
Fluorine-containing 4-quinolinemethanols as antimalarials
5
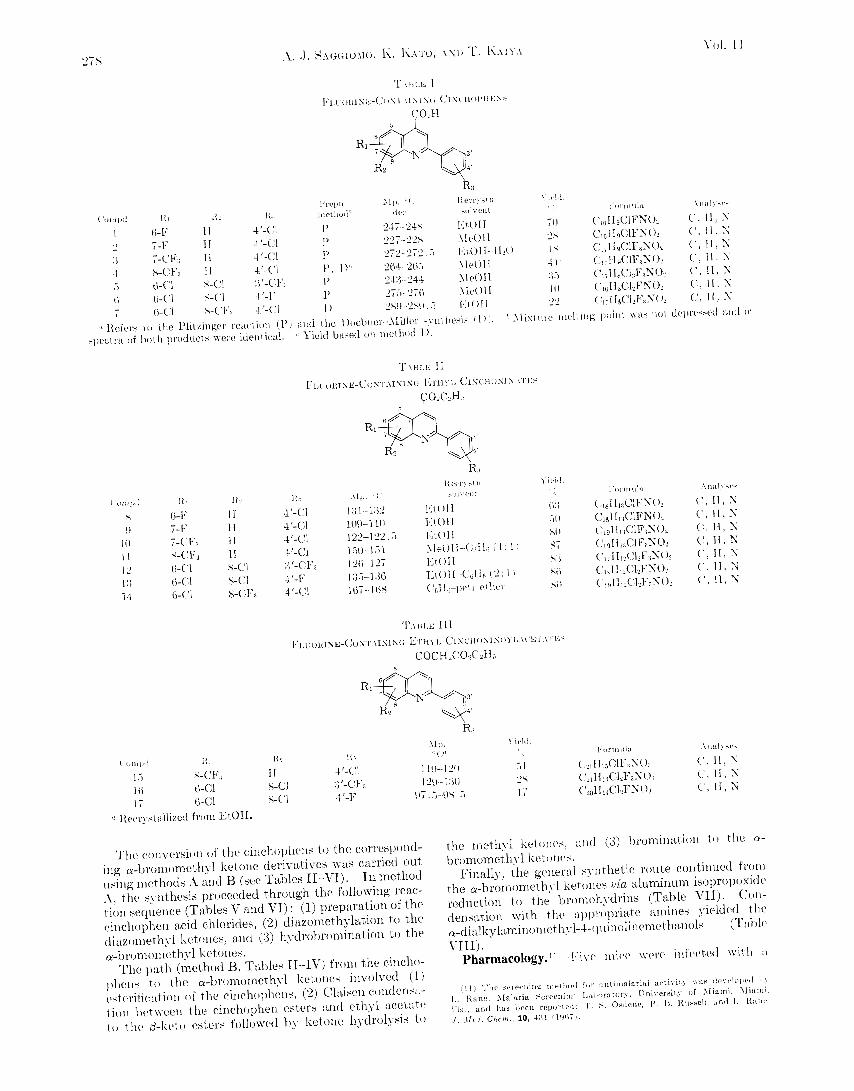
llurch 196s ASTIMALAI~IAL FLUOI~INATED ~-QUINOLISEMETHANOLS 277 To a solution of the foregoing ester (0.06 mole, 18.32 g) and ethyl 6-ben~amidocaproate~" (0.061 mole, 16.06 g) in 50 ml of dry C6TT6, NaSH2 (0.073 mole, 2.93 g) was added. The mixture was heated at 90' with vigorous stirring for 24 hr. iZfter cooling the mixture to .%", 32 ml of concentrated H2SO4 in 50 ml of H2O was added and refluxing was cont'inued for 65 hr. The C,& was then distilled off azeotropically and the residue was made alkaline with 30% aqueous NaOH keeping the temperature below 40". The mixture was then extracted with C6H6. After drying ( MgSOd) the solvent was removed under reduced pressure. The ir Fpectrum of the solid residiie indicated that the N-benzoyl group was not cleaved. The material was t'herefore suspended again in a solution of 30 ml of concentrated H2SO4 in 50 ml of IT20 and the mixt,ure was refluxed for 64 hr. After cooling it was made alkaline as before and extracted with CeH6. The dried CsFTe solution iipon concentration in vacuo left an oil to which 23 g of 487 HBr was added. Upon standing for a short while a yellow precipitate was obtained and filtered: the yield of 6-[6-methyl-2- ip-tolyl~cit~cho~ii~i~l]-n-amylamir~e dihydrobromide was 5.5 g (84'; based on recovered acid).': The aqueous alkaline phase was acidified with concentrated FICl and the resulting precipitate was filtered, washed with a little EtOH, and dried. The weight, of recovered 6-methyl-2- (p-tolyl)-4-cinchoninic acid from the unreacted ethyl ester was 7.8 g. The foregoing amine dihydrobromide (0.008 mole, 4 g) was dissolved in hot l8Yc HBr and treated rapidly with a solution of Br2 (0.008 mole, 1.28 g) in an equal volume of 487, HBr. The crude product was filtered and dispersed in 40 ml of boiling 95% EtOH, and H20 was added until a clear solution resulted. Cool- (15) This intermediate and the ones which follow en route to 60 mere used directly in the next synthetic step without characterization; c/. ref 9 and 14. ing gave a light yellow precipitate. Concentration of the mother liquor yielded some additional product'. The total yield of 6-bromo-6-[6-methyl-2-(p-tolyl)cittchonii1~l]-n-amylamine dihy- drobromide was 3.95 g (84(x). The foregoing product (1.5 g) was dissolved in 50 ml of 93% Et013 and 7 ml of 14% aqueous Na2C03was added. The mix- ture was shaken for 1 hr in a stoppered bottle and then hydro- genated over 20 mg of PtO2 in a Parr hydrogenation apparatus. The react,ion mixture was filtered and washed (Et,OH, hot CHC1,). The solvents were removed in vacuo. The residue was dissolved in hot CHC13 and filtered. Evaporation of the solvent left a brown residue. This was dissolved in absolute EtOH and the solution was saturated with dry HCI. After standing fnr a short while, Et,O was added and t,he precipitate was filt'ered to yield 0.5 g of t'he hydrochloride. A small amount, of this salt was converted into the free base 53. The ir spectra of the free base 53 and its hydrochloride salt were idelltical with those of t>he products obtained by catalytic reductions of the pyridyl ketone. 2-Aryl-4-quinolinecarboxylic Acids (Cinchoninic Acids) (I) (Table IV).-A11 of the substituted cinchophens reqiiired as starting material were synthesized by the Pfitzinger'e condensa- tion. In general, it was found that better yields were obtained when the mixtures of the appropriate isntins and substituted acetophenones in EtOH-KOH were refluxed for 30 hr; shorter periods of time gave poorer yields. Acknowledgment.-The authors wish to thank Professor A. Burger for fruitful discussion before and during the course of this work. (16) IT-. Pfitzinger, J. Prakt. Chen., 66, 283 (189i). Fluorine-Containing 4-Quinolinemethanols as Antimalarials' ANDREW J. SAGGIOMO, KAZUO KATO, AKD TOYO KAIYA Research Institute of Temple University, Phdadelphia, Pennsylvania 19144 Received September SO, 1967 Various fluorine-containing ~-dialkylaminomethyl-2-phenvl-4-quinolinemethanol derivatives have been pre- pared for evaluation against Plasmodium berghei in mice. Preliminary biological data indicate the fluorine com- pounds to be more potent at comparable doses than the corresponding chloro derivatives. a-Di-n-butyl- aminomethyl-2-(4-chlorophenvl)-7-trifluoromethvl-4-quinolinemethanol when administered to mice in a single . " subcut,aneous dose was curat,ive at 40 mg/kg. A high degree of antimalarial act'ivity was discovered in the 4-quinolinemethanol series during the World War I1 program supported by the government. Re- views2 of this work indicated that the most notable changes in activity in this series n-ere caused by sub- stituent variations in the aromat'ic rings. A consider- able number of the chlorine-subst'ituted a-dialkylamino- methyl-2-phenyl-4-quinolinemethanols showed pro- nounced antimalarial action. In the past two decades pharmacological investiga- tions have revealed that the replacement of chlorine and hydrogen in biologically active compounds by fluorine and fluorine-containing groups has provided in many cases highly potent fluorine-containing thera- peut,ic agents. We now report the synthesis and potent antimalarial activity of various fluorine-containing 4-quinoline- ' (1) This investigation was supported by the U. S. Army RIedical Res?arch and Development Command under Contract Dd-49-193-MD-2950 and is Contrihution KO. 290 from the Army Research Program on Malaria. (2) (a) F. T. Wiselople. ".i Survey of Antimalarial Drugs. 1941-1945," J. IT. Edwards. Ann Arbor. Ilich.. 1946: (h) G. R. Coatney, I\-. C. Cooper, N. U. Eddy. and J. Greenlierp, "Surveg. of .intimalarial Agents," Public Health Monograph No. 9, Washington, D. C., 1953. methanol derivatives. These compounds were pre- pared as part of a program to develop new and more- effective agents to combat drug-resistant malarial para- sites. Chemistry.-Our synthetic plan essentially paralleled those routes described previously for the preparation of 4-q~inolinemethanols.~~~ The general route to the fluorine-containing 4-quinolinemet'hanol derivatives commenced with the preparation of the appropriately substituted cinchophens (2-phenylcinchoninic acids). The latt'er (Table I) were obtained (a) from readily accessible anilines via the Sandmeyer isatin synthesis516 and the Pfitzinger r e a c t i o ~ i ~ * ~ ~ ~ and (b) through the Doebner-AIiller r e a c t i ~ n ~ ~ ~ ~ ~ ~ between the appropriate anilines, benzaldehydes, and pyruvic acid. (3) R. E. Lutz, el a?., J. An. Chem. Soc., 68, 1813 (1946). (4) S. Winstein, et a?., ibid., 68, 1831 (1946). (5) T. Sandmeyer, Hdu. Chim. Acta, 2, 234 (1919). (6) R. C. Elderfield in "Heterocyclic Compounds," Tol. 3, R. C. Elder- (7) IT. Pfitzinger, J. Prafct. Chem., 66, 283 (1897). (8) For excellent review. see ref 6, Vol. 3, p 222; Vol. 4, p 17. (9) 0. Doebner, Ann., 242, 265 (1887). (10) For a review, see ref 6, Vol. 4, p 25. field, Ed., John Wiley and Sons, Inc., New York. N. I-., 19.52, p 208.
Transcript of Fluorine-containing 4-quinolinemethanols as antimalarials

l lurch 196s ASTIMALAI~IAL FLUOI~INATED ~-QUINOLISEMETHANOLS 277
To a solution of the foregoing ester (0.06 mole, 18.32 g) and ethyl 6-ben~amidocaproate~" (0.061 mole, 16.06 g) in 50 ml of dry C6TT6, N a S H 2 (0.073 mole, 2.93 g) was added. The mixture was heated at 90' with vigorous stirring for 24 hr. iZfter cooling the mixture to .%", 32 ml of concentrated H2SO4 in 50 ml of H2O was added and refluxing was cont'inued for 65 hr. The C,& was then distilled off azeotropically and the residue was made alkaline with 30% aqueous NaOH keeping the temperature below 40". The mixture was then extracted with C6H6. After drying ( MgSOd) the solvent was removed under reduced pressure. The ir Fpectrum of the solid residiie indicated that the N-benzoyl group was not cleaved. The material was t'herefore suspended again in a solution of 30 ml of concentrated H2SO4 in 50 ml of IT20 and the mixt,ure was refluxed for 64 hr. After cooling i t was made alkaline as before and extracted with CeH6. The dried CsFTe solution iipon concentration in vacuo left an oil to which 23 g of 4 8 7 HBr was added. Upon standing for a short while a yellow precipitate was obtained and filtered: the yield of 6-[6-methyl-2- ip-tolyl~cit~cho~ii~i~l]-n-amylamir~e dihydrobromide was 5.5 g (84'; based on recovered acid).':
The aqueous alkaline phase was acidified with concentrated FICl and the resulting precipitate was filtered, washed with a little EtOH, and dried. The weight, of recovered 6-methyl-2- (p-tolyl)-4-cinchoninic acid from the unreacted ethyl ester was 7.8 g.
The foregoing amine dihydrobromide (0.008 mole, 4 g) was dissolved in hot l8Yc HBr and treated rapidly with a solution of Br2 (0.008 mole, 1.28 g) in an equal volume of 487, HBr. The crude product was filtered and dispersed in 40 ml of boiling 95% EtOH, and H20 was added until a clear solution resulted. Cool-
(15) This intermediate and the ones which follow en route to 60 mere used directly in the next synthetic step without characterization; c/ . ref 9 and 14.
ing gave a light yellow precipitate. Concentration of the mother liquor yielded some additional product'. The total yield of 6-bromo-6-[6-methyl-2-(p-tolyl)cittchonii1~l]-n-amylamine dihy- drobromide was 3.95 g (84(x).
The foregoing product (1.5 g) was dissolved in 50 ml of 93% Et013 and 7 ml of 14% aqueous Na2C03 was added. The mix- ture was shaken for 1 hr in a stoppered bottle and then hydro- genated over 20 mg of PtO2 in a Parr hydrogenation apparatus. The react,ion mixture was filtered and washed (Et,OH, hot CHC1,). The solvents were removed in vacuo. The residue was dissolved in hot CHC13 and filtered. Evaporation of the solvent left a brown residue. This was dissolved in absolute EtOH and the solution was saturated with dry HCI. After standing fnr a short while, Et,O was added and t,he precipitate was filt'ered to yield 0.5 g of t'he hydrochloride. A small amount, of this salt was converted into the free base 53.
The ir spectra of the free base 53 and its hydrochloride salt were idelltical with those of t>he products obtained by catalytic reductions of the pyridyl ketone.
2-Aryl-4-quinolinecarboxylic Acids (Cinchoninic Acids) (I) (Table IV).-A11 of the substituted cinchophens reqiiired as starting material were synthesized by the Pfitzinger'e condensa- tion. In general, i t was found that better yields were obtained when the mixtures of the appropriate isntins and substituted acetophenones in EtOH-KOH were refluxed for 30 hr; shorter periods of time gave poorer yields.
Acknowledgment.-The authors wish to thank Professor A. Burger for fruitful discussion before and during the course of this work.
(16) IT-. Pfitzinger, J. Prakt. C h e n . , 66, 283 (189i).
Fluorine-Containing 4-Quinolinemethanols as Antimalarials'
ANDREW J. SAGGIOMO, KAZUO KATO, AKD TOYO KAIYA
Research Inst i tute of Temple Universi ty , Phdadelphia, Pennsylvania 19144
Received September SO, 1967
Various fluorine-containing ~-dialkylaminomethyl-2-phenvl-4-quinolinemethanol derivatives have been pre- pared for evaluation against Plasmod ium berghei in mice. Preliminary biological data indicate the fluorine com- pounds to be more potent a t comparable doses than the corresponding chloro derivatives. a-Di-n-butyl- aminomethyl-2-(4-chlorophenvl)-7-trifluoromethvl-4-quinolinemethanol when administered to mice in a single . " subcut,aneous dose was curat,ive a t 40 mg/kg.
A high degree of antimalarial act'ivity was discovered in the 4-quinolinemethanol series during the World War I1 program supported by the government. Re- views2 of this work indicated that the most notable changes in activity in this series n-ere caused by sub- stituent variations in the aromat'ic rings. A consider- able number of the chlorine-subst'ituted a-dialkylamino- methyl-2-phenyl-4-quinolinemethanols showed pro- nounced antimalarial action.
In the past two decades pharmacological investiga- tions have revealed that the replacement of chlorine and hydrogen in biologically active compounds by fluorine and fluorine-containing groups has provided in many cases highly potent fluorine-containing thera- peut,ic agents.
We now report the synthesis and potent antimalarial activity of various fluorine-containing 4-quinoline-
'
(1) This investigation was supported by the U. S . Army RIedical Res?arch a n d Development Command under Contract Dd-49-193-MD-2950 and is Contrihution KO. 290 from the Army Research Program on Malaria.
(2) (a) F. T. Wiselople. ".i Survey of Antimalarial Drugs. 1941-1945," J. IT. Edwards. Ann Arbor. I l ich. . 1946: (h) G. R . Coatney, I\-. C. Cooper, N. U. Eddy. a n d J. Greenlierp, "Surveg. of .intimalarial Agents," Public Health Monograph No. 9, Washington, D. C., 1953.
methanol derivatives. These compounds were pre- pared as part of a program to develop new and more- effective agents to combat drug-resistant malarial para- sites.
Chemistry.-Our synthetic plan essentially paralleled those routes described previously for the preparation of 4-q~inolinemethanols.~~~ The general route to the fluorine-containing 4-quinolinemet'hanol derivatives commenced with the preparation of the appropriately substituted cinchophens (2-phenylcinchoninic acids). The latt'er (Table I) were obtained (a) from readily accessible anilines via the Sandmeyer isatin synthesis516 and the Pfitzinger r e a c t i o ~ i ~ * ~ ~ ~ and (b) through the Doebner-AIiller r e a c t i ~ n ~ ~ ~ ~ ~ ~ between the appropriate anilines, benzaldehydes, and pyruvic acid.
(3) R . E . Lutz, el a?., J . An. Chem. Soc., 68, 1813 (1946). (4) S. Winstein, et a?., ib id . , 68, 1831 (1946). ( 5 ) T. Sandmeyer, H d u . Chim. Acta, 2, 234 (1919). (6) R . C. Elderfield in "Heterocyclic Compounds," Tol. 3 , R . C. Elder-
(7) IT. Pfitzinger, J . Prafct. Chem., 66, 283 (1897). (8) For excellent rev iew. see ref 6, Vol. 3, p 222; Vol. 4, p 17. (9) 0. Doebner, Ann., 242, 265 (1887). (10) For a review, see ref 6 , Vol. 4, p 25.
field, Ed . , John Wiley and Sons, Inc., New York. N. I-., 19.52, p 208.
RZ

Rlarch 196s 279
TABLE IV FLUORINE-COXTAILYIKG 2-PHEXYL-k)UISOLYL 1\IErHYL KETOKES
COCH,
Ri
hIV! Cornpd R1 1% 2 1% 3 "C
18 7-CFr H 4'-C1 123 .j-124 19 8-CF3 H 4 '-C1 178-179 20 6-C1 8-C1 3'-CF7 14.5-146.5 21 6-C1 8-C1 4'-F 137-138 22 6-c1 8-CF3 4 ' 4 1 209-209.3
LIP, Kecrystn I ield, Coinpd R1 Rz R3 OC solvent 70
23 6-F I€ 4'-C1 157-1.X CsH, 86 24 7-F H 4'-C1 125-127 Ligroiii 68 23 A-CFJ H 4'-C1 194-196 C,Hs 7 3 26 6-C1 8-C1 3'-CF3 136-138 Ligroiii 90 27 6-C1 8-c1 4'-F 141-143 C6Hs 67 2X 6-C1 8-CF3 4'-CI 193-196 5 CaHs 94
Q Elemental analysis of these compounds was generally omitted. I r spectra of the acid chlorides indicated the C=O absorption band a t 5.7 p, whereas the ciiichoninic acids displayed the C=O peak at 5.9 ~ 1 .
R? Recr js tn Yieid, s o h ent 70 formula lnal,>es
EtOH .i 4 CisHii C lFJ i 0 C , TI, s Me&O-HiO 80 CisHnClF3SO C, H , S EtOH 36 CisHioClnFsNO C, 11, ?; EtOH 12 Ci7HioC12FSO C, H , N EtOAc 3 3 CisHioCllF3XO C, H , X
taining 4quinolinemethanol derivatives possess potent antimalarial action arid are curative in mice. Com- pound 45 was found to be significantly more active than the corresponding chloro derivative. None of the inter- mediates tested were active.
Photosensitization effects of 4quinolinemethanol derivatives were described in an earlier review.2a Re- cently, the phototoxic potency of a number of quinoline- methanols, including compounds 45 and 48, has been reported by Rothe and Jacobus.'*
Experimental Section13 The object of this research was to prepare as quickly as pussible
the pure target compounds in sufficient quantity for antimalarial t'esting. Consequently, the experimental conditions described herein do not necessarily represent the optimum. Occasioiially the isolation, extensive purification, or elemental analysis of an intermediate was omitted. Physical and analytical data are found in Tables I-VIII.
TAHLE VI FLUORIXE-CONT.UNIXG a-BROMOMETHYL 2-PHEXYL-4-QUINOLYL KETOXES
COCH2Br
Prepn hip, Recrlstn Yield, Cornpd R i R2 R3 methoda O C 601% ent 70 Tormula lnalgaeu
29* 6-F H 4 '-CI A 243-246 dec AcOH 8 tj C17H11Br2C1FN0 C , H, ?; 30* 7-F H 4'-C1 B 239-241 dec AcOH 53 C1;H1,Br2C1F?;0 C, H , ?; 31 7-CF3 H 4'-C1 B 169- 17 1 EtOH 9 3 C18HloBrClF3KO C , H , N 32' 8-CF3 H 4'-Cl A , B 162-164 ?rIeOH-HLO SSd C18HloBrC1FdS0 C, H, S 3 3 ~ 6-C1 8-CI 3'-CF3 A , B 168-169 EtOH 9.id C18H9BrCI2F3NO C , H, S 34 6-CI 8 4 1 4'-F A 173-174 EtOH 89 CI;H$BrCl,FNO C, H, N 35 6-C1 8-CF3 4'-C1 A , B 193-196 EtOH 8gd C I ~ H S B ~ C I I F ~ S O H , K; C"
a Refers to methods A and B in the discussion. b Isolated and analyzed as the hydrobromide salt. Prepared L K L methods A and B. e C: calcd, 46.68, l l ixture melting point was riot depressed and ir spectra of both products were identical.
found, 47.22. Yield based on method 9.
lethal dose of Plus??&iu.yy~ belyhei 3 days prior to ad- Materials.-& and 4-Fluoroaiiiliiie, 4-trifluoromethylaiiilitie, ministratior, of the ,,hemicai. ~ , ~ ~ t i ~ ~ ~ l ~ , the ,,hemicai 4-chloro-2-trifluoromethylaniline, 4-chloro-;S-nitrobeiizotrifl~ioride,
3'-trifluoromethylacetophenone, and 4'-fluoroacetophenone was administered subcutaneously in oil at, each of three were purchased from Pierce Chemical Co,, dose levels, namely at, 40, 160, arid 640 mg/kg. The 2- arid 3-trifluoromethylanilirie were obtained from Maumee mean survival time of infected control mice is 6.5 =I= 0.5 days. Extension in survival time of the chemically (12) W. E. Rothe and D. J. Jacobus, 154 th National LIeeting of the
t,reat,ed mice is interpreted as evidence of antimalarial activity. The number of mice surviving (out of five) a t 00 days post infection are considered cures.
preiimiIlary arltimalarial test data are reported in
American Chemical Society, Chicago, Ill., Sept 11-14, 1967, p 37. (13) Melting points were determined with a n electrically heated Tiiiele-
Dennis apparatus and are uncorrected. Elemental analyses were carried o u t by Schwarzkopf Alicroanalyticai Laboratory, IYoodside, X. Y.. and Micro-analysis, Inc. , Wilmington, Del. Where analyses are indicated only by symbols of the elements, analytical results obtained for those elements
Ta8ble IX. The results indicate that the fluorine-con- \vere witilin +0.4% of +,lie theoretical values.

"SO
H c;' OH c H 2x<R I R
Pftzinger Synthesis of 2-Phenylcinchoninic Acids. - ' l ' lrc , :ippri~priately siibrl itiitetl ihntiii (o.025 mole) and :iv tlerivntive (0.028 n i ~ l e j iri 1~:tOIl (:IO nil) cotit:iiiiiiig (1.5 inl ) \vert' refiiiscti for 7-+ frr. I.:tOTi was reinrived r i i i t i t * r rt~dr~treti prehsiire :iiitl ihc rc.sitliie \vas acidified wit ti i i lf; i , l lc ' l ,
flr~oiiiie-c.oiit:iiIiiiiy 9-~)t ie i i~lci i ivhoi i1i i~~ : i d s . Doebner-Miller Synthesis of 2-Phenylcinchoninic Acids:~
The following prcpar:ition of O-c.hloro-fi-trifliior(~iiieth?..l(.iii- c'hi~riiiric uc.id is :L l ) y i ( d prf~cediire. p-Chliirobeiizaltleh~(l(, i 17Y..j g, 1.27 miilea) was mixed nt 20' w i t h a srilutiori (if Laniiiio- .i-c.hlorol)eiizcJtrifliioi,i~~e (24!) g, 1 .27 niiilee) i i i 300 nil of Ad111. The f hirk hrwpeiisioii wa5 treated dropwke with I :it a rate that kept the iiitenial lernpemtiire a t 3-35', Stirriiig was c80iitinued for an additioiial 15 mill. Freshly dist>illed pyi,iivic* acid ((30 g, 1.01 nrole~) \V:W added iti 3 niin :tt 20' atid the rriis1,iii.e \\-as heated gradually 1):- rnantle to 120' (iiiteriial ternperatiirei ti ic~i7ig 1 hr . The cooled wspeii- s k i n was filtered and the precipitate, after being washed with cold AcOH, gave product', [rip 28!)".
Cinchoninic Esters.---The cinchoninic acids were heated n i t h Et013 in the presence of II,SOI for 1 hr. 011 cooling the e t h ~ ~ l est e m precipitated.
2-Phenyl-4-quinolyl Methyl Ketones.--A srispensioii of ( h e xppropriate cinchoninic ester18 (0.005 mole), anhydroirs E tOAi . (0.01 mole), aiid XaOEt (0.007:: inole) in predried CSIL (1.5 rnl)
,tdlizat ioii o f t I IP precipit
(-%.it P i " a prec4pitate formed.)

March 1YGS
RS R1% R2 \
Dose level, Cures*
Compd Ri Rz R3 R xngjkg (5 mice)
48 6421 8-C1 3'-CF3 n-CaI-1~ 40 3 160 1
640 J
160 1 640 3
45 i-CF, I1 4'-C1 n-CJ-ILl 40 5 160 )
640 3
64 0 4 e
40 6-C1 8-C1 3'-CFr n-CsHir 40 2.8c
d 7-C1 H 4'-C1 n-CdH9 160 9.7C
0 These preliminary test results were supplied by L. Rane, 11alaria Screening Laboratory, University of Miami, Miami, Fla. *Cure is defined as a survival of 60 days or more post- infection. ~Iricrease in mean survival time (AIST) in days. 1IST of infected control mice is 6.5 i 0.5 days. Survey no. SN 13710, ref 2. $One toxic death.
was refluxed for 16 hr. The reaction mixture was poured into 5y0 NaOH (20 ml) and ice to precipitate the sodium salt of the corresponding @-keto ester. In a few cases, the p-keto ester was isolated by treating the sodium salt with warm AcOH. The Claisen reaction mixtures or the isolated p-keto esters were heated with 3 0 4 5 % H2SOc a t refliix until CO, evolution ceased. After dilution with H20 , the resulting precipitates on recrystallization provided the methyl ketones.
Bromination of 2-Phenyl-4-quinolyl Methyl Ketone~.~-A solution of the methyl ketone (0.08 mole) in AcOH (655 ml) was slowly treated a t room temperature with 129 ml (1.1 moles) of 487, HBr. After the addition of sodium bromate (4.5 g, 0.03 mole) in H20 (47 ml) (dropwise during 20 min), the mixture was heated to 75" in 0.5 hr and at i 5 ' for O..i hr. Dilution of the
XATED ~-QUINOLINEMETHANOLS "1
Cinchoninic Acid Chlorides.-SOC12 and the acids were heated a t reflux for 3 hr. Excess reagent was removed under reduced pressure and the residue was slurried with CsHC. Recrystalliza- tion of the insoluble solid provided the acid chlorides. a-Bromomethyl2-Phenyl-4-quinolyl Ketones via Diazomethyl-
ation of the Cinchoninic Acid Chlorides.-The following syn- thesis of a-bromomethyl 6,~dichloro-Z-(3-trifluoromethyl- phenyl)-4-quinolyl ketone is a typical procedure. To a stirred mixture of 16.6 ml of 40% KOH and 55 ml of ether a t 5' was added i . 5 g (0.054 mole dry wt) of moist3 nitrosomethylurea*O at, such a rate (0.5 hr ) that the reaction temperature did not exceed 5'. The ether layer containing CHzN, was then decanted and dried over KOH pellets. To this well-stirred ethereal CH2K2 sohition (0.037 mole) a t -3" was added in 15 min the powdered cirichoniriic acid chloride ( 3 g, 0.012 mole) and the reaction was kept at 0-4" overnight. .4 mixture of 48% HBr (6 ml) in ether (6 ml) was added to the ether suspension of diazomethyl ketone. After stirring for 4 hr, the react,ion mixture was filtered and the solid was washed with HZO. Recrystallization provided the a-bromomethyl ketone.
Aluminum Isopropoxide Reduction of a-Bromomethyl 4- Quinolyl Ketones.-A stirred suspension of the a-bromoniethyl ketones (0.002 mole) and freshly prepared aluminum isopropoxide (0.007 mole) in 15 ml of anhydrous i-PrOH was slowly distilled uiider mild reflux until the distillate gave 110 further test for ace- tone with 3,4-dinitropheiivlh~.drazine (usually 1.5-2 hr). The solvent was removed under rediiced pressure and the residue was treated with lorc HCI. The precipitated bromohydrins were washed with H,O aiid dried. These compounds were used without further piirification in the coiidensation with dialkyl- amines.
a-(Di-n-butylaminomethyl)-2-phenyl-4-quino~inemethano~s.- The bromohydrins (0.002 mole) aiid di-n-butylamine (1.1 g, 0.008 mole) were heated at 75" for 12 hr. The reaction was cooled to room temperatiire, 21 ml of ether was added, and BiigNH2+Br- was filtered. The stirred filtrate was treated slowly with standardized ethereal HCl (0.0042 mole) and the precipitated BtiJH2 +C1- was removed. Further addition of an equivalent amoiint of et,hereal HCl gave the a-( di-n-butylarnino- methgl)-4-qriirioliriemethaiiol monohydrochlorides as almost- white salts which were then recrystallized.
The preparation of the di-n-hexylaminomethyl derivatives was carried oiit in a similar manlier. Excess di-n-hexylamine was removed by bteam distillatioii.
I r spectra of a-dialkylamiiiomethyl-4-qainoliiiemethariol monohydrochloride salt,s displayed OH absorption at 3.05- 3.10 p and NH + absorption as a doublet at 3.85 aud 3.94 p.
suspension with H 2 0 and recrystallizat,ion of the precipitate provided the a-byomomethyl ketones. Acknowledgment.-The technical assistance of
Pauline Schwartz arid Anthony Scotese is gratefully acknowledged. (19) The attempted conversion of ethyl 6-fluoro-2-(-l-ctiloroplienyl)-
cinchoninate to the p-keto ester employing NaOEt in CsHs resulted in dis- placement of the E a tom and isolation of ethyl 6-etl1oxy-2-(~-chloropheng.l)- cinchoninate (6%. Anal. (CzaHmCINOsj H. N; C: calcd, 67.51; foiind, (20) E. Arndt, "Organic Syntheses," Coll. Vol. 11, John Wiley and Sons. 67.05. Inc., New York, N. Y., 1943, p 461.


















