
Genome similarity of Baikal omul and sig
8
ISSN 1068-1620, Russian Journal of Bioorganic Chemistry, 2009, Vol. 35, No. 1, pp. 86–93. © Pleiades Publishing, Ltd., 2009. Original Russian Text © O.S. Bychenko, L.V. Sukhanova, S.S. Ukolova, T.A. Skvortsov, V.K. Potapov, T.L. Azhikina, E.D. Sverdlov, 2009, published in Bioorganicheskaya Khimiya, 2009, Vol. 35, No. 1, pp. 95–102. 86 INTRODUCTION The Baikal lake sig (Coregonus lavaretus baicalensis Dybovsky) is a deep-water bentophage inhabiting at the depths of 30–100 m and spawning in the lake. 2 The Baikal omul (C. autumnalis migratorius Georgi) is a pelagic planktosphage inhabiting the depths of 350– 400 m [1–4]. For a long time these fishes were referred to different species based on morphological signs. However, the recently performed analysis of mtDNA variability and isoenzymatic analysis showed the simi- larity of Baikal omul and true cisco, first of all, to C. lav- aretus L. species [5-8]. The next detailed phylogenetic analysis of the mtDNA structure of paleoarctic and neo- arctic sigs [9] as well as genealogical reconstruction of the Baikal sig family [10] provided the final conclusion on the belonging of the Baikal omul to the C. lavaretus L. species. Thus, the Baikal omul and the lake sig are close relatives and their divergence only occurred 10– 20 thousand years ago, probably, due to assimilation of new trophic niches [10]. Rather a short period of diver- gence of the Baikal sig and omul within the evolution implies that the comparison of their genomes may reveal genetic factors which in this case play a defining role upon the population divergence. The goal of our work was the search of genomic elements (both coding and noncoding) of omul and sig genomes differed in structure. RESULTS AND DISCUSSION We used the method of subtractive hybridization (SH) modified by us for comparison of sig and omul genomes. This method enables the sequences specific for genomic DNA of one organism, the so called tracer, which are absent in the DNA of another organism, a driver. The genomic structure of the sig family is unknown. However, analogously to Danio rerio and Tetraodon, whose full genome sequences have been determined [ZFIN, The Zebrafish Information Network, www.cns.fr/tetraodon], one can expect that they con- tain a large number of repeated elements which impede both the SH and the subsequent analysis of the clones obtained. This issue can be overcome by inclusion of the preliminary stage of “normalization” of the com- pared fragmented genomes widely used for cDNA SH schemes [11, 12]. This stage involves the formation of a rapidly renaturating fraction enriched with highly represented sequences, repeated genome elements in the case of genome comparison. Then this fraction is excluded from the further analysis. In this way the tracer is getting enriched with low represented genome sequences. Genome Similarity of Baikal Omul and Sig O. S. Bychenko a , L. V. Sukhanova b , S. S. Ukolova a , T. A. Skvortsov a , V. K. Potapov a , T. L. Azhikina a,1 , and E. D. Sverdlov a a Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117997 Russia b Institute of Limnology, Siberian Branch, Russian Academy of Sciences, ul. Lermontova 281, Irkutsk, 664033 Russia Received March 20, 2008; in final form, May 28, 2008 Abstract—Two members of the Baikal sig family, a lake sig (Coregonus lavaretus baicalensis Dybovsky) and omul (C. autumnalis migratorius Georgi) are close relatives that diverged from the same ancestor 10–20 thou- sand years ago. In this work, we studied genomic polymorphism of these two fish species. The method of sub- tractive hybridization (SH) did not reveal the presence of extended sequences in the sig genome and their absence in the omul genome. All the fragments found by SH corresponded to polymorphic noncoding genome regions varying in single nucleotide polymorphisms (SNPs) and short deletions. Many of them are mapped close to genes of the immune system and have regions identical to the Tc1-like transposons abundant among fish, whose transcription activity may affect the expression of adjacent genes. Thus, we showed for the first time that genetic differences between Baikal sig family members are extremely small and cannot be revealed by the SH method. This is another endorsement of the hypothesis on the close relationship between Baikal sig and omul and their evolutionarily recent divergence from a common ancestor. Key words: Coregonidae, speciation, genomic polymorphism, subtractive hybridization DOI: 10.1134/S1068162009010117 1 Corresponding author; phone: +7 (495) 330-6547; fax: +7 (495) 330-6538; e-mail: [email protected]. 2 Abbreviations: mtDNA, mitochondrial DNA; RFLP, restriction fragment length polymorphism; SH, subtractive hybridization.
-
Upload
o-s-bychenko -
Category
Documents
-
view
213 -
download
0
Transcript of Genome similarity of Baikal omul and sig

ISSN 1068-1620, Russian Journal of Bioorganic Chemistry, 2009, Vol. 35, No. 1, pp. 86–93. © Pleiades Publishing, Ltd., 2009.Original Russian Text © O.S. Bychenko, L.V. Sukhanova, S.S. Ukolova, T.A. Skvortsov, V.K. Potapov, T.L. Azhikina, E.D. Sverdlov, 2009, published in BioorganicheskayaKhimiya, 2009, Vol. 35, No. 1, pp. 95–102.
86
INTRODUCTION
The Baikal lake sig (
Coregonus
lavaretus
baicalensis
Dybovsky) is a deep-water bentophage inhabiting at thedepths of 30–100 m and spawning in the lake.
2
TheBaikal omul (
C
.
autumnalis
migratorius
Georgi) is apelagic planktosphage inhabiting the depths of 350–400 m [1–4]. For a long time these fishes were referredto different species based on morphological signs.However, the recently performed analysis of mtDNAvariability and isoenzymatic analysis showed the simi-larity of Baikal omul and true cisco, first of all, to
C
.
lav-aretus
L. species [5-8]. The next detailed phylogeneticanalysis of the mtDNA structure of paleoarctic and neo-arctic sigs [9] as well as genealogical reconstruction ofthe Baikal sig family [10] provided the final conclusionon the belonging of the Baikal omul to the
C
.
lavaretus
L. species. Thus, the Baikal omul and the lake sig areclose relatives and their divergence only occurred 10–20 thousand years ago, probably, due to assimilation ofnew trophic niches [10]. Rather a short period of diver-gence of the Baikal sig and omul within the evolutionimplies that the comparison of their genomes mayreveal genetic factors which in this case play a definingrole upon the population divergence. The goal of our
work was the search of genomic elements (both codingand noncoding) of omul and sig genomes differed instructure.
RESULTS AND DISCUSSION
We used the method of subtractive hybridization(SH) modified by us for comparison of sig and omulgenomes. This method enables the sequences specificfor genomic DNA of one organism, the so called tracer,which are absent in the DNA of another organism, adriver.
The genomic structure of the sig family is unknown.However, analogously to
Danio
rerio
and
Tetraodon
,whose full genome sequences have been determined[ZFIN, The Zebrafish Information Network,www.cns.fr/tetraodon], one can expect that they con-tain a large number of repeated elements which impedeboth the SH and the subsequent analysis of the clonesobtained. This issue can be overcome by inclusion ofthe preliminary stage of “normalization” of the com-pared fragmented genomes widely used for cDNA SHschemes [11, 12]. This stage involves the formation ofa rapidly renaturating fraction enriched with highlyrepresented sequences, repeated genome elements inthe case of genome comparison. Then this fraction isexcluded from the further analysis. In this way thetracer is getting enriched with low represented genomesequences.
Genome Similarity of Baikal Omul and Sig
O. S. Bychenko
a
, L. V. Sukhanova
b
, S. S. Ukolova
a
, T. A. Skvortsov
a
, V. K. Potapov
a
, T. L. Azhikina
a,
1
, and E. D. Sverdlov
a
a
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117997 Russia
b
Institute of Limnology, Siberian Branch, Russian Academy of Sciences, ul. Lermontova 281, Irkutsk, 664033 Russia
Received March 20, 2008; in final form, May 28, 2008
Abstract
—Two members of the Baikal sig family, a lake sig (
Coregonus lavaretus baicalensis
Dybovsky) andomul (
C. autumnalis migratorius
Georgi) are close relatives that diverged from the same ancestor 10–20 thou-sand years ago. In this work, we studied genomic polymorphism of these two fish species. The method of sub-tractive hybridization (SH) did not reveal the presence of extended sequences in the sig genome and theirabsence in the omul genome. All the fragments found by SH corresponded to polymorphic noncoding genomeregions varying in single nucleotide polymorphisms (SNPs) and short deletions. Many of them are mappedclose to genes of the immune system and have regions identical to the Tc1-like transposons abundant amongfish, whose transcription activity may affect the expression of adjacent genes. Thus, we showed for the first timethat genetic differences between Baikal sig family members are extremely small and cannot be revealed by theSH method. This is another endorsement of the hypothesis on the close relationship between Baikal sig andomul and their evolutionarily recent divergence from a common ancestor.
Key words: Coregonidae, speciation, genomic polymorphism, subtractive hybridization
DOI:
10.1134/S1068162009010117
1
Corresponding author; phone: +7 (495) 330-6547; fax: +7 (495)330-6538; e-mail: [email protected].
2
Abbreviations: mtDNA, mitochondrial DNA; RFLP, restrictionfragment length polymorphism; SH, subtractive hybridization.
RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY
Vol. 35
No. 1
2009
GENOME SIMILARITY OF BAIKAL OMUL AND SIG 87
In addition to the normalization stage we alsoexploited a methodical idea used for the RFLP analysisobtained by SH [13, 14]. The comparison of closelyrelated genomes shows that the greatest part of differ-ences is found in small fragments obtained by deletion–insertion and SNPs, whose detection is virtually impos-sible by routine SH schemes. The essence of themethod lies in parallel electrophoretic separation of twosets of restriction fragments obtained upon tracer anddriver fragmenting. The DNA pools of the same lengthswere compared in the process of SH, homologous frag-ments of different lengths being located in differentareas and not annealing to each other. In addition, withthe aim to exclude from consideration heteroduplexesformed by homologous fragments of similar lengths(and located in the same pool after electrophoretic sep-aration), we made a modification including treatment ofthe mixture with mung bean nuclease after the SH pro-cedure.
With the modifications described above, we per-formed subtraction of pools of genomic fragmentsshorter than 800 bp in length and obtained an arrangedlibrary enriched with sig specific genomic sequences.The library clones were analyzed by differentialhybridization with radioactive probes prepared from
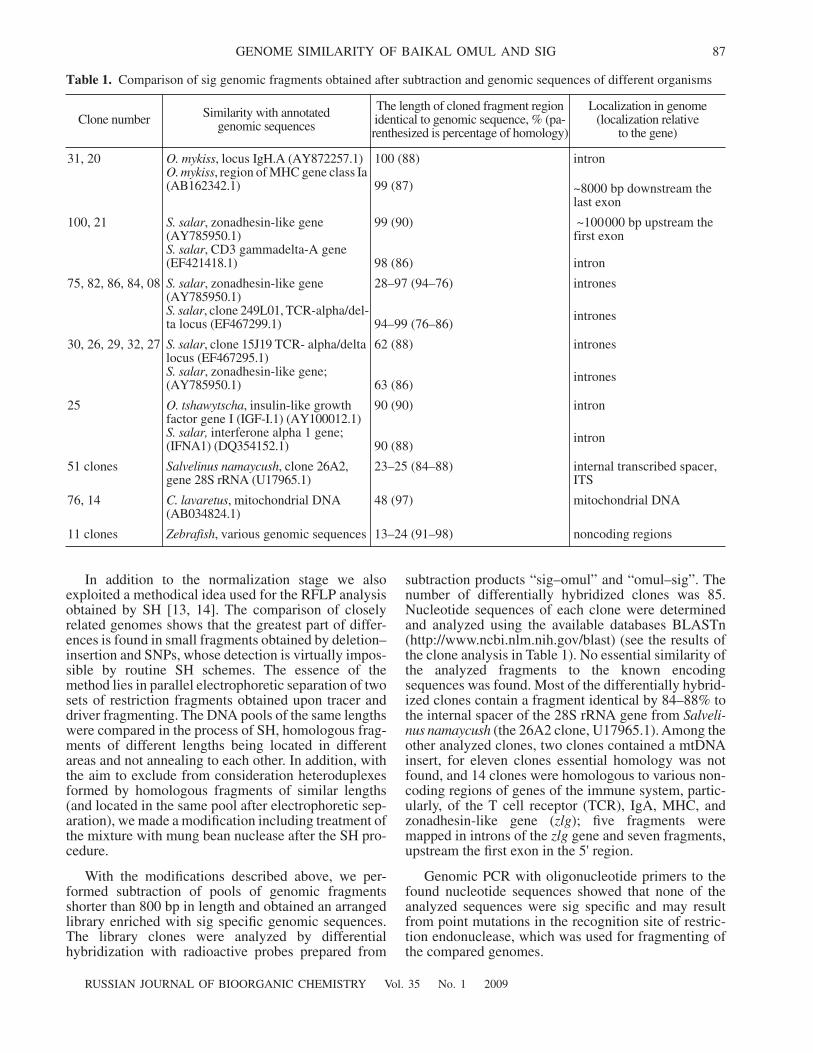
subtraction products “sig–omul” and “omul–sig”. Thenumber of differentially hybridized clones was 85.Nucleotide sequences of each clone were determinedand analyzed using the available databases BLASTn(http://www.ncbi.nlm.nih.gov/blast) (see the results ofthe clone analysis in Table 1). No essential similarity ofthe analyzed fragments to the known encodingsequences was found. Most of the differentially hybrid-ized clones contain a fragment identical by 84–88% tothe internal spacer of the 28S rRNA gene from
Salveli-nus
namaycush
(the 26A2 clone, U17965.1). Among theother analyzed clones, two clones contained a mtDNAinsert, for eleven clones essential homology was notfound, and 14 clones were homologous to various non-coding regions of genes of the immune system, partic-ularly, of the T cell receptor (TCR), IgA, MHC, andzonadhesin-like gene (
zlg
); five fragments weremapped in introns of the
zlg
gene and seven fragments,upstream the first exon in the 5' region.
Genomic PCR with oligonucleotide primers to thefound nucleotide sequences showed that none of theanalyzed sequences were sig specific and may resultfrom point mutations in the recognition site of restric-tion endonuclease, which was used for fragmenting ofthe compared genomes.
Table 1.
Comparison of sig genomic fragments obtained after subtraction and genomic sequences of different organisms
Clone number Similarity with annotated genomic sequences
The length of cloned fragment region identical to genomic sequence, % (pa-renthesized is percentage of homology)
Localization in genome (localization relative
to the gene)
31, 20
O. mykiss
, locus IgH.A (AY872257.1)
O. mykiss
, region of MHC gene class Ia (AB162342.1)
100 (88)
99 (87)
intron
~8000 bp downstream the last exon
100, 21
S. salar
, zonadhesin-like gene (AY785950.1)
S. salar
, CD3 gammadelta-A gene (EF421418.1)
99 (90)
98 (86)
~100000 bp upstream the first exon
intron
75, 82, 86, 84, 08
S. salar
, zonadhesin-like gene (AY785950.1)
S. salar
, clone 249L01, TCR-alpha/del-ta locus (EF467299.1)
28–97 (94–76)
94–99 (76–86)
intron
es
intron
es
30, 26, 29, 32, 27
S. salar
, clone 15J19 TCR- alpha/delta locus (EF467295.1)
S. salar
, zonadhesin-like gene; (AY785950.1)
62 (88)
63 (86)
intron
es
intron
es
25
O. tshawytscha
, insulin-like growth factor gene I (IGF-I.1) (AY100012.1)
S. salar,
interferone alpha 1 gene; (IFNA1) (DQ354152.1)
90 (90)
90 (88)
intron
intron
51
clones
Salvelinus namaycush
, clone 26A2, gene 28S
rRNA
(U17965.1)23–25 (84–88) internal transcribed spacer,
ITS
76, 14
C. lavaretus
, mitochondrial DNA (AB034824.1)
48 (97) mitochondrial DNA
11
clones
Zebrafish
, various genomic sequences 13–24 (91–98) noncoding regions

88
RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY
Vol. 35
No. 1
2009
BYCHENKO et al.
We carried out several experiments on nucleotidesequencing of RsaI restriction sites for several frag-ments determined by SH as differential. We used for theanalysis the fragments, which according to the similar-ity with genomic regions of
D. rerio, Salmo salar
, and
Oncorhynchus mykiss
, first, are located close to genes;and second, located close to each other, which enablesrapid determination of RsaI restriction sites betweenthem. For example, inserts of clones nos. 31 and 20were most highly homologous (91%) to the
O. mykiss
genomic region located between the third and forthexons of the
IgH.A
gene (GenBank deposition numberis AY872257.1) (Fig. 1
a
). In the
O. mykiss
genomehomologous sequences were separated with ~570 bp.We assumed that in sig and omul genomes the corre-sponding sequences are located close and, hence, canbe amplified with one mutual pair of primers. For veri-fication of this assumption we constructed a primer pair31_20 for/ 31_20 rev yielding an amplification productcontaining clone 31 and 20 sequences and the genomeregion between them. Indeed, amplification of sig andomul genomic DNA with this primer pair resulted inthe fragments of expected lengths of 1100 bp. The PCRproducts were cloned and nucleotide sequences were
determined for some of them. Numerous point muta-tion between DNA of different clones were found,which caused in one case (omul) variations in the RsaIrestriction site sequences (GTAC was replaced byATAA). The primary structures of the cloned sequencesvaried not only for sig and omul, but also within thespecies. A deletion of 38 bp in length was found for oneof the sig genome sequences.
Mutations in the RsaI restriction site can explainwhy the sequences presented in both species wereenriched when subtracted. After disappearance of therestriction site the formation of long tracer fragmentswas observed, which were not found among the driversequences of the same length.
Variations of sequences within the species for thefragment 31_20 were 3.7% and 2.7% for sig and omul,respectively; the mean value between sig and omul was3.5%. Since the intragenomic polymorphism percent-age of the tested organisms was approximately identi-cal to the differences between them, we can assume thatthese were not allele variants but duplicated loci of sigand omul genomes. To check this hypothesis, we per-formed real-time PCR for the 31_20 locus of the sigand omul DNA genomes with three primer pairs
(
a
)
Oncorhynchus mykiss,
locus
IgH.A
(AY872257.1)
(
b
)
exon
3
exon
4
31 20
31_20 for
31_20 rev
1100 bp
Salmo solar,
zonadhesin-like gene
(AY785950)
21 100
10 30 29 75 82 86
exon 1 exon 20 exon 21
21_100 for
21_100 rev
1100 bp
‹1
‹21200 bp
1500 bp
75_86 for 82 for
82 rev 75_86 rev
Fig. 1.
Mapping of sig specific sequences in the following genomic loci:
a
,
O. mykiss IgH.A
(AY872257.1) locus;
b
,
S. salar
zonad-hesin-like gene. Gray rectangles, found differential fragments with the corresponding library clone numbers; white arrows, uniqueprimers selected to them; black arrows, PCR genomic products obtained with these primers.
RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY
Vol. 35
No. 1
2009
GENOME SIMILARITY OF BAIKAL OMUL AND SIG 89
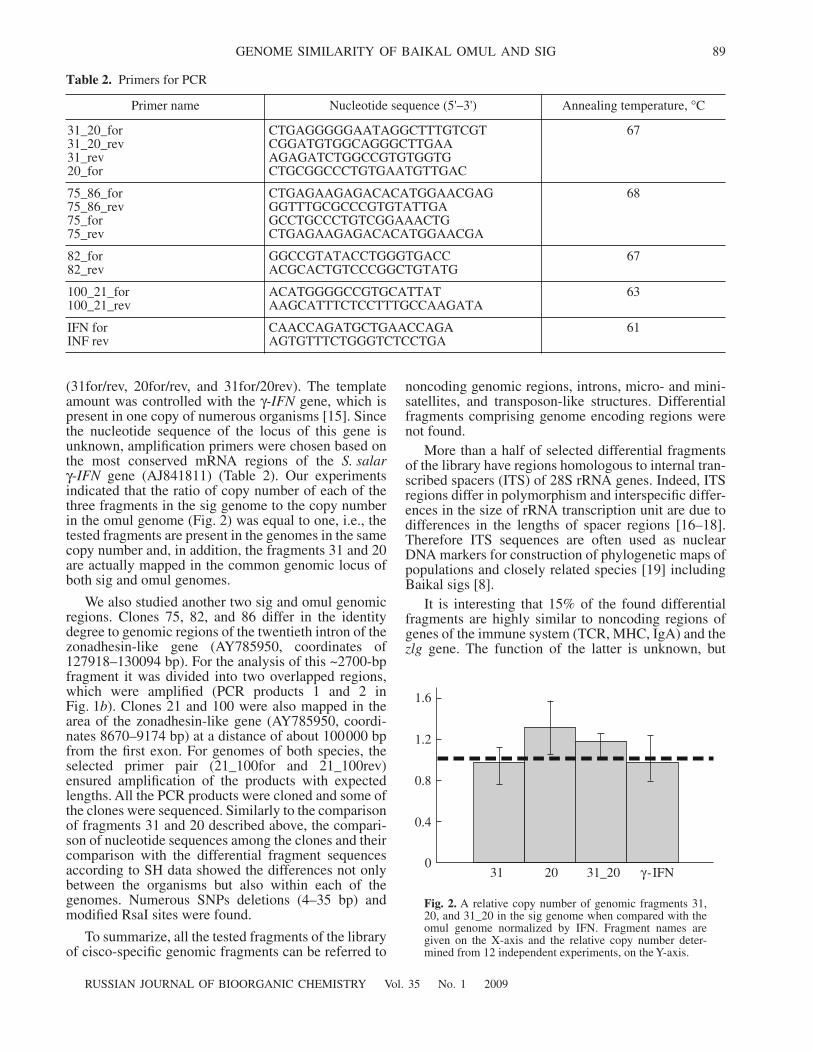
(31for/rev, 20for/rev, and 31for/20rev). The templateamount was controlled with the
γ
-IFN
gene, which ispresent in one copy of numerous organisms [15]. Sincethe nucleotide sequence of the locus of this gene isunknown, amplification primers were chosen based onthe most conserved mRNA regions of the
S. salar
γ
-
IFN
gene (AJ841811) (Table 2). Our experimentsindicated that the ratio of copy number of each of thethree fragments in the sig genome to the copy numberin the omul genome (Fig. 2) was equal to one, i.e., thetested fragments are present in the genomes in the samecopy number and, in addition, the fragments 31 and 20are actually mapped in the common genomic locus ofboth sig and omul genomes.
We also studied another two sig and omul genomicregions. Clones 75, 82, and 86 differ in the identitydegree to genomic regions of the twentieth intron of thezonadhesin-like gene (AY785950, coordinates of127918–130094 bp). For the analysis of this ~2700-bpfragment it was divided into two overlapped regions,which were amplified (PCR products 1 and 2 inFig. 1
b
). Clones 21 and 100 were also mapped in thearea of the zonadhesin-like gene (AY785950, coordi-nates 8670–9174 bp) at a distance of about 100000 bpfrom the first exon. For genomes of both species, theselected primer pair (21_100for and 21_100rev)ensured amplification of the products with expectedlengths. All the PCR products were cloned and some ofthe clones were sequenced. Similarly to the comparisonof fragments 31 and 20 described above, the compari-son of nucleotide sequences among the clones and theircomparison with the differential fragment sequencesaccording to SH data showed the differences not onlybetween the organisms but also within each of thegenomes. Numerous SNPs deletions (4–35 bp) andmodified RsaI sites were found.
To summarize, all the tested fragments of the libraryof cisco-specific genomic fragments can be referred to
noncoding genomic regions, introns, micro- and mini-satellites, and transposon-like structures. Differentialfragments comprising genome encoding regions werenot found.
More than a half of selected differential fragmentsof the library have regions homologous to internal tran-scribed spacers (ITS) of 28S rRNA genes. Indeed, ITSregions differ in polymorphism and interspecific differ-ences in the size of rRNA transcription unit are due todifferences in the lengths of spacer regions [16–18].Therefore ITS sequences are often used as nuclearDNA markers for construction of phylogenetic maps ofpopulations and closely related species [19] includingBaikal sigs [8].
It is interesting that 15% of the found differentialfragments are highly similar to noncoding regions ofgenes of the immune system (TCR, MHC, IgA) and the
zlg
gene. The function of the latter is unknown, but
Table 2.
Primers for PCR
Primer name Nucleotide sequence (5'–3') Annealing temperature,
°
C
31_20_for 31_20_rev 31_rev 20_for
CTGAGGGGGAATAGGCTTTGTCGT CGGATGTGGCAGGGCTTGAA AGAGATCTGGCCGTGTGGTG CTGCGGCCCTGTGAATGTTGAC
67
75_86_for 75_86_rev 75_for 75_rev
CTGAGAAGAGACACATGGAACGAG GGTTTGCGCCCGTGTATTGAGCCTGCCCTGTCGGAAACTGCTGAGAAGAGACACATGGAACGA
68
82_for 82_rev
GGCCGTATACCTGGGTGACC ACGCACTGTCCCGGCTGTATG
67
100_21_for 100_21_rev
ACATGGGGCCGTGCATTAT AAGCATTTCTCCTTTGCCAAGATA
63
IFN for INF rev
CAACCAGATGCTGAACCAGA AGTGTTTCTGGGTCTCCTGA
61
0
0.4
0.8
1.2
1.6
31 20 31_20
γ
-IFN
Fig. 2.
A relative copy number of genomic fragments 31,20, and 31_20 in the sig genome when compared with theomul genome normalized by IFN. Fragment names aregiven on the X-axis and the relative copy number deter-mined from 12 independent experiments, on the Y-axis.

90
RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY
Vol. 35
No. 1
2009
BYCHENKO et al.
based on the fact that it is mainly expressed in the fishintestine and shares considerable homology with genesof the immune system of the intestine mucosa (forexample, with
FCGBP
and mycine gene) [20], we canassume its involvement in the fish immunity formation.It is known that frequent rearrangement and variabilityare inherent for genes of the immune system. In ourcase all the found RFLP are located in noncoding generegions. As 5' and 3'' UTR regions occupy up to 90% ofthe zlg locus length numerous regulatory elements maybe located there, the mutations in which can alter genetranscription levels.
It is known that fishes, particularly,
S. salar
, bear agene of the major histocompatibility complex of class I,which is duplicated [21–23]. As our fragments mappedclose to genes of the immune system have a higherintragenomic divergence when compared with meanvalues of allele polymorphism of a tetraploid genome[22], one can propose that in one of the tested organ-isms these loci are also duplicated. However, our com-parative studies of the locus copy number in sig andomul genomes showed that their amounts in the com-pared genomes are the same. Probably, duplication ofthese loci occurred prior to divergence of omul and sigfrom the common ancestor and the fragments found bySH were localized in different duplicated regions,which can explain their abnormally high degree ofintragenomic divergence.
The found genomic fragments mapped close togenes of the immune system possess a distinguishingfeature. Nearly all of them have regions (9–37% of thefragment length), which share 65–85% homology withwidely abundant among fishes Tc1-like transposons(5% of the salmon genome) [24]. On the one hand, it isknown that expression of these mobile elements infishes is rather common and after their incorporationinto the genome rapid accumulation of mutationsoccurs [24]. On the other hand, there are numerousproofs of the involvement of noncoding RNA mobileelements in the regulation of gene expression at variouslevels [25–28]. The Tc1 expression of these mobile ele-ments is known to be especially activated by externalstressful stimuli. For example, a distinct correlationwas shown between the expression profiles of Tc1-liketransposons and genes involved in protective responseof the organism, signal transduction and transcriptionregulation for the rainbow trout [24]. It is likely that forBaikal sig and omul, transposon sequences play a rolein the activity of the found genes of the immune system.It is also possible that high phenotypic flexibility of thesig family is partially provided by activities of numer-ous transposon copies in their genome, particularly,upon their activation under stressful environmentalconditions, for example, upon cold snap.
As a whole, the obtained data indicate small differ-ences in sig and omul genomes. It may imply that omuland sig belong to two subpopulations of the same spe-cies. However, it is unclear how far the divergence of
these populations went. It can follow the way of regu-latory evolution, that is, the way of comparably minorchanges in the organism regulatory systems, whichnevertheless determine considerable differences ingene expression levels and tissue specificity of theirexpression. Recent studies of gene transcription carriedout in closely related organisms using the microarraytechnique showed that changes of transcription levelsof key regulatory genes may affect morphological signsto a much greater degree than transcriptional changesof structural genes [29–31]. It was proposed that varia-tions of gene expression between intact populationsmay be the ground for adaptation [32–34]. Probably forthis pair of the closely related sigs, transcriptionaldivergence due to SNPs and short deletions/insertionsin genomic DNA arises between isolated populationsfaster than scaled divergence at the genomic level.
EXPERIMENTAL
Growth and transformation of
E. coli
cells, electro-phoresis in agarose gel, and other manipulations withnucleic acids were performed using molecular biologi-cal procedures or protocols of suppliers. Isolation ofplasmid DNA was performed with a standard reagentkit Wizard Plus Minipreps DNA Purification System(Promega, United States) according to the supplier’sprotocol. The oligonucleotide synthesis was carried outon an automatic synthesizer ASM-102U DNA synthe-sizer (Biosset Ltd.). DNA sequencing was performedwith a reagent kit ABI PRISM
®
BigDye
TM
Terminatorv. 3.1 with a subsequent analysis of the products on anautomatic DNA sequencer ABI PRISM 3100-Avant.
Isolation of genomic sig and omul DNA.
Thematerial was collected in August 2005 in the region ofthe Small sea, the lake Baikal. Based on characteristicmorphometric signs one individual of sig (
C. lavaretusbaicalensis
Dybovsky) and one of omul (
C. autumnalismigratorius
Georgi) were selected. Genomic DNA wasisolated from fish white muscles using the standard pro-cedure of phenol–chloroform extraction [35].
Preparation of tracer and driver samples.
Sam-ples of sig and omul DNA (500 ng of each) were hydro-lyzed by RsaI restriction endonuclease (20 U, Fermen-tas, Lithuania) in the volume of 50
µl overnight. Therestricted DNA was precipitated with 96% ethanol andthe precipitate was washed with 70% ethanol and dis-solved in water (10 µl). Restriction fragments were sep-arated by the length in 1% agarose gel (APV, GreatBritain). The area corresponding to 200–800 bp frag-ments was cut off. From the agarose gel DNA was iso-lated using a QIAquick PCR Purification Kit (Qiagen,United States). The resulting fraction of sig fragmentedgenomic DNA was divided into two parts, and oligonu-cleotide adapters I and II were ligated to each of them(Table 3). Each of the adapters was obtained by anneal-ing of the corresponding oligonucleotide and a shorttemplate (200 pmol of each) in the TM buffer (10 mMTris–HCl, pH 7.8, and 10 mM MgCl2). Ligation was

RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY Vol. 35 No. 1 2009
GENOME SIMILARITY OF BAIKAL OMUL AND SIG 91
performed overnight at 16°C using T4 DNA ligase(Promega, United States) and the ligation mixture waspurified using a QIAquick PCR Purification Kit(Qiagen, United States).
Subtractive hybridization. Each of the sig restric-tion fragments with various ligated adaptors (Table 3)(tracer) was mixed with omul restriction fragments(driver) (700 ng) in 1× hybridization buffer (2 µl) con-taining 0.5 M NaCl, 50 mM HEPES, pH 8.3, 0.2 mMEDTA to give mixtures I and II. The mixture was cov-ered with mineral oil (20 µl), denatured for 10 min at95°C and hybridized for 4 h at 68°C. Denatured driver(2 µl, in 1×HB, 700 ng) and then hybridization mixtureII were added to mixture I. The resulting mixture washybridized overnight at 68°C, 1×HB buffer (100 µl)was added, and the mixture was kept at –20°C. An ali-quot of the hybridization mixture (20 µl) was incubatedfor 5 min at 72°C with Taq polymerase in the presenceof 0.2 mM the deoxyribonucleoside triphosphates(each of them) to complete overhanging ends, purifiedon PCR Purification Kit columns (Qiagen, UnitedStates), and treated with 5 U nuclease from mung beannuclease (Promega, United States) for 15 min at 37°Cin 1× reaction buffer (10 µl). The reaction was termi-nated by addition of 1 M Tris–HCl buffer (10 µl, pH 8).The resulting mixture (2 µl) was amplified withAdvantage2 Polymerase Mix (Clontech, United States)using an T7 external adapter primer (0.4 µM) under thefollowing conditions: at 95°C for 30 s; at 66°C for 30 s;and at 72°C for 90 s; 18 cycles. The 250-fold dilutedPCR product was reamplified with internal adapterprimers NotRsa and NotSrf (0.4 µM each) under thefollowing conditions: at 95°C for 30 s; at 68°C for 30 s;and at 72°C for 90 s; 15 cycles.
Preparation of the arranged library of differen-tial fragments and their analysis. The obtained PCRproduct was ligated with the pGEM-T vector using thesupplier’s recommendations (Promega, United States)
and cloned in E. coli (the DH-5α strain). The cloneswere sown in 96-well microbiological plates. Individ-ual clones were amplified with universal primers(5−3'): M13for GTAAAACGACGGCCAGTG andM13rev AACAGCTATGACCATG (95°C, 30 s; 68°C,30 s; 72°C, 1 min; 25 cycles). Amplification productswere transferred onto nitrocellulose membranes (APB,Great Britain) in two repeats according to the supplier’srecommendations. The membranes were used forhydridization in parallel to subtraction products “sig–omul” and “omul–sig” labeled using a Prime-a-GeneLabeling System (Promega, United States) (Fig. 3).Nucleotide sequences of differentially hydridizingclones were determined by the Sanger method. Homol-ogous sequences were searched using the GenBankNCBI database and the BLAST program(http://ncbi.nlm.nih. gov/BLAST).
The PCR procedure. The primers were constructedusing the Primer3 program (www.genome.wi.mit.edu/cgi_bin/primer/primer3) (Table 2). Their uniquenesswas checked using the BLAST program(http://ncbi.nlm.nih.gov/BLAST). The PCR was per-formed in the following mixture (25 µl): 20 ng ofgenomic DNA template, 10 pmol of each of the prim-ers, 0.02 mM each of deoxyribonucleoside triphos-phate, 1.5–2 mM MgCl2, 1–2.5 U Taq polymerase(BIOTAQ, Russia) in 1× buffer for Taq polymerase(16.6 mM (NH4)2SO4, 67 mM tricine, pH 9.1, 0.01%Tween-20). The following temperature regimen wasused: denaturation at 95°C for 2 min, one cycle; furtherdenaturation at 95°C for 45 s; annealing (depending onthe primer) for 40 s; elongation at 72°C for 50 s, 23–36cycles.
The real time PCR was performed with an EVAgreen RT-PCR kit (Sintol, Russia). The reaction wasconducted in a volume of 25 µl per 10 ng of genomicDNA template, 0.2 µM of each of the primers; the fol-lowing temperature regimen was used: 95°C for
Table 3. Oligonucleotides used in subtraction hybridization
Oligonucleotides Nucleotide sequence (5'–3')
Supression adapter I (obtained by annealing equimolar amounts of oligonucleotides T7NotSrf and Srf_10)
T7 NotSrf (44 bp) CTAATACGACTCACTATAGGGCTCGAGCGGCCGCCCGGGCAGGT
Srf_10 (10 bp) ACCTGCCCGG
Supression adapter II (obtained by annealing equimolar amounts of oligonucleotides T7NotRsa and Rsa_10)
T7 NotRsa (42 bp) CTAATACGACTCACTATAGGGCAGCGTGGTCGCGGCCGAGGT
Rsa_10 (10 bp) ACCTCGGCCG
Internal primer Not1Srf (22 bp) TCGAGCGGCCGCCCGGGCAGGT
Not1Rsa (22 bp) AGCGTGGTCGCGGCCGAGGT
External primer T7 (22 bp) CTAATACGACTCACTATAGGGC

92
RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY Vol. 35 No. 1 2009
BYCHENKO et al.
10 min; 95°C for 30 s; annealing of the appropriateprimer pairs (Table 2) for 30 s; 72°C for 1 min,40 cycles.
ACKNOWLEDGMENTS
The work was supported by the Russian Foundationfor Basic Research, project 05-04-486880; the program“Leading Schools of Russia”, project NS 2006.2003.4;and the Program of Presidium of Russian Academy ofSciences on Molecular and Cell Biology. DNAsequencing was performed in the Interinstitute SharedCenter “GENOME” of Institute of Molecular Biology(http://www.genome-centre.narod.ru/) partially sup-ported by RFBR, project 00-04-55000.
REFERENCES
1. Reshetnikov, Yu.S., Ekologiya i sistematika sigovykh ryb(Ecology and Systematics of Sig Fishes), Moscow:Nauka, 1980.
2. Reshetnikov, Yu.S., Voprosy Ikhtiologii, 1995, vol. 35,pp. 156–174.
3. Skryabin, A.G., Biologiya baikal’skikh sigov (Biology ofBaikal Sigs), Moscow: Nauka, 1969.
4. Skryabin, A.G., Ryby Bauntovskikh ozer Zabaikal’ya(Fishes of Baunt Lakes of Transbaikalie), Novosibirsk:Nauka, 1977.
5. Politov, D.V., Gordon, N.Yu., Afanasiev, K.A..,Altukhov, Yu.P., and Bickham, J.W., J. Fish Biol., 2000,vol. 57, suppl. A, pp. 51−71.
6. Politov, D.V., Gordon, N.Yu., and Makhrov, A.A., Biol-ogy and Management of Coregonid Fishes, 2002,vol. 57, pp. 21–34.
7. Sukhanova, L.V., Smirnov, V.V., and Kirilchik, S.V.,Biodiversity and dynamics of ecosystems in North Eur-asia, 2000, vol. 5, pp. 199–201.
8. Sukhanova, L.V., Smirnov, V.V., Smirnova-Zalumi, N.S.,Kirilchik, S.V., Griffiths, D., and Belikov, S.I., Arch.Hydrobiol. Spec. Issues Advanc. Limnol. Biology andManagement of Coregonid Fishes, 2002, vol. 97−106.
9. Politov, D.V., Bickham, J., and Patton, J.S., AnnalesZoologici Fennici, 2004, vol. 41, pp. 13–24.
10. Sukhanova, L.V., Smirnov, V.V., Kirilchik, S.V., andShimizu, I., Annales Zoologici Fennici, 2004, vol. 41,pp. 41–49.
11. Diatchenko, L., Lau, Y.F., Campbell, A.P., Chenchik, A.,Moqadam, F., Huang, B., Lukyanov, S., Lukyanov, K.,Gurskaya, N., Sverdlov, E.D., and Siebert, P.D., Proc.Natl. Acad. Sci. USA, 1996, vol. 93, pp. 6025–6030.
12. Gurskaya, N.G., Diatchenko, L., Chenchik, A., Siebert, P.D.,Khaspekov, G.L., Lukyanov, K.A., Vagner, L.L., Ermo-laeva, O.D., Lukyanov, S.A., and Sverdlov, E.D., Anal.Biochem., 1996, vol. 240, pp. 90–97.
13. Azhikina, T., Gvozdevsky, N., Botvinnik, A., Fushan, A.,Shemyakin, I., Stepanshina, V., Lipin, M., and Barry, C.,3rd, Sverdlov E, Res. Microbiol., 2006, vol. 157,pp. 282–290.
14. Rosenberg, M., Przybylska, M., and Straus, D., Proc.Natl. Acad. Sci. USA, 1994, vol. 91, pp. 6113–6117.
15. Robertsen, B., Fish Shellfish Immunol., 2006, vol. 20,pp. 172–191.
16. Dover, G., Nature, 1982, vol. 299, pp. 111–117.17. Hillis, D.M. and Dixon, M.T., The Quarterly Review of
Biology, 1991, vol. 66, pp. 411–453.18. Singer, M. and Berg, P., Genes and Genomes. A Chang-
ing Perspective. Mill Valley, California; University Sci-ence Books, 1991.
19. Harlton, C.E., Lyvesque, C.A., and Punja, Z.K., Phyto-pathology, 1995, vol. 85, pp. 1269–1281.
20. Hunt, P.N., Wilson, M.D., von Schalburg, K.R., David-son, W.S., and Koop, B.F., BMC Genomics, 2005, vol. 6,p. 165.
21. Dijkstra, J.M., Kiryu, I., Yoshiura, Y., Kumanovics, A.,Kohara, M., Hayashi, N., and Ototake, M., Immunoge-netics, 2006, vol. 58, pp. 152–167.
22. Lukacs, M.F., Harstad, H., Grimholt, U., Beetz-Sargent, M.,Cooper, G.A., Reid, L., Bakke, H.G., Phillips, R.B.,Miller, K.M., Davidson, W.S., and Koop, B.F., BMCGenomics, 2007, vol. 8, p. 251.
23. Shiina, T., Dijkstra, J.M., Shimizu, S., Watanabe, A.,Yanagiya, K., Kiryu, I., Fujiwara, A., Nishida-Umehara, C.,Kaba, Y., Hirono, I., Yoshiura, Y., Aoki, T., Inoko, H.,Kulski, J.K., and Ototake, M., Immunogenetics, 2005,vol. 56, pp. 878–893.
24. Krasnov, A., Koskinen, H., Afanasyev, S., and Molsa, H.,BMC Genomics, 2005, vol. 6, p. 107.
a — “sig–omul” b — “omul–sig”
Fig. 3. Examples of Southern hybridization of the sig genome fragment library with subtraction products: a, “sig–omul” (sig spe-cific genomic fragments) and b, “omul–sig” (omul specific genomic fragments). Examples of differential hybridization of siggenomic fragments are shown in circles.

RUSSIAN JOURNAL OF BIOORGANIC CHEMISTRY Vol. 35 No. 1 2009
GENOME SIMILARITY OF BAIKAL OMUL AND SIG 93
25. Chu, W.M., Ballard, R., Carpick, B.W., Williams, B.R.,and Schmid, C.W., Mol. Cell Biol., 1998, vol. 18, pp. 58–68.
26. Jensen, I., Larsen, R., and Robertsen, B., Fish ShellfishImmunol., 2002, vol. 13, pp. 367–378.
27. Rubin, C.M., Kimura, R.H., and Schmid, C.W., NucleicAcids Res., 2002, vol. 30, pp. 3253–3261.
28. Stevenson, D.S. and Jarvis, P., Curr. Biol., 2003, vol. 13,pp. R13–15.
29. Derome, N., Duchesne, P., and Bernatchez, L., Mol.Ecol., 2006, vol. 15, pp. 1239–1249.
30. Purugganan, M.D., BioEssays, 1998, vol. 20, pp. 700–711.
31. Rogers, S.M. and Bernatchez, L., Mol. Ecol., 2005,vol. 14, pp. 351–361.
32. Gibson, G. and Wagner, G., BioEssays, 2000, vol. 22,pp. 372–380.
33. Gibson, G. and Honeycutt, E., Curr. Opin. Genet. Dev.,2002, vol. 12, pp. 695–700.
34. Gibson, G. and Weir, B., Trends Genet., 2005, vol. 21,pp. 616–623.
35. Sambrook, J., Fritsch, E.F., and Maniatis, T., MolecularCloning: a Laboratory Manual (2nd Ed.), New York:;Cold Spring Harbor Laboratory Press, 1989.


















