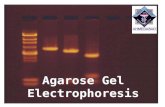
Gel electrophoresis
30
-
Upload
nasira-jaffry -
Category
Science
-
view
240 -
download
6
Transcript of Gel electrophoresis


Definition
Electrophoresis is a technique used to separate and sometimes purify macromolecules - especially proteins and nucleic acids - that differ in size, charge or conformation.

Purpose of Gel Electrophoresis
A method for separating DNA
Can be used to separate the size of
DNA
RNA
Protein
We will be using it to purify DNA, RNA AND PROTEINS.

Principle of gel electrophoresis:
Gel electrophoresis involves an electrical field.
The molecules to be separated are pushed by an electrical field through a gel that contains small pores
The molecules travel through the pores in the gel at a speed that is inversely related to their lengths.
This means that a small DNA molecule will travel a greater distance through the gel than will a larger DNA molecule

DNA and RNA are negatively charged molecules, they will be pulled toward the positively charged end of the gel.
IN case of proteins, first mix it with a detergent called sodium dodecyl sulfate. This treatment makes the proteins unfold into a linear shape and coats them with a negative charge, which allows them to migrate toward the positive end of the gel and be separated.

Types of gel electrophoresisAGAROSE GEL POLYACRYLAMIDE GEL
Poured Horizontally Poured Vertically
Separate large molecules Separate small molecules
Non-toxic neurotoxin
Mostly used for DNA separation Used for DNA or Protein separation
Staining step: before or after pouring the gel Ethidium bromide is mostly used
Staining step: after pouring the gel Coomassie blue stain is mostly used

Gel Electrophoresis of DNAMaterials
Ethidium Bromide (10mg/ml)
Gel Loading Dye, 6X, Blue-Orange (comes with Promega DNA ladder)
5X TBE (stock)
in a 1L beaker: 850 ml of dH2O, 20 ml of 0.5M EDTA (pH 8), 54g Tris base stir ,add 27.5 g Boric Acid, stir, pour into graduated cylinder and fill up to 1L with dH2O, autoclave

0.5M EDTA (pH 8.0)
46.5 EDTA in 200ml water
5 g of NaOH pellets to adjust pH to 8.0
EDTA won’t dissolve until pH is 8.0

Sample preparationGel electrophoresis requires some kind of DNA sample—a plasmid, a PCR product, a segment of a chromosome, etc
Whatever your sample is, it must be mixed with loading buffer (containing glycerol and dyes, as described above) before electrophoresis.
Add a volume of loading buffer equal to 1/5 the volume of your sample and mix it well before loading your sample on the gel.

How to pour and run a gel1. For one gel, you will need a total of 30 ml of a 1 %
agarose solution.
2. Prepare 300 ml of 10× TBE buffer by measuring 30ml into a 500-ml graduated cylinder, then filling thecylinder to the 300-ml mark with dH2O .
3. Pour 30 ml of the 1× TBE into the flask with youragarose. The remaining TBE will be your runningbuffer, which carries the current from one electrodeto the other.

4 -Heat the agarose in the microwave on high for 15 seconds.
5-Allow the melted agarose to cool on the bench for two minutes while you set up the casting tray.
6-Insert a gel plate into a casting tray as shown in Figure.


7-Pour the entire 30 ml of melted agarose into the gel casting tray. Insert a clean comb (which will form the
wells) near one end, and slide it up against the handles on the gel plate, as shown in Figure.


8-Let the agarose harden for at least 15 minutes. When the agarose has fully hardened, carefully remove the comb. Remove the gel plate, with the gel on it, from the casting tray and place it in the gel box.
9-Add running buffer until the gel is completely submerged: if you look from the side, it should be covered by a few millimeters of liquid. The gel is now ready for use.

10-Use a micropipette and a clean tip to transfer each of your DNA samples to a well.
11-Place the lid on the gel box and fit the power cords over the two electrodes.
12-Normally, you would want to stop the gel when the bromophenol blue (dark blue) dye line is near the bottom edge of the gel but has not yet started to run off. Stop it sooner if you need to see small DNA fragments. Turn off the power supply and disconnect the leads

13-Carefully remove the gel tray and gel from the gel box. Don't let the gel slide off! Slide the gel off the tray onto the UV light box of the photo documentation system

GEL ELECTROPHORESIS OF RNAMATERIALS
Gel Loading Buffer II -95% Formamide, 18mM EDTA, and 0.025% SDS, Xylene Cyanol, and Bromophenol Blue.
Gel Loading Solution -10X solution of 40% Sucrose, 0.17% Xylene Cyanol, and 0.17% Bromophenol Blue.

NorthernMax Formaldehyde Load Dye
NorthernMax-Gly Sample Loading Dye
An improved formulation used for RNA sample denaturation in any glyoxal gel protocol

Protocol
Prepare the gel.
Heat 1 g agarose in 72 ml water until dissolved, then cool to 60°C.
Add 10 ml 10X running buffer, and 18 ml 37% formaldehyde (12.3 M).
Pour the gel using a comb, assemble the gel in the tank, and add enough 1X running buffer to cover the gel by a few millimeters. Then remove the comb.

Prepare the RNA sample.
a. To 1-3 µg RNA, add 0.5-3X volumes Formaldehyde Load Dye.
To simply check the RNA on a denaturing gel, as little as 0.5X Formaldehyde Load Dye can be used, but to completely denaturate the RNA, e.g. for Northern blots, use 3X volumes of Formaldehyde Load Dye.

Ethidium bromide can be added to the Formaldehyde Load Dye at a final concentration of 10 µg/ml. Because ethidium bromide concentration affects RNA migration in agarose gels.
Heat denature samples at 65-70°C for 5-15 min

Electrophoresis
Load the gel and electrophorese at 5-6 V/cm until the bromophenol blue (the faster-migrating dye) has migrated at least 2-3 cm into the gel, or as far as 2/3 the length of the gel.
Results
Visualize the gel on a UV transilluminator

SDS-PAGE OF PROTEINMATERIALS
Stock arcylamide solution
Running gel buffer
Stacking gel buffer
10% ammonium persulfate (APS)
10% SDS
Sample buffer: tris HCL =0.6M Ph= 6.8, 5ml SDS= 0.5g, sucrose= 5g, β- mercapto blue =5ml all are mixed leaving bromophenol blue.

Electrophoresis buffer
Protein stain
De-stain solution


PROCEDURE:Clean the internal surface of the gel plate
Join the gel plate together to form cased
Mix following and transfer it to gel cassede.
Running gel buffer= 8ml
Water= 11.4 ml
Stock acrylamide= 20 ml
10% SDS= 0.4 ml
APS= 0.2 ml
TEMED=14μl

Butanol is added after setting the solution
Prepare the stacking gel solution and add 14μl TEMED and load it into gel cassade after dicarding butanol.
Insert comb to create well for polymerization
Assemble gel cassade in gel electrophoresis tank
Fill the top reservoir with gel electrophoresis buffer
Load the sample
Connect the power supply
continue the electrophoreses until bromophenol reaches the bottom

Dismantle the gel apparatus
Remove the gel and pass it through staining solution .
De-stain the gel to remove excess stain.

THANK YOU