
FDA Compliance Enforcement Actions: What you … · Abbreviated New Drug ... (510(k)) Substantial...
63
FDA Compliance Enforcement FDA Compliance Enforcement Actions: What you need to know Actions: What you need to know for clinical device trials for clinical device trials The 3 The 3 rd rd Annual FDA Regulatory and Compliance Symposium Annual FDA Regulatory and Compliance Symposium Track 3 Track 3 - - Pharma Product Development and Clinical Trials Pharma Product Development and Clinical Trials August 23, 2007 August 23, 2007 Cambridge, MA Cambridge, MA
Transcript of FDA Compliance Enforcement Actions: What you … · Abbreviated New Drug ... (510(k)) Substantial...

FDA Compliance Enforcement FDA Compliance Enforcement Actions: What you need to know Actions: What you need to know
for clinical device trialsfor clinical device trials
The 3The 3rdrd
Annual FDA Regulatory and Compliance Symposium Annual FDA Regulatory and Compliance Symposium Track 3Track 3--
Pharma Product Development and Clinical TrialsPharma Product Development and Clinical TrialsAugust 23, 2007August 23, 2007Cambridge, MACambridge, MA

The 3The 3rdrd
Annual FDA Regulatory Annual FDA Regulatory and Compliance Symposiumand Compliance Symposium
PRESENTED BY:PRESENTED BY:Sonali P. Gunawardhana M.P.H., J.D., Sonali P. Gunawardhana M.P.H., J.D., LL.M.LL.M.
Regulatory CounselRegulatory Counsel Food and Drug AdministrationFood and Drug Administration
Center for Devices and Radiological Center for Devices and Radiological HealthHealth
Office of ComplianceOffice of Compliance Division of Bioresearch MonitoringDivision of Bioresearch Monitoring

Devices vs. Drugs Devices vs. Drugs
How do studies with investigational How do studies with investigational devices differ from those with drugs and devices differ from those with drugs and biologics?biologics?
Nature of industryNature of industryStatutory distinctionsStatutory distinctionsRegulatory distinctionsRegulatory distinctionsResearch distinctionsResearch distinctions

Device FirmsDevice Firms
Entrepreneurial firms commonEntrepreneurial firms common93% have fewer than 100 employees93% have fewer than 100 employeesVenture capitalizedVenture capitalized
Diverse and specialized productsDiverse and specialized productsPrinciples of operation and intended usesPrinciples of operation and intended uses
Device Device ““developerdeveloper”” often involvedoften involvedMinimal clinical trial experienceMinimal clinical trial experienceRapid product cycles limiting testing timeRapid product cycles limiting testing time

Statutory DistinctionsStatutory Distinctions
Devices lack market exclusivity provisionsDevices lack market exclusivity provisionsWaxmanWaxman--Hatch (drugs)Hatch (drugs)Orphan drug (drugs/biologics)Orphan drug (drugs/biologics)
Differences in standards of approvalDifferences in standards of approval““SubstantialSubstantial”” adequate and welladequate and well--controlled trials (drug)controlled trials (drug)““ReasonableReasonable”” valid scientific evidence (device)valid scientific evidence (device)
Devices must down regulate Devices must down regulate FDAMA (1997) FDAMA (1997) ““least burdensomeleast burdensome”” provisionprovision

WellWell--controlled investigationscontrolled investigationsPartially controlled studiesPartially controlled studiesStudies and objective trials without matched Studies and objective trials without matched controlscontrolsWellWell--documented case histories by qualified documented case histories by qualified expertsexpertsReports of significant human experience with Reports of significant human experience with a marketed devicea marketed device
Valid Scientific Evidence*Valid Scientific Evidence*
* 21 CFR 860.7

Research ApplicationsResearch Applications
Investigational New Drug (IND) applicationInvestigational New Drug (IND) applicationCovers all research (drugs and biologics)Covers all research (drugs and biologics)21 CFR Part 31221 CFR Part 312
Investigational Device Exemption (IDE)Investigational Device Exemption (IDE)Covers significant risk researchCovers significant risk research
Implants, lifeImplants, life--threatening, or sightthreatening, or sight--threatening threatening
21 CFR Part 81221 CFR Part 812

Regulatory DistinctionsRegulatory Distinctions
IDE exempt studiesIDE exempt studiesIn vitro diagnostics (IVDs)In vitro diagnostics (IVDs)In commercial use before May 28, 1976In commercial use before May 28, 1976Consumer preference testingConsumer preference testingSolely for veterinary useSolely for veterinary usePost Approval StudiesPost Approval Studies

Marketing ApplicationsMarketing Applications
New Drug Application New Drug Application (NDA)(NDA)
InnovatorInnovator21 CFR Part 31421 CFR Part 314
Abbreviated New Drug Abbreviated New Drug Application (ANDA)Application (ANDA)
Substantial equivalenceSubstantial equivalence21 CFR Part 31421 CFR Part 314
Biologics Licensing Biologics Licensing Application (BLA)Application (BLA)
InnovatorInnovator21 CFR Part 60121 CFR Part 601
Premarket Approval Application Premarket Approval Application (PMA) (PMA)
New Use, Technology, or Class IIINew Use, Technology, or Class III21 CFR Part 81421 CFR Part 814
Premarket Notification (510(k))Premarket Notification (510(k))Substantial equivalenceSubstantial equivalence21 CFR Part 80721 CFR Part 807
Humanitarian Device Exemption Humanitarian Device Exemption (HDE)(HDE)
Similar to Orphan ProductSimilar to Orphan Product21 CFR Part 81421 CFR Part 814
In Vitro Diagnostics (IVDs)In Vitro Diagnostics (IVDs)21 CFR Part 80921 CFR Part 809

Product DistinctionsProduct Distinctions
vsvs..

Charging for Investigational Charging for Investigational ProductsProducts
Devices: Always have been able to Devices: Always have been able to charge in order to recoup the research charge in order to recoup the research cost. This request for reimbursement is cost. This request for reimbursement is generally submitted in the IDE.generally submitted in the IDE.Drugs: Special request is made for Drugs: Special request is made for reimbursement reimbursement –– this was not the norm in this was not the norm in the past but now there is a move towards the past but now there is a move towards making it easier for reimbursement.making it easier for reimbursement.

Combination ProductsCombination Products
Types of productsTypes of productsDrug/device, biologic/device, drug/biologic, or Drug/device, biologic/device, drug/biologic, or drug/device/biologicdrug/device/biologic
Products are assigned to lead Center based upon Products are assigned to lead Center based upon primary mode of actionprimary mode of action
Other Centers provide consulting reviewsOther Centers provide consulting reviewsProduct is required to follow regulation of lead Product is required to follow regulation of lead CenterCenterImportant to seek early consultationImportant to seek early consultation
FDAFDA’’s Office of Combinations Office of Combination ProductsProducts

Enforcement ActionsEnforcement Actions
REASONS WHY SOME OF THESE REASONS WHY SOME OF THESE ACTIONS ARE IMPLEMENTED:ACTIONS ARE IMPLEMENTED:Untitled Letters/Warning Letters Untitled Letters/Warning Letters Application Integrity Policy/ Integrity Application Integrity Policy/ Integrity HoldHoldNotice of Initiation of Disqualification Notice of Initiation of Disqualification Proceedings and Opportunity to Explain Proceedings and Opportunity to Explain (NIDPOE)(NIDPOE)

Compliance ToolsCompliance Tools
Untitled/Warning letterUntitled/Warning letterReRe--inspectioninspectionInformal conferenceInformal conference3rd party audits3rd party auditsRejection of site dataRejection of site dataDisqualificationDisqualification
CI, IRB, or GLPCI, IRB, or GLP
Invoke Application Invoke Application Integrity Policy or Integrity Integrity Policy or Integrity HoldHoldRevoke marketing or Revoke marketing or research permitresearch permitCivil Money PenaltiesCivil Money PenaltiesInjunctionInjunctionProsecutionProsecution

Untitled LettersUntitled Letters
■■
Untitled Letters are issued when Untitled Letters are issued when substantial violations are documented substantial violations are documented during inspection and requests voluntary during inspection and requests voluntary corrective action.corrective action.
■■Unlike Warning Letters, Untitled Letters Unlike Warning Letters, Untitled Letters are not posted on the FDA website.are not posted on the FDA website.

Warning LettersWarning Letters
The Warning Letter is the agencyThe Warning Letter is the agency’’s principal s principal means of notifying regulated industry of means of notifying regulated industry of violations (prior notice) and achieving prompt violations (prior notice) and achieving prompt voluntary correction.voluntary correction.
The Warning Letter clearly states that if there is The Warning Letter clearly states that if there is a failure to promptly achieve correction the a failure to promptly achieve correction the FDA may take enforcement action without any FDA may take enforcement action without any further notice.further notice.
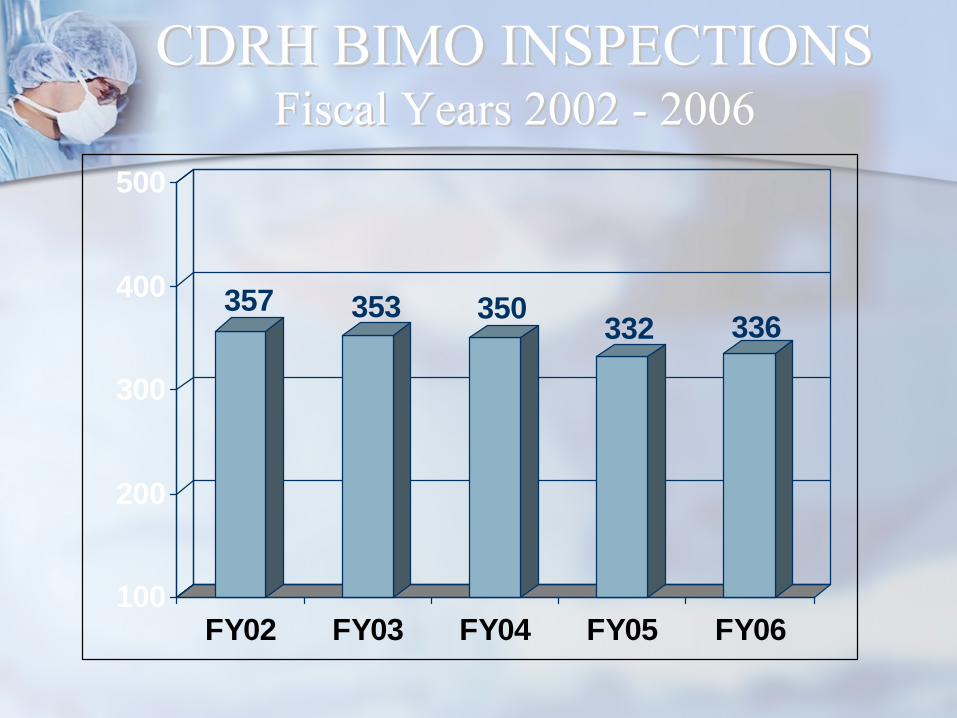
CDRH BIMO INSPECTIONSCDRH BIMO INSPECTIONS Fiscal Years 2002 Fiscal Years 2002 --
20062006
357 353 350 332 336
100
200
300
400
500
FY02 FY03 FY04 FY05 FY06

CDRH BIMO INSPECTIONSCDRH BIMO INSPECTIONS Fiscal Years 2002 Fiscal Years 2002 -- 20062006
Inspected Inspected EntityEntity 20022002 20032003 20042004 20052005 20062006
SponsorSponsor 7272 8181 7373 7070 5353
CICI 151151 170170 183183 183183 200200
IRBIRB 128128 8585 7373 4848 5959
GLPGLP 66 99 1919 3131 2424
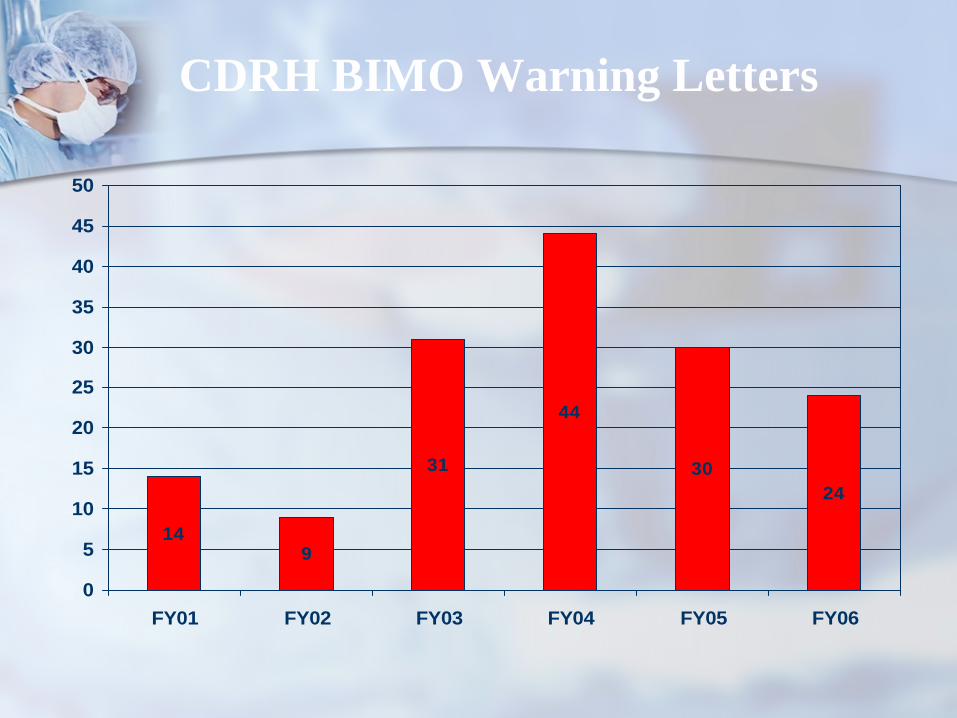
14
44
3024
9
31
0
5
10
15
20
25
30
35
40
45
50
FY01 FY02 FY03 FY04 FY05 FY06
CDRH BIMO Warning Letters
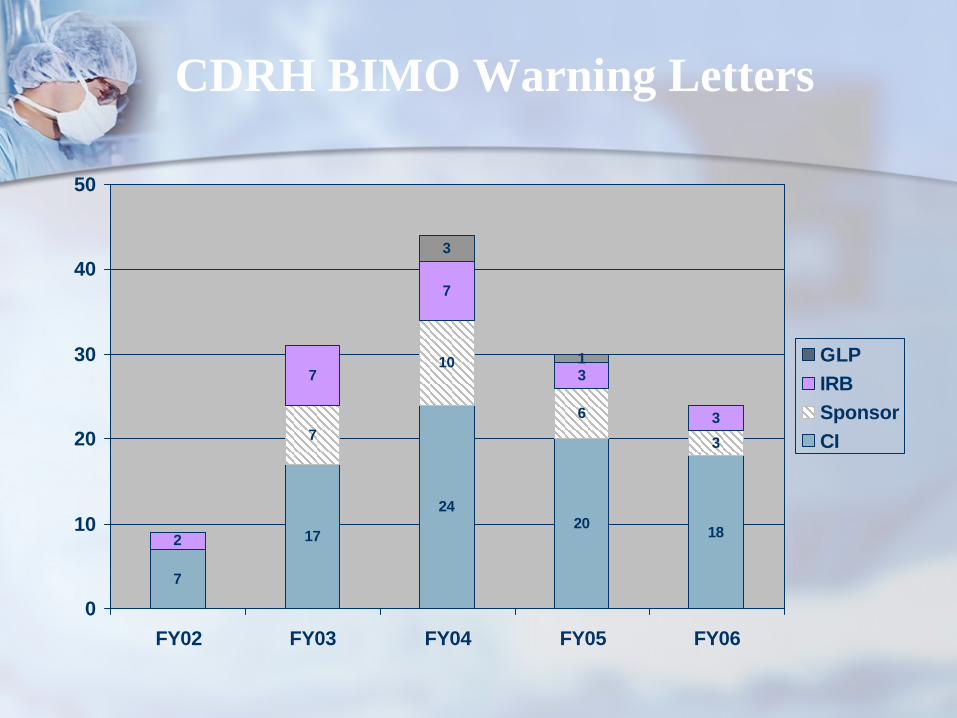
7
20 18
10
6
3
7
3
3
1
24
17
7
2
7
3
0
10
20
30
40
50
FY02 FY03 FY04 FY05 FY06
GLPIRBSponsorCI
CDRH BIMO Warning Letters

CDRH BIMO Compliance RatesCDRH BIMO Compliance Rates
13% 12%17%
24%
15%11%
0%
10%
20%
30%
40%
50%
60%
70%
10 Years FY02 FY03 FY04 FY05 FY06
NAIVAIOAI

CDRH BIMO OAI Rates CDRH BIMO OAI Rates (with & w/o (with & w/o ““For CauseFor Cause”” Inspections)Inspections)
13%
16%
9%
5%
10%
17%
24%
15%
11%
7%
0%
10%
20%
30%
10 Years FY03 FY04 FY05 FY06
OAI (NFC) OAI NFC = No “For Cause” inspections included

CDRH Sponsor Compliance RatesCDRH Sponsor Compliance Rates
19%
10%
24%
31%
15%11%
0%
10%
20%
30%
40%
50%
60%
70%
10 Years FY02 FY03 FY04 FY05 FY06
NAIVAIOAI

CDRH Sponsor Compliance RatesCDRH Sponsor Compliance Rates
19%
10%
24%
31%
11% 10%15%
0%
10%
20%
30%
40%
50%
60%
70%
10Years
FY02 FY03 FY04 FY05 FY06 FY06(NFC)
NAIVAIOAI

Sponsor Deficiencies Sponsor Deficiencies Fiscal Years 1999 Fiscal Years 1999 -- 20062006
FYFY19991999 2000 2000 20012001 20022002 20032003 20042004 2005 2005 20062006
Inadequate Inadequate monitoringmonitoring
65%65% 68%68% 65%65% 33%33% 37%37% 40%40% 24%24% 23%23%
Failure to secure Failure to secure investigator investigator compliancecompliance
27%27% 44%44% 27%27% 19%19% 24%24% 21%21% 15%15% 13%13%
Inadequate Inadequate device device accountabilityaccountability
23%23% 28%28% 19%19% 7%7% 19%19% 16%16% 18%18% 15%15%
Obtain FDA/IRB Obtain FDA/IRB approvalapproval
4%4% 18%18% 11%11% 8%8% 5%5%

CDRH Clinical Investigator CDRH Clinical Investigator Compliance RatesCompliance Rates
11%15% 17%
21%
11%17%
0%
10%
20%
30%
40%
50%
60%
70%
10 Years FY02 FY03 FY04 FY05 FY06
NAIVAIOAI

CDRH Clinical Investigator CDRH Clinical Investigator Compliance RatesCompliance Rates
11%15% 17%
21%17%
11%
7%
0%
10%
20%
30%
40%
50%
60%
70%
10Years
FY02 FY03 FY04 FY05 FY06 FY06(NFC)
NAIVAIOAI


Common Investigator Common Investigator DeficienciesDeficiencies
Follow investigational plan, investigator Follow investigational plan, investigator agreement, or protocolagreement, or protocolProtocol deviationsProtocol deviationsInadequate subject protection or informed Inadequate subject protection or informed consentconsentInadequate device accountabilityInadequate device accountabilityLack of FDA or IRB approvalLack of FDA or IRB approvalInadequate reporting of UADEs to Sponsor or Inadequate reporting of UADEs to Sponsor or IRBIRB
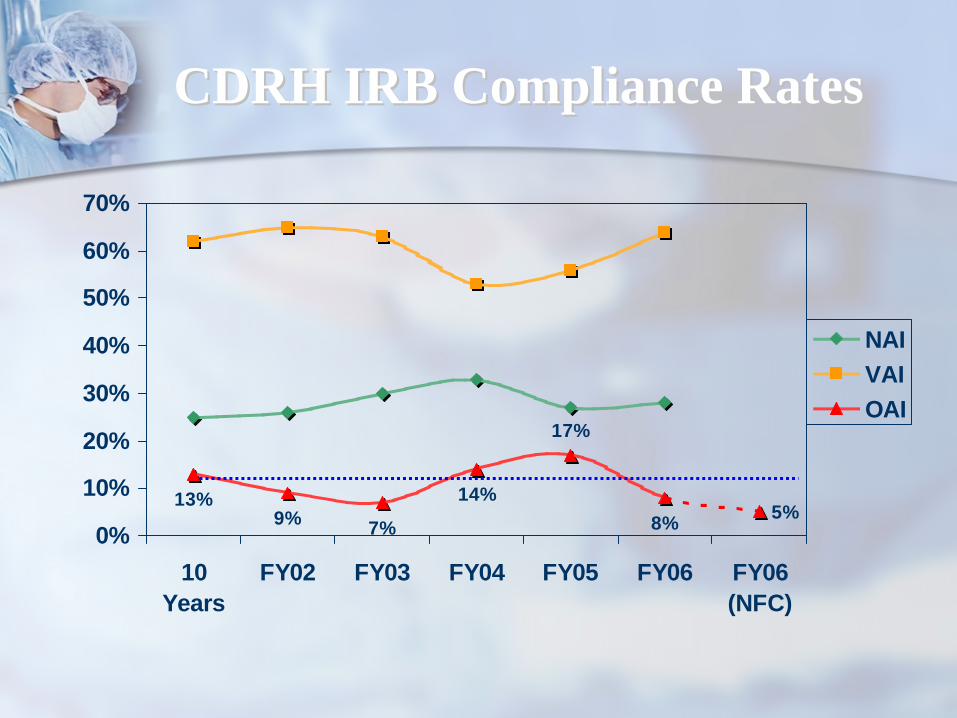
CDRH IRB Compliance RatesCDRH IRB Compliance Rates
13%9%
14%8% 5%
17%
7%0%
10%
20%
30%
40%
50%
60%
70%
10Years
FY02 FY03 FY04 FY05 FY06 FY06(NFC)
NAIVAIOAI

IRB DeficienciesIRB Deficiencies Fiscal Years 1999 Fiscal Years 1999 -- 20062006
FYFY19991999 20002000 20012001 20022002 20032003 20042004 20052005 20062006
Inadequate initial Inadequate initial &/or continuing &/or continuing reviewreview
64%64% 56%56% 39%39% 24%24% 25%25% 50%50% 37%37% 38%38%
Inadequate Inadequate minutesminutes
61%61% 42%42% 35%35% 11%11% 42%42% 28%28% 17%17% 20%20%
Lack of or Lack of or incorrect SR/NSR incorrect SR/NSR determinationdetermination
58%58% 42%42% 57%57% 10%10% 16%16% 34%34% 22%22% 7%7%
Inadequate Inadequate membership rostermembership roster
31%31% 22%22% 30%30% 13%13% 20%20% 21%21% 12%12% 12%12%
Addendum: FY06 – Lack of Quorum & Reporting Non-Compliance 12%

CDRH BIMO OAI CDRH BIMO OAI FollowFollow--up Inspections (as of 9/30/06)up Inspections (as of 9/30/06)
18%
30%
52%NAIVAIOAI
N = 64
Recidivist OAIs evenly distributed across program areas:
GLP = 17%
IRB = 25%
CI = 33%
S/M = 25%

CDRH BIMO Vulnerable CDRH BIMO Vulnerable Population InspectionsPopulation Inspections
10%
58%
32%
NAIVAIOAI
N = 164
OAI split among Sponsor (44%) and Clinical Investigator (56%) programs
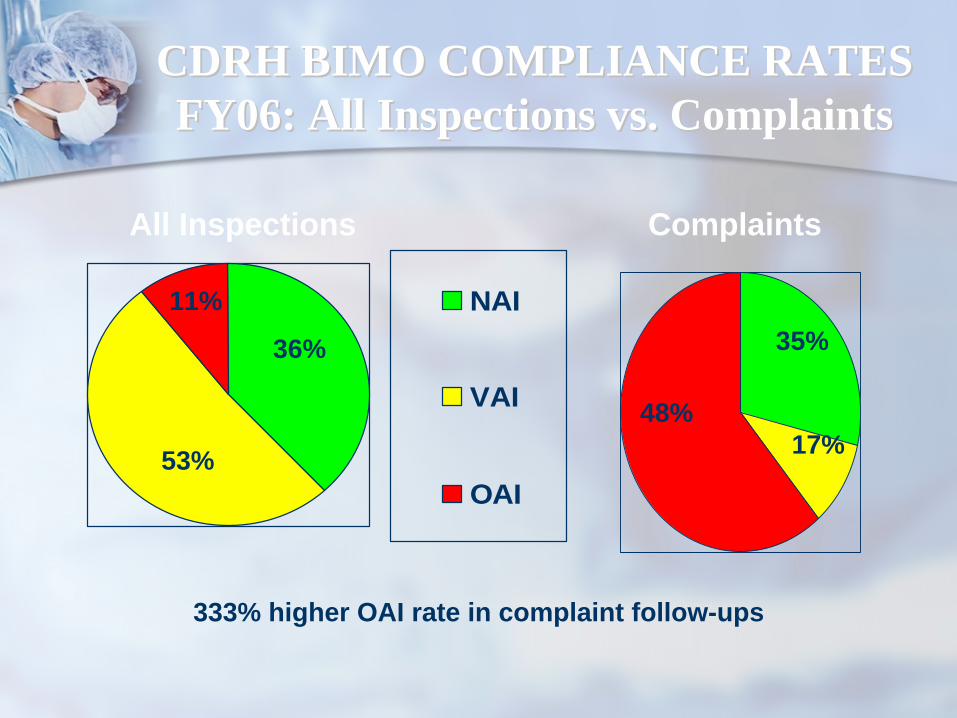
CDRH BIMO COMPLIANCE RATESCDRH BIMO COMPLIANCE RATES FY06: All Inspections vs. ComplaintsFY06: All Inspections vs. Complaints
NAI
VAI
OAI
11%
36%
53%
All Inspections Complaints
17%
35%
48%
333% higher OAI rate in complaint follow-ups

CDRH BIMO COMPLIANCE RATESCDRH BIMO COMPLIANCE RATES FY97FY97--06: All Inspections vs. Complaints06: All Inspections vs. Complaints
NAI
VAI
OAI
14%31%
55%
All Inspections Complaints
28%
26%
46%
230% higher OAI rate in complaint follow-ups over a 10 year period

What does AIP mean?What does AIP mean?

Application Integrity PolicyApplication Integrity Policy
■■What is What is ““Wrongful ActWrongful Act””??
■■What is an What is an ““Untrue Statement of Untrue Statement of Material FactMaterial Fact””??

Wrongful ActWrongful Act
“…“…A wrongful act is any act that may subvert A wrongful act is any act that may subvert the integrity of the review process. A wrongful the integrity of the review process. A wrongful act includes but is not limited to, submitting a act includes but is not limited to, submitting a fraudulent application, offering or promising an fraudulent application, offering or promising an illegal gratuity, or making an untrue statement illegal gratuity, or making an untrue statement of material fact. A wrongful act also includes of material fact. A wrongful act also includes submitting data that are otherwise due to, for submitting data that are otherwise due to, for example, a pattern of errors whether caused by example, a pattern of errors whether caused by incompetence, negligence, or a practice such as incompetence, negligence, or a practice such as inadequate standard operating procedures or a inadequate standard operating procedures or a systemsystem--wide failure to ensure the integrity of wide failure to ensure the integrity of data submissionsdata submissions…”…”

Untrue Statement of Material Untrue Statement of Material FactFact
“…“…An An ““untrue statement of material factuntrue statement of material fact””is a false statement, misstatement, or is a false statement, misstatement, or omission of fact. A determination that an omission of fact. A determination that an untrue statement is material is necessary untrue statement is material is necessary for purposes of invoking the AIPfor purposes of invoking the AIP…”…”MaterialityMateriality-- Under DevelopmentUnder DevelopmentAgentAgent-- Under DevelopmentUnder Development

Examples of Wrongful ActsExamples of Wrongful Acts
Submit Fraudulent ApplicationSubmit Fraudulent ApplicationOffer Bribe/Illegal GratuityOffer Bribe/Illegal GratuityMake Untrue Statement of Material FactMake Untrue Statement of Material FactSubmit Data Otherwise UnreliableSubmit Data Otherwise UnreliableOmitted DataOmitted DataManufactured DataManufactured DataAltered DataAltered DataOther Data Inconsistencies Other Data Inconsistencies

Examples of Data Integrity Examples of Data Integrity ProblemsProblems
Falsification of Specific Data or an Entire Falsification of Specific Data or an Entire SubmissionSubmissionOmission of Relevant and Important Data and Omission of Relevant and Important Data and InformationInformationInability to Account for Patient PopulationInability to Account for Patient PopulationInability to Account for Investigational DevicesInability to Account for Investigational DevicesFailure to Maintain Adequate Investigational Failure to Maintain Adequate Investigational RecordsRecordsUnreported Changes to the Investigational Unreported Changes to the Investigational DeviceDevice

ProcessProcess: : PrePre--Discovery StageDiscovery Stage
Tips from Anonymous/Known InformantTips from Anonymous/Known InformantCurrent/Former EmployeesCurrent/Former EmployeesFormer Business PartnersFormer Business PartnersPatientsPatientsOther Agencies (SEC, FTC, CMS)Other Agencies (SEC, FTC, CMS)Suspicious Data Found During Suspicious Data Found During Scientific/Clinical ReviewScientific/Clinical ReviewObservations During PreObservations During Pre--Approval InspectionApproval Inspection

Process: Inspection StageProcess: Inspection Stage
Inspection of Company/ SponsorInspection of Company/ SponsorInspection of Clinical SitesInspection of Clinical SitesInspections of CROInspections of CRO’’ssInspection of Clinical SitesInspection of Clinical Sites••
Data AuditData Audit••
System AuditSystem Audit••
Company Internal DocumentsCompany Internal Documents

Invoking the AIPInvoking the AIP
Pattern or Practice of Wrongful ConductPattern or Practice of Wrongful ConductSignificant Question of Data ReliabilitySignificant Question of Data ReliabilitySystemSystem--wide Failureswide FailuresDecision made by Center Director, The Decision made by Center Director, The Division of Bioresearch Monitoring and Division of Bioresearch Monitoring and The Office of Device Evaluation Integrity The Office of Device Evaluation Integrity Officer Officer

AgencyAgency’’s Actions Action
Defer Scientific ReviewDefer Scientific ReviewIssues Letter to ApplicantIssues Letter to ApplicantConducts Validity AssessmentConducts Validity Assessment
Scope, extent of problemScope, extent of problemInspectionInspectionAudit ReportAudit Report

ApplicantApplicant’’s Responsibilitiess Responsibilities
Cooperation with FDACooperation with FDAInternal Review (Audit)Internal Review (Audit)Independent Outside ConsultantIndependent Outside ConsultantIdentify/Remove IndividualsIdentify/Remove IndividualsSubmit CAPSubmit CAP
Commit to Safety, Efficacy and QualityCommit to Safety, Efficacy and QualityDescribe Ethics/Compliance ProgramsDescribe Ethics/Compliance ProgramsStandard Operating ProceduresStandard Operating ProceduresSteps to Address and Prevent Wrongful ActsSteps to Address and Prevent Wrongful Acts
Application Withdrawal, Patient Notification, Application Withdrawal, Patient Notification, Product Recall etc.Product Recall etc.

Global Industry IssuesGlobal Industry Issues
Systems to identity and/or address Systems to identity and/or address regulatory shortcomingsregulatory shortcomingsSystems to correct/prevent recurring Systems to correct/prevent recurring issuesissuesAccountable company cultureAccountable company cultureEnvironment of conflict of interestEnvironment of conflict of interest

FDA ResponsibilitiesFDA Responsibilities
Review of Corrective Action PlanReview of Corrective Action Plan
Field Onsite Inspection & RecommendationField Onsite Inspection & Recommendation
Headquarters ReviewHeadquarters Review
Letter to ApplicantLetter to ApplicantCenter DirectorCenter Director’’s Signature s Signature

Application Integrity ProgramApplication Integrity Program
““Fraud, Untrue Statements of Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Material Facts, Bribery, and Illegal Gratuities; Final Policy,Gratuities; Final Policy,””
56 F.R. 46191, 9/10/9156 F.R. 46191, 9/10/91http://www.fda.gov/ora/fr/fraud_ill_grat.htmlhttp://www.fda.gov/ora/fr/fraud_ill_grat.html

Application Integrity ProgramApplication Integrity Program
ApplicationApplication Integrity PolicyIntegrity PolicyRPM Chapter 10RPM Chapter 10
http://www.fda.gov/ora/compliance_ref/rpm_new2/rpm10aip.htmlhttp://www.fda.gov/ora/compliance_ref/rpm_new2/rpm10aip.html
““Points to Consider for Internal Points to Consider for Internal Reviews and Corrective Action Reviews and Corrective Action Operating PlansOperating Plans””
http://www.fda.gov/ora/compliance_ref/aip_points.htmlhttp://www.fda.gov/ora/compliance_ref/aip_points.html

Program Offices/ContactsProgram Offices/Contacts
ODE/OIVD Integrity OfficerODE/OIVD Integrity OfficerCarl DeMarco: 240Carl DeMarco: 240--276276--39933993Division of Bioresearch Monitoring, Division of Bioresearch Monitoring, Office of ComplianceOffice of ComplianceMichael Marcarelli: 240Michael Marcarelli: 240--276276--01250125Application Integrity Policy CommitteeApplication Integrity Policy CommitteeFDA Office of Criminal InvestigationsFDA Office of Criminal Investigations

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
Applies to Clinical InvestigatorsApplies to Clinical InvestigatorsSome clinical investigators may have Some clinical investigators may have already received a Warning Letter but in already received a Warning Letter but in some cases violations discovered on the some cases violations discovered on the first inspection are serious enough for the first inspection are serious enough for the Center to issue the NIDPOE.Center to issue the NIDPOE.

Disqualification Of Disqualification Of Clinical InvestigatorsClinical Investigators

Disqualification Of Disqualification Of Clinical InvestigatorsClinical Investigators
A NIDPOE letter informs the recipient clinical investigator A NIDPOE letter informs the recipient clinical investigator that that FDA is initiating an administrative proceeding to determine FDA is initiating an administrative proceeding to determine whether the clinical investigator should be disqualified from whether the clinical investigator should be disqualified from receiving investigational products pursuant to the Food and Drugreceiving investigational products pursuant to the Food and Drug
Administration's regulations. Generally, FDA issues a NIDPOE Administration's regulations. Generally, FDA issues a NIDPOE letter when it believes it has evidence that the clinical investletter when it believes it has evidence that the clinical investigator igator repeatedly or deliberately violated FDA's regulations governing repeatedly or deliberately violated FDA's regulations governing the the proper conduct of clinical studies involving investigational proper conduct of clinical studies involving investigational products or submitted false information to the sponsorproducts or submitted false information to the sponsor. .

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
Definition under CFR 812.119Definition under CFR 812.119
If FDA has information indicating that an investigator If FDA has information indicating that an investigator has repeatedly or deliberately failed to comply with the has repeatedly or deliberately failed to comply with the requirements of part 812, part 50, or part 56 of this requirements of part 812, part 50, or part 56 of this chapter, or has repeatedly or deliberately submitted chapter, or has repeatedly or deliberately submitted false information either to the sponsor of the false information either to the sponsor of the investigation or in any required report, the Center for investigation or in any required report, the Center for Devices and Radiological Health will furnish the Devices and Radiological Health will furnish the investigator written notice of the matter under investigator written notice of the matter under complaint and offer the investigator an opportunity to complaint and offer the investigator an opportunity to explain the matter in writing, or, at the option of the explain the matter in writing, or, at the option of the investigator, in an informal conferenceinvestigator, in an informal conference. .

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
If an explanation is offered and accepted by the If an explanation is offered and accepted by the Center for Devices and Radiological Health, the Center for Devices and Radiological Health, the disqualification process will be terminated. If disqualification process will be terminated. If an explanation is offered but not accepted by an explanation is offered but not accepted by the Center for Devices and Radiological Health, the Center for Devices and Radiological Health, the investigator will be given an opportunity for the investigator will be given an opportunity for a regulatory hearing under part 16 of this a regulatory hearing under part 16 of this chapter on the question of whether the chapter on the question of whether the investigator is entitled to receive investigational investigator is entitled to receive investigational devices.devices.

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
After evaluating all available information, including After evaluating all available information, including any explanation presented by the investigator, if the any explanation presented by the investigator, if the Commissioner determines that the investigator has Commissioner determines that the investigator has repeatedly or deliberately failed to comply with the repeatedly or deliberately failed to comply with the requirements of this part, part 50, or part 56 of this requirements of this part, part 50, or part 56 of this chapter, or has deliberately or repeatedly submitted chapter, or has deliberately or repeatedly submitted false information either to the sponsor of the false information either to the sponsor of the investigation or in any required report, the investigation or in any required report, the Commissioner will notify the investigator, the sponsor Commissioner will notify the investigator, the sponsor of any investigation in which the investigator has been of any investigation in which the investigator has been named as a participant, and the reviewing IRB that the named as a participant, and the reviewing IRB that the investigator is not entitled to receive investigational investigator is not entitled to receive investigational devices. The notification will provide a statement of devices. The notification will provide a statement of basis for such determination. basis for such determination.

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
Each investigational device exemption (IDE) Each investigational device exemption (IDE) and each cleared or approved application and each cleared or approved application submitted under this part, subpart E of part 807 submitted under this part, subpart E of part 807 of this chapter, or part 814 of this chapter of this chapter, or part 814 of this chapter containing data reported by an investigator who containing data reported by an investigator who has been determined to be ineligible to receive has been determined to be ineligible to receive investigational devices will be examined to investigational devices will be examined to determine whether the investigator has determine whether the investigator has submitted unreliable data that are essential to submitted unreliable data that are essential to the continuation of the investigation or essential the continuation of the investigation or essential to the approval or clearance of any marketing to the approval or clearance of any marketing application.application.

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
Consent AgreementsConsent AgreementsList specific responsibilities of the Clinical List specific responsibilities of the Clinical Investigator in terms of coming into Investigator in terms of coming into compliance.compliance.Can last for a specific amount of time or can be Can last for a specific amount of time or can be an agreement the disqualification is permanent.an agreement the disqualification is permanent.Can be viewed as a tool to bring the Clinical Can be viewed as a tool to bring the Clinical Investigator into compliance which in turn Investigator into compliance which in turn serves as a way to educate the Clinical serves as a way to educate the Clinical Investigator as to their regulatory responsibility Investigator as to their regulatory responsibility for the current and future clinical trials.for the current and future clinical trials.

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
What does disqualification mean for the What does disqualification mean for the Sponsor?Sponsor?
The data from the disqualified clinical site The data from the disqualified clinical site can not be used in their submission. can not be used in their submission. (Monetary and Ethical considerations)(Monetary and Ethical considerations)Sponsor is responsible for oversight of all Sponsor is responsible for oversight of all clinical investigators so there might be some clinical investigators so there might be some serious issues in terms of monitoring which serious issues in terms of monitoring which can lead to further regulatory action.can lead to further regulatory action.

Notice Of Initiation Of Disqualification Notice Of Initiation Of Disqualification Proceedings And Opportunity To ExplainProceedings And Opportunity To Explain
What does disqualification mean for the What does disqualification mean for the clinical investigator?clinical investigator?
Their name is added to a list on the FDA Their name is added to a list on the FDA website that indicates that they are website that indicates that they are disqualified from participation in any type of disqualified from participation in any type of clinical trial.clinical trial.Generally it means that they have incurred Generally it means that they have incurred legal fees and it can open them up to more legal fees and it can open them up to more eminent liability.eminent liability.Some might feel that it has had a negative Some might feel that it has had a negative impact on their reputations.impact on their reputations.

Web SitesWeb Sites
Device Advice
www.fda.gov/cdrh/devadvice
CDRH BIMO sitewww.fda.gov/cdrh/comp/bimo.html

Contact InformationContact Information
Sonali P. GunawardhanaSonali P. GunawardhanaFDA, CDRH, Office of ComplianceFDA, CDRH, Office of Compliance9200 Corporate Blvd9200 Corporate BlvdHFZHFZ--310310Rockville, MD 20850Rockville, MD 20850(240)276(240)[email protected]@fda.hhs.gov








![Responding to FDA FTC Enforcement Final [Read-Only] · Responding to FDA & FTC Enforcement ... product or product line affected) ... Nestle and Iovate consent decrees, POM Wonderful](https://static.fdocuments.net/doc/165x107/5b35cd077f8b9aad388c3f45/responding-to-fda-ftc-enforcement-final-read-only-responding-to-fda-ftc.jpg)









