
Facile and efficient preparation of phosphinate-functionalized aromatic diamines and their high-Tg...
14
Facile and Efficient Preparation of Phosphinate-Functionalized Aromatic Diamines and Their High-T g Polyimides CHIA WEI CHANG, 1 CHING HSUAN LIN, 1 PO WEN CHENG, 1 HANN JANG HWANG, 2 SHENGHONG A. DAI 1 1 Department of Chemical Engineering, National Chung Hsing University, Taichung, Taiwan 2 Department of Cosmetic Science, Chung Hwa University of Medical Technology, Tainan, Taiwan Received 30 October 2008; accepted 24 January 2009 DOI: 10.1002/pola.23320 Published online in Wiley InterScience (www.interscience.wiley.com). ABSTRACT: Two novel phosphorus-functionalized aromatic diamines, 1,1-bis(4-amino- phenyl)-1-(6-oxido-6H-dibenz \c,e [\1,2 [ oxaphosphorin-6-yl)ethane (1) and bis(4- aminophenyl)-(6-oxido-6H-dibenz \c,e [\1,2 [ oxaphosphorin-6-yl)phenylmethane (2), were prepared from 9,10-dihydro-oxa-10-phosphaphenanthrene-10-oxide, 4-ami- noacetophenone, or 4-aminobenzophenone in excess aniline using p-toluenesulfonic acid monohydrate as catalyst by an efficient, one-pot procedure. The effect of electron withdrawing/donating groups on the stabilization of the resulting carbocation seems critical for the success of the process and was discussed in detail. Based on diamines (1–2), a series of new polyimides, (5a–5d) and (6a–6d), were prepared, respectively. Polyimides (5a–5d) are flexible and creasable. In contrast, polyimides (6a–6d) are brittle because of the structure rigidity, according to the analysis based on the NMR temperature-dependent spectra of (2). Polyimides 5 displaying high T g (318–392 C), high moduli (3.39–4.49 GPa), low coefficient of thermal expansion (42–50 ppm/ C), and moderate thermal stability (T d 5 wt % at 426–439 C), are excellent high-T g and flame-retardant materials. V V C 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 2486–2499, 2009 Keywords: flame retardance; high-performance polymers; polyimides INTRODUCTION Aromatic polyimides are known for its high- temperature stability, oxidative and hydrolytic stability, and it can be applied in fields such as electronics, 1 optics, 2 membranes, 3,4 and compo- sites. 5 However, aromatic polyimides are difficult to process because of their high melting tem- perature, glass transition temperatures, and poor solubility in organic solvents. It has been reported that incorporating asymmetric 6,7 or bulky groups 5,8,9–11 into polymers can impart bet- ter organo-solubility because of the decreased packing density and crystallinity. The other bene- fit of the latter approach is that the T g of polymers can be maintained because of the restricted seg- mental mobility caused by the bulky pendant. It has also been reported that phosphorus-function- alized polymers, whether in the phosphine oxide form, 12–16 in the phosphonate 17,18 or in the phos- phinate form, 19–28 can improve organo-solubility, adhesion to metal, flame retardancy, and atomic Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 47, 2486–2499 (2009) V V C 2009 Wiley Periodicals, Inc. Additional Supporting Information may be found in the online version of this article. Correspondence to: C. Hsuan Lin (E-mail: linch@nchu. edu.tw) 2486
-
Upload
chia-wei-chang -
Category
Documents
-
view
214 -
download
2
Transcript of Facile and efficient preparation of phosphinate-functionalized aromatic diamines and their high-Tg...

Facile and Efficient Preparation ofPhosphinate-Functionalized Aromatic Diaminesand Their High-Tg Polyimides
CHIA WEI CHANG,1 CHING HSUAN LIN,1 PO WEN CHENG,1 HANN JANG HWANG,2 SHENGHONG A. DAI1
1Department of Chemical Engineering, National Chung Hsing University, Taichung, Taiwan
2Department of Cosmetic Science, Chung Hwa University of Medical Technology, Tainan, Taiwan
Received 30 October 2008; accepted 24 January 2009DOI: 10.1002/pola.23320Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: Two novel phosphorus-functionalized aromatic diamines, 1,1-bis(4-amino-phenyl)-1-(6-oxido-6H-dibenz \c,e[ \1,2[ oxaphosphorin-6-yl)ethane (1) and bis(4-aminophenyl)-(6-oxido-6H-dibenz \c,e[ \1,2[ oxaphosphorin-6-yl)phenylmethane(2), were prepared from 9,10-dihydro-oxa-10-phosphaphenanthrene-10-oxide, 4-ami-noacetophenone, or 4-aminobenzophenone in excess aniline using p-toluenesulfonicacid monohydrate as catalyst by an efficient, one-pot procedure. The effect of electronwithdrawing/donating groups on the stabilization of the resulting carbocation seemscritical for the success of the process and was discussed in detail. Based on diamines(1–2), a series of new polyimides, (5a–5d) and (6a–6d), were prepared, respectively.Polyimides (5a–5d) are flexible and creasable. In contrast, polyimides (6a–6d) arebrittle because of the structure rigidity, according to the analysis based on the NMRtemperature-dependent spectra of (2). Polyimides 5 displaying high Tg (318–392 �C),high moduli (3.39–4.49 GPa), low coefficient of thermal expansion (42–50 ppm/�C),and moderate thermal stability (Td 5 wt % at 426–439 �C), are excellent high-Tg andflame-retardant materials. VVC 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem
47: 2486–2499, 2009
Keywords: flame retardance; high-performance polymers; polyimides
INTRODUCTION
Aromatic polyimides are known for its high-temperature stability, oxidative and hydrolyticstability, and it can be applied in fields such aselectronics,1 optics,2 membranes,3,4 and compo-sites.5 However, aromatic polyimides are difficultto process because of their high melting tem-
perature, glass transition temperatures, andpoor solubility in organic solvents. It has beenreported that incorporating asymmetric6,7 orbulky groups5,8,9–11 into polymers can impart bet-ter organo-solubility because of the decreasedpacking density and crystallinity. The other bene-fit of the latter approach is that the Tg of polymerscan be maintained because of the restricted seg-mental mobility caused by the bulky pendant. Ithas also been reported that phosphorus-function-alized polymers, whether in the phosphine oxideform,12–16 in the phosphonate17,18 or in the phos-phinate form,19–28 can improve organo-solubility,adhesion to metal, flame retardancy, and atomic
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 47, 2486–2499 (2009)VVC 2009 Wiley Periodicals, Inc.
Additional Supporting Information may be found in theonline version of this article.
Correspondence to: C. Hsuan Lin (E-mail: [email protected])
2486

oxygen resistance. Generally, the glass transitiontemperature and thermal stability of phospho-nate-type polymers are generally not very highbecause of the plasticizing effect and low thermalstability of AOA(O¼¼)PAOA linkage. In the phos-phinate type, the A(O¼¼)PAOAC linkage, espe-cially in a cyclic form, is on the polymer sidechain. On heating, the cleavage of the phos-phinate pendant has little effect on the integrityof polymer main chain, so the side-chain phos-phinates are thermally more stable.28 However,although phosphine oxide and phosphinates-containing polymers display higher thermal sta-bility, except those reported by Liu et al.,23,27 atleast two synthetic procedures are required toprepare the phosphorus-functionalized diamines.Furthermore, the starting material—4,40-diami-nodiphenylbenzophenone is very expensive in Liuet al.’s work,23,27 limiting its application. Thiswork focuses on the facile and efficient prepara-tion of side-chain phosphinate-functionalized dia-mines. Herein, two novel diamines, 1,1-bis(4-aminodiphenyl)-1-(6-oxido-6H-dibenz\c,e[\1,2[oxaphosphorin-6-yl)ethane (1) and bis(4-aminodi-phenyl)-(6-oxido-6H-dibenz \c,e[\1,2[ oxaphos-phorin-6-yl)phenylmethane (2), were prepared byan effective and one-pot procedure. Based on di-amines (1–2), a series of polyimides (5a–5d, 6a–6d) with bulky biphenylene phosphinate pendantwere prepared from the condensation of (1–2) withvarious aromatic dianhydrides in N,N-dimethy-lacetamide (DMAc), followed by thermal imidiza-tion. The chemical structures, solubility, crystal-linity, thermal properties, and thermal stability ofthe polyimides were investigated. The referencedpolyimides (7a–7d) based on 4,40-diaminodiphenylmethane (DDM) were also prepared for propertycomparison.
EXPERIMENTAL
Materials
9,10-Dihydro-oxa-10-phosphaphenanthrene-10-oxide (DOPO, TCI), 4-aminoacetophenone (Acros),4-aminobenzophenone (Acros) aniline (Acros), p-toluenesulfonic acid monohydrate (p-TSA,SHOWA), 4,40-DDM (Aldrich) and calciumhydride (Acros) were used as received. Pyromel-litic dianhydride (PMDA, Acros) was dried at 170�C overnight before use. 3,30,4,40-Benzophenonete-tracarboxylic dianhydride (BTDA, Acros), 4,40-oxydiphthalic anhydride (Chriskev), and 3,30,4,40-
biphenyltetracarboxylic dianhydride (Chriskev)were recrystallized from acetic anhydride. DMAcwas purchased from TEDIA and purified by distil-lation under reduced pressure over calciumhydride (Acros) and stored over molecular sieves.The other solvents used are commercial products(HPLC grade) and used without further anypurification.
Characterization
NMR measurements were performed using a Var-ian Inova 600 NMR in DMSO-d6, and the chemi-cal shift was calibrated by setting the chemicalshift of DMSO-d6 as 2.49 ppm. The assignment ofindividual peak of (1–2) is assisted by the correla-tions shown in the 1H-1H COSY and 1H-13C HET-COR NMR spectra. IR spectra were obtained at1 cm�1 resolution in the standard wavenumberrange of 400–4000 cm�1 by Perkin-Elmer RX1infrared spectrophotometer. High-resolution massspectrometer measurements were performed by aFinnigan/Thermo Quest MAT 95XL mass spec-trometer by fast atom bombardment. Elementalanalysis was performed on an Elementar VarioEL III. Wide-angle X-ray diffraction measure-ments were performed at room temperature by aMAC Science DMAX2000 X-ray diffractometerwith monochromatic Cu Ka radiation (k ¼ 1.5418E, operating at 40 kV and 30 mA). The scanningrate was 3�/min over a range of 2h ¼ 5–40�. Differ-ential scanning calorimeter (DSC) scans wereobtained by a Perkin-Elmer DSC 7 in a nitrogenatmosphere at a heating rate of 20 �C/min. Tg wastaken as the midpoint of the heat capacity transi-tion between the upper- and lower-points of devia-tion from the extrapolated liquids and glass lines.Dynamic mechanical analysis (DMA) was per-formed by a Perkin-Eimer Pyris Diamond DMAat a heating rate of 5 �C/min. Thermo mechanicalanalysis (TMA) was performed by a SII TMA/SS6100 at a heating rate of 5 �C/min. Thermalgravimetric analysis (TGA) was performed by aSeiko Exstar 600 at a heating rate of 20 �C/min ina nitrogen atmosphere from 105 to 800 �C. Flameretardancy of polyimides was performed by a UL-94VTM vertical thin test. In the test, an 8 in. � 2in. sample is wrapped around a 1/200 mandrel, andthen taped on one end. The mandrel is removed,leaving a cone-shaped sample that is relativelyrigid. The two flame applications are 3 s insteadof 10 s. After the first ignition, the flame isremoved and the time for the polymer to self-
PHOSPHINATE POLYIMIDES 2487
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

extinguish (t1) is recorded. Cotton ignition wouldbe noted if polymer dripping occurs during thetest. After cooling, the second ignition is per-formed on the same sample and the self-extin-guishing time (t2) and dripping characteristics arerecorded. If t1 plus t2 is less than 10 s without anydripping, the polymer is considered to be a VTM-0material. If t1 plus t2 is in the range of 10–30 swithout any dripping, the polymer is consideredto be a VTM-1 material.
Synthesis of (1)
DOPO 10.81 g (0.05 mol), aniline 23.28 g (0.25mol), 4-aminoacetophenone 6.76 g (0.05 mol), andp-TSA 0.216 g (2 wt % of DOPO) were introducedinto a 250 mL round-bottom glass flask equippedwith a nitrogen inlet and a magnetic stirrer. Thereaction mixture was stirred at 130 �C for 24 h.After that, the precipitate was filtered and recrys-tallized from methanol, and then dried in a vac-uum oven at 100 �C for 8 h. Light-yellow crystal(80% yield) with a melting point of 255 �C (DSC)was obtained.
HR-MS (FAB) m/z: calcd. for C26H23O2N2P426.1497; ANAL., 427.1568 for C26H24O2N2P (M þ1þ). ELEM. ANAL.: calcd. for C26H23O2N2P: C,73.23%; H, 5.44%; N, 6.57%. Found: C, 73.29%; H,5.61%; N, 6.54%. IR(KBr) m: 3343, 3445 (NH2
stretch), m: 1583 (P-Ph), m: 1513 (NH2 bend), m:1215 (P¼¼O), m: 914 (PAO-Ph). 1H NMR (ppm,DMSO-d6), d ¼ 1.50 (3H, H6), 5.02 (4H, NH2),6.40 (2H, H20), 6.43 (2H, H2), 6.96 (2H, H3), 7.08(1H, H17), 7.11 (2H, H30), 7.14 (1H, H11), 7.18 (1H,H15), 7.30 (1H, H10), 7.36 (1H, H16), 7.64 (1H, H9),8.00 (1H, H14), 8.08 (1H, H8). 13C NMR (ppm,DMSO-d6), d ¼ 24.09 (C6), 51.42 (C5), 113.03 (C20),113.35 (C2), 119.00 (C11), 121.81 (C7), 123.27 (C8),123.82 (C15), 125.84 (C14), 125.95 (C13), 127.66(C10), 128.96 (C30), 129.84 (C12), 130.29 (C3),130.52 (C16), 131.90 (C17), 133.04 (C9), 135.86(C4,40), 146.92 (C10), 147.39 (C1), 150.72 (C18). Thesynthetic equation of diamine (1) is shown inScheme 1.
Synthesis of (2)
DOPO 10.81 g (0.05 mol), aniline 23.28 g (0.25mol), 4-aminobenzophenone 9.86 g (0.05 mol), andp-TSA 0.216 g (2 wt % of DOPO) were introducedinto a 250 mL round-bottom glass flask equippedwith a nitrogen inlet and a magnetic stirrer. Thereaction mixture was stirred at 130 �C for 24 h.After that, the solution was poured into the meth-anol. The precipitate was filtered and dried in avacuum oven at 100 �C for 8 h. Green-yellow pow-der (74% yield) with a melting point of 315 �C(DSC) was obtained.
HR-MS (FAB) m/z: calcd. for C31H25O2N2P488.1654; ANAL., 489.1729 for C31H26O2N2P (M þ1þ). ELEM. ANAL.: calcd. for C31H25O2N2P: C,76.22%; H, 5.16%; N, 5.73%. Found: C, 76.15%; H,5.22%; N, 5.69%. IR(KBr) m: 3335, 3450 (NH2
stretch), m: 1583 (P-Ph), m: 1512 (NH2 bend), m:1212 (P¼¼O), m: 919 (PAO-Ph). 1H NMR (ppm,DMSO-d6), d ¼ 5.06 (4H, NH2), 6.32 (2H, H2),6.45 (2H, H20), 6.84 (1H, H20), 6.88 (2H, H3), 7.03(1H, H18), 7.12 (2H, H30), 7.12 (2H, H7), 7.14 (1H,H19), 7.16 (1H, H14), 7.30 (1H, H13), 7.33 (1H, H9),7.35 (2H, H8), 7.62 (1H, H12), 7.81 (1H, H17), 8.02(1H, H11). 13C NMR (ppm, DMSO-d6), d ¼ 63.46(C5), 112.98 (C2,20), 118.87 (C20), 120.37 (C6),123.09 (C18), 123.28 (C11), 124.31 (C10), 124.95(C17), 126.27 (C16), 126.50 (C7), 127.47 (C19),128.77 (C13), 128.79 (C15), 129.88 (C14), 130.33(C9), 131.82 (C3,30), 131.98 (C8),133.04 (C12),136.46 (C21), 147.23 (C4,40), 150.23 (C1,10). Thesynthetic equation of diamine (2) is shown inScheme 1.
Scheme 1. Synthesis of diamines (1) and (2).
2488 CHANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

Preparation of Polyimides
Phosphorus-containing polyimides were preparedby reacting (1–2) with equal mole of dianhydrides(a–d). Polyimides syntheses are exemplified byspecific synthesis of 5a from the condensation of(1) and PMDA. The other polyimides were simi-larly prepared. To a 100-mL three-neck round-bottom flask equipped with a magnetic stirrer,nitrogen inlet, (1) 0.6397g (1.5 mmol), and CaH2-dried DMAc 3.87 g were added. After the mono-
mer had dissolved completely, the reactor wasplaced into an ice-bath cooling system. After thereaction mixture reached 3–5 �C, PMDA 0.3272 g(1.5 mmol) was added quickly and reacted at thattemperature for 12 h. Then, the viscous poly(amicacid) was casted on glass by an automatic film ap-plicator. The resulting poly(amic acid) thin filmswere dried at 80 �C overnight and imidized at 100�C (1 h), 200 �C (1 h), and 300 �C (1 h), respec-tively. Polyimides (7a–7d) based on 4,40-DDMwere also prepared by the same proceduredescribed earlier. The synthetic equations of poly-imides 5–7 are shown in Scheme 2.
RESULTS AND DISCUSSION
Synthesis and Reaction of (1–2)
Aromatic diamines (1–2) were prepared by thereaction of DOPO, 4-aminoacetophenone or 4-ami-nobenzophenone in excess aniline using p-TSA as
Scheme 2. Synthesis of polyimides 5 and 6.
PHOSPHINATE POLYIMIDES 2489
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

catalyst by one-pot procedure. The excess anilineplays the roles of reactant and reaction medium.Scheme 3 shows the proposed mechanism for thepreparation. In Scheme 3, DOPO attacks the car-bonyl group of 4-aminoacetophenone or 4-amino-benzophenone via a nucleophilic addition, formingan intermediate with a tertiary hydroxyl group.The tertiary hydroxyl group is then protonated byacid. Spontaneous dissociation of the protonatedhydroxyl group occurs to yield a tertiary carboca-
tion, which is stabilized by the resonance of theamino group, as shown in Scheme 4(a). Then, ani-line reacts with the resulting carbocation by anelectrophilic substitution, yielding (1–2). In thismechanism, the stability of carbocation is crucialto the reaction proceeding. To prove this idea,3-aminoacetophenone was chosen to replace 4-aminoacetophenone as the starting materialto prepare 1-(3-aminophenyl)-1-(4-aminophenyl)-1-(6-oxido-6H-dibenz\c,e[\1,2[ oxaphosphorin-
Scheme 3. Proposed mechanism for the preparation of (1–2).
Scheme 4. Synthesis of (3)–(6). [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
2490 CHANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

6-yl)ethane (4). As in Scheme 4(a), the reactionstopped at DOPO-aldehyde nucleophilic addition,and the electrophilic substitution of aniline did notproceed. As in Scheme 5(b), the amino group,which is on the meta position of the carbocation,can not resonate with the carbocation, and conse-quently can not stabilize the carbocation. As aresult, the carbocation did not form, and the elec-trophilic substitution of aniline did not occur.To further prove this idea, 4-nitroacetophenonewas chosen to replace 4-aminoacetophenone as thestarting material to prepare 1-(4-nitrophenyl)-1-(4-aminophenyl)-1-(6-oxido-6H-dibenz\c,e[\1,2[oxaphosphorin-6-yl)ethane (6). As in Scheme4(b), the reaction stopped at DOPO-aldehydenucleophilic addition, and the electrophilic substi-tution of aniline did not occur. As in Scheme 5(c),the partial positive charge induced by electro-withdrawing nitro group destabilizes the carbo-cation because of the repulsion of two positivecharges. As a result, the carbocation did not form,and the electrophilic substitution of aniline didnot occur.
Generally, bisphenols29–33 and diamines33–36
could be prepared by acid-catalyzed condensationof ketone or aldehyde compounds with excessphenol or aniline. Ketone/aldehyde compoundswith35,36 or without29,32–34 the electron-donatingphenolic-OH or amino groups can carry out thereaction. Especially, ketone compounds with anelectro-withdrawing nitro group can also carry
out the reaction.34,35 These results seem conflictwith our speculation, but actually they are not.Supporting Information Scheme S1 shows thepossible mechanism of stabilization of carbocationin the work of Shu and coworkers30 and Li et al.31
As in Supporting Information Scheme S1, afterone phenol reacted with the carbonyl group of 4-nitroacetophenone, the resulting carbocation wasstabilized by the phenolic-OH. Since the reso-nance between the phenolic-OH and the carboca-tion stabilized the carbocation, the electrophilicsubstitution of second phenol on the carbocationproceeds. However, in this work, according toScheme 4, the resulting carbocation has to be sta-bilized by electron-donating groups such as aminogroup from the starting material, so the proceed-ing of the reaction is strongly dependent on thestructure of starting material.
Characterization of (1–2)
Figure 1 shows the (a) 1H and (b) 13C NMR spec-tra of (1). In the 1H NMR spectrum, an aminopeak at 5.0 ppm and methyl peaks at around 1.5ppm was observed. The signals of methyl groupwere split into two peaks with a coupling constantof 17.4 Hz because of a 3JPAH coupling. Fourpeaks around 6.4 ppm, standing for H2 and H20
are observed. Clearly, H2 and H20 are not equiva-lent in chemical shift, and H2 and H20 were fur-ther split by H3 and H30, respectively, with a
Scheme 5. The resonance of (a) para-position amino group, (b) meta-position aminogroup, and (c) para-position nitro group with the tertiary carbocation.
PHOSPHINATE POLYIMIDES 2491
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

Figure 1. (a) 1H and (b) 13C NMR spectra of diamine (1).
2492 CHANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

coupling constant of around 8.0 Hz because of a3JHAH coupling. Similarly, H3 and H30 are notequivalent in chemical shift, and H3 and H30 werealso split because of the same reason. H2 and H20
are not equivalent in chemical shift because ofasymmetric orientation of the biphenylene phos-phinate pendant, according to the minimumenergy model of (1) (Fig. 2). One of the phenyleneis closer to the biphenylene phosphinate pendantthan the other. The closer one has larger chemicalshift because of the deshielding effect resultingfrom the ring current of biphenylene. Similarly,H3 and H30 are not equivalent in chemical shiftdue to the same reason. This speculation can beconfirmed by the temperature-dependent NMRspectra of (1), as shown in Figure 3. When thetemperature of DMSO-d6 solution of (1) wasincreased, the difference in chemical shiftbetween the signals of H2 and H20, H3, and H30
was reduced. The increase in temperatureapparently speeds up the rotation rate of thebiphenylene phosphinate pendant, making NMRspectrometer recorded a more average phenylenesignals. However, because of the temperaturelimitation of our NMR instrument, the tempera-ture at which H2-H20 and H3-H30 are equivalentwas not available. The peaks of Ar-H, assigned bythe correlations in the 1H-1H COSY spectrum(Supporting Information Fig. S1), were markedon Figure 1(a), further confirming the structure of
(1). In the 13C NMR spectrum, because of the P-C1J coupling, the signals of C5 were split into twopeaks at 51.1 and 51.7 ppm with a coupling con-stant of 92 Hz. The signals of C7 were split intotwo peaks at 122.6 and 123.4 ppm with a couplingconstant of 111.0 Hz due to the same reason. Thepeaks of Ar-C, assigned by the correlations in the1H-13C HETCOR NMR spectrum (Supporting In-formation Fig. S2), were marked in Figure 2(b),also confirming the structure of (1). In the 31PNMR spectrum, only one 31P NMR peak at 42.4ppm was observed, indicating both the purity andthe integrity (no ring opening or decomposition) ofthe biphenylene phosphinate pendant. The struc-ture of (2) was also evaluated by 1H, 13C, 1H-1HCOSY, 1H-13C HETCOR NMR spectra (Support-ing Information Figs. S3–S5), and the spectro-scopic data confirmed the structure of (2).
Polyimide Synthesis
Polyimides 5–6 were prepared by the reaction of(1–2) with various commercially available dianhy-drides (a–d) in DMAc to form poly(amic acid)s (3–4), followed by thermal imidization (Scheme 2).According to the principle of NMR, a larger chem-ical shift of an amino proton means a lower elec-tron density of nitrogen, corresponding to a lowerreactivity. The chemical shift of the amino groupof (1) is 5.0 ppm, which is larger than that (4.97ppm) of phosphorus-containing dietheramine (1,4-bis(4-aminophenoxy)-2-(6-oxido-6H-dibenz \c,e[\1,2[ oxaphosphorin-6-yl) phenylene),20 butsmaller than those (6.20 and 6.14 ppm) of phos-phorus-containing diesteramine (1,4-bis(4-amino-benzoyloxy)-2-(6-oxido-6H-dibenz \c,e[\1,2[oxaphosphorin-6-yl)phenylene naphthalene).18 Thissuggests that the reactivity of (1) is slightlysmaller than that of dietheramine, in which theelectron-donating ether increases the electron den-sity of the amino group, but much higher thanthat of diesteramine, in which the electron-with-drawing carbonyl group reduces the electron den-sity of the amino group. Because the reactivity of(1) is only slightly smaller than that of diether-amine,20 poly(amic acid)s with medium to high vis-cosity can be prepared. Supporting InformationFigure S6 shows the (a) 1H and (b) 13C NMR spec-tra of 5b. The assignment of each peak, assistedby the correlations shown in 1H-1H COSY and1H-13C HETCOR spectra (Supporting InformationFigs. S7–S8), confirms the structure of 5b. Onlyone 31P NMR peak at 36.9 ppm was observed, alsoindicating both purity and integrity of the
Figure 2. The minimum energy model of (1) calcu-lated by ChemOffice 2005. [Color figure can be viewedin the online issue, which is available at www.interscience.wiley.com.]
PHOSPHINATE POLYIMIDES 2493
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

biphenylene phosphinate pendant. In the IR spec-trum of 5b (Supporting Information Fig. S9), thedisappearance of amic acid absorptions at 1650–1700 cm�1(C¼¼ONH) and 2400–3400 cm�1
(C¼¼OOH), together with the appearance of char-acteristic imide absorption bands at 1778 cm�1
(C¼¼O asymmetric stretch), 1723 cm�1 (C¼¼O sym-
metric stretch), 1379 cm�1 (CAN stretch), 1118and 721 cm�1 (imide ring deformation) indicatethe transformation of o-carboxyl amide to theimide ring, therefore confirming the complete imid-ization of the poly(amic acid). Further, the absorp-tions of P-Ph absorption at 1583 cm�1, P¼¼Oabsorption at 1206 cm�1, and PAO-Ph absorption
Figure 3. Temperature-dependent 1H NMR spectra of (1).
2494 CHANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

at 921 cm�1 indicate that the biphenylene phos-phinate pendant did not cleavage by thermal imid-ization.
Properties of the Polyimides
Organosolubility and Crystallinity
The solubility properties of polyimides (5–6) andreference polyimide based on 4,40-DDM andBTDA are listed in Table 1. Compared with thereferenced polyimides (7a–7d), polyimides (5d,6d) show improved solubility. The bulky phosphi-nate pendant, which prevents the well-stacking ofpolymer chains and reduces the intermolecularinteractions, should be responsible for theimproved solubility. This speculation can be con-firmed by the difference in solubility of polyimides5 and 6 in DMSO and THF. Polyimides 6, whichcontain a phenyl substituent, showed better solu-bility than polyimides 5, which contain a lessbulky methyl substituent. The crystallinity of theprepared polyimides was evaluated by wide-angleX-ray diffraction, and the patterns show that allthe polyimides are amorphous. The bulky bi-phenylene phosphinate, which retards the well-stacking of polymer chains, should be responsiblefor the amorphous characteristic and improvedsolubility.
Film Quality and Thermal Properties
All the polyimides 5 are tough, flexible, and creas-able. However, polyimides 6 are brittle because
the structure might be too rigid. This speculationcan be confirmed by the temperature-dependentNMR spectrum of (2), as shown in Figure 4. Whenthe temperature of DMSO-d6 solution of (2) wasincreased, unlike compound (1), the difference inchemical shift between the signals of H2 and H20,and H3 and H30 is almost unchanged. Apparently,the increase in temperature doesn’t speed up therotation rate of the biphenylene phosphinatependant. This result indicates that diamine (2) isvery rigid, and consequently explains the brittlecharacteristic of polyimides 6. Because polyimides5 are flexible and cresable, DMA and TMA wereapplied to evaluate their thermal mechanicalproperties and dimensional stability. Figure 5shows the DMA curves of polyimides 5. The tem-perature corresponding to 1 GPa was as high as338, 306, 312, and 325 �C for 5a–5d, respectively,and the Tg obtained from the peak temperature oftan d is 392, 318, 326, and 343 �C for 5a–5d,respectively. Among the polyimides, 5a shows thehighest Tg because of the rigid pyromellitic moi-eties. In contrast, 5b displays the lowest Tg
because of the flexible ether linkage. As in Table 2,the Tgs of polyimides 5 are higher than that ofpolyimide 7, demonstrating the Tg-enhancementeffect of bulky pendant, which will hide the rota-tion of polymer chains. This indicates that intro-ducing the bulky phosphinate pendant does notsacrifice the glass transition even though the freevolume of polyimides 5 is much larger than thoseof polyimides 7. Besides, the Tgs of polyimides 5are much higher than those reported by Liuet al.,23 in which polyimides with two biphenylene
Table 1. Solubility Data of Polyimides 5–6a
Polymer
GPC Datab Solvent
Mn� 10�4 Mw � 10�4 PDIc NMP DMAc DMF DMSO m-Cresol THF CH2Cl2 CHCl3
5a NAd NA NA þ/�e þ/� þ/� � þ � � �5b 5.1 9.7 1.9 þh þ/� þh � þ/� � þ þ5c NA NA NA þh þ/� þ/� � þ/� � � �5d NA NA NA þh þ/� þ/� � þ � þ/� þ/�6a NA NA NA þ þ/� þ/� þ/� þ þ/� þ/� þ/�6b 3.0 6.3 2.1 þh þ/� þh þ/� þ/� þ/� þ þ6c NA NA NA þh þ/� þ/� þ/� þ/� þ/� þ/� �6d NA NA NA þh þ/� þ/� þ/� þ/� � þ/� þ/�
7a–7d NA NA NA � � � � � � � �a Solubility was tested with a 5 mg sample in 0.5 g of solvent at room temperature.bRelative to polystyrene standard, using DMF as eluent.c PDI ¼ Mw/Mn.d Partially soluble in DMF even on heating, so the GPC data is not available.eþ, soluble at room temperature; þh, soluble on heating; þ/�, partially soluble on heating; �, insoluble on heating.
PHOSPHINATE POLYIMIDES 2495
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

Figure 4. Temperature-dependent 1H NMR spectra of (2).
2496 CHANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
phosphinate pendants were prepared. The sym-metric distribution of the biphenylene phosphi-nate pendants makes the polyimides chain easierto rotate, explaining the lower Tg. In contrast, inthis case, the methyl and biphenylene phosphi-nate are completely asymmetric structures, mak-ing polymer chains difficult to rotate. The lower-ing of Tg by symmetric distribution of the bulkypendant can be supported by the fact that the Tg
of poly(vinyl dichloride) (Tg � �17 �C) is muchlower than that of poly(vinyl chloride) (Tg � 89�C). Another example is that the Tg of poly(vinyldifluoride) (Tg � �40 �C) is much lower than thatof poly(vinyl floride) (Tg � 40 �C). As in Table 2,the Tg values of polyimides 5 are higher thanthose of polyimides 6. Because the bulkiness ofsubstituents is in the order of biphenylene phos-phinate, phenyl, and methyl, the degree of asym-metry of polyimides 6 is not as large as that ofpolyimides 5, and this justifies the higher Tg ofpolyimides 5. The Tgs of polyimides 5 are alsomuch higher than those of poly(ether imides)reported in our previous work.24 In that work,although asymmetric bulky biphenylene phosphi-nate pendant was incorporated, the two flexibleether linkages lower the Tg. Figure 6 shows theTMA curves of polyimides 5. Tgs measured byTMA are in the range of 388–304 �C, which isslightly lower than those measured by DMA. CTE
of polyimides 5 at 100–250 �C are in the range of42–50 ppm/�C, which is comparable with those ofpolyimides 7 (43–54 ppm/�C), displaying gooddimensional stability of phosphinate-containingpolyimides.
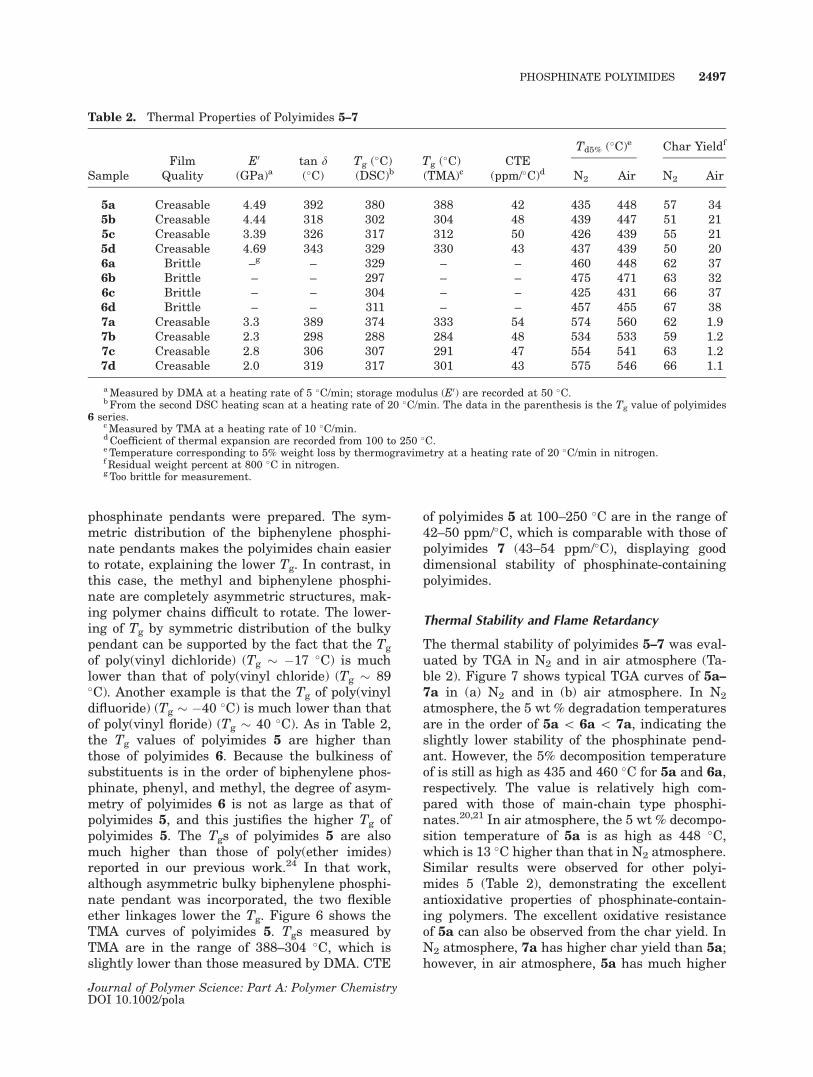
Thermal Stability and Flame Retardancy
The thermal stability of polyimides 5–7 was eval-uated by TGA in N2 and in air atmosphere (Ta-ble 2). Figure 7 shows typical TGA curves of 5a–7a in (a) N2 and in (b) air atmosphere. In N2
atmosphere, the 5 wt % degradation temperaturesare in the order of 5a \ 6a \ 7a, indicating theslightly lower stability of the phosphinate pend-ant. However, the 5% decomposition temperatureof is still as high as 435 and 460 �C for 5a and 6a,respectively. The value is relatively high com-pared with those of main-chain type phosphi-nates.20,21 In air atmosphere, the 5 wt % decompo-sition temperature of 5a is as high as 448 �C,which is 13 �C higher than that in N2 atmosphere.Similar results were observed for other polyi-mides 5 (Table 2), demonstrating the excellentantioxidative properties of phosphinate-contain-ing polymers. The excellent oxidative resistanceof 5a can also be observed from the char yield. InN2 atmosphere, 7a has higher char yield than 5a;however, in air atmosphere, 5a has much higher
Table 2. Thermal Properties of Polyimides 5–7
SampleFilm
QualityE0
(GPa)atan d(�C)
Tg (�C)(DSC)b
Tg (�C)(TMA)c
CTE(ppm/�C)d
Td5% (�C)e Char Yieldf
N2 Air N2 Air
5a Creasable 4.49 392 380 388 42 435 448 57 345b Creasable 4.44 318 302 304 48 439 447 51 215c Creasable 3.39 326 317 312 50 426 439 55 215d Creasable 4.69 343 329 330 43 437 439 50 206a Brittle –g – 329 – – 460 448 62 376b Brittle – – 297 – – 475 471 63 326c Brittle – – 304 – – 425 431 66 376d Brittle – – 311 – – 457 455 67 387a Creasable 3.3 389 374 333 54 574 560 62 1.97b Creasable 2.3 298 288 284 48 534 533 59 1.27c Creasable 2.8 306 307 291 47 554 541 63 1.27d Creasable 2.0 319 317 301 43 575 546 66 1.1
aMeasured by DMA at a heating rate of 5 �C/min; storage modulus (E0) are recorded at 50 �C.bFrom the second DSC heating scan at a heating rate of 20 �C/min. The data in the parenthesis is the Tg value of polyimides
6 series.cMeasured by TMA at a heating rate of 10 �C/min.dCoefficient of thermal expansion are recorded from 100 to 250 �C.e Temperature corresponding to 5% weight loss by thermogravimetry at a heating rate of 20 �C/min in nitrogen.f Residual weight percent at 800 �C in nitrogen.g Too brittle for measurement.
PHOSPHINATE POLYIMIDES 2497
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

char yield than 7a. As to the flame retardancy,according to UL-94VTM vertical thin test, the t1þ t2 is\2 s for 5a–5d, so they belong to the VTM-0 grade. In contrast, although Kapton is still aVTM-0 grade, the t1 þ t2 is[6 s, which is higherthan those of polyimides 5, demonstrating theirexcellent flame-retardant characteristic of phos-phinate-containing polyimides.
CONCLUSIONS
We have reported a one-pot process of preparingaromatic diamines (1–2) possessing bulky
Figure 6. TMA thermograms of polyimides 5.Figure 7. TGA curves of 5a–7a in (a) N2 and in (b)air atmosphere.
Figure 5. Dynamic mechanical analyses of polyimides 5.
2498 CHANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

biphenylene phosphinate pendant. A ‘‘nucleophilicaddition and then electrophilic substitution’’mechanism was proposed for the preparation of(1–2). In the mechanism, the stabilization of car-bocation is a crucial factor to the successful pro-ceeding of the reaction. When the carbocation isstabilized by strong electron-donating groupssuch as amino groups, the electrophilic substitu-tion of aniline can occur with high yield. Incontrast, if the carbocation is destabilized by anelectron-donating nitro groups, the electronic sub-stitution of aniline does not carry out. All the pol-yimides 5 are tough, flexible, and creasable. How-ever, polyimides 6 are brittle because of their highrigidity, as observed in the temperature-depen-dent NMR analysis of (2). The bulky biphenylenephosphinate pendant is not only responsible forthe improved solubility, but also for the high Tg
characteristic. The combination of flexibility,high glass transition temperature, high antioxida-tive stability, excellent flame retardancy, andimproved organosolubility, makes polyimides 5promising polymers for electronic applications,especially in the field of halogen-free flexibleprinted circuit boards, in which flame-retardancyand high-Tg is essential.
REFERENCES AND NOTES
1. Polyimides: Synthesis, Characterization, Applica-tion; Mittal, K. L., Ed.; Plenum Press: New York,1984; Vols. 1 and 2.
2. Terraza, C. A.; Liu, J. G.; Nakamura, Y.; Shiba-saki, Y.; Ando, S.; Ueda, M. J Polym Sci Part A:Polym Chem 2008, 46, 1510–1520.
3. Lee, C. H.; Wang, Y. Z. J Polym Sci Part A: PolymChem 2008, 46, 2262–2276.
4. Liaw, D. J.; Wang, K. L.; Chang, F. C.; Lee, K. R.;Lai, J. Y. J Polym Sci Part A: Polym Chem 2007,45, 2367–2374.
5. Wu, S.; Hayakawa, T.; Kakimoto, M., Oikawa, H.Macromolecules 2008, 41, 3481–3487.
6. Hsiao, S. H.; Lin, K. H. J Polym Sci Part A:Polym Chem 2005, 43, 331–341.
7. Chung, C. L.; Yang, C. P.; Hsiao, S. H. J PolymSci Part A: Polym Chem 2006, 44, 3092–3102.
8. Kricheldorf, H. R.; Fna, S. C.; Vakhtangishvili, L.;Schwarz, G.; Fritsch, D. J Polym Sci Part A:Polym Chem 2005, 43, 6272–6281.
9. Wu, S.; Hayakawa, T.; Kikuchi R.; Grunzinger S.J.; Kakimoto M. Macromolecules 2007, 40, 5698–5705.
10. Kim, Y. H.; Anh, S. K.; Kim, H. S.; Kwon, S. K. JPolym Sci Part A: Polym Chem 2002, 40, 4288–4296.
11. Liaw, D. J.; Liaw, B. Y. Macromolecules 1999, 32,7248–7250.
12. Watson, K. A.; Palmieri, F. L.; Connell, J. W. Mac-romolecules 2002, 35, 4968–4974.
13. Kwak, S. M.; Yeon, J. H.; Yoon, T. H. J Polym SciPart A: Polym Chem 2006, 44, 2567–2577.
14. Liu, Y. L.; Hsiue, G. H.; Lan, C. W.; Kuo, J. K.; Jeng,R. J.; Chiu, Y. S. J Appl Polym Sci 1997, 63, 875–882.
15. Jeong, K. U.; Kim, J. J.; Yoon, T. H. Polymer2001, 42, 6019–6030.
16. Sponton, M.; Ronda, J. C.; Galia, M.; Cadiz, V.J Polym Sci Part A: Polym Chem 2007, 45, 2142–2151.
17. Cheng, S. Z.; Chen, L.; Wang, Y. Z. J Polym SciPart A: Polym Chem 2008, 46, 5752–5759.
18. Canadell, J.; Mantecon, A.; Cadiz, V. J Polym SciPart A: Polym Chem 2007, 45, 1980–1992.
19. Canadell, J.; Mantecon, A.; Cadiz, V. J Polym SciPart A: Polym Chem 2007, 45, 1920–1930.
20. Banerjee, S.; Palit, S. K.; Maiti, S. J Polym SciPart A: Polym Chem 1994, 32, 219–227.
21. Liu, Y. L.; Hsiu, G. H.; Lee, R. H.; Chiu, Y. S. JAppl Polym Sci 1997, 63, 895–901.
22. Liou, G. S.; Hsiao, S. H. J Polym Sci Part A:Polym Chem 2001, 39, 1786–1799.
23. Liu, Y. L.; Hsu, C. Y.; Wu, C. S. J Appl Polym Sci2003, 89, 791–796.
24. Lin, C. H.; Lin, C. H. J Polym Sci Part A: PolymChem 2007, 45, 2897–2912.
25. Lin, C. H.; Wang, C. S. Polymer 2001, 42, 1869–1878.
26. Liu, Y. L. J Polym Sci Part A: Polym Chem 2002,40, 359–378.
27. Liu, Y. L.; Tsai, S. H. Polymer 2002, 43, 5757–5762.
28. Shieh, J. Y.; Wang, C. S. J Polym Sci Part A:Polym Chem 2002, 40, 369–378.
29. Liaw, D. J.; Liau, B. Y.; Lai, S. H. MacromolChem Phys 2001, 202, 807–813.
30. Leu, C. M.; Chang, Y. T.; Shu, C. F.; Teng, C. F.;Shiea, J. Macromolecules 2000, 33, 2855–2861.
31. Li, X.; Li, Y.; Tong, Y.; Shi, L.; Liu, X. Macromole-cules 2003, 36, 5537–5544.
32. Liaw, D. J.; Liau, B. Y. Macromol Chem Phys1999, 200, 1326–1332.
33. Liaw, D. J.; Liau, B. Y.; Chung, C. Y. MacromolChem Phys 2000, 201, 1887–1893.
34. Yi, M. H.; Huang, W.; Jin, M. Y.; Choi, K. Y. Mac-romolecules 1997, 30, 5606–5611.
35. Chang, Y. T.; Shu, C. F.; Le, C. M.; Wei, K. H.J Polym Sci Part A: Polym Chem 2003, 41, 3726–3735.
36. Ueda, M.; Nakayama, T. Macromolecules 1996,29, 6427–6431.
PHOSPHINATE POLYIMIDES 2499
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola


















