
FigS1 legend · Autoclave • 1 M Sodium phosphate buffer pH 7.0: 39% (v/v) 1M NaH2PO4; 61% (v/v)...
18
Transcript of FigS1 legend · Autoclave • 1 M Sodium phosphate buffer pH 7.0: 39% (v/v) 1M NaH2PO4; 61% (v/v)...






Reynoso-MappingT-DNAInsertionSitesProtocol
1
MappingofT-DNAinsertionsitesusingsequencecaptureandIllumina-MiseqMauricioA.Reynoso/Bailey-SerresLab,CenterforPlantCellBiology,UCRiverside
PurposeandBackgroundThepurposeof thismethod is to localizeT-DNA insertionsites in transgenicplants.Thisprocedurewastested with DNA from Oryza sativa L. ssp. Japonica (cv. Nipponbare). The protocol involves thehybridizationandcaptureofsequencescontainingT-DNAbordersusingbiotinylatedoligos.ThisallowsforanenrichmentofjunctionreadscarryingbothT-DNAandgenomicDNA(gDNA)sequences.Insertionsinmultiple(25ormore)transgenicplantscanbelocalizedinasinglesequencingrun.Thismethodincludesthebioinformaticanalyses.
Materials• 100mgyoungleaves• Bioruptorsonicator(Diagenode)orCovarisS2• AgencourtAMPureXPbeads(BeckmanCoulter)• Low-bindmicrocentrifugetube• SavantSpeedvac• DynabeadsM-270StreptavidinorDynabeadsMyOneStreptavidinC1(LifeTechnologies)• Illumina library preparation reagents (required for end-repairing, dA tailing, ligation and PCR
amplification).• Qubitflourometer(LifeTechnologies)• Agilent2100BioAnalyzer(AgilentTechnologies)• 1.5mLmicrocentrifugetubemagnet• Rotator/nutatormixer
Solutionsandbuffers• 40%PEG-8000• 5MNaCl• ElutionBuffer:10mMTris-HClpH7.5• 40XDenhart’ssolution:BovineSerumAlbumin0.8%;Ficoll4000.8%;Polyvinylpyrrolidone0.8%.
Dissolveinwater.Filtertosterilize.Aliquotandstoreat-20°C• 20XSalineSodiumCitrate (SSC)buffer:3MNaCl;0.3MNa-Citrate.Adjust topH7.0withHCl.
Autoclave• 1MSodiumphosphatebufferpH7.0:39%(v/v)1MNaH2PO4;61%(v/v)1MNa2HPO4
• 5 μg/μl Salmon Sperm DNA: use sterile forceps to collect the DNA and dissolve in water.Autoclavefor15mintofragmentDNA
• 2XHybridizationbuffer:0.5MSodiumphosphatebufferpH7.0;1%SDS;2mMEDTA;2XSSC,4XDenhardt’ssolution
• 2XBindandWashBuffer:10mMTris-HClpH7.5;2MNaCl;1mMEDTA;0.1%Tween-20• WashBuffer1:1XSSC;0.1%SDS• WashBuffer2:0.1XSSC;0.1%SDS• WashBuffer3:0.2XSSC• 0.15MNaOH• 1MTris-HClpH8.0
BiotinylatedandblockingoligosThe biotinylated oligos were designed for the T-DNA borders of H-TRAP and H-INTACT based on theexperience from Soichi and Isabelle Henry from Comai lab (UC Davis). There is a pair of 70 nt

Reynoso-MappingT-DNAInsertionSitesProtocol
2
oligonucleotidesforeachborderandtheyoverlap35ntwitheachother.The3’endoftheoligosRB1andRB2contain16ntand51ntmatchingwiththerightboder,respectively.The5’sequenceoftheoligosLB1andLB2contain67ntand32ntmatchingtheleftborder,respectively.Thebiotinylatedoligosaredilutedtoa3μMworkingsolution.>RB15’/5BiotinTEG/CTTGTGATATCACTAGTGCGGCCGCCTGCAGGTCGACCATATGGGAGAGCTCAAGCTTAGCTTGAGCTTG-3’>RB25’/5BiotinTEG/CCATATGGGAGAGCTCAAGCTTAGCTTGAGCTTGGATCAGATTGTCGTTTCCCGCCTTCAGTTTAAACTA-3’>LB15’/5BiotinTEG/TTGACGCTTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATT-3’>LB25’/5BiotinTEG/CGTTTTTAATGTACTGAATTAACGCCGAATTGAATTATCAGCTTGCATGCCGGTCGATCTAGTAACATAG-3’DNALibrariesmadefromdifferentplantswillsharetheuniversaladapterandapartoftheindexadapter.Duringthehybridizationtheseadapterscaninteractcausingcrosshybridizationandnon-specificbindingto the probes. To prevent this effect the hybridization mix contains an excess of adapter oligos thathybridize with the adapter region of molecules of the library protecting from cross hybridization. Theblocking oligos contain amodification at the 3’ end prevent amplification in the PCR enrichment thatfollows hybridization. These oligos should be designed to match the adapters used during librarypreparation.Theblockingoligosareusedat1mMconcentration.>UniversalOligoBlockerAATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT/3SpC3/-3'>IndexOligoBlocker'-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT/3SpC3/-3'ThesixNbasescorrespondtotheindexzone.IfusingeightntbarcodethisstretchshouldcontaineightNs.PrimersPrimersneededfortheenrichmentafterhybridization:>IlluminaP5AATGATACGGCGACCACCGA>IlluminaP7CAAGCAGAAGACGGCATACGAPrimersusedtocalculateefficiencyofthehybridization:>qRB_1685_fwdGCTTTTTTGTACAAACTTGTGAT>qRB_1796_revCTGAAGGCGGGAAACGACA>qLB_8408_fwdTGACGCTTAGACAACTTAATAACACAT>qLB_tNos_8527_RevGCGCGCGGTGTCATCT Theprimersareusedat10μMconcentration

Reynoso-MappingT-DNAInsertionSitesProtocol
3
DNAextraction1. Extract genomic DNA from transgenic plants using your regular lab protocol. 100mg of tissue
shouldgiveenoughmaterialforDNAlibrarypreparation.2. CheckDNAqualitybyelectrophoresisin1.2%agarosegels.3. QuantifyDNAconcentrationusingaNanodropspectrophotometer.
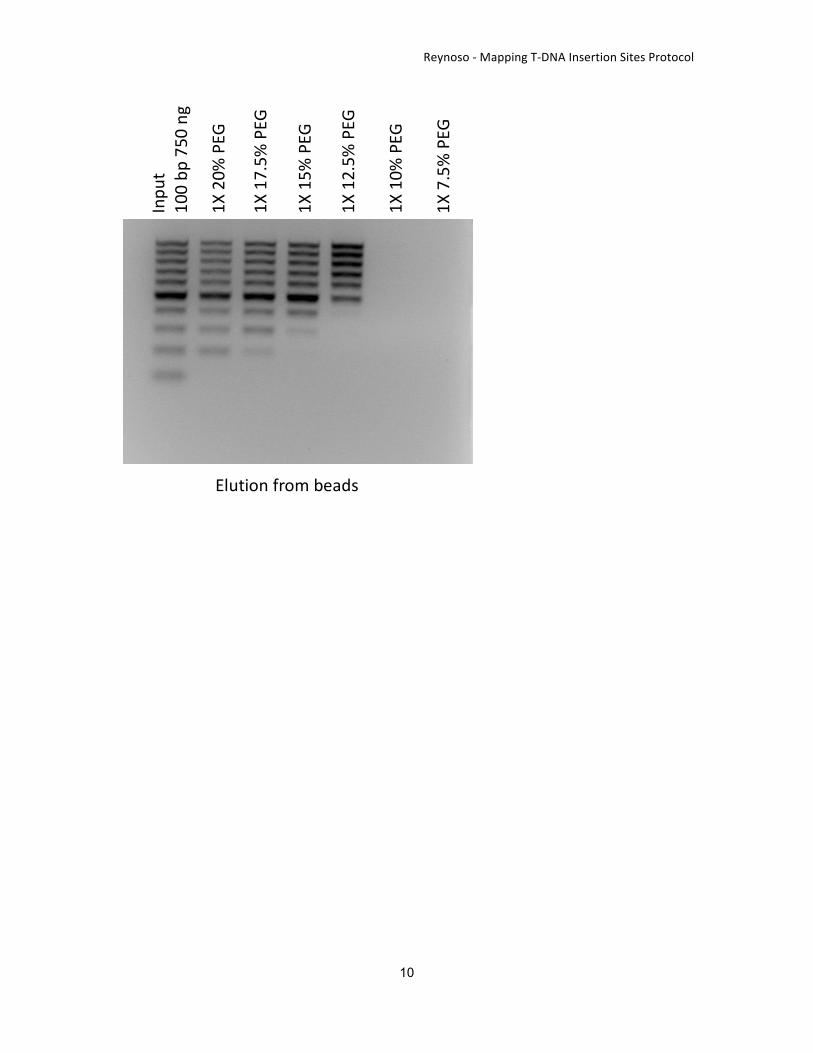
DNAfragmentationandpurificationFragmentationandpurificationofgenomicDNAsamplesfromtransgenicplants.FollowthenextstepsforeachDNAsample.Note: PEG content on AMPure XP beads binding buffers can be modified to obtain a distribution offragmentsrequiredforyourIlluminasequencing(seefigureattheendofthedocument).Inthiscaseweselected DNA fragments of approximately 400bp and sequenced using Illumina MiSeq to get 250bppaired-endreads.
1. DiluteonemicrogramofgenomicDNAto100μLusingDNAsefreewater.2. Sonicate at 4°C in a Bioruptor water bath sonicator for 25 min with power set to ‘low’ and
sonicationintervalssetto30secon/30secoff.Replacethewaterwithfresh4°Cwaterevery5minutes.
3. Wash15μLofAMPureXPbeads.Collecttothemagnetandresuspendin15μL20%PEG-8000and2.5MNaCl.Collectthebeadsagainandresuspendin100μLofasolution12%PEG-8000;2.5MNaCl. This bufferwill allow the binding of fragments bigger than 500 bp. (Note: amount ofbeadscanbeoptimized).
4. BindfragmentedDNAwiththeresuspendedAMPureXPbeadsfromthepreviousstepbyaddingtoalow-bindingmicrocentrifuge.Mixwellandincubatefor6minatroomtemperature.
5. Collectbeadstothemagnetandtransferthesupernatanttoanewtube.ThissolutioncontainsDNAfragmentsofsmallerthan500bp.
6. Wash15μLofAMPureXPbeads.Resuspendin15μL20%PEG-8000and2.5MNaCl.Collectthebeadstothemagnetandresuspendin13.33μLofasolutionof30%PEG-8000and1.25MNaCl.(Noteamountofbeadscanbeoptimized).
7. Add13.33μLofresuspendedbeadstothesupernatantofstep5.ThissolutionwillincreasetheconcentrationofPEG-8000to7.5%allowing for thebindingofDNAfragmentsbigger than400bp.
8. Incubatefor6minatroomtemperaturetoallowDNAbindingtoAMPureXPBeads.Collectthebeadstothemagnet.Washtwicefor30secwith150μL80%ethanol.Evaporatetheremainingethanolfor10minatroomtemperature.ElutewiththevolumerequiredinyourIlluminalibrarypreparationprotocolattheEnd-Repairstepoflibrarypreparation.
IlluminaLibraryPreparationFollow your library preparation protocol starting at the end repair step. Perform End-Repair and dAtailing.LigateadifferentbarcodeorindexforeachDNAsample.PurifytwicewithAMPureXPbeadstocaptureligatedfragmentsandavoidadapterdimers.Amplifyfragmentswith10cyclesofPCR.PurifywithAMPureXPbeads.Note:Concentrationofenzymesand incubationtimescanbemodified (increasedwithrespect toRNA-seqprotocol)toimprovetheefficiencyoflibrarypreparation.Qualitycontrolandlibrarypooling1.MeasuretheconcentrationofeachlibraryusingaQubit®Fluorometer(LifeTechnologies).
2.Run1μLoflibraryonabioanalyzer(Bio-RadExperion™orAgilent2100)usingaDNA1Kchip.Takenoteofconcentrationandtheaveragefragmentlengthoflibrary.
Optionally:PerformlibraryquantificationusingtheKAPALibraryQuantificationKit.
3.Basedontheconcentrationandaveragefragmentlengthforeachlibrary,poolthelibrariestoobtainatotalof500ngofDNA.

Reynoso-MappingT-DNAInsertionSitesProtocol
4
Forexample,whenmultiplexing25samples,addtoamicrocentrifugetube20ngofeachDNAlibrarytoobtain500ngofDNAforhybridization. It ispossibletoincreasethetotalamountofDNAto1μgwhenmultiplexingmoresamples.Note:Toevaluateefficiencyofthecaptureit ispossibletopool5%moreofDNAforeachsample(1ngmoreintheexample)andstorethatextravolumeat-20°Ctouseafterthecapture.4.Dryout500ngofpooledDNAlibrariesusingaSpeedvacconcentrator.AfterdryingtheDNA,resuspendin2μLofwater.HybridizationofpooledlibrariesThis part of the protocol ismodified from thexGen® Target Capture ProtocolProtocol forDNAProbeHybridizationandTargetCaptureUsinganIlluminaLibrary IDT Hybridizebiotinylatedoligostotargetsequencesinpooledlibraries.a.Combinethefollowinginalow-bind0.2mLPCRtube:3pmoleachbiotinylatedoligo(3pmol/μL;1μLperoligo) 4.0μL500ngIlluminalibrary(drieddown,andthenresuspendedinwaterto125ng/μL) 2.0μL10μgSalmonSpermDNA(5μg/μL) 2.0μL1mMUniversalOligoBlocker 1.0μL1mMIndexOligoBlocker 1.0μLTotalVolume 10μLb.Add10μL2XHybridizationBufferandvortex.c.Overlaywith40μLmineraloil.DONOTvortexafteroverlayingwithoil.
IMPORTANT:Itisessentialthatyouoverlaywithmineraloiltoavoidsignificantvolumeloss.d.Denaturethemixtureinathermalcyclerat95°Cfor5min,andthenslowlydecreasethetemperature(0.1°C/sec)to62°C.DONOTusetheheatedlidofthethermalcycler.e.Incubateat62°Cfor48hr.ThisreactionwillbeusedinStep2.e.(below).PurificationofcapturedDNA1.Bindhybridizedtargetsequencestostreptavidinbeads.a.AllowDynabeadsMyOneStreptavidinC1toequilibratetoroomtemperaturefor30min.b.Pipet50μLC1streptavidinbeads intoa fresh1.7mL low-bindmicrocentrifuge tubeandwash twicewith50μL2XBindandWashBuffer.Useapipettetoremovethebufferaftereachwash.c.CombinethefollowingtomakeBeadResuspensionBuffer2XBindandWashBuffer 50μLNuclease-FreeWater 30μLTotalVolume 80μLd.Resuspendthewashedbeadsin80μLBeadResuspensionBuffer(from1.c.).e.Recoverthehybridizationreaction(20μL)fromunderthemineraloil(stepe.;above),beingcarefulnottowithdrawanymineraloil,andaddtotheresuspendedbeads.Hint:A gel-loading pipette tipmakes it easy to remove the liquid fromunder themineral oil. To avoidtrappinganymineraloilinthetip,leaveasmallamountofpositivepressureinthetipuntilreachingthemineraloil/hybridizationsolution interface.Expel thesmallamountofoil in the tipbeforeremovingthehybridizationmixture.f.Placethetubeontuberotator/nutatormixerfor30minatroomtemperaturetobindthebiotinylatedcaptureprobestothestreptavidinbeads.2.WashthestreptavidinbeadstoremoveunboundDNA.

Reynoso-MappingT-DNAInsertionSitesProtocol
5
a.Brieflyspindownthetubefromstep1.f.(above)andplaceonamagneticseparationrack.Allowbeadstoseparatefromthesupernatant.Usingapipette,removethesupernatantcontainingunboundDNAanddiscard.b.Washthebeadswiththefollowingsolutions,sequentially:i.1000μLWashBuffer1for5min,62°Cwithrotationii.1000μLWashBuffer2for5min,62°Cwithrotationiii.1000μLWashBuffer2for5min,62°Cwithrotationiv.1000μLWashBuffer2for5min,RTwithrotationv.1000μLWashBuffer3for30sec,RTwithtubeonmagnet;keeptubeonmagnetEnsurethateachsolutionhasbeenpre-equilibratedtotheappropriatewashtemperature.Addthestatedvolumeofeachwashbuffer to thetubeandplaceonabenchtoptuberotator/nutatormixerfortheindicatedamountoftime.Pulsespin(1–3sec,800xg)tocollecttheliquidwithoutpelletingthebeads.Placetubeonmagneticholdertoattractthemagneticbeads.Removewashbufferusingapipette.
IMPORTANT!Afterthefinalwash(v),donotremovetubefrommagnet!RemoveALLoftheWashBuffer3fromthetubeafterthefinalwash.SSCpresent inthebuffermayreact with NaOH used in subsequent steps to produce a precipitate that will interfere withdownstreammanipulations.
c.Add50μL125mMNaOH(freshlydilutedfromamoreconcentratedstocksolution)andincubateatRTfor10min.Vortexevery2mintokeepbeadsinsuspension.d.Returnthetubetothemagnetfor1min.Leavethetubeonthemagnetwhileyouperformthenextsteps.e.Add50μL1MTris-HCl,pH8.8,toanew1.7mLlow-bindtube.f. Keeping the tube on themagnet, transfer the supernatant (d) to the tube of 1M Tris-HCl (e). ThisneutralizestheNaOHaddedtothehybridizationmixture.g.Add1.5X volumes (150μL)Agencourt®AMPure®XPbeads (Beckman-Coulter). Proceedaccording tothemanufacturer’sprotocol,elutingin22μLElutionBuffer.h.Transfer20μLofelutedproductintoanew1.7mLlow-bindtube,ensuringnobeadsarecarriedover.QuantificationoftheefficiencyofhybridizationThisstepisrecommendedbutcanbeomitted.PerformquantitativePCRonpooledlibrariesfrombeforeandafterhybridization.
a. Take1μLofelutedproductanddilute to10μL inElutionbuffer.ThisDNAcorresponds to5%fractionofthetotalelutedproduct.
b. Take5%oftheDNAfrompooledlibrariesfrominputofthehybridization.Intheexamplethatis25ngofpooledlibrariesas1/20(fractionofoutputusedforquantification)*500ng(input)=25ng.
c. QuantifytheabundanceofRBandLBendsforbothsamples.PerformqPCRwiththeprimersfortheRBandLBindicatedinmaterialsection.
d. UsedilutionsofavectorcontainingRBandLBtocalculateprimerefficiencyinqPCR.e. NormalizeCt valuesusingaprimerefficiency curveandcalculate the ratiosofefficiencyofRB
andLBcapture.Thiscanbeobtainedbydividingthenormalizedvaluesforthecapturedsamplestothoseobtainedfortheinputsample.
PCRenrichmentandqualitycontrol1.Post-CapturePCRThe eluate from step h allows retention of a small amount of captured library for troubleshooting, ifnecessary.Otherwise,thelibrarycanbeelutedinasmallervolumeandtheentireamountusedinthefinalPCRenrichment.Perform final PCR enrichment. This reaction can be done using the same PCR reagents used for PCRenrichmentduringthepreparationofthelibraries.

Reynoso-MappingT-DNAInsertionSitesProtocol
6
a.Preparethefollowingreactionmixinalow-bind0.2mLPCRtube:2XKAPAHiFi™HotStartReadyMix 25μL10μMIlluminaP5Primer 2.5μL10μMIlluminaP7Primer 2.5μLNuclease-FreeWater 4μLElutedcapturelibrary 16μLTotalVolume 50μLb.Brieflyvortexthetube,andthenspinbrieflyinamicrocentrifugetocollectthereactionmixtureatthebottomofthetube.c.Placereactionsinathermalcyclerandrunthefollowingprogram:
1. 98°C 5min2. 98°C 20sec3. 60°C 15sec4. 72°C5. Cycle14timestostep2
30sec
6. 72°C 5min7. 4°C Hold
d. Purify the fragments using 1.5X volume (75 μL) AMPure XP beads. Proceed according to themanufacturer’sprotocol,elutingin22μLElutionBuffer.e.Transfer20μLofelutedproductintoafresh1.7mLlow-bindtube,ensuringnobeadsarecarriedover.Note:someprotocolsuseasecondstepofhybridizationwiththePCRenrichedcaptureDNA,thiswasnottestedbutcanbeconsideredanalternativewhenobtainingalowefficiencyofcapture.2.Qualitycontrola.MeasuretheconcentrationofcapturedlibraryusingaQubit®Fluorometer(LifeTechnologies).b.Run1μLoflibraryonabioanalyzer(Bio-RadExperion™orAgilent2100)usingaDNA1Kchip.Takenoteoftheaveragefragmentlength.c.SubmitsampleforIlluminaMiSeq.(Weused250bppaired-endreads).SequencingandBioinformaticAnalysesTwoapproacheswere taken to analyze the 250bppaired-end reads, obtaining similar results. JérémieBazin fromBailey-Serres labhelpedon thedesignof theanalysis.Forbothcasespaired-end reads thatoverlappedwerecollapsedtoonelongerread.The firstmethod selects sequences that contain a fraction of the left or right border. These reads arelocallymapped to the genome, i. e. a part of the sequence align to the genome. Then, regionswith asignificant coverage of reads are selected to indicate potential insertions. Reads aligned to candidateregionsarealignedbacktotheT-DNAtodeterminewhichportionofitwasincorporatedinthegenome(indicatingthesiteofjunctionbetweenT-DNAandgDNA).1. Mergepaired-endreadscontainingoverlappingsequencestoobtainlongerreads###MERGEPAIRSINRusingFLASHmodule###mytargetsfq<-read.delim("your_directory/targets_lane1_fastq.txt",sep="\t")mypair1=mytargetsfq$FileName1mypair2=mytargetsfq$FileName2sample=mytargetsfq$SampleNamezip=paste0("flash",mypair1[1],"",mypair2[1],"-m12-M250-o",sample[1],"--suffix=flash")for(iinseq(along=mypair2)){

Reynoso-MappingT-DNAInsertionSitesProtocol
7
zip=paste0("flash",mypair1[i],"",mypair2[i],"-m12-M250-o",sample[i])system(zip)}2. Filterreadscontainingbordersequences####RUNINUNIXusingGREPmodule###foriin*extendedFrags.fastq;do grep --no-group-separator -B 1 -A 2 -E
"CTAGTGCGGCCGCCTGCAGGTCGACCATATGGGAGAGCTC|GAGCTCTCCCATATGGTCGACCTGCAGGCGGCCGCACTAG|TTATCAGCTTGCATGCCGGTCGATCTAGTAAC|GTTACTAGATCGACCGGCATGCAAGCTGATAA"$i>$i.grep;done
3. Mapreadscontainingbordersequencesagainstthegenome###inUNIXmakegenomefileaccessibleforBOWTIE2#####bowtie2-buildgenome.congenome.fa###ALIGNSELECTEDREADSFROMPREVIOUSSTEPInRusingBOWTIE2module####setwd("/your_directory")library(ShortRead);library(rtracklayer); library(GenomicRanges);
library(Rsamtools);library(GenomicAlignments);library(DESeq);library(edgeR);library(ShortRead);library(chipseq);library(lattice);library(ggbio)targets=read.delim("target.txt",header=T)input=paste0(targets$path,".grep")setwd("/your_directory")for(iinseq(along=input)){bowtie=paste0("bowtie2 --local -p 8 --very-sensitive -x /your_directory/genome.fa -U ", input[i]," |
samtoolsview-bS->",paste0(targets$SampleName[i],".bam"))system(bowtie)# -g: ignoreallalginmentswith>gmatches; -p:numberofthreadstouseforalignment
step;-i/-I:min/maxintronlengths;--segment-length:lengthofsplitreads(25isdefault)}4. Findregionswithcandidateinsertions####EXTRACTTHEPEAKSFOREACHSAMPLEANDWRITEATABLEinR####result=data.frame()for(iinseq(along=input)){alignedReads<-readGAlignments(paste0(targets$SampleName[i],".bam"))coverage<-coverage(alignedReads)#calculategenomiccoverageandreturnsanIRangesRleobjectpeakCutoff(coverage,fdr=0.0001)peaks<-slice(coverage,lower=100)peaks.sum<-peakSummary(peaks)#SummarizesthepeakdatainaRangedDataobject.df=as.data.frame(peaks.sum)df$sample=targets$SampleName[i]result=rbind(result,df)write.table(result,"insertions.xls",sep="\t")}5. AlignreadsofeachcandidateregiontothevectortodefinethesiteofjunctionbetweenT-DNAand
gDNA

Reynoso-MappingT-DNAInsertionSitesProtocol
8
##EXTRACTBAMFILESalignedtoinsertionswithSAMTOOLSsam_input<-read.delim("insertions.xls",sep="\t")chr=sam_input$spacefrom=sam_input$startto=sam_input$endsample=sam_input$samplefor(iinseq(along=chr)){ sam=paste0("samtools view -b ",sample[i],".bam ",chr[i],":",from[i],"-",to[i]," -o
",sample[i],"_ins",i,".bam")system(sam)}
##CONVERTBAMTOFASTQinUNIXusingBEDTOOLSmodules##targets=read.delim("insertions.xls",sep="\t")input=paste0(targets$sample,"_ins",targets$n,".bam")output=paste0(targets$sample,"_ins",targets$n,".fastq")for(iinseq(along=input)){bam2fastq=paste0("bedtoolsbamtofastq-i",input[i],"-fq",output[i])system(bam2fastq)#inUNIXmakevectorfileaccessibletoBOWTIE2aligner##bowtie2-buildvector.faTDNA.fa###ALIGNFASTQREADSFROMMAPPEDinRusingBOWTIE2module####setwd("/bigdata/mreynoso/data/illumina.bioinfo.ucr.edu/illumina_runs/299")library(ShortRead);library(rtracklayer); library(GenomicRanges);
library(Rsamtools);library(GenomicAlignments);library(DESeq);library(edgeR);library(ShortRead);library(chipseq);library(lattice);library(ggbio)targets=read.delim("insertions.xls",sep="\t")input=paste0(targets$sample,"_ins",targets$n,".fastq")output=paste0(targets$sample,"_ins",targets$n,"_vec.bam")setwd("/bigdata/mreynoso/data/illumina.bioinfo.ucr.edu/illumina_runs/299/")for(iinseq(along=input)){bowtie=paste0("bowtie2 --local -p8 --very-sensitive -xTDNA.fa -U ", input[i]," | samtools view -bS ->
",output[i])system(bowtie)#-g:ignoreallalginmentswith>gmatches;-p:numberofthreadstouseforalignment
step;-i/-I:min/maxintronlengths;--segment-length:lengthofsplitreads(25isdefault)}####EXTRACTTHEPEAKSFOREACHSAMPLESANDWRITEATABLEinR####targets=read.delim("insertions.xls",sep="\t")input=paste0(targets$sample,"_ins",targets$n,".fastq")output=paste0(targets$sample,"_ins",targets$n,"_vec.bam")name=paste0(targets$sample,"_ins",targets$n)result=data.frame()for(iinseq(along=input)){alignedReads<-readGAlignments(output[i])coverage<-coverage(alignedReads)#calculategenomiccoverageandreturnsanIRangesRleobjectpeaks<-slice(coverage,lower=100)peaks.sum<-peakSummary(peaks)#SummarizesthepeakdatainaRangedDataobject.df=as.data.frame(peaks.sum)df$sample=name[i]result=rbind(result,df)write.table(result,"insertions_in_vector.xls",sep="\t")

Reynoso-MappingT-DNAInsertionSitesProtocol
9
The secondmethod selects reads that do not align completely (end-to-end) to the genome to do theanalysis.Thosereadsarethenlocallymappedtothegenome,i.e.alignpartofthereadtothegenome.Then, regions with a significant coverage of reads are extracted. And finally, reads mapping to eachcandidateregionsarealignedtotheT-DNAtodeterminethepresenceofvectorsequencesandthesiteofjunctionbetweenT-DNAandgDNA.1. Selectreadsthatdonotaligncompletelytothegenome###SELECTREADSTHATDONOTMAPCOMPLETELYTOTHEGENOMEinRusingBOWTIE2module####setwd("/your_directory")library(ShortRead);library(rtracklayer); library(GenomicRanges);library(Rsamtools);library(GenomicAlignments);library(DESeq);library(edgeR);library(ShortRead);library(chipseq);library(lattice);library(ggbio)targets=read.delim("target.txt",header=T)input=targets$pathfor(iinseq(along=input)){bowtie=paste0("bowtie2 --end-to-end -p 8 -D 20 -R 3 -N 1 -L 20 -i S,1,0.50 -x/your_directory/genome.fa", " --un ", input[i], ".unaligned ","-U ", input[i]," | samtools view -bS - >",paste0(input[i],".bam"))system(bowtie)}Afterthispointtheanalysiscontinueswithsteps3,4and5describedforthepreviousapproachusingtheunalignedfastQfileproducedbythescriptabove.Note: It is important to verify that putative insertions do not map to sequences present in thetransformationvectorthatmaptothegenome(inourcaseOsRPL18andpromoters).TheinsertionsshouldbetestedbyPCRusingprimersthatamplifythejunctionbetweenT-DNAandgDNA.Primers qRB_1685_fwd and qLB_tNos_8527_Rev can be used for this purpose together with a primerdesignedforeachsideofinsertion,RBandLBrespectively.
Reynoso-MappingT-DNAInsertionSitesProtocol
10
Inpu
t 10
0bp
750
ng
1X20%
PEG
1X17.5%
PEG
1X15%
PEG
1X12.5%
PEG
1X10%
PEG
1X7.5%PEG
Elutionfrombeads

Kajala–rRNAdegradationprotocol
1
TargetedrRNAdegradationOriginal:GregSmaldonefromSingerlab,re-writtenbyKaisaKajalaoftheSiobhanBradylab,UCDavis.
PurposeandBackgroundDegraderRNAfromatotalRNAsamplewherepoly-ApurificationofmRNAcannotbedone;e.g.,INTACTnuclei.
Materials• Sampletissue/TotalRNA/RNeasyMicrokit(Qiagen)• TurboDNasekit(ThermoFisherAM2238)• AgencourtRNACleanXPbeads• ProbesdesignedagainstrRNAsequences(fornuclei;theseare5S,5.8S,18Sand25SrRNAs,probesat60bp
length,reversecomplementstherRNAsequences,orderedasamixat10nmolin1000ulofwater).Prepareaworkingmixwitheachprobeat1uM.
• HybridaseThermostableRNaseH(Epicenter#H39500)• RNAsefreePCRstrips• MagneticrackforPCRstrips• PCRmachine
Buffersandsolutions:• 70%ethanol• RNease-freewater• 5xHybridizationbufferH1(0.5Tris-HClpH7.0,1MNaCl;RNase-free;filtersterilizedandfrozen)• 10xHybridasebufferH2(0.5Tris-HClpH7.4,1MNaCl,200mMMgCl2;RNase-free;filtersterilizedandfrozen)
Procedure1. RNAisolationwithQiagenRNeasyMicrokitFornucleifromINTACT:
• PurifythenuclearRNAusingtheQiagenRNeasyMicrokit.Startbyadding350uloflysisbufferRLT(with10ulB-ME/1mladded)tothe20ulofpurifiednuclei.Vortexvigorouslyfor2min.
• Centrifugethelysateat1,000g(3,100rpm)for2minatRTtopelletthebeads.Useamagnettohelptransferthesupernatanttoanew1.5mltube,add350ulof70%ethanolandvortexseveraltimestomix.
• Pipettelysate/ethanolmixtureintoaRNeasyMinElutespincolumnrestingina2mlcollectiontubeandcentrifugeat10,000g(9,700rpm)for1minatRT.Discardflowthrough.
• Add350ulofbufferRW1tothecolumn.Centrifugeat10,000g(9,700rpm)for1minatRT.Discardflowthroughandmovethecolumntoanew2mlcollectiontube.
• Add500ulofbufferRPEtothecolumn.Centrifugeat10,000g(9,700rpm)for1minatRT.Discardflowthrough.• Add500ulof80%ethanoltothecolumn.Centrifugeat10,000g(9,700rpm)for1minatRT.Discardflowthrough
andmovethecolumntoanew2mlcollectiontube.• Openthecolumnlidandcentrifugeattopspeed(16,000g)for5minatRT.Discardtheflowthroughandplacethe
columnintoanew1.5mltube.• Add20ulRNase-freewaterontothecolumnmembraneandallowtostandfor1mnin.Centrifugeat16,000gfor
1minatRT.• StoreRNAat-80C.• QuantificationcanbedoneusingRiboGreenRNAquantitationkit,expectedyieldis100-500ng.

Kajala–rRNAdegradationprotocol
2
2. DNaseItreatment(eliminategenomicDNAcontamination)• UseDNaseIprotocolforTurboDNaseI• Addto20ulofRNA:
2ulof10xDNaseIreactionbuffer1uLDNaseI
• Incubate30minat37°C• Add2uLDNaseIinactivationreagent(vortexwellbeforeadding)andincubate5minatRT(vortexevery1min)• Spindown(2000gfor5min)andrecover20uLintoanewtube.
3. AgencourtRNACleanXPbeadcleanup.
• Add1.8volume(e.g.18ulfor10ulofRNA)ofRNACleanXPbeadstorxn• Spliteachreactionintoupto140uLaliquots• IncubateatRTfor10min• Ontomagnetfor5min• Removemostofthesupernatant(leave5ulbehindtoavoidpullingupbeads)• Leavetubesonmagnet,add200uL70%EtOH,letstandfor30s• Removeallthesupernatant• Repeat70%washasaboveoncemore(twowashestotal)• RemoveasmuchoftheEtOHaspossible• Airdrybeadsonmagnetfor10min(untilappearlightincolour)• Add15uLRNase-freewatertothefirstaliquotdriedbeadandmix• Ifyouhavealiquotedsamples,poolbacktogether.• IncubateatRTfor5min• Ontomagnetfor5min• Recover15uLeluate
4. NanoDropfor[RNA]and260/280ifyouwanttocrushyourdreams.
5. rRNAprobehybridization(bindDNAprobestorRNA)
• Startwith3ugtotalRNA(rxnvolumemustbe~6uL,3x1ugrxnsisokay).Orstartwithwhateveryouhave.• Addfollowingtogether:
1.2ul5xHybridizationbufferH1(0.5MTris-HCl(pH7.0),1MNaCl,RNase-free)1.0ulprobemixat1µM/oligo–thisconcentrationisfor1-3ugofRNA;bringdownforloweramounts.3.8ulRNA(makeupwithRNase-freewater)
• Incubateat95°Cfor2min,rampdownto45°Cat0.1°C/s,45°Cfor5min,holdat45°C6. Hybridase®(thermostableRNaseH)reaction(digestRNAofanRNA:DNAhybrid)
• Prepareamastermixforyoursamplesthatyoupreheatto45°C(inahotblock):1ul10xHybridasebufferH2(500mMTris-HClpH7.4,1MNaCl,200mMMgCl2)1ulHybridase(5U/uL)2ulRNase-freeH2O
• Add4uloftheMMtohybridizationreactionstillat45°C.• Incubateat45°Cfor30min.• Removetoice
7. DNaseItreatment(digestDNAoligosfrommRNApool)
• UseDNaseIprotocolforTurboDNaseI• Addto40ulofRNA:
4ulof10xDNaseIreactionbuffer2uLDNaseI
• Incubate30minat37°C

Kajala–rRNAdegradationprotocol
3
• Add2uLDNaseIinactivationreagent(vortexwellbeforeadding)andincubate5minatRT(vortexevery1min)• Spindown(2000gfor5min)andrecover43uLintoanewtube.
8. AgencourtRNACleanXPbeadcleanup.
• Add1.8volume(e.g.72ulfor40ulofRNA)ofRNACleanXPbeadstorxn• Spliteachreactionintoupto140uLaliquots• IncubateatRTfor10min• Ontomagnetfor5min• Removemostofthesupernatant(leave5ulbehindtoavoidpullingupbeads)• Leavetubesonmagnet,add200uL70%EtOH,letstandfor30s• Removeallthesupernatant• Repeat70%washasaboveoncemore(twowashestotal)• RemoveasmuchoftheEtOHaspossible• Airdrybeadsonmagnetfor10min(untilappearlightincolour)• Add10uLRNase-freewatertothefirstaliquotdriedbeadandmix• Ifyouhavealiquotedsamples,poolbacktogether• IncubateatRTfor5min• Ontomagnetfor5min• Recover10uLeluate


















