
Extraction et étude des propriétés physiques et...
84
I Extraction et étude des propriétés physiques et mécaniques des fibres d‟Alfa (Esparto grass) en vue d‟applications textiles Eva Rogge Tuteurs M. Abdelaziz Lallam (UHA), prof. dr. ir. Lieva van Langenhove (UGent), dr. ir. Vincent Nierstrasz (UGent) Accompagnateurs M. Mohamed Dallel (UHA), dr. Simone-Ileana Vasile (UGent) Masterproef ingediend tot het behalen van de academische graad van Master in de ingenieurswetenschappen : materiaalkunde Masterproef uitgevoerd in het kader van een ERASMUS-uitwisseling met UHA, Mulhouse, Frankrijk Ecole Nationale Supérieure d’Ingénieurs Sud-Alsace (UHA) Filière Textile et Fibres, sous la direction de prof. Jean-Yves Drean Faculteit Ingenieurswetenschappen (UGent) Vakgroep Textielkunde, sous la direction de prof. dr. Paul Kiekens Année universitaire 2009 – 2010
-
Upload
truongkiet -
Category
Documents
-
view
218 -
download
1
Transcript of Extraction et étude des propriétés physiques et...

I
Extraction et étude des propriétés physiques et mécaniques des fibres d‟Alfa (Esparto grass) en vue d‟applications textiles Eva Rogge Tuteurs M. Abdelaziz Lallam (UHA), prof. dr. ir. Lieva van Langenhove (UGent),
dr. ir. Vincent Nierstrasz (UGent)
Accompagnateurs M. Mohamed Dallel (UHA), dr. Simone-Ileana Vasile (UGent)
Masterproef ingediend tot het behalen van de academische graad van Master in de
ingenieurswetenschappen : materiaalkunde
Masterproef uitgevoerd in het kader van een ERASMUS-uitwisseling met UHA, Mulhouse,
Frankrijk
Ecole Nationale Supérieure d’Ingénieurs Sud-Alsace (UHA)
Filière Textile et Fibres, sous la direction de prof. Jean-Yves Drean
Faculteit Ingenieurswetenschappen (UGent)
Vakgroep Textielkunde, sous la direction de prof. dr. Paul Kiekens
Année universitaire 2009 – 2010

II
Préface et remerciements
L‟exécution d‟un projet de fin d‟études, ou tout simplement un stage, et l‟écriture du rapport
correspondant sont des missions importantes pour un étudiant. Après quatre ans et demi de
cours et d‟examens, le dernier semestre, réservé à ce projet, est une période complètement
différente. Finies les données et l‟information sous forme d‟une pile de papiers, à apprendre
pour les travaux personnels et les examens. Tout cela est remplacé par une période
d‟indépendance pendant laquelle l‟étudiant a la possibilité de faire état de ses propres
capacités sous forme d‟une recherche. Certes, cette indépendance n‟est pas absolue, le
tuteur et l‟accompagnateur sont là pour guider l‟étudiant dans le monde de la recherche, un
monde peu connu pour lui, et pour le guider à trouver son chemin. Cette période est
instructive dans différents domaines. L‟étudiant fait connaissance avec le monde de la
recherche, du travail et de la collaboration. Il apprend où il peut trouver l‟information
scientifique dont il a besoin pour mieux comprendre son sujet d‟étude et en utilisant les
appareils, il comprend mieux leur fonction et leur fonctionnement. Comme la recherche
n‟avance évidemment pas toujours comme voulue, l‟étudiant apprend à faire front aux
problèmes et à chercher des solutions avec les moyens disponibles. Et finalement, en
écrivant le mémoire, il développe sa langue scientifique et ses capacités à résumer
clairement une masse d‟informations, à présenter les résultats des expériences et à leurs
trouver des explications et des réponses et synthétiser tout cela en un seul rapport.
Comme un mémoire n‟est pas uniquement le résultat du travail de l‟étudiant lui-même,
j‟aimerais remercier les personnes qui m‟ont aidée et guidée.
D‟abord je voudrais remercier mon tuteur à l‟ENSISA, Mulhouse, France, M. Abdelaziz
Lallam pour m‟avoir offert la possibilité d‟intégrer le monde de la recherche, ainsi que pour
avoir répondu à mes questions multiples et m‟avoir donné ses avis et ses instructions pour la
suite des travaux.
Je voudrais également dire un grand merci à Mohamed Dallel, mon accompagnateur
quotidien, pour son soutien permanent et ses conseils tout au long de cette étude. Grâce à
son guidage, en me disant où et à qui je pouvais demander des conseils, j‟ai trouvé mon
chemin dans le bâtiment de l‟ENSISA et dans mon étude.
Je tiens à remercier également M. Stéphane Fontaine, responsable de l‟option « Eco-
conception » et Mme Laurence Schacher, responsable de la filière Textiles et Fibres de
l‟ENSISA et responsable des affaires étrangères (ERASMUS) parce que grâce à eux mon
stage à l‟ENSISA est devenu possible.
De plus j‟aimerais remercier toutes les personnes qui m‟ont aidée à la « Vakgroep
Textielkunde », UGent, Gand, Belgique, parce qu‟un programme ERASMUS n‟est possible
qu‟en cas d‟un accord entre deux universités. Je remercie également prof. dr. ir. Lieva Van
Langenhove, prof. dr. Kiekens, Katrien Hooreman et Judith Kenis pour avoir résolu tous les
problèmes en ce qui concerne l‟administration et mon programme ERASMUS.
Finalement je voudrais dire un très, très grand merci à Mme. Laurence Curie, enseignante
au CLAM à l‟UHA, à M. Abdelaziz Lallam, à Mohamed Dallel et à mon père, Rik Rogge, pour
la correction de mon rapport au niveau de la langue française et du contenu.

III
« A quelque chose malheur est bon » The author gives permission to make this master dissertation available for consultation and
to copy parts of this master dissertation for personal use.
In the case of any other use, the limitations of the copyright have to be respected, in
particular with regard to the obligation to state expressly the source when quoting results
from this master dissertation.
June 8, 2010

IV
Extraction et étude des propriétés physiques et mécaniques des fibres d‟Alfa (Esparto grass)
en vue d‟applications textiles
Eva Rogge
Masterproef ingediend tot het behalen van de academische graad van Master in de
ingenieurswetenschappen : materiaalkunde
Masterproef uitgevoerd in het kader van een ERASMUS-uitwisseling met UHA, Mulhouse,
Frankrijk
Tuteurs M. Abdelaziz Lallam (UHA), prof. dr. ir. Lieva van Langenhove (UGent),
dr. ir. Vincent Nierstrasz (UGent)
Accompagnateurs M. Mohamed Dallel (UHA), dr. Simone-Ileana Vasile (UGent)
Filière Textile et Fibres, sous la direction de prof. Jean-Yves Drean
Ecole Nationale Supérieure d‟Ingénieurs Sud-Alsace (UHA, Mulhouse, France)
Vakgroep Textielkunde, sous la direction de prof. dr. Paul Kiekens
Faculteit Ingenieurswetenschappen (UGent, Gent, Belgique)
Année universitaire 2009 – 2010
Résumé
La plante alfa est une plante sauvage qui pousse dans la région de la Méditerranée. Ses
tiges d‟alfa sont composées de filaments cellulosiques liés par de la lignine, des pectines et
de l‟hémicellulose.
Des fibres courtes sont obtenues par des méthodes d‟extraction agressives qui éliminent les
liants. Elles sont utilisées pour la production de papier ou le renforcement de composites.
Par contre, l‟extraction de fibres longues et souples pour la production de fils n‟est pas
encore développée et est donc le sujet de ce mémoire.
Le point de départ est un traitement de soude qui élimine la lignine. Ce traitement unique ne
suffit pas pour obtenir des fibres. Alors des pectinases sont utilisées pour l‟élimination des
pectines. Ce cycle double « soude – enzymes » ne donne toujours pas de résultats
satisfaisants, d‟où l‟addition d‟un traitement de soude supplémentaire. Finalement des fibres
longues sont obtenues. Elles sont un peu rigides à cause de la présence de lignine et de
pectines résiduelles, mais il est pourtant probable qu‟elles soient filables.
Une autre possibilité de production de fibres d‟alfa longues est la dissolution. Ce procédé à
base de NMMO est actuellement déjà utilisé dans l‟industrie à partir de cellulose de bois,
mais évidemment la cellulose d‟alfa peut également être dissoute.
Mots-clés
Fibres naturelles, alfa, Esparto grass (Stipa Tenacissima L.), extraction, dissolution

V
Extraction and study of the physical and
mechanical properties of Esparto grass fibres
in order to find textile applications
Eva Rogge
Supervisors: Abdelaziz Lallam, Lieva Van Langenhove, Vincent Nierstrasz, Mohamed Dallel,
Simone-Ileana Vasile
Abstract: The current article concerns the research
done in order to find on the one hand the best method
to extract fibres from Esparto grass stems, and on the
other hand the way to dissolve cellulose derived from
Esparto grass for spinning purposes.
Keywords: Natural fibres, Esparto grass, Stipa
Tenacissima L., extraction, dissolution
I. INTRODUCTION
In the past, natural fibres were the only available
fibres. Due to the development of chemistry, a wide
range of polymers that were easy and cheap to
produce became available. As a result of the
discovery that fibres could be produced from
polymers, synthetic fibres grew much more
important than the natural ones. Nowadays, because
of the polluting character of chemical and textile
processes, natural fibres have regained their
importance. Although even cotton, the most popular
natural fibre, is not very ecological as its cultivation
and production require a lot of water, insecticides
and pesticides. The search for alternatives led to
bamboo as well as to Esparto grass. The latter
grows in the dry and hot Mediterranean region. It
doesn’t need insecticides nor pesticides and only a
small amount of water. Because of those
advantages the Esparto grass culture is thus much
more ecologically friendly than cotton culture. If it
were possible to extract fibres to spin yarns and to
weave and knit, a new market would be born.
II. ESPARTO GRASS [1]
As Esparto grass is a plant with leaves looking like
stems, the name stem is used instead of leaves. The
plant, which can reach a height of 1m, has deep
roots with many secondary roots. Thanks to these
roots Esparto grass is able to limit the expansion of
the desert. The stem’s structure is complex. In
general cellulosic microfibrils or filaments are
linked together to form fibres. The stem itself
consists of many fibrebundles, which as a matter of
fact are a lot of fibres sticking together. The linking
components are lignin, pectins and hemicellulose.
Actual applications are the production of ropes and
baskets, cattle feed (the stems), non-wovens, high
quality paper and, at academic scale, the
reinforcement of composites (short fibres).
III. EXTRACTION
In this section the Esparto grass’ stems are treated
with chemical products to degrade and eliminate
the two main linking components, lignin and
pectins. As the objective is to produce fibres, the
hemicellulose doesn’t need to be eliminated
because it sticks the cellulosic filaments together to
form fibres.
A. Pre-treatments
First of all the stems are being carded mechanically
to refine their diameter. Afterwards they are
submerged in 35 g/l salt water during 24h at 60°C
or 12h at 80°C, to dissolve the waxes, a layer at the
surface to protect the plant against heat by limiting
the evaporation of water.
B. Extraction
The main chemical product used for extraction is
sodium hydroxide (NaOH), which eliminates the
lignin. Na2O4S2 is added as reducing agent to
protect the cellulose against oxidation (aka
degradation) by the NaOH. The varying parameters
are the temperature (50°C, 100°C, 130°C, 140°C
and >150°C), the NaOH concentration (0.25M,
0.5M, 1M and 2M) and the duration (1h and 2h),
which are chosen in function of the previous result.
By bleaching with a 9.6% NaOCl-solution the rigid
“fibres” get whiter, but not more flexible.
The replacement of the bleaching by an enzymatic
treatment with pectinases (5 ml/l, pH 7, 38°C, 2h)
improves the results and the fibres produced are
less rigid, even with NaOH-concentrations of

VI
0.25M at 140°C. Nevertheless this result isn’t really
acceptable because of the remaining rigidity.
A possible reason is the presence of non-removed
lignin in the inner structure of the stems. By
removing the pectins this lignin is now uncovered
which makes it possible to remove the lignin by a
second NaOH-treatment. Under the following
conditions this finally leads to the most acceptable
extracted fibres:
- NaOH: 0.25M, 140°C during 90 minutes
and with 1.5% Na2O4S2
- Pectinases: 2.5 ml/l, 38°C during 1h, pH 7
- NaOH: 0.25M, 140°C during 1h and with
0.5% Na2O4S2
C. Post-treatments
The fibres are being dried at 50°C - 60°C until they
are completely dry. The fibres cannot be dried in
the optimal way due to the lack of appropriate
equipment in the laboratory. So after drying the
fibres are stuck together and need to be separated
manually or with a brush. The length of the fibres is
about 5 cm.
D. Properties
Examination of the fibres with the SEM microscope
(Figure III.1) shows the presence of non-cellulosic
components and the non-alignment of some
cellulosic filaments, but makes also clear that those
filaments are not degraded. Of course this is a good
sign.
Figure III.1: SEM picture of the most acceptable fibres
Analysis by FTIR-ATR spectroscopy (Figure III.2)
shows the peaks for lignin (1650 cm-1, 1510-1450
cm-1, and close to 700 cm-1) and pectines (1730
cm-1), which confirms the SEM photos.
Figure III.2: FTIR-ATR spectrum of the fibres
A few tensile tests didn’t end up in reliable results,
especially not for natural fibres because of the
variance of diameter, composition, colour and
length. Anyhow some tests were done to obtain
some information about the mechanical properties
of the extracted Espartograss fibres. Force and
elongation were measured and transformed to
tension and relative elongation. An estimated
diameter of 135 µm was used for the calculations.
A big variance exists (Table III. 1) between the
results for the strength (σ), the relative elongation
(Δ L) and the Young’s modulus (E).
Table III. 1: Mechanical properties of Esparto grass fibres
parameter σ (MPa) Δ L (%) E (GPa)
mean value 63,83 3,12 2,05
standard deviation 16,80 0,63 0,77
variance coefficient 26,31 20,12 37,55
IV. DISSOLUTION
Another way to produce Esparto grass fibres is by
dissolving its cellulose in a solvent, followed by a
spinning process to produce filaments.
In textile industry the dissolution of cellulose is
widely being used, mainly under the name viscose.
Because of environmental reasons the solvent
NMMO is being preferred, like for the Lyocell
process.
The NMMO is dissolved in water, and the cellulose
is added. At temperatures between 70°C and 100°C,
with six times as much NMMO as cellulose under
constant stirring (500 rpm up to 900 rpm), the
excess of water is evaporated and finally a solution
is produced. The colour needs to be pale and
transparent, but in the beginning brown solutions
have been obtained. This indicates an un-wanted
degradation of the cellulose. This can be avoided by
adding 1% Na2O4S2 which gives a more transparent
and paler colour.
V. CONCLUSION
It is possible to extract Esparto grass fibres by using
chemical products, such as sodium hydroxide and
pectinases (a kind of enzymes), in a three step
process.
The dissolution of Esparto grass cellulose in an
aqueous NMMO-solution leads to a solution, which
can be used to spin filaments.
ACKNOWLEDGMENTS
The author wants to thank everyone who helped to
realise this dissertation.
REFERENCES
[1] Akchiche O., Messaoud Boureghda K. Esparto Grass
(Stipa Tenacissime L.), raw material of papermaking.
Chimija rastitel’nogo syr’ja, 2007, First Part, 4, pp. 25-30.
0
0,1
0,2
0,3
5001000150020002500300035004000
Intensity
k (1/cm)

Sommaire
LISTE DES ABREVIATIONS ET DES SYMBOLES 1
INTRODUCTION 3
CHAPITRE 1 : ETUDE BIBLIOGRAPHIQUE SUR L‟ALFA 4
1.1. LES FIBRES D‟ORIGINE NATURELLE 4
1.1.1. Les fibres de graines ou de fruits (Fibres of seeds and fruits) 4
1.1.2. Les fibres de feuilles (Leaf fibres) 4
1.1.3. Les fibres libériennes (Bast or stem fibres) 5
1.2. LA CELLULOSE : COMPOSANT PRINCIPAL DES FIBRES VEGETALES [1,2,3] 5
1.2.1. Généralités 5
1.2.2. Réactions chimiques 6
1.3. L‟ALFA 7
1.3.1. Généralités [4] 7
1.3.2. La morphologie de la plante 7
1.3.3. Structure et morphologie des fibres 8
1.3.4. Composition chimique 9
1.3.5. Les fibres d‟alfa comparées à d‟autres fibres naturelles aux points de vues
propriétés mécaniques 11
1.3.6. Applications actuelles 12
CHAPITRE 2 : EXTRACTION DES FIBRES D‟ALFA 14
2.1. INTRODUCTION 14
2.1.1. Généralités et objectif 14
2.1.2. Situation : l‟extraction des fibres courtes pour une pâte à papier 14
2.1.3. La recherche précédente : l‟extraction des fibres d‟alfa plus longues 15
2.1.4. Elimination des pectines avec des enzymes 16
2.2. MODE OPERATOIRE 17
2.2.1. Introduction 17
2.2.2. Les prétraitements 18
2.2.3. L‟extraction 20
2.2.4. Les post-traitements 29
2.2.5. Les propriétés et caractéristiques des fibres obtenues 31
2.3. CONCLUSION DU MODE OPERATOIRE 39
CHAPITRE 3 : DISSOLUTION DE LA CELLULOSE 41
3.1. L‟OBJECTIF DE LA DISSOLUTION : LE FILAGE DES FILAMENTS 41
3.1.1. Le filage par fusion (Figure 43) (angl. melt spinning) 41
3.1.2. Le filage à sec (Figure 44) (angl. dry spinning) 41
3.1.3. Le filage à humide (Figure 45) (angl. wet spinning) 41
3.1.4. Le filage à sec et par coagulation (angl. dry jet wet spinning) 42
3.2. LE PROCEDE VISCOSE 42
3.2.1. La pâte cellulosique (angl. cellulosic pulp) 43
3.2.2. L‟enflure de la pâte (angl. steeping) 43
3.2.3. Shredding 43
3.2.4. Mercérisation (angl. pre-ageing, mercerising) 43

3.2.5. Formation de xanthate (angl. xantation) 44
3.2.6. Dissolution du xanthate de cellulose et « ageing » 44
3.2.7. Filage par coagulation 44
3.3. LE PROCEDE LYOCELL 44
3.3.1. Généralités 44
3.3.2. En détail : la dissolution de cellulose 46
3.4. LA PARTIE EXPERIMENTALE 48
3.4.1. Introduction 48
3.4.2. La matière première 48
3.4.3. Les premiers essais 49
3.4.4. La première solution 49
3.4.5. Les solutions suivantes et le filage 50
3.5. RESUME ET CONCLUSION 52
CONCLUSION 54
APPENDICE A : APPAREILS ET OUTILS DANS LE LABORATOIRE 55
A.1. LES PLAQUES CHAUFFANTES 55
A.2. LES BALANCES 55
A.3. LA MACHINE A TEINTURE 56
A.4. LE PH-METRE 57
A.5. L‟IRTF (ANGL. FTIR) 58
A.6. LE MEB (ANGL. SEM) [24] 59
APPENDICE B : RESUME OBLIGATOIRE EN NEERLANDAIS 60
1. LITERATUURSTUDIE OVER ESPARTOGRAS 60
1.1. Inleiding 60
1.2. Cellulose, hoofdbestanddeel van plantaardige vezels [1,2,3] 60
1.3. Espartogras 60
2. EXTRACTIE VAN VEZELS 63
2.1. Inleiding 63
2.2. Extractie van espartograsvezels 63
2.3. Eigenschappen van de vezels 68
3. OPLOSSEN VAN ESPARTOCELLULOSE 70
APPENDICE C : LISTE DES FIGURES 72
APPENDICE D : LISTE DES TABLEAUX 74
BIBLIOGRAPHIE 75

1
Liste des abréviations et des symboles
AA angl. acrylic acid ; l‟acide acrylique
Ac angl. acetic anhydride ; l‟anhydride acétique
angl. anglais
c la vitesse de la lumière sous vide : 3*108 m/s [29]
conc. une concentration
Δ une différence entre deux valeurs
D.P. angl. degree of polymerisation ; le degré de polymérisation
Avec : Mtot : la masse totale du polymère
Mmm : la masse du monomère ou du groupe répétitif
ENSISA Ecole Nationale Supérieure des Ingénieurs de Sud-Alsace, composée de
l‟ENSISA Werner (les filières Textile & Fibres et Mécanique) et l‟ENSISA
Lumière (les filières Informatique & Réseaux, Systèmes & Signaux et
Systèmes de Production)
FTIR-ATR Fourier Transform InfraRed Spectroscopy – Attenuated Total Reflection
h la constante de Planck : 6.63*10-34 Js [29]
IR angl. infrared ; l‟infrarouge
IRTF-RAT la Spectroscopie InfraRouge à Transformée de Fourier – Réflexion Atténuée
Totale ; idem FTIR-ATR
k le nombre d‟onde ; l‟inverse de la longueur d‟onde (λ)
λ une longueur d‟onde
LPMT Laboratoire de Physique et Mécanique Textiles, à Mulhouse
MA angl. maleic anhydride ; l‟anhydride maléique
MH une masse humide : surtout la masse de fibres d‟alfa dans un état humide
MS une masse sèche : surtout la masse de fibres d‟alfa dans un état sec
MEB un microscope à balayage
M.W. angl. molecular weight ; la masse moléculaire basée sur le poids
∑
∑
∑
∑
Avec : ML : la masse moléculaire des chaînes ayant une longueur l
wL : la masse des molécules ayant une longueur l, ou bien le nombre
de molécules ayant une longueur l (nL) multiplié par ML
N une concentration sous forme de nombre de moles par litre d‟un liquide
NaOH l‟hydroxyde de sodium, nom commercial : la soude
NaOCl l‟hypochlorite de sodium, nom commercial : l‟eau de Javel
Na2O4S2 l‟hydrosulfite de sodium, nom commercial : le dithionite de sodium
NMMO angl. N-methylmorpholine-N-oxide ou 4-methylmorpholine-4-oxide; N-
méthylemorpholine-N-oxide
p une pression
p.e. par exemple
rpm rotations per minute; le nombre de rotations par minute
S angl. styrene; le styrène
T, temp. une température

2
UHA Université de Haut-Alsace
UV-VIS angl. UV-Visible light: rayons ultraviolets-visibles (longueur d‟onde dans le
spectre ultraviolet ou dans le spectre visible) ; utilisé dans la spectroscopie

3
Introduction
Dans ce rapport il s‟agit de la plante alfa et l‟extraction de fibres à partir de ses tiges. La
plante, qui pousse dans des régions sèches, a une grande importance écologique parce que
sa culture ne nécessite ni de quantités d‟eau énorme, ni d‟insecticides et de pesticides.
L‟extraction de fibres est démarrée à partir de la recherche préalable et des informations
trouvées dans la littérature. L‟objectif de l‟extraction est l‟élimination des liants non-
cellulosiques, tels que la lignine et les pectines. Beaucoup de manipulations ont été faites
dans le Laboratoire de Physique et Mécanique textiles (LPMT). Chaque fois l‟échantillon
obtenu est examiné et pour l‟essai suivant, les paramètres de travail sont adaptés en
fonction du résultat obtenu. Finalement des fibres ont été produites. Il ne s‟agit pas des
meilleures fibres possibles, mais ce résultat constitue quand même un bon point de départ
pour la suite de la recherche.
Une autre possibilité d‟obtenir des fibres d‟alfa est la dissolution de cellulose d‟alfa. Celle-ci
est produite lors des essais d‟extraction sous forme de fibres courtes et dégradées. Le
produit NMMO est choisi comme solvant parce que ce procédé est déjà connu dans
l‟industrie et a quelques avantages par rapport au procédé de viscose. Une solution de
cellulose d‟alfa est produite, mais faute de temps, le filage des filaments n‟a pas été réalisé.

4
CHAPITRE 1 : Etude bibliographique sur l’alfa
1.1. Les fibres d‟origine naturelle
Les fibres d‟origine naturelle sont extraites de la nature.
Il s‟agit
- des fibres végétales extraites de plantes, de fruits et d‟arbres comme le coton, le lin,
l‟agave, le jute, le chanvre, etc.
- des fibres animales extraites de poil d‟animaux comme le mouton, la chèvre, le
chameau, le lama, etc.
- des fibres minérales comme l‟amiante
Toutes les fibres naturelles sont biodégradables, ce qui veut dire que la nature peut les
dégrader jusqu‟à leurs composants de base sans intervention de l‟homme. Autrefois les
fibres naturelles étaient dépassées par les fibres synthétiques qui étaient moins chères et
plus faciles à produire et qui avaient des propriétés supérieures. A cause de la tendance
actuelle de respect croissant pour la nature, la biodégradabilité des fibres devient de plus en
plus importante. C‟est pourquoi les fibres naturelles biodégradables ont de nouveau gagné
de l‟ampleur par rapport aux fibres synthétiques non biodégradables.
Une caractéristique des fibres naturelles est la variation des propriétés mécaniques et
chimiques due à la variation de leur composition. L‟origine de cette variation de composition
est d‟une part le fait qu‟ils existent plusieurs variétés des plantes (p.e. plusieurs espèces de
coton) et des animaux (p.e. plusieurs espèces de laine) dont sont extraites les fibres et
d‟autre part le fait que même les fibres de coton d‟une plante particulière et les fibres de laine
d‟un seul mouton varient en longueur, épaisseur, rigidité, couleur, etc.
La fibre d‟alfa est extraite de la plante, il s‟agit donc d‟une fibre végétale. C‟est la raison pour
laquelle les fibres animales et minérales ne sont pas traitées plus en détail dans la suite de
ce mémoire de fin d‟études.
Les fibres végétales elles-mêmes sont divisées en trois catégories [1] :
- les fibres de graines ou de fruits, p.e. le coton et le coco
- les fibres de feuilles, p.e. le sisal, l‟abaca et l‟alfa
- les fibres libériennes ou fibres de tige, p.e. le lin, le jute, le chanvre et la ramie
1.1.1. Les fibres de graines ou de fruits (Fibres of seeds and fruits)
Ces fibres poussent sur des fruits ou des graines, elles sont courtes et fines. L‟exemple le
plus connu est le coton qui est devenu la fibre naturelle la plus importante au monde.
Récemment la quantité produite a été dépassée par celle de la fibre synthétique PET (le
téréphtalate de polyéthylène). La culture de coton demande beaucoup d‟eau et de soleil, de
pesticides, d‟insecticides et d‟engrais. Afin de réduire ces effets nuisibles sur
l‟environnement, les producteurs sont à la recherche d‟alternatives plus écologiques.
1.1.2. Les fibres de feuilles (Leaf fibres)
Ces fibres sont généralement plus longues que les fibres de graines, principalement à cause
des feuilles souvent très grandes. Outre des applications textiles (des fils pour des tissus et
des tricots), elles sont utilisées comme matière première dans la production de câbles et de
cordes. Les fibres cellulosiques issues d‟alfa brute sont des fibres de feuilles.

5
1.1.3. Les fibres libériennes (Bast or stem fibres)
Ces fibres se présentent sous forme de faisceaux de fibres ultimes liées entre-elles, formant
ainsi la tige. Celle-ci contient encore d‟autres composants, par exemple ceux transportant
l‟eau extraite du sol par les racines vers les feuilles et ceux qui sont responsables de la
liaison des fibres ultimes.
Pendant l‟extraction, les fibres ultimes sont obtenues par élimination des liants via un
procédé appelé rouissage, réalisé biologiquement dans les champs de culture ou
chimiquement dans des chambres de rouissage.
1.2. La cellulose : composant principal des fibres végétales [1,2,3]
1.2.1. Généralités
Le composant principal des fibres végétales est la cellulose. La formule brute est (C6H10O5)n.
La cellulose est un polymère naturel obtenu par la polymérisation d‟un dimère (le cellobiose,
Figure 2), lui-même issu de la dimérisation du monomère β-glucose (C6H12O6) (Figure 1),
selon les réactions suivantes :
(1.1)
(1.2)
La polymérisation de β-glucose, une polycondensation des groupes d‟hydroxyde (-OH) des
atomes de carbone numéro 1 et 4 avec la production d‟eau, donne le polymère cellulose
(Figure 3). Chaque molécule de glucose doit être orientée 180° par rapport aux voisines pour
que la polymérisation soit possible.
Par contre, la polymérisation de α-glucose, de nouveau par les groupes fonctionnels des
atomes de carbone 1 et 4, donne le polymère amidon (angl. starch) (Figure 4).
Contrairement à la polymérisation de β-glucose donnant la cellulose, les molécules de
β-glucose ne doivent pas être tournées l‟une contre l‟autre pour la polymérisation donnant
l‟amidon.
Figure 2 : Une molécule de cellobiose Figure 1 : Une molécule de β-glucose avec la numérotation des atomes de carbone
Figure 3 : La cellulose Figure 4 : L‟amidon

6
La différence entre la cellulose et l‟amidon est la liaison des molécules de glucose. Les
chaînes de cellulose sont faciles à organiser, donnant une structure cristalline, mais les
chaînes d‟amidon sont beaucoup plus difficiles à organiser. Pour cela des fibres de cellulose
plutôt fortes existent tandis que des fibres d‟amidon dans sa forme pure n‟existent pas.
Comme les chaînes de cellulose peuvent s‟aligner, des zones cristallines peuvent être
formées. Cela donne aux fibres finales, c‟est-à-dire après l‟extraction, une certaine rigidité
qui dépend du taux de cristallinité.
1.2.2. Réactions chimiques
Le glucose, et par conséquent la cellulose dérivée, est chimiquement réactive parce que
trois groupes d‟hydroxyde (-OH) sont présents dans chaque molécule de glucose, dont deux
alcools secondaires (atome de carbone numéro 2 et 3) et un alcool primaire (atome de
carbone numéro 6). Ces groupes participent aux réactions chimiques normales pour former
des esters et éthers. P.e. le nitrate de cellulose est devenu de grande importance chimique.
Exceptées ces réactions de type normales, la chaîne d‟atomes peut être cassée par une
attaque chimique. Alors le degré de polymérisation (D.P.) diminue et par conséquent les
propriétés mécaniques chutent de même.
1.2.2.1. Hydrolyse acide
Une hydrolyse acide produit de la hydrocellulose, qui est de la cellulose avec une chaîne
cellulosique plus courte. La valeur du pH est inférieure à 7 (d‟où le nom hydrolyse « acide »)
et la présence d‟eau est nécessaire pour la dégradation. Comme la chaîne est cassée, un
groupe d‟hydroxyde (-OH) à l‟atome de carbone numéro 1 apparaît au lieu de la liaison
d‟éther entre deux molécules de glucose. Ce groupe peut se transformer en un aldéhyde par
tautomérie (Figure 5).
1.2.2.2. Estérification
Les groupes d‟alcool (-OH) libres dans la chaîne de cellulose peuvent réagir avec des
acides. Une molécule d‟eau est éliminée par réaction et des esters sont formés. La réaction
est une modification de la cellulose sans dégradation nette. Un, deux ou trois groupes
peuvent réagir. Dans le cas de trois remplacements, le produit final est appelé le tri acétate
de cellulose. Dans les autres cas, il s‟agit d‟acétate de cellulose.
1.2.2.3. Oxydation
L‟oxydation est une réaction en présence de l‟oxygène. Avec la cellulose, l‟oxygène forme
une oxycellulose sous deux formes :
- L‟oxycellulose réduite, avec rupture de la chaîne cellulosique à cause de l‟attaque de
la liaison C-C par l‟oxygène et avec la formation de deux fonctions d‟aldéhyde (Figure
6).
Figure 5 : A gauche la hydrocellulose, à droite l‟aldéhyde obtenu par tautomérie

7
- L‟oxycellulose acide, dans laquelle le groupe alcoolique des atomes de carbone 2, 3
ou 6 est transformé en un aldéhyde et ensuite en un acide. Dans le cas des alcools
secondaires (2 et 3) l‟anneau du glucose est cassé (Figure 7), pour l‟alcool primaire
l‟anneau reste intact (Figure 8).
1.3. L‟alfa
1.3.1. Généralités [4]
Le nom latin d‟alfa est Stipa Tenacissima L., le nom anglais est Esparto grass ou Esparto.
La plante est une graminée et est un membre de la famille des herbes. C‟est une plante
permanente qui ne disparaît pas pendant l‟hiver et qui pousse indépendamment formant des
nappes. Grâce à la faible consommation d‟eau, l‟alfa est endémique dans la région
méditerranée d‟ouest, une région plutôt sèche.
La répartition territoriale est montrée dans le Tableau 1. Chaque année la quantité d‟alfa
poussant diminue à cause d‟actions humaines telles que l‟exploitation irrationnelle, le
surpâturage, les incendies, etc.
La plante a un intérêt écologique, économique et social. L‟alfa n‟a en effet pas besoin
d‟insecticides ni de pesticides nuisibles à l‟environnement et elle consomme très peu d‟eau.
La récolte et la transformation actuelle pour applications papetières offrent des emplois aux
habitants de la région, ainsi diminuant le taux de chômage et améliorant le niveau de vie.
Le jour où des applications textiles deviendront disponibles, de nouveaux marchés pourront
se développer, comme c‟était le cas lors de l‟industrialisation des fibres de bambou dans les
années 2000.
1.3.2. La morphologie de la plante
La plante est plutôt une grande herbe dure au lieu d‟une plante « normale » avec une grande
tige avec des ramifications et des feuilles. Par contre, beaucoup de tiges poussent en forme
de cercle (du vue d‟en haut) en se partageant la même racine (Figure 9). Les tiges peuvent
atteindre une hauteur d‟un mètre, et les racines une profondeur de plus d‟un mètre. Entre les
deux se trouve le rhizome (Figure 10). Les racines sont très ramifiées avec beaucoup de
Pays superficie (ha)
Algérie 4.000.000
Maroc 3.186.000
Tunisie 600.000
Libye 350.000
Espagne 300.000
Portugal peu
Figure 7 : L‟oxycellulose acide (C2 ou C3)
Figure 8 : L‟oxycellulose acide (C6)
Figure 6 : L‟oxycellulose réduite
Tableau 1 : La répartition territoriale de la plante alfa [5]

8
nœuds où de nouvelles racines secondaires commencent. De cette façon, l‟alfa est bien
ancrée dans le sol, ce qui est nécessaire dans les régions où elle pousse. Ces régions se
trouvent à la frontière du désert où le sol de sable n‟est pas un sol dur. Donc des racines
ramifiées sont nécessaires pour pouvoir y tenir et croître.
En même temps, le fait que l‟alfa stabilise bien le sable et le sol avec ses racines, donne une
fonction importante de cette plante, c‟est-à-dire elle sert à arrêter la désertification et à éviter
l‟érosion éolienne. Grâce à sa présence, le vent déplace peu le sable.
De plus, les tiges ou bien les feuilles proches du sol sont velues et cireuses. Quand le vent
souffle et transporte le sable et la poussière de sable, ces parties velues peuvent capter ces
particules et ainsi arrêter la désertification d‟une deuxième façon.
Les cires limitent l‟évaporation et cela est la raison pour laquelle l‟alfa peut résister à des
températures élevées et consomme peu d‟eau.
L‟alfa fleurit normalement de début mai jusqu‟à fin juin. L‟alfa « mûre » est récoltée
manuellement de juillet jusqu‟à novembre.
Elle est capable de résister à une grande variation de température. P.e. la nuit des
températures de -20°C ont été mesurées, mais pendant la journée en été la température
peut atteindre les 40°C [4].
1.3.3. Structure et morphologie des fibres
En général, la structure des fibres est hétérogène. Les plus petites parties dans les fibres
sont les filaments cellulosiques ou les fibrilles, ayant des longueurs de 2 à 5 mm et des
diamètres de 5 à 10 µm. Ces fibrilles se sont liées d‟une manière dense par l‟hémicellulose
en formant les fibres [6]. Leur coupe transversale a une forme irrégulière comme le montre la
photo prise avec un microscope optique (Figure 11). Les fibres ont un diamètre de 50 µm
environ (Figure 12). Ensuite les fibres se sont liées par la lignine et des pectines ce qui
donne les faisceaux de fibres, c‟est-à-dire les fibres techniques. Le lumen dont la fonction est
le transport de l‟eau dans la plante vivante, est étroit. La coupe transversale des faisceaux
de fibres montre que la section n‟est pas circulaire et que le diamètre est 200 µm environ
(Figure 13). La liaison des faisceaux de fibres donne finalement la tige (Figure 14).
Figure 9 : Représentation de la plante alfa Figure 10 : Photo de la plante alfa avec indication des parties principales

9
1.3.4. Composition chimique
Tout d‟abord il est intéressant de connaître la répartition des différents composants de l‟alfa.
O. Akchiche, A.B. Marchak et Y.G. Butko ont publié en 1987 des résultats d‟analyses
chimiques. Ils ont trouvé 74.5% d‟hydrocarbures, parmi lesquelles la cellulose,
l‟hémicellulose et les pectines, et 18.5% de lignine. Les taux des composants varient selon la
source :
1.3.4.1. La lignine [10]
La lignine est le composant liant dans les tiges d‟alfa et en général dans les plantes. C‟est le
liant entre les fibres formant des faisceaux ou des tiges. Sans la lignine, les tiges d‟alfa
n‟existeraient pas. L‟extraction des fibres consiste en la libération des fibres ultimes en
dégradant les composants non-cellulosiques comme la lignine. L‟élimination de la lignine est
appelée la délignification.
Matière [8] (%) [6] (%) [7] (%) [9] (%)
Cellulose 43,81 45 45 47,63
Lignine 18,76 23 24 17,71
Cendres 4,66 2 2 5,12
Silica 1,76
Hémicellulose/Pectines 28,4 25 24 22,15
Cires 5 5
Extraction et autres 2,61 7,39
Somme (%) 100 100 100 100
Figure 11 : Image microscope optique des fibres composées de filaments cellulosiques [6]
Tableau 2 : La composition d‟alfa
Figure 12 : Image MEB de la coupe transversale des fibres [7]
Figure 13 : Image MEB de la coupe transversale des faisceaux de fibres d‟alfa [7]
Figure 14 : Image MEB de la coupe transversale de la tige d‟alfa [6]

10
Une structure de base (Figure 15) de la lignine existe avec deux groupes fonctionnels qui
peuvent varier, ainsi réalisant plusieurs molécules différentes de lignine. Par conséquent en
réalité, le nom « la lignine » n‟est pas correct car il s‟agit de plusieurs molécules. Il est donc
mieux de parler « des lignines ». En considérant la structure de base, il est clair qu‟un
groupe aromatique est présent, et que les deux groupes qui varient sont un groupe
aromatique (-Ar) et aliphatique (-R). Mais pourtant la structure exacte reste inconnue [11].
Cependant des analyses de spectroscopie UV-VIS donnent une idée de la structure de la
lignine (Figure 16). Les liaisons covalentes sont relativement fortes, concluant à une bonne
résistance biologique et chimique. C‟est pour cela que l‟élimination de la lignine afin d‟obtenir
des fibres d‟alfa est difficile. Les propriétés mécaniques des lignines sont faibles.
1.3.4.2. L’hémicellulose [10]
L‟hémicellulose est présente avec la cellulose dans les parois des cellules des plantes. Sa
structure correspond fortement à celle de la cellulose. Néanmoins quelques différences
importantes existent ; p.e. la cellulose a une structure cristalline mais l‟hémicellulose est
amorphe et donc moins forte. De plus la cellulose est faite de β-glucose comme seul
monomère, mais pour la polymérisation d‟hémicellulose plusieurs monomères glucidiques,
comme le xylose, le mannose, le galactose, le rhamnose (un désoxy-hexose du mannose) et
l‟arabinose, sont possibles. Donc tout comme la lignine, l‟hémicellulose existe également
sous différentes formes, parmi lesquelles le xylane est la molécule la plus abondante. Ce qui
fait, que la cellulose est un composant désiré et non pas l‟hémicellulose.
1.3.4.3. Les pectines [10]
Les pectines font également partie des plantes, plus spécifiquement de leurs tiges et de
leurs fruits. Quant à l‟alfa, les pectines se trouvent dans les tiges.
La fonction des pectines est la liaison des faisceaux de fibres. Leurs structures ressemblent
beaucoup à celles des hémicelluloses. En outre, pendant la maturation des fruits, des
pectines sont souvent transformées en hémicelluloses, ce qui montre la similarité entre les
deux structures.
Les pectines sont donc, comme les hémicelluloses, composées de polysaccharides, c‟est-à-
dire des polymères formés à partir de monomères glucidiques. Une différence entre la
structure chimique des pectines et celle des hémicelluloses est le fait que les pectines
possèdent des groupes carboxyl (-COOH) tandis que l‟hémicellulose n‟en a pas.
De plus, l‟arrangement des pectines est similaire à celui de la cellulose, c'est-à-dire des
chaînes linéaires grâce à la rotation des monomères par rapport aux monomères voisins. La
Figure 15 : Structure de base de lignine
Figure 16 : Structures possibles de lignine [12]

11
combinaison des chaînes linéaires avec les groupes carboxyl rend possible les fortes
liaisons d‟hydrogène entre les chaînes.
1.3.4.4. Les cires [10]
Les cires sont des lipides qui se trouvent dans une fine couche à l‟extérieur des tiges. Les
cires ont plusieurs fonctions, comme la protection physique contre des conditions
environnementales défavorables et des insectes. Les structures peuvent être très
complexes, mais parmi les lipides les plus courants se trouvent les hydrocarbures (C21-C35),
les esters de cire (C34-C62), les cétones (C23-C33), les alcools (C22-C33) et les acides gras
(C16-C32) [13]. La structure et la composition de la couche des cires peuvent varier tout au
long de la plante et de la tige.
1.3.5. Les fibres d’alfa comparées à d’autres fibres naturelles aux points de
vues propriétés mécaniques
Le Tableau 3 donne les propriétés mécaniques en traction des principales fibres naturelles et
synthétiques. En ce qui concerne l‟alfa, il s‟agit des propriétés de l‟alfa technique, c‟est-à-dire
les faisceaux de fibres.
Pour les fibres (ultimes), il faut donc se contenter de valeurs moyennes estimées [14], la
densité étant 0.89 g/cm³, l‟élongation de rupture 5.8%, la tension de rupture 565 MPa et le
module d‟Young 22 GPa. Chaque des ces propriétés a des valeurs magnifiques, indiquant
les fibres élastiques, mais également fortes et très légères. Mais en comparant ces valeurs
avec celles de faisceaux d‟alfa ou de fibres de coton, il semble qu‟elles sont surestimées.
En ce qui concerne l‟élongation de rupture (angl. strain at break), la valeur d‟alfa technique
(1.5-2.4%) est proche de celle du jute (1.5-1.8%), du chanvre (1.6%) et du sisal (2-2.5%).
En ce qui concerne la tension de rupture (angl. specific stress at break) les 134-220 MPa de
l‟alfa technique sont proches des 191-398 MPa du coton.
Quant au module d‟Young (angl. specific Young modulus), qui est une indication pour la
rigidité, la valeur d‟alfa technique (13-17.8 GPa) est proche de celle du lin (18 GPa) et de la
ramie (17 GPa) et est supérieure à celle du coton (3.6-8.4 GPa) et l‟agave (4.2 GPa). La
ramie est la fibre végétale la plus forte avec un module d‟Young de 42-86 GPa.
En général les propriétés mécaniques en traction de l‟alfa technique approchent celles du
jute, du lin, du chanvre et du sisal.
Le Tableau 4 donne les résultats des essais de traction sur des faisceaux de fibres d‟alfa.
Les caractéristiques mécaniques varient beaucoup, ce qui est normal parce qu‟il s‟agit de
fibres naturelles. Figure 17 donne la courbe moyenne obtenue lors de ces essais. Pour une
Tableau 3 : Les propriétés mécaniques en traction des principales fibres naturelles et synthétiques [7]

12
tension de 200 MPa environ, une chute de tension due à des ruptures ou fissures des fibres
ultimes dans les fibres techniques a été constatée.
1.3.6. Applications actuelles
Aujourd‟hui l‟alfa est utilisée de deux façons.
D‟un côté la plante elle-même est utilisée
- pour produire des objets tels que paniers, tapis et cordes (applications artisanales)
- comme nourriture par les animaux sauvages comme la gazelle, le lièvre, etc.
- comme nourriture pour le bétail. Dans les régions où l‟alfa pousse, les paysans ne
peuvent pas toujours acheter toute la nourriture requise pour le bétail. Dans le cas
échéant, le bétail mange comme alternative les plantes d‟alfa qui ont une faible
valeur alimentaire. Comme l‟exploitation de l‟alfa est trop intensive par rapport à la
capacité de celle-ci pour se régénérer, le risque de surpâturage est réellement
présent.
D‟autre côté les fibres courtes sont utilisées
- pour des applications papetières dans la fabrication de papier de qualité supérieure,
de papier pour cigarettes et pour billets de banque
- pour des applications techniques comme des non-tissés et le renforcement des
composites. Pour obtenir des fibres d‟une telle courte longueur, des procédés
agressifs sont appliqués, voir 2.1.2.
A présent, des applications textiles, c‟est-à-dire des fils, des tissus, des tricots, etc.,
n‟existent pas encore sur le marché.
1.3.6.1. Les non-tissés
Pour les non-tissés, l‟alfa est souvent mélangée avec d‟autres fibres, soit naturelles comme
la laine, soit synthétiques comme le polypropylène (PP). La distribution des fibres différentes
peut varier selon les propriétés finales voulues.
1.3.6.2. Le renforcement des composites
Dans le secteur des composites, une évolution écologique est actuellement en cours. Des
fibres, autrefois synthétiques mais aujourd‟hui naturelles, sont ajoutées à une matrice pour
améliorer ses caractéristiques mécaniques et pour la renforcer. L‟ensemble forme le
composite, un matériel qui possède normalement les meilleures propriétés des deux
composants. Ce recours aux fibres naturelles se produit de plus en plus de nos jours afin de
réaliser des composites biodégradables davantage et donc avec moins d‟impact nuisible sur
l‟environnement. Un avantage supplémentaire: les fibres naturelles ne coûtent pas chères
comme quelques fibres synthétiques et elles sont abondantes.
Tableau 4 : Résultats des essais mécaniques sur des faisceaux de fibres d‟alfa [7]
Figure 17 : La courbe de traction sur des faisceaux de fibres d‟alfa [7]

13
La matrice des composites est souvent un polymère, et la recherche des polymères
biodégradables est en plein essor.
Une application possible de fibres naturelles dans le domaine du renforcement est l‟alfa sous
forme de fibres courtes. Le problème principal avec l„utilisation des fibres naturelles comme
matériel de renforcement est la cohésion et la liaison avec la matrice. Les fibres naturelles
sont hydrophiles, en revanche les polymères utilisés comme matrices sont souvent
hydrophobes. Par conséquent, les groupes fonctionnels ne se lient pas facilement. Ainsi le
composite perd ses propriétés mécaniques et se casse plus facilement au niveau de la
liaison. Des prétraitements peuvent diminuer l‟hydrophilie des fibres naturelles
(augmentation du caractère hydrophobe) et augmenter l‟hydrophilie des polymères
(diminution du caractère hydrophobe) pour qu‟une meilleure liaison soit possible. Les
prétraitements sont appliqués dans la plupart des cas sur les fibres de renforcement, parce
que les modifications des polymères peuvent influencer négativement la réalisation des
propriétés mécaniques désirées.
Bessadok et Al, 2006 [14] a testé l‟influence d‟actions chimiques sur les fibres d‟alfa, plus
particulièrement l‟impact causé par un traitement d‟anhydride maléique (MA), d‟acide
acrylique (AA), de styrène (S) et d‟anhydride acétique (Ac). Ces quatre possibilités peuvent
améliorer le caractère hydrophobe des fibres d‟alfa, ce qui veut dire que le taux de reprise
est réduit. Les meilleurs résultats sont obtenus avec les traitements S et Ac.
Une étude similaire [15] a été faite pour des fibres d‟agave et dans ce cas-ci, les traitements
avec S et MA donnaient les meilleurs résultats.

14
CHAPITRE 2 : Extraction des fibres d’alfa
2.1. Introduction
2.1.1. Généralités et objectif
L‟extraction de fibres est un procédé classique dans le textile. Souvent les fibres naturelles
se présentent sous une forme qui ne permet pas leur filage, donc il faut transformer par
extraction et purification la matière première ou brute en une forme filable.
Pour le coton, l‟extraction n‟est pas vraiment utile, parce que les fibres sont déjà présentes
en forme plus ou moins filable au moment de la récolte. Il faut quelques traitements pour
éliminer les graines et les autres impuretés, mais une vraie extraction n‟est pas nécessaire.
Par contre le lin est récolté sous forme de tiges qui contiennent des fibres techniques et des
liants. Une extraction est nécessaire dans ce cas-ci afin d‟obtenir des fibres ultimes qui sont
filables. Pendant l‟extraction les liants comme les pectines et la lignine sont éliminées et les
fibres techniques deviennent plus courtes. Ces fibres obtenues sont appelées les fibres
ultimes.
Pour chaque type de fibre, le procédé d‟extraction est différent parce que la matière brute est
composée différemment.
Dans le rapport actuel, il s‟agit de l‟extraction de fibres d‟alfa à partir d‟alfa brute. Il s‟agit plus
spécifiquement de la recherche concernant des fibres assez longues pour qu‟elles soient
filables. Une étude préliminaire sur les fibres courtes a déjà été réalisée [4,7,14]. Cela
constitue un bon point de départ pour la recherche actuelle.
2.1.2. Situation : l’extraction des fibres courtes pour une pâte à papier
Globalement le papier est produit comme un non-tissé par voie humide (angl. wet laid
process). Evidemment quelques différences existent entre les deux procédés, parce que le
papier est presque toujours plus dense que les non-tissés. Cela veut dire que le taux de
matière première dans la pâte, ici la cellulose, est plus élevé pour le papier.
La matière première est mélangée avec de l‟eau et des additifs comme des pigments, de la
colle, des fibres liantes, etc. Il est important pour la qualité du produit fini que la pâte formée
ainsi est la plus homogène possible. A ce but, elle est mixée et mélangée. Ensuite
l‟ensemble est étalé sur un support et l‟eau est évaporée. Puis, pour les non-tissés, le liage
est fait par un aiguilletage, une consolidation chimique ou thermique. La méthode utilisée est
déterminée en fonction des propriétés et caractéristiques finales demandées par le client.
Evidemment une combinaison des méthodes est également possible.
En ce qui concerne le papier, le liage se fait par une consolidation thermique en passant par
des calandres chaudes qui évaporent l‟eau et pressent en même temps le papier pour
diminuer l‟épaisseur [16].
Quant à la matière première, afin que les fibres ne se coagulent pas et ne forment pas de
nœuds, il faut qu‟elles soient assez courtes. Souvent elles sont extraites par des méthodes
agressives. La manière la plus fréquemment utilisée est l‟extraction chimique qui est basée
sur la dégradation des chaînes cellulosiques afin de diminuer le D.P. Le bois a longtemps
été la matière première pour la fabrication de papier. Mais dans le cadre du développement
durable et de la protection de la nature et des forêts, ces derniers temps davantage d‟autres
matières cellulosiques sont utilisées pour la production de papier. P.e. les plantes annuelles
dont le cycle de vie est une année. Une fois mortes, ces plantes peuvent être utilisées

15
comme matière première. D‟autres exemples sont la canne à sucre, l‟alfa, le sisal, le jute, le
lin et le bambou [9].
Un exemple d‟extraction chimique est le procédé Kraft (angl. Kraft pulping ou sulfate
process). Ce procédé est utilisé en général à délignifier de la matière cellulosique comme le
bois et l‟alfa. Il s‟agit d‟une cuisson chimique dans une solution aqueuse d‟hydroxyde de
sodium (NaOH, nom commercial : la soude) et de sulfure de sodium (Na2S) à une
température entre 170°C et 175°C pendant 2h environ [11]. Les anions d‟hydroxyde (OH-) et
sulfuryl (SH-) réagissent avec la lignine, l‟hémicellulose et les pectines en les dégradant par
hydrolyse. Les fragments courts de ces composants se dissolvent dans la solution. Le milieu
de réaction doit être basique car la cellulose est sensible à une hydrolyse acide (chapitre 1).
Quant à l‟alfa pour une pâte à papier, une méthode à base de soude est utilisée comme
extraction chimique. Dans l‟industrie, des concentrations de soude de 3N sont couramment
utilisées. 3N est une concentration élevée qui ne cause pas uniquement la délignification et
la dégradation de l‟hémicellulose, mais en outre la dégradation des chaines cellulosiques.
Pour des pâtes à papier, ce procédé de dégradations est souhaitable parce que des fibres
courtes sont nécessaires. Les fibres d‟alfa utilisées dans le papier ont une longueur
moyenne de 1.5 mm et un diamètre moyen de 12 m. En revanche pour l‟extraction de fibres
filables, la dégradation est indésirable.
En Tunisie et en Algérie une cuisson à base de soude est appliquée [12]. L‟alfa est mise
dans un réacteur avec 1.75N de soude à une température de 140°C à 170°C et une pression
élevée de 0.8 MPa (8 bar) pendant 13 à 15 minutes. Ensuite un blanchiment élimine la
quantité de lignine résiduelle. La pâte obtenue est alors prête pour la production de papier.
2.1.3. La recherche précédente : l’extraction des fibres d’alfa plus longues
Dans la littérature concernant l‟alfa, il s‟agit principalement de fibres courtes, seulement dans
un article une méthode d‟extraction de fibres longues est décrite [7].
La première étape est un traitement dans une solution de soude. La soude est une base
avec une masse moléculaire de 40.01 g/mole et elle se dissout facilement dans de l‟eau par
une réaction exothermique. La concentration utilisée dans le procédé est 3N (ou bien
120.03g dans 1l d‟eau), la durée est 2h sous une pression atmosphérique et à une
température de 100°C.
La deuxième étape est un blanchiment dans une solution d‟hypochlorite de sodium (NaOCl,
nom commercial : eau de Javel) de 40% pendant 1h. Puis l‟échantillon est lavé plusieurs fois
pour éliminer le mieux possible les traces de soude et d‟eau de Javel et ensuite séché
pendant 6h à une température de 60°C. Finalement une étape de cardage sépare et adoucit
les fibres. Les fibres d‟alfa ainsi obtenues sont utilisées pour le renforcement de composites.
De la recherche a déjà été faite au LPMT à Mulhouse, France, pendant un stage de fin
d‟études en 2008 [17]. Le procédé suivi était basé sur le procédé décrit ci-dessus. La suite
des manipulations consistait donc en un trempage, suivi par un traitement de soude, un
blanchiment dans de l‟eau de Javel et enfin un lavage et un séchage.
Pendant le trempage, les tiges d‟alfa sont immergées dans de l‟eau salée, afin d‟éliminer les
cires à l‟extérieur des tiges pour que la soude puisse mieux réagir lors d‟une étape suivante.

16
Le traitement avec la soude a été réalisé d‟abord avec une concentration de 3N comme
mentionnée dans l‟article [7]. Les résultats n‟étaient pas bons parce que des fibres d‟alfa
longues n‟ont pas pu être produites.
Ensuite les mêmes manipulations ont été répétées mais avec des concentrations moins
fortes, 1N et 2N, à une température de 100°C et avec une durée de 2h. De l‟hydrosulfite de
sodium (Na2O4S2, nom commercial : le dithionite de sodium) a été ajouté en tant que
réducteur afin de protéger la cellulose contre la dégradation par la soude.
Les résultats n‟étaient pas satisfaisants car les fibres étaient trop dégradées, d‟où la
nécessité d‟une recherche approfondie afin de trouver les conditions optimales.
2.1.4. Elimination des pectines avec des enzymes
2.1.4.1. Enzymes en général
Des enzymes sont des protéines et font partie des micro-organismes, des plantes, des
animaux et des hommes. Elles sont présentes partout dans la nature et sont des catalyseurs
biologiques. Chaque enzyme catalyse une réaction ou un substrat spécifique. Un catalyseur
accélère une réaction parce qu‟un autre « chemin » de réaction est suivi.
Le nom des enzymes est dérivé de la réaction catalysée, en ajoutant le suffixe –ase. Par
exemple la catalyse d‟une cellulose est réalisée par une cellulase, et celle d‟amylose par une
amylase.
En outre les enzymes sont identifiées par un numéro EC. « EC » est l‟abréviation d‟
« Enzyme Commission ». Ce numéro est attribué selon la réaction catalysée. Donc si deux
enzymes différentes catalysent la même réaction, elles auront le même numéro EC. Le
premier chiffre de l‟ensemble indique la classe de l‟enzyme (cinq classes ont été définies), le
deuxième chiffre indique la sous-classe et les derniers deux chiffres donnent plus
d‟information sur la réaction catalysée.
Les enzymes ont plusieurs avantages par rapport à un catalyseur chimique [18] :
- Elles accélèrent davantage la réaction et par rapport à une réaction non-catalysée, la
vitesse de réaction est 106 à 1023 fois plus élevée
- elles sont très spécifiques et fonctionnent avec un substrat particulier ou une réaction
particulière. Par conséquence la quantité de produits secondaires est minimale.
- les conditions de réaction sont plus modérées : une pression atmosphérique, une
température modérée et une valeur pH proche de 7 (un milieu quasiment neutre)
- après la réaction il suffit de changer un ou plusieurs paramètres pour que les
enzymes soient désactivées
Les enzymes utilisées dans le secteur textile ne sont pas nécessairement celles trouvées
dans la nature. Elles sont souvent extraites de leur micro-organisme et éventuellement
modifiées afin d‟augmenter leur réactivité, ce qui est appelé « enzyme engineering » (angl.).
Une des premières applications était le traitement de denim, les tissus en coton utilisés pour
les jeans. Les consommateurs aiment les jeans qui ont un aspect vieux avec quelques
taches pâles. Autrefois cet aspect était obtenu par lavage des tissus en présence de
zéolithes (des pierres ponces). Le procédé n‟était pas écologique et fréquemment, les
zéolithes endommageaient le tissu et les appareils. Aujourd‟hui le même effet est obtenu
grâce à des cellulases. Comme le composant principal de coton est la cellulose, les
cellulases sont capables de réagir avec le denim.

17
Un autre exemple d‟utilisation des enzymes est l‟addition aux détergents et aux poudres à
laver, afin de diminuer leur impact environnemental.
L‟état actuel des recherches a démontré que les enzymes pouvaient également réagir avec
des matériaux synthétiques comme des substrats, des fibres en polyamide (PA) ou en
polyester (PES). La possibilité de réaction avec le téréphtalate de polyéthylène (PET) est
très intéressante parce que ce polymère est utilisé en quantités énormes [19, 20]. Le but est
d‟améliorer l‟hydrophilie afin d‟optimaliser d‟une part les conditions de production et de
transformation, p.e. une diminution de la charge électrostatique et une amélioration du
potentiel de teinture, et d‟autre part les propriétés du PET, p.e. une augmentation du taux de
reprise.
2.1.4.2. Pectinase d’Aspergillus aculeatus (marque Sigma-Aldrich)
Lors de l‟extraction, chaque composant non-cellulosique doit être éliminé afin d‟obtenir des
fibres ayant un taux de cellulose le plus élevé possible. Parmi ces composants non-
cellulosiques sont les cires, la lignine et les pectines. En revanche l‟hémicellulose reste
importante parce qu‟elle est le liant entre les fibrilles de cellulose. Si l‟hémicellulose est
éliminée, c‟est-à-dire sans liant, des fibres longues ne sont plus possibles.
Le procédé Kraft est une façon d‟élimination des hémicelluloses, des pectines et de la lignine
en les dégradant. L‟inconvénient de ce procédé est que les fibres produites sont très courtes
à cause de l‟hémicellulose dégradée.
Les cires peuvent être éliminées par un trempage (section 2.2.2.2.) et la lignine par la soude
(section 2.2.3.1.). Quant aux pectines, des pectinases sont capables de les dégrader.
Comme il s‟agit d‟enzymes, ce traitement est plus écologique que le traitement Kraft.
Le nom « pectinase » est utilisé pour toutes les enzymes qui peuvent hydrolyser des liaisons
glycosidiques dans les pectines. Ces enzymes appartiennent à la classe des hydrolases
(classe numéro 3) parce qu‟elles hydrolysent. Leur sous-classe est la catégorie des
glycosydases (sous-classe numéro 2) parce qu‟elles hydrolysent des liaisons glycosidiques.
Leur numéro EC est donc toujours de la forme de 3.2.X.Y.
Un inconvénient de l‟utilisation des pectinases est que ces micro-organismes sont de même
capables d‟hydrolyser la liaison glycosidique de cellulose, ce qui veut dire que celle-ci sera à
son tour dégradée, un effet absolument indésirable. Comme mentionné dans le premier
chapitre, l‟hydrolyse de cellulose se passe surtout dans un milieu acide. Il est donc facile à
limiter et même éviter cette dégradation acide en tenant la valeur du pH égale à 7 (milieu
neutre) ou supérieure à 7 (milieu basique).
Les pectinases utilisées pendant l‟extraction de fibres d‟alfa viennent d‟Aspergillus aculeatus.
Leur température optimale est 38°C et leur pH optimal est 3.5 à 38°C, comme indiqué sur la
fiche livrée avec les enzymes.
2.2. Mode opératoire
2.2.1. Introduction
Les tiges d‟alfa utilisées ont un diamètre plus ou moins constant tout au long de la tige.
L‟homogénéité de la matière première est un paramètre important et aide à obtenir des
résultats homogènes, ce qui est toujours un objectif fondamental.
Le procédé complet d‟extraction de fibres d‟alfa est composé de trois parties principales : les
prétraitements, l‟extraction et les post-traitements. Les prétraitements sont le brossage
mécanique et le trempage, les post-traitements sont le séchage et la séparation des fibres.

18
La partie d‟extraction est composée de plusieurs étapes, basées principalement sur un
traitement de soude. Seulement celui-ci est utilisé à un premier instant, mais ne donne pas
de résultats satisfaisants. Une deuxième étape est donc ajoutée.
Il s‟agit au début d‟un blanchiment, appliqué sur les meilleurs échantillons du traitement de
soude. Les résultats ne sont toujours pas bons parce que la soude élimine seulement la
lignine et le blanchiment n‟élimine rien.
Pour pouvoir éliminer les pectines, le blanchiment est remplacé par un traitement
enzymatique. Des pectinases sont bien aptes à l‟élimination des pectines. L‟étape des
pectinases réussit en effet à améliorer les meilleurs résultats du traitement de soude.
Pourtant les fibres sont encore un peu rigides, ce qui pourrait être due à la présence de
lignine résiduelle.
Comme la soude est capable d‟éliminer la lignine, un traitement supplémentaire a donc été
appliqué sur les meilleurs échantillons déjà obtenus. Ce nouveau cycle d‟extraction « soude
– enzymes – soude » donne finalement les meilleures fibres.
2.2.2. Les prétraitements
2.2.2.1. Le brossage mécanique
Pendant les premiers essais d‟extraction, les tiges ne sont pas brossées mécaniquement
mais sont utilisées intégralement. Les résultats n‟étaient pas satisfaisants parce que la
matière était dure et rigide. En tout cas il n‟en était pas question de fibres.
Quand la matière première est plus fine dès le début, une amélioration des résultats est
envisageable. Les produits chimiques des étapes suivantes seront plus réactifs car une plus
grande surface est disponible pour la même quantité de matière.
Un brossage mécanique résout le problème de finesse parce qu‟avec des brosses
métalliques il est possible de fendre les tiges. Les brosses sont déplacées dans le sens
longitudinal des tiges et sont plutôt des peignes que des brosses. Comme les tiges sont
forcées à passer entre les dents du peigne, le diamètre des tiges est réduit et les
échantillons seront plus homogènes.
Les échantillons extraits à base de tiges brossées mécaniquement, sont plus fins et moins
rigides que ceux à base de tiges non-brossées, d‟où la décision d‟ajouter le brossage
comme prétraitement.
2.2.2.2. Le trempage
Un trempage est strictement dit une immersion dans un liquide. La matière trempée ici est
l‟alfa et le liquide est de l‟eau salée. La fonction du trempage dans le cycle d‟extraction de
fibres est d‟éliminer les cires, le sable et les poussières qui se trouvent à la surface des tiges.
Grâce à cette élimination, les tiges seront plus « ouvertes » aux traitements suivants.
L‟origine de l‟utilisation d‟eau salée remonte aux traitements anciens des producteurs d‟alfa.
Ils mettaient les tiges dans de l‟eau de mer pour les tremper. Comme cela fonctionnait bien,
le même procédé est appliqué aujourd‟hui dans le laboratoire mais avec une température
supérieure pour que le trempage se passe plus vite.
La concentration de sel dans l‟eau de mer n‟est pas identique partout dans le monde.
Comme l‟alfa pousse dans les pays autour de la Méditerranée, une imitation de l‟eau de la
Méditerranée est utilisée lors des essais.
Les concentrations trouvées varient entre 27g/l et 38g/l, donc une valeur de 35g/l est choisie.
Plus spécifiquement de l‟eau distillée et du chlorure de sodium (NaCl, nom commercial : le
sel de cuisine) sont utilisés.

19
Figure 18 : à droite le début du trempage, à gauche le trempage après quelques heures
En général la durée doit être suffisamment longue pour que les cires aient assez de temps
pour dissoudre dans l‟eau salée.
Dans la littérature les conditions suivantes sont mentionnées : 12h à 80°C ou 24h à 60°C.
Afin de connaître la différence entre les deux options, des tiges brossées sont séchées dans
un four pendant 24h à 60°C, ensuite trempées selon les différentes conditions données, puis
lavées avec de l‟eau distillée et enfin de nouveau séchées au four pendant 24h à 60°C. Les
tiges avant et après le trempage sont pesées et le taux de matière dissoute peut facilement
être calculé. Les résultats sont présentés dans Tableau 5.
Les séchages avant et après le trempage ont pour but d‟évaporer l‟eau dans les tiges de
sorte que dans les deux cas la masse exacte des tiges est déterminée (sans la présence de
l‟eau). De cette façon la différence de masse (Δ masse dans le Tableau 5) est exactement la
quantité de matière éliminée pendant le trempage. Le taux de matière dissoute (taux
masse dans le tableau) est le quotient de la différence de masse et la masse avant le
trempage.
Les valeurs obtenues sont inférieures aux valeurs théoriquement obtenues. Le taux moyen
de cires dans des tiges d‟alfa est 5% (Tableau 2). Donc soit la durée du trempage était trop
courte, soit les tiges utilisées contiennent moins de cires ou soit les cires ne sont pas toutes
éliminées et leur élimination demande plus qu‟un trempage. Cette dernière explication
semble le plus logique parce que le tableau indique que la différence entre les conditions de
trempage n‟est pas grande. Il est donc très probable que la situation s‟est stabilisée et que la
durée n‟était pas trop courte. Pour cette raison ainsi que pour des raisons pratiques, il est
décidé d‟utiliser un trempage de 24h à 60°C
Pendant le trempage l‟eau salée change de couleur (Figure 18). Au début la solution est
transparente, mais peu à peu la couleur devient jaune-verte et plus foncée.
Une remarque : Lors des essais, un seul bécher est placé sur la plaque chauffante mais pour
les photos pourtant deux béchers y sont placés afin de pouvoir montrer plus facilement le
changement de couleur.
trempage masse avant masse après masse taux masse
12h à 80°C 0,93 g 0,92 g 0,01 g 1,08 %
24h à 60°C 0,89 g 0,88 g 0,01 g 1,12 %
Tableau 5 : Influence des conditions de trempage

20
2.2.3. L’extraction
2.2.3.1. Le traitement de soude
La fonction de la soude est la délignification des tiges d‟alfa. Les tiges sont dures, longues et
épaisses. La lignine est, comme décrit au chapitre 1, le composant qui connecte ou colle les
fibrilles de cellulose pour ainsi former les tiges.
La soude dans le laboratoire se présente sous forme de pastilles et doit être dissoute pour
obtenir une solution aqueuse avec la concentration désirée.
Selon la température, la solution de soude est versée dans un bécher ou un biberon, c‟est-à-
dire un récipient de la machine à teinture (voir appendice A).
Les béchers sont utilisés pour des températures jusqu‟à 100°C sous pression
atmosphérique, en les plaçant dans un bain-marie remplie d‟eau et posée sur une plaque
chauffante. Dans ces conditions, l‟utilisation de béchers dans l‟eau est limitée au niveau de
la température.
Le bain d‟eau est utilisé pour obtenir une température homogène du bécher et de la solution
de soude. De cette façon, il est probable que la soude agit plus régulièrement ce qui donnera
un résultat plus homogène.
Pour obtenir des températures au-dessus de 100°C, des biberons sont utilisés. Ils sont
fermés et placés dans la machine à teinture, plus précisément dans un réservoir d‟huile.
L‟huile joue ici le rôle de l‟eau dans la première méthode. La température maximale de cette
méthode est 140°C, cela est en même temps la température maximale de la machine. En
outre il est possible de faire tourner les biberons de manière qu‟une force de frottement
apparaît. Celle-ci peut faciliter la délignification, mais peut également casser les tiges ou les
fibres lorsqu‟elles sont plus fines et plus fragiles.
Si des températures encore plus élevées sont désirées, la seule solution dans le laboratoire
est de placer les biberons fermés eux-mêmes, directement, sur les plaques chauffantes (voir
appendice A) qui chauffent à fond. Malheureusement avec cette méthode, la température et
la pression exactes sont inconnues parce que l‟indication sur les plaques est imprécise et
comme les biberons sont fermés, il est impossible de mesurer la température interne avec
un thermomètre. Un système pour mesurer la pression interne n‟est pas disponible.
Un autre inconvénient est le gradient de température des biberons, bien qu‟ils soient en
métal. Un avantage est la pression interne plus élevée que la pression atmosphérique. Cela
a une grande influence sur l‟activité de la soude parce que l‟enthalpie libre de la réaction
diminue avec la température et la pression (ΔG = ΔH - TΔS), pourvu que l‟enthalpie (ΔH) et
l‟entropie (ΔS) ne varient pas. Comme il s‟agit des paramètres thermodynamique, la
conclusion est que l‟activité de la soude augmente et que la réaction devient plus probable.
D‟où malheureusement le grand risque de dégradation des fibres. Donc cet avantage est en
même temps un inconvénient. Il est sûr que la pression interne est grande à ces
températures élevées, parce qu‟en ouvrant les biberons sans les refroidir, une petite
explosion se produit. Il faut donc refroidir les biberons avant de les ouvrir.
Pour avoir une vue globale des possibilités, il est utile de faire un schéma avec toutes les
combinaisons de paramètres. Les paramètres qui changent sont la concentration, la
température, la durée et la présence d‟un réducteur.
Un réducteur, également appelé un antioxydant, peut être ajouté afin de protéger les
filaments cellulosiques contre une dégradation (une oxydation) par la soude. Quelques
essais sont faits avec la même température (plus de 150°C), durée et concentration de

21
soude, mais alternant sans et avec l‟hydrosulfite de sodium (Na2O4S2, nom commercial : le
dithionite de sodium) comme réducteur. Le Na2O4S2 utilisé est un sel sous forme d‟une
poudre blanche et cristalline et bien soluble dans de l‟eau et des solutions aqueuses de
soude.
Les fibres produites en présence du réducteur sont moins dégradées. Il est donc clair que
l‟influence du dithionite de sodium n‟est pas négligeable et qu‟il vaut mieux l‟ajouter.
Quant aux autres paramètres :
- les températures utilisées sont 50°C, 100°C, 135°C - 140°C ou plus de 150°C (la
valeur exacte est inconnue)
- la concentration est 0.25N, 0.5N, 1N ou 2N
- la durée est 1h ou 2h
Cela donne trente-deux combinaisons possibles. Il ne faut pas les essayer toutes quand les
conditions pour un essai suivant sont basées sur le résultat de l‟essai précédent.
Tous les essais incluent un traitement de soude. Les échantillons rigides et jaunes ne sont
pas vraiment satisfaisants, d‟où le choix de blanchir les « fibres » avec une solution aqueuse
de 9.6% d‟eau de Javel pendant 1h. De cette façon la couleur devient plus blanche, mais la
rigidité reste identique parce que le blanchiment n‟élimine ni les pectines, ni la lignine.
La première manipulation est réalisée avec 2N de soude à 50°C pendant 1h. La température
modérée est choisie parce que la concentration de soude est élevée. Le résultat est très
rigide, donc la température est augmentée à 100°C pour la même concentration et durée.
Cela donne également un résultat très rigide, et la même manipulation est répétée pendant
2h au lieu de 1h. Comme cela donne toujours des résultats rigides, la température est
augmentée pour savoir son influence.
Les béchers sont remplacés par des biberons fermés sur des plaques chauffantes, d‟où une
température interne exacte inconnue mais supérieure à 150°C. La concentration de soude
choisie est 1N au lieu de 2N et la durée 1h. En même temps une manipulation à 100°C avec
1N de soude pendant 1h est exécutée pour connaître la différence entre ces deux résultats.
A 100°C un échantillon rigide est obtenu, comme prévu, mais à plus de 150°C une pâte de
fibres courtes et dégradées est formée. L‟augmentation de la concentration de réducteur
n‟améliore pas les résultats.
Les changements de température et de pression ont donc une grande influence. Au premier
instant il est intéressant de continuer à travailler à cette haute température et la même durée
en diminuant la concentration de soude jusqu‟à 0.5N. Cela donne également une pâte et la
concentration est encore diminuée à 0.25N. Pour la première fois avec cette haute
température, un mélange de pâte et de quelques fibres est obtenu. Le même traitement est
répété quelques fois avec de plus grandes concentrations de réducteur, mais des fibres ne
sont plus produites.
Il est donc possible que la durée d‟une heure est le seuil entre fibres et pâtes. Pour cette
raison des manipulations avec 0.25N et à plus de 150°C sont refaites pendant 55 minutes,
50 minutes, 45 minutes et 40 minutes. Les résultats ne sont pas réguliers et seulement après
45 minutes quelques fibres sont trouvées. Elles sont moins souples que celles produites
après 1h de soude.
Pour avoir une idée de la pureté et de la dégradation, les fibres de ces deux échantillons
sont examinées avec le microscope à balayage (MEB, angl. SEM, scanning electron
microscope).

22
Les fibres rigides contiennent encore des composants non-cellulosiques, ce qui est
clairement visible sur les photos (Figure 19). Les filaments cellulosiques ne sont pas libérés
complètement mais couvertes d‟une couche de matière non-cellulosique (Figure 20). Il est
constaté que cette couche n‟est pas lisse et plus ou moins épaisse à certains endroits
(Figure 21). Cela signifie que la soude ne réagit pas de façon homogène tout au long des
tiges. Une explication possible est que les tiges sont pliées dans les biberons, d‟où une
accessibilité réduite dans les plis pour la soude. Il faut donc étaler les tiges le plus
parallèlement possible dans les biberons.
Les fibres souples ne contiennent pas beaucoup de matière non-cellulosique et les filaments
deviennent visibles (Figure 22). En revanche, ces filaments et donc ces fibres sont en même
temps dégradées (Figure 23). Elles se présentent sous forme parallèle (Figure 24) mais
chaque filament a quelques « nœuds » qui indiquent une dégradation. Beaucoup de
« nœuds » sont visibles, donc il s‟agit d‟une dégradation importante.
Figure 19 : image MEB des fibres rigides
Figure 21 : image MEB des composants non-cellulosiques des fibres rigides
Figure 22 : image MEB des fibres souples
Figure 24 : image MEB des filaments cellulosiques non-dégradés
Figure 23 : image MEB de la dégradation des filaments cellulosiques
Figure 20 : image MEB des filaments cellulosiques des fibres rigides

23
A cause de la grande différence de résultat entre 100°C et plus de 150°C, quelques
manipulations avec une concentration de 0.25N et en plaçant les biberons dans la machine à
teinture, sont faites à des températures intermédiaires de 130°C et 140°C. D‟abord la durée
est 1h parce que cela permet de comparer les résultats obtenus à ces quatre températures.
A 130°C et 140°C, les échantillons sont rigides, mais moins rigides que ceux à 100°C (Figure
25). Lors des essais suivants avec la durée double de 2h, les résultats ne s‟améliorent pas.
Tableau 6 donne les résultats principaux des manipulations de soude, éventuellement
suivies par un blanchiment.
Tableau 6 : Les résultats principaux des traitements de soude des fibres d‟alfa
Comme indiqué, les échantillons sont rigides quand la température est 50°C ou 100°C,
même avec des concentrations hautes de soude et une durée longue. Par contre, pour des
manipulations à une température élevée, dans les biberons sous pression, les fibres sont
facilement dégradées et courtes, même avec des concentrations faibles et une durée courte.
Il est donc clair que la soude réagit mieux à une température élevée et sous pression.
Figure 25 : Influence de la température : de gauche à droite : 95°C, 130°C, 140°C et plus de 150°C.
Soude Réducteur Blanchiment Résultat
conc. temp. durée quantité
2N 50°C 1h X très rigide
2N 100°C 1h X rigide
2N 100°C 2h X rigide
1N 100°C 1h X rigide
1N > 150°C 1h fibres courtes, pâte
0,50N > 150°C 1h pâte
0,25N 100°C 1h rigide
0,25N 100°C 1h 1,0 % Ms rigide
0,25N 130°C 1h 1,0 % Ms rigide
0,25N 140°C 2h 0,5 % Ms rigide
0,25N 140°C 1h 1,5 % Ms rigide
0,25N > 150°C 1h 1x fibres, 1x pâte
0,25N > 150°C 1h 1,0 % Ms pâte
0,25N > 150°C 1h 1,5 % Ms pâte
0,25N > 150°C 55 min pâte
0,25N > 150°C 50 min 1,0 % Ms 1x moins de pâte, 1x pâte
0,25N > 150°C 45 min pâte avec quelques bonnes fibres
0,25N > 150°C 40 min rigide

24
Les fibres courtes formant une pâte sont fines. Dans ces conditions, il est probable que la
lignine et en même temps les hémicelluloses soient éliminées. De cette façon les fibrilles de
cellulose ne sont plus liées par les hémicelluloses et des fibres courtes sont obtenues. Il faut
donc trouver une méthode qui ne dégrade pas les hémicelluloses.
En revanche les échantillons rigides contiennent encore de la lignine et des pectines. Il faut
donc trouver un moyen pour améliorer la délignification et surtout pour éliminer les pectines.
Cela peut être réalisé avec des pectinases, un type d‟enzymes. Le blanchiment ne sera donc
plus fait car il ne contribue pas à l‟extraction, et un traitement enzymatique sera appliqué afin
d‟éliminer les pectines dans les tiges d‟alfa.
A cause des résultats dégradés et la mesure difficile de la température et de la pression, il
vaut mieux abandonner la méthode des biberons sur la plaque chauffante. L‟étape
supplémentaire des enzymes sera appliquée aux meilleurs échantillons rigides, c‟est-à-dire
ceux obtenus à 130 et 140°C.
2.2.3.2. Le remplacement du blanchiment par un traitement enzymatique
Comme conclu dans la section précédente un traitement unique de soude, éventuellement
suivi par un blanchiment, ne suffit pas. Soit les résultats sont rigides à cause de la lignine et
des pectines résiduelles, soit les résultats sont dégradés à cause de la température et de la
pression élevées.
Des pectinases sont aptes à éliminer les pectines, d‟où le choix de continuer avec les
meilleurs résultats rigides, c‟est-à-dire ceux obtenues avec 0.25N de soude à des
températures de 140°C pendant 1h et 2h. L‟objectif est d‟obtenir des fibres plus fines et plus
souples grâce à l‟élimination des pectines. Néanmoins les enzymes sont au début également
appliquées quelques fois aux échantillons traités à plus de 150°C ou à 100°C (avec 0.25N de
soude) pour vérifier que les pectinases fonctionnent.
La fiche des pectinases indique une activité maximale à 38°C avec une valeur du pH de 3.5.
Bien que cette valeur du pH puisse causer une hydrolyse acide des filaments cellulosiques
et ainsi une dégradation des fibres, elle est utilisée au début parce que l‟objectif est surtout
de constater si les pectinases fonctionnent bien.
La concentration utilisée est 5 ml/l et la durée monte jusqu‟à 5h pour examiner la capacité
des enzymes.
Les premiers traitements enzymatiques sont appliqués aux pâtes obtenues à plus de 150°C.
Comme la matière d‟alfa utilisée est déjà de la pâte, il n‟est pas surprenant que les
échantillons restent de la pâte. Puis le même traitement enzymatique est appliqué aux
échantillons rigides, traités à 100°C. Bien que leur résultat devienne un petit peu moins
rigide, il n‟en est pas question des fibres à la fin.
Pour les traitements appliqués aux meilleurs échantillons rigides, quelques paramètres sont
changés afin d‟éviter la dégradation des filaments cellulosiques :
- la valeur du pH choisie est 7
- la durée est d‟abord réduite à 4h et 5h mais il se produit toujours une dégradation.
Par conséquent elle est ensuite baissée jusqu‟à 2h et 2h30.
La même manipulation enzymatique est exécutée sur quatre échantillons différents, obtenus
avec 0.25N de soude à 140°C pendant 1h, 70 minutes, 90 minutes et 2h. Chaque fois une
amélioration du résultat est observée et les échantillons sont moins rigides.

25
Il est donc clair que les résultats sont meilleurs avec un traitement de pectinases que sans
aucun traitement (Figure 26), mais il est important de contrôler les paramètres.
L‟explication pour l‟amélioration peut être la suivante. Avec la soude la plupart de la lignine
est éliminée et avec les pectinases les pectines. En combinant les deux procédés, deux
composants non-cellulosiques sont éliminés pour une grande partie.
Néanmoins pour des températures maximales de 140°C, les résultats restent encore un peu
rigides et des fibres sous la forme désirée, c‟est-à-dire souples, lisses et longues, ne sont
pas obtenues. En supposant que les enzymes éliminent bien les pectines, il est probable que
la lignine ne soit pas complètement éliminée, ce qui rend les fibres rigides.
Le problème est que la structure des tiges n‟est pas connue comme c‟est le cas pour les
fibres de coton. Par conséquent, l‟ordre des couches de lignine et de pectines reste inconnu.
A l‟UHA la connaissance pour déterminer la structure interne des tiges n‟est pas disponible
parce qu‟il faut des experts dans ce secteur de biologie. La littérature ne parle pas de ce
sujet parce qu‟il y s‟agit surtout de fibres courtes obtenues par des méthodes agressives,
avec de fortes concentrations de soude (jusqu‟à 3N), qui détruisent la structure des tiges.
Il n‟est donc pas certain que la lignine se trouve uniquement à l‟extérieur. Si elle se trouve
également à l‟intérieur des tiges, elle n‟était pas accessible pendant le traitement de soude
car les pectines la couvraient. Maintenant, avec l‟addition d‟un traitement enzymatique qui
élimine les pectines, cette lignine intérieure est dévoilée.
Par conséquent un deuxième tour de soude peut l‟éliminer.
Tableau 7 donne les résultats principaux du cycle « soude – enzymes ».
Tableau 7 : Résultats principaux du cycle « soude – enzymes »
Une amélioration des meilleurs échantillons rigides, traités de soude à 140°C, est visible
grâce à un traitement enzymatique. Néanmoins les fibres sont encore trop rigides pour être
considérées comme satisfaisantes. A partir des meilleurs échantillons après ce double
traitement, une étape de soude supplémentaire peut continuer l‟amélioration de l‟extraction.
Figure 26 : Influence des enzymes : à gauche sans enzymes, à droite avec enzymes (5h, 38°C, pH 7). Le traitement de soude est identique pour les deux échantillons.
Soude Réducteur Enzymes Résultat
conc. temp. durée quantité conc. temp. durée pH
0,25N 100°C 1h 5,0 ml/l 38°C > 2h 3,5 rigide
0,25N 140°C 2h 0,5 % Ms 5,0 ml/l 38°C 2h 7 rigide
0,25N 140°C 90 min 1,0 % Ms 5,0 ml/l 38°C 5h 7 plutôt rigide
0,25N 140°C 75 min 1,0 % Ms 5,0 ml/l 38°C 4h 7 plutôt rigide
0,25N 140°C 1h 1,5 % Ms 5,0 ml/l 38°C 5h 7 plutôt rigide
0,25N > 150°C 1h 5,0 ml/l 38°C > 2h 3,5 pâte

26
2.2.3.3. L’addition d’un traitement de soude supplémentaire après les enzymes
Comme conclu dans la section précédente, il faut un traitement supplémentaire afin d‟obtenir
des résultats plus fins et moins rigides.
Il est évident que la recherche préalable n‟est pas refaite, mais que les meilleurs échantillons
obtenus sont sélectionnés et traités de nouveau. Ces échantillons sont produits sous les
conditions suivantes
- soude : 0.25N à 140°C pendant 1h à 2h avec 0.5% à 1.5% de réducteur
- enzymes : 5 ml/l à 38°C pendant 2h avec une valeur du pH de 7
Le premier échantillon choisi est produit avec 1h de soude et 2h d‟enzymes et subit un
nouveau traitement de soude. Pour faciliter cette manipulation, la température de 140°C et la
concentration de 0.25N du premier traitement sont de nouveau utilisées. Les seules
variables sont la durée et la concentration de réducteur.
A ce moment il faut distinguer le taux de réducteur dans les deux traitements. Comme la
matière première du premier est de l‟alfa sèche, la masse du réducteur est un pourcentage
de la masse de la matière sèche. Par contre la matière première du deuxième traitement de
soude est de l‟alfa humide, qui vient d‟être rincée après la manipulation des pectinases.
Dans ce cas-ci, la masse du réducteur est basée sur la masse de l‟alfa humide.
Donc pour le premier essai d‟un traitement supplémentaire, la durée choisie est 1h et le taux
de réducteur 0.5%. Le biberon est fait tourner en rond, résultant en une pâte cellulosique.
Comme les fibres ont l‟air fragile après le traitement enzymatique, il est probable que le
frottement causé par la rotation ait cassé les fibres. Pour éviter cela, les biberons ne sont
plus faits tourner en rond après ce premier essai.
Alors la même manipulation est répétée sans rotation des biberons, mais résulte de nouveau
en une pâte. La dégradation est donc également due aux conditions.
Alors premièrement la concentration des enzymes est diminuée à 2.5 ml/l pour réduire
l‟influence des pectinases très réactives. Cela donne un résultat plutôt rigide après quoi la
concentration de réducteur est diminuée jusqu‟à 0.25%. Même cela donne des résultats
rigides pour une durée de soude (deuxième fois) de 1h, 45 minutes et 30 minutes.
Apparemment une concentration de pectinases de 5 ml/l est trop élevée, tandis qu‟une
réduction à 2.5 ml/l donne des résultats rigides pour l‟instant. Néanmoins avec cette
concentration plus faible, le risque de dégrader les filaments cellulosiques diminue, d‟où le
choix de continuer les essais avec 2.5 ml/l.
Il faut donc changer un autre paramètre en plus afin de diminuer la rigidité. A ce but la durée
du premier cycle de soude est augmentée à 90 minutes (au lieu d‟une heure) pendant que
celle du deuxième cycle reste 1h. L‟échantillon obtenu est très hétérogène. Il contient des
fibres rigides, des fibres moins rigides et des fibres courtes, celles-ci formant une pâte.
L‟hétérogénéité est due
- d‟une part à l‟hétérogénéité des tiges d‟alfa, ce qui ne peut pas vraiment être
améliorée parce que cela fait partie de la nature des plantes et des caractéristiques
des fibres naturelles
- d‟autre part aux conditions de travail qui ne sont jamais constantes, une variation qui
pourrait influencer les résultats.
L‟augmentation de la durée du deuxième traitement de soude à 90 minutes donne un
résultat comparable.

27
Afin de produire un résultat plus homogène, l‟influence des enzymes est considérée. La
durée est diminuée de 2h à 1h, toujours avec une concentration de 2.5 ml/l et une valeur du
pH de 7. En même temps, pour éviter la production de fibres courtes, les quantités de
réducteur dans les deux traitements de soude sont augmentées à 1.5% pour le premier et
0.5% pour le deuxième.
Sous ces conditions le résultat devient effectivement plus homogène et la quantité de fibres
courtes est diminuée. Les fibres très rigides sont moins omniprésentes et quelques fibres de
5 à 6 cm sont visibles.
Evidemment il faut répéter ces manipulations pour exclure des résultats exceptionnellement
bons ou mauvais. De plus, il est utile de changer légèrement quelques paramètres de soude
pour voir la différence, du point de vue d‟améliorer ce résultat acceptable. Le traitement des
enzymes est délicat et n‟est pas changé. Il a déjà été remarqué précédemment qu‟un
changement de ses paramètres influence fortement le résultat et à ce moment seulement un
petit changement est souhaitable.
Une diminution de la durée du deuxième traitement de soude à 30 minutes donne un résultat
semblable mais le taux de fibres et faisceaux de fibres épaisses et rigides est plus élevé.
Une diminution de la température à 120°C et en même temps une augmentation de la
concentration de soude (0.5N) permet de constater l‟influence de la température et de la
concentration. Avec tous les autres paramètres inchangés, les résultats sont beaucoup plus
rigides pour 0.5N de soude à 120°C que pour 0.25N de soude à 140°C (Figure 27).
Une augmentation de la température à 130°C donne également un échantillon rigide. En
augmentant à ce point-ci la durée du traitement enzymatique à 2h, le résultat devient
comparable à celui obtenu avec 0.25N à 140°C.
Pour des raisons écologiques et économiques, il est préférable de travailler avec 0.25N de
soude à 140°C et 1h d‟enzymes, d‟où la décision que ce traitement donne les meilleures
fibres pour l‟instant (Figure 28). Les conditions sont donc :
- soude : 0.25N à 140°C pendant 90 minutes avec 1.5% de réducteur
- enzymes : 2.5 ml/l à 38°C pendant 1h avec une valeur du pH de 7
- soude : 0.25N à 140°C pendant 1h avec 0.5% de réducteur
Figure 27 : Influence de la température et la concentration : à gauche 120°C et 0.5N, à droite 140°C et 0.25N. Les autres paramètres sont identiques.

28
Tableau 8 et Tableau 9 donnent les paramètres et les résultats principaux des essais de type
« soude – enzymes – soude ».
Figure 28 : Le meilleur échantillon obtenu, toujours hétérogène
Soude Réducteur Enzymes
conc. temp. durée quantité conc. temp. durée pH
0,50N 120°C 90 min 1,5 % Ms 2,5 ml/l 38°C 1h 7
0,50N 120°C 90 min 1,5 % Ms 2,5 ml/l 38°C 1h 7
0,50N 130°C 90 min 1,5 % Ms 2,5 ml/l 38°C 1h 7
0,50N 130°C 90 min 1,5 % Ms 2,5 ml/l 38°C 2h 7
0,25N 140°C 90 min 1,5 % Ms 2,5 ml/l 38°C 1h 7
0,25N 140°C 90 min 1,5 % Ms 2,5 ml/l 38°C 1h 7
0,25N 140°C 90 min 1,0 % Ms 2,5 ml/l 38°C 2h 7
0,25N 140°C 1h 1,0 % Ms 2,5 ml/l 38°C 2h 7
0,25N 140°C 1h 0,5 % Ms 5,0 ml/l 38°C 2h 7
0,25N 140°C 1h 1,0 % Ms 2,5 ml/l 38°C 2h 7
0,25N 140°C 1h 1,0 % Ms 2,5 ml/l 38°C 2h 7
Tableau 8 : Valeurs des paramètres pour les deux premières étapes du cycle « soude – enzymes – soude »
Soude sans tourner Réducteur Résultat
conc. temp. durée quantité
0,50N 120°C 1h 0,5 % Mh rigide
0,50N 120°C 30 min 0,5 % Mh rigide
0,50N 130°C 1h 0,5 % Mh rigide
0,50N 130°C 1h 0,5 % Mh mixe: rigide + qqs fibres + pâte
0,25N 140°C 1h 0,50 % Mh le meilleur résultat
0,25N 140°C 30 min 0,50 % Mh un peu trop rigide
0,25N 140°C 90 min 0,25 % Mh mixe: rigide + qqs fibres + bp de pâte
0,25N 140°C 1h 0,25 % Mh rigide parce que sans tourner
0,25N 140°C 1h 0,5 % Mh pâte
0,25N 140°C 45 min 0,25 % Mh rigide avec plusieurs fibres
0,25N 140°C 30 min 0,25 % Mh rigide avec quelques fibres
Tableau 9 : Valeurs des paramètres et résultats pour la troisième étape du cycle « soude – enzymes – soude »

29
Le principal inconvénient de ce cycle est le temps nécessaire pour les trois étapes et ainsi le
temps nécessaire en total. L‟extraction elle-même a une durée de 3h30. En outre les fibres
sont lavées, pesées, etc à chaque étape. Donc la durée totale de l‟extraction est 4h au
minimum.
Dans le calcul de la durée totale, la durée des prétraitements et des post-traitements (voir les
sections suivantes) doivent également être prises en compte. La somme des durées donne
un procédé d‟extraction qui nécessite plus d‟un jour, ce qui est énorme dans l‟industrie. Cette
méthode donne donc les meilleurs résultats, mais une optimisation au niveau de la durée est
indispensable.
A la fin de toutes ces manipulations exécutées, il est clair qu‟un traitement enzymatique au
lieu d‟un blanchiment permet d‟obtenir un meilleur résultat. Pour améliorer sa rigidité, il faut
que la plupart de la lignine résiduelle soit éliminée. A ce but un deuxième traitement de
soude est appliqué après les pectinases. En changeant les paramètres d‟un nouvel essai en
fonction du résultat de l‟essai précédent, finalement des fibres sont produites. Il ne s‟agit pas
du meilleur résultat possible, mais c‟est en tout cas un bon point de départ pour
l‟optimisation. Figure 29 présente l‟évolution réalisée avec
- à gauche du carreau gauche le résultat après un traitement de soude
- à droite du carreau gauche un échantillon après l‟addition d‟un traitement
enzymatique
- au carreau droite le résultat final après une manipulation de soude supplémentaire
2.2.4. Les post-traitements
2.2.4.1. Le séchage
En général les fibres sont séchées dans un four à une température de 50°C à 60°C pendant
12h ou plus, jusqu‟à elles sont complètement sèches. Cette étape est une des étapes les
plus difficiles à exécuter, la difficulté étant la manière dans laquelle les fibres sont mises au
four pour sécher. Après l‟extraction, les échantillons sont mis dans un bécher pour lavage
avec de l‟eau distillée. Les fibres ne sont donc pas rangées parallèlement, mais forment une
pelote. Cette pelote doit être mise au four de manière que les fibres ne collent pas à elles-
mêmes ni pendant ni après le séchage. Sinon il devient difficile à les séparer (voir section
2.2.4.2.).
Figure 29 : Evolution réalisée au niveau d‟extraction des fibres d‟alfa

30
Au début, quand les fibres étaient rigides, il était facile de les étaler sur une pièce de papier
aluminium pour sécher. La pâte cellulosique obtenue plus tard était filtrée et séchée sur un
filtre à papier ou sur du papier aluminium. Depuis que le résultat est devenu un mélange de
fibres rigides, de fibres courtes et de fibres entre ces deux extrémités, il n‟est plus possible
de sécher un tel échantillon sur du papier aluminium parce qu‟il est nécessaire de séparer
les types de fibres obtenues pour le cardage et le filage mécanique. En essayant cela sur du
papier aluminium, les fibres humides se cassent facilement.
Alors un moule en silicone pour des petits gâteaux permet de répartir les fibres sur neuf
compartiments. De cette façon, les fibres rigides et courtes sont mises à part des fibres
moyennes. Evidemment il est impossible d‟avoir une séparation absolue et après le séchage
parce que les fibres courtes forment un liant entre les autres fibres. De plus, l‟alignement des
fibres est difficile, donc d‟autres fibres jouent également le rôle de liant. Cela signifie que
même avec cette méthode de séchage, il est impossible d‟obtenir des fibres individuelles
après le séchage. Certes, le moule facilite le séchage, mais n‟est pas la solution idéale. Un
jet d‟air chaud à faible vitesse circulant dans le four serait mieux, parce que ce jet pourrait
sécher les fibres en évitant la formation de nœuds. Un tel jet d‟air n‟est cependant pas
disponible dans le laboratoire, donc le séchage avec le moule est la meilleure alternative.
Une séparation des fibres sèches s‟avère nécessaire, sinon les échantillons sont plutôt des
non-tissés que des fibres individuelles. Comme chaque étape supplémentaire prend du
temps et coûte de l‟argent, il faudra optimiser dans le futur le procédé de séchage pour
qu‟une telle séparation soit réduite au minimum ou ne soit plus nécessaire du tout.
2.2.4.2. La séparation des fibres après séchage
Comme mentionné dans le paragraphe 2.2.4.1., la séparation des fibres est nécessaire pour
compenser le fait que le séchage ne se passe pas dans des conditions optimales et résulte
en un mélange de fibres de différentes qualités. Cette séparation consiste en un cardage des
fibres afin de les aligner. Comme les bonnes fibres sont fines, il faut trouver un moyen de
cardage (ou brossage) prudent pour que les fibres ne se cassent pas.
La séparation manuelle est la manière la plus prudente et la plus précise, mais prend
beaucoup de temps. Sans aucun doute cette méthode n‟est pas rentable dans le secteur
industriel, mais elle peut être utilisée au niveau de la recherche dans le laboratoire.
Une alternative est l‟usage d‟une brosse. Il est important d‟exercer une faible force de
frottement et de nettoyer les fibres doucement une fois séparées. La quantité de fibres
traitées à la fois avec la brosse dépasse celle du procédé manuel et donc le temps
nécessaire diminue.
Manuellement les meilleures fibres sont obtenues. Ce sont les plus propres et les moins
rigides parce que les fibres sont séparées et nettoyées l‟une après l‟autre. L‟inconvénient est
la durée de séparation et de nettoyage. Les fibres séparées par brossage sont moins
propres et plus rigides, mais ce procédé est plus performant. Les deux méthodes ont donc
des avantages et des inconvénients. Dans le futur il sera nécessaire de trouver une solution
qui combine les avantages des deux procédés.
Les meilleures fibres longues obtenues (voir 2.2.3.3.) sont séparées et brossées. Le résultat
est montré dans les photos suivantes (Figure 30 et Figure 31) :

31
2.2.5. Les propriétés et caractéristiques des fibres obtenues
2.2.5.1. MEB (angl. SEM)
Les meilleures fibres longues sont un peu rigides ce qui pourrait constituer une
caractéristique des fibres d‟alfa. Mais cela reste à confirmer par des photos prises avec le
MEB qui dans le cas échéant montreraient des fibres propres, c‟est-à-dire où les filaments
cellulosiques qui forment la fibre sont bien visibles et alignés parallèlement.
Si cela n‟est pas le cas, la rigidité est due à la présence de lignine et de pectines résiduelles,
d‟où la conclusion que les traitements n‟étaient pas assez efficaces.
Il est également utile d‟examiner la dégradation des filaments cellulosiques. Le but de
l‟extraction est d‟obtenir des fibres avec une dégradation minimale. Si les photos montrent
que les filaments, et en conséquence les fibres, sont dégradés, il faudra conclure que les
traitements étaient trop agressifs.
Pour démontrer l‟évolution déjà réalisée et l‟objectif de l‟extraction, quatre échantillons sont
examinés :
- une tige d‟alfa brute et non-traitée
- une tige d‟alfa brossée mécaniquement et trempée
- une des meilleures fibres longues obtenues par la méthode décrite précédemment
(voir section 2.2.3.3.)
- une fibre plus courte obtenue sous les mêmes conditions que les meilleures fibres
La photo de la tige d‟alfa est prise pour montrer le point de départ. Malheureusement la tige
s‟est fendue pendant la préparation de l‟échantillon d‟où la photo bizarre (Figure 32). La
partie supérieure montre la surface des tiges d‟alfa brute et la partie inférieure donne une
vue non-détaillée de l‟intérieur des tiges. La surface des tiges est relativement lisse. En tout
cas les filaments cellulosiques, les pierres de construction des fibres, sont invisibles car ils
sont couverts par des composants non-cellulosiques tels que la lignine, les pectines et les
cires.
La surface de la tige d‟alfa brossée mécaniquement et ensuite trempée dans de l‟eau salée,
ressemble à la surface de la tige d‟alfa brute (Figure 33). En comparant les photos de ces
deux échantillons avec la même magnification, la tige brossée et trempée est plus fine par
rapport à la tige brute. Cela est logique parce que le brossage raffine les tiges pour que l‟eau
salée, la soude et les enzymes puissent interagir plus efficacement. Un zoom est fait sur la
tige brossée et trempée (Figure 34). La surface reste lisse malgré que des lignes parallèles
soient peut-être vaguement visibles en haut et en bas de la photo.
Figure 30 : Fibres après séparation manuelle Figure 31 : Fibres après brossage

32
En comparant cette dernière photo avec la première d‟une des meilleures fibres (Figure 35)
extraites par un cycle « soude – enzymes – soude », avec la même magnification, il est clair
que les fibres obtenues sont beaucoup plus fines que les tiges trempées, ce qui est évident
parce que la tige est composée d‟un grand nombre de fibres. La photo de la fibre montre des
filaments cellulosiques alignés parallèlement, ce qui est favorable. Pourtant elle révèle
également que les fibres ne sont pas composées uniquement de tels filaments parce qu‟en
bas, une couche d‟une matière non-cellulosique est visible. Cette couche de lignine ou de
pectines n‟est pas alignée parallèlement aux filaments. En haut de la photo, quelques
filaments cellulosiques s‟étendent. Toujours en haut de la fibre, il reste encore une couche
fine qui rend la fibre raboteuse. Pour vérifier qu‟il ne s‟agit pas d‟une dégradation éventuelle
et indésirable des filaments, un zoom est nécessaire.
Cette photo plus détaillée (Figure 36) ne montre que des filaments non-dégradés, ce qui est
un bon résultat. Ils sont collés par les hémicelluloses, formant ainsi la fibre.
Un zoom additionnel sur la couche aperçue en haut de la fibre (Figure 37) montre des trous
qui indiquent une dégradation en cours. Cette couche entoure les filaments cellulosiques,
d‟où la conclusion qu‟il s‟agit plutôt d‟une matière non-cellulosique. Dans ce cas, la
dégradation est souhaitable parce qu‟elle élimine ce composant non-cellulosique.
En ce qui concerne la rigidité, il est clair après l‟interprétation des photos du MEB, qu‟elle est
due à la présence de lignine et de pectines résiduelles. Cela est confirmé par le fait que les
traitements qui produisent les meilleures fibres longues, donnent comme sous-produit
également des fibres plus courtes et davantage souples.
Comme l‟objectif de l‟extraction est d‟obtenir des fibres longues avec le taux de cellulose le
plus haut possible, il est intéressant de comparer les meilleures fibres obtenues avec ces
fibres plus courtes, fines et flexibles. La photo (Figure 38) montre que les fibres courtes sont
beaucoup plus propres que les « meilleures » fibres longues. Il reste peu de matière non-
cellulosique (Figure 39) et les filaments cellulosiques sont davantage alignés, sans qu‟ils
s‟étendent. Par conséquent, les fibres sont plus souples et lisses. De plus les filaments ne
sont pas dégradés, ce qui n‟était pas le cas avec les fibres examinées avec le MEB dans
section 2.2.3.1.
L‟objectif est d‟obtenir des fibres ayant une certaine longueur, mais qui ont de même la
souplesse et donc la structure, des fibres plus courtes examinées avec le MEB.
Malgré que les meilleures fibres obtenues soient encore un peu rigides, des essais
mécaniques et un microfilage, c‟est-à-dire un filage avec des quantités de fibres de l‟ordre de
50g à 200g, peuvent être effectués afin de déterminer les caractéristiques mécaniques et la
possibilité de filage.
Les fibres courtes et souples, obtenues comme sous-produit de la production des meilleures
fibres longues, peuvent être utilisées dans des mélanges avec d‟autres fibres naturelles
comme la laine et le coton.

33
Figure 32 : Image MEB de la tige d‟alfa brute Figure 33 : Image MEB de la tige d‟alfa brossée mécaniquement et trempée
Figure 36 : Image MEB des filaments cellulosiques bien visibles en non-dégradés
Figure 39 : Image MEB de la surface contenant peu de composants non-cellulosiques
Figure 34 : Image MEB d‟un zoom sur la tige d‟alfa brossée mécaniquement et trempée
Figure 35 : Image MEB d‟une des meilleures fibres longues
Figure 37 : Image MEB de la dégradation à la surface
Figure 38 : Image MEB d‟une des fibres courtes

34
2.2.5.2. IRTF-RTA (angl. ATR-FTIR)
Le nom de cette méthode d‟analyse est « la spectroscopie InfraRouge à Transformée de
Fourier par une Réflexion Totale Atténuée » (angl. Attenuated Total Reflection – Fourier
Transform InfraRed spectroscopy). En général la spectroscopie infrarouge est très puissante
et capable de déterminer les liaisons et les groupes fonctionnels dans un échantillon et ainsi
sa composition. L‟IRTF est une version de la spectroscopie infrarouge classique qui capte le
signal d‟une autre façon, de sorte que les mesures sont faites plus rapidement. Normalement
le mode de mesure est la transmission, ce qui signifie que le rayon infrarouge émis par la
source traverse complètement l‟échantillon. Par contre, l‟IRTF-RTA collecte la même
information par une méthode de réflexion. Le spectre obtenu donne l‟intensité du signal en
fonction du nombre d‟onde (k), c‟est-à-dire l‟inverse de la longueur d‟onde (λ) (2.1). Pour plus
d‟information voir appendice A.
Dans l‟échantillon à examiner, les atomes des liaisons différentes de la molécule vibrent à
une fréquence caractéristique. Le rayon infrarouge parcourt un intervalle de nombres d‟onde.
Ces nombres d‟onde correspondent à des fréquences (2.2 et 2.3). Lors du passage du rayon
infrarouge à travers l‟échantillon, un peu d‟énergie du rayon est absorbée quand la
fréquence des vibrations des liaisons est identique à celle du rayon infrarouge.
⁄ (2.1)
⇔ (2.2)
(2.2)
Le spectromètre détecte ces absorptions d‟énergie et enregistre la quantité absorbée pour
chaque nombre d‟onde correspondant L‟absorption d‟énergie est ensuite transformée en une
diminution de l‟intensité du rayon infrarouge. Avec cette intensité et le nombre d‟onde le
spectre est constitué.
Comme le spectre mesuré est celui de la combinaison air-échantillon, il faut également
enregistrer le spectre de l‟air. La soustraction de ces deux spectres donne celui de
l‟échantillon.
Le spectre est analysé en identifiant les pics parce que chaque pic correspond à une liaison
ou une configuration caractéristique [21]. En combinant l‟information de chaque pic, les
composants différents d‟un échantillon hétérogène peuvent être déterminés. La
spectroscopie infrarouge est un moyen puissant pour des analyses qualitatives.
Evidemment il existe également des inconvénients dont le fait qu‟une analyse quantitative,
c‟est-à-dire la détermination des taux de chaque composant, est impossible. La
spectroscopie infrarouge permet seulement de déterminer si un certain composant est
présent ou absent dans un échantillon à base de la présence ou l‟absence des pics
correspondants. De cette façon il est possible de comparer deux échantillons. Si un des
deux a un grand pic et le pic correspondant dans l‟autre spectre est presque invisible, il est
probable que le taux du composant considéré soit plus faible dans le deuxième échantillon.
Un deuxième inconvénient de la spectroscopie infrarouge est que seulement certaines
liaisons sont réactives sous un rayon infrarouge, parce qu‟il faut un moment dipôle, c‟est-à-
dire une répartition des électrons asymétrique.
Par contre, un avantage est qu‟il ne faut pas travailler sous vide car l‟oxygène et l‟azote dans
l‟atmosphère ne possèdent pas un tel moment dipôle et n‟absorbent ainsi pas de rayons
infrarouges.
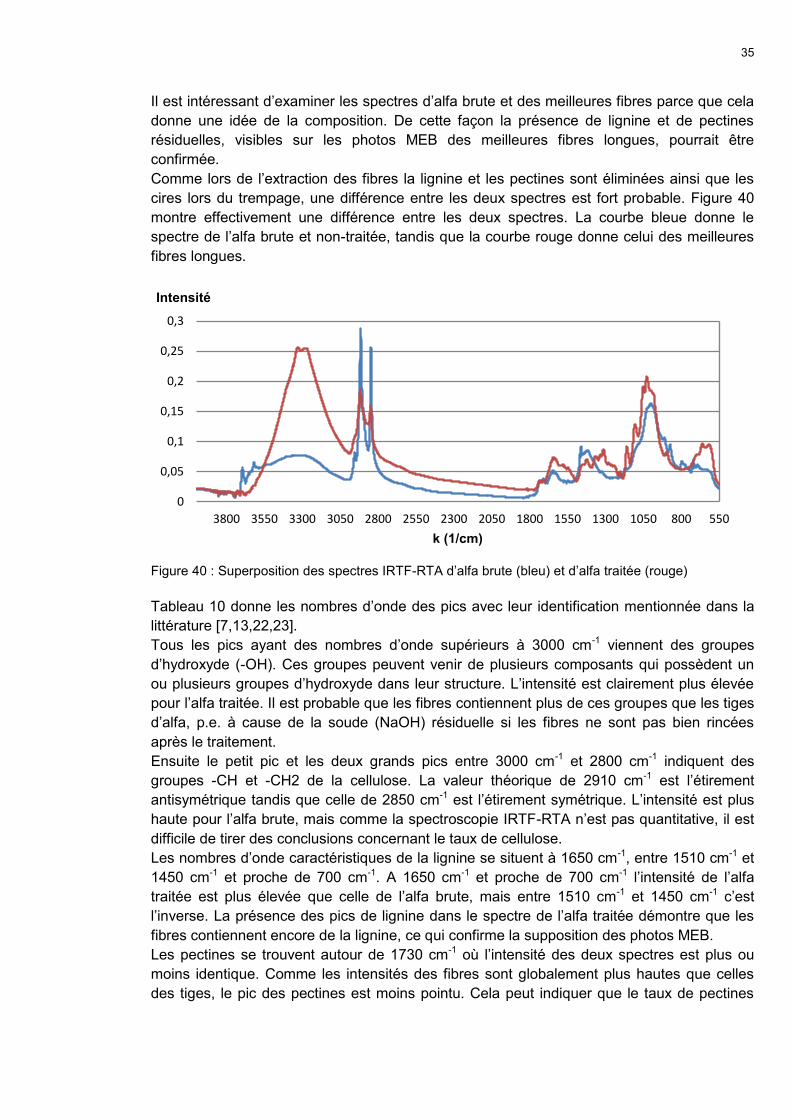
35
Il est intéressant d‟examiner les spectres d‟alfa brute et des meilleures fibres parce que cela
donne une idée de la composition. De cette façon la présence de lignine et de pectines
résiduelles, visibles sur les photos MEB des meilleures fibres longues, pourrait être
confirmée.
Comme lors de l‟extraction des fibres la lignine et les pectines sont éliminées ainsi que les
cires lors du trempage, une différence entre les deux spectres est fort probable. Figure 40
montre effectivement une différence entre les deux spectres. La courbe bleue donne le
spectre de l‟alfa brute et non-traitée, tandis que la courbe rouge donne celui des meilleures
fibres longues.
Tableau 10 donne les nombres d‟onde des pics avec leur identification mentionnée dans la
littérature [7,13,22,23].
Tous les pics ayant des nombres d‟onde supérieurs à 3000 cm-1 viennent des groupes
d‟hydroxyde (-OH). Ces groupes peuvent venir de plusieurs composants qui possèdent un
ou plusieurs groupes d‟hydroxyde dans leur structure. L‟intensité est clairement plus élevée
pour l‟alfa traitée. Il est probable que les fibres contiennent plus de ces groupes que les tiges
d‟alfa, p.e. à cause de la soude (NaOH) résiduelle si les fibres ne sont pas bien rincées
après le traitement.
Ensuite le petit pic et les deux grands pics entre 3000 cm-1 et 2800 cm-1 indiquent des
groupes -CH et -CH2 de la cellulose. La valeur théorique de 2910 cm-1 est l‟étirement
antisymétrique tandis que celle de 2850 cm-1 est l‟étirement symétrique. L‟intensité est plus
haute pour l‟alfa brute, mais comme la spectroscopie IRTF-RTA n‟est pas quantitative, il est
difficile de tirer des conclusions concernant le taux de cellulose.
Les nombres d‟onde caractéristiques de la lignine se situent à 1650 cm-1, entre 1510 cm-1 et
1450 cm-1 et proche de 700 cm-1. A 1650 cm-1 et proche de 700 cm-1 l‟intensité de l‟alfa
traitée est plus élevée que celle de l‟alfa brute, mais entre 1510 cm-1 et 1450 cm-1 c‟est
l‟inverse. La présence des pics de lignine dans le spectre de l‟alfa traitée démontre que les
fibres contiennent encore de la lignine, ce qui confirme la supposition des photos MEB.
Les pectines se trouvent autour de 1730 cm-1 où l‟intensité des deux spectres est plus ou
moins identique. Comme les intensités des fibres sont globalement plus hautes que celles
des tiges, le pic des pectines est moins pointu. Cela peut indiquer que le taux de pectines
0
0,05
0,1
0,15
0,2
0,25
0,3
550800105013001550180020502300255028003050330035503800
Intensité
k (1/cm)
Figure 40 : Superposition des spectres IRTF-RTA d‟alfa brute (bleu) et d‟alfa traitée (rouge)

36
dans les fibres est supérieur à celui dans les tiges. En tout cas, ce pic confirme la présence
de pectines résiduelles.
Entre 1450 cm-1 et 1000 cm-1 se trouvent d‟autres pics de cellulose. Globalement les
intensités pour l‟alfa traitée sont supérieures à celles de l‟alfa brute.
Les pics à 1360 cm-1 et 1320 cm-1 sont liés à la déformation de la cellulose et ceux à 1059
cm-1 et 1030 cm-1 à la vibration de la chaîne cellulosique.
Il est également utile de comparer le spectre des meilleures fibres avec celui de coton.
Comme le coton est composé en grande partie de cellulose, c‟est bon signe quand les deux
spectres se rapprochent, indiquant que le taux de cellulose est déjà plus élevé par rapport
aux tiges.
Les spectres pour cette comparaison sont enregistrés un autre jour avec une autre fibre
d‟alfa, donc le spectre d‟alfa traitée est légèrement différent. Par exemple, les pics entre
2400 cm-1 et 2300 cm-1 sont dus à des impuretés. A part ces impuretés, le spectre d‟alfa
traitée ressemble déjà à celui de fibres de coton nettoyé (Figure 41). Les pics entre
3000 cm-1 et 2800 cm-1 sont plus pointés. En revanche ceux entre 1800 cm-1 et 1500 cm-1
sont moins visibles que pour le coton.
alfa brute alfa traite théorie identification composant
3694 -OH cellulose, soude
3632 3329 3340 -OH cellulose, soude
3368 3278 -OH cellulose, soude
2954 2960 (CH) cellulose
2915 2917 2910 (CH2) cellulose
2848 2850 2850 (CH2) cellulose
1721 1737 1730 -C=O pectines
1656 1656 1650 -COOH lignine
1472 1510-1450 C-C=C lignine (?)
1461 1461 1510-1450 C-C=C lignine (?)
1457 1510-1450 C-C=C lignine (?)
1422 1426 1426 (CH2) cellulose
1368 1360 -OH cellulose
1314 1320 -OH cellulose
1163 1160 1160 C-O-C cellulose (?)
1106 C-O-C cellulose (?)
1050 1059 -OH secondaires cellulose
1029 1030 -OH primaires cellulose
718 env. 700 lignine
712 env. 700 lignine
694 env. 700 lignine
660
616
Tableau 10 : Identification des pics les plus importants des spectres IRTF-RTA [7,13,22,23]

37
2.2.5.3. Les essais mécaniques
Les essais mécaniques exécutés sur les fibres longues sont des essais de traction.
L‟appareil de traction est équipé du capteur le plus petit car il s‟agit de fibres fines et fragiles.
Les courbes de traction donnent la force en fonction de l‟allongement. Normalement la
contrainte est exprimée en fonction de la déformation relative. La conversion d‟un
allongement en une déformation relative est facile à calculer en divisant l‟allongement par la
longueur initiale. Cette longueur initiale est la distance entre les deux pièges de l‟appareil.
Elle est choisie au début et reste constante tout au long des essais mécaniques.
La force est transformée en une contrainte en la divisant par la surface de la fibre, mais le
problème est que le diamètre, et donc également la surface, des fibres est inconnu.
Néanmoins le diamètre est estimé pour que la conversion soit possible.
Vu la petite quantité de fibres, le nombre d‟essais mécaniques exécutés est minimal. A base
de tellement peu de données, il est impossible de tirer des conclusions fiables parce que des
résultats exceptionnellement bons ou mauvais pourraient influencer fortement les valeurs
moyennes.
Figure 42 donne une courbe de traction modèle qui exprime la force en fonction de
l‟allongement. La vitesse de déformation est 5 mm/min. Chaque courbe a une ou plusieurs
chutes de force dues à la fracture des filaments cellulosiques dont la fibre est composée. Les
fibres ne se cassent donc pas en une fois, mais peu à peu. Pour calculer le module d‟Young,
la contrainte est divisée par la déformation relative. Le module d‟Young est une
caractéristique du domaine élastique, donc il faut le calculer à partir des valeurs de
contrainte et de déformation contenues dans ce même domaine élastique. Comme le
diamètre est estimé, les valeurs du module d‟Young seront également estimées.
Les constantes pour la conversion de force en contrainte et d‟allongement en déformation :
- la distance entre les deux pièges : 40 mm (LI)
- le diamètre estimé : 135 µm
Le diamètre est estimé à partir de la photo MEB d‟une des meilleures fibres (Figure 35).
Comme la photo indique une échelle, il est possible de calculer la largeur, c‟est-à-dire le
Figure 41 : Comparaison des spectres IRTF-RTA de fibres d‟alfa et de coton
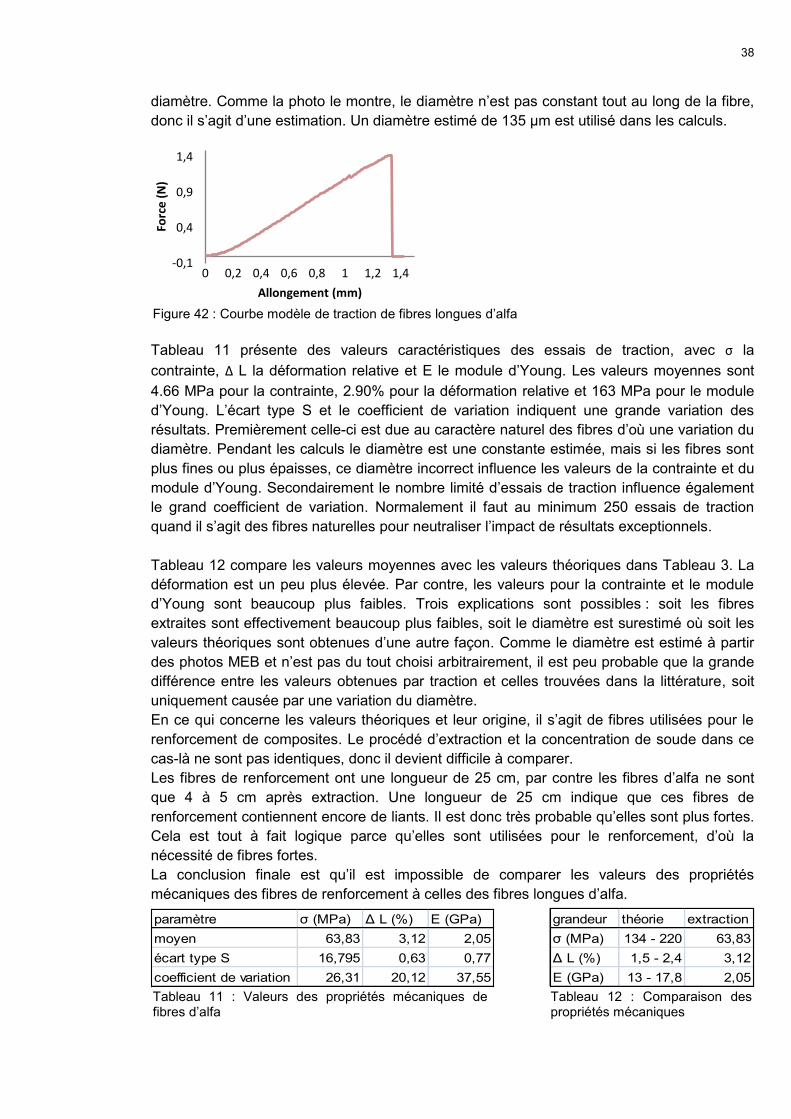
38
diamètre. Comme la photo le montre, le diamètre n‟est pas constant tout au long de la fibre,
donc il s‟agit d‟une estimation. Un diamètre estimé de 135 µm est utilisé dans les calculs.
Tableau 11 présente des valeurs caractéristiques des essais de traction, avec σ la
contrainte, Δ L la déformation relative et E le module d‟Young. Les valeurs moyennes sont
4.66 MPa pour la contrainte, 2.90% pour la déformation relative et 163 MPa pour le module
d‟Young. L‟écart type S et le coefficient de variation indiquent une grande variation des
résultats. Premièrement celle-ci est due au caractère naturel des fibres d‟où une variation du
diamètre. Pendant les calculs le diamètre est une constante estimée, mais si les fibres sont
plus fines ou plus épaisses, ce diamètre incorrect influence les valeurs de la contrainte et du
module d‟Young. Secondairement le nombre limité d‟essais de traction influence également
le grand coefficient de variation. Normalement il faut au minimum 250 essais de traction
quand il s‟agit des fibres naturelles pour neutraliser l‟impact de résultats exceptionnels.
Tableau 12 compare les valeurs moyennes avec les valeurs théoriques dans Tableau 3. La
déformation est un peu plus élevée. Par contre, les valeurs pour la contrainte et le module
d‟Young sont beaucoup plus faibles. Trois explications sont possibles : soit les fibres
extraites sont effectivement beaucoup plus faibles, soit le diamètre est surestimé où soit les
valeurs théoriques sont obtenues d‟une autre façon. Comme le diamètre est estimé à partir
des photos MEB et n‟est pas du tout choisi arbitrairement, il est peu probable que la grande
différence entre les valeurs obtenues par traction et celles trouvées dans la littérature, soit
uniquement causée par une variation du diamètre.
En ce qui concerne les valeurs théoriques et leur origine, il s‟agit de fibres utilisées pour le
renforcement de composites. Le procédé d‟extraction et la concentration de soude dans ce
cas-là ne sont pas identiques, donc il devient difficile à comparer.
Les fibres de renforcement ont une longueur de 25 cm, par contre les fibres d‟alfa ne sont
que 4 à 5 cm après extraction. Une longueur de 25 cm indique que ces fibres de
renforcement contiennent encore de liants. Il est donc très probable qu‟elles sont plus fortes.
Cela est tout à fait logique parce qu‟elles sont utilisées pour le renforcement, d‟où la
nécessité de fibres fortes.
La conclusion finale est qu‟il est impossible de comparer les valeurs des propriétés
mécaniques des fibres de renforcement à celles des fibres longues d‟alfa.
Tableau 12 : Comparaison des propriétés mécaniques
-0,1
0,4
0,9
1,4
0 0,2 0,4 0,6 0,8 1 1,2 1,4
Forc
e (
N)
Allongement (mm)
Figure 42 : Courbe modèle de traction de fibres longues d‟alfa
paramètre σ (MPa) Δ L (%) E (GPa)
moyen 63,83 3,12 2,05
écart type S 16,795 0,63 0,77
coefficient de variation 26,31 20,12 37,55
Tableau 11 : Valeurs des propriétés mécaniques de fibres d‟alfa
grandeur théorie extraction
σ (MPa) 134 - 220 63,83
Δ L (%) 1,5 - 2,4 3,12
E (GPa) 13 - 17,8 2,05

39
2.3. Conclusion du mode opératoire
L‟extraction de fibres d‟alfa à partir de tiges d‟alfa brute est un procédé en plusieurs étapes.
La structure de la tige est complexe, d‟où la nécessité de multiples traitements consécutifs
afin d‟éliminer tous les composants non-cellulosiques.
La première étape est un brossage mécanique qui raffine les tiges de sorte que l‟eau salée,
la soude et les enzymes puissent mieux interagir avec les tiges (voir sections 2.2.3.1.,
2.2.3.2 et 2.2.3.3.).
La deuxième étape consiste en un trempage dans de l‟eau salée, éliminant les cires. Après
ce trempage les tiges sont encore rigides et il n‟en est pas question de fibres.
La troisième étape est un traitement de sodium d‟hydroxyde. Ce produit est capable de
dégrader la lignine, qui lie les fibres et les bottes de fibres. Des concentrations et
températures élevées dégradent les fibres, d‟où le choix d‟une concentration faible (0.25N à
0.5N) qui, par contre, a comme inconvénient d‟augmenter la durée. Un traitement unique de
soude ne suffit pas pour obtenir des fibres, donc une étape supplémentaire est ajoutée.
Cette quatrième étape consiste en un traitement enzymatique. Les pectinases dégradent les
pectines, un liant comme la lignine. Ces enzymes sont très réactives d‟où une concentration
faible (2.5 ml/l d‟eau) et une durée limitée (1h) sous des conditions modérées. Après ce
traitement les fibres sont visibles, mais elles s‟avèrent toujours liées entre-elles.
La cinquième étape utilise de la soude pour éliminer une partie du liant résiduel. Des fibres
ayant une longueur de quelques centimètres sont obtenues. Elles sont encore un peu rigides
à cause de la présence de lignine et de pectines résiduelles.
Un des problèmes principaux est le résultat hétérogène. Trois types de fibres peuvent être
distingués :
- soit les fibres sont encore très rigides et très longues parce que la lignine et les
pectines ne sont pas bien éliminées. Dans ce cas il s‟agit plutôt de bosses de fibres.
- soit les fibres sont bonnes, avec une longueur de quelques centimètres et une
certaine rigidité. Ce type de fibres est acceptable et non-dégradé bien qu‟il reste
encore un peu de lignine et de pectines (voir les photos prises avec le MEB et les
spectres IRTF-RTA).
- soit les fibres sont courtes ou très courtes. De temps en temps elles sont dégradées
(voir les photos de MEB dans section 2.5.5.3.)
Premièrement cette variation est due au caractère naturel des fibres. Tout comme pour le
coton et la laine, l‟origine de la matière première est la nature. Les tiges d‟alfa sont
hétérogènes au niveau de longueur, de diamètre, de couleur, de structure interne, etc. Le
brossage mécanique réalise pourtant des diamètres plus homogènes.
Deuxièmement les conditions de travail variables peuvent influencer l‟homogénéité du
résultat. Il est possible que l‟eau salée, la soude et les enzymes puissent mieux atteindre
certaines parties des tiges que d‟autres, de manière que les cires, la lignine et les pectines
ne sont pas éliminées de façon homogène.
Chaque type de fibres a une propre destination possible. Les bottes de fibres pourraient
subir un deuxième traitement enzymatique et éventuellement un troisième traitement de
soude afin de produire des fibres longues et moins rigides. Cette possibilité n‟a ni encore été
examinée ni exécutée.

40
Les fibres courtes pourraient être utiles dans des mélanges de fibres, p.e. avec du coton ou
de la laine. Les fibres très courtes pourraient servir à produire de la pâte à papier ou à
dissoudre et filer comme dans le procédé viscose (voir chapitre 3).
Les meilleures fibres sont aptes à être transformées en fils par filage mécanique.
Un autre problème est la durée totale pour obtenir des fibres à partir des tiges d‟alfa brute.
Dans le futur il faudra donc optimiser les conditions de travail pour accélérer le procédé de
sorte qu‟il soit rentable.
Le microfilage des meilleures fibres n‟est pas encore réalisé. Pour faire cela, il faut collaborer
avec une entreprise spécialisée, actuellement très chargée, leur premier moment libre étant
en juillet. D‟ailleurs une certaine quantité de fibres est nécessaire mais doit encore être
produite.
Au moment où un fil d‟alfa sera produit, des essais de traction donneront ses propriétés
mécaniques qui détermineront son domaine d‟application, qui sera soit le textile technique en
cas de fils fortes, soit le textile classique.
La production d‟un tissu à partir de fils d‟alfa permettra à son tour de connaître les propriétés
du tissu.

41
CHAPITRE 3 : Dissolution de la cellulose
3.1. L‟objectif de la dissolution : le filage des filaments
Un procédé alternatif pour la production de fibres est le filage. Toutes les fibres synthétiques
comme les polyamides, les aramides et les polyesters sont obtenues par filage. Mais il est
également possible de générer des fibres cellulosiques de cette façon. Ces fibres ne sont
plus appelées naturelles, mais artificielles. Quelques exemples sont la viscose, le Lyocell,
l‟acétate et le triacétate.
Pour que le filage soit possible, une solution ou une quantité de polymère fusé est
indispensable. Le filage est, sous sa forme la plus simple, très similaire à la production de
spaghetti. Une masse plus ou moins liquide avec une haute viscosité est pressée contre une
plaque perforée (les filières ou les têtes de filage). Le matériel passe par les trous et forme
ainsi des filaments. Dans des conditions optimalisées, des filaments de longueur infinie
pourraient être produits. Dans des situations réelles, à la demande du client ou selon
l‟application finale, les filaments sont souvent coupés en petits morceaux d‟une longueur de
quelques centimètres.
Tout d‟abord, une solution stable doit être produite, soit par la fusion d‟un polymère, soit par
la dissolution de la matière première (une pâte cellulosique ou un polymère) dans un solvant.
La solution doit être filtrée avant le filage. Il s‟agit d‟une étape nécessaire afin d‟éviter
l‟obstruction des filières, un risque réel parce que les trous sont extrêmement fins.
Les têtes de filage sont souvent fabriquées en inox, mais les meilleures sont en platine.
Après le filage, l‟étirage améliore les propriétés mécaniques des fibres par l‟augmentation du
taux de cristallinité et la fixation thermique améliore la stabilité thermique des fibres.
Dans le secteur textile, quatre façons de filage sont utilisées fréquemment [24, 25].
3.1.1. Le filage par fusion (Figure 43) (angl. melt spinning)
La matière première est toujours un polymère fondu. Certains polymères ne peuvent pas
être utilisés car ils se dégradent avant fusion. Une pompe presse le polymère fondu contre
les filières. Les filaments formés sont coagulés par de l‟air froid. Le filage est suivi de post-
traitements comme l‟étirage, le coupage en petits morceaux, l‟ennoblissement...
Comme ce procédé se déroule sans solvant, le filage par fusion est le plus simple et a le
moindre impact environnemental.
3.1.2. Le filage à sec (Figure 44) (angl. dry spinning)
La matière première est un polymère qui n‟est pas facile à mettre en fusion. Ce polymère est
donc dissous dans un solvant dont la structure chimique dépend du type de polymère. Après
la formation des filaments, un jet d‟air chaud est soufflé à travers au sens inverse et évapore
le solvant. Les filaments coagulent et l‟air avec le solvant évaporé est récupéré afin de
recycler le solvant pour des raisons économiques.
3.1.3. Le filage à humide (Figure 45) (angl. wet spinning)
Un autre nom pour ce procédé est le filage par coagulation. Comme c‟est le cas pour le
filage à sec, la matière première est un polymère qui est difficile à mettre en fusion et qui est
donc dissous dans un solvant. Les filaments formés passent par un bain de coagulation qui
suit immédiatement vers les têtes de filage. Dans ce bain, les filaments coagulent et le

42
solvant est dissous. Ensuite, le contenu du bain est récupéré afin de recycler le solvant.
Cette méthode est utilisée seulement si les solvants sont non volatils.
3.1.4. Le filage à sec et par coagulation (angl. dry jet wet spinning)
Ce procédé est une combinaison des procédés de filage à sec et de filage par coagulation. Il
s‟agit donc également de polymères dissous dans un solvant. Après les têtes de filage, les
filaments sont refroidis par un jet d‟air froid et ensuite le solvant est dissous dans un bain de
coagulation.
3.2. Le procédé viscose
Le procédé viscose était le premier procédé mis au point pour produire des fibres artificielles
à base de cellulose. Le premier brevet d‟invention a été attribué à Cross, Bevan et Beadle en
Angleterre en 1893 [26]. Le nom initial du produit était « la soie artificielle » et depuis 1924
« la rayonne » est également employée comme nom.
La source principale de cellulose est le bois, mais évidemment d‟autres matières contenant
de la cellulose peuvent de même être utilisées. Un exemple sont les neps de coton qui
apparaissent lors du traitement de fibres de coton, où ils sont considérés comme des
déchets. Les neps peuvent cependant très bien être traités dans des pâtes cellulosiques.
La dissolution de la cellulose est difficile à cause des liaisons d‟hydrogène fortes qui existent
entre les chaînes et qui sont formées par les groupes fonctionnels d‟hydroxyde (-OH). Le
procédé viscose est donc complexe et de longue durée. Le schéma suivant donne les
étapes successives :
Figure 43 : Représentation schématique du filage par fusion
Figure 44 : Représentation schématique du filage à sec
Figure 45 : Représentation schématique du filage à humide

43
3.2.1. La pâte cellulosique (angl. cellulosic pulp)
La pâte cellulosique est la matière première authentique du procédé viscose. La manière la
plus fréquente pour obtenir une pâte est le procédé Kraft (voir section 2.1.2.).
Il est nécessaire que la longueur des fibres dans la pâte soit la plus homogène possible,
parce que des longueurs différentes provoquent des vitesses de réaction différentes dans les
étapes suivantes.
3.2.2. L’enflure de la pâte (angl. steeping)
La pâte est versée dans un bain et mixée avec de la soude. La cellulose est transformée en
cellulose alcaline et la pâte gonfle. Les groupes fonctionnels d‟hydroxyde sont transformés
en (-O-Na+) avec formation d‟eau (3.1). Les conditions sont [26] : 45°C à 55°C, 17% à 19%
de soude et une durée de 5 à 10 minutes. Après, l‟ensemble est pressé afin d‟éliminer
l‟excès d‟eau et de soude.
(3.1)
3.2.3. Shredding
Après le pressage, la pâte qui est devenue dense contient classiquement 30 à 36% de
cellulose et 13 à 17% de soude. Pendant cette étape, la pâte est étalée et ouverte pour
faciliter les étapes suivantes. Le D.P. de la cellulose alcaline est entre 750 et 850.
3.2.4. Mercérisation (angl. pre-ageing, mercerising)
Le D.P. de la cellulose alcaline est diminué jusqu‟à 270 à 350 par oxydation à une
température entre 40°C et 60°C et cela pendant entre 30 minutes et 5 h.
Figure 46 : Représentation schématique du procédé viscose [27]

44
3.2.5. Formation de xanthate (angl. xantation)
Dans cette étape la cellulose alcaline est transformée en sodium de xanthate de cellulose
(angl. sodium cellulose xanthate) selon la réaction 3.2. La durée varie entre 30 et 90 minutes
et la température se situe généralement entre 25°C et 37°C. Le CS2 est également
consommé dans des réactions secondaires avec de la soude. Les réactions 3.3 et 3.4
donnent le mécanisme simplifié.
(3.2)
(3.3)
(3.4)
3.2.6. Dissolution du xanthate de cellulose et « ageing »
Une fois le xanthate de cellulose est formé, le produit est dissous dans une solution aqueuse
et diluée de NaOH à une température de 8 à 12°C afin d‟augmenter la solubilité.
3.2.7. Filage par coagulation
Le bain de coagulation contient de l‟eau et de l‟acide sulfurique (H2SO4). L‟acide sulfurique
transforme le xanthate de cellulose en cellulose (3.5). Le mécanisme n‟est pas
complètement connu, parce que la réaction est une simplification du mécanisme réel. Les
produits secondaires formés pendant la formation de xanthate se dégradent en CS2, H2S
(toxique) et Na2SO4 pendant le filage (3.6 et 3.7).
(3.5)
(3.6)
(3.7)
3.3. Le procédé Lyocell
3.3.1. Généralités
Le Lyocell est la première fibre d‟une nouvelle génération de fibres cellulosiques. Les fibres
de cette génération utilisent un solvant plus écologique, recyclable et réutilisable. La durée
totale et le nombre d‟étapes du procédé diminuent par rapport au procédé viscose classique.
En 1984 les premières fibres sont produites avec le solvant organique N-méthylemorpholine-
N-oxide (NMMO, NMMNO, MMNO ou NMO). Depuis 1992 Courtaulds au Royaume-Uni et
depuis 1997 Lenzing AG en Autriche produisent les fibres Lyocell [28], ainsi qu‟Akzo Nobel
aux Pays-Bas. Le nom Lyocell est globalement accepté, mais il s‟agit plus précisément des
fibres produites par Lenzing AG. Celles de Courtaulds s‟appellent Tencel, celles de Akzo
Nobel s‟appellent Newcell.
Le schéma général de la production des fibres cellulosiques avec NMMO est donné dans la
figure suivante :

45
La cellulose est utilisée sous forme d‟une pâte et a un D.P. entre 400 et 1000. Elle est mise
dans une solution aqueuse de NMMO et dissout. Le gel obtenu a une couleur ambre foncé.
Il est filtré et filé par un filage à sec à une température entre 100 et 120°C [26]. Au moment
du refroidissement des filaments, le NMMO est récupéré et ensuite recyclé et réutilisé grâce
à un circuit fermé.
Un filage à sec par coagulation est également possible [22,27]. Dans la section à air, les
filaments sont étirés et ils sont coagulés dans le bain de coagulation. Le NMMO est dissous
dans ce bain et ainsi récupéré. Le filage par coagulation nécessite des faibles concentrations
de cellulose, ce qui n‟est pas rentable. De plus les propriétés des fibres sont inférieures à
celles de la viscose classique.
Le procédé est complété par quelques post-traitements.
Les avantages multiples du procédé Lyocell par rapport au procédé viscose
- la durée totale est inférieure à 8 heures, par contre le procédé viscose a une durée
supérieure à 40 heures
- le circuit est fermé et par conséquent le solvant peut être récupéré
- des produits secondaires toxiques tels que le H2S et le CS2 ne sont pas produits
- la concentration de cellulose dans la solution peut monter à des valeurs élevées de
25 jusqu‟à 35% [27]
- la vitesse de filage est plus grande : 150 à 300 m/min [27]
- les propriétés mécaniques des fibres sont meilleures grâce à un taux de cristallinité
plus élevé. Ceci est démontré p.e. par des essais de traction sur des fibres de Lyocell
et des fibres de viscose (Figure 48)
Quelques inconvénients
- le NMMO est fortement corrosif, donc du matériel en inox est recommandé
- le NMMO peut dégrader de façon exothermique à des températures supérieures à
150°C, ce qui peut causer une explosion
Figure 47 : Représentation schématique du procédé Lyocell [27]
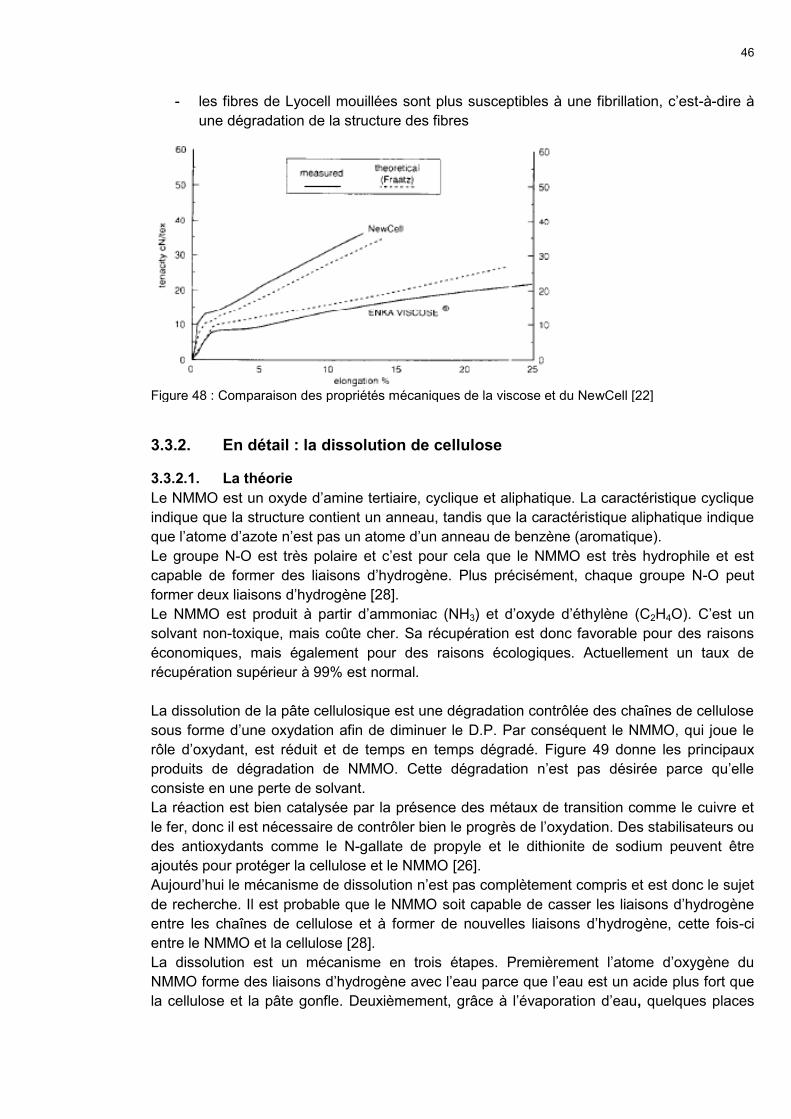
46
- les fibres de Lyocell mouillées sont plus susceptibles à une fibrillation, c‟est-à-dire à
une dégradation de la structure des fibres
3.3.2. En détail : la dissolution de cellulose
3.3.2.1. La théorie
Le NMMO est un oxyde d‟amine tertiaire, cyclique et aliphatique. La caractéristique cyclique
indique que la structure contient un anneau, tandis que la caractéristique aliphatique indique
que l‟atome d‟azote n‟est pas un atome d‟un anneau de benzène (aromatique).
Le groupe N-O est très polaire et c‟est pour cela que le NMMO est très hydrophile et est
capable de former des liaisons d‟hydrogène. Plus précisément, chaque groupe N-O peut
former deux liaisons d‟hydrogène [28].
Le NMMO est produit à partir d‟ammoniac (NH3) et d‟oxyde d‟éthylène (C2H4O). C‟est un
solvant non-toxique, mais coûte cher. Sa récupération est donc favorable pour des raisons
économiques, mais également pour des raisons écologiques. Actuellement un taux de
récupération supérieur à 99% est normal.
La dissolution de la pâte cellulosique est une dégradation contrôlée des chaînes de cellulose
sous forme d‟une oxydation afin de diminuer le D.P. Par conséquent le NMMO, qui joue le
rôle d‟oxydant, est réduit et de temps en temps dégradé. Figure 49 donne les principaux
produits de dégradation de NMMO. Cette dégradation n‟est pas désirée parce qu‟elle
consiste en une perte de solvant.
La réaction est bien catalysée par la présence des métaux de transition comme le cuivre et
le fer, donc il est nécessaire de contrôler bien le progrès de l‟oxydation. Des stabilisateurs ou
des antioxydants comme le N-gallate de propyle et le dithionite de sodium peuvent être
ajoutés pour protéger la cellulose et le NMMO [26].
Aujourd‟hui le mécanisme de dissolution n‟est pas complètement compris et est donc le sujet
de recherche. Il est probable que le NMMO soit capable de casser les liaisons d‟hydrogène
entre les chaînes de cellulose et à former de nouvelles liaisons d‟hydrogène, cette fois-ci
entre le NMMO et la cellulose [28].
La dissolution est un mécanisme en trois étapes. Premièrement l‟atome d‟oxygène du
NMMO forme des liaisons d‟hydrogène avec l‟eau parce que l‟eau est un acide plus fort que
la cellulose et la pâte gonfle. Deuxièmement, grâce à l‟évaporation d‟eau, quelques places
Figure 48 : Comparaison des propriétés mécaniques de la viscose et du NewCell [22]

47
sur les groupes N-O du NMMO sont libérées et donc des liaisons d‟hydrogène avec la
cellulose sont formées. Troisièmement la cellulose dissout.
3.3.2.2. La réalisation pratique
La pâte cellulosique (10 à 15%) est mise dans une solution diluée avec 50 à 60% de NMMO
et 20 à 30% d‟eau (la position « suspension » sur Figure 50). L‟ensemble est mixé à des
températures de 70°C jusqu‟à 90°C.
Comme la cellulose n‟est pas soluble dans cette solution, il faut changer la composition afin
d‟arriver dans la zone de solubilité (« solution area » sur Figure 50). A ce stade l‟excès d‟eau
est évaporé pendant l‟agitation jusqu‟à ce qu‟une solution isotrope soit obtenue contenant
environ 14% de cellulose, 76% de NMMO et 10% d‟eau (« spinning solution » sur Figure 50).
Une solution isotrope donne des fibres qui ont des propriétés mécaniques plus ou moins
identiques dans chaque direction. Les fibres seront plus fortes si les propriétés sont
anisotropes parce que de cette manière, une direction favorable avec de meilleures
propriétés mécaniques existera. De temps en temps dans le secteur textile il est donc
souhaitable d‟avoir une solution anisotrope.
Quant à la solution aqueuse de NMMO, le taux d‟eau doit être inférieur à 11% afin que la
solution soit anisotrope [27]. A cause de ce niveau bas d‟humidité, la viscosité est grande.
Celle-ci dépend également de la concentration de cellulose (la viscosité augmente avec la
concentration de cellulose) et du D.P. de la cellulose (la viscosité augmente avec la longueur
des chaînes cellulosiques et donc avec le D.P.).
Figure 51 donne la solubilité de cellulose avec un D.P. moyen dans une solution diluée de
NMMO à 100°C. La solubilité dépend fortement de la concentration d‟eau et le seuil maximal
est 15% à 17% d‟eau. Pour des concentrations d‟eau supérieures à 17%, l‟atome d‟azote du
NMMO forme des liaisons d‟hydrogène avec l‟eau, donc la cellulose ne dissout pas. Le seuil
Figure 49 : Deux produits de dégradation principaux
Figure 50 : Schéma des compositions possibles d‟un mélange de cellulose, de NMMO et d‟eau [27]

48
minimal pour que la dissolution soit possible est 4%. Pour ces concentrations d‟eau, la
température de dissolution est proche de celle de dégradation du NMMO, soit 150°C.
3.4. La partie expérimentale
3.4.1. Introduction
La dissolution de cellulose dans du NMMO est un procédé moins nuisible que la dissolution
dans du CS2. De plus, le nombre d‟étapes est diminué ce qui permet de réduire la durée du
traitement. Pour ces raisons le procédé NMMO est préféré. Comme le procédé fonctionne
déjà bien dans l‟industrie avec des pâtes cellulosiques classiques, il est probable qu‟il
donnera également de bons résultats avec une pâte à base de fibres courtes d‟alfa.
L‟objectif des expérimentations est triple
- mettre les fibres d‟alfa en solution
- réaliser un filage
- déterminer les propriétés mécaniques et les caractéristiques des fils obtenus par des
essais mécaniques et des études microscopiques
3.4.2. La matière première
Dans les exemples du procédé NMMO décrits dans la littérature [22,26,27], des pâtes
cellulosiques sont utilisées comme matière première. Il s‟agit de pâtes classiques qui sont
souvent achetées chez des entreprises spécialisées. Car le volume minimal à commander
dépasse largement la petite quantité de pâte requise pour les essais lors de la recherche à
Figure 51 : Solubilité de cellulose dans une solution de NMMO en fonction du taux de l‟eau [27]

49
l‟ENSISA, l‟achat n‟est pas une option. Il a alors été décidé de se servir des fibres courtes
produites avec les essais d‟extraction (chapitre 2).
Une exigence pour la matière première est qu‟elle consiste pour la plupart en cellulose. Les
autres composants des plantes comme la lignine, l‟hémicellulose et les pectines limitent et
ralentissent la dissolution de la cellulose dans le NMMO.
Pendant les essais d‟extraction le résultat était souvent des fibres/tiges rigides ou une pâte
de fibres courtes. Un résultat rigide indiquait que l‟extraction n‟était pas complète et que les
fibres contenaient encore des composants non-cellulosiques. Par contre une pâte de fibres
courtes indiquait que l‟extraction était trop forte. Les composants non-cellulosiques n‟étaient
plus présents, mais les fibres étaient déjà dégradées ce qui explique la petite longueur. Donc
ces fibres courtes sont une source idéale pour les essais de dissolution car de cette façon
les exigences sont remplies.
3.4.3. Les premiers essais
Pendant les premiers essais, les taux de composants trouvés dans la littérature sont
respectés. 0.5 g de cellulose d‟alfa et 3 g de NMMO sont mis dans un bécher avec
suffisamment d‟eau et un aimant afin de pouvoir mixer le contenu.
Les propriétés de la plaque chauffante sont une température maximale de 300°C, ce qui
n‟est pas précise et jamais obtenue en réalité dans un bécher, et une vitesse de rotation
maximale de 1100 rpm.
L‟échelle de température de la plaque chauffante n‟est pas détaillée, seulement les
températures de 50°C, 100°C, 150°C, 200°C, 250°C et 300°C sont indiquées. Il est donc
difficile à régler précisément la température à l‟aide de cette échelle. La solution est de
mesurer la température avec un thermomètre. Ainsi, il est constaté que la température réelle
est beaucoup plus basse que la température souhaitée (celle à laquelle la plaque a été
réglée). P.e. pour obtenir une température de 70°C, il faut mettre la plaque à 150°C ou
même 200°C. Une explication possible est que bien que la plaque soit sans aucun doute
chauffée à la température souhaitée, une perte de chaleur considérable se produit lors du
chauffage du bécher en verre borosilicate (Pyrex) et son contenu, et cela à cause des
conductivités de chaleur de verre (1.4 W/mK) et de l‟eau (0.6 W/mK) faibles par rapport au
fer (80 W/mK), à l‟acier (51.9 W/mK), et au cuivre (398 W/mK) [29].
Les essais sont effectués à une vitesse de rotation de 500 rpm et à une température entre
70°C et 90°C. A cause de cette température élevée l‟eau s‟évapore rapidement, donc de
temps en temps un peu d‟eau est ajoutée. De cette façon, la quantité d‟eau est tout le temps
suffisante et il devient facile à mixer la solution de NMMO aqueuse avec la cellulose. Au
début il est important que l‟eau soit présente abondamment pour faire gonfler la pâte
cellulosique. Plus tard l‟eau doit s‟évaporer afin d‟avoir un taux d‟eau assez faible pour
rendre possible la dissolution.
Malgré l‟observation régulière et le contrôle strict de la température et de la quantité d‟eau, la
cellulose ne dissout pas après cinq ou même six heures.
3.4.4. La première solution
Lors d‟un deuxième essai (Figure 52) 1g de NMMO est ajouté après 5h pour compenser la
quantité de NMMO évaporée et dégradée. Cette masse n‟a pas été calculée, donc cela ne
correspond probablement pas à la quantité optimale qu‟il faudra essayer de déterminer en
effectuant plus d‟essais dans le futur. En même temps la vitesse de rotation est augmentée

50
de 500 à 900 rpm. Sans addition d‟eau supplémentaire, une heure plus tard la solution
devient davantage homogène et la couleur devient plus foncée (Figure 53). Puis, tout à
coup, la pâte gonflée se transforme en un gel de couleur ambre foncé et collant et, à
première vue, avec une grande viscosité (Figure 54).
Le refroidissement à température ambiante qui suit, solidifie ce gel, et le réchauffement
postérieur change sa couleur en brun foncé ce qui indique probablement une dégradation
des chaînes cellulosiques à cause de la température trop élevée. Cette dégradation n‟est
évidemment pas souhaitable pour la qualité des fibres qui en seront filées.
La quantité de solution filable étant insuffisante pour un premier filage de filaments d‟alfa,
des « fibres » sont tirées avec une pincette et baignées dans l‟eau afin de les coaguler. Ainsi
les premières « fibres » d‟alfa sont obtenues (Figure 55).
3.4.5. Les solutions suivantes et le filage
Pour les essais de filage, il faut une solution non-dégradée. La dégradation de cellulose est
une oxydation et s‟exprime dans le gel par une couleur brune foncée. Une telle dégradation
est indésirable parce que les propriétés de la matière première détériorent. Comme les
propriétés des filaments sont déterminées principalement par celles de la matière première, il
est évident qu‟une oxydation des chaînes cellulosiques limite les propriétés des filaments.
2g de pâte cellulosique est mise dans un bécher avec 12g de NMMO, suffisamment d‟eau et
un aimant pour le mixage. La pratique semble plus difficile que ce que la théorie décrit, mais
avec 1g supplémentaire de NMMO, une solution est formée après deux fois 6h de mixage
avec une pause de 65h due au week-end intercalé entre ces deux opérations. Pendant cette
Figure 54 : La première solution de cellulose d‟alfa
Figure 52 : Début de la dissolution de fibres cellulosiques dans une solution aqueuse de NMMO
Figure 53 : Changement de couleur une fois que l‟eau s‟évapore
Figure 55 : Les premiers filaments d‟alfa

51
pause, la cellulose reste dans la solution aqueuse de NMMO sans mixage et à une
température ambiante.
La couleur de la nouvelle solution se ressemble à la couleur de caramel, tandis que la
première solution avait plutôt la couleur de chocolat noir. Cette différence de couleur indique
que la cellulose est moins oxydée, une amélioration donc par rapport à la première solution.
Le seul problème est la grande viscosité, la solution ne coule pas. La viscosité n‟est pas
mesurée et un essai de filage est tenté sans filtration préalable.
La machine de filage (Figure 57) est construite pour un filage à sec d‟un seul filament.
Pendant le premier essai de filage d‟alfa, l‟objectif est de trouver la valeur optimale des
paramètres de configuration de la machine plutôt que d‟obtenir un filament.
La solution est mise dans un petit four en haut de la machine de filage. En dessous du four
se trouve une tête de filage avec un seul trou dont le diamètre est 0.8 mm. La température
du four se règle facilement et précisément. Elle est mesurée en haut et en bas du four. Un
piston bougeant de haut en bas presse la matière filable avec une force réglable.
A peu près deux mètres en dessous du four, un cylindre est monté pour bobiner le filament.
En réglant la vitesse du cylindre, un étirage est réalisé en même temps.
Il faut donc déterminer les valeurs optimales de
- la température du four et donc de la matière à filer
- la force de pression du piston
- la vitesse de mouvement latéral du cylindre pour que le filament soit bien bobiné
- la vitesse de rotation du cylindre
Les températures de filage citées dans la littérature se situent entre 100°C et 120°C. Mais
avec le four à 120°C, la viscosité de la solution est encore trop élevée pour une solution avec
un bon écoulement. Même à 140°C la solution n‟est pas filable. En outre la couleur devient
plus foncée ce qui indique que la cellulose dégrade. Une raison possible de la haute
viscosité observée est le fait que le taux de NMMO dans la solution est plus haut que décrit
dans la littérature. Une température de 150°C est dangereuse à cause du risque de
dégradation exothermique du NMMO qui pourrait provoquer une explosion, donc l‟essai est
arrêté. Les valeurs optimales des paramètres de configuration de la machine de filage ne
sont pas trouvées.
Pour rendre possible le filage, il faut donc une solution qui d‟une part coule mieux et d‟autre
part est moins dégradée. La viscosité peut diminuer par l‟addition d‟une quantité de NMMO
plus précise. La couleur peut devenir moins brune et plus transparente par l‟addition d‟un
réducteur ou un antioxydant. L‟antioxydant utilisé dans le laboratoire est le dithionite de
sodium (Na2O4S2).
Au premier instant seulement le problème de la viscosité est considéré. Lors d‟un nouvel
essai, après 7h de mixage à 70°C, une température modérée afin d‟éviter la dégradation de
la cellulose ainsi que la température minimale citée dans la littérature, une solution de 1g de
cellulose d‟alfa et 6g de NMMO est formée (Figure 56). Elle est un peu plus fluide et colle
fortement. Malheureusement, sa couleur est toujours brune, indiquant une certaine
dégradation de la cellulose.
L‟amélioration de l‟écoulement est due au changement de la consistance de la solution.
Celle utilisée pour l‟essai de filage n‟était pas vraiment un gel. Le mixage et le chauffage sont
arrêtés tôt dès que le gel a commencé à se former afin d‟éviter la dégradation de la cellulose
dissoute. De plus l‟aimant ne pouvait pas bien mixer le contenu du bécher et l‟addition d‟1g

52
de NMMO a augmenté la viscosité. Pour ces trois raisons, la solution était incomplète et
mauvaise et ne coulait pas du tout.
En revanche la nouvelle solution s‟approche davantage d‟un gel. Grâce à une quantité
réduite de matière, l‟aimant arrive mieux à mixer le contenu du bécher. En outre la viscosité
du contenu a diminué grâce à la non-addition de NMMO au cours de la dissolution.
Finalement le mixage et le chauffage sont arrêtés plus tard, au moment où un véritable gel
est obtenu. Comme un gel coule toujours plus ou moins et la solution précédente ne coulait
pas, il est clair que l‟écoulement a amélioré.
Pour résoudre le problème de la dégradation et par conséquent de la couleur, un nouvel
essai avec 0.33g de cellulose d‟alfa, 2g de NMMO, suffisamment d‟eau et 1% de réducteur
(basé sur la masse de cellulose) est démarré. Après 7h de mixage à 70°C, une solution
beaucoup plus transparente (Figure 58) est produite. L‟utilisation de quantités de cellulose et
de NMMO tellement petites est due au fait qu‟il s‟agissait du dernier NMMO disponible. Cela
pourrait pourtant influencer l‟interprétation de la couleur obtenue. Il faut donc être très
prudent dans les conclusions.
3.5. Résumé et conclusion
La recherche sur et les essais de dissolution de cellulose d‟alfa ont été commencés plus tard
que ceux d‟extraction de fibres, au moment où l‟extraction avait déjà avancée mais pas
suffisamment. Pour cela la dissolution d‟alfa est devenue intéressante. Comme les procédés
de dissolution sont déjà connus et appliqués dans l‟industrie, beaucoup d‟information est
disponible dans la littérature, quoique la matière première soit une pâte cellulosique à base
de bois. Les fibres d‟alfa très courtes obtenues pendant les essais d‟extraction contiennent
également beaucoup de cellulose et peuvent donc servir comme matière première pour la
dissolution.
Le procédé de viscose est le procédé classique, mais a une longue durée. Le procédé
NMMO est plus court et a également d‟autres avantages, d‟où la décision d‟utiliser le NMMO.
La solution formée a l‟air d‟un gel, mais sa viscosité doit être contrôlée. Si elle est trop
élevée, des hautes températures de l‟ordre de 130°C à 140°C sont nécessaires pour obtenir
un bon écoulement, avec le risque de dégrader la cellulose dissout.
Un contrôle rigoureux de la quantité de NMMO (six fois plus de NMMO que de cellulose)
peut limiter la viscosité, tandis que le contrôle des températures de dissolution (70°C à 90°C)
Figure 56 : Solution sans antioxydant Na2O4S2
Figure 58 : Solution avec 1% d‟antioxydant Na2O4S2
Figure 57 : Machine à filage

53
et de filage (100°C à 120°C) et l‟addition d‟un réducteur (1% de Na2O4S2) permettent d‟éviter
l‟oxydation, c‟est-à-dire la dégradation, de la cellulose.
Malheureusement le NMMO était fini avant qu‟une bonne solution ait été formée dans une
quantité filable. Le filage n‟est donc pas réalisé et des filaments d‟alfa ne sont pas produits.
Par conséquent les propriétés mécaniques comme le module d‟Young, l‟élongation de
rupture et la tension de rupture des filaments d‟alfa ne sont pas encore connues.
Dans le futur il faudra donc essayer de filer le gel de cellulose d‟alfa dissoute et de
déterminer les propriétés mécaniques des filaments produits. A partir de ces propriétés, des
applications pourront être déterminées, soit pour les filaments longs, soit pour les filaments
coupés en fibres de quelques centimètres de longueur.

54
Conclusion
La partie expérimentale, c‟est-à-dire le mode opératoire, de ce stage consiste en deux
grandes parties. Premièrement l‟extraction de fibres d‟alfa longues à partir de tiges est
examinée. Deuxièmement la cellulose d‟alfa est dissoute dans le solvant NMMO afin
d‟obtenir une solution filable.
Globalement le cycle d‟extraction est composé de trois parties : les prétraitements,
l‟extraction elle-même et les post-traitements. Les prétraitements et post-traitements ne sont
pas les étapes les plus importantes, ils sont déjà décrits dans la littérature. Par contre, la
recherche lors de ce stage concernait principalement l‟extraction elle-même, avec l‟objectif
de trouver un moyen de produire des fibres. Après beaucoup d‟essais dans le laboratoire, les
meilleures fibres sont obtenues avec un traitement de soude, suivi d‟un traitement
enzymatique avec des pectines et terminé par un deuxième traitement de soude. Ces fibres
ont une longueur d‟environ cinq centimètres, mais sont encore un peu rigides à cause de la
présence de lignine et de pectines résiduelles, comme les pics des spectres IRTF-RTA et les
photos MEB montrent.
L‟objectif, qui est bien la production de fibres ayant une telle longueur, mais contenant moins
de composants non-cellulosiques afin d‟être moins rigides, n‟est pas réalisé. Néanmoins une
grande évolution et amélioration ont été réalisées en quatre mois.
La dissolution de cellulose d‟alfa est réalisée après que les essais d‟extraction. Le procédé
est bien décrit dans la littérature [18,19,20]. Le NMMO est choisi comme solvant grâce à ses
avantages par rapport au procédé de viscose classique. La couleur de la solution est un
indicateur important du degré de dégradation de la cellulose. Plus la couleur est pâle et
transparente, moins la cellulose est dégradée. L‟addition d‟un réducteur, c‟est-à-dire un
antioxydant, permet de limiter la dégradation.
La suite de ce travail et cette recherche pourrait être bien envisagée par la dissolution des
produits d‟extraction dans un solvant type NMMO et pourrait réaliser un filage à partir d‟une
solution concentrée.

55
Appendice A : Appareils et outils dans le laboratoire
A.1. Les plaques chauffantes
Les plaques chauffantes possèdent également un système magnétique de manière qu‟un
échauffement et un mixage sont possibles en même temps. La température de la plaque
peut varier entre 0°C et 300°C et la vitesse de rotation entre 0 rpm et 1100 rpm (Figure 59).
Comme chaque intervalle de température est 50°C, l‟échelle n‟est pas du tout précise. De
plus la largeur de chaque intervalle n‟est pas identique, donc l‟échelle n‟est pas linéaire, d‟où
les difficultés à régler la température. Pour ces raisons, il vaut mieux mesurer la température
du contenu du bécher chauffé sur la plaque avec un thermomètre.
A.2. Les balances
Les trois balances ne sont pas digitales, mais doivent être manipulées manuellement (Figure
60). La capacité maximale de deux balances est 200g et 1000g pour une troisième.
Seulement les balances jusqu‟à 200g sont utilisées parce qu‟elles sont plus précises. Elles
ont été fabriquées en 1955 et ont donc déjà 55 ans. Les balances sont très sensibles tant au
vent si elles ne sont pas bien fermées, qu‟à des vibrations causées par des passants et des
objets lourds posés brusquement sur la table où se trouvent les balances.
Comme visible dans la Figure 61, le bouton à gauche en haut peut être positionné à 0g ou à
100g. La position 0g est utilisée pour des masses entre 0g et 100g, tandis que la position
100g est utilisée pour des masses entre 100g et 200g. Le bouton à gauche en bas a un
intervalle de 10g. A droite en haut l‟intervalle est 1g et en bas 0.1g. Pour peser un objet, il
faut commencer par régler les intervalles les plus grands, donc d‟abord le 0/100g, ensuite le
bouton de 0/10/20g, etc.
La pesée d‟un objet se fait en ordre croissant de précision des quatre boutons. D‟abord la
centaine de la masse est déterminée avec le bouton 0/100g. Puis la dizaine de la masse est
déterminée avec le bouton de 0/10/20g, etc, et ensuite l‟unité avec le bouton 0/1/2g, etc, et
enfin le dixième avec le bouton 0/0.1/0.2g, etc.
Après avoir réglé tous les boutons, la masse est calculée comme la somme des quatre
valeurs indiquées. Ce total à la forme α.β g, un montant avec un décimal.
Figure 59 : Photo frontale d‟une plaque chauffante dans le laboratoire

56
Au milieu de la balance se trouvent deux échelles, allant de 0 jusqu‟à 100 à gauche et de 0
jusqu‟à 10 à droite. Normalement elles convergent en un point et la concaténation des
nombres de chaque échelle en ce point donne les trois décimaux suivants de la masse. P.e.
si le γδ à gauche et le ε à droite convergent, la masse totale est α.βγδε g.
A cause de toutes ces manipulations nécessaires, la durée de pesée est donc plus longue
par rapport aux balances digitales qui indiquent immédiatement la masse totale.
Un autre inconvénient de ces vieilles balances est qu‟il faut peser séparément le contenant
(le bécher) et son contenu (une quantité d‟un produit chimique). Il faut d‟abord peser la
masse du bécher. Ensuite, la masse totale à obtenir est la somme de la quantité de produit
voulue et la masse du bécher. Il faut donc peser deux fois. En revanche, les balances
modernes ont un bouton « tare ». Le bécher est placé sur la balance, et en pressant le
bouton « tare » la masse montrée est remise à 0g. De cette façon il faut peser uniquement la
masse du produit chimique nécessaire.
A.3. La machine à teinture
Comme les balances, la machine à teinture est vieille. Elle offre une seule vitesse de rotation
des biberons. Il est difficile à régler la température. Un écran avec deux aiguilles et une
échelle de température est utilisé pour le réglage (Figure 62). L‟aiguille rouge indique la
température demandée et l‟aiguille noire indique la température « réelle ». Il est possible que
la température réelle soit 20°C plus élevée que celle demandée, ce qui confirme le réglage
imprécis (Figure 63) mais est inacceptable. La température de l‟huile dans la machine peut
être mesurée exactement avec un thermomètre, et elle est plus proche de celle indiquée par
l‟aiguille rouge que celle indiquée par l‟aiguille noire.
Pour ces raisons, chaque variation de température souhaitée nécessite un nouveau réglage
de la machine. Les fibres obtenues ne sont pas dégradées à la température maximale de la
machine (140°C), d‟où le choix de continuer à travailler à cette température. Par conséquent,
cela permet d‟éviter les réglages supplémentaires.
Figure 60 : Une des vieilles balances dans le laboratoire
Figure 61 : Détail du système pour peser avec les quatre boutons et les deux échelles au milieu

57
A.4. Le pH-mètre
Un pH-mètre (Figure 64) est un appareil qui mesure la valeur du pH de solutions et de
liquides. La valeur du pH est mesurée à l‟aide d‟une sonde qui est composée d‟une électrode
de verre et d‟une électrode de référence qui se trouve dans une solution à l‟intérieur de la
sonde. L‟électrode de référence a un potentiel constant et connu et donc la valeur du pH de
sa solution est connue (pHR). Une différence de potentiel électrochimique entre les deux
électrodes est créée quand la sonde est mise dans une solution dont la valeur du pH doit
être mesurée.
La valeur du pH de la solution à analyser est calculée à base de la relation entre la valeur du
pH et la différence de potentiel électrochimique donnée par l‟équation 1. Les lettres a et b
sont des caractéristiques du pH-mètre et sont données par le fournisseur. La machine elle-
même fait les calculs et montre la valeur cherchée (pHI).
(1)
Il est nécessaire d‟étalonner le pH-mètre avec des solutions avec une valeur du pH connue.
Ces solutions de calibrage, également appelées des solutions tampons, doivent être les plus
récentes possible parce que leur valeur peut changer dans le temps. La composition des
solutions tampons est soit un acide faible (représentation schématique : AH) et son anion
(A-), soit une base faible (BOH) et son cation (B+). Dans le laboratoire les trois solutions
tampons les plus récentes possèdent une valeur du pH de 4 (acide), 7 (neutre) et 10
(basique), qui sont les trois valeurs d‟étalonnage classiques (Figure 65).
Figure 63 : Exemple d‟une température « réelle » plus élevée que la température choisie
Figure 62 : Réglage de la température
Figure 64 : pH-mètre avec la sonde à droite Figure 65 : Solutions tampons

58
A.5. L‟IRTF (angl. FTIR)
Le spectre infrarouge (IR) est la partie du spectre électromagnétique ayant une longueur
d‟onde (λ) entre 0.78 µm et 1000 µm, ce qui correspond à un nombre d‟onde (k) entre
10 cm-1 et env. 12820 cm-1. Ce spectre est divisé en trois parties
- l‟IR lointain avec λ entre 3 µm et 1000 µm et k entre 10 cm-1 et 3333.33 cm-1
- l‟IR moyen avec λ entre 1.4 µm et 3 µm et k entre 3333.33 cm-1 et 7142 cm-1
- l‟IR proche avec λ entre 0.78 µm et 1.4 µm et k entre 7142 cm-1 et 12820 cm-1
Le nombre d‟onde pendant les mesures varie entre 600 et 4000 cm-1, donc la longueur
d‟onde varie entre environ 2.5 µm et 16.67 µm.
La spectroscopie IR est un outil pratique et puissant pour déterminer la composition et les
composants d‟un échantillon. Le spectre obtenu donne l‟intensité d‟un signal ou l‟absorption
d‟énergie en fonction du nombre d‟onde (k). Chaque pic d‟intensité correspond à une
longueur d‟onde spécifique et à une liaison chimique particulière. L‟identification des pics est
faite avec des logiciels et des livres. Dans ces logiciels se trouvent également des spectres
de référence des polymères et des fibres naturelles qui permettent d‟identifier la matière
analysée.
IRTF est l‟abréviation de la spectroscopie « infrarouge à transformée de Fourier ». Il s‟agit
d‟une forme spéciale de la spectroscopie IR. La spectroscopie IR mesure la quantité
d‟énergie absorbée en fonction de la longueur d‟onde. Par contre, l‟IRTF utilise un signal qui
traverse l‟échantillon. Le signal final subit une transformation de Fourier et le spectre est
obtenu.
L‟avantage le plus important de l‟IRTF par rapport à l‟IR est que l‟information de chaque
nombre d‟onde est collectée en même temps. Ceci permet d‟obtenir plus rapidement un
spectre en répétant la mesure plusieurs fois (50-100 fois) jusqu‟au moment où le spectre
moyen ne change plus. Pour cette raison la plupart des mesures d‟IR sont faites avec des
appareils d‟IRTF.
L‟appareil à l‟ENSISA est un FTIR-ATR (angl. Fourier Transform IR spectroscopy-Attenuated
Total Reflection). Ce principe de fonctionnement est un peu différent de celui d‟un FTIR
conventionnel parce que le rayon IR ne traverse pas l‟échantillon, mais est réfléchi. La
représentation schématique du fonctionnement est montrée dans la Figure 66. Le principe
d‟une réflexion totale et atténuée est basé sur les changements dans un faisceau IR à cause
d‟une partie du faisceau qui entre et quitte l‟échantillon et qui influence ainsi le faisceau total
qui est réfléchi à l‟intérieur d‟un cristal dense. Un bon contact entre l‟échantillon et le cristal
est donc nécessaire pour un bon spectre. Quand le contact est mauvais, les changements
dans le rayon IR ne sont pas précis.
Figure 66 : Schéma du fonctionnement de FTIR-ATR avec plusieurs réflexions internes [30]

59
A.6. Le MEB (angl. SEM) [24]
Un MEB est un microscope électronique à balayage (angl. scanning electron microscope). Il
se sert d‟un faisceau d‟électrons pour obtenir une image d‟un échantillon.
En général, la longueur d‟onde de la source d‟électrons d‟un microscope est un des
paramètres qui détermine la résolution, c‟est-à-dire la distance minimale où deux points
peuvent être distingués. La plus grande la résolution, la plus petite cette distance minimale.
Un microscope optique utilise des photons, qui ont une longueur d‟onde dans le spectre
visible (380 nm à 780 nm). Sa résolution maximale n‟est pas grande (ordre µm) parce que
l‟énergie cinétique des photons n‟est pas grande. Par contre le MEB utilise des électrons
accélérés qui ont une grande énergie cinétique et donc une petite longueur d‟onde (2), d‟où
une résolution de l‟ordre de nanomètre (nm). Il est facile d‟accélérer des électrons grâce à
leur masse faible (9.11*10-31 kg) par rapport à p.e. la masse de protons (1.67262*10-27 kg) et
de neutrons (1.67492*10-27 kg) [29]. Grâce à cette résolution plus élevée, la magnification
maximale d‟un MEB peut être 250 fois plus grande que celle d‟un microscope optique.
⁄ (1)
Le MEB forme une image à base des interactions entre le faisceau d‟électrons et
l‟échantillon. Le mot « balayage » (angl. scanning) est utilisé car l‟image est obtenue point
par point (6 à 10 nm). Un risque avec le MEB est de détruire l‟échantillon parce que l‟énergie
cinétique des électrons est grande et le faisceau d‟électrons est focalisé à un point de la
surface.
Les échantillons examinés avec un MEB doivent être conductibles, dans le cas contraire une
couche d‟or ultrafine est déposée pour résoudre ce problème.
Le MEB a deux modes de fonctionnement, soit une image topographique (le contraste en
fonction du relief), soit une image de composition (le contraste en fonction du numéro
atomique des composants) est produite.

60
Appendice B : Résumé obligatoire en néerlandais
1. Literatuurstudie over espartogras
1.1. Inleiding
Natuurlijke vezels worden traditioneel opgedeeld in drie categorieën: plantaardige, dierlijke
en minerale vezels. Espartogras, ook wel alfagras of halfagras genoemd, is een plant en dus
zijn de corresponderende vezels uiteraard plantaardige vezels. Dit type vezels wordt
ingedeeld naar de oorsprong van de vezels [1]:
- zaad- en vruchtvezels als katoen en kokos
- bladvezels als sisal, abaca, agave en espartogras
- stengelvezels als linnen, jute, hennep en ramie
1.2. Cellulose, hoofdbestanddeel van plantaardige vezels [1,2,3]
Cellulose, het hoofdbestanddeel van plantaardige vezels, is een polymeer met als
brutoformule (C6H10O5)n. Het wordt gevormd door de polycondensatie van het dimeer
cellobiose, dat op zijn beurt gemaakt wordt door een 1-4 glucosidische binding van glucose
(C6H1206).
Cellulose kan ook chemische reacties ondergaan, zoals
- zure hydrolyse: een degradatie door reactie met water in een zuur milieu (pH<7) met
de vorming van hydrocellulose
- verestering: de hydroxidegroepen (-OH) reageren met een zuur
- oxidatie: zuurstof reageert met cellulose en vormt zo oxycellulose
1.3. Espartogras
1.3.1. Geografie en structuur
De Latijnse naam van espartogras is Stipa Tenacissima L., de Engelse Esparto grass en de
Franse alfa. De plant is een grassoort die voorkomt in het Middellandse zeegebied [5].
Dankzij een externe waslaag die het verdampen van water in de plant beperkt, is
espartogras zeer goed bestand tegen droogte. De bladeren zien eruit als stengels, maar
alhoewel ze daarvoor te fijn en niet vertakt genoeg zijn, wordt toch de term “stengel” gebruikt
in plaats van “blad”. Espartogras heeft het uitzicht van een verzameling stengels. Deze
kunnen tot een meter lang worden. Doordat de wortels diep en sterk vertakt zijn, verankert
de plant de grond. Dit zorgt ervoor dat de expansie van de woestijn wordt afgeremd. De plant
heeft weinig water en geen insecticiden, pesticiden noch meststoffen nodig, wat een
ecologisch voordeel is in vergelijking met de katoenteelt.
De interne structuur van espartogras is vrij complex. De elementaire bouwsteen zijn
microfibrillen of cellulosefilamenten die compact gebonden worden door hemicellulose en op
die manier vezels vormen [6]. De vezels hebben een onregelmatige dwarsdoorsnede met
een gemiddelde diameter van 50 µm (Figuur 1). Ze worden door lignine en pectines aan
elkaar gebonden tot vezelbundels die eveneens een onregelmatige dwarsdoorsnede hebben
en een gemiddelde diameter van 200 µm (Figuur 2). Het aaneenhechten van de
vezelbundels vormt de stengel (Figuur 3).

61
1.3.2. Chemische samenstelling
Zoals in vorige paragraaf vermeld, bestaan de stengels van espartogras niet enkel uit
cellulose, maar bevatten ze ook bindstoffen als lignine, pectines en hemicellulose en dient
een waslaag als bescherming tegen uitdroging. De chemische samenstelling van de stengels
van espartogras varieert naargelang de bron:
Lignine is het belangrijkste bindmiddel in stengels en planten. Om enkel de cellulose over te
houden, wordt de lignine bij de extractie verwijderd uit de stengels. De exacte structuur van
lignine is niet gekend, maar er wordt verondersteld dat er meerdere varianten bestaan
gebaseerd op één basisstructuur.
Hemicellulose heeft een structuur die goed lijkt op die van cellulose. Het belangrijkste
verschil is dat cellulose een homopolymeer is, terwijl hemicellulose een heteropolymeer is.
De monomeren zijn saccharides waardoor hemicelluloses polysaccharides zijn.
Pectines komen eveneens voor in espartograsstengels. Hun structuur lijkt goed op die van
hemicellulose. De pectinestructuur is echter meer lineair dan die van hemicellulose,
waardoor pectines sterker en kristallijner zijn. Een ander verschil is dat pectines
carboxylgroupen (-COOH) bevatten en hemicelluloses niet.
De wassen zijn vetten die zich aan de buitenkant van de stengels bevinden. Hun chemische
structuur kan erg complex zijn, maar bevat steeds een lange koolstofketen die tot zestig
koolstofatomen kan bevatten [10,13].
1.3.3. Mechanische eigenschappen
Tabel 2 geeft de mechanische eigenschappen weer bepaald door trektesten van espartogras
in vergelijking met andere vezels. De geteste espartograsvezels zijn lange vezels gebruikt
Figuur 1: SEM foto van de dwarsdoorsnede van een vezel [7]
Figuur 2: SEM foto van de dwarsdoorsnede van een vezelbundel [7]
Figuur 3: SEM foto van de dwarsdoorsnede van een stengel [6]
Bestanddeel [8] (%) [6] (%) [7] (%) [9] (%)
Cellulose 43,81 45 45 47,63
Lignine 18,76 23 24 17,71
As 4,66 2 2 5,12
Silica 1,76
Hemicellulose/Pectines 28,4 25 24 22,15
Was 5 5
Rest 2,61 7,39
Som (%) 100 100 100 100
Tabel 1: Samenstelling van espartograsstengels

62
voor het versterken van composieten. Globaal gezien benadert espartogras de
eigenschappen van jute, vlas, hennep en sisal.
Tabel 3 geeft de mechanische eigenschappen van espartograsvezelbundels. De gemiddelde
waarden wijken sterk van elkaar af, wat voornamelijk verklaard kan worden door het feit dat
het gaat om natuurlijke vezels. Figuur 4 geeft de bijhorende trek-rek curve. Ter hoogte van
een spanning van 200 MPa en een rek van 1% is er een plotse daling van de spanning
doordat op dat moment de cellulosefilamenten beginnen te breken.
1.3.4. Huidige toepassingen
Momenteel wordt espartogras onder twee vormen gebruikt. Enerzijds dient de plant in zijn
geheel voor het maken van gebruiksvoorwerpen als tapijten, manden en touwen en als
voedsel voor vee en wilde dieren, zoals de geit en de gazelle. Anderzijds worden via
agressieve extractiemethoden korte vezels van espartogras gemaakt. Deze worden gebruikt
voor het maken van kwaliteitspapier en sigarettenpapier. Verdere toepassingen zijn de
productie van vliesstoffen in combinatie met andere vezels zoals wol en polyester en de
versterking van composieten (op laboratoriumschaal).
Het probleem met natuurlijke vezels als versterking van een synthetische matrix is de
hechting tussen de twee fasen, omdat synthetische vezels typisch eerder hydrofoob zijn en
natuurlijke vezels eerder hydrofiel. De vezels gebruikt in composieten worden dan ook
speciaal behandeld om ze hydrofober te maken zodat de hechting sterker wordt [14].
De extractie en het gebruik van langere espartograsvezels wordt nog niet toegepast en
vormt dan ook het onderwerp van deze thesis.
Tabel 2: Mechanische eigenschappen van de belangrijkste natuurlijke en synthetische vezels. [16]
Tabel 3: Eigenschappen in trek van espartovezelbundels [7] Figuur 4: Bijhorende trek-rek curve [7]

63
2. Extractie van vezels
2.1. Inleiding
Met extractie van vezels wordt het proces, dat uit meerdere stappen kan bestaan, bedoeld
dat een niet mechanisch spinbaar materiaal omzet in een spinbaar materiaal. De extractie
van korte espartograsvezels voor de papier- en vliesstoffenindustrie wordt al toegepast en is
dan ook een goed uitgangspunt voor de extractie van langere vezels [4,7,14]. De extractie
van korte vezels gebeurt doorgaans via agressieve chemische methodes zoals het
Kraftproces [11]. Daarbij worden hoge concentraties natriumhydroxide (NaOH,
gebruiksnaam: natriumloog) en natriumsulfide (Na2S) gebruikt. Enerzijds worden de niet-
cellulose bestanddelen zoals lignine, pectines en hemicellulose gedegradeerd en opgelost,
anderzijds worden ook de celluloseketens gebroken waardoor de vezels korter worden.
In de literatuur worden voor de extractie van korte espartograsvezels concentraties van
1.75M (mol/liter water) tot 3M natriumhydroxide vermeld [12].
2.2. Extractie van espartograsvezels
2.2.1. Eerder onderzoek aan ENSISA
Slechts één document in de literatuur spreekt over langere vezels. Gezien de lengte (25 cm)
betreft het eigenlijk vezelbundels in plaats van vezels [7]. Bij eerder onderzoek aan ENSISA
werden proeven gedaan op basis van de gegevens in de tekst. Stengels van espartogras
werden behandeld met een 3M NaOH-oplossing en vervolgens gebleekt in een 40%
natriumhypochloriet-oplossing (NaOCl, gebruiksnaam: eau de javel, bleekwater), gewassen
in gedestilleerd water en gedroogd gedurende 24u op 60°C. De resultaten waren
allesbehalve goed. Een daling van de concentratie van de NaOH-oplossing tot 2M en 1M
leverde evenmin vezels op.
2.2.2. Inleiding op de extractie
Algemeen kan het volledige proces onderverdeeld worden in drie grote stappen: ten eerste
twee voorbehandelingen, nl. een mechanische kaarding en een onderdompeling in zout
water, vervolgens de extractie zelf, waarbij vertrokken wordt van een NaOH-behandeling. Op
basis van de resultaten wordt tenslotte een bleking toegepast. Aangezien dit geen vezels
oplevert, wordt de bleking vervangen door een enzymatische behandeling. De resultaten zijn
duidelijk beter, maar nog niet optimaal. Daarna wordt een tweede natriumhydroxide-
behandeling toegepast. Deze combinatie levert de beste resultaten op.
Tenslotte volgen op de extractie nog twee nabehandelingen, nl. het drogen en het kaarden
van de vezels.
2.2.3. Voorbehandelingen
De gebruikte espartograsstengels zijn allemaal vrij homogeen van diameter, dit met de
bedoeling vezels te extraheren die zo homogeen mogelijk zijn en een zekere soepelheid,
sterkte en lengte bezitten.
Enkele testen worden uitgevoerd met stengels die al dan niet vooraf mechanisch gekaard
zijn. De mechanisch gekaarde stengels leveren fijnere resultaten op, waardoor beslist wordt
deze voorbehandeling voortaan altijd toe te passen. De kaarde is een metalen kam. Doordat
de stengels tussen de tanden van de kam passeren, worden ze fijner en is hun diameter nog
homogener.

64
Na deze eerste voorbehandeling volgt een onderdompeling in zout water om de was en
onzuiverheden aan het oppervlak te verwijderen. Vroeger werd het espartogras voor een
gelijkaardige behandeling in zeewater ondergedompeld. Dit verklaart de keuze voor zout
water. De zoutconcentratie van de Middellandse zee wordt gesimuleerd met 35 g/l
natriumchloride (NaCl, gebruiksnaam: keukenzout). Om de was sneller op te lossen, wordt
het geheel verwarmd. Testen hebben uitgewezen dat de twee mogelijkheden die in de
literatuur vermeld worden, 24u op 60°C en 12u op 80°C, geen wezenlijk verschil opleveren
(Tabel 4). Praktisch gezien is een tijdspanne van 24u handiger, vandaar de keuze voor 24u
op 60°C. Gedurende de onderdompeling verandert de kleur van het water door de was die
erin oplost, zie Figuur 5 met rechts de beginsituatie en links de eindsituatie. Om dit verschil
in kleur te kunnen tonen, werden voor de foto twee bekers op de plaat gezet.
onderdompeling beginmassa eindmassa massa % massa
12h à 80°C 0,93 g 0,92 g 0,01 g 1,08 %
24h à 60°C 0,89 g 0,88 g 0,01 g 1,12 %
Tabel 4: Massaveranderingen tijdens onderdompeling
2.2.4. Extractie
2.2.4.1. Effect van NaOH: extractie op basis van natriumhydroxide
Het doel van een behandeling met natriumhydroxide is de degradatie en de eliminatie van de
lignine uit de stengels van het espartogras.
In het labo kunnen behandelingen onder atmosferische druk uitgevoerd worden door het
plaatsen van bekers met de NaOH-oplossing en de stengels in een waterbad. Het waterbad
dient om de temperatuur zo homogeen mogelijk te houden. De maximale temperatuur van
deze behandeling is 100°C.
Voor behandelingen tot 140°C wordt de verfmachine gebruikt, waarbij de reactie onder druk
verloopt door het sluiten van de verfbekers. Voor nog hogere temperaturen (>150°C) kunnen
de verfbekers ook rechtstreeks op een verwarmingsplaat geplaatst worden. De inwendige
temperatuur en druk zijn daarbij echter niet meetbaar, waardoor de reactieomstandigheden
onbekend zijn.
Tijdens de behandelingen bestaat een reële kans dat naast lignine ook cellulose
gedegradeerd (geoxideerd) wordt. Dat is niet de bedoeling en de toevoeging van een
reductans kan de degradatie van cellulose beperken of zelfs verhinderen. Testen met en
zonder natriumhydrosulfiet (Na2O4S2, gebruiksnaam: natriumdithioniet) als reductans tonen
aan dat de resultaten duidelijk minder gedegradeerd zijn als Na2O4S2 toegevoegd wordt.
Als mogelijke waarden voor de verschillende parameters wordt geopteerd voor:
- temperatuur: 50°C, 100°C, 130°C-140°C en >150°C (exacte waarde is onbekend)
- concentratie: 0.25M, 0.5M, 1M of 2M
- tijdsduur: 1u of 2u
Dit levert tweeëndertig combinaties op. Uiteraard worden ze niet allemaal uitgetest, maar
worden de temperatuur, concentratie en duur aangepast in functie van het resultaat van de
vorige extractiepoging.
Alle proeven bestaan uit een natriumhydroxidebehandeling van voorbehandelde stengels.
Omdat deze behandeling alleen gele en stijve resultaten oplevert, wordt een bleking met een
9.6% natriumhypochlorietoplossing (NaOCl, gebruiksnaam: eau de javel, bleekwater)
Figuur 5: Kleurverandering tijdens onderdompeling

65
toegepast. De monsters blijken achteraf bleker maar ze zijn nog even stijf aangezien
bleekwater geen lignine of pectines verwijdert.
De eerste extractiepoging met 2M NaOH bij 50°C gedurende 1u levert een stijf resultaat op.
Het verhogen van de temperatuur tot 100°C gedurende 2u blijkt niet te helpen. Vervolgens
worden gedurende 1u verfbekers geplaatst op een verwarmingsplaat bij 1M NaOH, wat korte
en gedegradeerde vezels oplevert. Bij de volgende proef produceert het verlagen van de
concentratie tot 0.25M voor de eerste keer enkele langere en veel korte vezels. De
reproduceerbaarheid is laag want het herhalen van de test geeft telkens pulp. Met een
kortere duur van 50 minuten worden geen vezels bekomen, maar bij 45 minuten, 0.25M
NaOH en een hoge maar onbekende temperatuur worden toch enkele vezels geëxtraheerd.
Ze zijn minder soepel dan degene gevonden na een 1u-durende extractie. Beide types vezel
worden onderzocht met de SEM. De 45 minuten-vezels bevatten veel niet-cellulose
materiaal en de cellulosefilamenten zijn niet duidelijk zichtbaar (Figuur 6). De foto‟s van de
1u-vezels tonen veel “knopen” in de cellulosefilamenten. Dit wijst op een ver gevorderde
degradatie (Figuur 7).
Omdat er een groot verschil is tussen de resultaten van behandelingen op 100°C en meer
dan 150°C, worden ook enkele extractieproeven gedaan op intermediaire temperaturen
(130°C en 140°C) met een 0.25M NaOH-oplossing gedurende 1u. Bij 130°C en 140°C zijn
de resultaten stijf, maar minder stijf dan op 100°C.
Dat de vezels gedegradeerd en kort zijn bij een hoge temperatuur, kan het gevolg zijn van
het feit dat de hemicellulose, die de cellulosefilamenten verbindt, eveneens gedegradeerd
wordt. De stijve monsters bevatten daarentegen nog pectines en soms nog wat lignine.
NaOH verwijdert immers enkel in grote mate de lignine; NaOCl verwijdert helemaal geen
bindmiddelen. Om nog meer bindingsstoffen uit de stengels te extraheren is dus een extra
behandeling nodig. Pectinases, een soort enzymen, zouden het resultaat kunnen verbeteren
doordat ze pectines afbreken.
Aangezien 130°C en 140°C de beste stijve resultaten opleveren, is het logisch om daarmee
verder te werken.
Tabel 5 geeft de belangrijkste reactiecondities en de resultaten van de NaOH-behandeling.
Figuur 6: SEM foto van de stijvere vezels Figuur 7: SEM foto van de soepelere vezels

66
2.2.4.2. Vervanging van de bleking door een enzymatische behandeling
Om verder te bouwen op de resultaten van de NaOH behandeling wordt nu een extra stap
toegevoegd. Om de pectines te elimineren worden pectinases gebruikt. Enzymen,
waaronder ook pectinases, zijn micro-organismen die een specifieke functie uitvoeren en
reageren als een biokatalysator. Ze werken het best onder milde omstandigheden. De hier
gebruikte pectinases zijn afkomstig van Aspergillus aculeatus. Hoewel hun ideale pH-waarde
3.5 is, wordt toch een pH van 7 gekozen om zure hydrolyse van de cellulosefilamenten te
vermijden. De temperatuur is 38°C zoals opgegeven door de producent (Sigma-Aldrich).
De beste resultaten met NaOH, gevonden bij 130°C en 140°C met een 0.25M NaOH-
oplossing gedurende 1u, 90 minuten en 2u, worden nu behandeld met een 5 ml/l
enzymatische oplossing gedurende 5u. Omdat dit gedegradeerde vezels oplevert, wordt de
duur van de behandeling daarna beperkt tot 2u. De monsters zijn na deze behandeling
duidelijk soepeler, maar nog steeds iets te stijf (Figuur 8). Deze verbetering is te danken aan
het feit dat de NaOH de lignine verwijdert en de pectinases de pectines. Dat de vezels nog
steeds te stijf zijn, kan komen doordat er nog lignine en/of pectines aanwezig zijn. Het is
mogelijk dat de lignine zich niet enkel aan de buitenkant van de espartograsstengel bevindt,
maar ook dieper van binnen (onder de pectines) en daardoor onbereikbaar is voor de eerder
toegepaste NaOH-oplossing Door de pectinasebehandeling kan deze lignine nu misschien
wel geëlimineerd worden. In dat geval zou een extra NaOH behandeling betere resultaten
moeten opleveren.
Tabel 6 geeft de belangrijkste reactieomstandigheden en resultaten weer.
NaOH Reductans Bleking Resultaat
conc. temp. duur hoeveelheid NaOCl
2M 50°C 1u X stijf
2M 100°C 2u X stijf
1M > 150°C 1u pulp van korte vezels
0,25M 130°C 1u 1,0 % Ms stijf
0,25M 140°C 1u 1,5 % Ms stijf
0,25M > 150°C 1u 1x vezels, 1x pulp
0,25M > 150°C 50 min 1,0 % Ms 1x pulp, 1x minder pulp
0,25M > 150°C 45 min pulp met enkele korte vezels
Tabel 5 : Belangrijkste omstandigheden en resultaten van een NaOH-behandeling
Tabel 6 : Belangrijkste resultaten van een “NaOH – enzymen” behandeling
NaOH Reductans Enzymen Resultaat
conc. temp. duur hoeveelheid conc. temp. duur pH
0,25M 140°C 2u 0,5 % Ms 5,0 ml/l 38°C 2u 7 stijf
0,25M 140°C 90 min 1,0 % Ms 5,0 ml/l 38°C 5u 7 eerder stijf dan soepel
0,25M 140°C 1u 1,5 % Ms 5,0 ml/l 38°C 5u 7 eerder stijf dan soepel
Figuur 8 : Invloed van de enzymen: links zonder enzymen, rechts met (5u, 38°C, pH 7)

67
2.2.4.3. Toevoeging van een extra NaOH-behandeling
Een tweede NaOH-behandeling wordt toegevoegd als derde extractiestap. Voor de eenvoud
worden dezelfde temperatuur en concentratie genomen als die van de eerste stap, nl. 140°C
en 0.25M. Na 1u levert dit korte vezels op, waarna de concentratie van de pectinases wordt
verlaagd tot 2.5 ml/l. Ook deze vezels zijn stijf. Een 90 minuten durende eerste NaOH-
behandeling levert een erg heterogeen resultaat op, met erg stijve vezels, maar ook met
minder stijve en korte vezels.
Om het resultaat homogener te maken wordt de duur van de pectinasebehandeling verlaagd
tot 1u in plaats van 2u. Om de productie van korte en gedegradeerde vezels te beperken,
wordt meer reductans (1.5% voor de eerste stap, 0.5% voor de derde stap) toegevoegd.
Deze drie stappen leveren uiteindelijk een homogener, reproduceerbaar resultaat op waarbij
vezels van 5 à 6 cm zichtbaar zijn.
De temperatuursinvloed wordt getest door te werken bij 120°C en 130°C met 0.5M NaOH.
De behandeling met 0.25M bij 140°C levert echter meer en soepelere vezels op.
De beste vezels worden dus geproduceerd door een drievoudige behandeling (Figuur 9):
- NaOH: 0.25M op 140°C gedurende 90 minuten met 1.5% reductans
- enzymen: 2.5 ml/l op 38°C gedurende 1u met een pH-waarde van 7
- NaOH: 0.25M op 140°C gedurende 1u met 0.5% reductans
De hoeveelheid reductans wordt in de eerste stap berekend ten opzichte van de massa van
de droge espartograsstengels en in de tweede stap ten opzichte van de natte massa van het
espartogras.
Tabel 7 geeft de belangrijkste reactieomstandigheden en resultaten van de cyclus “NaOH –
enzymen – NaOH” weer.
Tabel 7 : Opsomming van de belangrijkste resultaten van de cyclus “NaOH – enzymen – NaOH”
Figuur 9: Het beste resultaat, duidelijk heterogeen
NaOH Reductans Enzymen NaOH Reductans Resultaat
conc. temp. duur hoeveelheid conc. temp. duur pH conc. temp. duur hoeveelheid
0,50M 120°C 90' 1,5 % Ms 2,5 ml/l 38°C 1u 7 0,50M 130°C 1u 0,50 % Mh stijf
0,50M 130°C 90' 1,5 % Ms 2,5 ml/l 38°C 2u 7 0,50M 130°C 1u 0,50 % Mh heterogeen
0,25M 140°C 2u 1,0 % Ms 2,5 ml/l 38°C 1u 7 0,25M 140°C 90' 0,25 % Mh vooral pulp
0,25M 140°C 90' 1,5 % Ms 2,5 ml/l 38°C 1u 7 0,25M 140°C 1u 0,50 % Mh beste resultaat
0,25M 140°C 90' 1,5 % Ms 2,5 ml/l 38°C 1u 7 0,25M 140°C 30' 0,50 % Mh iets stijver resultaat
0,25M 140°C 1u 1,0 % Ms 2,5 ml/l 38°C 2u 7 0,25M 140°C 45' 0,25 % Mh stijf met enkele vezels

68
2.2.5. Nabehandelingen
De vezels worden na extractie gedroogd op 50°C à 60°C door ze uit te spreiden ofwel op
aluminiumfolie ofwel in een siliconen bakvorm voor cakes. Doordat beide manieren van
drogen niet ideaal zijn, gaan de vezels aan elkaar kleven. In deze toestand zijn ze niet te
spinnen, dus is een scheiding via een kaarding nodig. Dit kan manueel, wat tijdrovend is
maar de beste vezels oplevert (Figuur 10), of met een borstel, wat sneller gaat maar minder
zuivere vezels oplevert (Figuur 11).
De te stijve vezels zouden eventueel nog een enzymatische behandeling en zelfs een derde
NaOH-behandeling kunnen ondergaan. De kortere vezels (lengte 1 cm) zouden gebruikt
kunnen worden in vermengingen met synthetische of natuurlijke vezels zoals polyester, wol
of katoen. De korte vezels (lengte <1 cm) bevatten hoofdzakelijk cellulose en zijn bruikbaar
in pulp voor papier, non-wovens of in oplossingen (zie paragraaf 3). Op die manier wordt een
maximum aan vezelmateriaal gerecycleerd en hergebruikt.
2.3. Eigenschappen van de vezels
2.3.1. SEM (Scanning electron microscope)
De beste vezels, geëxtraheerd via de cyclus “NaOH – enzymen – NaOH”, worden
bestudeerd met de SEM. De foto‟s (Figuur 12) tonen enerzijds aan dat de vezels niet enkel
bestaan uit parallel geschikte cellulosefilamenten, maar dat er ook filamenten uitsteken en
dat er nog niet-cellulose materiaal aanwezig is. Dat laatste zou de stijfheid van de vezels
kunnen verklaren. Anderzijds blijkt uit de foto‟s dat de cellulosefilamenten niet gedegradeerd
zijn, wat uiteraard positief is.
Er zijn ook foto‟s genomen van de kortere en fijnere vezels (Figuur 13). Zij bevatten duidelijk
minder niet-cellulose materiaal. De filamenten steken niet uit en zijn niet gedegradeerd.
Het streefdoel is dus om de “zuivere” structuur van de korte vezels te bereiken bij de
lange(re) vezels.
Nochtans is het misschien mogelijk om de vezels te spinnen tot een garen. Er bestaan
gespecialiseerde firma‟s die een draad kunnen spinnen met 50g vezels. Wegens tijdsgebrek
bij de firma waarmee ENSISA samenwerkt, kan het spinnen ten vroegste midden juli
plaatsvinden.
Figuur 10: Manuele scheiding Figuur 11: Scheiding met een borstel

69
2.3.2. FTIR-ATR (Fourier Transform Infr a Red spectroscopy – Attenuated Total
Reflection)
IR spectroscopie is een krachtige methode om de bindingen in een materiaal en zelfs de
componenten in een heterogeen materiaal te bepalen. Aangezien elke binding een
specifieke eigenfrequentie, en dus ook een energie, een golflengte en een golfgetal (k) heeft,
kunnen de pieken in het IR-spectrum geïdentificeerd worden. Helaas is IR spectroscopie een
kwalitatieve en geen kwantitatieve methode, waardoor het onmogelijk is de hoeveelheid van
elke component in een heterogeen monster te bepalen. Enkel de aan- of afwezigheid van
een component kan vastgesteld worden al naargelang de corresponderende piek in het
spectrum wel dan niet zichtbaar is.
Figuur 14 toont de spectra van de espartograsstengels (blauw) en -vezels (rood). De pieken
bij golfgetallen hoger dan 3000 cm-1 zijn –OH groepen. De enorme stijging bij de vezels kan
te wijten zijn aan de aanwezigheid van NaOH ten gevolge van een slechte spoeling van de
vezels na extractie. Tussen 3000 cm-1 en 2800 cm-1 en tussen 1450 cm-1 en 1000 cm-1
bevinden zich de cellulosepieken. Bij 1650 cm-1, tussen 1510 cm-1 en 1450 cm-1 en in de
buurt van 700 cm-1 situeren zich de ligninepieken. De pectines zijn terug te vinden bij 1730
cm-1. De aanwezigheid van de pieken van lignine en pectines in het vezelspectrum wijst erop
dat er nog lignine en pectines aanwezig zijn. Dit is een bevestiging van de gelijkaardige
vaststelling aan de hand van de SEM foto‟s.
2.3.3. Trektesten
Om de mechanische eigenschappen van de langere vezels vast te stellen, worden trektesten
uitgevoerd. Normaal zijn voor natuurlijke vezels minstens 250 testen nodig om betrouwbare
Figuur 12: SEM foto van de langere en stijvere vezels
Figuur 13: SEM foto van de kortere en soepelere vezels
0
0,05
0,1
0,15
0,2
0,25
0,3
5509501350175021502550295033503750
Intensiteit
k (1/cm)
Figuur 14 : Superpositie van de FTIR-ATR spectra van espartograsstengels (blauw) en de langere vezels na extractie (rood)

70
waarden te verkrijgen. Het aantal uitgevoerde testen is hier echter minimaal, zodat het
moeilijk is betrouwbare conclusies te trekken.
De diameter van de vezels is onbekend en wordt aan de hand van de SEM foto (Figuur 12)
geschat op 135 µm. De kracht en het lengteverschil worden gemeten en omgezet in een
spanning en verlenging door te delen door resp. de dwarsdoorsnede en de initiële lengte.
Doordat de dwarsdoorsnede een geschatte waarde is, is de spanning dat eveneens. De
initiële lengte is de inklemlengte en bedraagt 40 mm.
De elasticiteitsmodulus is een kenmerk van het elastisch domein en is gelijk aan de helling in
dat domein bij de curven die de spanning weergeven in functie van de verlenging. Het betreft
hier eveneens geschatte waarden.
Figuur 15 geeft de modelcurve weer van de trektest uitgevoerd op de espartograsvezels. Als
gevolg van het breken van enkele cellulosefilamenten is een plotse daling van de spanning
te zien. Tabel 8 geeft de belangrijkste kenmerken weer voor de spanning (σ), de verlenging
(Δ L) en de elasticiteitsmodulus (E).
3. Oplossen van espartocellulose
Een andere mogelijkheid om espartograsvezels te produceren is het spinnen uit oplossing.
Hierbij wordt eerst cellulose opgelost in een solvent, waarna een droog- of natspinproces en
een strekking van de filamenten volgen. Door de bekomen filamenten op het einde van het
proces in stukken te snijden kunnen vezels van elke gewenste lengte geproduceerd worden.
Het oplossen van cellulosepulp op basis van hout is al veel gebruikt in de industrie, bv. voor
de productie van viscose. Viscose is geregenereerde cellulose. Dit houdt in dat de originele
cellulose wordt omgezet tot een ander product, gesponnen en daarna terug omgezet wordt
in zijn originele cellulosestructuur.
Het klassieke viscoseproces is vrij complex [26,27]. De cellulose wordt omgezet tot
cellulosexanthaat door reactie met NaOH en CS2. Het cellulosexanthaat wordt gesponnen en
daarna terug omgezet tot cellulose. Door de vele stappen en de noodzaak tot het rijpen van
de oplossing duurt het proces al gauw 40u. Bovendien worden daarbij toxische
nevenproducten gevormd (CS2, H2S) en is het proces, door de grote consummatie van
chemicaliën, milieuonvriendelijk.
parameter σ (MPa) Δ L (%) E (GPa)
gemiddelde 63,83 3,12 2,05
standaardafwijking 16,80 0,63 0,77
variatiecoëfficiënt 26,31 20,12 37,55
Tabel 8 : Mechanische eigenschappen van de espartograsvezels
0
0,5
1
1,5
0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6
Kra
cht
(N)
Lengteverschil (mm)
Figuur 15 : Modelcurve van de trektest op de langere espartograsvezels

71
Een oplossing werd ontwikkeld onder de naam “Lyocell” [26,27,28]. De Lyocellvezels, ook
Newcell en Tencel genoemd naargelang de producent, worden geproduceerd door een
eenvoudiger, sneller (hooguit 8u) en milieuvriendelijker (geen toxische nevenproducten)
proces op basis van het solvent N-methylmorfoline-N-oxide (NMMO). Door de aanwezigheid
van een atoom zuurstof kan de NMMO waterstofbindingen aangaan met zowel het water
waarin het opgelost wordt als met de cellulose, die opgelost wordt. Dit is een niet toxisch
solvent dat na het spinnen gerecupereerd, gezuiverd en hergebruikt kan worden via een
gesloten circuit. Doordat een kleine fractie NMMO degradeert, moet systematisch elke keer
een beetje NMMO toegevoegd worden.
De eerste etappe in het Lyocellproces is het oplossen van cellulosepulp in een waterige
NMMO oplossing. De tweede stap is het spinnen zelf. Als cellulosepulp wordt in de industrie
houtpulp gebruikt, maar het is evident dat net zo goed cellulose van espartogras gebruikt kan
worden. De kortste vezels bij de extractiepogingen bevatten bijna uitsluitend cellulose, en
zijn bijgevolg heel geschikt. Het oplossen gebeurt bij roeren op 70°C tot 100°C.
In het begin wordt veel water toegevoegd opdat de NMMO erin zou oplossen, om het roeren
te vergemakkelijken en de cellulose te laten zwellen. Onderzoek heeft echter aangetoond dat
de hoeveelheid water in de oplossing maximaal 17% mag bedragen opdat de cellulose zou
oplossen. Als de hoeveelheid water meer dan 17% van de totale massa bedraagt, gaat de
NMMO waterstofbindingen aan met het water i.p.v. met de cellulose, waardoor de oplossing
zwelt. Tijdens het oplossen wordt dan ook het teveel aan water verdampt, en na een vijftal
uren wordt een eerste oplossing gevormd (Figuur 16). 1g extra NMMO werd toegevoegd om
de gedegradeerde hoeveelheid te compenseren. De kleur is donkerbruin, terwijl de oplossing
eerder transparant zou moeten zijn. De donkere kleur kan het gevolg zijn van de ongewenste
degradatie van de cellulosevezels. Bij een lagere temperatuur van iets meer dan 70°C
ontstaat een nieuwe, karamelkleurige oplossing. De viscositeit is erg hoog en de oplossing
vloeit niet. Toch wordt een eerste spinpoging ondernomen, maar zelfs bij 140°C blijkt de
oplossing niet spinbaar. Hogere spintemperaturen worden niet uitgeprobeerd wegens het
gevaar op een exotherme ontbinding van de NMMO vanaf 150°C.
Om de viscositeit te beperken wordt de hoeveelheid NMMO preciezer afgewogen. Omdat de
hoeveelheid NMMO die degradeert minimaal is bij de kleine hoeveelheid cellulose die
gebruikt worden (1g tot 2g), wordt ook geen extra NMMO toegevoegd tijdens het oplossen.
De nieuwe oplossing heeft effectief een iets lagere viscositeit, maar is nog steeds bruin
(Figuur 17). Om de degradatie van de cellulose in de oplossing tegen te gaan wordt 1%
reductans (Na2O4S2, gebaseerd op de massa cellulose) toegevoegd tijdens het oplossen. De
kleur van de nieuwe oplossing is veel bleker en transparanter (Figuur 18). De hoeveelheid is
echter te weinig om te kunnen spinnen. Wegens gebrek aan tijd en product werd er geen
extra oplossing gemaakt en zijn dus geen filamenten gesponnen.
Figuur 16 : De eerste oplossing Figuur 17 : Zonder reductans Figuur 18 : Met reductans

72
Appendice C : Liste des figures
Les figures dans le « Extended abstract »
Figure III.1: SEM picture of the most acceptable fibres ......................................................................... VI
Figure III.2: FTIR-ATR spectrum of the fibres ........................................................................................ VI
Les figures dans le texte en français Figure 1 : Une molécule de β-glucose avec la numérotation des atomes de carbone ............................ 5
Figure 2 : Une molécule de cellobiose ..................................................................................................... 5
Figure 3 : La cellulose .............................................................................................................................. 5
Figure 4 : L‟amidon .................................................................................................................................. 5
Figure 5 : A gauche la hydrocellulose, à droite l‟aldéhyde obtenu par tautomérie .................................. 6
Figure 6 : L‟oxycellulose réduite .............................................................................................................. 7
Figure 7 : L‟oxycellulose acide (C2 ou C3) .............................................................................................. 7
Figure 8 : L‟oxycellulose acide (C6) ......................................................................................................... 7
Figure 9 : Représentation de la plante alfa .............................................................................................. 8
Figure 10 : Photo de la plante alfa avec indication des parties principales ............................................. 8
Figure 11 : Image microscope optique des fibres composées de filaments cellulosiques [6] ................. 9
Figure 12 : Image MEB de la coupe transversale des fibres [7] .............................................................. 9
Figure 13 : Image MEB de la coupe transversale des faisceaux de fibres d‟alfa [7] ............................... 9
Figure 14 : Image MEB de la coupe transversale de la tige d‟alfa [6] ..................................................... 9
Figure 15 : Structure de base de lignine ................................................................................................ 10
Figure 16 : Structures possibles de lignine [12] ..................................................................................... 10
Figure 17 : La courbe de traction sur des faisceaux de fibres d‟alfa [7] ................................................ 12
Figure 18 : à droite le début du trempage, à gauche le trempage après quelques heures ................... 19
Figure 19 : image MEB des fibres rigides .............................................................................................. 22
Figure 20 : image MEB des filaments cellulosiques des fibres rigides .................................................. 22
Figure 21 : image MEB des composants non-cellulosiques des fibres rigides ...................................... 22
Figure 22 : image MEB des fibres souples ............................................................................................ 22
Figure 23 : image MEB de la dégradation des filaments cellulosiques ................................................. 22
Figure 24 : image MEB des filaments cellulosiques non-dégradés ....................................................... 22
Figure 25 : Influence de la température : de gauche à droite : 95°C, 130°C, 140°C et plus de 150°C. 23
Figure 26 : Influence des enzymes : à gauche sans enzymes, à droite avec enzymes (5h, 38°C, pH 7).
Le traitement de soude est identique pour les deux échantillons. ......................................................... 25
Figure 27 : Influence de la température et la concentration : à gauche 120°C et 0.5N, à droite 140°C et
0.25N. Les autres paramètres sont identiques. ..................................................................................... 27
Figure 28 : Le meilleur échantillon obtenu, toujours hétérogène ........................................................... 28
Figure 29 : Evolution réalisée au niveau d‟extraction des fibres d‟alfa .................................................. 29
Figure 30 : Fibres après séparation manuelle ....................................................................................... 31
Figure 31 : Fibres après brossage ......................................................................................................... 31
Figure 32 : Image MEB de la tige d‟alfa brute ........................................................................................ 33
Figure 33 : Image MEB de la tige d‟alfa brossée mécaniquement et trempée ...................................... 33
Figure 34 : Image MEB d‟un zoom sur la tige d‟alfa brossée mécaniquement et trempée ................... 33
Figure 35 : Image MEB d‟une des meilleures fibres longues ................................................................ 33
Figure 36 : Image MEB des filaments cellulosiques bien visibles en non-dégradés ............................. 33
Figure 37 : Image MEB de la dégradation à la surface ......................................................................... 33
Figure 38 : Image MEB d‟une des fibres courtes ................................................................................... 33
Figure 39 : Image MEB de la surface contenant peu de composants non-cellulosiques ...................... 33

73
Figure 40 : Superposition des spectres IRTF-RTA d‟alfa brute (bleu) et d‟alfa traitée (rouge) ............. 35
Figure 41 : Comparaison des spectres IRTF-RTA de fibres d‟alfa et de coton ..................................... 37
Figure 42 : Courbe modèle de traction de fibres longues d‟alfa ............................................................ 38
Figure 43 : Représentation schématique du filage par fusion ............................................................... 42
Figure 44 : Représentation schématique du filage à sec ....................................................................... 42
Figure 45 : Représentation schématique du filage à humide ................................................................ 42
Figure 46 : Représentation schématique du procédé viscose [27] ........................................................ 43
Figure 47 : Représentation schématique du procédé Lyocell [27]......................................................... 45
Figure 48 : Comparaison des propriétés mécaniques de la viscose et du NewCell [22] ...................... 46
Figure 49 : Deux produits de dégradation principaux ............................................................................ 47
Figure 50 : Schéma des compositions possibles d‟un mélange de cellulose, de NMMO et d‟eau [27] 47
Figure 51 : Solubilité de cellulose dans une solution de NMMO en fonction du taux de l‟eau [27] ....... 48
Figure 52 : Début de la dissolution de fibres cellulosiques dans une solution aqueuse de NMMO ...... 50
Figure 53 : Changement de couleur une fois que l‟eau s‟évapore ........................................................ 50
Figure 54 : La première solution de cellulose d‟alfa .............................................................................. 50
Figure 55 : Les premiers filaments d‟alfa ............................................................................................... 50
Figure 56 : Solution sans antioxydant Na2O4S2 ..................................................................................... 52
Figure 57 : Machine à filage ................................................................................................................... 52
Figure 58 : Solution avec 1% d‟antioxydant Na2O4S2 ............................................................................ 52
Figure 59 : Photo frontale d‟une plaque chauffante dans le laboratoire ................................................ 55
Figure 60 : Une des vieilles balances dans le laboratoire ..................................................................... 56
Figure 61 : Détail du système pour peser avec les quatre boutons et les deux échelles au milieu ...... 56
Figure 62 : Réglage de la température .................................................................................................. 57
Figure 63 : Exemple d‟une température « réelle » plus élevée que la température choisie .................. 57
Figure 64 : pH-mètre avec la sonde à droite.......................................................................................... 57
Figure 65 : Solutions tampons ............................................................................................................... 57
Figure 66 : Schéma du fonctionnement de FTIR-ATR avec plusieurs réflexions internes [30] ............. 58
Les figures dans le résumé en néerlandais Figuur 1: SEM foto van de dwarsdoorsnede van een vezel [7] ............................................................. 61
Figuur 2: SEM foto van de dwarsdoorsnede van een vezelbundel [7] .................................................. 61
Figuur 3: SEM foto van de dwarsdoorsnede van een stengel [6] .......................................................... 61
Figuur 4: Bijhorende trek-rek curve [7] ................................................................................................... 62
Figuur 5: Kleurverandering tijdens onderdompeling .............................................................................. 64
Figuur 6: SEM foto van de stijvere vezels .............................................................................................. 65
Figuur 7: SEM foto van de soepelere vezels ......................................................................................... 65
Figuur 8 : Invloed van de enzymen: links zonder enzymen, rechts met (5u, 38°C, pH 7) ..................... 66
Figuur 9: Het beste resultaat, duidelijk heterogeen ............................................................................... 67
Figuur 10: Manuele scheiding ................................................................................................................ 68
Figuur 11: Scheiding met een borstel .................................................................................................... 68
Figuur 12: SEM foto van de langere en stijvere vezels ......................................................................... 69
Figuur 13: SEM foto van de kortere en soepelere vezels ...................................................................... 69
Figuur 14 : Superpositie van de FTIR-ATR spectra van espartograsstengels (blauw) en de langere
vezels na extractie (rood) ....................................................................................................................... 69
Figuur 15 : Modelcurve van de trektest op de langere espartograsvezels ............................................ 70
Figuur 16 : De eerste oplossing ............................................................................................................. 71
Figuur 17 : Zonder reductans ................................................................................................................. 71
Figuur 18 : Met reductans ...................................................................................................................... 71

74
Appendice D : Liste des tableaux
Les tableaux dans le « Extended abstract »
Table III. 1: Mechanical properties of Esparto grass fibres .................................................................... VI
Les tableaux dans le texte en français Tableau 1 : La répartition territoriale de la plante alfa [5] ........................................................................ 7
Tableau 2 : La composition d‟alfa ............................................................................................................ 9
Tableau 3 : Les propriétés mécaniques en traction des principales fibres naturelles et synthétiques [7]
............................................................................................................................................................... 11
Tableau 4 : Résultats des essais mécaniques sur des faisceaux de fibres d‟alfa [7] ............................ 12
Tableau 5 : Influence des conditions de trempage ................................................................................ 19
Tableau 6 : Les résultats principaux des traitements de soude des fibres d‟alfa .................................. 23
Tableau 7 : Résultats principaux du cycle « soude – enzymes » .......................................................... 25
Tableau 8 : Valeurs des paramètres pour les deux premières étapes du cycle « soude – enzymes –
soude »................................................................................................................................................... 28
Tableau 9 : Valeurs des paramètres et résultats pour la troisième étape du cycle « soude – enzymes –
soude »................................................................................................................................................... 28
Tableau 10 : Identification des pics les plus importants des spectres IRTF-RTA [7,13,22,23] ............. 36
Tableau 11 : Valeurs des propriétés mécaniques de fibres d‟alfa ......................................................... 38
Tableau 12 : Comparaison des propriétés mécaniques ........................................................................ 38
Les tableaux dans le résumé en néerlandais Tabel 1: Samenstelling van espartograsstengels .................................................................................. 61
Tabel 2: Mechanische eigenschappen van de belangrijkste natuurlijke en synthetische vezels. [16] .. 62
Tabel 3: Eigenschappen in trek van espartovezelbundels [7] ............................................................... 62
Tabel 4: Massaveranderingen tijdens onderdompeling. ........................................................................ 64
Tabel 5 : Belangrijkste omstandigheden en resultaten van een NaOH-behandeling ............................ 66
Tabel 6 : Belangrijkste resultaten van een “NaOH – enzymen” behandeling ........................................ 66
Tabel 7 : Opsomming van de belangrijkste resultaten van de cyclus “NaOH – enzymen – NaOH” ..... 67
Tabel 8 : Mechanische eigenschappen van de espartograsvezels ....................................................... 70

75
Bibliographie
[1] Cook J. Gordon, Handbook of textile fibres, 1. Natural fibres, Fifth edition, reprinted,
Merrow Technical Library, 1993, 170 p.
[2] Kiekens P., cours Vezelmaterialen, 2008.
[3] Peters R.H., Textile Chemistry I: The Chemistry of Fibres, First edition, Elsevier
publishing company, 1963, 477p.
[4] Akchiche O., Messaoud Boureghda K., Esparto Grass (Stipa Tenacissime L.), raw
material of papermaking. First Part, Chimija rastitel‟nogo syr‟ja, 4, 2007, 25-30.
[5] Portail de l‟agriculture marocaine, L‟alfa : Importance écologique et socio-
économique, Terre et Vie, 61-62, 2002, 1-3.
[6] Paiva M.C., Ammar I., Campos A.R., Cheikh R.B., Cunha A.M., Alfa fibres :
Mechanical, morphological and interfacial characterization, Composites Science and
Technology, 67, 2007, 1132-1138.
[7] Brahim S.B., Cheikh R.B., Influence of fibre orientation and volume fraction on the
tensile properties of unidirectional Alfa-polyester composite, Composites Science and
Technology, 67, 2007, 140-147.
[8] Harche M., Bounaga D., Etude comparative du tissu fibreux dans la feuille d‟alfa
« Stipa tenacissima L. », Bull Soc. – Hist. Nat. Alger., 1979, 113-120.
[9] Bouiri B., Amrani M., Production of dissolving grade pulp from Alfa, Bioresources,
5(1), 2009, 291-302.
[10] Peters R.H., Textile Chemistry II: Impurities in Fibres; Purification of Fibres, First
edition, Elsevier publishing company, 1967, 374p.
[11] Chakar F.S., Ragauskas A.J., Review of current and future softwood kraft lignin
process chemistry, Industrial Crops and Products, 20, 2004, 131-141.
[12] Nadji H., Diouf P.N., Benaboura A., Bedard Y., Riedl B., Stevanovic T., Comparative
study of lignins isolated from Alfa grass (Stipa tenacissima L.), Bioresource Technology, 100,
2009, 3585-3592.
[13] Dubis E.N., Dubis A.T., Morzycki J.W., Comparative analysis of plant cuticular waxes
using HATR FT-IR reflection technique, Journal of Molecular Structure, 511-512, 1999, 173-
179.
[14] Bessadok A., Roudesli S., Marais S., Follain N., Lebrun L., Alfa fibres for unsaturated
polyester composites reinforcement: Effects of chemical treatments on mechanical and
permeation properties, Composites, Part A, 40, 2009, 184-195.
[15] Bessadok A., Marais S., Roudesli S., Lixon C., Métayer M., Influence of chemical
modifications on water-sorption and mechanical properties of Agave fibres, Composites, Part
A, 39, 2008, 29-45.
[16] Adolphe D., Cours non-tissés, 2009.
[17] El Abdi L.K., Etude des fibres extraites de la plante Alfa, Rapport de stage de fin
d‟études - ENSISA Mulhouse, 2008, 32 p.
[18] Kiekens P., Cours fysische en chemische textieltechnologie, 2009.
[19] Gübitz G.M., Cavaco-Paulo A., Enzymes go big: surface hydrolysis and
fictionalisation of synthetic polymers, Trends in Biotechnology, 26(1), 2007, 32-38.
[20] Nierstrasz, V.A., Chapter 7: Enzymatic surface modification, In Wei Q, Surface
modification of textiles, Woodhead Publishing Ltd., 2009, 352 p.
[21] Coates J., Interpretation of Infrared Spectra, A Practical Approach, Encyclopedia of
Analytical Chemistry, Ed. R.A. Meyers, John Wiley & Sons, 2000, 10815-10837.

76
[22] Krüger R., Cellulosic filament yarn from the NMMO process, Lenzinger Berichte, 9/94,
1993, 49-52.
[23] Socrates G., IR and Raman Characteristic Group Frequencies, Tables and Charts, 3rd
Edition, John Wiley & Sons, 2001, 347 p.
[24] Drean J.Y., Cours de Matériaux avancés, 2009.
[25] Kiekens P., Cours de Geavanceerde vezels en afgeleide materialen, 2008.
[26] Woodings C. et al., Regenerated cellulose fibres, ed. Calvin Woodings, The Textile
Institute, Woodhead publishing limited, 2001, 329 p.
[27] Lewin M., Handbook of fiber chemistry, CRC Press, Third Edition, 2007, 1044 p.
[28] Rosenau T., Potthast A., Sixta H., Kosma P., The chemistry of side reactions and
byproduct formation in the system NMMO/cellulose (Lyocell process), Prog. Polym. Sci., 26,
2001, 1763-1837.
[29] Callister W.D. Jr., Materials Science and Engineering, an Introduction, Seventh
edition, John Wiley & Sonc, Inc., 2007, 712 p. plus appendixes.
[30] PerkinElmer Life and Analytical Sciences, Shelton CT (USA), FT-IR Spectroscopy
Attenuated Total Reflectance (ATR), Technical note, 2005, 5 p., www.perkinelmer.com.


















