
EXPANDED ACCESS WORKSHOP - CBI | Powering ... 31, 2017 POTUS Meeting with Pharmaceutical Executives...
46
EXPANDED ACCESS WORKSHOP Take a Deep Dive into the New Laws and Regulations to Prepare for Expanded Access Disclosures March 9, 2017 1 David J. Farber Partner King & Spalding LLP Preeya Noronha Pinto Partner King & Spalding LLP CBI Expanded Access Programs 2017
Transcript of EXPANDED ACCESS WORKSHOP - CBI | Powering ... 31, 2017 POTUS Meeting with Pharmaceutical Executives...

EXPANDED ACCESS
WORKSHOP
Take a Deep Dive into the New Laws and Regulations to Prepare for Expanded Access Disclosures
March 9, 2017
1
David J. Farber
Partner
King & Spalding LLP
Preeya Noronha Pinto
Partner
King & Spalding LLP
CBI Expanded Access Programs 2017

Expanded Access – The Basics
What’s Driving Policy Change?
What’s New from HHS/FDA, Congress and the States
Where Do We Go From Here?
Today’s Agenda
2

What is Expanded Access?
Program established by Congress and
regulated by FDA
Goal is to improve access to
investigational drugs by certain
patients for treatment purposes
Differs from a clinical
investigation where the primary
purpose is research(i.e., systematic
collection of data to determine safety
and/or effectiveness)
3

Authority for Expanded Access
1997 FDA Modernization Act (amended Section 561 of the FDCA)
21 C.F.R Part 312, Subpart I (FDA regulations have
permitted Expanded Access since 1987)
“Guidance for Industry, Expanded
Access to Investigational
Drugs for Treatment Use—Qs and As.”
FDA Guidance, June 2016
“Charging for Investigational Drugs Under an
IND—Qs and As.” FDA Guidance, June
2016
4

General Criteria for FDA Authorization
Patient must have a serious or immediately life-threatening
disease or condition
No comparable or satisfactory alternative therapy exists
Potential patient benefit justifies the potential risks, and those
risks are not unreasonable in the context of the disease or
condition
Providing the investigational drug will not interfere with or
compromise clinical investigations that could support
marketing approval
FDA must determine that:
5

Three Categories of Expanded Access
Individual Patients (including for
emergency use)
• 21 C.F.R. § 312.310
Intermediate-Size Patient Populations
• 21 C.F.R. § 312.315
Large Patient Populations
(Treatment IND or Treatment Protocol)
• 21 C.F.R. § 312.320
6

Individual Patient Expanded Access
Physician must determine that the probable risk is not greater than that of the disease
Manufacturer must agree to provide the investigational drug
FDA must determine that (1) the general criteria are met and (2) the patient cannot obtain the drug under another type of IND or protocol
IRB must review and approve
EXPANDED ACCESS
7

Submission/Request to FDA
Sponsor can submit an “individual patient access protocol” to its existing IND
• Physician is the investigator
Sponsor can submit an “individual patient access IND” and cross-reference information in existing IND
• Physician is the investigator
Physician can submit an “individual patient access IND” and cross-reference information in existing IND with permission from sponsor
• Physician is the sponsor and investigator
8

Individual Patient Emergency Use
Emergency use verbally approved by
FDA
All applicable criteria must be met
Physician or sponsor must agree to provide a written submission to FDA within 15 working
days of FDA authorization
IRB must be notified within 5 working days of
initiation of treatment
9

FDA Statistics – All Requests
10

FDA Statistics – Single Patient Requests
11
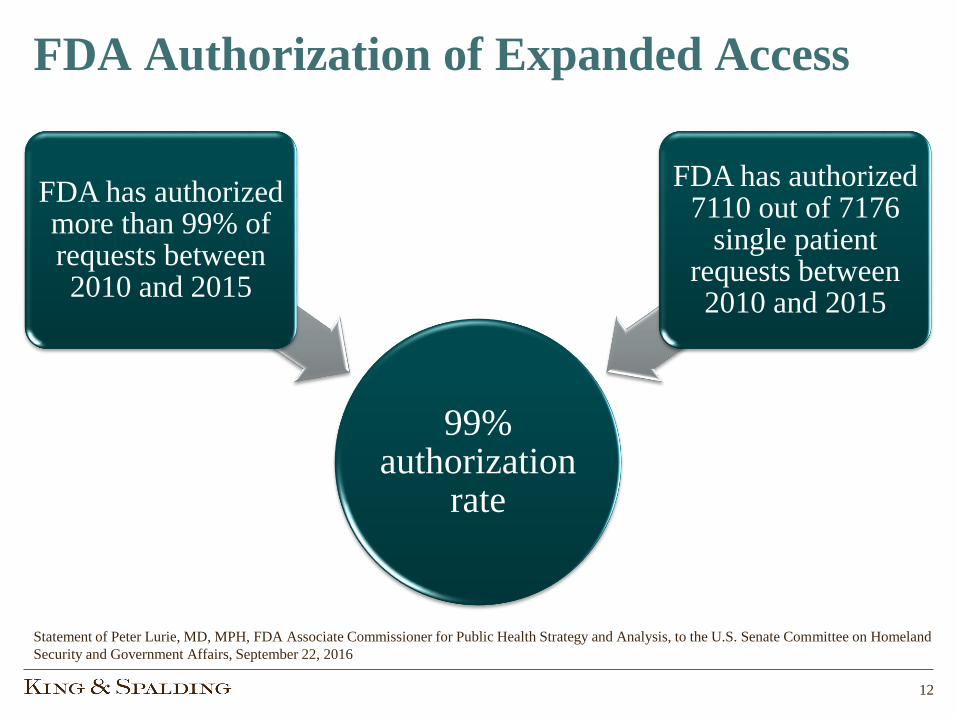
FDA Authorization of Expanded Access
99% authorization
rate
FDA has authorized more than 99% of requests between 2010 and 2015
FDA has authorized 7110 out of 7176
single patient requests between 2010 and 2015
12
Statement of Peter Lurie, MD, MPH, FDA Associate Commissioner for Public Health Strategy and Analysis, to the U.S. Senate Committee on Homeland
Security and Government Affairs, September 22, 2016

So What Is Driving Policy Change?
13

Josh Hardy Media Firestorm
14

Chimerix CEO Takes the Fall
15

Ongoing Media Focus
16
CNNSeptember 28, 2013 ·
Andrea Sloan is dying of ovarian cancer. She's
seeking "compassionate use" of a new drug that's
not FDA-approved, but a drug maker says it's too
risky. http://on.cnn.com/1fQkaEs

Ebola Raises More Questions
17

Trump Highlights the Issue
18
January 31, 2017 POTUS Meeting with Pharmaceutical Executives
“One thing that’s always disturbed me, they come up with a new drug for a
patient who’s terminal, and the FDA says, ‘We can’t have this drug used on the
patient,’ but they say that the patient within four weeks will be dead. They say,
‘Well, we still can’t approve the drug. And we don’t know if the drug works or it
doesn’t work, but we can’t approve the drug because we don’t want to hurt the
patient.’ But the patient is not gonna live more than four weeks.
So we’re gonna be changing a lot of – a lot of the rules.”

Pence Weighs in on “Right to Try”
February 7, 2017:
VP Pence meets with Right to Try advocates in the White House
19

Navigating the Issues
Clinical Data
Cost
Liability Protection &
Other Business Considerations
Population vs. Individual
Health
FDA
Ethics
20

Policy Changes and New Requirements
• State Right to Try Legislation
• 21st Century Cures Act
• Federal Right to Try Legislation pending
• ClinicalTrials.Govdisclosure requirements
• Revised request form for physicians
• Finalized draft guidances
• User-friendly website
FDA HHS
StatesCongress
21

FDA Reduces the Paperwork
• In February 2015, FDA issued Draft Guidance containing a new form (Form FDA 3926) for physicians to make expanded access requests for individual patients
• Form FDA 3926 streamlines the process for making such requests, which would otherwise need to be made on the general IND submission form (Form FDA 1571)
22

FDA Reduces the Paperwork
23

New Patient-Targeted “How To” Information
24

FDA’s New Final Guidances (June 2016)
25
“Expanded Access to Investigational Drugs for Treatment Use–Questions
and Answers”
Implementation of FDA’s regulations on expanded access to investigational drugs for
treatment use under an IND
How expanded access is defined, types of expanded access, when and how to request
expanded access, and what information should be included in requests
IRB review
The role of Form FDA 3926
“Charging for Investigational Drugs
Under an IND–Questions and Answers”
Criteria for charging for an investigational drug for expanded access for treatment use
Which costs can be recovered (i.e., direct costs such as cost of drug and shipping/handling, but
not indirect and administrative costs)
Circumstances under which FDA authorizes charging for an investigational drug in a clinical
trial

2008-2015: Recent Federal Policy Efforts2007-08
110th Congress
• H.R. 6270 and S. 3046 (June 2008)
• Bi-partisan, Democratic-led effort (Rep. Watson, D-CA)
• Required FDA to create new expanded access regime
• Permitted manufacturers, following Phase I, to apply for expanded access authority and to charge for drugs
• Provided manufacturer liability protection
2009-10
111th Congress
• H.R. 4732
• Same as previous bill, but no Senate counterpart
2011-12
112th Congress
• H.R. 6342
• Rep. Ron Paul (R-TX)
• Permitted expanded access upon informed consent of patient
• Prohibited FDA from regulating access, or even collecting information
• No Phase I requirement
2013-14
113th Congress
• H.R. 4475 – Rep. Morgan Griffith (R-VA) introduces updated version of Rep. Paul’s bill, with liability protection for manufacturers
• H.R. 5805 – Rep. Michael McCaul(R-TX) introduces Andrea Sloan CURE Act, requiring manufacturers to post contacts for requests and provide denials in writing, and requiring GAO and Task Force Reports to Congress
2015-16
114th Congress
• H.R. 790 – Rep. Griffith bill reintroduced
• H.R. 909 –Andrea Sloan CURE Act reintroduced
• H.R. 3102 –Right to Try Act of 2015
• 21st Century Cures Act (P.L. 114-255)
26

Overview of 21st Century Cures
27

CURES: Origins in the House
28
May 2014: House Energy & Commerce Committee “Call to Action”
• Focus on discovery, development and delivery
• Comments, ideas, 11 roundtables and hearings
January 2015: The first White Paper
April/May 2015: First drafts of House legislation
• Becomes H.R. 6
July 2015: Full House approves the bill
• Significantly modified from original proposals
• The politics – NIH Funding, FDA changes, and the Strategic Petroleum Reserve “pay-for”

CURES: The Senate’s Approach
29
• Individual sections move as stand-alone bills through HELP Committee, but don’t reach the Senate Floor
• Many ideas don’t even make it through Committee
• No consideration of NIH funding or the “pay-fors,” particularly the SPR, which has now been used for other legislation

CURES: It All Comes Together
30
Sept-Nov 2016: E&C, Ways & Means, and HELP Committees (with little input from Senate Finance) bring together a comprehensive bill
•Combines revised CURES, mental health, hospital outpatient, home infusion and numerous other provisions
•Addresses NIH funding with $4 billion authorization
November 30, 2016: H.R. 34 passes House
•Senator Grassley objects to “Sunshine” changes
December 7, 2016: Passed by Senate (and back through House)
December 13, 2016: Signed by President
Obama

Cures Section 3032
31

Section 3032: A Policy is Mandatory!
Manufacturer or distributor of investigational drugs “shall make available” an expanded access policy
• Mandatory requirement to have an expanded access policy, whether or not manufacturer permits expanded access for its products
Expanded access policy “shall be made public and readily available”
• Publicly available Internet posting is sufficient
• Policy may be generally applicable to all manufacturer’s products
Policy may be revised at any time
32

Section 3032: Content of Policy
Contact information to facilitate communication about expanded access requests
Procedures for making such requests
General criteria the manufacturer will use to evaluate requests for individual patients, and responses to requests
Length of time the manufacturer anticipates will be necessary to acknowledge receipt of requests
Hyperlink or other reference to the Expanded Access Record in ClinicalTrials.Gov
33

Section 3032: Effective Date
The later of:
February 11, 2017 (60 calendar days after
enactment of CURES)
The first initiation of a phase 2 or phase 3
study with respect to an investigational drug
34

New ClinicalTrials.gov Regulation
April 18, 2017: Deadline for
responsible parties to achieve
compliance
January 18, 2017: Final Rule
effective date
September 21, 2016: HHS issued
a Final Rule
November 19, 2014: HHS issued a Proposed Rule
on ClinicalTrials.gov
35
The statutory authority for the expanded access disclosure
requirements in the rule is Public Health Service Act § 402(j),
which requires the submission of information regarding
whether, for an applicable drug clinical trial of an unapproved
drug/biological product, expanded access is available under
FDCA § 561

General Requirements
• If the responsible party for an “applicable clinical trial” is both the sponsor and the manufacturer, and expanded access is available, there must be a separate Expanded Access Record containing details about how to obtain access to the investigational product
― Applies to all types of expanded access (individual, intermediate or large size)
― Expanded Access Record is assigned a unique NCT number that is linked to the clinical study record for the investigational product
36

What Information Is Required?
Brief Title Official Title Brief Summary
Study Type (which is “expanded
access” for this type of record)
Primary Disease or Condition
Intervention Name(s)
Other Intervention Names(s)
Intervention Description
Intervention Type (which is typically
“drug”)
Expanded Access Type
Eligibility Criteria Sex/Gender Age LimitsExpanded Access
StatusName of the
Sponsor
Responsible Party, by Official Title
Contact Information
Unique Protocol Identification
NumberSecondary ID
U.S. Food and Drug
Administration IND Number
Record Verification Date
Responsible Party Contact
Information
37
Only bolded data elements are
required for individual patient
expanded access records

When Must Information Be Submitted?
Expanded Access Record must be created no later
than 30 days after expanded access
becomes available
NCT number for Expanded Access Record must be
linked to the clinical trial record
no later than 30 days after the NCT number is assigned
Data elements must be updated within 30 days of
the change
38

Areas for Further Clarification…
Does CURES require a hyperlink to the clinical trial record or the
Expanded Access Record?
What if a manufacturer has not yet made a decision about whether
expanded access is available for an investigational product?
What if expanded access is available for an investigational product that is
not part of an “applicable clinical trial”?
39

More Transparency for Expanded Access
40

Status of State “Right to Try” Laws
41
Source: Goldwater Institute (righttotry.org/in-your-state/)
33 states
have adopted
Right to Try
laws, and 14
have pending
bills
Green = Passed Law
Blue = Introduced Legislation
Red = Vetoed

Do’s and Don’t’s of “Right to Try” Laws
What They DO:
• Generally, they aim to grant patient access to investigational treatments if:
• Patient is terminally ill
• Physician recommends treatment
• Patient provides informed consent, and
• Treatment has completed Phase I clinical safety/dose limitation trial
• They grant physicians and manufacturers liability protection against claims arising from adverse events caused by investigational treatments
What They DON’T DO:
• They don’t create a right for terminally ill patients to access life-saving treatments exempted from FDA rules
• State laws do not permit manufacturers to provide patients with access to unapproved drugs, when the federal law mandates “no person shall introduce or deliver for introduction into interstate commerce any new drug”
• They don’t address questions of patient welfare and public health
• They don’t provide free access to experimental treatments
• They don’t compel anyone or any company to fulfill a patient request
42

Some in Congress Trying to Build on State “Right to Try” Laws
Bills propose to federalize state RTT laws by
prohibiting FDA from blocking access if: (1)
intended to treat a patient who has been diagnosed
with a terminal illness; (2) is authorized by, and in
accordance with, State law; (3) drug is past Phase I
clinical trials
S.204 - TrickettWendler Right to Try Act of 2017
(Sen. Ron Johnson) - 44 co-sponsors
H.R.878 – Right to Try Act of 2017
(Rep. Andy Biggs) - 36 co-sponsors
43
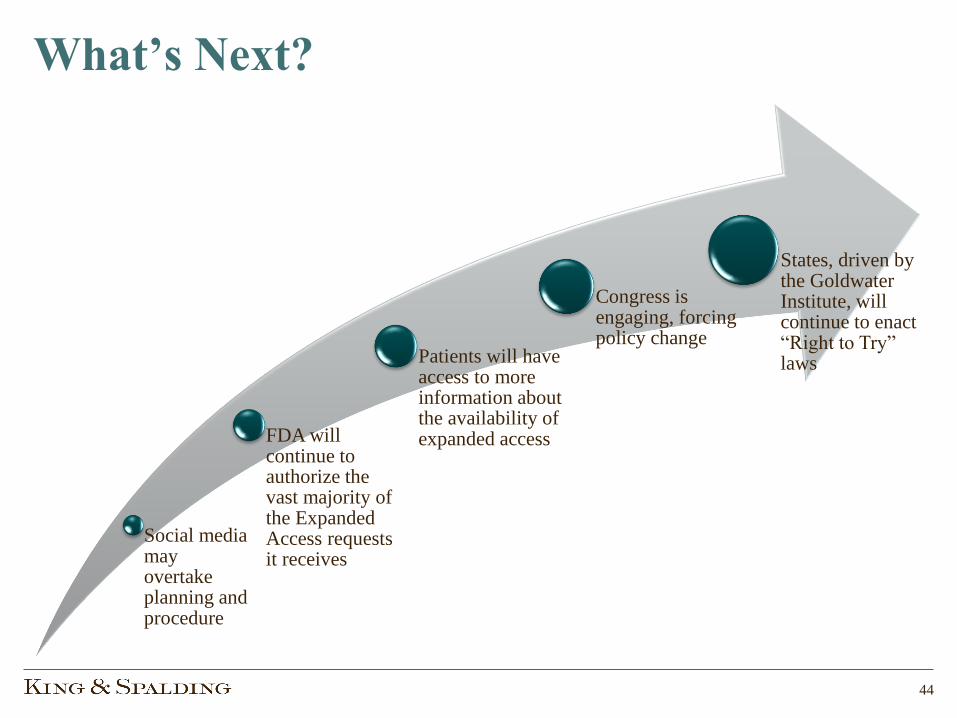
What’s Next?
Social media may overtake planning and procedure
FDA will continue to authorize the vast majority of the Expanded Access requests it receives
Patients will have access to more information about the availability of expanded access
Congress is engaging, forcing policy change
States, driven by the Goldwater Institute, will continue to enact “Right to Try” laws
44

Where Do We Go From Here?
How will patients and physician utilize the increased information that will be available from
manufacturers?
This is not a simple issue – there is as much opportunity to do harm than to do good
Is social media the best tool for such a nuanced issue? And how does one become a responsible
voice in the debate?
45

QUESTIONS?
THANK YOU!
46
David J. Farber
(202) 626-2941
Preeya Noronha Pinto
(202) 626-5547


















