
FACULTY OF MATHEMATICS AND NATURAL SCIENCES PHYSICAL … · 15 FACULTY OF MATHEMATICS AND NATURAL...
36
1 FACULTY OF MATHEMATICS AND NATURAL SCIENCES PHYSICAL AND THEORETICAL CHEMISTRY Local modes in vibrational (and rotational) spectroscopy (Picture courtesy of Python M)
Transcript of FACULTY OF MATHEMATICS AND NATURAL SCIENCES PHYSICAL … · 15 FACULTY OF MATHEMATICS AND NATURAL...

1
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Local modes in
vibrational (and
rotational)
spectroscopy
(Picture courtesy
of Python M)

2
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Where is
Wuppertal?

3
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
The Wuppertal monorail

4
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Vibrational energies of small molecules
Density o
f sta
tes

5
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
cm-1
0 4000 8000 cm-1
Spectrum?
n
I Simple and structured! Extremely complicated,
wall-to-wall lines?

6
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
H-X stretching vibrations modelled by
Morse oscillators:
Hamiltonian:
Eigenvalues:

7
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
HCAO model for H2X molecule stretch (Harmonically Coupled Anharmonic Oscillators)
Hamiltonian:
re
Diagonalized in basis set of products of
Morse-oscillator eigenfunctions
[1] Child MS, Lawton RT. Faraday Discuss Chem Soc 1981, 71:273–285.
[2] Mortensen OS, Henry BR, Mohammadi MA. J Chem Phys 1981, 75:4800–4808
[3] Child MS, Halonen L. Adv Chem Phys 1984, 57:1–58

8
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
HCAO matrices

9
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Vibrational
energies
patterns:
Normal mode,
Local mode…

10
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Vibrational intensities Empirical fact: Stretching part of
dipole moment function modelled
well by „Molecular Bond“ function
Favours vibrational transitions to states with all N
excitation quanta localized in one bond.
[1] HALONEN, L., 1998, Adv. chem. Phys., 104.

11
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Summary:
Excited stretching states form
„polyads“ with the structure
predicted by HCAO
Observed transitions from the vibrational ground
state end in lowest levels of polyad.
Results transferable to molecules with n > 2
equivalent H-X stretching vibrations.

12
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Example experiment: ICLAS (Intracavity Laser Absorption Spectroscopy)
[1] Campargue A: http://www.chem.uni-wuppertal.de/sphers/han-sur-
lesse/campargue/camparguenew.html
[2] Bertseva E, Kachanov AA, Campargue A. Chem Phys Lett 2002, 351:18–26.
• Extremely sensitive
• Absoption path lengths of order-of-magnitude 100 km

13
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Example experiment: ICLAS for H2S (Intracavity Laser Absorption Spectroscopy)
[1] Ding Y, Naumenko O, Hu S-M, Bertseva E, Campargue A.. J Mol Spectrosc 2003, 217:222–238.

14
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
[1] Ding Y, Naumenko O, Hu S-M, Bertseva E, Campargue A.. J Mol Spectrosc 2003, 217:222–238.
Example experiment: ICLAS for H2S (Intracavity Laser Absorption Spectroscopy)

15
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
1972 Dorney and Watson CH4 8-fold and 6-fold clusters
1978 Zhilinskii and Pavlichenkov H2O 4-fold clusters (Erb)
1978 Harter and Patterson Rotational energy surfaces and clusters
1991 Lehmann Local mode theory and clusters
1992 Kozin et al H2Se 4-fold clusters observed
1993 Kozin and Jensen H2Se 4-fold cluster theory (Erbs)
1994 Jensen and Bunker H2X 4-fold cluster symmetry
1996 Kozin et al H2Te 4-fold clusters (exp and theory)
1997 Jensen et al Review paper on 4-fold clusters
2000 Jensen Review paper on LMT and clusters
2012 Jensen Another review paper on LMT and clusters
Rotational Energy Level Clusters
[Mol. Phys. 98, 1253-1285 (2000)]
[WIREs Comput. Mol. Sci. (Wiley Interdisciplinary
Reviews) 2, 494–512 (2012)]

16
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
JJ,0
JJ,1
JJ-1,1
JJ-1,2
JJ-2,2
JJ-2,3
JJ-3,3
JJ-3,4
JJ-4,4
JJ,0
JJ,1
JJ-1,1
JJ-1,2
J
Ae=6.26, Be=6.11, Ce=3.09 cm-1
H2Te Rigid Rotor Energy Levels
[E(J
KaK
c)-
E(J
J0)]

17
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
J
JJ,0
JJ,1
JJ-1,1
JJ-1,2
JJ,0
JJ,1
JJ-1,1
JJ-1,2
JJ-2,2
JJ-2,3
JJ-3,3
JJ-3,4
JJ-4,4
Actual H2Te Energy Levels
[E(J
KaK
c)-
E(J
J0)]

18
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
1R 1L
2L 2R
1
2
1
1 1
2 2
2
JJ,0
JJ,1
JJ-1,1
JJ-1,2
in C2v(M)

19
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
[3] S.N. Yurchenko, W. Thiel, and P. Jensen, J. Mol. Spectrosc. 245, 126 (2007)
Programs: XY3 and TROVE
(Theoretical Rotation-Vibration Energies)
[1] H. Lin et al., J. Chem. Phys. 117, 11265 (2002)
[2] S.N. Yurchenko et al., Mol. Phys. 103, 359 (2005) and references given there.
XY3: Rotation-vibration energies for
pyramidal, ammonia-type
molecule in isolated electronic
state, calculated variationally.
TROVE: Rotation-vibration energies for
any molecule in isolated electronic
state, calculated variationally.

20
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
cc-pwCVTZ [1]
refined by fitting to experimental
vibrational term values [2]
[1] D. Wang,Q. Shi, and Q.-S. Zhu, J. Chem. Phys., 112, 9624 (2000)
[2] S.N. Yurchenko et al., Chem. Phys. 290, 59 (2003)
Variational rotation-vibration
calculations for PH3
Vibrational basis set:
6)(2 321 bendinv Vvvvv
J ≤ 80
Potential energy surface:

21
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Six-fold energy clusters
-6
-5
-4
-3
-2
-1
0
0 10 20 30 40 50 60 70 80
A1/A2
A2/A1
E
E
Cluster = A1 A2 2E in C3v(M)
J
[E(J
K)-
E(J
J)]

22
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
3 PCS
= (132)1 PCS
4 PCS
= (23)*1 PCS
6 PCS
= (13)*1 PCS
Cluster = A1 A2 2E in C3v(M)
2 PCS
= (123)1 PCS
5 PCS
= (12)*1 PCS
1 PCS
= E 1 PCS
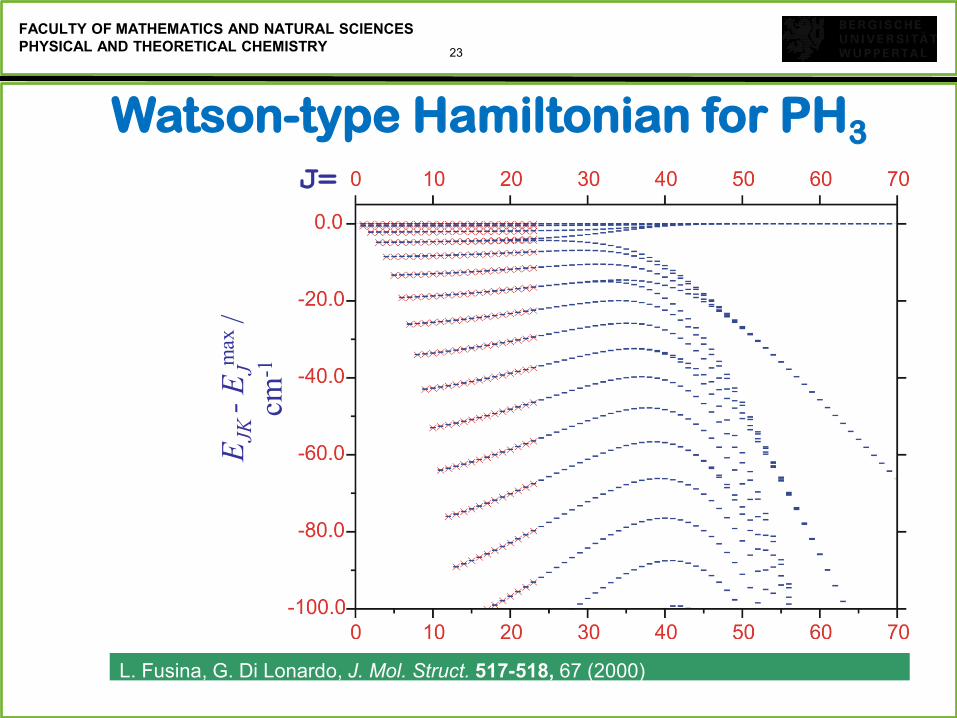
23
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
L. Fusina, G. Di Lonardo, J. Mol. Struct. 517-518, 67 (2000)
EJK
- E
Jmax
/
cm-1
Watson-type Hamiltonian for PH3
J=

24
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
• (,,) define orientation of molecule (xyz) relative to
laboratory (XYZ).
• (,) define orientation of Z axis relative to molecule (xyz).
Rotational coordinates
xyz is molecule-fixed; XYZ is space-fixed
x
y
z
x
y
z
X
Y
Z
Z
N
N

25
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
For states with m = J (and J large),
is the probability distribution
for the orientation of the angular
momentum relative to the molecule
),(,mJF
i
Integration over all vibronic coordinates and
For a rovibronic eigenstate
is the probability distribution for the orientation
of the Z axis relative to the molecule. Z
J ),(, mJFJ
dVF iimJ sin),(*
,

26
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
z z z
1
1
A
1
1
A
2
6
A
1
2
E
2
3
E
1
4
E 2
5
E
x y
z z z
y
y y
y y x
x x
x x
),(, mJF“Top cluster states” for J = m = 40,
vibrational ground state of PH3

27
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Then invert the unitary transformation to obtain
1
1
A =
Primitive cluster states j PCS
First symmetrize, e.g.
= 2
6
A
with similar expressions for the E functions....

28
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
1 PCS
-6
-5
-4
-3
-2
-1
0
0 10 20 30 40 50 60 70 80J=20
= 94º
z
y x
dVF iiJmJ sin),(*
,
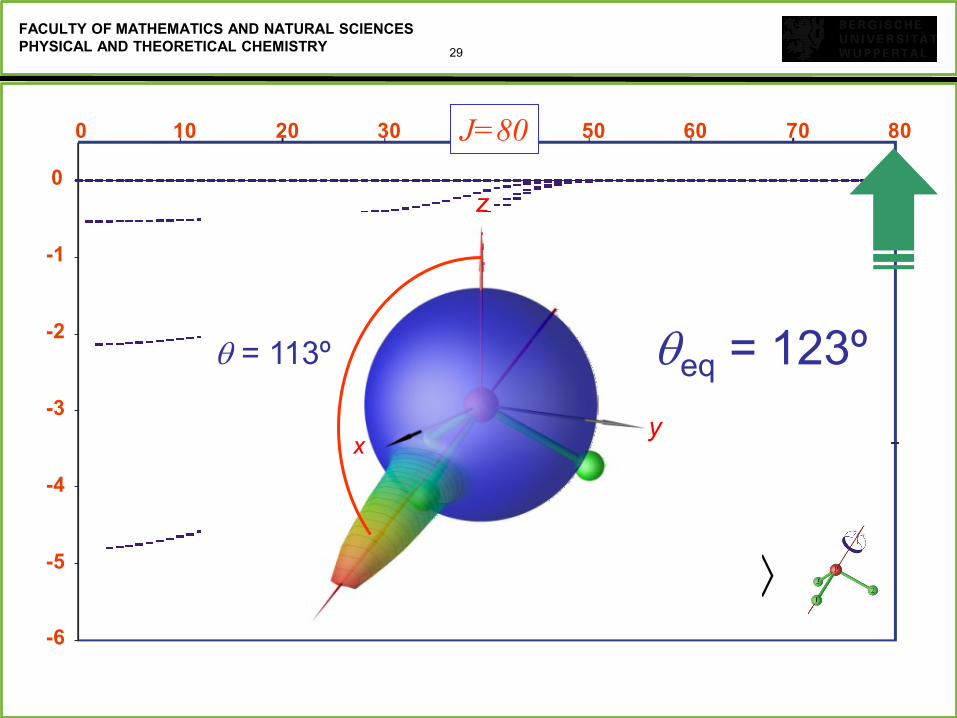
29
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
1 PCS
-6
-5
-4
-3
-2
-1
0
0 10 20 30 40 50 60 70 80J=80
= 113º
z
y x
eq = 123º

30
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
PH3 intensity calculations
Ab initio (CCSD(T)/aug-cc-pVTZ)
dipole moment surfaces
Rotation-vibration
wavefunctions from
the variational
calculation
Experimental data
from L. R. Brown,
R. L. Sams, I. Kleiner,
C. Cottaz, and L. Sagui,
J. Mol. Spectrosc. 215,
178-203 (2002)

31
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
PH3 cluster transitions
Large line strengths
at high J
Can the lower states
be populated somehow?

32
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Similar effects for
XHD2 or XH2D molecules?
?

33
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
XH2D: Cluster-free molecules
PH2D
BiH2D SbH2D
Reduced rotational term values

34
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
XHD2: Molecules with rotational energy cluster formation
PHD2
BiHD2 SbHD2
Reduced rotational term values

35
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Local mode phenomena manifest themselves as
• clustering of vibrational energy levels at high
vibrational excitation
• clustering of rotation–vibration energy levels at high
rotational excitation.
Thus local mode behaviour is
induced by both vibrational and
rotational excitation.
Conclusions

36
FACULTY OF MATHEMATICS AND NATURAL SCIENCES
PHYSICAL AND THEORETICAL CHEMISTRY
Thanks & Acknowledgments
The principal doer:
Sergei N. Yurchenko, Wuppertal, Ottawa,
Mülheim, Dresden
Other doers:
Miguel Carvajal, Huelva
Hai Lin, Denver
Serguei Patchkovskii, Ottawa
A fellow ponderer: Walter Thiel, Mülheim
Thanks for support from the European Commission,
the German Research Council (DFG), and the Foundation
of the German Chemical Industry (Fonds der Chemie).


















