
Violence and Conflict Negotiation: Exploring the Roles of - UMdrive

© Zeinab Ebrahimzadeh, 2019
Exploring the roles of phosphoinositides in the biology of the malaria parasite Plasmodium falciparum
Thèse
Zeinab Ebrahimzadeh
Doctorat en microbiologie-immunologie
Philosophiæ doctor (Ph. D.)
Québec, Canada

Exploring the roles of phosphoinositides in the biology of the malaria parasite Plasmodium falciparum
Thèse
Zeinab Ebrahimzadeh M. Sc.
Dave Richard, directeur de recherche

iii
Résumé
Plasmodium falciparum est un parasite appartenant au phylum Apicomplexa et est à
l’origine de la forme la plus sévère de la malaria. Dans les zones endémiques d'Afrique
subsaharienne, la plupart des victimes sont des enfants de moins de cinq ans. L’entrée de
P. falciparum dans sa cellule cible, le globule rouge, repose sur la sécrétion de protéines
par des organites spécialisés : les micronèmes, les rhoptries et les granules denses. Les
mécanismes de biogenèse de ces organites et la coordination de la libération de leur
contenu lors de l'invasion sont cependant pour la plupart inconnus. Il a été toutefois été
démontré que les protéines destinées à ces organites apicaux se concentrent dans des
microdomaines de l’appareil de Golgi, dont la composition en lipides et en protéines
détermine leur destination finale. À ce jour, les mécanismes de sélection et de transport
des protéines apicales vers les organites d'invasion ainsi que leurs mécanismes de
sécrétion durant l’invasion sont pour la plupart inconnus. Nous avons donc posé
l’hypothèse que les phosphoinositides (PI) et leurs protéines effectrices sont impliqués
dans ces processus chez P. falciparum.
Les PI sont sept lipides phosphorylés retrouvés de façon minoritaire dans les différentes
membranes cellulaires. Chaque membrane subcellulaire contient une espèce
caractéristique de PI qui peut être reconnue et liée spécifiquement par des protéines
effectrices. Une large gamme de processus biologiques sont régulés par les PI, tels le
trafic vésiculaire, les canaux ioniques, les pompes d’efflux et les transporteurs, ainsi que
certains processus endocytiques et exocytaires. Des études antérieures ont été en mesure
de détecter seulement cinq des sept espèces de PI chez P. falciparum. Dans le cadre d’un
premier projet, nous avons étudié la distribution de six PI, à savoir PI3P, PI4P, PI5P, PI
(4,5)P2, PI(3,4)P2 et PI(3,4,5)P3, chez P. falciparum. Pour ce faire, nous avons exprimé
chez le parasite des rapporteurs spécifiques correspondant à des domaines humains de
liaison aux PI, fusionnés à une protéine fluorescente. Cette méthode nous a permis de
confirmer des rapports antérieurs sur la localisation du PI3P dans la membrane de la
vacuole alimentaire, dans de petites vésicules près ou sur la membrane plasmique du
parasite ainsi qu’à l’apicoplaste. De plus, nous avons révélé pour la première fois la

iv
présence de PI5P chez P. falciparum et montré qu’il se localisait à la membrane
plasmique, au noyau et potentiellement dans le réticulum endoplasmique de transition.
Nous avons aussi montré que le PI4P est localisé dans la membrane plasmique ainsi que
dans l’appareil de Golgi et que le PI(4,5)P2 est présent dans la membrane plasmique tout
au long du cycle érythrocytaire. Cette carte de la distribution subcellulaire des PI
constitue un excellent outil pour mieux déchiffrer les rôles de ces lipides chez le parasite
P. falciparum.
Dans le cadre d’un second projet, nous avons caractérisé une protéine possédant un
domaine conservé chez les Apicomplexa, le domain d’homologie de la Pleckstrine, la
protéine PfPH2. En utilisant la stratégie de Knock-sideways pour inactiver
conditionnellement la protéine d’intérêt, nous avons montré que PfPH2 est impliquée
dans l’attachement initial du mérozoite à la surface du globule rouge. Cet effet est
directement lié à un défaut de sécrétion d'une population spécifique de micronèmes en
l’absence de la protéine PfPH2. Enfin, nous avons mis en évidence que le domaine PH
de PfPH2, lorsque exprimé sous forme de protéine recombinante, se lie aux PI avec une
grande spécificité. Pris ensemble, nos résultats démontrent le rôle essentiel des PI dans le
processus d’invasion et proposent un modèle mécanistique pour l'exocytose des
micronèmes.

v
Abstract
Plasmodium falciparum belongs to the phylum of Apicomplexa and causes the most severe
form of malaria. In endemic areas of sub-Saharan Africa, most of the victims are among
children under the age of five. P. falciparum relies on proteins released from sophisticated
invasion organelles called micronemes, rhoptries and dense granules to enter human
erythrocytes. The mechanism of biogenesis of invasion organelles and the coordinated
release of their contents during invasion are mostly unknown. It has been shown that
proteins targeted to the apical organelles accumulate in microdomains of the Golgi
apparatus with specific lipid and protein composition that determine the final destination of
their cargo. To date, the mechanisms of transport of the cargo molecules to the invasion
organelles and their release mechanism are mostly unknown. We proposed that
phosphoinositides (PIPs) and their effector proteins could be involved in these processes in
P. falciparum.
PIPs are seven minor phosphorylated lipids in cellular membranes. Each subcellular
membrane contains a characteristic species of PIPs that are specifically bound by PIP-
interacting proteins. A wide range of biological processes regulated by PIPs such as
vesicular trafficking, ion channels, pumps, and transporters and control both endocytic and
exocytic processes. Based on previous reports five out of seven PIP species have been
detected in P. falciparum. In my first project, we have studied the distribution of six PIPs
namely PI3P, PI4P, PI5P, PI(4,5)P2, PI(3,4)P2 and PI(3,4,5)P3 using expression of specific
reporters made up of human PIP-binding domains fused to a fluorescent protein. Here, we
have confirmed previous reports on PI3P localization to the food vacuole membrane, small
vesicles close/on the parasite plasma membrane and the apicoplast. Also, we have reported
for the first time the presence of PI5P in P. falciparum and showed that it localizes to the
PM, nucleus and potentially transitional ER. PI4P shows localization to the PM and Golgi
and PI(4,5)P2 localizes to the PM all over the erythrocytic cycle. The resulting map of the
subcellular distribution of PIPs will now be a great tool to further decipher the roles of
these lipids in P. falciparum,
In the second project, we have characterized a Pleckstrin Homology domain-containing
protein (PfPH2) conserved in all apicomplexan parasites. Using the knock sideways

vi
strategy to conditionally inactivate the protein, we show that PfPH2 is involved in an early
step of the invasion process, when the merozoites initially attach to red blood cells. We
further demonstrate that this is due to the abrogated secretion of a specific population of
micronemes. Finally, we reveal that recombinantly expressed PfPH2 binds PIPs with a
broad specificity. Taken together, our results present evidence for the role of PI in invasion
and propose a mechanistic model for the exocytosis of micronemes.

vii
Table of contents
Résumé ........................................................................................................................ iii
Abstract ........................................................................................................................ v
Table of contents ....................................................................................................... vii
List of Tables ................................................................................................................ x
List of Figures ............................................................................................................ xi
Abbreviation list ....................................................................................................... xii
Avant-Propos ........................................................................................................... xvi
Acknowledgments ................................................................................................. xvi
Contributions ....................................................................................................... xviii
Introduction ................................................................................................................. 1
Malaria ....................................................................................................................... 1
Etiology and Epidemiology .................................................................................... 1
Disease and Pathology of Plasmodium Infection ................................................... 4
Malaria vector ........................................................................................................ 4
Plasmodium falciparum life cycle ............................................................................. 5
Erythrocytic stage ................................................................................................... 5
Atypical organelles ................................................................................................. 8
Molecular bases of invasion ................................................................................. 10
Malaria Treatment .................................................................................................... 14
Diagnosis, Treatments and Resistance ................................................................. 14
Prevention and Vaccine Development ................................................................. 15
Drug Resistance and Discovery ........................................................................... 18
Phosphoinositides .................................................................................................... 21
PIP-binding proteins ............................................................................................. 23
Phosphoinositide species ...................................................................................... 25
Phosphoinositide metabolism .................................................................................. 31
PI kinases ............................................................................................................. 31
Phosphatases ........................................................................................................ 41
Chapter 1: Hypothesis and problem statement ................................................ 47
1.1 Hypothesis and objectives ............................................................................ 47
Chapter 2: A map of the subcellular distribution of phosphoinositides in the
erythrocytic cycle of the malaria parasite Plasmodium falciparum ...................... 50
Avant-propos ........................................................................................................... 50
Résumé ..................................................................................................................... 51
Article ......................................................................................................................... 52
Abstract .................................................................................................................... 53
Introduction .............................................................................................................. 54
Materials and methods ............................................................................................. 57

viii
Results and Discussion ............................................................................................ 59
Acknowledgments ................................................................................................... 68
References ................................................................................................................ 68
Tables .......................................................................................................................... 78
Figure legends .......................................................................................................... 79
Figures ..................................................................................................................... 82
Supplementary Table ............................................................................................... 86
Supplementary figure legends.................................................................................. 88
Supplementary figures ............................................................................................. 90
Chapter 3: A pan-apicomplexan phosphoinositide-binding protein acts in
malarial invasion-microneme exocytosis. ................................................................ 94
Avant-propos ........................................................................................................... 94
Résumé ..................................................................................................................... 95
Article ......................................................................................................................... 96
Abstract .................................................................................................................... 97
Introduction .............................................................................................................. 98
Results and Discussion .......................................................................................... 100
Conclusion ............................................................................................................. 108
Materials and Methods ........................................................................................... 108
References .............................................................................................................. 118
Competing interest ................................................................................................. 125
Materials and Correspondence ............................................................................... 125
Acknowledgments ................................................................................................. 125
Author contributions .............................................................................................. 125
Data availability ..................................................................................................... 126
Figure legends ........................................................................................................ 127
Expanded View Figure legends ............................................................................. 130
Figures ................................................................................................................... 132
Appendix ................................................................................................................ 141
Appendix figure legends ........................................................................................ 143
General Discussion, Conclusion and Perspectives ................................................ 153
A map of subcellular distribution of phosphoinositides in P. falciparum ............. 155
PI3P distribution ................................................................................................. 155
PI4P distribution ................................................................................................. 156
PI5P distribution ................................................................................................. 157
PI(4,5)P2 distribution ......................................................................................... 157
PI(3,4)P2 and PI(3,4,5)P3 distribution ............................................................... 158
Conclusion on the subcellular PIP distribution and general pitfalls .................. 159
A pan-apicomplexan phosphoinositide-binding protein acts in malarial invasion-
microneme exocytosis. ........................................................................................... 159
PfPH2 is a PH-containing protein with a relaxed PIP-binding specificity. ....... 159
PfPH2 localizes to a structure close to the apical tip of the merozoite .............. 160

ix
PfPH2 is essential for the erythrocytic cycle and its absence affects merozoite
invasion due to a default in microneme exocytosis............................................ 161
Conclusions on PfPH2 mechanism of action and future experiments ............... 164
References ................................................................................................................ 166

x
List of Tables
Table 1 Malaria vaccines in preclinical development or in clinical trial. .......................................................... 17
Table 2 Phosphoinositide kinases and phosphatases in P. falciparum compared to yeast and T. gondii. ..... 37

xi
List of Figures
Figure 1The malaria parasite life cycle. ............................................................................................................ 2
Figure 2Endemic area of malaria from 1900 to 2002. [6] .................................................................................. 3
Figure 3Erythrocytic cycle. ................................................................................................................................ 6
Figure 4Smear of erythrocytic stages under light microscope. ......................................................................... 6
Figure 5Atypical organelles of merozoite. ......................................................................................................... 9
Figure 6Invasion ligands and their receptors involved in the invasion of erythrocyte by Plasmodium falciparum ....................................................................................................................................................... 11
Figure 7An illustration of the seven known PIPs, and the enzymes involved in PI P metabolism. ................. 21
Figure 8A map of the subcellular localization of Pl in higher eukaryotic cells. ................................................ 23
Figure 9 PIP-recognizing effectors. ............................................................................................................... 24
Figure 10Kinases involved in phosphoinositide metabolism in yeast, mammalian cells and apicomplexan parasites. ........................................................................................................................................................ 32
Figure 11The role of the PI-PLC pathway and calcium signaling at different stages of the Plasmodium life cycle and downstream stage-specific effectors. ............................................................................................. 46

xii
Abbreviation list
A Anopheles
ACTs ART-based combination therapies
AMA1 Apical membrane antigen 1
ANTH AP180 N-terminal homology
AP Apicoplast
AP-1 Adaptator protein-1
AP-2 Adaptator protein-2
ARF1 ADP-ribosylationfactor 1
ARM Armadillo
ART Artemisinin
ARTs Artemisinin and its semi-synthetic derivatives
Atg Autophagy related protein
Atg14 Autophagy related protein14
Atg Autophagy related protein
BATS Barkor/Atg14(L) autophagosome targeting sequence
BIP Binding immunoglobulin protein
cKO conditional Knocking-Out
C2 Conserved region-2 of protein kinase C
Ca2+ Calcium
CDPKs Calcium-dependent protein kinases
CDPK1 Calcium-dependent protein kinase 1
CDPK5 Calcium-dependent protein kinase 5
CR1 Complement receptor 1
CyRPA GPI-anchored antigen
D Dense granule
DAG Diacylglycerol
DBL Duffy binding-like domain
DGK1 Diacylglycerol kinase-1
DHA Dihydroartemisinin
DHR-1 Dock homology region-1
DOC2.1 Double C2 domain protein
DOK5 Docking Protein 5
EBAs Erythrocyte binding antigens
EBA-140 Erythrocyte binding antigens 140 kDa
EBA-175 Erythrocyte binding antigens 175 kDa
EBA181 Erythrocyte binding antigens 181 kDa
EBL1 Erythrocyte-binding ligand 1
EE Early endosome
ENTH Epsin N-terminal homology

xiii
ENR Enoyl acyl carrier protein reductase
ER Endoplasmic reticulum
Fab1 Forms aploid and binucleate cells
FAPP1 Four-phosphate-adapter proteins 1
FAPP1 Four-phosphate-adapter proteins 2
FERM 4.1, ezrin, radixin, moiesin
FK506 a drug molecule (Immunosuppressor)
FV Food vauole
FYVE Conserved in Fab1, YOTB, Vac1 and EEA1
GAP45 Glideosome-associated protein 45 kDa
GAP50 Glideosome-associated protein 50 kDa
GlyA Glycophorin A
GlyB Glycophorin B
GlyC Glycophorin C
GOLPH3 Golgi phosphoprotein 3
GPI Glycosylphosphatidylinositol
GSK GlaxoSmithKline
GTP Guanosine triphosphate
heme Hemozoin
HMW High molecular weight
HPLC High-performance liquid chromatography
IMC Inner membrane complex
INPP4A Inositol polyphosphate 4-phosphatases A
INPP4B Inositol polyphosphate 4-phosphatases B
Ins(1,4,5)P3 or IP3 Inositol 1,4,5-trisphosphate
IPP Isopentenyl pyrophosphate
IPZ Imidazopyrazines
iRBC infected Red blood cell
iRBCM infected Red blood cell-membrane
KD Knock-down
KO Knock-out
KS Knocksideways
M Microneme
MSP1 Merozoite surface protein 1
MTM Myotubularin
MVBs Multivesicular bodies
P. Plasmodium
PA Phosphatidic acid
PAS Pre-autophagosomal structure
PDK-1 Phosphoinositide-dependent kinase 1
PDZ Postsynaptic density 95, disk large, zonula occludens

xiv
PCR Polymerase chain reaction
PEXEL Protein export element
PfATG8 P. falciparum autophagy protein 8
PfATG8 P. falciparum autophagy protein 18
PfEMP-1 P. falciparum erythrocyte membrane protein-1
PfFCP P. falciparum FYVE-containing protein
Pfs25 Post-fertilization antigen 25
Pfs230 Post-fertilization antigen 230
PfRH1 P. falciparum reticulocyte-binding like homolog 1
PfRH2a P. falciparum reticulocyte-binding like homolog 2a
PfRH2b P. falciparum reticulocyte-binding like homolog 2b
PfRH4 P. falciparum reticulocyte-binding like homolog 4
PfRH5 P. falciparum reticulocyte-binding like homolog 5
PfRIPR PfRH5 interacting protein
PH Pleckstrin homology domain
PI Phosphoinositides
PiK1 Phosphatidylinositol kinase
PIKs PI kinases
PI3Ks PI3-kinases
PI4Ks PI4-kinases
PI(4)K Phosphatidylinositol-4-OH kinase
PIKfyve Phosphoinositide kinase containing FYVE domain
PI3P Phosphoinositide 3-phosphate
PI4P Phosphoinositide 4-phosphate
PI5P Phosphoinositide 4-phosphate
PI(4,5)P2 Phosphoinositide 4,5-biphosphate
PI(3,5)P2 Phosphoinositide 3,5-biphosphate
PI(3,4)P2 Phosphoinositide 3,4-biphosphate
PI(3,4)P2 Phosphoinositide 3,4,5-triphosphate
PIPKs Phosphatidylinositol phosphate kinases or PIP kinases
PIP5Ks PIP 5-kinases
PIP4Ks PIP 4-kinases
PI4P5K Phosphatidylinositol 4-phosphate 5-kinase
PI-PLC PI-specific phospholipase C
PKA cAMP-dependent protein kinase A
PKC Protein kinase C
PKG Protein kinase G
PLCD Phospholipase C-delta
PM Plasma membrane
P4M PI(4)P binding of SidM/DrrA
PM-V Plasmepsin V

xv
PM-X Plasmepsin X
PM-IX Plasmepsin IX
PROPPINs β-propellers that bind PIs
PTB Phosphotyrosine binding
PTEN
Phosphatase and tensin homologue deleted on chromosome
10
PtIns Phosphatidylinositol
PV Parasitophorous vacuole
PVM Parasitophorous vacuole-membrane
PX Phox homology
R Rhoptry
Rab Ras-related
RAMA Rhoptry-associated
RAP1 Rhoptry associated protein1
RBC Red blood cell
RBCM Red blood cell-membrane
RDTs Rapid diagnostic tests
RHs Reticulocyte-binding like homologs
RON2 Rhoptry neck protein2
RON complex Rhoptry neck protein complex
SHIP SH2 domain-containing inositol 5-phosphatase
SERA Serine repeat antigen
SERA Serine repeat antigen5
SERA Serine repeat antigen6
SLI Selection-linked integration
SNARE Soluble NSF Attachment Protein REceptor
SP Signal peptide
Stt4 Staurosporine and temperature sensitive
SUB1 Subtilisin-like protease 1
SYLF SH3YL1, Ysc84p/Lsb4p, Lsb3p and plant FYVE proteins
TAPP1 Tandem PH domain-containing protein 1
TBVs Transmission-blocking vaccines
TGN Trans-Golgi network
T. Gondii Toxoplasma gondii
TgPH1 T. Gondii PH domain 1
TLC Thin-layer chromatography
Vps15 Vacuolar protein sorting 15
WHO World Health Organization

xvi
Avant-Propos
Acknowledgments
Ph.D. is a great opportunity from scientific and professional perspective but it can be very
difficult since it affects both personal and social life. However, I find myself very lucky and
glad for having nice companies that I encountered in the path. My supervisor, Dave, is one
of those that I grant my luck and this work would not been possible without his friendly
guidance, support and expert advice. His advice and support have been invaluable in
advancing my project. I am also grateful for the time he spent in revision of my thesis and
his kind consideration of my pregnancy and slow-moving! writing. Beside his professional
and science expertise, he is always a good friend to his student with a great sense of humor
that make the long hours of work, easy and cheerful in the lab.
I would like to thank the rest of my thesis committee: Prof. Josée Lavoie, Dr. Denis
Leclerc, and Dr. Christopher Fernández Prada, not only for their insightful comments and
encouragement, but also for the questions which incented me to widen my research from
various perspectives. I would especially like to thank Dr. Josée Lavoie as my teacher and
mentor, she has shown me, by her example, how a great scientist and person should be.
I take this opportunity to thank my fellow labmates for stimulating discussions, for the
sleepless nights we were spending together before deadlines, and for all the fun we had in
the last seven years. In particular, I am grateful that I had Dominic Gagnon as a collegue
for both his friendly attitude and professional work during my thesis. I do appreciate your
generous support and collaboration in all these years, something that indeed contributed to
the success of my work. I thank my fellow labmate, Stephanie Hallee for her cunning and
responsible personality. Also wish to express my deep gratitude to Dr. Angana Mukherjee
for her involvement in the second project and her invaluable comments and suggestions
which contributed greatly to the improvement of my thesis. I also want to thank all of those
whom I had the pleasure to work with during my PhD and my projects, Catherine, David,
Eamim, Mari-Eve and Audrey.
Quebec has definitely offered me a unique experience which is useless to count but
something unquestionable is the opportunity of meeting with amazing people from around
the world. I want to thank Carole Dumas for her kindness and unconditional support, Dr.

xvii
Ouafa Zghidi-Abouzid for her wise and kind advices and Dr. Prasad Padmanabhan for his
generosity and unique jokes.
Nobody would have been more important to me in pursuing my Ph.D studies than my
family. I would like to thank my mother, whose love and guidance is always with me. Also
my sister, Serva, which I am proud to have her always by my side. I would not forget the
part that my two children played in my PhD too. Avin and Aso, who are the source of
unending inspiration for me, came to this world in the hardest step of my life and filled my
life with joy and love. Special thanks are due to my ultimate love, Hiva for his continuous
support and understanding. I am lucky to have him by my side and I owe him my successes
and happiness.

xviii
Contributions
This doctoral thesis focuses on phosphoinositides’ localization and also characterization of
a phosphoinositide effector in Plasmodium falciparum. The thesis contains two main
projects, the first part provides a map of phosphoinositide distribution and the second part
presents a new molecular player in the invasion process of erythrocytic cells by
Plasmodium falciparum.
The presented thesis is divided into seven chapters. First chapter is a review of literature on
malaria parasite and phosphoinositide metabolism in the parasite and model organisms. The
introduction starts with the characterization of malaria, geographical distribution and the
general life cycle of the parasite. Continually, there are detailed description of parasite
morphological changes during erythrocytic cycle, atypical organelles and their role in
invasion erythrocytes. After, malaria treatment and prevention methods with their pitfalls
have been described and new developments in these areas have also been reviewed. In the
second part of the introduction, phosphoinositide structure, function and the enzymes
responsible for their metabolism and regulation are reviewed.
In the second chapter, the hypothesis and my objectives of my doctoral thesis have been
presented. In answer to my two objectives, a manuscript has been already published and a
manuscript is currently undergoing review.
The paper presented in Chapter 3, entitled “A map of the subcellular distribution of
phosphoinositides in the erythrocytic cycle of the malaria parasite Plasmodium
falciparum” has been written as a ''scientific paper''. The paper has been published in the
''International Journal for Parasitology'' (IJP) in January 2018.
In the manuscript presented in Chapter 4 entitled “A pan-apicomplexan
phosphoinositide-binding protein acts in malarial invasion-microneme exocytosis”. It
is a scientific paper and I am the first author. The article has been published in the
EMBO Reports in May 2019.
The final chapter is the discussion and general conclusions and perspectives on the two
projects.

1
Introduction
Malaria
Etiology and Epidemiology
Malaria has plagued humanity for millennia after finding evidence from as early as 2700 BC in
China to 400 BC in Greece, where it was described as periodic fevers and enlarged spleen [1].
From over a 100 Plasmodium species, only five are able to infect humans and cause malaria. These
include Plasmodium falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi. The relevance of
the malaria fevers to the presence of a parasite was first reported by Rastori in 1816. However, it
was not clear until Laveran in 1880 observed Plasmodium gametocyte stage parasites in the blood
of patients. Not long after, the method of transmission was revealed (1887-89) when Ross and
Grassi together linked the female Anopheline mosquito vector to malaria transmission [1, 2]. Later,
Shortt and Garnham in 1947 showed that a liver stage development comes before the erythrocytic
stage. An important step which demonstrates where parasite resides during 8-30 days after
infection [1]. Overall, the entire life cycle of Plasmodium was gradually unveiled over 100 years.
Our current knowledge of the Plasmodium life cycle is presented in the Fig. 1.1, which is common
among all species only with some differences in the details.
Malaria parasite belongs to the phylum Apicomplexa and shares two hosts: human and female
Anopheles mosquito. An infected female Anopheles mosquito takes a blood meal from a human
host and inoculates approximately 100 Plasmodium sporozoites into the dermis [3]. The
sporozoites travel through the dermis until they reach a blood vessel. They then migrate to the liver
where they invade, traverse and develop in hepatocytes via schizogony. Schizogony is an asexual
reproductive process used by some apicomplexans. During the process, the parasites undergo
nuclear division preceding cytokinesis. After a seven to ten day development, mature liver
schizonts rupture and release merozoites into the bloodstream where they start the next phase of
their life cycle called erythrocytic cycle. Merozoites invade erythrocytes and start a cycle of
maturation from a ring to a metabolically active, hemoglobin degrading trophozoite. This is
followed by another round of schizogony where they release new merozoites for reinvasion of new
erythrocytes. Some early erythrocytic stages undergo a different route of development into the
sexual gametocyte stages of the parasite. In P. falciparum, in particular, there are five
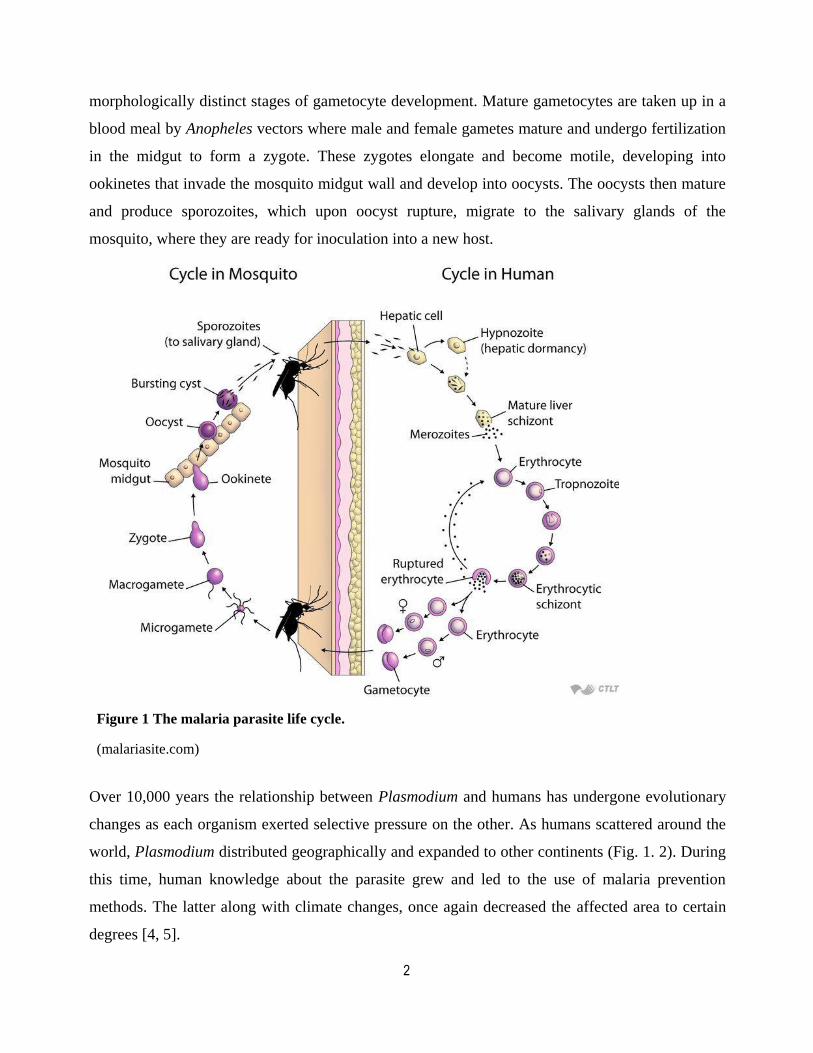
2
morphologically distinct stages of gametocyte development. Mature gametocytes are taken up in a
blood meal by Anopheles vectors where male and female gametes mature and undergo fertilization
in the midgut to form a zygote. These zygotes elongate and become motile, developing into
ookinetes that invade the mosquito midgut wall and develop into oocysts. The oocysts then mature
and produce sporozoites, which upon oocyst rupture, migrate to the salivary glands of the
mosquito, where they are ready for inoculation into a new host.
Figure 1 The malaria parasite life cycle.
(malariasite.com)
Over 10,000 years the relationship between Plasmodium and humans has undergone evolutionary
changes as each organism exerted selective pressure on the other. As humans scattered around the
world, Plasmodium distributed geographically and expanded to other continents (Fig. 1. 2). During
this time, human knowledge about the parasite grew and led to the use of malaria prevention
methods. The latter along with climate changes, once again decreased the affected area to certain
degrees [4, 5].

3
Figure 2 Endemic area of malaria from 1900 to 2002. [6]
In general the geographical area at risk for human malaria has been reduced, from around 53% of
the total earth land area to 27% [6]. Additionally, malaria deaths have also experienced a drop over
the past two decades. Fortunately, malaria-related child mortality has also decreased over 30% in
sub-Saharan Africa from 2004 [7, 8]. Similarly, a continuous fall has been observed in global
malaria deaths outside of Africa since 1990 [8]. A significant number of countries have also moved
toward malaria elimination from 2000. Among 106 countries with ongoing malaria transmission in
2000, 15 countries reached malaria elimination and more than 50 lowered the number of new
malaria cases to at least 75% by 2015 and eighteen countries reduced their malaria cases by 50-
75% [9].
Nevertheless, malaria still is a public health threat. Every year, 3.3 billion people are at risk of
malaria infection. Only in 2016, 216 million cases have been estimated which resulted in
approximately 445,000 deaths. Malaria transmission continues in South and South East Asia,
Central and South America, and Africa. Among which, the most deadly malaria outbreaks occur in
Sub-Saharan Africa due to P. falciparum [9]. Over the past decade, big steps have been made to
reduce mortality and morbidity, however, malaria control alone will not be sufficient. While the
available antimalarial interventions are effective and should be kept to control malaria (restrain
disease, prevent death and interrupt transmission), treatment methods need improvement due to the
new wave of resistance against current treatments. The ultimate goal in the control of malaria is to
design a vaccine that is effective against a broad range of malaria species. This needs a big

4
investment in both control and new drug strategies on the one hand and continued pressure on the
vector and parasite for future success and elimination efforts.
Disease and Pathology of Plasmodium Infection
Malaria causes symptoms that typically include fever, shivering, tiredness, vomiting, and
headaches [10]. Patients with uncomplicated malaria may present with fever, enlarged liver or
spleen, and mild jaundice or anemia. The symptoms of malaria are related to the asexual
erythrocytic stage. Along with the rupture of schizont and destruction of erythrocytes, numerous
known and unknown waste substances such as hemozoin and other toxic factors are released into
the blood stream which initiates an inflammatory response by the host immune system [10].
More severe disease occurs when malaria infection is complicated by organ failure or blood or
metabolic abnormalities which include cerebral malaria, severe anemia, hemoglobinuria, renal
failure and acute respiratory distress syndrome [11]. Severe symptoms are often the result of
parasite sequestration, often seen with late asexual stages of P. falciparum, which bind to
endothelial surfaces in capillaries and small blood vessels via the P. falciparum erythrocyte
membrane protein-1 (PfEMP-1), a protein present on the erythrocyte membrane. The result of such
a sequestration is the blocking of blood flow and oxygen deprivation in tissues [12]. Primary
symptoms usually begin ten to fifteen days after being bitten [13]. In those who have recently
survived an infection, reinfection usually causes milder symptoms. This partial resistance
disappears over months to years if the person has no continuing exposure to malaria [13]. Dormant
liver stage hypnozoite forms of the parasite are found in P. vivax and P. ovale infections and can
reactivate, resulting in relapse after patients have recovered from the illness months or years after
the original infection [14].
Malaria vector
More than 400 different species of Anopheles mosquito are recognized of which around 30
commonly transmit parasites of the genus Plasmodium [9]. Among them, Anopheles gambiae is
one of the best known, because of its major role in the transmission of P. falciparum to human
[15]. An important behavioral factor for a mosquito vector is the degree to which an Anopheles

5
species prefers to feed on humans (anthropophily) or animals. Both A. gambiae and A. funestus, the
primary malaria vectors in Africa, are strongly anthropophilic. Consequently, they are two of the
most efficient malaria vectors in the world [15, 16].
Plasmodium falciparum life cycle
As discussed above, the life cycle of Plasmodium species is overally similar (reviewed in early
introduction) with some differences in the duration of incubation time and of different sub-cycles.
Duration of the erythrocytic cycle in P. vivax, and P. falciparum is 48 hours. While in P. ovale and
P. malariae, it takes longer between 50 and 72 hours, respectively [17]. It is worth mentioning, in
P. vivax and P. ovale, hepatocytic stage can be dormant and persist in the liver cells for weeks, or
even years. Among all three different stages of Plasmodium infection, the erythrocytic stage has
driven the most attention due to its role in all malaria complications.
Erythrocytic stage
Invasion of erythrocyte begins when a merozoite attaches to the host plasma membrane and
penetrates into the erythrocyte using proteins released from the invasion organelles (Fig. 1. 3).
Invasion organelles are a set of secretory organelles namely micronemes, rhoptries and dense
granules which reside at the apical end of the merozoite [18]. The merozoite initial attachment is
weak and it is followed by merozoite rotation to its apical end, which results in a stronger bond
with host membrane [19]. This close contact is known as "tight-junction" which moves from the
apical to the posterior end of the merozoite during invasion, an active process played by the
parasite actin-myosin motor and proteases [20]. The invasion ligands mediating the tight-junction
are removed proteolitically via rhomboid family of serine proteases, a process known as "shedding"
event [21-23]. As the merozoite advances into the host cell; the parasite fabricates a
parasitophorous vacuole (PV). The PV isolates the parasite from the cytosol of the red blood cell
(RBC).
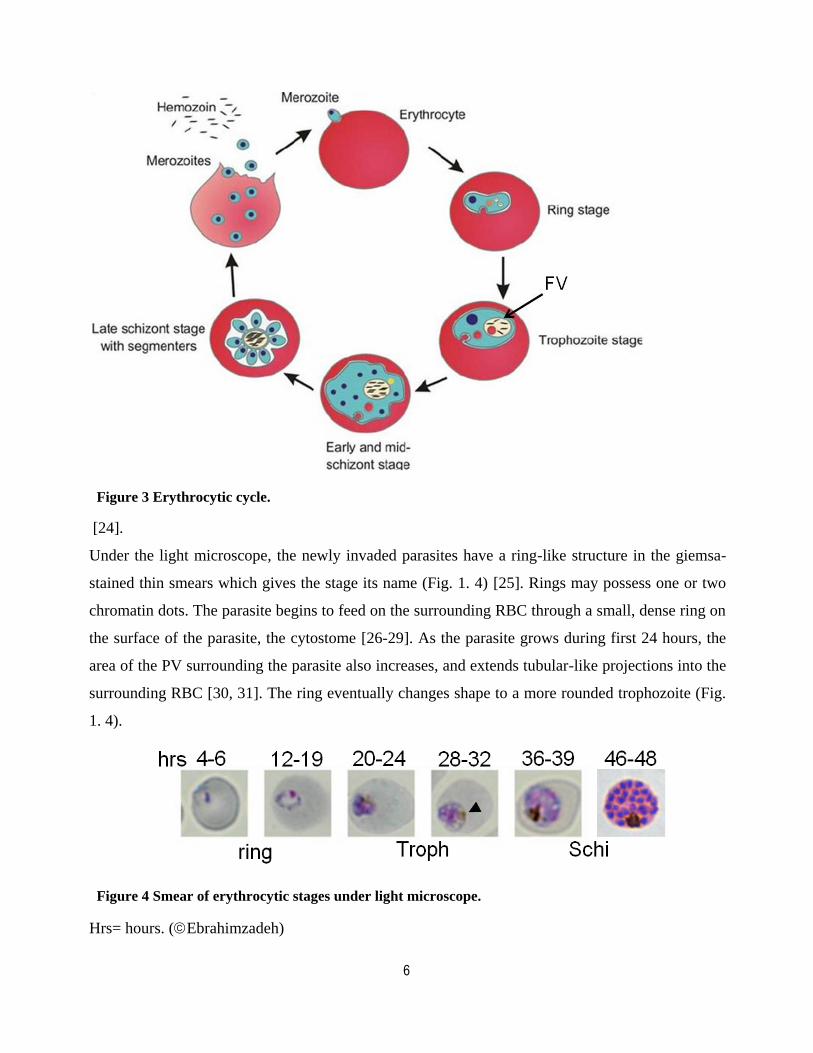
6
Figure 3 Erythrocytic cycle.
[24].
Under the light microscope, the newly invaded parasites have a ring-like structure in the giemsa-
stained thin smears which gives the stage its name (Fig. 1. 4) [25]. Rings may possess one or two
chromatin dots. The parasite begins to feed on the surrounding RBC through a small, dense ring on
the surface of the parasite, the cytostome [26-29]. As the parasite grows during first 24 hours, the
area of the PV surrounding the parasite also increases, and extends tubular-like projections into the
surrounding RBC [30, 31]. The ring eventually changes shape to a more rounded trophozoite (Fig.
1. 4).
Figure 4 Smear of erythrocytic stages under light microscope.
Hrs= hours. (Ebrahimzadeh)

7
A distinctive point between the ring and trophozoite stages is the appearance of a pigmented
vacuole within the parasite (Fig. 1. 3-arrow). The vacuole contains ingested hemoglobin with
brown crystals called hemozoin (heme) that originally accumulates within small vacuoles but later
fuse to form a single larger vacuole called food vacuole (FV) (Fig. 1. 4-arrowhead) [32]. The
parasite exports various parasite proteins into RBC which gradually alter its appearance and results
in some of known trophozoite stage manifestations, including Maurer's clefts and knob structures.
In the infected-red blood cell (iRBC) cytoplasm, Maurer's clefts are believed to act as a sorting
station to export parasite proteins toward RBC-membrane (RBCM) [33-35]. Later, the knob
structures are dense and rigid membranous protrusions that appear during trophozoite and schizont
stage on the iRBC-membrane. Owing to the hypervariability and the adhesive feature of parasite
antigens in the knob structures, the parasite is able to evade the host immune system by its
sequestration in capillaries and the formation of rosettes [36]. Sequestration allows the parasite to
avoid splenic clearance however it also leads to blood flow blockage, which is known to be one of
the cause of cerebral and placental malaria [37, 38]. Second, cytoadhesion to uninfected
erythrocytes, also called rosetting [39], makes it easy for new released merozoites to quickly
invade new host cells while hiding between uninfected erythrocytes. This is also a cause of
microvascular blockage and severe anemia [40, 41]. Therefore, knobs are central to the virulence of
P. falciparum. In general, during trophozoite stage the parasite increases protein synthesis and
enlarges in size and prepares for nuclear and organellar multiplication. Another special feature of
this stage is the branching of mitochondria and apicoplast in preparation for division [25]. The
synthesis of some of the molecules needed for parasite multiplication starts from the trophozoite
stage. In theory, a schizont is an intraerythrocytic parasite that is undergoing or has undergone
repetitive nuclear division. As the parasite approaches the end of the cycle, it continues to consume
hemoglobin. As a result, the parasite produces more hemozoin crystals and the FV grows bigger in
size [42]. The nucleus divides about four times or more to produce about 16-32 nuclei. Nuclear
division is endomitotic, division of chromosomes without nuclear division, a common feature in
unicellular eukaryotes. Therefore, the segregating chromosomes and the spindle apparatus remain
within the nuclear envelope throughout the process [26, 27, 43, 44].

8
Nuclear division is accompanied by multiplication of mitochondria, Golgi, and the apicoplast in the
cytoplasm. The Golgi apparatus consists of a single disc-shape cisterna which is originated from
nuclear envelope vesicles [43, 44]. It is believed apical organelles are the result of coated vesicles
initiated from the Golgi [43, 44] that fuse to create the two rhoptries [44], or they stay individually
to create micronemes or dense granules [25].
Subsequently, a cleavage furrow forms around each nascent merozoite containing a nucleus,
mitochondrion, Golgi and plastid. A constriction ring then separates each merozoite from the
residual body of the schizont containing the food vacuole. The separated merozoites group within
the parasitophorous vacuole. Finally, the PV-membrane (PVM) and RBCM are disrupted following
an increase in cGMP levels, which results in microneme secretion into the PV and onto the
merozoite surface [45]. A key protease, subtilisin-like protease 1 (SUB1) processes several
substrates that are important for downstream events [46]. Among these are members of the serine
repeat antigen (SERA) family. SERA5 and SERA6 are the most abundant SERAs in blood stages.
Recent work has revealed that both SUB1 and SERA6 are essential for successful egress. SUB1 is
required for PVM breakdown while SERA6 is needed to disrupt the RBCM [47]. Recent
observations also strongly suggest SUB1 is involved in proteolytic activation of several other
proteins, including SERA6 and the merozoite surface protein 1 (MSP1) [48, 49]. Breakdown of the
PVM and RBCM allows the merozoites to egress from iRBCs. The new liberated merozoites now
invade new RBCs. The synchronous release of merozoites and toxic material from the iRBCs are
responsible for the cyclical symptoms of the disease, including fever, chills, nausea, body aches
and headaches, which can lead to serious complications mentioned earlier.
Atypical organelles
Micronemes Micronemes are about 120 nm long and vary in shape and numbers between Plasmodium species
(Fig. 1. 5). They are enclosed in a bilayer membrane, and have a fine granular interior [18]. During
merozoite egress, micronemes release partially their content and their complete discharge happens
later during the invasion process. Microneme discharge happens presumably by membrane fusion
into the rhoptry duct/the plasma membrane (PM) and consequently to the exterior [50, 51].

9
Structural evidence suggests micronemes are originated from budding vesicles from the Golgi
apparatus [52].
Rhoptries These organelles are pear-shaped membrane-bound and found at the apical end of the merozoite
(Fig. 1. 5). Based on electron microscopy results, each rhoptry consists of two distinct parts: an
electron-dense rounded basal bulb and a less dense narrow duct [44]. Like for micronemes [44, 53],
rhoptries appear to be formed initially as small vesicles originating from the Golgi, which
eventually fuse together and grow in size [43, 44]. This has also been demonstrated in Toxoplasma
sp. [54]. The membrane surrounding the rhoptry is a bilayer membrane [44]. During invasion,
rhoptry ducts fuse with each other at the tips as well as with the merozoite plasma membrane [55],
and they crumble as they completely empty their content.
Figure 5 Atypical organelles of merozoite.
M= microneme, R= rhoptry, D= dense granule, AP= apicoplast. (Ebrahimzadeh)
Dense granules These organelles are spheroidal membranous vesicles, which in P. knowlesi are about 80 nm in
diameter [50, 56, 57], and of similar appearance in P. falciparum (Fig. 1. 5) [58-60]. They are

10
situated between the rhoptries and the merozoite nucleus, freely within the cytoplasm. After
invasion of a red blood cell, the dense granules move to the merozoite surface where they fuse with
the membrane and liberate their contents into the nascent parasitophorous vacuole, which originate
finger-like projections that extends into the red blood cytoplasm. [50, 56-60].
Apicoplast The discovery of the vestigial plastid (apicoplast) in apicomplexan parasites such as malaria and
Toxoplasma gondii gave us new insight into the origin of the phylum Apicomplexa. The first hints
on the existence of the apicoplast were images of circular, extra chromosomal DNA molecules in
P. lophurae, a malarial parasite of ducks, published by Kilejian [61]. Then Iain Wilson in
collaboration with an expert mitochondriologist, Donald Williamson, and researchers Malcolm
Gardner and Jean Feagin commenced to study malarial extra chromosomal DNAs. In 1991, the
group published a paper in Parasitology Today entitled "Have malaria parasites three genomes?"
[62]. This title was intentionally provocative. Two plant scientist, Geoffrey McFadden and Ross
Waller, which they did not typically read journals on parasitology, quite accidentally saw the paper.
As plant scientists, they were well aware of the dogma that only algae and plants have three
genomes. So, when they read the paper's title about malaria parasites having three genomes, this
was equivalent to saying that malarial parasites were plants [63]. We now know that the
Apicomplexan family originated from photosynthetic ancestors, probably similar to modern
dinoflagellate zooxanthellae [64, 65]. The apicoplast has four bounding membranes [66], a
characteristic of secondary endosymbiosis, in which the plastid is derived by eukaryote-eukaryote
endosymbiosis (Fig. 1. 5) [67]. We have also learned that the apicoplast contains an ensemble of
bacteria-like pathways to replicate and express its genome plus an anabolic capacity generating
fatty acids, heme and isoprenoid precursors. Apicoplasts are essential, and perturbing them, usually
results in parasite death, thus making apicoplast metabolism an attractive target for drugs (reviewed
in ref. [68]).
Molecular bases of invasion
Merozoites are probably the smallest form of all the Plasmodium spp, with dimensions close to a
large bacterium ( ≈1.6 μm long and 1.0 μm wide) [25]. This stage of the parasite is important

11
immunologically because it is exposed to the human immune system even if for as short as one
minute or so. Merozoites also share special features with other invasive forms of Plasmodium, the
sporozoite and ookinete form. The common characteristics of all are having a polarized
morphology and apical organelles, where invasion proteins are located [18]. This gives a good
opportunity for drug and vaccine designing to possibly target all three stages.
Invasion is a complex, multistep process and highly regulated. It is categorized into three stages:
pre-invasion, internalization, and post-invasion (Fig. 1. 6) [52]. In vitro the entire process, from
egress to the end of the invasion, is usually accomplished in less than a minute. Early interactions
of merozoite-erythrocyte appear to be mediated by MSP1 complex and is a random binding on
merozoite's sides (Fig. 1. 6). In this step, the interaction between invasion ligands and their
receptors is cumulative which cause the erythrocyte membrane to indent [69].
After the primary attachment, the merozoite reorients to its apical end where its invasive organelles
will be in close contact with the erythrocyte surface. Two family of proteins, the erythrocyte
binding antigens (EBAs) and the reticulocyte-binding like homologs (RHs), mediate reorientation
of merozoite [70, 71]. The EBAs and PfRhs are held in reserve until needed in
Figure 6 Invasion ligands and their receptors involved in the invasion of erythrocyte by
Plasmodium falciparum
[70].

12
micronemes and rhoptries, respectively. The EBA protein family includes EBA-175, EBL1, EBA-
140, and EBA-181 and they bind to erythrocyte receptors glycophorin (Gly) A, GlyB, GlyC, and
putative erythrocyte membrane protein band 4.1 respectively [72]. The binding of most EBA
ligands to their erythrocyte glycophorin receptors is dependent on sialic acid residues on these
glycoproteins.
The PfRHs family has five members: PfRH1, PfRH2a, and PfRH2b and PfRH4. The first three
members have no known receptors and PfRH4 binds to complement receptor 1 (CR1) [73]. The
fifth family member, PfRh5, has a different function downstream of the other family members (see
below). The EBAs and PfRHs are referred to as alternative pathway ligands because they are
functionally redundant and replaceable in part if not totally [73-75]. It has been shown the deletion
of EBA-175 in W2mef parasite strain results in the upregulation of PfRH4 which indicates to
functional substitution of EBA-175 [74]. Recent data suggest that alternative pathway ligands work
together with a combination of overlapping function and cooperation [71]. The parasite can
epigenetically silence or upregulate particular invasion-related genes, such as PfRH4 [74, 76, 77],
resulting in divergent ligand expression between isolates. Due to this plasticity, the parasite is able
to rapidly adjust to erythrocyte receptor polymorphisms in human populations [76, 78, 79].
Microneme and rhoptry content release occurs in multiple steps and it is speculated that EBAs and
PfRHs interactions with their respective receptors stimulate downstream invasion events [80].
Rhoptry release starts with early discharge during initial merozoite contact and ends when the
parasite has completed internalization into the host cell [81]. In the case of micronemes, there is
accumulating evidence suggesting they are composed of heterogeneous populations with specific
functions in egress and/or invasion[82]. The trigger for protein release from micronemes and
rhoptries is not well-known. There is some evidence hinting at changes in ion concentration like
potassium and Ca2+
during egress which trigger PfRH1 and PfEBA-175 release [83-85].
Interestingly, live cell imaging has revealed two calcium fluxes during the invasion. The first flux
in the merozoite is observed as a faint and weak signal upon egress. While the second flux is often
intense and punctate at the point of contact between the merozoite apex and its erythrocyte. The
timing of the second flux is immediately after merozoite contact with erythrocyte and before
proceeding to the invasion [69].

13
The most irregular member of PfRH family is PfRH5. Unlike other PfRHs, it is smaller in size, has
no transmembrane domain and is expressed in all parasite strains. The gene is refractory to
disruption attempts, a sign of its essentiality [86, 87]. It has two partner proteins, termed PfRH5
interacting protein (PfRIPR), and a GPI-anchored antigen (CyRPA). Through interaction with
PfRIPR and CyRPA, it is anchored to the merozoite surface [86, 88-90]. In this step, live cell
imaging on merozoites blocked by antibodies to PfRH5 or its erythrocyte receptor, basigin, shows
the complete reorientation of the merozoites [69, 88]. This is the distinction between the
attachment and entry of the parasite. Recently, a calcium-regulated phosphatase, calcineurin, has
been shown to play a role in host cell attachment too. Although it is not precisely clear how
calcineurin affects the attachment step, the findings show calcineurin is required for the
extracellular parasite to strongly attach to the host before intracellular entry [91, 92].
For entry, another step of rhoptry release is needed during which the rhoptry neck protein complex
(RON complex) is embedded in the erythrocyte membrane. The RON complex serves as a docking
site for apical membrane antigen 1 (AMA1) to hold on the erythrocyte membrane and forms the
tight-junction [93-96]. Once the tight-junction is established, the merozoite advances into the
erythrocyte membrane and the PV, mediated by rhoptry proteins, forms around the merozoite [55,
97]. Video microscopy of merozoites treated with antibodies against RON2 reveals there is an
important difference compared to the PfRH5 block [69, 98-100]. In addition to erythrocyte
deformation and merozoite reorientation, the RON2-antibody-treated merozoites cause
echinocytosis of their target erythrocyte, indicating RH5 complex triggers the echinocytosis of the
erythrocyte [69]. An interesting work of Volz et al. confirmed PfRH5/PfRIPR/CyRPA complex
binding to host receptor basigin is required for Ca2+ release and establishing the tight-junction
[88]. This finding plus the earlier calcium flux observations indicate a stage of rhoptry release
immediately upstream of the AMA1–RON2 interaction that is triggered by Ca2+ release results in
the tight-junction formation [69, 88].
In brief, the following order of events are characterized during recent advances in dissecting P.
falciparum invasion:(a) A weak, reversible interaction mediated by MSP1 complex results in slight
deformation in the binding area on the targeted erythrocyte. (b) Then release of the alternative-

14
pathway EBA/PfRH ligands which interact strongly and irreversibly with their receptors leading to
the merozoite reorientation. (c) Later, PfRH5 binds to the erythrocyte receptor basigin which
causes a further stage of rhoptry release indicated by a calcium flux at the parasite-host interface.
(d) Upon RON complex embedding in the erythrocyte membrane, AMA1-RON2 form a tight-
junction in the entry site [81] (e) Finally, as the merozoite invades the erythrocyte with the force of
actin-myosin motor, rhoptry proteins and lipids form the PV around the parasite, and protease
proteins degrade the used ligand-receptor bands in the tight-junction. In the end, the tight-junction
will be sealed and the parasite will be isolated in the PV from its surrounding erythrocyte [69].
Malaria Treatment
Diagnosis, Treatments and Resistance
Clinical diagnosis is based on the patient’s symptoms and on physical findings at examination. The
classic symptom of malaria is a cyclical occurrence of sudden coldness followed by shivering and
then fever and sweating [10]. The time of periodic cycle is different in Plasmodium species [17]. It
occurs every two days (tertian fever) in P. vivax and P. ovale infections, and every three days
(quatrain fever) for P. malariae. In case of P. falciparum infection, the recurrent fever is the
shortest and every 36-48 hours. Sometimes it is almost continuous fever and hard to diagnose from
other infections [17].
Malaria parasites can be identified by examining under the microscope [10, 101]. A drop of the
patient’s blood is spread out as a “blood smear” on a microscope slide. Both, thick and thin smears,
must be provided and examined by a laboratory technician. Prior to examination, the specimen is
stained by the Giemsa stain (or other available staining methods) to visualize the intracellular
parasites. The microscopy technique is still the gold standard for laboratory confirmation of
malaria. However the efficiency of the test varies, depends on the quality of the reagents, of the
microscope, and on the experience of the laboratorian [101]. The results of microscopy can be
deceiving especially in case of falciparum malaria. The degree of parasitemia or the parasite stage
can be underestimated due to partial antimalarial treatment or by sequestration of parasitized cells
deep into vascular walls. Therefore, double-checking of the infection by other methods, if
available, is recommended before proceeding to diagnostic.

15
Various test kits are available to detect antigens derived from malaria parasites and provide results
in 2-15 minutes. These “Rapid Diagnostic Tests” (RDTs) offer a useful alternative to microscopy
in situations where a reliable microscopic diagnosis is not available [10, 101, 102]. However, their
accuracy needs to be improved. Importantly, their cost in most of the malaria-affected areas are not
affordable.
Parasite nucleic acids can be detected using polymerase chain reaction (PCR) [10, 101, 102].
Although this technique is more sensitive than smear microscopy, it needs a standard healthcare
center in malaria endemic area. Even so, the PCR results are often not available quickly enough to
be of value to diagnose the type of infection. However, it is the best to detect the species of
malarial parasite after the primary diagnosis by either smear microscopy or RDT [101, 102].
Treatment of malaria depends on many factors, including the species of parasite(s) causing malaria,
the area of the world where the disease was contracted which could indicate which drugs the
parasites would be resistant to, disease severity, age, weight and if the patient is pregnant or not.
Malaria can be a severe, potentially fatal disease, especially when caused by P. falciparum, and
treatment should be initiated as soon as possible [103]. The current recommendation for treatment
is with artemisinin and its semi-synthetic derivatives (ARTs). For more efficiency, they are
prescribed with partner drugs in ART-based combination therapies (ACTs) [104]. Most of the
prescribed drugs are active only against the blood stage parasite and cannot block parasite
transmission. Moreover, the biggest challenge for malariologists is resistance to anti-malaria drugs.
Therefore, drug resistance testing, in case of availability, is recommended before doctors proceed
to any treatments.
Prevention and Vaccine Development
The first line of malaria elimination is prevention methods. Individual protection for mosquito
control like bednets, insecticides, and repellents are being recommended in endemic areas. Also,
protective clothing can help at times of the day when vectors are active [10]. Draining still waters
and using insecticides have an important impact on the vector control. Even so, disease elimination
is out of reach in many areas without a vaccine. The path to develop an effective malaria vaccine is
a very demanding process both due to parasite related-complexity and economically [105]. Malaria
parasites have a complex life cycle and the polymorphic nature of its antigens make it even more

16
difficult to find effective and universal candidates with required immunogenic property.
Economically, malaria affects mainly people in low-income countries leading to a lower interest in
the vaccine development investments.
An effective and durable vaccine for malaria must be able to target all three life stages of the
parasite: pre-erythrocytic stage, blood stage, and mosquito stage [106]. Though, such a vaccine
seems to be far from being available and existing; vaccine strategies are mostly targeting just one
stage of the parasite life cycle. Pre-erythrocytic vaccines target the clinically silent stages of
Plasmodium during sporozoite and liver stages intending to eliminate parasite at the first step of
parasite entrance. The advantage of such vaccines, in case of success, is prevention of disease and
most importantly interrupting parasite transmission. As of today, four candidates are in different
stages of clinical trials with the most promising being the RTS,S vaccine (Table. 1. 1) [106]. In the
last malaria vaccine symposium 2017, the World Health Organization (WHO) announced that the
RTS,S malaria vaccine completed its Phase III clinical trials and the pilot implementation programs
would begin in Ghana, Kenya, and Malawi in 2018 (Phase IV). The RTS,S vaccine is composed of
the repeat region of the circumsporozoite protein fused to the Hepatitis B virus surface antigen, and
is adjuvanted with AS01 adjuvant. Findings on RTS,S demonstrate it is safe and efficient in adults,
children, and young infants in sub-Saharan Africa [106]. More than 15,000 children were
vaccinated with RTS,S in 11 centers across seven African countries during the phase III trials
which is been estimated 829 clinical malaria episodes per 1000 children were prevented over 18
months of follow-up [106]. Though, RTS,S vaccine still shows moderate effects and its efficacy
declines over time [107].
Blood stage based vaccine development faces some restricting points such as antigenic
polymorphism of infected erythrocyte surface proteins, redundancy in the merozoite invasion
pathways (alternative pathways), and expressing conformationally correct parasite antigens [108].
Even so, exciting findings have recently identified two new protein candidates promising a high
level of specificity: PfRH5 and the AMA1-RON2 complex. RH5 is the first highly conserved target
from the merozoite which is susceptible to neutralizing antibody induced by vaccine [109]. It is an
invasion ligand which is common between alternative pathways [88]. In Aotus monkeys, PfRH5-
based vaccines induced antibody response and rose protection against a heterologous P. falciparum

17
challenge [110]. A first generation PfRH5-based vaccine is being also tested in clinical trials, but
more studies are required to improve vaccine efficacy, including identification of new RH5
epitopes [111] and a proper cell line for the large scale production of PfRH5 in a conformationally
correct form [112]. The AMA1-RON2 complex is another erythrocyte stage vaccine candidate that
targets merozoite invasion. The complex mediates merozoite invasion through tight-junction
formation that is a stage subsequent to PfPRH5. Initial reports demonstrate antibody response
raised against AMA1-RON2 complex peptide can protect against virulent P. yoelii and P.
falciparum infection in mice and Aotus monkeys, respectively [113]. The vaccine development is
still in preclinical trials.
Table 1 Malaria vaccines in preclinical development or in clinical trial.
[106]
The third category of vaccine strategies, transmission-blocking vaccines (TBVS), strive for parasite
transmission blocking via targeting parasites at the mosquito stage. Currently, Pfs25 (post-
fertilization antigen), Pfs230 (pre-fertilization antigen) and Pfs47 (an immune suppressor of A.
gambiae) showed good properties in preclinical studies as vaccine candidates for mosquito stage
Vaccine classification Current status
PfSPZ vaccine Whole organism Phase II
(radiation attenuation)
GAP vaccines Whole organism Phase I
(genetic attenuation)
RTS,S Subunit Phase IV
CVac Whole organism Phase I
(chemical attenuation)
Chemically attenuated Whole organism Preclinical
parasites
AMA1-RON2 Subunit Preclinical
Subunit Subunit
Pfs25 Subunit Phase I
Pfs230 Subunit Phase I
Pfs47 Subunit Phase IMos
quito
sta
ge
(TBV
s)
Parasite stage
Pre-
eryt
hroc
ytic
sta
geBl
ood
stag
e

18
[106]. Ongoing trials combine Pfs25 and Pfs230 vaccine antigens to test their efficiency and find
the best formulation for vaccine activity [114, 115]. In one case, both Pfs25 and Pfs230 conjugate
vaccines administered with AS01 adjuvant from the GlaxoSmithKline (GSK). Based on preclinical
studies this formulation might notably increase antibody titer and therefore serum functional
activity after vaccination [106].
Maintaining durable protection after immunization is one of the key limitations for the success of
malaria vaccines [106]. This could be due to the suppressing mechanisms of the parasite towards
host’s immune system and also poor immunogenic property of antigens. Combination of different
antigens from different stages, new methods of administration as well as the dosage and vaccine
schedules may potentially lead to a more durable and effective vaccine.
Drug Resistance and Discovery
The emergence of resistance to ARTs was reported several years ago and is now spread out in six
countries in South East Asia [116]. Malaria drug resistance is best described for P. falciparum as it
is the most deadly infection and a quick treatment is crucial for survival. The historical and genetic
origins of P. falciparum resistance to artemisinin, chloroquine, and sulfadoxine-pyrimethamine
have consistently been found within Western Cambodia [117]. Chloroquine-resistant P. falciparum
is the most widespread form in the world. Hence, combination therapies like antifolate, mefloquine,
and atovaquone were introduced, but later resistance against chloroquine combination therapies
also emerged [118]. This was coincident with the discovery of artemisinin in 1972 by a Chinese
scientist Tu Youyou who was awarded later half of the 2015 Nobel Prize in Medicine [119].
Artemisinin is extracted from Artemisia annua a herb employed in Chinese traditional medicine
[120]. Artemisinin and its derivatives are exceptionally fast acting against intra-erythrocytic
asexual blood-stage malaria parasites [120]. But with a big disadvantage; their very short half-life
in vivo (about one hour in human). As a result, they are co-administered with longer half-life
partner drugs, such as lumefantrine, amodiaquine, piperaquine, mefloquine, sulphadoxine-
pyrimethamine or pyronaridine as ACTs [104, 119].
The molecular basis for chloroquine, antifolate and atovaquone resistance has been well
established. Mutations in the target transporter or enzymes of these drugs allow for the parasite to

19
become resistant by a number of mechanisms, including increased efflux ability or inability to
inhibit the enzyme [119]. In the case of ARTs, the exact resistance mechanism is still elusive but
recent findings showed resistant parasites have mutations in Kelch13 gene affecting encoded
propeller, BTB/POZ domains [121, 122]. It has been shown that Kelch13 gene mutations are
associated with a slow parasite clearance rate after treatment with artemisinin derivatives [116,
121]. Following, scientists noticed a correlation between Kelch13 mutations and mutations in at
least four other genes (fd, arps10, mdr2, and crt). Whenever the kelch13 mutation was present in
the genome of a resistant parasite, the other four mutations almost invariably seemed to be there
too [121].
With the appearance of ART resistance, the need for new drug strategies is now more urgent than
ever. Many parasite pathways have been targeted due to their uniqueness including hemoglobin
digestion and the folate pathway [123, 124]. Mitochondrial function and respiration are also
interesting targets since the electron transport chain in Apicomplexa species differs from that of the
mammalian host mitochondria [125-127]. Inhibitors of the apicoplast, a plastid-like organelle
found in most Apicomplexa, which target the synthesis of crucial precursors like isopentenyl
pyrophosphate (IPP) are another route for anti-malarials [128]. Apicoplast pathways obviously do
not exist in the human host and there has been considerable excitement about targeting the
apicoplast pathways as a parasite Achilles' Heel for drug inventory with no or fewer side effects for
its infected host [68, 129, 130]. Plasmodium proteases also represent potential high-value targets
[49]. Partly, due to our knowledge of their enzymatic mechanisms and active site structures. More
importantly, they have been shown to be involved in a variety of pathways that are essential for
parasite survival [49]. For example, aspartic proteases called plasmepsins which are involved in
diverse cellular processes include interesting hits like Plasmepsin V, IX and X. In the ER lumen,
Plasmepsin V (PM-V) is involved in a specific cleavage downstream of an export signal. Most
Plasmodium exported proteins contain a protein export element (PEXEL) motif and its cleavage by
PM-V exposes a specific signal [131, 132] that is detected by a translocon at the parasitophorous
vacuole membrane [133]. PM-V is therefore a very promising target and its inhibition will likely
affect most extracellular functions of the parasite which are related to the parasite virulence.
Recently Nasamu A.S. et al. showed both PM-IX and PM-X are important for erythrocyte invasion
[134]. PM-X, in addition to invasion, controls egress and merozoite maturation via processing

20
SUB-I [134]. Of importance, the same group has identified compounds with potent antimalarial
activity targeting PM-X [134]. Plasmepsin X has also been suggested to play a role in midgut
transversal by ookinetes [135]. Therefore, a PM-X inhibitor has the potential of targeting two or
three stages. However, there are other parasite pathways that have yet to be explored for drug
discovery.
Currently, an area of focus for P. falciparum drug development is kinase inhibitors [136-139]. With
the success of kinase inhibitors as a treatment for other disease models such as cancers, much
research is going on to develop kinase inhibitors specifically targeting P. falciparum kinase
activity. Employing bioinformatic analysis, putative kinases and signaling pathways in P.
falciparum have been identified [140, 141]. Many parasite kinases have been found to be different
from mammalian kinases, which may indicate their usefulness as a drug target, specially kinases
that affect phosphoinositide metabolism [142-144]. Despite disappointing news of malaria
resistance to ACTs, phosphatidylinositol-4-OH kinase (PI(4)K) inhibitors have brought back hope
to the battle camp of malaria [145-149]. Fortunately, recent advances in malaria research in the
fields such as forward genetics and structure-activity relationship chemistry are powerful assistant
for drug designers to potentially target unique enzymes of P. falciparum. Along with that, basic
knowledge on Plasmodium spp. biology has considerably improved owing to recent progresses in
the fields of genetic modification methods. In particular, the development of conditional genetic
approaches in P. falciparum is allowing us for the first time to validate essential genes and study
their molecular functions. DiCre recombinase [150, 151] and selection-linked integration (SLI)
[152] are currently efficient available methods for P. falciparum. The DiCre recombinase system
allows for conditionally Knocking-Out (cKO) genes [153] while SLI system increases our ability to
select integrants in a shorter time compared to the traditionally used drug cycling method. Also SLI
system in combination with knocksideways permits mislocalization of native proteins [152].
Hopefully the new approaches will help to develop a better understanding of the basic biology of
the parasites which is vital to develop novel medical therapeutics.
21
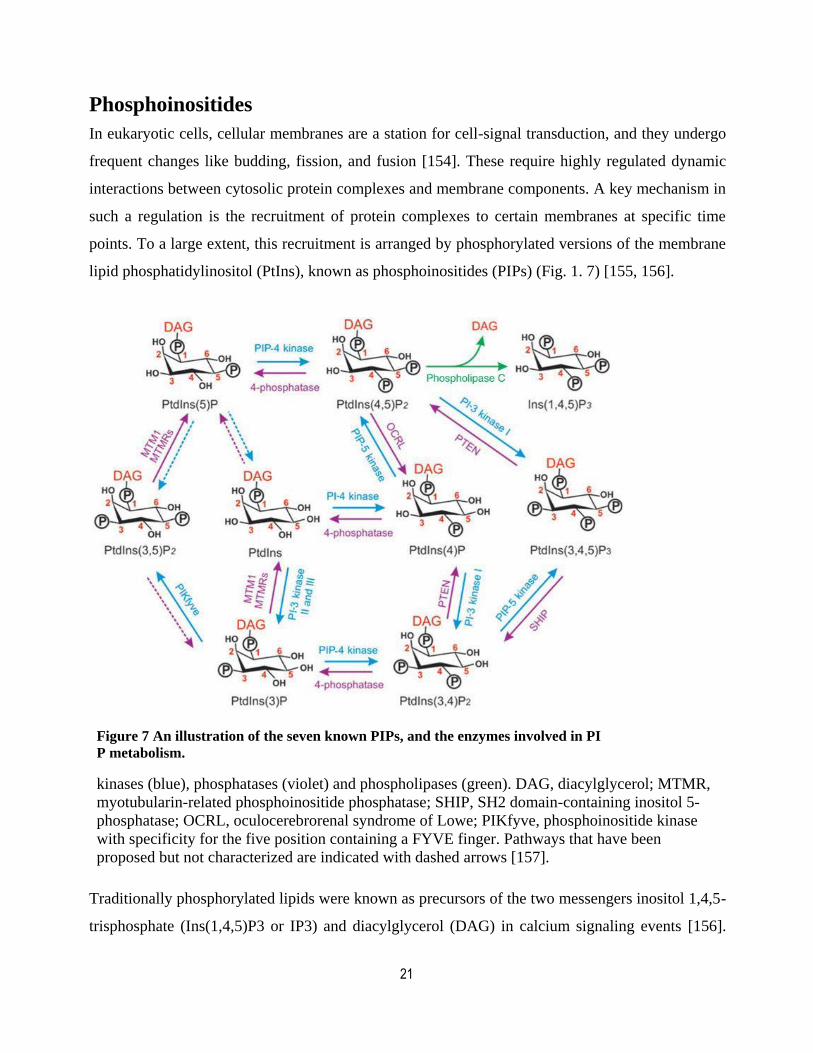
Phosphoinositides
In eukaryotic cells, cellular membranes are a station for cell-signal transduction, and they undergo
frequent changes like budding, fission, and fusion [154]. These require highly regulated dynamic
interactions between cytosolic protein complexes and membrane components. A key mechanism in
such a regulation is the recruitment of protein complexes to certain membranes at specific time
points. To a large extent, this recruitment is arranged by phosphorylated versions of the membrane
lipid phosphatidylinositol (PtIns), known as phosphoinositides (PIPs) (Fig. 1. 7) [155, 156].
Figure 7 An illustration of the seven known PIPs, and the enzymes involved in PI
P metabolism.
kinases (blue), phosphatases (violet) and phospholipases (green). DAG, diacylglycerol; MTMR,
myotubularin-related phosphoinositide phosphatase; SHIP, SH2 domain-containing inositol 5-
phosphatase; OCRL, oculocerebrorenal syndrome of Lowe; PIKfyve, phosphoinositide kinase
with specificity for the five position containing a FYVE finger. Pathways that have been
proposed but not characterized are indicated with dashed arrows [157].
Traditionally phosphorylated lipids were known as precursors of the two messengers inositol 1,4,5-
trisphosphate (Ins(1,4,5)P3 or IP3) and diacylglycerol (DAG) in calcium signaling events [156].

22
However, deeper investigations revealed the tiny lipids and their effector enzymes are the
regulators of vast cellular functions in eukaryotic cells such as signal transduction, cell motility,
cytoskeletal reorganization, DNA synthesis, cell cycle, adhesion, membrane transport,
permeability, and trafficking [155, 156]. They are especially the focus of many studies in drug
discovery field from cancers to infection [156].
Upon specific stimulation, PI-kinases and -phosphatases enrich or deplete certain subcellular
membranes from specific PIP (Fig. 1. 7) [158]. Most unicellular organisms like yeast produce only
five phosphoinositides PI3P, PI4P, PI5P, PI(3,5)P2, and PI(4,5)P2. Among these, only PI4P and
PI(4,5)P2 are essential for viability [156, 158]. Higher eukaryotes produce two more PIP isoforms
namely PI(3,4)P2 and PI(3,4,5)P3. The distribution of phosphoinositides establish organelle
identity known as the "PIP" code (Fig. 1. 8) [155, 158]. The PIP code is then “read” by specific
effector proteins, which contain domains with the ability to detect individual PIPs. These
interactions eventually lead to the recruitment of host proteins to specific intracellular
compartments [156].
Due to the importance of these tiny lipids, some studies started focusing on the role of PIPs in P.
falciparum. However, the molecular identity and the combination of the PIP profile in infected
RBC were not clear until recently. Uninfected RBCs only produce PI4P and PI(4,5)P2 when
labeled with 32
P-phosphate but after infection with P. falciparum, the production of PIPs
considerably changes in both PIPs level and diversity [159, 160]. In 2010, using a combination of
thin-layer chromatography (TLC) and high-performance liquid chromatography (HPLC), Tawk et
al. gave a detailed characterization of the PIPs profile in infected erythrocytes by P. falciparum
[160]. Their results showed that, in addition to PI4P and PI(4,5)P2, the infected erythrocytes
produce high levels of PI3P and low levels of PI(3,4)P2 and PI(3,4,5)P3 [160]. PI(3,5)P2 was not
detected and PI5P could not have been detected with the method used. The presence of PI(3,4,5)P3
was unexpected as unicellular organisms generally do not produce this lipid. Recently, PI(3,4,5)P3
production has been re-confirmed in both P. falciparum schizonts and in P. berghei ookinetes via
lipidome analysis [161]. Surprisingly, in T. gondii, only PI3P and PI4P have been identified in high
quantities [162]. However, unpublished observations of Wengelnik et al. have suggested the higher
phosphorylated species are present but only in small amounts [163].

23
Figure 8 A map of the subcellular localization of Pl in higher eukaryotic cells.
PIPs are concentrated in distinct pools of cytosolic membranes and serve as markers of various
cell compartments and regions. EE= early endosome; MVB= multivesicular bodies [155].
PIP-binding proteins
The identification of protein effectors of PIPs was a revolutionary step in the understanding of the
importance of the PIPs code in the regulation of different biological processes inside cells. In
particular, those with PIP-binding domains that can recognize individual PIPs specifically [155].
Since the recognition of the first protein module in 1994, the list of PIP-binding domains has grown
larger (Fig. 1. 9) [164].

24
Figure 9 PIP-recognizing effectors.
[154]
Presently, there are 16 modules that exhibit a wide range of affinities and selectivities toward lipid
derivatives in the membranes [154, 165]. These include PH (Pleckstrin homology domain), ANTH
(AP180 N-terminal homology), BATS (Barkor/Atg14(L) autophagosome targeting sequence), C2
(conserved region-2 of protein kinase C), DHR-1 (Dock homology region-1), ENTH (Epsin N-
terminal homology), FERM (4.1, ezrin, radixin, moiesin), FYVE (conserved in Fab1, YOTB, Vac1
and EEA1), GOLPH3 (Golgi phosphoprotein 3), P4M (PI(4)P binding of SidM/DrrA), PDZ
(postsynaptic density 95, disk large, zonula occludens), PROPPINs (β-propellers that bind PIPs),
PTB (phosphotyrosine binding), PX (Phox homology), SYLF (SH3YL1, Ysc84p/Lsb4p, Lsb3p and
plant FYVE proteins), and Tubby modules (reviewed in [154]). Among them, proteins containing
PH domains are one of the largest families of signaling proteins. PH domain-containing family are
important players of many biological processes such as membrane dynamics, protein trafficking,
intracellular signaling, cytoskeletal alteration and lipid metabolism [155, 158, 166-169]. Most of
the PH domains are promiscuous and can bind to a variety of PIPs but with different affinities.
Nevertheless, 10-20% of the PH domains show specificity for individual PIPs such as PI(3,4,5)P3,
PI(4,5)P2, and PI(3,4)P2 [169, 170]. Domains such as the FYVE domain and PX domain often
preferably bind to PI(3)P [171, 172]. Proteins with FYVE domains are implicated in the regulation
of endocytosis mostly [154, 173]. PX domains are found in many signaling and regulatory proteins
mediating essential biological processes such as endocytosis, protein sorting, membrane
trafficking, transcription, and cell polarity [154, 174]. Fluorescent molecular made up of PIP-
binding domains fused to a reporter protein are very useful tools to study PIP dynamics and allow
imaging of the PIP location in live cells [175].

25
Phosphoinositide species
The structure of PIPs are based on glycerol, a three-carbon alcohol with the formula CH 2 OH–
CHOH–CH 2 OH and an inositol ring. PtdIns synthase is responsible for the synthesis of PtdIns
which is located on the cytoplasmic face of the endoplasmic reticulum (ER), the Golgi,
mitochondria, and microsomes in both human [176] and yeast [177]. Partly, lipid phosphatases are
also involved in recycling PtdIns via dephosphorylation of mono-phosphoinositides [178, 179].
PI3P PI3P accounts for about 30% of the total PIPs in yeast and is as abundant as PI4P [180]. Contrarily,
in human cells, it represents less than 15% of monophosphorylated PIPs and is much less abundant
than PI4P [181]. PI3P is produced by the phosphorylation of PtdIns at position D3 of inositol or by
the dephosphorylation of PI(3,4)P2 or PI(3,5)P2 via 4- or 5-phosphatases [182]. In yeast and
mammalian cells, PI3P is enriched at endosomal membranes and membranes of the multivesicular
body (MVB) [173]. At the early endosomes, it plays a central role in recruiting endosomal effector
proteins which are involved in the endosomal sorting of proteins and the formation of the MVB
[183]. One of the physiological roles of PI3P is to serve as a precursor for PI(3,5)P2 and PI(3,4)P2
synthesis [181]. Furthermore, it is involved in autophagy through the formation of PI3P-enriched
regions at the ER via a PI3-kinase, a process termed pre-autophagosome formation [184, 185]. The
latter then serves as platforms to recruit autophagosomal proteins [186-188]. In fact, mutations
affecting the binding of human pre-autophagosome proteins to PI3P has been observed in cancers
and neurodegenerative diseases, indicating an important biological role of PI3P in the regulation of
autophagy [187]. Additional functions of PI3P include endocytic trafficking, macroautophagy,
phagocytosis, cytokinesis, and nutrient sensing [189].
The lipid has been studied extensively in apicomplexan parasites. Radioactive labeling experiments
revealed high levels of PI3P synthesis in P. falciparum-infected erythrocytes, approximately 30%
of the total PIP-monophosphates [160], or even higher, according to unpublished data from
Wengelnik et al. which is a condition more similar to yeast than mammalian cells [163]. Similarly,
in T. gondii, PI3P levels are relatively high [162]. In the malaria parasite, PI3P localizes to the food
vacuole, at the apicoplast and in the membranes of small vesicles clustered near the food vacuole
and/or the apicoplast [160]. A fluorescent PI3P reporter also localizes to the apicoplast in T. gondii
[162]. However, constitutive expression of the PI3P reporter was not tolerated in T. gondii and led

26
to the disturbance of apicoplast biogenesis. Consequently, the parasites lost their apicoplast and
died in a delayed death mode [190]. This finding brought up a probable role for PI3P in vesicular
trafficking toward the apicoplast and also an evolutionary link between phagosome formation, ER-
enriched PI3P membranes, and the apicoplast membrane. As mentioned before, the apicoplast is
originally derived from the phagocytic compartment during the secondary endosymbiosis event.
And the analysis of the outermost membrane of the apicoplast and the ER-enriched membranes
indicated the presence of autophagy proteins [191, 192]. An unusual function has also proposed for
PI3P in P. falciparum parasites as its involvement in protein export into parasite-host [193].
According to this finding, PI3P is present in the ER lumen and interacts with certain sequence
motifs that are important for the export of parasite proteins to the host erythrocyte [193]. Since
PIPs generally reside in the cytoplasmic leaflet of cellular membranes, the ER lumen is not a
common location for PIPs. This finding has been later challenged with a work that our lab has
contributed where it was showed that the PEXEL (an export motif) sequence does not bind PI3P
and that a PI3P-reporter containing PEXEL sequence was targeted into the PV, the default
secretory pathway, without accumulating in the ER lumen [194]. In a recent study, high PI3P levels
were found to be related to a specific mutation in Kelch13 in artemisinin resistance parasites.
Kelch13 mutation is one of the known molecular markers for artemisinin resistance in the malaria
field. It is hypothesized that Kelch13 regulates PfPI3Kinase (PfVps34) via ubiquitination and
control its abundance in the parasite [195]. Thus, Kelch13 modulates PI3P levels indirectly. Some
effector proteins containing PI3P-binding domain have been also studied in P. falciparum. A PI3P-
binding FYVE-containing protein (PfFCP) has been identified in the FV [196]. Recent findings on
an autophagy protein, PfATG18, also showed its importance for the apicoplast inheritance through
interaction with PI3P [197]. Its ortholog, TgATG18 binds to PI3P as well but it is not localized to
the apicoplast unexpectedly [197]. Another autophagy protein, PfAtg8, has been shown to localize
to the apicoplast [191]. The presence of autophagy proteins plus Vps34 suggests a role for PI3P in
apicoplast biology.
PI4P PI4P accounts for around 30% of the total PIPs in yeast and ≈ 45% in Human [181]. It is mostly
produced by the phosphorylation of PtdIns by PI4-kinases [198] and some parts are the result of the
dephosphorylation of diphosphorylated PIPs by 3- and 5-phosphatases [182]. In yeast, PI4P is

27
enriched mostly in the PM and the Golgi [199]. Some pools of PI4P are also found in endosomal
compartments and in the trans-Golgi [200]. The Golgi is a central station in the secretory pathway
where PI4P is involved in Golgi trafficking toward the PM [201], and the retrograde transport from
the Golgi to the endoplasmic reticulum [199]. Many proteins interacting with PI4P have been
localized to the Golgi [202] and most of them were shown to transport lipids in association with
PI4P [203]. At the ER, PI4P is hydrolyzed to PtdIns by the SAC1 phosphatase and the released
energy is, consequently, used for sterol transfer [204]. PI4P is also required for receptor sorting in
the early endosomes [205]. In addition, it is a key intermediate in the biosynthesis of PI(4,5)P2 at
the PM. The PH domain of FAPP1 and FAPP2 (four-adaptor-phosphate proteins 1 and 2) proteins,
Golgi resident proteins, are broadly used as PI4P-reporters in different organisms due to their
specificity for PI4P-binding [202, 206].
The intracellular distribution of PI4P is traced to foci and the parasite plasma membrane in P.
falciparum [145, 207]. In a study in P. berghei, parasites expressing a PI4P-reporter treated with
PI4K inhibitors showed redistribution of the PI4P-reporter signal from intracellular foci to the
parasite PM [145]. These parasites also showed a failure in membrane ingression during merozoite
formation, an evidence for the role of PI4P in cytokinesis in Plasmodium.
PI5P PI5P is the last identified monophosphorylated PIPs due to its low basal concentration in quiescent
mammalian cells, and due to technical difficulties in separating it from PI4P [208]. In basal
mammalian cell conditions, PI5P represents less than 10% of monophosphorylated PIPs [181] but
its level increases following specific stimuli. In mammalian cells, PI5P is mostly synthesized from
PtdIns by PIKfyve and some results from the dephosphorylation of P(3,5)P2 by 3-phosphatases
[209-211]. A fraction of PI5P is found in the nucleus, where it could be involved in stress
responses [212]. The PH domain of DOK (downstream of tyrosine kinase) proteins shows a strong
binding affinity to PI5P, and this binding activates the phosphorylation of DOK proteins in T cell
signaling [213]. Moreover, PI5P was also shown to localize in the PM and involved in PM and
endosomal functions [214, 215]. It has been shown the latter is employed by Shigella bacteria to
regulate membrane dynamics during host cell invasion. Shigella bacteria express a bacterial
phosphatase, IpgD, which dephosphorylates PI(4,5)P2 into PI5P [216, 217]. In addition, an

28
increase in the number of autophagosomes has been observed after PI5P addition to cells treated
with the Vps34 inhibitor wortmannin or knocked down for Vps34, indicating a link between PI5P
and autophagy [218].
To date, there is no indication for the presence of PI5P in P. falciparum, and the only detected
putative PIKfyve-like orthologue does not contain a FYVE domain.
PI(4,5)P2 PI(4,5)P2 is the most abundant PIPs in both yeast and human and it represents about 45% of total
PIPs [158]. PI(4,5)P2 is involved in a vast variety of biological processes including endocytosis,
phagocytosis, cell adhesion, and cell motility [219, 220]. PI(4,5)P2 is most abundantly present in
the cytosolic face of the PM, where it acts as an anchoring point and a major regulator of
membrane fusion events such as vesicle endocytosis and exocytosis [158, 182, 221]. It also
interacts with cytoskeletal proteins and bridges between the cytoskeleton and the PM [221]. In
yeast, PI(4,5)P2 is the result of the action of the PI5-kinase (Mss4) and the PI4-kinase (Stt4) at the
PM from PtdIns [182, 222]. Different effector proteins required for the internalization step of
endocytosis have ENTH, ANTH or PH domains that interact specifically with PI(4,5)P2 [158,
223]. PI(4,5)P2 has also been detected in the nucleus, where it is involved in the control of the
expression of targeted genes [224, 225]. Further functions for PI(4,5)P2 is coming from the fact
that the lipid is the precursor of intracellular signaling molecules IP3 and DAG through the action
of PIP-specific phospholipase C (PI-PLC).
In Plasmodium, PI(4,5)P2 is the most abundant PIPs [160]. It might be produced via
phosphorylation of PI4P by the single type I phosphatidylinositol 4-phosphate 5-kinase (PI4P5K)
[163, 226]. Most of our knowledge about the role of PI(4,5)P2 in Plasmodium comes from
calcium/PI(4,5)P2/PI-PLC signaling. Through this signaling cascade, PI(4,5)P2 is implicated in
many cellular functions such as ookinete motility, merozoite egress, and sporozoite gliding motility
[161, 227-229].

29
PI(3,4)P2 In human cells, PI(3,4)P2 counts for less than 10% of the total PIPs in quiescent cells while it is not
detectable in yeast. Upon cell stimulation, PI(3,4)P2 levels transiently increase via
dephosphorylation of PI(3,4,5)P3 by 5-phosphatases [181]. Alternatively, PI(3,4)P2 is synthesized
by the phosphorylation of PI4P via class II PI3K lipid kinases at the PM [182]. PI(3,4)P2 acts as a
secondary messenger by recruiting protein kinases such as Akt (protein kinase B) and PDK1
(phosphoinositide-dependent kinase 1) through their PH domain [230]. A transforming mutation in
the PH domain of Akt1 is associated with several cancers [231]. The link between PI(3,4)P2 and
the PI3K/Akt signaling pathway suggests that this PIP could be involved in numerous biological
processes, such as controlling the cell cycle, cell survival, and clathrin-dependent endocytosis
[232]. The balance between PI(3,4)P2 and PI(3,4,5)P3 is essential in modulating signaling
pathways downstream of Akt [233]. Therefore, the interaction between lipid kinases and
phosphatases keeps this balance and deregulations of molecular players could lead to numerous
problems [234]. Among the various protein domains binding PI(3,4)P2, only the PH domains of
TAPP1 (tandem PH domain-containing protein 1) interact specifically with PI(3,4)P2 [235, 236].
In Plasmodium, PI(3,4)P2 has been detected at low levels by metabolic labeling. However, there is
no evidence of its function or localization yet.
PI(3,4,5)P3 In yeast, there is no detectable PI(3,4,5)P3 and in human, it represents less than 5% of total PIPs in
quiescent cells. However, following various stimuli, its intracellular levels rapidly and transiently
increase up to 100-fold [237]. PI(3,4,5)P3 is mainly localized in the PM and is synthesized by class
I PI3-kinases from PI(4,5)P2. Small pools of PI(3,4,5)P3 can be synthesized in other subcellular
membranes in response to agonists [181]. It is a signaling molecule in many signaling pathways
and its regulation is critical. PTEN (phosphatase and tensin homologue deleted on chromosome
10), a PI-phosphatase, is among its regulators. It has been categorized as a tumor suppressor since
mutations in the pten gene are linked to many cancers [238]. PI(3,4,5)P3 regulates important
cellular functions such as cell proliferation and cell survival, cytoskeleton dynamics, cell motility,
membrane trafficking and apoptosis [202, 239].

30
In Plasmodium, PI(3,4,5)P3 has been detected in low levels [160, 161]. Apicomplexan parasites
only possess a single PI3-kinase (class III) which is known to synthesize only PI3P in other
organisms. Phosphorylation assays using the immunoprecipitated PfPI3-kinase revealed that the
enzyme was able to phosphorylate PtdIns, PI4P, and PI(4,5)P2 [240]. However, a recent work has
challenged this result and suggested that trace levels of PI(3,4)P2 and PI(3,4,5)P3 in P. falciparum
are produced by other related kinases found in the P. falciparum genome [241].
PI(3,5)P2 PI(3,5)P2 is a rare PIP and represents less than 5% of the total PIPs in both yeast and human. In
response to osmotic stress, PI(3,5)P2 synthesis is stimulated, and its intracellular level increases
20-fold compared to non-stressed cells [242]. In yeast, PI(3,5)P2 is synthesized from PI3P to
generate PI(3,5)P2 via PI3P 5-kinase Fab1 [182]. It is mostly enriched in vesicular and tubular
domains in late endosomes [158, 243]. At the endosomes, yeast epsins Ent3 and Ent5 interact with
PI(3,5)P2 through their ENTH domain and are required for endosomal sorting of ubiquitylated
cargos and endosomal recycling of SNARES [244-246]. To date, the yeast Atg18/Svp1 and Hsv2
proteins which are involved in autophagy show the highest affinity and specificity for PI(3,5)P2 in
vitro [247, 248]. In human, PI(3,5)P2 synthesis is similar to yeast, and it is catalyzed by PIKfyve,
the sole PI3P 5-kinase [249]. PI(3,5)P2 plays an essential role in protein sorting at the late
endosomes/MVB, lysosomal homeostasis or signaling pathway regulations [181, 250].
In P. falciparum, PI(3,5)P2 is not detected. Although a PIKfyve-like protein is present in
Plasmodium genomes, it does not contain a FYVE domain in contrast to PIKfyve of all other
organisms, and there is no evidence that it is capable of PI(3,5)P2 production [160]. In T. gondii,
the single PIKfyve has been studied and revealed enlargement of the apicoplast upon its
conditional disruption [190]. Comparable results were obtained after depletion of an ATG18
homologue, an autophagy protein in T. gondii [197]. TgATG18 was also described to bind both
PI(3,5)P2 and PI3P in lipid blots and liposome binding assays. Similar observations have been seen
with PI3P depletion following PI3K inhibition [251]. Altogether, these data suggest a common role
of TgPIKfyve, ATG18 and PI3K in apicoplast homeostasis and inheritance along with PI3P and
PI(3,5)P2 lipids. In another study, using a pull-down approach in T. gondii, an interactor protein of
PI(3,5)P2 was detected and denoted TgPH1 [251]. However, it binds PI3P in vivo and, similarly,

31
co-localized with common PI3P reporters like the PX domain of p40phox and FYVE domain of
EEA1. Thus far, there is no detectable function of TgPH1 in apicoplast homeostasis or parasite
survival. Yet, PI(3,5)P2 is left to be detected in T. gondii [190] and P. falciparum.
Phosphoinositide metabolism
Three types of enzymes are involved in phosphoinositide metabolism: PI kinases, PI phosphatases
and PI lipases [156]. The PI kinases (PIKs) convert PtdIns to PtdIns monophosphate and PtdIns
monophosphate to PtdIns pyrophosphate in separated activities called “PI-kinases” and “PIP
kinases” [156]. Based on the place of action on the inositol ring PIKs can be categorized into 3
types: PI3-kinases (PI3Ks), PI4-kinases (PI4Ks), and PI-phosphate (PIP) kinases (PIP5Ks and
PIP4Ks).
PI kinases
Phosphatidylinositol-3 kinases (PI3Ks) There are three PI3K classes (I, II and III) which can phosphorylate the D3-position hydroxyl of
the D-myo-inositol head group and generate specific phosphoinositide forms [252]. In vivo, class I
PI3K syntheses PI3P, PI(3,4)P2, and PI(3,4,5)P3. Class II and III PI3K are both able to synthesize
PI3P from PtdIns whilst class II PI3K also synthesize PI(3,4)P2 [232, 252-254]. The PI3K family
appears restricted to eukaryotes, and only the class III PI3K is conserved from yeast to human
[252]. The homologue of class III PI3K in yeast is vesicle protein sorting 34 (Vps34) and exists in
a cytosolic and a vesicular form (Fig. 1. 10) [186]. A phylogenetic study suggests the co-evolution
of Vps34/PI3KIII and its regulatory subunit Vps15 in most eukaryotes [255]. Vps34 is the key
molecular player in trafficking in the endosomal/lysosomal system, the membrane invaginations in
multivesicular bodies (MVB) and the various forms of autophagy [156, 184, 247, 256, 257]. The
essential role of Vps34 in vesicular trafficking is to ensure the proper sorting of proteins to a
prevacuolar compartment and, subsequently, to the vacuole by the production of PI3P [183, 258].
In each of these functions, different proteins are associated with the Vps34p-Vps15 complex
through interaction with its product PI3P [257]. In human, the lipid kinase activity of Vps34 leads
to the formation of PI3P-enriched regions at the ER [185, 259]. This results in the recruitment of
endosomal effector proteins with FYVE domains, PX domains, and other less characterized PIP-
binding domains via PI3P [156, 183, 260]. Consequently, a protein complex comprising Vps34,

32
Vps15, Beclin1 and ATG14 forms at the ER membranes, which is the core for autophagosome
formation. Chemical inhibitors such as Wortmannin and LY294002 have been useful tools to
evaluate PI3K functions. They can block the activities of all PI3K enzymes and induce cell cycle
arrest and cell death [259]. Some new specific inhibitors targeting PI3K function in cancer cells
have been also developed [259].
Based on genome analysis prediction, the P. falciparum genome encodes only one PI3K
(PF3D7_0515300) (Table- 2). Since PfPI3K is related to class III PI3K it is denoted by PfVps34
[160, 162, 190, 240]. PfVps34 is much bigger compared to its yeast homologue.
Figure 10 Kinases involved in phosphoinositide metabolism in yeast, mammalian cells and
apicomplexan parasites.
At left, it is the chemical structure of phosphatidylinositol with the hydroxyl-groups that can be
phosphorylated highlighted in red. The kinases responsible for phosphoinositides synthesis are
indicated on the arrows. The color code on the top right indicating their organisms of origin. In the
bottom right phosphoinositide-specific phospholipase C (PI-PLC) and its products (in yellow box)
are mentioned. Solid redlines are represented pathways that have been confirmed in apicomplexan
parasites and black dotted lines and dotted boxes representing to reactions and products described
in other organisms which have not currently any proof in apicomplexan parasites. Finally, red
dotted lines used for hypothetical reaction pathways in Plasmodium. DAG, diacylglycerol; IP3,
inositol 3-phosphate; Fab1, forms aploid and binucleate cells; Lsb6, Las Seventeen binding; Mss4,
multicopy suppressor of Stt4 mutation; Pik1, phosphatidylinositol kinase; PIKfyve, FYVE domain-

33
containing phosphoinositide kinase; Stt4, staurosporine and temperature sensitive; Vps34, vacuolar
protein sorting [163].
In addition to the main PI3K domains, PfVps34 also contains several different repetitive peptide
motifs of six to eight amino acids [160, 240]. A comparative analysis of PI3K sequences in
Plasmodium showed a high conservation between the main domains, but not the repetitive
sequences [160, 240]. Previously, It has been shown PfVps34 can produce all three forms of D3
phosphorylated products (PI3P, PI(3,4)P2, PI(3,4,5)P3) in vitro [240] but recently a new study
showed it is not true. Thus far, PfVps34 and its product PI3P have been shown to be involved in
hemoglobin endocytosis [240], apicoplast biogenesis [162, 190], autophagy and artemisinin
resistance [195].
In a study, immunoprecipitation-purified PfVps34 showed a significant decrease in its activity in
the presence of PIK inhibitors [240]. Employment of the latter on P. falciparum parasites resulted
in parasite growth defect by the disturbance of the trafficking of hemoglobin-containing vesicles
revealing a role for PfVps34 in the endocytosis of hemoglobin from the host cytoplasm to the FV
[240]. Localization studies demonstrated endogenous PfVps34 localized to the FV, the FV
membrane, vesicular structures concentrated near the PVM/PM and erythrocyte membrane during
the blood stage. The localization profile provides additional support for the function of PfVps34 in
hemoglobin metabolism [160, 240, 261]. However, Wengelnik et al. have challenged the FV and
erythrocyte localization. They reasoned both localizations are very surprising for a generally
cytosolic enzyme, and they also considered the possibility of cross-reactivity of used antisera with
other parasite proteins in this experiment [163].
PfVps34 is also reported to be found at the apicoplast and its product (PI3P), in addition to the
apicoplast, is enriched at the FV membrane and nearby single-membrane vesicles [160]. Over-
expression of a PI3P reporter, GFP-2xFYVE, in T. gondii parasites led to the accumulation of
vesicles containing apicoplast peripheral membrane proteins around the apicoplast. This eventually
resulted in the loss of the apicoplast most likely due to the deleterious effect of the abundance of
the PI3P reporter which might compete with natural ligands of PI3P; therefore, interfering with
vesicular transport of lipids or proteins to the apicoplast [162]. Consistently, conditional depletion

34
of TgVps34 disturbed apicoplast biogenesis and led to apicoplast loss [162, 190]. The parasites
lacking their apicoplast also lost their ability to divide their DNA, which eventually resulted in
parasite death [162, 190]. Despite that, over-expression of a tagged version of TgVps34 under the
control of the tubulin promoter led to a scatter punctate staining in the cytosol (possibly at the ER)
but not at the apicoplast. Moreover, the gene was refractory to gene disruption attempts in T. gondii
[190], P. berghei and P. falciparum indicating its essentiality ([163]- unpublished data), [160, 190,
226].
Another role for PfVps34 and its product PI3P has been proposed in the autophagy process. Their
localization status at the FV membrane, the ER [262] and apicoplast is consistent with the role of
Vps34 in autophagy in yeast and mammals. Interestingly, PfAtg8, an autophagy protein has been
frequently observed to localize to the apicoplast which coincides with PfVps34 and PI3P
localization [191]. Consequently, the apicoplast membrane is proposed as the site of phagophore
formation [263]. On the other hand, due to the lack of some autophagy effector proteins in
Plasmodium genomes, post-translational modifications of ATG proteins are suspected to play a
role in regulating autophagy [264]. Accordingly, cAMP-dependent protein kinase A (PKA) is
proposed to play a regulatory role. As the phosphorylation status of Vps34 and some other
autophagy proteins collected from PlasmoDB are at the typical PKA sites in infected red blood
cells [264]. Another evidence that points to the Vps34 role in the autophagy process arose from the
characterization of TgPROP1 and TgPROP2, homologues to ATG18/WIPI [265]. The two proteins
contain WD-repeat that can bind PI3P, the Vps34 product, for their recruitment to vesicular
structures upon stress and are known to be important for the autophagic process.
The versatile functions of PfVps34 remain to be fully elucidated in P. falciparum as recently a new
function proposed from Mbengue A. et al. work on artemisinin-resistant parasite strains. In a cell-
based screen study to find new PfVps34 inhibitors, they identified three more inhibitors in addition
to Wortmannin and LY29400. One of them was dihydroartemisinin (DHA), the active form of the
artemisinin family of potent anti-malarial drugs [195]. Unexpectedly, they observed a correlation
between PI3P level and point mutations in Kelch13 in the artemisinin-resistant strains of both
laboratory produced and clinical parasites [195]. A specific mutation in Kelch13 (C580Y) found to
be associated with an increased level of PfVps34 (about ~2.5 fold) compared with the wild-type

35
strains [195]. Kelch13 is hypothesized to be a part of E3 ligase and modulates PI3P levels through
modification of PfVps34 (ubiquitination) and, thus, enzyme abundance in the parasite [195]. The
same team reported recently PI3P localization to the ER lumen and adjacent vesicles, both by cryo-
IEM/PI3P antibody and PI3P-fluorescent fused reporters [262]. Interestingly, PI3P enriched
regions at the ER also include Kelch13 protein [262]. Altogether, these findings indicate a new
mechanism of resistance by PI3P-vesicle amplification and PfVps34 level augmentation in the
artemisinin resistance parasites with Kelch 13 mutants [262].
Phosphatidylinositol-4 kinases (PI4Ks) The phosphatidylinositol 4-kinases (PI4Ks) phosphorylate the D4 position of the inositol ring of
PtdIns and produce PI4P. Humans have two type II PI4Ks (α and β) and two type III enzymes (α
and β) that have different domain organization (Fig. 10). PI4Ks are conserved in all the eukaryotes
from yeast to human [23]. Type II PI4K, the most abundant PI4K in mammalian cells and mainly
localized in the trans-Golgi network (TGN) and endosomes, produces more than half of the Golgi
PI4P [266-268] and is involved in many cellular pathways, including PI(4,5)P2 synthesis,
membrane trafficking, signal transduction, phagocytosis, and the exo-endocytic cycle of synaptic
vesicles [156, 266, 269, 270]. The most well-known function of PI4KII is the regulation of vesicle
trafficking toward the PM from the Golgi/TGN by the production of PI4P. The latter includes the
recruitment of the clathrin adaptor AP-complex to the membrane [266]. Its yeast homologue has
been implicated in the regulation of actin polymerization and in endosome mobility, but is not an
essential gene for yeast survival [271]. Homologues of type III PI4 kinases are Stt4p and Pik1p and
both are essential genes in yeast [272]. Stt4p and Pik1 correspond to the human PI4K IIIα and
PI4K IIIβ enzymes, respectively. Stt4p functions are mainly in the cytoplasm and provide the PI4P
substrate for the Mss4p, a PI4P-5 kinase that synthesizes the cytoplasmic pool of PI(4,5)P2 [273].
PI4K IIIα like Stt4p localizes to the PM. While Pik1p was identified to function mainly at the
Golgi and nucleus [274]. In the Golgi, PI4K IIIβ/Pik1 regulates trafficking in the secretory pathway
along with the four-phosphatase-adaptor proteins 1 and 2 (FAPP1 and FAPP2) by synthesizing
PI4P [275, 276]. The PH domain of FAPP protein is commonly used to detect PI4P lipid in live
cells [277]. Disturbance with FAPP expression leads to transport inhibition between TGN and the
PM. In downstream, PI4K simultaneously recruits Rab11 to the membrane [278]. Inhibition of this
interaction abolishes Rab11 localization to the Golgi and blocks transport from the Golgi to the PM

36
[279]. Golgi to the PM [279]. New functions have also been discovered in the regulation of
autophagy, where Pik1p was shown to regulate Atg9p trafficking from the trans-Golgi to the pre-
autophagosomal structure (PAS) [274]. PI4Ks type III are sensitive to PI3K inhibitors such as
Wortmannin [280]. They regulate PI(4,5)P2 pools which are sensitive to hormone signaling. Thus,
they control the intracellular concentrations of second messengers like IP3, DAG, and Ca2+
indirectly [281].

37
Table 2 Phosphoinositide kinases and phosphatases in P. falciparum compared to yeast and T.
gondii.
[163]
*The color code corresponds to observed phenotypes: red, essential; blue, fitness; green, dispensable; black,
not analysed
In P. falciparum, there are three genes that code for phosphatidylinositol 4-kinases: PI4KII
(PF3D7_0311300), PI4KIIIɑ (PF3D7_0311300), and PI4KIIIβ (PF3D7_0509800) (Table. 2). A
knock-out screen performed in P. berghei demonstrated that only PI4KIIIβ is likely essential
Name Annotaion Yeast P. falciparum T.gondii References
PI3K/Vps34 PI3 kinase Vps34 PF3D7_0515300 TGME49_215700 Tawk et al., 2010, 2011;
875aa 2133aa 2935aa Vaid et al., 2010;
Daher et al., 2015
PIKfyve PI3-phosphate 5-kinase PF3D_1412400 TGME49_258960 Daher et al., 2015
3216aa 5839aa
TGME49_258920
5839aa
PI4K II PI4 kinase II Lsb6p PF3D_0311300 TGME49_276170
607aa 953aa 1228
PI4K IIIα PI4 kinase IIIα Sttp4 PF3D_0419900 TGME49_226690
1900aa 5035aa 6780aa
PI4K IIIβ PI4 kinase IIIβ Pik1 PF3D_0509800 TGME49_296010 Kruger et al., 2010;
1066aa 1559aa 1583aa McNamara et al., 2013;
Paquet et al., 2017
PI4P5K (A) PI4-phosphate 5-kinase PF3D_0110600 TGME49_230490 Leber et al., 2009;
1710aa 1293 Brochet et al., 2014
TGME49_245730
2144aa
PI-PLC PI-specific phospholipase PF3D_1013500 TGME49_248830 Fang et al., 2008;
1385aa 1097aa Raabe et al., 2011a, 2011b;
Bullen et al., 2016
SAC1 (A) Inositol-phosphate 5-phosphatase PF3D_1354200 TGME49_316230 Thiearult and Richard, 2017
803aa 1006aa
SAC-like (B) Inositol-phosphate phosphatase PF3D_0705500 TGME49_238400
2814aa 2708aa
SAC-like (C) Inositol 5-phosphatase PF3D_0802500 TGME49_256630
1419aa 2264aa

38
during the asexual stage and the knock-out of two other genes can be tolerated, although they
showed a considerably slower growth compared to their control parasite lines [226]. Most of the
information on the PI4K role in Plasmodium comes from PfPI4KIIIβ. With 1559 amino acids, it
contains armadillo (ARM) repeats in addition to several other structural features characteristic of
PtdIns kinases [282]. ARM repeats are involved in protein-protein interactions in other systems
[282]. Orthologs of PI4KIIIβ are found in all Plasmodium species and are extensively conserved at
the amino acid level, including 97% identity in the catalytic domain between the P. falciparum and
P. vivax orthologs [145]. It has been shown the Pik domain of PfPIKIIIβ is capable of
complementing a yeast temperature-sensitive pik1 mutant and localizes to the Golgi and the
nucleus as its endogenous homologue [282]. In P. berghei, PI4KIIIβ is described to be involved in
ookinete gliding motility [161]. Specific point mutations in PI4KIIIβ have been shown to be related
to a significant decrease in gliding. Later on, the same group found out these sites are regulatory
phosphorylation sites by protein kinase G (PKG) upon gametocyte activation [161].
A study on new antimalarials namely imidazopyrazines (IPZ) has also led to the discovery of
several mutations in PI4KIIIβ in laboratory-generated resistant strains. The mechanism of
resistance to IPZ was consequently demonstrated to act through their association with cytokinesis
in the late stage of Plasmodium parasite [145]. McNamara et al. observed a disruption of plasma
membrane ingression around the developing daughter merozoites due to PI4KIIIβ inhibition in
parasites treated by an IPZ (specifically KDU407). Using a parasite line expressing a PI4P reporter,
they showed that this was the result of an alteration in PI4P pools from an unknown compartment
to the PM. The discovery of PI4K inhibitors in Plasmodium has been very fruitful also in the
Plasmodium treatment field. For example, IPZ not only show a strong potency against blood-stage
field isolates of P. falciparum and P. vivax, but also IPZ molecules like KDU691, GNF179, and
BQR695 inhibit parasite-derived PfPI4KIIIβ at very low nanomolar concentrations [145]. In
addition, some IPZ derivatives such as GNF179 also eliminate quiescent rings which are resistant
to dihydroartemisinin (DHA) [283]. Its analog, KAF156, has demonstrated efficacy in early
clinical trials against a broad range of stages of the Plasmodium life cycle, including liver and
asexual/sexual blood stages as well as parasites bearing Kelch13 (K13) propeller mutations
(reviewed in [283]). These two compounds opened up an opportunity of using PI4K inhibitors as a
combination drug in malaria treatment against dormant rings resulting from DHA treatment and

39
more importantly in blocking malaria transmission. Two other notable PI4K inhibitors are
MMV390048 [145] and BRD73842 [284]. They have been shown to have inhibitory activity
against strains resistant to other antimalarials in all stages of the parasite life cycle excluding
MMV390048 which is not active against liver-dormant stage [147].
Phosphatidylinositol phosphate kinases or PIP-Kinases (PIPKs) PIP kinases are soluble peripherally membrane-bound proteins that have a highly conserved
catalytic core but vary only in their C-terminal [156]. They also do not have considerable
homology with other protein kinases [259]. Phosphatidylinositol phosphate kinases (PIPKs) are
classified into three different categories based on their substrate specificity and sequence similarity
(Fig. 10) [156, 259]. Type I and II PIPKs both produce PI(4,5)P2 though using two different routes.
It is important to mention that the majority of the PI(4,5)P2 pool is produced by type I PIPKs
[259]. Type III PIPK, on the other hand, is responsible for PI(3,5)P2 production [156]. Type I
PIPKs (PIP 5-kinases (PIP5K) synthesize PI(4,5)P2 via phosphorylation of D5 position of PI4P at
the inositol ring [285]. Whereas type II (PIP 4-kinases (PIP4K)) use PI5P to produce PI(4,5)P2
[156, 286]. In mammalian cells, three isoforms exist for type I and II PIPKs [156, 259]. The
relative tissue distribution and the cellular localization of the various isoforms are unique. Type I
PIP5Ks isoforms are found mostly in the PM [287, 288] which is found to be uniquely responsible
for the generation of the PI(4,5)P2 pools linked to Ins(1,4,5)P3 synthesis and Ca2+
signaling in
HeLa cells [289]. PIPKI’s main function has been associated with endocytosis via binding to the
AP-2 adaptor and clathrin [290] and by promoting actin polymerization at the PM [291]. The only
homologue of type I PIPKs in yeast is Mss4 which is also important for actin organization and
membrane morphogenesis [222, 292]. Mss4 also has a role in the late stages of secretion. PIPKII is
found in the cytosol, the nucleus and mainly the cis-Golgi [293, 294]. Recent findings on type II
PIPKs revealed its main function is to control PI5P levels via converting PI5P into PI(4,5)P2 [156,
259]. In this capacity, PIPKIIs are implicated in the regulation of various processes like
proliferation, secretion, ion channel function, stress responses and insulin sensitivity [295, 296].
In Plasmodium, there is a single PIP5K which is likely an essential gene in P. berghei (Table. 2)
[161, 226]. Interestingly, in addition to a PPI 5-kinase domain in the C-terminal, it has also an N-
terminal domain of the neuronal calcium sensor family [297]. The 5-kinase activity of the enzyme

40
was validated using recombinant expression of the Plasmodium PI-kinase domain. Expectedly, the
recombinant protein is as well triggered by the small G protein ADP-ribosylation factor 1 (ARF1)
both from P. falciparum and from mammals [297]. It has been speculated the N-terminal calcium
sensor domain in this enzyme could be a sign of regulation of PI(4,5)P2 production by calcium
[163].
The type III PIPK (PI3P5K) family member is PIKfyve, a single-copy gene that is conserved from
yeast to mammals during evolution [259]. PIKfyve phosphorylates PI3P to produce PI(3,5)P2
[209]. In mammalian cells, the disruption of PI3P5K function leads to a major disturbance in the
dynamics of cellular membranes which results in vacuolation and enlargement of early and late
endosomes [259]. Similarly, its yeast homologue Fab1 is necessary for normal vacuolar
morphology and functions [298, 299]. As to its function, it has been reported PI3P5K localizes to
early endosomes and vacuolar membranes [300-302]. Fab1 acts in a larger signaling complex that
contains Vac14, Vac7, Atg18 [303] and Fig4, a phosphatase that dephosphorylates PI(3,5)P2 into
PI3P [304, 305]. Findings conclude Vac14, Vac7, and Fig4 are regulators of Fab1 and their
coordinated activity control PI(3,5)P2 levels in the multivesicular body (MVB) sorting pathway
and autophagy [248, 298, 300, 306, 307].
In P. falciparum, there is a putative homologue for PIKfyve and its regulator ArPIKfyve (Vac14)
but it is not analyzed yet. PfPIKfyve does not have FYVE domain as its yeast and mammals
homologue. In T. gondii, conversely, the PIKfyve gene encodes a 5639 amino acid protein with an
N-terminal FYVE and a C-terminal 5-kinase domain [190]. Epitope tagging of endogenous
TgPIKfyve did not permit detection or localization of the protein. Over-expression of TgPIKfyve
under control of the tubulin promoter demonstrated a dot-like staining in the cytoplasm and some
co-localization with the apicoplast [190]. The gene is essential as the gene knock-out attempt failed
and conditional disruption of TgPIKfyve resulted in enlarged apicoplast showing its critical role in
apicoplast homeostasis. Interestingly, knock-down of TgArPIKfyve led to the same defect of
apicoplast homeostasis [190].

41
Phosphatases
Phosphoinositide 3-phosphatases All phosphoinositide 3-phosphatases contain a phosphatase domain containing a catalytic motif
called CX5R in their active sites. The PIP 3-phosphatases are classified into two families: PTEN
(phosphatase and tensin homologue deleted on chromosome 10) and multiple members of the
myotubularin family. PTEN protein is localized in both the cytosol and the nucleus [308]. The most
important function of PTEN is the regulation of Akt-kinase via PI(3,4,5) dephosphorylation
specifically at the D3 position on the inositol ring. Therefore, PTEN controls cell survival signaling
through negative control of Akt activation and its downstream signaling pathway [259, 309, 310].
Loss of PTEN protein function is been observed in some human cancers [311, 312]. Hence, it is
also known as a tumor suppressor [259].
The myotubularin (MTM) family consists of 15 members containing a characteristic phosphatase
domain. Myotubularin phosphatases favor PI3P and/or PI(3,5)P2 as their substrate [259]. They are
involved in many processes including cell proliferation and differentiation, autophagy, cytokinesis,
and cytoskeletal and cell junction dynamics [313].
Phosphoinositide 4-phosphatases Thus far, only four proteins have been identified with 4-phosphatase activity in mammalian cells:
two inositol polyphosphate 4-phosphatases A and B (INPP4A and INPP4B) and two
transmembrane proteins denoted as TMEM55A and TMEM55B (Table. 4) [156, 259]. INPP4s
dephosphorylate the D4 position of PI(3,4)P2, whereas the TMEM55 proteins dephosphorylate the
D4 position of PI(4,5)P2 inositol ring [156].
INPP4A is localized to the endosomes in quiescent cells and in the PM in stimulated cells [364].
Upon activation, INPP4A promotes receptor-mediated endocytosis [314]. However, INPP4B
shows a diffused distribution in the cytoplasm. Both INPP4A and INPP4B are shown to be a
negative regulator of Akt signaling [315]. INPP4B is been also suggested being a tumor suppressor
in human cancers [316].

42
TMEM55s show cytosolic and late endosomal membrane localization [317]. Nonetheless, DNA
damage induces TMEM55B translocation from the cytosol to the nucleus [318]. As a result, an
increase in PI5P level is observed, suggesting that TMEM55B plays a role in the control of nuclear
levels of PI5P [319]. This is believed to be related to the p53-dependent apoptosis through PI5P-
ING2 interaction.
Phosphoinositide 5-phosphatases
This enzyme family consists of 10 mammalian and five yeast members [320]. In yeast, INPP5
types I-IV family proteins contain a central catalytic ‘5- phosphatase’ domain, except for 5-
phosphatase I [238, 321]. Therefore, only types II-IV hydrolyze phosphoinositide substrates as well
as soluble inositol phosphates. The type II enzymes include the synaptojanins, OCRL1, INPP5B,
INPP5J, and SKIP. The type III INPP5 enzymes are SHIP1, SHIP2, and the sole type IV enzyme is
INPP5E. INPP4s are involved in a variety of cellular events, such as protein trafficking,
phagocytosis, and synaptic vesicle recycling [238, 322, 323]. Several other enzymes can act as
phosphoinositide 5-phosphatases.
Sac family phosphatases There are five Sac1 domain-containing proteins in both human and yeast [320]. In mammalian
cells, the Sac domain is found in the synaptojanins and in the Sac family phosphatases Sac1, Sac2
and Sac3 [324, 325]. Sac domain containing proteins sometimes show PI-phosphatase activity to
different PIP species [259]. For instance, mammalian Sac1 acts on PI3P, PI4P and PI(3,5)P2 but
not on PI(4,5)P2 as yeast Sac1p [259]. In mammals, the Sac1 protein is found associated with both
the ER and the TGN [326]. Through control of PI4P levels, Sac1 directly modulates growth-
dependent secretion at the Golgi [327]. In yeast, loss of function of Sac1 causes a variety of cellular
defects [320] such as actin cytoskeleton disorganization, drug sensitivities, vacuolar function, cell
wall maintenance and ATP uptake into the ER [328-332]. Sac3 and its yeast homologue Fig4p are
the PI-5-phosphatase that specifically convert PI(3,5)P2 back to PI(3)P [305, 333]. As mentioned
above, Sac3/Fig4p is part of a complex with the PI(3)P-5-kinase Fab1p and the scaffold protein
Vac14p. This complex is involved in regulating the subcellular levels of PI(3,5)P2 [303, 306].

43
Bacterial PI-phosphatases Several studies on host-bacterial infections have clearly shown a common mechanism of infection
via the exploitation of the host PIP metabolism by bacterial virulence factors [148, 149]. One way
by which several bacterial pathogens do this is by injecting PIP metabolizing enzymes like PI
phosphatases into the host cell. The enterobacteria Salmonella enterica injects the PI phosphatase
SigD/SopB into the host and this is essential for enteropathogenicity [334]. Similarly, Shigella
flexneri [335] and Mycobacterium tuberculosis [336], by secreting PI phosphatases, exploit their
host cells. The growing body of data demonstrates the importance of this strategy employed by
bacterial pathogens [337, 338].
PI phosphatases in apicomplexan parasites In P. falciparum, there are three homologue sequences to the Sac family including Sac1 (A), Sac-
like (B) and Sac-like (C) (Table. 2). Only Sac1 has been studied recently [339] and was found to be
localized to the ER and transitional ER. It is likely essential according to the unsuccessful knock-
out effort. In systematic knock-out analysis, PbSac-like B was found to be essential, and the PbSac-
like knock-out resulted in a slower growth [226]. And finally, there are four putative Sac family
phosphatases in T. gondii [340]. Among them, the homologue of Sac-like B and C are essential,
Sac1 A shows slower growth, and Sac-like D is dispensable for parasite survival.
PLC and PI signaling Phospholipase C, PLC is the enzyme that produces two signaling molecules IP3 and DAG via the
hydrolysis of the phosphodiester bond at the SN3 position of PI(4,5)P2 (Fig. 10) [156]. IP3 is
responsible for the liberation of calcium ions from the internal stores such as the ER while DAG
activates protein kinase C (PKC) [341, 342]. PLC is an important player in receptor-regulated
signal transduction [343]. PLCs are detected both in membranes and soluble fractions, and they
mostly work in the PM [156]. Each PI-PLC contains a common basic core such as a PH domain,
four EF-hands, and a C2 domain [344]. PLC recruitment, as well as its regulation, are mediated
through interactions of effector molecules with the PH domain and the C2 domain [156]. A PLC
homologue, Plc1, has also been identified in yeast [345, 346]. Yeast deficient in the plc1 gene
demonstrate growth defects, impaired cell wall integrity and decreased osmotic resistance [346].
The PH domain of Plc1 has been structurally characterized and exhibits binding to P(4,5)P2, which

44
has been employed by many studies to investigate the roles of PI(4,5)P2 and its localization [347-
349].
Many critical events, such as gametocyte activation [350-353], ookinete motility [161], egress
[161, 354] and sporozoite gliding motility [227, 228] are controlled via PLC/PI(4,5)P2/Ca2+
signaling in the Plasmodium life cycle (Fig. 11). A single plc gene has been studied in both
Plasmodium and T. gondii [51, 352, 353, 355] which is essential for parasite survival and localizes
to the PM. In T. gondii, PLC is also detected in the parasite cytosol and apical accumulations [51].
It has been shown that gametocyte activation is the result of PI(4,5)P2 hydrolysis by the calcium-
dependent PI-PLC activity [350, 351]. Moreover, recent investigations in P. berghei gametocytes
revealed redistribution of a PI(4,5)P2-reporter from the PM to the cytosol upon gametogenesis
stimulation suggesting a link between PI(4,5)P2 cleavage and calcium-dependent PI-PLC signaling
in preparation for gametogenesis [353].
In T. gondii, PLC/PI(4,5)P2/Ca2+ is implicated in the regulated exocytosis of the invasion
organelles [51]. During invasion, following external stimuli, PLC produces IP3 and DAG. IP3
causes Ca2+ release from internal stores and CDPKs activation. In parallel, DAG is converted to
phosphatidic acid (PA) by Diacylglycerol kinase-1 [51]. This PA is then bound by a specific
binding protein (TgAPH) on the microneme surface which leads to microneme exocytosis [51,
356]. Consequently, PLC localizes to the invasion organelles in extracellular parasites [51]. In P.
falciparum a rise in cytosolic Ca2+, potentially through the phospholipase C (PLC) pathway,
triggers microneme release [84]. Since P. falciparum APH (PfAPH) also binds selectively to PA
both on PIPstrips and in liposome assays [51], exocytosis of the invasion organelles might follow
the same principles. In other words, the PLC/PI(4,5)P2/Ca2+ pathway and following PA sensing
events might be a conserved mechanism in apicomplexan. Therefore, it is proposed that PfAPH
interacting with PA, bring the microneme and PM to a closed position to fuse together [51, 356] via
the involvement of SNARE-like proteins such as DOC2.1 [357, 358]. It is important to note that
there is no predicted PKC orthologue in apicomplexan parasites [163].

45
PKG/Ca2+/PIP signaling in Plasmodium The key regulatory molecule in the PI-PLC/PI(4,5)P2/Ca2+ signaling is protein kinase G (PKG).
PKG modulates PIP metabolism events that lead to the synthesis of IP3 and eventually Ca2+
release (Fig. 11) [161]. To synthesize PI(4,5)P2 from phosphatidylinositol, enzymes like PI4KIIIβ
and PI4P5K are required to be activated. Consequently, the resulting PI(4,5)P2 is hydrolyzed by
PI-specific phospholipase C (PI-PLC) [156]. Eventually, the release of Ca2+ from intracellular
stores activates specific Ca2+-dependent protein kinases (CDPKs) depending on the parasite stage.
There is good evidence for the regulation of PI kinases, PI-PLC and CDPKs by phosphorylation
through a cascade involving protein kinase G (PKG) [161, 228]. Pieces of evidence for PKG in
controlling this signaling cascade are also arising in many other processes. Redistribution of an
epitope-tagged version of P. berghei PI4P5K from the cell periphery of ookinetes to the cytosol by
PKG inhibitor suggests its regulatory role through the subcellular localization of PI4P5K [161]. In
gliding ookinetes, PKG inhibition also causes a specific decrease in the relative abundance of PIP-
mono- and -bisphosphates. Similarly, in P. falciparum schizonts, a decrease in PIP-mono-, bi-, and
trisphosphates, and an increase in the PIPs were observed upon PKG stimulation which lead to the
premature egress of the merozoites [161]. Moreover, the presence of an N-terminal domain of the
neuronal calcium sensor family in PfPI4P5K could be an indication that PI(4,5)P2 production is
also controlled by calcium [297].
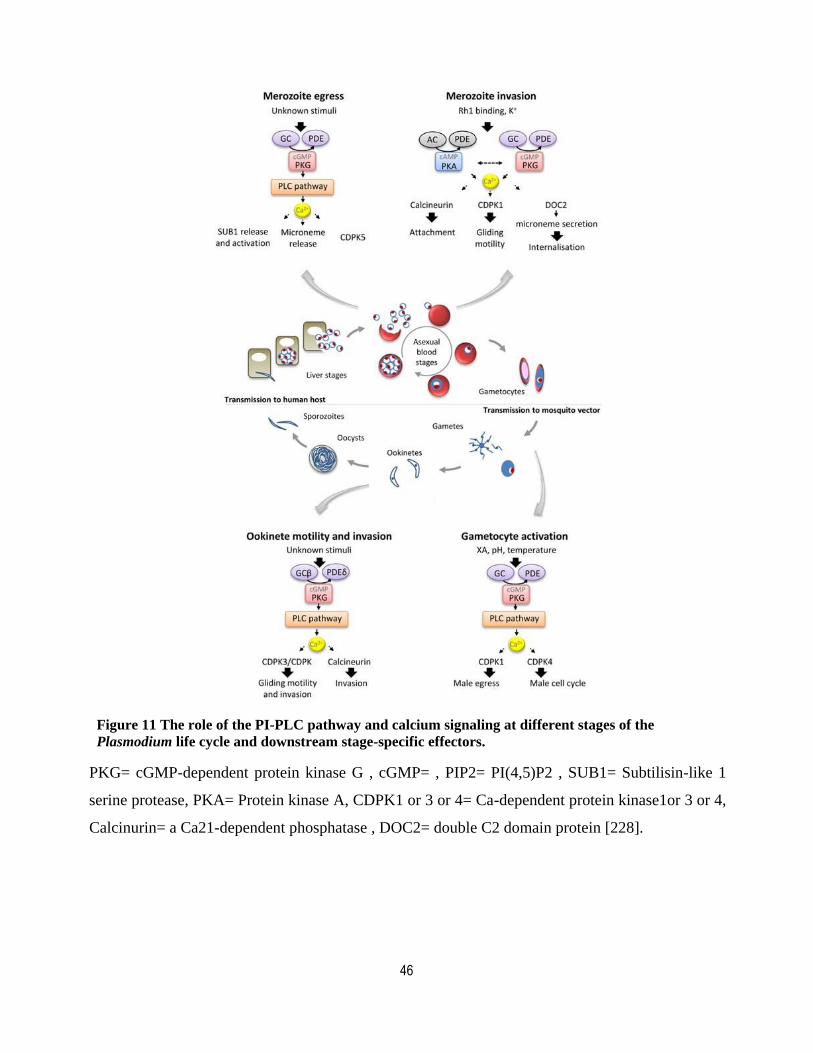
46
Figure 11 The role of the PI-PLC pathway and calcium signaling at different stages of the
Plasmodium life cycle and downstream stage-specific effectors.
PKG= cGMP-dependent protein kinase G , cGMP= , PIP2= PI(4,5)P2 , SUB1= Subtilisin-like 1
serine protease, PKA= Protein kinase A, CDPK1 or 3 or 4= Ca-dependent protein kinase1or 3 or 4,
Calcinurin= a Ca21-dependent phosphatase , DOC2= double C2 domain protein [228].

47
Chapter 1: Hypothesis and problem statement
Malaria affects more than 200 million people each year and is the most widespread parasitic
disease confronted. The new wave of resistance to current medicines has already arisen from
South-East-Asia, while the malaria vaccine, RTS,S, is still far from being fully effective.
Accordingly, there is an urgent need for new therapeutic methods and vaccine strategy.
Plasmodium is an intracellular parasite and evades the human immune system by hiding and
exploiting erythrocytic cells. The parasite is only exposed to the host immune system for a very
short period of time, once before migrating from the dermis to the hepatocytic cells and once when
newly formed merozoites are released into the blood stream to find a new host cell. Therefore,
many protein candidates for vaccine-based strategies are proteins found in the apical organelle
which are molecular players involved in erythrocyte invasion. Similarly, understanding of the
pathways involved in the biogenesis of these organelles could potentially lead to new drug targets
since without them, the parasite cannot invade. Despite this, the mechanism governing the
secretory pathway toward apical organelles and their biogenesis are still poorly known. Work from
our lab has proposed a Golgi-to-apical organelle protein-sorting model in asexual blood stage
parasites. In this model the presence of specific microdomains inside the Golgi apparatus has been
suggested that determine the final destination of secretory proteins. Based on this theory, the
microdomains have a specific composition of lipid and protein, which license the selection of
proteins destined for the different cell compartments. A specific signal in the microdomains
recruits cytosolic effector proteins, which lead to vesicle budding and the departure of cargo
protein to its next destination. However, the lipid and protein composition of such microdomains
are mostly unknown.
1.1 Hypothesis and objectives
In 2009, Richard et al. have demonstrated that the rhoptry-associated membrane antigen, (RAMA)
through an interaction with the N-terminus of rhoptry-associated protein 1 (RAP1), recruited
RAP1, 2 and 3 into rhoptry-destined protein complexes [359]. Disruption of this interaction
resulted in the mistargeting of RAP1 to the PV, the default secretory pathway. The role of RAMA
in recruiting these proteins is reminiscent of escorter proteins in higher eukaryotic cells that target
cargo proteins to different membrane destinations through their interaction with special adaptor

48
proteins. These data suggested that one way that proteins could be differentially targeted to the
apical complex would be by clustering of protein complexes in membrane microdomains.
However, how these microdomains would be differentiated from one another and the mechanisms
behind the actual trafficking from the Golgi to the apical organelles is currently unknown. In
eukaryotes, there are seven different phosphoinositides, which define the identity of subcellular
membranes and act as a code that can be read by PIP effector proteins. Uninfected erythrocytic
cells only produce two PIP species, PI4P and PI(4,5)P2. This changes dramatically after infection
by P. falciparum since five PIP species are detected in iRBCs which imply the importance of these
tiny phospholipids for parasite survival. Recently, an interesting study from Bullen et al. proposed
that in Toxoplasma, PIP signaling plays a role in invasion through induction of microneme
exocytosis [51]. Based on their model, following external stimuli, TgPI-PLC activity increased and
led to IP3 and DAG production. Consequently, IP3 production induces calcium release, which in
turn stimulates calcium-dependent kinases that starts a cycle of phosphorylation of their
downstream substrates. Concurrently, the generated DAG is phosphorylated to PA, which is
detected by a PH-containing protein called TgAPH. TgAPH is on the microneme membrane and
has been shown to be essential for microneme exocytosis. P. falciparum possesses a homologue of
TgAPH but whether it plays the same role is currently unknown. Consistently with this, TgPI-PLC
is observed in merozoite apex [51] which suggests that PIPs might also play a role in RBC invasion
by P. falciparum. Our working hypotheses were thus:
1. Phosphoinositides are involved in defining the identity of subcellular membranes in the malaria
parasite P. falciparum and therefore play a central role in the differential trafficking of proteins to
the apical complex.
2. Effectors of phosphoinositides are among the molecular players involved in protein trafficking
toward the apical organelles and they might play essential roles in their biogenesis and their
exocytosis.
To test the hypotheses, I pursued the following specific objectives:
1. To develop fluorescent probes to monitor the intracellular distribution of phosphoinositides in P.
falciparum.

49
2. To characterize a putative PIP-binding protein and investigate its potential role in trafficking of
proteins to the apical complex and the invasion process.

50
Chapter 2: A map of the subcellular distribution of
phosphoinositides in the erythrocytic cycle of the malaria
parasite Plasmodium falciparum
Avant-propos
The paper presented in Chapter 3, entitled “A map of the subcellular distribution of
phosphoinositides in the erythrocytic cycle of the malaria parasite Plasmodium falciparum”
has been written as a ''scientific paper''. I performed and analyzed all the experiments. I also
designed some of the experiments and wrote the manuscript. Angana Mukherjee supervised the
project and wrote the manuscript. Dave Richard designed and supervised the project, and wrote the
manuscripts. The paper has been published in the ''International Journal for Parasitology'' (IJP) in
January 2018.

51
Résumé
Bien qu’ils représentent un faible pourcentage des lipides cellulaires des cellules eucaryotes, les
phosphoinositides (PIPs) sont impliqués dans divers processus essentiels tels le trafic intracellulaire
et la transduction de signal. Au centre de leurs diverses fonctions se retrouve la distribution
différentielle des différentes espèces de PIP dans des compartiments membranaires spécifiques,
grâce l’actions de différentes kinases, phosphatases et lipases. Malgré leur importance dans le cycle
de vie du parasite de la malaria, la distribution subcellulaire de la plupart des espèces de PIP est
encore inconnue. Les travaux présentés ici montrent la localisation subcellulaire de plusieurs
espèces de PIP tout au long du cycle érythrocytaire de Plasmodium falciparum. En effet, nous
avons montré que le PI3P se trouve principalement à l’apicoplaste et à la membrane de la vacuole
alimentaire, et que le PI4P est associé à l'appareil de Golgi et à la membrane plasmique. Quant au
PI (4,5) P2, il est plutôt détecté au niveau de la membrane plasmique et à l’intérieur de structures
sphériques en forme de cavité. Finalement, nous avons montré que le PI5P se localise à la
membrane plasmique, au noyau et potentiellement dans le reticulum endoplasmique de transition.
Notre carte de la distribution subcellulaire des différentes espèces de PIP chez P. falciparum est un
outil important qui permettra de mieux comprendre la dynamique de ces lipides chez ce parasite
morte.

52
Article
A map of the subcellular distribution of phosphoinositides in the erythrocytic
cycle of the malaria parasite Plasmodium falciparum
Zeinab Ebrahimzadeh, Angana Mukherjee, Dave Richard*
Centre de recherche en infectiologie, CRCHU-Université Laval, 2705 Boul. Laurier
Québec (QC), Canada, G1V 4G2
*Corresponding author. Dave Richard, Tel.: 1-418-525-4444 ext 47975; fax: 1-418-654-2715.
E-mail address: [email protected] (DR)
Note: Supplementary data associated with this article.

53
Abstract
Despite representing a small percentage of the cellular lipids of eukaryotic cells, phosphoinositides
(PIPs) are critical in various processes such as intracellular trafficking and signal transduction.
Central to their various functions is the differential distribution of PIP species to specific
membrane compartments through the actions of kinases, phosphatases and lipases. Despite their
importance in the malaria parasite lifecycle, the subcellular distribution of most PIP species in this
organism is still unknown. We here localize several species of PIPs throughout the erythrocytic
cycle of Plasmodium falciparum. We show that PI3P is mostly found at the apicoplast and the
membrane of the food vacuole, that PI4P associates with the Golgi apparatus and the plasma
membrane and that PI(4,5)P2, in addition to being detected at the plasma membrane, labels some
cavity-like spherical structures. Finally, we show that the elusive PI5P localizes to the plasma
membrane, the nucleus and potentially to the transitional endoplasmic reticulum (ER). Our map of
the subcellular distribution of PIP species in P. falciparum will be a useful tool to shed light on the
dynamics of these lipids in this deadly parasite.
Keywords: Malaria, Phosphoinositide, Subcellular distribution

54
Introduction
Despite recent progress in reducing mortality and morbidity, malaria still takes a tremendous toll
on human health. The disease is caused by five species of the genus Plasmodium with Plasmodium
falciparum as the causative agent of the most virulent form of malaria. In 2015, there were 438,000
deaths from malaria, mostly children in sub-Saharan Africa [360]. The symptoms of human malaria
are caused by the asexual red blood cell (RBC) stages of the parasite. Within the RBC, the parasite
develops in its own compartment surrounded by the parasitophorous vacuolar membrane (PVM).
During this symptomatic phase, the intraerythrocytic parasite modifies the host RBC to make it
suitable for its survival and growth. The ~48 h asexual life cycle of a parasite is complex, with
three successive distinct morphological stages (ring, trophozoite and schizont). Ultrastructural
studies have indicated that ring stage parasites are cup shaped [26, 43]. At this stage, host cell
modification is at its peak with the parasite exporting remodeling and virulence proteins into the
RBC [361]. The trophozoite stage, from approximately 24 to 36 h after invasion, is characterised
by rapid parasite growth, repeated DNA replication and the appearance of hemozoin (inert remnant
of digested hemoglobin) in the food vacuole. Finally, the parasite enters the schizont stage (~36-48
h) during which individual nuclei and associated organelles are partitioned to produce daughter
parasites during a specialised type of cytokinesis called schizogony, and generates up to 32
daughter merozoites [362]. The host cell then ruptures and releases the merozoites which may then
go on to invade new RBCs. The parasite, apart from possessing the classically observed organelles
of eukaryotic cells, contains some more atypical compartments, probably driven by its need to
survive in an unusual biological niche. These include the apicoplast, a four-membrane bounded
plastid-like organelle, the food vacuole, a lysosome-like compartment that contains proteolytic
enzymes that degrade host cell hemoglobin internalised by the parasite, and finally specialised
secretory organelles forming the apical complex (micronemes, rhoptries and dense granules),
located at the apical pole of the merozoite and playing critical roles during host cell invasion.
Uncovering the principles governing the biogenesis of these various cellular compartments is of
great interest because they likely represent sources of new potential targets for the development of
antimalarial therapeutic drugs.
Phosphoinositides are phospholipids found on the cytosolic surface of a variety of intracellular
membranes in eukaryotic cells. They contain an inositol head group that can be reversibly

55
phosphorylated at three positions. Although they account for less than 1% of total cellular lipids,
they are of paramount significance in a variety of processes such as signal transduction, cell
motility, cytoskeletal reorganisation, DNA synthesis, cell cycle, adhesion, membrane transport,
permeability and trafficking [158, 363, 364]. Phosphorylation and dephosphorylation of inositol
head groups by different kinases and phosphatases result in seven different phosphatidyl inositol
phosphate (PIP) species including three phosphatidylinositol monophosphates (PI3P, PI4P and
PI5P), three phosphatidylinositol biphosphates (PI(3,4)P2, PI(4,5)P2 and PI(3,5)P2) and one
phosphatidylinositol triphosphate (PI(3,4,5)P3 [365]. Each of these seven PIPs has a unique
subcellular membrane distribution (see below). Furthermore, within a given membrane, the
localization of specific PIPs can be heterogeneous. Altogether, these enzymatic reactions lead to a
specific PIP code, where certain subcellular membranes are enriched or depleted of specific PIPs,
creating a membrane identity (reviewed in [155]).
To shed light on the respective roles and localisation of the different species of PIPs, live-cell
imaging studies have provided relatively clear maps of the intracellular PIP distribution in several
types of eukaryotic cells (reviewed in [155]) [366]. PI3P largely resides in early endosomes [173]
and contributes to endosomal maturation, cargo protein degradation and cell signaling [367-369],
and autophagy [370]. Some pools of specific PIPs can be found at more than one location in a cell.
For example, PI4P is highly enriched at the Golgi membrane where it is involved in trafficking
events and a deficiency in PI4P has been shown to affect Golgi structure and function (reviewed in
[371]) while another pool is found at the plasma membrane where it acts as a precursor for the
synthesis of PI(4,5)P2 [372-374]. PI(4,5)P2 exists predominantly on the plasma membrane
although it can also be delivered from the Golgi complex by membrane carriers [375, 376].
PI(4,5)P2 is implicated in a number of cell surface related events such as exocytosis, endocytosis,
phagocytosis, cell motility, cell adhesion, microtubule capture, regulation of integral membrane
proteins and has a central role in cell signalling through its degradation into inositol 1,4,5
triphosphate (IP3) and diacylglycerol (DAG) by the action of Phospholipase C (reviewed in [158]).
Unlike constitutive PIPs, others such as PI5P, PI(3,4)P2 and PI(3,4,5)P3 are rapidly and transiently
produced in response to the activation of cell surface receptors and other stimuli. PI(4,5)P2 is the
precursor for the synthesis of PI(3,4,5)P3, critical in signalling pathways involved in cell
proliferation, migration, chemotaxis, phagocytosis and micropinocytosis (reviewed in [377, 378]),

56
which, together with PI(3,4)P2, accumulate in the plasma membrane but only after specific
signalling activation [156, 379]. PI(3,4)P2 is also found at the early endocytic pathway and
although poorly characterised, specific roles of PI(3,4)P2 have been described in both clathrin-
dependent and independent endocytosis [232, 380]. There are only small amounts of PI5P in
resting cells and so its function remains poorly characterised but after certain types of stimuli, PI5P
accumulates at the plasma membrane and in the nucleus and nuclear PI5P has been proposed to act
as a stress response element [319, 381]. As for PI(3,5)P2, it is found to be enriched in the late
compartments of the endosomes where it may regulate endosomal operations (fission and fusion)
that maintain endomembrane homeostasis and proper performance of the trafficking pathways
emanating from or traversing endosomes in yeast, mammalian and plant cells [298, 301, 382-384].
Despite their central role in the cellular biology of several types of eukaryotic cells, comparatively
little is known about the role of PIPs in the malaria parasite P. falciparum. Normal mature
mammalian RBCs have only small amounts of detectable PIPs, however upon infection with P.
falciparum, the phosphoinositide profile undergoes profound changes with important increases in
PI3P, PI4P and PI(4,5)P2, and the detection of some small amounts of PI(3,4)P2 and PI(3,4,5)P3
[160, 385]. The latter finding is interesting since these two PIP species are usually not detected in
other unicellular organisms where only class III PI3-kinases are present [160, 386]. What functions
the individual PIPs might play has recently become the focus of a number of studies. PI3P is
thought to play a role in processes such as hemoglobin uptake to the food vacuole [159, 160, 196,
240, 387], biogenesis of the apicoplast [160, 162], resistance to artemisinin [195] and export of
proteins to the erythrocyte [388] although the latter data have recently been challenged [194].
Inhibition of a P. falciparum PI4kinase with imidazopyrazines and quinoxalines has revealed that
PI4P was likely critical for proper plasma membrane ingression during schizogony [145]. Much of
what is known with regards to PI(4,5)P2 in malaria seems to be related to its involvement in
calcium signalling cascades as a substrate for Phospholipase C in processes such as male
gametocyte exflagellation [350, 351], gametocyte activation [353], synchronization of the
erythrocytic cycle by the hormone melatonin [389, 390], sporozoite gliding motility [227],
merozoite egress [45, 229] and potentially subsequent erythrocyte invasion [391]. Finally,
PI(3,5)P2 has not been detected in P. falciparum-infected RBCs (iRBCs) [160] and PI5P has also
not yet been reported for any Plasmodium spp.

57
In an effort to gain a deeper understanding of the multiple roles that PIPs are likely to play in the P.
falciparum erythrocytic cycle, we undertook a comprehensive analysis of the subcellular
localisation of each individual PIP species throughout the asexual blood stages. Our results show
that the distribution of most PIPs is quite dynamic between the different steps of the cycle. We
confirm that PI3P is found at the digestive vacuole membrane and the apicoplast and reveal that
PI4P is found at the Golgi apparatus and, similar to PI(4,5)P2, at the plasma membrane.
Furthermore, we demonstrate the presence of PI5P in P. falciparum and show that it localises to the
plasma membrane and potentially the transitional endoplasmic reticulum. Our map will be a useful
tool in the further unravelling of the numerous roles played by PIPs in the malaria parasite P.
falciparum.
Materials and methods
This study was approved by the Canadian Blood Services (CBS) research ethics board, project
number 2015.001 and by the Centre Hospitalier Universtaire (CHU) de Québec institutional
research board, Canada, project number 2015–2230, B14-12-2230, SIRUL 104595. Written
consent was obtained by the CBS for all study participants.
2.1. Parasite culture
Plasmodium falciparum 3D7 parasites were originally obtained from David Walliker at Edinburgh
University, Scotland. Plasmodium falciparum asexual stage parasites were cultured under standard
conditions in RPMI-HEPES medium at 4% hematocrit (human erythrocytes of O+ group) and 0.5%
(w/v) AlbumaxTM
(Invitrogen, Canada) [392]. Parasites were kept at 37°C in a gas mixture of 5%
oxygen, 5% carbon dioxide and 90% nitrogen. Synchronisation was performed using sorbitol as
previously described [393].
2.2. Parasite transfection
Tightly synchronous ring stage parasites were transfected with 100 µg of purified plasmid DNA
(Promega) as described previously[394]. The transfectants were selected using blasticidin (BSD) at
2.5 µg/ml. Co-transfections were performed with 100 µg of each plasmid and parasites were
selected with BSD together with 10 nM WR99210 (Jacobus Pharmaceuticals, USA) or 5 nM
DSM1 (MR-4), depending on the selectable marker.

58
2.3. Construct generation
To create the PIP sensors, PBDs were PCR amplified with flanking restriction sites from human
cDNA (PXP40
, PHFAPP
, PHBTK
) or from a series of PIP sensor plasmids previously designed for use
in Arabidopsis thaliana (2xPH PLCδ
and PHTAPP1
) [366]. In the case of the DOK5 PH domain, PCR
was done on a plasmid containing the domain fused to GST [395]. Most PIP sensors were cloned in
the pGFP-glmS vector from Prommana et al. [396]. This plasmid allows the expression of genes of
interest with GFP fused at their C-terminus. For the triple hemagglutinin (HA) tagged PHFAPP
and
PHDOK5
, the domains were cloned using 5’ AvrII and 3’Xho1 in a pLN-GFP vector where the GFP
had been replaced by a 3HA tag [397]. Primer sequences and restriction sites used are listed in
Supplementary Table S1.
2..4 Fluorescence imaging
The PBD expressing cell lines were imaged using a GE Healthcare Applied Precision Deltavision
Elite microscope with a sCMOS camera and the obtained images analyzed with the SoftWorx
software. For live cell imaging, infected RBCs were incubated with 0.1 μg/ml of DAPI (Invitrogen)
for 10 min at 37˚C. For IFAs, parasites were fixed either in solution using 4% paraformaldehyde-
0.0075% glutaraldehyde (ProSciTech, Australia) [398] or on slides using 100% methanol. After
blocking in 3% BSA (Sigma Aldrich, Germany) the cells were incubated for 1 h with rabbit
polyclonal anti-ERD2 1:2000 [399]; mouse monoclonal anti-HA 1:2000; mouse monoclonal anti-
RAP1 1:2000 [400]; rabbit polyclonal anti-RON4 1:1000 [359, 401]; rabbit anti-AMA1 1:1000
[402]. Bound antibodies were then visualised with Alexa Fluor-594 anti-rabbit IgG and Alexa
Fluor-488 anti-mouse IgG diluted 1:1000. Parasites were mounted in Vectashield (Vecta
Laboratories, USA) containing with 0.1 μg/ml of DAPI. Images represent single optical slices
except where indicated.
2.5. Western blot analysis
Mixed-stage P. falciparum parasite cultures were saponin-lysed and resuspended in SDS protein
sample buffer. Proteins were separated on 10% SDS-PAGE gels and transferred onto
polyvinylidene fluoride membranes (Millipore, USA). Monoclonal mouse anti-GFP (Roche
Canada, clones 7.1 and 13.1), rabbit polyclonal anti-GFP (Abcam, USA, AB6556) or rabbit

59
polyclonal anti-mCherry (Abcam; AB167453) were used at a 1/500 dilution in 1% skimmed milk
(w/v). Appropriate horseradish peroxidase- coupled secondary antibodies were used and
immunoblots were revealed by chemiluminescence (ECL, Amersham Biosciences, Canada).
Results and Discussion
3.1. Generation of a set of PIP biosensors expressed throughout the P. falciparum asexual blood
stages
Proteins involved in various aspects of PIP metabolism often have very well-defined lipid binding
domains through which they interact with specific species of PIPs. Sensors made up of isolated
lipid-binding domains with the ability to recognize a single type of PIP paired with fluorescent
reporters have been used to determine the subcellular distribution of PIPs in a variety of living cells
[156, 366, 373]. Here, in order to localise the various PIP species in P. falciparum asexual blood
stages, we selected six different PIP binding domains (PBDs) with extensive literature supporting
their PIP specificity (Supplementary Table S2). We did not choose any PBD binding to PI(3,5)P2
as this species was not detected in P. falciparum iRBCs and a P. falciparum homologue of
PIKfyve/Fab kinase, the enzyme responsible for the synthesis of PI(3,5)P2 has not yet been
identified [159, 160]. Our initial strategy was to generate a set of PIP sensors where each PBD
would be fused separately to GFP and to mCherry, cloned in plasmid vectors with different
selectable markers (at least two different selectable markers, for a total of four plasmids for each
PBD) to allow co-transfection and subsequent colocalization analysis of multiple PIP species in the
same cell. However, this turned out to be much more complicated than we anticipated. In the end,
most of the data obtained were with a plasmid containing the blasticidin deaminase selectable
marker. In addition, for some of the PBDs, we never managed to obtain good expression levels
with either GFP, mCherry or mCitrine fusion. All our sensors were placed under the control of the
well characterised constitutive P. falciparum Heat shock protein 86 promoter [403, 404]. A caveat
of using PIP sensors is that these will compete with native PIP effectors, which might result in
dominant negative effects. One method that has been used to decrease the likelihood of this
happening is through the use of mild promoters [175, 373]. In our experience with P. falciparum
transgenics, this has never been critical since the parasite seems very capable of decreasing the
expression of any transgene to non-detrimental levels, with no measurable decrease in parasite

60
growth. This translates into a smaller number of parasites with detectable levels of fluorescence or
even no fluorescence at all if the transgene is not tolerated (see Section 3.2).
3.2. Subcellular localisation of PI3P
To determine the localisation of PI3P, we chose the Phox (PX) domain of the mammalian P40phox
protein that has been successfully used in various organisms to visualize PI3P [366, 405]
(Supplementary Table S2). In ring stage parasites, the GFP signal was thin, rod shaped and
somewhat away from the nucleus (Fig. 1A). As the parasite matured to the trophozoite and schizont
stages, the sensor was detected at the membrane surrounding the food vacuole, recognisable by the
dark crystals of hemozoin in the bright field image (Fig. 1B, C). As previously reported by Tawk
and colleagues (2010) using a PI3P sensor made of 2xFYVE domains, some regions of the food
vacuole membrane had more intense GFP labelling that sometimes appeared as small circles or rod
shaped structures (Fig. 1B, Supplementary Fig. S1A, arrows). Some parasites also displayed
cytosolic vesicle-like structures at or near the parasite membrane (Supplementary Fig. S1A,
arrowheads). Western blot was performed on parasite extracts to control for the integrity of the
sensor and this revealed a major band around the expected size of 46.4 kDa for PXP40
-GFP and a
very faint band around 26 kDa, potentially some degradation product (Fig. 1D). Several lines of
evidence strongly support a critical role for the sole PfPI3K in hemoglobin transport from the host
cell cytoplasm to the parasite food vacuole: localisation studies using a specific antibody revealed
that PfPI3K is found inside the food vacuole and in vesicular structures near the parasite
membrane/PVM [240]; incubation of P. falciparum parasites with the PI3K inhibitors wortmannin
and LY294002 results in a significant reduction of parasite growth potentially through a defect in
the transport of hemoglobin from the host cell to the food vacuole [240, 406, 407]. Whether the
PI3P found as these locations results directly from the activity of PfPI3K is not known, however.
From the trophozoite stage, a bright spot of fluorescence not associated with the food vacuole was
also seen and it increased in number as the parasite went through schizogony (Fig. 1B, C,
arrowheads). In a few late trophozoites and schizonts, we sometimes observed the PXP40 –GFP
signal in a more complex, branched form, reminiscent of the apicoplast (Supplementary Fig. S1B)
[408]. The PI3P focus had previously been shown to represent the apicoplast [160] and to further
confirm this, we co-transfected a PXP40
-mCherry sensor in a parasite line expressing GFP fused to

61
Enoyl acyl carrier protein reductase (ENR), an apicoplast resident protein [66, 409]. Observation of
doubly-labelled cells was a very rare event but when it occurred, the signal from ENR-GFP was
always colocalizing with the foci of PXP40
, confirming an enrichment of PI3P at the apicoplast
membrane (Supplementary Fig. S2). Further support for the critical role of PI3P in apicoplast
homeostasis comes from an elegant study in the related apicomplexan Toxoplasma gondii where
conditional ablation of TgPI3K expression resulted in altered morphology of the organelle and
delayed parasite death, proving the essential contribution of this lipid kinase in preserving
apicoplast integrity [190]. Whether the same is true in Plasmodium remains to be demonstrated.
Control parasites transfected with GFP alone showed a cytosolic distribution throughout the blood
stages (Supplementary Fig. S3).
In conclusion, our results confirm that PI3P is found at numerous intracellular locations throughout
the P. falciparum asexual blood stages where it likely plays critical roles in a variety of processes
such as apicoplast biogenesis and hemoglobin endocytosis. Of interest, PI3P levels do not fluctuate
greatly in most eukaryotic cells so the fact that P. falciparum increases PI3P levels up to four-fold
in proportion compared with total phosphoinositides is very unusual [160]. Targeting the enzymes
responsible for the generation of PI3P might therefore represent an attractive antimalarial strategy
and recent results suggesting that PfPI3K might be a target of artemisinins supports this [195].
3.3. Subcellular localisation of PI4P
To investigate the subcellular distribution of PI4P we used the well-characterised Pleckstrin
homology (PH) domain of the human Four-phosphate-Adaptor Protein 1 (FAPP1) [235]. We
initially transfected parasites with a sensor containing a single FAPP1 PH domain but capturing
images turned out to be difficult as the signal faded out quickly (results not shown). To increase the
avidity of our PI4P sensor, we used two FAPP1 PH domains in tandem as this was shown to
provide a better and more stable signal in other systems [275, 366]. In ring stage parasites, the
sensor labelled the plasma membrane together with an intense focus of GFP staining (Fig. 2A). As
the ring matured to the trophozoite stage, distinct intracellular foci arose, in addition to the plasma
membrane signal. This distribution was also observed by McNamara and colleagues using the PI4P
probe GFP-PHOsh2
[145]. Some cytosolic signal could also be observed in some parasites which is
sometimes indicative of the degradation of a portion of the sensor resulting in the presence of a sole

62
GFP protein (Fig. 2B). However, western blot with an anti-GFP antibody on parasite extracts
revealed only a single band at the expected size of approximately 80 kDa and no degradation
product (Fig. 2E). In schizonts, each individual merozoite contained one focus together with some
plasma membrane labelling (Fig. 2C). Ruptured merozoites seemed to have lost the plasma
membrane signal and intriguingly, in a few of them, the fluorescence seemed to cap one end of the
parasite (Fig. 2D, arrows). A few studies have shown that in vitro, PHFAPP
also has the ability to
bind to PI(4,5)P2 although with lower affinity than PI4P [203, 410]. It is therefore possible that
some of the plasma membrane labelling is due to PI(4,5)P2 (see below). The recently characterised
PI4P-specific binding domain of the Legionella pneumophila SidM protein could potentially help
in quantifying the true amount of PI4P present at the membrane [411, 412]. A role for PI4P
generated by Golgi apparatus-localised PI4Ks during regulation of membrane trafficking events
and cytokinesis has been shown in a variety of systems such as mammalian cells [276, 413, 414],
Drosophila [415] and yeast [416]. To determine whether the focus of fluorescence could represent
the Golgi, we performed IFAs with an antibody to Endoplasmic reticulum Retention Defective
protein 2 (ERD2), a marker of this structure [399, 417-419]. When observing parasites that had
been fixed with paraformaldehyde-glutaraldehyde, we noticed that the fluorescence signal was no
longer found as dots and at the plasma membrane but instead was redistributed throughout the
cytosol, suggesting that the centrifugation steps required in this fixation protocol had an impact on
PI4P location (results not shown). To circumvent this, we reduced the time between harvesting and
fixation by using methanol fixation on parasite smears. For this analysis, we used parasites
transfected with a FAPP PH domain fused to a triple HA tag since our anti-GFP antibody does not
work on methanol fixed slides. Analysis of the transfectants showed that the distribution of the foci
mirrored what was seen in live parasites, however the plasma membrane labelling was somewhat
less obvious although some overlap with the plasma membrane marker Merozoite surface protein 1
(MSP1) could still be observed (Supplementary Fig. S4 versus Fig. 2). This might potentially be
explained by the fact that methanol fixation is known to deplete phospholipids [420].
Colocalisation experiments revealed that PHFAPP
-3HA overlapped extensively with Golgi ERD2
throughout the erythrocytic cycle (Fig. 3A, B: Pearson’s correlation: Rings: 0.63±0.03,
Trophozoites: 0.63±0.03, Schizonts: 0.60± 0.03). As a control for proteins residing in separate
organelles, we used ERD2 and Rhoptry associated protein 1 (RAP1) and obtained a coefficient of
0.12±0.01. To assess if the PHFAPP
-3HA signal localised to the apical complex, we next performed

63
IFAs with antibodies labelling different organelles forming this structure. The micronemes contain
effectors involved in diverse processes such as merozoite egress, formation of the tight junction
and as adhesions and evidence suggest that they form a heterogeneous population [19, 421]. IFAs
performed with antibodies to Apical membrane antigen 1 (AMA1) and Erythrocyte binding antigen
175 (EBA175), markers of two different micronemal populations, failed to show any strong
overlap with PHFAPP
-3HA (Fig. 3Ca,b). The rhoptry is the most prominent secretory organelle in
the parasite and it is subdivided into two regions, the bulb, which is the larger lipid-rich region of
the organelle and the apical duct known as the rhoptry neck [422]. IFA using an anti-RON4
antibody, a marker of the rhoptry neck [401], and co-transfection of the 2xPHFAPP
-GFP line with
mCherry-tagged Rhoptry associated membrane antigen (RAMA), a marker of the bulb, showed an
absence of colocalization in both cases (Fig. 3Cc, d). Quantification revealed that the R coefficients
between PHFAPP
and any of the apical markers was not statistically different from the negative
control ERD versus RAP1 (Fig. 3D: PHFAPP
-3HA vs AMA1, 0.15±0.03; PHFAPP
-3HA versus
EBA175, 0.13±0.02; PHFAPP
-3HA versus RON4, 0.10±0.02; PHFAPP
-GFP versus mCherry-RAMA,
0.17±0.03; ERD2 versus RAP1, 0.12±0.02). Plasmodium falciparum possesses three putative
PI4kinases and incubation of parasites with the antimalarial compounds imidazopyrazines and
quinoxalines, targeting the PfPI4KIIIβ homologue, led to a major defect in cytokinesis due to
perturbed membrane biogenesis and ingression around developing merozoites [145]. This is in line
with other systems where Golgi-generated PI4P plays a critical role in regulating vesicular
trafficking in the late secretory pathway and in cytokinesis (Audhya et al., 2000; Polevoy et al.,
2009). PfPI4KIIIβ is found spread throughout the cytosol in trophozoites but foci of concentrated
fluorescence, hypothesised to be the Golgi apparatus, are seen in schizont stages [145]. It is
tempting to speculate that PfPI4KIIIβ might be the enzyme responsible for the generation of the
Golgi-associated PI4P we detect in schizont stages and the fact that the PIP4 foci disappear in
parasites treated with imidazopyrazines supports our observation. However, the broad cytosolic
distribution of PfPI4KIIIβ in trophozoites makes it unlikely to be involved in the synthesis of the
Golgi PI4P we detect in earlier stages. As to the origin of the plasma membrane associated PI4P
pool seen throughout the erythrocytic cycle, it could be generated in situ by other PI4Ks or perhaps
by dephosphorylation of PI(4,5)P2 [423], or coming from Golgi-derived PI4P containing vesicles,
or both possibilities, as seen in other systems [424]. In addition to serving as a precursor for
PI(4,5)P2 synthesis, plasma membrane PI4P contributes directly to a number of signalling

64
pathways in mammalian cells (reviewed in [425]). Through its contribution to the total negative
charge unique to the cytoplasmic leaflet of the plasma membrane, PI4P might also be involved in
the recruitment of polybasic motifs-containing effectors requiring an enrichment in polyanionic
lipids [423, 426, 427]. Recently, several studies have uncovered a critical role for PI4P in the
bidirectional transfer of certain lipid types such as sterols and phosphatidylserine between the
endoplasmic reticulum (ER) and the plasma membrane at what are called membrane contact sites.
This action is performed by lipid transfer proteins of the oxysterol-binding protein-related protein
family that take advantage of the energy created by the PI4P gradient between the ER and the
plasma membrane [428-430], reviewed in [431]. We have identified at least one homologue of
oxysterol-binding proteins in P. falciparum (PF3D7_1131800, www.plasmodb.org) and proteomics
analyses have detected that it is expressed in schizont stage parasites [432, 433]. Whether it plays a
similar role remains to be investigated. It is interesting to note that although not much is known
with regards to membrane contact sites in P. falciparum, there is evidence that they occur between
the ER and the apicoplast, and between the mitochondria and the apicoplast [434-436].
Furthermore, the extensive branching of the parasite ER in trophozoites and developing schizonts
make contact sites between the organelle and the plasma membrane a possibility [434].
In conclusion, our results show that PI4P is found at the Golgi apparatus and at the plasma
membrane in all stages of the erythrocytic cycle.
3.4. Subcellular localisation of PI5P
Being the last identified species, PI5P is the most enigmatic of the PIPs, even though it is not the
least abundant in cells (reviewed in [437]). The pathways regulated by PI5P in mammalian cells
and its effectors/binding proteins are still poorly understood, and monitoring the intracellular
localisation of PI5P has been a challenge to date. The PI5P pool was long thought to be confined to
the nucleus, where it is involved in various processes such as protection against UV stress,
apoptosis, transcriptional control and the cell cycle [381] [318, 438](reviewed in [156, 437]). Only
recently have extranuclear pools of PI5P been identified in the plasma membrane, ER, Golgi [208]
and endosomes [439]. Since nothing is known about PI5P in Plasmodium parasites, we sought to
determine whether it could be detected in P. falciparum by using a sensor containing the PH
domain of mammalian Downstream of tyrosine kinase 5 (DOK5), a well described domain with

65
high affinity and specificity for PI5P [213, 395]. Examination of the PHDOK5
-GFP transgenics
revealed that PI5P was detected throughout the asexual lifecycle of the parasite. In ring stages, the
GFP signal seemed to be more pronounced in certain regions of the plasma membrane, together
with some diffused cytoplasmic signal (Fig. 4Aa, arrow and Supplementary Fig. S5A, star). An
anti-GFP western blot revealed a single band around the expected size of 41.3 kDa and no
degradation product (Fig. 4Da). As the parasite matured to the trophozoite stage, the plasma
membrane associated signal seemed to become concentrated in some regions whilst a significant
portion remained cytosolic (Fig. 4Ab, arrow). In some instances, some PI5P signal was found very
close to, and sometimes overlapping with, DAPI in the nucleus (Supplementary Fig. S5B). At >40
h in late schizonts, we no longer saw colocalization with the nucleus (Fig. 4Ac). In the developing
daughter merozoites, the signal seemed to be enriched as a focus close to the nucleus of the
daughter cells with some fluorescence sometimes wrapping around the DAPI (Fig. 4Ac and
Supplementary Fig. S5C, arrowheads) and sometimes almost capping the apical tip (Fig. 4Ac, star).
To try to identify the structure corresponding to the foci of fluorescence in schizonts, we performed
IFAs on a parasite line expressing PHDOK5
-3HA. In most individual developing merozoites, the
signal partially overlapped with the Golgi marker ERD2 (Fig. 4B). Quantification of the
colocalization revealed an R coefficient of 0.50±0.02 which is significantly less than the 0.60± 0.03
obtained for PHFAPP
-3HA versus ERD2 (Fig. 4C). The PHDOK5
pattern of fluorescence is
reminiscent what is observed with PfSEC13p, a marker of the transitional ER [440]. To determine
whether PI5P colocalized with PfSEC13p, parasites were transfected with plasmids expressing
PHDOK5
-GFP and PfSEC13p-mCherry but unfortunately we never recovered doubly-labelled
parasites. Perhaps this is due to some sort of steric hindrance between the fusion proteins which
prevented binding of both proteins at the same time. Of note, as we saw with the PI4P probe, the
plasma membrane associated fluorescence detected in live cells was much less prominent by IFA
although it could still be observed in some cells. The detection of PI5P leads to the intriguing
question as to how it is generated in malaria parasites. Recent reports have provided evidence that
its direct synthesis from PI by the enzyme PIKfyve is the major route in mammalian cells [211,
441, 442]. However, P. falciparum does not have a direct PIKfyve homologue. PfPI3K, with its
demonstrated relaxed specificity [240] or perhaps some of the other PfPIkinases might play a role.
Another way through which PI5P was proposed to be generated is by the dephosphorylation of
PI(4,5)P2 by myotubularin/myotubularin-related phosphatases but again, this class of enzymes is

66
not found in P. falciparum. Four putative PI-phosphatases are found in the P. falciparum genome
but so far, nothing is known about either their subcellular localisation or their substrate specificity
[443].
In conclusion, our data suggest that PI5P is indeed found in the malaria parasite asexual blood
stages and that it localises to the plasma membrane, to a structure potentially corresponding to the
transitional ER in schizonts, and sometimes in the nucleus. This varied subcellular distribution of
PI5P suggests that it might potentially play pleitrophic roles in the malarial erythrocytic cycle.
3.5. Subcellular localisation of PI(4,5)P2
PI(4,5)P2 is by far the most abundant PIP, corresponding to 70% of total PIPs in P. falciparum
asexual stages [160]. To determine the location of PI(4,5)P2, parasites were transfected with a
fusion of the phospholipase C δ PH domain and a fluorescent reporter [348]. When using a single
PH domain, the signal was too faint to be imaged, whether it was fused to GFP, mCitrine or
mCherry (results not shown) as was previously described in other cell types [203, 372]. To increase
the avidity of our probe, we then used a sensor containing two PLCδ PH domains in tandem.
Analysis of the mCherry-2xPHPLCδ
parasite line revealed a very strong labelling of the plasma
membrane at all stages of the cycle (Fig. 5A), confirmed by the extensive colocalization with
MSP1 (Fig. 5B), an abundant merozoite surface protein expressed from the late trophozoite stage
onwards [444]. It is worth noting that unlike the other sensors we used, mCherry-2xPHPLCδ
proved
to be stable enough to perform IFAs with paraformaldehyde/glutaraldehyde fixation. The integrity
of the sensor was next confirmed by western blot on parasite extracts and a single band of the
expected size of approximately 100 kDa was observed (Fig. 5C). We often observed large vesicle-
like structures at the plasma membrane in ring stages that increased in numbers as the parasites
transitioned to trophozoites and schizonts (Fig. 5Aa, b; Supplementary Fig. S6, arrows). These
could potentially represent cytostomes, endocytic structures involved in hemoglobin transport from
the host cytoplasm to the parasite digestive vacuole [28, 435, 445]. Furthermore, a cavity was
previously shown to originate from an invagination of the PVM and the parasite plasma membrane
and to contain host cell cytosol, however the structure disappeared as the parasites entered
schizogony [446]. Moreover, a similar kind of spherical structure had also been shown to remain
connected to the RBC cytoplasm and was hypothesised to be involved in hemoglobin endocytosis

67
[447]. Whether the mCherry-2xPHPLCδ
labelled features we saw in early stage parasites are the
same as those in schizonts is unknown but it is assumed that hemoglobin uptake is much less
important once cell division has started [448]. Whatever cellular processes occur at these
structures, our results suggest that there is no massive decrease in PI(4,5)P2 levels present on their
membranes, at least as detectable using our sensor. Whether this is because the lipid plays no role,
that its function does not require its modification or that it is resynthesized as it is consumed
remains to be seen. Finally, in very late schizonts, once cytokinesis is completed, each daughter
merozoite displays a very uniformly labelled plasma membrane (Fig. 5Ac, d). Control parasites
transfected with mCherry alone showed a cytosolic distribution throughout the blood stages
(Supplementary Fig. S7).
As described earlier, in other systems, plasma membrane-localised PI(4,5)P2 is implicated in
several cell surface related actions (reviewed in [158]). Plasmodium falciparum possesses a PI-
PLC [159] and inhibitor studies have implicated it in processes such as gametocyte activation and
differentiation [351, 353], sporozoite motility [227], synchronization of the erythrocytic cycle
[449], schizont egress [229] and invasion of merozoites (Vaid et al., 2008), although the latter
finding was subsequently questioned (Jones et al., 2009). Recently, a PH domain-containing
ccalcium-dependent protein kinase (PfCDPK7) was shown to specifically bind to PI(4,5)P2 and to
localise to vesicles in close proximity to the parasite ER [450]. Close inspection of some of our
2xPHPLCδ
parasites sometimes showed some minor fluorescence close to the DAPI-stained nucleus
but it was certainly not as defined as the PfCDPK7 pattern published by Kumar and colleagues
(2014) (Fig. 5Aa, Max proj panel, arrowhead). Perhaps the tandem nature of our sensor precludes
its efficient binding to these intracellular vesicles. In addition, it has been reported that PH domains
with different affinities to the same PIP species sometimes show slightly different intracellular
localisation [366]. Of note, in other eukaryotes, although the major pool of PI(4,5)P2 is locally
produced at the plasma membrane by the action of a PIP5K on PI4P [451], small subcellular pools
have also been found at the Golgi, the ER and endosomes [424, 452].
Our data show that PI(4,5)P2 is found all over the parasite plasma membrane and at some vesicular
structures potentially corresponding to cytostomes or the previously described cavity [446].

68
In summary, our data, summarised in Fig. 6, reveals that the distribution of most PIP species is
quite stable during the erythrocytic cycle of P. falciparum except for PI5P which is found
potentially associated to the transitional ER (tER) only in schizonts but otherwise at the plasma
membrane. Remarkably, the repartition of most PIPs in the parasite is quite similar to what was
previously observed in other eukaryotic cell types (Table 1). Of interest, despite some minor
amounts of PI(3,4)P2 and PI(3,4,5)P3 previously detected in schizonts [160], only a broad
cytosolic signal identical to the fluorescent protein alone was detected when we used sensors
specific to each of these PIP species (results not shown). It will now be of great interest to identify
the mechanisms behind the specific localisation of each PIP, whether through the action of kinases,
phosphatases and lipases or through the activity of lipid transfer proteins. This will in turn shed
some light on the various roles likely played by these critical but tiny lipids.
Acknowledgments
We would like to thank Philip Shaw (National Center for Genetic Engineering and Biotechnology,
Thailand) for the gift of the original pGFP-glmS plasmid, Yvon Jaillais (Université Claude Bernard
Lyon 1, France) the PIPline plasmids and Jacques Nunes (Centre de Recherche en Cancérologie e
Marseille, France) for plasmid GST-PH DOK5. We also thank Jacobus Pharmaceuticals for
WR99210 and Dominic Gagnon for technical assistance. The following reagents were obtained
through MR4 as part of the BEI Resources, National Institute of Allergy and Infectious Diseases,
National Institutes of Health, USA: Polyclonal Anti-Plasmodium falciparum PfERD2 (antiserum,
Rabbit), MRA-1; plasmid pLN-ENR-GFP, MRA-846, contributed by David A. Fidock; DMS1,
MRA-1161. We would also like to acknowledge the Canadian Blood Services for providing human
erythrocytes. The authors declare no competing financial interests. This study was funded through
a Canadian Institutes for Health Research (CIHR) operating grant MOP 130359. DR was a Fonds
de la Recherche du Québec-Santé Junior 1 fellow.
References
Abu Bakar, N., Klonis, N., Hanssen, E., Chan, C., Tilley, L., 2010. Digestive-vacuole genesis and
endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum. J Cell
Sci 123, 441-450.

69
Adjalley, S.H., Lee, M.C.S., Fidock, D.A., 2010. A method for rapid genetic integration into
Plasmodium falciparum utilizing mycobacteriophage Bxb1 integrase. Meth Mol Biol
(Clifton, N.J.) 634, 87-100.
Agarwal, S., Singh, M.K., Garg, S., Chitnis, C.E., Singh, S., 2013. Ca(2+) -mediated exocytosis
of subtilisin-like protease 1: a key step in egress of Plasmodium falciparum merozoites.
Cell Microbiol 15, 910-921.
Aikawa, M., 1966. The fine structure of the erythrocytic stages of three avian malarial parasites,
Plasmodium fallax, P. lophurae, and P. cathemerium. Am J Trop Med Hyg 15, 449-471.
Aikawa, M., Hepler, P.K., Huff, C.G., Sprinz, H., 1966. The feeding mechanism of avian malarial
parasites. J Cell Biol 28, 355-373.
Aikawa, M., Huff, C.G., Sprinz, H., 1967. Fine structure of the asexual stages of Plasmodium
elongatum. J Cell Biol 34, 229-249.
Alves, E., Bartlett, P.J., Garcia, C.R., Thomas, A.P., 2011. Melatonin and IP3-induced Ca2+
release from intracellular stores in the malaria parasite Plasmodium falciparum within
infected red blood cells. J Biol Chem 286, 5905-5912.
Balla, A., Tuymetova, G., Tsiomenko, A., Varnai, P., Balla, T., 2005. A plasma membrane pool
of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III
alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol
Cell 16, 1282-1295.
Balla, T., 2013. Phosphoinositides: Tiny Lipids With Giant Impact on Cell Regulation. Physiol
Rev 93, 1019-1137.
Balla, T., Várnai, P., 2009. Visualization of cellular phosphoinositide pools with GFP-fused protein-
domains, in: Bonifacino, J.S., Hartford, J.B., Lippincott-Schwartz, J., Yamada, K.M., (Eds.) Current
Protocols in Cell Biology. Wiley, New York. Chapter 42:24.4:24.4.1–24.4.27.
Beraldo, F.H., Mikoshiba, K., Garcia, C.R., 2007. Human malarial parasite, Plasmodium
falciparum, displays capacitative calcium entry: 2-aminoethyl diphenylborinate blocks the
signal transduction pathway of melatonin action on the P. falciparum cell cycle. J Pineal
Res 43, 360-364.
Bhattacharjee, S., Speicher, K.D., Stahelin, R.V., Speicher, D.W., Haldar, K., 2012. PI(3)P-
independent and -dependent pathways function together in a vacuolar translocation
sequence to target malarial proteins to the host erythrocyte. Mol Biochem Parasitol 185,
106-113.
Boddey, J.A., O'Neill, M.T., Lopaticki, S., Carvalho, T.G., Hodder, A.N., Nebl, T., Wawra, S.,
van West, P., Ebrahimzadeh, Z., Richard, D., Flemming, S., Spielmann, T., Przyborski, J.,
Babon, J.J., Cowman, A.F., 2016. Export of malaria proteins requires co-translational
processing of the PEXEL motif independent of phosphatidylinositol-3-phosphate binding.
Nat Commun 7, 10470.
Brill, J.A., Hime, G.R., Scharer-Schuksz, M., Fuller, M.T., 2000. A phospholipid kinase regulates
actin organization and intercellular bridge formation during germline cytokinesis.
Development 127, 3855-3864.

70
Brombacher, E., Urwyler, S., Ragaz, C., Weber, S.S., Kami, K., Overduin, M., Hilbi, H., 2009.
Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-
phosphate-binding effector protein of Legionella pneumophila. J Biol Chem 284, 4846-
4856.
Brown, J.R., Auger, K.R., 2011. Phylogenomics of phosphoinositide lipid kinases: perspectives
on the evolution of second messenger signaling and drug discovery. BMC Evol Biol 11, 4.
Bruns, J.R., Ellis, M.A., Jeromin, A., Weisz, O.A., 2002. Multiple roles for phosphatidylinositol
4-kinase in biosynthetic transport in polarized Madin-Darby canine kidney cells. J Biol
Chem 277, 2012-2018.
Cantley, L.C., 2002. The phosphoinositide 3-kinase pathway. Science 296, 1655-1657.
Carey, A.F., Singer, M., Bargieri, D., Thiberge, S., Frischknecht, F., Ménard, R., Amino, R.,
2014. Calcium dynamics of Plasmodium berghei sporozoite motility. Cell Microbiol ,
16(5):768-783.
Christoforidis, S., McBride, H.M., Burgoyne, R.D., Zerial, M., 1999. The Rab5 effector EEA1 is
a core component of endosome docking. Nature 397, 621-625.
Chung, J., Torta, F., Masai, K., Lucast, L., Czapla, H., Tanner, L.B., Narayanaswamy, P., Wenk,
M.R., Nakatsu, F., De Camilli, P., 2015. INTRACELLULAR TRANSPORT.
PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma
membrane contacts. Science 349, 428-432.
Collins, C.R., Hackett, F., Strath, M., Penzo, M., Withers-Martinez, C., Baker, D.A., Blackman,
M.J., 2013. Malaria Parasite cGMP-dependent Protein Kinase Regulates Blood Stage
Merozoite Secretory Organelle Discharge and Egress. PLoS Pathog 9, e1003344.
Corvera, S., D'Arrigo, A., Stenmark, H., 1999. Phosphoinositides in membrane traffic. Curr Opin
Cell Biol 11, 460-465.
Counihan, N.A., Kalanon, M., Coppel, R.L., de Koning-Ward, T.F., 2013. Plasmodium rhoptry
proteins: why order is important. Trends Parasitol, 29(5):228-236.
Cowman, A.F., Crabb, B.S., 2006. Invasion of red blood cells by malaria parasites. Cell 124, 755-
766.
Czech, M.P., 2003. Dynamics of phosphoinositides in membrane retrieval and insertion. Ann Rev
Physiol 65, 791-815.
Daher, W., Morlon-Guyot, J., Sheiner, L., Lentini, G., Berry, L., Tawk, L., Dubremetz, J.-F.,
Wengelnik, K., Striepen, B., Lebrun, M., 2014. Lipid kinases are essential for apicoplast
homeostasis in Toxoplasma gondii. Cell Microbiol , 17(4):559-578.
Dalal, S., Klemba, M., 2015. Amino acid efflux by asexual blood-stage Plasmodium falciparum
and its utility in interrogating the kinetics of hemoglobin endocytosis and catabolism in
vivo. Mol Biochem Parasitol 201, 116-122.
de Lartigue, J., Polson, H., Feldman, M., Shokat, K., Tooze, S.A., Urbe, S., Clague, M.J., 2009.
PIKfyve regulation of endosome-linked pathways. Traffic 10, 883-893.
De Matteis, M.A., Di Campli, A., Godi, A., 2005. The role of the phosphoinositides at the Golgi
complex. Biochim Biophys Acta 1744, 396-405.

71
de Saint-Jean, M., Delfosse, V., Douguet, D., Chicanne, G., Payrastre, B., Bourguet, W.,
Antonny, B., Drin, G., 2011. Osh4p exchanges sterols for phosphatidylinositol 4-
phosphate between lipid bilayers. J Cell Biol 195, 965-978.
Di Paolo, G., De Camilli, P., 2006. Phosphoinositides in cell regulation and membrane dynamics.
Nature 443, 651-657.
DiDonato, D., Brasaemle, D.L., 2003. Fixation methods for the study of lipid droplets by
immunofluorescence microscopy. J Histochem Cytochem 51, 773-780.
Doughman, R.L., Firestone, A.J., Anderson, R.A., 2003. Phosphatidylinositol phosphate kinases
put PI4,5P(2) in its place. J Membr Biol 194, 77-89.
Dowler, S., Currie, R.A., Campbell, D.G., Deak, M., Kular, G., Downes, C.P., Alessi, D.R., 2000.
Identification of pleckstrin-homology-domain-containing proteins with novel
phosphoinositide-binding specificities. Biochem J., 351, 19-31.
Elabbadi, N., Ancelin, M.L., Vial, H.J., 1994. Characterization of phosphatidylinositol synthase
and evidence of a polyphosphoinositide cycle in Plasmodium-infected erythrocytes. Mol
Biochem Parasitol 63, 179-192.
Elmendorf, H.G., Haldar, K., 1993. Identification and localization of ERD2 in the malaria
parasite Plasmodium falciparum: separation from sites of sphingomyelin synthesis and
implications for organization of the Golgi. EMBO J 12, 4763-4773.
Francia, M.E., Striepen, B., 2014. Cell division in apicomplexan parasites. Nat Rev Microbiol.
12(2):125-136.
Gary, J.D., Wurmser, A.E., Bonangelino, C.J., Weisman, L.S., Emr, S.D., 1998. Fab1p is
essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and
membrane homeostasis. J Cell Biol 143, 65-79.
Gillooly, D.J., Morrow, I.C., Lindsay, M., Gould, R., Bryant, N.J., Gaullier, J.M., Parton, R.G.,
Stenmark, H., 2000. Localization of phosphatidylinositol 3-phosphate in yeast and
mammalian cells. EMBO J 19, 4577-4588.
Gilson, P.R., Nebl, T., Vukcevic, D., Moritz, R.L., Sargeant, T., Speed, T.P., Schofield, L.,
Crabb, B.S., 2006. Identification and stoichiometry of glycosylphosphatidylinositol-
anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol
Cell Proteo: MCP 5, 1286-1299.
Godi, A., Di Campli, A., Konstantakopoulos, A., Di Tullio, G., Alessi, D.R., Kular, G.S., Daniele,
T., Marra, P., Lucocq, J.M., De Matteis, M.A., 2004a. FAPPs control Golgi-to-cell-surface
membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 6, 393-404.
Godi, A., Di Campli, A., Konstantakopoulos, A., Di Tullio, G., Alessi, D.R., Kular, G.S., Daniele,
T., Marra, P., Lucocq, J.M., De Matteis, M.A., 2004b. FAPPs control Golgi-to-cell-
surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol 6, 393-404.
Godi, A., Pertile, P., Meyers, R., Marra, P., Di Tullio, G., Iurisci, C., Luini, A., Corda, D., De
Matteis, M.A., 1999. ARF mediates recruitment of PtdIns-4-OH kinase-beta and
stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol 1, 280-287.
Gozani, O., Karuman, P., Jones, D.R., Ivanov, D., Cha, J., Lugovskoy, A.A., Baird, C.L., Zhu, H.,
Field, S.J., Lessnick, S.L., Villasenor, J., Mehrotra, B., Chen, J., Rao, V.R., Brugge, J.S.,

72
Ferguson, C.G., Payrastre, B., Myszka, D.G., Cantley, L.C., Wagner, G., Divecha, N.,
Prestwich, G.D., Yuan, J., 2003a. The PHD finger of the chromatin-associated protein
ING2 functions as a nuclear phosphoinositide receptor. Cell 114, 99-111.
Gozani, O., Karuman, P., Jones, D.R., Ivanov, D., Cha, J., Lugovskoy, A.A., Baird, C.L., Zhu, H.,
Field, S.J., Lessnick, S.L., Villasenor, J., Mehrotra, B., Chen, J., Rao, V.R., Brugge, J.S.,
Ferguson, C.G., Payrastre, B., Myszka, D.G., Cantley, L.C., Wagner, G., Divecha, N.,
Prestwich, G.D., Yuan, J., 2003b. The PHD finger of the chromatin-associated protein
ING2 functions as a nuclear phosphoinositide receptor. Cell 114, 99-111.
Grüring, C., Heiber, A., Kruse, F., Ungefehr, J., Gilberger, T.-W., Spielmann, T., 2011.
Development and host cell modifications of Plasmodium falciparum blood stages in four
dimensions. Nature Comm 2, 165.
Guittard, G., Gérard, A., Dupuis-Coronas, S., Tronchère, H., Mortier, E., Favre, C., Olive, D.,
Zimmermann, P., Payrastre, B., Nunès, J.A., 2009. Cutting edge: Dok-1 and Dok-2
adaptor molecules are regulated by phosphatidylinositol 5-phosphate production in T cells.
J Immunol 182, 3974-3978.
Guittard, G., Mortier, E., Tronchère, H., Firaguay, G., Gérard, A., Zimmermann, P., Payrastre, B.,
Nunès, J.A., 2010. Evidence for a positive role of PtdIns5P in T-cell signal transduction
pathways. FEBS Lett 584, 2455-2460.
Hammond, G.R., Machner, M.P., Balla, T., 2014. A novel probe for phosphatidylinositol 4-
phosphate reveals multiple pools beyond the Golgi. J Cell Biol 205, 113-126.
Hammond, G.R., Schiavo, G., Irvine, R.F., 2009. Immunocytochemical techniques reveal
multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem J 422, 23-35.
Hammond, G.R.V., Fischer, M.J., Anderson, K.E., Holdich, J., Koteci, A., Balla, T., Irvine, R.F.,
2012. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane
identity. Science 337, 727-730.
Hanssen, E., Goldie, K.N., Tilley, L., 2010. Ultrastructure of the asexual blood stages of
Plasmodium falciparum. Meth Cell Biol 96, 93-116.
Healer, J., Crawford, S., Ralph, S., McFadden, G., Cowman, A.F., 2002. Independent
translocation of two micronemal proteins in developing Plasmodium falciparum
merozoites. Infect Immun 70, 5751-5758.
Healer, J., Triglia, T., Hodder, A.N., Gemmill, A.W., Cowman, A.F., 2005. Functional analysis of
Plasmodium falciparum apical membrane antigen 1 utilizing interspecies domains.
Infection and Immunity 73, 2444-2451.
Heo, W.D., Inoue, T., Park, W.S., Kim, M.L., Park, B.O., Wandless, T.J., Meyer, T., 2006.
PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma
membrane. Science 314, 1458-1461.
Hopkins, J., Fowler, R., Krishna, S., Wilson, I., Mitchell, G., Bannister, L., 1999. The plastid in
Plasmodium falciparum asexual blood stages: a three-dimensional ultrastructural analysis.
Protist 150, 283-295.

73
Hotta, C.T., Gazarini, M.L., Beraldo, F.H., Varotti, F.P., Lopes, C., Markus, R.P., Pozzan, T.,
Garcia, C.R., 2000. Calcium-dependent modulation by melatonin of the circadian rhythm
in malarial parasites. Nat Cell Biol 2, 466-468.
Howe, R., Kelly, M., Jimah, J., Hodge, D., Odom, A.R., 2013. Isoprenoid biosynthesis inhibition
disrupts Rab5 localization and food vacuolar integrity in Plasmodium falciparum. Eukary
Cell 12, 215-223.
Hsu, V.W., Shah, N., Klausner, R.D., 1992. A brefeldin A-like phenotype is induced by the
overexpression of a human ERD-2-like protein, ELP-1. Cell 69, 625-635.
Ikonomov, O.C., Sbrissa, D., Delvecchio, K., Xie, Y., Jin, J.P., Rappolee, D., Shisheva, A., 2011.
The phosphoinositide kinase PIKfyve is vital in early embryonic development:
preimplantation lethality of PIKfyve-/- embryos but normality of PIKfyve+/- mice. J Biol
Chem 286, 13404-13413.
Jean, S., Kiger, A.A., 2014. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci 127,
923-928.
Jones, D.R., Bultsma, Y., Keune, W.J., Halstead, J.R., Elouarrat, D., Mohammed, S., Heck, A.J.,
D'Santos, C.S., Divecha, N., 2006. Nuclear PtdIns5P as a transducer of stress signaling: an
in vivo role for PIP4Kbeta. Mol Cell 23, 685-695.
Kanai, F., Liu, H., Field, S.J., Akbary, H., Matsuo, T., Brown, G.E., Cantley, L.C., Yaffe, M.B.,
2001. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell
Biol 3, 675-678.
Katso, R., Okkenhaug, K., Ahmadi, K., White, S., Timms, J., Waterfield, M.D., 2001. Cellular
function of phosphoinositide 3-kinases: implications for development, homeostasis, and
cancer. Annu Rev Cell Dev Biol 17, 615-675.
Kumar, P., Tripathi, A., Ranjan, R., Halbert, J., Gilberger, T., Doerig, C., Sharma, P., 2014.
Regulation of Plasmodium falciparum development by calcium-dependent protein kinase
7 (PfCDPK7). J Biol Chem 289, 20386-20395.
Kutateladze, T.G., 2010. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol
6, 507-513.
Lambros, C., Vanderberg, J.P., 1979. Synchronization of Plasmodium falciparum erythrocytic
stages in culture. J Parasitol 65, 418-420.
Langreth, S.G., Jensen, J.B., Reese, R.T., Trager, W., 1978. Fine structure of human malaria in
vitro. J Protozool 25, 443-452.
Lemmon, M.A., Ferguson, K.M., O'Brien, R., Sigler, P.B., Schlessinger, J., 1995. Specific and
high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain.
Proc Natl Acad Sci USA 92, 10472-10476.
Levine, T.P., Munro, S., 2002. Targeting of Golgi-specific pleckstrin homology domains involves
both PtdIns 4-kinase-dependent and -independent components. Curr Biol 12, 695-704.
Lewis, M.J., Pelham, H.R., 1990. A human homologue of the yeast HDEL receptor. Nature 348,
162-163.

74
Lewis, M.J., Pelham, H.R., 1992. Ligand-induced redistribution of a human KDEL receptor from
the Golgi complex to the endoplasmic reticulum. Cell 68, 353-364.
Li, H., Marshall, A.J., 2015. Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and
effector proteins: A distinct branch of PI3K signaling. Cell Signal 27, 1789-1798.
Marti, M., Baum, J., Rug, M., Tilley, L., Cowman, A.F., 2005. Signal-mediated export of proteins
from the malaria parasite to the host erythrocyte. J Cell Biol 171, 587-592.
Martin, S.K., Jett, M., Schneider, I., 1994. Correlation of phosphoinositide hydrolysis with
exflagellation in the malaria microgametocyte. J Parasitol 80, 371-378.
Mbengue, A., Bhattacharjee, S., Pandharkar, T., Liu, H., Estiu, G., Stahelin, R.V., Rizk, S.S.,
Njimoh, D.L., Ryan, Y., Chotivanich, K., Nguon, C., Ghorbal, M., Lopez-Rubio, J.J.,
Pfrender, M., Emrich, S., Mohandas, N., Dondorp, A.M., Wiest, O., Haldar, K., 2015. A
molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature
520, 683-687.
McIntosh, M.T., Vaid, A., Hosgood, H.D., Vijay, J., Bhattacharya, A., Sahani, M.H., Baevova, P.,
Joiner, K.A., Sharma, P., 2007. Traffic to the malaria parasite food vacuole: a novel
pathway involving a phosphatidylinositol 3-phosphate-binding protein. J Biol Chem 282,
11499-11508.
McLaughlin, S., Murray, D., 2005. Plasma membrane phosphoinositide organization by protein
electrostatics. Nature 438, 605-611.
McNamara, C.W., Lee, M.C.S., Lim, C.S., Lim, S.H., Roland, J., Nagle, A., Simon, O., Yeung,
B.K.S., Chatterjee, A.K., McCormack, S.L., Manary, M.J., Zeeman, A.-M., Dechering,
K.J., Kumar, T.R.S., Henrich, P.P., Gagaring, K., Ibanez, M., Kato, N., Kuhen, K.L.,
Fischli, C., Rottmann, M., Plouffe, D.M., Bursulaya, B., Meister, S., Rameh, L., Trappe,
J., Haasen, D., Timmerman, M., Sauerwein, R.W., Suwanarusk, R., Russell, B., Rénia, L.,
Nosten, F., Tully, D.C., Kocken, C.H.M., Glynne, R.J., Bodenreider, C., Fidock, D.A.,
Diagana, T.T., Winzeler, E.A., 2013. Targeting Plasmodium PI(4)K to eliminate malaria.
Nature. Dec 12;504(7479(:248-253.
Militello, K.T., Dodge, M., Bethke, L., Wirth, D.F., 2004. Identification of regulatory elements in
the Plasmodium falciparum genome. Mol Biochem Parasitol 134, 75-88.
Nkrumah, L.J., Muhle, R.A., Moura, P.A., Ghosh, P., Hatfull, G.F., Jacobs, W.R., Jr., Fidock,
D.A., 2006. Efficient site-specific integration in Plasmodium falciparum chromosomes
mediated by mycobacteriophage Bxb1 integrase. Nat Methods 3, 615-621.
Ogwan'g, R., Mwangi, J., Gachihi, G., Nwachukwu, A., Roberts, C.R., Martin, S.K., 1993. Use of
pharmacological agents to implicate a role for phosphoinositide hydrolysis products in
malaria gamete formation. Biochem Pharmacol 46, 1601-1606.
Olkkonen, V.M., 2015. OSBP-Related Protein Family in Lipid Transport Over Membrane
Contact Sites. Lipid Insights 8, 1-9.
Pandey, R., Mohmmed, A., Pierrot, C., Khalife, J., Malhotra, P., Gupta, D., 2014. Genome wide
in silico analysis of Plasmodium falciparum phosphatome. BMC genomics 15, 1024-1022.
Pease, B.N., Huttlin, E.L., Jedrychowski, M.P., Talevich, E., Harmon, J., Dillman, T., Natarajan,
K., Doerig, C., Chakrabarti, R., Gygi, S.P., Chakrabarti, D., 2013. Global Analysis of

75
Protein Expression and Phosphorylation of Three Stages of Plasmodium falciparum
Intraerythrocytic Development. J Proteome Res. 12(9):4028-4045.
Petiot, A., Ogier-Denis, E., Blommaart, E.F., Meijer, A.J., Codogno, P., 2000. Distinct classes of
phosphatidylinositol 3'-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem 275, 992-998.
Posor, Y., Eichhorn-Gruenig, M., Puchkov, D., Schoneberg, J., Ullrich, A., Lampe, A., Muller,
R., Zarbakhsh, S., Gulluni, F., Hirsch, E., Krauss, M., Schultz, C., Schmoranzer, J., Noe,
F., Haucke, V., 2013. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-
bisphosphate. Nature 499, 233-237.
Prommana, P., Uthaipibull, C., Wongsombat, C., Kamchonwongpaisan, S., Yuthavong, Y.,
Knuepfer, E., Holder, A.A., Shaw, P.J., 2013. Inducible Knockdown of Plasmodium Gene
Expression Using the glmS Ribozyme. PloS One 8, e73783.
Raabe, A.C., Wengelnik, K., Billker, O., Vial, H.J., 2011. Multiple roles for Plasmodium berghei
phosphoinositide-specific phospholipase C in regulating gametocyte activation and
differentiation. Cell Microbiol 13, 955-966.
Ralph, S.A., van Dooren, G.G., Waller, R.F., Crawford, M.J., Fraunholz, M.J., Foth, B.J., Tonkin,
C.J., Roos, D.S., McFadden, G.I., 2004. Tropical infectious diseases: metabolic maps and
functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol 2, 203-216.
Ramel, D., Lagarrigue, F., Pons, V., Mounier, J., Dupuis-Coronas, S., Chicanne, G., Sansonetti,
P.J., Gaits Iacovoni, F., Tronchère, H., Payrastre, B., 2011. Shigella flexneri infection
generates the lipid PI5P to alter endocytosis and prevent termination of EGFR signaling.
Science Signal 4, ra61-ra61.
Richard, D., Kats, L.M., Langer, C., Black, C.G., Mitri, K., Boddey, J.A., Cowman, A.F., Coppel,
R.L., 2009. Identification of rhoptry trafficking determinants and evidence for a novel
sorting mechanism in the malaria parasite Plasmodium falciparum. PLoS Pathog 5,
e1000328.
Richard, D., MacRaild, C.A., Riglar, D.T., Chan, J.-A., Foley, M., Baum, J., Ralph, S.A., Norton,
R.S., Cowman, A.F., 2010. Interaction between Plasmodium falciparum apical membrane
antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte
invasion process of malaria parasites. J Biol Chem 285, 14815-14822.
Roy, A., Levine, T.P., 2004. Multiple pools of phosphatidylinositol 4-phosphate detected using
the pleckstrin homology domain of Osh2p. J Biol Chem 279, 44683-44689.
Rutherford, A.C., Traer, C., Wassmer, T., Pattni, K., Bujny, M.V., Carlton, J.G., Stenmark, H.,
Cullen, P.J., 2006. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve)
regulates endosome-to-TGN retrograde transport. J Cell Sci 119, 3944-3957.
Sarkes, D., Rameh, L.E., 2010. A novel HPLC-based approach makes possible the spatial
characterization of cellular PtdIns5 Pand other phosphoinositides. Biochem J 428, 375-
384.
Sbrissa, D., Ikonomov, O.C., Filios, C., Delvecchio, K., Shisheva, A., 2012. Functional
dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P2 by means of the
PIKfyve inhibitor YM201636. Am J Physiol Cell Physiol 303, C436-446.

76
Schofield, L., Bushell, G.R., Cooper, J.A., Saul, A.J., Upcroft, J.A., Kidson, C., 1986. A rhoptry
antigen of Plasmodium falciparum contains conserved and variable epitopes recognized
by inhibitory monoclonal antibodies. Mol Biochem Parasitol 18, 183-195.
Shewan, A., Eastburn, D.J., Mostov, K., 2011. Phosphoinositides in Cell Architecture, in:Simons,
K. Cold Spring Harbor Perspectives in Biology.3(8):a004796.
Shisheva, A., 2013. PtdIns5P: news and views of its appearance, disappearance and deeds. Arch
Biochem Biophys 538, 171-180.
Simon, M.L.A., Platre, M.P., Assil, S., van Wijk, R., Chen, W.Y., Chory, J., Dreux, M., Munnik,
T., Jaillais, Y., 2013. A multi-colour/multi-affinity marker set to visualize
phosphoinositide dynamics in Arabidopsis. Plant J 77, 322-337.
Simonsen, A., Lippe, R., Christoforidis, S., Gaullier, J.M., Brech, A., Callaghan, J., Toh, B.H.,
Murphy, C., Zerial, M., Stenmark, H., 1998. EEA1 links PI(3)K function to Rab5
regulation of endosome fusion. Nature 394, 494-498.
Slomianny, C., 1990. Three-dimensional reconstruction of the feeding process of the malaria
parasite. Blood Cells 16, 369-378.
Stefan, C.J., Audhya, A., Emr, S.D., 2002. The yeast synaptojanin-like proteins control the
cellular distribution of phosphatidylinositol (4,5)-bisphosphate. Mol Biol Cell 13, 542-
557.
Struck, N.S., Herrmann, S., Langer, C., Krueger, A., Foth, B.J., Engelberg, K., Cabrera, A.L.,
Haase, S., Treeck, M., Marti, M., Cowman, A.F., Spielmann, T., Gilberger, T.W., 2008.
Plasmodium falciparum possesses two GRASP proteins that are differentially targeted to
the Golgi complex via a higher- and lower-eukaryote-like mechanism. J Cell Sci 121,
2123-2129.
Su, X.Z., Wellems, T.E., 1994. Sequence, transcript characterization and polymorphisms of a
Plasmodium falciparum gene belonging to the heat-shock protein (HSP) 90 family. Gene
151, 225-230.
Tan, J., Brill, J.A., 2013. Cinderella story: PI4P goes from precursor to key signaling molecule.
Crit Rev Biochem Mol Biol.,.49(1)33-58.
Tawk, L., Chicanne, G., Dubremetz, J.-F., Richard, V., Payrastre, B., Vial, H.J., Roy, C.,
Wengelnik, K., 2010. Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium,
localizes to the food vacuole membrane and the apicoplast. Eukaryot Cell 9, 1519-1530.
Tawk, L., Dubremetz, J.-F., Montcourrier, P., Chicanne, G., Merezegue, F., Richard, V.,
Payrastre, B., Meissner, M., Vial, H.J., Roy, C., Wengelnik, K., Lebrun, M., 2011.
Phosphatidylinositol 3-monophosphate is involved in Toxoplasma apicoplast biogenesis.
PLoS Pathog 7, e1001286.
Tonkin, C.J., van Dooren, G.G., Spurck, T.P., Struck, N.S., Good, R.T., Handman, E., Cowman,
A.F., McFadden, G.I., 2004. Localization of organellar proteins in Plasmodium
falciparum using a novel set of transfection vectors and a new immunofluorescence
fixation method. MolBiochem Parasitol 137, 13-21.
Trager, W., Jensen, J.B., 1976. Human malaria parasites in continuous culture. Science 193, 673-
675.

77
Treeck, M., Sanders, J.L., Elias, J.E., Boothroyd, J.C., 2011. The Phosphoproteomes of
Plasmodium falciparum and Toxoplasma gondii Reveal Unusual Adaptations Within and
Beyond the Parasites Boundaries. Cell Host Microbe 10, 410-419.
Vaid, A., Ranjan, R., Smythe, W.A., Hoppe, H.C., Sharma, P., 2010. PfPI3K, a
phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host
erythrocyte and is involved in hemoglobin trafficking. Blood 115, 2500-2507.
Vaid, A., Thomas, D.C., Sharma, P., 2008. Role of Ca2+/calmodulin-PfPKB signaling pathway in
erythrocyte invasion by Plasmodium falciparum. J Biol Chem 283, 5589-5597.
van Dooren, G.G., Marti, M., Tonkin, C.J., Stimmler, L.M., Cowman, A.F., McFadden, G.I.,
2005. Development of the endoplasmic reticulum, mitochondrion and apicoplast during
the asexual life cycle of Plasmodium falciparum. Mol Microbiol 57, 405-419.
van Gisbergen, P.A., Li, M., Wu, S.Z., Bezanilla, M., 2012. Class II formin targeting to the cell
cortex by binding PI(3,5)P(2) is essential for polarized growth. J Cell Biol 198, 235-250.
Vanhaesebroeck, B., Leevers, S.J., Ahmadi, K., Timms, J., Katso, R., Driscoll, P.C., Woscholski,
R., Parker, P.J., Waterfield, M.D., 2001. Synthesis and function of 3-phosphorylated
inositol lipids. Ann Rev Biochem 70, 535-602.
Várnai, P., Balla, T., 2006. Live cell imaging of phosphoinositide dynamics with fluorescent
protein domains. Biochim Biophys Acta 1761, 957-967.
Vial, H.J., Ancelin, M.L., Philippot, J.R., Thuet, M.J., 1990. Biosynthesis and dynamics of lipids
in Plasmodium-infected mature mammalian erythrocytes. Blood Cells 16, (2-3):531-555.
Viaud, J., Mansour, R., Antkowiak, A., Mujalli, A., Valet, C., Chicanne, G., Xuereb, J.M.,
Terrisse, A.D., Severin, S., Gratacap, M.P., Gaits-Iacovoni, F., Payrastre, B., 2015.
Phosphoinositides: Important lipids in the coordination of cell dynamics. Biochimie.
Jun;125:250-258.
von Filseck, J.M., Mesmin, B., Bigay, J., Antonny, B., Drin, G., 2014. Building lipid ‘PIPelines’
throughout the cell by ORP/Osh proteins. Biochem Soc Trans 42, 1465-1470.
Waller, R.F., Reed, M.B., Cowman, A.F., McFadden, G.I., 2000. Protein trafficking to the plastid
of Plasmodium falciparum is via the secretory pathway. EMBO J 19, 1794-1802.
Watt, S.A., Kular, G., Fleming, I.N., Downes, C.P., Lucocq, J.M., 2002. Subcellular localization
of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of
phospholipase C delta1. Biochem J 363, 657-666.
Wengelnik, K., Vial, H.J., 2007. Characterisation of the phosphatidylinositol synthase gene of
Plasmodium species. Res Microbiol 158, 51-59.
WHO, 2015. World Malaria Report. World Health Organization, Geneva, Switzerland.
Yeung, T., Gilbert, G.E., Shi, J., Silvius, J., Kapus, A., Grinstein, S., 2008. Membrane
phosphatidylserine regulates surface charge and protein localization. Science 319, 210-
213.
Zhang, Y., McCartney, A.J., Zolov, S.N., Ferguson, C.J., Meisler, M.H., Sutton, M.A., Weisman,
L.S., 2012. Modulation of synaptic function by VAC14, a protein that regulates the
phosphoinositides PI(3,5)P(2) and PI(5)P. EMBO J 31, 3442-3456.
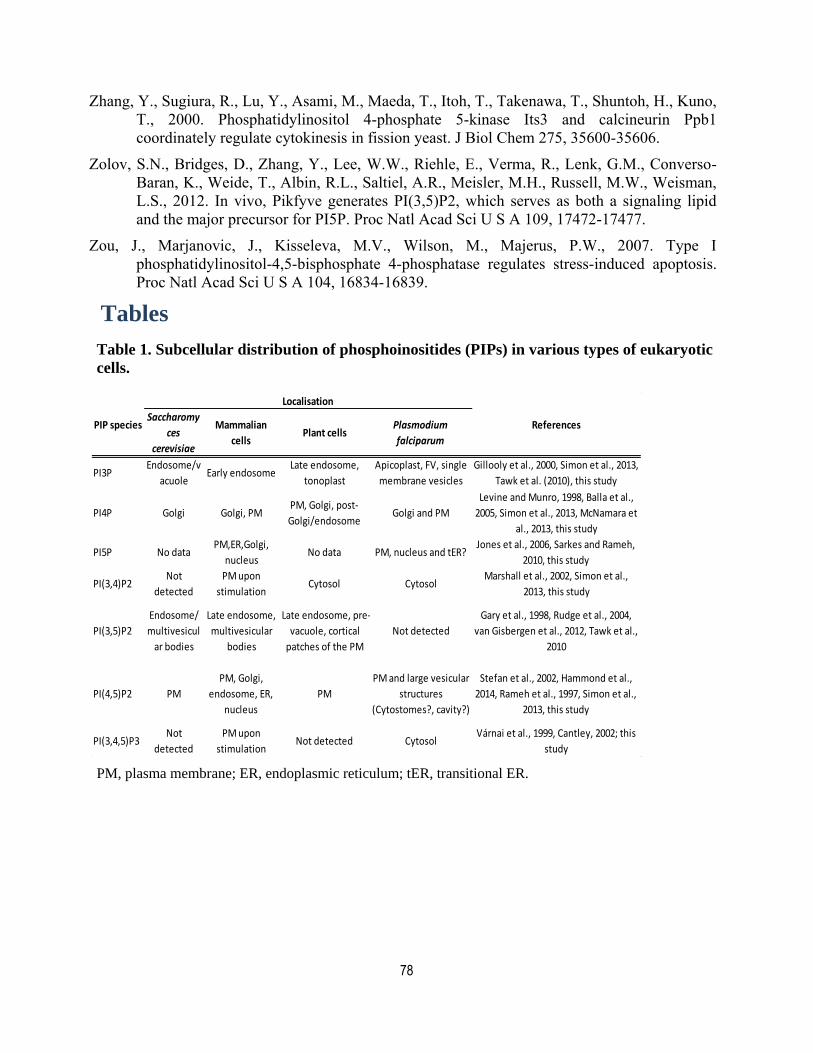
78
Zhang, Y., Sugiura, R., Lu, Y., Asami, M., Maeda, T., Itoh, T., Takenawa, T., Shuntoh, H., Kuno,
T., 2000. Phosphatidylinositol 4-phosphate 5-kinase Its3 and calcineurin Ppb1
coordinately regulate cytokinesis in fission yeast. J Biol Chem 275, 35600-35606.
Zolov, S.N., Bridges, D., Zhang, Y., Lee, W.W., Riehle, E., Verma, R., Lenk, G.M., Converso-
Baran, K., Weide, T., Albin, R.L., Saltiel, A.R., Meisler, M.H., Russell, M.W., Weisman,
L.S., 2012. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid
and the major precursor for PI5P. Proc Natl Acad Sci U S A 109, 17472-17477.
Zou, J., Marjanovic, J., Kisseleva, M.V., Wilson, M., Majerus, P.W., 2007. Type I
phosphatidylinositol-4,5-bisphosphate 4-phosphatase regulates stress-induced apoptosis.
Proc Natl Acad Sci U S A 104, 16834-16839.
Tables
Table 1. Subcellular distribution of phosphoinositides (PIPs) in various types of eukaryotic
cells.
PM, plasma membrane; ER, endoplasmic reticulum; tER, transitional ER.
Saccharomy
ces
cerevisiae
Mammalian
cellsPlant cells
Plasmodium
falciparum
PI3PEndosome/v
acuoleEarly endosome
Late endosome,
tonoplast
Apicoplast, FV, single
membrane vesicles
Gillooly et al., 2000, Simon et al., 2013,
Tawk et al. (2010), this study
PI4P Golgi Golgi, PMPM, Golgi, post-
Golgi/endosomeGolgi and PM
Levine and Munro, 1998, Balla et al.,
2005, Simon et al., 2013, McNamara et
al., 2013, this study
PI5P No dataPM,ER,Golgi,
nucleusNo data PM, nucleus and tER?
Jones et al., 2006, Sarkes and Rameh,
2010, this study
PI(3,4)P2Not
detected
PM upon
stimulationCytosol Cytosol
Marshall et al., 2002, Simon et al.,
2013, this study
PI(3,5)P2
Endosome/
multivesicul
ar bodies
Late endosome,
multivesicular
bodies
Late endosome, pre-
vacuole, cortical
patches of the PM
Not detected
Gary et al., 1998, Rudge et al., 2004,
van Gisbergen et al., 2012, Tawk et al.,
2010
PI(4,5)P2 PM
PM, Golgi,
endosome, ER,
nucleus
PM
PM and large vesicular
structures
(Cytostomes?, cavity?)
Stefan et al., 2002, Hammond et al.,
2014, Rameh et al., 1997, Simon et al.,
2013, this study
PI(3,4,5)P3Not
detected
PM upon
stimulationNot detected Cytosol
Várnai et al., 1999, Cantley, 2002; this
study
PIP species
Localisation
References

79
Figure legends
Fig. 1. Subcellular localization of phosphatidylinositol 3-monophosphate (PI3P) in
Plasmodium falciparum. (A) In ring stage parasites, the PXP40
-GFP sensor is found as a single
structure close to the nucleus. (B) In trophozoites (Troph), the signal now surrounds the food
vacuole and an intense dot close to the plasma membrane (arrowhead) is also observed. In addition,
some large vesicular structures were often seen in close proximity to the food vacuole membrane
(arrow). (C) In schizonts (Schiz), the number of fluorescent dots increased along with the number
of nuclei. The food vacuole membrane and the large vesicles seen in trophozoites were still present
(arrowheads and arrow, respectively). (D) Western blot of mixed stages parasite extracts confirms
expression of the full-length PXP40
-GFP sensor. Blue, DAPI stained nuclei; BF, bright field. Scale
bar: 5 m.
Fig. 2. Subcellular localization of phosphatidylinositol 4-monophosphate (PI4P) in
Plasmodium falciparum. (A) In ring stage parasites, the 2xPHFAPP
-GFP sensor is found around the
plasma membrane and as a focus on fluorescence. (B) In trophozoites (Troph), in addition to the
plasma membrane associated signal, two foci of intense fluorescence are now seen on each side of
the DAPI stained nucleus. (C) As the parasite proceeds through schizogony (Schiz), each
individual merozoite potentially possesses a single spot of GFP, together with some plasma
membrane labeling. (D) In free merozoites (Mero), the plasma membrane GFP is no longer seen
whilst the focus is still there. In certain cases, the signal seemed to cap one side of the merozoite
(arrows). (E) Western blot of mixed stages parasite extracts confirms expression of the full-length
2xPHFAPP
-GFP sensor. Blue, DAPI stained nuclei; BF, bright field. Scale bar: 5 m.
Fig. 3. Phosphatidylinositol 4-monophosphate is found at the Golgi apparatus throughout the
Plasmodium falciparum erythrocytic cycle. (A) The PHFAPP
-3HA sensor colocalizes extensively
with the Golgi marker Endoplasmic reticulum Retention Defective protein 2 (ERD2) in rings (a),
trophozoites (Troph) (b), schizonts (Schiz) (c) and free merozoites (Mero) (d). (B) Quantification
of the colocalization between PHFAPP
-3HA and ERD2 at each stage of the erythrocytic cycle.
ERD2-Rhoptry associated protein 1 (RAP1) is used as a control for proteins residing in different
organelles. The coefficients were calculated by the intensity correlation of Alexa fluor 488 and
594. Each dot on the graph represents an individual cell. Horizontal line represents the mean. The

80
mean ± S.E.M. is represented for each condition. ****P <0.0001. Number of cells: PHFAPP
versus
ERD2: Ring, n=15; Troph, n=14; Schiz, n= 15; ERD2 versus RAP1, n=20. An unpaired t-test was
used to calculate the P value. (C) PHFAPP
sensors do not colocalize with micronemal markers Apical
membrane antigen 1 (AMA1) (a) and Erythrocyte binding antigen 175 (EBA175) (b), the rhoptry
neck marker RON4 (c) and the rhoptry bulb marker RAMA (d). (D) Quantification of the
colocalization between PHFAPP
-3HA and the apical markers. Number of cells: PHFAPP
-3HA versus
AMA1, n=7; PHFAPP
-3HA versus EBA175, n=7; PHFAPP
-3HA versus RON4, n=7; 2xPHFAPP
-GFP
versus mCherry-RAMA, n=10; ERD versus RAP1, n=20. NS, non-significant in unpaired t-test.
(E) Western blot of mixed stage parasite extracts confirms expression of the full-length PHFAPP
-
3HA sensor. Blue, DAPI stained nuclei; BF, bright field; mC-RAMA, mCherry-RAMA. Scale bar:
5 m.
Fig. 4. Subcellular localization of phosphatidylinositol 5-monophosphate (PI5P) in
Plasmodium falciparum. (A) In ring (a) and trophozoite (Troph) (b) stage parasites, the PHDOK5
-
GFP sensor is found in the cytosol and at the plasma membrane with an enriched region (arrow). In
schizonts (Schiz) (c), the GFP signal was often seen as capping the apical region of individual
merozoites (star) and sometimes as a single dot per merozoite (arrowhead). (B) IFA with an anti-
ERD2 antibody shows that the PHDOK5
-3HA sensor overlaps partially with the Golgi in schizonts.
Blue, DAPI stained nuclei; BF, bright field. Scale bar: 5 m. (C) Quantification of colocalization
shows that PHDOK5
-3HA overlaps significantly less with the Golgi marker ERD2 than PHFAPP
-
3HA. Horizontal line represents the mean. The mean ± S.E.M. is represented for each condition. *P
<0.05. Number of cells: PHDOK5
-3HA versus ERD2, n=15; PHFAPP
-3HA versus ERD2, n=15. An
unpaired t-test was used to calculate the P value. (D) Western blot of mixed stages parasite extracts
confirms expression of the full-length (a) PHDOK5
-GFP and (b) PHDOK5
-3HA sensors.
Fig. 5. Subcellular localization of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) in
Plasmodium falciparum. (A) The mCherry-2xPHPLCδ
sensor labels the parasite plasma membrane
throughout the erythrocytic cycle. In rings (a) and trophozoites (Troph) (b), the mCherry signal was
additionally labeling a cavity and some membrane protrusions (arrows). Maximum projection
shows more than one structure in the ring. (B) IFA with an anti-merozoite surface protein 1
(MSP1) antibody shows that the PHPLCδ
sensor overlaps extensively with the plasma membrane in

81
early (a) and late (b) schizonts (Schiz). (C) Western blot of mixed stages parasite extracts confirms
expression of the full-length mCherry-2xPHPLCδ
sensor. Blue, DAPI stained nuclei; mC, mCherry;
BF, bright field; Max proj, Maximum projection. Scale bar: 5 m.
Fig. 6. Subcellular distribution of phosphoinositide (PIP) species during the erythrocytic cycle
of Plasmodium falciparum. PI3P is found at the apicoplast (AP), food vacuole (FV) membrane
and vesicles (V). PI4P is found at the plasma membrane (PM) and the Golgi apparatus (G). PI5P is
found at the PM in all stages but also potentially at the transitional endoplasmic reticulum (tER) in
schizonts. PI(4,5)P2 is found at the PM and large vesicular structures potentially corresponding to
the cytostome/cavity (C). RBCm, red blood cell membrane; N, nucleus. Note that the ring and the
schizont have been magnified for clarity, and that the distinct regions of PM labeling for PI4P and
PI(4,5)P2 do not represent distinct domains and are just for clarity.
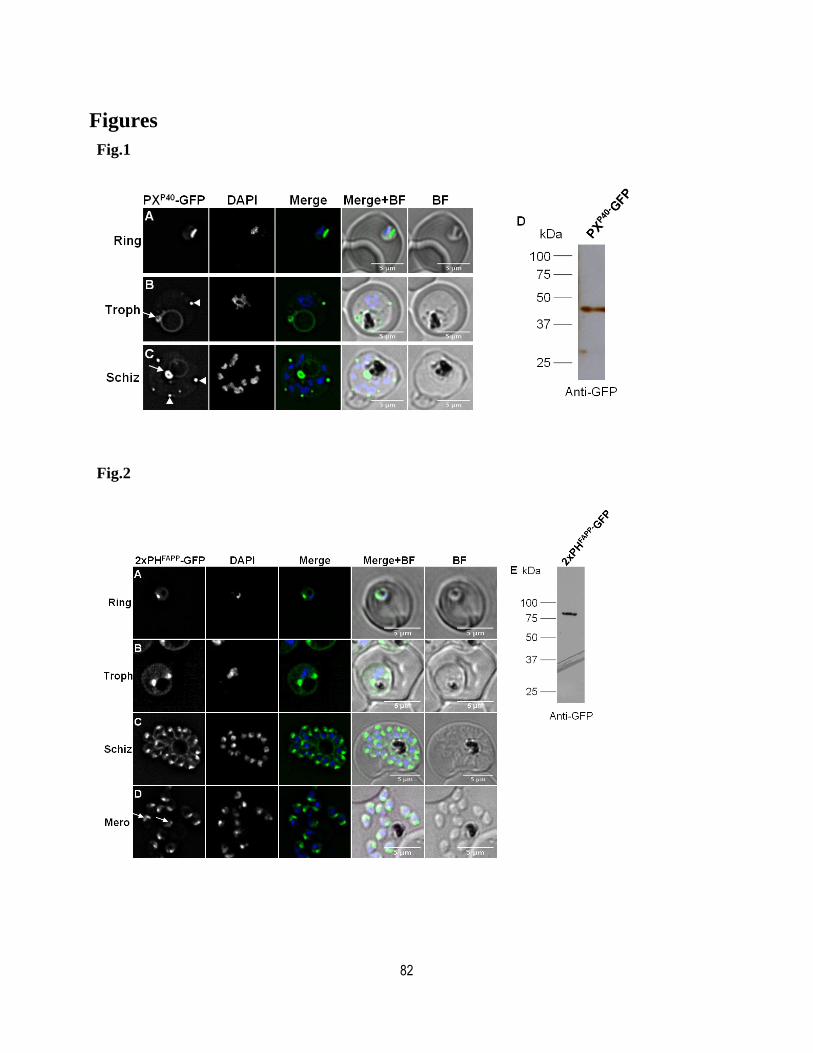
82
Figures
Fig.1
Fig.2
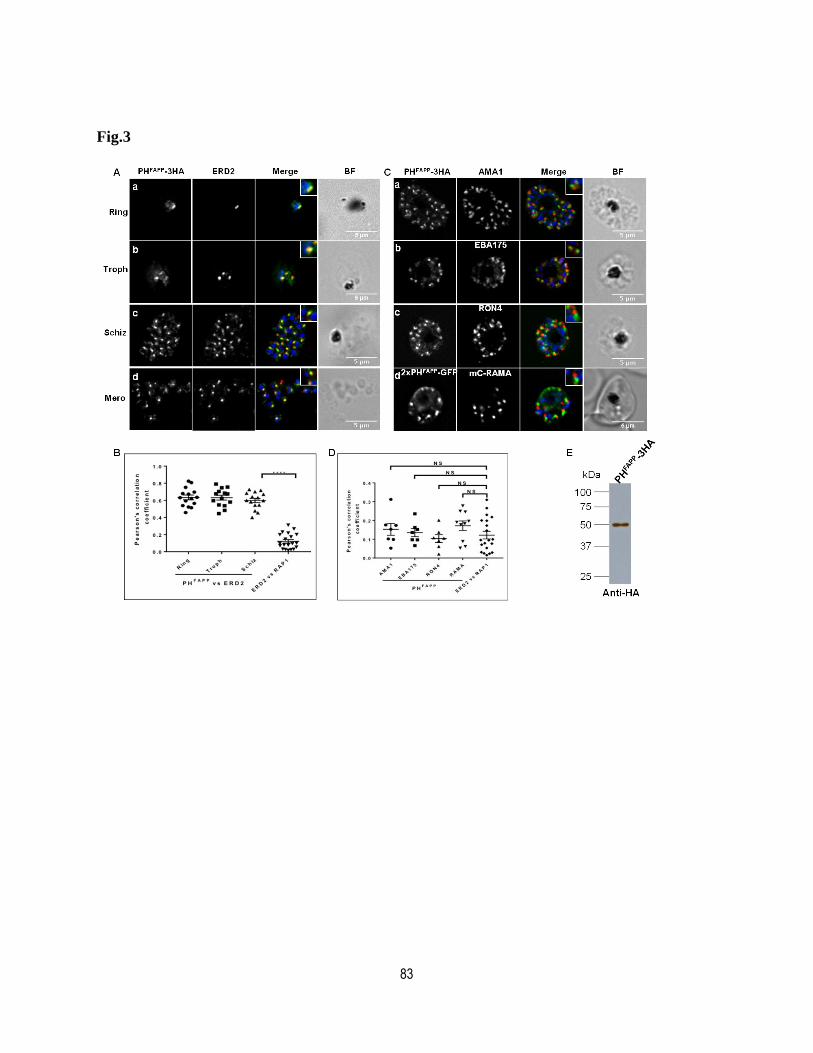
83
Fig.3
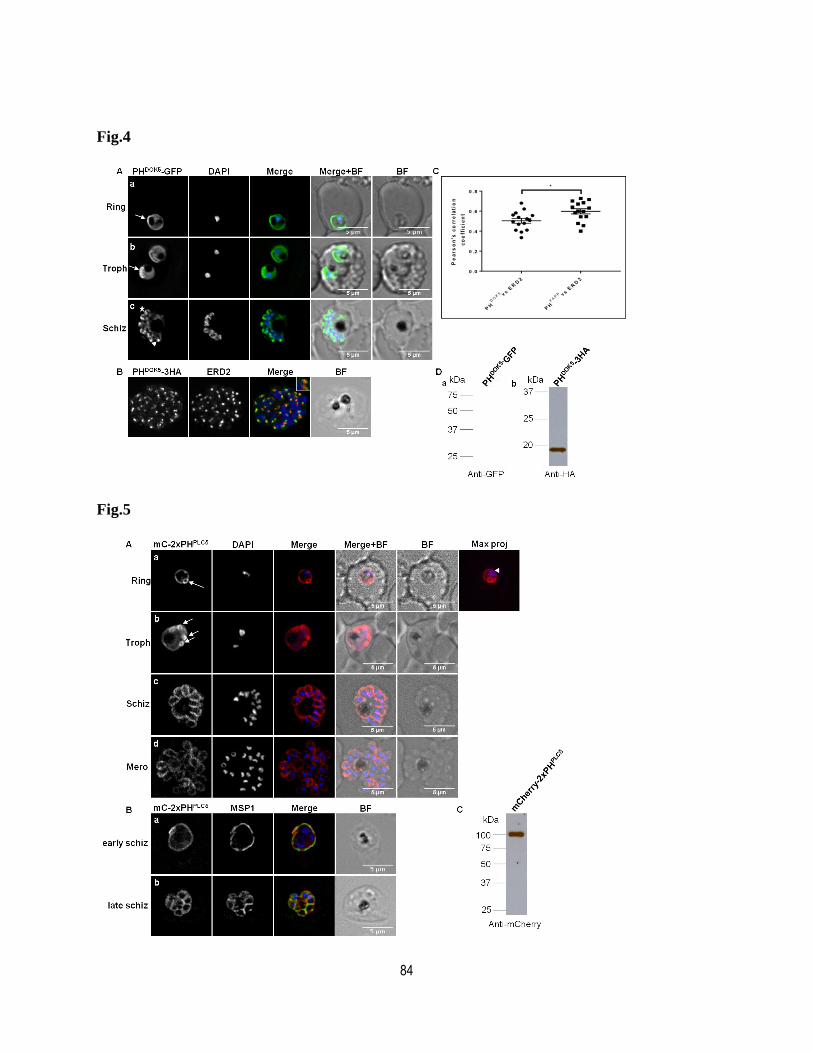
84
Fig.4
Fig.5
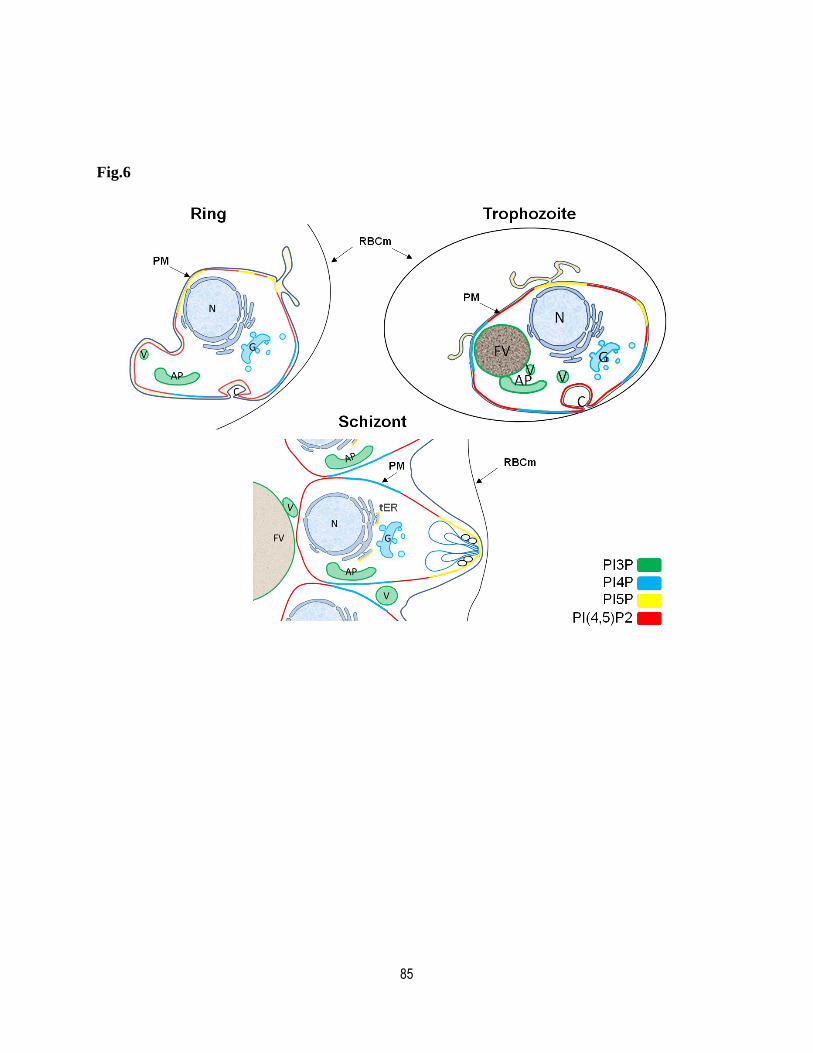
85
Fig.6

86
Supplementary Table
Table S1: List of primers and plasmids used in this study
a: pglmS-Rep20-Hsp86 allows the expression of a fusion protein using the Heat shok protein 86
promoter. pLN-Cam allows the expression of a fusion protein through the Calmodulin promoter.
PIP sensor Primers Sequence (5'->3') Expression Systema
Enzymes
Selectable
marker
5- AGATCTTAT ATATAATGG CTGTGG CCC AGC AGCTG -3
5- GGTACCTGA GTC ATA GGG CGA CTG G -3
5- AGATCTTAT ATATAATGG CTGTGG CCC AGC AGCTG -3
5- GGTACCTGA GTC ATA GGG CGA CTG G -3
5- AGATCT TAT ATATAATGG AGG GGGTGT TGT ACA AGT GGA CCA AC -3
5- GGTACC CCT TGT ATC AGT CAA ACATGC TTT -3
5-TAT ATATAATGGCTTCCAATTTTAATGACATAG-3
5-GGTACCGATCCGTGTTCCTACACACTCCATCTG-3
5-CCTAGGTATATATAATGGTGAGCAAGGGCGAGGAGC-3
5-CTGCAGCTAACCGGGGGGATGCTCAG-3
5-AGATCTTATATATAGCAGGCTTAACCATGCTAGACCCTTTG-3
5-CAACTATGTATAATAAAGTTGCCTCGAGCTACTGGATGTTGA-3
5- AGATCT TAT ATATAATGG CCG CAGTGA TTCTGG AGA G -3
5- GGT ACC AGGTTT TAA GCT TCC ATT CCT GT -3
5-CCTAGGTATATATAATGAGTAAAGGAGAAGAACTTTTCACTG-3
5-CTCGAGCTACTGGATGTTGAGCTCCTTCAGG-3
5-ACGCGTATATATAATGGTGAGCAAGGGCGAGGAG-3
5-CTGCAGTTACCTAGGCTTGTACAGCTCGTC
5-ACGCGTATATATAATGGTGAGCAAGGGCGAGGA-3
5-CTGCAGTTACCTAGGCTTGTACAGCTCGTC-3
5-CCTAGGTATATATAATGG AGG GGGTGT TGT ACA AGT GGA CCA AC -3
5-CTCGAGCCTTGTATCAGTCAAACATGCTTT -3
5-CCTAGGTATATATAATGGCTTCCAATTTTAATGACATAG-3
5-CTCGAGGATCCGTGTTCCTACACACTCCATCTG-3
BSD
BSD
BSD
BSD
BSD
BSD
BSD
yDHODH
BSD
BSD
BSD
BSD
BglII-XhoI
BglII-KpnI
AvrII-XhoI
MluI-AvrII-stop-PstI
AvrII-XhoI
AvrII-XhoI
BglII-KpnI
BglII-KpnI
BglII-KpnI
BglII-KpnI
BglII-KpnI
AvrII-PstI
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pLN-Cam
pLN-Cam
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
pglmS-Rep20-Hsp86
PHBTK
-GFP
GFP
mCITRINE
Cherry
PHFAPP
-3HA
PHDOK5
-3HA
PHPX40
-GFP
PHPX40
-Cherry
2xPHFAPP
-2xGFP
PHDOK5
-GFP
mCITRINE-PHTAPP1
2xCherry-2xPHPLC

87
Table S2: Phosphoinositide binding domains used in this study.
h, human; r, rat
Domain Gene Protein PIP detected Amino acids Reference
PX p40phox PX40 (h) PtdIns(3,4)P2 1-140Kanai, F., Liu, H., Field, S.J., Akbary, H., Matsuo, T., Brown, G.E.,
Cantley, L.C., Yaffe, M.B., 2001. Nat Cell Biol 3, 675-678.
phosphatidylinositol-
four-phosphate-
adaptor-protein-1
FAPP1(h) PtdIns(4)P 1-99.Dowler, S., Currie, R.A., Campbell, D.G., Deak, M., Kular, G.,
Downes, C.P., Alessi, D.R., 2000. Biochem J., 351, 19-31.
docking protein 5 DOK5(h) PtdIns(5)P 1-116
Guittard, G., Gérard, A., Dupuis-Coronas, S., Tronchère, H.,
Mortier, E., Favre, C., Olive, D., Zimmermann, P., Payrastre, B.,
Nunès, J.A., 2009. J Immunol 182, 3974-3978.
tandem PH-domain-
containing protein-1TAPP1(h) PtdIns(3,4)P2 182-305
Kimbar, W.A., Trinkle-Mulcahy, L., Cheung, P.C.F., Deak, M.,
Marsden, L.J., Kieloch, A., Watt, S., Javier, R.T., Gray, A.,
Downes, C.P., Lucocq, J.M., Alessi, D.R., 2002. Biochem J, 361
(Pt3) 525-536
phospholipase Cδ1 PLCδ1(r) PtdIns(4,5)P2 1-175Lemmon, M.A., Ferguson, K.M., O'Brien, R., Sigler, P.B.,
Schlessinger, J., 1995. Proc Natl Acad Sci USA 92, 10472-10476.
Bruton
agammaglobulinemia
tyrosine kinase
Btk1(h) PtdIns(3,4,5)P3 1-177Salim, K., Bottomley, M.J., Querfurth, E., Zvelebil, M.J., Gout, I.,
Scaife, R., Margolis, R.L., Gigg ,R., Smith, C.I., Driscoll ,P.C.,
Waterfield, M.D., Panayotou, G., 1996. EMBO J. 15(22):6241-6250
PH

88
Supplementary figure legends
Supplementary Fig. S1. More examples of the subcellular localization of phosphatidylinositol
3-monophosphate (PI3P) in Plasmodium falciparum. (A) An early schizont stage parasite shows
numerous large vesicular structures close to the food vacuole (arrows) together with some smaller
vesicles close to the plasma membrane (arrowheads). (B) In a later schizont, the PXP40
-GFP sensor
labels a reticulated structure reminiscent of the apicoplast. Blue, DAPI stained nuclei; BF, bright
field. Scale bar: 5 m.
Supplementary Fig. S2. The foci of phosphatidylinositol 3-monophosphate (PI3P) colocalize
with the apicoplast in Plasmodium falciparum. Imaging of parasites expressing PXP40
-mCherry
and a fusion of the apicoplast protein Enoyl acyl carrier protein reductase (ENR) with GFP shows
strong overlap between the PI3P foci and the apicoplast in trophozoites (A), and early schizonts (B,
C). Note that very few doubly-transfected parasites were observed and none in ring or in late
schizont stages. Blue, DAPI stained nuclei; mC, mCherry; BF, bright field. Scale bar: 5 m.
Supplementary Fig. S3. Subcellular localization of GFP in Plasmodium falciparum. (A) The
GFP is found throughout the cytosol with some enrichment close to the nucleus (arrows) in rings
(A), trophozoites (Troph) (B) and schizonts (Schiz) (C). Blue, DAPI stained nuclei; BF, bright
field. Scale bar: 5 m.
Supplementary Fig. S4. Additional images of phosphatidylinositol 4-monophosphate (PI4P)
in Plasmodium falciparum. (A) IFA showing PHFAPP
-3HA sensor shows a similar pattern to
PHFAPP
-GFP in Fig. 2 (B) IFA showing some overlap between PHFAPP
-3HA and the parasite
plasma membrane marker merozoite surface protein 1 (MSP1) in early (a) and in late schizonts
(Schiz) (b). Blue: DAPI stained nuclei; BF: bright field; Troph, trophozoite. Scale bar: 5 m.
Supplementary Fig. S5. Additional images of phosphatidylinositol 5-monophosphate (PI5P)
in Plasmodium falciparum. (A) Another example of a parasite with a zone of the plasma
membrane enriched with the PHDOK5
-GFP sensor (star). (B) A trophozoite (Troph) with an
accumulation of the PHDOK5
-GFP sensor in the nucleus and in two spots very close to it (arrow).

89
(C) An example of a schizont (Schiz) where the foci of fluorescence are closely associated with the
DAPI stained nucleus in individual merozoites. In some merozoites, the signal is not concentrated
in a spot but instead looks more like a slightly curved line around the nucleus (arrowhead). Blue,
DAPI stained nuclei;BF, bright field. Scale bar: 5 m.
Supplementary Fig. S6. An example of the phosphatidylinositol 4,5-bisphosphate PI(4,5)P2
large vesicular structures in schizonts in Plasmodium falciparum. Early schizont stage parasites
with large vesicular structures associated with the parasite plasma membrane (arrows). Blue, DAPI
stained nuclei; mC, mCherry; BF, bright field; Max proj, Maximum projection. Scale bar: 5 m.
Supplementary Fig. S7. Subcellular localization of mCherry in Plasmodium falciparum. (A)
The mCherry protein is found throughout the cytosol with some enrichment close to the nucleus
(arrows) in rings (A), trophozoites (Troph) (B) and schizonts (Schiz) (C). Blue, DAPI stained
nuclei; BF, Bright field. Scale bar: 5 m.

90
Supplementary figures
Supplementary Fig.1
Supplementary Fig.2

91
Supplementary Fig.3
Supplementary Fig.4
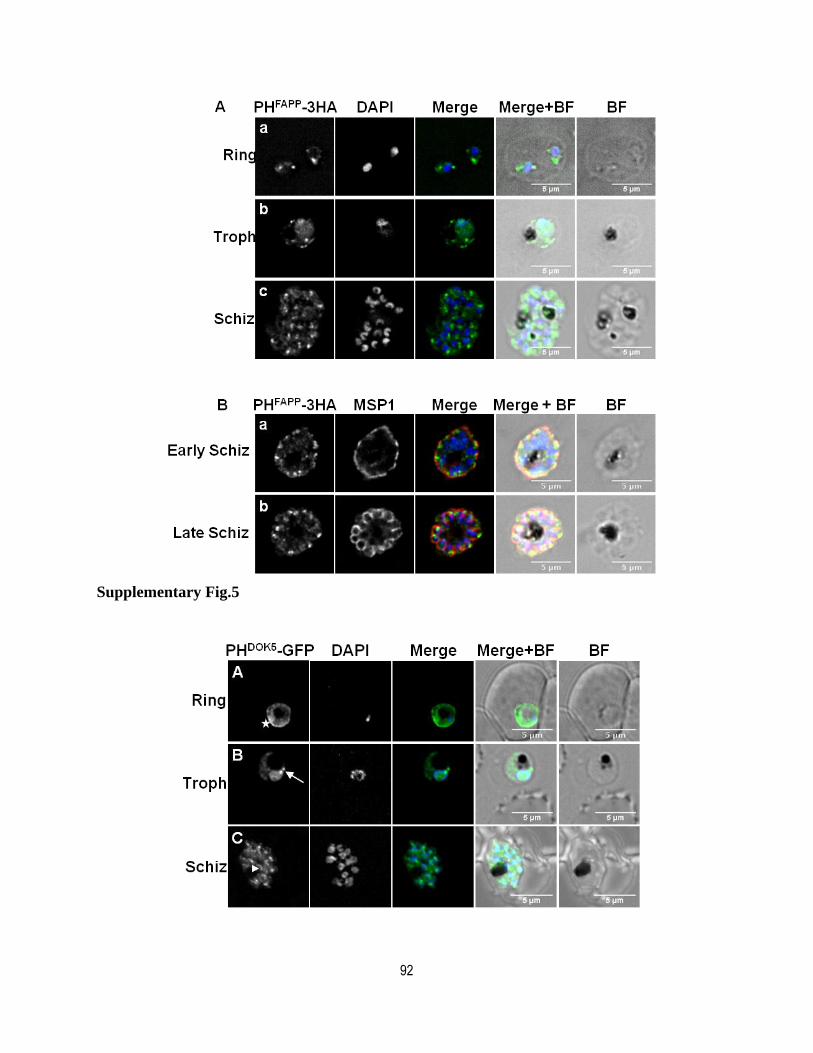
92
Supplementary Fig.5
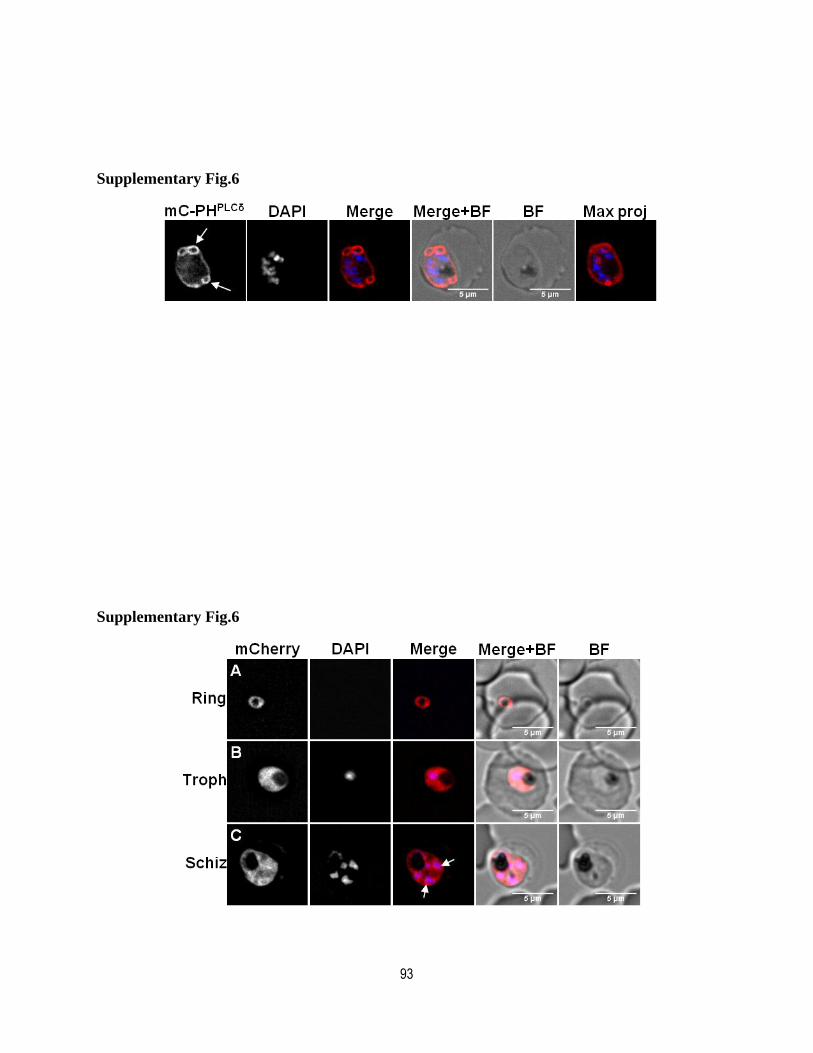
93
Supplementary Fig.6
Supplementary Fig.6

94
Chapter 3: A pan-apicomplexan phosphoinositide-
binding protein acts in malarial invasion-microneme
exocytosis.
Avant-propos
In the article presented in Chapter 4 entitled “A pan-apicomplexan phosphoinositide-binding
protein acts in malarial invasion-microneme exocytosis”. It is a scientific paper and I am the
first author. I did most of the parasite manipulation, phenotypic analysis of the results related to
the knocksideways (Fig. 1A, 3, 4A, B&C, EV1, 2, 3 S4, S5, S6, S7A) and designed some
experiments. I also contributed to the writing of the manuscript. Angana Mukherjee performed the
invasion assays with enzyme-treated erythrocytes (Fig.4F, S8) and contributed to the writing of the
manuscript. Marie-Ève Crochetière and Audrey Sergerie analyzed the secretion of PfAMA1 by
IFA and performed the secretion assay with free merozoites. (Fig. 2, 4A, EV2B, 3B, S3). Souad
Amiar performed the protein-lipid interaction analyses and contributed to the writing of the
manuscript (Fig. 1, S1, 2). L. Alexa Thompson and Joel B. Dacks performed the bioinformatics
analysis (Fig.5, EV4, S8) and contributed to the writing the manuscript, David Gaumond and
Dominic Gagnon performed parasite manipulations. R.V.S. interpreted results and contributed to
the writing of the manuscript (Fig.3B). Dave Richard conceptualized the study, designed
experiments, supervised the project, interpreted results, contributed to some of experiments
(Fig.4D, S7B) and wrote the manuscript. The article has been published in the EMBO Reports in
May 2019.

95
Résumé
L'invasion des globules rouges par le parasite de la malaria Plasmodium falciparum, est une étape
essentielle dans le développement de la maladie. Par conséquent, les acteurs moléculaires
impliqués dans l'invasion des cellules hôtes représentent des cibles importantes pour le
développement d'inhibiteurs et le de vaccins anti-malariaux. Le processus d’invasion du globule
rouge par les mérozoïtes est médié par la sécrétion de protéines contenues dans les organites du
complexe apical. Peu de choses sont connues quant aux mécanismes derrières l’exocytose de leur
contenu. Ici, nous avons identifié une protéine contenant un domaine conservé de liaison aux
phosphoinositides et avons montré que celle-ci est importante pour l’attachment et les étapes
subséquentes d’invasion de l'érythrocyte par le mérozoïte. De plus, nous avont montré que retirer la
protéine de son site d’action, en utilisant la technique de knock sideways empêche la sécrétion
d’une population spécifique de protéines de micronèmes liées à l’invasion. Nos résultats
fournissent donc des preuves du rôle important des phosphoinositides dans le processus d’invasion
par le parasite de la malaria et apporte un éclairage nouveau quant aux mécanismes de sécrétion
différentielle des populations de protéine de micronèmes, un concept qui pourrait être applicable
aux autres parasites de la famille Apicomplexa.

96
Article
A pan-apicomplexan phosphoinositide-binding protein acts in malarial invasion-
microneme exocytosis.
Zeinab Ebrahimzadeh1, Angana Mukherjee
1,4, Marie-Ève Crochetière
1,4, Audrey Sergerie
1,4, Souad
Amiar2, L. Alexa Thompson
3, Dominic Gagnon
1, David Gaumond
1, Robert V. Stahelin
2, Joel B.
Dacks3 and Dave Richard
1*
1 : Centre de recherche en infectiologie, CRCHU de Québec-Université Laval, 2705 Boul. Laurier
Québec (QC), Canada, G1V 4G2
2: Department of Medicinal Chemistry and Molecular Pharmacology and the Purdue Institute of
Inflammation, Immunology and Infectious Disease, Purdue University, West Lafayette, IN 47907 USA
3: Division of Infectious Disease, Department of Medicine, Faculty of Medicine and Dentistry,
University of Alberta.
4: These authors contributed equally to this work.
*Corresponding author. Dave Richard, Tel.: 1-418-525-4444 ext 47975; fax: 1-418-654-2715.
E-mail address: [email protected] (DR)
Running title: Malaria microneme exocytosis

97
Abstract
Invasion of human red blood cells by the malaria parasite Plasmodium falciparum is an essential
step in the development of the disease. Consequently, the molecular players involved in host cell
invasion represent important targets for inhibitor design and vaccine development. The process of
merozoite invasion is a succession of steps underlined by the sequential secretion of the organelles
of the apical complex. However, little is known with regards to how their contents is exocytosed.
Here, we identify a phosphoinositide-binding protein conserved in apicomplexan parasites and
show that it is important for the attachment and subsequent invasion of the erythrocyte by the
merozoite. Critically, removing the protein from its site of action by knock sideways
preferentially prevents the secretion of certain types of micronemes. Our results therefore provide
evidence for a role of phosphoinositide lipids in the malaria invasion process and provide further
insight into the secretion of microneme organelle populations, which is potentially applicable to
diverse apicomplexan parasites.

98
Introduction
With more than 216 million cases and 445,000 deaths in 2016, malaria still represents a devastating
disease[1]. The widespread occurrence of antimalarial drug resistance and the lack of a
commercialized vaccine highlight the need for novel therapeutics. Plasmodium spp., the etiological
agents of the disease, are obligate intracellular parasites and their invasion of human red blood cells
(RBCs) is an essential part of their lifecycle. The invasion process debuts with the initial
recognition of the erythrocyte membrane by merozoite surface proteins[2-4] after which the
merozoite reorientates so that its apical tip becomes juxtaposed to the RBC membrane. Tight
attachment of the parasite then occurs through the binding of parasite invasion ligands to RBC
surface receptors. These ligands are the erythrocyte-binding-like (EBAs) and reticulocyte binding-
like (RHs) proteins to their cognate receptors[5-8]. Subsequently, a tight junction is formed
through an interaction between the apical membrane antigen 1 (AMA1) and the rhoptry neck
(RON) complex[9-11]. Through an acto-myosin molecular motor, the parasite pulls itself into a
parasitophorous vacuole in which it will reside [12-14].
Several of the effector proteins involved in the process of merozoite invasion are stored in the
apical complex organelles and released in a controlled fashion[15]. How apical organelles are
secreted by the merozoite is poorly known but evidence suggests that calcium and cGMP
signaling are implicated[16,17]. Indeed, studies have shown that microneme discharge through
the activation of calcium-dependent protein kinases (CDPKs)[18,19] and the cyclic GMP-
dependent protein kinase (PKG)[20,21] is required for egress of merozoites from the schizont.
Of interest, it was proposed that the disparate roles of PKG throughout the malaria parasite
lifecycle could potentially be explained by its regulation of phosphoinositide metabolism and its
effect on calcium signalling and potentially vesicular trafficking[17]. Exposure to low potassium
levels as found in human plasma leads to a rise in intracellular calcium that then triggers the
secretion of the micronemal proteins PfAMA1 and PfEBA175 and the subsequent interaction of
the latter with glycophorin A on the RBC surface then results in the exocytosis of the rhoptries
[22,23]. The interaction of PfRH1 with its as yet unknown receptor also results in an increase in
intracellular calcium and secretion of PfEBA175[24]. The ability of the PLC inhibitor U73122 to
abrogate microneme secretion suggests that the P. falciparum PLC homologue is implicated in the
process[22].

99
Recent work in the related apicomplexan parasite Toxoplasma gondii further suggested that
recognition of phosphatidic acid produced through the action of TgPI-PLC by an acylated
Pleckstrin-homology protein (TgAPH) present on the surface of the micronemes led to their
exocytosis and parasite egress. The authors further showed that recombinantly expressed P.
falciparum APH also bound to PA but whether it plays an equivalent role in microneme exocytosis
is unknown [25,26]. Finally, the snare-like C2 domain-containing protein PfDOC2.1 was shown to
be required for the secretion of the micronemal protein PfEBA175 and the rhoptry neck protein
PfRH2a[27,28].
Until recently and despite early evidence suggesting that the P. falciparum micronemes were
composed of heterogenous populations with specific functions in egress and/or invasion[29],
most studies extrapolated results obtained while studying one micronemal protein (most often
PfAMA1) to apply to all the others. Of interest, this heterogeneity of micronemes is also conserved
in T. gondii[30,31]. A few studies have now started to take this into consideration. For example,
recent results have shown that PfCDPK1 knockdown parasites have no defect in egress but cannot
invade erythrocytes due to a specific defect in the secretion of PfEBA175-containing micronemes
but not of PfAMA1 micronemes[32]. Absalon and colleagues revealed that the signaling cascade
containing PfPKG and PfCDPK5 led to the secretion of PfAMA1 on the surface of merozoite
before egress but not of PfEBA175 which remains in the micronemes and led them to suggest the
inclusion of additional subsets of egress-specific micronemes [18]. The mechanisms underlying
this differential exocytosis are currently unknown.
Intriguingly, despite representing only a minor fraction of the total lipids of eukaryotic
membranes, phosphoinositides (PIPs) are critical components involved in a variety of cellular
processes and recent work has shown that it was also the case for apicomplexan parasites
(reviewed in [33]). More specifically for P. falciparum, roles for PIPs have been shown in
cytokinesis and merozoite formation[34], apicoplast biogenesis and inheritance[35-37],
hemoglobin endocytosis[38], merozoite egress[17], gametocyte activation [39-41] and ookinete
motility[17], the latter two occurring in the mosquito vector, and finally in resistance to
artemisinin[42,43]. In line with these varied functions, the determination of the subcellular
distribution of several species of PIPs in the malaria parasite asexual erythrocytic stages showed

100
localizations to structures such as the Golgi apparatus, the plasma membrane, the food vacuole
membrane, the endoplasmic reticulum and the apicoplast[34,35,44,45].
While hydrolysis of PI(4,5)P2 by the Toxoplasma gondii PI-phospholipase C is critical for the
invasion of this parasite (see below,[25]), a direct role for PIPs in the malaria merozoite invasion
process has not yet been described.
We here identify a P. falciparum PH domain-containing protein that is important for the secretion
of micronemes containing PfEBAs. This protein has a relaxed phosphoinositide binding ability and
we show, using knock sideways (KS), that it is required for merozoite attachment and invasion.
Furthermore, we show bioinformatically that this protein is present in diverse apicomplexans
suggesting that it plays a role in pathogenesis beyond Plasmodium. Our results therefore provide
further insight into the secretion of micronemal populations.
Results and Discussion
The Pleckstrin homology domain of PF3D7_1337700 has a relaxed phosphoinositide
binding specificity
As part of our efforts to investigate potential roles for PIPs in the invasion process, we identified
PF3D7_1337700 (www.plasmodb.org), a putative PIP-binding protein containing a PH domain.
(Figure 1A). Less than 10% of the characterized PH domains possess the ability to bind PIPs but a
common feature of PIP binding PH domains is the presence of a basic sequence motif KXn
(K/R)XR involved in the binding to the head group of the inositol moiety[46]. Inspection of the
PF3D7_1337700 PH domain sequence revealed that such a motif was present
(84KANIFYIYKLR94, Figure 1A). To determine whether the PH domain had the capacity to bind
to PIPs, we recombinantly produced the WT PH domain and a version where residues K84, K92
and R94 were mutated to alanines, fused to an N-terminal glutathione-S-transferase tag.
Coomassie staining of the purified proteins revealed a band at the expected size of 38 kDa along
with a 26 kDa degradation product likely corresponding to GST alone (Appendix Figure S1).
Incubation of the GST-WT PH domain with PIP-Strips showed that the protein interacts
significantly with PI(3)P, whereas the well-established PLC PH domain bound PI(4,5)P2

101
[47](Figure 1B). The WT PH domain also showed some residual interaction with PI(4)P,
PI(5) (Figure 1B).The PH domain triple mutant was used to confirm the specificity of this
domain to phosphoinositides (PIPs). Lipid overlay showed the ability of the PH domain mutant to
interact the same PIPs than the wildtype with higher affinity to PI(3)P (Figure 1B).
PIP strips contain lipids spotted on nitrocellulose, which cannot recapitulate a membrane bilayer.
To determine if the PH domain and triple mutant could interact with PIPs in a membrane bilayer,
we employed a liposome-binding assay using a 5% molar ratio of PIPs or DPPA (phosphatidic
acid) containing liposomes. The liposome-binding assay indicated the selective binding of WT
PH domain to the phosphonoisitol monophosphate species. Indeed, as shown in Figure 1C, about
20% of the protein was found in the pellet fraction of PI(3)P, PI(4)P, or PI(5)P. The WT PH
domain also exhibited significant binding to PI(4,5)P2 and a small amount of detectable binding to
PI(3,4,5)P3 and PA containing vesicles. However, no binding of WT PH domain was observed
for two other phosphonoisitol bisphosphate containing membranes (PI(3,4)P2 and PI(3,5)P2)
(Figure 1C, top panel). In contrast, the triple mutation of the PH domain showed a clear loss of
the ability to bind liposomes containing different PIPs (Figure 1C, bottom panel). Taken together,
these data indicate that the WT PH domain can interact robustly with several PIP species in lipid
bilayers, whereas the triple mutant had a greatly diminished ability to interact with PIP
containing membranes. Since the liposome-binding assay showed that the triple mutant lost the
ability of a stable binding to specific PIP species, this suggests that the basic sequence motif
identified (K84, K92 and R94) may be involved in the binding to the PIP head group in the lipid
bilayer. To confirm this hypothesis, we performed surface plasmon resonance (SPR) analysis to
determine the binding affinity of both WT and the PH domain triple mutation to PI(3)P-containing
lipid vesicles. We employed lipid vesicles on the surface of a L1 sensor chip. The SPR assay
confirmed the ability of the WT PH domain to bind lipid vesicles containing PI(3)P with an
apparent disassociation constant (Kd) of 630 nM (Figure 1D and Appendix Figure S2). In contrast,
the PH domain triple mutant had weak binding to PI(3)P containing vesicles and we were not able
to determine an apparent Kd for the triple mutant (Figure 1D and Appendix Figure S2). The
apparent PI(3)P membrane binding affinity of the WT PH domain is on par with other well
characterized PIP binding domains[48]. Promiscuous PIP binding has been described for 67% of
yeast PH domains and their specific subcellular distribution requires coincidence detection of

102
additional factors such as another protein or membrane curvature for example [49]. These results
demonstrate that PF3D7_1337700 is a true PIP-binding protein and we renamed it PfPH2
following the uncharacterized PfPH1 named after a T. gondii PI(3,5)P2-binding protein [50].
PfPH2 is likely essential for the asexual erythrocytic cycle
To further characterize PfPH2, we endogenously tagged its C-terminus with GFP by single
cross-over recombination using the recently developed selection-linked integration (SLI)
strategy[51]. To allow the functional analysis of PfPH2 by KS (see below), a double FK506
binding protein domain (2xFKBP) tag was also appended[51] (Figure EV1A). Proper
integration of the vector and the absence of a WT allele were verified by polymerase chain
reaction (PCR) demonstrating that we had successfully tagged the pfph2 gene (Figure EV1B).
Time course analysis of PfPH2-2xFKBP-GFP expression by Western blot using an anti-GFP
antibody on parasite protein extracts taken throughout the asexual erythrocytic cycle (from the ring
through to the schizont stage) revealed a single band at the expected size of around 133 kDa for the
PfPH2-2xFKBP-GFP fusion protein in schizont stage parasites. An antibody against the
constitutive protein PfHSP70 was used as staging control (Figure EV1C). Immunofluorescence
assays (IFA) of PfPH2-2xFKBP-GFP parasites showed a faint punctate signal in late schizont
stages that did not colocalize with any of the markers investigated (micronemes: PfAMA1,
PfEBA175 and PfEBA140 (Figure EV2A), rhoptry bulb: PfRAP1 or neck: PfRON4, RH1, RH4
AND RH5 (Figure EV2B), dense granules: PfRESA (Figure EV2C) or the Golgi apparatus:
PfERD2 (Figure EV2D). To try to get a less crowded view than with schizonts, IFAs were also
performed on free merozoites recently egressed. Again, no strong colocalization could be seen
between PfPH2 and any of the markers (Figure EV3A and B). However, the PfPH2 signal often
seemed to be more apical (Figure EV3Aii and iii and B). To look at this in a more quantitative
manner, we calculated the distance between the farthest edge of the DAPI and PfPH2, PfRON4
and PfEBA175 and used this to infer how apical a protein potentially is i.e. the bigger the
distance between the marker and the DAPI, the more apical a protein likely is. These data show
that PfPH2 is farther from the DAPI than PfEBA175 (91.341.78 vs 75.601.72 pixels, Figure 2
Ai and B) and potentially more than PfRON4 (90.832.83 vs 83.192.49 pixels) though the
latter difference was not statistically significant (Figure 2Aii and B). PfRON4 is a well described

103
marker of the rhoptry neck so our results suggest that PfPH2 localizes to a structure close to the
apical tip of the merozoite.
To investigate the essentiality of PfPH2 for the asexual erythrocytic cycle, we first tried to knock
out (KO) its gene by single cross-over recombination using the SLI for targeted gene disruption
strategy (SLI-TGD)[51]. The fact that we could never detect proper integration of the vector by
PCR and the persistence of the WT allele in three KO attempts suggest that PfPH2 might be
required for the asexual stage growth (unpublished observations). This is further supported by a
recent whole genome functional screen using saturation mutagenesis by the piggyBac
transposon[52]. To gain insight into the role of PfPH2 in the asexual blood stages, we performed
KS, a strategy that allows the conditional removal of a protein of interest from its site of
action[53]. To do so, we transfected the PfPH2-2xFKPB-GFP parasite line with an episome
expressing a nuclear mislocalizer consisting of a triple nuclear localization signal fused to a
double FKBP12-rapamycin binding domain and the cherry fluorescent protein (3xNLS-2xFRB-
cherry)[51]. We next tested the ability of the mislocalizer to translocate PfPH2-2xFKPB-GFP to
the nucleus in the presence of rapamycin (Rapa), thus removing it from its normal site of action.
As seen in figure 3Ai, in absence of Rapa, the mislocalizer colocalizes with the DAPI stained
nucleus whilst PfPH2-2xFKPB-GFP shows its normal punctate pattern. When adding Rapa at the
ring stage, before the expression of PfPH2 is turned on, and letting the parasites mature to late
schizonts, a substantial proportion of the GFP signal was now observed in the nucleus instead of
the apical foci (Figure 3Aii). We quantified the levels of KS by calculating Pearson’s correlation
coefficient for PfPH2-2xFKPB-GFP vs the mislocalizer and also vs the DAPI staining, in the
presence and absence of Rapa. Our results show that the addition of Rapa does lead to a
significant increase in the colocalization between GFP and both the mislocalizer (0.240.02 vs
0.580.02) and the DAPI (0.210.01 vs 0.500.01) at the population level though there is some
variability when looking at individual cells (Appendix Figure S3). This shows that we succeeded in
performing KS on PfPH2.
We next determined the effect of the PfPH2 KS on the ability of the parasite to proliferate. To do
this, tightly synchronous PfPH2-2xFKPB-GFP+3xNLS-2xFRB-cherry (PfPH2- GFP+mislocalizer)
parasites were incubated with or without Rapa and growth was monitored over 2 cycles. We first
checked the effect of adding Rapa at different times and found that growth inhibition was

104
maximal when the compound was added at the ring stage and that adding it shortly before egress
decreased the effect by around 50% (unpublished observations). We hypothesize that this is due to
the fact that PfPH2 is potentially strongly attached to an apical structure so that it cannot easily be
“extracted” by the mislocalizer. For efficient mislocalization to occur, PfPH2 would need to be
captured as it is synthesized. Based on this, all subsequent experiments were performed with
Rapa added at the ring stage. As a control, we used the PfPH2-2xFKPB-GFP without the
mislocalizer (PfPH2-GFP). As shown in Figure 3Bi and ii, the KS resulted in an around 65%
decrease in parasitemia over one reinvasion cycle and up to 86% after the second reinvasion cycle
whilst Rapa had no effect on the control line without mislocalizer. This suggests that PfPH2 is
required for optimal proliferation of asexual stages.
Having confirmed that the KS was dependent on the presence of the mislocalizer and that the
concentration of Rapa used in our assays was not toxic in itself, the remaining experiments were
only performed with the PfPH2+mislocalizer line incubated with or without Rapa. We first
looked at the integrity of various subcellular structures by IFA and could see no obvious
difference in the PfPH2-GFP+mislocalizer incubated with Rapa (referred to as the KS line
onwards) (Appendix Figure S4). Next, to check whether the reduced parasitemia was due to a
failure of the parasites to egress from the RBC, the number of schizonts and rings was followed
over a 16-hour period, at every 4 hours, by Giemsa staining of parasite smears. As seen in Figure
3C and Appendix Figure S5, there was no difference in the evolution of schizont rupture between
the KS and the control line however, there was an important decrease in the number of rings
formed. These data show that egress proceeds normally in the KS line. We next looked at
whether the KS line produced fewer merozoites per schizont by counting DAPI-stained cells by
fluorescence microscopy but again, no difference was seen (Figure 3D) which suggests that a
defect in merozoite invasion was potentially the cause of the reduced parasitemia. To directly
address this, invasion assays were performed with +Rapa or -Rapa PfPH2+mislocalizer parasites
and this revealed a 672% decrease in the formation of new rings (Figure 3E). To determine
what step of the invasion process was affected, we monitored the ability of the merozoites to
bind to RBCs in the presence of the actin polymerization inhibitor cytochalasin D, which blocks
entry but not attachment (Appendix Figure S6 for schematic of the experiment)[54,55]. This

105
showed that the KS had a 3510% reduction in its ability to attach (Figure 3E). Globally,
this suggests that a major consequence of the KS of PfPH2 is a default in the ability of the
merozoites to strongly bind to erythrocytes and to proceed to invasion.
KS of PfPH2 potentially more strongly affects the secretion of a subset of microneme
proteins.
The sequential secretion of apical complex organelles is central to invasion with the
effectors contained in each set playing specific roles at distinct steps[15,55,56]. The lack of
a measurable egress defect in the PfPH2 KS line indicates that secretion of the egress
protease Subtilisin 1 from the exonemes[57] occurred normally. To investigate whether
PfPH2 had a role in microneme secretion, we first looked at the translocation of PfAMA1
to the merozoite surface in late stage schizonts with fully mature daughter merozoites and
saw no difference (Figure 4A). This shows that the population of micronemes
containing PfAMA1, recently referred to as egress-related[18], is properly secreted in
the PfPH2 KS line. Several of the invasion effectors are cleaved off from the merozoite
surface and released into the culture supernatant as invasion proceeds[58]. In order to
investigate whether PfPH2 had a role in microneme secretion, equal numbers of tightly
synchronous PfPH2 KS and control schizonts were let to rupture and reinvade over a 12-
hour period and the resulting culture supernatant was collected and probed with antibodies
against a variety of micronemal proteins. To control for schizont number and parasite
egress we normalized the secreted apical protein levels to PfSERA5, a parasitophorous
vacuole protein that is released upon schizont rupture[57]. As seen in Figure 4B, the
shedding of PfEBA175 and PfEBA140 was much reduced in the presence of Rapa.
Quantification of three independent biological replicates revealed decreases of around 90%
for PfEBA175 and 79% for PfEBA140 (Figure 4C and Appendix Figure S7). To verify that
the decrease of PfEBA175 in the supernatant correlated with an increase in the
unprocessed form in free merozoites, we next quantified the amount of unprocessed
PfEBA175 using purified merozoites stimulated with the calcium ionophore A23187 to
induce microneme secretion[22]. The results show that indeed, a higher amount of

106
unprocessed PfEBA175 is seen in Rapa+ merozoites (1.390.10 for Rapa+ vs 0.450.23
for Rapa-, normalized ratio to PfHSP70 levels) (Figure 4D and E).
Though we could not detect a defect in PfAMA1 translocation to the merozoite surface in
schizonts, the protein was decreased by around 24% in the supernatant (Figure 4B and C
and Appendix Figure S6). This is likely due to the reduced invasion of the KS merozoites
i.e less PfAMA1 is used up and released in the supernatant since fewer merozoites
successfully attach and invade. Taken together these results suggest that PfPH2 perhaps
plays a more critical role in the secretion of micronemes containing EBA ligands than of
PfAMA1-containing micronemes. Of interest, it was recently shown that PfCDPK1 was
specifically implicated in the secretion of PfEBA175[32] so it is tempting to speculate that
PfPH2 could potentially be regulated by phosphorylation through this kinase.
The interaction between parasite ligands and their cognate receptors on the RBC surface
defines the host cell tropism of Plasmodium parasites[59]. In P. falciparum, an interesting
dichotomy is based on the dependency, or lack thereof, of parasite ligands to the
presence of sialic acid on RBC receptors. Since PfEBA175 and PfEBA140 are involved in
the sialic acid-dependent pathway and 3D7 PfEBA175 KO parasites were previously
shown to be severely impaired in their capacity to invade erythrocytes treated with
chymotrypsin[60], we expected that their severely decreased levels in the PfPH2 KS line
would impact its host cell tropism. However, invasion assays performed in enzyme-
treated erythrocytes showed that there was no difference in the sensitivity to either trypsin,
chymotrypsin or neuraminidase treatment between the control and KS line suggestive of
a more general invasion defect (Appendix Figure S8). Perhaps the depletion of
PfEBA175 in the PfPH2 KS line is not severe enough to completely phenocopy the 3D7
PfEBA175 KO parasites. Interestingly, the decrease in invasion obtained with the PfPH2
KS line was slightly higher than any of the enzymatic treatments though the difference was
not statistically significant compared to chymotrypsin (Figure 4F). These data could
mean that, at least for the 3D7 parasite line that we have used in our study, decreasing
the levels of EBA ligands has a more profound impact on invasion than removing sialic
acid on erythrocyte receptors. This might be potentially explained by the fact that the
invasion ligands functionally interact with one another as was previously shown[61]. What

107
precise role PfPH2 plays in microneme exocytosis is unknown but it might interact with
the calcium-activated snare-like PfDOC2.1 previously suggested to be involved in the
membrane fusion event between PfEBA-175 containing micronemes and the parasite
plasma membrane[28].Since PfRH1 has been shown to be required for PfEBA175
exocytosis[24], another possibility could be that PfPH2 is implicated in the secretion of
PfRH1 instead of PfEBAs directly.
PfPH2 is Apicomplexan Specific and the PIP-Binding Sequence Motif is Partially
Conserved.
Finally, to assess the extent to which PfPH2 is a broader feature of apicomplexan
invasion/attachment, we took an informatics approach. BLASTp and HMMer homology
searching was undertaken to search for orthologues in a wide variety of taxa, with at
least two organisms from each of the six supergroups and with emphasis on apicomplexan
organisms and their outgroups (Dataset EV1). Initial BLASTp searches using the full
length PfPH2 protein as a query generated multiple positive results in organisms both
within and outside the apicomplexa phylum. Notably most of these protein hits were from
a variety of different protein families that also contained PH domains. In order to
mitigate the possibility of false positives due to the conserved PH domain, we
performed the homology searching using the query with the PH domain removed. This
only retrieved positive hits from proteomes of apicomplexan organisms (Dataset EV1).
To mitigate the possibility of false negatives due to sequence divergence, we further
searched the predicted proteomes using HMMer. We identified orthologues of PfPH2 in
all apicomplexan genomes examined, notably including C. parvum (Figure 5A and
B). All orthologues also shared the domain structure of an N-terminal PH domain followed
by a putative SMC-N domain and were within ~25% of the size of the P. falciparum
protein. By contrast, we were unable to identify orthologues in any of the outgroup taxa
that we searched. Searching by tBLASTn into the nuclear scaffolds of these outgroup
taxa, in order to mitigate false negatives due to mis-prediction of the protein similarly
failed to identify and PfPH2 orthologues (Table EV1). These outgroups include the
chromerids which is the closest sister lineage to apicomplexans, indicating the protein was

108
gained within the apicomplexan phylum, potentially as an adaptation to a parasitic specific
lifestyle.
We next wanted to determine if the PIP-binding KXn(K/R)XR sequence motif identified
within the PH domain of PfPH2 is conserved between all orthologues to give some sense
of functional homology (Figure EV4). The initial lysine residue was largely conserved, as
was a terminal positively charged residue R/H/K in all orthologues with the exception of
those from T. annulata and B. microti. This raises the possibility that the method of
microneme secretion by means of PfPH2 binding PIP residues may be conserved across
apicomplexans. Interestingly, results from a genome-wide knock out screen suggests that,
contrarily to PfPH2, the T. gondii orthologue is not essential for in vitro growth[62].
Conclusion
In conclusion, our work has uncovered a potential role for phosphoinositide lipids in the
process in merozoite invasion of red blood cells. The presence of PfPH2 proteins in diverse
relatives may indicate that this mechanism could have broader relevance to understanding
apicomplexan invasion mechanisms. Determining whether PfPH2 is a substrate of
PfCDPK1 and identifying proteins interacting with PfPH2 are now logical steps to further
our understanding of the cascade of effectors involved in the secretion of micronemes.
Materials and Methods
The study was approved by the Canadian Blood Services (CBS) research ethics board,
project number 2015.001 and by the CHU de Québec IRB, project number 2015–2230,
B14-12-2230, SIRUL 104595. Written consent was obtained by the CBS for all study
participants. All experiments were performed in accordance with relevant guidelines and
regulations.

109
Production and purification of the recombinant PfPH2 PH domains
The PH domain of PfPH2 (amino acids 70 to 175) was amplified on 3D7 WT mixed
stages cDNA using primers 5’BamH1-210-PfPH2 and 3’Xho1-525-PfPH2 (WT version)
and 5’BamH1-210-PfPH2mut and 3’Xho1-525-PfPH2 (mutated version) and subsequently
cloned in pGEX-6P3 (GE Healthcare) to be expressed as a recombinant GST fusion
following standard procedures. pGST-PfPH2-PHWT, pGST-PfPH2-PHK84A/K92A/R94A and
empty vector were transformed into Escherichia coli BL21 (DE3) competent cells and
bacterial cultures were grown at 37 ◦C and further induced at an OD600 of 0.6-0.65
with 0.5 mM isopropyl -D-1- thiogalactopyranoside overnight at 16 ◦C. Bacterial
pellets were resuspended in Gibco PBS buffer (pH 7.4) containing 1 mM EDTA, 50 mM
DTT, 100 µg/ml lysozyme and and 1x Thermo Scientific Halt protease inhibitor cocktail,
and then sonicated four times for 30 s at 4°C. Further, protein solubilization was performed
with 0.1% Triton-100X for 15 min at 4°C then centrifuged at 30,000 g for 25 min at 4°C.
Proteins were purified with Sigma glutathione- agarose according to the manufacturer’s
instructions, eluted with 50 mM Tris (pH 9.0), and 10 mM reduced glutathione, and
analyzed by SDS/PAGE and Coomassie staining for purity check.
For lipid overlay assay, the proteins were washed three times with cold 50 mM Tris (pH
8.0) with Amicon Ultra 30K MWCO to remove free glutathione. For SPR studies, the
proteins were buffer exchanged in 20 mM HEPES containing 160 mM NaCl, pH 7.4. For
Liposome-Binding assays, extensive washes were performed to remove any residual
detergent post-agarose beads that may interfere with the lipid binding of liposome stability.
However, the extensive washes resulted in reduction of protein yield and stability during
the binding experiments. To overcome this issue, an additional solubilization step of the
protein from inclusion bodies was performed prior to sonication as described
previously[63]. The protein was then washed with cold 50 mM Tris (pH 8.0) with an
Amicon Ultra 30K MWCO to remove free glutathione.
Protein-lipid overlay assay
Lipid overlay assay was carried out using PIP StripsTM
(Echelon Biosciences) and the
protein- lipid binding experiment were performed according to the manufacturer’s

110
instructions[64]. Briefly, the PIP strips were blocked overnight with PBS buffer
containing 1% milk and 0.1% Tween 20 at 4°C. The membranes were then incubated with
2 µg/ml of recombinant protein (GST-PfPH2-PHWT, GST-PfPH2-PHK84A/K92A/R94A, GST-
alone) or 0.5 µg/ml of GST-PLC-PH (Echelon Biosciences) in the same buffer for 1 h
at room temperature. After three washes with the same buffer, the membranes were
incubated for 1 h at room temperature with anti-GST antibody (Bethyl Laboratories
1:10,000). After three more washes with the same buffer, the membranes were incubated
for 1 h at room temperature with HRP-conjugated Anti-rabbit antibody (Abcam 1:10,000)
followed by three washes. The bound proteins were detected with Clarity™ Western
ECL kit from Bio-Rad Laboratories.
Liposome-Binding assay
The preparation of liposomes was performed based on the previously described
method[65]. Briefly, the chloroform mixture of lipid POPC:POPE:TopFluor-PC
(8.9:1:0.1 mol:mol:mol ratio), POPC:POPE:POPS:TopFluor-PC (7.9:1:2:0.1) or
POPC:POPE:POPS:X:TopFluor-PC (7.4:1:2:0.5:0.1), where X is one of tested PIPs or
DPPA, were mixed and dried under a stream of lipid nitrogen. The dried lipid films were
resuspended in liposome buffer (250 mM raffinose pentahydrate, 20 mM HEPES
containing 160 mM NaCl, pH 7.3) to hydrate the lipid films for 15 min at 37°C. The
multilamellar vesicles formed were extruded through 100 nm polycarbonate membrane to
form large unilamellar vesicles. The liposomes were then resuspended in 3 times volume of
liposome-binding buffer (20 mM HEPES, pH 7.3, containing 160 mM NaCl) and
centrifuged at 50,000 g for 15 min at 22°C using a Beckman TLA-55 rotor. The pelleted
liposomes were resuspended in corresponding volume of liposome-binding buffer. The
protein was centrifuged for 5 min at 16,000 g to pellet any possible aggregation and the
protein in the soluble fraction was diluted in liposome-binding buffer. For the liposome-
binding assay, 50 µM of lipid vesicles were mixed with 500 ng of protein and
incubated for 30 min at room temperature with continuous and gentle shaking. The
mixture was then centrifuged at 75,000 g for 30 min at 22°C using a Beckman TLA-100
rotor. Supernatants (SN) corresponding to the soluble fraction were separated from the

111
pellet (P) (lipid-bound fraction) and analyzed with Western blot using an anti-GST
antibody (Bethyl Laboratories 1:10,000) and HRP-conjugated Anti-rabbit antibody (Abcam
1:10,000). The protein fractions were detected with a Clarity™ Western ECL kit from Bio-
Rad Laboratories
Surface plasmon resonance analysis
SPR-based lipid binding experiments were performed at 25 °C on a Biacore™ X100 (GE
Healthcare) as described previously[66]. Briefly, the L1 sensor chip (GE Healthcare) was
equilibrated in running buffer (20 mM HEPES containing 160 mM NaCl, pH 7.4). Large
unilamellar vesicles (LUVs) with ~50 nm diameter were prepared from POPC/POPE (9:1
mol:mol-control LUVs) or POPC/POPE/PIP (8.5:1:0.5 mol:mol-active LUVs) by the
extrusion method in SPR running buffer at a final concentration of 0.5 mM. The L1 sensor
chip was then coated by injecting lipid vesicles at 5 µl/min for a response of 6000-7000
resonance unites (RU) either with control LUVs or active LUV preparations. Lipid coating
was then stabilized by injecting 4 µl of 50 mM NaOH three times at 30 µl/min following
lipid coating followed by a blocking with 0.1 mg/ml fatty acid free BSA at 10 µl/min for
three times 100 min. SPR measurements were done at the flow rate of 5 µl/min with
increasing protein concentrations (from 10 nM to 2000 nM) in freshly prepared SPR
running buffer with a contact time of 600 sec to give the protein association time required
time to reach saturation. Data were analyzed with BiaEval Software (GE Healthcare) and
plotted with Kaleidagraph. The apparent Kd of vesicle binding was determined using a non-
linear least squares analysis using the equation Req= Rmax/(1+ Kd/[C]). Where Req was
measured in response units (RU) and was plotted versus protein concentration, [C], for
protein concentration injected in each experiment. Rmax is the theoretical maximum RU
response and Kd is the apparent membrane affinity[67].
Parasite Culture
P. falciparum 3D7 asexual stage parasites (from David Walliker, Edinburgh University)
were cultured under standard conditions in RPMI-HEPES medium at 4% hematocrit

112
(human erythrocytes of O+
group) and 0.5% (w/v) AlbumaxTM
(Invitrogen) and kept at
37°C in a gas mixture of 5.0% oxygen, 5.0% carbon dioxide and 90% nitrogen[68].
Vector Construction and Transfection
To tag the endogenous PfPH2 with 2xFKBP-GFP, around 500 bp of the C-terminus of
PfPH2 was amplified with primers 5'Not1-1700-PfPH2 and 3'AvrII-stopless-PfPH2 and
cloned in frame with 2xFKBP-GFP in pSLI-2xFKBP-GFP-hDHFR digested NotI-
AvrII[51]. Parasites were transfected and integrants were selected as described previously
with some modifications[51]. Briefly, P. falciparum 3D7 parasites were transfected
with 100 µg of purified pSLI-PfPH2-2xFKBP-GFP plasmid. Positive selection for
transfectants was achieved using 5nM WR99210 (WR). Then drug resistant parasites
were split into 3 separate wells with 2-4% parasitemia and went under another round of
selection using 400µg/ml neomycin (NEO) to select for integrants. After parasite re-
emergence (after around 10 days) WR was put back in the culture medium. Genomic
DNA was prepared from NEO and WR resistant parasites. Integration was monitored by
PCR using the forward 5'upstream-1250-PfPH2-F (primer 1) and the reverse 3'90-GFP-R
primer (primer 2) for 5' integration and 5'pARL-F (primer 3) with the reverse 3’-3UTR-
PfPH2-R primers (primer 4) for the 3' integration. Primer 1 was used primer 4 to detect the
WT version of the gene. (see Appendix Table S1 for primer sequences).
To generate the parasite line for the knock sideways, the PfPH2-2xFKBP-GFP line was
transfected with 100 µg of purified p3xNLS-FRB-mCherry-BSD plasmid and selected 2
µg/ ml blasticidin (Sigma-Aldrich) to obtain the PfPH2-GFP+mislocalizer line.
Western Blotting
For the time-course of expression analysis, parasites were synchronized twice at an 18-
hour interval (between 18 and 22 hours ring stages), with a 0.3 M alanine-10mM HEPES
solution (as described in [69]). Synchronous parasites were then harvested by saponin
lysis at 8, 30, 36 and

113
42h post-reinvasion, the pellets solubilized in SDS protein sample buffer and separated
on a 7.5% SDS-PAGE gel under reducing conditions and transferred to PVDF
membranes (Millipore). The antibodies rabbit polyclonal anti-PfHSP70 (SPC-186C;
StressMarq Bioscience Inc., 1:20000) [70] and mouse monoclonal anti-GFP, (Roche,
1:1000, JL8), were diluted in 0.1% (v/v) Tween 20-phosphate-buffered saline with 1%
(w/v) skim milk. Appropriate HRP-coupled secondary antibodies were used and
immunoblots were revealed by ECL (Amersham Biosciences). For all expression analyses,
proteins extracted from an equal number of cells were used for each time point.
Fluorescence Imaging
Fluorescence images of parasites were captured using a GE Applied Precision Deltavision
Elite microscope with 100x 1.4NA objective and with a sCMOS camera and
deconvolved with the SoftWorx software. Chromatic calibration of the microscope was
performed prior to imaging experiments. For immunofluorescence assays, parasites were
fixed on slides using 4% paraformaldehyde (ProSciTech)[71]. After blocking in 3%
bovine serum albumin (Sigma Aldrich) the cells were incubated for 1 hour with rabbit
polyclonal anti-PfERD2 (1:2000)[72], mouse monoclonal anti-PfRON4 (1:2000)[73],
mouse monoclonal anti-PfRAP1 (1:2000)[74], mouse monoclonal anti-PfRESA
(1:2000)[75], rabbit anti-PfEBA175 (1:1000)[76], rabbit anti- PfEBA140 (1:1000)[61],
rabbit anti-PfAMA1 (1:2000)[29], rabbit anti-PfGAP45 (1:2000)[70] and rabbit anti-
PfMSP1 (1:1000)[77]. Bound antibodies were then visualized with Alexa Fluor-
594 anti-rabbit IgG and Alexa Fluor-488 anti-mouse IgG diluted 1:1000. Parasites were
mounted in Vectashield (Vecta Laboratories) containing 0.1 μg/ml 4', 6–diamidino-2-
phenylindole (Dapi, Invitrogen). Images shown represent a single optical slice from a
deconvolved z-stack.
To measure the distance between the Dapi and the different markers, a line was first drawn
across the center of the merozoite, always starting at the extreme end of the DAPI. In the
resulting line intensity profile (Plot Profile, Fiji), the relative distance in pixel was defined
as the maximum-intensity point of the other channel (green = PfPH2, red = EBA175 or

114
RON4), considering the 0 is always the extreme end of the Dapi. The distance was then put
in GraphPad Prism 7 and a One-way ANOVA was performed as a statistical test.
Growth assays
Tightly synchronous ring stage PfPH2-2xFKBP-GFP+mislocalizer parasites were
seeded at 0.2% parasitemia and grown +/- 250 nM rapamycin. After 28, 60 and 120 hours
in culture, the cells were harvested and analyzed by fluorescence-activated cell sorting
(FACS) on a BD FACSCanto A to calculate the parasitemia as previously described[78].
Briefly, the cells were stained with SYBRGold (Invitrogen-Molecular Probe) and then fixed
with 1% paraformaldehyde for 1 hour. 100 000 events were acquired on the FACSCanto
A using the FACSDiva software. The data were analyzed with the FlowJo software. The
percentage of survival was obtained by normalizing to untreated parasites in the same
experiment, which was taken as 100% survival. Uninfected red blood cells were used to
determine the threshold for FITC signal. Experiments were performed with a minimum of 3
biological replicates.
Merozoite number assay
Tightly synchronous ring stage PfPH2-2xFKBP-GFP+mislocalizer parasites were grown
+/- 250 nM rapamycin until they reached the schizont stage after which 10 µM of the
protease inhibitor E64 was added for 4-6 hours to prevent schizont rupture. When the
majority of parasites had developed into well-segmented merozoites, the number of
merozoite nuclei per schizont was counted. Only single parasite-infected red blood cells
with one food vacuole were counted in this experiment. Nuclei of a total of 20 parasites
were counted per condition and the experiment was repeated in 4 biological replicates.
Monitoring of schizont rupture
Tightly synchronized ring stage PfPH2-2xFKBP-GFP+mislocalizer parasites which were
grown +/- 250 nM rapamycin until the schizont stage, Percoll-purified and then
plated at a 2% parasitemia and incubated at 37°C with or without rapamycin. Parasites

115
were smeared every 4hours for 16 hours and the number of schizonts and newly formed
rings was counted. The experiment was performed in biological triplicates.
Attachment/Invasion assay
Tightly synchronized PfPH2-2xFKBP-GFP + mislocalizer parasites which were grown +/-
250 nM rapamycin until the schizont stage, Percoll-purified and then plated at a 2%
parasitemia in 48 well plates containing 100 µl of a pre-made mix of medium plus drugs
and 2% hematocrit. For the attachment assay, 1 µM cytochalasin D was used to prevent
invasion following merozoite attachment. Plates were shaken for 3 min and then
incubated for 12 hours. For the merozoite attachment assays, the number of merozoites
bound to red blood cells was counted while for the invasion assay, the number of rings
was counted. All counts were performed on Giemsa-stained parasite smears. All
experiments were performed in biological triplicates.
Microneme secretion assay
Tightly synchronous ring stage PfPH2-2xFKBP-GFP+mislocalizer parasites were
incubated +/- 250 nM rapamycin, until they reached the schizont stage at which point they
were let to rupture and reinvade overnight and the invasion supernatant collected. The
resulting supernatants were solubilized in SDS sample-buffer to extract the proteins and
analyzed by Western blot. The antibodies used were: rabbit anti-PfSERA5 (1:5000)[79],
rabbit anti-PfEBA175 (1:2500)[76], rabbit anti-PfEBA140 (1:1000)[61] and rabbit anti-
PfAMA1[29], were diluted in 0.1% (v/v) Tween 20-phosphate-buffered saline with 4%
(w/v) skim milk. Appropriate HRP-coupled secondary antibodies were used and
immunoblots were revealed by ECL (Amersham Biosciences). The signal for each
antigen was normalized to PfSERA5 to control for schizont number and egress
efficiency. To determine the amount of full length PfEBA175 remaining in merozoites
after induction of microneme exocytosis, free merozoites of PfPH2-2xFKBP-
GFP+mislocalizer parasites incubated +/- 250 nM rapamycin were prepared as described
previously[80]. Purified merozoites were incubated for 30 min at room temperature with
10uM of the calcium ionophore A23187 to induce microneme secretion[22] after which

116
they were pelleted and resuspended in SDS sample buffer and analyzed by Western blot
using rabbit anti- PfEBA175 (1:2500)[76]. A rabbit polyclonal anti-PfHSP70 (SPC-186C;
StressMarq Bioscience Inc., 1:20000) [70] was used as a loading control. To analyze the
amount of unprocessed PfEBA175, free merozoites were purified according to [55,80] and
incubated for 15 minutes at 37 οC with 10 μM of the calcium ionophore A23187 to
stimulate microneme exocytosis. The merozoites were then pelleted, resuspended in
SDS sampled buffer and analyzed by Western blot.
Invasion assays in enzyme-treated erythrocytes
Invasion assays in enzyme-treated RBCs were performed as described previously with
modifications[81,82]. Uninfected RBCs were washed with incomplete RPMI (iRPMI)
and 100 μl was treated with 10 volumes of 66 mU/ml neuraminidase (Sigma), 1mg/ml
chymotrypsin (Sigma), 1mg/ml trypsin (Sigma) or iRPMI (vehicle) for 1 hour at 37°C.
All samples were washed with iRPMI and then trypsin and chymotrypsin samples were
treated with 0.5mg/ml soybean trypsin inhibitor (Sigma) at 37°C for 10 minutes. All
samples were then extensively washed (3-4x) in iRPMI. Ring infected RBCs were
synchronized with sorbitol once at 0-4hpi and then 16 hours later. 34-38hpi infected
RBCs (iRBC) from the same cycle were then purified on a 65%-35% Percoll gradient, the
purified iRBC pellet was washed 3-5 times in iRPMI and mixed with enzyme-treated
RBCs at 2% hematocrit and 0.2-0.5% parasitemia and plated in 96 well plate in media
containing +/- rapamycin (250nM). Following incubation in standard parasite culture
conditions for 30-36 hours, parasitemia was measured by flow cytometry in the FITC
channel after cells were stained with SYBRGold and fixed with 1% paraformaldehyde
for 1 hour. The experiments were performed in at least 3 biological replicates.
Statistical analysis
Prism 7(GraphPad) was used for all statistical analyses. Depending on the assay, one-way
ANOVA or two-tailed unpaired t-tests were performed. A minimum of 3 biological
replicates was done for each experiment. A P value of < 0.05 was considered statistically
significant.

117
Homology Searching
PfPH2 (PF3D7_1337700) was used a query for searches using BLAST[83] into the protein
databases of Acanthamoeba castellani, Arabidopsis thaliana, Babesia microti,
Bigelowiella natans, Cyclospora cayetanensis, Cryptosporidum parvum, Cystiospora
suis, Chromera velia, Dictyostelium discoideum, Eimeria necatrix, Guillardia theta,
Hammondia hammondi, Homo sapiens, Neospora caninum, Plasmodium falciparum,
Plasmodium vivax, Perkinsus marinus, Phytophthora sojae, Saccharomyces cerevisiae,
Symbiodinium minutum, Theileria annulata, Trypanosoma brucei, Toxoplasma gondii,
Tetrahymena thermophila, Trichomonas vaginalis and Vitrella brassicaformis using
inhouse scripts. Potential orthologues were then used as queries for a homology search
using BLAST into the P. falciparum protein database. Candidates were considered
positive orthologues when both forward and reverse BLAST hits generated an E- value
below a 0.05 threshold, if the candidate protein in question retrieved the relevant query
with e-values two orders of magnitude better compared to the next non-redundant hit, and
if the top reverse BLAST hit was PF3D7_1337700. HMMer (v3.2.1)[84] searches were
also conducted using a HMMer profile made of PF3D7_1337700 known orthologues into
the protein databases of the organisms listed above. The HMMer profile was generated
using MUSCLE alignment software (v3.8.31) [85]and Fetch software (v5.7.7 downloaded
from https://fetchsoftworks.com) was used to search into the protein databases. Potential
orthologues were validated using the above procedure. BLAST and HMMer searches
were performed for both the full length PF3D7_1337700 query as well as
PF3D7_1337700 without the PH domain (amino acids 71-172). In the case where
searches were performed without the PH domain, CDD on NCBI
(https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) identified the location of the PH
domain and Emboss:extractseq (http://www.bioinformatics.nl/cgi-bin/emboss/extractseq)
was used to trim the PH domain from the query. Conserved domain structures of the
orthologues were identified using Phyre v2.0
(http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index). tBLASTn searches were also
performed using PF3D7_1337700 without the PH domain (amino acids 71-172) as the

118
query into the nuclear scaffolds of the non-apicomplexan taxa, with homology assessed
using the above criteria.
References
1. WHO (2017) World Malaria
Report.
2. Beeson JG, Drew DR, Boyle MJ, Feng G, Fowkes FJ, Richards JS (2016) Merozoite
surface proteins in red blood cell invasion, immunity and vaccines against malaria. FEMS
Microbiol Rev 40: 343-372
3. Sanders PR, Gilson PR, Cantin GT, Greenbaum DC, Nebl T, Carucci DJ,
McConville MJ, Schofield L, Hodder AN, Yates JR, et al. (2005) Distinct protein
classes including novel merozoite surface antigens in Raft-like membranes of
Plasmodium falciparum. The Journal of biological chemistry 280: 40169-40176
4. Das S, Hertrich N, Perrin AJ, Withers-Martinez C, Collins CR, Jones ML,
Watermeyer JM, Fobes ET, Martin SR, Saibil HR, et al. (2015) Processing of
Plasmodium falciparum Merozoite Surface Protein MSP1 Activates a Spectrin-Binding
Function Enabling Parasite Egress from RBCs. Cell Host Microbe 18: 433-444
5. Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M,
Mboup S, Ndir O, Kwiatkowski DP, Duraisingh MT, et al. (2011) Basigin is a receptor
essential for erythrocyte invasion by Plasmodium falciparum. Nature 480: 534-537
6. Crick AJ, Theron M, Tiffert T, Lew VL, Cicuta P, Rayner JC (2014) Quantitation of
Malaria Parasite-Erythrocyte Cell-Cell Interactions Using Optical Tweezers. Biophysj 107: 846-853
7. Tham W-H, Healer J, Cowman AF (2012) Erythrocyte and reticulocyte binding-like
proteins of Plasmodium falciparum. Trends in parasitology 28: 23-30
8. Volz JC, Yap A, Sisquella X, Thompson JK, Lim NT, Whitehead LW, Chen L,
Lampe M, Tham WH, Wilson D, et al. (2016) Essential Role of the
PfRh5/PfRipr/CyRPA Complex during Plasmodium falciparum Invasion of Erythrocytes.
Cell Host Microbe 20: 60-71
9. Collins CR, Withers-Martinez C, Hackett F, Blackman MJ (2009) An inhibitory
antibody blocks interactions between components of the malarial invasion machinery.
PLoS Pathog 5: e1000273
10. Lamarque M, Besteiro S, Papoin J, Roques M, Vulliez-Le Normand B, Morlon-
Guyot J, Dubremetz J-F, Fauquenoy S, Tomavo S, Faber BW, et al. (2011) The RON2-
AMA1 interaction is a critical step in moving junction-dependent invasion by
apicomplexan parasites. PLoS pathogens 7: e1001276
11. Srinivasan P, Beatty WL, Diouf A, Herrera R, Ambroggio X, Moch JK, Tyler JS,
Narum DL, Pierce SK, Boothroyd JC, et al. (2011) Binding of Plasmodium merozoite

119
proteins RON2 and AMA1 triggers commitment to invasion. Proceedings of the National
Academy of Sciences of the United States of America, 10.1073/pnas.1110303108
12. Baum J, Richard D, Healer J, Rug M, Krnajski Z, Gilberger T-W, Green JL,
Holder AA, Cowman AF (2006) A conserved molecular motor drives cell invasion
and gliding motility across malaria life cycle stages and other apicomplexan parasites.
The Journal of biological chemistry 281: 5197-5208
13. Pinder JC, Fowler RE, Dluzewski AR, Bannister LH, Lavin FM, Mitchell GH,
Wilson RJ, Gratzer WB (1998) Actomyosin motor in the merozoite of the malaria
parasite, Plasmodium falciparum: implications for red cell invasion. Journal of cell
science 111 ( Pt 13): 1831-1839
14. Field SJ, Pinder JC, Clough B, Dluzewski AR, Wilson RJ, Gratzer WB (1993) Actin
in the merozoite of the malaria parasite, Plasmodium falciparum. Cell Motil Cytoskeleton
25: 43-48
15. Zuccala ES, Gout AM, Dekiwadia C, Marapana DS, Angrisano F, Turnbull L, Riglar
DT, Rogers KL, Whitchurch CB, Ralph SA, et al. (2012) Subcompartmentalisation of
proteins in the rhoptries correlates with ordered events of erythrocyte invasion by the
blood stage malaria parasite. PLoS One 7: e46160
16. Lourido S, Moreno SN (2015) The calcium signaling toolkit of the Apicomplexan
parasites Toxoplasma gondii and Plasmodium spp. Cell Calcium 57: 186-193
17. Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K,
Schwach F, Chappell L, Gomes AR, Berriman M, et al. (2014) Phosphoinositide
Metabolism Links cGMP- Dependent Protein Kinase G to Essential Ca2+ Signals at Key
Decision Points in the Life Cycle of Malaria Parasites. PLoS biology 12: e1001806
18. Absalon S, Blomqvist K, Rudlaff RM, DeLano TJ, Pollastri MP, Dvorin JD (2018)
Calcium- Dependent Protein Kinase 5 Is Required for Release of Egress-
Specific Organelles in Plasmodium falciparum. MBio 9:
19. Dvorin JD, Martyn DC, Patel SD, Grimley JS, Collins CR, Hopp CS, Bright AT,
Westenberger S, Winzeler E, Blackman MJ, et al. (2010) A plant-like kinase in
Plasmodium falciparum regulates parasite egress from erythrocytes. Science (New York,
NY) 328: 910-912
20. Collins CR, Hackett F, Strath M, Penzo M, Withers-Martinez C, Baker DA,
Blackman MJ (2013) Malaria Parasite cGMP-dependent Protein Kinase Regulates Blood
Stage Merozoite Secretory Organelle Discharge and Egress. PLoS pathogens 9: e1003344
21. Taylor HM, McRobert L, Grainger M, Sicard A, Dluzewski AR, Hopp CS,
Holder AA, Baker DA (2010) The malaria parasite cyclic GMP-dependent protein kinase
plays a central role in blood-stage schizogony. Eukaryot Cell 9: 37-45
22. Singh S, Alam MM, Pal-Bhowmick I, Brzostowski JA, Chitnis CE (2010) Distinct
external signals trigger sequential release of apical organelles during erythrocyte invasion
by malaria parasites. PLoS pathogens 6: e1000746
23. Dawn A, Singh S, More KR, Siddiqui FA, Pachikara N, Ramdani G, Langsley G,
Chitnis CE (2014) The Central Role of cAMP in Regulating Plasmodium falciparum
Merozoite Invasion of Human Erythrocytes. PLoS pathogens 10: e1004520

120
24. Gao X, Gunalan K, Yap SSL, Preiser PR (2013) Triggers of key calcium signals
during erythrocyte invasion by Plasmodium falciparum. Nature communications 4: 2862
25. Bullen HE, Jia Y, Yamaryo-Botte Y, Bisio H, Zhang O, Jemelin NK, Marq JB,
Carruthers V, Botte CY, Soldati-Favre D (2016) Phosphatidic Acid-Mediated Signaling
Regulates Microneme Secretion in Toxoplasma. Cell Host Microbe 19: 349-360
26. Darvill N, Dubois DJ, Rouse SL, Hammoudi PM, Blake T, Benjamin S, Liu B,
Soldati-Favre D, Matthews S (2018) Structural Basis of Phosphatidic Acid Sensing by
APH in Apicomplexan Parasites. Structure, 10.1016/j.str.2018.05.001
27. Paul AS, Saha S, Engelberg K, Jiang RH, Coleman BI, Kosber AL, Chen CT, Ganter M,
Espy N, Gilberger TW, et al. (2015) Parasite Calcineurin Regulates Host Cell
Recognition and Attachment by Apicomplexans. Cell Host Microbe 18: 49-60
28. Farrell A, Thirugnanam S, Lorestani A, Dvorin JD, Eidell KP, Ferguson DJP,
Anderson- White BR, Duraisingh MT, Marth GT, Gubbels M-J (2012) A DOC2 Protein
Identified by Mutational Profiling Is Essential for Apicomplexan Parasite Exocytosis.
Science (New York, NY)335: 218-221
29. Healer J, Crawford S, Ralph S, McFadden G, Cowman AF (2002) Independent
translocation of two micronemal proteins in developing Plasmodium falciparum
merozoites. Infection and Immunity 70: 5751-5758
30. Kremer K, Kamin D, Rittweger E, Wilkes J, Flammer H, Mahler S, Heng J,
Tonkin CJ, Langsley G, Hell SW, et al. (2013) An Overexpression Screen of
Toxoplasma gondii Rab- GTPases Reveals Distinct Transport Routes to the Micronemes.
PLoS pathogens 9: e1003213
31. Gaji RY, Flammer HP, Carruthers VB (2011) Forward targeting of Toxoplasma
gondii proproteins to the micronemes involves conserved aliphatic amino acids. Traffic
(Copenhagen, Denmark) 12: 840-853
32. Kumar S, Kumar M, Ekka R, Dvorin JD, Paul AS, Madugundu AK, Gilberger T,
Gowda H, Duraisingh MT, Keshava Prasad TS, et al. (2017) PfCDPK1 mediated
signaling in erythrocytic stages of Plasmodium falciparum. Nat Commun 8: 63
33. Wengelnik K, Daher W, Lebrun M (2018) Phosphoinositides and their functions in
apicomplexan parasites. Int J Parasitol 48: 493-504
34. McNamara CW, Lee MCS, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung
BKS, Chatterjee AK, McCormack SL, et al. (2013) Targeting Plasmodium PI(4)K
to eliminate malaria. Nature, 10.1038/nature12782
35. Tawk L, Chicanne G, Dubremetz J-F, Richard V, Payrastre B, Vial HJ, Roy C,
Wengelnik K (2010) Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium,
localizes to the food vacuole membrane and the apicoplast. Eukaryotic cell 9: 1519-1530
36. Walczak M, Ganesan SM, Niles JC, Yeh E (2018) ATG8 Is Essential Specifically
for an Autophagy-Independent Function in Apicoplast Biogenesis in Blood-Stage
Malaria Parasites. MBio 9:

121
37. Bansal P, Tripathi A, Thakur V, Mohmmed A, Sharma P (2017) Autophagy-Related
Protein ATG18 Regulates Apicoplast Biogenesis in Apicomplexan Parasites. MBio 8:
38. Vaid A, Ranjan R, Smythe WA, Hoppe HC, Sharma P (2010) PfPI3K, a
phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host
erythrocyte and is involved in hemoglobin trafficking. Blood 115: 2500-2507
39. Raabe AC, Wengelnik K, Billker O, Vial HJ (2011) Multiple roles for Plasmodium
berghei phosphoinositide-specific phospholipase C in regulating gametocyte activation
and differentiation. Cellular Microbiology 13: 955-966
40. Martin SK, Jett M, Schneider I (1994) Correlation of phosphoinositide hydrolysis
with exflagellation in the malaria microgametocyte. The Journal of parasitology 80: 371-
378
41. Ogwan'g R, Mwangi J, Gachihi G, Nwachukwu A, Roberts CR, Martin SK (1993)
Use of pharmacological agents to implicate a role for phosphoinositide hydrolysis
products in malaria gamete formation. Biochem Pharmacol 46: 1601-1606
42. Mbengue A, Bhattacharjee S, Pandharkar T, Liu H, Estiu G, Stahelin RV, Rizk SS,
Njimoh DL, Ryan Y, Chotivanich K, et al. (2015) A molecular mechanism of
artemisinin resistance in Plasmodium falciparum malaria. Nature 520: 683-687
43. Bhattacharjee S, Coppens I, Mbengue A, Suresh N, Ghorbal M, Slouka Z, Safeukui
I, Tang HY, Speicher DW, Stahelin RV, et al. (2018) Remodeling of the malaria
parasite and host human red cell by vesicle amplification that induces artemisinin
resistance. Blood 131: 1234-1247
44. Ebrahimzadeh Z, Mukherjee A, Richard D (2018) A map of the subcellular
distribution of phosphoinositides in the erythrocytic cycle of the malaria parasite
Plasmodium falciparum. Int J Parasitol 48: 13-25
45. Boddey JA, O'Neill MT, Lopaticki S, Carvalho TG, Hodder AN, Nebl T, Wawra S,
van West P, Ebrahimzadeh Z, Richard D, et al. (2016) Export of malaria proteins
requires co-translational processing of the PEXEL motif independent of
phosphatidylinositol-3-phosphate binding. Nat Commun 7: 10470
46. Lemmon MA (2007) Pleckstrin homology (PH) domains and phosphoinositides.
BiochemSoc Symp, 10.1042/BSS074008181-93
47. Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V,
McLaughlin S, Rebecchi MJ (1995) The pleckstrin homology domain of phospholipase
C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer
membranes. Biochemistry 34: 16228-16234
48. Ceccato L, Chicanne G, Nahoum V, Pons V, Payrastre B, Gaits-Iacovoni F, Viaud J
(2016) PLIF: A rapid, accurate method to detect and quantitatively assess protein-lipid
interactions. Sci Signal 9: rs2
49. Yu JW, Mendrola JM, Audhya A, Singh S, Keleti D, DeWald DB, Murray D, Emr
SD, Lemmon MA (2004) Genome-wide analysis of membrane targeting by S. cerevisiae
pleckstrin homology domains. Mol Cell 13: 677-688

122
50. Daher W, Morlon-Guyot J, Alayi TD, Tomavo S, Wengelnik K, Lebrun M (2016)
Identification of Toxoplasma TgPH1, a pleckstrin homology domain-containing
protein that binds to the phosphoinositide PI(3,5)P2. Mol Biochem Parasitol 207: 39-44
51. Birnbaum J, Flemming S, Reichard N, Soares AB, Mesen-Ramirez P, Jonscher E,
Bergmann B, Spielmann T (2017) A genetic system to study Plasmodium falciparum
protein function. Nat Methods 14: 450-456
52. Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner
IF, Casandra D, Mayho M, et al. (2018) Uncovering the essential genes of the
human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360:
53. Haruki H, Nishikawa J, Laemmli UK (2008) The anchor-away technique: rapid,
conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925-932
54. Miller LH, Aikawa M, Johnson JG, Shiroishi T (1979) Interaction between
cytochalasin B- treated malarial parasites and erythrocytes. Attachment and junction
formation. J Exp Med 149:172-184
55. Riglar DT, Richard D, Wilson DW, Boyle MJ, Dekiwadia C, Turnbull L, Angrisano
F, Marapana DS, Rogers KL, Whitchurch CB, et al. (2011) Super-resolution dissection
of coordinated events during malaria parasite invasion of the human erythrocyte. Cell
Host Microbe9: 9-20
56. Weiss GE, Gilson PR, Taechalertpaisarn T, Tham WH, de Jong NW, Harvey KL,
Fowkes FJ, Barlow PN, Rayner JC, Wright GJ, et al. (2015) Revealing the sequence and
resulting cellular morphology of receptor-ligand interactions during Plasmodium
falciparum invasion of erythrocytes. PLoS Pathog 11: e1004670
57. Yeoh S, O'Donnell RA, Koussis K, Dluzewski AR, Ansell KH, Osborne SA,
Hackett F, Withers-Martinez C, Mitchell GH, Bannister LH, et al. (2007) Subcellular
discharge of a serine protease mediates release of invasive malaria parasites from host
erythrocytes. Cell 131:1072-1083
58. Carruthers VB, Blackman MJ (2005) A new release on life: emerging concepts in
proteolysis and parasite invasion. Mol Microbiol 55: 1617-1630
59. Cowman AF, Tonkin CJ, Tham WH, Duraisingh MT (2017) The Molecular
Basis of
Erythrocyte Invasion by Malaria Parasites. Cell Host Microbe 22:
232-245
60. Duraisingh MT, Maier AG, Triglia T, Cowman AF (2003) Erythrocyte-binding
antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-
dependent and -independent pathways. Proc Natl Acad Sci U S A 100: 4796-4801
61. Lopaticki S, Maier AG, Thompson J, Wilson DW, Tham W-H, Triglia T, Gout A,
Speed TP, Beeson JG, Healer J, et al. (2011) Reticulocyte and erythrocyte binding-like
proteins function cooperatively in invasion of human erythrocytes by malaria parasites.
Infection and Immunity79: 1107-1117

123
62. Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij
JPJ, Carruthers VB, Niles JC, et al. (2016) A Genome-wide CRISPR Screen in
Toxoplasma Identifies Essential Apicomplexan Genes. Cell 166: 1423-1435 e1412
63. Massiah MA, Wright KM, Du H (2016) Obtaining Soluble Folded Proteins from
Inclusion Bodies Using Sarkosyl, Triton X-100, and CHAPS: Application to LB and M9
Minimal Media. Curr Protoc Protein Sci 84: 6 13 11-16 13 24
64. Shirey CM, Scott JL, Stahelin RV (2017) Notes and tips for improving quality of
lipid- protein overlay assays. Anal Biochem 516: 9-12
65. Julkowska MM, Rankenberg JM, Testerink C (2013) Liposome-binding assays to
assess specificity and affinity of phospholipid-protein interactions. Methods Mol Biol
1009: 261-271
66. Adu-Gyamfi E, Johnson KA, Fraser ME, Scott JL, Soni SP, Jones KR, Digman MA,
Gratton E, Tessier CR, Stahelin RV (2015) Host Cell Plasma Membrane
Phosphatidylserine Regulates the Assembly and Budding of Ebola Virus. J Virol 89:
9440-9453
67. Del Vecchio K, Stahelin RV (2016) Using Surface Plasmon Resonance to
Quantitatively
Assess Lipid-Protein Interactions. Methods Mol Biol 1376:
141-153
68. Trager W, Jensen JB (1976) Human malaria parasites in continuous culture.
Science 193:673-675
69. Ansorge I, Benting J, Bhakdi S, Lingelbach K (1996) Protein sorting in Plasmodium
falciparum-infected red blood cells permeabilized with the pore-forming protein
streptolysin O. Biochem J 315 ( Pt 1): 307-314
70. Jones ML, Kitson EL, Rayner JC (2006) Plasmodium falciparum erythrocyte
invasion: a conserved myosin associated complex. Mol Biochem Parasitol 147: 74-84
71. Hayashi M, Taniguchi S, Ishizuka Y, Kim HS, Wataya Y, Yamamoto A, Moriyama Y
(2001) A homologue of N-ethylmaleimide-sensitive factor in the malaria parasite
Plasmodium falciparum is exported and localized in vesicular structures in the cytoplasm
of infected erythrocytes in the brefeldin A-sensitive pathway. J Biol Chem 276: 15249-
15255
72. Elmendorf HG, Haldar K (1993) Identification and localization of ERD2 in the
malaria parasite Plasmodium falciparum: separation from sites of sphingomyelin
synthesis and implications for organization of the Golgi. The EMBO journal 12: 4763-
4773
73. Richard D, MacRaild CA, Riglar DT, Chan J-A, Foley M, Baum J, Ralph SA,
Norton RS, Cowman AF (2010) Interaction between Plasmodium falciparum apical
membrane antigen 1 and the rhoptry neck protein complex defines a key step in the
erythrocyte invasion process of malaria parasites. The Journal of biological chemistry
285: 14815-14822
74. Schofield L, Bushell GR, Cooper JA, Saul AJ, Upcroft JA, Kidson C (1986) A
rhoptry antigen of Plasmodium falciparum contains conserved and variable epitopes

124
recognized by inhibitory monoclonal antibodies. Molecular and biochemical parasitology
18: 183-195
75. Kemp DJ, Coppel RL, Anders RF (1987) Repetitive proteins and genes of malaria. Annu RevMicrobiol 41: 181-208
76. O'Donnell RA, Hackett F, Howell SA, Treeck M, Struck N, Krnajski Z, Withers-
Martinez C, Gilberger TW, Blackman MJ (2006) Intramembrane proteolysis mediates
shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J Cell Biol
174: 1023-1033
77. Wilson DW, Fowkes FJI, Gilson PR, Elliott SR, Tavul L, Michon P, Dabod E,
Siba PM, Mueller I, Crabb BS, et al. (2011) Quantifying the Importance of MSP1-19 as
a Target of Growth-Inhibitory and Protective Antibodies against Plasmodium
falciparum in Humans. PloS one 6: e27705
78. Hallee S, Counihan NA, Matthews K, de Koning-Ward TF, Richard D (2018) The
malaria parasite Plasmodium falciparum Sortilin is essential for merozoite formation and
apical complex biogenesis. Cell Microbiol, 10.1111/cmi.12844e12844
79. Stallmach R, Kavishwar M, Withers-Martinez C, Hackett F, Collins CR, Howell SA,
Yeoh S, Knuepfer E, Atid AJ, Holder AA, et al. (2015) Plasmodium falciparum SERA5
plays a non- enzymatic role in the malarial asexual blood-stage lifecycle. Mol Microbiol
96: 368-387
80. Boyle MJ, Wilson DW, Richards JS, Riglar DT, Tetteh KKA, Conway DJ, Ralph SA,
Baum J, Beeson JG (2010) Isolation of viable Plasmodium falciparum merozoites to
define erythrocyte invasion events and advance vaccine and drug development.
Proceedings of the National Academy of Sciences of the United States of America 107:
14378-14383
81. Dvorin JD, Bei AK, Coleman BI, Duraisingh MT (2010) Functional diversification
between two related Plasmodium falciparum merozoite invasion ligands is determined by
changes in the cytoplasmic domain. Mol Microbiol 75: 990-1006
82. Reed MB, Caruana SR, Batchelor AH, Thompson JK, Crabb BS, Cowman AF (2000)
Targeted disruption of an erythrocyte binding antigen in Plasmodium falciparum is
associated with a switch toward a sialic acid-independent pathway of invasion. Proc
Natl Acad Sci U S A97: 7509-7514
83. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ
(1997) Gapped BLAST and PSI-BLAST: a new generation of protein database
search programs. Nucleic Acids Res 25: 3389-3402
84. Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:
755-763
85. Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced
time and space complexity. BMC Bioinformatics 5: 113
86. Slapeta J, Morin-Adeline V (2011) Apicomplexa Levine 1970. Sporozoa Leucart
1879. Version 18 May 2011. http://tolweb.org/Apicomplexa/2446/2011.05.18 in The Tree
of Life Web Project, http://tolweb.org/.

125
Competing interest
The authors declare no competing interests.
Materials and Correspondence
Requests for materials should be addressed to Dave Richard.
Acknowledgments
We would like to thank Tobias Spielman for the pSLI plasmids and Robin Anders,
James Beeson, Michael Blackman, Alan Cowman and Julian Rayner for antibodies. We
also thank Jacobus Pharmaceuticals for WR99210. The following reagent was obtained
through MR4 as part of the BEI Resources, National Institute of Allergy and Infectious
Diseases, National Institutes of Health, USA: Polyclonal Anti-Plasmodium falciparum
PfERD2 (antiserum, Rabbit). We would also like to acknowledge the Canadian Blood
Services for providing human erythrocytes. The authors declare no competing financial
interests. This study was funded through a Canadian Institutes for Health Research
(CIHR) operating grant MOP 130359 to DR. DR is a Fonds de la Recherche du uébec-
Santé unior 2 fellow. Work in the Dacks Lab is supported by a Discovery Grant from the
Natural Sciences and Engineering Research Council of Canada (RES0021028). JBD is
the Canada Research Chair (Tier II) in Evolutionary Cell Biology.
Author contributions
Z.E. performed most of the knock sideways parasite manipulation and phenotypic analysis
and contributed to the writing of the manuscript. A.M. performed the invasion assays with
enzyme- treated erythrocytes and contributed to the writing of the manuscript. M.E.C and
A.S. analyzed the secretion of PfAMA1 by IFA and performed the secretion assay with
free merozoites. SA performed the protein-lipid interaction analyses and contributed to the
writing of the manuscript. L.A.T. and J.B.D. performed the bioinformatics analysis and
contributed to the writing the manuscript, DGau and DGag performed parasite

126
manipulations. R.V.S. interpreted results and contributed to the writing of the
manuscript. D.R. conceived the study, designed experiments, interpreted results and wrote
the manuscript.
Data availability
The data supporting the findings of this study are available within the paper and its
Appendix and are also available from the corresponding author upon request.

127
Figure legends
Figure 1: PF3D7_1337700 is a phosphoinositide-binding protein with a relaxed
specificity. A) Schematic of PF3D7_1337700 showing the PH domain with the
conserved PIP-binding motif.
B) Lipid blots showing that the WT PH domain and triple mutant PH domain bind
several species of PIPs. A negative (GST) and positive control (PLC-PH) are also shown
for respective lipid binding.
C) Liposome-binding assays of GST-tagged WT and triple mutant PH domains. GST-
tagged PfPH2-PH proteins (500 ng) were incubated with 50 µM liposomes composed of
POPC:POPE, POPC:POPE:POPS, or liposomes containing 5% molar ratio of one of seven
PIP species or DPPA (POPC:POPE:POPS:DPPA or POPC:POPE:POPS:PIPs). kDa
indicates molecular weight, P: pellet fraction, SN: supernatant fraction.
D) SPR analysis of the WT PfPH2-PH domain and PH domain triple mutant
demonstrates binding to lipid vesicles (POPC:POPE) containing 5 mol% PI(3)P. The
response values shown (determined by subtracting the binding signal from control lipid
vesicles) were plotted versus PH domain concentration to determine the apparent affinity
(Kd) of vesicle binding.
Figure 2: PfPH2 localizes close to the apical tip.
A) IFA showing the distance between the farthest edge of the DAPI and PfPH2 and (Ai)
PfEBA175 and (Aii) PfRON4. Two images are shown for EBA175 to demonstrate how
much variety can be observed between different cells. Green: PfPH2. Red: EBA175
(Ai) or RON4 (Aii). Blue: DAPI stained nucleus. Scale bar: 0.5 µm.
B) Quantification of the distances reveals that the distance between PfPH2 and the
DAPI is significantly bigger than PfEBA175 and the latter but not with PfRON4. ****: p-
value<0.0001. ns: non significant. One-way ANOVA. Horizontal lines: Median. Box
limits: 25tth
to 75th
percentile. Whiskers: Min to max values. Error bars: stander error of the
mean.

128
Figure 3: PfPH2 is important for attachment to and subsequent invasion of the red
blood cell.
A) Live cell microscopy showing that in the presence of 250 nM Rapa, PfPH2-
2xFKBP-GFP (GFP) is translocated from the apical pole to the nucleus in the presence of
the nuclear mislocalizer. Scale bar represents 5m. Blue: DAPI stained nucleus. BF: bright
field.
B (Bi) Growth curve analysis showing that the KS of PfPH2 severely decreases the
asexual
replication of the parasite. The PfPH2-GFP parasite line (black square) not transfected with
the nuclear mislocalizer is used as a control to show that the reduced growth is dependent
on the presence of both Rapa and the mislocalizer. Full line: -Rapa. Dashed line: +Rapa.
Mean SEM of 3 biological replicates are shown. (Bii) Data from (Bi) represented as the
percentage of growth of parasites incubated with Rapa compared to their control without
Rapa. Mean SEM of 3 biological replicates are shown.
C) Time course of schizont rupture (red) and subsequent new ring formation (blue)
showing that the PfPH2 KS line has no egress defect but an important decrease in the
formation of new rings. Results from one experiment representative of 3 biological
replicates are shown. Full line: -Rapa. Dashed line: +Rapa.
D) Quantification of the number of merozoites formed per schizont. n= 4 biological
replicates with 20 schizonts counted per condition. n.s: non significant. Unpaired t-test. p-
value= 0.1524. Error bars: Standard error of the mean.
E) Merozoite attachment and invasion is decreased in the PfPH2 KS line. Attachment was
measured by incubating parasites with 1 µM cytochalasin D. CytD: cytochalasin D. n=3
biological replicates for both attachment and invasion assays. One-way ANOVA
followed by Fisher’s LSD test. Error bars: Standard error of the mean.
Figure 4: PfPH2 is required for the secretion of PfEBA-micronemes.
A) IFA showing that the PfPH2 KS line has no defect in the exocytosis of PfAMA1
containing micronemes as measured by the translocation of PfAMA1 from the apical tip to

129
the merozoite surface. n= 3 biological replicates with 150 cells counted for each condition.
n.s.: non significant in a 2-tailed t-test. p-value=0.9061. Error bars: Standard error of the
mean. Scale bar: 5 µm
B) Western blot showing the proteolytic shedding of micronemal proteins in the
supernatant following merozoite invasion. The parasitophorous vacuole protein PfSERA5 is
used as a control for schizont rupture. Sn: supernatant.
C) Densitometric analysis of reveals that the PfPH2 KS line has a severe defect in the
shedding of PfEBA175 and PfEBA140 but not PfAMA1. Data normalized to
PfSERA5 and then expressed as a percentage of the control incubated without Rapa. Data
from 3 independent biological replicates. s: significant. An unpaired t-test was performed
and showed a significant difference of invasion between the PfAMA1 and both PfEBA175
(p-value=0.0047) and PfEBA140 (p-value=0.0019). Error bars: Standard error of the mean.
D) Western blot showing that the amount of unprocessed PfEBA175 associated with free
merozoites after stimulation of microneme exocytosis. Biorep: Bioreplicate
E) Densitometric analysis reveals that free merozoites of the PfPH2 KS line have
increased levels of unprocessed PfEBA175. Data normalized to PfHSP70. Data from
3 biological replicates is shown. p-value=0.0201. Unpaired t-test. Error bars: Standard
error of the mean.
F) Comparison of the effect of the PfPH2 KS and enzymatic treatments on invasion.
Values represent % of invasion relative to -Rapa PfPH2 line in normal untreated
erythrocytes. Rapa=rapamycin. s: significant. ns: non significant. Data from 3 biological
replicates is shown. An unpaired t-test was performed and showed a significant difference
of invasion between the PfPH2 KS and trypsin (p-value=0.0041) and neuraminidase (p-
value=0.0069). Error bars: Standard error of the mean. Untr: Untreated. Ezn: Enzyme. T:
trypsin. C: Chymotrypsin. NM: Neuraminidase. Rapa: Rapamycin. Biorep: Bioreplicate
Figure 5: PfPH2 is conserved throughout apicomplexan parasites.
A) Schematic of PfPH2 showing the PH domain with the conserved PIP-binding motif.

130
B) PfPH2 proteins are conserved but restricted to apicomplexans. This cartoon
illustrates the presence of PfPH2 orthologues mapped onto the tree of major apicomplexan
representatives with genome sequences available. Relationships between the taxa are based
on [86]. The phylogenetic distribution of PfPH2 orthologues demonstrates apicomplexan
specificity. Positive orthologues were identified in all apicomplexans sampled (red) and
could not be identified denoted by an asterisk in Ciliates (green), Dinoflagellates (purple)
or the Chromerids outgroup to the Apicomplexa (yellow), as demonstrated by the E-values
for the candidate orthologue into the P. falciparum proteome (Column Reverse
BLAST E-values). Relationships based on[86]. Conserved domain structure in all
orthologues is shown based on Phyre v2.0. Black arrow represents the gain of PfPH2 at
the base of Apicomplexa coincident with the shift to parasitism. Orange box; PH domain.
Blue box; SMC_N superfamily domain.
Expanded View Figure legends
Figure EV1: Generation of the PfPH2-2xFKBP-GFP parasite line.
A) Schematic showing the tagging strategy by single cross-over recombination using SLI.
B) PCR on parasite genomic DNA showing the proper integration of the tagging vector at
(5’ junction: primers P1 and P3, 3’junction: primers P2 and P4) and the disappearance of
the WT allele (primers P1 and P4) in all 3 SLI attempts.
C) Time course of expression of parasite protein extracts taken throughout the erythrocytic
cycle shows that PfPH2 is expressed in schizonts. PfHSP70 is used as a control for a
constitutive protein. Cartoon shows the time points in hours post invasion (hpi). 1: 8 hpi, 2:
30 hpi, 3: 36 hpi, 4: 42 hpi.
Figure EV2: Localization of PfPH2 in schizonts.
A–D IFA showing that PfPH2 does not colocalize with markers of the micronemes:
PfAMA1, PfEBA175 and PfEBA140) (A), of the rhoptries: PfRAP1, PfRON4, PfRH1,

131
PfRH4 and PfRH5 (B), of the dense granules: PfRESA (C) or of the Golgi apparatus:
PfERD2 (D). Scale bar represents 5m. Blue: DAPI stained nucleus. BF: bright field.
Figure EV3: Localization of PfPH2 in free merozoites.
A, B IFA showing that PfPH2 does not colocalize with markers of the micronemes:
PfAMA1 (secreted on the parasite surface in free merozoites), PfEBA175 and
PfEBA140 (A) or of the rhoptry markers PfRON4, PfRAP1, PfRH1, and PfRH5 (B).
Scale bar represents 5m. Blue: DAPI stained nucleus.
Figure EV4. Alignments of the KXn(K/R)XR binding motif in Apicomplexan
orthologues of PfPH2.
Alignments of a portion of the PH domains from PF3D7_1337700 identified orthologues
in Apicomplexa are shown. The three arrows outline the alignments of the sequence
motif from PF3D7_1337700 with similar amino acid positions of the other proteins.
Notably the first and terminal positions of the KXn(K/R)XR motif show conservation.
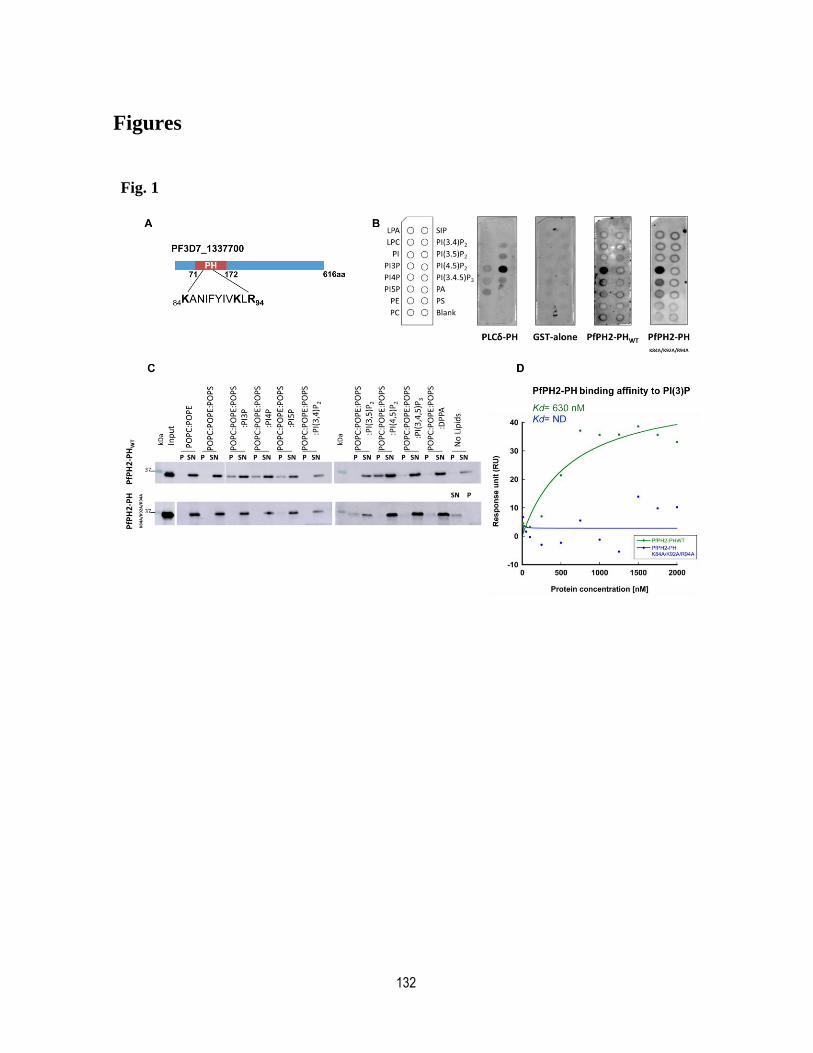
132
Figures
Fig. 1

133
Fig. 2
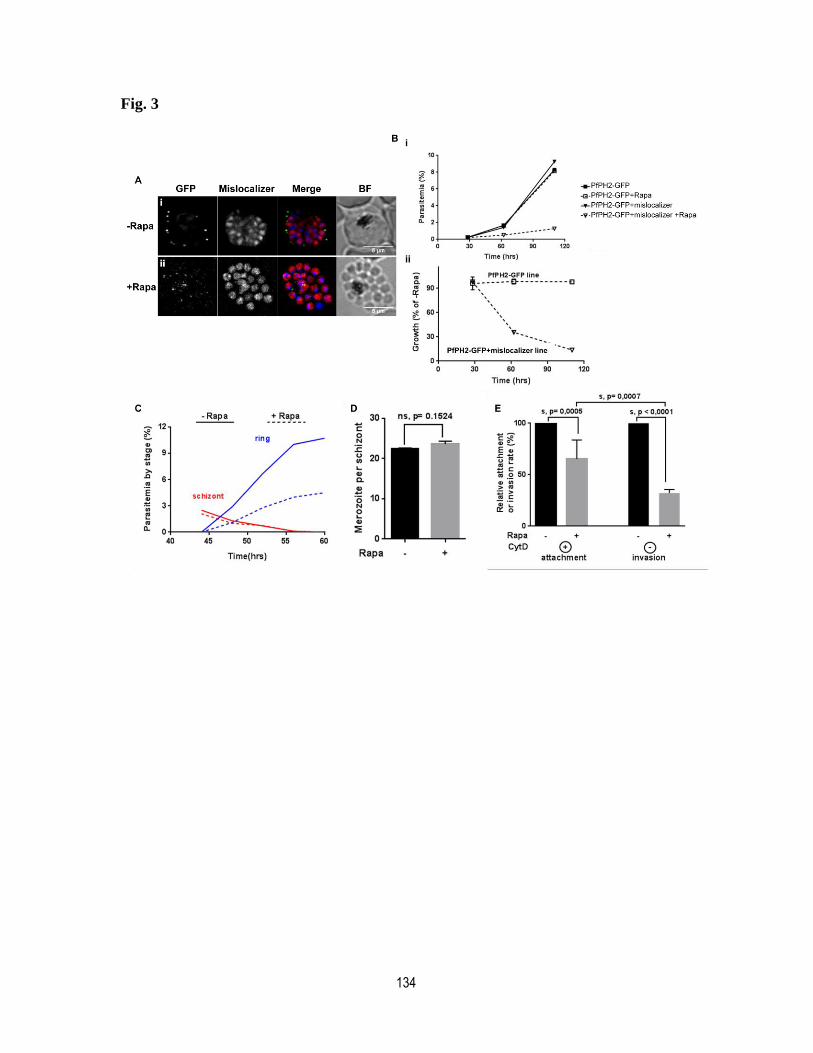
134
Fig. 3

135
Fig. 4
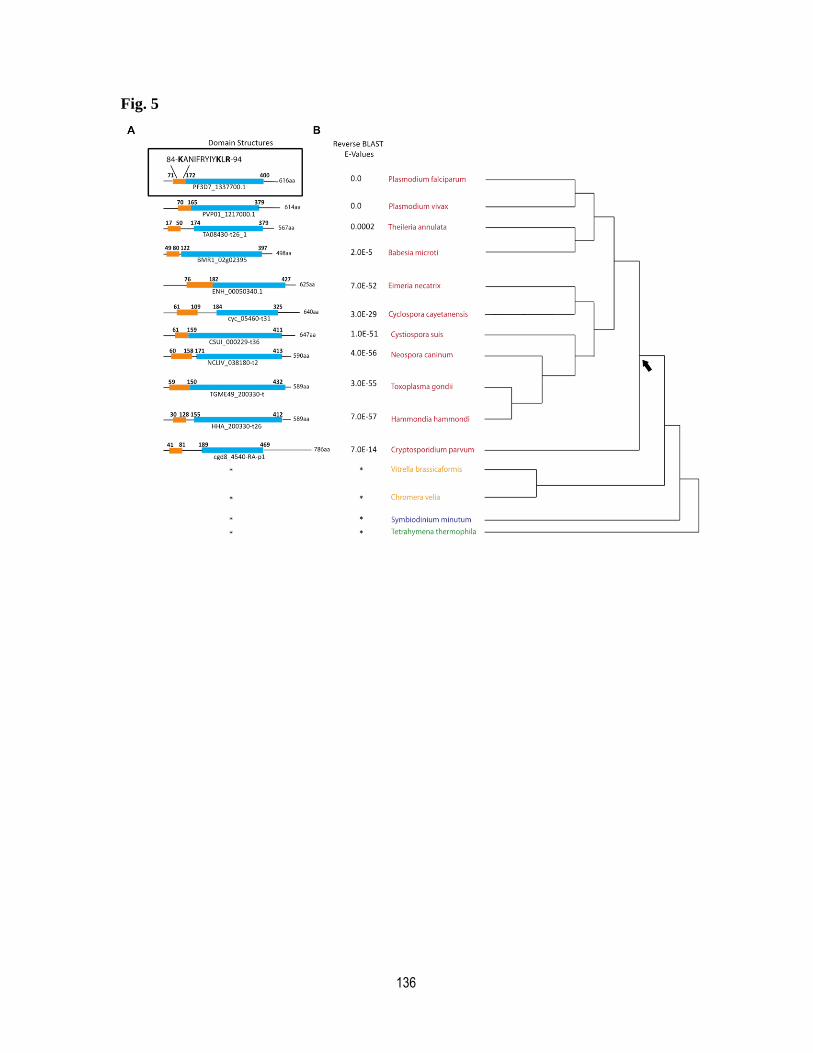
136
Fig. 5
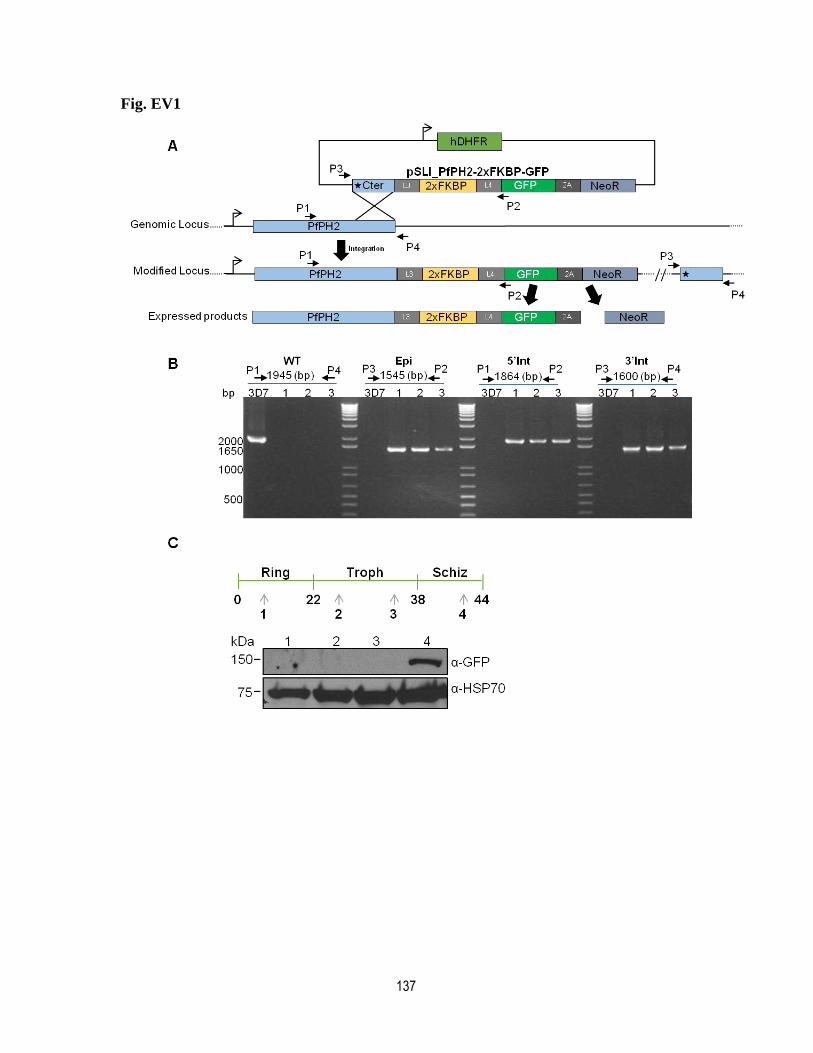
137
Fig. EV1

138
Fig. EV2
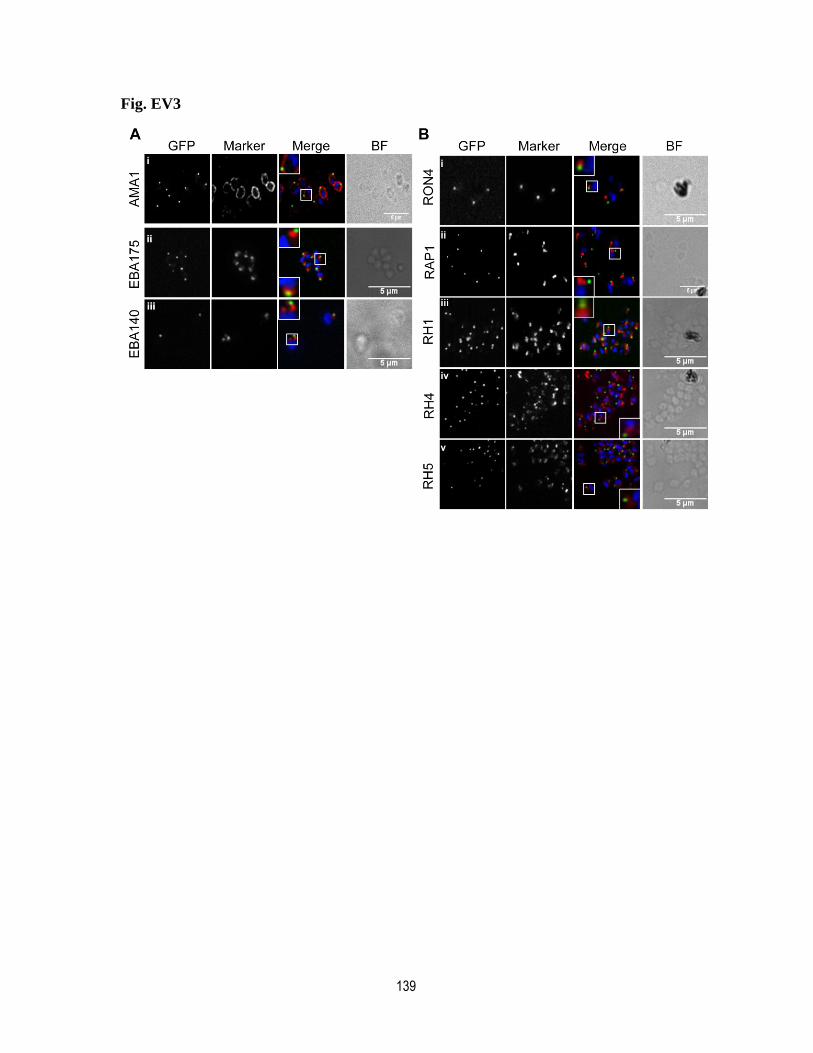
139
Fig. EV3

140
Fig. EV4
P.falciparum 1 WERYYCFIKANIFYYKLRGDYKPHVIFLLP.vivax 1 WVKCFCFVKSNFFYYKERGDYRPSIIFLLT.annulata 1 WKELNLEIRGNCLLCDASDECGPVMGWILB.microti 1 WEENNVEIRGNALFYPQYDQCVIGRGYLLE.necatrix 1 WLKRYCEIRGNLLLYTSHAEAAFEGAYMLC.cayetanensis 1 WLKRYCELKGNLLLYAPHSDAAFEGAFMLN.caninum 1 WVKRFCTVKANLLGFAPHSDAPFEGAYLLT.gondii 1 WVKRFCTVKANLFGFAPHSDAPFEGAYLLH.hammondi 1 WVKRFCTVKANLFGFAPHSDAPFEGAYLLC.suis 1 WIKRFCVVKANMLGFAPHSDAPFEGAYLLC.parvum 1 WEQYYFNLKGGMMFTSKKDGSTLEVVYVL

141
Appendix
-Appendix Table S1. Page 141
-Appendix figure titles. Page 142 to 143
-Appendix figures. Page 144 to 151
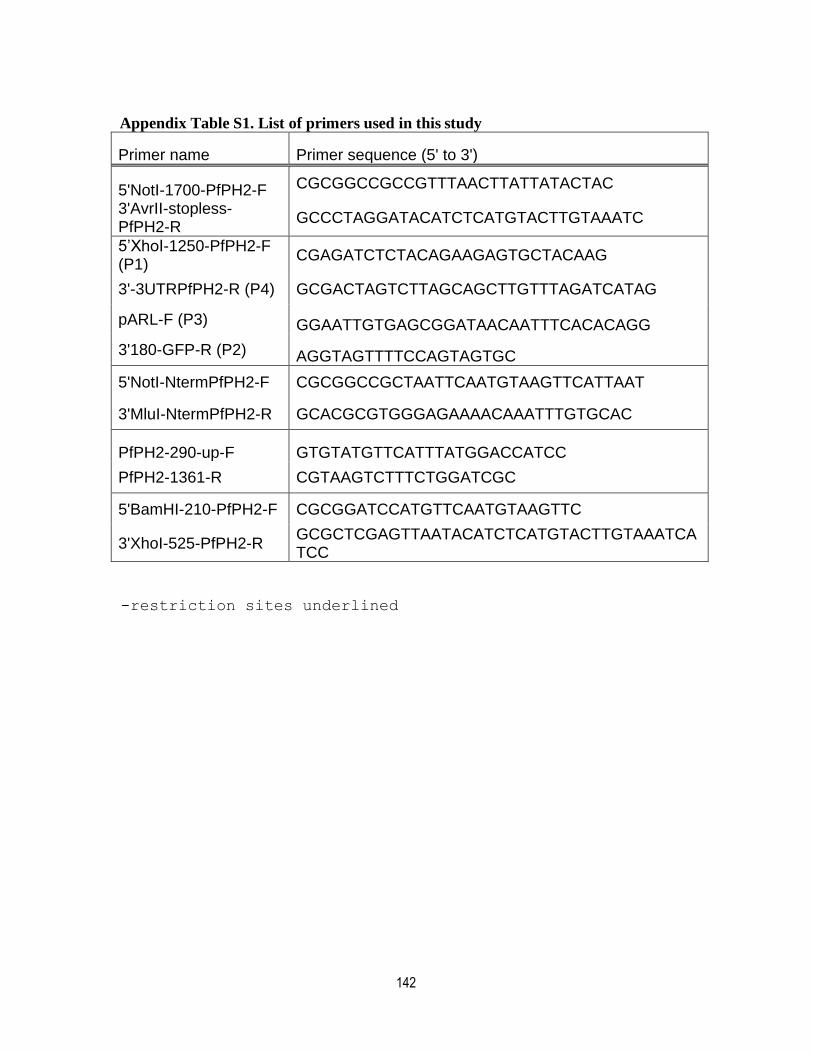
142
Appendix Table S1. List of primers used in this study
Primer name Primer sequence (5' to 3')
5'NotI-1700-PfPH2-F CGCGGCCGCCGTTTAACTTATTATACTAC
3'AvrII-stopless-PfPH2-R
GCCCTAGGATACATCTCATGTACTTGTAAATC
5’XhoI-1250-PfPH2-F (P1)
CGAGATCTCTACAGAAGAGTGCTACAAG
3'-3UTRPfPH2-R (P4) GCGACTAGTCTTAGCAGCTTGTTTAGATCATAG
pARL-F (P3) GGAATTGTGAGCGGATAACAATTTCACACAGG
3'180-GFP-R (P2) AGGTAGTTTTCCAGTAGTGC
5'NotI-NtermPfPH2-F CGCGGCCGCTAATTCAATGTAAGTTCATTAAT
3'MluI-NtermPfPH2-R GCACGCGTGGGAGAAAACAAATTTGTGCAC
PfPH2-290-up-F GTGTATGTTCATTTATGGACCATCC
PfPH2-1361-R CGTAAGTCTTTCTGGATCGC
5'BamHI-210-PfPH2-F CGCGGATCCATGTTCAATGTAAGTTC
3'XhoI-525-PfPH2-R GCGCTCGAGTTAATACATCTCATGTACTTGTAAATCATCC
-restriction sites underlined

143
Appendix figure legends
Fig. S1: Purification of the recombinant GST fusion of the PH domain of
PF3D7_1337700. Coomassie stained gel showing soluble GFP-PH fusion protein at the
expected size of around 40 kDa. The lower 26 kDa band likely represents a degradation
product of GST alone.
Fig. S2: Generation of the PfPH2-2xFKBP-GFP parasite line. A) Schematic showing
the tagging strategy by single cross-over recombination using SLI. B) PCR on parasite
genomic DNA showing the proper integration of the tagging vector at (5’ junction: primers
P1 and P3, 3’junction: primers P2 and P4) and the disappearance of the WT allele (primers
P1 and P4) in all 3 SLI attempts. C) Time course of expression of parasite protein extracts
taken throughout the erythrocytic cycle shows that PfPH2 is expressed in schizonts.
PfHSP70 is used as a control for a constitutive protein. Cartoon shows the time points in
hours post invasion (hpi). 1: 8 hpi, 2: 30 hpi, 3: 36 hpi, 4: 42 hpi.
Fig. S3: IFA showing that the PfPH2 knock sideways does not noticeably impact the
appearance of various subcellular structures by IFA. A) PfRAP1: rhoptry bulb. B)
PfRON4: rhoptry neck. C) PfRESA: dense granules. D) PfAMA1: egress-related
micronemes E) PfEBA175: invasion-related micronemes. F) PfMSP1: Merozoite surface.
G) PfGAP45: Inner membrane complex. Scale bar represents 5m. Blue: DAPI stained
nucleus.
Fig. S4: Bioreplicates for the egress assay, related to figure 3C.
Fig. S5: Schematic explaining the attachment-invasion assay related to figure 3E. In
normal conditions (-Rapa, -CytD), the molecular motor is functional, the merozoites invade
the erythrocytes and become rings. Counting the number of rings in -Rapa vs +Rapa shows
the decrease in invasion (i.e. less rings are formed) in the PfPH2 KS. When adding the actin
polymerization inhibitor cytochalasin D, the merozoites attach to the surface of the red
blood cell (red arrows) but since the molecular motor is not functional, they cannot proceed

144
to invasion. This allows the uncoupling of the attachment and invasion steps. Counting the
number of merozoites bound to the surface of red blood cells in -Rapa vs +Rapa, both in
the presence of CytD, shows the decrease in merozoite attachment (i.e. less merozoites are
found associated with the red blood cell surface) in the PfPH2 KS. Rapa: Rapamycin.
CytD: Cytochalasin D.
Fig. S6: Bioreplicates for the microneme secretion assay, related to figure 4B.
Fig. S7: Invasion assays using enzyme-treated erythrocytes show that host cell tropism
of the PfPH2 KS line is not affected. Data expressed as the % of invasion compared to the
control without enzyme treatment. T= trypsin. C= chymotrypsin. NM= neuraminidase. n= 3
biological replicates with at least 3 technical replicates for each condition. n.s; non-
significant in a multiple t-test.
Figure S8. Alignments of the KXn(K/R)XR binding motif in Apicomplexan
orthologues of PfPH2. Alignments of a portion of the PH domains from PF3D7_1337700
identified orthologues in Apicomplexa are shown. The box outlines the alignments of the
sequence motif from PF3D7_1337700 with similar amino acid positions of the other
proteins. Notably the first and terminal positions of the KXn(K/R)XR motif show
conservation.
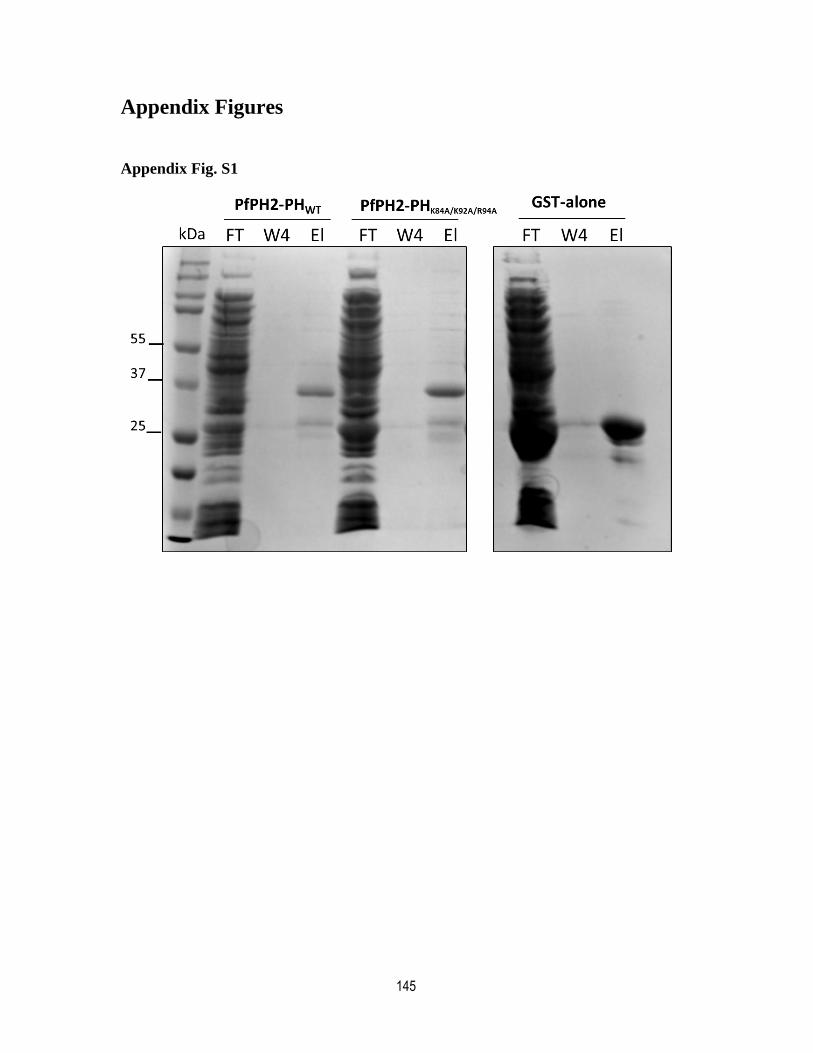
145
Appendix Figures
Appendix Fig. S1

146
Appendix Fig. S2

147
Appendix Fig. S3
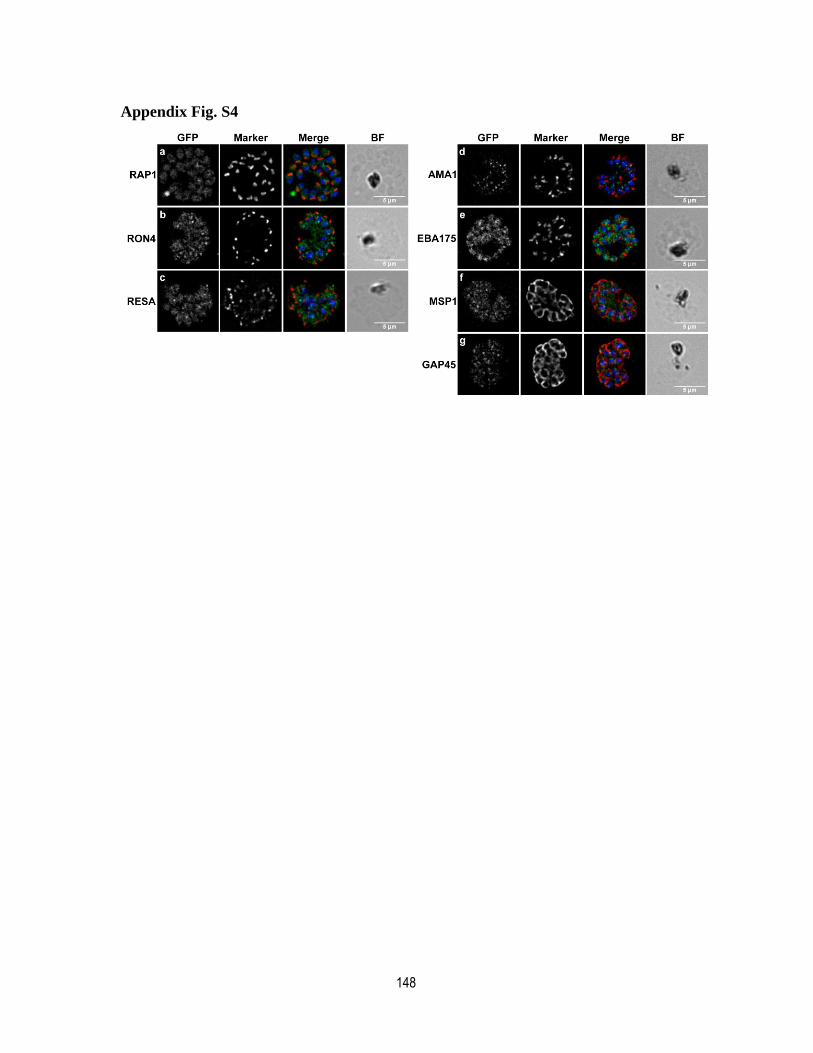
148
Appendix Fig. S4
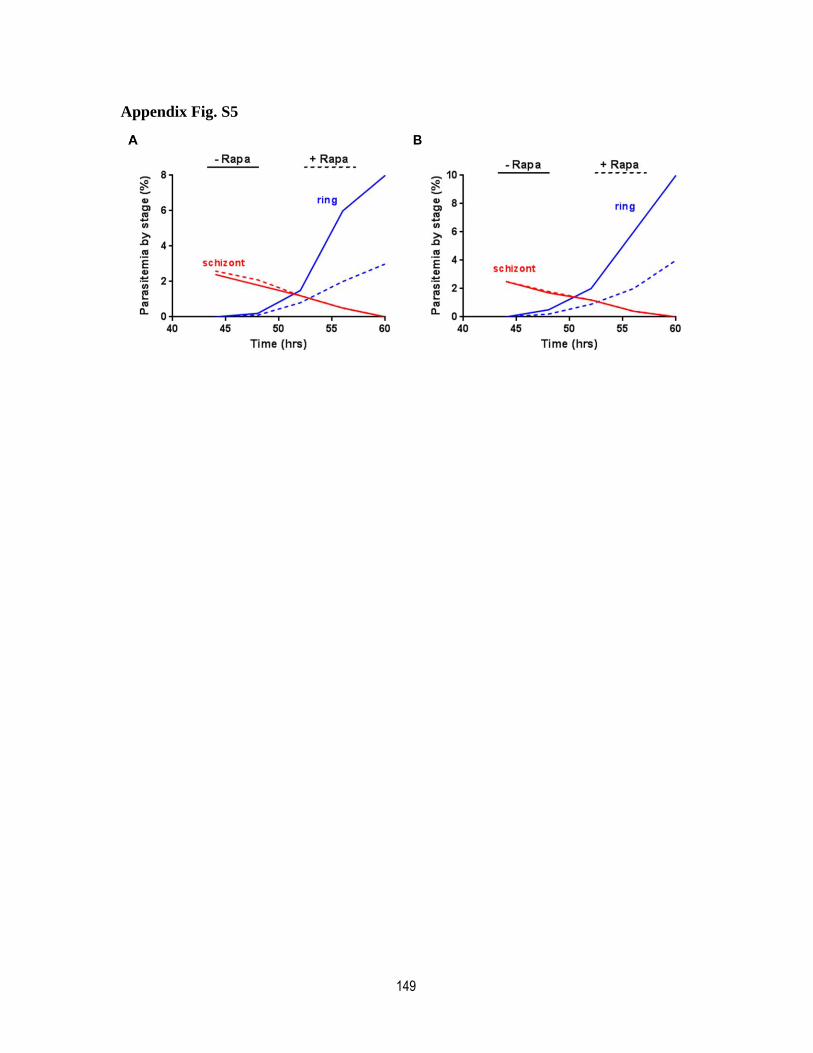
149
Appendix Fig. S5
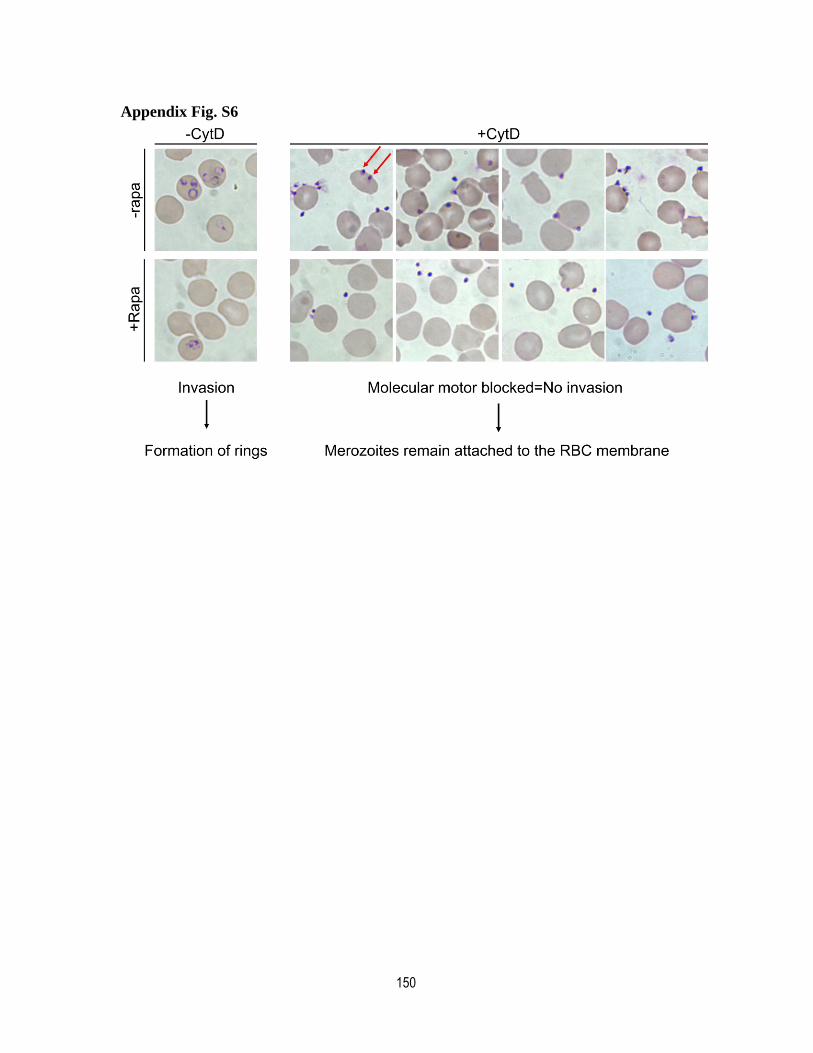
150
Appendix Fig. S6
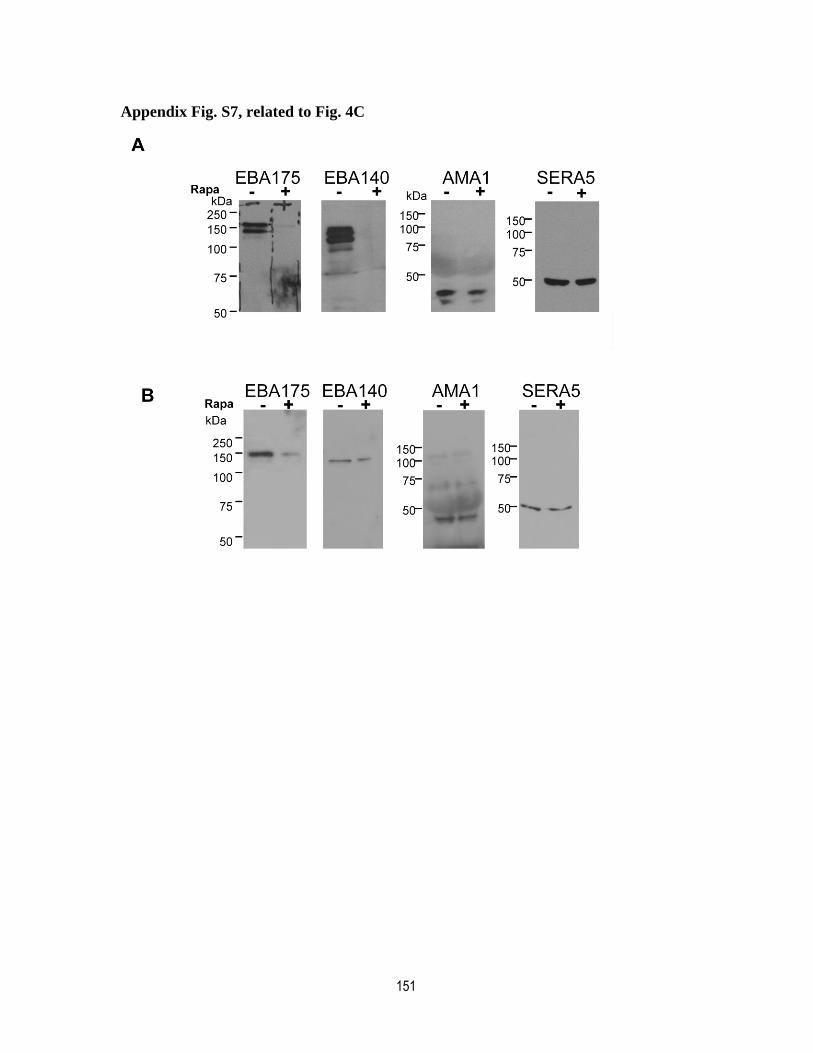
151
Appendix Fig. S7, related to Fig. 4C
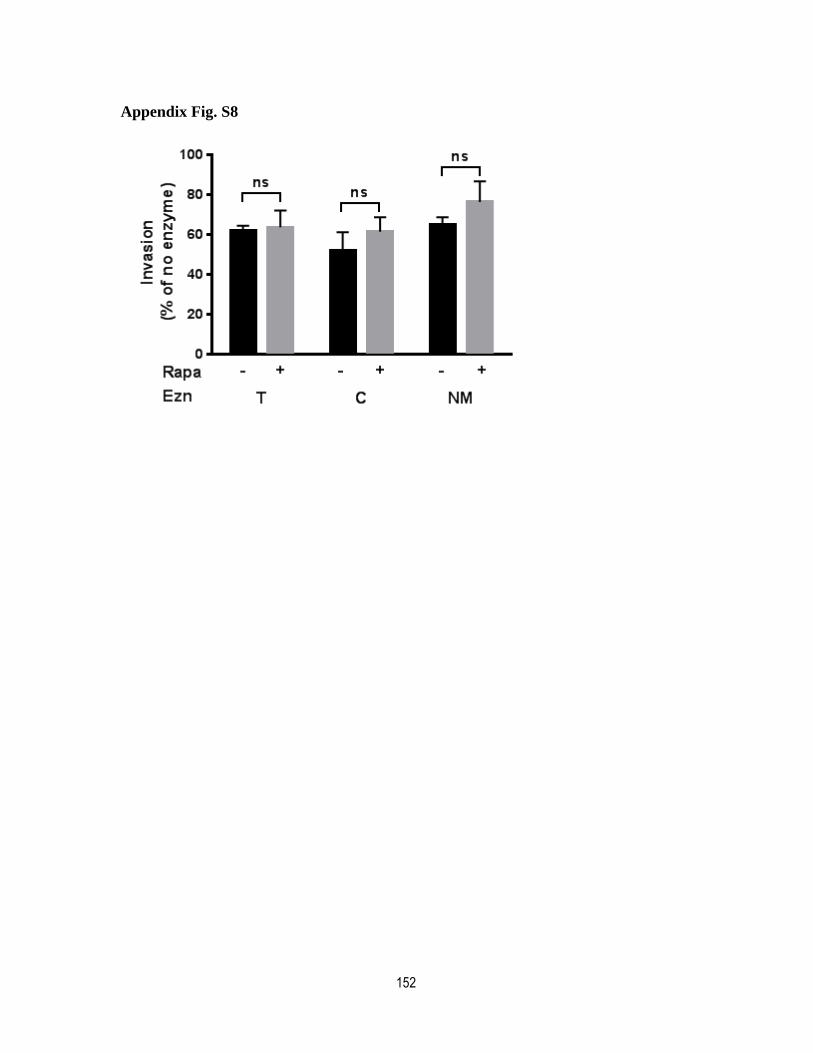
152
Appendix Fig. S8

153
General Discussion, Conclusion and Perspectives
The apical organelles are a set of secretory organelles that contain proteins whose function
is necessary for invasion of the red blood cell by the parasite [445]. Therefore, a
disturbance in their secretion results in egress or invasion defects. Due to the uniqueness of
these organelles and their essential role in parasite survival, a better understanding of the
mechanisms behind the biogenesis and transport of proteins to these organelles are
necessary since identification of the molecular players has the potential to be used for
therapeutic purposes and vaccine design strategies. The Plasmodium genome contains
homologues of many coat, adaptor and regulatory proteins that are involved in vesicle
traffic and protein transport. There are also Plasmodium specific proteins which might play
a role in the protein transport machinery that is specific to the parasite. However, the
protein transport machinery and biogenesis of these organelles are poorly understood. PIPs
have been shown to play important roles in the Plasmodium life cycle. Recent findings
have revealed the role of PIPs in gametocyte activation [353], merozoite egress [45, 229],
resistance to artemisinin [262], cytokinesis [145], and male gametocyte exflagellation [350,
351], most of which were related to the hydrolysis of PI(4,5)P2. As mentioned earlier, in P.
falciparum, a model of shuttling cargo molecules has been proposed by Richard et al. that
posits the existence of specific microdomains in the Golgi membrane [359]. Based on the
model, proteins destined to the PM or apical organelles accumulate into distinct
microdomains with specific lipid compositions. Then, specific escorters that are exposed to
the cytosolic face of microdomains recruit coat and adaptor proteins to form a shuttle
vesicle. Our lab has also shown a rhoptry protein, RAMA, interacts with another rhoptry
protein RAP1 to escort specifically to the rhoptry destined vesicles. However, RAMA does
not have a cytosolic domain that recruits vesicle components to the sub-domain.
Subsequently, recent work from our lab has shown that the more general sorting escorter is
a homologue of Sortiline. Inside the Golgi lumen, PfSortiline interacts either directly or
indirectly with the receptor of the protein cargo inside the Golgi lumen to induce vesicle
budding [453, 454]. Interestingly, the conditional knockdown of PfSortiline leads to
disturbed protein transport that disrupts the biogenesis of apical organelles and the IMC.
PfSortiline has been shown to bind to RAMA directly in the case of rhoptry proteins and

154
also to be involved in sorting of micronemes and dense granule and potentially IMC
proteins [455]. In the cytosolic face of eukaryotic membranes apart from the cytosolic
domain of Sortiline, PIPs also act as a recruiting signal for the vesicle components. Many
PIP effectors either have a PIP-binding or kinase domain, most of which are not
characterized in P. falciparum. In addition, several of the parasite’s putative PIP effectors
don't show any homology to known proteins in the vesicle traffic machinery of the other
organisms other than the predicted PIP-binding domain. Therefore, some functions of PIPs
and their effector molecules might be specific to Plasmodium given the existence of unique
cellular compartments, specifically in the case of apical organelles. The goal of my work
was to generate a map of the distribution of PIPs inside the parasite and to identify PIP
interacting partners implicated in the biogenesis of the invasion organelles or their secretion
during the process of invasion.
We hypothesized that phosphoinositides are part of the protein transport machinery in P.
falciparum and create a distinct membrane identity in different subcellular membranes. The
results obtained from the PIP mapping in P. falciparum will be discussed in the section 5.
1.
For my second project, I have characterized the protein of PF3D7_1337700 which is an
apicomplexan specific gene with unknown function. Apart from a PH domain there is no
other known domains, therefore, based on this and the fact that the protein is the second
studied Plasmodium protein with a PH domain we renamed it to PfPH2[251]. As PfPH2
has a PIP-binding domain we proposed that it might be involved in the membrane transport
or PIP-signaling. At the end of this work, we will show that PfPH2 is an essential gene that
is important for the exocytosis of a specific population of micronemes. This will be
discussed in the section 5. 2.

155
A map of subcellular distribution of phosphoinositides in P.
falciparum
PI3P distribution
In eukaryotic cells, PI3P localizes on early endosome membranes and is involved in
endocytosis and vesicular trafficking toward lysosomes [156]. In P. falciparum, our results
confirmed what has been previously reported by Tawk et al. [160] and showed PI3P
localizes to the food vacuole membrane, vesicles at or the vicinity of the food vacuole and
the PM, and the apicoplast. However, we did not detect any trace of PI3P at the ER vicinity
despite the recent evidence of PI3P-enriched regions at the ER [262]. This could be
explained by this fact that the PX domain that we used here as PI3P binding reporter for
some reason cannot detect the PI3P pools at the ER or it is possible that the level of PI3P is
so low that is out of the range of our detection tool. In yeast, PI3P production is mostly the
result of the kinase Vsp34 and the lipid phosphatases Fig4 (also known as Sac3),
Sjl2/Inp52, Sjl3/Inp53 and Sac1[238]. Lipid phosphatases all possess a SAC catalytic
domain able to dephosphorylate PtdIns(3,5)P2 in PtdIns3P. So far the only homologue
identified is a PfSAC1 phosphoinositide-phosphatase recently characterized in our lab
[456]. Although, its phosphatase activity is unknown, it localizes to the ER and Golgi
where it might be involved in the synthesis of PI3P pool at the ER. The lipid counts for
30% of total monophosphorylated PIPs in the parasite. The unusually high level of the lipid
suggests that it might have critical functions for the erythrocytic cycle especially during late
stages of parasite development [160]. A recent example of a such role has been revealed
recently in the resistance mechanism to ARTs [262]. It has been shown that parasite strains
resistant to ARTs have increased levels of PI3P. This might be related to the role of
PI3P/Vsp34 in response to the drug stress as in yeast, the deletion of the VPS34 gene
results in reduced resistance to numerous stress factors (temperature, pH and some drugs)
[182]. So far, evidence of a role for PI3P has been found in important parasite pathways
such as hemoglobin transport [240, 406, 407], autophagy [197, 264], apicoplast
homeostasis [160, 162], and apicoplast inheritance [190, 197]. Moreover, PI3P/Vsp34
might use the apicoplast as the pre-phagosome station [190, 197] as many homologues of
ATG proteins (autophagy proteins) are present in apicomplexans [264]. One of such
homologues is PfATG18 which has been identified in both P. falciparum and T. gondii.

156
PfATG binds to PI3P (and PI(3,5)P2) in lipid dot blot and liposome binding assays [197].
Although there is no evidence for the existence of PI(3,5)P2 so far in P. falciparum.
However, conditional ablation of TgVsp34 expression in T. gondii [162] and the depletion
of the only TgPIKfyve both result in apicoplast loss and delay death, suggesting PI3P might
be used as a substrate to produce PI(3,5)P2. Because of the critical importance of PI3P in
the parasite life cycle, targeting the enzyme responsible for PI3P production could be an
interesting antimalarial strategy.
PI4P distribution
The IFA results obtained from the parasite expressing a PI4P-tracker in our laboratory
showed PI4P localizes to the Golgi apparatus and the PM during all life stages of the
parasite. Upon addition of imidazopyrazines to the parasites expressing a different PI4P-
tracker, McNamara et al. observed a redistribution of the PI4P signal from an internal foci
to the parasite PM [145]. The affected parasites eventually showed a cytokinesis defect and
perturbed membrane ingression around the developing merozoite which resulted in
drastically decrease in the parasitemia in the next cycle. Similar to other systems [156],
disturbance in PI4P leads to a major defect in cytokinesis. Plasmodium has three putative
PI4kinases and McNamara et al. demonstrated that the target enzyme of imidazopyrazines
and quinoxalines is the PI4KIIIβ homologue. Their microscopy results also revealed that
PfPI4KIIIβ localizes through the cytosol and apical ends of nascent merozoites during
trophozoite and late schizont stage, respectively. Therefore, we speculate that PI4P-residing
Golgi is the result of PfPI4KIIIβ activity and that the lipid likely then delivered to the PM
via vesicular traffic. Furthermore, IPZ molecules like KDU691, GNF179, and BQR695
inhibit parasite-derived PfPI4KIIIβ in a very low nanomolar activity compared to any
human lipid kinase which is an advantage to use as a drug against the parasite [145]. These
results also reveal that the PI4K target of KDU691 is different from KAF156 (or its analog
GNF179) [283]. Since KDU691 is potent against ring stage and can only kill dormant rings
while KAF156 inhibits ring stage parasite in addition to schizont and trophozoite stage.
Having different PI4K inhibitors is an advantage to study parasite PI4K function. As for the
role of PI3K/kelch13 in the ART resistance mechanism, PI4K target of GNF179, might

157
play a regulatory function in the control or reactivation of the quiescent rings through
regulation of PIP metabolism.
PI5P distribution
Similar to PI3P, the lipid is present in many compartments throughout the cell, including
nucleus, PM, Golgi, ER, and endosomes [437]. The available data on its role is mostly
obtained from the nuclear pool which is involved in various processes such as protection
against UV stress, apoptosis, transcriptional control and the cell cycle [437]. PI5P lipid has
not been reported so far in Plasmodium, however, this could be simply due to limitations in
current technologies being used to detect PIPs [163]. As to our findings with a PI5P-tracker
we demonstrated, for the first time, pools of PI5P in the PM throughout the erythrocytic
cycle, potentially transitional ER during schizont stage and sometimes in the nucleus. PI5P
synthesis in mammalian cells is the result of the interplay of PIKfyve activity on PtdIns and
3-phosphatase activity on PI(3,5)P2 for which the first is the main route [211].
Nevertheless, the only detected PIKfyve-like orthologue does not contain a FYVE domain
and there is no evidence of its 5-kinase activity, and as mentioned earlier PI(3,5)P2 is also
not detected in Plasmodium. Moreover, there is no evidence of the substrate activity of four
annotated phosphatases. Assuming the presence of PI5P in Plasmodium, it is therefore
tempting to consider that PIKfyve-like orthologue can produce PI5P directly via
phosphorylation of PtdIns. PI5P in P. falciparum might act as a second messenger in
different signaling pathways such as regulation of cell cycle in nucleus and actin
remodeling in the PM in response to parasite stimuli during different stages.
PI(4,5)P2 distribution
PI(4,5)P2 is the most abundant PIP in mammals and yeast [156]. Although, PI(4,5)P2 is
mostly in the PM there are small pools in the Golgi, ER and endosomes. Most of its
functions are related to cell surface related actions. Our observation with the PI(4,5)P2
tracker showed the lipid localizes to the parasite PM during all the erythrocytic cycle
showing no variation in its localization. This might be the result of its structural nature that
is needed continuously and synthesized and consumed in the same time. We have also

158
observed lipid localization to large vesicular-structures on the parasite PM. Since PI(4,5)P2
is mediating endocytosis events in other systems, these structures might be involved in
endocytosis and possibly the hemoglobin ingestion [458]. A small pool of PI(4,5)P2 was
also detected close to the nucleus in early stages of the parasites. Whether there is a relation
between this PI(4,5)P2 pool and a PH domain-containing calcium-dependent protein kinase
(PfCDPK7) that localizes to vesicles in close proximity to the parasite ER is left to be
investigated [450]. PfCDPK7 was shown to specifically bind to PI(4,5)P2. In P.
falciparum, there are several PH domain/ENTH-containing protein and two PX/PDZ-
containing proteins but there is no evidence so far of their ability for PIP-binding. Most of
the evidence for the role of PI(4,5)P2 in P. falciparum comes primarily from studies using
inhibitors of the single PfPI-PLC enzyme [159] which is implicated in many essential
processes such as gametocyte activation [351, 353], ookinete motility [227],
synchronization of the erythrocytic cycle [449], schizont egress [229] and invasion of
merozoites [391]. However, a role for PfPI-PLC in merozoite invasion was later argued
[459].
PI(3,4)P2 and PI(3,4,5)P3 distribution
In eukaryotes, PI(3,4)P2 and PI(3,4,5)P3 are synthesized in response to cell stimuli and
localize to the PM [156]. Very low amounts of these two lipids have been detected in the
infected RBCs [160] though our attempts to localize PI(3,4)P2 and PI(3,4,5)P3 in P.
falciparum were not successful. In our hands, only a diffused cytosolic signal was
visualized like what we observed with GFP- or mCherry-alone all over the erythrocytic
cycle. In the case of PI(3,4,5)P3, it was even rare to find parasites that expressed the
reporter protein. It is likely that these lipids need special stimuli which might not have been
present in our in vitro experiments in contrast to the stable signal that we observe from the
structural lipids such as PI3P, PI4P and PI(4,5)P2. Such signals could be required for
gametocytogenesis for example. It is also possible that the signals were so quick that we
were not able to capture the time of their actions. Looking at parasite development using
live-cell imaging could potentially reveal transient PI(3,4)P2 and PI(3,4,5)P3 containing
structures. Another possibility would be to use different PI(3,4)P2- and PI(3,4,5)P3-binding
domains that might help to visualize these lipids.

159
Conclusion on the subcellular PIP distribution and general pitfalls
Although PIP tracker expressing parasite lines were generally stable and showed no
difficulty in their expression, the PIP tracker signal easily disappeared after processing the
samples such as centrifugation steps which made microscopy studies difficult. To avoid this
problem we used methanol-fixation for IFAs, which does not require centrifugation step.
Also, double-transfectants with Plasmodium organellar-markers were in general a rare
occasion and in the few successful ones, the parasite did not look healthy.
Surprisingly, PIP distribution in P. falciparum is a reflection of what has been observed
with other systems. As P. falciparum has unusual organelles like apicoplast and invasion
organelles we expected to record some differences compared to model organisms like yeast
and human (Fig. 3. 6). We were interested in determining whether invasion organelles were
enriched in specific PIPs however none of the probes we have used labelled these
structures. A different strategy such as BioID might help to better clarify the PIP
composition of the parasite’s various subcellular features. BioID is based on the fusion of
the promiscuous biotin ligase BirA to a protein of interest (PIP-binding domains in our
case). Following its expression in cells, addition of biotin in the culture medium results in
the biotinylation of proteins proximal to the BirA fusion. These can then be identified by
proteomics. Identification of proteins from a particular organelle when using specific PIP-
binding domain-BirA probe could mean that a particular PIP species is enriched. This
strategy could also potentially allow the identification of new interactors of PIPs in P.
falciparum.
A pan-apicomplexan phosphoinositide-binding protein acts in
malarial invasion-microneme exocytosis.
PfPH2 is a PH-containing protein with a relaxed PIP-binding
specificity.
The aim of my second project was to characterize a PH-containing protein, namely PfPH2
(PF3D7_1337700). It is a hypothetical protein conserved throughout apicomplexans. It
does not have a transmembrane domain or a known secretion signal, but it does contain a
basic sequence motif (KXn (K/R)XR), responsible for binding to the inositol head group

160
[460]. Here, we showed that the recombinant PH domain of PfPH2 alone is able to interact
with different PIP species both by lipid blot assays and liposome binding assays. The
results of liposome binding assay seems reliable, as PIP strip cannot mimic membrane
bilayer and showed stronger interaction with PI(4,5)P2 and monophosphate species and
some residual interaction with PI(3,4,5)P3 and PA containing vesicles. We observed a clear
loss of binding ability of a triple mutant version of the basic motif where residues K84, K92
and R94 were mutated to alanines, suggesting the role of the basic sequence motif (KXn
(K/R)XR) in binding to the inositol head group[460]. Moreover, comparison of binding
affinity of the wild type version and its mutant, incubated with PI3P-containing lipid
vesicles reaffirmed our hypothesis. All together, our results demonstrate that
PF3D7\1337700 is a genuine PIP-binding protein. A relaxed PIP-binding ability is known
for a majority of PH domains [461]. For a specific PIP-interaction, the PH domain of
PfPH2 might need auxiliary domains that are hidden in 3D-conformation of native protein
or the presence of partner proteins which is referred as coincidence detection to lead to a
specific subcellular distribution. To elucidate more its PIP-binding ability, it would be
necessary to study the full-length protein with and without the PH domain as well. In
addition, it would be interesting to perform pull-down assays using the full-length
protein/the PH domain alone. Analysing these protein interactions might lead us to the
PfPH2 potential partners and elucidating its molecular mechanism.
PfPH2 localizes to a structure close to the apical tip of the
merozoite
To analyze the localization of PfPH2, we tagged the protein at its C-ter by 2xFKBP-GFP
and detected a band of ~133 kDa consistent with the tagged-protein size at the late schizont
parasites. Fluorescence microscopy of 2xFKBP-GFP-tagged PfPH2 parasite line showed a
punctate pattern of GFP signal in the late schizont, reminiscent of apical organelles pattern.
Interestingly, IFA attempts revealed no colocalization of PfPH2-2xFKBP-GFP signal with
apical organelles or any organellar markers tested. However, the PfPH2 signal is always
farther from the DAPI than PfEBA175 and potentially more than PfRON4, though the
difference with PfRON4 was not statistically significant.. To clarify the exact localization
of PfPH2 we need to produce a specific PfPH2 antibody. A PfPH2-specific antibody will

161
be very useful to reassert the IFAs with untagged parasite line and also perform immuno-
electron microscopy (IEM) experiments.
PfPH2 is essential for the erythrocytic cycle and its absence affects
merozoite invasion due to a default in microneme exocytosis.
To investigate the function of PfPH2 we performed knock-sideways (KS) analysis on
PfPH2-2xFKBPGFP line that allows dislocating the protein from its site of action and
mistargeting it to the nucleus in presence of mTOR inhibitor, rapamycin. The proliferation
of rapamycin-treated parasites, the KS line, showed around 65% decrease in the first cycle
and up to 86% in the second cycle compared to the control line. Therefore, we observed
few rings formed in the next cycle compared to the control line. At the same time, all our
three knockout attempts failed, suggesting an essential role for the protein. Consequently,
we analyzed different cellular processes including organelle formation, nuclear division,
egress, and invasion to find out what stage of life cycle has been impaired. The analysis of
the IFAs and egress assay proved that the absence of PfPH2 does not affect the events
before egress such as nuclear division, merozoite formation, organellar integrity and egress
itself but potentially affects the merozoite invasion.
The invasion process is a rapid and timely controlled process during which invasion
organelles secrete their contents in orderly steps. Invasion can be divided in the attachment
of the merozoite and entry into the erythrocyte. Using an actin inhibitor we were able to
stop merozoites at the entry and study their attachment which revealed a significant
decrease in the attachment of the KS line. This observation suggests lack of PfPH2 from its
site of action affects merozoite attachment to the erythrocyte which is necessary for entry.
A micronemal protein (AMA1) and the RON complex are responsible for a tight junction
that along with a motor-myosin mediate the parasite entry. In absence of PfPH2, the normal
translocation of AMA1 to the merozoite surface confirmed that the entry process was not
affected. Merozoites engage in a stable attachment with the erythrocyte membrane using
two different routes regarded as the alternative pathways. The EBAs and RHs family
proteins, which mediate the attachment would be held in reserve in micronemes and
rhoptries respectively, until they are needed. However, during the invasion, their secretion
is triggered to the merozoite surface leading to the interactions with their receptors on the

162
erythrocyte membrane after which they are cleaved and shed into culture supernatant via
parasite proteases during invasion. It has been shown that some of the invasion-receptor
interactions like Rh1 are related to Ca2+-dependent release of microneme proteins
specifically EBA175 [85].The order of secretion of each set of ligands is critical and well-
documented during invasion in P. falciparum. AMA1 is among the first set of invasion
ligands, secreting into merozoite surface and its secretion appeared normal in both control
and KS line, based on the IFAs results. To further investigate their secretion, we performed
western blot on the collected supernatant of the KS and the control line cultures and detect
their presence via antibodies specific to each ligand. The results showed significant
decrease in shedding of EBA ligands into the supernatant of KS line (EBA-175~90% and
EBA-140 ~79%) indicating a defect in secretion of microneme proteins. Micronemes
release their content upon receiving a signal. It has been shown that the trigger is a change
in the level of intracellular Ca2+, which activates a cascade of events that lead to
microneme discharge via fusion of microneme membrane with the parasite PM. PfPH2
function appear to be necessary for microneme discharge but the exact mechanism of action
of this protein in this process still need to be elucidated. AMA1, also showed decrease but
with a lesser degree (around 24%) suggesting that, secretion of microneme proteins is
generally affected but at different degrees. Moreover, we observed the decrease of EBA175
in the supernatant correlated with an increase in the unprocessed form in free merozoites of
KS line. Notably, there was no increase in the unprocessed AMA1 pointing to a defect in
EBA175 secretion alone. The discrepancy in exocytosis of the contents of same organelle
can't be explained unless we assume there are different populations of micronemes.
Recently, some evidence arose about the presence of at least two different populations of
micronemes and AMA1 has been suggested to be related to the egress-related micronemes
[82]. Here, our results also suggest the possibility of two populations of micronemes since
we observed a significant decrease in EBAs ligands but not in AMA1. The 24% decrease in
AMA1 shedding can be also explained by the fact that in the KS line, a low amount of
successful invasion events happens compared to the control line leading to less shedding of
AMA1. Therefor we suggest the possibility of a new group of micronemes that are
invasion-related and contains EBA family proteins which display a defect in their
exocytosis due to the absence of PfPH2. Lately, it has been shown inhibition of a PM

163
resident protein, PfCDPK1, in the young and mature merozoites results in microneme
discharge arrest [462]. PfCDPK1 is a calcium-dependent protein kinase and specifically
involved in the EBA-175 secretion [84, 463]. After a rise in merozoite intracellular Ca2+,
the kinase starts a cascade of phosphorylation that leads to invasion. Hence, we speculate
PfPH2 might be a substrate of PfCDPK1 which is regulated through phosphorylation. This
could be determined through use of phospho-specific antibody or by immunoprecitation
techniques and looking into the phosphorylated peptides
EBA family proteins bind to a specific group of erythrocyte receptors, which has sialic acid
(SA) branches in their structure and are sensitive to neuraminidase treatments. They differ
from RH-receptors that are resistant to such treatments. Plasmodium species have different
preferences in choosing the erythrocyte receptors during the merozoite invasion of RBCs
[73-75]. Therefore, EBAs and RHs are referred as alternative pathway ligands or SA-
dependent and SA-independent pathway ligands, respectively. For example, the deletion of
EBA-175 in W2mef parasite strain results in the upregulation of PfRh4 which indicates the
functional substitution of EBA-175 [74]. The parasite can epigenetically silence or
upregulate particular invasion-related genes, such as PfRh4 [74, 76, 77], resulting in
divergent ligand expression between isolates. Due to this plasticity, the parasite is able to
rapidly adjust to erythrocyte receptor polymorphisms in human populations [76, 78, 79].
Considering the severe decrease in EBA-175 and EBA-140 level, we anticipated that if we
treat erythrocytes with neuramidase enzymes to remove sialic-acid groups, the drastic
diminution of EBAs due to absence of PfPH2 in KS line should affect the host cell tropism.
However, the invasion assays performed on different enzyme treated erythrocytes showed
no difference in the sensitivity to either trypsin, chymotrypsin or neuraminidase treatment
between the control and KS line. This suggests that absence of PfPH2 leads to a more
general invasion defect. Even, the invasion rate decrease in KS line was generally higher
than control line and in some cases this was significant. This indicates that the effect of
PfPH2 mislocalization is more severe than removing sialic acid groups from erythrocyte
receptors. This could be potentially explained by considering overlap of alternative
pathway ligands in function and cooperating together during invasion as it has been shown,
previously [71].

164
Conclusions on PfPH2 mechanism of action and future
experiments
The exact function of PfPH2 on microneme exocytosis is not clear yet. A recent work has
shown a PH-containing domain in the related apicomplexan parasite T. gondii, TgAPH, is
involved in phosphatidic acid (PA) sensing during microneme exocytosis events. Since
PfPH2 contain PH domain and showed PIP-binding ability, it is tempting to speculate that
PfPH2, like TgAPH, is involved in the fusion of invasion-related microneme membranes
with the PM. An interesting candidate that might interact with PfPH2 is a calcium-activated
snare-like protein named PfDOC2.1 which has a role in the fusion events. It has been
shown PfDOC2.1 knockdown impairs merozoite invasion and blocks microneme secretion.
Importantly it is specifically required for Ca2+-dependent release of EBA175 from the
micronemes as well as PfCDPK1 and Rh1 [357]. Consequently, PfCDPK1 might be
implicated in regulation of PfPH2 likely via its kinase activity. Also, Rh1 interaction could
be a common point in a complex signalling network that might connect the alternative
pathways and PfCDPK1 by triggering Ca+ signal after its interaction. Whether PfPH2 is a
substrate to PfCDPK1 and would be phosphorylated during the activation process needs to
be further investigated.
Based on our results and what is discussed on the potential roles of other factors, we
propose a model for differential exocytosis of the micronemes. During late schizont stage,
PA sensing of P. falciparum homologue of TgAPH in the merozoite apical tips potentially
leads to the liberation of AMA1 from egress-related micronemes. Later, upon egress and
exposure of the merozoites to the extracellular environment, a signaling cascade starts
through PfCDPK1 by phosphorylation of its substrates that results in the secretion of EBA-
containing invasion micronemes. One of the PfCDPK1 substrates is potentially the PfPH2
protein, which might be involved in the exocytosis of invasion-related micronemes through
interaction with an unspecified PIP species. In this model, absence of PfPH2 leads to a
deficient attachment of merozoites to the erythrocyte membrane that leads to a drastic
decrease in the invasion rate.
To elucidate the exact function of PfPH2, it is necessary to identify its interacting partners.
Also, whether PfPH2 is one of the substrates of PfCDPK1 may clarify the cascade of

165
events that control the microneme exocytosis. Moreover, solubility assays can provide
some information on the protein position in vivo. If the protein is membrane bound it could
suggest it interacts with the membranes through the PIPs or other phosphorylated forms
most likely via its PH domain. As the growth defect is a more intense defect compared to
what we see with SA-dependent ligands, it would be recommended to analyze secretion of
other invasion ligands. Finally, in a broader view, the function of PfPH2 in P. falciparum
might be a common role in the exocytosis of micronemes and might help to better
understand the invasion mechanism in all apicomplexans.

166
References
1. Cox, F.E.G., History of the discovery of the malaria parasites and their vectors. Parasites & Vectors, 2010. 3(1): p. 5.
2. Schlagenhauf, P., Malaria: from prehistory to present. Infectious Disease Clinics of North America, 2004. 18(2): p. 189-205.
3. Jin, Y., C. Kebaier, and J. Vanderberg, Direct microscopic quantification of dynamics of Plasmodium berghei sporozoite transmission from mosquitoes to mice. Infection and immunity, 2007. 75(11): p. 5532-5539.
4. Cotter, C., et al., The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet, 2013. 382(9895): p. 900-911.
5. Feachem, R.G.A., et al., Shrinking the malaria map: progress and prospects. The Lancet. 376(9752): p. 1566-1578.
6. Hay, S.I., et al., The global distribution and population at risk of malaria: past, present, and future. The Lancet Infectious Diseases, 2004. 4(6): p. 327-336.
7. Murray, C.J.L., et al., Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet. 379(9814): p. 413-431.
8. Murray, C.J.L., et al., Global, regional, and national incidence and mortality for HIV, tuberculosis, and malaria during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. The Lancet. 384(9947): p. 1005-1070.
9. WHO, World Malaria Report. 2016.
10. Sadanand, S., Malaria: An Evaluation of the Current State of Research on Pathogenesis and Antimalarial Drugs. The Yale Journal of Biology and Medicine, 2010. 83(4): p. 185-191.
11. CDC, Malaria - About Malaria - Disease. . 2015.
12. Pasternak, N.D. and R. Dzikowski, PfEMP1: An antigen that plays a key role in the pathogenicity and immune evasion of the malaria parasite Plasmodium falciparum. The International Journal of Biochemistry & Cell Biology, 2009. 41(7): p. 1463-1466.
13. Caraballo H, K.K., Emergency department management of mosquito-borne illness: Malaria, dengue, and west nile virus. Emergency Medicine Practice, 2014. 16(5): p. 1-23.
14. Hulden, L. and L. Hulden, Activation of the hypnozoite: a part of Plasmodium vivax life cycle and survival. Malaria Journal, 2011. 10: p. 90-90.
15. Killeen, G.F., et al., Eliminating malaria vectors. Parasites & Vectors, 2013. 6(1): p. 172.
16. CDC, Malaria: Anopheles biology. 2015.
17. Bartoloni, A. and L. Zammarchi, Clinical Aspects of Uncomplicated and Severe Malaria. Mediterranean Journal of Hematology and Infectious Diseases, 2012. 4(1): p. e2012026.
18. Preiser, P., et al., The apical organelles of malaria merozoites: host cell selection, invasion, host immunity and immune evasion. Microbes and Infection, 2000. 2(12): p. 1461-1477.
19. Cowman, A.F. and B.S. Crabb, Invasion of red blood cells by malaria parasites. Cell, 2006. 124(4): p. 755-766.
20. Keeley, A. and D. Soldati, The glideosome: a molecular machine powering motility and host-cell invasion by Apicomplexa. Trends Cell Biol, 2004. 14(10): p. 528-32.

167
21. Harris, P.K., et al., Molecular identification of a malaria merozoite surface sheddase. PLoS pathogens, 2005. 1(3): p. 241-251.
22. Dowse, T.J., et al., Apicomplexan rhomboids have a potential role in microneme protein cleavage during host cell invasion. International Journal for Parasitology, 2005. 35(7): p. 747-756.
23. M. Santos, J., A. Graindorge, and D. Soldati-Favre, New insights into parasite rhomboid proteases. Molecular and Biochemical Parasitology, 2012. 182(1): p. 27-36.
24. Juliane Wunderlich, P.R., John Pius Dalton The malaria digestive vacuole Frontiers in Bioscience, 2012: p. 1424-1448.
25. Bannister, L.H., et al., A Brief Illustrated Guide to the Ultrastructure of Plasmodium falciparum Asexual Blood Stages. Parasitology Today, 2000. 16(10): p. 427-433.
26. Aikawa, M., C.G. Huff, and H. Sprinz, Fine structure of the asexual stages of Plasmodium elongatum. J Cell Biol, 1967. 34(1): p. 229-49.
27. Aikawa, M., Morphology of plasmodia. 1980.
28. Slomianny, C., Three-dimensional reconstruction of the feeding process of the malaria parasite. Blood Cells, 1990. 16(2-3): p. 369-78.
29. Aikawa, M., C.G. Huff, and H. Spinz, Comparative feeding mechanisms of avian and primate malarial parasites. Mil Med, 1966. 131(9): p. Suppl:969-83.
30. B C Elford, G.M.C., D J P Ferguson, Parasite-regulated membrane transport processes and metabolic control in malaria-infected erythrocytes. Biochemical Journal, 1995. 308(2): p. 361-374.
31. Atkinson CT1, A.M., Ultrastructure of malaria-infected erythrocytes. Blood Cells, 1990. 16(2-3): p. 351-68.
32. Francis, S.E., et al., HEMOGLOBIN METABOLISM IN THE MALARIA PARASITE PLASMODIUM FALCIPARUM. Annual Review of Microbiology, 1997. 51(1): p. 97-123.
33. Marti, M., et al., Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science (New York, NY), 2004. 306(5703): p. 1930-1933.
34. Hiller, N.L., et al., A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science (New York, NY), 2004. 306(5703): p. 1934-1937.
35. Mundwiler-Pachlatko, E. and H.-P. Beck, Maurer's clefts, the enigma of Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America, 2013. 110(50): p. 19987-19994.
36. Kirchgatter, K. and H.A. Del Portillo, Clinical and molecular aspects of severe malaria. Anais da Academia Brasileira de Ciências, 2005. 77: p. 455-475.
37. Kraemer, S.M. and J.D. Smith, A family affair: var genes, PfEMP1 binding, and malaria disease. Current Opinion in Microbiology, 2006. 9(4): p. 374-380.
38. Abdi, A.I., et al., Global selection of Plasmodium falciparum virulence antigen expression by host antibodies. Scientific Reports, 2016. 6: p. 19882.
39. Chen, Q., et al., Identification of <em>Plasmodium falciparum</em> Erythrocyte Membrane Protein 1 (PfEMP1) as the Rosetting Ligand of the Malaria Parasite <em>P. falciparum</em>. The Journal of Experimental Medicine, 1998. 187(1): p. 15.
40. Uyoga, S., et al., Transfer of 4-hydroxynonenal from parasitized to non-parasitized erythrocytes in rosettes. Proposed role in severe malaria anemia. British Journal of Haematology, 2012. 157(1): p. 116-124.

168
41. Gomes, P.S., et al., Immune Escape Strategies of Malaria Parasites. Frontiers in Microbiology, 2016. 7: p. 1617.
42. Nagao, E., O. Kaneko, and J.A. Dvorak, Plasmodium falciparum-Infected Erythrocytes: Qualitative and Quantitative Analyses of Parasite-Induced Knobs by Atomic Force Microscopy. Journal of Structural Biology, 2000. 130(1): p. 34-44.
43. Langreth, S.G., et al., Fine structure of human malaria in vitro. J Protozool, 1978. 25(4): p. 443-52.
44. Bannister, L.H., Ultrastructure of rhoptry development in Plasmodium falciparum erythrocytic schizonts. 2000: p. 1-15.
45. Collins, C.R., et al., Malaria Parasite cGMP-dependent Protein Kinase Regulates Blood Stage Merozoite Secretory Organelle Discharge and Egress. PLoS pathogens, 2013. 9(5): p. e1003344.
46. Yeoh, S., et al., Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell, 2007. 131(6): p. 1072-1083.
47. Thomas, J.A., et al., A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nature Microbiology, 2018. 3(4): p. 447-455.
48. Das, S., et al., Processing of Plasmodium falciparum Merozoite Surface Protein MSP1 Activates a Spectrin-Binding Function Enabling Parasite Egress from RBCs. Cell Host Microbe, 2015. 18(4): p. 433-44.
49. Deu, E., Proteases as antimalarial targets: strategies for genetic, chemical, and therapeutic validation. The Febs Journal, 2017. 284(16): p. 2604-2628.
50. Bannister, L.H. and G.H. Mitchell, The fine structure of secretion by Plasmodium knowlesi merozoites during red cell invasion. The Journal of protozoology, 1989. 36(4): p. 362-367.
51. Bullen, H.E., et al., Phosphatidic Acid-Mediated Signaling Regulates Microneme Secretion in Toxoplasma. Cell Host Microbe, 2016. 19(3): p. 349-60.
52. Dvorak, J.A., et al., Invasion of erythrocytes by malaria merozoites. Science, 1975. 187(4178): p. 748.
53. Jaikaria, N.S., et al., Biogenesis of rhoptry organelles in Plasmodium falciparum. Molecular and Biochemical Parasitology, 1993. 57(2): p. 269-279.
54. Hager, K.M., et al., The nuclear envelope serves as an intermediary between the ER and Golgi complex in the intracellular parasite Toxoplasma gondii. Journal of Cell Science, 1999. 112(16): p. 2631.
55. Aikawa, M., et al., Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite. The Journal of cell biology, 1978. 77(1): p. 72-82.
56. Bannister, L.H., et al., Structure and invasive behaviour of Plasmodium knowlesi merozoites in vitro. Parasitology, 1975. 71(3): p. 483-491.
57. Torii, M., et al., Release of merozoite dense granules during erythrocyte invasion by Plasmodium knowlesi. Infection and Immunity, 1989. 57(10): p. 3230-3233.
58. Aikawa, M., et al., Pf155/RESA antigen is localized in dense granules of Plasmodium falciparum merozoites. Experimental parasitology, 1990. 71(3): p. 326-329.
59. Trager, W., et al., Transfer of a dense granule protein of Plasmodium falciparum to the membrane of ring stages and isolation of dense granules. Infection and Immunity, 1992. 60(11): p. 4656-4661.

169
60. Culvenor, J.G., K.P. Day, and R.F. Anders, Plasmodium falciparum ring-infected erythrocyte surface antigen is released from merozoite dense granules after erythrocyte invasion. Infection and Immunity, 1991. 59(3): p. 1183.
61. Kilejian, A., Circular mitochondrial DNA from the avian malarial parasite Plasmodium lophurae. Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis, 1975. 390(3): p. 276-284.
62. Wilson, R.J.M., et al., Have malaria parasites three genomes? Parasitology Today, 1991. 7(6): p. 134-136.
63. McFadden Geoffrey, I. and F. Waller Ross, Plastids in parasites of humans. BioEssays, 2005. 19(11): p. 1033-1040.
64. Moore, R.B., et al., A photosynthetic alveolate closely related to apicomplexan parasites. Nature, 2008. 451: p. 959.
65. Janouškovec, J., et al., A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proceedings of the National Academy of Sciences, 2010. 107(24): p. 10949.
66. Ralph, S.A., et al., Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol, 2004. 2(3): p. 203-16.
67. Gould, S.B., R.F. Waller, and G.I. McFadden, Plastid evolution. Annu Rev Plant Biol, 2008. 59: p. 491-517.
68. McFadden, G.I. and E. Yeh, The apicoplast: now you see it, now you don’t. International journal for parasitology, 2017. 47(2-3): p. 137-144.
69. Weiss, G.E., et al., Revealing the sequence and resulting cellular morphology of receptor-ligand interactions during Plasmodium falciparum invasion of erythrocytes. PLoS Pathog, 2015. 11(2): p. e1004670.
70. Harvey, K.L., P.R. Gilson, and B.S. Crabb, A model for the progression of receptor–ligand interactions during erythrocyte invasion by Plasmodium falciparum. International Journal for Parasitology, 2012. 42(6): p. 567-573.
71. Lopaticki, S., et al., Reticulocyte and erythrocyte binding-like proteins function cooperatively in invasion of human erythrocytes by malaria parasites. Infection and Immunity, 2011. 79(3): p. 1107-1117.
72. Lanzillotti, R. and T.L. Coetzer, The 10 kDa domain of human erythrocyte protein 4.1 binds the Plasmodium falciparum EBA-181 protein. Malaria Journal, 2006. 5(1): p. 100.
73. Tham, W.-H., et al., Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proceedings of the National Academy of Sciences of the United States of America, 2010. 107(40): p. 17327-17332.
74. Stubbs, J., et al., Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science (New York, NY), 2005. 309(5739): p. 1384-1387.
75. Duraisingh, M.T., et al., Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proceedings of the National Academy of Sciences of the United States of America, 2003. 100(8): p. 4796-4801.
76. Cortés, A., et al., Epigenetic Silencing of Plasmodium falciparum Genes Linked to Erythrocyte Invasion. PLOS Pathogens, 2007. 3(8): p. e107.

170
77. Gaur, D., et al., Upregulation of expression of the reticulocyte homology gene 4 in the Plasmodium falciparum clone Dd2 is associated with a switch in the erythrocyte invasion pathway. Molecular and Biochemical Parasitology, 2006. 145(2): p. 205-215.
78. Persson, K.E.M., et al., Variation in use of erythrocyte invasion pathways by Plasmodium falciparum mediates evasion of human inhibitory antibodies. The Journal of Clinical Investigation, 2008. 118(1): p. 342-351.
79. Persson, K.E.M., et al., Erythrocyte-binding antigens of Plasmodium falciparum are targets of human inhibitory antibodies and function to evade naturally acquired immunity. Journal of immunology (Baltimore, Md. : 1950), 2013. 191(2): p. 785-794.
80. Gaur, D., D.C.G. Mayer, and L.H. Miller, Parasite ligand–host receptor interactions during invasion of erythrocytes by Plasmodium merozoites. International Journal for Parasitology, 2004. 34(13): p. 1413-1429.
81. Zuccala, E.S., et al., Subcompartmentalisation of Proteins in the Rhoptries Correlates with Ordered Events of Erythrocyte Invasion by the Blood Stage Malaria Parasite. PloS one, 2012. 7(9): p. e46160.
82. Absalon, S., et al., Calcium-Dependent Protein Kinase 5 Is Required for Release of Egress-Specific Organelles in Plasmodium falciparum. mBio, 2018. 9(1).
83. Singh, S., K.R. More, and C.E. Chitnis, Role of Calcineurin and Actin Dynamics in Regulated Secretion of Microneme Proteins in Plasmodium falciparum Merozoites during Erythrocyte Invasion. Cellular Microbiology, 2013.
84. Singh, S., et al., Distinct external signals trigger sequential release of apical organelles during erythrocyte invasion by malaria parasites. PLoS pathogens, 2010. 6(2): p. e1000746.
85. Gao, X., et al., Triggers of key calcium signals during erythrocyte invasion by Plasmodium falciparum. Nature communications, 2013. 4: p. 2862.
86. Baum, J., et al., Reticulocyte-binding protein homologue 5 – An essential adhesin involved in invasion of human erythrocytes by Plasmodium falciparum. International Journal for Parasitology, 2009. 39(3): p. 371-380.
87. Hayton, K., et al., Erythrocyte Binding Protein PfRH5 Polymorphisms Determine Species-Specific Pathways of Plasmodium falciparum Invasion. Cell host & microbe, 2008. 4(1): p. 40-51.
88. Volz, J.C., et al., Essential Role of the PfRh5/PfRipr/CyRPA Complex during Plasmodium falciparum Invasion of Erythrocytes. Cell Host Microbe, 2016. 20(1): p. 60-71.
89. Reddy, K.S., et al., Multiprotein complex between the GPI-anchored CyRPA with PfRH5 and PfRipr is crucial for Plasmodium falciparum erythrocyte invasion. Proceedings of the National Academy of Sciences of the United States of America, 2015. 112(4): p. 1179-1184.
90. Chen, L., et al., An EGF-like protein forms a complex with PfRh5 and is required for invasion of human erythrocytes by Plasmodium falciparum. PLoS pathogens, 2011. 7(9): p. e1002199.
91. Paul, A.S., et al., Parasite calcineurin regulates host cell recognition and attachment by apicomplexans. Cell host & microbe, 2015. 18(1): p. 49-60.
92. Philip, N. and Andrew P. Waters, Conditional Degradation of Plasmodium Calcineurin Reveals Functions in Parasite Colonization of both Host and Vector. Cell Host & Microbe, 2015. 18(1): p. 122-131.
93. Cao, J., et al., Rhoptry neck protein RON2 forms a complex with microneme protein AMA1 in Plasmodium falciparum merozoites. Parasitology international, 2009. 58(1): p. 29-35.

171
94. Besteiro, S., et al., Export of a Toxoplasma gondii rhoptry neck protein complex at the host cell membrane to form the moving junction during invasion. PLoS pathogens, 2009. 5(2): p. e1000309.
95. Tonkin, M.L., et al., Host cell invasion by apicomplexan parasites: insights from the co-structure of AMA1 with a RON2 peptide. Science (New York, NY), 2011. 333(6041): p. 463-467.
96. Besteiro, S., J.F. Dubremetz, and M. Lebrun, The moving junction of apicomplexan parasites: a key structure for invasion. Cellular Microbiology, 2011. 13(6): p. 797-805.
97. Miller, L.H.e.a., Interaction between cytochalasin B-treated malarial parasites and erythrocytes. Attachment and junction formation. The Journal of Experimental Medicine, 1979. 149(1): p. 172-184.
98. Weiss, G.E., B.S. Crabb, and P.R. Gilson, Overlaying Molecular and Temporal Aspects of Malaria Parasite Invasion. Trends Parasitol, 2016. 32(4): p. 284-95.
99. Leykauf, K., et al., Protein kinase a dependent phosphorylation of apical membrane antigen 1 plays an important role in erythrocyte invasion by the malaria parasite. PLoS pathogens, 2010. 6(6): p. e1000941.
100. Treeck, M., et al., Functional analysis of the leading malaria vaccine candidate AMA-1 reveals an essential role for the cytoplasmic domain in the invasion process. PLoS pathogens, 2009. 5(3): p. e1000322.
101. CDC, Malaria diagnosis. Available from: https://www.cdc.gov/malaria/diagnosis_treatment/diagnosis.html. 2015.
102. Landier, J., et al., The role of early detection and treatment in malaria elimination. Malaria Journal, 2016. 15: p. 363.
103. CDC, Malaria treatment. Available from:. 2015.
104. Tilley, L., et al., Artemisinin Action and Resistance in Plasmodium falciparum. Trends Parasitol, 2016. 32(9): p. 682-96.
105. Bustamante, L.Y., et al., Synergistic malaria vaccine combinations identified by systematic antigen screening. Proceedings of the National Academy of Sciences of the United States of America, 2017. 114(45): p. 12045-12050.
106. Coelho, C.H., et al., Advances in malaria vaccine development: report from the 2017 malaria vaccine symposium. npj Vaccines, 2017. 2(1): p. 34.
107. Epstein, J.E. and T.L. Richie, The whole parasite, pre-erythrocytic stage approach to malaria vaccine development: a review. Current opinion in infectious diseases, 2013. 26(5): p. 420-428.
108. Satchwell, T.J., Erythrocyte invasion receptors for Plasmodium falciparum: new and old. Transfusion Medicine, 2016. 26(2): p. 77-88.
109. Douglas, A.D., et al., The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nature communications, 2011. 2: p. 601.
110. Douglas, Alexander D., et al., A PfRH5-Based Vaccine Is Efficacious against Heterologous Strain Blood-Stage <em>Plasmodium falciparum</em> Infection in <em>Aotus</em> Monkeys. Cell Host & Microbe. 17(1): p. 130-139.
111. Wright, K.E., et al., Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature, 2014: p. 1-16.

172
112. Hjerrild, K.A., et al., Production of full-length soluble Plasmodium falciparum RH5 protein vaccine using a Drosophila melanogaster Schneider 2 stable cell line system. Scientific Reports, 2016. 6: p. 30357.
113. Srinivasan, P., et al., A malaria vaccine protects Aotus monkeys against virulent Plasmodium falciparum infection. npj Vaccines, 2017. 2(1): p. 14.
114. Talaat, K.R., et al., Safety and Immunogenicity of Pfs25-EPA/Alhydrogel®, a Transmission Blocking Vaccine against Plasmodium falciparum: An Open Label Study in Malaria Naïve Adults. PLOS ONE, 2016. 11(10): p. e0163144.
115. MacDonald, N.J., et al., Structural and Immunological Characterization of Recombinant 6-Cysteine Domains of the Plasmodium falciparum Sexual Stage Protein Pfs230. The Journal of Biological Chemistry, 2016. 291(38): p. 19913-19922.
116. Dondorp, A.M., et al., Artemisinin Resistance in Plasmodium falciparum Malaria. The New England journal of medicine, 2009. 361(5): p. 455-467.
117. White, N.J., Artemisinin resistance—the clock is ticking. The Lancet. 376(9758): p. 2051-2052.
118. Bloland, P.B., Drug Resistance in Malaria. World Health Organization., 2001.
119. Ouji, M., et al., Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite, 2018. 25: p. 24.
120. Dondorp, A.M., et al., Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet, 2010. 376(9753): p. 1647-1657.
121. Miotto, O., et al., Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet, 2015. 47(3): p. 226-34.
122. Ariey, F., et al., A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature, 2014. 505(7481): p. 50-55.
123. Fong, K.Y. and D.W. Wright, Hemozoin and antimalarial drug discovery. Future medicinal chemistry, 2013. 5(12): p. 1437-1450.
124. Kronenberger, T., I. Schettert, and C. Wrenger, Targeting the vitamin biosynthesis pathways for the treatment of malaria. Future Medicinal Chemistry, 2013. 5(7): p. 769-779.
125. Olszewski, K.L., et al., Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature, 2010. 466: p. 774.
126. van Dooren, G.G., L.M. Stimmler, and G.I. McFadden, Metabolic maps and functions of the Plasmodium mitochondrion. FEMS Microbiology Reviews, 2006. 30(4): p. 596-630.
127. Tarr, S.J., R.E.R. Nisbet, and C.J. Howe, Transcript Level Responses of Plasmodium falciparum to Antimycin A. Protist, 2012. 163(5): p. 755-766.
128. Qidwai, T., et al., Exploring Drug Targets in Isoprenoid Biosynthetic Pathway for Plasmodium falciparum. Biochemistry Research International, 2014. 2014: p. 12.
129. Walczak, M., et al., ATG8 Is Essential Specifically for an Autophagy-Independent Function in Apicoplast Biogenesis in Blood-Stage Malaria Parasites. mBio, 2018. 9(1): p. e02021-17.
130. Yeh, E. and J.L. Derisi, Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum. PLoS biology, 2011. 9(8): p. e1001138.

173
131. Boddey, J.A., et al., An aspartyl protease directs malaria effector proteins to the host cell. Nature, 2010. 463(7281): p. 627-631.
132. Russo, I., et al., Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature, 2010. 463(7281): p. 632-636.
133. Gilson, P.R., et al., Host cell remodelling in malaria parasites: a new pool of potential drug targets. International Journal for Parasitology, 2017. 47(2): p. 119-127.
134. Nasamu, A.S., et al., Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science, 2017. 358(6362): p. 518.
135. Li, F., et al., Plasmodium falciparum ookinete expression of plasmepsin VII and plasmepsin X. Malaria Journal, 2016. 15(1): p. 111.
136. Bullard, K.M., R.K. DeLisle, and S.M. Keenan, Malarial Kinases: Novel Targets for In Silico Approaches to Drug Discovery, in In Silico Models for Drug Discovery, S. Kortagere, Editor. 2013, Humana Press: Totowa, NJ. p. 205-229.
137. Veronica, M.Z., C. Marina, and C.W. Norman, Targeting Protein Kinases in the Malaria Parasite: Update of an Antimalarial Drug Target. Current Topics in Medicinal Chemistry, 2012. 12(5): p. 456-472.
138. Hallyburton, I., et al., Screening a protein kinase inhibitor library against Plasmodium falciparum. Malaria Journal, 2017. 16: p. 446.
139. Brown James, R., et al., Kinase Inhibitors Among Hits from Malaria Cellular Screens.
140. Anamika, n., N. Srinivasan, and A. Krupa, A genomic perspective of protein kinases in Plasmodium falciparum. Proteins: Structure, Function, and Bioinformatics, 2004. 58(1): p. 180-189.
141. Wilkes, J.M. and C. Doerig, The protein-phosphatome of the human malaria parasite Plasmodium falciparum. BMC Genomics, 2008. 9(1): p. 412.
142. Carvalho, T., C. Doerig, and L. Reininger, Nima- and Aurora-related kinases of malaria parasites. Biochimica et biophysica acta, 2013.
143. Dorin‐Semblat, D., et al., Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Molecular Microbiology, 2007. 65(5): p. 1170-1180.
144. Haste, N.M., et al., Exploring the Plasmodium falciparum cyclic-adenosine monophosphate (cAMP)-dependent protein kinase (PfPKA) as a therapeutic target. Microbes and infection / Institut Pasteur, 2012. 14(10): p. 838-850.
145. McNamara, C.W., et al., Targeting Plasmodium PI(4)K to eliminate malaria. Nature, 2013.
146. Dembele, L., et al., The Plasmodium PI(4)K inhibitor KDU691 selectively inhibits dihydroartemisinin-pretreated Plasmodium falciparum ring-stage parasites. Scientific Reports, 2017. 7(1): p. 2325.
147. Luth, M.R., et al., Using in Vitro Evolution and Whole Genome Analysis To Discover Next Generation Targets for Antimalarial Drug Discovery. ACS Infectious Diseases, 2018. 4(3): p. 301-314.
148. Dechering, K.J., et al., Modelling mosquito infection at natural parasite densities identifies drugs targeting EF2, PI4K or ATP4 as key candidates for interrupting malaria transmission. Scientific Reports, 2017. 7: p. 17680.
149. Zeeman, A.-M., et al., PI4 Kinase Is a Prophylactic but Not Radical Curative Target in Plasmodium vivax-Type Malaria Parasites. Antimicrobial Agents and Chemotherapy, 2016. 60(5): p. 2858-2863.

174
150. Collins, C.R., et al., Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Molecular microbiology, 2013.
151. Yap, A., et al., Conditional expression of apical membrane antigen 1 in Plasmodium falciparumshows it is required for erythrocyte invasion by merozoites. Cellular Microbiology, 2014. 16(5): p. 642-656.
152. Birnbaum, J., et al., A genetic system to study Plasmodium falciparum protein function. Nature Methods, 2017. 14: p. 450.
153. de Koning-Ward, T.F., P.R. Gilson, and B.S. Crabb, Advances in molecular genetic systems in malaria. Nature Reviews Microbiology, 2015. 13: p. 373.
154. Kutateladze, T.G., Molecular Analysis of Protein–Phosphoinositide Interactions. Current topics in microbiology and immunology, 2012. 362: p. 111-126.
155. Kutateladze, T.G., Translation of the phosphoinositide code by PI effectors. Nature chemical biology, 2010. 6(7): p. 507-513.
156. Balla, T., Phosphoinositides: Tiny Lipids With Giant Impact on Cell Regulation. Physiological reviews, 2013. 93(3): p. 1019-1137.
157. Rusten, T.E. and H. Stenmark, Analyzing phosphoinositides and their interacting proteins. Nature Methods, 2006. 3: p. 251.
158. Di Paolo, G. and P. De Camilli, Phosphoinositides in cell regulation and membrane dynamics. Nature, 2006. 443(7112): p. 651-657.
159. Elabbadi, N., M.L. Ancelin, and H.J. Vial, Characterization of phosphatidylinositol synthase and evidence of a polyphosphoinositide cycle in Plasmodium-infected erythrocytes. Molecular and biochemical parasitology, 1994. 63(2): p. 179-192.
160. Tawk, L., et al., Phosphatidylinositol 3-phosphate, an essential lipid in Plasmodium, localizes to the food vacuole membrane and the apicoplast. Eukaryotic cell, 2010. 9(10): p. 1519-1530.
161. Brochet, M., et al., Phosphoinositide Metabolism Links cGMP-Dependent Protein Kinase G to Essential Ca2+ Signals at Key Decision Points in the Life Cycle of Malaria Parasites. PLoS biology, 2014. 12(3): p. e1001806.
162. Tawk, L., et al., Phosphatidylinositol 3-monophosphate is involved in toxoplasma apicoplast biogenesis. PLoS pathogens, 2011. 7(2): p. e1001286.
163. Wengelnik, K., W. Daher, and M. Lebrun, Phosphoinositides and their functions in apicomplexan parasites. International Journal for Parasitology, 2018. 48(7): p. 493-504.
164. Harlan, J.E., et al., Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature, 1994. 371: p. 168.
165. Moravcevic, K., C.L. Oxley, and M.A. Lemmon, Conditional peripheral membrane proteins: facing up to limited specificity. Structure, 2012. 20(1): p. 15-27.
166. Kutateladze, T.G., Translation of the phosphoinositide code by PI effectors. Nat Chem Biol. 6(7): p. 507-13.
167. Lietha, Phosphoinositides – The Seven Species: Conversion and Cellular Roles. Encyclopedia in Life Sciences, 2011. 108(3): p. 698-705

175
168. Lemmon, M.A. and K.M. Ferguson, Signal-dependent membrane targeting by pleckstrin homology (PH) domains. The Biochemical journal, 2000. 350 Pt 1: p. 1-18.
169. DiNitto, J.P. and D.G. Lambright, Membrane and juxtamembrane targeting by PH and PTB domains. Biochim Biophys Acta, 2006. 1761(8): p. 850-67.
170. Lemmon, M.A. and K.M. Ferguson, Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J, 2000. 350 Pt 1: p. 1-18.
171. Gillooly, D.J., et al., Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J, 2000. 19(17): p. 4577-88.
172. Simonsen, A., et al., The role of phosphoinositides in membrane transport. Curr Opin Cell Biol, 2001. 13(4): p. 485-92.
173. Gillooly, D.J., et al., Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. The EMBO journal, 2000. 19(17): p. 4577-4588.
174. Seet, L.-F. and W. Hong, The Phox (PX) domain proteins and membrane traffic. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 2006. 1761(8): p. 878-896.
175. Balla, T. and P. Várnai, Visualization of cellular phosphoinositide pools with GFP-fused protein-domains. Current protocols in cell biology / editorial board, Juan S. Bonifacino ... [et al.], 2009. Chapter 24: p. Unit 24.4.
176. Antonsson, B.E., Purification and characterization of phosphatidylinositol synthase from human placenta. Biochemical Journal, 1994. 297(Pt 3): p. 517-522.
177. Jùn-ichi, N. and Y. Satoshi, Molecular cloning of the gene encoding CDPdiacylglycerol–inositol 3-phosphatidyl transferase in Saccharomyces cerevisiae. European Journal of Biochemistry, 1984. 143(2): p. 251-256.
178. Laporte, J., et al., Myotubularins, a large disease-associated family of cooperating catalytically active and inactive phosphoinositides phosphatases. Human Molecular Genetics, 2003. 12(suppl_2): p. R285-R292.
179. Liu, Y., et al., Functional studies of the mammalian Sac1 phosphoinositide phosphatase. Advances in enzyme regulation, 2009. 49(1): p. 75-86.
180. Auger, K.R., et al., Phosphatidylinositol 3-kinase and its novel product, phosphatidylinositol 3-phosphate, are present in Saccharomyces cerevisiae. J Biol Chem, 1989. 264(34): p. 20181-4.
181. Payrastre, B., et al., Phosphoinositides : key players in cell signalling, in time and space. Cellular Signalling, 2001. 13(6): p. 377-387.
182. De Craene, J.-O., et al., Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. International Journal of Molecular Sciences, 2017. 18(3): p. 634.
183. Henne, William M., Nicholas J. Buchkovich, and Scott D. Emr, The ESCRT Pathway. Developmental Cell, 2011. 21(1): p. 77-91.
184. Obara, K. and Y. Ohsumi, PtdIns 3-Kinase Orchestrates Autophagosome Formation in Yeast. Journal of Lipids, 2011. 2011: p. 498768.
185. Hamasaki, M., et al., Autophagosomes form at ER–mitochondria contact sites. Nature, 2013. 495: p. 389.
186. Axe, E.L., et al., Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. The Journal of Cell Biology, 2008. 182(4): p. 685-701.

176
187. Proikas-Cezanne, T., et al., WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci, 2015. 128(2): p. 207-17.
188. Polson, H.E.J., et al., Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy, 2010. 6(4): p. 506-522.
189. Backer, Jonathan M., The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochemical Journal, 2016. 473(15): p. 2251.
190. Daher, W., et al., Lipid kinases are essential for apicoplast homeostasis in Toxoplasma gondii. Cellular Microbiology, 2014: p. n/a-n/a.
191. Kitamura, K., et al., Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLoS One, 2012. 7(8): p. e42977.
192. Eickel, N., et al., Features of autophagic cell death in Plasmodium liver-stage parasites. Autophagy, 2013. 9(4): p. 568-580.
193. Bhattacharjee, S., et al., Endoplasmic reticulum PI(3)P lipid binding targets malaria proteins to the host cell. Cell, 2012. 148(1-2): p. 201-212.
194. Boddey, J.A., et al., Export of malaria proteins requires co-translational processing of the PEXEL motif independent of phosphatidylinositol-3-phosphate binding. Nat Commun, 2016. 7: p. 10470.
195. Mbengue, A., et al., A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature, 2015. 520(7549): p. 683-7.
196. McIntosh, M.T., et al., Traffic to the malaria parasite food vacuole: a novel pathway involving a phosphatidylinositol 3-phosphate-binding protein. The Journal of biological chemistry, 2007. 282(15): p. 11499-11508.
197. Bansal, P., et al., Autophagy-Related Protein ATG18 Regulates Apicoplast Biogenesis in Apicomplexan Parasites. MBio, 2017. 8(5).
198. De Matteis, M.A., D. Corda, and A. Luini, The Golgi complex. FEBS letters, 2009. 583(23): p. 3731.
199. Audhya, A., M. Foti, and S.D. Emr, Distinct roles for the yeast phosphatidylinositol 4-kinases, Stt4p and Pik1p, in secretion, cell growth, and organelle membrane dynamics. Mol Biol Cell, 2000. 11(8): p. 2673-89.
200. D'Angelo, G., et al., The multiple roles of PtdIns(4)P -- not just the precursor of PtdIns(4,5)P2. Journal of cell science, 2008. 121(Pt 12): p. 1955-1963.
201. Mizuno-Yamasaki, E., et al., Phosphatidylinositol 4-phosphate controls both membrane recruitment and a regulatory switch of the Rab GEF, Sec2p. Developmental cell, 2010. 18(5): p. 828-840.
202. Lemmon Mark, A., Phosphoinositide Recognition Domains. Traffic, 2003. 4(4): p. 201-213.
203. Levine, T.P. and S. Munro, Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr Biol, 2002. 12(9): p. 695-704.
204. Mesmin, B., et al., A Four-Step Cycle Driven by PI(4)P Hydrolysis Directs Sterol/PI(4)P Exchange by the ER-Golgi Tether OSBP. Cell, 2013. 155(4): p. 830-843.
205. Henmi, Y., et al., PtdIns4KIIα generates endosomal PtdIns(4)P and is required for receptor sorting at early endosomes. Molecular Biology of the Cell, 2016. 27(6): p. 990-1001.
206. Lenoir, M., et al., Structural Basis of Dynamic Membrane Recognition by trans-Golgi Network Specific FAPP Proteins. Journal of Molecular Biology, 2015. 427(4): p. 966-981.

177
207. Roy, A. and T.P. Levine, Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. The Journal of biological chemistry, 2004. 279(43): p. 44683-44689.
208. Sarkes, D. and L.E. Rameh, A novel HPLC-based approach makes possible the spatial characterization of cellular PtdIns5 Pand other phosphoinositides. The Biochemical journal, 2010. 428(3): p. 375-384.
209. Sbrissa, D., O.C. Ikonomov, and A. Shisheva, PIKfyve, a Mammalian Ortholog of Yeast Fab1p Lipid Kinase, Synthesizes 5-Phosphoinositides: EFFECT OF INSULIN. Journal of Biological Chemistry, 1999. 274(31): p. 21589-21597.
210. Tronchère, H., et al., Production of Phosphatidylinositol 5-Phosphate by the Phosphoinositide 3-Phosphatase Myotubularin in Mammalian Cells. Journal of Biological Chemistry, 2004. 279(8): p. 7304-7312.
211. Zolov, S.N., et al., In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A, 2012. 109(43): p. 17472-7.
212. Bulley, S.J., et al., Exploring phosphatidylinositol 5-phosphate 4-kinase function. Advances in Biological Regulation, 2015. 57: p. 193-202.
213. Guittard, G., et al., Cutting edge: Dok-1 and Dok-2 adaptor molecules are regulated by phosphatidylinositol 5-phosphate production in T cells. The Journal of Immunology, 2009. 182(7): p. 3974-3978.
214. Viaud, J., et al., Phosphatidylinositol 5-phosphate regulates invasion through binding and activation of Tiam1. Nature communications, 2014. 5: p. 4080.
215. Boal, F., et al., TOM1 is a PI5P effector involved in the regulation of endosomal maturation. Journal of Cell Science, 2015. 128(4): p. 815.
216. Pendaries, C., et al., PtdIns(5)P activates the host cell PI3‐kinase/Akt pathway during <em>Shigella flexneri</em> infection. The EMBO Journal, 2006. 25(5): p. 1024.
217. Niebuhr, K., et al., Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. EMBO J, 2002. 21(19): p. 5069-78.
218. Vicinanza, M., et al., PI(5)P Regulates Autophagosome Biogenesis. Molecular Cell, 2015. 57(2): p. 219-234.
219. Mattila, P.K., et al., Missing-in-metastasis and IRSp53 deform PI(4,5)P(2)-rich membranes by an inverse BAR domain–like mechanism. The Journal of Cell Biology, 2007. 176(7): p. 953-964.
220. Kolay, S., U. Basu, and P. Raghu, Control of diverse subcellular processes by a single multi-functional lipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)<em>P</em><sub>2</sub>]. Biochemical Journal, 2016. 473(12): p. 1681.
221. Martin, T.F.J., Role of PI(4,5)P(2) in Vesicle Exocytosis and Membrane Fusion. Sub-cellular biochemistry, 2012. 59: p. 111-130.
222. Desrivières, S., et al., MSS4, a Phosphatidylinositol-4-phosphate 5-Kinase Required for Organization of the Actin Cytoskeleton in Saccharomyces cerevisiae. Journal of Biological Chemistry, 1998. 273(25): p. 15787-15793.
223. De Craene, J.-O., et al., Evolutionary analysis of the ENTH/ANTH/VHS protein superfamily reveals a coevolution between membrane trafficking and metabolism. BMC Genomics, 2012. 13: p. 297-297.

178
224. Osborne, S.L., et al., Nuclear PtdIns(4,5)P<sub>2</sub> assembles in a mitotically regulated particle involved in pre-mRNA splicing. Journal of Cell Science, 2001. 114(13): p. 2501.
225. Li, W., R.S. Laishram, and R.A. Anderson, The Novel Poly(A) Polymerase Star-PAP is a Signal-Regulated Switch at the 3′-end of mRNAs. Advances in biological regulation, 2013. 53(1): p. 64-76.
226. Bushell, E., et al., Functional Profiling of a Plasmodium Genome Reveals an Abundance of Essential Genes. Cell, 2017. 170(2): p. 260-272.e8.
227. Carey, A.F., et al., Calcium dynamics of Plasmodium berghei sporozoite motility. Cellular Microbiology, 2014: p. n/a-n/a.
228. Brochet, M. and O. Billker, Calcium signalling in malaria parasites. Molecular Microbiology, 2016. 100(3): p. 397-408.
229. Agarwal, S., et al., Ca(2+) -mediated exocytosis of subtilisin-like protease 1: a key step in egress of Plasmodium falciparum merozoites. Cell Microbiol, 2013. 15(6): p. 910-21.
230. Edlich, C., et al., Structure and Phosphatidylinositol-(3,4)- Bisphosphate Binding of the C-Terminal PH Domain of Human Pleckstrin. Structure, 2005. 13(2): p. 277-286.
231. Carpten, J.D., et al., A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature, 2007. 448: p. 439.
232. Posor, Y., et al., Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature, 2013. 499(7457): p. 233-7.
233. Hers, I., E.E. Vincent, and J.M. Tavaré, Akt signalling in health and disease. Cellular Signalling, 2011. 23(10): p. 1515-1527.
234. Woolley, J.F., I. Dzneladze, and L. Salmena, Phosphoinositide signaling in cancer: INPP4B Akt(s) out. Trends in Molecular Medicine, 2015. 21(9): p. 530-532.
235. Dowler, S., et al., Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. The Biochemical journal, 2000. 351(Pt 1): p. 19-31.
236. Thomas, C.C., et al., Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain-containing protein 1 (TAPP1): molecular basis of lipid specificity. The Biochemical journal, 2001. 358(Pt 2): p. 287-294.
237. Milne, S.B., et al., A targeted mass spectrometric analysis of phosphatidylinositol phosphate species. Journal of lipid research, 2005. 46(8): p. 1796-1802.
238. Liu, Y. and V.A. Bankaitis, Phosphoinositide phosphatases in cell biology and disease. Progress in lipid research, 2010. 49(3): p. 201-217.
239. Hilpelä, P., M.K. Vartiainen, and P. Lappalainen, Regulation of the Actin Cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3, in Phosphoinositides in Subcellular Targeting and Enzyme Activation, H. Stenmark, Editor. 2004, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 117-163.
240. Vaid, A., et al., PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood, 2010. 115(12): p. 2500-2507.
241. Hassett, M.R., et al., Heterologous Expression, Purification, and Functional Analysis of Plasmodium falciparum Phosphatidylinositol 3'-Kinase. Biochemistry, 2017. 56(33): p. 4335-4345.
242. Dove, S.K., et al., Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature, 1997. 390: p. 187.
243. Takatori, S., et al., Phosphatidylinositol 3,5-Bisphosphate-Rich Membrane Domains in Endosomes and Lysosomes. Traffic, 2015. 17(2): p. 154-167.

179
244. Friant, S., et al., Ent3p Is a PtdIns(3,5)P<sub>2</sub> Effector Required for Protein Sorting to the Multivesicular Body. Developmental Cell, 2003. 5(3): p. 499-511.
245. Eugster, A., et al., Ent5p Is Required with Ent3p and Vps27p for Ubiquitin-dependent Protein Sorting into the Multivesicular Body. Molecular Biology of the Cell, 2004. 15(7): p. 3031-3041.
246. Chidambaram, S., J. Zimmermann, and G.F. von Mollard, ENTH domain proteins are cargo adaptors for multiple SNARE proteins at the TGN endosome. Journal of Cell Science, 2008. 121(3): p. 329.
247. Baskaran, S., et al., Two-Site Recognition of Phosphatidylinositol 3-Phosphate by PROPPINs in Autophagy. Molecular cell, 2012. 47(3): p. 339-348.
248. Dove, S.K., et al., Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. The EMBO Journal, 2004. 23(9): p. 1922-1933.
249. Shisheva, A., PIKfyve: PARTNERS, SIGNIFICANCE, DEBATES AND PARADOXES. Cell biology international, 2008. 32(6): p. 591-604.
250. Odorizzi, G., M. Babst, and S.D. Emr, Fab1p PtdIns(3)P 5-Kinase Function Essential for Protein Sorting in the Multivesicular Body. Cell, 1998. 95(6): p. 847-858.
251. Daher, W., et al., Identification of Toxoplasma TgPH1, a pleckstrin homology domain-containing protein that binds to the phosphoinositide PI(3,5)P2. Molecular and Biochemical Parasitology, 2016. 207(1): p. 39-44.
252. Vanhaesebroeck, B., et al., The emerging mechanisms of isoform-specific PI3K signalling. Nature Reviews Molecular Cell Biology, 2010. 11: p. 329.
253. Backer, J.M., The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J, 2008. 410(1): p. 1-17.
254. Falasca, M. and T. Maffucci, Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochemical Journal, 2012. 443(3): p. 587.
255. Lecompte, O., O. Poch, and J. Laporte, PtdIns5P regulation through evolution: roles in membrane trafficking? Trends in biochemical sciences, 2008. 33(10): p. 453-460.
256. Babst, M., et al., Endosome-Associated Complex, ESCRT-II, Recruits Transport Machinery for Protein Sorting at the Multivesicular Body. Developmental Cell, 2002. 3(2): p. 283-289.
257. Kihara, A., et al., Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol, 2001. 152(3): p. 519-30.
258. Stack, J.H. and S.D. Emr, Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J Biol Chem, 1994. 269(50): p. 31552-62.
259. Sasaki, T., et al., Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res, 2009. 48(6): p. 307-43.
260. Cheever, M.L., et al., Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nature Cell Biology, 2001. 3: p. 613.
261. Hain, A.U.P. and J. Bosch, Autophagy in Plasmodium, a multifunctional pathway? Vol. 8. 2013.
262. Bhattacharjee, S., et al., Remodeling of the malaria parasite and host human red cell by vesicle amplification that induces artemisinin resistance. Blood, 2018. 131(11): p. 1234-1247.
263. Voss, C., et al., Overexpression of Plasmodium berghei ATG8 by Liver Forms Leads to Cumulative Defects in Organelle Dynamics and to Generation of Noninfectious Merozoites. MBio, 2016. 7(3).

180
264. Latré de Laté, P., et al., Apicomplexan autophagy and modulation of autophagy in parasite-infected host cells. Biomedical Journal, 2017. 40(1): p. 23-30.
265. Nguyen, H.M., et al., Characterisation of two Toxoplasma PROPPINs homologous to Atg18/WIPI suggests they have evolved distinct specialised functions. PLoS ONE, 2018. 13(4): p. e0195921.
266. Wang, Y.J., et al., Phosphatidylinositol 4 Phosphate Regulates Targeting of Clathrin Adaptor AP-1 Complexes to the Golgi. Cell, 2003. 114(3): p. 299-310.
267. Craige, B., et al., Phosphatidylinositol-4-Kinase Type II Alpha Contains an AP-3–sorting Motif and a Kinase Domain That Are Both Required for Endosome Traffic. Molecular Biology of the Cell, 2008. 19(4): p. 1415-1426.
268. Jović, M., et al., Endosomal sorting of VAMP3 is regulated by PI4K2A. Journal of cell science, 2014: p. jcs.148809.
269. Pizarro‐Cerdá, J., et al., Type II phosphatidylinositol 4‐kinases promote Listeria monocytogenes entry into target cells. Cellular Microbiology, 2007. 9(10): p. 2381-2390.
270. Coppolino, M.G., et al., Inhibition of Phosphatidylinositol-4-phosphate 5-Kinase Iα Impairs Localized Actin Remodeling and Suppresses Phagocytosis. Journal of Biological Chemistry, 2002. 277(46): p. 43849-43857.
271. Shelton, S.N., et al., Saccharomyces cerevisiae contains a Type II phosphoinositide 4-kinase. Biochemical Journal, 2003. 371(2): p. 533.
272. Boura, E. and R. Nencka, Phosphatidylinositol 4-kinases: Function, structure, and inhibition. Experimental Cell Research, 2015. 337(2): p. 136-145.
273. Audhya, A. and S.D. Emr, Stt4 PI 4-Kinase Localizes to the Plasma Membrane and Functions in the Pkc1-Mediated MAP Kinase Cascade. Developmental Cell, 2002. 2(5): p. 593-605.
274. Strahl, T., et al., Yeast phosphatidylinositol 4-kinase, Pik1, has essential roles at the Golgi and in the nucleus. J Cell Biol, 2005. 171(6): p. 967-79.
275. Godi, A., et al., FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nature cell biology, 2004. 6(5): p. 393-404.
276. Godi, A., et al., ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol, 1999. 1(5): p. 280-7.
277. Gloor, Y., et al., Interaction between Sec7p and Pik1p: The first clue for the regulation of a coincidence detection signal. European Journal of Cell Biology, 2010. 89(8): p. 575-583.
278. Burke, J.E., et al., Structures of PI4KIIIβ complexes show simultaneous recruitment of Rab11 and its effectors. Science, 2014. 344(6187): p. 1035.
279. de Graaf, P., et al., Phosphatidylinositol 4-kinasebeta is critical for functional association of rab11 with the Golgi complex. Mol Biol Cell, 2004. 15(4): p. 2038-47.
280. Balla, T., Phosphatidylinositol 4-kinases. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, 1998. 1436(1): p. 69-85.
281. Balla, A., et al., Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Molecular Biology of the Cell, 2008. 19(2): p. 711-721.
282. Krüger, T., C.P. Sanchez, and M. Lanzer, Complementation of Saccharomyces cerevisiaepik1ts by a phosphatidylinositol 4-kinase from Plasmodium falciparum. Molecular and biochemical parasitology, 2010. 172(2): p. 149-151.

181
283. Dembele, L., et al., Imidazolopiperazines Kill both Rings and Dormant Rings in Wild-Type and K13 Artemisinin-Resistant Plasmodium falciparum In Vitro. Antimicrobial Agents and Chemotherapy, 2018. 62(5): p. e02235-17.
284. Herman, J.D., et al., The Cytoplasmic Prolyl-tRNA Synthetase of the Malaria Parasite is a Dual-Stage Target for Drug Development. Science translational medicine, 2015. 7(288): p. 288ra77-288ra77.
285. Ishihara, H., et al., Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J Biol Chem, 1998. 273(15): p. 8741-8.
286. Rameh, L.E., et al., A new pathway for synthesis of phosphatidylinositol-4,5-bisphosphate. Nature, 1997. 390(6656): p. 192-6.
287. Schill, Nicholas J. and Richard A. Anderson, Two novel phosphatidylinositol-4-phosphate 5-kinase type Iγ splice variants expressed in human cells display distinctive cellular targeting. Biochemical Journal, 2009. 422(Pt 3): p. 473-482.
288. Giudici, M.-L., et al., The intracellular localisation and mobility of Type Igamma phosphatidylinositol 4P 5-kinase splice variants.
289. Wang YJ, L.W., Wang J, Xu K, Dong P, Luo X, Yin HL. , Critical role of PIP5KIgamma 87 in InsP3-mediated Ca2signaling. J Cell Biol 2004. 167: p. 1005–1010.
290. Thieman, J.R., et al., Clathrin Regulates the Association of PIPKIγ661 with the AP-2 Adaptor β2 Appendage. Journal of Biological Chemistry, 2009. 284(20): p. 13924-13939.
291. El Sayegh, T.Y., et al., Phosphatidylinositol-4,5 Bisphosphate Produced by PIP5KIγ Regulates Gelsolin, Actin Assembly, and Adhesion Strength of N-Cadherin Junctions. Molecular Biology of the Cell, 2007. 18(8): p. 3026-3038.
292. Homma, K., et al., Phosphatidylinositol-4-phosphate 5-Kinase Localized on the Plasma Membrane Is Essential for Yeast Cell Morphogenesis. Journal of Biological Chemistry, 1998. 273(25): p. 15779-15786.
293. Clarke, J.H., P.C. Emson, and R.F. Irvine, Localization of phosphatidylinositol phosphate kinase IIγ in kidney to a membrane trafficking compartment within specialized cells of the nephron. American Journal of Physiology - Renal Physiology, 2008. 295(5): p. F1422-F1430.
294. Yoo, S.H., et al., Localization and projected role of phosphatidylinositol 4-kinases IIα and IIβ in inositol 1,4,5-trisphosphate-sensitive nucleoplasmic Ca(2+) store vesicles. Nucleus, 2014. 5(4): p. 341-351.
295. Rozenvayn, N. and R. Flaumenhaft, Protein kinase C Mediates Translocation of Type II Phosphatidylinositol 5-Phosphate 4-Kinase Required for Platelet α-Granule Secretion. Journal of Biological Chemistry, 2003. 278(10): p. 8126-8134.
296. Luoh, S.-W., N. Venkatesan, and R. Tripathi, Overexpression of the amplified Pip4k2β gene from 17q11–12 in breast cancer cells confers proliferation advantage. Oncogene, 2003. 23: p. 1354.
297. Leber, W., et al., A unique phosphatidylinositol 4-phosphate 5-kinase is activated by ADP-ribosylation factor in Plasmodium falciparum. International Journal for Parasitology, 2009. 39(6): p. 645-653.
298. Gary, J.D., et al., Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol, 1998. 143(1): p. 65-79.

182
299. Yamamoto, A., et al., Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Molecular Biology of the Cell, 1995. 6(5): p. 525-539.
300. Bonangelino, C.J., et al., Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol, 2002. 156(6): p. 1015-28.
301. Rutherford, A.C., et al., The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci, 2006. 119(Pt 19): p. 3944-57.
302. Ikonomov, O.C., et al., PIKfyve Controls Fluid Phase Endocytosis but Not Recycling/Degradation of Endocytosed Receptors or Sorting of Procathepsin D by Regulating Multivesicular Body Morphogenesis. Molecular Biology of the Cell, 2003. 14(11): p. 4581-4591.
303. Jin, N., et al., VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. The EMBO Journal, 2008. 27(24): p. 3221-3234.
304. Gary, J.D., et al., Regulation of Fab1 Phosphatidylinositol 3-Phosphate 5-Kinase Pathway by Vac7 Protein and Fig4, a Polyphosphoinositide Phosphatase Family Member. Molecular Biology of the Cell, 2002. 13(4): p. 1238-1251.
305. Rudge, S.A., D.M. Anderson, and S.D. Emr, Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell, 2004. 15(1): p. 24-36.
306. Botelho, R.J., et al., Assembly of a Fab1 Phosphoinositide Kinase Signaling Complex Requires the Fig4 Phosphoinositide Phosphatase. Molecular Biology of the Cell, 2008. 19(10): p. 4273-4286.
307. Dove, S.K., et al., Vac14 Controls PtdIns(3,5)<em>P</em><sub>2</sub> Synthesis and Fab1-Dependent Protein Trafficking to the Multivesicular Body. Current Biology, 2002. 12(11): p. 885-893.
308. Gil, A., A. Andrés-Pons, and R. Pulido, Nuclear PTEN: a tale of many tails. Cell Death And Differentiation, 2006. 14: p. 395.
309. Lin, H.-K., et al., Regulation of Androgen Receptor Signaling by PTEN (Phosphatase and Tensin Homolog Deleted on Chromosome 10) Tumor Suppressor through Distinct Mechanisms in Prostate Cancer Cells. Molecular Endocrinology, 2004. 18(10): p. 2409-2423.
310. Okumura, K., et al., Cellular transformation by the MSP58 oncogene is inhibited by its physical interaction with the PTEN tumor suppressor. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(8): p. 2703-2706.
311. Knobbe, C.B., et al., The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene, 2008. 27(41): p. 5398-415.
312. Song, M.S., L. Salmena, and P.P. Pandolfi, The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol, 2012. 13(5): p. 283-96.
313. Hnia, K., et al., Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends in Molecular Medicine, 2012. 18(6): p. 317-327.
314. Shin, H.W., et al., An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol, 2005. 170(4): p. 607-18.
315. Ivetac, I., et al., The Type Iα Inositol Polyphosphate 4-Phosphatase Generates and Terminates Phosphoinositide 3-Kinase Signals on Endosomes and the Plasma Membrane. Molecular Biology of the Cell, 2005. 16(5): p. 2218-2233.
316. Rynkiewicz, N.K., et al., INPP4A/INPP4B and P-Rex proteins: Related but different? Advances in Biological Regulation, 2012. 52(1): p. 265-279.

183
317. Ungewickell, A., et al., The identification and characterization of two phosphatidylinositol-4,5-bisphosphate 4-phosphatases. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(52): p. 18854-18859.
318. Zou, J., et al., Type I phosphatidylinositol-4,5-bisphosphate 4-phosphatase regulates stress-induced apoptosis. Proc Natl Acad Sci U S A, 2007. 104(43): p. 16834-9.
319. Gozani, O., et al., The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell, 2003. 114(1): p. 99-111.
320. Hsu, F. and Y. Mao, The structure of phosphoinositide phosphatases: Insights into substrate specificity and catalysis. Biochimica et biophysica acta, 2015. 1851(6): p. 698-710.
321. Majerus, P.W., M.V. Kisseleva, and F.A. Norris, The Role of Phosphatases in Inositol Signaling Reactions. Journal of Biological Chemistry, 1999. 274(16): p. 10669-10672.
322. Astle Megan, V., et al., Regulation of phosphoinositide signaling by the inositol polyphosphate 5‐phosphatases. IUBMB Life, 2008. 58(8): p. 451-456.
323. Whisstock, J.C., et al., The Structure and Function of Catalytic Domains Within Inositol Polyphosphate 5‐Phosphatases. IUBMB Life, 2008. 53(1): p. 15-23.
324. Hughes, W.E., F.T. Cooke, and P.J. Parker, Sac phosphatase domain proteins. Biochemical Journal, 2000. 350(Pt 2): p. 337-352.
325. Guo, S., et al., SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem, 1999. 274(19): p. 12990-5.
326. Rohde, H.M., et al., The human phosphatidylinositol phosphatase SAC1 interacts with the coatomer I complex. J Biol Chem, 2003. 278(52): p. 52689-99.
327. Blagoveshchenskaya, A., et al., Integration of Golgi trafficking and growth factor signaling by the lipid phosphatase SAC1. J Cell Biol, 2008. 180(4): p. 803-12.
328. Novick, P., B.C. Osmond, and D. Botstein, Suppressors of Yeast Actin Mutations. Genetics, 1989. 121(4): p. 659-674.
329. Hughes, W.E., et al., Mutations in the Saccharomyces cerevisiae gene SAC1 cause multiple drug sensitivity. Yeast, 1999. 15(11): p. 1111-1124.
330. Tahirovic, S., M. Schorr, and P. Mayinger, Regulation of Intracellular Phosphatidylinositol-4-Phosphate by the Sac1 Lipid Phosphatase. Traffic, 2004. 6(2): p. 116-130.
331. Schorr, M., et al., The phosphoinositide phosphatase Sac1p controls trafficking of the yeast Chs3p chitin synthase. Current Biology, 2001. 11(18): p. 1421-1426.
332. Kochendörfer, K.U., et al., Sac1p plays a crucial role in microsomal ATP transport, which is distinct from its function in Golgi phospholipid metabolism. The EMBO Journal, 1999. 18(6): p. 1506-1515.
333. Duex, J.E., et al., Phosphoinositide 5-Phosphatase Fig4p Is Required for both Acute Rise and Subsequent Fall in Stress-Induced Phosphatidylinositol 3,5-Bisphosphate Levels. Eukaryotic Cell, 2006. 5(4): p. 723-731.
334. E., G.E., et al., A secreted effector protein of Salmonella dublin is translocated into eukaryotic cells and mediates inflammation and fluid secretion in infected ileal mucosa. Molecular Microbiology, 1997. 25(5): p. 903-912.
335. Niebuhr, K., et al., Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S.flexneri effector IpgD reorganizes host cell morphology. The EMBO Journal, 2002. 21(19): p. 5069-5078.

184
336. Vergne, I., et al., Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(11): p. 4033-4038.
337. Toulabi, L., et al., Identification and Structural Characterization of a Legionella Phosphoinositide Phosphatase. The Journal of Biological Chemistry, 2013. 288(34): p. 24518-24527.
338. Hsu, F., et al., Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proceedings of the National Academy of Sciences of the United States of America, 2012. 109(34): p. 13567-13572.
339. Theriault, C. and D. Richard, Characterization of a putative Plasmodium falciparum SAC1 phosphoinositide-phosphatase homologue potentially required for survival during the asexual erythrocytic stages. Sci Rep, 2017. 7(1): p. 12710.
340. Sidik, S.M., et al., A Genome-Wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell, 2016. 166(6): p. 1423-1435.e12.
341. Wang, J., et al., Phosphorylation of GIT1 tyrosine-392 is required for PLCγ activation and podosome formation in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology, 2010. 30(10): p. 1976-1982.
342. Fukami, K., et al., Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Progress in Lipid Research, 2010. 49(4): p. 429-437.
343. Harden, T.K., et al., Mechanism of activation and inactivation of Gq/phospholipase C-beta signaling nodes. Chem Rev, 2011. 111(10): p. 6120-9.
344. Meldrum, E., P.J. Parker, and A. Carozzi, The PtdIns-PLC superfamily and signal transduction. Biochim Biophys Acta, 1991. 1092(1): p. 49-71.
345. Yoko-o, T., et al., The putative phosphoinositide-specific phospholipase C gene, PLC1, of the yeast Saccharomyces cerevisiae is important for cell growth. Proc Natl Acad Sci U S A, 1993. 90(5): p. 1804-8.
346. Flick, J.S. and J. Thorner, Genetic and biochemical characterization of a phosphatidylinositol-specific phospholipase C in Saccharomyces cerevisiae. Mol Cell Biol, 1993. 13(9): p. 5861-76.
347. Ferguson, K.M., et al., Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell, 1995. 83(6): p. 1037-1046.
348. Lemmon, M.A., et al., Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proceedings of the National Academy of Sciences of the United States of America, 1995. 92(23): p. 10472-10476.
349. Stauffer, T.P., S. Ahn, and T. Meyer, Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P<sub>2</sub> concentration monitored in living cells. Current Biology, 1998. 8(6): p. 343-346.
350. Ogwan'g, R., et al., Use of pharmacological agents to implicate a role for phosphoinositide hydrolysis products in malaria gamete formation. Biochem Pharmacol, 1993. 46(9): p. 1601-6.
351. Martin, S.K., M. Jett, and I. Schneider, Correlation of phosphoinositide hydrolysis with exflagellation in the malaria microgametocyte. The Journal of parasitology, 1994. 80(3): p. 371-378.
352. Raabe, A., et al., Genetic and transcriptional analysis of phosphoinositide-specific phospholipase C in Plasmodium. Experimental parasitology, 2011.

185
353. Raabe, A.C., et al., Multiple roles for Plasmodium berghei phosphoinositide-specific phospholipase C in regulating gametocyte activation and differentiation. Cellular Microbiology, 2011. 13(7): p. 955-966.
354. Agrawal, S., et al., An apicoplast localized ubiquitylation system is required for the import of nuclear-encoded plastid proteins. PLoS Pathog, 2013. 9(6): p. e1003426.
355. Fang, J., N. Marchesini, and Silvia N.J. Moreno, A <em>Toxoplasma gondii</em> phosphoinositide phospholipase C (<em>Tg</em>PI-PLC) with high affinity for phosphatidylinositol. Biochemical Journal, 2006. 394(2): p. 417.
356. Darvill, N., et al., Structural Basis of Phosphatidic Acid Sensing by APH in Apicomplexan Parasites. Structure, 2018.
357. Farrell, A., et al., A DOC2 Protein Identified by Mutational Profiling Is Essential for Apicomplexan Parasite Exocytosis. Science (New York, NY), 2012. 335(6065): p. 218-221.
358. Jean, S., et al., Plasmodium falciparum double C2 domain protein, PfDOC2, binds to calcium when associated with membranes. Experimental Parasitology, 2014. 144: p. 91-95.
359. Richard, D., et al., Identification of rhoptry trafficking determinants and evidence for a novel sorting mechanism in the malaria parasite Plasmodium falciparum. PLoS pathogens, 2009. 5(3): p. e1000328.
360. WHO, World Malaria Report. 2015.
361. Marti, M., et al., Signal-mediated export of proteins from the malaria parasite to the host erythrocyte. The Journal of cell biology, 2005. 171(4): p. 587-592.
362. Francia, M.E. and B. Striepen, Cell division in apicomplexan parasites. Nature reviews Microbiology, 2014.
363. Corvera, S., A. D'Arrigo, and H. Stenmark, Phosphoinositides in membrane traffic. Curr Opin Cell Biol, 1999. 11(4): p. 460-5.
364. Shewan, A., D.J. Eastburn, and K. Mostov, Phosphoinositides in Cell Architecture. Cold Spring Harbor perspectives in biology, 2011.
365. Vanhaesebroeck, B., et al., Synthesis and function of 3-phosphorylated inositol lipids. Annual review of biochemistry, 2001. 70(1): p. 535-602.
366. Simon, M.L.A., et al., A multi-colour/multi-affinity marker set to visualize phosphoinositide dynamics in Arabidopsis. The Plant Journal, 2013. 77(2): p. 322-337.
367. Simonsen, A., et al., EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature, 1998. 394(6692): p. 494-8.
368. Christoforidis, S., et al., The Rab5 effector EEA1 is a core component of endosome docking. Nature, 1999. 397(6720): p. 621-5.
369. Jean, S. and A.A. Kiger, Classes of phosphoinositide 3-kinases at a glance. Journal of cell science, 2014. 127(Pt 5): p. 923-928.
370. Petiot, A., et al., Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem, 2000. 275(2): p. 992-8.
371. De Matteis, M.A., A. Di Campli, and A. Godi, The role of the phosphoinositides at the Golgi complex. Biochim Biophys Acta, 2005. 1744(3): p. 396-405.
372. Hammond, G.R., G. Schiavo, and R.F. Irvine, Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem J, 2009. 422(1): p. 23-35.

186
373. Várnai, P. and T. Balla, Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochimica et biophysica acta, 2006. 1761(8): p. 957-967.
374. Balla, A., et al., A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell, 2005. 16(3): p. 1282-95.
375. Czech, M.P., Dynamics of phosphoinositides in membrane retrieval and insertion. Annual review of physiology, 2003. 65: p. 791-815.
376. Stefan, C.J., A. Audhya, and S.D. Emr, The yeast synaptojanin-like proteins control the cellular distribution of phosphatidylinositol (4,5)-bisphosphate. Mol Biol Cell, 2002. 13(2): p. 542-57.
377. Cantley, L.C., The phosphoinositide 3-kinase pathway. Science, 2002. 296(5573): p. 1655-7.
378. Katso, R., et al., Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol, 2001. 17: p. 615-75.
379. McLaughlin, S. and D. Murray, Plasma membrane phosphoinositide organization by protein electrostatics. Nature, 2005. 438(7068): p. 605-11.
380. Li, H. and A.J. Marshall, Phosphatidylinositol (3,4) bisphosphate-specific phosphatases and effector proteins: A distinct branch of PI3K signaling. Cell Signal, 2015. 27(9): p. 1789-98.
381. Jones, D.R., et al., Nuclear PtdIns5P as a transducer of stress signaling: an in vivo role for PIP4Kbeta. Mol Cell, 2006. 23(5): p. 685-95.
382. de Lartigue, J., et al., PIKfyve regulation of endosome-linked pathways. Traffic, 2009. 10(7): p. 883-93.
383. Zhang, Y., et al., Modulation of synaptic function by VAC14, a protein that regulates the phosphoinositides PI(3,5)P(2) and PI(5)P. EMBO J, 2012. 31(16): p. 3442-56.
384. van Gisbergen, P.A., et al., Class II formin targeting to the cell cortex by binding PI(3,5)P(2) is essential for polarized growth. J Cell Biol, 2012. 198(2): p. 235-50.
385. Vial, H.J., et al., Biosynthesis and dynamics of lipids in Plasmodium-infected mature mammalian erythrocytes. Blood Cells, 1990. 16(2-3): p. 531-55; discussion 556-61.
386. Brown, J.R. and K.R. Auger, Phylogenomics of phosphoinositide lipid kinases: perspectives on the evolution of second messenger signaling and drug discovery. BMC Evolutionary Biology, 2011. 11(1): p. 4.
387. Wengelnik, K. and H.J. Vial, Characterisation of the phosphatidylinositol synthase gene of Plasmodium species. Research in microbiology, 2007. 158(1): p. 51-59.
388. Bhattacharjee, S., et al., PI(3)P-independent and -dependent pathways function together in a vacuolar translocation sequence to target malarial proteins to the host erythrocyte. Molecular and biochemical parasitology, 2012. 185(2): p. 106-113.
389. Beraldo, F.H., K. Mikoshiba, and C.R. Garcia, Human malarial parasite, Plasmodium falciparum, displays capacitative calcium entry: 2-aminoethyl diphenylborinate blocks the signal transduction pathway of melatonin action on the P. falciparum cell cycle. J Pineal Res, 2007. 43(4): p. 360-4.
390. Hotta, C.T., et al., Calcium-dependent modulation by melatonin of the circadian rhythm in malarial parasites. Nat Cell Biol, 2000. 2(7): p. 466-8.
391. Vaid, A., D.C. Thomas, and P. Sharma, Role of Ca2+/calmodulin-PfPKB signaling pathway in erythrocyte invasion by Plasmodium falciparum. The Journal of biological chemistry, 2008. 283(9): p. 5589-5597.

187
392. Trager, W. and J.B. Jensen, Human malaria parasites in continuous culture. Science, 1976. 193(4254): p. 673-5.
393. Lambros, C. and J.P. Vanderberg, Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol, 1979. 65(3): p. 418-20.
394. Gilberger, T.-W., et al., The cytoplasmic domain of the Plasmodium falciparum ligand EBA-175 is essential for invasion but not protein trafficking. The Journal of cell biology, 2003. 162(2): p. 317-327.
395. Guittard, G., et al., Evidence for a positive role of PtdIns5P in T-cell signal transduction pathways. FEBS letters, 2010. 584(11): p. 2455-2460.
396. Prommana, P., et al., Inducible Knockdown of Plasmodium Gene Expression Using the glmS Ribozyme. PloS one, 2013. 8(8): p. e73783.
397. Adjalley, S.H., M.C.S. Lee, and D.A. Fidock, A method for rapid genetic integration into Plasmodium falciparum utilizing mycobacteriophage Bxb1 integrase. Methods in molecular biology (Clifton, N.J.), 2010. 634: p. 87-100.
398. Tonkin, C.J., et al., Localization of organellar proteins in Plasmodium falciparum using a novel set of transfection vectors and a new immunofluorescence fixation method. Molecular and biochemical parasitology, 2004. 137(1): p. 13-21.
399. Elmendorf, H.G. and K. Haldar, Identification and localization of ERD2 in the malaria parasite Plasmodium falciparum: separation from sites of sphingomyelin synthesis and implications for organization of the Golgi. The EMBO journal, 1993. 12(12): p. 4763-4773.
400. Schofield, L., et al., A rhoptry antigen of Plasmodium falciparum contains conserved and variable epitopes recognized by inhibitory monoclonal antibodies. Molecular and biochemical parasitology, 1986. 18(2): p. 183-195.
401. Richard, D., et al., Interaction between Plasmodium falciparum apical membrane antigen 1 and the rhoptry neck protein complex defines a key step in the erythrocyte invasion process of malaria parasites. The Journal of biological chemistry, 2010. 285(19): p. 14815-14822.
402. Healer, J., et al., Functional analysis of Plasmodium falciparum apical membrane antigen 1 utilizing interspecies domains. Infection and Immunity, 2005. 73(4): p. 2444-2451.
403. Su, X.Z. and T.E. Wellems, Sequence, transcript characterization and polymorphisms of a Plasmodium falciparum gene belonging to the heat-shock protein (HSP) 90 family. Gene, 1994. 151(1-2): p. 225-30.
404. Militello, K.T., et al., Identification of regulatory elements in the Plasmodium falciparum genome. Mol Biochem Parasitol, 2004. 134(1): p. 75-88.
405. Kanai, F., et al., The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nature cell biology, 2001. 3(7): p. 675-678.
406. Howe, R., et al., Isoprenoid biosynthesis inhibition disrupts Rab5 localization and food vacuolar integrity in Plasmodium falciparum. Eukaryotic cell, 2013. 12(2): p. 215-223.
407. Dalal, S. and M. Klemba, Amino acid efflux by asexual blood-stage Plasmodium falciparum and its utility in interrogating the kinetics of hemoglobin endocytosis and catabolism in vivo. Mol Biochem Parasitol, 2015. 201(2): p. 116-22.
408. Waller, R.F., et al., Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. The EMBO journal, 2000. 19(8): p. 1794-1802.

188
409. Nkrumah, L.J., et al., Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods, 2006. 3(8): p. 615-21.
410. Roy, A. and T.P. Levine, Multiple pools of phosphatidylinositol 4-phosphate detected using the pleckstrin homology domain of Osh2p. J Biol Chem, 2004. 279(43): p. 44683-9.
411. Brombacher, E., et al., Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J Biol Chem, 2009. 284(8): p. 4846-56.
412. Hammond, G.R., M.P. Machner, and T. Balla, A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol, 2014. 205(1): p. 113-26.
413. Bruns, J.R., et al., Multiple roles for phosphatidylinositol 4-kinase in biosynthetic transport in polarized Madin-Darby canine kidney cells. J Biol Chem, 2002. 277(3): p. 2012-8.
414. Godi, A., et al., FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat Cell Biol, 2004. 6(5): p. 393-404.
415. Brill, J.A., et al., A phospholipid kinase regulates actin organization and intercellular bridge formation during germline cytokinesis. Development, 2000. 127(17): p. 3855-64.
416. Zhang, Y., et al., Phosphatidylinositol 4-phosphate 5-kinase Its3 and calcineurin Ppb1 coordinately regulate cytokinesis in fission yeast. J Biol Chem, 2000. 275(45): p. 35600-6.
417. Hsu, V.W., N. Shah, and R.D. Klausner, A brefeldin A-like phenotype is induced by the overexpression of a human ERD-2-like protein, ELP-1. Cell, 1992. 69(4): p. 625-35.
418. Lewis, M.J. and H.R. Pelham, Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell, 1992. 68(2): p. 353-64.
419. Lewis, M.J. and H.R. Pelham, A human homologue of the yeast HDEL receptor. Nature, 1990. 348(6297): p. 162-3.
420. DiDonato, D. and D.L. Brasaemle, Fixation methods for the study of lipid droplets by immunofluorescence microscopy. J Histochem Cytochem, 2003. 51(6): p. 773-80.
421. Healer, J., et al., Independent translocation of two micronemal proteins in developing Plasmodium falciparum merozoites. Infection and Immunity, 2002. 70(10): p. 5751-5758.
422. Counihan, N.A., et al., Plasmodium rhoptry proteins: why order is important. Trends in parasitology, 2013.
423. Hammond, G.R.V., et al., PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science (New York, NY), 2012. 337(6095): p. 727-730.
424. Viaud, J., et al., Phosphoinositides: Important lipids in the coordination of cell dynamics. Biochimie, 2015.
425. Tan, J. and J.A. Brill, Cinderella story: PI4P goes from precursor to key signaling molecule. Critical reviews in biochemistry and molecular biology, 2013.
426. Heo, W.D., et al., PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science, 2006. 314(5804): p. 1458-61.
427. Yeung, T., et al., Membrane phosphatidylserine regulates surface charge and protein localization. Science, 2008. 319(5860): p. 210-3.
428. von Filseck, J.M., et al., Building lipid ‘PIPelines’ throughout the cell by ORP/Osh proteins. Biochemical Society transactions, 2014. 42(5): p. 1465-1470.

189
429. de Saint-Jean, M., et al., Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J Cell Biol, 2011. 195(6): p. 965-78.
430. Chung, J., et al., INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science, 2015. 349(6246): p. 428-32.
431. Olkkonen, V.M., OSBP-Related Protein Family in Lipid Transport Over Membrane Contact Sites. Lipid Insights, 2015. 8(Suppl 1): p. 1-9.
432. Pease, B.N., et al., Global Analysis of Protein Expression and Phosphorylation of Three Stages of <i>Plasmodium falciparum<i> Intraerythrocytic Development. Journal of Proteome Research, 2013.
433. Treeck, M., et al., The Phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii Reveal Unusual Adaptations Within and Beyond the Parasites' Boundaries. Cell Host and Microbe, 2011. 10(4): p. 410-419.
434. van Dooren, G.G., et al., Development of the endoplasmic reticulum, mitochondrion and apicoplast during the asexual life cycle of Plasmodium falciparum. Mol Microbiol, 2005. 57(2): p. 405-19.
435. Aikawa, M., The fine structure of the erythrocytic stages of three avian malarial parasites, Plasmodium fallax, P. lophurae, and P. cathemerium. Am J Trop Med Hyg, 1966. 15(4): p. 449-71.
436. Hopkins, J., et al., The plastid in Plasmodium falciparum asexual blood stages: a three-dimensional ultrastructural analysis. Protist, 1999. 150(3): p. 283-95.
437. Shisheva, A., PtdIns5P: news and views of its appearance, disappearance and deeds. Arch Biochem Biophys, 2013. 538(2): p. 171-80.
438. Gozani, O., et al., The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell, 2003. 114(1): p. 99-111.
439. Ramel, D., et al., Shigella flexneri infection generates the lipid PI5P to alter endocytosis and prevent termination of EGFR signaling. Science signaling, 2011. 4(191): p. ra61-ra61.
440. Struck, N.S., et al., Plasmodium falciparum possesses two GRASP proteins that are differentially targeted to the Golgi complex via a higher- and lower-eukaryote-like mechanism. Journal of cell science, 2008. 121(Pt 13): p. 2123-2129.
441. Sbrissa, D., et al., Functional dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P2 by means of the PIKfyve inhibitor YM201636. Am J Physiol Cell Physiol, 2012. 303(4): p. C436-46.
442. Ikonomov, O.C., et al., The phosphoinositide kinase PIKfyve is vital in early embryonic development: preimplantation lethality of PIKfyve-/- embryos but normality of PIKfyve+/- mice. J Biol Chem, 2011. 286(15): p. 13404-13.
443. Pandey, R., et al., Genome wide in silico analysis of Plasmodium falciparum phosphatome. BMC genomics, 2014. 15(1): p. 1024-22.
444. Gilson, P.R., et al., Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics, 2006. 5(7): p. 1286-99.
445. Hanssen, E., K.N. Goldie, and L. Tilley, Ultrastructure of the asexual blood stages of Plasmodium falciparum. Methods in cell biology, 2010. 96: p. 93-116.
446. Grüring, C., et al., Development and host cell modifications of Plasmodium falciparum blood stages in four dimensions. Nature communications, 2011. 2: p. 165.

190
447. Abu Bakar, N., et al., Digestive-vacuole genesis and endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum. J Cell Sci, 2010. 123(Pt 3): p. 441-50.
448. Aikawa, M., et al., The feeding mechanism of avian malarial parasites. J Cell Biol, 1966. 28(2): p. 355-73.
449. Alves, E., et al., Melatonin and IP3-induced Ca2+ release from intracellular stores in the malaria parasite Plasmodium falciparum within infected red blood cells. J Biol Chem, 2011. 286(7): p. 5905-12.
450. Kumar, P., et al., Regulation of Plasmodium falciparum development by calcium-dependent protein kinase 7 (PfCDPK7). Journal of Biological Chemistry, 2014. 289(29): p. 20386-20395.
451. Doughman, R.L., A.J. Firestone, and R.A. Anderson, Phosphatidylinositol phosphate kinases put PI4,5P(2) in its place. J Membr Biol, 2003. 194(2): p. 77-89.
452. Watt, S.A., et al., Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J, 2002. 363(Pt 3): p. 657-66.
453. Hallee, S., et al., Evidence that the Plasmodium falciparum Protein Sortilin Potentially Acts as an Escorter for the Trafficking of the Rhoptry-Associated Membrane Antigen to the Rhoptries. mSphere, 2018. 3(1).
454. Hallée, S., et al., The malaria parasite Plasmodium falciparum Sortilin is essential for merozoite formation and apical complex biogenesis. Cellular Microbiology, 2018. 20(8): p. e12844.
455. Hallee, S., et al., Repurposing of the conserved endolysosomal escorter Sortilin into a central player of the regulated secretory pathway of the malaria parasite Plasmodium falciparum. Cell Host and Microbe, 2018.
456. Thériault, C. and D. Richard, Characterization of a putative Plasmodium falciparum SAC1 phosphoinositide-phosphatase homologue potentially required for survival during the asexual erythrocytic stages. Scientific reports, 2017. 7(1): p. 12710-12710.
457. Polevoy, G., et al., Dual roles for the Drosophila PI 4-kinase four wheel drive in localizing Rab11 during cytokinesis. J Cell Biol, 2009. 187(6): p. 847-58.
458. Elliott, D.A., et al., Four distinct pathways of hemoglobin uptake in the malaria parasite Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America, 2008. 105(7): p. 2463-2468.
459. Jones, M.L., C. Cottingham, and J.C. Rayner, Effects of calcium signaling on Plasmodium falciparum erythrocyte invasion and post-translational modification of gliding-associated protein 45 (PfGAP45). Mol Biochem Parasitol, 2009. 168(1): p. 55-62.
460. Lemmon, M.A., Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp, 2007(74): p. 81-93.
461. Yu, J.W., et al., Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol Cell, 2004. 13(5): p. 677-88.
462. Bansal, A., et al., Characterization of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1) and its role in microneme secretion during erythrocyte invasion. J Biol Chem, 2013. 288(3): p. 1590-602.
463. Kumar, S., et al., PfCDPK1 mediated signaling in erythrocytic stages of Plasmodium falciparum. Nature Communications, 2017. 8: p. 63.


















