
Exploring the reactivity of S-cis-methylaziridine aldehyde · ii Exploring the reactivity of...
52
Exploring the reactivity of S-cis-methylaziridine aldehyde by Genevieve Canzonieri A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Genevieve Canzonieri, 2013
-
Upload
dangnguyet -
Category
Documents
-
view
216 -
download
0
Transcript of Exploring the reactivity of S-cis-methylaziridine aldehyde · ii Exploring the reactivity of...

Exploring the reactivity of S-cis-methylaziridine aldehyde
by
Genevieve Canzonieri
A thesis submitted in conformity with the requirements for the degree of Master of Science
Department of Chemistry University of Toronto
© Copyright by Genevieve Canzonieri, 2013

ii
Exploring the reactivity of S-cis-methylaziridine aldehyde
Genevieve Canzonieri
Master of Science
Department of Chemistry
University of Toronto
2013
Abstract
In 2006, an amphoteric molecule containing both an aldehyde and an unprotected amine was
reported in the Yudin group by Dr. Ryan Hili. The unprotected aziridine aldehyde exists as
homochiral dimers. Furthermore, due to the reversibility of the hemiacetal formation, the free
aldehyde is available to undergo a wide array of reactions including an Ugi multicomponent
reaction to give the final peptide macrocycle. Thus far, the mechanistic pathway involved in the
Ugi reaction between S-trans aziridine aldehyde dimer and L-amino acids in the presence of tert-
butyl isocyanide has given high diastereoselectivity whereas low diastereoselectivity is observed
if the aziridine dimer is of the opposite stereochemistry. Herein, preliminary results show that a
S-cis aziridine aldehyde with either a D or L-secondary amino acid gives high diastereoselectivity
showing that the reaction is under Felkin-Ahn control.

iii
Acknowledgments
I would like to thank Dr. Andrei Yudin for his support and guidance throughout my Master’s
degree. I would also like to thank Dr. Ronald Kluger for taking the time to read my thesis. All of
the members of Yudin group have been a pleasure to work with, particularly Dr. Conor Scully
for his thoughtful insights and discussions about chemistry. Also, much appreciation to Dr.
Jennifer Hickey and Mr. Serge Zaretsky for taking the time to help me troubleshoot some
experimental details and working with the HPLC. In addition, I’d like to thank former summer
student Spencer Ler for all of his help in showing me some of the chemistry when I first started.
I am grateful to many of the departmental staff such as Anna Liza Villavelez and Kevin Ahn for
their graduate assistance. I am also grateful for the financial support I received from the Leslie
Gladstone Cook Fellowship.
I would also like to acknowledge Dmitry Pichugin in the NMR facility for his continuous efforts
in helping me set up different NMR experiments as well as Dr. Timothy Burrow for his NMR
guidance.
I also owe thanks to friends I made in the department for many cheerful moments and including
me in some of their group outings. Finally, I would like to extend many thanks to my family for
their ongoing support and encouragement even during the toughest of times.

iv
Table of Contents
Acknowledgments .......................................................................................................................... iii
Table of Contents ........................................................................................................................... iv
List of Tables .................................................................................................................................. v
List of Figures ................................................................................................................................ vi
List of Schemes ............................................................................................................................. vii
List of Appendices ....................................................................................................................... viii
1 Introduction .......................................................................................................................... 1
2 Results and Discussion ......................................................................................................... 3
2.1 Secondary amino acid versus primary amino acid derived piperazinones ......................... 5
2.2 Reactivity of L-Proline terminated peptides with cis-S methyl aziridine aldehyde ........... 13
2.3 Is S- cis- methylaziridine dimer in equilibrium with its monomeric species? .................. 15
3 Conclusion ............................................................................................................................... 17
4 Experimental procedures ...................................................................................................... 17
4.1 Protocols for unprotected aziridine aldehydes ............................................................. 18
4.2 Protocols for piperazinones ............................................................................................ 21
5 References ................................................................................................................................ 24

v
List of Tables
Table 1.1 Summary of terminology of compounds……………………………………………….3
Table 1.2 Synthesis and NMR analysis of piperazinones………………………………….……12

vi
List of Figures
Figure 1.1 The Ugi four-component condensation (U-4CC)……………………………………..1
Figure 1.2 Piperazinone of relative stereochemistry……………………………………….….….6
Figure 1.3 S-primary amino acid versus proline derived iminium ion………………………..….7
Figure 1.4 S cis-methyl aziridine aldehyde and D-Pro…………………………...…………….…7
Figure 1.5 S cis-methyl aziridine aldehyde and L-Pro…………………………………………….8
Figure 1.6 Crude LC-MS spectrum of piperazinone derived from D- Pro and S -cis aziridine
aldehyde dimer………………………………………………………………………….…………9
Figure 1.7 Crude LC-MS spectrum of piperazinone derived from L- Pro and S -cis aziridine
aldehyde dimer……………………………………………………………………………….……9
Figure 1.8 Cyclization involving substrates with matched and mismatched α-stereocenters….10
Figure 1.9 S cis-methyl aziridine aldehyde and N-benzyl tyrosine………………………….….11
Figure 1.10 X-Ray crystal of piperazinone with phenylalanine with relative stereochemistry
S/R/S……………………………………………………………………………………………..12
Figure 1.11 Aziridine aldehyde and a secondary amine form the aminal that is in equilibrium
with its ring open form……………………………………………………………………….…..13
Figure 1.12 N- unprotected aziridine aldehyde monomeric species reported by Reinhoudt and
colleagues in 1983………………………………………………………………………………..16
Figure 1.13. gCOSY of cis methyl aziridine aldehyde………………………………………….16

vii
List of Schemes
Scheme 1.1 Aziridine aldehyde dimer/monomer equilibrium…………………………………....2
Scheme 1.2 The disrupted Ugi 4CC with unsubstituted aziridine dimer and L-Pro…………..….6

viii
List of Appendices
Appendix I: 1H,
13C and NOESY NMR……………………………………………………26

1
1 Introduction
The Ugi four-component condensation (U-4CC) was first reported in 1959 by Ivar Ugi. 1
In this reaction, an α-aminoacyl amide is made from an aldehyde, an amine, a carboxylic acid
and an isocyanide (see Figure below).
Figure 1.1 The Ugi four-component condensation (U-4CC)
Extensive use and application of the Ugi 4CC in the scientific community has been implemented
ever since2 for its ability in enabling the union of several functional groups into one product, a
pseudo-peptide. To add a transformative dimension using chemistry developed in the Yudin lab,
Dr. Ryan Hili showed the utility of the aziridine aldehyde dimer in a rerouted Ugi
condensation3f,4
, taking advantage of its bifunctional nature3, whereby the aziridine N-H acts as a
nucleophile and the aldehyde is the electrophile (see Scheme 1.1 below). The properties of
unprotected aziridine aldehydes have been thoroughly explored.5-8
Dimerization to the five
membered ring is thought to proceed through a hemiaminal intermediate 2 and subsequent
nucleophilic attack by the adjacent hydroxyl to the aldehyde functionality produces a stable
product 3. The kinetically formed 5-membered hetereocycle is also the product of
thermodynamic control6 in which a highly favourable hydrogen bond interaction (Scheme 1.1)
was detected by X-ray analysis.5

2
Scheme 1.1 Aziridine aldehyde dimer/monomer equilibrium
As previously reported,5 the computed dissociation process for the unsubstituted S-aziridine
aldehyde 3 to the monomeric species 1, in the gas phase, was calculated to be
Erel=+23.3kcal/mol, further supporting the high energetic barrier between the two.
In the rerouted Ugi 4CC,3f
condensation of the amine (from an amino acid) with the aldehyde
(from open dimer) forms the imine, followed by formation of the mixed anhydride and exocyclic
attack by the acyl aziridine generates the final cyclic product (refer to Scheme 1.2).
Thus far, the mechanistic pathway involved in the Ugi reaction between the S- trans aziridine
dimer with a L secondary amino acid in the presence of tert-butyl isocyanide has given high
diastereoselectivity whereas low diastereoselectivity is observed if the aziridine dimer is of the
opposite stereochemistry.9 Yet, what remains to be explored will be discussed herein with focus
on the reactivity of cis S-methyl aziridine aldehyde.

3
2 Results and Discussion
Exploration of the reactivity and stability of cis aziridine aldehyde in an Ugi 4CC will be
discussed herein. Previous work with aziridine aldehyde has been done using the trans
configuration and therefore no prior knowledge of its cis congener was addressed. For reference
of terminology used throughout this thesis, a summary and short description can be found in the
table below.
Table 1.1 Summary of terminology of compounds
Terminology Description Compound Structure
Unsubstituted aziridine
aldehyde
-
Methyl aziridine aldehyde Methyl group is cis from the
aldehyde
Hydroxy-methyl aziridine
aldehyde
Hydroxymethyl group is trans
from the aldehyde

4
NBn-Phe piperazinone -
NBn-Tyr(OtBu) piperazinone -
Peptide aminal -
Piperazinone derived from L-
Pro
Ring opened aziridine with
thiobenzoic acid

5
2.1 Secondary amino acid versus primary amino acid derived piperazinones
Dr. Ryan Hili from the Yudin group, previously described the utility of unprotected amino
aldehydes, in the form of homodimeric aziridine aldehydes,3 in an intercepted Ugi condensation.
In this reaction, the dimer was used as the substrate (i.e. aziridine NH is the nucleophile and
aldehyde is the electrophile). The success of the process relies upon the orthogonality between
the aldehyde and aziridine allowing for high chemoselectivity of iminium ion formation at the
amino acid N-terminus.5
Piperazinones can be prepared via an Ugi 4CC using the aziridine aldehyde in place of an
aldehyde and an amine. Subsequent α-addition of the isocyanide on 4 occurs exclusively at the
si-face, which is also sterically governed by the unreacted monomer, meanwhile, the second
equivalent of monomer quickly dimerizes with another monomer. A mixed anhydride 5
generated forming an active ester that is attacked by the exocyclic amine generating the final
acyl aziridine ring 6.4,5,9

6
Scheme 1.2 The disrupted Ugi 4CC with unsubstituted aziridine dimer and L-Pro
Previous seminal work by Kim10
and Dyker11
showed that primary amino acids in an Ugi
condensation lead to R-stereochemistry of the newly formed methine center 6b whereas imines
derived from a secondary amino acid such as proline provided the opposite stereochemistry. This
can be explained by facial preference of isocyanide attack exclusively at the si-face whereas 1, 3
allylic strain drives the attack on the re-face of the primary amino derived imine.5,9
Figure 1.2 Piperazinone of relative stereochemistry S/S/S in the order starting from a going
clockwise. The newly formed methine center is labeled b.

7
Figure 1.3 S-primary amino acid versus proline derived iminium ion
As part of a study aimed at exploring the stereoselectivity of S-cis aziridine aldehyde with
secondary amino acids (D and L Proline) in the Ugi 4CC, a variety of 6 membered rings or
piperazinones have been synthesized (refer to Table 1.2). The stereochemistry of the major
isolated diastereomer of the aziridine-containing product was determined using through-space
NMR experiments such as nuclear Overhauser effect (nOe) spectroscopy and comparing 3J
coupling between the exocyclic proton Hb and the adjacent aziridine proton. The nOe
interactions between the protons are shown in the figures below.
Figure 1.4 S cis-methyl aziridine aldehyde and D-Pro.

8
Figure 1.5 S cis-methyl aziridine aldehyde and L-Pro.
An interesting observation was made when cylization of S-cis aziridine aldehyde with D-Pro gave
one diastereomer 7 with relative stereochemistry S/R/R. The proton of the newly formed methine
center was anti to the adjacent aziridine proton with a 3J = 9 Hz which is consistent with a 180⁰
dihedral angle (see Table 2). This agrees with a 1, 2-diaxial relationship between the protons
referred to above. Furthermore, a nOe interaction is observed between the Ha and Hb protons
confirming a syn relationship (see Figure 1.4). However, this is not the case with R trans
aziridine aldehyde (substituted with iBu) and L-Pro. In fact, low stereoselectivity is observed with
both diastereomers isolated with the assigned relative stereochemistry of: (R/S/S) and (R/R/S).9
This low diastereoselectivity can be explained using a Felkin-Ahn model (see Figure 1.8). In a
mismatched case, the Felkin- Ahn model competes with the proline stereochemistry. In a
matched case, the preferred side of isocyanide addition is matched with the facial preference
inherent to proline (hydrogen is smaller than carboxylate).5,9
In comparison, when L-Pro underwent cyclization with S-cis aziridine aldehyde, one
diastereomer 8 was also isolated showing high diastereoselectivity with both enantiomers. The
relative stereochemistry of S/S/S was confirmed with a nOe interaction between Ha and Hb and
thus, a syn relationship was noted. Furthermore, a 3J= 7 Hz between the exocyclic proton Hb and

9
min1 2 3 4 5 6
0
200000
400000
600000
800000
1000000
1200000
1400000
MSD1 TIC, MS File (D:\GENEVIEVE\GC02-25B.D) ES-API, Pos, Scan, Frag: 50, "TIC"
min1 2 3 4 5 6
0
250000
500000
750000
1000000
1250000
1500000
1750000
2000000
MSD1 TIC, MS File (D:\GENEVIEVE\DEF_LC 2013-03-27 11-19-32\GC02-29-1HR.D) ES-API, Pos, Scan, Frag: 50, "TIC"
the adjacent aziridine proton is consistent with what would be expected with a 60⁰ dihedral angle
or an axial-equatorial relationship (see Figure 1.5).
Figure 1.6 Crude LC-MS spectrum of piperazinone derived from D- Pro and S -cis aziridine
aldehyde dimer (RT=retention time)
Figure 1.7 Crude LC-MS spectrum of piperazinone derived from L- Pro and S -cis aziridine
aldehyde dimer (RT=retention time)
Crude D-Pro with cis
dimer
[M+1]+=266.2
RT=3.117min
Crude L-Pro+ cis
dimer
[M+1]+= 266.2
RT=3.257min

10
Certainly, a given homochiral aziridine aldehyde dimer leads to different stereochemistry in
reactions with primary versus secondary amine-containing amino acids, yet is matched with the
same stereoisomer of the corresponding amino acid (see Figure 1.3). As alluded to earlier, there
is a difference in the way the corresponding iminium ions are activated in the open dimer
environment. In the case of proline, the aminal 9a, b is in equilibrium with the iminium ion 10a,
b respectively. As shown in Figure 1.8, α- addition of tert-butyl isocyanide is under Felkin-Ahn
control in the matched case.
Figure 1.8 Cyclization involving substrates with matched and mismatched α-stereocenters.
In the mismatched scenario, the Felkin-Ahn model competes with the proline control of
stereochemistry, leading to the observed low diastereoselectivity.

11
Furthermore, the N-substituted α-amino acids also significantly influence the degree of
diastereoselectivity during nucleophilic additions to the aldehyde group.5 Therefore, with larger
substituents on the nitrogen, facial control is governed by the Felkin-Ahn model, similar to the
observed reactivity ascribed to proline- derived imines. Nevertheless, our previous work was
done with S-trans-aziridine aldehyde.3-9
Assumptions aside, a piperazinone 11 derived from S
cis-methyl aziridine aldehyde and N-benzyl-tyrosine showed a syn relationship between the
exocyclic proton Hb and the α-proton of the amino acid Ha. This was supported by nOe
interactions as shown in Figure 1.9. Thus, the resulting stereochemistry is S/S/S similar to what
would be observed with a secondary amino acid. This finding was important in further
establishing our knowledge about the driving factors that influence the stereochemistry of the
newly formed methine center.
Figure 1.9 S cis-methyl aziridine aldehyde and N-benzyl tyrosine.
The crystal structure obtained by Dr. Ryan Hili of the piperazinone containing TBDMS dimer, L-
Phenylalanine and tert-butyl isocyanide showed an (R) stereocenter at the newly formed
exocyclic center (see Figure 1.10 below).

12
Figure 1.10 X-Ray crystal of piperazinone with phenylalanine with relative stereochemistry
S/R/S.
As established previously, observed reactivity between S-trans aziridine aldehyde and secondary
versus a primary amino acid lies in the difference in conformation of the imine that is formed.
For an L-primary amino acid, 1, 3-allylic strain would bias isocyanide attack from the top face
(re- face) while the si-face is the preferred side of attack in the case of an imine derived from a
secondary amino acid (see Figure 1.3).
Table 1.2 Synthesis and NMR analysis of piperazinones
Entry R Aziridine
Stereochemistry
/Configuration
Amino
acid
1H δ
(ppm)
1H J
(Hz)
Product
(diastereomer)
1 H S/trans L-Pro 3.65 6.1 S/S/S
2 Me S/cis L-Pro 3.50 7.0 S/S/S
3 Me S/cis D-Pro 3.05 9.1 S/R/R
4 Me S/cis NBn-
Tyr(OtBu)
3.89 8.4 S/S/S
5 CH2-OH S/trans NBn-Phe 3.59 7.0 S/S/S

13
Reaction conditions: aziridine aldehyde dimer (0.5eq), amino acid (1.0 eq), isocyanide (1.0 eq), TFE (0.1M), rt, 1h. R refers to the substituent on the aziridine 3-position, trans from the aldehyde. Aziridine stereochemistry refers to the α-position. Chemical shifts refer to the proton of the newly formed methine center.
In conclusion, the S cis-methyl aziridine aldehyde with L-Pro gives the cis diastereomer with
relative stereochemistry of S/S/S; where the α-proline proton has a syn relationship with the
newly formed exocyclic proton that is generated after attack of tert-butyl isocyanide. In contrast,
the piperazinone derived from D-Pro and S- cis-methyl aziridine aldehyde gives the trans
diastereomer with S/R/R stereochemistry which lies in agreement of a 1, 2-diaxial hydrogen
relationship. It was also found that a N-substituted amino acid behaved similarly with the S-cis
aziridine aldehyde as did the secondary amino acids with S-trans aziridine aldehyde.
2.2 Reactivity of L-Proline terminated peptides with cis-S methyl aziridine aldehyde
In recent studies, the aziridine aldehyde dimer dissociation was found to be the rate determining
step in reductive amination.6,7
Experiments that suggested this was the evidence of hemiaminal
formation 13 with pyrrolidine and aziridine aldehyde in trifluoroethanol (TFE) as shown below:
Figure 1.11 Aziridine aldehyde and a secondary amine form the aminal that is in equilibrium
with its ring open form.

14
As described by earlier works, 5-7
the role of TFE is important in the dissociation of the dimeric
aziridine aldehyde by facilitating its acyclic dimer formation via disruption of intramolecular
hydrogen bonding between the free aziridine N-H and the hemiaminal. As previously reported, 6
the chemoselectivity and stereoselectivity is mainly driven by the open dimer intermediate
whereby the aminal carbon and the NH aziridine are assembled through reversible covalent
bonds. The resulting aminal is not observed with primary amines but rather the imine is trapped
and formed in situ by reduction with cyanoborohydride.6
In addition, the stereoselectivity observed between two different α-stereocenters, where the
aziridine aldehyde is of the oppositive stereochemistry of the N-terminal peptide, occurs as a
mismatched case where the observed product is an aminal.5,9
Interestingly, there seems to be an
added factor not considered previously. In cases where a cis S-aziridine aldehyde and an S-
proline terminated peptide were reacted under Ugi conditions, there was no observable cyclic
peptide but rather the formation of a different product that prevented attack by the isocyanide.
Also, this product was observed by LC-MS to have formed even without the addition of
isocyanide and only the cis S-aziridine aldehyde. After its purification and isolation, it was found
to have formed the aminal. A similar finding was formerly alluded to as an “enthalpic sink” in
the mismatch seen with trans R-aziridine aldehyde and L-Pro terminated peptides.5
Needless to say, evidence of whether or not a matched case is seen with a D-Pro terminated
peptide has yet to be explored in order to fully assess the reactive species that is precluding
macrocylization from occurring with L-Pro at the N- termini.

15
2.3 Is S- cis- methylaziridine dimer in equilibrium with its monomeric species?
The formation of aminal with S-proline terminated peptides prompted a closer assessment of the
cis S-aziridine aldehyde. Why is there disfavourable reactivity with this dimer with peptides
when trans S-aziridine aldehyde yielded high selectivity for the formation of the macrocycle in
an Ugi 4CC? Furthermore, the formation of the aminal is a stable product which suggests a by-
product of the aziridine aldehyde is formed in situ, disallowing an Ugi 4CC to take place. More
interestingly, this “by-product” was shown to be in equilibrium with the dimeric species after
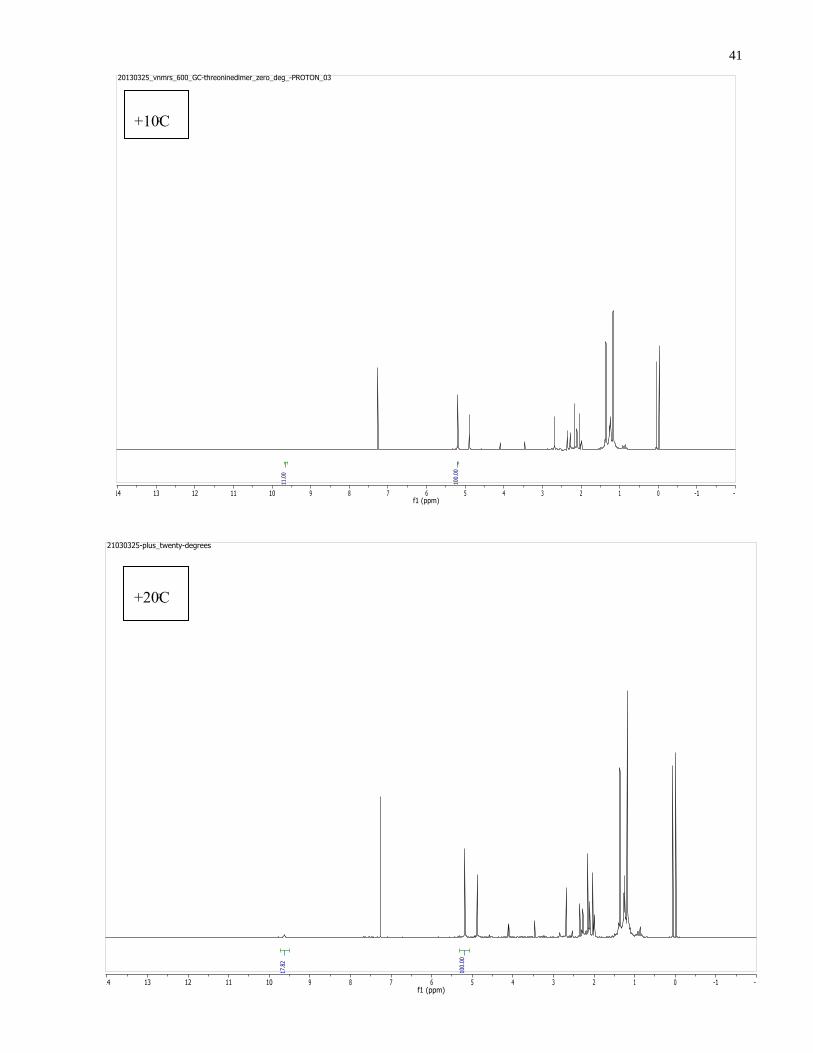
running variable temperature NMRs descending from +25⁰C to -20⁰C and ascending to + 30⁰C.
Four distinct peaks disappeared below 0⁰C and re-emerged above 0⁰C on the spectrum with the
following chemical shifts: 9.64 (s, 1H), 2.85 (d, J = 4.7 Hz, 1H), 2.54 (m, 1H), 1.28 (d, J = 5.7
Hz, 1H) (see Figure 1.13).
Upon initial inspection of the relative chemical shifts, it seemed to be the monomeric species of
the aziridine aldehyde. However, lack of a carbon correlation with the peak at 9.64ppm pointed
to the possibility of it being the N-H of the aziridine ring. After doing a D2O shake where a drop
of D2O was added to the sample dissolved in CDCl3, the proton at 9.64 ppm disappeared
suggesting one of two possibilities: a) that this peak has an exchangeable proton (N-H) or b) that
the aldehyde underwent nucleophilic addition by water thus resulting in the formation of a
hydrate species.
The only report of an N- unprotected aziridine aldehyde monomeric species in the literature was
reported by Reinhoudt13
in 1983, which was a by-product of decomposition during purification.
The aldehyde peak shows up at 9.56ppm with no 13
C NMR peak provided.

16
Figure 1.12 N- unprotected aziridine aldehyde monomeric species reported by Reinhoudt and
colleagues in 1983.
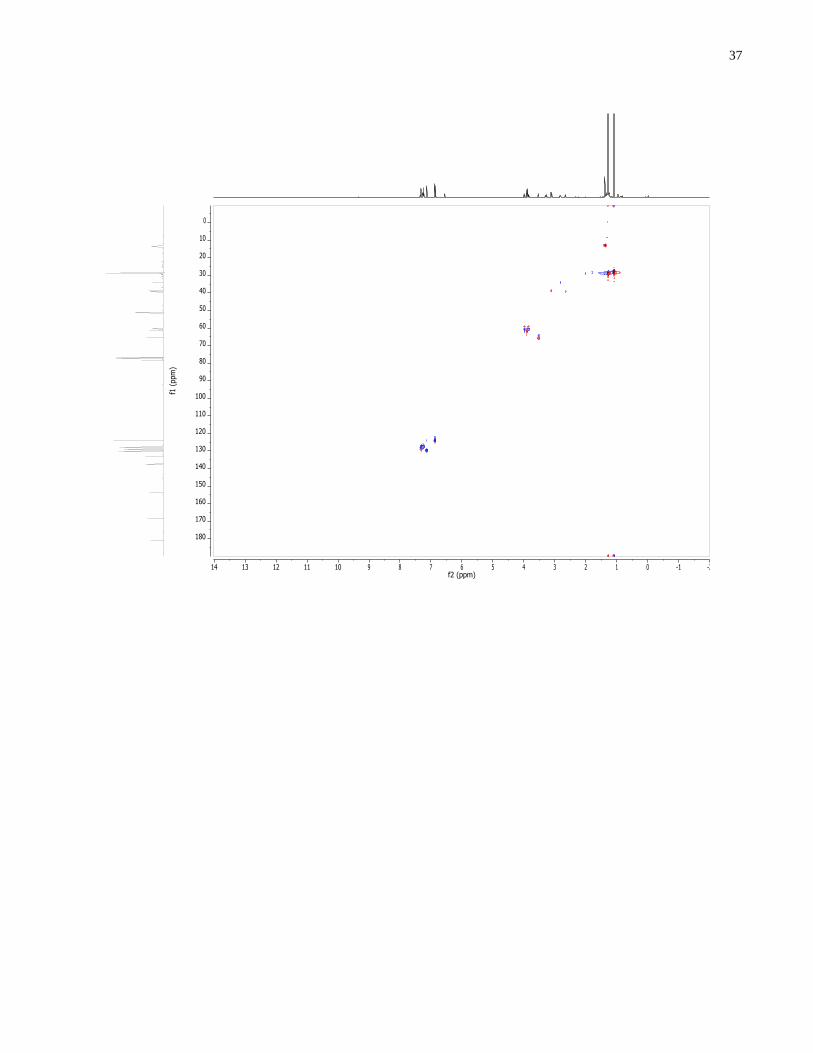
Figure 1.13. gCOSY of cis methyl aziridine aldehyde. The circled peaks and arrow show the
presence of another species related to the dimer that disappear and reappear below and above
zero degrees, respectively (see Appendix for VT NMRs).

17
That being said, because the aziridine alehyde dimer is of the cis configuration the above NMR
results perhaps suggest a lower kinetic barrier with its monomeric species than seen with trans
aziridine aldehyde.
3 Conclusion
In summary, differences observed in the stereoselectivity involving S-cis dimer versus S-trans
dimer with secondary amino acids were explored. Herein, preliminary results show that using a
S-cis aziridine dimer with either a D or L derived secondary amino acid gives high
diastereoselectivity. These novel findings provide evidence towards the reactivity of the resulting
iminium ion during alpha addition of the isocyanide. Interestingly enough, a peptide containing a
N-terminated L-Proline gave a “mismatched” case where the final product formed was an aminal
precluding any macrocyclization from occurring. Furthermore, the present results gathered also
show presence of free aziridine ring or a breakdown by-product of cis methyl aziridine dimer in
temperatures above 0⁰C while this species disappears at lower temperature below 0
⁰C in CDCl3
demonstrating that this species is in equilibrium with aziridine aldehyde dimer.
4 Experimental procedures
General Information: Anhydrous toluene and dichloromethane were purchased and used as
received. All other solvents including tert-butyl isocyanide and 2, 2, 2-trifluoroethanol (TFE)
were of reagent grade quality.

18
Chromatography: Flash column chromatography was carried out using Silicycle 230-400 mesh
silica gel and thin layer chromatography (TLC) was performed on EMD pre-coated glass back
TLC plates (TLC Silica Gel 60 F254, 0.25mm) and visualized using a UV lamp (254nm) and
stained with either potassium permanganate (KMnO4) or ninhydrin where applicable.
Nucleur magnetic resonance spectra: 1H NMR and
13C NMR spectra were recorded on either
the Bruker 400, 500 or 600 MHz spectrometers. The internal standard was TMS and 13
C NMR
spectra were referenced to either CDCl3 (77.23ppm), DMSO (39.52) or MeOD (49.00). Peak
multiplets were designated using the following abbreviations: s, singlet; d, doublet; t, triplet, q,
quartet; m, multiplet; dd, doublet of doublet, ddd, doublet of double of doublet; td, triplet of
doublets.
RP-HPLC/MS: Low resolution mass spectra (ESI) were collected on an Agilent Technologies
1200 series HPLC paired to a 6130 Mass Spectrometer. Compounds were resolved on an Agilent
Poroshell 120 EC-C18, 2.7 µm, 4.6 x 50 mm2 column at room temperature with a flow of 5
mL/min. The gradient consisted of eluents A (0.1% Formic acid in double distilled water) and B
(0.1% Formic acid in HPLC-grade acetonitrile).
4.1 Protocols for unprotected aziridine aldehydes
(2R, 4R, 5S, 6S)-6-(hydroxymethyl)-2-((2S, 3S)-3-(hydroxymethyl) aziridin-2-yl)-3-oxa-1-
azabicyclo[3.1.0]hexan-4-ol
Prepared according to the literature procedure. 3a
(2R,4R,5S)-2-((S)-aziridin-2-yl)-3-oxa-1-azabicyclo[3.1.0]hexan-4-ol
Prepared according to the literature procedure. 3a

19
(2S,3R)-propyl 2-amino-3-hydroxybutanoate
To chilled (-10 °C) and stirring 1-propanol (200 mL) was added SOCl2 (6.7 mL, 92.4 mmol)
drop-wise over 30 minutes. L-Threonine (10 g, 84 mmol) was then added and the mixture was
heated to 70 °C and allowed to stir under reflux for 4 hours. The reaction was allowed to cool
before precipitating the ester as a thick, white oil with diethyl ether and stored overnight in the
freezer. After decanting the ether, the recovered ester was dissolved in concentrated NH4OH
(15mL) and the free base was extracted 4 times with DCM (~200mL). The mixture was dried
over Na2SO4, and concentrated under low vacuum at 3 C to give quantitative yield of crude
free base. The product was used for the subsequent step without further purification.
(2S,3S)-propyl 3-methylaziridine-2-carboxylate
(2S,3S)-propyl 3-methylaziridine-2-carboxylate (15.5g, 96.6 mmol) was dissolved in anhydrous
DCM 3 m ) and the solution cooled to -1 C. Triphenyl phosphine (25g, 96.6 mmol) was
then added, and the flask equipped with an addition funnel. DIAD (19mL, 96.6 mmol) was
added drop-wise over 1 hour along the side of the flask, under a nitrogen atmosphere. Following
the addition, the reaction was allowed to warm to room temperature and stirred for 16 hours. The
solvent was evaporated under low pressure, hexanes added, and left in the fridge for 72 hours to
precipitate triphenylphosphine oxide. The product was purified by Kugelrohr distillation (0.15
mmHg) and fractions collected at 60, 75, 80, 85, 90, 95 and 100°C. 1H NMR indicated that
fractions 90-100 °C were the cleanest but crude NMR showed a few extra peaks and thus it was
further purified by flash column chromatography using a gradient 100% hexanes to 50% hexanes

20
and ethyl acetate providing the product as a clear solution (1.75g, 12.4%).1H NMR (400 MHz,
Chloroform-d) δ 4.13 t, J = 6.7 Hz, 2H), 2.63 (d, J = 6.1 Hz, 1H), 2.30 (p, J = 5.8 Hz, 1H), 1.69
(hept, J = 6.9 Hz, 2H), 1.30 (d, J = 5.7 Hz, 3H), 0.96 (t, J = 7.4 Hz, 3H).13
C NMR (101 MHz,
Chloroform-d) δ 171.1, 67. , 35.1, 33.8, 22.1, 13.3, 1 .4.
(2R,4R,5S,6S)-6-methyl-2-((2S,3S)-3-methylaziridin-2-yl)-3-oxa-1-azabicyclo[3.1.0]hexan-4-
ol
In a flame dried 100mL Schlenk flask equipped with a stir bar was placed (2S,3S)-propyl 3-
methylaziridine-2-carboxylate (1g, 6.9 mmol) in 20mL of anhydrous toluene under nitrogen
atmosphere. The solution was cooled to -78 C. 1.5M DI A in toluene 9.8m , 14.7mmol) was
added dropwise along the wall of the vessel over 1.5 hrs. nce the addition was complete the
reaction was left stirring at -78 C for 4 hrs until reaction was done as shown by TLC (100%
EtOAc, Rf =0.1).
Subsequently, the reaction was diluted with diethyl ether (50mL) at a rate of 2mL/min.
he reaction was quenched with water .6m ) over 1min, followed by 1M a .6m ) and
water .6m ). he reaction was left stirring for an additional 5min at -78 C and later warmed to
C and slowly brought to room temperature. After 30min of stirring at RT, MgSO4 was added
and the aluminum salts were filtered and rinsed with 1:1 MeOH: DCM (200mL). The filtrate was
transferred to falcon tubes, centrifuged and concentrated in vacuo. The crude oil was purified by
flash column chromatography (28:2:1:1 EtOAc: ACN: MeOH: water) providing the product as a
white powder (0.238g, 41%).1H NMR (500 MHz, Chloroform-d) δ 5.19 s, 1 ), 4.91 s, 1 ),
2.69 (d, J = 5.5 Hz, 1H), 2.35 (s, 1H), 2.29 (s, 1H), 2.11 (s, 1H), 1.37 (d, J = 6.0 Hz, 3H), 1.19
(d, J = 6.3 Hz, 3H).13
C NMR (126 MHz, CDCl3) δ 95.5, 91. , 47.4, 36.9, 33.4, 30.7, 12.2, 6.8.

21
4.2 Protocols for piperazinones
General procedure for synthesis of piperazinones was prepared according to literature4
(3S,5S,6S,7S)-3,4-dibenzyl-N-(tert-butyl)-7-(hydroxymethyl)-2-oxo-1,4
diazabicyclo[4.1.0]heptane-5-carboxamide
ESI MS [M+H]+ expected: 421.2, experimental: 422.2; LC/MS retention time 5.690 min.
1H NMR (500 MHz, Chloroform-d) δ 7.3 – 7.26 (m, 2H), 7.26 – 7.12 (m, 8H), 3.90 (d, J = 1.2
Hz, 2H), 3.82 – 3.75 (m, 1H), 3.67 – 3.61 (m, 2H), 3.59 (d, J = 7.0 Hz, 1H), 3.46 – 3.33 (m, 1H),
3.18 (dd, J = 7.0, 2.8 Hz, 1H), 2.90 – 2.80 (m, 1H), 2.70 – 2.66 (m, 1H), 1.21 (s, 9H).
13C NMR (126 MHz, Chloroform-d) δ 184.9, 168.3, 138.3, 136.9, 129.5, 128.8, 128.5, 128.2,
64.7, 60.5, 60.3, 57.5, 51.2, 42.8, 38.5, 36.8, 28.2
(1S,3aS,8S,8aS)-N-(tert-butyl)-1-methyl-3-oxooctahydroazirino[1,2-a]pyrrolo[1,2-
d]pyrazine-8-carboxamide
ESI MS [M+H]+ expected: 265.2, experimental: 266.2; LC/MS retention time 3.257 min.

22
1H NMR (500 MHz, Chloroform-d) δ 6.11 s, 1 ), 3.5 d, J = 7.0 Hz, 1H), 3.26 – 3.22 (m, 1H),
3.06 (dd, J = 7.0, 4.9 Hz, 1H), 2.96 – 2.90 (m, 1H), 2.68 (qd, J = 6.2, 4.9 Hz, 1H), 2.17 – 1.98
(m, 2H), 1.93 – 1.80 (m, 3H), 1.40 (d, J = 6.2 Hz, 3H), 1.35 (s, 9H).
13C NMR (126 MHz, Chloroform-d) δ 18 .7, 168. , 64.8, 64.0, 55.3, 51.3, 40.8, 40.2, 28.8, 21.7,
21.7, 13.2.
ESI MS [M+H]+ expected: 267.2, experimental: 268.2; LC/MS retention time 2.659 min.
1H NMR (400 MHz, Chloroform-d) δ 3.6 dddd, J = 9.2, 6.7, 4.4, 2.1 Hz, 1H), 3.30 (t, J = 8.2
Hz, 1H), 3.16 (d, J = 6.9 Hz, 1H), 3.07 (ddd, J = 9.0, 7.5, 3.9 Hz, 1H), 2.47 (q, J = 8.6 Hz, 1H),
2.08 – 1.96 (m, 3H), 1.87 (dddd, J = 14.5, 12.6, 7.7, 4.5 Hz, 3H), 1.30 (s, 9H), 1.02 (t, J = 7.4
Hz, 3H).
13C NMR (101 MHz, Chloroform-d) δ 174.3, 169.2, 69.5, 65.8, 54.6, 51.7, 29.8, 28.8, 24.3, 23.7,
22.5, 11.4.
(1S,3aR,8R,8aS)-N-(tert-butyl)-1-methyl-3-oxooctahydroazirino[1,2-a]pyrrolo[1,2-
d]pyrazine-8-carboxamide
ESI MS [M+H]+ expected: 265.2, experimental: 266.2; LC/MS retention time 3.117 min.

23
1H NMR (500 MHz, Chloroform-d) δ 6.53 s, 1 ), 3.65 dd, J = 9.4, 4.2 Hz, 1H), 3.39 (dd, J =
9.0, 5.1 Hz, 1H), 3.05 (d, J = 9.1 Hz, 1H), 3.04 – 3.01 (m, 1H), 2.97 (td, J = 9.1, 6.4 Hz, 1H),
2.84 (qd, J = 6.1, 5.0 Hz, 1H), 2.36 – 2.26 (m, 1H), 2.06 – 1.92 (m, 2H), 1.89 – 1.76 (m, 1H),
1.38 (s, 9H), 1.27 (d, J = 6.1 Hz, 3H).
13C NMR (126 MHz, Chloroform-d) δ 187. , 167.6, 77.4, 77.3, 77.1, 76.9, 66.0, 56.0, 51.3, 47.2,
37.8, 37.7, 29.9, 28.9, 24.9, 10.6.
(3S,5S,7S)-4-benzyl-3-(4-(tert-butoxy)benzyl)-N-(tert-butyl)-7-methyl-2-oxo-1,4-
diazabicyclo[4.1.0]heptane-5-carboxamide
ESI MS [M+H]+ expected: 477.3, experimental: 478.3; LC/MS retention time 5.314 min.
1H NMR (400 MHz, Chloroform-d) δ 7.34 – 7.19 (m, 5H), 7.15 – 7.09 (m, 2H), 6.87 – 6.82 (m,
2H), 6.54 (s, 1H), 3.98 (d, J = 15.7 Hz, 1H), 3.89 (d, J = 8.4 Hz, 1H), 3.84 (d, J = 15.7 Hz, 1H),
3.52 (dd, J = 8.7, 3.5 Hz, 1H), 3.27 (dd, J = 14.2, 8.7 Hz, 1H), 3.10 (dd, J = 8.4, 4.7 Hz, 1H),
2.82 (dd, J = 14.2, 3.5 Hz, 1H), 2.64 (qd, J = 6.2, 4.6 Hz, 1H), 1.36 (d, J = 6.2 Hz, 3H), 1.27 (s,
9H), 1.07 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 18 .8, 168.6, 153.8, 137.7, 133.3, 13 . , 128.9, 127.8, 124.1,
78.3, 65.3, 61.4, 60.3, 51.1, 39.2, 38.7, 34.0, 28.8, 28.2, 13.3.

24
5 References
(1) (a) Ugi, I., Meyr, R., Fetzer, U.,Steinbruckner, C. Angew. Chem. 1959, 71, 386. (b) Ugi,
I., Steinbruckner, C. Angew. Chem. 1960, 72, 267. (c) Ugi, I. and Steinbrückner, C. Chem.
Ber. 1961, 94, 2802–2814.
(2) (a) Failli, A., Nelson, V., Immer, H., Gotz, M. Can. J. Chem. 1979, 57, 3257-3261. (b)
Domling, A., Ugi, I. Angew. Chem. Int. Ed. 2000, 39, 3168-3210
(3) (a)Hili, R., Yudin, A. K. J. Am. Chem. Soc. 2006, 46, 14772-14773. (b) Yudin, A. K.,
Hili, R. Chem. Eur. J., 2007, 23, 6538-6542. (c) Hili, R., Yudin, A. K. Amphoteric amino
aldehydes enable rapid assembly of unprotected amino alcohols. Angew. Chem. Int. Ed.
2008, 22, 4188-4191. (d) Baktharaman, S., Hili, R., Yudin, A. K. Aldrichimica
Acta, 2008, 41, 109-119. (e) Hili, R., Baktharaman, S., Yudin, A. K. Eur. J. Org. Chem.
2008, 31, 5201-5213.(f) Hili, R., Yudin, A. K. J. Am. Chem. Soc. 2009, 45, 16404-16406.
(g) Hili, R., Rai, V., Yudin, A.K. J. Am. Chem. Soc. 2010, 132, 2889-91.
(4) Rotstein, B. H., Rai, V., Hili, R., Yudin, A. K. Nat. Prot. 2010, 11, 1813-1822.
(5) Hili, R.M. Unprotected Amino Aldehydes in Organic Synthesis. Ph.D. Thesis, University
of Toronto, Toronto, Ontario, December 2009.
(6) Assem, N., Hili, R., He, Z., Kasahara, T., Inman, B. L., Decker, S., Yudin, A. K. J. Org.
Chem. 2012, 77, 5613-5623.
(7) Assem, N.M. Reactivity of dimeric aziridine aldehydes and their utility in the synthesis of
peptidomimetics. Ph.D Thesis, University of Toronto, Toronto, Ontario, July 2012.
(8) Decker, S.M. Synthetic application of N-H aziridine containing compounds. MSc. Thesis,
University of Toronto, Toronto, Ontario, October 2010.
(9) Rotstein, B.H. Unprotected aziridine aldehydes in isocyanide based multicomponent
reactions.Ph.D Thesis, University of Toronto, Toronto, Ontario, June 2012.
(10) Park, S., Keum, G., Kang, S., Koh, H., Kim, Y., Lee, D. Tetrahedron Lett. 1998, 39,
7109-7112.

25
(11) Dyker, G. Angew. Chem. Int. Ed. 1997, 36, 1700-1702
(12) Pennings, M.L.M., Reinhoudt, D.N., Harkema, S.,Van Hummel, G.J. J. Org.
Chem. 1983, 48, 486-491.

26
Appendix I: 1H,
13C and NOE NMR spectra

27

28

29

30
Hb
Irradiate Ha

31

32

33

34

35
Irradiate
Ha
Hb

36
37

38
Irradiate
Hb
Ha Hc

39

40
-2 C
C
41
+10⁰
C
+20⁰
C
1 C
2 C

42
3 C

43

44


















