
Genomics: Gene prediction and Annotations Kishor K. Shende Information Officer Bioinformatics...
49
Genomics: Gene prediction and Annotations Kishor K. Shende Information Officer Bioinformatics Center, Barkatullah University Bhopal
-
Upload
christiana-gardner -
Category
Documents
-
view
217 -
download
0
Transcript of Genomics: Gene prediction and Annotations Kishor K. Shende Information Officer Bioinformatics...

Genomics: Gene prediction and Annotations
Kishor K. Shende
Information Officer
Bioinformatics Center,
Barkatullah University Bhopal
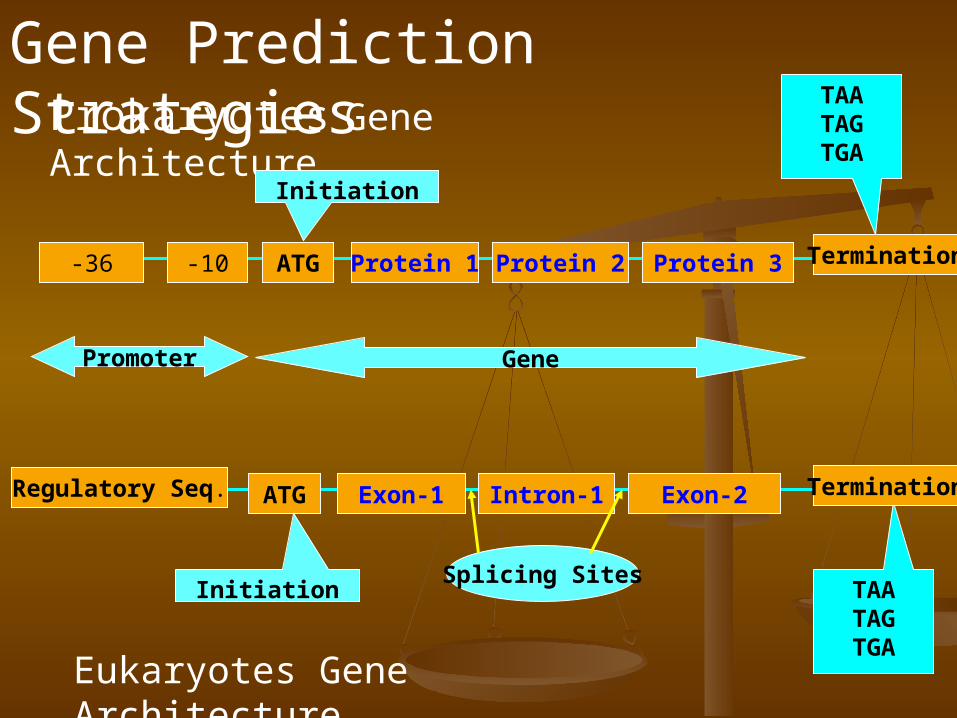
Prokaryotes Gene Architecture
ATG-10-36 Protein 1 Protein 2 Protein 3 Termination
TAATAGTGA
Promoter Gene
Initiation
ATGRegulatory Seq. Exon-1 Intron-1 Exon-2
TAATAGTGA
Initiation
Termination
Eukaryotes Gene Architecture
Splicing Sites
Gene Prediction Strategies
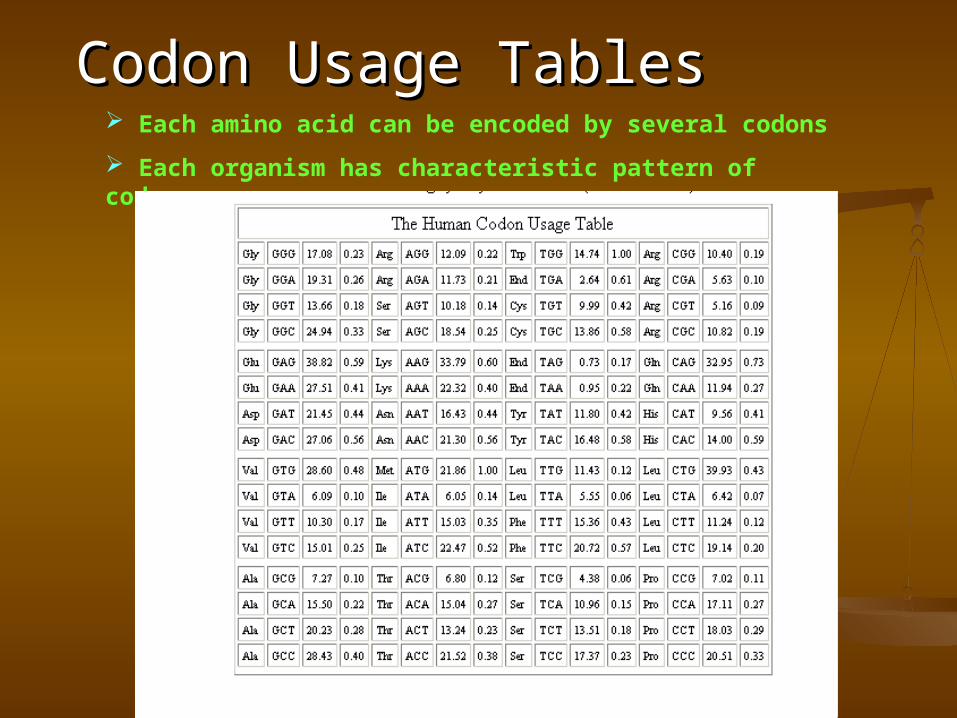
Codon Usage TablesCodon Usage Tables Each amino acid can be encoded by several codons
Each organism has characteristic pattern of codon usage

Problems in Gene Prediction Distinguishing Pseudogenes from Genes
Exon-Intron Structure in Eukaryotes, Exon flanking regions – not very well conserved
Alternative Splicing – Shuffling of Exons
Genes can overlap each other and occur on different strand of DNA

Gene Identification
1. Homology Based Gene prediction Sequence Similarity Search against gene database using BLAST and FAST searching tools
EST (Expressed Sequence Tags) similarity search
2. Ab initio Gene Prediction Prokaryotes
- ORF finding
Eukaryotes
- Promoter prediction
- Start-Stop codon prediction
- Splice site Prediction (Exon-Intron and Intron –Exon)
- PolyA signal prediction
1. Homology Based Gene prediction Sequence Similarity Search against gene database using BLAST and FAST searching tools
EST (Expressed Sequence Tags) similarity search
2. Ab initio Gene Prediction Prokaryotes
- ORF finding
Eukaryotes
- Promoter prediction
- Start-Stop codon prediction
- Splice site Prediction (Exon-Intron and Intron –Exon)
- PolyA signal prediction

ORF Finding in ProkaryotesEasier due to ………..
Small Genome have high gene density (Haemophilus influenza – 85% genic)
No Introns or Few Introns
Operons
- One Transcript, many genes
Open Reading Frames (ORF)
- Contigous set of codons, start with Met-codon, ends with stop codon
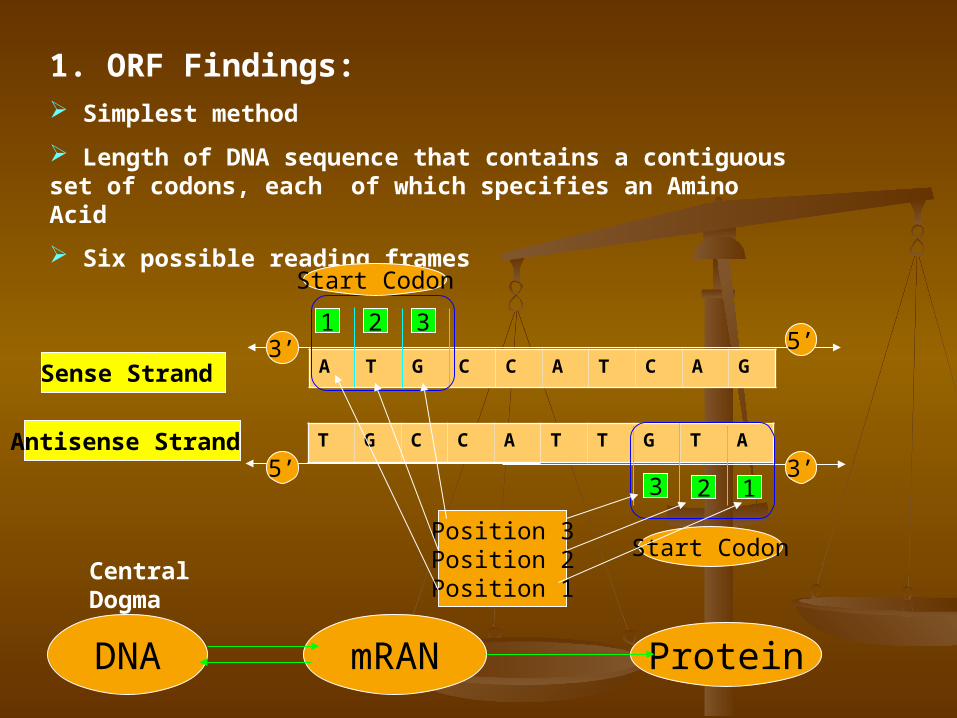
1. ORF Findings: Simplest method
Length of DNA sequence that contains a contiguous set of codons, each of which specifies an Amino Acid
Six possible reading frames
Sense Strand
Antisense Strand
A T G C C A T C A G
T G C C A T T G T A
5’
3’
3’
5’
123
1 2 3
Start Codon
Start CodonPosition 3Position 2Position 1
DNA mRAN Protein
Central Dogma
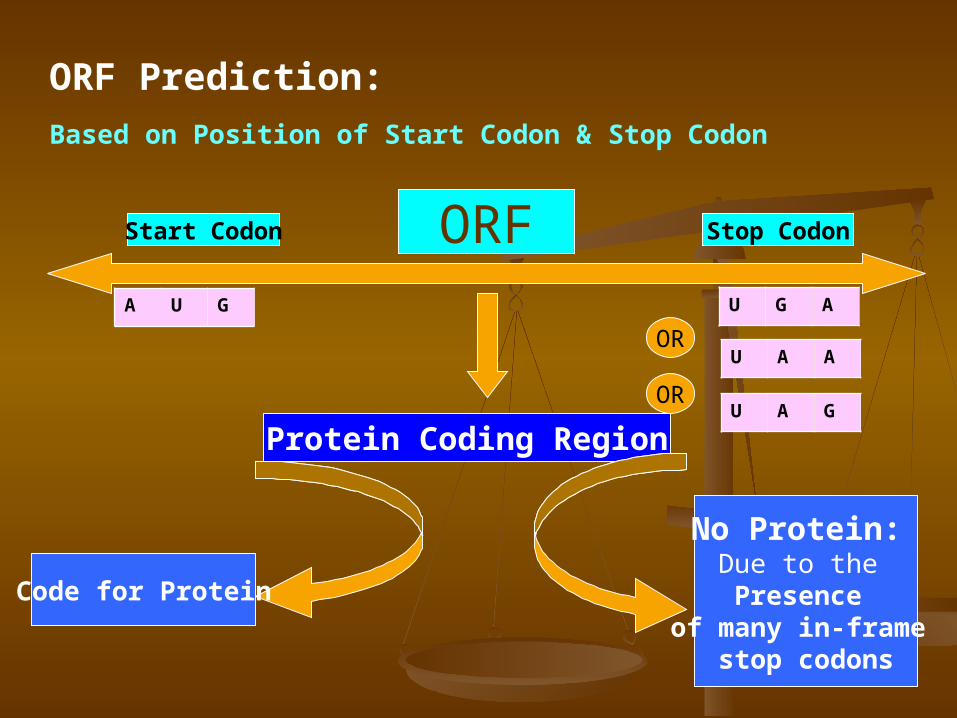
ORF Prediction: Based on Position of Start Codon & Stop Codon
Start Codon Stop Codon
A U G
U A G
U A A
U G A
OR
OR
ORF
Protein Coding Region
Code for Protein
No Protein: Due to the Presence
of many in-frame stop codons

Example of ORFExample of ORF
There are six possible ORFs in each sequence for both directions of transcription.

Difficulty in ORF Prediction:1. Prokaryotes & Viruses: Presence of multiple genes on mRNA
and Overlapping genes in which two different proteins may be encoded in different reading frames of the same mRNA
2. Eukaryotes: Protein coding region (Exon) is followed by non-coding region (Intron)
3. Differential mRNA splicing create different mRNA, hence different proteins
4. Variation in Genetic Code from Universal code
Reliability of ORF Prediction: Characteristics of ORF regions
1. Ordered list of specific codons that reflects the evolutionary origin of the gene and constraints associated with gene expressions
2. Characteristics pattern of use of synonymous codons i.e. codons that stands for same Amino Acid
3. In Eukaryotes strong preferences for codon pairs at Intron-Exon or Exon-Intron junction
4. High genome content of GC have a strong bias of G & C in the third codon positions
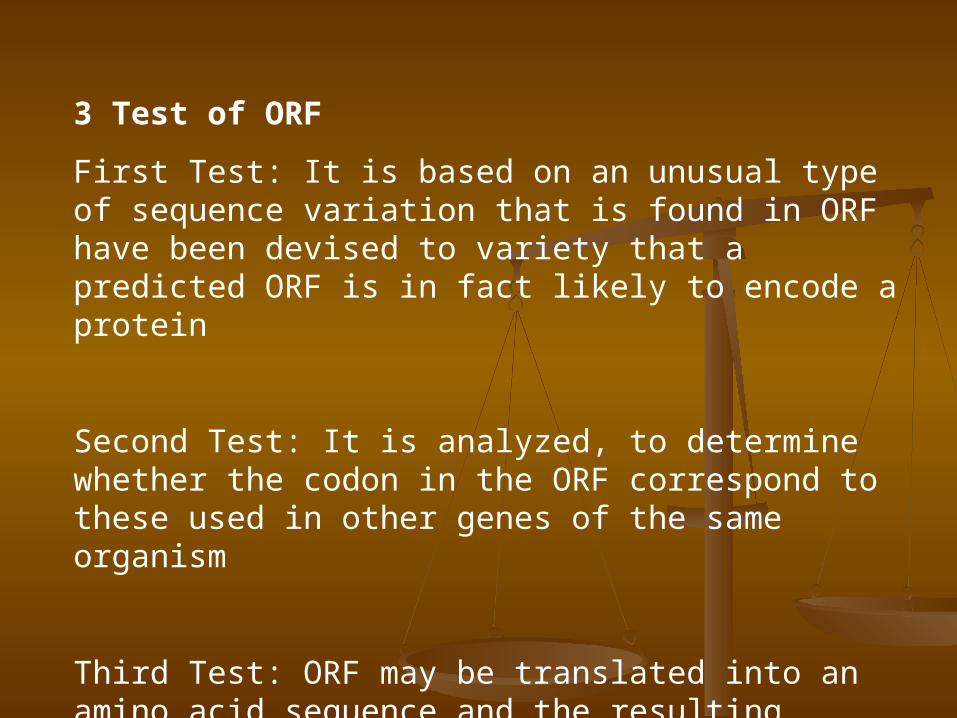
3 Test of ORF
First Test: It is based on an unusual type of sequence variation that is found in ORF have been devised to variety that a predicted ORF is in fact likely to encode a protein
Second Test: It is analyzed, to determine whether the codon in the ORF correspond to these used in other genes of the same organism
Third Test: ORF may be translated into an amino acid sequence and the resulting sequence then compound to the databases of existing sequence

Repeated Sequence Elements and Nucleosome Structure
1. Eukaryotic DNA is wrapped around histon-protein complexes
2. Some base pairs in the major or minor grooves of the DNA molecules face the nucleosome surface
3. Other pair face outside of the structures
4. Nucleosome located in the promoter regions are remodeled in a manner that can influence the availability of binding sites for regulatory proteins making them more or less available
Hidden Morkov Model (HMM) of Eukaryotic Internal Exon
Computational Background: Repeated patterns of sequence have been found in the Introns and Exons and near the start site of Transcriptuion of Eukaryotic genes
Bending Pattern: Bending is influenced by
1. Repeated pattern i.e. not T, A or G, G
2. AA/TT dinucleotide

Ab initioAb initio gene prediction gene predictionPredictions are based on the observation Predictions are based on the observation
that gene DNA sequence is not random:that gene DNA sequence is not random:
-- Gene-coding sequence has Gene-coding sequence has start and stop start and stop codons.codons.
- Each species has a characteristic pattern Each species has a characteristic pattern of of synonymous codon usagesynonymous codon usage..
- Non-codingNon-coding ORFs are very ORFs are very short.short.- Gene would correspond to the Gene would correspond to the longest ORF.longest ORF.
These methods look for the characteristic These methods look for the characteristic features of genes and score them high.features of genes and score them high.

Ab initio gene prediction methodsAb initio gene prediction methods
GeneScanGeneScan – – Fourier transform of DNA sequence to Fourier transform of DNA sequence to find characteristic patterns.find characteristic patterns.
GeneParser GeneParser – – predicts the most likely combination predicts the most likely combination of exons/introns. Dynamic programming.of exons/introns. Dynamic programming.
GeneMarkGeneMark – – mostly for prokaryotes, Hidden mostly for prokaryotes, Hidden Markov Models. Also for EukaryotesMarkov Models. Also for Eukaryotes
Grail IIGrail II – – predicts exons, promoters, Poly(A) sites. predicts exons, promoters, Poly(A) sites. Neural network plus dynamic programming.Neural network plus dynamic programming.

Gene Preference Score : Important indicator of coding region
Observation:Observation: frequencies of codons and codon pairs in coding and non-frequencies of codons and codon pairs in coding and non-coding regions are different.coding regions are different.
Given a sequence of codons:Given a sequence of codons:
and assuming independence, the probability of finding coding regionand assuming independence, the probability of finding coding region ::
The probability of finding sequence “The probability of finding sequence “CC” in non-coding regions:” in non-coding regions:
The gene preference score:The gene preference score:
))(
)(log(
0 CP
CPGPS
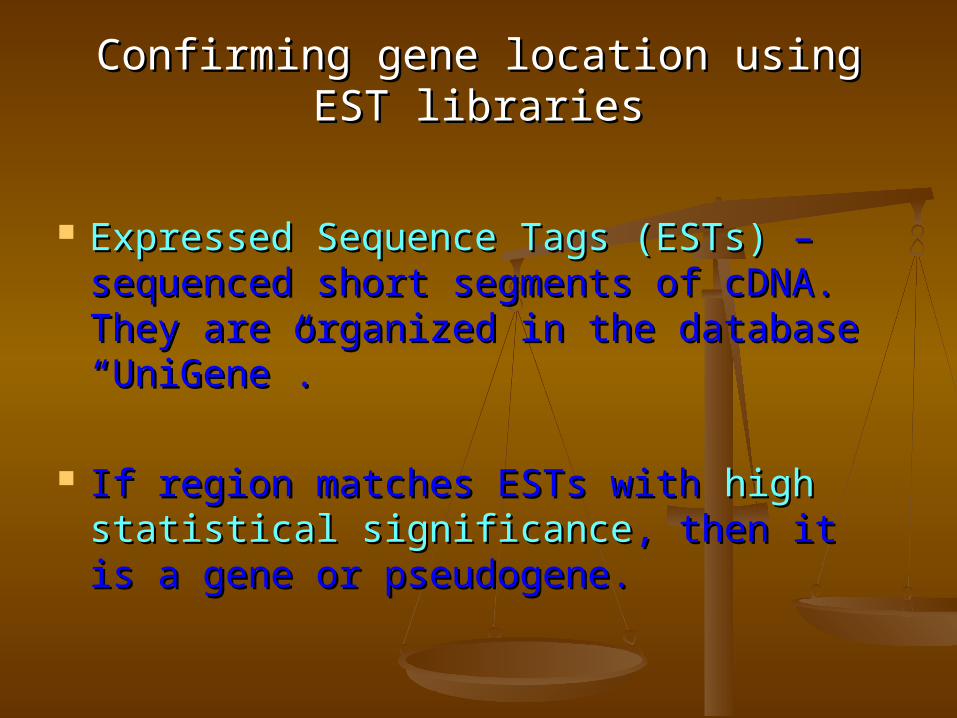
Confirming gene location using EST librariesConfirming gene location using EST libraries
Expressed Sequence Tags (ESTs)Expressed Sequence Tags (ESTs) – – sequenced short segments of cDNA. sequenced short segments of cDNA. They are organized in the database They are organized in the database “UniGene”.“UniGene”.
If region matches ESTs with If region matches ESTs with high high statistical significancestatistical significance, then it is a gene , then it is a gene or pseudogene.or pseudogene.

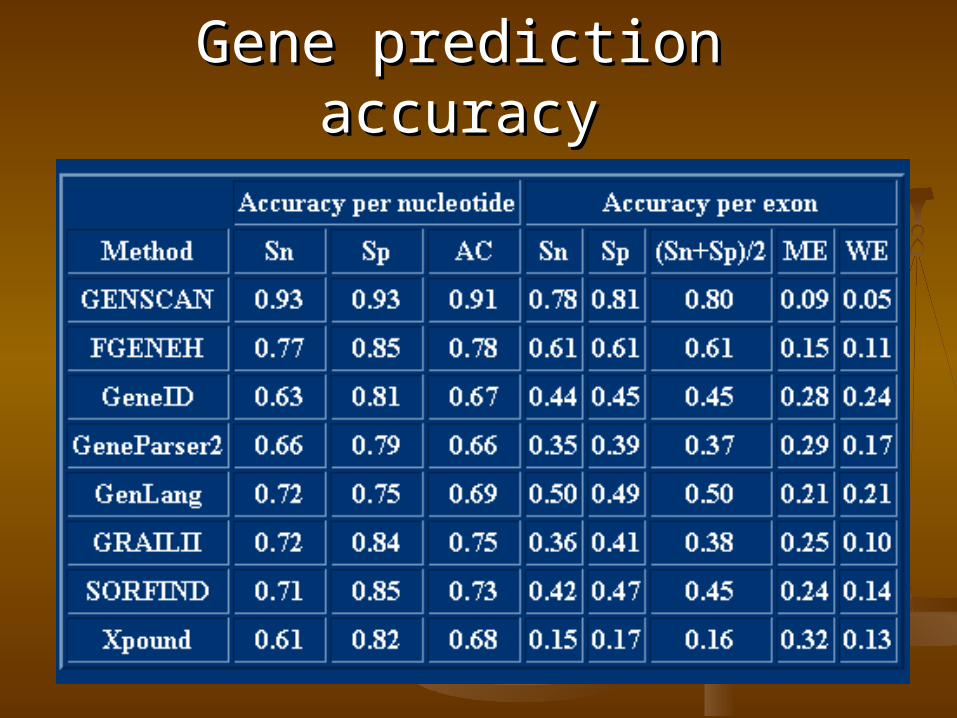
Gene prediction accuracyGene prediction accuracy
True positives (TP)True positives (TP) – nucleotides, which – nucleotides, which are correctly predicted to be within the are correctly predicted to be within the gene.gene.
Actual positives (AP)Actual positives (AP) – nucleotides, which – nucleotides, which are located within the actual gene.are located within the actual gene.
Predicted positives (PP)Predicted positives (PP) – nucleotides, – nucleotides, which are predicted in the gene.which are predicted in the gene.
Sensitivity = TP / APSensitivity = TP / AP
Specificity = TP / PPSpecificity = TP / PP
Gene prediction accuracyGene prediction accuracy

Common Difficulties of Gene PredictionCommon Difficulties of Gene Prediction
First and last exons difficult to annotate First and last exons difficult to annotate because they contain UTRs.because they contain UTRs.
Smaller genes are not statistically Smaller genes are not statistically significant so they are thrown out.significant so they are thrown out.
Algorithms are trained with sequences from Algorithms are trained with sequences from known genes which biases them against known genes which biases them against genes about which nothing is knowngenes about which nothing is known..

Genome Analysis for Gene Prediction
Genome analysisGenome analysis
Genome – the sum of genes and intergenic Genome – the sum of genes and intergenic sequences of haploid cell.sequences of haploid cell.
The value of genome sequences lies in their annotationThe value of genome sequences lies in their annotation
Annotation – Characterizing genomic features Annotation – Characterizing genomic features using computational and experimental using computational and experimental methodsmethods
Genes: levels of annotationGenes: levels of annotation Gene Prediction – Where are genes?Gene Prediction – Where are genes? What do they encode?What do they encode? What proteins/pathways involved in?What proteins/pathways involved in?
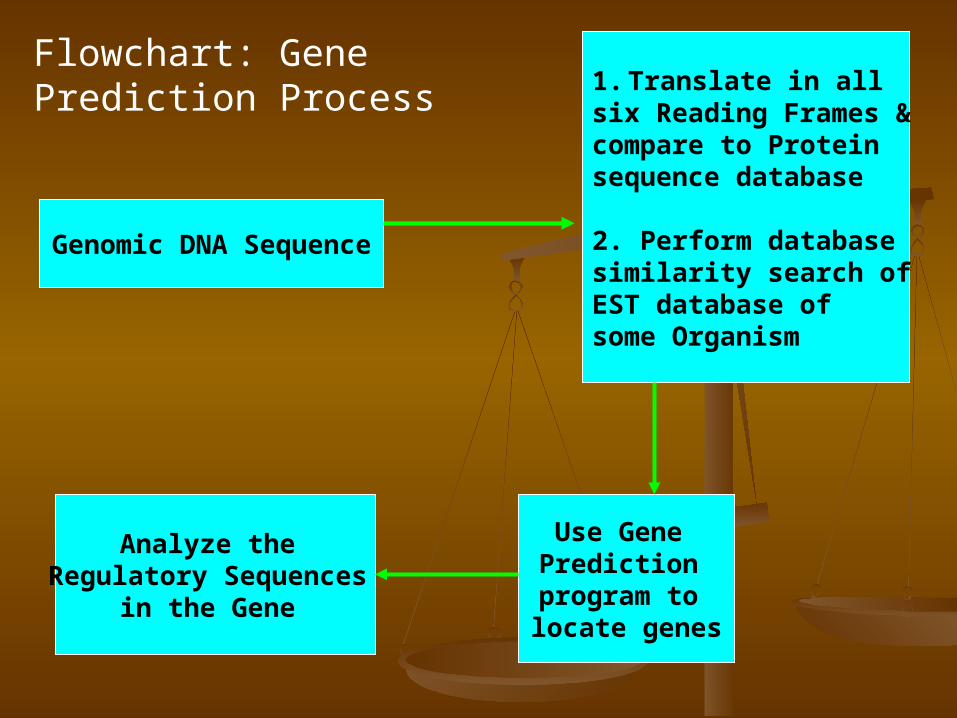
Flowchart: Gene Prediction Process
Genomic DNA Sequence
1. Translate in all six Reading Frames & compare to Protein sequence database
2. Perform database similarity search of EST database of some Organism
Use Gene Prediction program to locate genes
Analyze the Regulatory Sequences
in the Gene
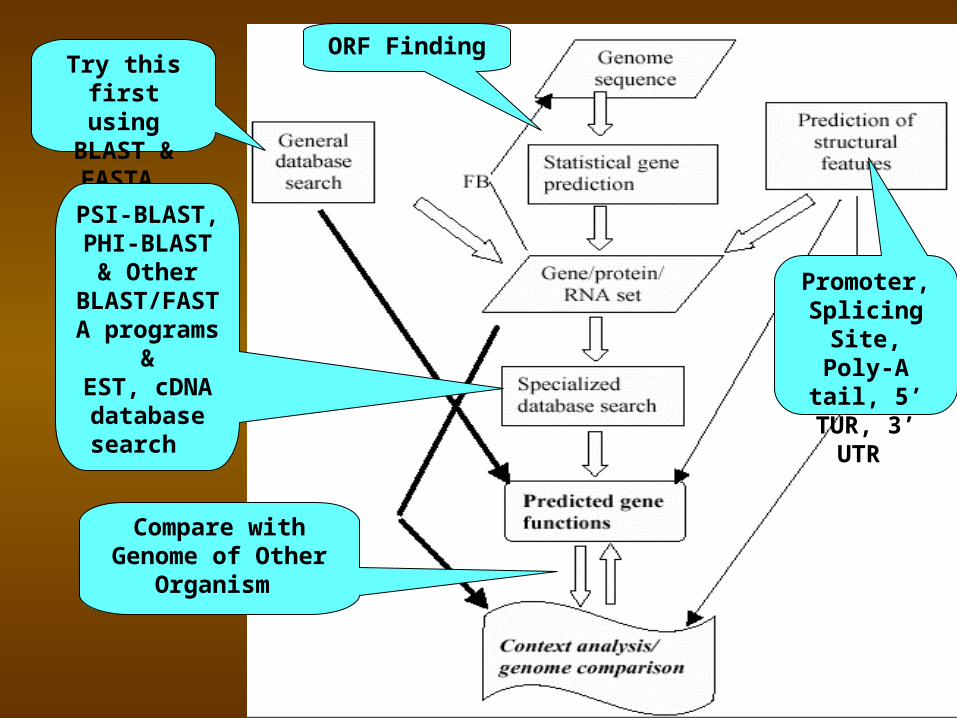
Try this first using BLAST & FASTA
ORF Finding
Promoter, Splicing
Site, Poly-A tail, 5’ TUR, 3’
UTR
PSI-BLAST, PHI-BLAST
& Other BLAST/FAS
TA programs
&EST, cDNA database search
Compare with Genome of Other
Organism
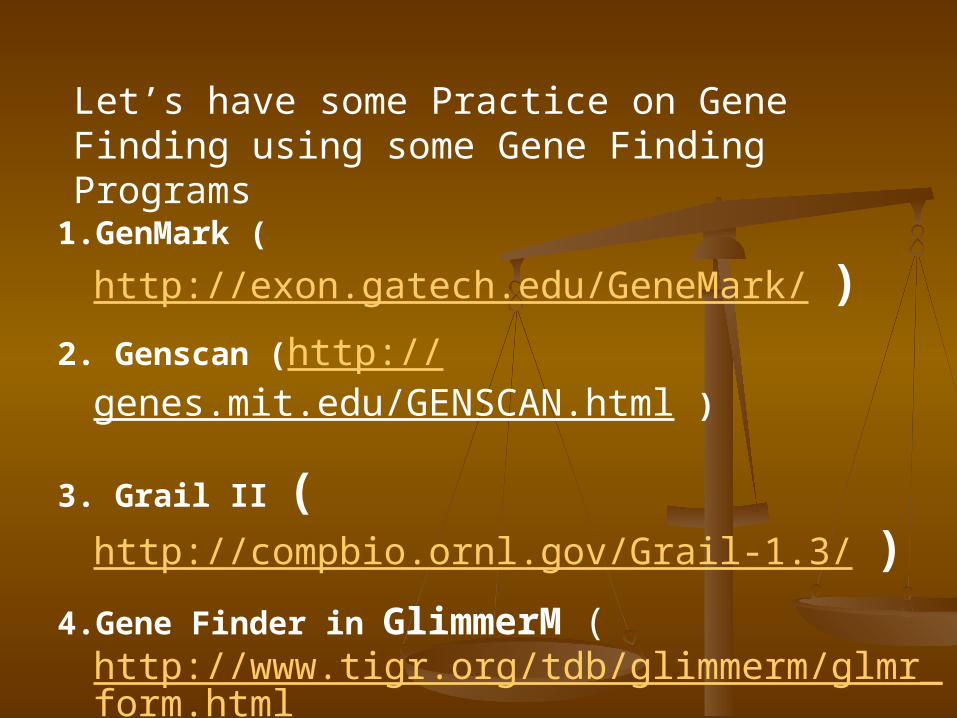
Let’s have some Practice on Gene Finding using some Gene Finding Programs
1.GenMark (http://exon.gatech.edu/GeneMark/
)2. Genscan (http://
genes.mit.edu/GENSCAN.html )
3. Grail II (http://compbio.ornl.gov/Grail-1.3/ )
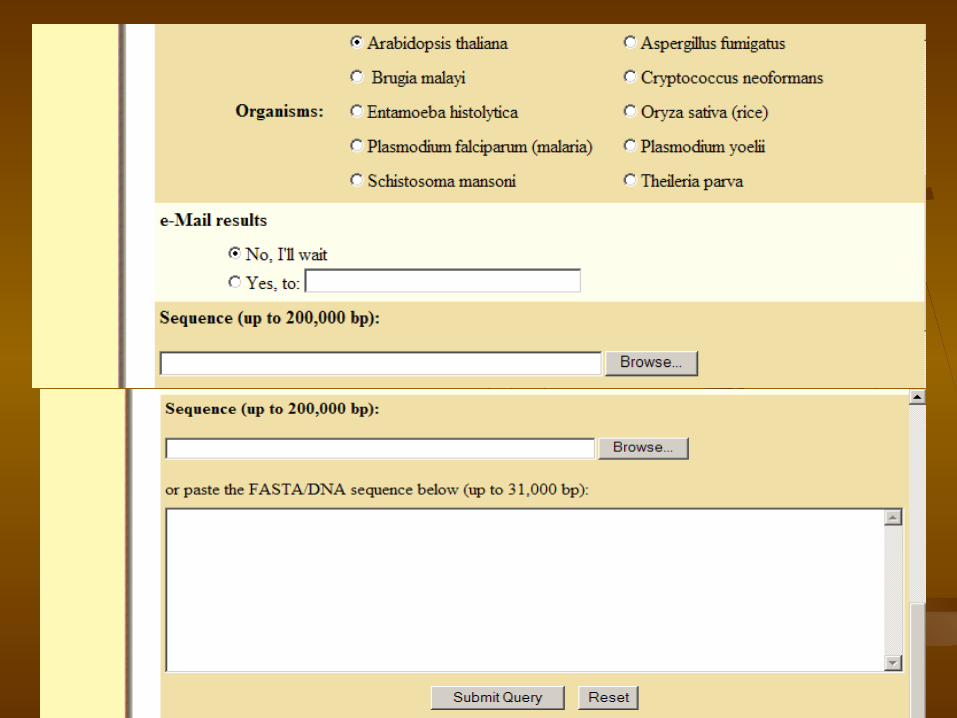
4.Gene Finder in GlimmerM (http://www.tigr.org/tdb/glimmerm/glmr_form.html )

HMMgene - Prediction of genes in vertebrate and C. elegans Gene Discovery Page FramePlot - protein-coding region prediction tool for high GC-content bacteria tRNAscan-SE Search for transfer RNA genes in genomic sequence NETGENE - Predict splice sites in human genes ORF Finder BCM Gene Finder Grail Genemark Genie: A Gene Finder Based on Generalized Hidden Markov Models GENSCAN - predict complete gene structures Splice Site Prediction by Neural Network Procrustes GenePrimer GenLang MZEF Gene Finder Webgene - Tools for prediction and analysis of protein-coding gene structure MAR-Finder - Nuclear matrix attachment region prediction Glimmer bacterial/archael gene finder

Promoter Region, Transscription Factor and Signals1. TRANSFAC - Transcription Factor database
TFD Transcription Factor Database TransTerm - A Translational Signal Database PLACE - a database of plant cis-acting regulatory DNA elements NNPP: Promoter Prediction by Neural Network FastM/ModelInspector TFSEARCH MatInd and MatInspector Transcription Element Search Software (TESS) CorePromoter (Core-Promoter Prediction Program) Gene Express - analysis of genomic regulatory sequences Signal Scan PromoterInspector Promoter Scan II Pol3scan TargetFinder - finds DNA-binding proteins.

Overview Overview GENE PREDICTION TOOLSGENE PREDICTION TOOLS
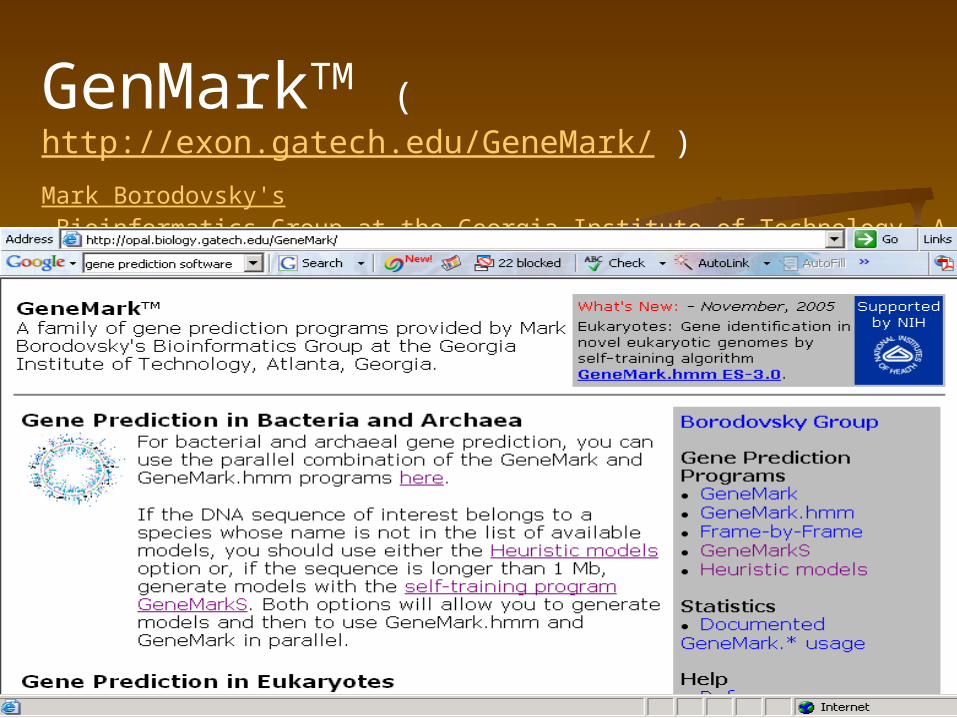
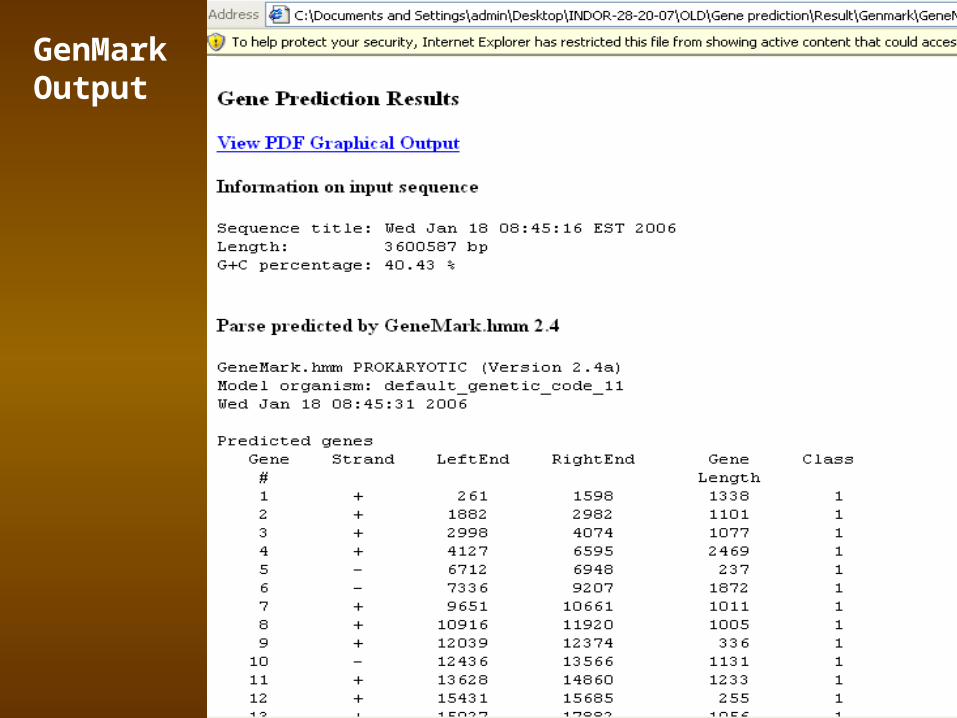
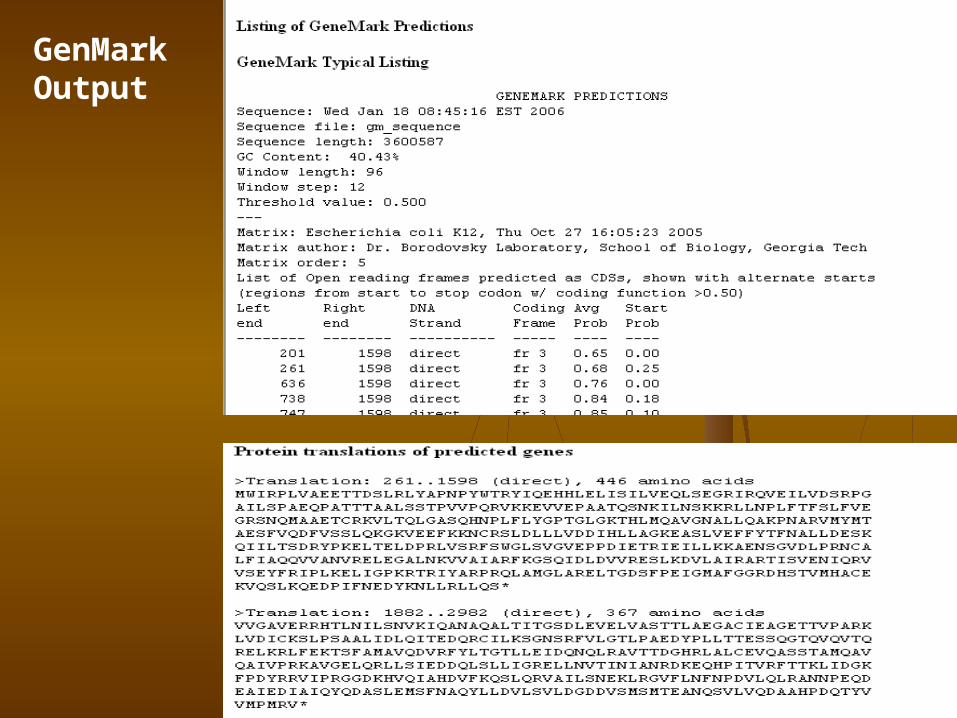
GenMarkTM (http://exon.gatech.edu/GeneMark/ )Mark Borodovsky's Bioinformatics Group at the Georgia Institute of Technology, Atlanta, Georgia

GeneMark.hmm for Prokaryotes (Version 2.4)
Reference:
Lukashin A. and Borodovsky M., GeneMark.hmm: new solutions for gene finding, NAR, 1998, Vol. 26, No. 4, pp. 1107-1115
Bacterial and archaeal gene prediction, you can use the parallel combination of the GeneMark and GeneMark.hmm programs
Heuristic Approach for Gene Prediction in Prokaryotes If the DNA sequence of interest belongs to a species whose name is not in the list of available models, use the Heuristic models option
Self Training Program of Genmarks If the sequence is longer than 1 Mb, generate models with the self-training program GeneMarkS
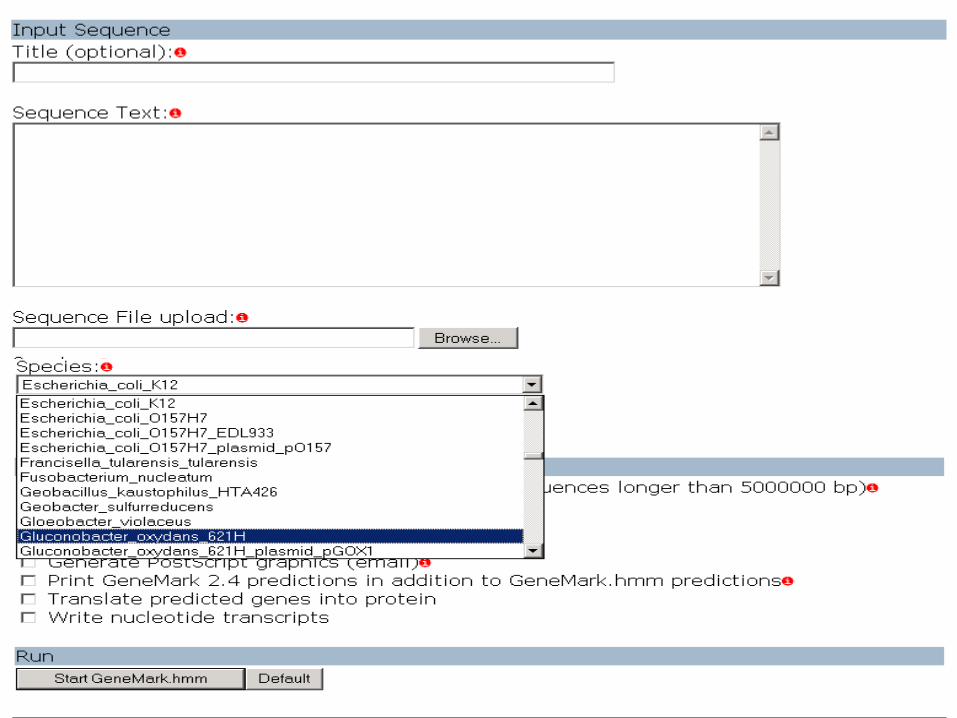

Gene Prediction in Eukaryotes
Eukaryotic gene prediction: Use the parallel combination of the GeneMark and GeneMark.hmm
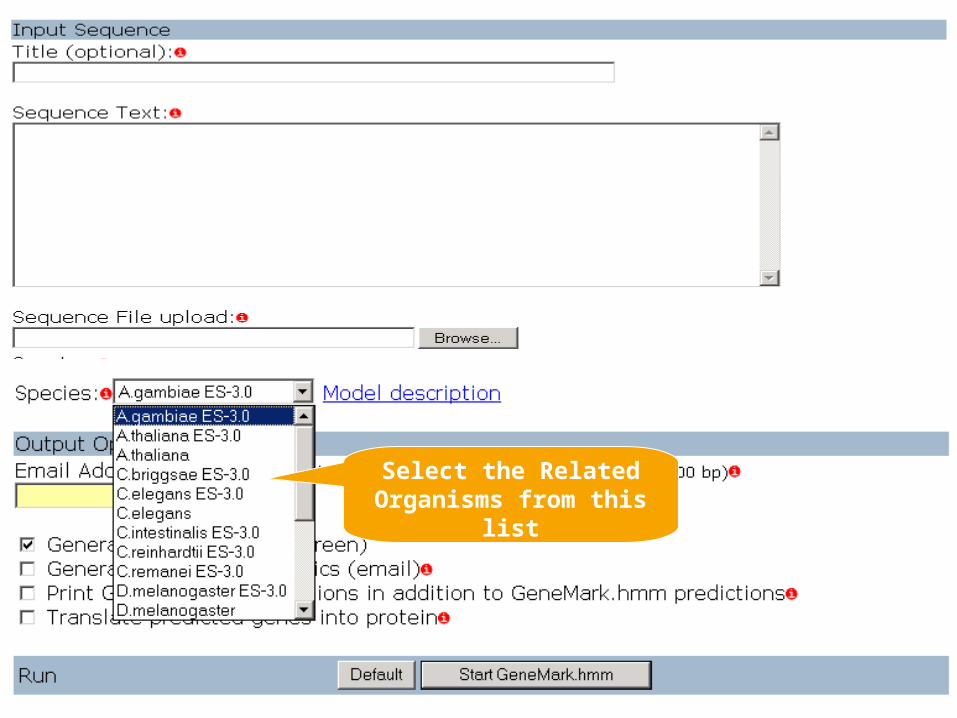
Select the Related Organisms from this
list

Gene Prediction in EST and cDNA
To analyze ESTs and cDNAs
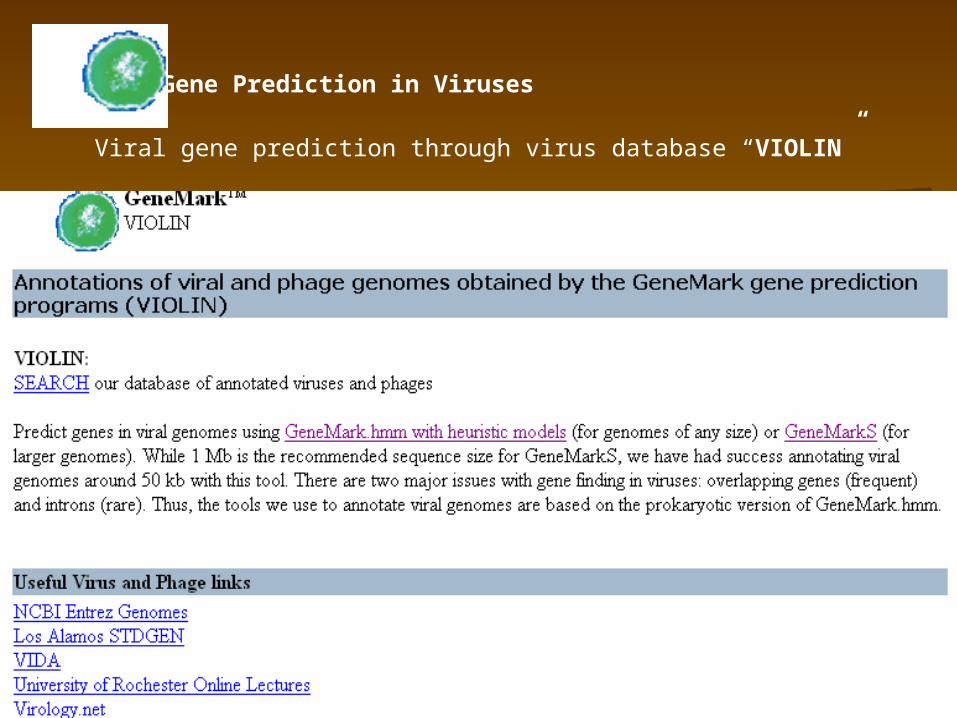
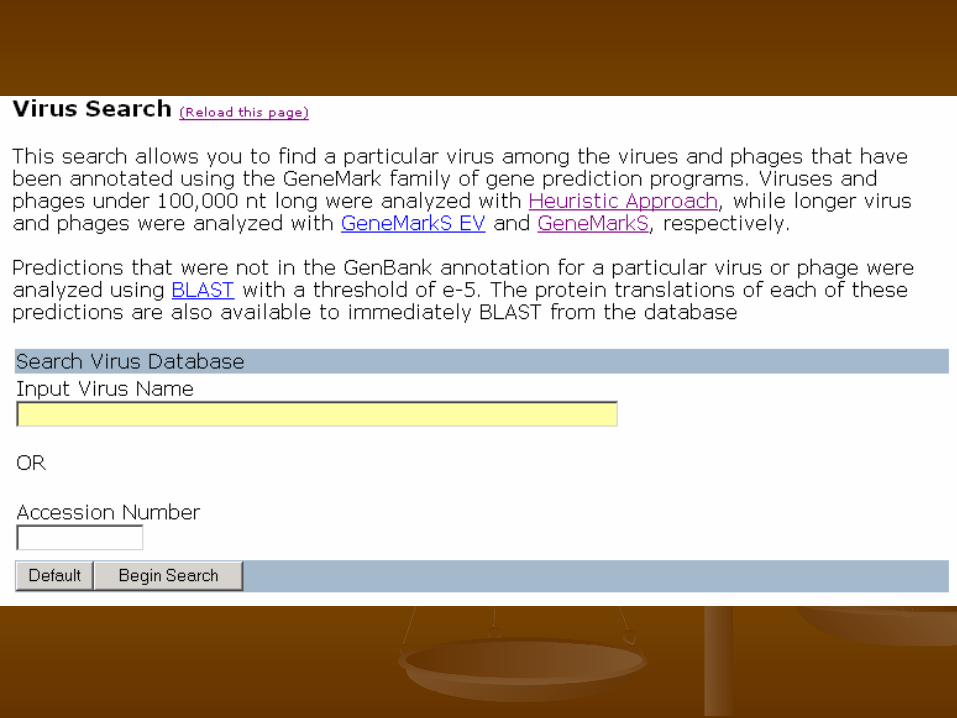
Gene Prediction in Viruses
Viral gene prediction through virus database “VIOLIN”
GenMark Output
GenMark Output
New GENSCAN Web Server at MIT
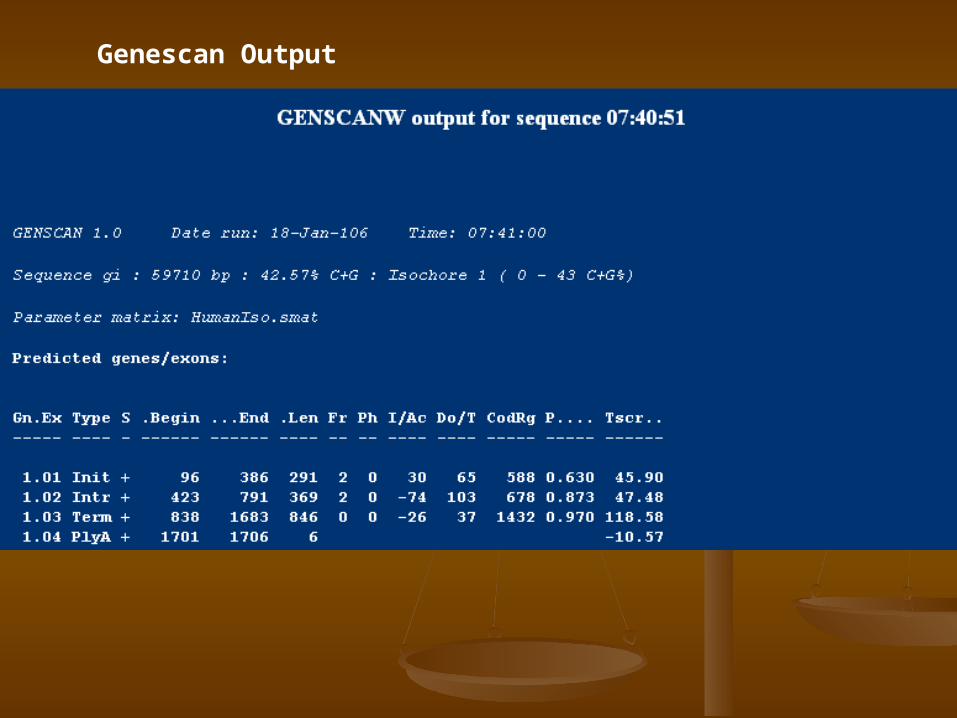
Genescan Output
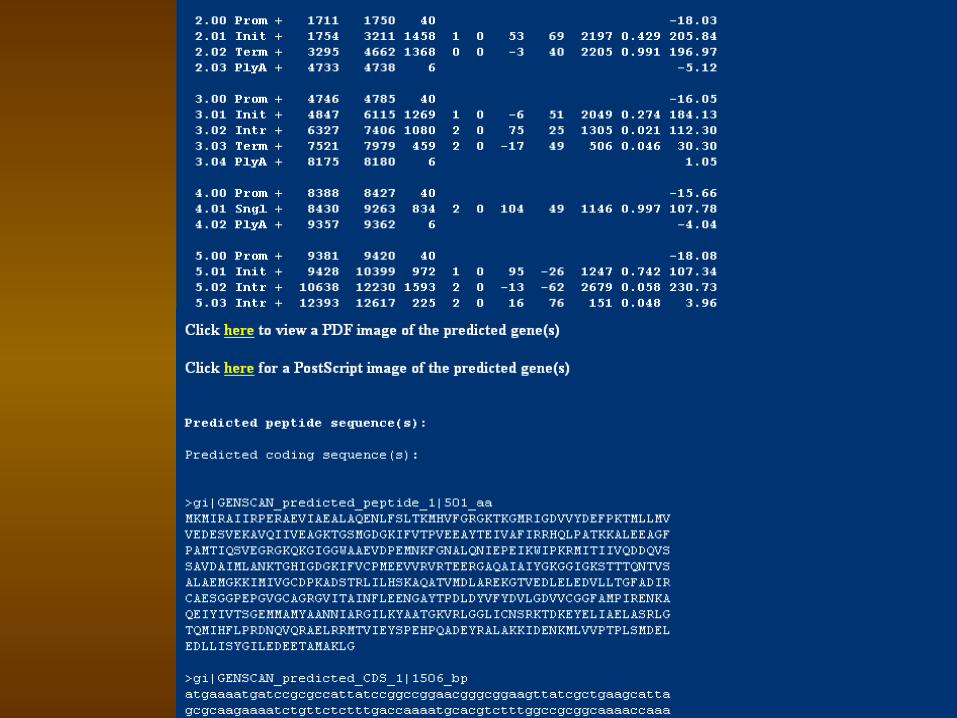
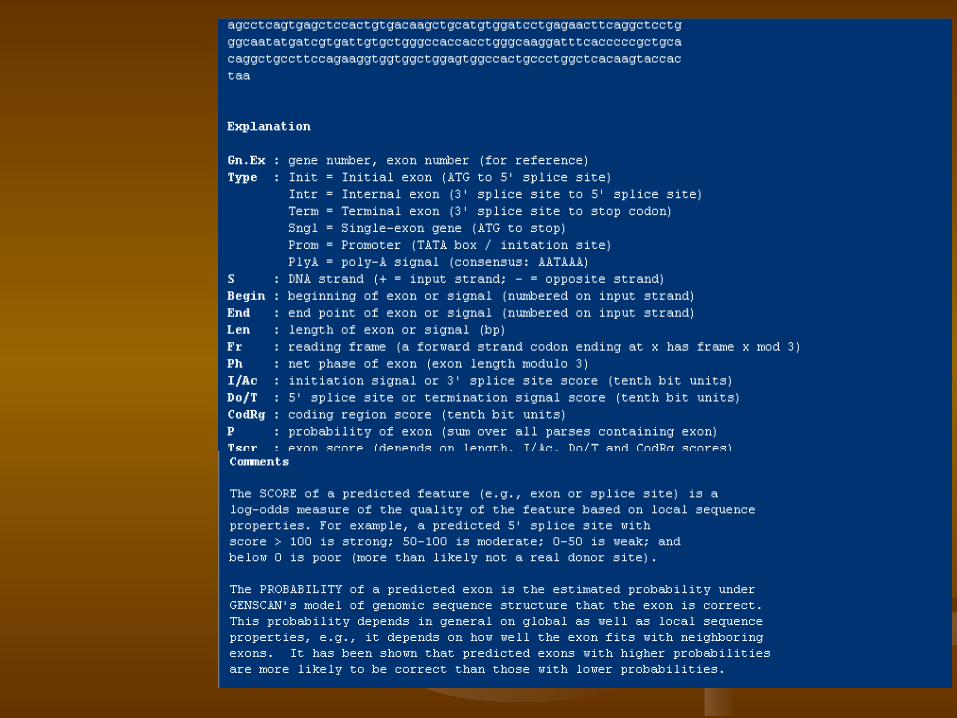

1.Locate protein coding genes within DNA sequence,
2.Locate EST/mRNA alignments, 3.Locate certain types of promoters,
polyadenylation sites, CpG islands, and repetitive elements.
GrailEXP
GrailEXP is a gene finder………….1.EST alignment utility 2.exon prediction program, 3.a promoter/polya recognizer, 4.a CpG island finer, 5.a repeat masker,
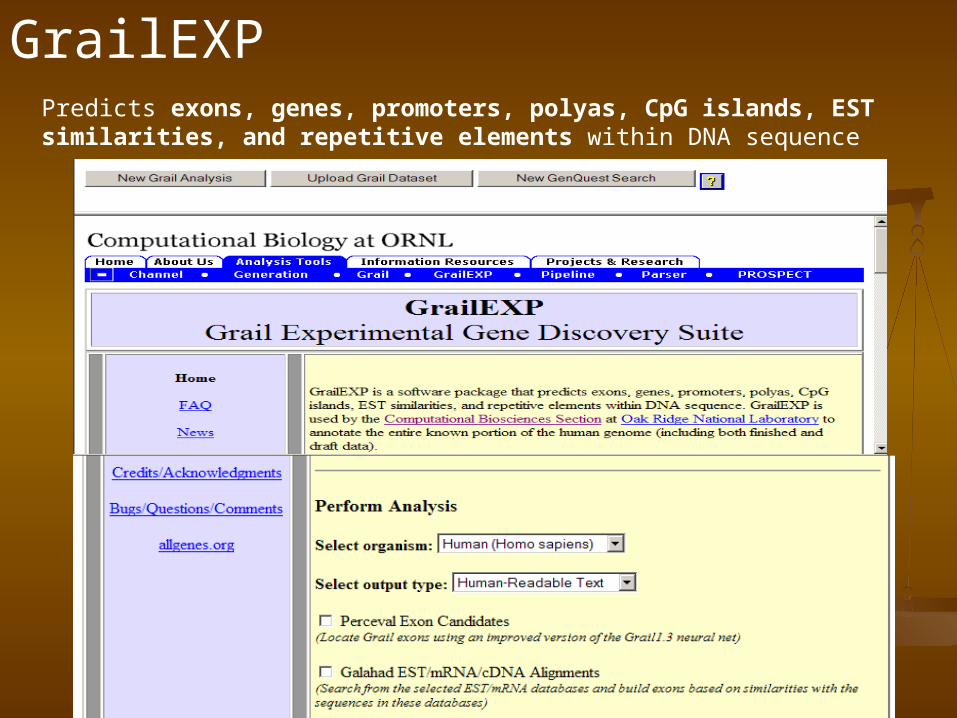
GrailEXPPredicts exons, genes, promoters, polyas, CpG islands, EST similarities, and repetitive elements within DNA sequence
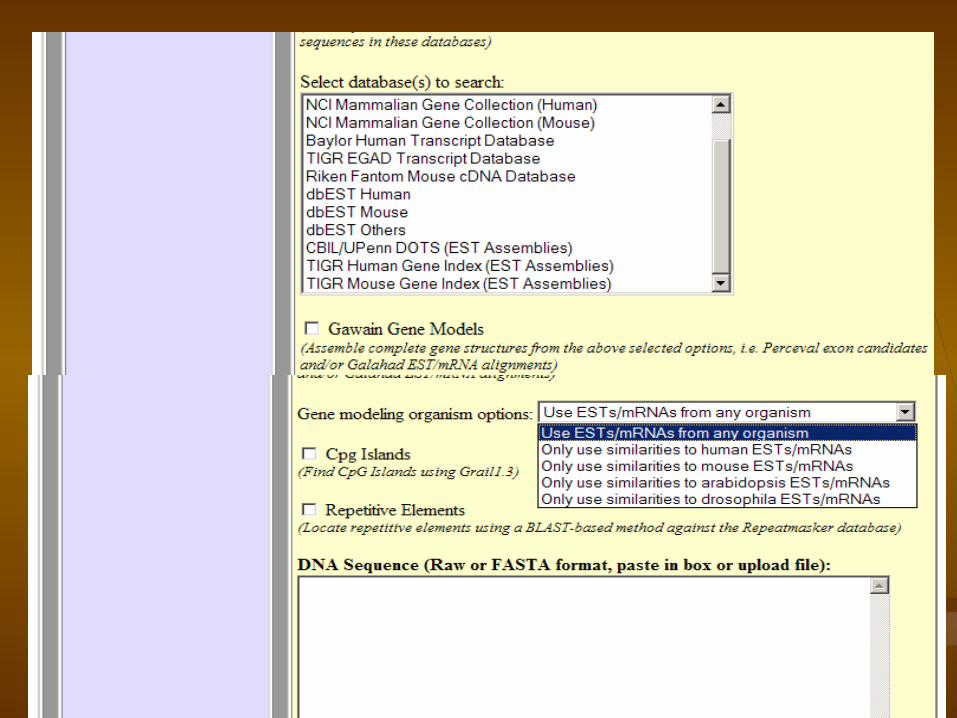

GlimmerM: http://www.tigr.org/tdb/glimmerm/glmr_form.html
A system for finding genes in microbial DNA, especially the genomes of bacteria and archaea.Glimmer (Gene Locator and Interpolated Markov Modeler) uses interpolated Markov models (IMMs) to identify the coding regions and distinguish them from noncoding DNA.
GlimmerHMM: For Eukaryotic Organisms
Genesplicer: Fast, flexible system for detecting splice sites in the genomic DNA of various eukaryotes.
GLimmerM Gene Finder

Kishor K. Shende
Information Officer
Bioinformatics Center,
Barkatullah University Bhopal


















