
fl-Lactamase inhibitors
7
453 Biochem J. (1988) 251, 453-459 (Printed in Great Britain) fl-Lactamase inhibitors The inhibition of serine fl-lactamases by specific boronic acids Ian E. CROMPTON,* Bill K. CUTHBERT,t Gordon LOWEt$ and Stephen G. WALEY* *Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford OXI 3RE, and tDyson Perrins Laboratory, University of Oxford, South Parks Road, Oxford OXI 3QY, U.K. Many ,-lactamases have active-site serine residues, and are competitively inhibited by boronic acids. Hitherto, the boronic acids used have lacked any structural resemblance to the substrates of /1-lactamases. Phenylacetamidomethaneboronic acid, trifluoroacetamidomethaneboronic acid and 2,6-dimethoxy- benzamidomethaneboronic acid have now been synthesized. The first of these contains the side-chain moiety of penicillin G, and the last that of methicillin. The pH-dependence of binding of the first inhibitor to fl-lactamase I from Bacillus cereus revealed pK values of 4.7 and 8.2 for (presumably) active-site groups in the enzyme. The kinetics of inhibition were studied by cryoenzymology and by stopped-flow spectrophotometry. These techniques provided evidence for a two-step mechanism of binding of the first two boronic acids mentioned above to ,-lactamase I, and for benzeneboronic acid to a ,1-lactamase from Pseudomonas aeruginosa. The slower step is probably associated with a change in enzyme conformation as well as the formation of an 0-B bond between the active-site INTRODUCTION ,8-Lactamases are mechanistically interesting and clinically important enzymes. Their clinical importance stems from being a major cause of resistance of bacterial pathogens to the widely used fi-lactam antibiotics. The mechanistic interest of fl-lactamases derives from their resemblance to the serine proteinases in the deployment of an active-site serine residue, but differing in that the co-operating residues appear to be lysine and glutamic acid, rather than histidine and aspartic acid (Herzberg & Moult, 1987). Again, f8-lactamases resemble proteinases in catalysing the hydrolysis of an amide bond, but differ in that the basic residue has to be deprotonated in proteinases but (apparently) protonated in 8-lactamases (Herzberg & Moult, 1987). Inhibitors of,-lactamases may be useful clinically and for crystallographic (or n.m.r.) studies. Boronic acids occupy a special place among /J-lactamase inhibitors. They are virtually the only active-site-directed inhibitors of ,-lactamases that are not themselves ,-lactams. ,J- Lactams have drawbacks for crystallographic studies, as they are usually hydrolysed and the products will diffuse away, but boronic acids are, in effect, stable inhibitors,. and will remain bound at the active site. Moreover, boronic acids that can diffuse through the outer membane are conceivably clinically useful 8-lactamase inhibitors. The boronic acids that have hitherto been studied as ,f- lactamase inhibitors (Dobozy et al., 1971; Kiener & Waley, 1978; Beesley et al., 1983) owe their affinity largely to the boronic acid group, and lack an appropriate side chain. We now report the synthesis of phenyl- acetamidomethaneboronic -acid (BzB) (I) and the related compounds trifluoroacetamidomethaneboronic acid (TFB) (II) and 2,6-dimethoxybenzamidomethaneboronic serine hydroxy group and the boronic acid. (I) Ph-CH2-CONH-CH2-B(OH)2 (II) CF3-CH2-CONH-CH2-B(OH)2 (III) 2,6-(MeO)2C6H3-CONH-CH2-B(OH)2 acid (MeB) (III), and their interaction with the class A ,8-lactamase I from Bacillus cereus. The use of low- temperature techniques, or rapid reaction methods at ordinary temperatures, has enabled us to detect two-step binding. The marked effects of pH on binding have also been characterized. The interaction of benzeneboronic acid (PhB) with a class C ,8-lactamase also takes place by a two-step mechanism. Our results confirm the potential usefulness of boronic acids in structural studies of ,- lactamase action. MATERIALS AND METHODS ,1-Lactamase I from Bacillus cereus 569H/9 was prepared as described previously (Davies et al., 1974; Baldwin et al., 1980). The use of a Millipore Pellikon cassette system to 'desalt' and concentrate the eluate from Celite greatly speeded up the preparation. ,J- Lactamase from the constitutive mutant of Pseudomonas aeruginosa 1822 S/H (Flett et al., 1976; Berks et al., 1982) was prepared by affinity chromatography (Cart- wright & Waley, 1984). N.m.r. spectra were recorded on a Bruker AM250 or AM500 spectrometer. 1H- and 13C-n.m.r. chemical shifts are in p.p.m. downfield from external tetramethylsilane ([2H]chloroform) as reference. '1B-n.m.r. chemical shifts are in p.p.m. downfield from external boron trifluoride ([2H]chloroform) as reference. Mass spectra were recorded on a V. G. Micromass 30FD spectrometer under electron-impact (e.i.) or ammonia chemical-ionization (NH3 c.i.) conditions. Abbreviations used: BzB, phenylacetamidomethaneboronic benzamidomethaneboronic acid; PhB, benzeneboronic acid. t To whom correspondence should be addressed. acid; TFB, trifluoroacetamidomethaneboronic acid; MeB, 2,6-dimethoxy- Vol. 251
-
Upload
truongkiet -
Category
Documents
-
view
228 -
download
1
Transcript of fl-Lactamase inhibitors

453Biochem J. (1988) 251, 453-459 (Printed in Great Britain)
fl-Lactamase inhibitorsThe inhibition of serine fl-lactamases by specific boronic acids
Ian E. CROMPTON,* Bill K. CUTHBERT,t Gordon LOWEt$ and Stephen G. WALEY**Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford OXI 3RE,and tDyson Perrins Laboratory, University of Oxford, South Parks Road, Oxford OXI 3QY, U.K.
Many ,-lactamases have active-site serine residues, and are competitively inhibited by boronic acids.Hitherto, the boronic acids used have lacked any structural resemblance to the substrates of /1-lactamases.Phenylacetamidomethaneboronic acid, trifluoroacetamidomethaneboronic acid and 2,6-dimethoxy-benzamidomethaneboronic acid have now been synthesized. The first of these contains the side-chain moietyof penicillin G, and the last that of methicillin. The pH-dependence of binding of the first inhibitor tofl-lactamase I from Bacillus cereus revealed pK values of 4.7 and 8.2 for (presumably) active-site groups in theenzyme. The kinetics of inhibition were studied by cryoenzymology and by stopped-flow spectrophotometry.These techniques provided evidence for a two-step mechanism of binding of the first two boronic acidsmentioned above to ,-lactamase I, and for benzeneboronic acid to a ,1-lactamase from Pseudomonasaeruginosa. The slower step is probably associated with a change in enzyme conformation as well as theformation of an 0-B bond between the active-site
INTRODUCTION
,8-Lactamases are mechanistically interesting andclinically important enzymes. Their clinical importancestems from being a major cause of resistance of bacterialpathogens to the widely used fi-lactam antibiotics. Themechanistic interest of fl-lactamases derives from theirresemblance to the serine proteinases in the deploymentof an active-site serine residue, but differing in that theco-operating residues appear to be lysine and glutamicacid, rather than histidine and aspartic acid (Herzberg &Moult, 1987). Again, f8-lactamases resemble proteinasesin catalysing the hydrolysis of an amide bond, but differin that the basic residue has to be deprotonated inproteinases but (apparently) protonated in 8-lactamases(Herzberg & Moult, 1987).
Inhibitors of,-lactamases may be useful clinically andfor crystallographic (or n.m.r.) studies. Boronic acidsoccupy a special place among /J-lactamase inhibitors.They are virtually the only active-site-directed inhibitorsof ,-lactamases that are not themselves ,-lactams. ,J-Lactams have drawbacks for crystallographic studies, asthey are usually hydrolysed and the products will diffuseaway, but boronic acids are, in effect, stable inhibitors,.and will remain bound at the active site. Moreover,boronic acids that can diffuse through the outer membaneare conceivably clinically useful 8-lactamase inhibitors.The boronic acids that have hitherto been studied as ,f-
lactamase inhibitors (Dobozy et al., 1971; Kiener &Waley, 1978; Beesley et al., 1983) owe their affinitylargely to the boronic acid group, and lack an appropriateside chain. We now report the synthesis of phenyl-acetamidomethaneboronic -acid (BzB) (I) and the relatedcompounds trifluoroacetamidomethaneboronic acid(TFB) (II) and 2,6-dimethoxybenzamidomethaneboronic
serine hydroxy group and the boronic acid.
(I) Ph-CH2-CONH-CH2-B(OH)2(II) CF3-CH2-CONH-CH2-B(OH)2(III) 2,6-(MeO)2C6H3-CONH-CH2-B(OH)2
acid (MeB) (III), and their interaction with the class A,8-lactamase I from Bacillus cereus. The use of low-temperature techniques, or rapid reaction methods atordinary temperatures, has enabled us to detect two-stepbinding. The marked effects of pH on binding have alsobeen characterized. The interaction of benzeneboronicacid (PhB) with a class C ,8-lactamase also takes place bya two-step mechanism. Our results confirm the potentialusefulness of boronic acids in structural studies of ,-lactamase action.
MATERIALS AND METHODS,1-Lactamase I from Bacillus cereus 569H/9 was
prepared as described previously (Davies et al., 1974;Baldwin et al., 1980). The use of a Millipore Pellikoncassette system to 'desalt' and concentrate the eluatefrom Celite greatly speeded up the preparation. ,J-Lactamase from the constitutive mutant of Pseudomonasaeruginosa 1822 S/H (Flett et al., 1976; Berks et al.,1982) was prepared by affinity chromatography (Cart-wright & Waley, 1984).
N.m.r. spectra were recorded on a Bruker AM250 orAM500 spectrometer. 1H- and 13C-n.m.r. chemical shiftsare in p.p.m. downfield from external tetramethylsilane([2H]chloroform) as reference. '1B-n.m.r. chemical shiftsare in p.p.m. downfield from external boron trifluoride([2H]chloroform) as reference.Mass spectra were recorded on a V. G. Micromass
30FD spectrometer under electron-impact (e.i.) orammonia chemical-ionization (NH3 c.i.) conditions.
Abbreviations used: BzB, phenylacetamidomethaneboronicbenzamidomethaneboronic acid; PhB, benzeneboronic acid.
t To whom correspondence should be addressed.
acid; TFB, trifluoroacetamidomethaneboronic acid; MeB, 2,6-dimethoxy-
Vol. 251

I. E. Crompton and others
Preparation of acylaminomethaneboronic acidsDibutyl iodomethaneboronate is a common inter-
mediate for the preparation of acylaminomethane-boronic acids either by the method of Linquist & Nguyen(1977) or by that of Matteson et al. (1981). It wasprepared via iodomethylmercuric iodide by the methodof Matteson & Cheng (1968), and had &B ([2H]-chloroform) 28.0 p.p.m and showed characteristicbroadening of the CH2B peak in the 'H-n.m.r. spectrumdue to 11B quadrupolar scalar relaxation.Attempts to get dibutyl iodomethane boronate to
react with phenylacetamide by the method of Linquist& Nguyen (1977) were unsuccessful. It was thereforeinstead converted into dibutyl NN-bis(trimethylsilyl)-aminomethaneboronate by the method of Matteson et al.(1981).
(i) Phenylacetamidomethaneboronic acid (BzB) (I).Dibutyl NN-bis(trimethylsilyl)aminomethaneboronate(194 mg, 0.59 mmol) was vigorously stirred duringaddition of phenylacetyl chloride (0.60 ml, 5.0 mmol) at-78 'C. The solution was allowed to warm to 20 'C over2 h; then, after cooling to -78 'C, methanol (2 ml,50 mmol) was added. After 30 min at 20 'C, water (10 ml)was added and the whole was stirred for 16 h (pH 1).Washing with diethyl ether (3 x 15 ml) and removal ofwater left a clear oil (66 mg, 59 %). N.m.r.: 3H (2H20)500 MHz 7.15 (SH, m, C6HA), 3.53 (2H, s, CH2Ph)and 2.20 p.p.m. (2H, s, CH2B); 4C (2H20) 250 MHz176.7 (s, C0O), 129-127 (aromatic), 38.5 (t, CH2Ph)and 32.5 p.p.m. (v. broad, CH2B); &B (2H20) 80 MHz18.1 p.p.m. (s). M.s.: m/z (NH3 c.i.) 150 (PhCH2-CONH2-CH2 ).
(ii) Trifluoroacetamidomethaneboronic acid (TFB) (II).Dibutyl NN-bis(trimethylsilyl)aminomethaneboronate(0.5 ml, 1.3 mmol) in dichloromethane (1 ml) wasvigorously stirred during addition of trifluoroaceticanhydride (2.0 ml, 14.0 mmol) at -78 'C. The solutionwas allowed to warm to 20 'C over 2 h, and evaporated;then, after cooling to -78 'C, methanol (2 ml, 50 mmol)was added. After 30 min at 20 'C, water (10 ml) wasadded and the whole was stirred for 2 h (pH1). Washingwith diethyl ether (3 x 15 ml) and freeze-drying left awhite powder (61 mg, 27 %). N.m.r.: 8H (2H20)250 MHz 2.77 p.p.m. (2H, s, CH2B) 6B (2H20)80 MHz 29.1 p.p.m. (s).
(iii) 2,6-Dimethoxybenzamidomethaneboronic acid(MeB) (III). Dibutyl NN-bis(trimethylsilyl)amino-methaneboronate (0.5 ml, 1.3 mmol) in dichloromethane(1 ml) was vigorously stirred during addition of 2,6-dimethoxybenzoyl chloride (1.5 g, 7.5 mmol) in dichloro-methane (3 ml) at -78 'C. The solution was allowed towarm to 20 'C over 2 h, and evaporated; then, aftercooling to -78 'C, methanol (2 ml, 50 mmol) wasadded. After 30 min at 20 'C, water (10 ml) was addedand the whole was stirred for 2 h (pH 1). Washing withdiethyl ether (3 x 15 ml) and freeze-drying left a whitesolid (121 mg, 39 %). N.m.r.: &H (2H20) 250 MHz 6.7(3H, m, C6H3), 3.75 (6H, s, 2 x OCH3) and 2.70 p.p.m.(2H, s, CH2B); &B (2H20/[2Hjmethanol) 80 MHz 28.7p.p.m. (s); 8C (2H20/[2Hjmethanol) 63 MHz 171.2(C=0), 160-105 (aromatics, 4 signals), 56.9 (OCH03) and33.0 p.p.m. (broad, CH2B). M.s.: m/z (e.i.) 267 (M+).
Measurement of Ilactamase activityThe activity of fl-lactamase I was measured with
nitrocefin (O'Callaghan et al., 1972) as substrate by thechange in absorbance at 500-580 nm, with either a Cary219 or a Cecil CE 373 spectrophotometer. The activity ofthe Pseudomonas /J-lactamase was measured withcephalosporin C [recrystallized as described by Newton &Abraham (1956)] as substrate, the change in absorbanceat 260-280 nm being recorded.
CryoenzymologyThe apparatus was as described previously (Bicknell &
Waley, 1985); solutions were mixed by stirring slowly byhand for 1 min with a plastic rod of square cross-section.The volume of added solution containing enzyme was nogreater than 20,u, and the temperature had returned toits original value within 3 min. The inhibition of f,-lactamase I by boronic acids, with nitrocefin (25-50 #M)as substrate, at -30 °C was studied with 50 %-saturatedammonium acetate (the solution saturated at 4 °C wasdiluted with an equal volume of water) (Cartwright &Waley, 1987). The Pseudomonas ,J-lactamase was addedas a solution in 50% (v/v) ethylene glycol to thesubstrate in 40% (v/v) dimethyl sulphoxide, the enzymebeing insufficiently soluble in the latter mixture. Theconcentration of the substrate (cephalosporin C) was1 mm, about 0.6 x Km.The enzymes were shown to be stable during catalysis
by the method of Selwyn (1965).
Stopped-flow spectrophotometryExperiments were carried out at 25 °C in a Hi-Tech
SF-42 (Hi-Tech Scientific, Salisbury, Wilts., U.K.)instrument, with automatic data acquisition. Use ofsignal averaging to improve the ratio of signal to noisewas important in several experiments. The hydrolysis ofcephalosporin C (40 /LM, about 0.8 x Km) by the Pseudo-monas fl-lactamase was carried out in 0.1 M-NaCl/20 mM-Mops/NaOH buffer, pH 7. The hydrolysis ofnitrocefin (50 uM, about 0.7 x Km) by /J-lactamase I wascarried out in 0.5 M-NaCl/20 mM-Mes/NaOH buffer,pH 6.
pH-dependence of the inhibition constantThese experiments with ,8-lactamase I were carried out
at 30 °C in the following buffers, in 0.5 M-NaCl: Mes/NaOH (pH 5-6.5), Mops/NaOH (pH 6.5-8) and tri-ethanolamine/HCl (pH 8-9). Initial rates were deter-mined from progress curves (Duggleby, 1985) and fittedto Hanes plots to obtain Km(app /kcat. Parallel plots inthe presence or absence of inhibitor confirmed thatinhibition was competitive.
RESULTSpH-dependence of inhibition of &Iactamase by boronicacids
Inhibition constants for the effect of BzB (I) on thehydrolysis of nitrocefin by ,-lactamase I were determinedas follows. Progress curves were used to obtain initialrates by the method of Duggleby (1985), in which non-linear regression is used, with-the -final absorbance beingtreated as an unknown parameter. The non-linearregression was again. used to fit. the data. to theMichaelis-Menten equation; the program of Duggleby
14988
454
Inhibition of serine /1-lactamases by boronic acids
4.75'
0.QL
pH
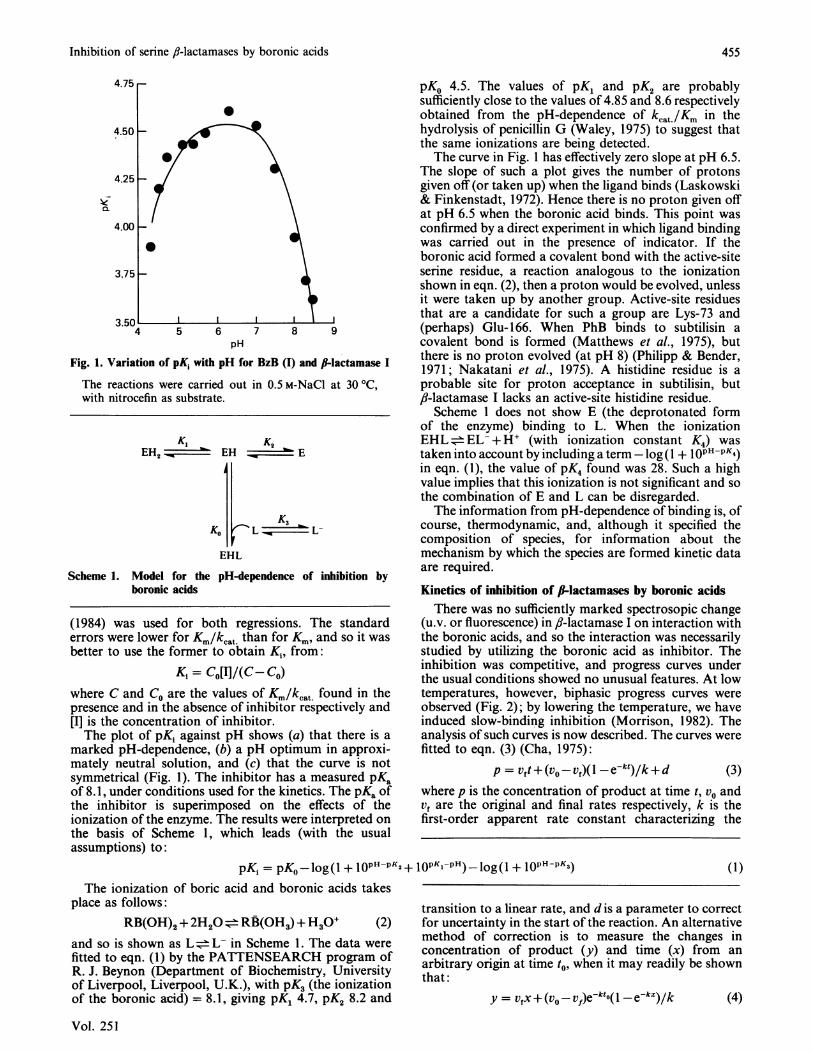
Fig. 1. Variation of pKI with pH for BzB (I) and I-lactamase I
The reactions were carried out in 0.5 M-NaCl at 30 °C,with nitrocefin as substrate.
K, K2EH, _ EH ~ E
Ko
EHL
Scheme 1. Model for the pH-dependence of inhibition byboronic acids
(1984) was used for both regressions. The standarderrors were lower for Km/kcat than for Ki,m and so it wasbetter to use the former to obtain K,, from:
Ki = CO[I]/(C- CO)where C and CO are the values of Km/kcat found in thepresence and in the absence of inhibitor respectively and[I] is the concentration of inhibitor.The plot of pK, against pH shows (a) that there is a
marked pH-dependence, (b) a pH optimum in approxi-mately neutral solution, and (c) that the curve is notsymmetrical (Fig. 1). The inhibitor has a measured pKaof 8.1, under conditions used for the kinetics. The pKa ofthe inhibitor is superimposed on the effects of theionization of the enzyme. The results were interpreted onthe basis of Scheme 1, which leads (with the usualassumptions) to:
pKo 4.5. The values of pK, and pK2 are probablysufficiently close to the values of 4.85 and 8.6 respectivelyobtained from the pH-dependence of kcat/Km in thehydrolysis of penicillin G (Waley, 1975) to suggest thatthe same ionizations are being detected.The curve in Fig. 1 has effectively zero slope at pH 6.5.
The slope of such a plot gives the number of protonsgiven off (or taken up) when the ligand binds (Laskowski& Finkenstadt, 1972). Hence there is no proton given offat pH 6.5 when the boronic acid binds. This point wasconfirmed by a direct experiment in which ligand bindingwas carried out in the presence of indicator. If theboronic acid formed a covalent bond with the active-siteserine residue, a reaction analogous to the ionizationshown in eqn. (2), then a proton would be evolved, unlessit were taken up by another group. Active-site residuesthat are a candidate for such a group are Lys-73 and(perhaps) Glu-166. When PhB binds to subtilisin acovalent bond is formed (Matthews et al., 1975), butthere is no proton evolved (at pH 8) (Philipp & Bender,1971; Nakatani et al., 1975). A histidine residue is aprobable site for proton acceptance in subtilisin, butfl-lactamase I lacks an active-site histidine residue.Scheme 1 does not show E (the deprotonated form
of the enzyme) binding to L. When the ionizationEHL= EL- + H+ (with ionization constant K4) wastaken into account by including a term - log( + lOPHPK4)in eqn. (1), the value of pK4 found was 28. Such a highvalue implies that this ionization is not significant and sothe combination of E and L can be disregarded.The information from pH-dependence of binding is, of
course, thermodynamic, and, although it specified thecomposition of species, for information about themechanism by which the species are formed kinetic dataare required.
Kinetics of inhibition of fl-lactamases by boronic acidsThere was no sufficiently marked spectrosopic change
(u.v. or fluorescence) in fl-lactamase I on interaction withthe boronic acids, and so the interaction was necessarilystudied by utilizing the boronic acid as inhibitor. Theinhibition was competitive, and progress curves underthe usual conditions showed no unusual features. At lowtemperatures, however, biphasic progress curves wereobserved (Fig. 2); by lowering the temperature, we haveinduced slow-binding inhibition (Morrison, 1982). Theanalysis of such curves is now described. The curves werefitted to eqn. (3) (Cha, 1975):
p = Vft+ (Vo-Vf)(l -e-kt)/k+ d (3)where p is the concentration of product at time t, vo andVf are the original and final rates respectively, k is thefirst-order apparent rate constant characterizing the
pKi = pKo log(1 + 1OPHPK2+ lOPKiPH)_log(l + 1OPH-PK3)The ionization of boric acid and boronic acids takes
place as follows:RB(OH)2 + 2H20 =RB(- H3) + H30+ (2)
and so is shown as L L- in Scheme 1. The data werefitted to eqn. (1) by the PATTENSEARCH program ofR. J. Beynon (Department of Biochemistry, Universityof Liverpool, Liverpool, U.K.), with pK3 (the ionizationof the boronic acid) = 8.1, giving pK1 4.7, pK2 8.2 and
transition to a linear rate, and d is a parameter to correctfor uncertainty in the start of the reaction. An alternativemethod of correction is to measure the changes inconcentration of product (y) and time (x) from anarbitrary origin at time t, when it may readily be shownthat:
y = Vfx + (v0 - vf)ekto(1- ekx)/k (4)
Vol. 251
(1)
455
I. E. Crompton and others
0.14
0.12
(a)
0.10 H
04 r (b)
-iZL
0~
25 50Time (min)
75
0.2 I
0 40 80 120Time (ms)
160 200
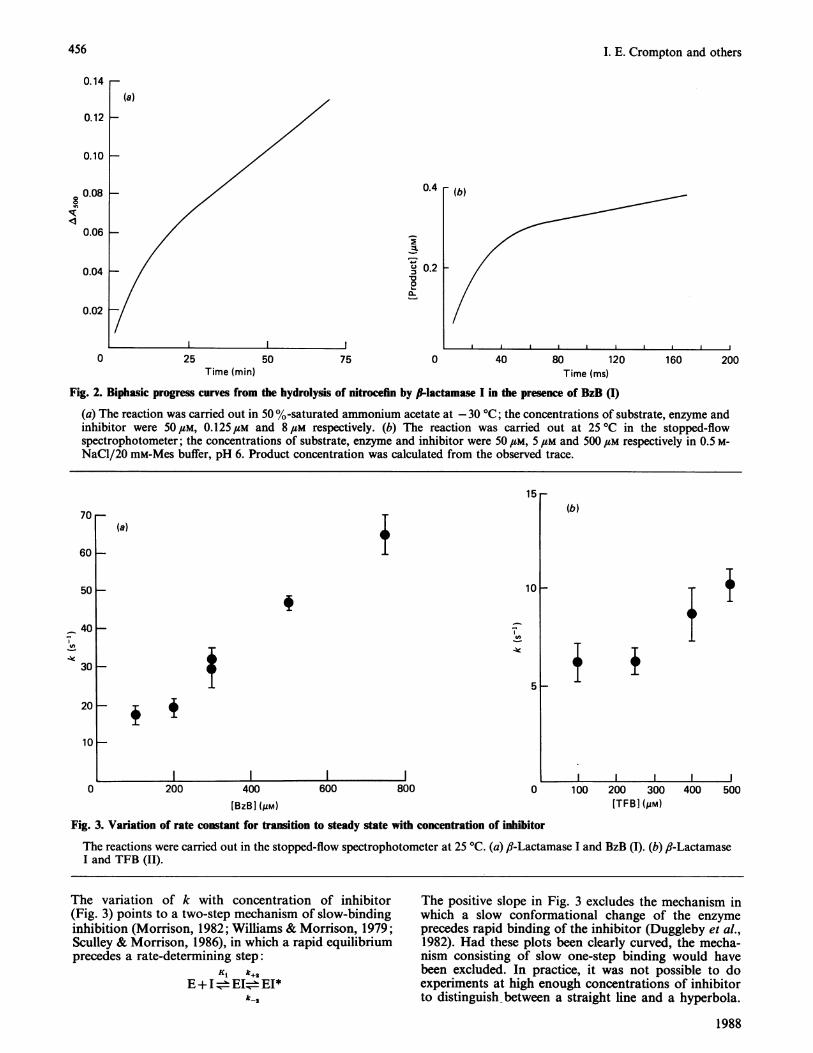
Fig. 2. Biphasic progress curves from the hydrolysis of nitrocefin by Ilactamase I in the presence of BzB (1)(a) The reaction was carried out in 50 %-saturated ammonium acetate at -30 °C; the concentrations of substrate, enzyme andinhibitor were 50 FM, 0.125#M and 8#M respectively. (b) The reaction was carried out at 25 °C in the stopped-flowspectrophotometer; the concentrations of substrate, enzyme and inhibitor were 50 FM, 5 FM and 500 FM respectively in 0.5 M-NaCI/20 mM-Mes buffer, pH 6. Product concentration was calculated from the observed trace.
15-r
4
200 400
[BzB] (#iM)
600
(b)
101
4
I
800
5
0 100 200 300[TFB] (pM)
400 500
Fig. 3. Variation of rate constant for transition to steady state with concentration of inhibitor
The reactions were carried out in the stopped-flow spectrophotometer at 25 'C. (a) ,-Lactamase I and BzB (I). (b) f,-LactamaseI and TFB (II).
The variation of k with concentration of inhibitor(Fig. 3) points to a two-step mechanism of slow-bindinginhibition (Morrison, 1982; Williams & Morrison, 1979;Sculley & Morrison, 1986), in which a rapid equilibriumprecedes a rate-determining step:
Ki k+2
E+I=±EI=EI*k-2
The positive slope in Fig. 3 excludes the mechanism inwhich a slow conformational change of the enzymeprecedes rapid binding of the inhibitor (Duggleby et al.,1982). Had these plots been clearly curved, the mecha-nism consisting of slow one-step binding would havebeen excluded. In practice, it was not possible to doexperiments at high enough concentrations of inhibitorto distinguish between a straight line and a hyperbola.
1988
0.08 _-
0.06 H
0.04 H
0.02 -/
0
(a)70
60 _
50 H
40 H
30 H
20 H
10
0
456
Inhibition of serine fl-lactamases by boronic acids
However, one-step slow binding is not a chemicallysatisfactory mechanism for interaction of protein with aligand: the encounter rate must be large. Additionally,v0 showed dependence on i in accordance with eqn. (6).The overall dissociation constant, K.*, is then related tothe dissociation constant of the first step, Ki, by:
K* = Ki. k -2k+2+k-2
non-linear regression. Then v0 and vf were used to obtainKi and K1* from eqns. (6) and (7):
Vmax [S]- [S]+ Km(l + i/Ki)
Vmax.[S][S] + Km(l + i/Ki*)
(6)
(7)(5)
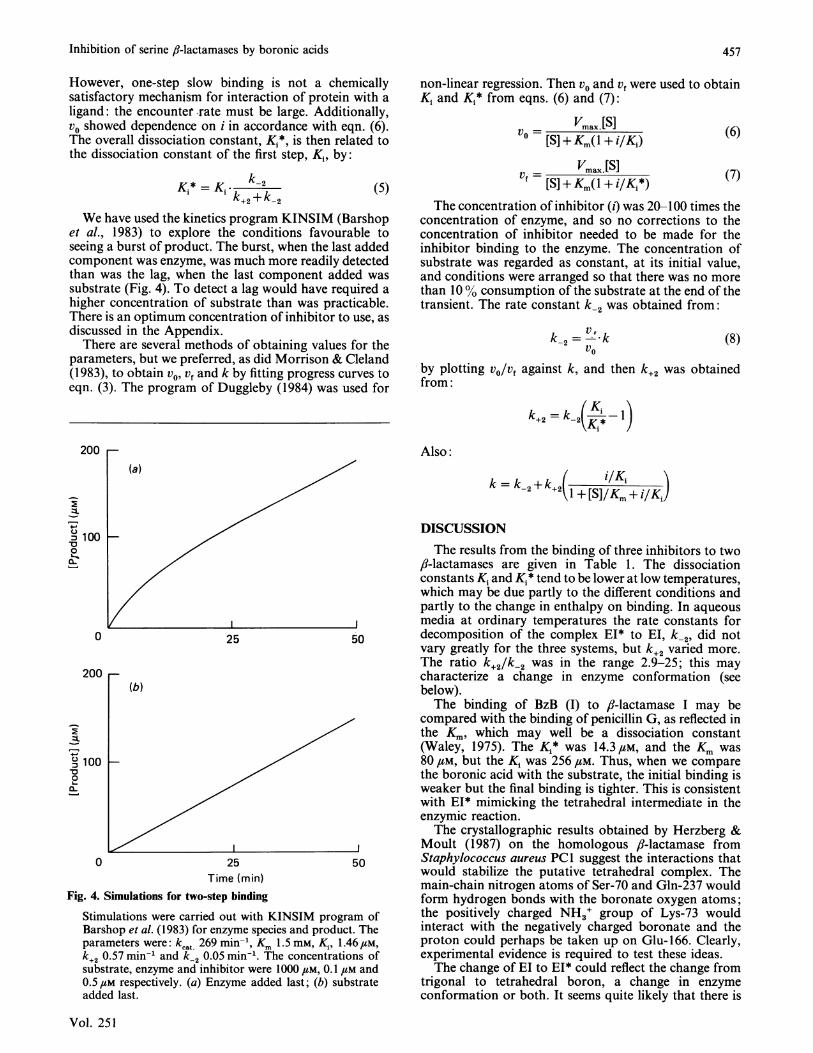
We have used the kinetics program KINSIM (Barshopet al., 1983) to explore the conditions favourable toseeing a burst of product. The burst, when the last addedcomponent was enzyme, was much more readily detectedthan was the lag, when the last component added wassubstrate (Fig. 4). To detect a lag would have required ahigher concentration of substrate than was practicable.There is an optimum concentration of inhibitor to use, asdiscussed in the Appendix.
There are several methods of obtaining values for theparameters, but we preferred, as did Morrison & Cleland(1983), to obtain v0, vf and k by fitting progress curves toeqn. (3). The program of Duggleby (1984) was used for
The concentration of inhibitor (i) was 20-100 times theconcentration of enzyme, and so no corrections to theconcentration of inhibitor needed to be made for theinhibitor binding to the enzyme. The concentration ofsubstrate was regarded as constant, at its initial value,and conditions were arranged so that there was no morethan 100 consumption of the substrate at the end of thetransient. The rate constant k2 was obtained from:
V-k2= V.V.k2vO (8)
by plotting v0/v, against k, and then k+2 was obtainedfrom:
+2 __KI - )
200
(a)
Z1.
m 100'a0
0 25
200(b)
2.Z.)
, 100 _-00,L
0 25Time (min)
Fig. 4. Simulations for two-step binding
50
50
Stimulations were carried out with KINSIM program ofBarshop et al. (1983) for enzyme species and product. Theparameters were: kcat 269 min-', Km 1.5 mm, K,, 1.46,UM,k+2 0.57 min-' and k-2 0.05 min-1. The concentrations ofsubstrate, enzyme and inhibitor were 1000 /LM, 0.1 LM and0.5,M respectively. (a) Enzyme added last; (b) substrateadded last.
Also:
2++2(1 + [S]/Km + iKi)
DISCUSSIONThe results from the binding of three inhibitors to two
/?-lactamases are given in Table 1. The dissociationconstants Ki and K1* tend to be lower at low temperatures,which may be due partly to the different conditions andpartly to the change in enthalpy on binding. In aqueousmedia at ordinary temperatures the rate constants fordecomposition of the complex EI* to El, k-2, did notvary greatly for the three systems, but k+2 varied more.The ratio k+2/k-2 was in the range 2.9-25; this maycharacterize a change in enzyme conformation (seebelow).The binding of BzB (I) to ,-lactamase I may be
compared with the binding of penicillin G, as reflected inthe K., which may well be a dissociation constant(Waley, 1975). The Kj* was 14.3,M, and the Km was80 /M, but the K, was 256 /M. Thus, when we comparethe boronic acid with the substrate, the initial binding isweaker but the final binding is tighter. This is consistentwith EI* mimicking the tetrahedral intermediate in theenzymic reaction.The crystallographic results obtained by Herzberg &
Moult (1987) on the homologous ,-lactamase fromStaphylococcus aureus PCI suggest the interactions thatwould stabilize the putative tetrahedral complex. Themain-chain nitrogen atoms of Ser-70 and Gln-237 wouldform hydrogen bonds with the boronate oxygen atoms;the positively charged NH3+ group of Lys-73 wouldinteract with the negatively charged boronate and theproton could perhaps be taken up on Glu-166. Clearly,experimental evidence is required to test these ideas.The change of El to EI* could reflect the change from
trigonal to tetrahedral boron, a change in enzymeconformation or both. It seems quite likely that there is
Vol. 251
457
458 I. E. Crompton and others
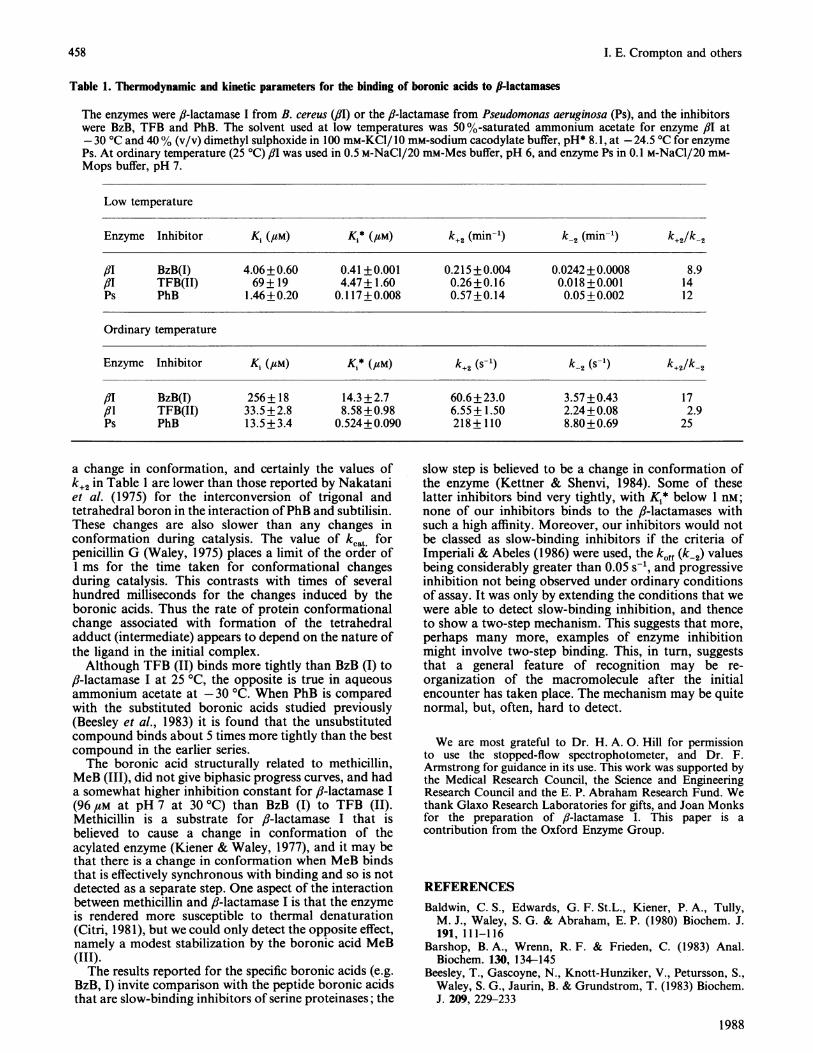
Table 1. Thermodynamic and kinetic parameters for the binding of boronic acids to II-lactamases
The enzymes were fl-lactamase I from B. cereus (flu) or the fi-lactamase from Pseudomonas aeruginosa (Ps), and the inhibitorswere BzB, TFB and PhB. The solvent used at low temperatures was 50 %-saturated ammonium acetate for enzyme flu at-30 °C and 40% (v/v) dimethyl sulphoxide in 100 mM-KCI/10 mM-sodium cacodylate buffer, pH* 8.1, at -24.5 °C for enzymePs. At ordinary temperature (25 °C) ,81 was used in 0.5 M-NaCI/20 mM-Mes buffer, pH 6, and enzyme Ps in 0.1 M-NaCl/20 mM-Mops buffer, pH 7.
Low temperature
Enzyme Inhibitor K1 (,UM) K1* (#M) k+2 (min-') k_2 (min-') k+2/k-2
,81 BzB(I) 4.06+0.60 0.41 +0.001 0.215+0.004 0.0242+0.0008 8.9/3I TFB(II) 69+ 19 4.47+1.60 0.26+0.16 0.018+0.001 14Ps PhB 1.46+0.20 0.117+0.008 0.57+0.14 0.05+0.002 12
Ordinary temperature
Enzyme Inhibitor K, (,UM) Ki* (4aM) k+2 (s-') k-2 (S-1) k+2/k-2
fIT BzB(I) 256+ 18 14.3+2.7 60.6+23.0 3.57+0.43 17/31 TFB(II) 33.5 +2.8 8.58 +0.98 6.55+ 1.50 2.24+0.08 2.9Ps PhB 13.5+3.4 0.524+0.090 218+110 8.80+0.69 25
a change in conformation, and certainly the values ofk+2 in Table 1 are lower than those reported by Nakataniet al. (1975) for the interconversion of trigonal andtetrahedral boron in the interaction ofPhB and subtilisin.These changes are also slower than any changes inconformation during catalysis. The value of kcat forpenicillin G (Waley, 1975) places a limit of the order ofI ms for the time taken for conformational changesduring catalysis. This contrasts with times of severalhundred milliseconds for the changes induced by theboronic acids. Thus the rate of protein conformationalchange associated with formation of the tetrahedraladduct (intermediate) appears to depend on the nature ofthe ligand in the initial complex.
Although TFB (II) binds more tightly than BzB (I) to,?-lactamase I at 25 °C, the opposite is true in aqueousammonium acetate at -30 'C. When PhB is comparedwith the substituted boronic acids studied previously(Beesley et al., 1983) it is found that the unsubstitutedcompound binds about 5 times more tightly than the bestcompound in the earlier series.The boronic acid structurally related to methicillin,
MeB (III), did not give biphasic progress curves, and hada somewhat higher inhibition constant for fl-lactamase I(96#M at pH 7 at 30 C) than BzB (I) to TFB (II).Methicillin is a substrate for ,J-lactamase I that isbelieved to cause a change in conformation of theacylated enzyme (Kiener & Waley, 1977), and it may bethat there is a change in conformation when MeB bindsthat is effectively synchronous with binding and so is notdetected as a separate step. One aspect of the interactionbetween methicillin and ,-lactamase I is that the enzymeis rendered more susceptible to thermal denaturation(Citri, 1981), but we could only detect the opposite effect,namely a modest stabilization by the boronic acid MeB(III).The results reported for the specific boronic acids (e.g.
BzB, I) invite comparison with the peptide boronic acidsthat are slow-binding inhibitors of serine proteinases; the
slow step is believed to be a change in conformation ofthe enzyme (Kettner & Shenvi, 1984). Some of theselatter inhibitors bind very tightly, with K1* below 1 nM;none of our inhibitors binds to the ,3-lactamases withsuch a high affinity. Moreover, our inhibitors would notbe classed as slow-binding inhibitors if the criteria ofImperiali & Abeles (1986) were used, the koff (k-2) valuesbeing considerably greater than 0.05 s-', and progressiveinhibition not being observed under ordinary conditionsof assay. It was only by extending the conditions that wewere able to detect slow-binding inhibition, and thenceto show a two-step mechanism. This suggests that more,perhaps many more, examples of enzyme inhibitionmight involve two-step binding. This, in turn, suggeststhat a general feature of recognition may be re-organization of the macromolecule after the initialencounter has taken place. The mechanism may be quitenormal, but, often, hard to detect.
We are most grateful to Dr. H. A. 0. Hill for permissionto use the stopped-flow spectrophotometer, and Dr. F.Armstrong for guidance in its use. This work was supported bythe Medical Research Council, the Science and EngineeringResearch Council and the E. P. Abraham Research Fund. Wethank Glaxo Research Laboratories for gifts, and Joan Monksfor the preparation of ,-lactamase I. This paper is acontribution from the Oxford Enzyme Group.
REFERENCESBaldwin, C. S., Edwards, G. F. St.L., Kiener, P. A., Tully,M. J., Waley, S. G. & Abraham, E. P. (1980) Biochem. J.191, 111-116
Barshop, B. A., Wrenn, R. F. & Frieden, C. (1983) Anal.Biochem. 130, 134-145
Beesley, T., Gascoyne, N., Knott-Hunziker, V., Petursson, S.,Waley, S. G., Jaurin, B. & Grundstrom, T. (1983) Biochem.J. 209, 229-233
1988

Inhibition of serine /J-lactamases by boronic acids 459
Berks, M., Redhead, K. & Abraham, E. P. (1982) J. Gen.Microbiol. 128, 155-159
Bicknell, R. & Waley, S. G. (1985) Biochemistry 24, 6876-6887Cartwright, S. J. & Waley, S. G. (1984) Biochem. J. 221,
505-512Cartwright, S. J. & Waley, S. G. (1987) Biochemistry 26,
5329-5337Cha, S. (1975) Biochem. Pharmacol. 24, 2177-2185Citri, N. (1981) in Beta-Lactam Antibiotics (Mitsuhashi, S.,
ed.), pp. 225-232, Springer-Verlag, New YorkDavies, R. B., Abraham, E. P. & Melling, J. (1974) Biochem. J.
143, 115-127Dobozy, O., Mile, I., Ferecz, I. & Csanyi, V. (1971) Acta
Biochim. Biophys. Acad. Sci. Hung. 6, 97-105Duggleby, R. G. (1984) Comput. Biol. Med. 14, 447-455Duggleby, R. G. (1985) Biochem. J. 228, 55-60Duggleby, R. G., Attwood, P. V., Wallace, J. C. & Keech,
D. B. (1982) Biochemistry 21, 3364-3370Flett, F., Curtis, N A. C. & Richmond, M. H. (1976) J.
Bacteriol. 127, 1585-1586Herzberg, 0. & Moult, J. (1987) Science 236, 694-701Imperiale, B. & Abeles, R. H. (1986) Biochemistry 25,
3760-3767Kettner, C. A. & Shenvi, A. B. (1984) J. Biol. Chem. 259,
15106-15112Kiener, P. A. & Waley, S. G. (1977) Biochem. J. 165, 279-285Kiener, P. A. & Waley, S. G. (1978) Biochem. J. 169, 197-204Laskowski, M. & Finkenstadt, W. R. (1972) Methods Enzymol.
17, 193-227Linquist, R. N. & Nguyen, A. C. (1977) J. Am. Chem. Soc. 99,
6435-6437Matteson, D. S. & Cheng, T.-C. (1968) J. Org. Chem. 33,
3055-3060Mattheson, D. S., Sadhu, K. M. & Lienhard, G. E. (1981)
J. Am. Chem. Soc. 103, 5241-5242Matthews, D. A., Alden, R. A., Birktoft, J. J., Freer, S. T. &
Kraut, J. (1975) J. Biol. Chem. 250, 7120-7126Morrison, J. F. (1982) Trends Biochem. Sci. 7, 102-105Morrison, J F. & Cleland, W. W. (1983) Biochemistry 22,
5507-5513
Nakatani, H., Uehara, Y. & Hiromi, K. (1975) J. Biochem.(Tokyo) 78, 611-616
Newton, G. G. F. & Abraham, E. P. (1956) Biochem. J. 62,651-665
O'Callaghan, C. H., Morris, A., Kirby, S. M. & Shingler,A. H. (1972) Antimicrob. Agents Chemother. 1, 283-288
Philipp, M. & Bender, M. L. (1971) Proc. Natl. Acad. Sci.U.S.A. 68, 478-480
Sculley, M. J. & Morrison, J. F. (1986) Biochim. Biophys. Acta874, 44-53
Selwyn, M. J. (1965) Biochim. Biophys. Acta 105, 193-195Waley, S. G. (1975) Biochem. J. 149, 547-551Williams, J. W. & Morrison, J. F. (1979) Methods Enzymol.
63, 437-467
APPENDIXConditions for a high burst numberThe size of the burst in slow-binding inhibition may be
defined in terms of the dimensionless burst number, B,as: v -v
k.eoUse of eqn. (8) in the text then gives:
B = vf[1 -(v,/v0)]/(k-2 . eo)Then use of eqns. (6) and (7) in the text followedby partial differentiation leads to the condition that(aB/ai) = 0 is that:
iopt. = Ki*(1 + [S]/Km)Moreover, the largest burst number is then:
Vmax. i(l/Ki*1-/Ki\opt. 4k2 e0' 1 + i/Ki
If i > K, and K,* << K, then:
Bopt. = kcat./4k-2
Received 14 August 1987/19 October 1987; accepted 4 December 1987
Vol. 251


















