
Fusion Kinases Identified by Genomic Analyses of Sporadic ...Kazuhito Sato1,2, Masahito...
13
Translational Cancer Mechanisms and Therapy Fusion Kinases Identified by Genomic Analyses of Sporadic Microsatellite Instability–High Colorectal Cancers Kazuhito Sato 1,2 , Masahito Kawazu 3 , Yoko Yamamoto 1 , Toshihide Ueno 2 , Shinya Kojima 2 , Genta Nagae 4 , Hiroyuki Abe 5 , Manabu Soda 2 , Takafumi Oga 2 , Shinji Kohsaka 3 , Eirin Sai 3 , Yoshihiro Yamashita 2 , Hisae Iinuma 6 , Masashi Fukayama 5 , Hiroyuki Aburatani 4 , Toshiaki Watanabe 7,† , and Hiroyuki Mano 2,8 Abstract Purpose: Colorectal cancers with microsatellite instability– high (MSI-H) status, due to mismatch repair deficiency, are associated with poor patient outcomes after relapse. We aimed to identify novel therapeutic targets for them. Experimental Design: We performed MSI analyses of over 2,800 surgically resected colorectal tumors obtained from consecutive patients treated in Japan from 1998 through June 2016. Whole-exome sequencing, transcriptome sequencing, and methylation analyses were performed on 149 of 162 tumors showing MSI in BAT25 and BAT26 loci. We analyzed patient survival times using Bonferroni-adjusted log-rank tests. Results: Sporadic MSI-H colorectal cancers with promoter methylation of MLH1 (called MM) had a clinicopatholog- ical profile that was distinct from that of colorectal cancers of patients with germline mutations (Lynch syndrome, LS-associated) or somatic, Lynch-like mutations in mis- match repair genes. MM tumors had more insertions and deletions and more recurrent mutations in BRAF and RNF43 than LS-associated or Lynch-like MSI-H tumors. Eleven fusion kinases were exclusively detected in MM MSI-H colorectal cancers lacking oncogenic KRAS/BRAF missense mutations and were associated with worse post-relapse prognosis. We developed a simple method to identify MM tumors and applied it to a validation cohort of 28 MSI-H colorectal cancers, identifying 16 MM tumors and 2 fusion kinases. Conclusions: We discovered that fusion kinases are frequently observed among sporadic MM MSI-H colorectal cancers. The new method to identify MM tumors enables us to straightforwardly group MSI-H patients into candidates of LS or fusion kinase carriers. Introduction Approximately 10% of all colorectal cancers exhibit a micro- satellite instability–high (MSI-H) status, in which the number of mono-, di-, or tri-nucleotide repeats in microsatellite sequences are frequently altered (1–3). Most patients with MSI-H colorectal cancers have poor prognosis after relapse according to consensus molecular subtyping (4, 5), supporting the concept that MSI-H cancers are resistant to 5-fluorouracil (6). The considerable num- ber of mutations resulting from DNA mismatch repair (MMR) deficiency has hampered the identification of driver oncogenes that play essential pathogenic roles in MSI-H colorectal cancers. MSI-H colorectal cancers have been conventionally divided into hereditary or sporadic. To identify cases potentially involving Lynch syndrome (LS), we have to rely on patients' clinical infor- mation, such as family history or age. It is well known that MSI-H colorectal cancers are tightly linked to the CpG island methylator phenotype (CIMP; refs. 7, 8), which is characterized by the cancer- specific methylation of a definite set of CpG islands. Rational patient stratification based on the precise recognition of the etiology is essential for the appropriate management of patients with MSI-H colorectal cancers. To address these issues, we conducted an integrated molecular characterization of 149 MSI-H colorectal tumors. Materials and Methods Ethics Patients with colorectal cancer gave written informed consent prior to their participation in the study. This project was approved by the institutional ethics committees of The University of Tokyo 1 Department of Surgical Oncology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan. 2 Department of Cellular Signaling, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan. 3 Department of Medical Genomics, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan. 4 Genome Science Division, Research Center for Advanced Science and Tech- nologies, The University of Tokyo, Tokyo, Japan. 5 Department of Pathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan. 6 Department of Surgery, Teikyo University School of Medicine, Tokyo, Japan. 7 Department of Surgical Oncology and Vascular Surgery, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan. 8 National Cancer Center Research Institute, Tokyo, Japan. Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). † Deceased. Corresponding Author: Masahito Kawazu, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan, Phone: 81- 3-3547-5201: E-mail: [email protected] doi: 10.1158/1078-0432.CCR-18-1574 Ó2018 American Association for Cancer Research. Clinical Cancer Research Clin Cancer Res; 25(1) January 1, 2019 378 on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574
Transcript of Fusion Kinases Identified by Genomic Analyses of Sporadic ...Kazuhito Sato1,2, Masahito...

Translational Cancer Mechanisms and Therapy
Fusion Kinases Identified by Genomic Analysesof Sporadic Microsatellite Instability–HighColorectal CancersKazuhito Sato1,2, Masahito Kawazu3, Yoko Yamamoto1, Toshihide Ueno2, Shinya Kojima2,Genta Nagae4, Hiroyuki Abe5, Manabu Soda2, Takafumi Oga2, Shinji Kohsaka3, Eirin Sai3,Yoshihiro Yamashita2, Hisae Iinuma6, Masashi Fukayama5, Hiroyuki Aburatani4,Toshiaki Watanabe7,†, and Hiroyuki Mano2,8
Abstract
Purpose: Colorectal cancers with microsatellite instability–high (MSI-H) status, due to mismatch repair deficiency, areassociatedwith poor patient outcomes after relapse.We aimedto identify novel therapeutic targets for them.
Experimental Design: We performed MSI analyses of over2,800 surgically resected colorectal tumors obtained fromconsecutive patients treated in Japan from 1998 through June2016. Whole-exome sequencing, transcriptome sequencing,and methylation analyses were performed on 149 of 162tumors showing MSI in BAT25 and BAT26 loci. We analyzedpatient survival times using Bonferroni-adjusted log-ranktests.
Results: Sporadic MSI-H colorectal cancers with promotermethylation of MLH1 (called MM) had a clinicopatholog-ical profile that was distinct from that of colorectal cancersof patients with germline mutations (Lynch syndrome,
LS-associated) or somatic, Lynch-like mutations in mis-match repair genes. MM tumors had more insertions anddeletions and more recurrent mutations in BRAF and RNF43than LS-associated or Lynch-like MSI-H tumors. Elevenfusion kinases were exclusively detected in MM MSI-Hcolorectal cancers lacking oncogenic KRAS/BRAF missensemutations and were associated with worse post-relapseprognosis. We developed a simple method to identify MMtumors and applied it to a validation cohort of 28 MSI-Hcolorectal cancers, identifying 16 MM tumors and 2 fusionkinases.
Conclusions: We discovered that fusion kinases arefrequently observed among sporadic MM MSI-H colorectalcancers. The new method to identify MM tumors enables usto straightforwardly group MSI-H patients into candidatesof LS or fusion kinase carriers.
IntroductionApproximately 10% of all colorectal cancers exhibit a micro-
satellite instability–high (MSI-H) status, in which the number ofmono-, di-, or tri-nucleotide repeats in microsatellite sequences
are frequently altered (1–3). Most patients with MSI-H colorectalcancers have poor prognosis after relapse according to consensusmolecular subtyping (4, 5), supporting the concept that MSI-Hcancers are resistant to 5-fluorouracil (6). The considerable num-ber of mutations resulting from DNA mismatch repair (MMR)deficiency has hampered the identification of driver oncogenesthat play essential pathogenic roles in MSI-H colorectal cancers.
MSI-H colorectal cancers have been conventionally dividedinto hereditary or sporadic. To identify cases potentially involvingLynch syndrome (LS), we have to rely on patients' clinical infor-mation, such as family history or age. It is well known that MSI-Hcolorectal cancers are tightly linked to the CpG island methylatorphenotype (CIMP; refs. 7, 8), which is characterized by the cancer-specific methylation of a definite set of CpG islands. Rationalpatient stratification based on the precise recognition of theetiology is essential for the appropriate management of patientswith MSI-H colorectal cancers.
To address these issues, we conducted an integrated molecularcharacterization of 149 MSI-H colorectal tumors.
Materials and MethodsEthics
Patients with colorectal cancer gave written informed consentprior to their participation in the study. This project was approvedby the institutional ethics committees of The University of Tokyo
1Department of Surgical Oncology, Graduate School of Medicine, The Universityof Tokyo, Tokyo, Japan. 2Department of Cellular Signaling, Graduate School ofMedicine, The University of Tokyo, Tokyo, Japan. 3Department of MedicalGenomics, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan.4Genome Science Division, Research Center for Advanced Science and Tech-nologies, The University of Tokyo, Tokyo, Japan. 5Department of Pathology,Graduate School of Medicine, The University of Tokyo, Tokyo, Japan.6Department of Surgery, Teikyo University School of Medicine, Tokyo, Japan.7Department of Surgical Oncology and Vascular Surgery, Graduate School ofMedicine, The University of Tokyo, Tokyo, Japan. 8National Cancer CenterResearch Institute, Tokyo, Japan.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
†Deceased.
Corresponding Author: Masahito Kawazu, Graduate School of Medicine, TheUniversity of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan, Phone: 81-3-3547-5201: E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-18-1574
�2018 American Association for Cancer Research.
ClinicalCancerResearch
Clin Cancer Res; 25(1) January 1, 2019378
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

(TheHumanGenome,Gene Analysis Research Ethics Committee;G10063 and G3546) and Teikyo University (#14–197), and thestudy was conducted in accordance with the Declaration ofHelsinki.
Sample collectionSurgically resected colorectal tumors (n ¼ �2,800) were
obtained from consecutive patients treated at The University ofTokyo Hospital and Teikyo University Hospital between 1998and June 2016. To validate a fusion-detection strategy, we added185 primary colorectal cancers, collected at The University ofTokyo Hospital from July 2016 through April 2017.
Microsatellite instability testingTumor tissues and corresponding normal mucosae were
obtained from surgically resected specimens and were eithersnap-frozen in liquid nitrogen immediately after resection andstored at �80�C or immersed in RNAlater Tissue Protect Tubes(Qiagen) overnight at 4�C followed by storage at�20�Cuntil use.GenomicDNAwas extracted from tissue sampleswith theDNeasyBlood and Tissue Kit or the QIAamp DNAMini Kit (Qiagen) andanalyzed by PCR at two microsatellite loci, BAT25 and BAT26,using the labeled primers indicated in Supplementary Table S1.PCR products were electrophoresed on an ABI PRISM 3100,an ABI PRISM 3700, or a 3130xl Genetic Analyzer (AppliedBiosystems), and fluorescent signals were analyzed using GeneS-can 3.7, GeneScan 3.5, or GeneMapper 4.0, and Genotyper 2.1,Genotyper 3.6, or PeakScanner 1.0 (all fromApplied Biosystems),in accordance with the manufacturer's instructions.
Comprehensive genomic and epigenomic analysesGenomicDNA from149MSI-H tumors (148 adenocarcinomas
and 1 adenoma in 146 patients, all chemotherapy-na€�ve, 1 casewith preoperative radiotherapy) and corresponding paired-nor-mal tissues consisting of adjacent, histologically normal tissuesresected at the time of surgery was subjected to whole-exomesequencing (WES) with HiSeq2500 (Illumina). We analyzed the
genome-wide DNA methylation profiles of 93 tumors with anInfinium Human MethylationEPIC BeadChip (Illumina) andperformed whole-transcriptome sequencing (RNA-seq) from111 MSI-H with HiSeq2500 (Illumina).
DNA methylation analysesGenome-wide DNA methylation analysis. Infinium HumanMethylationEPIC BeadChip (Illumina) was used in accordancewith the manufacturer's protocol. Beta values were normalizedusing BMIQ function in the R package wateRmelon (http://www.bioconductor.org/packages/release/bioc/html/wateRmelon.html). For downstream analysis, we selected probes thatwere designed on promoter-associated sites of autosomes.Then, we performed consensus clustering with 3,073 probes(probes with variance ranked in the top 1%) using the Rpackage ConsensusClusterPlus (http://www.bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html).
Bisulfite sequencing of MLH1 promoter. Genomic DNA wassubjected to bisulfite conversion using an EpiTect Bisulfite Kit(Qiagen). Converted DNA fragments were amplified by PCRusing a Kapa HiFi Uracilþ Kit (Kapa Biosystems) with the primersets indicated in Supplementary Table S1. Amplified PCR pro-ducts were subjected to sequencing using the MiSeq system(Illumina) to determine the proportion of methylated alleles ateach cytosine residue.
MLH1 promoter methylation assay with methylation-sensitiverestriction enzyme. MLH1 promoter methylation was assessed byPCR after digestion of genomic DNA with methylation-sensitiverestriction enzyme. Genomic DNA (200 or 25 ng) was digested ina volume of 20 or 10 mL by Anza 22 SmaI (Thermo FisherScientific), followed by heat inactivation of restriction enzyme,in accordancewith themanufacturer's instructions.DigestedDNA(20 or 5 ng) was subjected to 25 or 27 cycles ofmultiplex PCR in atotal volume of 25 mL using primeSTAR GXL DNA polymerase(Takara Bio), in accordance with the manufacturer's instructions.BRAFwas used as a positive control. The primer sets are indicatedin Supplementary Table S1.Methylation statuswas determinedby2% agarose gel electrophoresis of 12.5 mL of PCR products.
Whole-exome sequencing including mutation call,copy-number analysis, and signature analysis
Genomic DNA was isolated from each sample and underwentenrichment of exonic fragments using SureSelect HumanAll ExonKit v5 (Agilent Technologies). Massively parallel sequencing ofisolated fragments was performed with a HiSeq2500 (Illumina)using the paired-end option. Paired-end whole-exome sequenc-ing (WES) reads were independently aligned to the humanreference genome (hg38) using BWA (9), Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), and NovoAlign(http://www.novocraft.com/products/novoalign/). Somaticmutations were called using MuTect (http://www.broadinstitute.org/cancer/cga/mutect), SomaticIndelDetector (http://www.broadinstitute.org/cancer/cga/node/87), and VarScan(http://varscan.sourceforge.net). Mutations were discarded if:(i) the read depth was <20 or the variant allele frequency (VAF)was <0.1, (ii) they were supported by only one strand of thegenome, or (iii) they were present in normal human genomes ineither the 1000 Genomes Project dataset (http://www.internatio
Translational Relevance
Microsatellite instability–high (MSI-H) colorectal cancershave been conventionally divided into hereditary (Lynch syn-drome, LS) and sporadic categories. This report provides arational basis for further classification of sporadicMSI-H colo-rectal cancers into those with somatic mutations in mismatchrepair genes (Lynch-like, LL) and those with promoter meth-ylation of MLH1 (MM). There were significant differencesbetween the LS/LL and MM groups in clinicopathologicalproperties including tumor localization, number of inser-tions/deletions, and recurrent mutations of KRAS/APC andBRAF/RNF43. Such a classification would enable precise man-agement of patients with MSI-H colorectal cancer. Fusionkinases were detected only in MM MSI-H colorectal cancerslacking oncogenic KRAS or BRAF mutations and were associ-ated with worse prognosis after relapse. A new, convenientmethod for detectingMM tumorsmakes it possible to straight-forwardly identifyLScandidatesorMSI-Htumors likely tocarryfusion kinases that are therapeutic targets of kinase inhibitors.
Actionable Fusion Kinases in MSI-H CRCs
www.aacrjournals.org Clin Cancer Res; 25(1) January 1, 2019 379
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

nalgenome.org/) or our in-house database. Gene mutations wereannotated by SnpEff (http://snpeff.sourceforge.net). Copy-num-ber status was analyzed using our in-house pipeline, whichdetermines the logR ratio (LRR) as follows: (i) we selected SNPpositions in the 1000 Genomes Project database that were in ahomozygous state (VAF� 0.05 or� 0.95) or a heterozygous state(VAF 0.4–0.6) in the genomes of respective normal samples, (ii)normal and tumor read depths at the selected position wereadjusted based on GþC percentage of a 100-bp window flankingtheposition (10), (iii)we calculated the LRR¼ log2
tini, whereni and
ti are normal and tumor-adjusted depths at position i, and (iv)each representative LRR was determined by the median of amoving window (1 Mb) centered at position i.
LRR of the copy number of both alleles, that of themajor allele,and that of the minor allele were determined for every region ofthe genome. The P values for gain or loss of respective genomicregions were determined from the LRRs with the permutation test(100,000 iterations) following the algorithm used in GISTIC(11, 12). Q values were calculated from the P values using theR package q value (http://github.com/jdstorey/qvalue).
Mutational signatures were analyzed using the Wellcome TrustSanger Institute Mutational Signature Framework (http://jp.mathworks.com/matlabcentral/fileexchange/38724-wtsi-mutational-signature-framework). The optimal number of signatures wasdetermined in accordance with the signature stabilities and aver-age Frobenius reconstruction errors.
Immunohistochemical analysis of mismatch repair proteinsImmunohistochemistry of mismatch repair proteins (MLH1,
MSH2, MSH6, and PMS2) was performed on whole sections ofeach tumor. Formalin-fixed paraffin-embedded tumor blockswere sliced into 3-mm-thick sections, which were then immunos-tained with the Ventana Benchmark automated immunostainer(Roche). Primary antibodies used were mouse monoclonal anti-MLH1 antibody (clone ES05, dilution 1:50; Leica), mousemono-clonal anti-MSH2 antibody (clone FE11, dilution 1:50; Dako),rabbit monoclonal anti-MSH6 antibody (clone EPR3945, dilu-tion 1:200; GeneTex), and mouse monoclonal anti-PMS2 anti-body (clone EPR3947, no dilution; Roche).
Immunostained slides were blindly evaluated by a board-certified pathologist (Hiroyuki Abe). Nuclear staining was eval-uated in each tumor. Epithelial cells in the proliferative zone ofnon-neoplastic colonic mucosa and lymphocytes in the germinalcenter of lymph follicles were used as internal positive controls.
Pathway analysisThe Database for Annotation, Visualization and Integrated
Discovery web-based tool (https://david.ncifcrf.gov) was used toidentify pathways. Pathways defined in Gene Ontology (limitedto the "biological pathway" category; http://www.geneontology.org), KEGG pathway (http://www.genome.jp/kegg/), BioCarta(https://cgap.nci.nih.gov/Pathways/BioCarta_Pathways), and theReactome Pathway Database (http://www.reactome.org) wereused for the analysis.
Transcriptome sequencing, expression analysis, and detectionof fusion genes
Total RNA was extracted with RNA Bee reagent (Tel-Test Inc.)and treated with DNase I (Qiagen) using an RNeasy Mini Kit(Qiagen). RNA integrity was evaluated using either a Bioanalyzer
(Agilent Technologies) or TapeStation (Agilent Technologies).RNAwith a high RNA integrity number underwent RNA-seq usinga NEBNext Ultra Directional RNA Library Prep Kit (New EnglandBioLabs) in which complementary DNA (cDNA) was preparedfrom polyA-selected RNA. RNA with a low RNA integrity numberwas subjected to RNA-seq using a TruSeq RNAAccess Library PrepKit (Illumina) in which cDNA was generated with random pri-mers. Prepared RNA-seq libraries underwent next-generationsequencing of 120 bp from both ends (paired-end reads). Theexpression level of each genewas calculated usingDESeq2 (http://bioconductor.org/packages/release/bioc/html/DESeq2.html)with VST transformation, and gene fusions were detected using adeFuse pipeline (https://bitbucket.org/dranew/defuse) and STAR(https://github.com/alexdobin/STAR).
Cloning of fusion genesComplementary DNAs for wild-type, mutant, and fusion pro-
teins were amplified by reverse transcription PCR (RT-PCR) fromRNA samples and then ligated into the pMXs retroviral vector(Cell Biolabs). The sequences of all cDNAswere verified by Sangersequencing. Primer sequences are provided in SupplementaryTable S1.
3T3 cell transformation assayHuman embryonic kidney (HEK) 293T cells and mouse 3T3
fibroblasts were obtained from the ATCC and maintained inDMEM-F12 supplemented with 10% FBS (both from Life Tech-nologies). Cell lines were propagated for less than 3 months afterinitial plating. Cultured cells were tested for mycoplasma con-tamination using a MycoAlert Mycoplasma Detection Kit(Lonza). To obtain infectious virus particles, recombinant vectorswere introduced together with an ecotropic packaging plasmid(Takara Bio) into HEK293T cells by transfection. For the focusformation assay, 3T3 cells were infected with ecotropic recombi-nant retroviruses and cultured for 12 days in DMEM-F12 supple-mented with 5% calf serum. Cell numbers were counted using aluminometer and the CellTiter-Glo Luminescent Cell Viabilityassay (Promega). Linsitinib (S1091), entrectinib (S7998), regor-afenib (S1178), and PLX7904 (S7964) were obtained fromSelleck Chemicals (Houston).
Animal experimentAll animal experimental procedures were approvedby the Insti-
tutional Animal Care and Use Committee of The University ofTokyo.We also adhered to the standards articulated in the NC3Rsguidelines (Animal Research: Reporting of In Vivo Experiments).
3T3 cells expressing SLC12A2–INSR and RUFY1-RET were sub-cutaneously inoculated into the flank of 6- to 8-week-old femalenude mice (BALB/c-nu/nu; Charles River Laboratories Japan, Inc.)at 2 � 106 cells/200 mL of PBS. Mice carrying implanted tumorswere divided randomly into two or three groups after confirmingtumor growth in each experiment. Kinase inhibitors, dissolved inN-methyl-2-pyrrolidone (Nacalai Tesque) and polyethylene glycol(PEG; Sigma-Aldrich), were applied orally once daily as indicated.Linsitinib (LCL #L-5814; LC Laboratories) was applied at a dose of25 mg/kg body weight (8 tumors in 4 mice). Regorafenib(AK106990; Ark Pharm) was applied at a dose of 100 mg/kg(10 tumors in 5 mice) or 10 mg/kg (8 tumors in 4 mice). Tumordiameter was measured using callipers, and tumor volume wasdetermined by calculating the volume of an ellipsoid using thefollowing formula: length � width2 � 0.5.
Sato et al.
Clin Cancer Res; 25(1) January 1, 2019 Clinical Cancer Research380
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574
Statistical analysisNumerical variables were summarized by median and range.
Comparisons of numerical variables between groups werecarried out using a nonparametric approach (Wilcoxon rank-sum test, two-tailed). Comparisons of the distribution of cat-egorical variables in different groups were performed using theFisher's exact test. The survival curve was established by theKaplan–Meier method and compared by the log-rank test withBonferroni adjustment. Cox proportional hazards model wasalso used to evaluate the effects of multiple variables. Statisticalanalysis was performed using the computing environment R(version 3.2.3).
Data availabilityRaw sequencing datawere deposited in the JapaneseGenotype-
Phenotype Archive (http://trace.ddbj.nig.ac.jp/jga), which ishosted by the DNA Data Bank of Japan (13) under accessionnumber JGAS00000000113 (NBDC number: hum0094).
ResultsDemographic characteristics
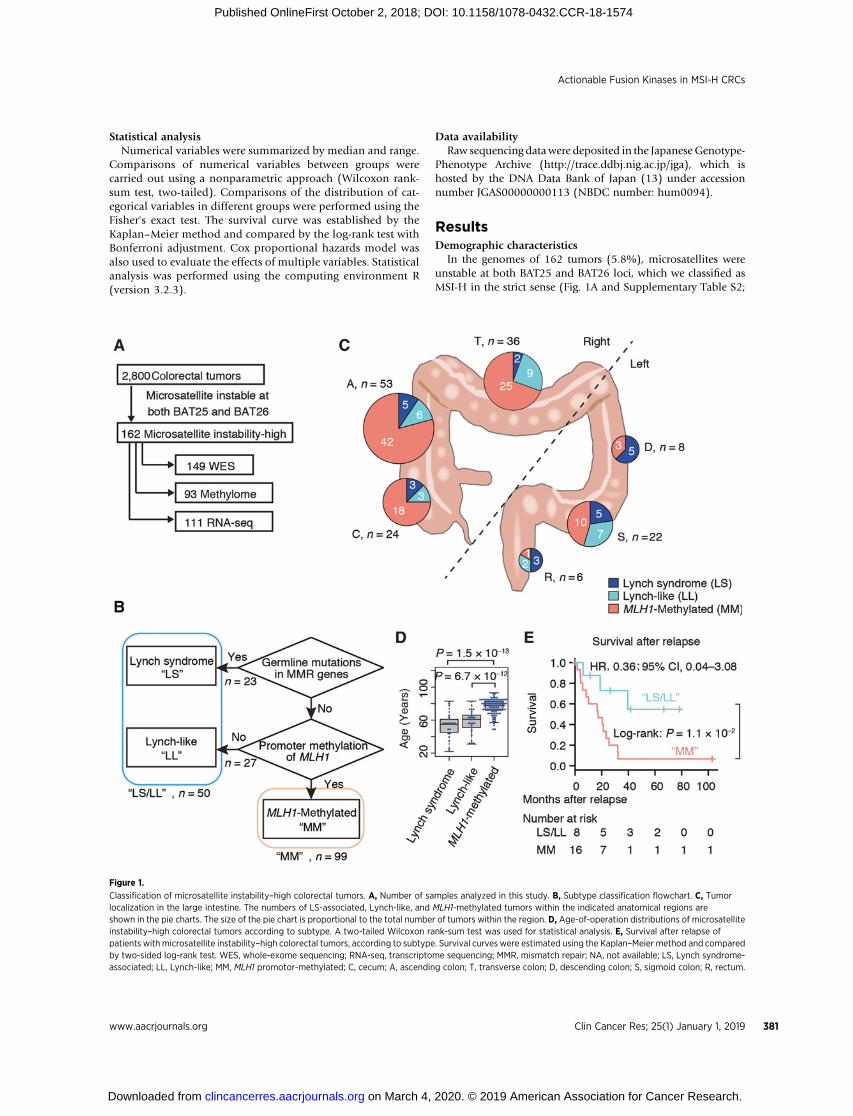
In the genomes of 162 tumors (5.8%), microsatellites wereunstable at both BAT25 and BAT26 loci, which we classified asMSI-H in the strict sense (Fig. 1A and Supplementary Table S2;
Figure 1.
Classification of microsatellite instability–high colorectal tumors. A, Number of samples analyzed in this study. B, Subtype classification flowchart. C, Tumorlocalization in the large intestine. The numbers of LS-associated, Lynch-like, and MLH1-methylated tumors within the indicated anatomical regions areshown in the pie charts. The size of the pie chart is proportional to the total number of tumors within the region. D, Age-of-operation distributions of microsatelliteinstability–high colorectal tumors according to subtype. A two-tailed Wilcoxon rank-sum test was used for statistical analysis. E, Survival after relapse ofpatients with microsatellite instability–high colorectal tumors, according to subtype. Survival curves were estimated using the Kaplan–Meier method and comparedby two-sided log-rank test. WES, whole-exome sequencing; RNA-seq, transcriptome sequencing; MMR, mismatch repair; NA, not available; LS, Lynch syndrome-associated; LL, Lynch-like; MM, MLH1 promotor-methylated; C, cecum; A, ascending colon; T, transverse colon; D, descending colon; S, sigmoid colon; R, rectum.
Actionable Fusion Kinases in MSI-H CRCs
www.aacrjournals.org Clin Cancer Res; 25(1) January 1, 2019 381
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

ref. 14). The frequency of MSI-H was lower than expected (6),probably because only two mononucleotide repeat markers wereused, while the probability of including MSI-low tumors was low(15–17). Clinicopathological characteristics of the MSI-H casesare summarized in Supplementary Table S3.
Classification of MSI-H colorectal cancerTo clarify the association between MSI-H status and the
CIMP (7, 8), consensus clustering was performed using datafrom methylation profile analyses. Sixty-four tumors (69%)that were clustered in the CIMP branch were also found in asubset of 65 tumors in which MLH1 expression was silencedbecause of promoter methylation (Supplementary Fig. S1),confirming almost complete overlap between these two clus-ters. In this report, we define CIMP based on the methylationstatus of the MLH1 promoter in order to avoid ambiguitycaused by the different clustering methods. Promoter methyl-ation of MMR genes other than MLH1, namely, MSH2, MSH6,PMS1, MSH3, PMS2, and MLH3, was not detected (Supple-mentary Fig. S1).
To complement the genome-widemethylation profile analysis,the promoter region ofMLH1 (chr3: 36,993,202–36,993,864) inthe genome of 149 samples was analyzed by bisulfite sequencing.The methylation status determined by bisulfite sequencing waswell correlated with that determined by genome-wide analysis(Supplementary Fig. S2). We refer to the MLH1-silenced MSI-Hcolorectal cancers as MLH1 promoter-methylated (MM) tumors(Fig. 1B).
Of the 23 tumor samples from patients with LS in our cohort,11 LS tumors underwent methylation profile analysis, and wefound that none of these tumors clustered in the CIMP branch(Supplementary Fig. S1). Bisulfite sequencing also revealed thatpromoter methylation of MLH1 was absent in all 23 LS tumors.Neither germline mutations in MMR genes nor promoter meth-ylation of MLH1 was observed in 27 tumor samples of MSI-Hcolorectal cancers, 17 of which were subjected to methylationprofile analysis. Because the 17 tumors clustered together with LStumors in consensus clustering, we refer to these samples as"Lynch-like (LL)" tumors (Fig. 1B, Supplementary Fig. S1). Asshown in the flowchart in Fig. 1B, our cohort of patients withMSI-H colorectal cancer was classified into three subgroups withdistinct genomic/epigenomic profiles: LS (n ¼ 23), LL (n ¼27), and MM (n ¼ 99) (Fig. 1B).
Distinct clinical features of LS/LL tumors and MM tumorsInterestingly, althoughMMtumorsweremore likely to develop
in the "right" side of the large intestine (consisting of the cecum,and the ascending and transverse colon), a finding that waspreviously reported concerning MSI-H colorectal cancers in gen-eral (1), LS/LL tumors were evenly distributed across the largeintestine (Fig. 1C). This observation is consistent with the hypoth-esis that aberrant DNA methylation may develop in the specificmicrobiological environment of the "right" side of the largeintestine (18, 19). Notably, and justifying our classificationapproach, we found that the age of the patients at operationdiffered between the subtypes [one-wayANOVA, F(2,143)¼54,P< 2.0 � 10�16; Fig. 1D]. The patients with MM tumors had worsesurvival after relapse than those with LS/LL tumors (log-rank test,P ¼ 0.013; Fig. 1E), although the difference was not statisticallysignificant after adjusting for age and stage at operation (Coxproportional hazard model, HR, 0.36; 95% CI, 0.04–3.08).
Further analysis with a larger population is required to evaluatethe difference in prognosis.
Mutations in MMR genesBecause MSI-H colorectal cancers often harbor disruptive
somatic mutations within MMR genes that can in turn be affectedby MMR deficiency, it is difficult to distinguish causative muta-tions in MMR genes from resultant mutations. Because MLH1promoter methylation is regarded as the primary cause of MMRdeficiency (20), we considered that somatic mutations in MMRgenes detected inMLH1 promoter-methylated tumors were likelyresultant ones, and we excluded them from the causal MMR genemutation panel of LS/LL (Fig. 2). The excluded somaticmutationswere as follows: MLH1 [(S193fs; n ¼ 1), MSH2(L229fs; n ¼ 3),MSH6(R248fs) (n ¼ 4), and MSH6(P1087fs; n ¼ 41; located on(A)6, (A)7, (A)7, and (C)8 repeats, respectively]. Mutations inMMR genes that were truncated or reported to be pathogenicwere regarded as being causative (Supplementary Table S4).
Next, we investigated how the function of MMR genes with aheterozygousmutation in LS was abolished (Fig. 2A, Supplemen-tary Fig. S3). Somatic uniparental disomy (UPD) in three MMRgenes was observed in 12 LS tumors (MLH1, n ¼ 8; PMS2, n ¼ 3;and MSH2, n ¼ 1). Additional somatic single-nucleotide varia-tions (SNV) or insertions/deletions (indels) in MMR genes wereobserved infive LS tumors (MSH6, n¼ 2;PMS2, n¼ 2; andMSH2,n ¼ 1). An LS tumor with a heterozygous germline mutation inPMS2 lost the wild-type allele, and additional pathogenic muta-tions were not identified in the remaining five LS tumors.
We further sought to identify the alterations ofMMRgenes in LLtumors. Sixteen LL tumors (16/27, 59%) harbored somatic muta-tions within MMR genes, as previously reported (21). Six tumorshad somatic SNVs/indels inMLH1 and one tumor had a somaticSNV in MSH2, both of which were accompanied by UPD of therespective gene. Biallelicmutations inMSH2were observed infivetumors. Heterozygous somatic mutations in MLH1 and MSH2were observed in three tumors and one tumor, respectively,without additional detectable events. However, in the remaining11 tumors, we did not identify pathogenic mutations in MMRgenes that would account for the MSI-H status, although sixvariants of uncertain significance (VUS), listed in SupplementaryTable S4, are not included in Fig. 2A.
We validated these results by immunohistochemistry (IHC) ofMMR proteins, obtaining concordant results in 91% (32/35) ofcases for which both the genotype data and protein expressiondata were available (Fig. 2). In seven cases without genotype data,the results of IHC revealed probable disrupted MMR genes.
Somatic SNVs and indelsWith >20-fold coverage of at least 85% of the target regions,
whole-exome sequencing (WES) identified a median of 962(range, 103–6,973) somatic nonsynonymous SNVs and amedianof 130 (range, 5–308) somatic indels in coding regions. Thenumber of recurrent (>5% frequency) indels (n¼ 684) was largerthan that of recurrent (>5% frequency) SNVs (n ¼ 5). Thefrequencies of SNVs did not differ significantly between LL/LStumors and MM tumors (P ¼ 0.49), whereas the frequencies ofindels were higher in MM tumors than those found in LS/LLtumors (P ¼ 0.0028, Wilcoxon rank-sum test; SupplementaryFig. S4).
To identify mutations that are likely relevant to MSI-H colo-rectal cancer oncogenesis under conditionswith a large number of
Sato et al.
Clin Cancer Res; 25(1) January 1, 2019 Clinical Cancer Research382
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

backgroundmutations, the identified somatic variants were strat-ified into three tiers (Supplementary Fig. S4): Tier 1 (n¼ 78,187 in18,646 genes), consisting of all somatic variants detected; Tier 2 (n¼ 46,502 in 10,748 genes), consisting of those Tier 1 variationsthat were either also detected in RNA-seq analysis or presentrecurrently (�10 times) in the Catalogue of Somatic Mutationsin Cancer database (http://cancer.sanger.ac.uk/cosmic/; Supple-mentary Table S5); and Tier 3 (n¼ 3,346 in 458 genes), consistingof those Tier 2 variations present in genes found in the CancerGene Census (22) or that were reported to be recurrent (�30%frequency) in a previous study of MSI-H colorectal cancer (23).
The 40 genes most frequently affected in Tier 3 are well-recognized targets in theMSI-H status, includingACVR2A, SEC63,TGFBR2, andRNF43 (shown in Fig. 3A).We found that therewere209 frequently altered genes (>20% of samples) within Tier 2variants (Supplementary Table S5). We also obtained a list of 860significantly (Q < 0.01) mutated genes using MutSigCV (Supple-mentary Table S6, Supplementary Fig. S4; ref. 24). Notably, 153(73%) of the 209 most frequently altered Tier 2 genes wereincluded in the list of significantlymutated genes, partly justifyingthe stratification strategy used in this study. Mutations in KRAS,APC, and TCF7L2 were more prevalent in LS/LL tumors than inMM tumors (P ¼ 5.7 � 10�9, 6.5 � 10�8, and 1.1 � 10�3,
respectively), whereas mutations in BRAF and RNF43 were moreprevalent in MM tumors than in LS/LL tumors (P ¼ 5.5 � 10�12
and 9.4 � 10�8, respectively; Fig. 3A, Supplementary Table S7).Mutations encoding BRAF(V600E) were not observed among LS/LL tumors, in accordance with the well-established notion thatBRAF mutations and germline mutations in MMR genes aremutually exclusive (25, 26). We did not observe statisticallysignificant differences in mutation frequencies between LS andLL tumors (Supplementary Table S7).
Through signature analysis of SNVs (27), four mutationalsignatures (termed Signatures A, B, C, and D) were detected (Fig.3A and B). Signatures A, B, and C exhibited profiles similar tosignatures associated with MMR deficiency, whereas Signature Dwas an age-related signature (28). Signature C, which may bederived from G-T mismatch, was abundant in MM tumors; themedian percentage of signature Cmutations in LS/LL tumors was13.9% (interquartile range: 7.04%–24.4%), while that in MMtumors was 24.0% (interquartile range: 19.0%–29.8%; Fig. 3C).Signature A,whichmay be derived fromG-T andA-Cmismatches,was enriched in tumors with PMS2 disruptions; the medianpercentage of signature A mutations in tumors with defectivePMS2 was 73.2% (interquartile range: 65.0%–87.3%), whilethat in the other tumors was 26.4% (interquartile range:
Figure 2.
Mutations in DNA mismatch repair genes and immunohistochemical analysis of DNA mismatch repair proteins. A, Mutational status of DNA mismatch repairgenes is shown in association with the results of immunohistochemical analysis of DNA mismatch repair proteins. The columns in the table denote samples and therows denote gene alleles (top) or proteins (bottom). B, Representative images of the immunohistochemical analysis. SNV, single-nucleotide variation; indel,insertion or deletion; UPD, uniparental disomy.
Actionable Fusion Kinases in MSI-H CRCs
www.aacrjournals.org Clin Cancer Res; 25(1) January 1, 2019 383
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

21.5%–31.3%; Fig. 3D). These data may be useful for furtherexploration of the etiology of MSI-H colorectal cancers.
Somatic copy-number alterationsA previous study reported that somatic copy-number altera-
tions (CNA) are infrequent inMSI-H colorectal cancers comparedwith those in MSS colorectal cancers (29). In this study, allele-
specific copy number (CN) analysis using WES data revealed 41recurrent (Q < 0.01) CNAs, 9 of which were involved in UPD(Fig. 3E, Supplementary Table S8). Notably, these CNA profilesdiffered between LS/LL and MM tumors. UPD encompassingTGFBR2 (3p24.1), MLH1 (3p22.2), and CTNNB1 (3p22.1) wasobserved specifically in LS/LL tumors, and these genes have beenlinked to the pathogenesis ofMSI-H colorectal cancer. In contrast,
Figure 3.
Summary of mutations inmicrosatellite instability–highcolorectal tumors. A, The 40 mostfrequently mutated genes with theirmutation status color-coded for eachpatient. The frequency of synonymousor nonsynonymous substitutions andinsertions/deletions is shown at thetop, and the percentages of themutational signatures are shown atthe bottom. The frequencies ofmutations in each gene according tosubtype are shown on the left. B, Thefour mutational signatures identifiedin this study. C, The percentage ofSignature C according to subtype.D, The percentage of Signature Aaccording to PMS2 mutational status.E, Allele-specific copy-numberalterations in microsatelliteinstability–high colorectal tumors. Theprofiles of allele-specific copy-numberalterations according to subtype areshown. Red and blue lines indicate theQ-value for gains of major allele andlosses ofminor allele, respectively. They-axis indicates the frequency of theobserved gain or loss. Representativegenes affected by respective copy-number alteration are shown. A two-tailed Wilcoxon rank-sum test wasused for statistical analysis. LS,Lynch syndrome; LL, Lynch-like;MM, MLH1-methylated.
Sato et al.
Clin Cancer Res; 25(1) January 1, 2019 Clinical Cancer Research384
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

we found that a CN gain within the long arm of chromosome 8was recurrent in MM tumors. In addition, UPD within the shortarm of chromosome 6 involving the major histocompatibilitycomplex class I and class II genes was prevalent in both LS/LL andMM tumors, whichmay contribute to the avoidance of antitumorimmunity by tumor cells.
Affected pathwaysTo better understand genetic alterations in theMSI-H colorectal
cancer genome in our cohort, we performed pathway analysisusing the Database for Annotation, Visualization and IntegratedDiscovery web-based tool (https://david.ncifcrf.gov; refs. 30, 31).Of the 209 most frequently altered Tier 2 genes, four molecularpathways were identified, specifically, DNA damage-sensing andhistone H3 methylation-associated pathways, as well as Wntsignaling andRAS/RAF/mitogen-activated protein kinase (MAPK)pathways (Fig. 4, Supplementary Table S9). A similar result wasobtained using the 860 significantly mutated genes identified
withMutSigCV (Supplementary Table S10), partly supporting ourstratification of mutated genes.
Oncogenic alterations in MSI-H colorectal cancerConsidering that activating kinase fusion events are extremely
rare in colorectal cancer and do not seem to cluster in a particulargenomic or epigenomic subtype of the disease (32), we did notanticipate the existence of recurrent fusion genes; surprisingly,using RNA-seq, we detected in-frame fusion transcripts encodingfusion-type kinases in 11 out of 111 tumors (9.9%). These 11tumors carrying fusion kinases were all found in theMM subtype,appearing at a frequency of 11% (11/99). Considering themutualexclusivity of oncogenic KRAS/BRAF missense mutations andfusion kinases, clinically actionable fusion kinases includingnovel insulin receptor gene fusion accounted for 55% (11/20)ofMMtumors lacking oncogenicKRAS/BRAFmissensemutations(Fig. 5A). In addition, we identified oncogenic mutants of ERBB2(n¼ 3), ERBB3 (n¼ 5),HRAS (n¼ 2), RRAS2 (n¼ 1), and RAC1
Figure 4.
Frequently affected pathways. Major components of the pathways extracted from the top 209 Tier 2 genes using The Database for Annotation, Visualizationand Integrated Discovery web-based tool algorithm are shown. The frequencies of single-nucleotide variations and insertions/deletions in the genes areexpressed as a percentage of all cases according to subtype. Warm colors denote activated genes, and cold colors denote inactivated genes. The frequencies arecolor-scaled. LS, Lynch syndrome; LL, Lynch-like; MM, MLH1-methylated.
Actionable Fusion Kinases in MSI-H CRCs
www.aacrjournals.org Clin Cancer Res; 25(1) January 1, 2019 385
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

Figure 5.
Oncogenic fusion proteins in microsatellite instability–high colorectal tumors. A, Mutation plot of oncogenes with transforming potential. Mutations withtransforming capacity are shown in darker colors, whereas mutations of unknown significance are shown in lighter colors. B, Schematic diagrams of thefusion proteins identified in this study. Amino acid numbering on the fusion proteins refers to the sequences of the wild-type proteins. A novel fusion kinase(SLC12A2–INSR) is shown in orange. A novel fusion partner (RUFY1) of RET fusion kinase is shown in green. C, The transcript fusion point of SLC12A2–INSRcomplementary DNA determined by Sanger sequencing. D, A schematic representation of the gene rearrangement generating the SLC12A2–INSR fusion gene.The sequence of the genomic fusion point determined by Sanger sequencing is shown at the bottom. E, The response to kinase inhibitors of 3T3 cells expressingfusion proteins. Cells were treatedwith the indicated drugs at the indicated concentrations. Cellularity wasmeasured 9 days after treatment and is plotted relative tothat of cultures treated with the lowest concentration. Error bars indicatemodel-based standard errors. In the graphs showing the data obtained for cells expressingTPM3-NTRK1, KANK1-NTRK3, or EML4-NTRK3, the control v-Ras data are identical. A two-tailed student t test was used for statistical analysis. CC, coiled coil.
Sato et al.
Clin Cancer Res; 25(1) January 1, 2019 Clinical Cancer Research386
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

(n ¼ 1; Supplementary Fig. S5, Supplementary Table S11),although these variants were not necessarily mutually exclusiveregarding KRAS, BRAF, and fusion kinase genes.
The detected fusion kinases involved INSR (n¼ 1), RET (n¼ 2),NTRK1 (n ¼ 2), NTRK3 (n ¼ 2), and BRAF (n ¼ 4; Fig. 5B). Theexpression of these fusion transcripts was confirmed by reverse-transcription PCR (Fig. 5C, Supplementary Table S12).We furtheridentified the genomic fusion points of 10 fusion genes, includingSLC12A2–INSR, all of which were confirmed to be somatic (Fig.5D, Supplementary Figs. S6 and S7).
Excluding AKAP9–BRAF, which was predicted to be 4,253amino acids in length and technically difficult to manipulate,and TRIM24–BRAF, which has been well described previously(33), tumorigenicity of the fusion proteins identified in our studywas confirmed using a 3T3 transformation assay (SupplementaryFig. S5). Luciferase reporter assay indicated that these fusionproteins activated the mitogen-activated protein kinase pathway(Supplementary Fig. S5). In addition, small-molecular inhibitorsthat suppress the activity of the kinases identified in our fusionproducts significantly attenuated themalignant transformationof3T3 cells in a concentration-dependent manner (Fig. 5E). Nota-bly, linsitinib, which is usually used for the main purpose ofantagonizing IGF1R signaling (34), inhibited the growth of 3T3cells expressing SLC12A2–INSR. Although it may be difficult totarget oncogenic BRAF fusion proteins with specific BRAF inhi-bitors that are currently available, it is anticipated that tumorswith BRAF fusion proteins could be targeted with a combinationof MAPK kinase and PI3K inhibitors (35–37).
It has been reported that alectinib and entrectinib inhibited thein vivo growth of the Lc-2/ad lung cancer cell line carrying CCDC6-RET and the KM12 colorectal cancer cell line carrying TPM3–NTRK1, respectively (38, 39). Because we could not identify MSI-H colorectal cancer cell lines harboring SLC12A2–INSR orRUFY1–RET, we evaluated the therapeutic efficacy using an invivo mouse model, in which 3T3 cells transformed by the fusionkinases were subcutaneously inoculated. Linsitinib and regorafe-nib substantially suppressed growth of the transformed 3T3 cellsexpressing SLC12A2–INSR and RUFY1–RET, respectively (Sup-plementary Fig. S8).
Comparison with MSSWe analyzed theMSS colorectal cancer RNA-seq database from
the Cancer Genome Atlas Network (29). Twenty-six paired-endand 181 single-end RNA sequences (median 36,213,120 readsper sample, 76 bp) were available. Only one fusion kinase (HLA-A-RET) was detected in 189 patients. To adjust the conditions ofanalysis, the 36 randomly selected mega reads per sample fromour RNA-seq data (median 206,727,876 reads per sample,120 bp) were trimmed down to 76 bp and subjected to analysis,identifying all of the fusion transcripts described above except forthat encoding ARMC10-BRAF. Taken together, oncogenic fusionkinases were more prevalent in MSI-H colorectal cancers than inMSS colorectal cancers.
Effective strategy for detection of fusion kinasesDespite the considerable number of fusion kinases in MSI-H
colorectal cancers, it is expensive to perform next-generationsequencing for all MSI-H samples in clinical practice. In addition,bisulfite-treated DNA and particular devices such as pyrosequen-cer are required for precise MLH1 promoter methylation assays.To effectively detect fusion kinases, we first developed a simple
method to detect MLH1 promoter methylation utilizing methyl-ation-sensitive restriction enzyme. As shown in Fig. 6, the meth-ylation status determined by our MLH1 promoter methylationassay was well correlated with that determined by genome-wideanalysis. Tumors in a validation cohort were subjected to MSItesting, and the methylation status of MSI-H tumors was deter-mined by our MLH1 methylation assay. Subsequently, we con-ducted RNA-seq in MM MSI-H colorectal cancers lacking onco-genic KRAS/BRAFmissensemutations. We succeeded in detectingtwo fusion kinases (NCOA4-RET and CUL1–BRAF; ref. 40) out offour MMMSI-H colorectal cancers lacking oncogenic KRAS/BRAFmissense mutations (Supplementary Fig. S9). We used BRAF as acontrol for PCR because we detected CNAs in neither chromo-some 7, including BRAF as previously reported (29) nor the SmaIsite.
PrognosisIt has been recognized that mutation in BRAF is a poor prog-
nostic factor in metastatic or relapsed MSI-H colorectal cancer(4, 41, 42, 43). We analyzed the survival data in our cohort, giventhe considerable number of fusion kinases observed in MSI-Hcolorectal cancers lacking oncogenic KRAS/BRAF missense muta-tions, as mentioned above. When VUS was included in the wild-type group, patients harboring BRAF (V600E) had worse overallsurvival than patients in the BRAF-wild-type group (P ¼ 2.3 �10�2; Supplementary Fig. S10A) and patients in the BRAF-wild-type non-fusion group (P ¼ 9.5 � 10–3; Supplementary Fig.S10B). The survival after relapse of patients harboring fusionkinases was shorter than that of patients harboring oncogenicKRAS mutations (P ¼ 8.2 � 10�3; Supplementary Fig. S10C).
DiscussionIn this study, we performed a comprehensive analysis of genetic
alterations inMSI-H colorectal cancer, revealing several important
Figure 6.
Precise and simple detection of MLH1-methylated tumors. A comparisonbetween extracted data of the degree of methylation in the MLH1 promoterregion of 13 tumors by genome-wide methylation array (see SupplementaryFig. S1C; top) and detection of MLH1 methylated tumors using our MLH1methylation assay (bottom). Amplicon of BRAF is a loading control andused as a template of Sanger sequencing for detecting BRAF(V600E)mutation.M, 50-bp DNA ladder marker; RKO, positive control; DLD1, negative control;NTC, no template control.
Actionable Fusion Kinases in MSI-H CRCs
www.aacrjournals.org Clin Cancer Res; 25(1) January 1, 2019 387
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

pathological aspects ofMSI-H colorectal cancers that have clinicalimplications.
First, we showed that MSI-H colorectal cancers could be clas-sified into three subtypes. These classifications were validatedthrough several observations: (i) there was a greater number ofindels in theMMsubtype than in the LS/LL subtype; (ii) one of themutational signatures associated with MMR deficiency wasenriched in the MM subtype; (iii) the frequencies of mutationsin several genes differed between the LS/LL andMMsubtypes; (iv)the LS/LL and MM subtypes exhibited distinctive CNA profiles;and (v) fusion genes encoding oncogenic kinases were enriched inthe MM subtype. However, a caveat of our findings is that the LLsubtype may contain misdiagnosed LS tumors because someforms of germline mutations in MMR genes are difficult to detect(44). Although it is not clear whether the LS and LL subtypes havedistinctive clinicopathological characteristics, further investiga-tion is warranted.
Second, we showed that the classification described abovehas important clinical implications. We also propose a feasiblemethod using a methylation-sensitive restriction enzyme forthe detection of MM tumors. Importantly, we chose thegenomic region, in which the methylation status is most strictlyassociated with the silencing of MLH1 based on the genome-wide methylation analysis (Supplementary Fig. S2). Among theMSI-H colorectal cancers, MM tumors that lack KRAS or BRAFmutation are expected to carry a targetable fusion kinase withhigh probability. Determination of the MM subtype mayhelp to identify patients who can benefit from an intensivesearch for fusion kinases. Despite the remaining ambiguitybetween LS and LL subtype classifications, recognition of theLL subtype is also clinically valuable in practice whenconsidering patients of whom germline mutations in MMRgenes may escape detection.
Third, we identified oncogenic fusion genes that have trans-forming activity and are potential therapeutic targets. Using ourMLH1 methylation assay and considering their mutual exclusiv-ity, it is possible to develop a simple diagnostic sequencingstrategy for the efficient detection of LS and fusion kinases toprovide improved personalized therapeutic opportunities forpatients withMSI-H colorectal cancer. AlthoughMSI-H colorectalcancers carry multiple driver oncogenes, knockdown of TPM3–NTRK1 in MSI-H colorectal cancer cells reduced their prolifera-tion, further supporting the role of NTRK1 fusions as clinicallyactionable (45).
Fourth, we detected those biological pathways that were affect-ed by many mutations. A unique finding of our study was theidentification of the DNA damage-sensing pathway based on thegenetic alterations in MSI-H colorectal cancer genomes, which isof particular interest given that the DNA double-strand breakrepair machinery is closely associated with MMR machinery (46)
and that chromosomal aberrations are relatively infrequent inMSI-H colorectal cancers (29). A precise understanding of thefunctional consequences of mutations in the DNA damage-sens-ing pathway in MSI-H colorectal cancers may lead to the devel-opment of novel diagnostic and/or therapeutic approaches forpatients with MSI-H colorectal cancer.
Taken together, our genomic and epigenomic analyses ofMSI-Hcolorectal cancer provide clinically relevant findings that mayimpact on patient management and care in association withcancer predisposition, molecular-targeted therapy, and cytotoxicchemotherapy.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: K. Sato, T. Watanabe, H. ManoDevelopment of methodology: K. Sato, Y. YamamotoAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): K. Sato, Y. Yamamoto, G. Nagae, H. Abe, M. Soda,S. Kohsaka, Y. Yamashita, H. Iinuma, M. Fukayama, H. Aburatani, T. Watanabe,H. ManoAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): M. Kawazu, T. Ueno, S. Kojima, M. SodaWriting, review, and/or revision of the manuscript: K. Sato, M. Kawazu,Y. Yamamoto, H. Abe, M. Soda, S. Kohsaka, Y. Yamashita, H. Iinuma,M. Fukayama, T. Watanabe, H. ManoAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): T. Oga, E. Sai, M. Fukayama, T. Watanabe,H. ManoStudy supervision: M. Kawazu, H. Aburatani, T. Watanabe
AcknowledgmentsThe authors thank Miki Tamura, Reina Takeyama, Akane Maruyama, Junko
Tamura, Riyo Kakimoto, Dr. Takamitsu Kanazawa, Dr. Yuzo Nagai, andDr. Takashi Kobunai for their technical assistance, and Dr. Yoichi Furukawafor his advice and discussion. We are grateful to all of the patients and familieswho contributed to this study.We thank allmembers of the Colorectal Group ofThe University of Tokyo and Teikyo University for their support of this research.We also thank Edanz Group (www.edanzediting.com/ac) for editing a draft ofthis article. This study was supported in part by grants-in-aid for ScientificResearch (KAKENHI, grant numbers 17J00386, 26430106, 16K07143, and16H02672) from the Japan Society for the Promotion of Science (JSPS) andby grants from Leading Advanced Projects for Medical Innovation (LEAP,JP17am0001001; to H. Mano) and from the Project for Cancer Research andTherapeutic Evolution (P-CREATE, JP17cm0106502; to M. Kawazu, M. Soda,H. Aburatani, and T. Watanabe) from the Japan Agency for Medical Researchand Development. K. Sato is a research fellow of JSPS.
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received May 20, 2018; revised July 31, 2018; accepted September 27, 2018;published first October 2, 2018.
References1. Thibodeau S, Bren G, Schaid D. Microsatellite instability in cancer of the
proximal colon. Science 1993;260:816–9.2. Aaltonen L, Peltomaki P, Leach F, Sistonen P, Pylkkanen L, Mecklin J, et al.
Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
3. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastro-enterology 2010;138:2073–2087.e3.
4. Sinicrope FA, Shi Q, Allegra CJ, Smyrk TC, Thibodeau SN, Goldberg RM,et al. Association of DNA mismatch repair and mutations in BRAF and
KRAS with survival after recurrence in stage III colon cancers: a second-ary analysis of 2 randomized clinical trials. JAMA Oncol 2017;3:472–80.
5. Guinney J, Dienstmann R, Wang X, de Reyni�es A, Schlicker A, Soneson C,et al. The consensus molecular subtypes of colorectal cancer. Nat Med2015;21:1350–6.
6. Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethyla-tion in drug resistance to 5-fluorouracil in colorectal cancer cell lines.Int J Cancer 2003;106:66–73.
Clin Cancer Res; 25(1) January 1, 2019 Clinical Cancer Research388
Sato et al.
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

7. Toyota M, Ho C, Ahuja N. Identification of differentially methylatedsequences in colorectal cancer by methylated CpG island amplification.Cancer Res 1999;59:2307–12.
8. Weisenberger DJ, Siegmund KD, CampanM, Young J, Long TI, Faasse MA,et al. CpG island methylator phenotype underlies sporadic microsatelliteinstability and is tightly associated with BRAF mutation in colorectalcancer. Nat Genet 2006;38:787–93.
9. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60.
10. Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detectionof copy number variants using read depth of coverage. Genome Res2009;19:1586–92.
11. Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al.Assessing the significance of chromosomal aberrations in cancer:methodology and application to glioma. Proc Natl Acad Sci U S A 2007;104:20007–12.
12. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G.GISTIC2.0 facilitates sensitive and confident localization of the targets offocal somatic copy-number alteration in human cancers. Genome Biol2011;12:1–14.
13. Kodama Y, Mashima J, Kosuge T, Katayama T, Fujisawa T, Kaminuma E,et al. The DDBJ Japanese genotype-phenotype archive for genetic andphenotypic human data. Nucleic Acids Res 2015;43:D18–22.
14. Jass JR, Walsh MD, Barker M, Simms LA, Young J, Leggett BA. Distinctionbetween familial and sporadic forms of colorectal cancer showing DNAmicrosatellite instability. Eur J Cancer 2002;38:858–66.
15. Wright CM, Dent OF, Newland RC, Barker M, Chapuis PH, Bokey EL, et al.Low level microsatellite instability may be associated with reduced cancerspecific survival in sporadic stage C colorectal carcinoma. Gut 2005;54:103–8.
16. Lee SY, Kim D-W, Lee HS, Ihn MH, Oh H-K, Min BS, et al. Low-levelmicrosatellite instability as a potential prognostic factor in sporadic colo-rectal cancer. Medicine 2015;94:e2260.
17. Bocker T, R~aschoff J. Diagnostic microsatellite instability: definition andcorrelation with mismatch repair protein expression. Cancer Res 1997;57:4749–56.
18. Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, et al.Fusobacterium in colonic flora and molecular features of colorectalcarcinoma. Cancer Res 2014;74:1311–8.
19. Mima K, Cao Y, Chan AT, Qian ZR, Nowak JA, Masugi Y, et al. Fusobacter-iumnucleatum in colorectal carcinoma tissue according to tumor location.Clin Transl Gastroenterol 2016;7:e200.
20. Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidenceand functional consequences of hMLH1 promoter hypermethylation incolorectal carcinoma. Proc Natl Acad Sci U S A 1998;95:6870–5.
21. Mensenkamp AR, Vogelaar IP, Van Zelst-Stams WAG, Goossens M,Ouchene H, Hendriks-Cornelissen SJB, et al. Somatic mutations in MLH1and MSH2 are a frequent cause of mismatch-repair deficiency in lynchsyndrome-like tumors. Gastroenterology 2014;146:643–646.
22. Futreal P, Coin L,Marshall L, DownT,Hubbard T,Wooster T, et al. A censusof human cancer genes. Nat Rev Cancer 2004;4:177–83.
23. Kim T, Laird PW, Park PJ. The landscape of microsatellite instability incolorectal and endometrial cancer genomes. Cell 2013;155:858–68.
24. LawrenceMS, Stojanov P, Polak P, Kryukov G V.Cibulskis K, Sivachenko A,et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8.
25. Bessa X, Ballest�e B, Andreu M, Castells A, Bellosillo B, Balaguer F, et al. Aprospective, multicenter, population-based study of BRAF mutationalanalysis for lynch syndrome screening. Clin Gastroenterol Hepatol2008;6:206–14.
26. Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, BoardmanLA, et al. BRAF Mutations in colon cancer are not likely attributable todefective DNA mismatch repair. Cancer Res 2003;63:5209–12.
27. Alexandrov LB, StrattonMR.Mutational signatures: the patterns of somaticmutations hidden in cancer genomes. Curr Opin Genet Dev 2014;24:52–60.
28. Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutationalsignatures in human cancers. Nat Rev Genet 2014;15:585–98.
29. The Cancer Genome Atlas Network. Comprehensive molecular character-ization of human colon and rectal cancer. Nature 2012;487:330–7.
30. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysisof large gene lists using DAVID bioinformatics resources. Nat Protoc2009;4:44–57.
31. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools:paths toward the comprehensive functional analysis of large gene lists.Nucleic Acids Res 2009;37:1–13.
32. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J.Consensus molecular subtypes and the evolution of precision medicine incolorectal cancer. Nat Rev Cancer 2017;17:79–92.
33. Nakaoku T, Tsuta K, Ichikawa H, Shiraishi K, Sakamoto H, Enari M, et al.Druggable oncogene fusions in invasive mucinous lung adenocarcinoma.Clin Cancer Res 2014;20:3087–93.
34. MulvihillMJ, Cooke A, Rosenfeld-FranklinM, Buck E, Foreman K, LandfairD, et al. Discovery of OSI-906: a selective and orally efficacious dualinhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem2009;1:1153–71.
35. Kim HS, Jung M, Kang HN, Kim H, Park CW, Kim SM, et al. OncogenicBRAF fusions in mucosal melanomas activate the MAPK pathway and aresensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene2017;36:3334–45.
36. Hutchinson KE, LipsonD, Stephens PJ, Otto G, Lehmann BD, Lyle PL, et al.BRAF fusions define a distinct molecular subset of melanomas withpotential sensitivity to MEK Inhibition. Clin Cancer Res 2013;19:6696–702.
37. TurskiML, Vidwans SJ, Janku F, Garrido-Laguna I,Munoz J, Schwab R, et al.Genomically driven tumors and actionability across histologies: BRAF-mutant cancers as a paradigm. Mol Cancer Ther 2016;15:533–47.
38. KodamaT, Tsukaguchi T, SatohY, YoshidaM,WatanabeY, KondohO, et al.Alectinib Shows Potent Antitumor Activity against RET-Rearranged Non-Small Cell Lung Cancer. Mol Cancer Ther 2014;13:2910–8.
39. Ardini E, Menichincheri M, Banfi P, Bosotti R, DePonti C, Pulci R, et al.Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiplemolecularly defined cancer indications.Mol Cancer Ther 2016:15:628–39.
40. Grisham RN, Sylvester BE, Won H, McDermott G, DeLair D, Ramirez R,et al. Extreme outlier analysis identifies occult mitogen-activated proteinkinase pathway mutations in patients with low-grade serous ovariancancer. J Clin Oncol 2015;33:4099–105.
41. Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, et al. Impact of BRAFmutation and microsatellite instability on the pattern of metastatic spreadand prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
42. Godstein J, Tran B, Ensor J, Gibbs P, Wong HL, Wong SF, et al. Multicenterretrospective analysis of metastatic colorectal cancer (CR) with high-levelmicrosatellite instability (MSI-H). Ann Oncol 2014;25:1032–8.
43. Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, FisherD, et al. Mismatch repair status and BRAF mutation status in metastaticcolorectal cancer patients: a pooled analysis of theCAIRO, CAIRO2, COIN,and FOCUS studies. Clin Cancer Res 2014;20:5322–30.
44. Taylor CF, Charlton RS, Burn J, Sheridan E, Taylor GR. Genomic deletionsin MSH2 or MLH1 are a frequent cause of hereditary non-polyposiscolorectal cancer: Identification of novel and recurrent deletions by MLPA.Hum Mutat 2003;22:428–33.
45. Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al.Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer.Nat Med 2013;19:1469–72.
46. Spies M, Fishel R. Mismatch repair during homologous and homeologousrecombination. Cold Spring Harb Perspect Biol 2015;7:a022657.
www.aacrjournals.org Clin Cancer Res; 25(1) January 1, 2019 389
Actionable Fusion Kinases in MSI-H CRCs
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574

2019;25:378-389. Published OnlineFirst October 2, 2018.Clin Cancer Res Kazuhito Sato, Masahito Kawazu, Yoko Yamamoto, et al.
High Colorectal Cancers−Microsatellite Instability Fusion Kinases Identified by Genomic Analyses of Sporadic
Updated version
10.1158/1078-0432.CCR-18-1574doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2018/10/02/1078-0432.CCR-18-1574.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/25/1/378.full#ref-list-1
This article cites 46 articles, 18 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/25/1/378.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/25/1/378To request permission to re-use all or part of this article, use this link
on March 4, 2020. © 2019 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 2, 2018; DOI: 10.1158/1078-0432.CCR-18-1574


















