
Freeware for Sequence Analyses files... · Some data formats/ file extensions .ab1 Raw DNA data...
57
Freeware for Sequence Analyses Michael Kube
Transcript of Freeware for Sequence Analyses files... · Some data formats/ file extensions .ab1 Raw DNA data...

Freeware for Sequence Analyses
Michael Kube

1656 genomes in June 2011
(Kube0711)

������������ �����������
(Kube0711)

General need for tools and databases!

General information on bioinformatic tools & databases
http://www.biomedcentral.com/bmcbioinformatics/
http://www.ploscompbiol.org/
http://bioinformatics.oxfordjournals.org/
http://nar.oxfordjournals.org/
Database issue, January 2011
(Kube0711)

Some useful links providing information on bioinformatical software
Mac http://www.apple.com/science/profiles/osxporting/ and Seqanswers overview Windows http://molbiol-tools.ca/molecular_biology_freeware.htm http://www.bioinformatics.org/softwaremap/?form_cat=2 Linux and other operating systems http://seqanswers.com/wiki/Software
Percentage of windows applications (64 bit) will increase within the next years!
(Kube0711)

The Input- Read, Reads, Next Generation Sequencing
Scale and objective make a difference in the strategy in bacterial genomics Small Projects: 1-20 Sanger reads Medium projects: 100-200’000 Sanger reads High Throughput Projects: 10’000- 100’000’000 reads
provided by different platforms in different data formats
data pipelines and adapted
software packages�
Most of the large projects cannot be handled on Windows or Mac systems due to the demands on
software, RAM and CPUs! Linux is needed as operating system!�
(Kube0711)
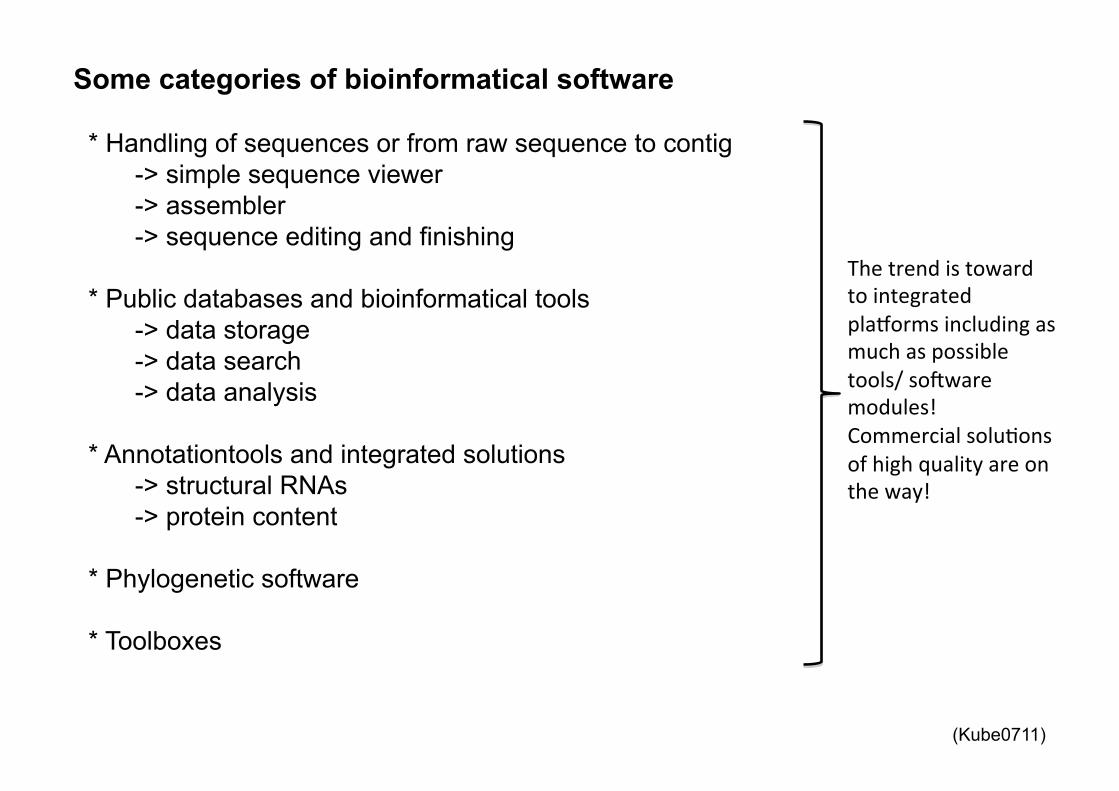
Some categories of bioinformatical software
* Handling of sequences or from raw sequence to contig -> simple sequence viewer -> assembler -> sequence editing and finishing
* Public databases and bioinformatical tools
-> data storage -> data search -> data analysis
* Annotationtools and integrated solutions
-> structural RNAs -> protein content
* Phylogenetic software * Toolboxes
����������������������������������� �������������� ����������������������������� ��������� ���������������������� �����!������������!��
(Kube0711)

PCR templates

Tools for PCR primer design Overview of the primer design software
-> in general, e.g. - http://molbiol-tools.ca/PCR.htm - http://www.science.co.il/biomedical/primer-tools.asp -> specific/ unique - http://www.ncbi.nlm.nih.gov/tools/primer-blast/
And something nice to calculate the optimal annealing temperature considering the content of the PCR mix from NEB - http://www.neb.com/nebecomm/tech_reference/TmCalc/Default.asp (Kube0711)

Inspect sequences

Sequence viewer and simple processing tools Some software assigned for this purpose: DNA Chromatogram Explorer Lite (www.dnabaser.com) FinTV (www.geospiza.com) CLC Sequence Viewer (www.clcbio.com) etc. Common windows software not assigned but can be used: MEGA5 (www.megasoftware.net) Bioedit (www.mbio.ncsu.edu/bioedit/bioedit.html) Common scientific platform including features for data processing and editing: GAP4/5 of the staden package (sourceforge.net/projects/staden) p g ( g p j )
Unprocessed data formats can be used as input, format conversion/export functions and editing functions are included. Software is limited to low scale/small projects except GAP, which allows -> sequence assembly
-> the handling of medium size projects and HT-projects in combination with other software�
Unprocessed data formats can be used as input. Data format conversion/export and editing functions are included.�
(Kube0711)

�������������������� �������������� ����� "#���������#��� ��
DNA Chromatogram Explorer Lite
��������������$���� �%��������&����'���� ��
Finc
hTV

http://www.clcbio.com/
CLC sequence viewer -> allows the import of large data sets
(Kube0711)

Processing & Assembly

Sequence assembly- some terms Sequence data Read: one DNA sequence derived from one trace chromatogram Trace: abbreviation for one trace chromatogram after gel or capillary electrophoresis Assembly Singlet: a sequence that represents a single object (-> stays alone) Contig: consists of several objects (sequences) which are connected in an alignment,
but each sequence present in a database is a contig (old definition: “A contig is a set of gel readings that are related to one another by overlap of their sequences. All gel readings belong to one and only one contig, and each contig contains at least one gel reading. The gel readings in a contig can be summed to form a contiguous consensus sequence and the length of this sequence is the length of the contig.”, by
James Bonfield) Debris: sequences too short (after trimming), megahubs, singlets (during the assembly),
low coverage alignments (threshold) Consensus sequence:
The consensus sequence is calculated from multiple sequence alignment taking in account the quality value for each base.
(Kube0711)

Some data formats/ file extensions .ab1 Raw DNA data taken from a scientific instrument and output from Applied Biosystems' Sequencing Analysis Software; encodes an electropherogram, DNA base sequence and associated tags (e.g. instrument, key, value etc.)
.scf SCF format files are used to store data from DNA sequencing instruments. Each file contains the data for a single reading (trace sample points, called sequence, positions of the bases relative to the trace sample points, numerical estimates of the accuracy of each base (same data are stored within the database format .exp).
.fasta (.fas .fa) >gi|304309652:19179-20701 Gamma proteobacterium HdN1, complete genome�AGAGTTTGATCATGGCTCAGATTGAACGCTGGCGGCAGGCCTAACACATGCAAGTCGAGCGGTAACAGGC�CTTCGGGTGCTGACGAGCGGCGGACGGGTGAGGATAGCGCAGGAATCTGCCTTGTAGTGGGGGATAGTCC�
.fastq @3926�ATAAGTTGTCATCACGCCTCTCCTTTCTGGTATGTGATCTGAGTATTGGCGATATTTTAAATATGTTTATGGAATTATCTGGATAATATGGCACCTAAACG�+3926�ceY\addcddeeedeaaeaeeed^eeceeeeeeeeddccdeacdde\eeee\ede\\ccb^dZ`d`dbcc\`T_Y__`c^ca\ccddd`\a``T_b`LTa`� Internal extensions or data formats .cons (only internal) .staden (software specific data format), .aux (“old” storage format of GAP4)�
(Kube0711)

Examples: Assembly software Phrap*: Sanger derived reads, considering read-pairs (requires high RAM and CPU resources) MIRA*: Sanger and NGS reads, considering read-pairs (requires high RAM and CPU resources), still one of the most powerful de novo assemblers CLC Assembly engine (commercial solution, free trial) Sanger and NGS reads, considering read-pairs (low requirements on RAM and CPU, fast) Newbler: Sanger and NGS reads, considering read-pairs Velvet NGS reads, considering read-pairs (low requirements on RAM and CPU) And several other (each months a new one) SSAKE, SHARCGS, VCAKE, Celera Assembler, Euler, ABySS, AllPaths, and SOAPdenovo � References performance comparison see Zhang et al., 2011; PMID: 21423806 assembly algorithms see Miller et al., 2010; PMID: 20211242) *MIRA (Mimicking Intelligent Read Assembly; Chevreux, B., Wetter, T. and Suhai, S. (1999): Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB) 99, pp. 45-56.) *Gordon, David. "Viewing and Editing Assembled Sequences Using Consed", in Current Protocols in Bioinformatics,A. D. Baxevanis and D. B. Davison, eds, New York: John Wiley & Co., 2004, 11.2.1-11.2.43.
(Kube0711)

Some assembly storage formats Becoming the default for a lot of assembly software .sam (and .bam) SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments. -> SAM is a tab-delimited text format (easy to understand, to parse, to generate, to check but slow to parse) -> store all the alignment information generated by various alignment programs -> easily generated by alignment programs or converted from existing alignment formats -> compact in file size; -> allows most of operations on the alignment to work on a stream without loading the whole alignment into memory -> allows the file to be indexed by genomic position to efficiently retrieve all reads aligning to a locus (Li et al., 2009; PMID: 19505943) BAM ⇒ BAM, the binary equivalent to SAM is used in intensive data processing.
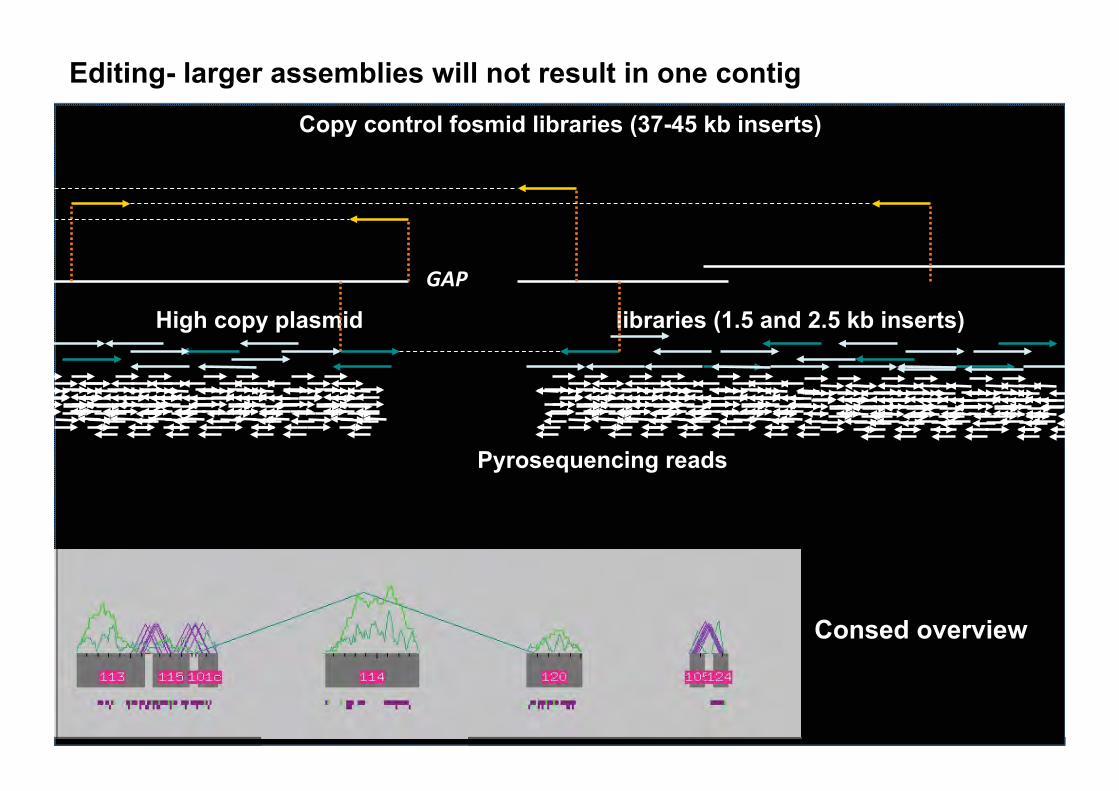
Editing- larger assemblies will not result in one contig Copy control fosmid libraries (37-45 kb inserts)
High copy plasmid libraries (1.5 and 2.5 kb inserts)
Pyrosequencing reads
���
����
Consed overview

Platforms for editing of the assembled sequence (Freeware) GAP4/5 of the staden package (http://staden.sourceforge.net/) Bonfield JK, Whitwham A. Gap5--editing the billion fragment sequence assembly. Bioinformatics. 2010 Jul 15;26(14):1699-703. PMID: ()*+,--(
Advantages: Easy to install and to handle Disadvantages: Large scale projects need external assemblers Readpair information is not visualized No autofinisher function Software environment is limited on Windows GAP5 development is in progress

Platforms for editing of the assembled sequence (Freeware) Consed (www.phrap.org/consed/consed.html)
Advantages: best software available Disadvantages: installation is a horrible needs external assemblers uses ACE format as input resulting in several limits and pitfalls no Windows version
Gordon, D., C. Abajian, and P. Green. 1998. Consed: A Graphical Tool for Sequence Finishing. Genome Research. 8:195-202 Gordon, D., C. Desmarais, and P. Green. 2001. Automated Finishing with Autofinish. Genome Research. 11(4):614-625.

Commercial solutions
Keep in mind! Commercial solutions show a reduced performance and are limited in the functions of the editor in comparison so far! This situation will hopefully change within the next years. Some examples running on windows: ⇒ DNAstar ⇒ Sequencher ⇒ CLC workbench etc.
(Kube0711)

Data analysis

Some important resources for sequence analysis NCBI-Software http://www.ncbi.nlm.nih.gov/guide/sequence-analysis/
EBI-Tools http://www.ebi.ac.uk/Tools/
emboss http://emboss.sourceforge.net/
Center for Biological Sequence Analysis (CBS) http://www.cbs.dtu.dk/index.shtml
(Kube0711)

http://blast.ncbi.nlm.nih.gov/Blast.cgi
(Kube0711)
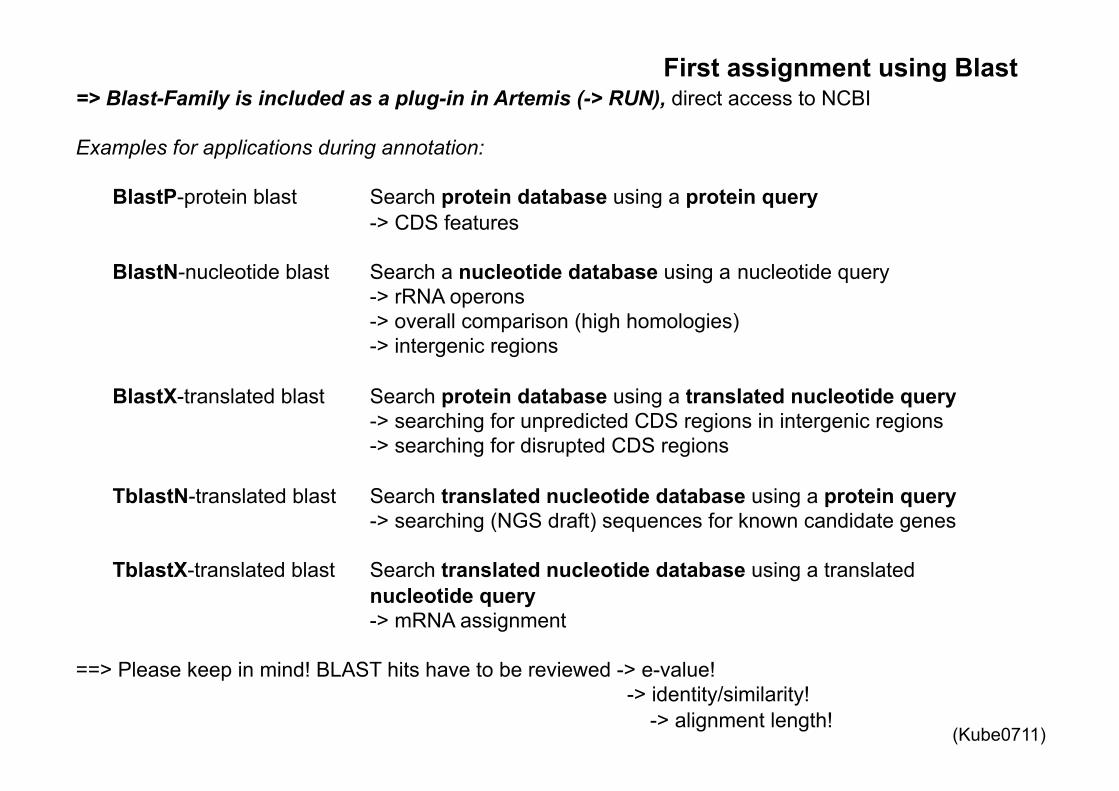
First assignment using Blast => Blast-Family is included as a plug-in in Artemis (-> RUN), direct access to NCBI Examples for applications during annotation:
BlastP-protein blast Search protein database using a protein query -> CDS features
BlastN-nucleotide blast Search a nucleotide database using a nucleotide query -> rRNA operons -> overall comparison (high homologies) -> intergenic regions BlastX-translated blast Search protein database using a translated nucleotide query -> searching for unpredicted CDS regions in intergenic regions -> searching for disrupted CDS regions
TblastN-translated blast Search translated nucleotide database using a protein query -> searching (NGS draft) sequences for known candidate genes
TblastX-translated blast Search translated nucleotide database using a translated nucleotide query -> mRNA assignment
==> Please keep in mind! BLAST hits have to be reviewed -> e-value!
-> identity/similarity! -> alignment length!
(Kube0711)

Structural RNAs- rRNA operon: predict or align
http://greengenes.lbl.gov/cgi-bin/nph-index.cgi
����������� �����'��
http://www.cbs.dtu.dk/services/RNAmmer/
(Kube0711)

Structural RNAs- tRNAs
����������������������./0����"12��
⇒ tRNAscan-SE does not perform tRNA detection itself, but instead negotiates the flow of information between three independent tRNA prediction programs, performs some post-processing and outputs the results ⇒ identifies 99-100% of transfer RNA genes in a DNA sequence (less than one false positive per 15 gigabases) ⇒ uses Sprinzl tRNA database (weak point, e.g. for mitochondrial genomes)
Specify source type (e.g. bacterial)�
(Kube0711)

Non-coding RNA (ncRNA), coding a functional RNA product ⇒ �������������������� �������3�����
��./0����������� ������� ����� ������������������ ������������������������ �4������������ ��56+4�6(4�674�6*�����6-�./0�8����������� ����5��� ����./08������
(Kube0711)
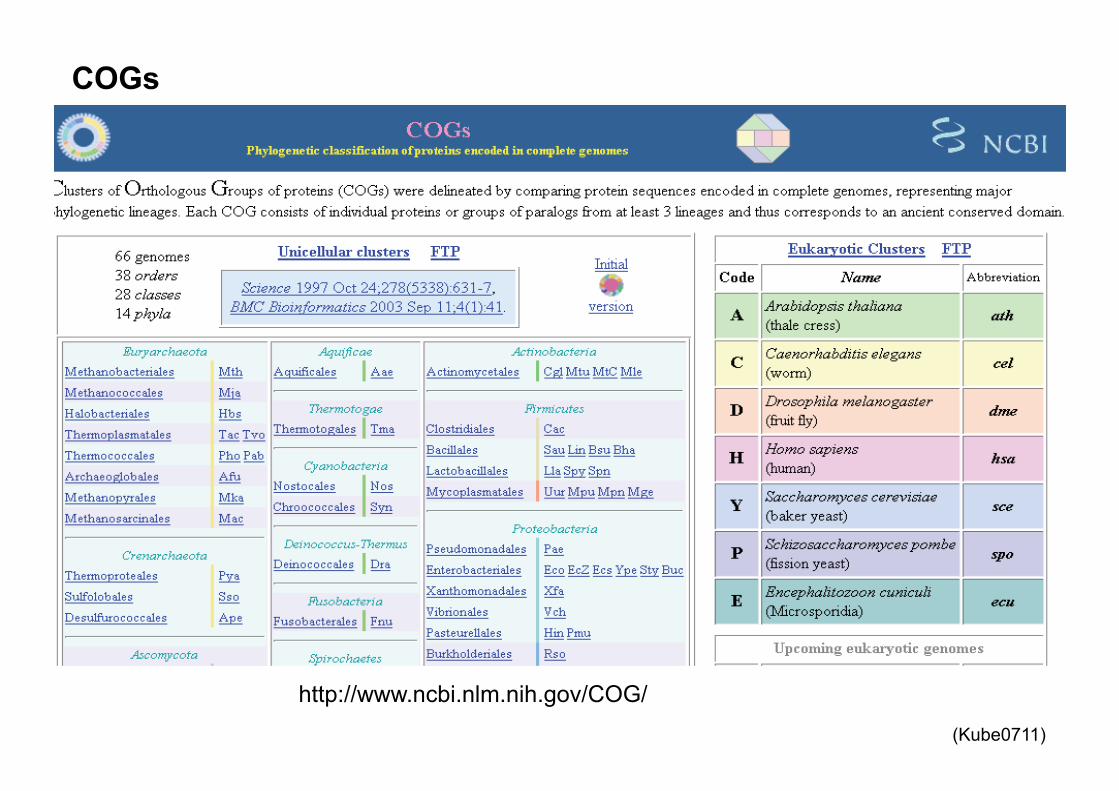
http://www.ncbi.nlm.nih.gov/COG/
COGs
(Kube0711)
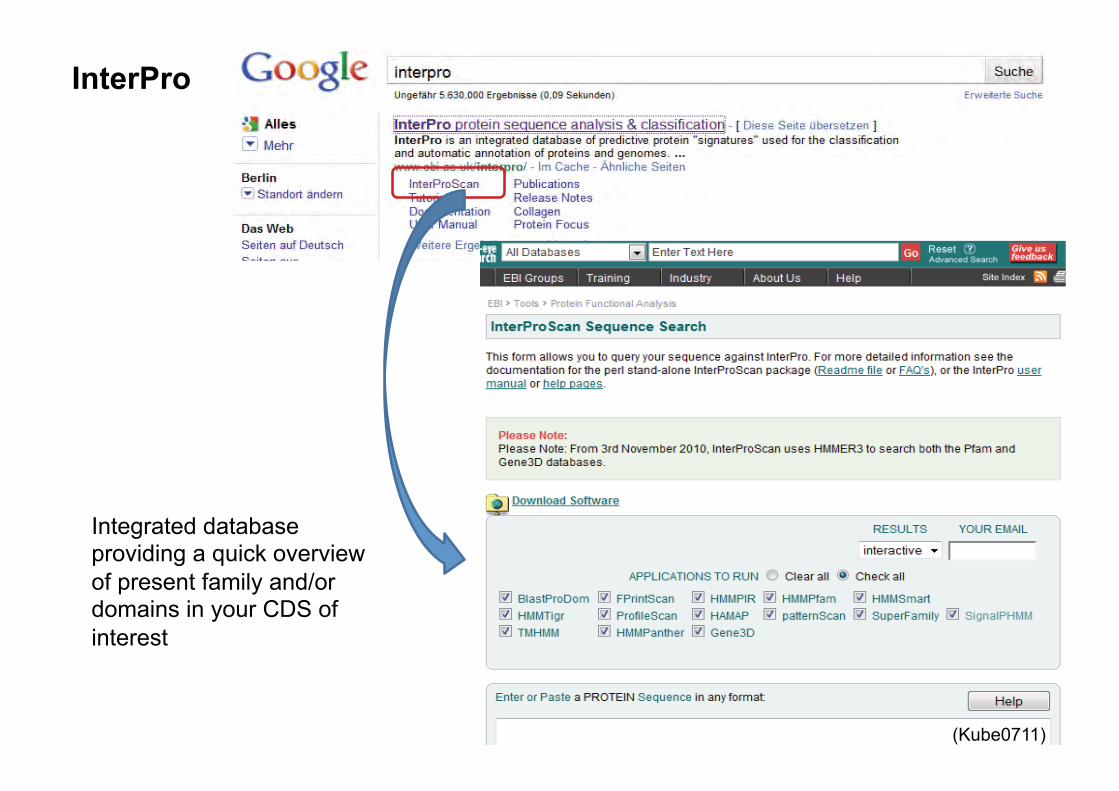
InterPro
Integrated database providing a quick overview of present family and/or domains in your CDS of interest
(Kube0711)

Functional assignment of protein coding genes using InterPro - InterPro (InterProScan, single Fasta usage) for protein domains and families, includes HMM SignalP search (use SignalP for improved search, if necessary http://www.cbs.dtu.dk/services/SignalP/) �
(Kube0711)

Efficient searches for pathways by EC no., protein name, pathway etc.
(Kube0711)

Resources for functional reconstruction: Metacyc- experimental elucidated pathways
http://metacyc.org/ ; related resources http://ecocyc.org/ (E. coli K-12 MG1655), http://biocyc.org/
(Kube0711)

�������������!���� ���9����������� ���!���� ��9���������������!���� ��
Or focused
(Kube0711)

Ove
rvie
w o
f pat
hway
s an
d fu
nctio
nal c
lass
es fo
r ge
nom
es: h
ttp://
img.
jgi.d
oe.g
ov/c
gi-b
in/p
ub/m
ain.
cgi
6������ ������� ����������������
(Kube0711)

������������������������� �������
����������� �
(Kube0711)
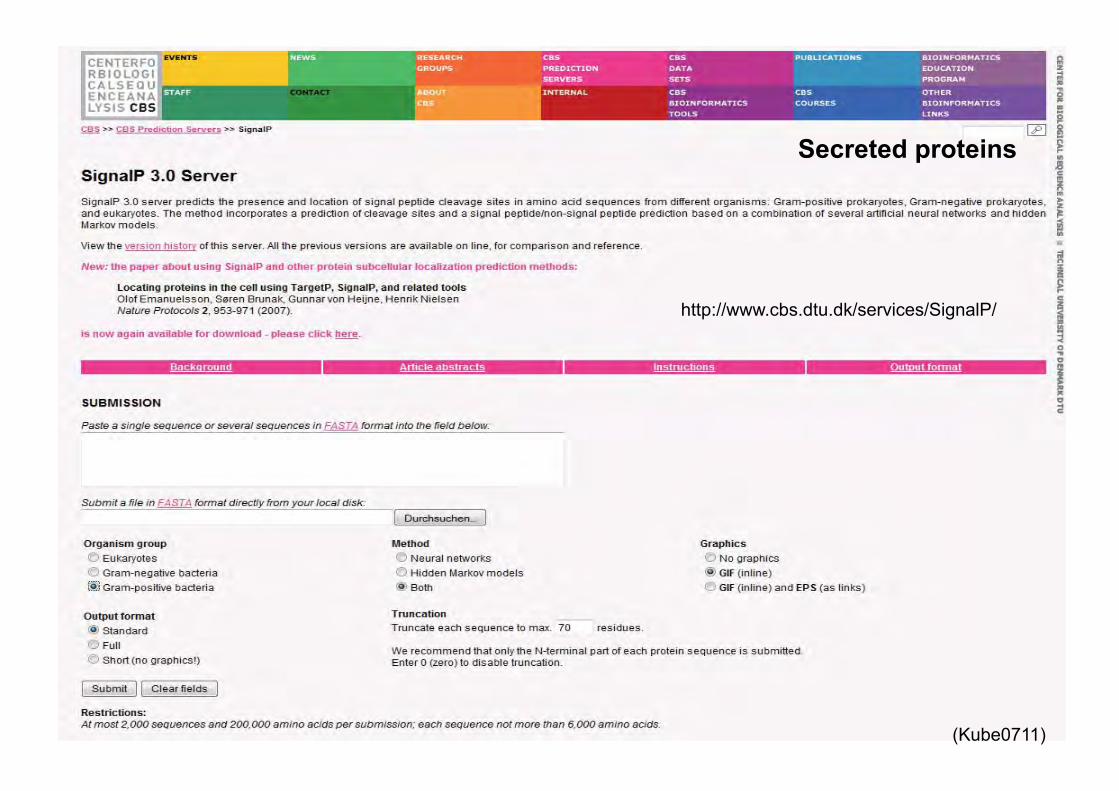
Secreted proteins
http://www.cbs.dtu.dk/services/SignalP/
(Kube0711)

Prediction of transmembrane and signal peptides
������������������������
(Kube0711)

Packages for general data analysis- EMBOSS
EMBOSS is "The European Molecular Biology Open Software Suite". EMBOSS is a free Open Source software analysis package specially developed for the needs of the molecular biology (e.g. EMBnet) user community. The software automatically copes with data in a variety of formats and even allows transparent retrieval of sequence data from the web. ⇒ Hundreds of software tools!
⇒ Usage: Specific problems e.g. pattern search.
������ ������������������

http://ugene.unipro.ru/
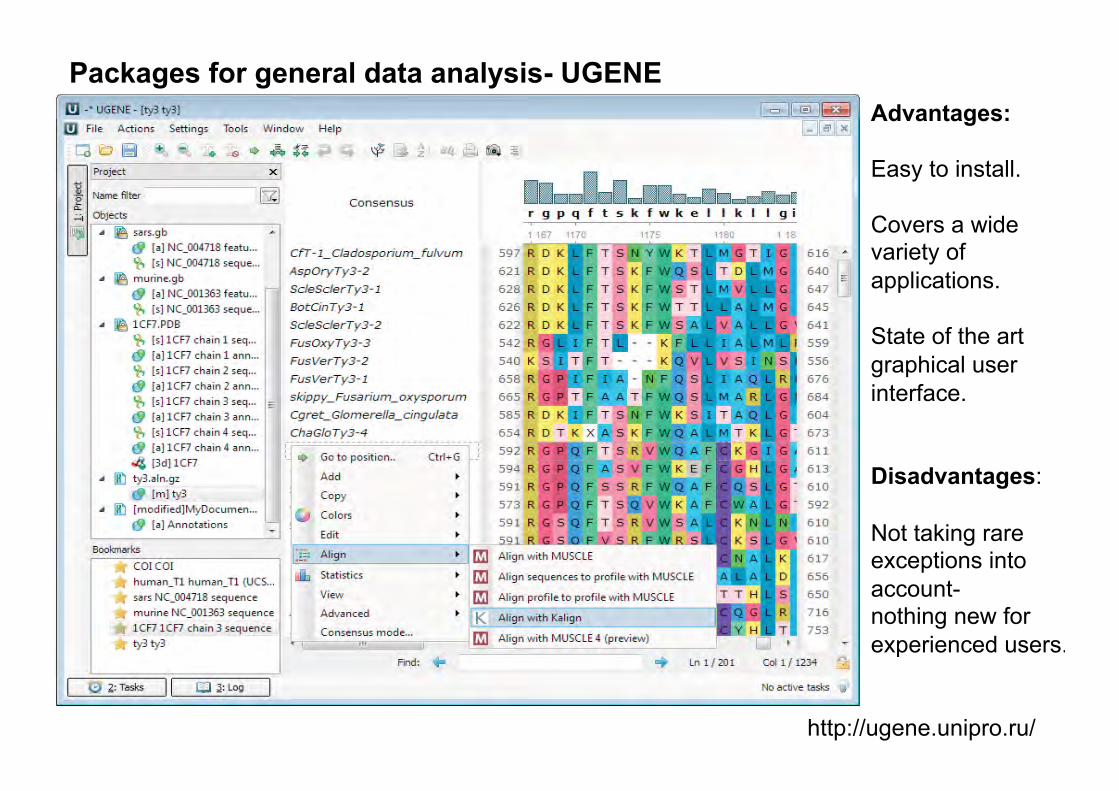
http://ugene.unipro.ru/
Packages for general data analysis- UGENE Advantages: Easy to install. Covers a wide variety of applications. State of the art graphical user interface. �Disadvantages: Not taking rare exceptions into account- nothing new for experienced users.

Artemis- Annotation platform
�������������������3�1������0�� ����
⇒ ��� ������������������������������������������!�����������"����
:.22;0.2�
(Kube0711)

Genome comparison via ACT (artemis comparison tool)
http://www.sanger.ac.uk/Software/ACT/ http://www.webact.org/WebACT/generate
- in silico fragmented genome sequence is aligned via BLAST to a reference - genome sequences and BLAST results are opened in ACT - ACT supports a graphical overview and an editor (similar to artemis) - assigned regions are illustrated by connecting lines - problems result from the BLAST approach (unspecific hits)
Full functionality by big_blast Online, limited options
(Kube0711)

������������������������� ��'��
<��'�������!�� �����=�����!������������ �������� ����� ����������������������"�����'��������!�'������������������ ���������'��������
������������ ���������������������������������������
�������������������� "��"���������3�����������5�����������������&������������������� ������������������ �������� �8����"��!��������������� �$��������������������������� "��"������ ����
����������������������3������������"�>������������������������������ ���������������� ����� ����
�������������������������������?����������������!���@<<�
Mauve
(Kube0711)
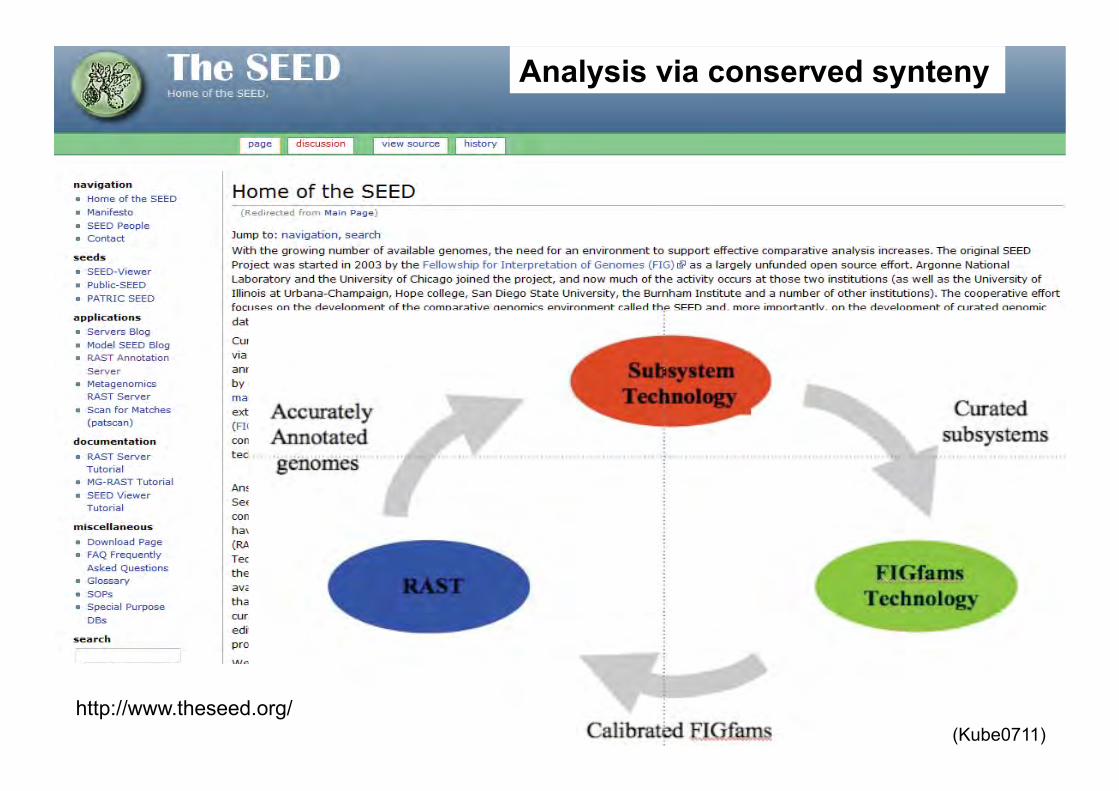
http://www.theseed.org/
Analysis via conserved synteny
(Kube0711)

Example: Searching for Genes in SEED
(Kube0711)

Example: Searching for Genes in SEED
(Kube0711)
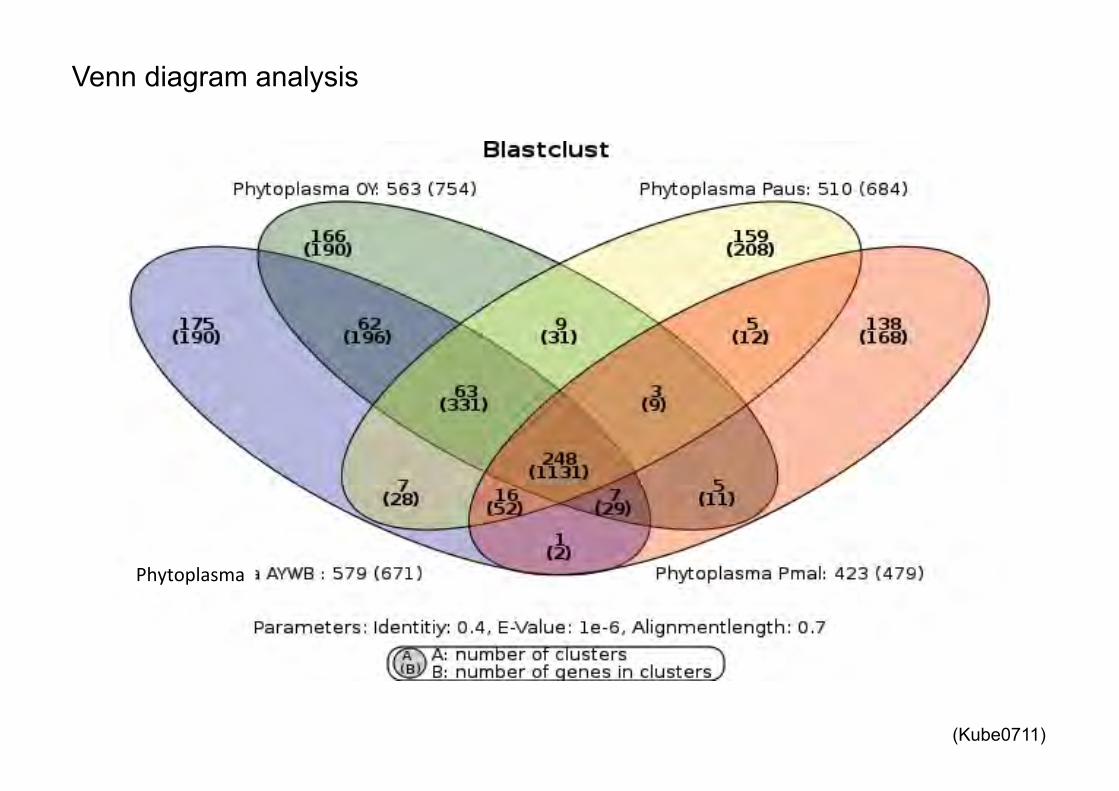
Venn diagram analysis
%�!������ ��
(Kube0711)

�������$������� ���$� ����
Calculation of the core, pan and dispensable Genome for the deduced Proteins
(Kube0711)

Phylogenetic marker

Phylogenetic markers They are used to infer a correct organismal phylogeny, using orthologous genetic loci, in which common ancestry of two sequences can be traced back to a speciation event. General requirements on the sequences - identification (and amplification) of suitable homologous part of the genomes to be compared - exclude events such as duplication or horizontal gene transfer General requirements on the provided information - homologous sites/ positions represent sites derived from the same ancestral organism - presence of informative sites provided by mutations (substitution, insertion and deletion)
=> Single copy genes encoding core functions of the organism were often used for
phylogenetic studies - most of them are under a similar selective pressure (comparable) - higher probability for conserved regions usable for the design of universal or degenerated primers and to anchor the multiple alignment
(Kube0711)

Selection of phylogenetic marker genes using protein encoding sequences- a systematic approach Strategy - selection of a taxonomic group (source data set depends on the phylogenetic question) - extract protein data from complete genome - determination of the genes/deduced proteins shared by the group - limitation of the genes/deduced proteins to single copy genetic elements - limitation to genes/deduced proteins with a minimal size (single locus or multiple locus analysis) - limitation to genes/deduced proteins with a functional description ⇒ resulting in a set of about 30 protein encoding sequences (single locus analysis) for bacteria, which are used alone or in combination with other bacteria (e.g. GroEL, Mitrovic et al., in press) ⇒ multi locus analyses sum up the phylogenetic signal of several proteins and thereby weaken the noise, which is derived from single genetic elements (e.g. Witek et. al., 2009; PMID:19654049) ==> The establishment of a “new” phylogenetic marker is a rare event today!
New means for the majority of markers just new established within a taxonomic group.
(Kube0711)

The next step- Phylogenomics Strategy - based on (whole) genome content - two major strategies (combination possible)
* sequence-based methods -> based on the combination/sum of the phylogenetic signal of shared genes/proteins (information dominated by InDels, e.g. supertree)
*based on whole-genome features -> methods based on gene order -> methods based on gene content �
Objectives - robust phylogeny up to phylum level - application and results depend on data available for analysis References: Delsuc et al., 2005 (PMID: 15861208)
(Kube0711)

Thanks!
Michael Kube
MPI for Molecular Genetics & Humboldt University, Berlin (Germany)


















