
Formation of ilmenite-type CoTiO3 on TiO2 and its performance in oxidative dehydrogenation of...
6
Short Communication Formation of ilmenite-type CoTiO 3 on TiO 2 and its performance in oxidative dehydrogenation of cyclohexane with molecular oxygen Bipul Sarkar a , Chandrashekar Pendem a , L.N. Sivakumar Konathala a , Takehiko Sasaki b , Rajaram Bal a, ⁎ a Catalytic Conversion & Processes Division, CSIR-Indian Institute of Petroleum, Dehradun 248005, India b Department of Complexity Science and Engineering, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwanoha, Kashiwa-shi, Chiba 277-8561, Japan abstract article info Article history: Received 20 May 2014 Received in revised form 18 June 2014 Accepted 19 June 2014 Available online 27 June 2014 Keywords: Ilmenite-type CoTiO 3 Oxidative dehydrogenation Anatase TiO 2 CoTiO 3 catalyst Cyclohexane to benzene Ilmenite-type CoTiO 3 supported on TiO 2 was prepared and their catalytic activities were tested for oxidative dehydrogenation of cyclohexane to produce benzene. The catalyst was characterized by XRD, FE-SEM, HR- TEM, XPS, TPR and XANES. XRD reveals the formation of ilmenite-type CoTiO 3 on TiO 2 . The catalyst showed 87.8% cyclohexane conversion with 89.2% benzene selectivity at 450 °C in a continuous fixed bed down flow reactor at atmospheric pressure. The influence of different reaction parameters like temperature, gas hour space velocity (GHSV), Co loading was studied in detail. © 2014 Elsevier B.V. All rights reserved. 1. Introduction Dehydrogenation of hydrocarbons is an important commercial conversion process because of the great demand for dehydrogenated hydrocarbons, required to manufacture various chemical products such as detergents, high octane motor fuels, pharmaceutical products, plastics, and synthetic rubbers [1]. Direct dehydrogenation of alkane to alkene is an endothermic reaction which required relatively high temperature to obtain high yield. However, high reaction temperature favours high thermal cracking to lower alkane and coke formation, resulting a decrease in product yield and rapid catalyst deactivation [2]. Therefore to overcome those limitations, one approach is the introduction of molecular oxygen in the dehydrogenation reaction. The introduction of oxygen, effectively oxidized the hydrogen and helps to shift the equilibrium to obtain the desired product range at a lower temperature [3,4]. Hence, the development of more suitable oxidative dehydrogenation (ODH) catalyst is the challenge for the hydrocarbon industry [1]. Industrially cyclohexane synthesis is based on hydrogenation of benzene and finally this largely produced cyclo- hexane is then converted stepwise to adipic acid, a common intermedi- ate for the production of nylon. However, it is unusual to consider dehydrogenation of cyclohexane to benzene which could adapt by industry. Worldwide production of benzene reaches 42 million tonnes per annum and predicted to grow further in coming years [5]. During the large-scale production of benzene via catalytic cracking of straight chain fossil hydrocarbons and subsequent purification of benzene from the ‘heart-cut’ tower [6], significant quantity of impurity persists [7,8]. Common impurities found in benzene are water, cyclohexane, cyclohexene, toluene, methylcyclohexane, etc. Removal of those impurities requires high temperature, so it should be beneficial to suppress the extra energy so that the production cost will reduce. Hence the catalytic low temperature oxidative dehydrogenation of cyclohexane to benzene can provide the solution. However, the catalysts for oxidative dehydrogenation of cyclohex- ane are very limited; Fe/Pt/group IIIA metal catalyst has been reported for ODH reaction [9,10]. Pt based catalyst has also been used for dehy- drogenation of cyclohexane [11,12], but rapid gas phase dissociation of benzene on Pt(III) forms unwanted bi-product. The probable mecha- nistic pathway has also been investigated by Koel et al.; according to their study, the dehydrogenation of cyclohexane been predicted to follow the sequential dehydrogenation mechanism [13] with the forma- tion of metastable η 3 -C 6 H 9 as allylic intermediate. The oxidative dehydrogenation (ODH) of cyclohexane has also been reported with vanadate [14], zeolites [15,16], supported and unsupported molybdena [17] based catalyst. Kunugi et al. found that transition metals like Fe, Mo, and Co supported over molecular sieve catalysed oxidative dehydrogenation of cyclohexane to ben- zene with good conversion and selectivity, even at lower oxygen Catalysis Communications 56 (2014) 5–10 ⁎ Corresponding author. Tel.: +91 135 2525797; fax: +91 135 2660202. E-mail address: [email protected] (R. Bal). http://dx.doi.org/10.1016/j.catcom.2014.06.021 1566-7367/© 2014 Elsevier B.V. All rights reserved. Contents lists available at ScienceDirect Catalysis Communications journal homepage: www.elsevier.com/locate/catcom
Transcript of Formation of ilmenite-type CoTiO3 on TiO2 and its performance in oxidative dehydrogenation of...

Catalysis Communications 56 (2014) 5–10
Contents lists available at ScienceDirect
Catalysis Communications
j ourna l homepage: www.e lsev ie r .com/ locate /catcom
Short Communication
Formation of ilmenite-type CoTiO3 on TiO2 and its performance inoxidative dehydrogenation of cyclohexane with molecular oxygen
Bipul Sarkar a, Chandrashekar Pendem a, L.N. Sivakumar Konathala a, Takehiko Sasaki b, Rajaram Bal a,⁎a Catalytic Conversion & Processes Division, CSIR-Indian Institute of Petroleum, Dehradun 248005, Indiab Department of Complexity Science and Engineering, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwanoha, Kashiwa-shi, Chiba 277-8561, Japan
⁎ Corresponding author. Tel.: +91 135 2525797; fax: +E-mail address: [email protected] (R. Bal).
http://dx.doi.org/10.1016/j.catcom.2014.06.0211566-7367/© 2014 Elsevier B.V. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 20 May 2014Received in revised form 18 June 2014Accepted 19 June 2014Available online 27 June 2014
Keywords:Ilmenite-type CoTiO3
Oxidative dehydrogenationAnatase TiO2
CoTiO3 catalystCyclohexane to benzene
Ilmenite-type CoTiO3 supported on TiO2 was prepared and their catalytic activities were tested for oxidativedehydrogenation of cyclohexane to produce benzene. The catalyst was characterized by XRD, FE-SEM, HR-TEM, XPS, TPR and XANES. XRD reveals the formation of ilmenite-type CoTiO3 on TiO2. The catalyst showed87.8% cyclohexane conversion with 89.2% benzene selectivity at 450 °C in a continuous fixed bed down flowreactor at atmospheric pressure. The influence of different reaction parameters like temperature, gas hourspace velocity (GHSV), Co loading was studied in detail.
© 2014 Elsevier B.V. All rights reserved.
1. Introduction
Dehydrogenation of hydrocarbons is an important commercialconversion process because of the great demand for dehydrogenatedhydrocarbons, required to manufacture various chemical productssuch as detergents, high octane motor fuels, pharmaceutical products,plastics, and synthetic rubbers [1]. Direct dehydrogenation of alkaneto alkene is an endothermic reaction which required relatively hightemperature to obtain high yield. However, high reaction temperaturefavours high thermal cracking to lower alkane and coke formation,resulting a decrease in product yield and rapid catalyst deactivation[2]. Therefore to overcome those limitations, one approach is theintroduction of molecular oxygen in the dehydrogenation reaction.The introduction of oxygen, effectively oxidized the hydrogen andhelps to shift the equilibrium to obtain the desired product range at alower temperature [3,4]. Hence, the development of more suitableoxidative dehydrogenation (ODH) catalyst is the challenge for thehydrocarbon industry [1]. Industrially cyclohexane synthesis is basedon hydrogenation of benzene and finally this largely produced cyclo-hexane is then converted stepwise to adipic acid, a common intermedi-ate for the production of nylon. However, it is unusual to consider
91 135 2660202.
dehydrogenation of cyclohexane to benzene which could adapt byindustry. Worldwide production of benzene reaches 42 million tonnesper annum and predicted to grow further in coming years [5]. Duringthe large-scale production of benzene via catalytic cracking of straightchain fossil hydrocarbons and subsequent purification of benzenefrom the ‘heart-cut’ tower [6], significant quantity of impurity persists[7,8]. Common impurities found in benzene are water, cyclohexane,cyclohexene, toluene, methylcyclohexane, etc. Removal of thoseimpurities requires high temperature, so it should be beneficial tosuppress the extra energy so that the production cost will reduce.Hence the catalytic low temperature oxidative dehydrogenation ofcyclohexane to benzene can provide the solution.
However, the catalysts for oxidative dehydrogenation of cyclohex-ane are very limited; Fe/Pt/group IIIA metal catalyst has been reportedfor ODH reaction [9,10]. Pt based catalyst has also been used for dehy-drogenation of cyclohexane [11,12], but rapid gas phase dissociationof benzene on Pt(III) forms unwanted bi-product. The probable mecha-nistic pathway has also been investigated by Koel et al.; according totheir study, the dehydrogenation of cyclohexane been predicted tofollow the sequential dehydrogenationmechanism [13]with the forma-tion of metastable η3C6H9 as allylic intermediate.
The oxidative dehydrogenation (ODH) of cyclohexane has alsobeen reported with vanadate [14], zeolites [15,16], supported andunsupported molybdena [17] based catalyst. Kunugi et al. foundthat transition metals like Fe, Mo, and Co supported over molecularsieve catalysed oxidative dehydrogenation of cyclohexane to ben-zene with good conversion and selectivity, even at lower oxygen

Fig. 1. (A) XRD pattern of the as-prepared pure TiO2 (B) 2.5%Co–TiO2 and (C) 5%Co–TiO2.
6 B. Sarkar et al. / Catalysis Communications 56 (2014) 5–10
pressure [18]. The ODH of cyclohexane over Ru and Ru–Sn catalystwas studied [19] and found that 16.5% cyclohexane was achieved at300 °C. Tyo et al. reported ODH of cyclohexane over Co3O4 andfound that the selectivity toward benzene is dependent on the sizeof Co-oxide particle and operating temperature [20]. It is also report-ed in the literature that larger particles favourably produce cyclohexenewhereas particle with smaller size predominantly gives benzene athigher temperatures [20]. Nano-octahedra and nanocubes of FeOx andAu/Fe3O4 nanocrystals with a cubic inverse spinel structure are activecatalysts for the oxidative dehydrogenation of cyclohexane at 200 °C[21].
In this work, we report the preparation of ilmenite-type CoTiO3 onanatase TiO2 and we found that the catalyst shows 87.8% cyclohexaneconversion with 89.2% benzene selectivity at 450 °C.
2. Experimental
2.1. Materials
Titanium(IV) isopropoxide (Sigma-Aldrich Co.) was used as thesource of titanium. The following analytical grade chemicals wereused without further purification. Poly(ethylene glycol) (average Mn380–420) and cobalt(II) nitrate hexahydrate were purchased fromSigma-Aldrich Co. Ethanol and cetyltrimethylammonium bromide(CTAB) were purchased from Merck KGaA, Darmstadt, Germany.Double distilled water was prepared with a BOROSIL® water distilla-tion unit. Commercial TiO2 and CoO were purchased from Sigma-Aldrich Co.
2.2. Synthesis of Co supported catalyst
In the typical preparation method, 10 g (35.2 mmol) titanium(IV)isopropoxide was dissolved in 150 ml distilled water containing 10 mlpoly(ethylene glycol) with vigorous stirring to get a homogeneoussolution and finally the resultant mixture was autoclaved in a stainlessvessel at 180 °C for 24 h for crystallization. The white precipitate wascollected by filtration, washed thoroughly with water and ethanol anddried overnight. Thematerialwas calcined at 650 °C in air to get anataseTiO2.
Co-supported catalystwas prepared as follows: 0.247 g (0.84 mmol)Co(NO3)2·H2O was dissolved in 20 ml ethanol. Equivalent amount ofcetyltrimethylammonium bromide (CTAB) was added at 50 °C andstirred for 30 min to get a homogeneous solution. Finally, 1 g preparedTiO2 was added to the resultant mixture and kept at 50 °C for around2 h. The resultant gel was dried overnight at 100 °C in an oven andcalcined at 550 °C for 3 h in air. The prepared catalyst is denoted as(Co wt.%)Co–TiO2. For comparison, Co–TiO2 catalyst was also preparedby conventional impregnation and co-precipitation method usingcommercial TiO2 (~21 nm particle size) nano-powder.
2.3. Oxidative dehydrogenation of cyclohexane
The oxidative dehydrogenation of cyclohexane was carried out in afixed-bed down flow reactor at atmospheric pressure. Typically,200 mg of catalyst was placed in between two quartz wool plugged inthe centre of the 6mmquartz reactor and dehydrogenation of cyclohex-ane was carried out in a temperature range of 300–500 °C. The gashourly space velocity (GHSV) was varied between 5000 ml g−1 h−1 to30,000ml g−1 h−1with amolar ratio of C6H12:O2:Heof 1:1:8. Cyclohex-ane was introduced to the reactor system by bubbling helium, whereasbubbler was heated by circulating hot water through the outer jacket ofthe bubbler. Gas lines from the outlet of the reactor were heated with aheating tape in order to avoid any condensation in between the lines.The reaction products were analysed using an online gas chromatogra-phy (Agilent 7890A)fittedwith a FID& TCDdetector using two differentcolumns HP Pona (for analysing C6H12, C6H6 etc.) and PoraPack-Q (for
analysingO2 and CO2). Cyclohexane conversions and product selectivitywere calculated as:
Conversion %ð Þ ¼ Moles of cyclohexane reactedð ÞMoles of cyclohexane in feedð Þ x 100
Selectivity %ð Þ ¼ Moles of product definedð ÞMoles of cyclohexane reactedð Þ � 100:
2.4. Characterization techniques
X-ray diffraction (XRD) of TiO2 and Co-TiO2 catalyst was performedwith a Rigaku-Geigerflex diffractometer using Cu Kα radiation (λ =0.15406 nm), and the data was collected in the 2θ range of 2°–60° at astep size of 0.02°. The microstructures of the catalyst surfaces of thesamples were observed using scanning electron microscopy-energydispersive X-ray spectroscopy (SEM-EDS) (FEI Quanta 200 F), fieldemission scanning electron microscopy (FE-SEM) (FEI Quanta 200 F),and transmission electron microscopy (TEM) (JEM-2100, JEOL). Sam-ples were prepared by mounting an ethanol-dispersed sample on alacey carbon Formvar coated Cu grid with an accelerating voltage of200 kV. XANES spectra of the Co–TiO2 catalyst were carried out at thephoton factory in the High Energy Accelerator Research Organization(KEK-IMMS-PF). The measurement was made at transition mode andspectra were taken at BL-7C and BL-9C. The electron storage ring wasoperated at 2.5GeV and450mA, synchrotron radiation from the storagering was monochromatized by a Si (111) channel cut crystal. Ionizedchamber, which were used as detectors for incident X-ray (Io)and transmitted X-ray (I), were filled with Ar (15%)–N2 mixture gasand Ar (100%) respectively. Temperature Programmed Reduction(TPR) were carried out in a Micromeritics, Auto Chem II 2920 instru-ment in the temperature range of 20°–1000 °C with a heating rate of10 °C/min using helium as the carrier gas. The elemental analysis ofthe samples was carried out by X-ray photoelectron spectroscopy(XPS) (K-Alpha, Thermo Scientific Corp.). The monochromatized X-rayMg Kα radiation (1253.6 eV) was used. The core levels were calibratedby reference to the first component of the C 1s core level peak(unfunctionalized hydrocarbons) set at 284.6 eV.
3. Results and discussion
3.1. Catalyst characterization
Fig. 1 shows the XRD diffraction patterns of the pure TiO2 and Cosupported TiO2 catalysts. From the XRD pattern (after the reduction at

Fig. 3. Photoelectron spectra of Co 2p prepared by simple impregnationmethod; (A) freshand (B) spent catalyst.
7B. Sarkar et al. / Catalysis Communications 56 (2014) 5–10
550 °C) it is found that all the catalyst consist of anatase TiO2 (JCPDSCard No-89-4921) with the XRD peaks at 2θ values of 25.3° (101) and48.1° (200) which are the characteristic peaks of anatase TiO2. We alsonoticed that when loading was less than 5 wt.%, peaks for Co2O3 at37.2° was also appeared in the catalyst. The peaks at 32.8°, 35.5° and40.6° indicate the formation of ilmenite-type CoTiO3 oxide (JCPDSCard No-77-0153) in the 5%Co–TiO2 catalyst. The representative SEMimages of the catalyst are shown in Fig. 2A and S1 (Supplementarycontent). Themorphologies of the prepared catalyst are totally differentfrom that of the catalyst prepared by conventional impregnation meth-od (Fig. S2, Supplementary content). The presence of Co was confirmedby energy dispersive X-ray analysis (EDX) (Fig. S3, Supplementary con-tent). The elemental mapping of the catalyst reveals a homogeneousdistribution of Co on the surface (Fig. S1, Supplementary content). Therespective TEM images of 5%Co–TiO2 been shown in Fig. 2B–D. Where,from the TEM images, it is clear that the size of the 5%Co–TiO2 particleis around 50–70 nm. The lattice fringes with a D-spacing for 3.8 Å and2.7 Å correspond to the (110) and (121) planes of CoTiO3 which werealso observed and shown in Fig. 2C–D and Fig. S4 (Supplementarycontent). As we increase the Co loading form 5 to 10%; the size of theCoTiO3 particle increases (80–100 nm) compare to 5%Co–TiO2 catalyst(Fig. S5; Supplementary content).
The Ti 2p XPS spectra of 5%Co–TiO2 (fresh and spent) are shown inFig. S6 (Supplementary content). From the deconvoluted XPS spectrathe Ti 2p3/2 binding energy peak (BE) at 458.4 eV was obtained alongwith its satellite peak at BE of 464.4 eV (2p3/2) for both fresh andspent catalyst which confirm the presence of Ti2+ [22,23]. Fig. 3shows the XPS binding energy (BE) of Co 2p3/2 in the fresh and spentcatalyst, where A and B represent the spectra (in deconvoluted form)of the fresh and spent catalyst respectively. The Co 2p3/2 binding energy
Fig. 2. SEM and TEM images of 5%Co–TiO2 catalyst; (A) is the SEM im
in the XPS spectra for the fresh and spent catalyst were 780.0 eV and780.3 eV respectively, which shows the presence of Co2+ in the surface[22]. XPS spectra also indicate that the oxidation state Co did not changeduring the oxidation process.We believe that the Co2+ of CoTiO3 acts asthe active species in the oxidative dehydrogenation of cyclohexane tobenzene in the presence of molecular oxygen. The reducibility and theinteraction between the active phases with the support TiO2 were stud-ied by conducting temperature programed reduction (TPR) experimentand it is shown in Fig. S7. As the TiO2 has a very high, negative free
age of the catalyst and (B–D) is the TEM image of the catalyst.

Table 1Catalytic performance of various nano-composite catalysts on the selective dehydrogena-tion of benzene.
Sl no. Catalyst Cyclohexaneconversione
Benzeneselectivityf
1 TiO2 21.9 55.22 TiO2
a 9.7 23.83 5%Co–TiO2
b 32.5 56.14 5%Co–TiO2 87.8 89.25 5%Co–TiO2
c 1.3 62.16 5%Co–TiO2
d 10.5 93.37 Mg–V–O 19.6 42.88 Nanolith 6.3 54.7
Catalyst: 0.2 g, cyclohexane to oxygen: 1:1, reaction time: 1 h.a Commercial TiO2.b CoTiO3 on commercial TiO2.c Prepared via in situ hydrothermal method.d Reaction carried out without O2.e Conversion and;f Selectivity are in mole %.
8 B. Sarkar et al. / Catalysis Communications 56 (2014) 5–10
energy of formation, it is very hard to reduce TiO2 below 1000 °C [24]hence no peakwas observed for the support TiO2. However two distinctpeaks were observed between 200 °C to 500 °C for both 5%Co and10%Co loaded TiO2 catalyst. The first peak is attributes to the reductionof Co2+ to Coδ+ and the 2nd peak (shouldered with the first peak)was attributes to Coδ+ to metallic Co, respectively for 5%Co–TiO2 [25,26]. It is well known that for Co supported on TiO2, higher temperatureis required for the reduction of larger cobalt particle compared to thesmaller particles [27]. We also observed that for 10%Co–TiO2 catalyst,the particle size of CoTiO2 is bigger compared to that of 5%Co–TiO2 cat-alyst (Fig. S5; Supplementary content).Which is clearly visualized in thecase of 5%Co loaded catalyst where the reduction peaks (at 360 °C and403 °C) appears earlier than that of the 10% Co loaded catalyst (at390 °C and 435 °C). Co K-edge XANES spectra for the 5%Co-TiO2 catalystwere recorded and shown in Fig. 4. A high degree of similarity wasfound between previously reported Co-edge XANES for CoTiO3 andour prepared 5%Co-TiO2 catalyst [28]. A strong absorption band around7725 eV was observed in XANES spectra for 5%Co–TiO2 (for fresh andused catalyst) which corresponds to CoTiO3 present in the catalyst. Itstrongly indicates the formation of (+2) oxidation state of Co in the cat-alyst which very strongly resembles that of CoTiO3, but is significantlydifferent from that of CoO [29]. A strong absorption band at around7730 eV was also found in the as-synthesised material, which ensuredthe presence of Co3O4 in the pre-calcined material [30].
3.2. Catalytic activity
The catalytic performance of the various Co catalysts for the oxida-tive dehydrogenation of cyclohexane is summarized in Table 1. Titaniaprepared by hydrothermal method shows 21.9% cyclohexane conver-sion with 55.2% benzene selectivity (entry 1). Compare to that, com-mercially available TiO2 showed only 9.7% conversion with 23.8%benzene selectivity. These results clearly indicate that both CoTiO3
phase and nanocrystalline TiO2 is necessary for the high conversionand selectivity. Co loaded on commercial TiO2 nanopowder shows32.5% cyclohexane conversion with 56.1% benzene selectivity (entry3). Whereas, as prepared 5%Co–TiO2 shows high cyclohexane conver-sion of 87.8% with very high (89.2%) benzene selectivity (entry 4);in the absence of O2, the catalyst shows 10.5% cyclohexane conversionalthough the selectivity of benzene reaches a maxima of 93.3% (entry6). But the catalyst suffers heavy coke decomposition which causesrapid deactivation with time-on-stream. The same catalyst prepared
Fig. 4. Plots of time-resolved XANES for (A) uncalcined material (B) fresh and (C) spentcatalyst.
via in situ hydrothermal method (entry 5) shows a negligible conver-sion of 1.3%; which probably because of the encapsulation of activemetal particle in to the bulk TiO2. The activity of our 5%Co–TiO2 catalystis very high compared to the Mg–V–O and nanolith catalysts (entries 7,8) reported in the literature [31,32] as listed in the table. The effect ofsupport material is also been investigated and the results are shown inFig. S8. It is observed, that the basic or amphoteric support enhancedthe reaction better than the acidic support. With TiO2 support, 87.8%cyclohexane conversion achieved whereas, with more acidic Al2O3
only 1.6% cyclohexane conversion was found. The selectivity towardbenzene is also found to increase with basic supports. A steady increasein cyclohexane conversion was noted as the temperature increases(Fig. 5A) from 300 °C to 500 °C. However, benzene selectivity wasfound to rich at maxima 93.3% at around 400 °C and then drops downto 68.7% at 500 °C; as the rapid oxidation of benzene to COx dominatesafter 450 °C.
3.3. Effect of Co loading
Wehave examined the effect of Co loading as a function of cyclohex-ane conversion and benzene selectivity; the results are shown in Fig. 5B.It was observed that cyclohexane conversion increases and benzeneselectivity decreases with Co loading; when the Co loading was 2.5%the cyclohexane conversion was 65.1% with 89.9% benzene selectivity.The increase in Co loading from 2.5 to 15%; the cyclohexane conversionincreases from 65.1% to 91.7% and beyond 10% Co loading the conver-sion remain almost constant. With 5%Co loading a cyclohexane conver-sion of 87.8% with benzene selectivity of 89.2% was observed. Thecatalyst shows 91.2% cyclohexane conversion with 57% benzene selec-tivity when Co loading was 10%. We assumed that with the increase inCo loading formation of cobalt oxide (Co2O3) (as detected by XRD)dominates rather than CoTiO3 formation, which decreases the benzeneselectivity.
3.4. Influence of gas hour space velocity (GHSV)
The effects of gas hour space velocity (GHSV) were studied and theresults are shown in Fig. 5C. It was found that with the increase inGHSV the conversion decreases rapidly, whereas, the benzene selectiv-ity increases very slowly.
3.5. Influence of time on steam
The influence of time on steamwas investigated and found that theactivity of the 5%Co–TiO2 catalyst remains constant for 20 h (Fig. 5D).Based on the result it can be conclude that electron deficient CoTiO3
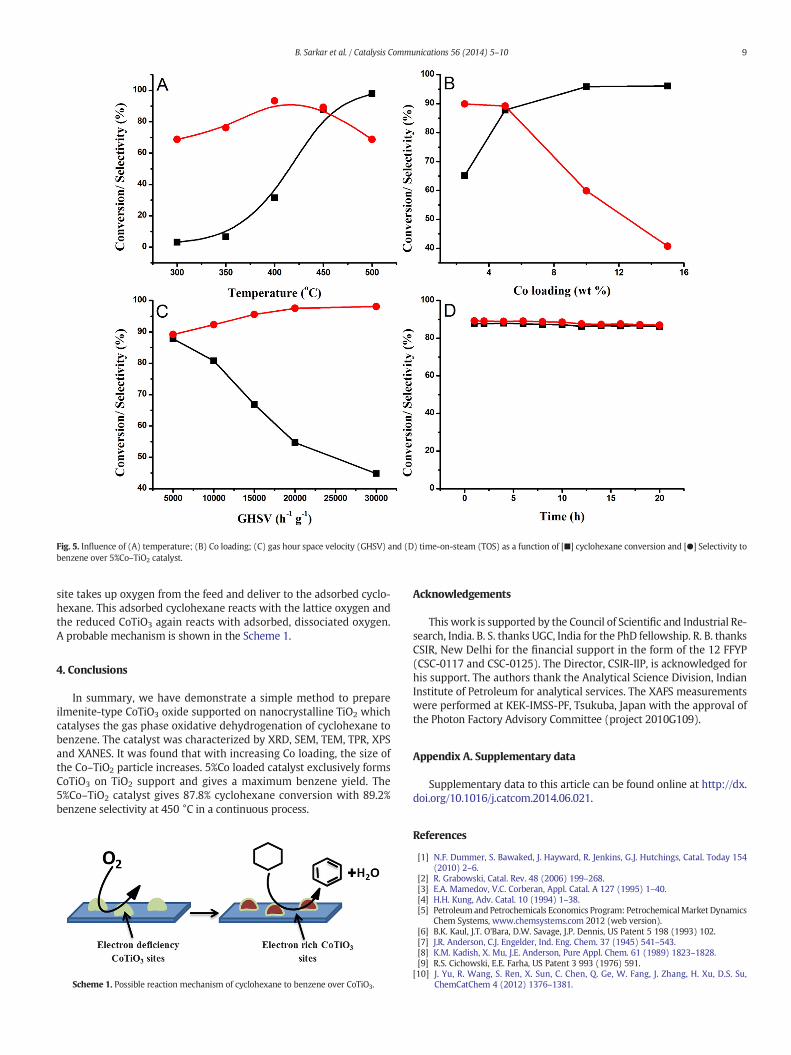
Fig. 5. Influence of (A) temperature; (B) Co loading; (C) gas hour space velocity (GHSV) and (D) time-on-steam (TOS) as a function of [■] cyclohexane conversion and [●] Selectivity tobenzene over 5%Co–TiO2 catalyst.
9B. Sarkar et al. / Catalysis Communications 56 (2014) 5–10
site takes up oxygen from the feed and deliver to the adsorbed cyclo-hexane. This adsorbed cyclohexane reacts with the lattice oxygen andthe reduced CoTiO3 again reacts with adsorbed, dissociated oxygen.A probable mechanism is shown in the Scheme 1.
4. Conclusions
In summary, we have demonstrate a simple method to prepareilmenite-type CoTiO3 oxide supported on nanocrystalline TiO2 whichcatalyses the gas phase oxidative dehydrogenation of cyclohexane tobenzene. The catalyst was characterized by XRD, SEM, TEM, TPR, XPSand XANES. It was found that with increasing Co loading, the size ofthe Co–TiO2 particle increases. 5%Co loaded catalyst exclusively formsCoTiO3 on TiO2 support and gives a maximum benzene yield. The5%Co–TiO2 catalyst gives 87.8% cyclohexane conversion with 89.2%benzene selectivity at 450 °C in a continuous process.
Scheme 1. Possible reaction mechanism of cyclohexane to benzene over CoTiO3.
Acknowledgements
This work is supported by the Council of Scientific and Industrial Re-search, India. B. S. thanks UGC, India for the PhD fellowship. R. B. thanksCSIR, New Delhi for the financial support in the form of the 12 FFYP(CSC-0117 and CSC-0125). The Director, CSIR-IIP, is acknowledged forhis support. The authors thank the Analytical Science Division, IndianInstitute of Petroleum for analytical services. The XAFS measurementswere performed at KEK-IMSS-PF, Tsukuba, Japan with the approval ofthe Photon Factory Advisory Committee (project 2010G109).
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.catcom.2014.06.021.
References
[1] N.F. Dummer, S. Bawaked, J. Hayward, R. Jenkins, G.J. Hutchings, Catal. Today 154(2010) 2–6.
[2] R. Grabowski, Catal. Rev. 48 (2006) 199–268.[3] E.A. Mamedov, V.C. Corberan, Appl. Catal. A 127 (1995) 1–40.[4] H.H. Kung, Adv. Catal. 10 (1994) 1–38.[5] Petroleumand Petrochemicals Economics Program: PetrochemicalMarket Dynamics
Chem Systems, www.chemsystems.com 2012 (web version).[6] B.K. Kaul, J.T. O'Bara, D.W. Savage, J.P. Dennis, US Patent 5 198 (1993) 102.[7] J.R. Anderson, C.J. Engelder, Ind. Eng. Chem. 37 (1945) 541–543.[8] K.M. Kadish, X. Mu, J.E. Anderson, Pure Appl. Chem. 61 (1989) 1823–1828.[9] R.S. Cichowski, E.E. Farha, US Patent 3 993 (1976) 591.
[10] J. Yu, R. Wang, S. Ren, X. Sun, C. Chen, Q. Ge, W. Fang, J. Zhang, H. Xu, D.S. Su,ChemCatChem 4 (2012) 1376–1381.

10 B. Sarkar et al. / Catalysis Communications 56 (2014) 5–10
[11] D.P. Land, C.L. Pettiette-Hall, R.T. McIver, J.C. Hemminger, J. Am. Chem. Soc. 111(1989) 5970–5972.
[12] D.H. Parker, C.L. Pettiette-Hall, Y. Li, R.T. McIver, J.C. Hemminger, J. Phys. Chem. 96(1992) 1888–1894.
[13] B.E. Koel, D.A. Blank, E.A. Carter, J. Mol. Catal. A 131 (1998) 39–53.[14] M.C. Kung, H.H. Kung, J. Catal. 128 (1991) 287–291.[15] B. Coughlan, M.A. Keane, Catal. Lett. 4 (1990) 223–234.[16] I. Mochida, T. Jitsumatsu, A. Kato, T. Seiyama, J. Catal. 36 (1975) 361–370.[17] E.C. Alyea, M.A. Keane, J. Catal. 164 (1996) 28–35.[18] T. Kubo, H. Arai, H. Tominage, T. Kunugi, J. Chem. Soc. Jpn. 45 (1972) 613–616.[19] M.A. Sanchez, V.A.Mazzieri,M.R. Sad, R. Grau, C.L. Pieck, J. Chem. Technol. Biotechnol.
86 (2011) 447–453.[20] E.C. Tyo, C. Yin, M.D. Vece, Q. Qian, G. Kwon, S. Lee, B. Lee, J.E. DeBartolo, S. Seifert, R.
E. Winans, R. Si, B. Ricks, S. Goergen, M. Rutter, B. Zugic, M.F. Stephanopoulos, Z.W.Wang, R.E. Palmer, M. Neurock, S. Vajda, ACS Catal. 2 (2012) 2409–2423.
[21] S. Goergen, C. Yin, M. Yang, B. Lee, S. Lee, C. Wang, P. Wu, M.B. Boucher, G. Kwon, S.Seifert, R.E. Winans, S. Vajda, M.F. Stephanopoulos, ACS Catal. 3 (2013) 529–539.
[22] C.D.Wagner,W.M.Riggs, L.E. Davis, J.F.Moulder, G.E.Muilenberg,Handbook of X-rayPhotoelectron Spectroscopy, Perkin-Elmer Corp., Eden Prairie, USA, 1979.
[23] Handbook of the Elements and Native Oxides, XPS International, 754 Leona Lane,Mountain View, California 94040, USA, 1999.
[24] S. Watanabe, X. Ma, C. Song, J. Phys. Chem. C 113 (2009) 14249–14257.[25] A.R. Belambe, R. Oukaci, J.G. Goodwin, J. Catal. 166 (1997) 8–15.[26] Y. Ji, Z. Zhao, A. Duan, G. Jiang, J. Liu, J. Phys. Chem. C 113 (2009) 7186–7199.[27] B. Sarkar, P. Prajapati, R. Tiwari, R. Tiwari, S. Ghosh, S.S. Acharyya, C. Pendem, R.K.
Singha, L.N.S. Konathala, J. Kumar, T. Sasaki, R. Bal, Green Chem. 14 (2012) 2600–2606.[28] K.H. Cats, I.D. Gonzalez-Jimenez, Y. Liu, J. Nelson, D. van Campen, F. Meirer, A.M.J.
van der Eerden, F.M.F. de Groot, J.C. Andrews, B.M. Weckhuysen, Chem. Commun.39 (2013) 4622–4624.
[29] S.A. Chambers, S.M. Heald, T. Droubay, Phys. Rev. B 67 (100401) (2003) 1–4.[30] Y. Iwama, N. Ichikuni, K.K. Bando, J. Shimazu, Appl. Catal. A 323 (2007) 104–109.[31] M. Jin, Z.M. Cheng, Catal. Lett. 131 (2009) 266–278.[32] H. Feng, J.W. Elam, J.A. Libera, M.J. Pellin, P.C. Stair, Chem. Eng. Sci. 64 (2009)
560–567.


















