). The predominance of cellulose in
the plant kingdom makes it the major constituent of biomass, an abundant renewable
resource and a significant contributor to the global carbon cycle.
Its distribution together with its unique properties, has made the use of cellulose
widespread. Humans have been exploiting the predominance of this natural material for
centuries and have invented several applications for cellulose-derived materials, making
cellulose-based products a quintessential part of our daily lives. Its unique properties of
high tensile strength, flexibility and recalcitrance, make it an ideal material for lumber,
paper, textiles and many commodities. The extreme dependence of mankind on cellulosic
products continues in present day society. This has put a severe burden on forestry and

2
agriculture and has lead to long-term adverse effects on the planet like deforestation and
loss of indigenous flora.
An obstacle in the use of plant-based cellulosic material, is that for many of its
applications, a pure form of cellulose is desired. But, the lignin and hemicellulosic
network of plant cell walls are not easily removed or circumvented. Furthermore,
conversion of cellulose is impeded by the inherent recalcitrance of cellulose. Purification
of cellulose therefore, involves several steps of harsh alkaline treatments and/or the use of
sulfites to digest the lignin and free the cellulose away from cell wall polymers. This
process of purifying cellulose from plant material is a highly energy-demanding, water-
consuming and polluting process (Canadian Environmental Protection Act Priority
Substances List Assessment 1991). A new term, "paper pollution", has been coined to
refer to the environmental hazards of the pulp-milling process, which is the third largest
industrial contributor to pollution, causing land, water and air pollution (Teruyama, Itoh
et al. 1990). The complete abolishment of the use of cellulosic materials is not likely,
thus, sustainable and environmentally-friendly methods for cellulose purification are of
relevance.
In addition to its traditional uses in paper and lumber industries, at present the
major focus on cellulose is due to its potential use as the starting material for bioethanol
production. Cellulose is envisioned as a future source for biofuels, in the form of
cellulosic ethanol which is touted to substitute petroleum as a transport fuel (Sticklen
2008). Cellulosic is derived from non-edible portions of renewable feed stocks like corn
stover and straw, or from non-food sources like agricultural wastes, bagasse, sugarcane
and wood (2007). In addition to being environment-friendly, it provides a greater net

3
energy benefit and a lower green house emission than corn-based ethanol (Energy
Conservation Board). Current research on cellulose is largely directed towards
genetically-engineering plants in order to obtain large quantities of cellulosic biomass
without using much land space. Such approaches include but are not restricted to,
augmenting the rate of cellulose synthesis in the plant cell walls (Andersson-Gunneras,
Mellerowicz et al. 2006), making cellulosic material less crystalline (Abramson,
Shoseyov et al. 2010) and modifying the chemistry of other wall polymers like lignin and
hemicellulose with the aim of weakening their matrix and making cellulose more
accessible (Ragauskas, Williams et al. 2006; Chen and Dixon 2007; Abramson, Shoseyov
et al. 2010).
Finding alternatives to the process of chemical pulping, altering the properties of
cellulose and enhancing its biosynthesis require an in depth understanding of the plant
cell wall architecture as well as knowledge of the biochemical pathways leading towards
the cell wall polymer synthesis and incorporation. Specifically, this endeavor necessitates
a thorough inquiry into the structure, mechanical properties and the process of cellulose
biogenesis. My dissertation work, though not directly applicable to the production of
biofuels, is directed towards understanding the cellulose biosynthetic machinery. The
enzymes and structural proteins contributing to the biosynthesis of cellulose, have a
profound impact in the chemistry and morphology of the final product. Thus, mastering
the technology to derive enhanced cellulose production in plant cells requires that we
acquire adequate information on the process of cellulose biosynthesis. This would not
only help to understand how nature produces this polymer, but also enable us to modify
or engineer the properties of cellulose for various applications.

4
Chemical structure of cellulose
The properties of any polymer are largely dictated by its chemical constituents
and structure. As shown in Figure 1.1, cellulose is a linear polymer of β-1,4-linked
glucose residues. This makes the basic unit of cellulose, a dimer glucose, called
cellobiose, where each glucose is rotated at an angle of 180C with respect to its
neighboring residue. As shown in Figure1.1b, the straight chains of cellulose, have a
directionality conferred upon by the anomeric carbon. The end of the cellulose chain with
the unlinked anomeric carbon is the reducing end and one with an exposed C-4 is the
non-reducing end. This directionality is a critical factor in determining the mechanism of
polymer synthesis and extension, which will be discussed in this chapter in the section on
“Cellulose biosynthetic pathway and mechanism of cellulose synthesis”.

5
Figure 1.1 Chemical structure of cellulose
a) Glucose is a six-carbon sugar with two forms based on the orientation of the hydroxyl
group of the anomeric carbon, C-1. The α-form has the hydroxyl group on the opposite
side of the ring from the -CH2OH group and ß-glucose has the hydroxyl group on the
same side as the -CH2OH group.
b) Cellobiose is a dimer of ß-D-glucose where C-1 of one sugar is linked to the C-4 of the
other by an acetal linkage.
c) Cellulose is a linear polymer of ß-1,4-linked glucose units.

6
Another feature of cellulose conferred upon it by the β-1,4 linkage, that
differentiates cellulose from other glucan polymers like starch (α-1,4 linked branched,
helical polymer) and callose (β-1,3 linkage), is the formation of extended, straight,
unbranched chains with hydroxyl groups at C-2, C-3 and C-5 positions available for
formation of intra- and inter-chain hydrogen bonds and van der Waals interactions
(Brown and Montezinos 1976; Montezinos and Brown 1976). These forces govern
hierarchical associations of cellulose fibers to highly energy-minimized, para-crystalline
forms that are insoluble yet very flexible and possess the tensile strength equivalent to
steel (Niklas 1992), making it the strongest organic molecule on density basis
(Yamanaka, Watanabe et al. 1989).
Physical properties of cellulose
The basic unit of a cellulose fiber aggregate is an termed as a microfibril, a name
that was originally given for the thinnest strands of cellulose observed under electron
microscope (Hakoshima, Itoh et al. 1990). Cellulose microfibrils are classified based on
their unit size, spectral properties, degree of polymerization and orientation of fibers. The
size of the microfibrils, varies from 2-10 nm in plants to 30nm in Spirogyra (Hahne,
Herth et al. 1983; Hausser and Herth 1983).
The cellulose chains can be arranged in different orientations in the microfibril,
giving rise to different crystalline forms (Brown and Montezinos 1976; Montezinos and
Brown 1976; Montezinos and Brown 1976) of cellulose, known as cellulose I and
cellulose II allomorphs (Kaihoh, Itoh et al. 1990). Majority of the cellulose in nature
occurs as the cellulose I allomorph (Chiou, Chen et al. 1990; Kaihoh, Itoh et al. 1990).
Using specific silver staining for reducing ends and cellobiohydrolase-mediated

7
digestion, it was shown that all the glucan chains in cellulose I are oriented parallel to
each other, with reducing ends of all the chains pointed in one direction (Herth 1983).
Anti-parallel arrangement of the cellulose chains gives rise to the cellulose II allomorph,
the most thermodynamically stable form, which is a rare occurrence in nature (Brown
1996). Other than the marine alga Halocystis (Mulisch, Herth et al. 1983) and Gram
positive bacterium Sarcina (Canale-Parola 1970), which produce the cellulose II
allomorph naturally, this form of cellulose is known to be produced exclusively in cases
where the ordered arrangement of cellulose fiber is perturbed due to dye addition (Chang
and Itoh 1990), mercerization (Chanzy and Roche 1975), strong alkaline treatment or
mutations in cellulose producing proteins (Kuga, Takagi et al. 1993; Chen and Brown
1996). An extra hydrogen bond for each glucose residues contributes to the extreme
stability of cellulose II (Itoh, Oneil et al. 1984).
Their distinctly specific spectral signatures help in differentiating the allomorphs
and sub-allomorphs of cellulose. Cellulose I can be further categorized into two
allomorphs, Iα and Iβ (Kaihoh, Itoh et al. 1990; Ohchi, Itoh et al. 1990). Fiber diffraction
and 13
C-NMR studies on these sub-allomorphs, revealed that cellulose Iα has a single
chain, triclinic unit cell, which further confirms the parallel chain model and Iβ is a
monoclinic unit cell with two chains (Brown and Montezinos 1976; Chiou, Chen et al.
1990). Based on a crude estimates, (Kaihoh, Itoh et al. 1990), Acetobacter cellulose and
cotton cellulose contain approximately 60-70% Iα and Iβ respectively. Thus the Iα
form is predominant in prokaryotic cellulose, in which cellulose biosynthesis is part of
the cell cycle and the Iβ form predominates in plant cellulose where secondary cell wall
cellulose is produced after cell division is completed and the wall therefore possesses a

8
complex architecture. Electron diffraction pattern of the entire length of microfibrils of
the alga Microdictyon tenuis revealed regions that were purely Iα or Iβ or a mixture of the
two forms (Chiou, Chen et al. 1990). This led to the conclusion that all naturally-
occurring celluloses are a mixture of Iα and Iβ-allomorphs in varying ratio (Chiou, Chen
et al. 1990). Since Iα-cellulose is meta-stable and more reactive than Iβ cellulose (Chiou,
Chen et al. 1990), the presence of both forms in different ratios accounts for the
differential reactivity of native celluloses obtained from different sources, leading to
substantial variation in the crystal packing and hydrogen bonding patterns, that influences
their physical properties (Chiou, Chen et al. 1990).
Though there is considerable interspecies variation in the composition and size of
cellulose, the allomorphic distribution and dimensions of the microfibrils remain
consistent for a given species at a particular stage of its life cycle. Electron micrograph
images show that the dimensions of the microfibrils are reflected by the number of chains
in each microfibril, which is in turn determined by the number and pattern of the
cellulose synthesizing complex subunits involved in the synthesis (Brown and
Montezinos 1976; Zaar 1979; Okuda, Tsekos et al. 1994; Reiss, Katsaros et al. 1996).
The structural information of cellulose is crucial for its characterization as well as for
speculating on and developing models for the mechanisms of its synthesis. Similarly, the
process and factors involved in cellulose biosynthesis, leave an imprint on the final
morphology and pattern of cellulose formed.
G. HANSENII AS THE MODEL ORGANISM FOR CELLULOSE SYNTHESIS

9
Other than plants, may species across the various kingdoms of life possess
cellulose biosynthetic ability. Cellulose synthesis is a rare but existent phenomenon in
animals, belongibg to the family of urochordates (Toyoshima, Onda et al. 1990). The
mycetozoan Dictyostelium discoidum (Tsuji, Itoh et al. 1990) and the protozoans of
Acanthamoeba species (Wanibe, Yokoyama et al. 1990) also exhibit cellulose synthetic
abilities. Although not a component of the wall, cellulose synthesis as part of primary
metabolism is observed in bacterial species such as Acetobacter xylinum (Ross, Mayer et
al. 1991), Rhizobium leguminosarum (Kitagawa, Kanamori et al. 1990; Mukai, Toba et
al. 1990), Klebsiella pneumoniae (Nomura, Harino et al. 1990), Sarcina ventricle(Ross,
Mayer et al. 1991), Agrobacterium tumifaciens (Matthysse, White et al. 1995),
Salmonella typhimurium (Hatta, Baba et al. 1990) and Escherichia coli (Hatta, Baba et al.
1990) and cyanobacteria (Ayaki, Fujikawa et al. 1990). Some prevalent organisms that
have been used a model systems for studies on cellulose synthesis are Gossypium
hirsutum (cotton), Oryza sativa (rice), Arabidopsis thaliana (plant), Physcomitrella
(moss), Valonia (algae), and Acetobacter xylinum, Gluconacetobater hansenii (bacteria).
Among the bacterial species, organisms of the Acetobacter / Gluconacetobacter
genus stand out due to their ability to synthesize and extrude copious amounts of highly
pure ribbons of cellulose. One among these is G. hansenii, that forms the subject of study
in this dissertation. In a recent taxonomic shuffle (Lisdiyanti, Navarro et al. 2006), some
strains of Acetobacter xylinum were placed under the genus Gluconacetobater. Therefore,
the present day nomenclature of some of the cellulose producing strains of Acetobacter
xylinum is Gluconacetobacter hansenii. This change in nomenclature has resulted in the
strain used in this work, ATCC 23769 being classified as G. hansenii.

10
G. hansenii, a Gram negative, obligate aerobic bacterium has been considered for
several years, as an archetype for cellulose synthesis-related studies. A single cell can
polymerize 200,000 glucose molecules per second (Hestrin and Schramm 1954), which
are extruded in the form of a 100 nm wide, flat ribbon of cellulose along the longitudinal
axis of the cell (Brown, Willison et al. 1976; Akiyama, Yamada et al. 1990) and remain
attached to the cells during cell division (Marx-Figini 1982; Ring 1982). When cultured
under static conditions, the cellulose released forms a thick mat that accumulates at the
air-liquid interface of the culture medium (Schramm and Hestrin 1954). This visible film,
known as a pellicle, floats on the surface and completely covers the culture medium
(Schramm and Hestrin 1954; Schramm and Hestrin 1954). It is composed of cellulose
fibers enmeshed with bacterial cells (Schramm and Hestrin 1954). When cultivated under
agitated conditions, the increased oxygen tension facilitates faster growth of cells and the
cellulose produced is observed as round balls (Schramm and Hestrin 1954).
Cellulose production is directly proportional to the cell density of the culture and
as much as 50% of the available carbon is assimilated into the cellulose (Schramm and
Hestrin 1954; Kamide, Matsuda et al. 1990). The bacterial population contains several
strain variants that overproduce cellulose and form a thicker aggregate in shaking culture
and a thicker film in the stationary medium (Williams and Cannon 1989). These
populations are transient and revert back to their cellulose synthesizing ability upon
transfer into a static culture. Several generations of sub-culturing is required to obtain
permanent cellulose non-producing mutants (Valla and Kjosbakken 1982).
There have been many attempts to understand the utility of an extracellular
polysaccharide matrix. The most common notion is that the pellicle functions as a

11
flotation device to keep the aerobic cells in close proximity to the atmosphere (Schramm
and Hestrin 1954; Cook and Colvin 1980). Another interesting observation is that when,
co-cultured with molds and other bacterial species on fruits, the cellulosic film provided
competitive advantage to the Acetobacter cells over others in the ability to colonize the
substrate and protected the cells from being invaded by other species (Williams and
Cannon 1989). When observed under an electron microscope, these pellicles displayed a
regular arrangement of tunnel-like lacunae along the surface of microfiber aggregates,
suggesting the possibility of higher order in the organization of cellulose microfibrils
(Thompson, Carlson et al. 1988).
Bacterial cellulose: properties and uses
G. hansenii cellulose fibers contain microfibrils with average diameter of 20-40Å
(Akiyama, Yamada et al. 1990). Bacterial cellulose is 64% 1α (Kaihoh, Itoh et al. 1990;
Yoshida, Morisaki et al. 1990). Being an extracellular, inert and highly pure form of
cellulose, it is an easy material to isolate and study as compared to the plant cellulose.
Other obvious advantages of using a bacterial system versus a plant system are: non-
requirement of specialized culture conditions, faster growth cycle and ease of mutant
generation and isolation.
The unique combination of high mechanical strength, extreme flexibility, in
addition to its insolubility, biocompatibility, elasticity, mechanical strength and resistance
to degradation, has made bacterial cellulose an ideal material of choice for biomedical
applications (Ross, Mayer et al. 1991; Czaja, Kawecki et al. 2004; Czaja, Romanovicz et
al. 2004). Bacterial cellulose is an anisotropic network of fibers, with a high degree of
hydration, making it a suitable scaffold for seeding epithelial cells after severe skin injury

12
and therefore the material of choice to serve as an artificial skin graft for burn victims
(Czaja, Kawecki et al. 2004; Czaja, Romanovicz et al. 2004). This skin-substitute could
be used in future, in lieu of an autograft (Cheung, Neikirk et al. 1990). Native and
modified bacterial cellulose have been used as scaffolds for tissue engineering of
cartilage (Yamaguchi, Ohsawa et al. 1990), blood vessels (Tobita, Kusama et al. 1990)
and cardiac valves (Kuno, Kamisaki et al. 1990). Bacterial cellulose also finds use in
electronic paper (Shah and Brown 2004) and acoustic diaphragms in earphones (Becker,
Itoh et al. 1990). Thus, bacterial cellulose synthesis has implications much beyond its
assumed role as a model to study plant cellulose synthesis and could be a potential
replacement for the latter in many of its uses. Since, many of its applications are in the
biomedical and specialty material sectors, the knowledge of the mechanism of bacterial
cellulose synthesis would be an important contribution to the basic as well as applied
sciences.
It was using A. xylinum, that the major breakthroughs in the study of cellulose
synthesis and characterization were achieved. Some of these milestones, which are
discussed in detail in subsequent sections, are listed as follows,
1. The first successful isolation and cloning of a cellulose synthase gene (Saxena,
Lin et al. 1990). The first plant gene encoding cellulose synthase was identified based in
its homology to the bacterial gene (Pear, Kawagoe et al. 1996).
2. Demonstration of high rates of in vitro cellulose synthetic activity (Glaser 1958; Aloni,
Delmer et al. 1982)
3. Identification of a four gene operon encoding for proteins involved in cellulose
synthesis (Wong, Fear et al. 1990)

13
4. Determination of conserved residues within the catalytic domains of cellulose synthase
protein (Saxena, Lin et al. 1990; Saxena, Henrissat et al. 1995)
5. Identification of c-di-GMP as a regulator for cellulose synthesis (Ross, Weinhouse et
al. 1987)
6. Demonstration of Calcofluor as a dye to bind to and alter cellulose properties (Haigler,
Brown et al. 1980)
7. Model for cellulose biogenesis as a coupled process of polymerization and
crystallization (Benziman, Haigler et al. 1980)
8. Electron microscopic observation of a linear array of complexes involved in active
cellulose synthesis (Brown, Willison et al. 1976; Akiyama, Yamada et al. 1990)
9. Simulation of the assembly of higher plant cell walls (Yamaguchi, Ohsawa et al. 1990)
Some of the above-mentioned findings will be elaborated in the subsequent sections.
Visualization of the cellulose-synthesizing complexes
Biosynthesis of cellulose has been studied using various tools and from different
perspectives. One of the earliest techniques employed to characterize cellulose synthesis
was microscopy. Roelofsen (Roelofsen 1958) proposed as early as 1958 that enzyme
complexes located at the growing tip of the cellulose chain, were involved cellulose
synthesis and polymerization. Using freeze- fracture electron microscopy, Brown et. al.
(Brown, Willison et al. 1976) observed a single linear array of particles involved in
cellulose synthesis on the outer membrane (OM) of A. xylinum. The site of emergence of
the cellulose fibers on the surface of the cells is called a terminal complex (TC), named
so because freeze-fracture electron microscopic analysis revealed the globular protein
complexes on plant cell walls at the termini of cellulose fibrils (Brown and Montezinos

14
1976; Montezinos and Brown 1976)as predicted by Roelofsen (Roelofsen 1958;
Montezinos and Brown 1976).
After the discovery of the proteins involved in cellulose synthesis, it was possible
to use immunological techniques to ascertain that the TC observed by microscopic
methods in the past were indeed cellulose synthases (Kimura, Laosinchai et al. 1999).
Sodium dodecyl sulfate-solubilized freeze fracture replica labeling (SDS-FRL) was used
to visualize the A. xylinum TC labeled with antibodies generated against cellulose
synthase proteins (Kimura, Chen et al. 2001). The gold-labeled antibodies revealed that a
linear row of 12-nm particles was localized in the inner side of the OM, which
correspond to the ring-shaped pores observed in the exoplasmic side of the OM. These
antibodies convincingly proved that the TCs contained proteins involved in cellulose
biogenesis.
TCs show a great diversity in their arrangement in different organisms. Freeze-
fracture studies on plant and algal cell walls revealed that the TCs were organized in the
form of a six-membered rosette in land plants and green algae (Kiermayer and Sleytr
1979), (Giddings, Brower et al. 1980). In many algae, the TCs are arranged in the form of
larger, rectangular arrays synthesizing cellulose ribbons (Katsaros, Reiss et al. 1996;
Reiss, Katsaros et al. 1996). Linear arrays of TCs are observed in prokaryotic bacteria
and certain red and brown algae (Zaar 1979). The correlation between the size of a
complex and the dimensions of the emerging cellulose were calculated by Herth (Herth
1983). These studies were corroborated by evidence presented by Okuda et. al. (Okuda,
Tsekos et al. 1994), by reviewing different types of TC arrangement and cellulose sizes.
It has been observed, as could be predicted intuitively, that the cellulose chains emerging

15
from linear TCs have a flat ribbon-like morphology, where as those from plant rosettes
are cylindrical in shape (Itoh 1990; Itoh and Kimura 2001). Thus, it can inferred that the
diversity of microfibril dimensions, seen across species and kingdoms, arises from the
number of cellulose chains constituting the fibril, which in turn is governed by the
number and arrangement pattern of the TCs. Factors that determine the ordering of TCs
into a linear or a rosette pattern, can be identified only through biochemical and structural
analysis of the proteins constituting them.
Nucleotide derivatives that drive cellulose synthesis: Uridine diphosphate glucose
(UDP-glucose) and cyclic di guanosine monophosphate (c-di-GMP)
A significant contribution to the understanding of cellulose synthesis was the
Nobel-winning discovery of sugar-nucleotides as “high energy molecules” by Leloir et al.
(Caputto, Leloir et al. 1950; Murai, Saito et al. 1990). Leloir et al. (Leloir, Olavarria et al.
1959; Leloir and Goldemberg 1960; Leloir 1961)elucidated the role of sugar-nucleotide
UDP-glucose in the synthesis of glycogen and thereby showed that biological
polysaccharide synthesis reactions were not the reversal of degradation reactions, as was
assumed previously (Caputto, Leloir et al. 1950; Leloir and Cardini 1957; Murai, Saito et
al. 1990). Using glycogen synthesis as an example it was shown that all other
polysaccharide synthesis reactions are in fact transfer reactions where the sugar from the
sugar-nucleotide is transferred to the polymer which increases in length with each
addition and the enzymes catalyzing these reactions were termed as glycosyl transferases
(Leloir 1961). In case of bacterial cellulose, the UDP-glucose for cellulose synthesis is
provided by UDP-glucose pyrophosphorylase (Swissa, Aloni et al. 1980).
Cyclic diguanylate (c-di-GMP) : the unique activator of cellulose synthesis

16
The first-ever demonstration of in vitro cellulose synthesis activity was achieved
as early as 1958 by Glaser (Glaser 1958), in particulate membrane fractions prepared
from G. hansenii cells. However, it was in 1982 that high rates of synthesis in cell-free
extracts were obtained by the Benziman group (Aloni, Delmer et al. 1982). However the
most importan contribution of this work was that it led to the discovery of c-di-GMP
(Ross, Weinhouse et al. 1987).
Cyclic nucleotides (cAMP and cGMP) have been known to be crucial
components of prokaryotic and eukaryotic signal transduction pathways (Karpen 2004).
Cyclic di-GMP was included in the list of second messengers after its biological role was
elucidated by Benziman and co-workers, as the factor that allosterically activates
cellulose synthesis in A. xylinum (Ross, Mayer et al. 1990). Eventually, several workers
revealed the involvement of c-di-GMP in regulation of a wide array of cellular functions
that influence virulence, pilus formation, cell cycle, antibiotic secretion and biofilm
formation (Dow, Fouhy et al. 2006; Fouhy, Lucey et al. 2006; Jenal and Malone 2006;
Ryan, Fouhy et al. 2006; Cotter and Stibitz 2007; Pratt, Tamayo et al. 2007; Tamayo,
Pratt et al. 2007; Wolfe and Visick 2008). In general, it is noted that c-di-GMP is
involved in quorum sensing and favors switching of a motile, planktonic lifestyle to a
sessile on (Romling, Gomelsky et al. 2005). At low concentrations. c-di-GMP promotes a
motile phenotype and at high concentrations it stimulates a sessile, biofilm-associated
lifestyle (Cotter and Stibitz 2007; Pratt, Tamayo et al. 2007; Wolfe and Visick 2008).
Thus, the discovery of c-di-GMP is a milestone not just in the field of cellulose synthesis
but also in all areas of bacterial signaling (Romling, Gomelsky et al. 2005; Ryan, Fouhy
et al. 2006). Intracellular concentrations of c-di-GMP are maintained by the action of two

17
types of enzymes, diguanylate cyclases (Dgc) and phosphodiesterases (Pde) (Tal, Wong
et al. 1998; Ausmees, Mayer et al. 2001; Ryan, Fouhy et al. 2006). Under cellular
conditions, Dgc and Pde coordinately control c-di-GMP levels (Figure 1.2) to affect a
target protein which acts as a switch to control the bacterial behaviour (Jenal and Malone
2006; Pratt, Tamayo et al. 2007; Tamayo, Pratt et al. 2007). In case of Acetobacter, the
target protein is cellulose synthase which is activated by c-di-GMP, thereby promoting
the process of cellulose synthesis (Ross, Mayer et al. 1990).

18
Figure 1.2 Turnover of cyclic di-GMP in bacterial cells The cellular levels of c-di-
GMP are maintained by the concerted action of two enzymes: diguanylate cyclase (DGC)
and phosphodiesterase (PDE). These activity of these enzymes are regulated based on the
environmental conditions. The DGCs have a conserved GGDEF motif in their active site
(Ausmees, Mayer et al. 2001) and produce cyclic di-GMP by cyclization of two
molecules of GTP (Paul, Weiser et al. 2004; Ryjenkov, Simm et al. 2006). Degradation
of cyclic di-GMP molecules into two GMPs is mediated by PDE. These enzymes contain
an EAL or a HD-GYP motif in their active site. The enzymes with EAL domain linearize
the c-diGMP to form 5’pGpG, (Schmidt, Ryjenkov et al. 2005; Tamayo, Tischler et al.
2005) which is further cleaved to form two GMP molecules by non-specific PDEs
(Christen, Christen et al. 2005; Romling, Gomelsky et al. 2005). The HD-GYP domain-
containing proteins break the phosphodiester linkage in cyclic di-GMP first to form 5’-
pGpG which is further cleaved to into two (Leloir, Olavarria et al. 1959; Leloir and
Goldemberg 1960; Leloir 1961) GMPs by the same enzyme (Ryan, Fouhy et al. 2006).

19
In vitro cellulose synthesis in presence of its regulator
A systematic work by Bureau and Brown in 1984 (Bureau and Brown 1987),
showed for the first time that the cellulose synthetic activity was localized in the CM of
Acetobacter. The TM of the bacterium was separated into CM and OM fractions by
sucrose-density gradient centrifugation after solubilizing the membrane fractions with
lysozyme and trypsin(Bureau and Brown 1987). A cellulose synthesis assay was
performed by incubating the membrane preparations in presence of 14
C-labelled UDP-
glucose and Mg2+
. The insoluble radioactive product was separated by filtration and
measured using liquid scintillation. The product was characterized by enzymatic
hydrolysis using cellobiohydrolase and endoglucanase, methylation analysis followed by
gas chromatography, high performance gel permeation chromatography and X-ray
diffraction. The degree of polymerization of the resultant product was 5270 and the
crystalline nature determined by X-ray diffraction was found to be that of cellulose II
(Bureau and Brown 1987). The Km of this reaction which followed Michaelis-Menten
kinetics, was 2.0 x 10-4
mM and the Vmax was found to be 52.4 nmol glucose
incorporation per mg of protein per minute. Similar values were obtained for digitonin-
solubilized whole membrane fractions, by Aloni et. al. (Aloni, Delmer et al. 1982). The
cellulose synthase activity observed predominantly in the CM had an optimum
temperature of 30ºC and pH of 8.3. The enzyme activity in the membrane-bound and
digitonin-solubilized form was found to be Mg-dependent, and inhibited by uridine
mono- (Ki = 0.7mM), di- and triphosphates (Ki = 0.14mM) and guanyl nucleotides: pGpG
and GpG (Ross, Mayer et al. 1990). Following the lead of cell-free cellulose synthesis in
bacteria, in vitro cellulose synthesis was achieved in cotton fibers (Okuda, Li et al. 1993),

20
(Kudlicka, Brown et al. 1995), aspen cell suspension cultures (Colombani, Djerbi et al.
2004) , mung bean (Kudlicka and Brown 1997) and blackberry (Lai-Kee-Him, Chanzy et
al. 2002).
Identification of c-di-GMP as the regulator (Ross, Weinhouse et al. 1987) and
demonstration of in vitro activity (Bureau and Brown 1987) facilitated purification of
proteins (Lin, Brown et al. 1990; Mayer, Ross et al. 1991) and determination of the
cellulose synthase genes (Wong, Fear et al. 1990). Cellulose synthase was purified using
the product entrapment method (Lin, Brown et al. 1990), that was successfully employed
to isolate chitin synthase by Kang et. al. (Kang, Elango et al. 1984). This technique
involves incubation of detergent-solubilized membranes in a reaction mixture as
described above, and subsequent centrifugation to obtain the cellulose synthase entrapped
within an insoluble cellulosic pellet. When the reaction mixture lacks either UDP-glucose
or c-di-GMP, no synthase activity is retrieved in the pellet, proving the identity of the
enzyme recovered from the pellet to be a cellulose synthase. Moreover, treatment with
cellulase released 50% of the enzyme activity in the soluble portion. This also reflects the
high affinity of the enzyme for the product formed.
Identification of the genes involved in cellulose synthesis
Using the product entrapment method, up to 350-fold purification of the enzyme
could be obtained which was further used for isolating a highly pure synthase protein.
The pure enzyme was found to be composed of an 83 and a 93kDa polypeptide (Lin
1989). These polypeptides were used variously for antibody generation for
immunological and localization studies (Chen and Brown 1996) as well for development
of radiolabelled probes to identify protein binding characteristics and molecular weight

21
determination (Lin, Brown et al. 1990). But most importantly, the peptide sequences
derived (Mayer, Ross et al. 1991) were used to design oligonucleotide probes to clone
and sequence the gene encoding for cellulose synthase (Saxena, Lin et al. 1990) and
deduce the operon harboring it (Wong, Fear et al. 1990).
Figure 1.3 Structure of cellulose synthase operon in related strains of Acetobacter Cellulose synthase genes have been variously named as acsA (Acetobacter cellulose
synthase, axCesA (for Acetobacter xylinum cellulose synthase) and bcsA (bacterial
cellulose synthase). G. hansenii strains ATCC 23769 and ATCC 53582 possess a single
open reading frame for the cellulose synthse gene (acsAB) (Kawano, Tajima et al. 2002).
In the strain 1306-3, the gene contains two open reading frames (acsA and acsB)
characterized by Wong et al., whereas in G. xylinus strain NBRC 3222, the cellulose
synthase gene contains three open reading frames (Ogino, Azuma et al. 2011). In the
strain ATCC 1306-3, in which the operon structure was first studied, the initiation codon
(97 bp upstream of the cellulose synthase gene and the termination codon (26 bp
downstream of acsD gene) for the operon, are indicated by the triangle and stem-loop
structure respectively.

22
THE BACTERIAL CELLULOSE SYNTHASE OPERON
Genes involved in the synthesis of bacterial polysaccharides are usually organized
as an operon that encodes for proteins mediating the various steps in the synthetic process
(Vazquez, Moreno et al. 1999; Whitney, Hay et al. 2011). In case of cellulose synthesis,
the enzyme cellulose synthase converts UDP-glucose to cellulose in a single step (Lin,
Brown et al. 1990). This enzyme is encoded as part of an operon, referred to as the
Acetobacter cellulose synthase (acs) operon (Wong, Fear et al. 1990). The operon
structure of cellulose synthase was elucidated by Wong et. al. (Wong, Fear et al. 1990) by
genetic complementation of cellulose non-producing mutants of the strain A. xylinum
1306-3. The operon was further characterized by Saxena et al. (Saxena, Kudlicka et al.
1994) in 1994 to elucidate the function of the protein encoded by each gene of the
operon, using site-directed insertional mutagenesis of ATCC53582 strains (Saxena,
Kudlicka et al. 1994). As shown in Figure. 1.3, the Acetobacter cellulose synthase (Acs)
operon consists of four genes that were found to be transcribed into a polycistronic
mRNA, with the site of transcription initiation located 97 bp upstream of the acsA gene.
These genes encode for four proteins: AcsA (84.4kDa). AcsB (85.3kDa), AcsC (141kD)
and AcsD (17.3kDa). Sequence comparisons with initiation codons of the acs, ald and alh
genes revealed that a highly conserved GGACGNG sequence is located 2-6 bases 5' of
the AcsA start site (Wong, Fear et al. 1990). Based on similar homologous regions in the
sequences upstream of the three genes, it was inferred that the transcription initiation site
is represented by the sequence CATCGCTG which is located between -11 bp and -4 bp
upstream of acsA. The transcription termination site is a 26 bp region at the 3' end of the
acsD gene containing an inverted repeat sequence that has the potential to form a stem-

23
loop structure which serves as the signal for transcription termination in bacteria. In the
strains ATCC53582 and ATCC 23769, the acsA and acsB genes are fused to form one
gene (Kawano, Tajima et al. 2002), while in other strains like NBRC 3288 (Ogino,
Azuma et al. 2011), the ORF is split into three genes (Figure 1.3).
AcsAB
Although acsA and acsB were initially considered as two separate genes, it was
found later that depending on the strains of Acetobacter used for study, acsA and acsB
were either found as two separate ORFs or a single gene referred to as acsAB (Figure
1.3) In the Acetobacter xylinum strains ATCC23769 (now changed to G. hansenii) and
ATCC53582, cellulose synthase is encoded by a single gene encoding a 168 kDa protein,
whereas in 1306-3, BPR 2001, JCM 7664, there are two genes encoding for the different
regions of the protein.
Comparing the operon of catalytic A. xylinum ATCC 53582 with that of A.
xylinum ATCC 1306-3 revealed that the AcsA and AcsB polypeptides share ~81%
similarity to the N-terminal and C-terminal of the AcsAB protein (Saxena, Kudlicka et al.
1994). Using hydrophobic cluster analysis (HCA), where secondary structure prediction
of a protein is combined with the alignment to homologous proteins, conserved residues
in cellulose synthase protein were identified (Saxena, Brown et al. 1995). Cellulose
synthase belongs to the glycosyl transferase family 2 (GT2) and contains a DXXD motif
and another single highly-conserved aspartate residue and followed by QXXRW motif
(Saxena, Brown et al. 1995; Saxena and Brown 1997). Collectively this signature motif
of GTs is referred to as the D,D,D, Q/RXXRW motif . Attempts to replace the aspartate
residues by site-directed mutagenesis, resulted in loss of catalytic activity, providing the

24
reason behind the asparatate being conserved across species . This motif is conserved not
only in cellulose synthases of all cellulose-producing organisms but is also common to
GTs like chitin synthase, hyaluronan synthase and glycosyl ceramide synthase (Saxena,
Brown et al. 1995; Saxena, Henrissat et al. 1995). Close examination of the deduced
amino acid sequence shows that this sequence is found in the AcsA or the N-terminal half
of the AcsAB protein. Using photoaffinity labeling of the purified protein with (-32
P)-
azido-UDP-glucose, Lin et al. (Lin, Brown et al. 1990)identified this protein to be the 83
kDa subunit of cellulose synthase (Lin, Brown et al. 1990). Based on this and the
identification of the DXXD motif to be crucial for binding UPD-glucose, the acsAB gene
was considered to encode for the catalytic domain of the protein.
The C-terminus of the AcsAB protein or the AcsB protein is presumed to contain
sites for c-di-GMP binding (Kimura, Chen et al. 2001), thus serving as the regulatory
domain of the protein. Kimura et al (Kimura, Laosinchai et al. 1999) used the 93 kDa
polypeptide obtained from product entrapment to generate antibodies and localize this
protein in the membrane fraction of the A. xylinum cells. However, after the discovery of
PilZ domain as the cyclic di-GMP binding motif (Amikam and Galperin 2006; Ryjenkov,
Simm et al. 2006) the function of AcsB as the regulatory domain was disproved
(Amikam and Galperin 2006). This is because of the fact that based on alignment studies,
the PilZ domain is found in the C-terminus of the AcsA protein and in the center of the
AcsAB protein (Consortium 2012). Thus, currently the exact role played by the AcsB
protein is unknown.
The amino acid sequence of the AcsAB protein shows 11 transmembrane
domains (Saxena, Kudlicka et al. 1994). Data from this prediction further confirms the

25
results of the in vitro cellulose synthesis studies described earlier (Bureau and Brown
1987) and proves that AcsAB forms a integral membrane protein. The cytoplasmic
localization of the cellulose synthetic activity and thereby the AcsAB, was also
demonstrated to be localized in the cytoplasmic membrane of A. xylinum cells using
sucrose density gradient centrifugation for separation of membrane fractions and
subsequent assay of the fractions with radiolabelled UDP-(14
C)-glucose as substrate
(Bureau and Brown 1987).
AcsC
The acsC gene codes for a 138kDa polypeptide. GTG in lieu of ATG, is the start
codon in acsC gene and this codon overlaps the termination codon of the acsAB gene.
Though the exact role played by the gene product of the acsC has not been
experimentally proved, its sequence homology to bacterial membrane channels and
porins, suggests that the protein is involved in extrusion of the cellulose chains. Sequence
based-prediction tools also show that it contains an N-terminal signal sequence for OM
localization. Further the sequence reveals that the protein contains seven tetratricopeptide
repeat (TPR, COG4783) motifs, which constitute approximately 20% of the protein, and
a conserved motif for post-translational modification and protein turnover (COG3118)
(Marchler-Bauer, Anderson et al. 2005). The presence of TPR domains in many proteins
is critical for their role in membrane transport and occurs in multiple copies in many
proteins involved in binding with other proteins or ligands (Das, Cohen et al. 1998;
Blatch and Lassle 1999). Hence, the presence of a TPR motif in AcsC may in fact, be
crucial for its role as the probable OM pore for cellulose secretion. This is further
exemplified in the significant homology of this protein to VirB10 from A. tumefaciens

26
(47% similar, 23% identical) and Tra2 region of E. coli protein Trb1 (49% similar, 29%
identical), which are known to interact with other proteins and form pore structures for
secretion of macromolecules (Saxena, Kudlicka et al. 1994). From mutagenesis studies, it
was established that AcsC is required for in vivo cellulose synthesis but not for the in
vitro cellulose production (Saxena, Kudlicka et al. 1994). It is evident that a protein
whose function is to form a pore in the membrane of the cells for extrusion of the
cellulose fibers, would not be necessary when the cellulose is produced under cell-free
conditions.
AcsD
The acsD gene encodes for a 17.3kDa protein whose role in the process of
cellulose synthesis is largely unknown. Saxena et. al. (Saxena, Kudlicka et al. 1994)
characterized this protein using TnphoA-mediated site-directed insertions of kanamycin
Genblock in the acsD gene (AcsD::Km). Unlike the wild type cells, the kanamycin
resistant cells produced vastly reduced amounts of cellulose, under static as well as
agitated culture conditions(Saxena, Kudlicka et al. 1994). Though the cellulose pellicle
produced under static growth conditions by the AcsD::Km cells was very thin compared
to the thick cellulosic mat of the wild-type cells, it was composed of cellulose II
allomorph. Similar to wild-type cells, the cells from the stationary culture of AcsD::Km
showed a linear array of intra-membranous particles (Saxena, Kudlicka et al. 1994).
However, under agitated culture by AcsD-deficient cells was a mixture of both cellulose I
and cellulose II allomorphs, as revealed by X-ray diffraction analysis high-magnification
observation of the product (Saxena, Kudlicka et al. 1994). AcsD protein is also very
unique because it is the only protein encoded by the operon whose crystal and solution

27
structure has been deduced (Hu 2008; Hu, Gao et al. 2010). The localization and structure
of AcsD and its possible role in the crystallization of cellulose ribbons are discussed in
detail in Chapter III and IV of this dissertation.
The acs operon is flanked by genes that modulate cellulose synthesis
Other then the proteins encoded by the acs operon, other proteins are shown to be
involved in the process of cellulose synthesis (Koo, Song et al. 1998; Koo, Song et al.
1998; Kawano, Tajima et al. 2008)The acs operon is flanked 5’ and 3’ ends by genes
encoding an endoglucanase (cmcax) and a β-glucosidase (bglxA) (Standal, Iversen et al.
1994). Surprising though it may seem, expression of these cellulases has been shown to
augment the rate and quantity of cellulose production (Tonouchi, Thara et al. 1995; Koo,
Song et al. 1998).
The cmcax gene upstream of the acs operon encodes for an endoglucanase
belonging to GT family 8. Cmcax is an abbreviation for carboxymethylcellulase from
Acetobacter xylinum. The protein has a molecular weight of 42 kDa and shows an N-
terminal 21 amino acid signal sequence for secretion. Overexpression of this protein, as
well as its addition to the culture medium has shown to enhance cellulose production
after incubation for 3 days, but this effect was not observed if the enzyme was added after
7 days (Kawano, Tajima et al. 2002). Though Acetobacter culture reaches stationary
phase after five days, with cellulose production peaking at the third day, the
endoglucanase expression was found to be elevated after five days of culture (Kawano,
Tajima et al. 2008), and this seems to contradict its role as an enhancer of cellulose
synthesis (Kawano, Tajima et al. 2002). The cellulose hydrolyzing activity of
endoglucanase, but not its ability to bind cellulose, serves to enhance cellulose production

28
(Tonouchi, Thara et al. 1995). It was shown that addition of Cmcax to cultures causes
dispersion of cellulose fibers as shown in TEM images (Tonouchi, Thara et al. 1995).
Haigler et. al. (Haigler 1982) proposed that since the rate-determining step in cellulose
polymerization and crystallization is the assembly of the microfibers, disruption of this
assembly by endoglucanase causes accelerated cellulose synthesis (Haigler 1982).
Presence of a protein in plants, homologous to endoglucanase (KORRIGAN), in close
proximity to the cellulose biosynthetic proteins, (Nicol, His et al. 1998; Robert, Bichet et
al. 2005)further emphasizes the significance of the role of cellulose hydrolyzing activity
in the process of cellulose synthesis.
The bglxA gene downstream of the acs operon encodes for a glucosidase
belonging to GT family-3 (Tajima, Nakajima et al. 2001). It has been suggested that the
-glucosidase in A. xylinum functions to condense glucose units in the media to form a
gentiobiose that serves activate the endogluconase activity in the cultures, which in turn
accentuates cellulose production (Kawano, Tajima et al. 2008). In general, glucosidases
hydrolyze cellobiose and smaller cello-oligosaccharides to produce glucose units and also
catalyze the reverse reaction of addition of residues to cellulose chains. However, many
glucosidases also function as transglycosidases (Kono, Kawano et al. 1999). This implies
that BglxA might serve to maintain steady levels intracellular glucose and cello-
oligosaccharides as substrates for cellulose synthase.
Dgc and PdeA
The Acetobacter genome contains three homologous cdg (cyclic diguanylate)
operons that are each composed of a pdeA gene upstream of the dgc gene (Tal, Wong et
al. 1998). These encode for three orthologous isozymes of Dgcs and Pdes with

29
homologous GGDEF and EAL domain organizations (Tal, Wong et al. 1998). The N-
termini of the Dgc and PdeA proteins also contain oxygen-sensitive domains (Tal, Wong
et al. 1998; Ryan, Fouhy et al. 2006). In addition to this, the cdg1 operon contains
oxygen-regulated transcription activator gene (cdg1a) (Ryan, Fouhy et al. 2006).
Presence of oxygen-sensitive motifs in the proteins which control the turnover of the
activator of cellulose synthesis (c-di-GMP), suggests that oxygen tension plays a crucial
role in regulation of this process (Schmidt, Ryjenkov et al. 2005; Tamayo, Tischler et al.
2005; Ryan, Fouhy et al. 2006). As expected, inactivation of the Dgc gene, leads to
decreased cellulose production (Tal, Wong et al. 1998; Delmer 1999). Cyclic-di-GMP
binds to the PilZ domain of this protein (Amikam and Galperin 2006), which contains the
conserved RxxxR and D/NxSxxG amino acid motifs. Although the PilZ domain functions
as the effector of c-di-GMP, the mechanism of c-di-GMP-dependent regulation is largely
unknown. Structural studies with PilZ domains of Vibrio cholerae and Pseudomonas
aeruginosa have revealed that it undergoes a drastic conformational change upon binding
to c-di-GMP (Benach, Swaminathan et al. 2007; Cotter and Stibitz 2007; Ramelot, Yee et
al. 2007). X-ray crystal structure of the dimeric PilZ bound c-di-GMP, directs us to the
possibility that the molecular surface created upon binding is available for binding to
other target proteins, thereby translating the inter-domain changes induced by ligand-
binding to the downstream regulatory effects (Benach, Swaminathan et al. 2007).

30
The cellulose biosynthetic pathway and mechanism of cellulose synthesis
The steps involved in the pathway of cellulose biosynthesis were deduced using
tracer studies using 14
C-glucose The major steps in the pathway are as follows:
1) Transport of glucose across the cell membrane.
2) Phosphorylation of glucose to glucose-6-phosphate by glucokinase.
3) Isomerization of glucose-6-phosphate to glucose-1-phosphate by
phosphoglucomutase.
4) Conversion of glucose-1-phosphate to UDP-glucose by UDP-glucose
phosphorylase.
5) Polymerization of glucose into cellulose chains by the action of cellulose synthase.
Cellulose synthesis is an irreversible, energy consuming reaction, wherein all the
enzymes except cellulose synthase are shared by other pathways in the cell. Being the
unique enzyme in the cellulose synthesis pathway that catalyzes the committed step,
cellulose synthase activity is the primary candidate for strict regulatory control.
Although the pathways leading to cellulose synthesis have been efficiently identified, the
mechanism of glucose addition to the growing cellulose chain is still shrouded in
mystery. It is known that UDP-glucose binds to the globular region of cellulose synthase,
which is presumed to be cytoplasmic based on inferences from prediction tools
(http://www.cbs.dtu.dk/services/TMHMM/). But some key questions remain unanswered.
Are there more than one binding sites for UDP-glucose in the protein? How does the
inversion of one glucose residue with respect to the other, take place? Is there a primer
involved? How does the allosteric regulator cyclic di-GMP affect binding and catalysis?
Is the polymer elongated from the reducing end or the non-reducing end?

31
Several hypothesis have been formulated to explain the mechanism of glycosyl
residue incorporation into the cellulose chain. Though, identification of the mechanism
underlying the process of cellulose synthesis remains a major challenge in the field, it is
clearly known that the enzyme, cellulose synthase utilizes UDP-glucose and not
cellobiose as the natural substrate (Ross, Mayer et al. 1991). This aspect of cellulose
biosynthesis, wherein the repeating unit of the polymer is not the same as the monomers
that bind to the synthesizing enzyme, is the most intriguing of all questions in the area of
cellulose biogenesis.
Several mechanisms have been proposed to determine the series of events that
account for the formation of β-1,4 linkages, release of the product from the enzyme active
site, and extrusion of the result in polymer. Saxena et al.(Saxena, Brown et al. 1995)
proposed that at the enzyme active site, UDP-glucose molecules bind to two distinct
pockets, such that they are rotated at an angle of 180C with respect to one another and
this dimer is continuously fed to the growing cellulose chain from the reducing end.
Delmer (Delmer 1999) modified this model to one in which the catalytic site is large
enough to allow the rotation of glucose units, thereby facilitating inversion of each
glucose within the binding pocket, during polymerization (Delmer 1999). In an extension
to this model, the catalytic subunits from two enzymes are proposed to be organized to
form a dimerized active site participating in the addition of two glucose residues at a time
(Albersheim, Darvill et al. 1997).
Many workers have proposed the involvement of a lipid-intermediate (Carpita and
Vergara 1998). The lipid portion of this intermediate tethers to the membrane while
glucose residues are added to form a chain of cellulose in the cytoplasm . Once a

32
threshold length of the chain is reached, it is flipped to transport the chains outside the
cells (Cooper and Manley 1975). Evidence for the presence of a lipid-intermediate was
provided by Han and Robyt (Han and Robyt 1998) based on pulse-chase experiment with
14C-labelled UDP-glucose, to reveal that the elongation of cellulose chain occurs from its
reducing ends. According to their model, nucleophilic addition of glucose from a
lipopyrophosphoryl-glucosyl intermediate, to the cellulose chain, which is linked by a α-
linkage to a lipid pyrophosphate, results in a β-1,4 linkage. This model accounts for the
elongation of the cellulose chains from the reducing ends as seen in other bacterial
polysaccharides like O-antigen polysaccharide and cell wall peptidomurein (Han and
Robyt 1998; Han and Robyt 1998)
Glucan polymers like starch and glycogen have a glycoprotein precursor, so it is
logical to assume that cellulose synthesis could also require a protein-linked glycan. This
assumption is supported by a pulse-chase experiment done in Acetobacter whole cells, by
Swissa et. al. (Swissa, Aloni et al. 1980). In spite of several initial evidences into the
nature of the cellulose synthase or the polymer itself, a glycosylated cellulose synthase
has not been isolated and the type of glycosylation is not known. Similarly, the enzymes
involved in the lipopyrophosphorylation of UDP-glucose have not been isolated from
bacterial membranes. Thus, the details of the exact mechanism of processive addition of
glucose units and formation of the cellulose chain are still obscure and subject to
speculation.

33
STATEMENT OF THE PROBLEM
The details of the mechanism of cellulose synthase-catalyzed reaction are largely
unknown and in all probabilities common to both the plant and bacterial systems. Once
the cellulose is synthesized, it serves diverse roles in the two kingdoms. In plants,
cellulose is incorporated as part of the cell wall, whereas in bacteria, it is released outside
the cells as an extracellular polysaccharide. Due the differences in the cell wall
architecture and the site of deposition, another essential inquiry specific to the bacterial
system is, the process of extrusion of the polymer. The lack of understating the
mechanism of synthesis as well extrusion is due to the dearth of information regarding
the structure of the protein components of the cellulose synthetic machinery. There is
considerable evidence that the proteins encoded by the cellulose-synthesis operon are
involved in the synthesis and extrusion of the polymer (Saxena, Kudlicka et al. 1994).
However, there is no experimental evidence to prove their association with one another
to form the cellulose synthesis and extrusion complex. If there is indeed there is a set of
proteins that serve to extrude the synthesized polymer outside the cells, then such a
protein complex is yet to be identified. Biochemical and structural characterization of the
proteins should therefore be the starting point of any further attempts to understand the
whole system of cellulose synthesis.

34
SUMMARY
My work uses the two-pronged approach of characterizing the known proteins
encoded by the operon as well as sequencing the genome to identify other proteins that
contribute towards cellulose synthesis. The major research contributions from my work
on characterizing cellulose synthesis are:
Sequencing the genome of G. hansenii ATCC 23769.
Localization and solution structure determination of the protein involved in
crystallization of bacterial cellulose.
Identification of the in vivo processing of cellulose synthase protein leading to cleavage
into two polypeptides.
Developing a procedure for in-vitro bacterial cellulose synthase assay using zymogram
method.
Heterologous expression and solubilization of the non-membrane bound regions of the
cellulose synthase protein.
Chapter II of this dissertation describes the whole genome sequencing of G. hansenii that
exists as a draft genome in the public domain of the National Center for Biotechnology
Information (NCBI). The genome sequence is made possible because of a combination of
454-titanuium FLX sequencing and SOLid sequencing methods. This is the first fully
sequenced genome of a cellulose producing Acetobacter species. Therefore, it provides a
good reference database for mapping the genomes of other related bacterial species and
strains of Acetobacteriaceae. The sequenced genome was significant for our needs due to
its contribution as the reference database towards our proteomic and mass spectrometric

35
analysis of cellulose synthetic proteins. A concise version of this chapter has been
published in the following journal article:
Iyer PR, Geib SM, Catchmark J, Kao T-H, Tien M. Genome Sequence of a Cellulose-
Producing Bacterium, Gluconacetobacter hansenii ATCC 23769. (2010) Journal of
Bacteriology 192:16, 4256-4257.
In Chapter III, we investigated the localization of the AcsD protein. At the time of
our study, exact role of this protein, its structure and its localization were not known and
could not be inferred from its sequence. Being the smallest protein encoded by the
operon, it was easy to clone and express the gene and purify the protein heterologously.
This facilitated antibody generation against this protein as well as its structural
characterization as described in Chapter IV. The antibody against AcsD was used to
locate the protein in the different cellular compartments. Cytoplasmic membrane (CM),
outer membrane (OM), cytoplasm and periplasm were isolated from the cells and their
purity ascertained using marker enzyme assays. When proteins from these subcellular
fractions were subjected to a western blot using anti-AcsD antibody, it was found that the
protein is localized in the periplasmic region of the cell. Though, a simple experiment,
this work has put the missing piece in the puzzle of secretion machinery in its right place.
A modified form of Chapter III has been published in the journal article:
Iyer PR, Catchmark JM, Brown RM, Tien M. Biochemical localization of a protein
involved in synthesis of Gluconacetobacter hansenii cellulose (2010). Cellulose 18 (3):
739-747.
In Chapter IV, we tried to further characterize the AcsD protein, in terms of its
structure. Around this time, Hu et. al. (Hu 2008) studied and released the crystal structure

36
of this protein. We therefore attempted to solve its solution structure. This work is an
addition to the crystal structure because the proteins is studied under the conditions in
which it exists in the cellular environment. We found using gel filtration, analytical
ultracentrifugation and small angle X-ray scattering (SAXS) that the protein indeed exists
as an octamer in solution, as shown in its crystal structure.
Chapter V of this dissertation describes an important finding with regards to the
processing of the cellulose synthase (AcsAB) protein. Although, encoded by a single
gene, this protein is processed into three parts, as revealed by Western blots using
specific antibodies against different regions of the AcsAB protein. Our attempts at
purifying the polypeptides and the exact location of the cleave have not seen much
success. However, based on the molecular weight, we have proposed the sequence of the
resultant products.
The sixth chapter of this thesis presents a consolidated view of attempts at isolation of the
cellulose synthase complex from the Acetobacter cells. Though not purified to its most
elemental components, we have a partially purified complex of proteins that associate
with one another and can be purified using in-gel methods. Native polyacrylamide gels
were used to obtain a core complex of proteins that retained their activity after detergent
solubilization followed by electrophoresis and chromatography. Using blue native gels,
we have isolated the complex, that contains all the proteins encoded by the cellulose
synthase operon as well as other proteins relevant to the pathway.

37
CHAPTER II
WHOLE GENOME SEQUENCING OF GLUCONACETOBACTER HANSENII
ATCC 23769
INTRODUCTION
The synthesis of cellulose occurs by a polymerization reaction catalyzed by the
enzyme cellulose synthase, which utilizes UDP-glucose as the substrate (Aloni 1983;
Ross, Mayer et al. 1991). Since glucose units are polymerized to form the cellulose
chains, the chemical composition of the cellulose thus produced is the same in all
cellulose-producing organisms (Haworth 1932). However, the morphological properties
of the polymer are unique to each species (Brown, Willison et al. 1976; Herth 1983; Itoh
and Brown 1984) and these differences in crystallinity and dimensions of the polymer are
largely attributed to the great diversity in the pattern and organization of the complexes
that serve as sites of synthesis and extrusion of cellulose (Brown, Willison et al. 1976;
Giddings, Brower et al. 1980; Herth 1983; Itoh and Brown 1984; Tsekos 1999). although
it is known that these complexes harbor the cellulose synthase protein itself (Kudlicka
and Brown 1997; Itoh and Kimura 2001), their other proteins components of these
complexes are yet to be completely identified. It is speculated that in addition to the
cellulose synthase, these complexes contain the proteins that contribute to the process of
cellulose extrusion (Saxena, Kudlicka et al. 1994; Delmer 1999).
Our primary goal is to characterize the process of cellulose synthesis in G.
hansenii and in doing so, we aim to determine the composition and organization and of

38
the cellulose synthesis complex in this bacterium. In our attempts to isolate and
characterize the components of the cellulose synthesis complex using mass spectrometry
(MS) and sequencing techniques, we were hindered by the absence of a completely
sequenced genome. So far, advances in studying the biogenesis of bacterial cellulose
have been restricted to the proteins that are encoded by the acs- operon and its
neighboring genes (Saxena, Lin et al. 1990; Saxena and Brown 1995; Tajima, Nakajima
et al. 2001). Therefore, it is not possible to know whether more proteins other those
encoded by the Acetobacter cellulose synthesis operon (acs) participate and exert an
effect on the process of cellulose synthesis. Public databases (NCBI, EXPASY) are
replete with many versions of cellulose synthesis-related proteins that were discovered at
different time points by different authors (Lin, Brown et al. 1990; Wong, Fear et al. 1990;
Mayer, Ross et al. 1991; Nakai, Moriya et al. 1998). This further complicates the
identification of newer proteins using MS-based techniques. We therefore sequenced the
genome of the cellulose producing species Gluconacetobacter hansenii ATCC 23769, to
explore the genetic blue-print further and unravel more factors contributing to this
process.
The sequenced genome was used for proteomic studies using mass spectrometry.
In this chapter, I will also be describing the results from the MudPIT (Multidimensional
Protein Identification Tool) analysis of the G. hansenii membrane compartments. This
analysis provided a complete proteomic profile of the total membrane (TM), cytoplasmic
membrane (CM) and the outer membrane (OM). We have specifically concentrated on
the comparison of the proteins related to cellulose synthesis in all of these compartments.

39
Figure 2.1 Steps involved in a genome sequencing project. Sequencing the genome of
an organism involves both biological as well as computational aspects. The biological
work consists of isolation of the genomic DNA and fragmenting it into several pieces of
suitable sizes for subsequent amplification to generate several clonal copies of each
fragment (Duan 2010) which are sequenced using pyrosequencing platforms . These
sequences, recorded in a file, are ready for further computational analysis(Duan 2010).
The first step is to align and arrange the random sequences in a proper orientation in
order to reconstruct the original full-length genomic DNA. This is called assembling the
genome. The assembled genome is mapped to that of a neighboring organism, if
available. This step can be skipped if the genome is sequenced de novo (Jarvie and
Harkins 2008; Duan 2010). The sequence is then converted to coherent information
through gene finding (Delcher, Harmon et al. 1999) and annotation . The files generated
from the assembly and annotation are submitted to the databank , where the project
becomes accessible to public.

40
STEPS IN GENOME SEQUENCING
Genome sequencing employs a combination of molecular biology,
instrumentation and computational techniques, to deduce the sequence of the genomic
DNA of an organism (Koonin 2001; Lesk 2007). This process essentially comprises of
sequencing short fragments of genomic DNA, assembling them as one large scaffold,
determining the open reading frames (ORFs), annotating these genes and finally
converting it into a data repository (Duan 2010). This entire process is therefore regarded
as the journey of a DNA sample from a test-tube to database (Lesk 2007). The steps
involved in the process of sequencing are shown in Figure 2.1. Though sequencing as a
technique requires interdisciplinary skills (Lesk 2007), our endeavor in this direction was
motivated by the desire to probe further into the biochemistry of cellulose synthesis.
General outline of sequencing protocol employed in the 454-sequencing platform
The 454-sequencing technology was developed by CuraGen (Andrew 2003) for
high-throughput DNA sequencing at a low cost. Presently owned by Roche , the 454-
sequencing technology employs large-scale pyrosequencing method to sequence large
stretches of DNA at a rapid rate . The latest version of the GS-FLX Titanium platform is
capable of sequencing 600 megabases of DNA within 10 hours . The general workflow
consists of DNA fragmentation , library generation, emulsion PCR , sequencing and
analysis of the sequence (Duan 2010), all which are described in the subsequent section.

41
Preparation of single stranded DNA library
The DNA is sheared into random fragments by nebulization . Pieces that are
between 500-800bp are then selected. After DNA repair, short adaptors are ligated to the
3’ and 5’ ends of the blunt-ended DNA fragments . These adaptors serve as universal
primers for amplification of the DNA fragment as well as sequencing .
Amplification of the library by emulsion PCR
The fragmented DNA ligated to a common primer/adaptor are immobilized on
DNA-capture beads such that each bead carries one unique single stranded DNA
fragment. The DNA fragments amplified by emulsion PCR (emPCR) by emulsifying the
beads in a mixture containing all the reagents required for PCR . Amplification generates
single-molecule replicates of each fragment called “polonies' on the surface of each bead
(Mitra, Shendure et al. 2003), that contains millions of clonal copies of a particular
library . Each clonal library-bound bead is segregated from the other by depositing each
bead in a well of a titanium-coated Pico Titer Plate (PTP) .
Sequencing by synthesis (Pyrosequencing)
The amplified clonal fragments are sequenced by the 454-platform using the
"sequencing by synthesis" method (Nyren 2007). Here, each base incorporated in the
growing DNA strand, is detected from the pyrophosphate released at each step of
incorporation. Hence, the name "pyrosequencing" (Ronaghi, Karamohamed et al. 1996;
Ronaghi, Uhlen et al. 1998). Each well of the PTP with beads containing the clonal
library, is loaded with DNA polymerase, ATP sulphurylase and luciferase (Ronaghi,
Karamohamed et al. 1996; Ronaghi, Uhlen et al. 1998). In each cycle, four nucleoside
triphosphates are added to the reaction sequentially, in a predetermined order. When the

42
complementary nucleotide is linked to the primer, the reaction catalyzed by DNA
polymerase releases a pyrophosphate (PPi). ATP sulphurylase stoichiometrically converts
this released PPi into ATP in presence of adenosine 5’ phosphosulphate .
ATP quantitatively fuels the reaction catalyzed by luciferase that converts luciferin to
light- producing oxyluciferin (Gould and Subramani 1988).
The unincorporated nucleotides and ATP are continuously degraded by the
enzyme apyrase and the system is ready for another nucleotide addition. As the
complementary strand is extended, each new incorporated base is recorded by the charge
coupled device camera (CCD) which records the bioluminescence. The intensity of the
chemiluminescent signal generated is proportional to the number of bases incorporated .
The 454 data analysis software converts the signal intensity after incorporation of
a base in each well, into the type and number of nucleotide. Signals from all the wells are
recorded simultaneously to generate sequences from all of the libraries . These
collections of short fragments (700-800bp) of sequences are called as "reads" . Read
length is the number of contiguous sequences that can be determined in one sequencing
attempt (Lesk 2007). The read length generated after every sequence round, determines
the limits for the subsequenct assembly procedure (Pop and Salzberg 2008). Since the
DNA is initially fragmented to generate short stretches that can be efficiently sequenced
ATP sulphurylase
AMP-SO3H + PPi ATP + SO42-
Luciferase
Luciferin + ATP ADP + Oxyluciferin

43
by the pyrosequecing technology, the reads thus generated have to be re-organized like
pieces from a jigsaw puzzle (Lesk 2007). The presence of several copies of the genes
helps to paste the sequences wherever they are missing, but some regions of the genome
are too GC rich to be sequenced efficiently (Szybalski 1993). Such regions in the
sequence contribute to “gaps” (Lesk 2007). The process of sequencing to close these gaps
is called "finishing" (Lesk 2007). These gaps are either resolved using primer-walking
(Strauss, Kobori et al. 1986; Kaiser, MacKellar et al. 1989) or more rounds of sequencing
(Jarvie 2006) and mapping (Lesk 2007) .
Paired-end library generation
A recommended method for closing the gaps is to complement the sequencing
results obtained from the de novo sequencing with a paired-end library, and sequencing
the fragments using the "sequencing by ligation" principle (Jarvie and Harkins 2008). A
paired-end library is generated by shearing the DNA and selecting for either 3 kb, 8 kb or
20 kb fragments which will determine the distance between the paired-end tags (Jarvie
and Harkins 2008). The clonal amplification steps using emPCR are similar to the ones
previously described (2007). The process of paired-end library generation is explained in
detail in Figure 2.2.

44
Figure 2.2 Generation of DNA library by paired-end method. The genomic DNA is
methylated to prevent EcoR1 mediated cleavage and subjected to fragmentation. The DNA
fragments are ligated on the 3' and 5' ends to biotinylated-hairpin adaptors that contain non-
methylated EcoRI sites and Mme1 site. Exonuclease digestion removes all the DNA species
which do not contain hairpin adaptors. EcoRI-mediated digestion, removes the terminal hairpin
structures of the adaptors and exposes the cohesive ends on either side of the fragments, which
self-ligate. This enables circularization of the DNA fragment around the two remaining portion
of the two adaptors (now called linkers). The 8kb circular DNA contains a 44-mer linker
between the fragmented DNA. The DNA circles are linearized by nebulization into
approximately 250bp fragments. Those fragments that contain the biotinylated linker (Bio) are
selected for, by immobilizing them to a streptavidin bead (SA). These fragments contain a 44bp
linker flanked by 100bp DNA on either side . The two 100 bp fragments were located 8kb apart
in the original DNA sample. These fragments are again linked to longer paired-adaptors that
provide priming regions for subsequent amplification as well as sequencing (Jarvie and Harkins
2008).

45
SOLid sequencing
The sequence of the paired-end library is determined by the "Sequencing by ligation"
method (2007; Pop and Salzberg 2008) . This procedure exploits the property of DNA ligase,
which can repair single-strand cleavage by adding nucleotides complementary to the template
strand and thereby extending one strand of DNA (Lehman 1974). This technology was
commercialized by Applied Biosystems in 2007 and adapted in a platform called “Support
Oligonucleotide Ligation Detection” (SOLid) (Duan 2010).
A paired-end library, is sequenced both from the forward and the reverse directions,
using the DNA ligase and it provides 35 bp reads from either ends of the DNA (Pop and
Salzberg 2008). In this method, the DNA library is immobilized on a bead using the universal
paired-end primer and this serves as the template. A set of eight color-coded di-base (two
nucleotide) probes, compete for ligating to the template strand and extend the sequencing primer.
Each di-base is represented by one of the four colors: blue, red, green or yellow . In the first
round of sequencing all 16 possible the probes are added to the reaction. When the 3' end of the
probe is complementary to the sequence immediately adjacent to the sequencing primer,
annealing occurs. DNA ligase mediates the ligation and the unbound probes are washed away.
The un-extended reactions are prevented from further annealing cycles by selective de-
phosphorylation. The probes are cleaved with silver nitrate to remove the fluorescent tag and the
last three bases. The ligation reaction is repeated to obtain another 5-mer that contains two
nucleotides at 3' end, that are complementary to the template. A round of 15 cycles generates a
75 bp sequence, after which the extended sequences are melted off the template and another
primer is hybridized, such that it is one base closer to the bead. The same pool of probes is used
again to determine the sequence. The process of resetting the primer and sequencing is repeated

46
five times. This ensures that each nucleotide in the sequence is probed twice and therefore the
chance of sequencing errors is minimized. For sequencing from the reverse side, a 3'
hydroxylated primer is ligated to the adaptor of the template and 5' phosphorylated probes are
annealed to the primer. Subsequent steps in sequencing are performed similar to that of the
forward template .
The result is displayed as a color-space sequence of di-bases. Since the adaptor sequence
is known, the 1st base pair of the probe, annealed to the primer can be inferred. From here the
entire color-coded sequence can be converted to a nucleotide sequence. The di-base system
serves as a built-in accuracy check for the sequencing. This method, unlike the pyrosequencing
technique, is not hampered by homopolymer repeat regions .
ANALYSIS OF SEQUENCE DATA USING SOFTWARE TOOLS
Assembly
Sequence assembly involves aligning and merging the reads generated by the sequencer in order
to reconstruct the original sequence. If we look at the entire genome as a book then, these reads
are likes shreds of all the pages from that book. The process of assembly is analogous to taking
these shreds of several copies of a book and trying to paste the pieces of paper back together to
regenerate one copy of the original book.
Generation of contigs
The assembler developed by Roche, to merge the reads generated by the 454-
sequencing platform is called as GS (Genome Sequencer) assembler or Newbler (New
Assembler) . Newbler can assemble reads generated by a single or multiple platforms .
When a genome is sequenced by both 454 and paired-end methods, the assembler
identifies reads as either linker-positive (454-pyrosequencing) or linker-negative (paired-

47
end, SOLid sequencing) (2010). As a first step, the linker-negative reads are assembled
into a de novo shotgun assembly (Staden 1979). These are catalogued into contigs which
are long and contiguous sequences of DNA obtained by aligning and merging shorter
reads, by identifying the best sequence overlaps between them (Jarvie 2006; Jarvie and
Harkins 2008). The contigs can be filtered on the basis of minimum sequence length
desired, thereby eliminating very short and redundant sequences. The contigs generated
by merging overlapping regions of DNA, result in sequences that are aligned without any
regard to their order or position (Jarvie 2006; 2010).
Scaffolds
Although shotgun sequencing (Staden 1979) affords superiority in terms of longer
read lengths (2007), it is through the paired-end library reads that the contigs can be
properly ordered and oriented into larger concatenated assemblies, called scaffolds
(Jarvie and Harkins 2008). This is done in the second round of assembly, where the
assembler considers the linker-positive reads and identifies matching regions between the
reads and the known contigs, using pair-wise alignment. When the end-tags of a paired-
end read map uniquely to two different contigs, then these contigs are linked into a
scaffold, if the known distance between the paired-ends (8kb) is reflected in the sequence
contained in the two contigs (Jarvie and Harkins 2008) .
The assembly by Newbler thus produces two levels of sequence data
(Jarvie 2006). Scaffolds are larger stretches of sequences, where the contigs are oriented
and ordered based on the uniquely mapped paired-end halves (2010). Based on the size of
the insert generated when both the halves of a paired-end map to the same contig, the
insert size is determined and this is used to derive the distance between adjacent contigs

48
(2010). This information is essential to find out if the gap is real or an artifact of
sequencing. The gaps between contigs are represented by a series of Ns in the sequence
(2010).
If the gaps in the assembly are unresolved even after multiple approaches and
rounds of shotgun sequencing, then such gaps are filled by primer walking (Strauss,
Kobori et al. 1986), a procedure in which primers are designed to the nucleotide
sequences flanking the gap, and the DNA is amplified by PCR (Strauss, Kobori et al.
1986). The amplified portion is sequenced and manually aligned to the gap region using
an alignment software (Tamura, Dudley et al. 2007). If the gap is not completely closed,
the process of primer-design for the newly generated ends and amplification is continued
until the complete sequence length covering the gap, is obtained (Lesk 2007). Based on
the final application of the sequenced genome, some persistent and difficult-to-sequence
regions are left behind as gaps. For most biological applications, a genome that is 90%
gapless, is considered to be sufficient for further inquiry (Lesk 2007).
Gene prediction and annotation
Once the genome has been assembled into consenses contigs or scaffolds, it is
ready for gene predicitions and annotation (Lesk 2007; Aziz, Bartels et al. 2008; Pop and
Salzberg 2008). Annotation of a sequence catalogues the sequences and gives access to
the information contained in it, which would otherwise be just a long linear list of
nucleotide bases. The method of predicting a gene involves using a program like
GLIMMER (Gene Locator and Interpolated Markov Modeler) to scan the sequence and
identify ORFs based on predictions from a variable Markov model algorithm (Delcher,
Harmon et al. 1999). All possible reading frames in both strands are scanned individually.

49
Those candidate gene sets that score beyond the threshold set by the algorithm are
predicted as coding sequences (Salzberg, Delcher et al. 1998; Delcher, Harmon et al.
1999).
MATERIALS AND METHODS
Isolation of genomic DNA
The genetic material of G. hansenii was isolated using the Genomic DNA
purification kit A1120, Promega, Wisconsin). The procedure given in the instruction
manual accompanying the kit (2010) was modified slightly to accommodate the culture
duration and conditions for G. hansenii. Briefly, the bacterial cell culture was started by
inoculating 5 ml of Schramm-Henstrin (SH) medium with cells from a frozen glycerol
stock. After 1 day of growth, the culture was supplemented with 0.1% (w/v) cellulase
(from Aspergillus niger, Sigma-Aldrich). After 72 hours of growth, the cell pellet was
obtained by centrifugation at 16,000 × g for 2 minutes and was re-suspended in 600 µl of
"nuclei lysis solution" and incubated at 80°C for 5 minutes. The solution was cooled to
room temperature and 3µl of ribonuclease was added to the suspension and incubated at
37°C for 30 minutes. The sample was again brought to room temperature and 200 µl of
“protein precipitation solution” was mixed with it by vortexing at high-speed for 20
seconds. After incubation on ice for 5 minutes, the sample was centrifuged at 16,000 × g
for 3 minutes. The supernatant containing the DNA was transferred to a microcentrifuge
tube containing 600 µl of isopropanol. Upon gentle mixing, thread-like strands of DNA
become visible. This DNA was recovered by centrifugation at 16,000 × g for 2 minutes.
The supernatant was decanted and 600 µl of 70% ethanol was added to the DNA pellet to
wash it by gently inverting the tube several times. The residual ethanol was removed after

50
centrifugation. The DNA pellet was allowed to air-dry for 15 minutes. DNA rehydration
solution (100 µl) was added to the tube and incubated at 65°C for 1 hour. The rehydrated
DNA was stored at 4°C.
The concentration of the isolated DNA was determined by reading the absorbance
at 260 nm, of 1µl of the sample using a Nanodrop spectrophotometer. The samples
submitted for the de novo 454-sequencing and for paired-end sequencing contained 25 μg
of DNA, and 22 μg of DNA respectively, as measured by Nanodrop spectrophotometer .
Lane 1 Lane 2
20 kb
10 kb
Lane 1 Lane 2
20 kb
10 kb
Figure 2.3 Agarose gel electrophoresis of genomic DNA. 5µl of the genomic DNA
isolated from the G. hansenii cells was analyzed on a 1% agarose gel. The genomic DNA
can be seen as a clear band (indicated by an arrow, Lane 1) that migrates above the 20kb
band in the “Fermentas Gene-ruler 1kb (SM#0333)” molecular weight ladder (Lane 2).
The large molecular weight of the isolated DNA and the absence of smaller bands
indicated that the DNA isolation procedure yielded a high-quality, non-fragmented
genomic DNA sample suitable for the purposes of sequencing.

51
Processing and analysis of the sequenced data using software tools
The sequence data output from the 454-sequencer was provided in the form of a
.sff (Standard Flowgram Format) file, which contained light information on the intensity
of signal for each read. The sequence contained in this file were assembled using the GS
assembler, Newbler. Newbler was run in a 64-bit version in the Linux operating system,
using a command-line interface. The command to access a file and assembles its reads is:
“runAssembly aceto.sff”. The output directory created was given a name automatically
by the software which shows the time and date of the assembly performed. For instance,
the assembly folder generated after combined assembly of the 454- and paired-end reads,
was called P_2010_03_01_09_04_14_runAssembly”. This directory contained the
following sub directories. Containing the following files after completion of assembly:
Newblermetrics.txt: Each Newbler metrics file provides information about the number
of scaffolds, and the number of contigs.
Contigs.fna: Fasta file of all reads
Contigs.qual: quality scores of bases within a contig
Scaffolds,fna: fasta file of all contigs within a scaffold. Gaps represented by a series of
Ns
Scaffolds.qual: quality file of all contigs in a scaffold.
The 454 reads and paired-end reads were assembled individually, in addition to
assembling the two reads together. The contigs and the scaffolds generated by these

52
assemblies were carefully analyzed. The largest contigs and scaffold was identified using
the Newbler metrics file. Since the largest scaffold was as large as the bacterial genome
size, the gaps (Ns), in this scaffold were examined. The Newbler assembler, often adds a
gap of 21 nucleotides and these were located and, being artifacts of the assembly
process, were deleted. The remaining large and small gaps were closed by PCR.
Primer walking: Primers were designed for sequences flanking the gap regions. PCR
was performed according to standard procedures using HF Phusion Taq polymerase
(Thermo Scientific) PCR reagents. The PCR product from each reaction was subjected to
agarose gel electrophoresis and for verifying the size of the amplified product. Along
with 5 µl specific primers, 5 µl of PCR products were sent to the "Nucleic acid
Sequencing Center", in University Park, PSU.
The sequences obtained were aligned with the contigs using MEGA 4.1 (Beta)
(Tamura, Dudley et al. 2007) and the gaps were closed by substituting the "Ns" with the
sequenced region. Since some gaps were very large, several rounds of primer design,
PCR and sequencing were done to fill in the nucleotides that were missing. The gaps that
were persistent and could not be closed in spite of several rounds of PCR were left in the
sequence as a series of Ns. The fasta file of the largest scaffold, in which several gaps had
been replaced with meaningful sequences, was now considered as a high quality draft
assembly (Chain, Grafham et al. 2009) and this was used as the dataset for subsequent
steps of gene prediction and annotation.
Annotation: The high quality draft assembly was annotated using multiple platforms.
Firstly, we used the Glimmer software tool for gene-predictions (Delcher, Harmon et al.
1999), in order to obtain a private genome repository for analyzing our in-house

53
proteomic data. The assembled genome was also uploaded and analyzed using the Rapid
Annotation Service Tool (RAST) (Aziz, Bartels et al. 2008), for obtaining a genome
viewer to study the operon structure and features of the regions flanking the operon and
to enable comparative analysis of the genome and the genes with those of the related
species. However, in order to generate a public genome database, we submitted all the
files in the format described by NCBI to the NCBI-owned, Prokaryotic Genomes
Automatic Annotation Pipeline (PGAAP) pipeline .
Sample preparation for MudPIT analysis
Membrane fractions were obtained as described in the Materials and Methods
section of chapter III. A total of 1 mg of protein from each membrane compartment was
subjected to in-solution digestion, using the methods provided by the "Proteomics Core
facility", Hershey, PSU. Briefly, Protein concentrations of the samples from each
membrane compartment were determined using the Bradford method (Bradford 1976).
The volume of sample from each compartment, containing 1 mg of protein, was
subjected precipitation by mixing with 100% (w/v) trichloroacetic acid to the final
concentration of 30% (v/v) and freezing the sample. After thawing, these samples were
centrifuged at 10,000 x g for 25 minutes to obtain the proteins in the pellet. The protein
pellet was washed in 80 % ice-sold acetone and allowed to dry in the fume-hood. The
pellet was resuspended in 100 µl of 100 mM Tris-Cl buffer, pH 7.8, containing 6 M Urea.
To this suspension, 5 µl of reducing agent (30 mg dithiothreitol in 100 mM Tris- Cl, pH
7.8) was added to obtain a final concentration of 10 mM DTT. The reduction was
allowed to proceed for 1 h. The reduced protein sample was alkylated by adding 20 µl of
alkylating reagent (36 mg of iodoacetamide in 100 mM Tris-Cl buffer, pH 7.8), to obtain

54
a final concentration of 10 mM iodoacetamide. The protein sample was alkylated for 1 h
at room temperature. The sample was again reduced as before, to remove any
unconsumed iodoacetamide. To finally digest the protein, 100 µl of Trypsin is added to
the sample from a stock of 20 ng/ µl. The sample is allowed to incubate for 16 hours at
37°C. The digested sample was dried in a vacuum concentrator and reconstituted in 100
µl distilled water. The drying and reconstitution was repeated three times and the sample
is sent for MudPIT analysis to the Proteomics Core facility, Hershey, PSU. This strategy
of multidimensional chromatography involves, use of biphasic column made of
polysulfoethyl sspartamide, which is packed at its distal end with a reverse-phased resin,
such as C18 resin. The proximal end of the column is packed with a strong-cation
exchange (SCX) resin. The digested peptide mixtures are introduced onto the SCX resin
at the rate of 1ml/ min, and fractions are eluted with a stepped-salt gradient. The elutions
from the SCX flow into the C18-column, from where the fractions are eluted into the
mass spectrometer, by applying a gradient of acetonitrile. After regeneration of the
reverse phase resin, another fraction is released from the SCX-resin using an increased
salt gradient. This cycle is repeated until the SCX-resin is exhausted. The elutions from
the C18-column are mixed with a flow of MALDI matrix solution and spotted onto a
stainless steel MALDI target plate. MALDI target plates (15 per experiment) were
analyzed in a data-dependent manner on an ABI 4800 MALDI TOF-TOF. After
acquisition of MS/MS spectra for all the spots in all the plates, protein identification and
quantitation were performed using the Paragon algorithm as implemented in Protein Pilot
3.0 software (version 2.01), from ABI/MDS-Sciex. All the identified peptides were
enlisted in an excel file, only identifications with a ProteinPilot Unused Score of > 1.3

55
(>95% confidence interval) were accepted. The MudPIT analysis and subsequent MS/MS
analysis was performed by Anne Stanley, Proteomics Core Facility, Hershey, PSU).
RESULTS
Assembly metrics
A combinatorial sequencing approach generated 489,201 reads containing
162,0766,26 bp from the shotgun library and 195,088 reads containing 592,174,90 bp
from 8-kb paired-end library. Together these reads contained a total of 221,294,116 bp.
These reads were assembled using the Newbler assembler, producing 88 large contigs
(>500 bp) and a chromosome-sized scaffold of 3,646,142 bp with an average coverage of
50.5X. This scaffold contained exclusively chromosomal DNA and no plasmid
sequences.
Finishing, annotation and databank entry
The gaps in the large scaffold were filled by primer-walking and subsequent
sequencing of the PCR products. The resulting high-quality draft assembly, consisting of
a large scaffold with 71 contigs, was annotated using the Prokaryotic Genomes
Automatic Annotation Pipeline (PGAAP) service of the National Institute of
Biotechnology Information (NCBI). The gene predictions in the pipeline were made
using Genemark and Glimmer (Salzberg, Delcher et al. 1998; Delcher, Harmon et al.
1999). The genome can be accessed from NCBI. It has been given the accession number
PRJNA43711. The genome viewer can also be accessed from the RAST database (Aziz,
Bartels et al. 2008).

56
Genome features
The chromosomal sequence of G. hansenii 23769 contains 3,547,122 bp, with a G
+ C content of 59%. The genome contains a total of 3,351 genes, of which 3,308 are
protein-encoding genes, which account for 84% of the genome. All the genes contain the
prefix "GXY". There are 43 genes for transfer RNAs and 2 ribosomal RNA loci.
Genome features were also analyzed using the RAST subsystem technology
version 4.0 (Aziz, Bartels et al. 2008), which reconstructs metabolic networks and the
resulting annotation data is viewable through the SEED-viewer. Based on the RAST
analysis, the genome contains 14 genes encoding enzymes involved in fatty acid
synthesis, 17 heme and siroheme biosynthesis related coding sequences and bacterial
chemotaxis associated cheA, cheB and cheR genes (Aziz, Bartels et al. 2008). All major
subsystems are depicted in the pie graph in Figure 2.4.
Features relevant to cellulose synthesis
The genome contains the genes encoding proteins involved in cellulose synthesis
in an operon consisting of acsAB (GXY_04277), acsC (GXY_04282), and acsD
(GXY_04292), as previously shown by Saxena et. al. (Saxena, Kudlicka et al. 1994) and
Wong et. al. (Wong, Fear et al. 1990). Interestingly, there are two additional copies of
acsAB, GXY_08864 and GXY_14452 which share 40% and 46% identity, respectively,
with the acsAB in the operon. Three sets of the acsAB and AcsC protein were reported
for the strain ATCC 7664 (Umeda, Hirano et al. 1998), but for the strain used in the
present study (ATCC 23769), only two of these three sets of acsAB genes have been
reported (Saxena and Brown 1995). There are two acsC copies, GXY_08869 and
GXY_014472, in the genome, which have 28% and 30% identity to the acsC gene in the

57
operon. Each copy of acsAB is adjacent to a copy of acsC. The distance between acsAB,
GXY_08864 and acsC, GXY_08869 is 17bp. acsAB GXY_14452 and acsC GXY_14472
are separated by 3299 base pairs. However, acsD is only present in the operon and is not
duplicated elsewhere in the genome. The genome also contains three copies of the
diguanylate cyclase genes, as reported by Tal et. al. (Tal, Wong et al. 1998) at loci GXY_
01169, GXY_016414 and GXY_01393.
Proteomic analysis of the membrane compartments
In addition to identifying unique features of the genome, we have utilized this
genome for proteomic studies of the G. hansenii membrane compartments. MudPIT is a
specialized form of LC/MS analysis in which is suited for the analysis of membrane
proteins. The MudPIT analysis of the TM, OM and CM compartment generated
enormous amount of proteomic data. The
All the files containing the raw data for peptides as well as the proteins, along with
important prameters like coverage, confidence and scores in the supplementary files are
contained in the storage disk provided with the dissertation (MudPIT/ TM/
TM_protein.xls, MudPIT/ TM/ TM_peptide.xls, MudPIT/ TM/ TM_protein.xls, MudPIT/
CM/ CM_peptide.xls, MudPIT/ OM/ OM_protein.xls, MudPIT/ OM/ OM_peptide.xls).
The MudPIT results were analyzed only in the context of the proteins that are
involved in the cellulose synthesis pathway. Table 2.1 lists the all the cellulose-
biosynthetic proteins in the TM, CM and OM fractions of these cells. Among the Acs
proteins, AcsC is seen in the OM compartment and only one unique peptide
corresponding AcsD is seen in the CM and the OM compartment. TM contains four
unique peptides of AcsD. Our results in chapter III identify the localization of AcsD In
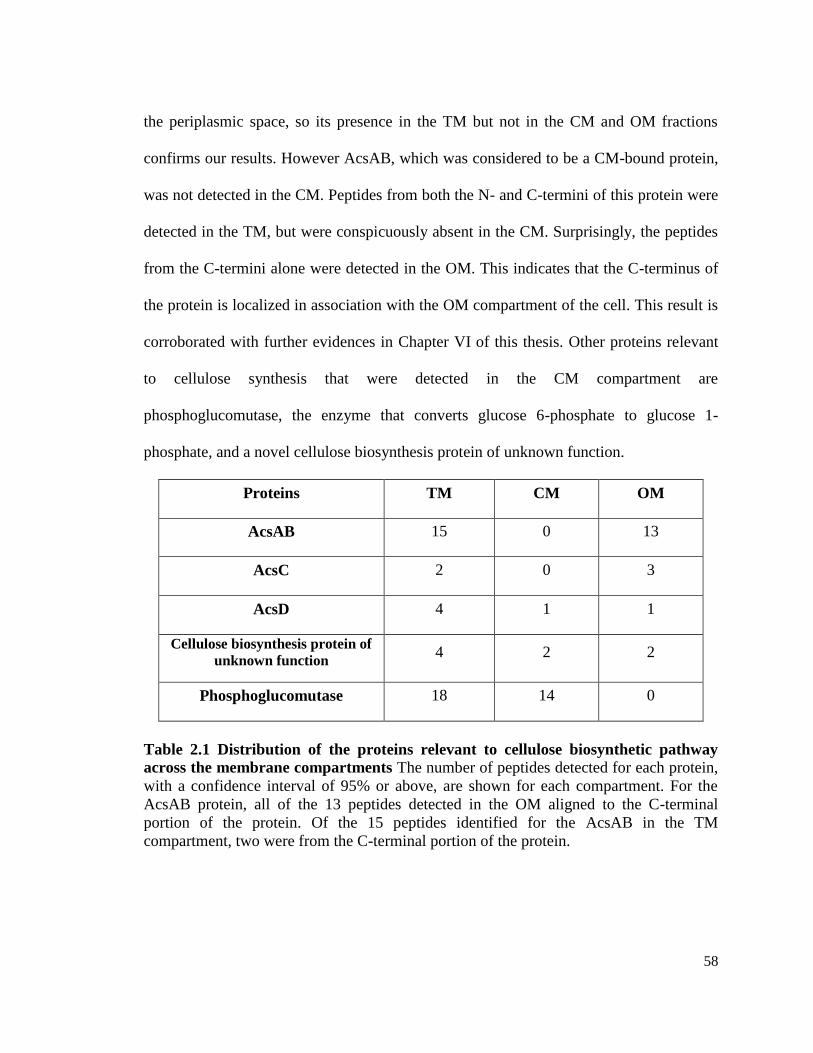
58
the periplasmic space, so its presence in the TM but not in the CM and OM fractions
confirms our results. However AcsAB, which was considered to be a CM-bound protein,
was not detected in the CM. Peptides from both the N- and C-termini of this protein were
detected in the TM, but were conspicuously absent in the CM. Surprisingly, the peptides
from the C-termini alone were detected in the OM. This indicates that the C-terminus of
the protein is localized in association with the OM compartment of the cell. This result is
corroborated with further evidences in Chapter VI of this thesis. Other proteins relevant
to cellulose synthesis that were detected in the CM compartment are
phosphoglucomutase, the enzyme that converts glucose 6-phosphate to glucose 1-
phosphate, and a novel cellulose biosynthesis protein of unknown function.
Proteins TM CM OM
AcsAB 15 0 13
AcsC 2 0 3
AcsD 4 1 1
Cellulose biosynthesis protein of
unknown function 4 2 2
Phosphoglucomutase 18 14 0
Table 2.1 Distribution of the proteins relevant to cellulose biosynthetic pathway
across the membrane compartments The number of peptides detected for each protein,
with a confidence interval of 95% or above, are shown for each compartment. For the
AcsAB protein, all of the 13 peptides detected in the OM aligned to the C-terminal
portion of the protein. Of the 15 peptides identified for the AcsAB in the TM
compartment, two were from the C-terminal portion of the protein.

59
Fig
ure
2.4
Su
bsy
stem
cata
logu
e of
the
gen
es
The
gen
es e
nco
ded
by G
. hanse
nii
can
be
clas
sifi
ed b
ased
on v
ario
us
crit
eria
. T
he
pie
dia
gra
m d
istr
ibute
s th
e gen
es b
ased
on
thei
r ro
les
in t
he
cell
ula
r m
etab
oli
sm.
This
cla
ssif
icat
ion i
s bas
ed o
n t
he
assu
mp
tion
that
all
the
enco
ded
gen
es a
re e
xp
ress
ed a
nd a
bio
chem
ical
dem
onst
rati
on o
f th
e p
rote
in f
un
ctio
n i
s re
quir
ed t
o c
onfi
rm t
he
exis
tence
of
the
met
aboli
c pat
hw
ays.

60
DISCUSSION
Characterization of any biochemical process requires the complete knowledge of
not just the proteins participating in the pathway but also the other factors that regulate,
interact and contribute to the process indirectly. Biochemical techniques like
electrophoresis, cross-linking and chromatography can be used to isolate the proteins that
interact with the operon-encoded cellulose synthase and related proteins. However, in
order to identify these isolated proteins, we need to use tools like mass spectrometry and
protein sequencing. These techniques in turn need a database of sequences to serve as a
reference. Thus, to identify all the components of the cellulose secretion machinery, it
was important to have a database of a sequenced genome.
The relevance of a sequenced genome is that, it allows us to look at all the factors
influencing the cellulose-producing lifestyle of Acetobacter, instead of myopically
concentrating on only the proteins that are encoded by the acs operon. We have used the
sequenced genome as a reference database and determined the proteomic profile of the
membrane compartments of this bacterium by MudPIT (Multidimensional Protein
Identification Tool) analysis. Although, a large amount of proteomic data has been
generated using MudPIT, we have solely searched for the cellulose-synthesis related
proteins and found some insightful results. The assumption so far in the field of cellulose
synthesis is that the AcsAB protein is an integral membrane protein (Bureau and Brown
1987), localized in the CM of the bacterial cells. This was based on the detection of
cellulose biosynthetic activity in the CM of the A. xylinum cells. We found that all the
peptides corresponding to cellulose synthase were identified in the OM fraction. These
peptides, when aligned to the whole-length protein of 1550 amino acid residues, match to

61
the N-terminal end of the protein. None of these peptides match to the catalytic domain of
the protein. This finding is relevant because, we have presented evidence for the
association of the N-terminal end of the cellulose synthase protein to the OM
compartment. This result will be further discussed in Chapter V. AcsC protein is
localized in the OM, as expected from its N-terminal signal sequence for translocation to
the OM. The peptides corresponding to AcsC are absent in the analysis of CM fractions.
In addition to the Acs proteins, phosphoglucomutase, a cytoplasmic enzyme involved in
the UDP-glucose synthesis is found associated with the TM and CM compartments. Our
results direct us towards the distribution and organization of the proteins involved in the
cellulose synthesis complex. The organization of this complex was further studied and is
described in the subsequenct chapters.
The proteomic data obtained for the membrane compartments, can be analyzed
further, with respect to other pathways in the bacterial cells. Other than proteomic studies
this sequenced genome is also being used for identifying protein-protein interactions
contributing to cellulose synthesis using Yeast-two-hybrid system (Fields and Song 1989;
Iyer, Burkle et al. 2005) and studies involving random mutagenesis that influence
cellulose production (both are unpublished work, by Deng, Y. and Kao, T-H ). The
results from MudPIT and other mass spectrometric analysis for identifying the proteins
components of the cellulose synthase complex, are elaborated in the Chapter VI.
In the wake of the genomic era, an additional tool to understand the significance
and evolution of cellulose biosynthetic ability, is through phylogenetic analysis. In a
recent search for the phylogenetic origin of horizontally transferred genes in plants, 15
genes were found to be common between Bacteria and Plantae (Price, Chan et al. 2012).

62
It was shown that the plants acquired the genes for thiamine-pyrophosphate-dependent
pyruvate decarboxylase family protein, through a horizontal gene transfer event from
species of Proteobacteria. Among these bacterial species were G. hansenii 23769 and the
cellulose non-producing G. diazotrophicus, which in an ancient HGT event, has
contributed to the acquisition of the thiamine pyrophosphate dependent pyruvate
decarboxylase gene, by the plant genome. This protein is involved in an alcohol
fermentation pathway (Price, Chan et al. 2012). The maximum likelihood phylogenetic
tree included sequences from the G. hansenii genome as well of as those of G.
diazotrophicus, and A. pasteurensis. Thus, in this study, the genome sequence has
contributed towards attempts at understanding the evolution of the plant photosynthetic
machinery.
CONCLUSIONS
We have sequenced the genome of G. hansenii 23769. this genome is can be
accessed using the accession number PRJNA43711, in NCBI. Using this genome, we
have obtained the proteomic profile of the membrane compartments of this bacterium.
We have identified that although peptides from the full-length AcsAB protein were
detected in the MS-analysis of the TM, the C-terminus of the AcsAB protein is associated
with only the OM compartment. The N-terminus of this protein was not identified as in
either the CM or OM. AcsC is present in the OM as expected and AcsD tends to associate
largely with the OM.

63
CHAPTER III
LOCALIZATION OF THE ACSD PROTEIN IN THE PERPIPLASM OF
G. HANSENII CELLS
INTRODUCTION
The acs operon has been shown to encode for the proteins which participate in the
process of cellulose synthesis (Wong, Fear et al. 1990). The roles of acs-operon encoded
proteins have been determined by using site-directed mutagenesis (Saxena, Kudlicka et
al. 1994) and inferred by sequence analysis (Saxena, Lin et al. 1990; Saxena, Brown et al.
2001) and limited biochemical characterization (Bureau and Brown 1987; Lin, Brown et
al. 1990; Chen and Brown 1996). As mentioned in Chapter I of this dissertation, AcsAB
harbors the active site for glucose polymerization and is localized in the cytoplasmic
membrane (CM) (Bureau and Brown 1987; Lin, Brown et al. 1990). Even though the role
of AcsC has not been proven experimentally, the sequence shows a 21 amino N-terminal
signal for outer membrane (OM) localization (Gattiker, Michoud et al. 2003). The
presence of several tetratricopeptide repeat domains (Gattiker, Michoud et al. 2003) and
the homology of AcsC to several bacterial porins (Saxena, Kudlicka et al. 1994) suggests
that it serves as the pore in the outer membrane (OM) of G. hansenii cells through which
cellulose is secreted (Saxena, Kudlicka et al. 1994).
Using mutants developed by TnphoA-mediated site-directed insertions, both
AcsA and AcsC proteins were found to be required for cellulose synthesis in vivo
(Saxena, Kudlicka et al. 1994). Upon disruption of the AcsD gene by insertional
mutagenesis, the mutant produced has a partial Cel- phenotype on agar plates and
produces 40% less cellulose compared to the wild-type cells (Saxena, Kudlicka et al.

64
1994). Also, the cellulose produced was composed of both the cellulose I and cellulose II
allomorphs (Saxena, Kudlicka et al. 1994). It was therefore inferred that AcsD is required
for maximal cellulose production and influences the crystalline nature of the cellulose
produced (Saxena, Kudlicka et al. 1994). Since in vitro cellulose synthesis was
unhampered by the absence of AcsD, it could be gathered that this protein was not an
essential requirement for cellulose biogenesis but was involved in the structural assembly
of the final product. This lead to the speculation that this protein could be either localized
in extracellular compartment, tethered to the OM or in the periplasmic space (Endler,
Sanchez-Rodriguez et al. 2010). In both these scenarios, the protein is capable of acting
on the cellulosic chains emerging from the CM-bound cellulose synthase. However,
unlike AcsAB or AcsC, the sub-cellular localization of AcsD has neither been proved
experimentally nor can be inferred from its sequence. Though the structure of AcsD has
been recently elucidated by X-ray diffraction pattern (Hu 2008), absence of information
about the localization of the protein makes it difficult to understand its interactions with
the other proteins encoded by the operon, as well as the precise mechanism by which the
protein contributes to the process of cellulose extrusion.
In this study, sub-cellular fractions of G. hansenii cells were probed using Western
Blot with antibodies developed against AcsD. We found that AcsD resides in the
periplasmic region of the bacterial cells. If indeed the acs operon-encoded proteins form a
complex that mediates the synthesis and secretion of the polymer, then knowledge of the
organization of the protein components of this complex and their interaction, is crucial to
understanding the mechanism of cellulose synthesis and extrusion. Therefore, elucidating
the sub-cellular localization of AcsD is a step towards speculating on the model of a

65
cellulose biosynthesis system.
MATERIALS AND METHODS
Materials
Ni–NTA resin was purchased from Qiagen (Cat#301210). p-nitrophenyl phosphate
(N1127) and L-malate were products of Sigma–Aldrich. PCR primers were ordered from
Integrated DNA technologies.
Bacterial strains and culture conditions
G. hansenii ATCC 23769 cells were cultured in Schramm-Hestrin medium (SH)
(Schramm and Hestrin 1954) at 30°C in a rotary shaker. All cellular incubations were
performed in SH media in the presence of 0.1% (w/v) cellulase (Trichoderma reesei
cellulase, Sigma–Aldrich).
AcsD cloning, expression and protein purification
Single colony PCR (Woodman 2008) was used for amplification of AcsD gene
using primers (5'- ggatccacaatttttgagaaa -3' and 5'-ctcgagggtcgcggaact-3'). The
primers encode for restriction sites BamH1 and Xho1 respectively. The PCR product was
analyzed in a 1% agarose gel (Figure 3.1a) from which the band corresponding to the
471bp fragment was provided. The gene was ligated into pGEM-T vector and was
transformed into JM109 cells. Transformation cultures were plated onto LB (Luria–
Bertani) plates supplemented with 50 µg/mL ampicillin (LB-Amp plate) and 0.5mM
IPTG and 80 lg/mL X-gal. White colonies from the plate were picked and used for
plasmid isolation. The plasmid was purified using the Qiagen plasmid mini-prep
procedure. Presence of the restriction sites for BamH1 and Xho1 enabled digestion of
pGEM-T-ligated gene (Figure 3.1b) as well as the pET-21a vector, with which the gene

66
was subsequently ligated. The ligated gene was transformed in competent BL-21 DE3
cells. Ligations and transformations were performed using the procedure given in the
pGEM vector manual (pGEM-T and pGEM-T Easy vector systems, Technical manual
No.042, Promega). The transformants were identified by plating 100µl of the
transformation culture on an LB-ampicillin plate. The presence of the AcsD gene insert in
pET-21a was verified by sequencing the ligated vector using specific primers designed
for amplification.
a b
acsD gene
471 bp506 bp
1 2
acsD gene
471 bp506 bp
1 2
acsD gene
471 bp506 bp
1 21 2 3
pGEM
(cloning
vector)
acsD
(gene
insert)
1kb
500
bp
1 2 3
pGEM
(cloning
vector)
acsD
(gene
insert)
1 2 3
pGEM
(cloning
vector)
acsD
(gene
insert)
1kb
500
bp
1kb
500
bp
Figure 3.1 Agarose gel electrophoresis of PCR product and pGEM ligated AcsD
a) Amplification of AcsD gene from A.xylinum cells: The gene was obtained from A.
xylinum culture by colony PCR. Lane 1 shows the PCR product and lane 2 shows the
molecular weight ladder.
b) Cloning of the acsD gene: The amplified PCR product was cloned into pGEM-T
vector and restriction digested by BamH1 and EcoR1 enzymes. b) The restriction
digested gene and pGEM vector are observed as bands between 400-500 bp and 3 kb
respectively in Lane 1. The digested product was ligated into pET-21a and transformed
into BL-21 (DE3). Lane 2 and 3 contain the molecular weight standards.

67
Protein expression and purification
The AcsD protein was heterologously expressed and purified using standard
procedures. Briefly, a single colony was picked from the LB-Ampicillin plate, used for
identification of transformants, and inoculated in 5 mL LB medium supplemented with
2.5µl of 100 mg/mL ampicillin. The culture was incubated at 37°C for 16 h, with
vigorous shaking at 200 rpm. This overnight grown culture was used as the inoculum for
1 L of LB medium supplemented with 500µl of ampicillin, and was again incubated
under similar condition as before. The absorbance of the culture was monitored at 30 min
intervals until an absorbance at 600 nm reached 0.6. At this stage, IPTG was added to a
final concentration of 1mM and the culture was allowed to continue for an additional 4h.
Cells were harvested by centrifugation at 1,500 x g for 30 min and the cell pellet was
frozen overnight at -20°C. The frozen cell pellet was thawed, resuspended in lysis buffer
composed of 50mM NaH2PO4 pH 8.0 and 300mM NaCl and sonicated for 5 min with 15
s pulses. The lysate was centrifuged at 2,300 x g for 30min to obtain the protein in the
supernatant. Protein purification was performed as per the instructions in the Qiagen
handbook for Ni-NTA columns (The QiaExpressionist Handbook). Protein concentration
was quantified by the Bradford assay using bovine serum albumin as a standard
(Bradford 1976).
Antibody preparation
Purified AcsD (1 mg) was sent to Covance research products for preparation of
polyclonal antibodies from rabbits. The antibodies were affinity purified by transferring
pure AcsD onto a nitrocellulose membrane using a modification of the method described
by Robinson et al. (Robinson, Anderton et al. 1988). Briefly, the pure protein is subjected

68
to SDS–PAGE and transferred onto a nitrocellulose membrane. The band corresponding
to the AcsD protein is detected by staining with Ponceau S stain (0.25% Ponceau S, 40%
methanol, 15% acetic acid). The band visible on the nitrocellulose membrane is cut out
and destained in TBS buffer (10 mM Tris, pH 8.0, 150 mM sodium chloride, 0.05%
Tween-20). The strip is incubated for 1 h in 6 mL crude serum diluted with 4 mL of TBS
buffer. The non-specifically bound proteins are removed by three washes in TBST (TBS
buffer containing 0.5% Tween-20). The bound antibody is eluted by dipping the strips for
10 min in 3mL 0.2 M glycine. The nitrocellulose strip is washed three times in TBST. To
adjust the pH of the strip to 7.5, 100 µl of 3M Tris-Cl pH 8.0, is added to the strip and the
whole process starting from the 1 h incubation, is repeated 6 times to obtain affinity-
purified antibodies.

69
a b
1 2 3 1 2 3 1 2 3 4
17 kDa
1 2 3 4
17 kDa
Fig. 3.2 Overexpression and Purification of recombinant AcsD a) Lane 1 contains proteins from un-induced cell pellet. Induction with IPTG causes
overexpression of the AcsD protein as shown in Lane 2. The molecular weight of the
protein band is a little above 17 kDa as the heterologously expressed protein contains six
histidines at its C-terminal.
b) AcsD was purified by Ni-Sepharose chromatography. The Coomassie blue stained
SDS–PAGE gels shows the flow through from loading the crude cell extract (lane 1). The
column was washed (lane 2) and then eluted with imidazole (lane 4). Lane 3 shows the
molecular weight markers. Purified AcsD was then used to obtain rabbit polyclonal
antibodies.
Preparation of membrane fractions
The cell pellet was obtained by centrifugation of a 48-hour grown culture. The
total membrane fraction (TM) containing the CM and OM (along with components
entrapped in the periplasm) from G. hansenii cells were obtained using the procedure
described by Myers and Myers (Myers and Myers 1992) as modified by Ruebush et al.
(Ruebush, Brantley et al. 2006). Briefly, cell pellet from the culture at mid-log phase was
obtained by centrifugation at 1500 x g for 20 min. The cells were resuspended in 24 mL
of TS buffer (25% sucrose in 10 mM Tris–Cl pH 8.0) per gram wet weight. The
suspension was constantly stirred at room temperature for 15 min, after which sequential

70
additions of the following components were made at 15 min intervals: one-tenth volume
of lysozyme (0.64 mg/ml lysozyme), one-tenth volume of EDTA to obtain a final
concentration of 5mM (20 mg/ ml), a final concentration of 0.3% (w/v) Brij58, a final
concentration of 12 mM MgCl2 from a 15 mM M stock and finally a few crystals of
deoxyribonuclease. The resulting suspension was centrifuged at 1500 g for 30 min to
remove cell debris and whole cells. The supernatant composed of membrane fractions
was centrifuged for 2 h at 177,500 x g (Ruebush, Brantley et al. 2006) (50,000 rpm) in a
Beckman Ti-70 rotor to obtain the TM pellet. The pellet was resuspended in 10 ml of 10
mM Tris–Cl buffer pH 8.3 and the protein content of this pellet was measured by Lowry
et al. (Lowry, Rosebrough et al. 1951).
To isolate the CM and OM, 6 mL of TM was loaded on a 25–55% sucrose step-gradient
tube and spun at 82,500 x g (25,000 rpm) for 17 h in a Beckman SW 28 rotor. The OM
was obtained as a thick band around the 55% sucrose concentration and the CM band was
obtained at the density of 35%. The fractions were collected as one ml aliquots using a 1
mL pipette. The TM, CM and OM were stored as 10% glycerol stocks at -70°C.
Preparation of periplasmic and cytoplasmic fractions
The periplasmic and cytosolic fractions were isolated by modification of methods
described by Thomas et al. (Thomas, Daniel et al. 2001) and Streeter and Le Rudulier
(Streeter and Le Rudulier 1990). Cells were pelleted by centrifugation of 50 mL of
actively growing culture at 1,500 x g for 10 min at 4°C. The cell pellet was resuspended
in 7.5 mL TES buffer (50 mM Tris pH 8.0, 20% sucrose and 0.1 mM EDTA) containing
0.8 mg/mL lysozyme and incubated at room temperature for 10 min. Cells were
harvested by centrifugation as before and resuspended in 2 mL of 5 mM ice-cold

71
magnesium chloride. After incubation for 10 min on ice, centrifugation at 8,000 × g
yielded periplasm in the supernatant and spheroplast in the pellet. The spheroplasts were
resuspended in 2 mL HEPES-saline buffer (50 mM HEPES, 150 mM KCl), pH 7.2 and
sonicated for 15 min at 15% pulse to release the cytoplasm, which was obtained in the
supernatant after centrifugation at 250,000 × g for 30 min.
The purity of the membrane fractions was assessed by assaying for associated marker
enzyme levels for each cellular compartment. Succinate dehydrogenase activity was
assayed according to the methods described by Anwar et al. (Anwar, Brown et al. 1983).
The reaction mixture contained 60 mM sodium phosphate (pH 7.2), 10 mM potassium
cyanide, 10 µg phenazime methosulfate, 20 µg dichlorophenol-indolphenol (DCIP), 25
mM sodium succinate and cellular fraction containing 100 µg protein in a total volume of
1 mL. The decrease in absorbance at 600 nm was monitored for 10 min at 25°C and
specific activity calculated using extinction coefficient of DCIP, e = 13 mM-1
cm-1
(Fox,
Borneman et al. 1990). Alkaline phosphatase activity was assayed by modification of the
procedure described by Garen and Levinthal (Garen and Levinthal 1960). The increase in
410 nm absorbance corresponding to hydrolysis of p-nitrophenyl phosphate (PNP) to p-
nitrophenol was monitored in a reaction mixture composed of 0.1 mL of the cellular
fraction, 1 mM PNP and 1 M Tris–Cl buffer pH 8.0. The change in absorbance was
recorded for 5 min and the specific activity was calculated using the extinction
coefficient (e) of p-nitrophenol as 16.7 mM-1
cm-1
(Halford 1971). For malate
dehydrogenase (de Maagd and Lugtenberg 1986), the decrease in 340 nm absorbance due
to oxidation of NADH, was monitored in a 1.1 mL assay mixture composed of 50 mM N-
2-hydroxyethylpiperazine-N'-2'-ethanesulfonic acid (HEPES) pH 7.2, 0.3 mM NADH,

72
100 µL of the subcellular fraction. The reaction was initiated by addition of 25 µL of 10
mM oxaloacetate. Specific activity of the enzyme was calculated using extinction
coefficient for NADH, e = 6.2 mM-1
cm-1
.
Detection of AcsD using Western blot
The protein content in all the fractions was determined by using the method
described by Lowry et al. (Lowry, Rosebrough et al. 1951). A total of 10 µg of the
protein was loaded into wells of a 15% polyacrylamide gel and blotted into a
nitrocellulose membrane. Western blotting was carried out using 1:300 dilution of anti-
AcsD as the primary antibody and a 1:10,000 dilution of the anti-rabbit IgG conjugated
with alkaline phosphatase as the secondary antibody. 5-Bromo-4-chloro-3-
indolylphosphate/nitro blue tetrazolium (BCIP/NBT) substrate was used for visualization
of antibody-bound protein bands.
RESULTS
Purification of AcsD
The entire length of the 471bp acsD gene was amplified by colony PCR. The
resultant DNA was sequenced, ligated to pET-21a vector such that the 3' end of the gene
encoded for 6 histidine residues. The vector containing the acsD gene was transformed
into BL-21 (DE3) cells. Addition of IPTG enabled enhanced expression of AcsD with a
histidine-tag at the C-terminus as shown in Figure 3.2a. Sonication and subsequent
centrifugation of the cell pellet in lysis buffer, released the protein in the supernatant
fraction. The protein is therefore not strongly membrane-associated and is a soluble
protein. This soluble fraction was used for purification of AcsD using Ni-affinity column.
Purity (greater than 95%) was assessed by subjecting the protein to SDS–PAGE followed

73
by Coomassie blue staining (Fig. 3.2b). In addition to having the correct molecular
weight (17kDa), the purified protein was identified as AcsD by cross-reactivity with the
anti-histidine tag antibody on Western blot.
Specificity of anti-AcsD antibody
The purified AcsD protein was used to obtain polyclonal antibodies from rabbit.
The specificity of the antibody is shown in Fig. 3.3. Only one cross-reactive band was
observed in Western blot when the anti-AcsD antibody was used to probe the whole cell
extracts of the G. hansenii (Figure 3.3). Since the anti-AcsD antibody is highly specific, it
was suitable for localization studies of AcsD in G. hansenii cellular compartments.
1 2 31 2 3
Figure 3.3 Determining the specificity of anti-AcsDantibody. Total cell extracts were
subjected to SDS–PAGE and proteins visualized by Coomassie blue (lane 2) Lane 1 is
molecular weight marker. Lane 3 shows the band obtained after the whole cell proteins
were transferred to nitrocellulose membrane and visualized by Western blot with the
antibody.

74
Subcellular fractionation
We isolated the membrane fraction (and subsequently separated it into CM and
OM), the periplasmic fraction and the cytosol. The relative purity of the periplasmic
fraction, the TM fractions and the cytoplasm fractions was assessed by marker enzyme
assays for each of the respective fractions (Table 1). The cytoplasmic marker enzyme,
malate dehydrogenase, exhibited an activity ratio of 6.25 between the cytoplasm and the
periplasm. An activity ratio of 34 was observed for the CM marker enzyme, succinate
dehydrogenase for the CM to periplasm. Our results from the marker enzyme assays,
shown in Table 3.1 indicate that for all four fractions (CM, OM, periplasm and
cytoplasm), there is little contamination between the fractions (ratio of specific activities
is at least 5.6). More importantly, our results show that the periplasmic sample is largely
free of cytosolic and membrane components.
Detection of AcsD in the periplasmic fraction
Upon establishing the purity of each fraction, we then performed experiments to
determine the localization of AcsD. First, the periplasm, cytoplasm and TM fractions
were subjected to SDS–PAGE (Fig. 3.4). The protein profile shown in Fig. 3.4a shows
that the each fraction has a unique protein band profile, indicating the lack of similarity
between the fractions and of the purity of each subcellular fraction.
All the cellular fractions were subjected to SDS–PAGE, followed by electrotransfer
to a nitrocellulose membrane, which was analyzed by Western blotting with anti-AcsD
antibody (Fig. 3.4b). A single band at 17 kDa corresponding to the molecular weight of
AcsD, was observed in periplasm. An intense band was also detected in the TM fraction
(Figure 3.4b). Because TM fraction preparations will also contain proteins from the

75
periplasm (entrapped by protein-protein interactions), our results remain consistent with
AcsD localized in the periplasm. This was further confirmed when the TM fraction was
separated into its component parts of the OM and the CM using sucrose-density
ultracentrifugation. Western blot analysis again revealed localization in the periplasm.
Neither the OM nor the CM fraction contained appreciable amount of AcsD when
compared to the periplasmic fraction (Fig. 3.4).
The 17 kDa band corresponding to the one obtained in the Western blot, in the lane
containing the periplasmic fraction was excised from the Coomassie-stained gel. This
band was digested with trypsin and analyzed by LC–MS. (The procedure for trypsin-
digestion is elaborated in the Chapter VI of this thesis, which mainly focuses on the MS-
related work.) The results of the MS analysis (Table 3.2), clearly indicated that the 17kDa
band is AcsD. The TM fraction also showed a similar size band which would not be
surprising due to entrapment of the periplasmic fraction during membrane isolation
procedure.

76
Table 3.2 Protein identification by LC-MS of trypsin-digested 17kDa gel band.
The individual scores for the peptides are indicated with brackets along with the peptide
sequence. Individual scores >42 indicate identity or extensive homology (p<0.05).
Figure 3.4 Subcellular fractionation and detection of AcsD.
a) SDS-PAGE profile of proteins from the subcellular fractions of G. hansenii.
Each lane contained 10 µg of protein from the specified cellular compartment and subjected to
SDS-PAGE. The band in periplasmic fraction, that was excised and sent for MS-analysis is
indicated by an arrow. Cytop: Cytoplasm. Perip: Periplasm.
b) Western blot of the cellular compartments using anti-AcsD antibody.
Using anti-AcsD antibody, the proteins separated by SDS-PAGE were transferred onto a
nitrocellulose membrane and Western blotted using anti-AcsD antibody. Bands corresponding to
the AcsD protein can be seen only in the periplasmic and TM fractions.
Entry Description Mascot
Score
MW Peptides Coverage
(%)
ACSD_ACEX
Y
Cellulose
synthase operon
protein D
353 17432 R.DVDAEDLNAVPR.
Q (91)
R.WVTSQAGAFGDY
VVTR.D (149)
9.2
11.8
CM OM Cytop. Perip. TM
34
52
17
26
42
72
MW CM OM Cytop. Peri.
CM OM Cytop. Perip. TM
TM

77
MTIFEKKPDFTLFLQTLSWEIDDQVGIEVRNELLREVGRGMGTRIMPPPC
QTVDKLQIELNALLALIGWGTVTLELLSEDQSLRIVHENLPQVGSAGEPS
GTWLAPVLEGLYGRWVTSQAGAFGDYVVTRDVDAEDLNAVPRQTIIM
YMRVRSSAT
Figure 3.5 The amino acid sequence of AcsD
The N-terminal domain shows twin lysin residues (bold font) that indicate that the protein is
possible candidate for secretion into the extracellular space using Sec-dependent transport.
DISCUSSION
Cellulose is synthesized by G. hansenii in the form of an extracellular ribbon of
crystalline microfibrils. It has been shown that the enzyme machinery forms a linear complexes
arranged along the longitudinal axis of the cells (Brown and Montezinos 1976). The
biochemistry behind the process of cellulose synthesis and secretion is yet to be completely
understood. However, it has been shown that the polymerization of glucose occurs in a single-
enzymatic step from UDP-glucose to cellulose, catalyzed by cellulose synthase (Swissa, Aloni et
al. 1980). This enzyme is encoded by the first gene of the acs operon, called acsAB (Wong, Fear
et al. 1990; Saxena, Kudlicka et al. 1994). The other proteins (AcsC and AcsD) encoded by the
operon are presumed to be instrumental in the assembly and export of the polymer across the cell
membranes (Saxena, Kudlicka et al. 1994). Since, both AcsAB and AcsC are membrane-
associated (Saxena, Kudlicka et al. 1994), to determine the organization of the cellulose
synthesis and extrusion system, it was essential to know if the AcsD protein is a membrane
protein or a cytoplasmic protein.
Determination of the role of AcsD in cellulose synthesis was first addressed by

78
mutagenesis studies by Saxena et. al. (Saxena, Kudlicka et al. 1994). These workers showed that
cells that lacked a functional AcsD, could still produce cellulose. However, these mutants
exhibited a lowered rate of cellulose synthesis in vivo. But the in vitro rate (with isolated
membrane fractions) is not altered in the absence of this protein. The cellulose that is made by
the mutants lacking AcsD, however has altered crystallinity, being a mixture of both cellulose I
and cellulose II allomorphs (Saxena, Kudlicka et al. 1994). This forces us to conclude that the
protein comes into contact with the nascent cellulose chains and influences their assembly. This
also indicates that the protein acts downstream of cellulose synthase in the process of cellulose
extrusion.
Prior to our work, no other localization studies have been done for AcsD. Using
conventional Western blot analysis and subcellular fractionation, we have shown for the first
time that AcsD is localized in the periplasm. AcsD sequence lacks the known signals such as the
twin arginine motif (RR) required for transportation of proteins to the periplasm (Yahr and
Wickner 2001; Palmer 2007). Examination of AcsD sequence (Figure 3.5) shows that the N-
terminal of the protein contains twin-lysine residues, which is reminiscent of several proteins
transported by the Sec-dependent pathway (Eitan 2007). However, we cannot conclude that the
protein is transported by this pathway, based on the presence of the lysine residues alone.
Interestingly, commonly-used methods for computational prediction of periplasmic
proteins like pSORTb (Gardy, Laird et al. 2005) provide no clue about the cellular localization of
AcsD. However, there has been evidence of other proteins devoid of such signals peptides like
the Brucella abortus catalase (Sha, Stabel et al. 1994) which have been localized in the
periplasm. Like AcsD, the periplasmic localization of this protein also cannot be deciphered
using pSORTb. Based on the organization of proteins in other non-protein efflux systems

79
such as the AcrA/AcrB/TolC system (Gerken and Misra 2004), we presume that AcsD protein
serves as a periplasmic protein channel for transport of the newly-synthesized cellulose chain.
Our view is corroborated by evidence of weak interactions between of AcsD with cello-
oligomers (Hu, Gao et al. 2010). A schematic consistent with this arrangement is shown in
Chapter IV.
It has been proposed previously that cellulose synthesis is a cell-directed process and
several levels of regulations operate between the polymerization of glucose residues and
emergence of twisted ribbons of cellulose (Benziman, Haigler et al. 1980; Haigler 1982).
Localization of AcsD in the periplasm together with its role in defining the crystalline character
of the emerging cellulose fibers, leads us to the conclusion that the most probable role of AcsD
to provide a channel through the periplasm for the nascent cellulose strand. For it to function as a
channel necessitates that some part of AcsD be associated with AcsAB and/ or AcsC. Further
studies on the interactions between the proteins would clearly accentuate the evidences presented
herein for periplasmic localization of AcsD.
CONCLUSION
We have successfully cloned and heterologously expressed the AcsD protein. The
specific antibody raised against this protein was used to determine its sub-cellular localization in
the G. hansenii cells. The AcsD protein is localized in the periplasm. This work is the first
attempts towards understanding the organization of the proteins of the cellulose synthesis
complex, across the bacterial membrane.

80
CHAPTER IV
DETERMINATION OF THE SOLUTION-STRUCTURE OF ASCD
INTRODUCTION
The previous chapter of this thesis describes our attempts at determining the sub-
cellular localization of AcsD. Using specific antibodies against AcsD, we have found that
the protein is localized in the periplasm of the Gram negative bacterium G. hansenii. This
chapter describes our studies with the pure AcsD protein in the determination its
structure.
Hu et al.(Hu 2008; Hu, Gao et al. 2010) elucidated the crystal structure of this
protein at the time that our study was initiated. These workers found that the protein
crystallizes as an octamer (Hu 2008; Hu, Gao et al. 2010). They also succeeded in
obtaining the solution-structure of this protein, while our attempts in this area were
underway (Hu, Gao et al. 2010). The AcsD protein forms an octameric complex. The
octameric assembly is composed of a tetramer of dimers and forms a cylindrical structure
with a 4-fold axis of symmetry and a central pore. Each AcsD monomer is oriented in the
complex, in such a way that the N-termini of all the monomers are directed towards the
center, and the C-termini radiate outside the cylinder. Four monomers in the upper layer
of this octameric complex interact with four monomers in lower layer with the two layers
shifted at an angle of 50ºC with respected one another . This creates four dimer-dimer
interfaces that create four spiral interstices in the wall of the cylinder. Each of these
passageways serve as a site of interaction for a glucan chain (Hu 2008; Hu, Gao et al.
2010).

81
The octamer is cylindrical in conformation and is composed of two stacked
tetrameric complexes. The cylinder has a height of 62 Å, an outer diameter of 90 Å and
an inner diameter of 65 Å. The top layer consisting of four monomers, is twisted at angle
of 50ºC with respect to the bottom layer. The N-termini of each monomer in the
octameric AcsD cylinder, form four inner passageways Each of these passageways is a
site for association of a cellopentaose chain. There are two equally-probable and opposite
orientations in which each cellopentaose can align with respect the dimeric interface.
(Hu, Gao et al. 2010).
This preliminary crystallographic analysis paved the way for our work on the
solution-structure determination of this protein. Though, it can be argued that the crystal
structure provides enough material to study the protein, our attempt at solution-structure
determination was directed towards studying the oligomerization behavior of this protein
under conditions close to the cellular environment. In doing so, we wanted to enquire if
the oligomerization was indeed a preferred state of this protein, or was it a consequence
of the best orientation assumed by the protein in its unit cell, during the crystal lattice
formation. We determined the molecular weight of the predominant species of AcsD
protein that exists in solution by employing analytical ultracentrifugation (AUC),
dynamic light scattering (DLS) and gel filtration. All these experiments, described herein,
indicated that the AcsD protein assembles as an octamer in solution.
We studied the solution-structure of this protein using small angle X-ray
scattering (SAXS) and found that the octamer associates in the form of a dimer of
tetramers with a central pore. Putting together the periplasmic localization and the
cylindrical structure of this protein, a model for cellulose secretion complex is described.

82
Since solution structure determination of a protein using SAXS is not a routine
experiment in biochemistry laboratories, the underlying principles and the methods
followed for the experiment and analysis, will be briefly described in the following part
of this section.
Principles behind structural analysis by SAXS
Biological SAXS experiments are performed by exposing a solution of
macromolecules to a high-intensity of collimated X-ray photon beam (Koch, Vachette et
al. 2003). The incident X-rays are scattered by the electrons of the macromolecule at
different angles. The elastic scattering pattern of the radiation contains information about
the distribution of electron densities within the macromolecule. Therefore, the scattering
pattern of this radiation is registered in the form of circles of finite width and plotted in as
a function of the scattering angle, Q (Putnam, Hammel et al. 2007).
Q =4/*sin(/2) Equation 4.1
This scheme of events is depicted in Figure 4.1
The ability of a macromolecule like protein, to scatter X-rays depends on the
concentration of the protein in solution, which determines the electron density of the
protein. The difference between the electron density of the proteins and that of the
aqueous buffer it is contained in, is referred to, as the excess scattering length density or
contrast. The average electron density of protein and water are ~0.44 e-/Å
3 and ~0.33 e
-
/Å3, respectively (Putnam, Hammel et al. 2007).

83
Figure 4.1 Small angle X-ray scattering experiment: The sample in the exposure capillary is
irradiated by X-rays. The incident X-rays are scattered by the electrons in the sample to an angle
of 2. The detector converts the 2D scattering into a 1D scattering profile, by means of radial
integration.*This figure is adapted from Putnam et al. (Putnam, Hammel et al. 2007).
This results in a very minimal contrast, which is only 10% of what can be
achieved if the protein were in vacuo (Putnam, Hammel et al. 2007). However, in vacuo
analysis of protein structure is both impractical as well as far removed from cellular
conditions. A measurable signal from the protein molecule against an almost equally
intense signal of the aqueous buffer, is obtained by subtracting the scattering due to
buffer from the total scattering data for the protein in the buffer. The data obtained from
the SAXS experiment is thus a 1D-scattering curve that represents the scattering of a
single particle averaged over all orientations. From this scattering profile, it is possible to
calculate the aggregation state and dimensions of the macromolecule.

84
Data analysis: The important parameters that can be deduced from the scattering curve
are: the radius of gyration (Rg) and the pair distribution function P(r). The Rg also known
as the second moment of inertia, refers to the mass distribution of a macromolecule
around its center of gravity. Analysis of the SAXS scattering curve at low intensities can
be used for approximation of the Rg of a protein assuming that the scattering due to
proteins is to be equal to that of a spherical particle. This was calculated by the French
scientist Andre Guinier (Guinier 1955). The Guinier approximation is represented in the
form of a plot of natural logarithm of the measured intensities against Q2. In case, of the
proteins, Guinier approximation is derived in the region near the beam-stop where the Q
x Rg < 1.3. The extrapolated Y-intercept of this plot gives the value of I(0), which is the
intensity at zero scattering angle, called the forward scattering (Guinier 1955).
I(Q) = I (0) exp {(-1/3)Rg2Q
2}3 Equation 4.2
The Fourier transform of the I(Q) scattering curve yields the pair distribution
function P(r) function (Glatter 1977), which describes the paired-set of distances between
all of the electrons in a protein. Since this function describes all the paired-distances of
electrons in the macromolecule, small changes in the relative positions of these electrons
leads to large changes in P(r) distribution, thereby detecting conformational changes in
the protein. The Rg of a molecule can be calculated from the P(r) function. Unlike the Rg
obtained from the Guinier plot, the one derived from P(r) function is not restricted to low
angles of scattering, but takes into consideration all of the data in real space. Using
Debye equation (Debye 1915) the P(r) functions can be related to the distribution of a
single macromolecule to the scattering intensity as a function of scattering angle, Q
(Glatter 1977).

85
I(0) = 4 0 Dmax
P(r) dr Equation 4.3
As a corollary to this, if the shape and oligomerizarion of the protein are known a
priori, then the scattering profile of the molecule can me calculated and P(r) distribution
can be computed from the atomic position in the model. The point of intersection of the
P(r) distribution curve with the X-axis is the Dmax, the maximum diameter of the
particle. All the available and known parameters of the protein, such as the scattering
profiles, oligomerization state, symmetry and stoichiometry, are fed into a program called
GASBOR (Svergun 2001b), which reconstructs the 3D shape of the protein in the form of
a bead-model. Since there are multiple structures that can have the same scattering
profile, the modeling program narrows down on the appropriate structure by discarding
all values of the scattering that do not match the limits set by Dmax. Using the prior
knowledge of symmetry of the molecule the GASBOR program creates a model of
sphere of diameter equal to the Dmax and populates it with number of beads equal to the
number of amino acids in the protein. The adaptation of these principles for determining
the structure of AcsD will be discussed in the subsequent sections.
MATERIALS AND METHODS
AcsD overexpression and purification: The procedures for protein expression and
purification have been described in Chapter III. The protein was further purified in a
High trap QHP column of resin volume 5ml (GE Healthcare), to obtain a highly pure
sample for DLS and AUC experiments (Figure 4.2). The protein obtained after Ni-NTA
purification (25 ml of 0.5 mg /ml) was dialyzed against 50 mM Tris Cl pH 8.4 (Buffer
A). The protein was loaded into the column in Buffer A and eluted with a linear gradient
of 0 to 100% buffer B (50 mM Tris-Cl pH 8.4, 1M NaCl) at the rate of 1ml per min for

86
100 minutes. The AcsD protein elutes as a major peak (900 mAU) at approximately 90
ml with a smaller shoulder (200 mAU). The sample collected at the peak (1.3 mg/ ml)
and shoulder were subjected to SDS-PAGE and staining (Figure 4.2 b).
Sample preparation for AUC: AcsD purified using Ni-NTA affinity column (Figure
3.2), was in the elution buffer, which contained 50 mM NaH2PO4, 300 mM NaCl and 25
mM imidazole. pH 8.2. This protein was dialyzed thoroughly in the same buffer without
the imidazole. 1 ml of AcsD at a concentration of 2 mg/ml was submitted for
sedimentation velocity (SV) ultracentrifugation. A 35 ml sample of the dialysate buffer
(50mM NaH2PO4 and 300mM NaCl, pH 8.2) was also supplied.
Analytical ultracentrifugation: Using Sednterp program, the following physical
constants for the protein were calculated MW = 17,375 Da, 20° =0.7408 ml/g. The
buffer density and viscosity were calculated to be 1.0176 g/ml and 0.010576 poise at
20°C, respectively. Sedimentation velocity experiment was conducted at 20°C and
50,000 rpm using interference optics with a Beckman-Coulter XL-1 analytical
ultracentrifuge. Double sector synthetic boundary cells equipped with sapphire windows
were used to match the sample and reference menisci. The rotor was equilibrated under
vacuum at 20°C and after an equilibration period of ~1 hour at 20°C the rotor was
accelerated to 50,000rpm. The interference scans were acquired at 60 second intervals for
approximately 7 hours. Sample dilutions for the analysis were prepared as shown in
Table 6.1

87
Figure 4.2 Anion exchange chromatography of AcsD
a) The major peak at 90 ml elution volume is followed immediately by a small shoulder.
The peaks are eluted at 60% buffer B. b) The fractions collected at the peak of the elution
and the shoulders were subjected to SDS-PAGE. Lanes 1contains the molecular weight
ladder. Lane 2 is an empty lane. Lane 3 and 4 contain 20 μl of the peak fraction collected
at the shoulder.
a
1 2 3 4

88
Sample preparation for DLS: AcsD protein concentration was measured using the
absorbance at 280 nm using extinction coefficient of 26470 M-1
cm-1
. The concentrations
used for DLS experiment were obtained by dilution from a stock of 8 mg/ml in 50 mM
Tris Cl pH 8.4 with 5% glycerol.
DLS experiment: All measurements were carried using the Viscotek-DLS equipment in
The X-ray crystallography facility, University Park. The Omnisize software linked to the
instrument automatically generated a correlation curve for the sample based on its
scattering profile. The size-range for calculating the hydrodynamic radius (Rh) was
restricted between 1nm to 1000nm. Based on the Rh value the MW of the proteins is
estimated by the software assuming the molecule is a perfect sphere. The data obtained
from these measurements are used to arrive at the approximate, and not the exact
molecular weight, of the protein.
Sample preparation for SAXS: AcsD protein (50 ml containing 6.5 mg of protein) was
subjected to anion exchange, as described above. The protein fraction collected at the
peak was concentrated a using a 10K concentrator (Amicon) to obtain a final
concentration of 5.2 mg in a volume of 200 μl. Half the sample volume (100 μl ) was
further purified by gel filtration. The other half was use for in-line gel filtration with
SAXS experiment. Protein concentrations were measured by reading the absorbance at
280 nm and using the extinction coefficient of 23760 M-1
cm-1
.
Gel filtration: Gel filtration was carried out using a Superdex 200 column (24 ml, GE
Healthcare). The sample (100 μl of 26 mg AcsD) was loaded using a 100 μl loop. The
buffer (50 mM Tris pH 8.4, 300mM NaCl and 5% glycerol) was passed through the
column at the flow rate of 0.5 ml/min, for the duration of 48 minutes to obtain allow

89
elution of the protein in 1 column volume of buffer. The 2ml volume protein eluted at the
peak (1.2 AU), was collected and concentrated to 1 ml protein at 3 mg/ml.
SAXS experiment: The AcsD protein was purified using anion exchange and gel
filtration chromatography was used for the SAX experiment. Three kinds of samples
were used for the analysis, pure AcsD, pure AcsD incubated at room temperature for 24
hours and .
Data acquisition: All the SAXS experiments were performed at the Advanced Photon
Source (APS), Argonne National Laboratory (ANL) using the Biophysics Collaborative
Access Team (BioCAT) undulator beamline 18-ID. The scattering profile was obtained at
very low angle of the beam (1.03 Å). These beams are directed towards an exposure
capillary into which the sample is drawn. The measurements are carried out by 100 µl of
AcsD sample between the capillary and the sample tube at the rate of 50 µl/s. 10-20
exposures were taken for each sample. Each exposure lasted for 1 s. Measurements of
empty capillary as well as capillary with buffer were taken to serve as blanks. The empty
capillary was exposed for 20 shots and 10 shots each of the buffer and the protein sample
were taken. The scattering profile due to the capillary, buffer and protein are shown in
Figure 4.6. The scattering due to buffer and the capillary has to be subtracted from the
solution scattering profile. This is achieved by subtracting the scattering due to the
capillary, from that of the protein and then subtracting the capillary scattering from the
buffer, using the equation:
(Protein - Buffer)-Capillary - x (Buffer - Protein)
x is a fraction which is approximately equal to 0.993 and is proportional to the amino
acid composition, concentration of the protein and the partial specific volume. Since the

90
sequence of the protein and the concentration are known, we can calculate the partial
specific volume (υ = ml/g), which gives us the fraction of the total volume occupied by
the protein alone.
SAXS data analysis The scattering data were analyzed using IGOR software and the
programs contained in the ATSAS package. Rg was calculated using the Guinier
program using the IGOR BioCAT macros. The P(r) distribution was calculated using
GNOM software (Semenyuk 1991). The P(r) distribution was used by the GASBOR
program (Svergun 2001b) for generating 50 bead models. With the a priori knowledge of
the crystal structure, the model was built with an imposed symmetry of p42. The models
were superimposed for comparison using Supcomb (Kozin 2001) and the average
structure was arrived at using the DAMAVER (Volkov 2003) program. The average
structure was further refined using the damfilt software and this structure was overlaid
with the existing crystal structure of the AcsD protein.

91
RESULTS
DLS experiment: This technique was valuable in monitoring the aggregation pattern of
the protein over a range of concentrations. We observed that the protein showed a
consistent molecular weight between 140 kDa and 153 kDa, across concentrations from
0.5 mg/ml to 8 mg/ml (Figure 4.3). The molecular weight of the major and minor species,
for AcsD at different concentrations, are given in Figure 4.3. It can be seen from the
graph and the accompanying data, that the mass distribution remains consistent across
various concentrations of the protein, giving a value for molecular weight between 141-
153 kDa. This signifies the presence of a species equal to an octamer. DLS gave us our
first indication to probe further into the oligomeric associations of AcsD.
This preliminary technique was valuable in monitoring the aggregation pattern of the
protein over a range of concentrations. We observed that the protein showed a consistent
molecular weight between 140 kDa to 153 kDa, across concentrations from 0.5 mg /ml to
8 mg/ml (Figure 4.3). It can be seen from the graph and the accompanying data, that the
mass distribution remains consistent across various concentrations of the protein, giving a
value for molecular weight between 141- 153 kDa. This signifies, presence of a species
equal to an octamer. DLS gave us our first indication to probe further into the oligomeric
associations of AcsD.

92
AcsD concentration: 0.5 mg/ml
Peak % Area Rh (nm) Position Std Dev % RSD MW (kD)
1 97.7 4.93 5.01 0.35 7.1 143.28
2 1.8 22.42 21.38 1.16 5.2 5137.64
3 0.5 180.71 169.82 17.64 9.8 7.11E+05
AcsD concentration: 1 mg/ml
Peak % Area Rh (nm) Position Std Dev % RSD MW (kD)
1 98.3 4.91 5.01 0.23 4.6 141.95
2 1.7 34.1 32.36 1.77 5.2 1.38E+04

93
AcsD concentration: 8mg /ml
Figure 4.3 DLS analysis of AcsD: The plot of mass distribution of the AcsD protein is
displayed after selction for the range between 1 nm – 1000nm. The amplitude of the
hydrodynamic radius of the protein sphere is indicated at the peak. Based on this Rh
value, the molecular weight of the protein is estimated by the software (Omnisize). a. At
the concentration of 0.5 mg/ml, the protein has shydrodynamic radius of 4.93 and a
molecular weight of 143.28 kDa. b. At a concentration of 1 mg/ml, the protein has a Rh
value of 4.91, corresponding to a molecular weight of 141.95 kDa. c. At a very
concentration of 8 mg/ml the DLS analysis gives a Rh of 5.12 nm and molecular weight
of 156.625 kDa.
Peak % Area Rh (nm) Position Std Dev % RSD MW (kD)
1 97.6 5.12 5.01 0.42 8.3 156.62
2 1.8 52.59 48.98 5.82 11.1 3.85E+04
3 0.6 153.01 153.11 21.59 14.1 4.80E+05

94
Gel filtration
The AcsD purified by Ni- NTA was further purified by gel filtration, for purposes
of structural studies (SAXS) as well as to determine the aggregation behavior of the
protein. The elution profile of the protein (Figure 4.4), shows a major peak which elutes
at 13.2 ml. When analyzed by a semilog plot with standard protein elutions, this
corresponds to the molecular weight of 136 kDa. This value again gives us an octameric
complex of AcsD. We proceeded to determine the accurate molecular weight of this
complex by sedimentation velocity experiment using the pure protein.
Figure 4.4 Gel filtration profile of AcsD. The plot of absorbance (in milli absorbance
units) versus elution volume shows that the protein elutes at a volume of 13.2 ml. The
inset shows a semi-log plot of elutions of protein with known molecular weights.

95
Sedimentation velocity experiments
The data from the SV run were analyzed using three modeling programs, viz.
DcDt, Sedfit, and Sedphat. The DcDt + program provides a model independent
sedimentation distribution, g(s*) analysis using a time derivative of the concentration
profile. An overlay of the normalized distribution plots for the three highest
concentrations of AcsD is shown in Figure 4.5. The protein concentrations were
determined by integration of the g(s*) profile. The three concentrations used were 0.29
mg/ml, 0.71 mg/ml and 1.12 mg/ml. the lowest concentration was not used since the
concentrations derived from integrating the g (s*) was only 70% of the estimated values.
The three curves overlay with one another. This indicates that the over the range of
concentrations covered by the samples, there are no reversible reactions. If there are
larger aggregates formed, there would have been a noticeable shift in the sedimentation
coefficient towards a higher value upon increasing the concentration of the sample.
However, we observe that in the curve for the highest concentration of the protein (1.21
mg/ml), although there is a slight increase in the amount of material sedimenting at a
higher S value, the lack of a prominent shift in the peak, indicates that this is a reversible
aggregation. The protein predominantly exists as a single species in solution.

96
Figure 4.5 Sedimentation coefficient distributions calculated from a sedimentation
velocity experiment
a) An overlay showing the normalized distribution plots for AcsD The sedimentation velocities have been corrected to standard conditions of water as
solvent and temperature 20C. Protein concentrations shown in the figure were derived
by integration of g (s*) profile. The plots superimpose, suggesting matching behavior of
the sample over a range of concentrations. There is no indication of self association as
there is not a considerable shift towards a higher S value. The arrow shows the slight
increase in the sedimentation coefficient in the highest concentration of AcsD used (1.21
mg/ml), but this is not a prominent shift, obviating the possibility of formation of
irreversible aggregates.
Analysis 2: Sedfit program
Sedfit, version 11.71 was used as the direct boundary modeling program for individual
concentrations of AcsD, to generate continuous sedimentation coefficient distribution c(s)
plots. The c(s) analysis was done at a resolution of 0.05S, using maximum entropy
regularization with 95% confidence limit. As shown in Figure 4.5b, the c(s) distribution
plots are sharpened relative to other analysis methods, because the broadening effects of

97
diffusion are removed by use of an average value for the frictional coefficient that is
obtained as a fitted parameter. The plot is consistent with the g(s*) data from DcDt+, in
that the prominent peak is at 6.75 S.
The plot of c(s)data at a scale of 10X that of the full scale plot (Figure 4.5c),
shows that there is a small amount of material (1.5-3%) sedimenting slower than the peak
and approximately 15% of the material sedimenting faster than the main peak. This is in
agreement again with the g(s*) results as the change in the amount of a higher order
aggregate is too small to suggest that it is a irreversible aggregate formed as a
consequence of higher concentration.
Using model-based numerical solutions to the Lamm equation, multiple sets of
the sedimentation velocity data were analyzed by the boundary modeling program for
global analysis, Sedphat version 6.50. Data analysis by Sedphat program was done with a
model of a hybrid local continuous distribution and a single global discrete species. This
model was used because, in the sedimentation behavior of AcsD, there are two species in
solution. There is a main non-interacting species that we are interested in characterizing,
along with smaller aggregates that should be accounted for, in order to not bias the
analysis of the species of interest. The values for the globally fitted parameters are a
sedimentation coefficient of 6.75 S, and a molecular weight of 145 kDa with a of 95%
confidence range of 141 to 149. The best fit value for the molecular weight is in close
agreement with the expected value of 139 kDa for an octamer of AcsD. This confirms
that AcsD exists as an octamer in solution.

98
b c
Figure 4.5
b) Continuous sedimentation coefficient distribution c(s). Analysis of sedimentation plot of
three continuous sedimentation coefficient distribution c(s), normalized to the concentrations
derived from integration of the c(s) plot. The three concentrations used are 0.275 mg/ml (green
curve), 0.699 mg/ml (blue curve) and 1.116 mg/ml (black curve). The values for sedimentation
coefficients have all been corrected to standard conditions, S(20,w). A small amount of material
sediments at approximately 9 S (indicated by arrow), but the major peak is at roughly 6.5 S.
c) 10X magnification of the normalized c(s) plot This magnification allows discerning the
minor peaks. We observe that 1.5-2.5% of material (shown in encircled peak), sediments slower
than the main peak and 14 - 20% of the material (peak indicated by arrow) sediments at a faster
rate. The main peak is at approximately 6.5S

99
Determination of the AcsD structure using SAXS
Since the existence of a stable octameric complex of AcsD had been demonstrated by more than
one method, we further explored the structure of AcsD in solution using small angle X-ray
scattering. As a first step in this experiment, the protein scattering profile was obtained along
with the scattering profile of the buffer it was contained in, and the capillary that held the sample
during exposure. An example of scattering curves obtained for the empty capillary, buffer and
the AcsD protein is shown in Figure 4.6a.
a
Figure 4.6a: Experimental scattering profiles: The scattering curve is plotted with
intensity in a logarithmic scale as the X-axis as a function of momentum transfer (Q). The
units of Q are the inverse of wavelength units. So it is measured as Å-1
The scattering
curve of the empty capillary (green) and buffer (blue) drop more rapidly than that of the
protein (red), due to the greater Rg value of the latter.
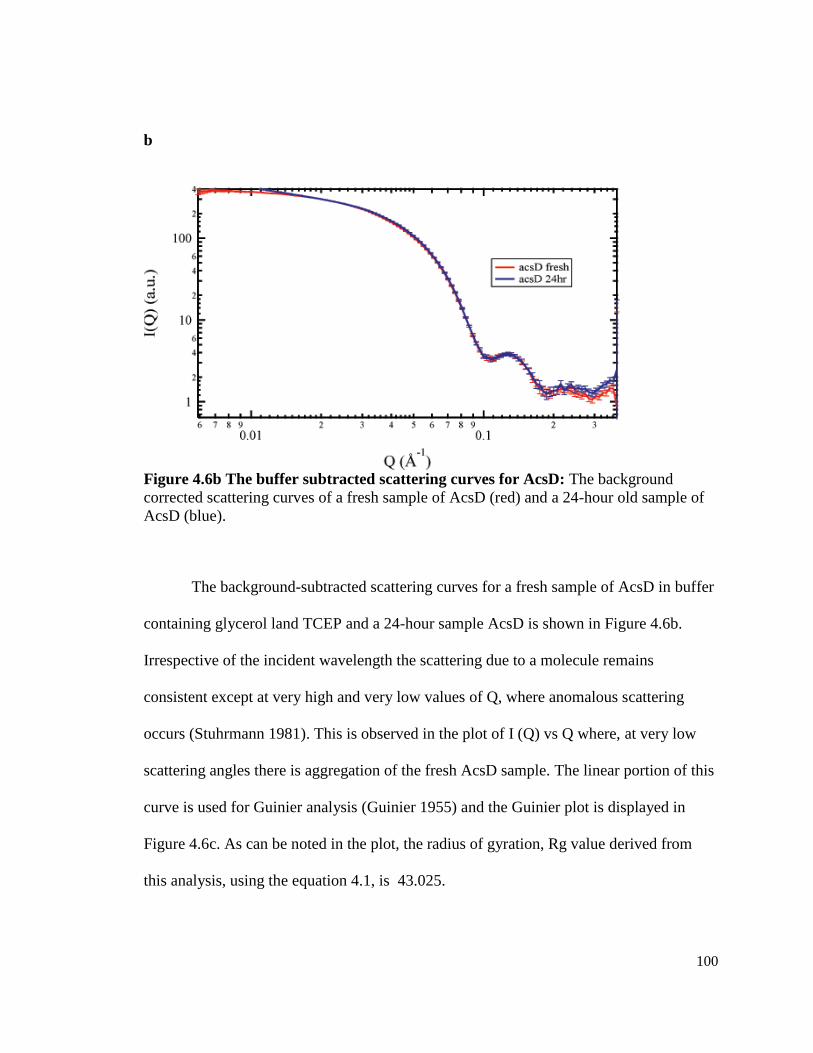
100
b
Figure 4.6b The buffer subtracted scattering curves for AcsD: The background
corrected scattering curves of a fresh sample of AcsD (red) and a 24-hour old sample of
AcsD (blue).
The background-subtracted scattering curves for a fresh sample of AcsD in buffer
containing glycerol land TCEP and a 24-hour sample AcsD is shown in Figure 4.6b.
Irrespective of the incident wavelength the scattering due to a molecule remains
consistent except at very high and very low values of Q, where anomalous scattering
occurs (Stuhrmann 1981). This is observed in the plot of I (Q) vs Q where, at very low
scattering angles there is aggregation of the fresh AcsD sample. The linear portion of this
curve is used for Guinier analysis (Guinier 1955) and the Guinier plot is displayed in
Figure 4.6c. As can be noted in the plot, the radius of gyration, Rg value derived from
this analysis, using the equation 4.1, is 43.025.

101
c
Figure 4.6c. A represention of the linear region of the fresh AcsD sample used for
deriving the Guinier distribution.
In addition to obtaining the Rg value using the linear portion of the scattering
curve, the P(r) function was obtained by Fourier transformation of the scattering data
using the GNOM program. The representative pair-distribution plot shown in Figure 4.7,
gives the Dmax value at 175. This Dmax defined as the largest linear dimension of the
particle (Putnam, Hammel et al. 2007). The Dmax of 175, amino acids residue number of
1256 (=8 x 156), and symmetry of p42, was imposed for reconstructing the 3D structure
of AcsD octamer, using the GASBOR program (Svergun 2001b). This program generates
several bead models of all possible structures that can be obtained by populating the
sphere of diameter equal to 175 Å with 1256 atoms with a symmetry of 4 x 2. This value
of p42 symmetry was used based on the information obtained from the crystal structure

102
which is tetramer of dimers. Some of these representative models are shown in Figure
4.8a.
Figure 4.7: Plot for pair distribution function derived using GNOM: The scattering
curve of AcsD was Fourier transformed by GNOM program (Semenyuk 1991) to obtain
the pair distribution function. The P(r) function is zero at r=0 and r > Dmax. The largest
value of r at which P(r) is zero is considered Dmax.
The C-terminal end of the proteins are found extending in all structures and all
possible orientation of these chains are observed in all the models. The protein associates
in form of a cylinder with an upper and lower layer, surrounding a central pore. The
average structure was derived from 50 independent models, using the DAMVER program
(Figure 4.8). DAMFILT software finds the regions in the protein structures and identifies
regions of highest densities and selects for the filtered structure
(www.emblhamburg.de/ExternalInfo/Research/Sax/). This structure when superimposed
with the available crystal structure of the protein, agrees well with the latter (Figure
4.8c). However, when the filtered structure is superimposed with the average structure,
the former is of a greater dimension (Figure 4.8d). This is due to the fact that it is the

103
representation of all the models hence contains all the orientation of the atoms and
therefore a much larger distribution of particles is observed.
From all the structures obtained using the SAXS analysis, the protein associates in
a manner similar to that shown in the crystal structure. When the two structures were
compared as shown in Figure 4.9, the tilt observed between the two tetrameric stacks can
also be seen in the SAXS structure in both the top view and the side view. Our data
confirm that the protein exists as an octameric complex in-solution and similar to the
crystal structure, the essential features of the protein, central pore and the twist in the
molecule, are seen in our analysis as well.

104
a b
Figure 4.8: Representative bead models of AcsD generated by the GASBOR program : 3D
shape reconstruction of AcsD was by imposing a p42 symmetry (derived from crystal structure)
and residues of amino acids in the AcsD protein. a) Two of the many globally distributed bead
models are shown. inside a sphere of diameter equal to the Dmax (175) and number of beads
equal to the number of amino acid residues. b) Average structure (pink beads) and the filtered
structure of the protein (blue beads).
Top view
Side view

105
c
d
c) Filtered structure of the AcsD octamer is (blue beads) superimposed with the crystal
structure (red ribbon). d) The average structure is superimposed with the filtered structure
and crystal structure shown in (b) to show the comparatively large dimensions of the
former, since it takes into account all possible orientations of the protein in space.
*The ribbon structure of the AcsD crystal was obtained from the pdb database (Hu Gao et
al 2010)
b
a
d

106
a
b
Figure 4.9: Comparison the contours of the solution structure and the crystal
structure of AcsD: The solution structure is represented in blue burred image and the
crystal structure is represented in bead model with each monomeric chain depicted in a
different color a. Top view: The central pore in the crystal structure is seen as a cavity in
the solution structure. It can be seen that the lower layer of tetramers is slightly offset
against the upper layer. The tilt in the alignment of the two layers is indicated by arrows.
b. Side view: The slight slant in the alignment of the dimeric subunits in the crystal
structure is reproduced in the solution structure. The structures were made using Sculptor
software (sculptor.biomachina.org). The crystal structure file (2z9e) was downloaded
from the protein databank (www.rcsb.org).

107
DISCUSSION
The cylindrical octamer organized as a tetramer of dimers with a central hole, is
the structure arrived at, by us in our SAXS analysis. In addition to SAXS, all our
characterization techniques, for the determination of in-solution oligomerization and for
structural analysis of the AcsD protein, converge towards identifying that the protein
exists as an octamer in solution. This structure is composed of residues arranged in two
layers, in such a way that the interfaces between the dimers form four tilted passageways
that are shown to interact with four glucan chains. The AcsD structure determined from
our SAXS analysis shows, these spiral passageways in form of notches between the
dimeric units shown in the Figure 4.9. The presence of these tilted interaction sites for
glucan chains are presumed to contribute to the spinning the glucan chains and
assembling them together. The involvement of AcsD in forming the passage for cellulose
extrusion has been suggested previously (Hu, Gao et al. 2010). The structure of the
octameric complex, with a central pore, confirms the assumption that AcsD could serve
in the passage of the glucan chain outside the bacterial cells.
It is known that the there are no homologues of this protein in the plant kingdom.
However, AcsD protein sequence is the most conserved of all the acs operon-encoded
protein, among all the cellulose-synthesizing bacteria. Thus, the contribution of this
protein towards the unique properties should be explored further. With our initial finding
of the periplasmic localization together with this structural characterization, we propose
that the AcsD protein, serves as the channel in the periplasm through which the newly-

108
synthesized cellulose fibers pass as they are enter the periplasm (Figure 7.1, Chapter VII).
The twists in the internal walls of the AcsD cylinder that interact with the cellulose fibers,
The structure of this protein not only enables passage of the glucan chains through the
aqueous periplasm but also imposes a spin in the glucan chains and serve to assemble the
fibers. It has also been shown that cellulose synthesis, assembly and crystallization are
cell-directed process and occur simultaneously (Haigler, Malcolmbrown et al. 1980). It
has also been observed that the bacterial cellulose has an inherent twist in its structure
(Colvin 1961).Our model serves to explain the mechanism behind this twist in bacterial
cellulose as well as corroborating the observation that deletion of this protein results in a
lower yield of cellulose and an alteration in its crystalline nature (Saxena, Kudlicka et al.
1994; Hu, Gao et al. 2010). Our model agrees well with the earlier finding by Saxena et
al. (Saxena, Kudlicka et al. 1994) that, in the absence of this protein, cellulose synthesis
by the CM-bound AcsAB is unaffected and cellulose extrusion from the OM pore, is still
possible with the passageway through periplamic space is lost. This means that the glucan
chains are not assembled well to facilitate their extrusion through the porin. This results
in extrusion of a mass of unassembled fibers leading to loss in crystallinity and some of
the fibers tend to accumulate in the periplasmic space. Consequently, instead of being
bundled and directed towards the OM, this results in reduction of the amount of cellulose
extruded.
Considering that all the Acs proteins are encoded by genes, which are part of a
single operon, the octamerization of the AcsD, poses a question about the oligomerization
status of the other Acs proteins. Since the antibodies against all the four proteins have

109
been generated, as part of my work for this thesis, the quantification of these proteins, can
inform us about the stoichiometry of these proteins in the G. hansenii cells.
CONCLUSIONS
We have shown that the AcsD protein exists as an octamer in solution and
assumes a cylindrical structure, with a central pore. Although the crystal structure of this
protein has been determined before our work, we have obtained a fair idea of how the
protein exists under solution conditions. The various arrangement of the end chains in the
structure suggests the possibility that the protein is capable of assuming different
conformations in solution, a fact that cannot be seen in the crystal structure of the protein.
However, the arrangement of the monomers around a central pore in the form of two
stacked tetrameric rings, is in agreement with the crystal structure of the protein.
Facilities that were used for this work:
1. Use of the Advanced Photon Source at Argonne National Laboratory was supported by
the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences,
under Contract No. DE-AC02-06CH11357 and project ID APS ESAF 18-ID-2010.
2. Analytical Ultracentrifugation was performed at the Biotechnology Bioservices Center
in the University of Connecticut. The samples were shipped, on ice, and all the analysis
was done by Lary, J. W.

110
CHAPTER V
BIOCHEMICAL CHARACTERIZATION OF ACSAB PROTEIN,
THE CELLULOSE SYNTHASE OF G. HANSENII 23769
INTRODUCTION
Bacterial cellulose synthase is composed of a catalytic domain and (Lin, Brown et
al. 1990) a regulatory domain (Mayer, Ross et al. 1991). The regulatory domain binds to
cyclic di GMP, which has been shown to be the allosteric activator of this protein (Ross,
Weinhouse et al. 1987; Mayer, Ross et al. 1991). The catalytic domain binds to the
substrate UDP-glucose and polymerizes glucose units processively to form a cellulose
chain. This subunit contains the D,D,D and QXXRW motif (Saxena, Brown et al. 1995;
Saxena and Brown 1997; Saxena and Brown 2000; Saxena, Brown et al. 2001), which is
a characteristic feature, conserved in all glycosyltransferases that use a nucleotide sugar
as a glycosyl donor (Saxena, Brown et al. 1995). In most bacterial species, the different
subunits of cellulose synthase are encoded by two or more genes. In Enterobacteriaceae
species like Salmonella typhimurium (Hatta, Baba et al. 1990) and Escherichia coli
(Hatta, Baba et al. 1990; Perna, Plunkett et al. 2001), the ORFs bcsA and bcsB (bcs:
bacterial cellulose synthase) encode the catalytic and regulatory subunits of the protein,
respectively. In case of Gluconacetobacter, it has been shown that G. xylinus NBRC
3288 (Ogino, Azuma et al. 2011) contains three ORFs coding for the cellulose synthase,
while the strains JCM7664 (Umeda, Hirano et al. 1999) and 1306-3(Wong, Fear et al.
1990) contain acsA and acsB as two distinct ORFs encoding for cellulose synthase.
However, in the strains ATCC 23769 and ATCC 53582, the genes are fused to form one
open reading frame (ORF).

111
Cellulose synthase purified using product entrapment from A. xylinum 53582, was
shown to be composed of 93 kDa and 83 kDa polypeptides (Lin 1989). Photolabeling
studies using radioactive UDP-glucose as the substrate demonstrated that the 83 kDa
polypeptide is the substrate-binding domain of the enzyme (Lin, Brown et al. 1990). The
gene encoding this catalytic subunit of the protein was identified by sequencing the
protein (Wong, Fear et al. 1990) and analysis of the cellulose-deficient mutants (Wong,
Fear et al. 1990). Based on biochemical and sequencing data, the 93kDa protein was
proposed to be the cyclic di GMP-binding protein and was suggested to be associated
with the CM (Bureau and Brown 1987; Saxena, Kudlicka et al. 1994). However, using
freeze-fracture labeling techniques with antibodies raised against the 93 kDa protein, this
protein is localized in the protoplasmic fracture (PF) face of the OM and concluded to be
a CM protein (Kimura, Chen et al. 2001).
Mayer et al. (Mayer, Ross et al. 1991) showed that in strain 1306-21, cellulose
synthase is composed of three major peptides of molecular weight 90, 67 and 54 kDa.
The gene encoding the 90 kDa polypeptide was cloned and expressed in E. coli (Mayer,
Ross et al. 1991). It was found in Western blots using these antibodies generated against
these polypeptides that the 67 and 54 kDa peptides were cleavage products of the 90 kDa
polypeptide, proving that there is a possibility of post-translational processing of the
cellulose synthases. AcsB/ BcsB was found to bind to the activator cyclic diGMP, which
led to the general agreement that the B subunit is the regulatory domain (Amikam and
Benziman 1989; Mayer, Ross et al. 1991). However, recently this regulatory role for
AcsB has been questioned (Amikam and Galperin 2006).

112
In case of the strain of Acetobacter ATCC 23769, which has been the center of
our work, the acs operon harbors a single gene (acsAB) encoding cellulose synthase. This
feature of fused-ORF is also seen in the other two cellulose synthase genes in the genome
which are not part of an operon. The polypeptide sequence of the translated product
shows that the encoded protein contains 1550 amino acids and has a molecular weight of
168,161 kDa. This protein is predicted to contain 10 transmembrane helices (TMHs) and
also two large globular non-membrane-bound regions (Figure 5.1).
In this work we have heterologously expressed and purified the non-membrane
bound regions of the protein. Using specific antibodies against these regions, we have
found that the protein is processed into three polypeptides. The polypeptide of 45 kDa
containing the catalytic domain localizes in the CM, but the larger polypeptide (95 kDa)
localizes in the OM. We have also shown that this polypeptide does not contain the PilZ
domain. Instead, the short stretch of 34 kDa region between the AcsA and AcsB subunits
harbors the regulatory, cyclic di-GMP-binding PilZ domain. In addition to determining
this processing pattern, we have also determined the organization of the Acs proteins in
the membrane compartment.

113
1 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 1500 1550
CesA 133 364
355 471 555 653
Glycosyltransferase PilZ
P1 P2
Figure 5.1 Depiction of the heterologously expressed regions of the AcsAB protein
The polypeptide sequence of the AcsAB protein was analyzed using the TMHMM
software tool (Krogh, Larsson et al. 2001) in order to identify the transmembrane-bound
and non-membrane-bound regions. The AcsAB protein sequence is depicted in the form
of a black line with the transmembrane helical regions as white boxes. The conserved
cellulose synthase (CesA), glycosyltransferase and PilZ domains, identified using the
Uniprot software (Consortium 2012), are indicated in grey boxes. The sequences
between 129 - 397 and 568 -1510, do not contain any membrane spanning domains.
Polypeptide sequences selected from these non-membrane spanning regions were
heterologously expressed and used for antibody generation, are shown as AcsAB1 and
AcsAB2. The peptide regions, P1 and P2 were selected for synthesis and antibody
generation. The numbers flanking the lines and boxes, show the position of N- and C-
terminal position residues of the polypeptides with respect to the full-length protein.
MATERIALS AND METHODS
Cloning and heterologous expression of the acsAB gene regions
Two major non-membrane-bound regions of the AcsAB protein were identified
using prediction tools, TMHMM (Krogh, Larsson et al. 2001) and HMMTOP (Tusnady
and Simon 1998; Tusnady and Simon 2001) as shown in Figure 6.1. The procedures for
cloning and transformation of the gene regions coding for the non-membrane-bound parts
129 397
AcsAB
610
581 600 721 740
1550 AcsAB2

114
of the protein (depicted in Figure 5.1), was performed using the procedures described in
Chapter III for AcsD expression. Using the primer pairs (5'-gctagcatgttccagacgatcgcgccg-
3' and 5’-ctcgagccgctggcccccatgacag-3’), containing the Nhe1 and Xho1 restriction site,
the acsAB1 was amplified from G. hansenii cells, by colony PCR (Figure 5.2). Similarly
primers, 5'-gctagcgccgcggtaaagatgtcatgg-3' and 5’-ctcgagcgacttgcgcctct-3’, were used to
amplify the acsAB2 gene fragment. The PCR products was ligated into the pGEM-T easy
vector and digested with Nhe1 and Xho1 restriction enzymes, to enable cloning into the
pET-21a vector (Novagen).
Expression of the AcsAB soluble regions
Expression vectors were transformed into the BL-21 (DE3) cells and plated on
LB-ampicillin plates to isolate colonies containing the plasmid. A single colony was
inoculated from this plate into a culture volume of 5ml in a shaker flask and cultured
overnight at 30C and 220 rpm. This culture was used to inoculate a liter of LB 50 μg/ml
of ampicillin. The cultures were grown at 37 C at 220 rpm until the absorbance at 600
nm reached 0.4-0.6. At this point, the protein expression was induced by adding IPTG to
a final concentration of 1 mM and continuing the culture for 3h. Samples of induced and
un-induced culture (20 μl) were subjected to SDS-PAGE to confirm protein expression
(Figure 6.3). The cells were harvested by centrifugation at 5,000 × g for 15 min at 4 C.
The cell pellet was resuspended in 5 ml of lysis buffer (100mM NaH2PO4, 10 mM Tris-
Cl, 8 M Urea pH 8.0) per gram of wet weight and lysed by stirring for 60 min at room
temperature. The lysate was centrifuged at 2,500 × g for 30 min at room temperature to
pellet the cell debris. For SDS-PAGE, 20 μl of the supernatant was boiled in 5 × SDS-
sample buffer and subjected to electrophoresis.

115
Protein purification using denaturing method
The protein concentration of the supernatant obtained after cell-lysis, was
determined by Bradford (Bradford 1976). The supernatant was mixed with 50% Ni-NTA
slurry at a concentration of 1ml of resin per 10 mg of protein. The lysate was allowed to
equilibrate with the resin by incubation on a rotary shaker for 60 min at room
temperature. The lysate-resin mixture was loaded onto a column which was washed with
an equal volume of wash buffer (100 mM NaH2PO4, 10 mM Tris-Cl, 8 M Urea pH 6.3).
and proteins were eluted by passing 0.5 ml of elution Buffer 1 (100 mM NaH2PO4, 10
mM Tris-Cl, 8 M Urea pH 5.8) four times and elution Buffer 2 (100 mM NaH2PO4, 10
mM Tris-Cl, 8 M Urea pH 4.5) four times through the column. All the fractions (flow
through, wash and elutions) were analyzed by SDS-PAGE and saved in -20C.
Expression and purification of AcsAB2 was performed in a similar manner (Figure 6.3
b).

116
Figure 5.2 Agarose gel of amplified products of acsAB gene regions.
PCR amplification of the 828 bp acsAB1 (Lane 2) and 2443 bp acsAB2 (Lane 2) gene sequences
was done from G. hansenii cells using colony PCR. Molecular weight ladder was loaded in lane
1 and lane 3 is an empty lane.
5
06 bp
1000 bp
3000 bp
1636 bp
828 bp
2443 bp bp
500 bp
1 2 3 4

117
a b
Figure 5.3 SDS-PAGE of heterologously-expressed AcsAB polypeptides
a) SDS-PAGE analysis of with protein fractions from AcsAB1 expression and purification: 1: Uninduced cell pellet; 2: Induced cell pellet; 3, 4: Flow through after Ni-NTA purification; 5,
6: Washes from Ni-NTA column; 7, 8, 9: Elutions containing pure AcsAB1 at 31kDa; 10:
Molecular weight ladder.
b) SDS-PAGE analysis of with protein fractions from AcsAB2 expression and purification: 1: Molecular weight ladder; 2: Uninduced cell pellet, 3: Empty lane; 4: Induced cell pellet. 5, 6:
Flow through after Ni-NTA purification; 7: Wash from Ni-NTA column; 8, 9: Elutions
containing pure AcsAB2.
Antibody generation, purification and Western blot
The polypeptides were subjected to SDS-PAGE and the bands were cut from the
gel and sent to Covance research products for antibody generation in rabbits. Antibodies
were also generated against two peptide regions in the protein, that were predicted to be
antigenic as well as non-homologous to any other protein in the cells. The anti-AcsAB1
and anti-AcsAB2 antibodies were affinity purified as described in Chapter III. Peptide
antibodies were affinity purified by using Sulfolink agarose G-resin following the
procedures given in the Sulfolink immobilization kit for peptides (Thermo Scientific,
Pierce Research products). All antibodies were stored as 1ml aliquots at -70C. Western
blots were performed using standard procedures using 20 µg of TM or whole cell
proteins.

118
Sucrose density gradient centrifugation
TM was isolated as described in Chapter III. A total of 12 mg of TM protein was
loaded onto a 35-ml 25%-55% discontinuous sucrose gradient. The gradient was
centrifuged for a duration of 16 hours at 177,500 × g. The fractions from the gradient
were collected manually and stored at -70C as 3 ml aliquots. In order to locate the Acs
proteins in these fractions, 50 μl of each fraction was boiled in 10 μl of 5× SDS-sample
buffer, and subjected to SDS-PAGE followed by Western blotting using anti- AcsAB1,
anti-AcsAB2, anti-C and anti-D antibody.
RESULTS
Predicting the soluble regions of AcsAB protein
The amino acid sequence of AcsAB was analyzed by the TMHMM software
(http://www.cbs.dtu.dk/services/TMHMM/). The software predicted two major non-
membrane-bound regions and ten TMHs in the protein. The outcome of this prediction is
shown in Figure 5.1. Based on the predictions, there are two major regions devoid of
TMHs. These are between amino acids 129 - 397 and 568 - 1510. The region from 129 -
397 contains the conserved QRVRW motif for glycosyltransferases (Saxena, Brown et al.
2001) as well as the domain (DXD, D) (Saxena, Brown et al. 1995; Saxena and Brown
1997; Saxena, Brown et al. 2001), residues involved in substrate binding.
We amplified the portions of the acsAB gene, which encode for the predicted
non-membrane bound parts of the protein and heterologously expressed and purified
these polypeptides. The Coomassie-stained gel showing the SDS-PAGE analysis of the
crude and purified proteins is shown in Figure 5.3. The two heterologously expressed

119
regions of the protein are a 31 kDa AcsAB1, that contains the regions between amino
acids 129 - 397 and a 102 kDa-AcsAB2, which contains amino acid sequence from 610 –
1550. Both these polypeptides were expressed with a C-terminal hexa-histidine tag.
These purified polypeptides were then used as antigens to obtain polyclonal antibodies to
be used in Western blots, to locate cellulose synthase proteins in the G. hansenii whole
cell and TM.
Western blot using anti-AcsAB1 and anti-AcsAB2 antibodies
The antibodies against AcsAB1 and AcsAB2 were expected to cross-react with a
single 168 kDa-protein, in Western blot of G. hansenii whole cell and TM proteins.
However, as shown in Figure 5.4a, the anti-AcsAB1 antibody identifies a protein with
molecular weight between 45-52 kDa, in the Western blot. The precise molecular weight
of the this polypeptide was calculated to be 46 kDa, by plotting the logarithm of the
molecular weight against the distance of migration of the protein bands, using the
commercial molecular weight marker as the standard (Figure 5.5). The anti-AcsAB2
antibody binds to a 95 kDa band in whole cells. In case of the AcsB2 fragment, the
molecular weight directly matched to the 95 kDa band, as seen in Figure 5,4b, and
therefore a graph was not used to calculate the molecular weight. This indicates that the
AcsAB protein is cleaved into at least two polypeptide fragments of molecular weights
46 kDa (AcsA) and 95 kDa (AcsB). However, their molecular weight does not add up to
168 kDa, which is the calculated molecular weight of the entire protein encoded by
acsAB gene.
Western blots using anti-peptide antibodies

120
In order to further understand the cleavage pattern of the protein, antibodies were
generated against two synthesized-peptides corresponding to the selected regions of
AcsAB protein (Figure 5.1). The anti-peptide antibody was generated in order to find if
there are other processed fragments of this protein that had been left undetected by the
anti-AcsAB1 and anti-AcsAB2 antibodies. The two synthesized peptides correspond to
twenty amino acid-regions between residues 581- 600 and 721 - 740, in the full-length
protein (shown as blue lines in Figure 5.1). One of the peptides (P1), spans the region
between 581 to 600, in the AcsAB protein, that lies between the expressed-AcsAB1 (129
- 397) and AcsAB2 (610 - 1550) polypeptides. The other peptide (P2), spans the region
from 721 to 740 within the AcsAB2 polypeptide (610 - 1550). The antibody generated
these peptides (anti-P1P2) was expected to potentially identify the sequence not
identified by the anti-AcsAB1 and anti-AcsAB2 antibodies. Both peptides were injected
in one rabbit and therefore, the anti-serum contains antibodies against both the peptides.
The antiserum was affinity purified to using both P1 and P2 peptides.
When the affinity purified anti-P1P2 antibody was employed in a Western blot
against whole cellular proteins, the predominant bands seen on the membrane were of
molecular weights in 95 kDa and 34 kDa, as shown in the Figure 5.4c. This antibody
does not cross-react with heterologously expressed AcsAB1 but detects the AcsAB2 band
in the Western blot. This indicates that the 95 kDa protein recognized by anti-AB2 and
anti-P1P2 antibody are the same polypeptide, but the 34 kDa band recognized by anti-
P1P2 antibody lies in between the 45 kDa and the 95 kDa polypeptide sequences.

121
a b c
Figure 5.3 Western blot using specific polypeptide and peptide antibodies
a) Anti-AcsAB1 antibody recognizes a band between 42 kDa and 52 kDa in both whole
cells (Lane 2) and TM (Lane 3). Lane 1 is the molecular weight ladder. Lane 4 contains
the heterologously-expressed and purified AcsAB1.
b) Anti-AcsAB2 antibody binds to a protein of molecular weight 95 kDa, in both whole
cells (Lane 1) and TM (Lane 2). This band. Lane 1 contains the molecular weight ladder.
Lane 4 contains the heterologously-expressed AcsAB2 protein as a positive control.
c) Anti-peptide antibodies recognize a band at 95 kDa and a 34 kDa band in whole cells
(lane 2). The anti-peptide antibodies do not bind to pure AcsAB1 protein (lane 3) but
recognize the 95 kDa AcsAB2 band (lane 4).
y = -0.0305x + 2.8835
R2 = 0.9928
0
0.5
1
1.5
2
2.5
3
0 10 20 30 40 50 60
Distance (mm)
Lo
g M
W

122
Figure 5.5 Graph for Molecular weight determination of processed AcsA polypeptide.
Standards loaded in the gel were from the commercially available molecular weight ladder
(Fermentas Spectra MulticolorBroad Range Protein ladder). The molecular weights of the ladder
were 260k Da, 135 kda, 95 kDa, 72 kDa, 53 kDa, 42 kDa, 34 kDa, 26 kDa, and 17 kDa.
Substituting the value of x in the line equation for the distance of migration of the cellular AcsA
band (30mm) (lanes 1 and 2 in Figure 5.4a), we get the number 1.967, which corresponds to the
molecular weight of 46 kDa. The distance of migration of the pure AcsA protein (45.5 kDa) was
also calculated by substituting the value of x in the equation. The calculated molecular weight of
the pure protein (31.3 kDa) matched that of the molecular weight derived from the protein
sequence (31.5 kDa).
Sucrose density gradient centrifugation
Cellulose synthase protein has been localized previously to the CM compartment of the
bacterial cells (Bureau and Brown 1987; Kimura, Chen et al. 2001). We wanted to explore if
both AcsA as well as AcsB polypeptides were localized in this membrane compartment. We
therefore subjected the TM to sucrose density gradient in order to separate the CM and OM
fractions. After the high-speed ultracentrifugation at 82,500 x g, the TM is fractionated into CM
and OM compartments which distribute themselves in the sucrose gradient based on their
density. The CM was a slightly yellow band at 35% density and OM is seen as a denser, white
band at 45% density. When aliquots from the sucrose density gradient were collected, majority
of the CM band was in second aliquot and the major portion of the OM was in aliquots 8 and 9.
When all the fractions were subjected to Western blot, AcsA and AcsC are seen
predominantly in the CM and OM respectively, as was expected. AcsD is distributed throughout
the gradient. AcsB is also found through-out the length of the gradient, but a greater amount of
the protein, as indicated by the thickness of the cross-reacting band in the Western blot, is in the
fractions where AcsC is present. This implies that the AcsB is not in the same membrane
compartment as AcsA. This protein is either periplasmic with a strong association with the OM,
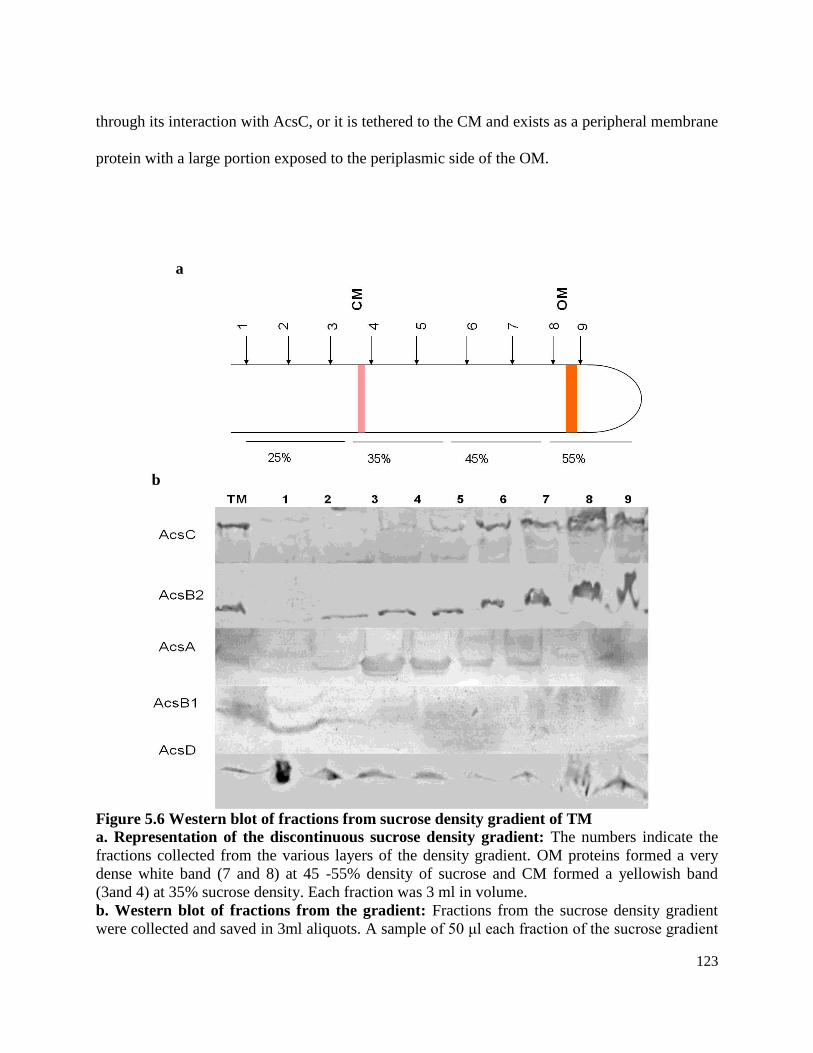
123
through its interaction with AcsC, or it is tethered to the CM and exists as a peripheral membrane
protein with a large portion exposed to the periplasmic side of the OM.
a
b
Figure 5.6 Western blot of fractions from sucrose density gradient of TM
a. Representation of the discontinuous sucrose density gradient: The numbers indicate the
fractions collected from the various layers of the density gradient. OM proteins formed a very
dense white band (7 and 8) at 45 -55% density of sucrose and CM formed a yellowish band
(3and 4) at 35% sucrose density. Each fraction was 3 ml in volume.
b. Western blot of fractions from the gradient: Fractions from the sucrose density gradient
were collected and saved in 3ml aliquots. A sample of 50 μl each fraction of the sucrose gradient

124
was subjected to SDS-PAGE. Western blot using specific antibodies against Acs A, B,C, D
proteins, shows that the AcsC protein is seen predominantly in the OM compartment (lanes 7 and
8) and AcsA in CM fraction (lanes 2, 3). A dense band of AcsB was observed in the fractions in
which AcsC was present, indicating the association of the AcsB protein with the OM. TM
(control lane) contains a 10 μg TM protein to serve as positive control.
DISCUSSION
Our results using antibodies against the different regions of cellulose synthase show that
there are predominantly three polypeptides generated by the processing of this protein. These
polypeptides are of molecular weight 46 kDa, 34 kDa and 95 kDa. We have named these
processed polypeptides as AcsA, AcsB1 and AcsB2 respectively. Since such a processing of
cellulose has not been shown before, we searched for the cellulose synthse sequences from other
bacterial species to locate subunits of the protein that are similarly processed. We found that in
Escherichia coli str B171 (Perna, Plunkett et al. 2001; Aziz, Bartels et al. 2008) and
Xanthomonas campestris sp. (Thieme, Koebnik et al. 2005; Aziz, Bartels et al. 2008), the
cellulose synthase is encoded by three ORFs. The three ORFs encode 3 polypeptide regions of
the protein. The first of the three genes codes for the catalytic subunit containing the D, D, D
and QXXRW motifs. The protein encoded by the second gene contains the PilZ domain and the
third gene codes for the largest polypeptide whose function is unknown, though it is classified as
a BscB protein.
We further explored the sequence of G. hansenii cellulose synthase in the light of the
calculated molecular weights of the polypeptides and tried to find if the protein is processed in a
manner identical to the ones from the E. coli (Perna, Plunkett et al. 2001) and X. campestris
(Thieme, Koebnik et al. 2005). Heterologously expressed AcsAB1 is a 31 kDa protein that spans
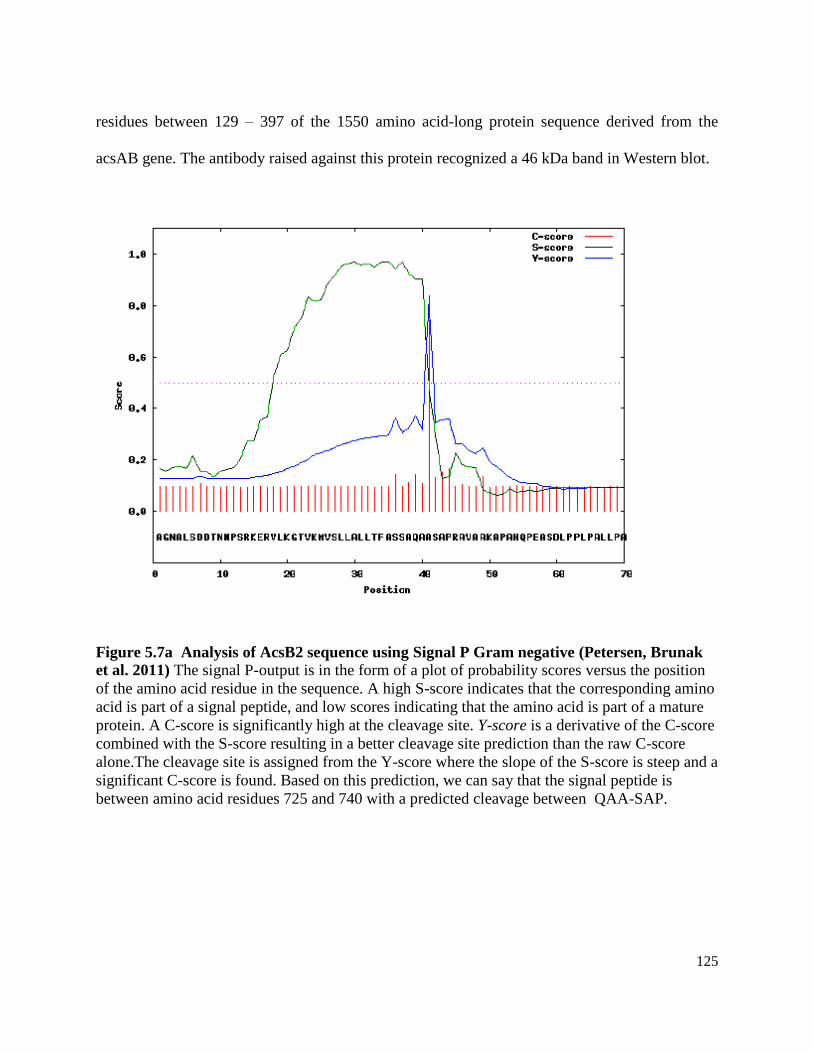
125
residues between 129 – 397 of the 1550 amino acid-long protein sequence derived from the
acsAB gene. The antibody raised against this protein recognized a 46 kDa band in Western blot.
Figure 5.7a Analysis of AcsB2 sequence using Signal P Gram negative (Petersen, Brunak
et al. 2011) The signal P-output is in the form of a plot of probability scores versus the position
of the amino acid residue in the sequence. A high S-score indicates that the corresponding amino
acid is part of a signal peptide, and low scores indicating that the amino acid is part of a mature
protein. A C-score is significantly high at the cleavage site. Y-score is a derivative of the C-score
combined with the S-score resulting in a better cleavage site prediction than the raw C-score
alone.The cleavage site is assigned from the Y-score where the slope of the S-score is steep and a
significant C-score is found. Based on this prediction, we can say that the signal peptide is
between amino acid residues 725 and 740 with a predicted cleavage between QAA-SAP.

126
Figure 5.7b Lipo P prediction of a signal sequence in the AcsB polypeptide (Rahman,
Cummings et al. 2008) When the sequence of AcsB (642 - 1550) polypeptide derived from our
calculations, was analyzed by the LipoP software, the signal peptide (SP1) was predicted to be
between residues 736 - 745. The Y-axis shows probability of the signal peptide region in
logarithmic scale and the X-axis is the number of amino acid residue in the sequence of the
polypeptide. Since this software can predict only signal peptides in the first 30 amino acid
stretch, when the entire sequence starting from 694th amino acid residue was analyzed, it could
not detect the signal peptide.
Based on the results from the Western blot using the anti-peptide and anti-AcsAB2
antibodies, we conclude that AcsB1 is the regulatory domain containing from residues 409 to
693, with a calculated molecular weight of 32 kDa. This subunit harbors the PilZ domain (572 -
647) (Amikam and Galperin 2006; Ryjenkov, Simm et al. 2006) that binds to the c-di-GMP.
AcsB2 portion of the protein extends from amino acid residue 694 to 1550, with an exact
molecular weight of 91,633 kDa. These are predicted sites of the processing of the AcsAB
protein, based on molecular weight and cleavage pattern of similar proteins from other organisms
(Perna, Plunkett et al. 2001; Aziz, Bartels et al. 2008) and experimental evidences are required
for exact identification of the N- and C- termini of each polypeptide.
A
A
A
Q
P S
Q
A
R
A
A
V
K
725 735 745 755 765 775 785 Amino acid residues

127
In order to determine the exact site of cleavage and the sequences of these resultant
polypeptides we tried to isolate the 95 kDa band from the G. hansenii whole cells and determine
its sequence by N-terminal sequencing methods. However, we were unable to isolate sufficient
protein for required for the sequencing. Since the exact role of AcsB2 polypeptide is not known,
we used the specific antibodies generated against the AcsB2 region of the protein to locate this
protein in the membrane compartments. We found that this protein co-localizes with the AcsC
protein in the OM compartment. Our results are in consensus with the freeze-fracture study by
Kimura et al. (Kimura, Chen et al. 2001), which revealed that the 93 kDa-polypeptide associates
with the PF face of the OM. Though it was concluded from this finding that the protein is
localized in the CM, we believe that their results are directing towards the possibility of the
AcsB2 region to be in association or exposed to the OM.
To corroborate our finding from the sucrose density gradient, we analyzed AcsB2
polypeptide to find if there is a possibility of this polypeptide to exist in the periplasm or in
association with the OM-bound AcsC. When the sequence was analyzed using Signal P
(Petersen, Brunak et al. 2011) (Figure 5.7a) and Lipo P (Rahman, Cummings et al. 2008) (Figure
5.7b) software tools, we found that both the prediction software identify a signal peptide of 25
kDa in the sequence between the residues 725 to 740, with a cleavage site between residues 740 -
741. This implies that the AcsB2 polypeptide is either anchored to the CM with a large portion
exposed to the periplasm or is released into the periplasm. Another analysis software PSLpred
(Bhasin, Garg et al. 2005) predicts the substrate binding regions (133- 364) and PilZ domain
(555- 653) of the AcsAB sequence to be cytoplasmic, but identifies the of AcsB2 region (694 to
1550), to be periplasmic. More studies are required to exactly predict the nature of the signal
peptide and verify that it serves as a translocation signal. However, all the predictions

128
corroborate our finding, that the AcsB2 polypeptide is largely associated with the OM with a
major portion in the periplasm.
The existence of more than one polypeptide subunits in the cellulose synthases from all
bacterial species (Perna, Plunkett et al. 2001; Thieme, Koebnik et al. 2005; Ye, Lan et al. 2010),
proves that the functional cellulose synthase enzyme requires catalytic and regulator domains to
be harbored in distinct poylpeptides. Therefore, the functional and evolutionary significance of
the fused gene should be explored in G. hansenii strains 23769 and 53582 (Lin, Brown et al.
1990; Kawano, Tajima et al. 2002) given the fact that the enzyme still requires three polypeptide
subunits. There is much to be understood regarding the structure of the cellulose synthase and the
localization and organization of all the processed subunits of this proteins and how they come
together to contribute to the functioning of this enzyme. But, our data from this work leads us to
explore the cellulose synthase sequence in a new light, for structural studies as well as
organization of the cellulose synthase complex.
CONCLUSIONS
1. The cellulose synthase in the strain G. hansenii 23769, encoded by a single gene is processed
into three polypeptides of molecular weights 45 kDa, 34 kDa and 95 kDa. The sizes of these
polypeptides suggest that the AscA contains the substrate binding and catalytic regions. AcsB1
harbors the PilZ region and AcsB2 contains no conserved motifs in the sequence, and therefore
its role in the process of cellulose synthesis is yet to be understood.
2. Though catalytic activity of the cellulose synthase resides in the CM as shown before, the
AcsB2 portion of the protein is predominantly exposed to the periplasm.

129
CHAPTER VI
ISOLATION OF THE CELLULOSE SYNTHASE COMPLEX USING
ELECTROPHORETIC TECHNIQUES
INTRODUCTION
The bacterial cell envelope is a unique structure that performs several crucial
functions. Being the outermost boundary of the bacterial cells, it serves as a barrier that
protects the cell from external stress (Seltmann and Holst 2002; Talaro 2007). It also
provides the site for signal transduction (Greie, Hebestreit et al. 2003; Galperin 2005) and
synthesis reactions (Seltmann and Holst 2002; Talaro 2007). It performs the major task of
communicating with the surrounding milieu through selective transport of substances to
and from the cells (Seltmann and Holst 2002; Talaro 2007). The cell membrane is a
selectively permeable structure with specific carrier-mechanisms for import of nutrients
(Erni 2001) and export of metabolic products (Seltmann and Holst 2002; Talaro 2007).
Cellulose is one such metabolic product, which is released in to the extracellular
environment by the cells of the Gram negative bacterium, G. hansenii (Schramm and
Hestrin 1954). While it is not known why bacteria secrete cellulose, it is well known that
microbes secrete polysaccharides (Whitney, Hay et al. 2011) supposedly for support of
the microbial community (Ma and Wood 2009). The cell envelope of a Gram
negative bacterium (Brock 1999), is composed of two concentric phospholipid bi-layers,
the CM and the OM, separated by an aqueous, peptidoglycan-containing periplasmic
space (Filloux, Bally et al. 1990; Seltmann and Holst 2002; Saier 2006). Transport of
molecules in these bacteria therefore requires proteins that span the membrane
compartments and provide a passageway across these three layers (Filloux, Bally et al.

130
1990; Saier 2006). The transport systems required for toxins (Sheps, Zhang et al. 1996),
ions (Stintzi, Barnes et al. 2000; Greie, Hebestreit et al. 2003; Debut, Dumay et al. 2006;
Dumay, Debut et al. 2006) and proteins (Delepelaire and Wandersman 2001) have been
extensively studied and categorized into several types based on the structure and
organization of the secretion or assimilation system (Boos and Eppler 2001; Greie,
Hebestreit et al. 2003; Quinaud, Ple et al. 2007). However, much remains to be
understood about the synthesis and secretion mechanism of bacterial polysaccharides like
cellulose.
The cellulose synthase AcsAB was initially considered as an integral membrane
protein in the CM (Saxena, Lin et al. 1990; Saxena, Henrissat et al. 1995). But our work,
described in the previous chapter, has shown that AcsAB is processed into three
polypeptides (Chapter V) which span the CM and the periplasm. We have also shown
that AcsD is a periplasmic, octameric-protein with a central pore (Iyer 2010). Based on
its N-terminal signal sequence and secondary structure-predictions, AcsC is the outer-
membrane porin (Saxena, Kudlicka et al. 1994). It contains seven TPR motifs
(Consortium 2012) that are considered as regions for protein-proteins interactions and
serve as multi-protein scaffold in polysaccharide export complex (Whitfield and
Mainprize 2010; Whitney, Hay et al. 2011). The hypothesis that the Acs proteins form a
complex, has been proposed in the past (Chen and Brown 1996; Endler, Sanchez-
Rodriguez et al. 2010). The existence of a secretion system for other polysaccharides
(Whitney, Hay et al. 2011), also provides us clues towards the nature and composition of
the cellulose secretion complex. However, such a complex has so far, not been isolated
and characterized.

131
In order to explore the exact composition, structure and organization of the
cellulose synthesis and extrusion machinery, I initiated studies to purify this complex. I
also aimed to identify the core components of the cellulose synthase complex. We have
used a combinatorial approach of membrane protein solubilization using mild detergents,
and subsequent isolation by gel electrophoresis. We have compared the efficiency of two
detergents, dodecyl maltoside (DDM) and Triton X-100, in solubilizing total membrane
proteins of G. hansenii and identified that DDM is more efficient in isolating the
cellulose synthase complex as well in preserving its enzymatic activity. Our results from
blue-native gel (BN-PAGE) shows that all the proteins encoded by the acs-operon are
associated in a complex. This experiment also reveals that other proteins involved in the
cellulose biosynthetic pathway, from glucose 6-phosphate to UDP-glucose are also
associated with this complex.
MATERIALS AND METHODS
Bacterial culture conditions
A G. hansenii cell pellet was obtained from a 60 L culture of the bacterial cells in
SH medium (Schramm and Hestrin 1954), grown for 48 h at 30C. The culture
preparation was carried out in the “Shared Fermentation Facility”, Huck Institute of the
Life Sciences. After 48 h of culture, under agitated conditions, 320 g of cell paste was
obtained by centrifugation. This cell paste was stored at -70°C. All the membranes used
in this study were prepared with this frozen cell paste as the source of Acetobacter cells.
Solubilization of the TM proteins
TM preparations were obtained using the procedure described by Ruebush et al.
(Ruebush, Brantley et al. 2006) and were stored in 10mM Tris-Cl buffer, pH 8.2 in 10%

132
glycerol at -70°C. Isolation of the TM and its separation into CM and OM fractions were
performed according Chapter III. Protein concentrations were determined by method
described by Bradford (Bradford 1976).
For BN-PAGE experiments, membrane proteins were solubilized using the
methods of Wittig et al. (Wittig, Braun et al. 2006). TM aliquots containing 20 mg of
protein (6.6 ml) were diluted five-fold in 10 mM Tris-Cl, pH 8.6 and centrifuged at
100,000 × g for 30 min. The pellet obtained was resuspended in solubilization buffer (50
mM sodium chloride, 2 mM amino hexanoic acid and 1 mM imidazole pH 8.2) to obtain
a protein concentration of 10 mg/ ml. Dodecylmaltoside was added to obtain a final
concentration of 1g per g protein. With Triton X-100, it was added to obtain a final
concentration of 1.5 g per g protein. The samples were allowed to solubilize for 10
minutes, on ice. Glycerol was added to a final concentration of 5% (w/v). Coomassie G-
250 was added to give a protein to dye ratio of 8:1. When DDM was used as the
detergent, 50 µl of the Coomassie stock was added and 100 µl was added, if Triton X-100
was used. A similar procedure, as described above, was used when 20 mg of CM proteins
were subjected to BN-PAGE. In all the detergent-mediated solubilizations, DDM (final
concentration 1% w/v) and Triton X-100 (final concentration 1.5% w/v) were used in
concentrations much above their CMC values of 0.01% and 0.015%, respectively .
Native gel electrophoresis of the solubilized TM proteins
For zymogram experiments, proteins were solubilized using the same final
concentrations of detergent as described above. Solubilized TM, containing 120 µg
protein in final volume of 40 µl, was subjected to native polyacrylamide gel

133
electrophoresis (Harwood 2000) at 4°C, 5-10 mA constant current until the dye front
reached the bottom of the gel.
Zymography
After electrophoresis, individual lanes from the native gel were incubated for 16
hours at room temperature, the lanes were incubated in 10 ml of TME (50 mM Tris-Cl
pH 8.0, 10 mM MgCl2 and 1 mM EDTA) containing final concentrations of 20 µM UDP-
glucose, 1 µM c-di GMP, 20 mM MgCl2 and 5 mM CaCl2. The composition of the
reaction mixture was adapted from the buffer for an in vitro cellulose synthase assay, by
Mayer et al. (Mayer, Ross et al. 1991). For negative control, the gel lane was incubated
in a reaction mixture devoid of UDP-glucose. After overnight incubation, the gel lanes
were washed for 10 minutes in deionized water followed by staining in 10ml of 0.01%
Calcofluor for 10 minutes. Enzyme activity was confirmed by visualization of
Calcofluor-bound cellulose as a green-fluorescent band under UV light (Monheit, Cowan
et al. 1984).
Blue native polyacrylamide gel electrophoresis (BN-PAGE)
The procedure for BN-PAGE was adapted from the method described by Wittig
et. al. (Wittig, Braun et al. 2006). This procedure is briefly described in this section.
Gel composition: Acrylamide solution used for the first dimension of BN-PAGE
was prepared by mixing 48 g of acrylamide and 1.5 g of bis-acrylamide in 100 ml of
water. This is referred to as AB-3 mix (Hjerten 1962). The composition of the gel buffers
is provided in Table 6.1b. The gel was assembled using custom-made glass plates (16 cm
x 17.5 cm) of and spacers (0.1cm). A 4-15% gradient gel was poured between the glass
plates using a gradient maker. Once the gradient gel was polymerized, the 3.5% stacking

134
gel was poured and a custom-made comb with 5 wells of dimensions 2 cm x 2.5 cm x
0.1 cm, was placed in it. After polymerization, the comb is removed and the wells are
overlaid with 1X gel buffer (Table 6.1b). The gel is covered with wet paper towels and
plastic wrap and saved in 4°C until further use.
Electrophoresis conditions: The anode chamber and cathode chambers were
filled with respective buffers. The composition of these buffers is provided in Table 6.1b.
A volume of 500 µl of solubilized TM sample was loaded into each well. BN-PAGE was
conducted at 4°C with the power supply set at 100 V until the samples entered the gel.
Electrophoresis was continued beyond this point at a voltage of 500 V with current
limited to 15 mA. The undiluted cathode buffer (Table 6.1b) was replaced by the diluted
cathode buffer after the dye front reached one-third of the total gel length.
Electroblotting of proteins from first dimension BN gel: After electrophoresis,
the gel was carefully removed from the cast and the lanes were separated from one
another using a gel cutter. The individual lanes were then subjected to a second
dimension denaturing PAGE or Western blot. Western transfer was carried out at a
constant current of 30 mA for 16 hours.
Second dimension SDS-PAGE: The gel strip from the first dimension was
soaked for 60 min in 100 mM Tris-Cl pH 8.0 containing 1% SDS and 1%
mercaptoethanol. The strip was washed briefly with water, placed horizontally between
two gel plates, and assembled such that the gel strip was towards the bottom of the plate.
This was done to ensure ease of pouring a gel without inducing air bubble-formation. The
4% acrylamide solution was poured first to form a layer of stacking gel immediately
above the native gel piece and after polymerization of this gel, the resolving gel was cast.

135
After polymerization, the cast was turned the right way up, to obtain the BN-gel strip on
the top. The gel was subjected to electrophoresis at initially a constant voltage of 100 V
and the voltage was increased to a 300 V after the dye front reached the resolving gel.
Once the dye front reached the bottom of the resolving gel, the electrophoresis was
stopped and the gel was stained overnight with a solution of colloidal Coomassie stain
(Neuhoff, Arold et al. 1988).
Table 6.1a: Composition of polyacrylamide gradient BN-gel*
Stacking gel Gradient Resolving gel
3.5% acrylamide 4% acrylamide 15% acrylamide
AB-3 mix 0.44 ml 1.5ml 4.45 ml
Gel buffer 3X 2.0 ml 6.0 ml 5 ml
Glycerol - - 3.0 g
Water 3.4 ml 10.4 ml 2.55ml
10%APS 50 µl 100 µl 100 µl
TEMED 5 µl 10 µl 10 µl
Total volume 6 ml 18 ml 15 ml

136
Table 6.1b: Buffers for BN-PAGE*
Cathode
buffer
Dilute cathode
buffer
Anode
buffer Gel buffer
Tricine (mM) 50 50 - -
Imidazole (mM) 7.5 7.5 25 75
Coomassie blue G-250
(%) 0.02 0.002 - -
6-aminohexanoic
acid (M) - - - 1.5
*Compositions of the gel and buffers are adapted from the methods described by Wittig
et al. (Wittig, Braun et al. 2006).
In-gel digestion of gel bands and spots: The bands visible after the first
dimension BN-PAGE or the stained gel spots from second dimension PAGE, were
analyzed by LC/MS. In-gel digestions were done according to the instructions provided
by the Proteomics core facility, Hershey, PSU. Briefly, the procedure involved excision
of a spot or band from the gel and dicing it using a clean unused razor blade into
approximately 1-3 mm pieces. The pieces from individual spots/ band were taken in a
labeled Eppendorf tube and destained with 200 µl of 100 mM ammonium bicarbonate in
50% acetonitrile, for 10 min at 37ºC. The gel pieces were dehydrated by incubation in 50
µl of acetonitrile for 10 minutes followed by air-drying in a laminar hood for 30 minutes.
Proteins in the gel were reduced in 100 µl of 10 mM dithiothreitol in 25 mM ammonium
bicarbonate, pH 8.0, for 30 minutes, at room temperature. The reduced proteins were
alkylated by incubation for 30 minutes, in a solution of 100 µl of 20 mM iodoacetamide
in 25mM ammonium bicarbonate (pH 8.0). The gel pieces were again dehydrated as

137
described above, prior to adding a 20µl of 200 ng/µl of Promega sequencing grade
trypsin in 25 mM ammonium bicarbonate. The gel pieces were allowed to soak the
trypsin by keeping the tubes at 37ºC for 30 minutes, after which more ammonium
chloride was added to completely cover the gel slices, and the tubes were incubated at
37ºC. After 16 h of incubation, the tubes were spun for 30 seconds to collect all the gel
pieces at the bottom and the trypsin-containing solution was transferred into an
autosampler vial. The gel pieces were then sonicated in a solution of 50µl of 50%
acetonitrile and 5% formic acid, to extract the digested peptides. This extract was added
to the first extract in the vial. The samples were dried in a vacuum concentrator to a final
volume of approximately 10µl and submitted for LC-MS analysis in the Proteomics and
mass spectrometric core facility, University Park, PSU.

138
Figure 6.1. Zymogram of detergent-solubilized G. hansenii TM.
a. DDM-solubilized TM were subjected to electrophoresis under non-denaturing
conditions in a gel composed of 3% stacking and 6% resolving acrylamide. After
incubation in a reaction buffer containing UDP-glucose and c-diGMP, staining with
Calcofluor results in a fluorescent smear at the top of resolving gel (Lane 1). The staining
of the gel in colloidal Coomassie stain shows protein throughout the gel length (Lane 2).
b. Electrophoresis of the DDM-solubilized TM in a 4-15% gradient acrylamide gel
results in a more discrete band after Calcofluor staining (Lane 2). Similar band of lower
intensity (indicated by arrow) is observed when an equivalent amount of TM protein is
solubilized with Triton X-100 (Lane 2). However, when the gel lane is incubated in a
reaction mixture without UDP-glucose, no fluorescent band is seen in the same region
after Calcofluor staining (Lane 1).
b
1 2 3 1 2
a

139
RESULTS
Zymogram of in vitro cellulose synthesis
Native-gel electrophoresis of solubilized TM was performed with a 3% stacking
and 6% resolving gel (Figure 6.1a). A total of 120 µg of TM protein was loaded in each
well. After electrophoresis, one lane of the gel was incubated with UDP-glucose and
cyclic di-GMP in Tris-buffer pH 7.0, as described in Materials and Methods After
overnight incubation, at room temperature, the gel was stained with Calcofluor which
visualized a cellulose band (Lane 1). The adjacent lane from the gel (Lane 2) was
visualized for protein with Coomassie. The two visualization methods are consistent
with a large moleccular complex actively catalyzing cellulose synthesis after
electrophoretic separation..
Due to the instability of the 3% portion of the gel, the electrophoresis was again
performed with a 4-15% gradient gel (Figure 6.1b). Unlike the smear obtained in a 6%
gel, proteins migrated in a more discrete pattern in the 4-15% gradient gel. No cellulose
was detected in the absence of UDP-glucose (lane 1). Solubilization of the same amount
of protein using Triton X-100 results in a fainter band after zymogram reaction and
Calcofluor staining (lane 3).
Selection of an efficient detergent for BN-PAGE
The zymogram described above suggested that an active cellulose-synthesizing
complex is stable to electrophoresis. As such, we used BN-PAGE techniques to attempt
identification of the proteins involved in this complex. In order to identify the better
detergent of the two, both TM and CM were solubilized with DDM and Triton X-100, as
described in the "Materials and Methods". The comparative images of all four second

140
dimension denaturing gel are shown in Figure 6.2. There are six major complexes in
these gels as indicated by the five rows formed by the vertically aligned spots. The most
number of spots are obtained in the gel containing TM proteins solubilized with DDM
(Figure 6.2a). When this gel is compared to that of Triton X-100 solubilized TM (Figure
6.2b), the number and intensity of spots was found to be greatly reduced, however, the
separation of the proteins still follows the same pattern, in the form of six major
complexes. When the second dimension gel with CM proteins, is compared to the that of
the TM, several spots in each vertical row are absent, indicating that those spots
correspond to OM proteins in TM. The triton-X-100 solubilized CM does not show any
discernable protein spot. We aimed to identify the profile of CM and OM proteins by
analyzing the spots in the CM and TM gels. MS-based analysis of the trypsin-digested
spots from the second dimension gel were uninformative, due to abundance of keratin
and/ or very low confidence of proteins detected. Although the second dimension SDS-
PAGE did not give any information using MS-analysis, we used the gel profiles as
indicators to identify DDM as the more efficient detergent.
BN-PAGE of DDM-solubilized TM
We performed BN-PAGE of DDM-solubilized TM. A representative gel from the
first dimension of a BN-gel is shown in Figure 6.3. Adjacent second and third lane from
the same gel were visualized by Western blot using anti-AcsA and anti-AcsD antibody.
Both Western blots showed a band in the same position of the blots.

141
Figure 6.2 Comparison of the second dimension gel profiles
a.TM solubilized with DDM, b: CM solubilized with DDM, e. TM solubilized with
Triton X-100 and d. CM solubilized with Triton X-100. The orientation of the gel
from BN PAGE is indicated by the horizontal arrow with the arrowhead pointing towards
the bottom of the BN-gel. The vertical arrows indicate the proteins that migrate along a
linear path, indicating that these proteins are part of a complex
b
d
a b
a
c
a
d
a

142
The BN-PAGE band aligning with the bands in the Western blots, was subjected
to In-gel digestion followed by LC/MS analysis. The proteins identified by LC-MS are
listed in the Table 6.2. This band contains proteins relevant to the cellulose-biosynthetic
pathway (Swissa, Aloni et al. 1980), in addition to other proteins in the cell. Of the
operon-encoded proteins, only cellulose synthase is detected by this method. AcsD
protein was detected in the Western blot, but not detected in MS analysis. Since
antibodies are more sensitive and accurate than MS-based detection methods, presence
of AcsD in the gel band is well-supported. Thus, other than AcsC, (for which the
antibody was not available at the time of this work), we observe all the proteins that were
known to be involved in cellulose synthesis, in this BN-gel band.
Figure 6.3 BN PAGE of G. hansenii TM a) Lane 1 of the first dimension BN-gel shows
several bands.
a) When the BN-gel lane is lined up with the Western blotted lanes from the same gel, it can
be seen that the cross-reacting bands in the Western blot align against each other, as well as
with, one of the bands in the first dimension gel.
a
1 2 3

143
Table 6.2 Proteins detected after LC-MS of the BN-gel band
The band in the BN-PAGE gel (shown in figure 6.2b) was in-gel digested and analyzed
by LC-MS. The G. hansenii 23769 genome was used as the reference database. PLGS
(Protein Lynx Globak server) is a statistical measure of accuracy of the peptide
assignement with a higher score indicating a higher confidence of proteins identity
(Rosenegger et al. 2010). Proteins that are known to be involved in the cellulose
synthesis pathway are indicated in bold font.
Accession number Protein name PLGS score Coverage (%)
EFG85019.1
putative
phosphoribosylaminoimidazole
carboxylase catalytic subunit
6785.32 20.89
EFG83091.1 S-adenosyl L-homocysteine
hydrolase 5711.04 32.33
EFG82973.1 hypothetical protein GXY 15499 4678.76 33.70
EFG85152.1 putative glutamate synthase
NADPH large chain precursor 1918.27 46.30
EFG85151.1 putative oxidoreductase 1630.00 52.90
EFG85173.1 outer membrane protein OmpA 1345.23 47.19
EFG84008.1 UTP glucose 1 phosphate
uridylyltransferase 964.02 36.64
EFG85627.1 6 phosphogluconate
dehydrogenase like protein 813.62 41.14
EFG84192.1 phosphoglucomutase 580.85
40.87
BAC82543.1 cellulose synthase subunit AB 469.35 13.35
EFG85976.1 putative phosphoketolase 436.035 17.12
EFG85920.1 chaperone clpB 421.8425 55.23
EFG83882.1 succinate CoA transferase 266.56 41.70
EFG85957.1 hypothetical protein GXY 00199 225.61 20.3
EFG84948.1 import inner membrane translocase
subunit Tim44 170.62 10.13

144
DISCUSSION
Protein-protein interaction studies heavily rely on the availability of specific
identification techniques like Western blot (using antibodies against proteins of known
sequences), protein sequencing and mass-spectrometry. All these techniques in turn
require a database of known protein sequences and therefore by extension, require a fully
sequenced genome of the organism. In order to generate a genomic database for
proteomic studies, sequenced the genome of A. xylinum 23769 (described in Chapter II ).
Other than genome sequencing, another important contribution to all our
experiments described in this chapter, is the generation of antibodies against the acs-
operon-encoded proteins. Since we are specifically interested in the proteins contributing
to cellulose biogenesis antibodies were made against AcsA, AcsB, AcsC and AcsD
proteins. Our intention was to identify all the protein components of the cellulose
synthase complex; the more abundant and the unknown ones by mass spectrometry and
the known targets using Western Blot.
Since the acs operon-encoded proteins are predominantly membrane-localized, as
shown in Chapter I and Chapter IV, we have used selective detergent solubilization to
mildly separate the membrane proteins without disrupting the protein-protein interactions
in complexes. It has been shown that BN-PAGE allows high degree of separation of
membrane proteins under native conditions. The detergents enable solubilization of the
proteins in a manner that the integrity of protein complexes is not compromised, but
individual complexes are separated from one-another (Schagger and von Jagow 1991;
Wittig, Braun et al. 2006). Coomassie G-250 dye in the sample buffer remains tightly
bound to the proteins and imparts a negative charge to the complexes and serves to

145
maintain the otherwise hydrophobic membrane protein- complexes in a soluble form
(Schagger and von Jagow 1991; Camacho-Carvajal, Wollscheid et al. 2004). Both Triton
X-100 and DDM have been used as detergent of choice for BN-PAGE of membrane
proteins (Wittig, Braun et al. 2006). We found that when TM proteins that are solubilized
with DDM were subjected to BN-PAGE, distinct bands corresponding to each protein
complex can be seen (Figure 6.2a). Using specific antibodies against the AcsD and AcsA
proteins, we located the band in this BN-gel, that contained the cellulose synthase
complex. MS analysis of the BN-gel band, identified proteins from cytoplasmic as well
as membrane compartments (Table 6.2). The cytoplasmic enzymes detected in the MS-
analysis are phosphoglucomutase, UDP-glucose phosphorylase and cellulose synthase
(Swissa, Aloni et al. 1980). Phosphoglucomutase is the enzyme that moves the phosphate
group in glucose 6-phosphate from C-6 to C-2, to form glucose 1-phosphate. Glucose 1-
phosphate is converted to UDP-glucose, by the enzyme UDP-gluocose phosphorylase.
UDP-glucose is the nucleotide sugar substrate for the CM-bound cellulose synthase, that
poymerizes the sugar into cellulose chains (Swissa, Aloni et al. 1980).
Our finding suggests that the cellulose synthase complex contains, in addition to
the proteins encoded by the acs operon (AcsA and AcsD proteins detected by Western
blot), proteins involved in the synthesis of the substrate, UDP-glucose. This implies that
in G. hansenii, the cellulose complex is composed of proteins involved in synthesis as
well as secretion of the polymer. This feature is shared by the alginate synthase complex,
which contains, all the cytoplasmic proteins involved in synthesis of nucleotide sugar as
well as membrane proteins involved in alginate synthesis as part of a large secretion
complex.

146
Other than the proteins known to be involved in cellulose biogenesis, some other
proteins are also seen in this band. At this point we can only speculate about their
contribution to the cellulose synthesis pathway. OmpA protein has recently been shown
reduce cellulose production on hydrophobic surfaces through induction of stress response
system, in Escherichia coli cells (Ma and Wood 2009). The relevance of this proteins in
G. hansenii cellulose synthesis is yet to be understood.
In second dimension gels, it can be seen that there are proteins migrate as five
major complexes in the TM and CM gels. From comparing the profiles of these gels, it
can be seen that for both TM as well as CM solubilization, more protein spots are visible
on the gel, when the TM is solubilized with DDM. We found that the in-gel digestion and
subsequent LC-MS of the spots selected from the gels, gave us very poor quality data
with very low confidence. None of the proteins detected in these spots corresponded to
proteins known to be involved in cellulose synthesis. Thus, MS-based analysis of the BN-
PAGE data has been the biggest bottle-neck to our complete understanding of the
cellulose synthase complex.
Another form of native gel technique employed to characterize the cellulose
synthase complex was the zymogram method. We have used zymograms as a means of
visualizing the cellulose synthetic activity of the detergent-solubilized TM. This assay
detects the protein of interest by the virtue of its enzymatic activity by incubating the gel
with a suitable substrate and using a product-specific stain as an indicator (Lantz and
Ciborowski 1994; Martinez, Alarcon et al. 2000). When detergent-solubilized TM were
subjected to native gel electrophoresis and incubated in the cellulose synthesis enzyme
reaction mixture, the band gives fluorescence after staining with the cellulose-binding dye,

147
Calcofluor (Figure 6.1). There are several proteins in this band that are not associated with
cellulose synthesis and are merely present due to their comparable molecular weights. But,
more importantly, our results prove that bacterial cellulose synthase activity can be assayed
by zymogram method, without the use of radioactive reagents. Here again, DDM serves as
a detergent of choice, as solubilization of the same concentration of protein (120 µg) with
this detergent gives a brighter fluorescence after Calcofluor staining, than that obtained
after solubilization with Triton X-100.
It has also been shown previously that membrane fractions from A. xylinum when
incubated in presence of cyclic di GMP and UDP-glucose, synthesize cellulose (Bureau
and Brown 1987) and in vitro cellulose synthesis is possible even after inactivation of
AcsD and AcsC (Saxena, Kudlicka et al. 1994). With this study have proved that, these
proteins are nevertheless structurally associated with cellulose synthesis complex.
CONCLUSIONS
An efficient method for solubilization of the total membrane has been developed,
that ensures retention of the cellulose synthase enzymatic activity in vitro as well as
maintains the protein-protein interactions of the Acs complex. Our work has shown that:
i) BN-PAGE of DDM-solubilized membranes is an efficient method for isolation of the
minimal complex of proteins related to the cellulose biosynthetic pathway.
ii) Cellulose synthesis and secretion machinery is composed of a protein complex
spanning the cytoplasm and the membrane compartments. This complex is composed of
proteins encoded by the operon and those involved in the pathway from glucose 6-
phosphate to cellulose.

148
CHAPTER VII
SUMMARY AND FUTURE DIRECTIONS
My doctoral work has opened up several areas where research can be continued to
obtain more insights into the biochemistry of cellulose synthesis. One important
contribution to the study of bacterial cellulose synthesis, is the generation of antibodies
against proteins encoded by the cellulose synthase operon. The antibodies have
contributed towards some very insightful discoveries in the course of my work.
The antibodies against different regions of the cellulose synthase protein, revealed
the nature of processing of the cellulose synthase protein in vivo. We have shown that the
protein is processed into three polypeptides. The 45 kDa N-terminal polypeptide, AcsA,
contains the catalytic domain of the protein with substrate binding (Saxena and Brown
1997; Saxena and Brown 2000) and conserved glycosyl transferase motifs (Saxena and
Brown 1997; Saxena and Brown 2000). The PilZ motif is contained in the 34 kDa
polypeptide and is considered as the regulatory region of the enzyme. We have named it
AcsB1, to distinguish it from the C-terminal 95 kDa subunit, AcsB2, whose function is
unknown.
These antibodies have also led to the identification of the subcellular localization
of the protein subunits. We have found that, although AcsA is CM-bound, the AcsB2
protein is largely periplasmic in nature and contains a signal peptide (Rahman,
Cummings et al. 2008) which enables the transport of AcsB2, to this subcellular
compartment. MudPIT analysis and sucrose density gradient centrifugation, also provide
evidence for the association of this protein to the OM. With these findings, we have
shown that, the cellulose synthase in G. hansenii 23769, is composed of multiple

149
subunits, similar to the cellulose synthase in other bacterial species (Perna, Plunkett et al.
2001; Thieme, Koebnik et al. 2005). Thus, in spite of being encoded by a single gene, the
cellulose synthase from this organism contains the same multi-subunit architecture like
that of other bacterial species, which are encoded by more than one gene (Hatta, Baba et
al. 1990; Ogino, Azuma et al. 2011).
This finding directs us to enquire into the evolutionary significance of the single
gene in G.hansenii. This is also crucial for understanding the structure of this protein.
With the heterologously expressed AcsAB1 region, I have attempted solubilization and
crystallization trials, with initial success. The methods which led to crystal formation
were not amenable to scale-up. However, with the knowledge of the exact region of
cleavage of AcsAB protein, a new set of primers can be designed, that would amplify the
gene regions that code for the processed fragments of the protein. This would mimic the
cellular polypeptide in G. hansenii and could be a better candidate for structure
determination.
The Anti-AcsAB1 antibody can also be used for quantitation of this protein, for
purposes of arriving at the stoichiometry of AcsA and AcsD proteins in the cell, as well
as deriving the enzyme kinetics for cellulose synthase. In addition to the anti-cellulose
synthase antibodies, the anti-AcsD antibody helped to determine the periplasmic
localization of the AcsD protein.
Other than the antibodies, a major contribution of my work has been sequencing
the genome of G. hansenii 23769. This sequenced genome has been the quintessential
reference database for our proteomic experiments. MudPIT analysis of the TM, OM and
CM, has given us the proteomic profiles of these membrane compartments. These

150
proteome data are available for further enquiry into the components of these
compartments. We have solely concentrated on the cellulose synthesis-related proteins in
this proteome data and have found that the proteins involved in cellulose synthesis are
distributed in the CM and OM compartments unequally. We find a greater signal for the
AcsB protein from the OM compartment confirming our findings from Chapter V.
This genome has also been used for identification of proteins from BN-PAGE
bands. It has also contributed towards identification of binding partners in the cellulose
synthesis and extrusion complex. We now know that similar to the other polysaccharide
extrusion systems (Pecina, Pascual et al. 1999; Svanem, Skjak-Braek et al. 1999;
Vazquez, Moreno et al. 1999; Keiski, Harwich et al. 2010; Whitney, Hay et al. 2011), the
complex contains cytoplasmic proteins involved in the biochemical pathway from the
precursor of the sugar-nucleotide substrate to the cellulose. This complex also contains
structural proteins which span the membrane compartments, serving as passageways for
assembly and release of the cellulose fibers.
Combining the significant findings from all the work described in my dissertation,
a model for the bacterial cellulose synthesis and extrusion complex, as shown in Figure
7.1. According to this model, the AcsA protein is the CM-bound cellulose synthase that
contains its catalytic region exposed towards the cytoplasm, where it binds to the
substrate UDP-glucose. This UDP-glucose is formed in close proximity to the synthase
because our data suggest that it is associated with phosphoglucomutase and UDP-glucose
phosphorylase. These enzymes catalyze the formation of glucose 1-phosphate and UDP-
glucose respectively. Their close association with the cellulose secretion machinery
serves to channel cellular glucose towards the cellulose-synthase complex. Cellulose

151
synthase is activated by binding of cyclic di-GMP to the PilZ domain. Our findings show
that this PilZ domain is also exposed to the cytoplasmic side of the CM. This is in
agreement with the known cytoplasmic distribution of the cyclic di-GMP to which the
PilZ domain binds (Amikam and Galperin 2006). This, so far, is the putative organization
of the elements involved in the synthesis of cellulose. The mechanism of transport of the
synthesized cellulose into the periplasm is unknown and yet to be determined. However,
once in the periplasm, data suggest that the cellulose fibers are channeled into the pore
formed by the AcsD octamer via the AcsB protein that serves as a scaffold between the
CM and the OM. The passageway through the AcsD pore which has interaction sites for
cellulose chains, assembles the cellulose fibers into a bundle. This bundle is twisted due
to the tilted nature of dimer-dimer interfaces that serve as interaction sites. This twist in
the fibers condenses them and serves to crystallize the chains into microfibrils. The
assembled microfibrils are extruded through the porin-like AcsC protein, located in the
OM. Many of the findings that have contributed to this model have been made as part of
my work towards this dissertation. This model can be further refined with the knowledge
of the stoichiometry of the constituent proteins and their structural characterization.

152
Figure 7.1 Working model for the cellulose synthesis complex This model shows
cytoplasmic proteins phosphoglucomutase (blue box) and UDP-glucose
pyrophosphorylase (purple box) in association with the CM-bound AcsA and AcsB1.
UDP-glucose is produced from glucose 1-phosphate by phosphoglucomutase and UDP-
glucose pyrophosphorylase. This serves as the substrate for cellulose synthesis by AcsA.
Both AcsA and AcsB1 contain transmembrane helical and globular region. The globular
region in AcsA exposed to the cytoplasm, contains the UDP-glucose-binding site,
whereas the cytoplasm-exposed region of the AcsAB1 contains the cyclic di-GMP- The
AcsB2 protein is tethered to the CM but largely exposed to the periplasm with close
contact with the AcsC protein. The cellulose fibers produced by the AcsA protein are
directed into the pore formed by octameric AcsD, which is localized in the periplasm.
These microfibrils are assembled in the periplasm and are extruded through the OM-
porin, AcsC.

153
REFERENCES "Analysis software: Point and click tools for assembly, mapping and amplicon variant
analysis. GS-de novo assembler." 454-Sequencing, Roche.
"GS FLX Titanium." General Library Preparation Manual, Roche 454 Life Sciences Oct
2008.
"GS FLX+ System." http://454.com/products/gs-flx-system/index.asp.
"High Fidelity Phusion Taq
http://www.finnzymes.fi/pdf/f531_f532_phusion_high_fidelity_pcr_master_mix_pro
dinfo_low.pdf."
"Introduciton to emPCR, GS FLX emPCR Method Manual." Roche, 454 Life Sciences Dec
2007.
"Nanodrop Products, Thermo Scientifc." http://www.nanodrop.com/.
"NCBI Prokaryotic Genomes Automatic Annotation Pipeline."
http://www.ncbi.nlm.nih.gov/genomes/static/Annotation_pipeline_README.txt
"Overview of SOLID sequencing chemistry." Applied Biosystems,
http://www.appliedbiosystems.com/absite/us/en/home/applications-
technologies/solid-next-generation-sequencing/next-generation-systems/solid-
sequencing-chemistry.html.
"Principle of pyrosequencing techonology."
http://www.pyrosequencing.com/DynPage.aspx?id=7454.
"www.piercenet.com." Thermo Scientific, Pierce Protein Research Products
(2007). "GS FLX Paired-end DNA library method manual." Roche, 454 Life Sciences.
(2007). "WorldOfCorn2007.pdf." Cellulosic ethanol has certain unique properties that
make it a far superior fuel than corn-based ethanol. World of corn[online],
http://www.ncga.com/WorldOfCorn/main/.
(2010). "How Newbler works?" http://contig.wordpress.com/.
(2010). "Wizard genomoc DNA purification kit " Promega, Instruction Manual.
Abramson, M., O. Shoseyov, et al. (2010). "Plant cell wall reconstruction toward improved
lignocellulosic production and processability." Plant Science 178(2): 61-72.
Akiyama, K., N. Yamada, et al. (1990). "Long-Lasting Enhancement of Ibotenate-
Stimulated Polyphosphoinositide Hydrolysis in the Amygdala/Pyriform Cortex of
Deep Prepiriform Cortical Kindled Rats." Japanese Journal of Psychiatry and
Neurology 44(2): 455-457.
Albersheim, P., A. Darvill, et al. (1997). "Do the structures of cell wall polysaccharides
define their mode of synthesis?" Plant Physiology 113(1): 1-3.
Aloni, Y., Cohen, Y., Benziman, M., Delmer, D. P. (1983). "Solubilization of UDP-
glucose:1,4-D-glucan 4-D-glucosyltransferase (cellulose synthase) from
Acetobacter xylinum." Journal of Biological Chemistry 258: 4419-4423.
Aloni, Y., D. P. Delmer, et al. (1982). "Achievement of high rates of in vitro synthesis of 1,4-
beta-D-glucan - Activation by cooperative Interaction of the Acetobacter xylinum
enzyme-system with GTP, Polyethylene-Glycol, and a protein factor." Proceedings
of the National Academy of Sciences of the United States of America-Biological
Sciences 79(21): 6448-6452.
Amikam, D. and M. Benziman (1989). "Cyclic diguanylic acid and cellulose synthesis in
Agrobacterium tumefaciens." Journal of Bacteriology 171(12): 6649-6655.
Amikam, D. and M. Y. Galperin (2006). "PilZ domain is part of the bacterial c-di-GMP
binding protein." Bioinformatics 22(1): 3-6.

154
Andersson-Gunneras, S., E. J. Mellerowicz, et al. (2006). "Biosynthesis of cellulose-enriched
tension wood in Populus tremula: global analysis of transcripts and metabolites
identifies biochemical and developmental regulators in secondary wall
biosynthesis.(vol 45, pg 144, 2005)." Plant Journal 46(2): 349-349.
Andrew, P. (2003). "Company says it mapped genes of virus in one day." The New York
Times.
Anwar, H., M. R. Brown, et al. (1983). "Isolation and characterization of the outer and
cytoplasmic membranes of Pseudomonas cepacia." J Gen Microbiol 129(2): 499-507.
Ausmees, N., R. Mayer, et al. (2001). "Genetic data indicate that proteins containing the
GGDEF domain possess diguanylate cyclase activity." FEMS Microbiology Letters
204(1): 163-167.
Ayaki, T., K. Fujikawa, et al. (1990). "Induced Rates of Mitotic Crossing over and Possible
Mitotic Gene Conversion Per Wing Anlage Cell in Drosophila-Melanogaster by X-
Rays and Fission Neutrons." Genetics 126(1): 157-166.
Aziz, R. K., D. Bartels, et al. (2008). "The RAST Server: rapid annotations using
subsystems technology." BMC Genomics 9: 75.
Becker, P. L., T. Itoh, et al. (1990). "Acetylcholine Can Elevate [Ca-2+]I in Voltage-
Clamped Smooth-Muscle Cells Via Release from Internal Stores." Biophysical
Journal 57(2): A157-A157.
Benach, J., S. S. Swaminathan, et al. (2007). "The structural basis of cyclic diguanylate
signal transduction by PilZ domains." Embo Journal 26(24): 5153-5166.
Benziman, M., C. H. Haigler, et al. (1980). "Cellulose biogenesis: Polymerization and
crystallization are coupled processes in Acetobacter xylinum." Proceedings of the
National Academy of Sciences of the United States of America-Biological Sciences
77(11): 6678-6682.
Bhasin, M., A. Garg, et al. (2005). "PSLpred: prediction of subcellular localization of
bacterial proteins." Bioinformatics 21(10): 2522-2524.
Blatch, G. L. and M. Lassle (1999). "The tetratricopeptide repeat: a structural motif
mediating protein-protein interactions." Bioessays 21(11): 932-939.
Boos, W. and T. Eppler (2001). "Chapter 4: Prokaryotic binding protein-dependent ABC
transporters." Microbial transport systems. Winkelmann, G. (Ed.) Wiley-VCH: 77-
114.
Bradford, M. M. (1976). "A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding." Anal Biochem
72: 248-254.
Brock, T. D. (1999). "Milestonesi n microbiology: 1546-1940." ASM Press, Washingtom
D.C. 2nd Edition: 215-218.
Brown, R. M. (1996). "The biosynthesis of cellulose." Journal of Macromolecular Science-
Pure and Applied Chemistry A33(10): 1345-1373.
Brown, R. M., Jr., J. H. Willison, et al. (1976). "Cellulose biosynthesis in Acetobacter
xylinum: visualization of the site of synthesis and direct measurement of the in vivo
process." Proc Natl Acad Sci U S A 73(12): 4565-4569.
Brown, R. M. and D. Montezinos (1976). "Cellulose Microfibrils - Visualization of
Biosynthetic and Orienting Complexes in Association with Plasma-Membrane."
Proceedings of the National Academy of Sciences of the United States of America
73(1): 143-147.
Bureau, T. E. and R. M. Brown (1987). "Invitro Synthesis of Cellulose-II from a
Cytoplasmic Membrane-Fraction of Acetobacter xylinum." Proceedings of the
National Academy of Sciences of the United States of America 84(20): 6985-6989.

155
Camacho-Carvajal, M. M., B. Wollscheid, et al. (2004). "Two-dimensional Blue native/SDS
gel electrophoresis of multi-protein complexes from whole cellular lysates: a
proteomics approach." Molecular & cellular proteomics : MCP 3(2): 176-182.
Canadian Environmental Protection Act Priority Substances List Assessment, R. N.,
Effluents from Pulp Mills Using Bleaching (1991).
Canale-Parola, E. (1970). "Biology of the sugar-fermenting Sarcinae." Bacteriol Rev 34(1):
82-97.
Caputto, R., L. F. Leloir, et al. (1950). "Isolation of the coenzyme of the galactose
phosphate-glucose phosphate transformation." Journal of Biological Chemistry
184(1): 333-350.
Carpita, N. and C. Vergara (1998). "Botany - A recipe for cellulose." Science 279(5351):
672-673.
Chain, P. S. G., D. V. Grafham, et al. (2009). "Genome Project Standards in a New Era of
Sequencing." Science 326(5950): 236-237.
Chang, C. Y. and T. Itoh (1990). "Microwave Active-Filters Based on Coupled Negative-
Resistance Method." Ieee Transactions on Microwave Theory and Techniques
38(12): 1879-1884.
Chanzy, H. D. and E. J. Roche (1975). "Fibrous Mercerization of Valonia Cellulose."
Journal of Polymer Science Part B-Polymer Physics 13(9): 1859-1862.
Chen, F. and R. A. Dixon (2007). "Lignin modification improves fermentable sugar yields
for biofuel production." Nature Biotechnology 25(7): 759-761.
Chen, H. P. and R. M. Brown (1996). "Immunochemical studies of the cellulose synthase
complex in Acetobacter xylinum." Cellulose 3(2): 63-75.
Cheung, P., D. P. Neikirk, et al. (1990). "Optically Controlled Coplanar Wave-Guide Phase
Shifters." Ieee Transactions on Microwave Theory and Techniques 38(5): 586-595.
Chiou, S. H., S. W. Chen, et al. (1990). "Comparison of the Gamma-Crystallins Isolated
from Eye Lenses of Shark and Carp - Unique Secondary and Tertiary Structure of
Shark Gamma-Crystallin." Febs Letters 275(1-2): 111-113.
Christen, M., B. Christen, et al. (2005). "Identification and characterization of a cyclic di-
GMP-specific phosphodiesterase and its allosteric control by GTP." Journal of
Biological Chemistry 280(35): 30829-30837.
Colombani, A., S. Djerbi, et al. (2004). "In vitro synthesis of (1 -> 3)-beta-D-glucan (callose)
and cellulose by detergent extracts of membranes from cell suspension cultures of
hybrid aspen." Cellulose 11(3-4): 313-327.
Colvin, J. R. (1961). "Twisting of bundles of bacterial cellulose microfibrils." Polymer
Chemistry 49(152): 473-477.
Consortium, T. U. (2012). "Reorganizing the protein space at the Universal Protein
Resource (UniProt)." Nucleic Acids Research 40(D1): D71-D75.
Cook, K. E. and J. R. Colvin (1980). "Evidence for a beneficial influence of cellulose
production on growth of Acetobacter xylinum in liquid medium." Current
Microbiology 3(4): 203-205.
Cooper, D. and R. S. J. Manley (1975). "Cellulose synthesis by Acetobacter xylinum .3.
Matrix, primer and lipid requirements and heat-stability of cellulose-forming
enzymes." Biochimica Et Biophysica Acta 381(1): 109-119.
Cotter, P. A. and S. Stibitz (2007). "c-di-GMP-mediated regulation of virulence and biofilm
formation." Current Opinion in Microbiology 10(1): 17-23.
Czaja, W., M. Kawecki, et al. (2004). "Application of bacterial cellulose in treatment of
second and third degree burns." Abstracts of Papers of the American Chemical
Society 227: U303-U303.

156
Czaja, W., D. Romanovicz, et al. (2004). "Structural investigations of microbial cellulose
produced in stationary and agitated culture." Cellulose 11(3-4): 403-411.
Das, A. K., P. T. W. Cohen, et al. (1998). "The structure of the tetratricopeptide repeats of
protein phosphatase 5: implications for TPR-mediated protein-protein
interactions." Embo Journal 17(5): 1192-1199.
de Maagd, R. A. and B. Lugtenberg (1986). "Fractionation of Rhizobium leguminosarum
cells into outer membrane, cytoplasmic membrane, periplasmic, and cytoplasmic
components." J Bacteriol 167(3): 1083-1085.
Debut, A. J., Q. C. Dumay, et al. (2006). "The iron/lead transporter superfamily of
Fe3+/Pb2+ uptake systems." Journal of Molecular Microbiology and Biotechnology
11(1-2): 1-9.
Debye, P. (1915). "Zerstreuung von röntgenstrahlen. Scattering from non-crystalline
substances." Annals of Physics 46: 809-823.
Delcher, A. L., D. Harmon, et al. (1999). "Improved microbial gene identification with
GLIMMER." Nucleic Acids Research 27(23): 4636-4641.
Delepelaire, P. and C. Wandersman (2001). "Chapter 7: Protein export and secretion in
Gram negative bacteria." Microbial transport systems. Winkelmann, G. (Ed.)
Wiley-VCH: 165-208.
Delmer, D. P. (1999). "Cellulose biosynthesis: Exciting times for a difficult field of study."
Plant Physiology and Plant Molecular Biology 50: 245-276.
Dow, J. M., Y. Fouhy, et al. (2006). "The HD-GYP domain, cyclic Di-GMP signaling, and
bacterial virulence to plants." Molecular Plant-Microbe Interactions 19(12): 1378-
1384.
Duan, J., Heikkila, J. J., Glick, B.R. (2010). "Sequencing a bacterial genome: An overview."
Current Research Technology and Educational Topics in Microbiology and
Microbial Biotechnology A Mendez Vilas (Ed.): 1443-1551.
Dumay, Q. C., A. J. Debut, et al. (2006). "The copper transporter (Ctr) family of Cu+
uptake systems." Journal of Molecular Microbiology and Biotechnology 11(1-2): 10-
19.
Eitan, B. (2007). "The Periplasm: Co- and posttranslational protein targetting to the
SecYEG translocon in Escherichia coli." ASM Press, Washington D.C.
Endler, A., C. Sanchez-Rodriguez, et al. (2010). "Glycobiology: Cellulose squeezes
through." Nature chemical biology 6(12): 883-884.
Energy Conservation Board, T. "www. seco.cpa.state.tx.us/re_ethanol_cellulosic.htm."
Erni, B. (2001). "Glucose transport by the bacterial phosphotransferase system (PTS): An
interface between energy- and signal transduction." Microbial transport systems.
Winkelmann, G. (Ed.) Wiley-VCH: 115-138.
Fields, S. and O. Song (1989). "A novel genetic system to detect protein-protein
interactions." Nature 340(6230): 245-246.
Filloux, A., M. Bally, et al. (1990). "Protein secretion in gram-negative bacteria: transport
across the outer membrane involves common mechanisms in different bacteria."
The EMBO Journal 9(13): 4323-4329.
Fouhy, Y., J. F. Lucey, et al. (2006). "Cell-cell signaling, cyclic di-GMP turnover and
regulation of virulence in Xanthomonas campestris." Research in Microbiology
157(10): 899-904.
Fox, B. G., J. G. Borneman, et al. (1990). "Haloalkene Oxidation by the Soluble Methane
Monooxygenase from Methylosinus-Trichosporium Ob3b - Mechanistic and
Environmental Implications." Biochemistry 29(27): 6419-6427.

157
Galperin, M. Y. (2005). "A census of membrane-bound and intracellular signal
transduction proteins in bacteria: bacterial IQ, extroverts and introverts." BMC
microbiology 5: 35.
Gardy, J. L., M. R. Laird, et al. (2005). "PSORTb v.2.0: expanded prediction of bacterial
protein subcellular localization and insights gained from comparative proteome
analysis." Bioinformatics 21(5): 617-623.
Garen, A. and C. Levinthal (1960). "A fine-structure genetic and chemical study of the
enzyme alkaline phosphatase of E. coli. I. Purification and characterization of
alkaline phosphatase." Biochimica Et Biophysica Acta 38: 470-483.
Gattiker, A., K. Michoud, et al. (2003). "Automated annotation of microbial proteomes in
SWISS-PROT." Computational biology and chemistry 27(1): 49-58.
Gerken, H. and R. Misra (2004). "Genetic evidence for functional interactions between
TolC and AcrA proteins of a major antibiotic efflux pump of Escherichia coli."
Molecular Microbiology 54(3): 620-631.
Giddings, T. H., D. L. Brower, et al. (1980). "Visualization of Particle Complexes in the
Plasma-Membrane of Micrasterias-Denticulata Associated with the Formation of
Cellulose Fibrils in Primary and Secondary Cell-Walls." Journal of Cell Biology
84(2): 327-339.
Glaser, L. (1958). "Synthesis of Cellulose in Cell-Free Extracts of Acetobacter-Xylinum."
Journal of Biological Chemistry 232(2): 627-636.
Glatter, O. (1977). "A new method for evaluation of small angle X-ray scattering data."
Journal of Applied Crystallography 10: 415-421.
Gould, S. J. and S. Subramani (1988). "Firefly luciferase as a tool in molecular and cell
biology." Analytical Biochemistry 175(1): 5-13.
Greie, J.-C., G. D. Hebestreit, et al. (2003). "Chapter 2. Energy-transducing ion pumps in
bacteria: structure and function of ATP synthases." Microbial transport systems.
Winkelmann, G. (Ed.) Wiley-VCH: 23-46.
Guinier, A. F., F. (1955). "Small angle scattering of X-rays." New York: Wiley
Interscience.
Hahne, G., W. Herth, et al. (1983). "Wall Formation and Cell-Division in Fluorescence-
Labeled Plant-Protoplasts." Protoplasma 115(2-3): 217-221.
Haigler, C. H. (1982). "Biogenesis of cellulose I microfibrils occurs by cell-directed self-
assembly in Acetobacter xylinum." Cellulose and other natural polymer systems.
Plenum publishing Corp. (New York).
Haigler, C. H. (2007). "Substate supply for cellulose synthesis and its stress sensitivity in the
cotton fiber." Cellulose: Molecular and structural biology, Springer. Brown, R. M.
and Saxena, I. M.: 147-168.
Haigler, C. H., R. M. Brown, Jr., et al. (1980). "Calcofluor white ST Alters the in vivo
assembly of cellulose microfibrils." Science 210(4472): 903-906.
Haigler, C. H., R. Malcolmbrown, et al. (1980). "Calcofluor White St Alters the Invivo
Assembly of Cellulose Microfibrils." Science 210(4472): 903-906.
Hakoshima, T., T. Itoh, et al. (1990). "Crystallization and Preliminary-X-Ray Investigation
of Nonspecific Complexes of a Mutant Ribonuclease-T1 (Y45w) with 2'amp and
2'ump." Journal of Molecular Biology 216(3): 497-499.
Halford, S. E. (1971). "Escherichia coli alkaline phosphatase. An analysis of transient
kinetics." Biochem J 125(1): 319-327.
Han, N. S. and J. F. Robyt (1998). "Biosynthesis of Acetobacter xylinum cellulose:
Mechanism of cellulose chain elongation." Abstracts of Papers of the American
Chemical Society 215: U108-U108.

158
Han, N. S. and J. F. Robyt (1998). "The mechanism of Acetobacter xylinum cellulose
biosynthesis: direction of chain elongation and the role of lipid pyrophosphate
intermediates in the cell membrane." Carbohydrate Research 313(2): 125-133.
Harwood, A. J. (2000). Native Polyacrylamide Gel Electrophoresis
The Nucleic Acid Protocols Handbook. R. Rapley, Humana Press: 73-75.
Hatta, Y., M. Baba, et al. (1990). "Changes of Pulmonary-Function in Patients Treated with
Bone-Marrow Transplantation after Total-Body Irradiation." Acta Haematologica
Japonica 53(6): 923-930.
Hausser, I. and W. Herth (1983). "The Ca-2+-Chelating Antibiotic, Chlorotetracycline
(Ctc), Disturbs Multipolar Tip Growth and Primary Wall Formation in
Micrasterias." Protoplasma 117(2): 167-173.
Haworth, W. N. (1932). "Molecular structure of cellulose and of amylose." Nature 129: 365-
365.
Herth, W. (1983). "Arrays of Plasma-Membrane Rosettes Involved in Cellulose Microfibril
Formation of Spirogyra." Planta 159(4): 347-356.
Herth, W. (1983). "Taxol Effects Cytoskeletal Microtubules, Flagella and Spindle Structure
of the Chrysoflagellate Alga Poterioochromonas." Protoplasma 115(2-3): 228-239.
Hestrin, S. and M. Schramm (1954). "Synthesis of cellulose by Acetobacter xylinum. II.
Preparation of freeze-dried cells capable of polymerizing glucose to cellulose."
Biochem J 58(2): 345-352.
Hjerten, S. (1962). "Chromatographic separation according to size of macromolecules and
cell particles on columns of agarose suspensions." Archives of Biochemistry and
Biophysics 99: 466-475.
http://www.cbs.dtu.dk/services/TMHMM/.
Hu, S., Gao, Y., Tajima, K., Yao, M., Yoda, T., Shimura, D., Satoh, Y., Kawano, S., Tanaka,
I., Munekata, M. (2008). "Purification, crystallization and preliminary X-ray
studies of AxCesD required for efficient cellulose biosynthesis in Acetobacter
xylinum." Protein and Peptide Letters 15(1): 115-117.
Hu, S. Q., Y. G. Gao, et al. (2010). "Structure of bacterial cellulose synthase subunit D
octamer with four inner passageways." Proceedings of the National Academy of
Sciences of the United States of America 107(42): 17957-17961.
Itoh, T. (1990). "Cellulose Synthesizing Complexes in Some Giant Marine-Algae." Journal
of Cell Science 95: 309-319.
Itoh, T. and R. M. Brown (1984). "The Assembly of Cellulose Microfibrils in Valonia-
Macrophysa Kutz." Planta 160(4): 372-381.
Itoh, T. and S. Kimura (2001). "Immunogold Labeling of terminal cellulose-synthesizing
complexes." Journal of Plant Research 114(1116): 483-489.
Itoh, T., R. M. Oneil, et al. (1984). "Interference of Cell-Wall Regeneration of Boergesenia-
Forbesii Protoplasts by Tinopal Lpw, a Fluorescent Brightening Agent."
Protoplasma 123(3): 174-183.
Iyer, K., L. Burkle, et al. (2005). "Utilizing the split-ubiquitin membrane yeast two-hybrid
system to identify protein-protein interactions of integral membrane proteins."
Science's STKE : signal transduction knowledge environment 2005(275): pl3.
Iyer, P. R., Catchmark, J. M, Brown, N.R. (2010). "Biochemical localization of a protein
invovled in synthesis of Gluconacetobacter hansenii cellulose." Cellulose
Unpublished.
Jarvie, T. (2006). "Whole genome assembly using paired-end reads in E. coli, B.
licheniformis, and S. cerevisiae." Genome Sequencer System, Roche, 454 Life
Sciences.

159
Jarvie, T. and T. Harkins (2008). "De novo assembly and genomic structural variation
analysis with genome sequencer FLX 3K long-tag paired end reads." BioTechniques
44(6): 829-831.
Jenal, U. and J. Malone (2006). "Mechanisms of cyclic-di-GMP signaling in bacteria."
Annual Review of Genetics 40: 385-407.
Kaihoh, T., T. Itoh, et al. (1990). "The Degradation Pathway of 1,2,3,4-Tetrazine."
Chemical & Pharmaceutical Bulletin 38(12): 3191-3194.
Kaiser, R. J., S. L. MacKellar, et al. (1989). "Specific-primer-directed DNA sequencing
using automated fluorescence detection." Nucleic Acids Res 17(15): 6087-6102.
Kamide, K., Y. Matsuda, et al. (1990). "Effect of Culture Conditions of Acetic-Acid Bacteria
on Cellulose Biosynthesis." British Polymer Journal 22(2): 167-171.
Kang, M. S., N. Elango, et al. (1984). "Isolation of Chitin Synthetase from Saccharomyces-
Cerevisiae - Purification of an Enzyme by Entrapment in the Reaction-Product."
Journal of Biological Chemistry 259(23): 4966-4972.
Karpen, J. W. (2004). "Ion channel structure and the promise of bacteria: Cyclic
nucleotide-gated channels in the queue - Commentary." Journal of General
Physiology 124(3): 199-201.
Katsaros, C., H. D. Reiss, et al. (1996). "Freeze-fracture studies in brown algae: Putative
cellulose-synthesizing complexes on the plasma membrane." European Journal of
Phycology 31(1): 41-48.
Kawano, S., K. Tajima, et al. (2002). "Effects of endogenous endo-beta-1,4-glucanase on
cellulose biosynthesis in Acetobacter xylinum ATCC23769." Journal of bioscience
and bioengineering 94(3): 275-281.
Kawano, S., K. Tajima, et al. (2002). "Effects of endogenous endo-beta-1,4-glucanase on
cellulose biosynthesis in Acetobacter xylinum ATCC23769." Journal of Bioscience
and Bioengineering 94(3): 275-281.
Kawano, S., K. Tajima, et al. (2008). "Regulation of endoglucanase gene (cmcax) expression
in Acetobacter xylinum." Journal of bioscience and bioengineering 106(1): 88-94.
Kawano, S., K. Tajima, et al. (2002). "Cloning of cellulose synthesis related genes from
Acetobacter xylinum ATCC23769 and ATCC53582: comparison of cellulose
synthetic ability between strains." DNA research : an international journal for
rapid publication of reports on genes and genomes 9(5): 149-156.
Keiski, C. L., M. Harwich, et al. (2010). "AlgK is a TPR-containing protein and the
periplasmic component of a novel exopolysaccharide secretin." Structure 18(2): 265-
273.
Kiermayer, O. and U. B. Sleytr (1979). "Hexagonally ordered rosettes of particles in the
plasma-membrane of micrasterias-denticulata breb and their significance for
microfibril formation and orientation." Protoplasma 101(1-2): 133-138.
Kimura, S., H. P. Chen, et al. (2001). "Localization of c-di-GMP-Binding protein with the
linear terminal complexes of Acetobacter xylinum." Journal of Bacteriology
183(19): 5668-5674.
Kimura, S., W. Laosinchai, et al. (1999). "Immunogold labeling of rosette terminal
cellulose-synthesizing complexes in the vascular plant Vigna angularis." Plant Cell
11(11): 2075-2085.
Kitagawa, H., M. Kanamori, et al. (1990). "Multiple Spinal Ossified Arachnoiditis - a Case-
Report." Spine 15(11): 1236-1238.
Koch, M. H., P. Vachette, et al. (2003). "Small-angle scattering: a view on the properties,
structures and structural changes of biological macromolecules in solution." Q Rev
Biophys 36(2): 147-227.

160
Kono, H., S. Kawano, et al. (1999). "Structural analyses of new tri- and tetrasaccharides
produced from disaccharides by transglycosylation of purified Trichoderma viride
beta-glucosidase." Glycoconjugate Journal 16(8): 415-423.
Koo, H. M., S. H. Song, et al. (1998). "Evidence that a beta-1,4-endoglucanase secreted by
Acetobacter xylinum plays an essential role for the formation of cellulose fiber."
Bioscience Biotechnology and Biochemistry 62(11): 2257-2259.
Koo, H. M., S. H. Song, et al. (1998). "Expression and characterization of CMCax having
beta 1,4-endoglucanase activity from Acetobacter xylinum." Journal of
Biochemistry and Molecular Biology 31(1): 53-57.
Koonin, E. V. (2001). "Computational Genomics, NIH " National Center for Biotechnology
Information, National Library of Medicine.
Kozin, M. B. S., D. I. (2001). "Autaomated matching of high- and low- resolution structural
models." Journal of Applied Crystallography 34: 33-41.
Krogh, A., B. Larsson, et al. (2001). "Predicting transmembrane protein topology with a
hidden Markov model: application to complete genomes." Journal of Molecular
Biology 305(3): 567-580.
Kudlicka, K. and R. M. Brown (1997). "Cellulose and callose biosynthesis in higher plants
.1. Solubilization and separation of (1->3)- and (1->4)-beta-glucan synthase activities
from mung bean." Plant Physiology 115(2): 643-656.
Kudlicka, K., R. M. Brown, et al. (1995). "Beta-Glucan Synthesis in the Cotton Fiber .4. In-
Vitro Assembly of the Cellulose-I Allomorph." Plant Physiology 107(1): 111-123.
Kuga, S., S. Takagi, et al. (1993). "Native folded-chain Cellulose II." Polymer 34(15): 3293-
3297.
Kuno, N., Y. Kamisaki, et al. (1990). "Inhibition of Cyclic-Amp Accumulation by Alpha-2-
Adrenoceptors in the Rat Cerebral-Cortex." European Journal of Pharmacology
176(3): 281-287.
Lai-Kee-Him, J., H. Chanzy, et al. (2002). "In vitro versus in vivo cellulose microfibrils from
plant primary wall synthases: Structural differences." Journal of Biological
Chemistry 277(40): 36931-36939.
Lantz, M. S. and P. Ciborowski (1994). "Zymographic techniques for detection and
characterization of microbial proteases." Methods Enzymol 235: 563-594.
Lehman, I. R. (1974). "DNA ligase: structure, mechanism, and function." Science
186(4166): 790-797.
Leloir, L. F. (1961). "Biosynthesis of Glycogen, Starch and Other Polysaccharides." Harvey
Lectures(56): 23-&.
Leloir, L. F. and C. E. Cardini (1957). "Biosynthesis of Glycogen from Uridine Diphosphate
Glucose." Journal of the American Chemical Society 79(23): 6340-6341.
Leloir, L. F. and S. H. Goldemberg (1960). "Synthesis of Glycogen from Uridine
Diphosphate Glucose in Liver." Journal of Biological Chemistry 235(4): 919-923.
Leloir, L. F., J. M. Olavarria, et al. (1959). "Biosynthesis of Glycogen from Uridine
Diphosphate Glucose." Archives of Biochemistry and Biophysics 81(2): 508-520.
Lesk, A. M. (2007). "Mapping, sequencing, annotation and databases." Introduction to
genomics, Oxford University Press, New York: 202-262.
Lesk, A. M. (2007). "Preface." Introduction to genomics, Oxford University Press, New
York.
Lin, F. C., R. M. Brown, Jr., et al. (1990). "Identification of the uridine 5'-diphosphoglucose
(UDP-Glc) binding subunit of cellulose synthase in Acetobacter xylinum using the
photoaffinity probe 5-azido-UDP-Glc." J Biol Chem 265(9): 4782-4784.

161
Lin, F. C., Brown, R. M. (1989). "Purification of cellulose synthase from Acetobacter
xylinum." In: Schuerch C, ed. Cellulose and wood - chemistry and technology. New
York: John Wiley and Sons: 473-492.
Lisdiyanti, P., R. R. Navarro, et al. (2006). "Reclassification of Gluconacetobacter hansenii
strains and proposals of Gluconacetobacter saccharivorans sp. nov. and
Gluconacetobacter nataicola sp. nov." Int J Syst Evol Microbiol 56(Pt 9): 2101-2111.
Lowry, O. H., N. J. Rosebrough, et al. (1951). "Protein measurement with the Folin phenol
reagent." J Biol Chem 193(1): 265-275.
Ma, Q. and T. K. Wood (2009). "OmpA influences Escherichia coli biofilm formation by
repressing cellulose production through the CpxRA two-component system."
Environmental microbiology 11(10): 2735-2746.
Marchler-Bauer, A., J. B. Anderson, et al. (2005). "CDD: a conserved domain database for
protein classification." Nucleic Acids Research 33: D192-D196.
Martinez, T. F., F. J. Alarcon, et al. (2000). "Improved detection of amylase activity by
sodium dodecyl sulfate-polyacrylamide gel electrophoresis with copolymerized
starch." Electrophoresis 21(14): 2940-2943.
Marx-Figini, M. (1982). "The control of molecular weight and molecular weight
distribution." Cellulose and other natural polymer systems. Plenum publishing
Corp. (New York) Brown, R. M. Jr (ed.): 243-271.
Matrone, G., G. H. Ellis, et al. (1946). "A Modified Norman-Jenkins method for the
determination of cellulose and its use in the evaluation of feedstuffs." Journal of
Animal Science 5(3): 306-312.
Matthysse, A. G., S. White, et al. (1995). "Genes required for cellulose synthesis in
Agrobacterium tumefaciens." Journal of Bacteriology 177(4): 1069-1075.
Mayer, R., P. Ross, et al. (1991). "Polypeptide composition of bacterial cyclic diguanylic
acid-dependent cellulose synthase and the occurrence of immunologically cross-
reacting proteins in higher plants." Proceedings of the National Academy of
Sciences of the United States of America 88(12): 5472-5476.
Meier, H. (1964). "General chemistry of cell walls and distribution of the chemical
constituents across the wall. ." The formation of wood in forest trees Academic
press New york(Zimmermann (ed)): 137-151.
Mitra, R. D., J. Shendure, et al. (2003). "Fluorescent in situ sequencing on polymerase
colonies." Analytical biochemistry 320(1): 55-65.
Monheit, J. E., D. F. Cowan, et al. (1984). "Rapid detection of fungi in tissues using
calcofluor white and fluorescence microscopy." Arch Pathol Lab Med 108(8): 616-
618.
Montezinos, D. and R. M. Brown (1976). "Surface Architecture of Plant-Cell - Biogenesis of
Cell-Wall, with Special Emphasis on Role of Plasma-Membrane in Cellulose
Biosynthesis." Journal of Supramolecular Structure 5(3): 277-290.
Montezinos, D. and R. M. Brown (1976). "Visualization of Cellulose Microfibril Assembly
in Association with Plasma-Membrane." Plant Physiology 57(5): 57-57.
Mukai, T., T. Toba, et al. (1990). "Structural Investigation of the Capsular Polysaccharide
from Lactobacillus-Kefiranofaciens K1." Carbohydrate Research 204: 227-232.
Mulisch, M., W. Herth, et al. (1983). "Chitin Fibrils in the Lorica of the Ciliate
Eufolliculina-Uhligi - Ultrastructure, Extracellular Assembly and Experimental
Inhibition." Biology of the Cell 49(2): 169-178.
Murai, S., H. Saito, et al. (1990). "A Simple and Rapid Hplc-Ecd Assay of Several
Neurotransmitters and Related-Compounds in the Same Sample of Mouse Discrete
Brain-Areas." European Journal of Pharmacology 183(2): 417-418.

162
Myers, C. R. and J. M. Myers (1992). "Localization of cytochromes to the outer membrane
of anaerobically grown Shewanella putrefaciens MR-1." J Bacteriol 174(11): 3429-
3438.
Nakai, T., A. Moriya, et al. (1998). "Control of expression by the cellulose synthase (bcsA)
promoter region from Acetobacter xylinum BPR 2001." Gene 213(1-2): 93-100.
Neuhoff, V., N. Arold, et al. (1988). "Improved staining of proteins in polyacrylamide gels
including isoelectric focusing gels with clear background at nanogram sensitivity
using Coomassie Brilliant Blue G-250 and R-250." Electrophoresis 9(6): 255-262.
Nicol, F., I. His, et al. (1998). "A plasma membrane-bound putative endo-1,4-beta-D-
glucanase is required for normal wall assembly and cell elongation in Arabidopsis."
EMBO Journal 17(19): 5563-5576.
Niklas, K. J. (1992). "Plant Biomechanics: an engineering appraoch to plant form and
function." The University of Chicago Press Chicago: 607.
Nomura, M., H. Harino, et al. (1990). "Anomalous Change in Temperature during the
Pressure-Induced Phase-Transition of Ki." Japanese Journal of Applied Physics
Part 1-Regular Papers Short Notes & Review Papers 29(11): 2456-2459.
Nyren, P. (2007). "The history of pyrosequencing." Methods in molecular biology 373: 1-14.
Ogino, H., Y. Azuma, et al. (2011). "Complete Genome Sequence of NBRC 3288, a Unique
Cellulose-Nonproducing Strain of Gluconacetobacter xylinus Isolated from
Vinegar." Journal of Bacteriology 193(24): 6997-6998.
Ohchi, H., T. Itoh, et al. (1990). "Inhouse Private Exchange System for Isdn." Sharp
Technical Journal(47): 39-44.
Okuda, K., L. K. Li, et al. (1993). "Beta-Glucan Synthesis in the Cotton Fiber .1.
Identification of Beta-1,4-Glucan and Beta-1,3-Glucan Synthesized Invitro." Plant
Physiology 101(4): 1131-1142.
Okuda, K., I. Tsekos, et al. (1994). "Cellulose Microfibril Assembly in Erythrocladia-
Subintegra Rosenv - an Ideal System for Understanding the Relationship between
Synthesizing Complexes (Tcs) and Microfibril Crystallization." Protoplasma 180(1-
2): 49-58.
Palmer, T. (2007). "The periplasm: The Tat protein export pathway." ASM Press,
Washingtom D.C.
Paul, R., S. Weiser, et al. (2004). "Cell cycle-dependent dynamic localization of a bacterial
response regulator with a novel di-guanylate cyclase output domain." Genes &
Development 18(6): 715-727.
Pear, J. R., Y. Kawagoe, et al. (1996). "Higher plants contain homologs of the bacterial celA
genes encoding the catalytic subunit of cellulose synthase." Proceedings of the
National Academy of Sciences of the United States of America 93(22): 12637-12642.
Pecina, A., A. Pascual, et al. (1999). "Cloning and expression of the algL gene, encoding the
Azotobacter chroococcum alginate lyase: purification and characterization of the
enzyme." Journal of Bacteriology 181(5): 1409-1414.
Perna, N. T., G. Plunkett, 3rd, et al. (2001). "Genome sequence of enterohaemorrhagic
Escherichia coli O157:H7." Nature 409(6819): 529-533.
Petersen, T. N., S. Brunak, et al. (2011). "SignalP 4.0: discriminating signal peptides from
transmembrane regions." Nature Methods 8(10): 785-786.
Pop, M. and S. L. Salzberg (2008). "Bioinformatics challenges of new sequencing
technology." Trends in genetics : TIG 24(3): 142-149.
Pratt, J. T., R. Tamayo, et al. (2007). "PilZ domain proteins bind cyclic diguanylate and
regulate diverse processes in Vibrio cholerae." Journal of Biological Chemistry
282(17): 12860-12870.

163
Price, D. C., C. X. Chan, et al. (2012). "Cyanophora paradoxa genome elucidates origin of
photosynthesis in algae and plants." Science 335(6070): 843-847.
Putnam, C. D., M. Hammel, et al. (2007). "X-ray solution scattering (SAXS) combined with
crystallography and computation: defining accurate macromolecular structures,
conformations and assemblies in solution." Q Rev Biophys 40(3): 191-285.
Quinaud, M., S. Ple, et al. (2007). "Structure of the heterotrimeric complex that regulates
type III secretion needle formation." Proceedings of the National Academy of
Sciences of the United States of America 104(19): 7803-7808.
Ragauskas, A. J., C. K. Williams, et al. (2006). "The path forward for biofuels and
biomaterials." Science 311(5760): 484-489.
Rahman, O., S. P. Cummings, et al. (2008). "Methods for the bioinformatic identification of
bacterial lipoproteins encoded in the genomes of Gram-positive bacteria." World
Journal of Microbiology & Biotechnology 24(11): 2377-2382.
Ramelot, T. A., A. Yee, et al. (2007). "NMR structure and binding studies confirm that
PA4608 from Pseudomonas aeruginosa is a PilZ domain and a c-di-GMP binding
protein." Proteins-Structure Function and Bioinformatics 66(2): 266-271.
Reiss, H. D., C. Katsaros, et al. (1996). "Freeze-fracture studies in the brown alga
Asteronema rhodochortonoides." Protoplasma 193(1-4): 46-57.
Ring, G. J. (1982). "A study of polymerization kinetics of bacterial cellulose through gel-
permeation chromatography." Brown, R. M. Jr (ed.) Cellulose and other natural
polymer systems. Plenum Publishing Corp., New York.
Robert, S., A. Bichet, et al. (2005). "An Arabidopsis endo-1,4-beta-D-glucanase involved in
cellulose synthesis undergoes regulated intracellular cycling." Plant Cell 17(12):
3378-3389.
Robinson, P. A., B. H. Anderton, et al. (1988). "Nitrocellulose-Bound Antigen Repeatedly
Used for the Affinity Purification of Specific Polyclonal Antibodies for Screening
DNA Expression Libraries." Journal of Immunological Methods 108(1-2): 115-122.
Roelofsen, A. (1958). "Cell wall structure as related to surface growth." Acta Botanica
Neerlandica 7: 77-89.
Romling, U., M. Gomelsky, et al. (2005). "C-di-GMP: the dawning of a novel bacterial
signalling system." Molecular Microbiology 57(3): 629-639.
Ronaghi, M., S. Karamohamed, et al. (1996). "Real-time DNA sequencing using detection of
pyrophosphate release." Analytical biochemistry 242(1): 84-89.
Ronaghi, M., M. Uhlen, et al. (1998). "A sequencing method based on real-time
pyrophosphate." Science 281(5375): 363, 365.
Ross, P., R. Mayer, et al. (1991). "Cellulose Biosynthesis and Function in Bacteria."
Microbiological Reviews 55(1): 35-58.
Ross, P., R. Mayer, et al. (1990). "The Cyclic Diguanylic Acid Regulatory System of
Cellulose Synthesis in Acetobacter-Xylinum - Chemical Synthesis and Biological-
Activity of Cyclic-Nucleotide Dimer, Trimer, and Phosphothioate Derivatives."
Journal of Biological Chemistry 265(31): 18933-18943.
Ross, P., H. Weinhouse, et al. (1987). "Regulation of Cellulose Synthesis in Acetobacter
xylinum by cyclic diguanylic acid." Nature 325(6101): 279-281.
Ruebush, S. S., S. L. Brantley, et al. (2006). "Reduction of soluble and insoluble iron forms
by membrane fractions of Shewanella oneidensis grown under aerobic and
anaerobic conditions." Appl Environ Microbiol 72(4): 2925-2935.

164
Ryan, R. P., Y. Fouhy, et al. (2006). "Cell-cell signaling in Xanthomonas campestris involves
an HD-GYP domain protein that functions in cyclic di-GMP turnover." Proceedings
of the National Academy of Sciences of the United States of America 103(17): 6712-
6717.
Ryan, R. P., Y. Fouhy, et al. (2006). "Cyclic Di-GMP signaling in bacteria: Recent advances
and new puzzles." Journal of Bacteriology 188(24): 8327-8334.
Ryjenkov, D. A., R. Simm, et al. (2006). "The PilZ domain is a receptor for the second
messenger c-di-GMP - The PilZ domain protein YcgR controls motility in
enterobacteria." Journal of Biological Chemistry 281(41): 30310-30314.
Saier, M. H. (2006). "Protein secretion and membrane insertion systems in gram-negative
bacteria." Journal of Membrane Biology 214(1-2): 75-90.
Saier, M. H. (2006). "Protein secretion systems in Gram negative bacteria." Microbe 1(9):
414-419.
Salzberg, S. L., A. L. Delcher, et al. (1998). "Microbial gene identification using
interpolated Markov models." Nucleic Acids Research 26(2): 544-548.
Saxena, I. M. and R. M. Brown (1995). "Identification of a 2nd gellulose synthase gene
(Acsa II) in Acetobacter xylinum." Journal of Bacteriology 177(18): 5276-5283.
Saxena, I. M. and R. M. Brown (1997). "Identification of cellulose synthase(s) in higher
plants: Sequence analysis of processive beta-glycosyltransferases with the common
motif 'D, D, D35Q(R,Q)XRW'." Cellulose 4(1): 33-49.
Saxena, I. M. and R. M. Brown (2000). "Cellulose synthases and related enzymes." Current
Opinion in Plant Biology 3(6): 523-531.
Saxena, I. M., R. M. Brown, et al. (2001). "Structure-function characterization of cellulose
synthase: relationship to other glycosyltransferases." Phytochemistry 57(7): 1135-
1148.
Saxena, I. M., R. M. Brown, et al. (1995). "Multidomain architecture of glycosyl
transferases - Implications for mechanism of action." Journal of Bacteriology
177(6): 1419-1424.
Saxena, I. M., B. Henrissat, et al. (1995). "Analysis of genes involved in cellulose
biosynthesis - from sequence comparisons to mechanism of glycosyl transfer." Plant
Physiology 108(2): 9-9.
Saxena, I. M., K. Kudlicka, et al. (1994). "Characterization of genes in the cellulose-
synthesizing operon (Acs Operon) of Acetobacter xylinum - Implications for cellulose
crystallization." Journal of Bacteriology 176(18): 5735-5752.
Saxena, I. M., F. C. Lin, et al. (1990). "Cloning and sequencing of the cellulose synthase
catalytic subunit gene of Acetobacter xylinum." Plant Molecular Biology 15(5): 673-
683.
Schagger, H. and G. von Jagow (1991). "Blue native electrophoresis for isolation of
membrane protein complexes in enzymatically active form." Analytical
Biochemistry 199(2): 223-231.
Schmidt, A. J., D. A. Ryjenkov, et al. (2005). "The ubiquitous protein domain EAL is a
cyclic diguanylate-specific phosphodiesterase: Enzymatically active and inactive
EAL domains." Journal of Bacteriology 187(14): 4774-4781.
Schramm, M. and S. Hestrin (1954). "Factors affecting production of cellulose at the
air/liquid interface of a culture of Acetobacter xylinum." J Gen Microbiol 11(1):
123-129.
Schramm, M. and S. Hestrin (1954). "Synthesis of cellulose by Acetobacter xylinum. I.
Micromethod for the determination of celluloses." Biochem J 56(1): 163-166.

165
Seltmann, G. and O. Holst (2002). "Chapter 1: Introduction " The bacterial cell wall,
Springer: 3-8.
Seltmann, G. and O. Holst (2002). "Chapter 3: Periplasmic space and rigid layer." The
bacterial cell wall, Springer: 103-132.
Semenyuk, A. V. S., D. I. (1991). "GNOM – a program package for small-angle scattering
data processing." Journal of Applied Crystallography 24: 537-540.
Sha, Z., T. J. Stabel, et al. (1994). "Brucella abortus catalase is a periplasmic protein
lacking a standard signal sequence." J Bacteriol 176(23): 7375-7377.
Shah, J. and R. M. Brown (2004). "Microbial cellulose and the development of electronic
paper." Abstracts of Papers of the American Chemical Society 227: U305-U305.
Sheps, J. A., F. Zhang, et al. (1996). Bacterial toxin transport: The hemolysin system.
Membrane Protein Transport. S. R. Stephen, JAI. Volume 3: 81-118.
Staden, R. (1979). "A strategy of DNA sequencing employing computer programs." Nucleic
acids research 6(7): 2601-2610.
Standal, R., T. G. Iversen, et al. (1994). "A new gene required for cellulose production and a
gene encoding cellulolytic activity in Acetobacter xylinum are colocalized with the
Bcs operon." Journal of Bacteriology 176(11): 3443-3443.
Sticklen, M. B. (2008). "Plant genetic engineering for biofuel production: towards
affordable cellulosic ethanol (Retracted article. See vol 11, pg 308, 2010)." Nature
Reviews Genetics 9(6): 433-443.
Stintzi, A., C. Barnes, et al. (2000). "Microbial iron transport via a siderophore shuttle: A
membrane ion transport paradigm." Proceedings of the National Academy of
Sciences 97(20): 10691-10696.
Strauss, E. C., J. A. Kobori, et al. (1986). "Specific-primer-directed DNA sequencing."
Analytical Biochemistry 154(1): 353-360.
Streeter, J. G. and D. Le Rudulier (1990). "Release of periplasmic enzymes from Rhizobium
leguminosarum bv phaseoli bacteroids by lysozyme is enhanced by pretreatment of
cells at low pH." Current Microbiology 21(3): 169-173.
Stuhrmann, H. B. (1981). "Anomalous small angle scattering." Quarterly Reviews in
Biophysics 14: 433-460.
Svanem, B. I., G. Skjak-Braek, et al. (1999). "Cloning and expression of three new
Aazotobacter vinelandii genes closely related to a previously described gene family
encoding mannuronan C-5-epimerases." Journal of Bacteriology 181(1): 68-77.
Svergun, D. I., Petoukhov, M. V. & Koch, M. H. J. (2001b). "Determination of domain
structure of proteins from X-ray solution scattering." Biophysical Journal 76: 2879-
2886.
Swissa, M., Y. Aloni, et al. (1980). "Intermediary Steps in Acetobacter-Xylinum Cellulose
Synthesis - Studies with Whole Cells and Cell-Free Preparations of the Wild-Type
and a Cellulose-Less Mutant." Journal of Bacteriology 143(3): 1142-1150.
Szybalski, W. (1993). "From the double-helix to novel approaches to the sequencing of large
genomes." Gene 135(1-2): 279-290.
Tajima, K., K. Nakajima, et al. (2001). "Cloning and sequencing of the beta-glucosidase
gene from Acetobacter xylinum ATCC 23769." DNA Research 8(6): 263-269.
Tal, R., H. C. Wong, et al. (1998). "Three cdg operons control cellular turnover of cyclic di-
GMP in Acetobacter xylinum: Genetic organization and occurrence of conserved
domains in isoenzymes." Journal of Bacteriology 180(17): 4416-4425.
Talaro, K. P. (2007). "Chapter 4: An introduction to cell and prokaryotic cell structure and
function." Foundations in Microbiology: Basic Principles McGraw Hill
International Edition(6th edition): 89-122.

166
Tamayo, R., J. T. Pratt, et al. (2007). "Roles of cyclic diguanylate in the regulation of
bacterial pathogenesis." Annual Reviews in Microbiology 61: 131-148.
Tamayo, R., A. D. Tischler, et al. (2005). "The EAL domain protein VieA is a cyclic
diguanylate phosphodiesterase." Journal of Biological Chemistry 280(39): 33324-
33330.
Tamura, K., J. Dudley, et al. (2007). "MEGA4: Molecular Evolutionary Genetics Analysis
(MEGA) software version 4.0." Molecular biology and evolution 24(8): 1596-1599.
Teruyama, T., T. Itoh, et al. (1990). "Interfacial Reactions of Off-Stoichiometric Ybacuo
Films on Mgo and Si Substrates." Journal of the Electrochemical Society 137(1):
336-339.
Thieme, F., R. Koebnik, et al. (2005). "Insights into genome plasticity and pathogenicity of
the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by
the complete genome sequence." Journal of Bacteriology 187(21): 7254-7266.
Thomas, J. D., R. A. Daniel, et al. (2001). "Export of active green fluorescent protein to the
periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli." Mol
Microbiol 39(1): 47-53.
Thompson, N. S., J. A. Carlson, et al. (1988). "Tunnel Structures in Acetobacter xylinum."
International Journal of Biological Macromolecules 10(2): 126-127.
Tobita, K., Y. Kusama, et al. (1990). "A Parasitic Effect in Neutral Particle Diagnostic
Using a Helium Probing Beam." Japanese Journal of Applied Physics Part 1-
Regular Papers Short Notes & Review Papers 29(4): 760-764.
Tonouchi, N., N. Thara, et al. (1995). "Addition of a small amount of an endoglucanase
enhances cellulose production by Acetobacter xylinum." Bioscience Biotechnology
and Biochemistry 59(5): 805-808.
Toyoshima, H., K. Onda, et al. (1990). "Mbe Growth of (Inas)M(Gaas)N Superlattices on
Gaas Substrates Monitored by Rheed Oscillations and Its Application to N-
Algaas/(Inas)M/Gaas 2degfets." Institute of Physics Conference Series(112): 79-84.
Toyoshima, H., K. Onda, et al. (1990). "Mbe Growth of (Inas)M(Gaas)N Superlattices on
Gaas Substrates Monitored by Rheed Oscillations and Its Application to N-
Algaas/(Inas)M/Gaas 2degfets." Gallium Arsenide and Related Compounds 1990
112: 79-84.
Tsekos, I. (1999). "The sites of cellulose synthesis in algae: Diversity and evolution of
cellulose-synthesizing enzyme complexes." Journal of Phycology 35(4): 635-655.
Tsuji, H., T. Itoh, et al. (1990). "Expansive Laminoplasty for Lumbar Spinal Stenosis."
International Orthopaedics 14(3): 309-314.
Tusnady, G. E. and I. Simon (1998). "Principles governing amino acid composition of
integral membrane proteins: Application to topology prediction." Journal of
Molecular Biology 283(2): 489-506.
Tusnady, G. E. and I. Simon (2001). "The HMMTOP transmembrane topology prediction
server." Bioinformatics 17(9): 849-850.
Umeda, Y., A. Hirano, et al. (1998). "Conversion of CO2 into cellulose by gene
manipulation of microalgae: Cloning of cellulose synthase genes from Acetobacter
xylinum." Advances in Chemical Conversions for Mitigating Carbon Dioxide 114:
653-656.
Umeda, Y., A. Hirano, et al. (1999). "Cloning of cellulose synthase genes from Acetobacter
xylinum JCM 7664: implication of a novel set of cellulose synthase genes." DNA
research : an international journal for rapid publication of reports on genes and
genomes 6(2): 109-115.

167
Valla, S. and J. Kjosbakken (1982). "Cellulose-Negative Mutants of Acetobacter-Xylinum."
Journal of General Microbiology 128(Jul): 1401-1408.
Vazquez, A., S. Moreno, et al. (1999). "Transcriptional organization of the Azotobacter
vinelandii algGXLVIFA genes: characterization of algF mutants." Gene 232(2):
217-222.
Volkov, V. V. S., D. I. (2003). "Uniqueness of ab initio shape determination in small-angle
scattering." Journal of Applied Crystallography 36(860-864).
Wanibe, Y., H. Yokoyama, et al. (1990). "Expansion during Liquid-Phase Sintering of Iron-
Copper Compacts." Powder Metallurgy 33(1): 65-69.
Whitfield, C. and I. L. Mainprize (2010). "TPR motifs: hallmarks of a new polysaccharide
export scaffold." Structure 18(2): 151-153.
Whitney, J. C., I. D. Hay, et al. (2011). "Structural basis for alginate secretion across the
bacterial outer membrane." Proceedings of the National Academy of Sciences of the
United States of America 108(32): 13083-13088.
Williams, W. S. and R. E. Cannon (1989). "Alternative Environmental-Roles for Cellulose
Produced by Acetobacter-Xylinum." Applied and Environmental Microbiology
55(10): 2448-2452.
Wittig, I., H. P. Braun, et al. (2006). "Blue native PAGE." Nature protocols 1(1): 418-428.
Wolfe, A. J. and K. L. Visick (2008). "Get the message out: Cyclic-Di-GMP regulates
multiple levels of flagellum-based motility." Journal of Bacteriology 190(2): 463-
475.
Wong, H. C., A. L. Fear, et al. (1990). "Genetic organization of the cellulose synthase
operon in Acetobacter xylinum." Proc Natl Acad Sci U S A 87(20): 8130-8134.
Woodman, M. E. (2008). "Direct PCR of intact bacteria (colony PCR)." Curr Protoc
Microbiol Appendix 3: Appendix 3D.
Yahr, T. L. and W. T. Wickner (2001). "Functional reconstitution of bacterial Tat
translocation in vitro." EMBO J 20(10): 2472-2479.
Yamaguchi, K., A. Ohsawa, et al. (1990). "Structure of a 1,2,3,5-Tetrazin-4-One." Acta
Crystallographica Section C-Crystal Structure Communications 46: 261-263.
Yamaguchi, K., A. Ohsawa, et al. (1990). "Structure of a 1,3-Dicyanoazimine." Acta
Crystallographica Section C-Crystal Structure Communications 46: 821-823.
Yamanaka, S., K. Watanabe, et al. (1989). "The Structure and Mechanical-Properties of
Sheets Prepared from Bacterial Cellulose." Journal of Materials Science 24(9):
3141-3145.
Ye, C., R. Lan, et al. (2010). "Emergence of a new multidrug-resistant serotype X variant in
an epidemic clone of Shigella flexneri." Journal of clinical microbiology 48(2): 419-
426.
Yoshida, J., T. Morisaki, et al. (1990). "Carcinoma in Adenoma of the Ampulla of Vater
Synchronous with Cancer of the Sigmoid Colon." Digestive Diseases and Sciences
35(2): 271-275.
Zaar, K. (1979). "Visualization of pores (Export Sites) Correlated with Cellulose
Production in the Envelope of the Gram-Negative Bacterium Acetobacter-
Xylinum." Journal of Cell Biology 80(3): 773-777.

VITA
Publications
1. Sato S, Feltus FA, Iyer PR, Tien M. (2009) The first genome-level transcriptome of the
wood-degrading fungus Phanerochaete chrysosporium grown on red oak. Curr Genet. 55 (3):
273-86.
2. Iyer PR, Geib SM, Catchmark J, Kao TH, Tien M (2010) Genome sequence of a cellulose-
producing bacterium, Gluconacetobacter hansenii ATCC 23769. J Bacteriol. 192 (16):4256-7.
3. Iyer PR, Catchmark J, Brown NR, Tien M. (2011) Biochemical localization of a protein
involved in synthesis of Gluconacetobacter hansenii cellulose. Cellulose 18 (3): 739-47.
Presentations
1. “Biochemistry of bacterial cellulose synthesis: How do bacteria spin the yarn?” 243rd ACS
National Meeting, Division of Cellulose and Renewable Materials, San Diego, California (March
25-29, 2012).
2. “Isolation and characterization of proteins involved in Gluconacetobacter hansenii cellulose
synthesis” Division of Cellulose and Renewable Materials, 241st ACS National Meeting &
Exhibition in Anaheim, California (March 27-31, 2011).
1. “Biochemical characterization of the cellulose synthase complex from Gluconacetobacter
hansenii” Energy Frontier Research Center, The Pennsylvania State University (November,
2010).
2. Poster “Biochemical and structural characterization of AcsD, a protein involved in bacterial
cellulose synthesis”. Graduate Exhibition. The Pennsylvania State University (March, 2010).
3. “Whole genome sequencing of Gluconacetobacter hansenii 23769” Energy Frontier Research
Center Retreat, The Pennsylvania State University (May, 2010).
4. Poster :“Protein-protein interactions in Acetobacter xylinum cellulose synthase complex” 12th
Annual Environmental Chemistry Student Symposium, The Pennsylvania State University
(March 27 – 28, 2009).
5. Poster :“Study of proteins involved in bacterial cellulose synthesis” Graduate Exhibition, The
Pennsylvania State University (March 29, 2009).
6. “Characterization of Acetobacter xylinum Cellulose Synthase” Institute of Biological
Engineering, Santa Clara (March 19 – 21 2009).
Awards and Scholarships
1. Awarded second position in poster presentation Environmental Chemistry Student Symposium
(2010)
2. Awarded first position in poster presentation in Environmental Chemistry Student Symposium
(2009)
3. Recipient of the Huck Institutes of Life Sciences Fellowship at The Pennsylvania State
University (2006-2007)
4. Recognized and awarded as the “Best Outstanding Student” by Srimad Andavan College of
Arts and Sciences, Tiruchirappalli, India (2001)
5. Awarded Merit scholarship for the best academic performance by Department of
Biochemistry, Srimad Andavan College of Arts and Sciences, Trichy, India (2001).
6. Won Second Prize in “Chemquiz’97”, an inter-college chemistry quiz event organized by the
Pune University Department of Chemistry, Pune, India (1997).


















