
Evaluation of the cellular and molecular effects of HCV ...
176
Evaluation of the cellular and molecular effects of HCV non- structural proteins on mitochondrial mediated apoptotic pathway By Farakh Javed 2009-NUST-DirPhD-V&I-43 Atta-ur-Rahman School of Applied Biosciences National University of Sciences & Technology Islamabad-Pakistan 2016
Transcript of Evaluation of the cellular and molecular effects of HCV ...

Evaluation of the cellular and molecular effects of HCV non-
structural proteins on mitochondrial mediated apoptotic
pathway
By
Farakh Javed
2009-NUST-DirPhD-V&I-43
Atta-ur-Rahman School of Applied Biosciences
National University of Sciences & Technology
Islamabad-Pakistan
2016

Evaluation of the cellular and molecular effects of HCV non-structural
proteins on mitochondrial mediated apoptotic pathway
By
Farakh Javed
2009-NUST-DirPhD-V&I-43
A dissertation submitted in the partial fulfillment of the requirement for the
degree of
Doctor of Philosophy
IN
VIROLOGY AND IMMUNOLOGY
Supervisor
Dr. Sobia Manzoor
Atta-ur-Rahman School of Applied Biosciences
National University of Sciences & Technology
Islamabad-Pakistan
2016

Dedicated
To
My PARENTS & my Son
Shahmir sheikh

TABLE OF CONTENTS
TITLE Page No.
Acknowledgements i
List of Abbreviations iii
List of Tables vi
List of Figures vii
Abstract xi
Chapter -1
INTRODUCTION
1.1 Introduction 01
1.2 Aims and objectives of the study 07
Chapter -2
LITERATURE REVIEW
2.1 Anatomy and physiology of Liver 07
2.2 Hepatitis 09
2.3 Viral hepatitis 09
2.3.1 Hepatitis A virus 10
2.3.2 Hepatitis B virus 10
2.3.3 Hepatitis C virus 11
2.3.4 Hepatitis D virus 12
2.4.5 Hepatitis E virus 12
2.4 The discovery of Hepatitis C virus 13
2.5 Epidemiology of HCV 14
2.6 HCV transmission 15

2.7 Treatment of HCV 16
2.8 HCV genomic organization 17
2.9 HCV replication and role of viral proteins 20
2.9.1 Core protein 21
2.9.2 Envelop proteins 23
2.9.3 P7 23
2.9.4. Nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, NS5B) 24
2.9.4.1 NS2 24
2.9.4.2 NS3 24
2.9.4.3 NS4A 25
2.9.4.4 NS4B 25
2.9.4.5 NS5A 26
2.9.4.6 NS5B 26
2.10 Apoptosis and HCV 27
2.11 Apoptosis 29
2.12 Apoptosis molecular mechanism 30
2.12.1 Bcl-2 Family 32
2.12.2 Caspases executors of apoptosis 34
Chapter -3
MATERIALS AND METHODS
3.1 Materials 37
3.1.1 Expression vectors and host cells 37
3.1.2 Chemicals and other consumables 37
3.1.3 Enzymes 37
3.1.4 Antibodies 38

3.1.5 Antibiotics 39
3.1.6 Molecular weight markers 39
3.1.7 Reagents 39
3.1.8 Media 39
3.1.9 Buffers 40
3.1.10 Instrumentations 44
3.2 Methodology 44
3.2.1 Primer designing 44
3.2.2 Amplification of NS3, NS3-4A and NS4A genes 44
3.2.3 Amplification of NS3 gene using pFLAG-CMV2 primers 46
3.2.4 Amplification of NS3 gene using pEGFP-C1 primers 47
3.2.5 Amplification of NS3-4A gene using pFLAG-CMV2 primers 48
3.2.6 Amplification of NS3-4A gene using pEGFP-C1 primers 49
3.2.7 Amplification of NS4A gene using pFLAG-CMV2 primers 50
3.2.8 Amplification of NS4A gene using pEGFP-C1 primers 51
3.2.9 Agarose gel electrophoresis 52
3.2.10 Purification of DNA products from agarose gel 52
3.2.11 Construction of pEGFP-C1 recombinant plasmids 53
3.2.12 Construction of pFLAG-CMV2 recombinant plasmids 53
3.2.13 Selection of positive clones 54
3.2.14 Large scale preparation of plasmid DNA 54
3.2.15 Cell culture and transfection 55
3.2.16 Preparation of whole cell lysate for SDS page 55
3.2.17 SDS-poly acrylamidegel electrophoresis 56
3.2.18 Western Blotting 57

3.2.19 Immunofluorescence Microscopy 58
3.2.20 Crystal violet cell viability assay 59
3.2.21 Subcellular Fractionation 59
3.2.22 Mitochondrial Superoxide Estimation 60
3.2.23 Measurement of Mitochondrial Complex I Enzyme Activity 60
3.2.24 Hoechst Staining 61
3.2.25 Statistical analysis 61
Chapter -4
RESULTS
4.1 Amplification and construction of recombinant vectors 62
4.2 Sequencing of GFP-tagged recombinantvectors 66
4.3 Sequencing of FLAG-tagged recombinantvectors 70
4.4 Western blot analysis of FLAG-tagged HCV NS3, NS3-4A
and NS4A proteins 74
4.5 Western blot analysis of GFP-tagged HCV NS3, NS3-4A
and NS4A proteins 76
4.6 Immunofluorescence analysis of GFP tagged recombinant
vectors 78
4.7 Immunofluorescence analysis of FLAG tagged recombinant
vectors 81
4.8 NS3-4A and NS4A proteins induces cell death 84
4.9 Intracellular localizations of NS3, NS3-4A and NS4A proteins. 88
4.10 NS3-4A and NS4A proteins induces mitochondrial
fragmentation/fission 91
4.11 BAX translocation and Bcl-XL regulation in NS3-4A and NS4A
expressing cells 94
4.12 NS4A and NS3-4A increased mitochondrial superoxide
generation in Huh-7 cells 98
4.13 Cytochrome c translocation in NS4A and NS3-4A
expressing cells 101
4.14 Effects of NS4A and NS3-4A proteins on the mitochondrial

oxidative Phosphorylation system 103
4.15 Activation of caspase-3, 7, 9 and cleavage of poly
(ADP-ribose) Polymerase (PARP) in NS4A and NS3-4A expressing
cells 107
4.16 Chromatin condensation in NS4A and NS3-4A expressing
cells 113
Chapter -5 116
Discussion
Chapter -6
References 123

i
Acknowledgements
All the acclamation is for all Almighty “Allah”, Who bestowed the mankind
with knowledge and wisdom. All the respect and honor for Holy Prophet
Muhammad (PBUH) for enlighting with the essence of faith in Allah and guiding
the mankind, the true path for life.
I feel pleasure to place on record my deep sense of thankfulness and
indebtedness to my honorable supervisor Dr. Sobia Manzoor, Assistant professor,
ASAB-NUST, for her kind supervision, dexterous, incentive teaching, scholarly and
sympathetic attitude and help throughout this research and in preparation of this
dissertation.
I am honored to express my deep sense of gratitude and indebtedness to respecter
Prof. Dr. Aleem Siddiqui, University of California, San Diego, USA, who provided
me a golden opportunity to conduct my partial research work in his lab for a period of
seven months. I am really thankful to him for his valuable suggestions that make my
research work more strengthen.
These lines provide me the opportunity to rightly acknowledge, unmatched
sincerity of Dr. Peter John, principal, ASAB-NUST, who encouraged and helped me
to conduct and complete this research work.
I would like to extend my heartiest gratitude to Dr. Ghulam Hussain Syed and
Dr. Mohsin khan, UCSD, USA, for their kind guidance, cooperation and technical
support in my research work. I acknowledge the cooperation, moral encouragement,
inspiration and valuable suggestions of my research project Guidance and
Examination Committee (GEC) members honorable Dr. Aneela Javed, Assistant
professor, ASAB-NUST, Dr. Muhammad Faraz Bhatti, Assistant Professor,
ASAB-NUST and Dr. Ali Raza Awan, Assistant Professor, Department of
Biochemistry, University of veterinary and Animal Sciences, Lahore.
I also would like to thank my class fellows and my lab fellows Sana Gul, Fahed
Parvaiz, Shamim Bhatti, Zia-ur-rehman, Fazal Jalil, Sadia, Yasir Waheed,
Muhammad Imran, Rehan Zafar and Babar Aslam for their cooperation during my
work.

ii
I feel obligatory to express my thanks to my friends and lab Juniors Muqddas
Tariq, Madiha Khalid, Laeeque Ahmed, Huma Tariq, Abbas Raza, Mustafeez Babar,
Naghmana Kanwal, Yasmeen Badshah, Waseem Ashraf, Beenish Bashir,
Khushbakht Hanif, Yusara Naveed, Maheen Bokhari, Sibgha Noor and Tabinda
Hussain. I also acknowledge the cooperation and sympathetic behavior of all Faculty
and Staff members, ASAB-NUST.
I would like to heartiest and profound thanks to my friends in University of
Haripur, Dr. Aamer Ali Khattak, Dr. Sadiq Noor Khan, Dr. Darima Ashfaq, Miss
Afshan Saleem, Miss Javeria Asghar and Miss Naureen Aurangzeb.
I must acknowledge the financial support of Higher Education Commission of
Pakistan (HEC). I am also grateful to UCSD, USA and ASAB-NUST, Pakistan
for providing support and technical resources to conduct this research.
And at last but not the least, I deem it a great honor and privilege to record
profound sense of gratitude and extend my compliments to my affectionate parents,
sisters (Sadia Asad and Nadia Sajjad) and brothers (Sheikh Jibran and
Sheikh Waqas) for their encouragement and inspiration for higher ideas, moral and
their love and affection, amicable attitude and their countless prays for my glorious
success about my pursuits throughout my life. I would do no more than to reaffirm my
eternal devotion to all the members of my family.
I would like to express my whole love to my sweet nephews, niece and son Nafih,
Muneeb, Taha, Aman, Meerab and lovely Shahmir.
“I do appreciate all of those who remembered me in their prayers and encouraged me
throughout my life and education carrier”.
Farakh Javed Shiekh

iii
LIST OF ABBREVIAT`IONS
Ab Antibody
Ag Antigen
APS Ammonium persulfate
BCA Bicinchoninic acid assay
bp Base pair
BSA Bovine serum albumin
CPE Cytopathic effect
C-terminal Carboxy terminal
DMEM Dulbecco’s modified eagle medium
DNA Deoxyribonucleic acid
DTT Dithiothreitol
EDTA Ethylenediamine tetracetic acid
ELISA Enzyme linked immunosorbent assay
EMEM Eagle’s minimum essential medium
ER Endoplasmic reticulum
FBS Fetal bovine serum
GAPDH Glyceraldehyde -3-phosphate dehydrogenase
HBSS Hank’s balanced salt solution
HCC Hepatocellular carcinoma
HCV Hepatitis C virus
HIS Hyper immune sera
HPI Hours post infection
HRP Horse radish peroxidase

iv
IFA Immunofluorescence assay
IFN Interferon
IPTG Isopropyl β-D-thio-galactopyranoside
IRES Internal ribosomal entry site
Kan Kanamycin
kb Kilo base pairs
kDa Kilo dalton
LB Luria bertani broth
mAb Monoclonal antibody
NS3 Non-structural protein 3
NS4A Non-structural protein 4A
N-terminal Amino terminal
OD Optical density
ORF Open reading frame
PAGE Polyacrylamide gel electrophoresis
PARP Poly (ADP-ribose) polymerase
PBS Phosphate buffer saline
PBST Phosphate buffered saline with Tween-20
PCR Polymerase chain reaction
PI Propidium iodide
PMSF Phenylmethyl sulfonyl fluoride
RNA Ribose nucleic acid
ROS Reactive oxygen species
RT-PCR Reverse transcriptase polymerase chain reaction
SD Standard deviation

v
SDS Sodium dodecyl sulphate
SDS-PAGE Sodium dodecyl sulphate-polyacrylamide gel
TBS Tris buffer saline
TBST Tris buffer saline with Tween-20
TEMED N, N, N', N'-Tetramethylethylenediamine
WHO World Health Organization
β-ΜΕ Beta mercaptoethanol

vi
LIST OF TABLES
Table No. Title Page No.
Table 3.1 List of antibiotics 39
Table 3.2 List of primers used in the study 45
Table 3.3 Composition of different SDS-PAGE gels 57

vii
LIST OF FIGURES
Figure No. Title
Page No.
Figure 2.1 Anatomy of liver
08
Figure 2.2 Hepatitis C virus (HCV): model structure. 19
Figure 2.3 Genome organization of hepatitis C virus
19
Figure 2.4 Replication cycle of HCV
22
Figure 2.5 Mitochndrail mediated apoptosis pathway induced by HCV
proteins
28
Figure 2.6 Diagrammatic illustration of the main molecular pathways
leading to apoptosis
33
Figure 4.1 1% agarose gel electrophoresis shows the results of double
digestion of recombinant pEGFP-C1 plasmids.
63
Figure 4.2 1% agarose gel electrophoresis shows the results of double
digestion of recombinant pFLAG-CMV2 plasmids.
64
Figure 4.3 Maps of recombinant pEGFP-C1 and pFLAG-CMV2
constructs
65
Figure 4.4 Representative chromatograms and sequence homology of
NS3 gene
67
Figure 4.5 Representative chromatograms and sequence homology of
NS3-4A gene
68
Figure 4.6 Representative chromatograms and sequence homology of 69

viii
NS4A gene
Figure 4.7 Representative chromatograms and sequence homology of
NS3 gene
71
Figure 4.8 Representative chromatograms and sequence homology of
NS3-4A gene
72
Figure 4.9 Representative chromatograms and sequence homology of
NS4A gene
73
Figure 4.10 Western blot analyses of FLAG tagged NS3, NS3-4A and
NS4A proteins
75
Figure 4.11 Western blot analysis of GFP tagged NS3, NS3-4A and NS4A
proteins
77
Figure 4.12 (A) Immunofluorescence analysis of recombinant pEGFP-C1
plasmids
79
Figure 4.12 (B) Graphical representation of immunofluorescence analysis of
recombinant pEGFP-C1 vectors.
80
Figure 4.13 (A) Immunofluorescence analysis of recombinant pFLAG-CMV2
plasmids
82
Figure 4.13 (B) Graphical representation of immunofluorescence analysis of
recombinant pFLAG-CMV2
83
Figure 4.14 Cell death induced by NS4A and NS3-4A proteins
85
Figure 4.15 Cell death induced by NS4A and NS3-4A proteins
87
Figure 4.16 Intracellular localizations of NS3, NS3-4A and NS4A proteins
89

ix
Figure 4.17 NS3-4A and NS4A induces mitochondrial fragmentation
90
Figure 4.18 (A) NS3-4A and NS4A induces BAX translocation to
mitochondria, up regulation of BAX and down regulation of
Bcl-XL proteins expression
92
Figure 4.18 (B) Graphical representation of NS3-4A and NS4A induces
mitochondrial fragmentation
93
Figure 4.19 (A) NS3-4A and NS4A increased the mitochondrial superoxide
generation
96
Figure 4.19 (B) Western blot analysis of pro-apoptotic protein BAX 97
Figure 4.20 (A) NS3-4A and NS4A increased the mitochondrial superoxide
generation
99
Figure 4.20 (B) Graphical representation of NS3-4A and NS4A increased the
mitochondrial superoxide generation
100
Figure 4.21 Cytochrome c translocation in NS4A and NS3-4A expressing
cells
102
Figure 4.22 (A) Graphical representation of OXPHOS Complex I activity
between whole HCV genome (HCVcc) and Huh-7 cells
(mock)
104
Figure 4.22 (B) Graphical representation of the effect of HCV individual
proteins on the reduction of OXPHOS Complex I activity
105
Figure 4.22 (C) Western blot analysis of HCV proteins used for the assay. β-
actin was used as an internal loading control
106
Figure 4.23 (A) Western blot analysis of caspase-9 in NS3-4A and NS4A
expressing cells
109

x
Figure 4.23 (B) Western blot analysis of caspase-7 in NS3-4A and NS4A
expressing cells
110
Figure 4.23 (C) Western blot analysis of caspase-3 in NS3-4A and NS4A
expressing cells
111
Figure 4.23 (D) Western blot analysis of caspase-3 in NS3-4A and NS4A
expressing cells
112
Figure 4.24 (A) Chromatin condensation in NS4A and NS3-4A expressing
cells
114
Figure 4.24 (B) Graphical representation of chromatin condensation in NS3-4A
and NS4A expressing cells
115

xi
ABSTRACT
Presently about 170 million of the world population are suffering with Hepatitis C
virus (HCV) that is the major cause of liver diseases, which leads to liver fibrosis,
cirrhosis and hepatocellular carcinoma. Approximately 10 % of the Pakistani
population is infected with HCV, while genotype 3a is the most prominent genotype
with the prevalence rate between 75-90 %. Genetic heterogeneity is the only main
reason to compare the apoptotic pathway in different HCV genotypes. Present study
illustrates that HCV non-structural proteins NS3-4A and NS4A of genotype 3a
induces apoptosis by mitochondrial-mediated, caspase-3 dependent pathway as
confirmed by Hoechst staining. Findings of present study reveal that NS3-4A and
NS4A induces cell death in Huh-7 cells. Moreover, our results revealed that NS3-4A
and NS4A was not only localized on endoplasmic reticulum but also on the
mitochondria. Bax a pro-apoptotic protein translocated to the mitochondria in NS3-
4A and NS4A expressing cells, while up-regulated expression of Bax and down-
regulated expression of anti-apoptotic Bcl-xL protein was also observed with
increased level of cytosolic cytochrome c. High level of mitochondrial superoxide
generation, mitochondrial fragmentation and reduction of OXPHOS Complex I
activity was also observed. In addition, protein immunoblot assays were done which
showed that NS3-4A and NS4A triggers a cascade mechanism of activation starting
from caspase-9, then caspase-7 and caspase-3 ends on cleavage of poly (ADP-ribose)
polymerase PARP. Collectively findings of the present study suggest that HCV NS3-
4A and NS4A alone may possibly induce apoptosis through Bax-triggered,
mitochondrial-mediated, caspase-3 dependent pathway.

Introduction

Chapter 1 Introduction
1
INTRODUCTION
Hepatitis C virus (HCV) isolated in 1989, is a blood borne pathogen causing of acute
and chronic liver disease worldwide. HCV infection is the main source of viral
hepatitis, steatosis, cirrhosis and liver cancer (Moradpour, et al., 2007). World Health
Organization (WHO) declared that 3 % of world’s population is suffering from this
deadly virus (Scheel and Rice, 2013). Each year about 3 to 4 million new cases of
HCV are reported. In Pakistan, approximately 10 % of the population is chronically
infected with HCV out of which 85 % is of genotype 3a (Butt, et al., 2011).
Due to significant genetic heterogeneity, the virus can be classified into six genotypes,
but the major ones are genotype 1-3. These genotypes are further divided into more
than 50 subtypes that differ in their nucleotide sequences by 10-30 % while a
nucleotide variation of 30-50 % is found among genotypes (Hoofnagle, 2002). Source
of this variation is HCV genome coded its error prone RNA polymerase with high
mutation rate (Timm and Roggendorf, 2007).
HCV, a member of flaviviridae family belongs to genus hepacivirus and have a
single-stranded RNA genome with 9.6-kb-long size, which encodes a polyprotein of
about 3,010 amino acids (Alter, et al., 1992). HCV genome contains a single large
open-reading frame (ORF) flanked by non-coding regions at 5' and 3' ends (Tang and
Grise, 2009). The polyprotein is post-translationally cleaved by both viral/cellular
proteases to produce about 10 polypeptides that include structural (core, E1, E2, p7)
and six mature nonstructural (NS2, NS3, NS4A, NS4B, NS5A, NS5B) proteins with
diversity in their functions (Ivanov, et al., 2013). C gene encodes the core (capsid)
protein, which form the viral particle, include the core which forms the viral
nucleocapsid protein and E1, E2 encodes envelope glycoproteins E1 & E2. A short

Chapter 1 Introduction
2
membrane peptide p7 separates both Structural and non-structural proteins from each
other and putative roles of p7 include cation channel activity, productive infection,
virion maturation and egress. (Penin, et al., 2004).
HCV non-structural protein NS2 is a small protein that interacts with its
adjacent protein forming NS2/NS3 protease catalyzing site. Three residues
(His143, Glu163 and Cys184) have been explicitly found to be involved in proteolytic
activity (Grakoui et al., 1993).
69 kDa HCV non-structural protein NS3 is a multifunctional protein and is
indispensable for HCV replication. NS3 has protease and helicase activities which are
essential replicative components of HCV (Kolykhalov, et al., 2000; Chevaliez and
Pawlotsky, 2006; Lam and Frick, 2006). NS3 protein’s N-terminal domain is an
integral part of NS2-NS3 proteinase. NS3 protein has a serine protease activity with
assistance of cofactor non-structural protein NS4A permits its stabilization and
localization at the endoplasmic reticulum (ER) membrane (Bartenschlager, Lohmann
et al., 1995). Another important function of NS3-4A serine protease is to cleave its
own NS2-NS3 junction with all other (NS3/ NS4A, NS4A/NS4B, NS4B/NS5A and
NS5A/NS5B) intersections (Brass, et al., 2006; Tang and Grise, 2009). Whereas on
the other hand C-terminal domain of NS3 is a superfamily 2 RNA helicase/ NTPase,
which unwinds RNA-RNA substrates, resolving secondary structures during RNA
replication and also take part in assembly of viral particle (Ma, et al., 2008; Tang and
Grise, 2009).
HCV nonstructural protein NS4A a 7 kDa is a small multifunctional protein with 54
amino acids residues. NS4A act as an essential co-factor for the NS3 protease enzyme
(Tang and Grise 2009; Joyce and Tyrrell 2010). NS4A has three domain i.e.

Chapter 1 Introduction
3
membrane anchorage, hydrophobic N terminal domain directs NS3/4A complex to the
mitochondrial outer membrane and endoplasmic reticulum, hydrophobic central
domain involved in the activation of NS3 that works as a cofactor peptide and its
acidic C terminal domain promotes helicase directed ATP hydrolysis at the time of
RNA replication (Wolk, et al., 2000; Beran, et al., 2009; Zaidi, et al., 2012). HCV
non-structural protein NS4b is a highly hydrophobic protein that strongly interacts
with lipid moieties and thereby favors viral replication (Palomares-Jerez et al.,
2012).
NS5A is a phosphorylated protein with 56 kDa (basal form) and 58 kDa
(hyperphosphorylated form) molecular weight. It is located in the cytoplasm where it
is found to be in association with endoplasmic reticulum via its amphipathic α-helix
and induces viral replication (Brass et al., 2002; Bartenschlager and Lohmann, 2000).
NS5A has a prominent role in HCV induced disease progression primarily
because of interferon resistance. HCV non-structural protein NS5b is an RNA
dependant RNA polymerase (RdRp) that make use of negative stranded RNA as
the template favoring the synthesis of negative stranded RNA (genomic RNA).
NS5b of HCV shares a common crystal structure of RdRp showing right hand with
the palm, thumb and finger domains (Butcher et al., 2001).
As a result of high genetic mutability among all HCV genotypes they differ in their
biology, transmission dynamics, persistence, disease development and sensitivity to
therapeutics (Simmonds, 2005; Feld and Hoofnagle, 2005; Gottwein, et al., 2010).
High prevalence of genotype 3a is reported worldwide, especially in several countries
of South America and Asia including Pakistan. In Pakistan, type 3 is the prominent
genotype with the prevalence rate 75–90 % (Qureshi, et al., 2009).

Chapter 1 Introduction
4
Lack of cheap and efficient in vitro cell culturing facilities, no animal model to study
HCV pathogenesis, molecular pathways and screening of candidate antiviral drugs are
the main limiting factors in the study of HCV(Couto and Kolykhalov, 2006; Butt, et
al., 2011; Tariq, et al., 2012). Although the complete mechanism of HCV associated
pathogenesis is not completely understood yet although a significant digits of reports
proposed a potential role of apoptosis in HCV infections (Deng, et al., 2008; Malhi
and Gores, 2008). The apoptotic process is thought to be crucial in establishing the
persistent viral infections, viral clearance, antiviral immunity and hepatocellular
carcinoma (Jang, et al., 2014).
Apoptosis can be better explained as “ Scheduled cell death having a sequale of
events” coming after one another in a progrmmed and coordinated manner after any
damage or diseased condition of cell. The possible outcomes of cell death as a result
of apoptotis include cell deformate like shrinkage of cell size, condensation of
chromatin, DNA disintegration, apoptotic body development, vacuolization of
cytoplasmic and cell breakdown (Zhao, et al., 2012). Mitochondria are very
important organelles in cell performing many essential functions and play crucial role
during the induction of apoptosis through increasing permeabilization of
mitochondrial membrane during oxidative stress (Ivanov, et al., 2013). This leading
role of mitochondria has been categorize in three phases: Initiation phase; proapototic
messengers accumulation, responsible for membrane permeabilization (MP), second
decision phase; event of mitochondrial membrane permeabilization (MMP) third and
last degradation phase; commencement of caspases and hydrolases activities (Mordon
and Blanchemaison, 2008; Sala, et al., 2008).
Apoptic cell death occurs through intrinsic and extrinsic pathways. Receptor mediated
extrinsic pathway starts with binding of tumor necrosis factor (TNF)-α, Fas ligand

Chapter 1 Introduction
5
and glucocorticoids to their specific receptors and leads to the activation of its
subsequent messengers e.g., Caspase-8 activation (Bantel and Schulze-Osthoff, 2003;
Chou, et al., 2005; Jang, et al., 2014). This pathway can results in two consequences;
cell survival or cell death.
Intrinsic pathway is initiated as a result of ROS production. It is based on the
transition in membrane permeabilization of mitochondria as a result of proapoptotic
signals which in turn cause the release of inter membrane space proteins (such as
cyto-chrome c, apoptosis inducing factor (AIF), Endo G and Smac/DIABLO (Second
mitochondria-derived activator of caspase /direct IAP binding protein with a low pI)
in the cytosol and form apoptosome having ATP and APAF-1 (Fischer, et al., 2007).
Activation of Caspase-9 eventuates following the formation of apoptosome (Galle, et
al., 1994). Caspase-9 promotes the activation of effector Caspase-3, PARP poly
(ADP-ribose) polymerase and downstream apoptotic events (Owen, et al., 1994;
Kumar, 2007).
There are different reports suggesting apoptotic and anti-apoptotic functions of
different HCV proteins, for instance NS3, NS4A, core and E2 are to induce apoptosis
(Prikhod'ko, et al., 2004; Benali-Furet, et al., 2005; Nomura-Takigawa, et al., 2006;
Lee, et al., 2007). Clones of HCV genotype 2a in human hepatocellular carcinoma
cell line have reported to be an efficient replication system for virus and proved HCV
induces apoptosis in the cell culture system (Deng, et al., 2008). In Chronic HCV
infection apoptosis is related to be involved in liver damage. However, extensive
mechanism is still unclear (Valva, et al., 2010). Infection with hepatitis C virus causes
hepatocellular death by tempts mitochondrial mediated death signaling pathways
(Deng, et al., 2008).

Chapter 1 Introduction
6
Many findings provide ample evidence of involvement of various structural and non-
structural proteins in the induction of extrinsic as well as intrinsic apoptosis pathways.
HCV non-structural protein NS4A and NS4B has been studied with reference to the
induction of mitochondrial mediated apoptotic pathway. Among HCV non-structural
proteins, complex of NS3-4A proteins is the most potent proteins that can damage
host cellular machinery for viral and disease propagation, that can favor
oxidative stress, production of reactive oxygen species with demolition of
mitochondria. However, there is no conclusive study so far that describes potential
role of NS3-4A protein in the induction through Bax-triggered, mitochondrial-
mediated, caspase-3 dependent pathway. Therefore, the present study was designed to
investigate, the cellular and molecular effects of HCV non-structural NS3-4A protein
in the induction of mitochondrial death pathway.
1.2 Aims and objectives of the study
Aims and objectives of the current study are:
• Establish a cell culture based system expressing HCV non-structural proteins.
• Evaluate protein expression of different key cellular genes involved in
mitochondrial mediated apoptosis in transiently expressing cell culture based
system (cell line containing HCV non-structural proteins).
• Investigate the HCV induced mitochondrial mediated apoptotic pathway.

Review of
Literature

Chapter 2 Review of literature
7
REVIEW OF LITERATURE
2.1 Anatomy and Physiology of Liver
A vital lobular organ of the human body located in upper right quadrate of
abdominal cavity “Liver” consist of four lobes. Maintenance of homeostasis,
energy metabolism, detoxification of toxic substances, processing, storage and
distribution of dietary products like lipoproteins, purines, ketones bodies and
glucose are the key roles of liver in human body (Malarkey et al., 2005). When
different types of functional cells combined to one structural unit in liver then this
unit is called as Acinus. Hepatocytes (liver cells) make up 70-80 % of liver
volume, cells of bile duct, Kupffer cells, Endothelial cells, Oval cells, Pit cells and
Hepatic stellate cells (HSC) form remaining 20-30 % of liver. Liver parenchymal
cells cover the whole liver and thus have high rate of metabolic activity. Liver
epithelial cells form threads and junctions between them allow the fluids and
nutrients movement between neighboring cells. Endothelial cells form Hepatic
sinusoids which have large endocytic activity. Mediators like Interferon,
Interleukin 1, 6 and nitric oxides secreted by these endothelial sinusoids act as
paracrine secretions (Kmiec, 2001; Malik, Selden, and Hodgson, 2002).
Macrophages of liver i.e. Kupffer cells have immunomodulatory functions. These
cells play important role in the antigen/bacterial destruction, removal of debris and

Chapter 2 Review of literature
8
Figure 2.1: Anatomy of Liver. Liver is composed of various types of functional
cells. Liver anatomy showing the location of different cells types. Major portion of
liver consist of hepatocytes, rest is made up of endothelial cells. Blood cells
migrate through the fenestrations of liver. Adapted from (Saladin, 2012).

Chapter 2 Review of literature
9
dead cells from the blood stream by producing hydrolytic enzymes, hydrogen
peroxide, superoxide anions and eicosanoids. Pit cells are intrahepatic leukocytes
of liver believed to have Natural killer activity. Pit cells involve in cytotoxic role
against viral infections and tumors (Wake, 2004).
2.2 Hepatitis
Hepatitis is a Latin word which is derived from two words Hepa means “liver” and
titis means “Inflammation, so hepatitis is the term used for liver inflammation.
There are many types of hepatitis like drug induced hepatitis, alcohol, toxins,
chemicals, parasitic, bacterial and viral hepatitis. Viral hepatitis is a major health
issue around the world, especially of many Asian countries (Lu et al., 2003).
Depending on the severity of disease, hepatitis has two clinical conditions acute
and chronic. Acute is condition in which disease is progressive and short term
which later on convert to chronic condition while, chronic is long term and more
aggressive lead to severe symptoms.
2.3 Viral hepatitis
The term viral hepatitis is coined for the viruses targeting hepatocytes (liver cells),
therefore, also known as hepatotropic viruses. This group of viruses is not new
rather it is as old at the beginning of mankind. It includes a diverse group of
viruses named as hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis C
virus (HCV), hepatitis D virus (HDV), hepatitis E virus (HEV), hepatitis F virus
(HFV) and hepatitis G virus (HGV). After infection, they can move from acute to
chronic infections. As very little is known about HFV and HGV, so hepatitis A
through E will be discussed here. Each of these viruses infects and damages the
liver causing the classic symptoms of jaundice with elevated level of liver

Chapter 2 Review of literature
10
enzymes. HAV and HEV use fecal oral route for its transmission and infection
mostly resolve after acute stage. HBV, HCV and HDV conversely spread via blood
transfusion, sexual, vertical and parenteral, can lead to both acute and chronic
infection. All this viral hepatitis can be diagnose by testing patient’s blood by
serological and molecular tests (Schiff, 2007).
2.3.1 Hepatitis A virus
HAV belongs from Picornaviridae family and this virus cause acute viral hepatitis
which is the most common viral hepatitis worldwide (Feinstone et al., 1975).
Route of transmission for the spread of HAV is mainly by the fecal-oral route via
contaminated food and water. HAV is endemic in poor and socioeconomically
weak countries with lack of good hygienic food, poor sanitation, clean drinking
and irrigating water resources. Transmission through injection drug user, sexual
contact, blood transfusion and needle stick injury are rarely reported. Mother to
fetus HAV transmission is less frequently reported (Mohebbi et al., 2012; Franco
et al., 2012).
2.3.2 Hepatitis B virus
Presently about 350 million peoples around the globe are HBV carriers. It is
double stranded DNA virus replicate through reverse transcription and is a member
of hepadnavirus family. Mostly transmission through injection drug user, sexual
contact, blood transfusion and needle stick injury and mother to fetus transmission
of HBV is less primary mode of infection. Identification of Hepatitis B surface
Antigens (HBsAg) of HBV can be done by immunochromatographic technique,
enzyme linked immune sorbent assay (ELISA) and final confirmation of this viral
infection can be by amplification of HBV DNA by polymerase chain reaction

Chapter 2 Review of literature
11
(PCR). But sometime the viral DNA amplification from patient’s serum shows
negative results but HBV still there in hepatocytes, this (false negative) dormancy
condition is termed as HBV occult infection (Arababadi et al., 2012).
2.3.3 Hepatitis C virus
In 1989 HCV virus was discovered and was known as non-heptatitis A, non-
heptatitis B virus (Farci et al., 2002). HCV is positive sense single stranded RNA,
enveloped virus belongs to genus Hepacivirus and family Flaviviridae. Based on
genotypic analysis HCV genome has been classified into six main genotypes.
Based on nucleotide sequences these genotypes 30-33 % on average differs from
each other and closely related subtypes which show less heterogeneity of 20-25 %
in nucleotide sequences like genotypes 1a and 1b. Error-prone synthesis of RNA in
HCV infected patient produces many variants of HCV known as quasispecies.
HCV is a fatal blood borne pathogen targeting hepatocytes. At present nearly 160
million peoples are HCV infected and in many cases it remain asymptomatic acute
infection but 50 % – 80 % of unclear infected cases progress to a persistent state of
viral replication and results in hepatitis (Farci et al., 2002). HCV molecular
organization, life cycle, replication and pathogenesis will be explicate below in
details.
2.3.4 Hepatitis D virus
In 1977 HDV was discovered (Rizzetto et al., 1977) and termed as “delta agent”.
HDV is circular single stranded RNA virus, 36-nm particle size and show gross
similarities with main viruses, viral satellites and viroids of plant origin. HBV,

Chapter 2 Review of literature
12
HCV and HDV have common routes of transmission. This is defective RNA virus
can only infect persons that have HBV infection which play the role of carrier host.
HBV and HDV together cause progressive chronic liver disease which could lead
to cirrhosis and cancer of liver in about 80 % of patients that have both HBV and
HDV infection. This virus is significant cause of severe acute liver injury in many
countries of the world (Kumar et al., 1992). Prevalence of HDV infection in
HBsAg positive individuals are about 16.6 % in Pakistani population. The major
source of HDV is the poor hygienic conditions, yet there is some decline in the
HDV cases in Pakistan. The study showed that these individuals infected with
HBV may have either HDV super infection or co-infection of with HBV (Mumtaz,
2005).
2.3.5 Hepatitis E virus
HEV is RNA based positive polarity single stranded genome virus without envelop
is about 32 to 34 nanometers in size. It is the only member of the Hepevirus genus
in the family of Hepeviridae. Like HAV the transmission and distribution of this
hepatitis virus depend on the geographic location. In 1983 HEV was for the first
time isolated from the facial sample of volunteer who was experimentally infected
with HEV (Balayan et al., 1983). This virus is generally categorized in four groups
i.e. 1, 2, 3 and 4 with single serotype. In about 96 % of HEV infected cases,
increased level of serum bilirubin and elevated liver enzymes have been observed.
But neither HAV nor HEV infection progress to chronic case and the mortality rate
in HEV infection is high as compared to HAV in pregnant women especially (Zhao
et al., 2009; Li et al. 2006).

Chapter 2 Review of literature
13
2.4 The discovery of Hepatitis C virus
Acute viral hepatitis has history of over 2,000 years described by Hippocrates.
Within last thirty years epidemiology, Biology, Immunology, Pathology, Genetics
and clinical feature different viral hepatitis have been identified as hepatitis has a
very long history in humans. Previously it was speculated that hepatitis might be
cause by an infectious agent, after many decades with the development of advance
complicated research tools and methodology researcher discovered different viral
hepatitis A, B, and C. Detection of hepatitis infection in blood transfusion
recipients lead towards the discoveries of these viruses. Patients with increased
serum liver enzymes level with symptoms of fever, jaundice, vomiting and nausea
were documented before 1940s, after almost 150 years of first ever human blood
transfusion (Beeson, 1943). Clinical history and infection pattern in viral hepatitis
gave a clue that more than one type of virus is responsible of hepatitis instead of
"serum" hepatitis. In 1947 for the first time for the hepatitis A and hepatitis B
terms were brought in to differentiate between the two different forms of
hypothesized hepatitis (MacCallum, 1947).
With the advancement in diagnosis of viral infection, in 1974 serological
diagnostic procedure was discovered for HAV and HBV infection and so their
contribution in spread of post transfusion hepatitis was cleared (Prince et al.,
1974). Since the serum screening tests for hepatitis A and B in donor blood was
not screened it was obvious that there is still another unrevealed infectious agent
liable for mass post transfusion hepatitis infections, so for the first time ever “non-
A, non-B hepatitis” (NANBH) terminology was used (Alter et al., 1975; Feinstone
et al., 1975). With the continuous research progress for about decade, while

Chapter 2 Review of literature
14
digging for this NANBH's causative agent, during that period other viruses causing
hepatitis i.e. HDV and HEV were discovered (Balayan et al., 1983; Bonino et al.,
1984; Rizzetto et al., 1977). Since 1940 or longer that virus might exist but in 1989
it was identified for the first time. Chiron Corporation in USA unambiguously
identified and did genetic mapping of HCV by using very advance tool i.e.
molecular biology and recombinant DNA technology (Shepard et al., 2005).
2.5 Epidemiology of HCV
HCV infection is a global health problem and victimizing 180 million people
worldwide out of which 130 million are chronic carrier (Shepard et al., 2005).
Countries like Canada, Australia, USA, Japan and in most of European countries
incidence of HCV infection is about > one percent. While approximately 2 %
population of Africa and Southeast Asia infected with HCV (Nakamura et al.,
2008). According to epidemiology survey of HCV about 10 million Pakistanis are
suffering from HCV infection (Raja and Janjua, 2008) and provincial HCV
surveillance data revealed that Punjab province is ranked highest in HCV
prevalence. And in male population incidence of this infection is higher as
compared to female. More than half of the HCV positive population belong from
age group of 40-50 years (Idrees and Riazuddin, 2008). Healthy blood donors
show high prevalence of HCV infection and different studies conducted at national
level reported about 4 % of blood donor are sub-clinically infected with HCV
(Waheed et al., 2009). According to Arif et al. (2008) hemophilic and thalassemic
population showed about 49% HCV infection and in health care workers HCV
infection percent incidence is almost 5 % (Hamid et al., 2004). Every year Pakistan
adds approximately 0.37 million new cases of HCV infections (Ali et al., 2009).

Chapter 2 Review of literature
15
2.6 HCV transmission
In past HCV main mode of transmission was through blood transfusion, now HCV
first and foremost spread by means of direct contact to HCV infected blood, body
fluid and blood products (Burns et al., 2003). HCV infections in children and HCV
positive antibodies in their parents clearly depict that transmission of HCV to them
probably by exposure to infected blood or contaminated syringes (Strickland et al.,
2005). According to CDC (center for disease control) HCV transmission as
compared to Human Immuno deficiency (HIV) via sexual intercourse is
infrequently but from the saliva and semen its virions have been isolated (Schultz,
2005). There is high rate of HCV infection in individual with multiple sex partners,
sex workers and homosexual however, there are very less cases of sexual
transmission have been reported so far. In case of intravenous drug user, sharing of
shaving razors, toothbrushes are also potential sources of HCV transmission
(Pasquier et al., 2003; Schultz, 2005). Hospital acquired infection of HCV from
patient to healthcare worker and from patient to patient is very common in which
needle-stick injury contributes about 10 % in the transmission of HCV infections
(Foley et al., 2009). Unsterilized razors, scissors, brushes at barber shops, piercing
of different body parts and tattooing helps in spreading of HCV infection (Burns et
al., 2003).
Transmission via hemodialysis (HD) is very rare in developed countries but this
rate is quite high in under developed and in developing countries, in which HCV
infected HD machines and biologically unhygienic/infectious practices of health
care workers are in common practices (Bracho et al., 2005; Nemati et al., 2009). In
semen, cervical smear, viginal fluids, saliva, nasal secretion and breast milk HCV

Chapter 2 Review of literature
16
RNA has been also identified (Lock et al., 2006; Aaron et al., 2008). Mother to
child (vertical transmission) are slightly high i.e. 5-8 % in pregnant women with
increased viral load (HIV/ HCV co-infected). Role of mosquito in the spread of
HCV is still unclear. But few studies reported the presence of HCV RNA in
mosquito fed on HCV positive blood (Chang et al., 2001).
2.7 Treatment of HCV
HCV treatment regimens are constantly improving with time and have HCV
replication inhibiting capabilities and with improvement. First approve drug for the
treatment of HCV was recombinant interferon alfa-2b. Interferons are basically
cytokines that have antiviral and anti-proliferative activity consequent of multiple
gene expression as virus enters the host body. By this way viral replication inhibits
and induces anti-proliferative effect on the cell (Neumann et al., 1998). In
combination ribavirin increases treatment response rates in HCV infected patients
when treatment with interferon and ribavirin in combination given for 24 weeks.
But failure to this response is also observed in non respondent and relapse patients
(Chevaliez et al., 2007). But treatment of HCV with Peg-IFN has is more effective
but that in combination with ribavirin the treatment response is more robust. Those
patients that were treated with ribavirin only show poor treatment response. When
there is mutation at 415 codon amino acid in NS5B domain (RNA-dependent RNA
polymerase) ribavirin shows resistance. Genetic diversity of HCV affect antiviral
treatment response in HCV infected patients (Hayashi and Takehara, 2006).
2.8 HCV Genomic organization
HCV viral genome is described as a positive sense single-stranded RNA of almost
9.6 kilo base approximately 10000 nucleotides (Takamizawa et al., 1991). HCV

Chapter 2 Review of literature
17
genome encodes a single long open reading frame (ORF) encoding a polypeptide
of 3010 amino acids residues (Fig 2.2) (Grakoui, 1993). ORF of HCV translates
through a ~340 nucleotide and 5’ un-translated regions (UTRs) which performs
entry site for internal ribosome (IRES). In the absence of translation initiation
factors RNA binds with 40S ribosomal subunit in such a way that P site become
direct neighborhood for initiation codon (Hellen and Sarnow, 2001). It allows
ribosomes to bind ORF’s start codon. Cellular and viral proteases enzymes cleave
newly formed HCV polypeptide into three main structural proteins i.e. for core
protein/nucleocapsid C, envelop glycoprotein1 E1 and for envelop glycoprotein2
with six non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B) is E2
(Kato, 1993).
Very less is known about NS1 but it is thought that its functions are linked with
E2. NS2 translate in trans-membrane proteins whereas 630 amino acid protein NS3
having three enzymatic activities domains, 180 residues peptide with N terminal
and C terminal have serine protease, helicase and nucleoside triphosphate activities
respectively. Unwinding of viral genome and cleavage of different non-structural
proteins (NS3-4A, NS4A-4B, NS5A-5B) at different junction sites by NS3
protease (Bartenschlager et al., 1993).
NS4 plays the role of co-factor for NS3 protease which is indispensable for
polyprotein processing (Failla et al., 1994). NS5a are interferon-resistance protein
whereas NS5b play role of RNA-dependent RNA polymerase indispensable for
viral replication (Behrens et al., 1996).

Chapter 2 Review of literature
18
Figure 2.2: Hepatitis C virus (HCV): model structure. Adopted from
http://www.the-scientist.com/?articles.view/articleNo/24336/title/
Culturing-Hepatitis-C/
Figure 2.3: Genome Organization of Hepatitis C virus.

Chapter 2 Review of literature
19
2.9 HCV replication and role of viral proteins
HCV replication mainly take place in hepatocytes but there are many evidences
that it can replicate in macrophages, B and T cells in vitro cell lines (Esteban et al.,
1996), in gut epithelial cells (Deforges et al., 2004) and nerons (Forton et al.,
2004). HCV cell cycle starts with the attachment of this deadly virus with host cell
by specific interaction between host cell surface a receptor protein (CD81 a
member of tetra-spanin) cell surface protein express on all cells, and protein (E2
glycoprotein evident from in vitro studies) on the HCV surface (Pileri et al., 1998).
Furthermore, the virus has ability to enter in host cell via low density lipoprotein
receptors binding (Monazahian et al., 1999). While after many structural
rearrangements in E1 fusion peptide take part in fusion of membrane of the E1
which facilitates in viral particle entry into the host cytoplasm (Flint et al., 1999).
As virus binds to its receptor, internalization completes with the release of
nucleocapsid into the cytoplasm. After decapsidation, translation for polypeptide
and replication occur in cytoplasm. Protein translation directly takes place as HCV
RNA is positive polarity RNA and this RNA function as mRNA as well. This
translation is cap independent unlike other cellular RNAs where cap binding to
ribosome machinery is prerequisite for translation. Translation initiation of HCV
RNA starts by ribosomal binding of 5/ end of IRES. Rough endoplasmic reticulum
(RER) is the main site for HCV RNA translation for synthesis of single
polypeptide. For the production of structural and non structural proteins co/post
cleavage of this single polyprotein take place via cellular and viral proteases. For
the replication of HCV genome, synthesis of negative strand RNA take place by
RNA-dependent RNA polymerase (NS5B) which will later on serve as template

Chapter 2 Review of literature
20
for the synthesis of positive sense RNA (El-Hage and Luo, 2003; Gosert et al.,
2003). Viral non structural and proteins of host cell form a membranous web
known as replication complex nearer to perinuclear membranes is the site for
replication and post-translational process. Process like encapsidation of viral
genome and enveloping of nucleocapsids take place in ER while final processing
and mature at in Golgi apparatus after that newly formed virions release by
exocytosis in pericellular spaces (Penin et al., 2004).
2.9.1 Core protein
Core protein contains HCV first 191 amino acids, 3 domain (on the basis of
hydrophobicity) basic protein which forms viral nucleocapsid. In the serum of
HCV infected patient 21 kDa core proteins has been isolated. This protein is
involved in activation of many signaling pathways by interacting with different
host cellular proteins. The significant role of core protein is encapsidation of viral
genome and assists its replication (Imran et al., 2012). Cytosolic membrane-bound
this core protein has been linked with ER, mitochondria, nucleus and lipid droplets,
Involved in steatosis and in liver carcinogenesis (Hope et al., 2002; Lerat et al.,
2002). Cell functions can be influence as HCV core protein interacts with different
host.

Chapter 2 Review of literature
21
Figure 2.4: Replication cycle of HCV. (1) Binding of virus to receptors. (2)
Endocytosis. (3) Uncoating. (4) Translation of genomic RNA and proteolytic
processing of the polyprotein associated with the endoplasmic reticulum. (5) RNA
replication complex formation in the membranous web. (6) Virions
morphogenesis(genome replication). (7) Encapsidation of genomic RNA and
assembly of virus particles through interactions between the ER and lipid droplets
(maturation). (8) Release of virus particles from the cell (Exocytosis). Adapted
from New and experimental therapies for HCV (Arema and Jacobson, 2009).

Chapter 2 Review of literature
22
2.9.2 Envelop proteins
HCV consist of highly glycosylated two envelop proteins (E1 and E2) having
significant role in HCV entry in host cell. These E1 and E2 proteins are 35 and 72
kDa correspondingly. Envelope proteins are considered to involve in cell-mediate
cell entry by identifying receptor proteins of cellular membrane probably for
membrane fusion. E1 and E2 proteins leads polyprotein precursor to the ER moves
to ER lumen and after cleavage these remain fixed on the inner side of ER lumen.
Two hypervariable regions have been recognized in E2 protein which is
responsible for mutation, possibly neutralizing antibodies target (Ashfaq et al.,
2011).
2.9.3 P7 protein
A polypeptide found between E2 and NS2 genes, a 63-amino acid is known as P7.
It is a trans-membrane protein found on ER with two domains one connected
through cytoplasmic loop and other is adjusted in the direction of ER luman. It is
clear that C-terminal this trans-membrane domin of P7 can perform a signal
sequence direct NS2 translocation to cleave by host cell signal peptidases in ER
lumen. P7 proteins also form ion channels causing HCV infection’s pathogenic
effect. P7 HCV protein has similar features like a group of proteins known as
viroporins and this protein is crucial for viral particle assembly and release (Ashfaq
et al., 2011).

Chapter 2 Review of literature
23
2.9.4 Nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B)
2.9.4.1 NS2
A trans-membrane protein 21-23 kDa NS2 is indispensable for in vitro and in
vivo for viral replication cycle completion (Khromykh and Westaway, 1997). N-
terminal amino acids are highly hydrophobic with 3-4 trans-membrane helices
anchored in ER membrane. Some part of C-terminal of NS2 and NS2/3 show auto
protease activity collectively with of N-terminal NS3. Chelating agents (EDTA)
studies showed lose of protease activity of NS2-3 metalloprotease have exogenous
zinc medicated protease. This zinc element is important for NS3 structural
stabilization present at its active site (Lorenz et al., 2006).
2.9.4.2 NS3
A multifunctional NS3 67 kDa protein’s C-terminal show NTPase/helicase
whereas N-terminal performs serine protease activity (Gallinari et al., 1998). NS3
and NS4A are ER membrane bounded proteins (Wolk et al., 2000). N-terminus of
NS3 protease of HCV is involved in cleavage between NS3-4A, 4A-4B, 4B-5A
and 5A-5B. NTPase/helicase enzymatic activity of the NS3 is crucial for HCV
RNA replication. Presumed functions of this protein may be unwinding of double
stranded RNA intermediates during replication, RNA secondary structures removal
or to split nucleic acid binding proteins from genome. Current advancement in
molecular mechanisms understanding NS3 could be use novel antiviral
strategy (Serebrov and Pyle, 2004).

Chapter 2 Review of literature
24
2.9.4.3 NS4A
It is 54 amino acids residues containing HCV protein serve as cofactor for NS3.
Deletion analysis of this protein’s showed that N-terminal is involved in directing
NS3 to the ER membrane (Wolk et al., 2000). NS4A and NS3 interaction is
stimulated between NS3 core residues and NS4A C-terminal. This interaction
favors active site activation of NS3 which leads to more proficient protease
cleavage (Kim et al., 1996). NS5A phosphorylation takes place by NS4A and also
directly interacts with NS5A. Amino acids deletion from the central region of
NS5A protein revealed NS4A dependent phosphorylation of NS5A (Asabe et al.,
1997).
2.9.4.4 NS4B
Small 27 kDa hydrophobic protein NS4B, have significant role in employment
many viral proteins (Lundin et al., 2006). This protein directly interacts with
NS4A and indirectly with NS3 and NS5A. It is ER localized integral membrane
protein and ER membrane also contains non-structural proteins (Lin et al., 1997).
Electron microscopy data on NS4B protein of HCV revealed that it from a
structure known as membranous web by changing ER membrane morphology
(Gretton et al., 2005). It is evident from studies that replication complex
formation takes place here as all viral proteins were localized to ER (Egger et al.,
2002). Oncogenic and cytopathic effect of NS4B in transgenic mice’s liver is still
unclear (Wang et al., 2006).

Chapter 2 Review of literature
25
2.9.4.5 NS5A
A phospho-protein NS5A is hydrophilic in nature without any transmembrane
domains, performs different tasks for HCV like activation of cell signaling
pathways, interferon responses and viral replication (Macdonald et al., 2004).
Some studies reported its linked with other HCV proteins purposed it as part of
replication complexes (Neddermann et al., 1999). NS5A mutations are critical for
maintaining replicon cell line and HCV replication (Lohmann et al., 1999). NS5A
gain its popularity due to its crucial part in interferon response modulating it also
contain interferon resistant associated region which impart resistance in HCV virus
against treatment with it (Gale et al., 1997). It is suggested that NS5A has
interactions with numerous proteins of cell signaling pathways and influencing
their function. NS5A can alter the 3 main pathways of MAPK (regulate growth and
activation) which are responsible for host cell mitogenic signaling. Both pro-
apoptotic and anti-apoptotic pathways are also modulated by NS5A proteins.
NS5A is also associated in altering phosphatidylinositol 3-kinase signalling and
ROS pathways that are consider to cause transformation in hepatocytes and leading
to hepatocellular carcinoma (Macdonald et al., 2004).
2.9.4.6 NS5B
A 65 kDa NS5B protein is a tail anchored protein which substitute for RNA
dependent RNA polymerase so have crucial role in HCV new genome
synthesis (Behrens et al., 1996). This polymerase activity was discovered after
sequence analysis of an amino acid motif GDD (Yamashita et al., 1998). NS5B
structural organization is distinctive polymerase (right hand shape) with encircled

Chapter 2 Review of literature
26
active site having sub domains of finger, palm, and thumb (Lesburg et al., 1999).
RNA genome serve as template for synthesis of negative strand of a
complementary and the succeeding synthesis positive strand RNA of genomic
from this negative sense RNA strand intermediate. Playing central part in HCV
replicase, now NS5B is potential biomarker for new pharmaceutical (antiviral)
discoveries (De Francesco and Migliaccio, 2005).
2.10 Apoptosis and HCV
Apoptosis of hepatocyte is manifested in liver related injury either by metabolic
diseases, alcoholic, autoimmune, drug induced or viral hepatitis. For the normal
cellular homeostasis apoptosis plays a critical role by removing damaged,
abnormal proliferated and aged cells (Shin et al., 1998; Favaloro et al., 2012).
Sometime certain stimuli cause collapse apoptosis mechanism leads to form
various in tumor which are resistant to cytotoxic therapy (Pitot, 1993). Apoptosis
in mammalian cells can be induced via two major pathways 1st death receptor
pathway (extrinsic pathway) and 2nd
apoptosis pathway (intrinsic pathway) is
mitochondrial mediated in response to viral proteins, DNA damage and oxidative
stress. In mitochondrial mediated apoptosis expression Bax, Bad, Bak pro-
apoptotic genes and Bcl-2 or Bcl-xL anti-apoptotic protein express leading to PM
(Fischer, 2007) as shown in figure 2.7.

Chapter 2 Review of literature
27
Figure 2.5: Interference of HCV proteins with the apoptosis cascade. Pro and
anti-apoptotic effects of HCV proteins converge at the mitochondria (e.g., NS2,
NS3/4A, NS5A, E2, core), partly indirectly via p53 (NS5A, core) and activation
of PKB/Akt, c-Jun kinase JNK (core) or NFkB (NS5A). Adapted from (Fischer,
Baumert et al. 2007).

Chapter 2 Review of literature
28
HCV proteins perform mitochondrial mediated apoptosis by playing role as
antiapoptotic or proapototic proteins, it all depends on the expression system and
experimental condition (Marusawa et al., 1999). NS3 and NS5a protein of HCV
have anti-apoptotic while core protein has regulatory properties (Macdonald et al.,
2004). It is evident from many studies that hepatocytic apoptosis is involved in
pathogenic effect of HCV infection. It is thought that core protein of HCV restrains
c-myc, TNF-α, cisplatin or Fas mediated apoptosis but mechanism behind this
HCV core protein involvement is still not fully known. Due to unavailability of
effective tissue culture for HCV or animal model, the study of hepatocyte
apoptosis induced by HCV infection (Zekri et al., 2011).
2.11 Apoptosis
Employment of complex signalling mechanism for the removal disease or
damaged condition of cells leads by series of steps in a programmed and codinated
manner or we can say programmed cell death coordinnated with sequale of events.
There are many stimuli and causative agents that are consider to take part in the
event of apoptosis. This phenomena starts with discrepancy in the normal ongoing
functions in the cell. The most important of which is irregularity in redox condition
of the cell. Broadly apoptotic stimuli can be categories in two main groups; stress
and physiological stimuli Physiological stimuli like (viral, irradiation, UV light and
bacterial infections) lead to death by cell surface receptors (TNF or CD95) and
initiate mitochondria induced stress apoptosis (Gulbins, 2003).
The leading and vital organelle in performing cellular important functions and even
in apoptosis is mitochondria. Whenever cell faces oxidative stress condition the
membrane permeability of mitochondria increases leading to induction of

Chapter 2 Review of literature
29
apoptosis. The major function of mitochondria in apoptosis can be categories in 3
parts: Initiation, decision and degradation phases. In initial phase gathering of
proapototic messengers implicated in MP, in decision phase event of MMP take
place and in degradation phase caspases and hydrolases activation happen (Mordon
and Blanchemaison, 2008; Sala et al., 2008).
Mitochondria is target organelle for different stress responses type of like reactive
oxygen species, ceramide, fatty acids their oxidation products, superoxides, nitric
oxide, hydrogen peroxides etc these pro-apoptotic signals leads to mitochondrial
MP as a results in release of many mitochondrial proteins in cytosol which ends on
mitochondrial induced apoptosis. Metabolites and enzymes concentration for
instance NADPH oxidases persuade the strength of MMP (Wang et al., 2008).
Mitochondria stop pro-apototic proteins from performing their duties in cytoplasm
so we can call it as act as gatekeeper. Protein like cytochrome, caspase 9,
mitochondrial activator of caspases, apoptosis-inducing factor, high temperature
requiring proteins and endonuclease G release from mitochondria as result of
marked increase in leading apoptosis (Gulbins, 2003).
2.12 Apoptosis molecular mechanism
Ubiquitous form of cell death (Apoptosis) takes place in hepatic diseases.
Apoptosis is a form of cell death occurring in human liver diseases.
Morphologically apoptotic cell show distinctive features cytoplasmic shrinkage,
fragmentation of nucleus, condensation of chromatin, plasma membrane blebbing
and apoptotic bodies. There are two ways of hepatocytes apoptosis first death
receptor (apoptosis via extrinsic pathway) secondly by cellular perturbations

Chapter 2 Review of literature
30
(apoptosis via intrinsic pathway) and both congregate on mitochondria (Goldman,
1994).
i) The extrinsic pathway
Extrinsic mean outside, cell surface transmembrane proteins act as death receptor
in extrinsic pathway, these belongs from receptor super-family of tumor necrosis
factor and nerve growth factor. These are distinct on basis of their specificity to
ligand i.e. Fas ligand, tumor necrosis factor alpha or tumor necrosis factor related
apoptosis inducing ligand. These cell surface proteins have N-terminus
extracellular that binds with ligands and intracellular C-terminal domain containing
a conserve region which is called as death domain. This pathway activate when an
external ligand such as tumor necrosis factor, Fas ligand, glucocorticoids,
cytokines binds with their specific receptor leading to the down regulation of
secondary messengers with the activation of caspase 8. Caspase 8 cleaves
proapoptotic BH3 only protein (Bcl-2 family) Bid to tBid proteolytically, leads to
activation of Bax and Bak results in mitochondria pore formation. The outcome of
this pathway can be either cell survival or death as a result of apoptosis at all
depends on signals availability and induced pathways. Activation of transcription
factor (NFkB) can also occur due the balance between them and give a cell
survival signal (Owen et al., 1994).
ii) The Intrinsic (Mitochondrial mediated apoptosis) pathway
Intrinsic mean starts from inside, this type of apoptosis activates in response to loss
of cell-survival factors, DNA damage and different severe intracellular stresses
receive and induce by cell’s membrane bound organelle like lysosomes and ER. C-
jun N terminus kinase activator of intrinsic pathway of apoptosis can be activated

Chapter 2 Review of literature
31
by DNA damage and steatosis. All these processes transduced by proteins of Bcl-2
family (pro-apoptotic and anti-apoptotic proteins) come together on mitochondria
so called as mitochondrial or Bcl-2-regulated apoptic pathway (Dreschers and
Bock, 2003). It starts with the production of ROS modification in MMP as a result
of proapoptotic signals induces the release of many intermembrane space proteins
e.g Endo G, apoptosis inducing factor, cyto-chrome c etc induces apoptosome
formation which eventuates activates Caspase 9 (Galle et al., 1994). Caspase 9
additional activates caspase 7 and caspese 3 (effectors caspases) ending with the
start of membrane degradation, apoptotic bodies formation and fragmentation of
DNA (Owen et al., 1994). Figure 2.6 below in brief represents the types and
mechanism apoptotic pathways.
2.12.1 Bcl-2 Apoptosis Regulator Family
Bcl-2 is apoptosis regulator family proteins which govern MMP and can perform
pro-apoptotic proteins (BAD, Bak, Bok, Bax etc) or can play as anti-apoptotic
proteins (Bcl-w, Bcl-2 proper, Bcl-xL). Mitochondrial integrity is control by
protein of Bcl-2 family present on the outer membrane of mitochondrial. About 24
years ago B-cell lymphoma-2 (Bcl-2) consequence of up-regulation in follicular B
cell lymphoma, it was declared that Bcl-2 doesn’t promotes proliferation in tumors
rather it restrains apoptosis (Hockenbery et al., 1993). During research it is
discovered that in this family there are about 25 members and on the bases of
homology to certain extend this Bcl-2 family is segregate in two sub-families i.e
anti-apoptotic and pro-apoptotic subfamilies. Over-expression of this protein is

Chapter 2 Review of literature
32
Figure 2.6: Diagrammatic illustration of the main molecular pathways
leading to apoptosis. In the extrinsic pathway upon ligand binding to specific
receptors the DISC complex is formed and caspase 8 activated. In the intrinsic
pathway release of cyt c from the mitochondria result in the formation of the
apoptosome and activation of caspase 9. Caspase 8 and 9 then activate downstream
caspases such as caspase 3 resulting in cell death. The two pathways are connected
through the cleavage of the BH3 only protein BID. Adapted from (Favaloro et al.,
2012).

Chapter 2 Review of literature
33
attributable to translocation of chromosome as it has been observed in many
hematological malignancies (Reed et al., 2005). In different model systems for
example glucocorticoid-induced apoptosis of lymphoma cells, Bcl-2 had proved its
role as inhibitor of apoptosis.
2.12.2 Caspases executors of Apoptosis
Caspases cysteine proteases play important function in inflammation, necrosis and
apoptosis and belong to family cysteine-aspartic or we can say cysteine-
dependent aspartly-directed proteases. These proteases are found in many
membranous organelles and cytoplasm. Based on structural analogy and substrate
specificity, 14 different caspases have been found (Kaufmann and Earnshaw,
2000). These are also known as executioner based on their significant role in cells
for apoptosis and synchronized at post-translational stage. Produced in inactive
form known as pro-caspases with two subunits (smaller and larger) but the effector
caspases has very smaller (Chandra et al., 2000). The caspases contains domain
that facilitates them to activate after interacting with other molecules which
ultimately ends on effector caspase. On C-terminal, there is aspartate amino acid
residue a point for cleavage (Fan et al., 2005).
Two terms auto-activation or sometime proteolytic cascade are used for caspase
activation which occur when proteolytic cleavage of aspartate residues not less
than ultimately separates two subunits transforming inactive form into active form
(Ozben, 2007). There are three caspases activation pathways: 1. Cytotoxic T
lymphocytes and Natural killer cells release granzyme B which are responsible for
caspase-3/-7 activation. 2. TRAIL receptors, Fas, and TNF receptor activate
caspase -8/ -10. 3. Cytochrome c and the Bcl-2 protein family (as in case of HCV

Chapter 2 Review of literature
34
infection induced apoptosis) regulated via apoptosome results in activation of -9
caspase. Human twelve caspases -3, to -10 may expose to have a major
contribution in programmed cell death. Based on task and caspases activations in
signaling pathway they have been categorized in 3 classes: first apoptotic
activators -2, 8, 9, 10 (Initiator caspases), second apoptotic executors -3, 6, 7
(Effector caspases) and caspase -1, 4, 5, 11, 12, 13, 14 (Inflammatory caspases).
Activation of effector caspases starts when pro-apoptotic stimuli activate initiator
caspases and start their proteolytic cleavage. These caspases then perform their
functions by deterioration of cell and expanding the proteins cleavage, obstructing
apoptotic activity by releasing apoptotic inhibitors. All catalysis enzymes and
regulatory proteins become free progressing in collapse of enzyme activity. Chief
effector caspase (8, 9) are caspases and effector caspase (3, 6, 7) destroying
proteolytic enzymes leading cells towards apoptosis (Pop and Salvesen, 2009; Li
and Yuan, 2008).
There are many contradictory studies that reported many HCV pro-apoptotic or
anti-apoptotic proteins are directly involved in apoptosis. In some reports it is
declare that core proteins, E1, E2, NS3, NS4A, and NS5A and NS5B HCV
proteins activate apoptosis. Some studies reported that these HCV proteins work as
anti-apoptotic. Apoptosis induced by HCV by two ways external and internal
pathways. Hepatocyte apoptosis occur in patients with HCV chronically infected
patients. TNF-R1, TRAIL-R1, Fas and TRAIL-R2 express on liver cells (Tatsuo
Kanda et al., 2013).
In case of hepatocytes hepatic apoptosis the role of Fas and TNF-R1 are well
documented on the bases of both in vitro and in vivo agonist antibodies anti-Fas

Chapter 2 Review of literature
35
and TNFa induced hepatotoxicity. TRAIL induced hepatotoxicity is still under
discussion but it was thought that minor role in apoptosis progression however its
expression is caused by DNA damage. HCV non-structural NS3 protein induces
host cell transformation and tumor suppressor p53 and the N-terminus of NS3
forms a complex which in turn inactivates actinomycin D-induced apoptosis. Many
research studies have revealed that HCV proteins interact with host cells proteins
and thought that induce hepatocarcinogenesis (Tatsuo Kanda et al., 2013).

Material &
Methods

Chapter 3 Materials and methods
37
3.1 Materials and Methods
3.1.1 Expression vectors and host cells
Human hepatoma cell line (Huh-7) used in this study was grown in high-glucose
DMEM (Gibco) supplemented with 10 % fetal bovine serum (Hyclone), 1 % MEM
non-essential amino acids (Gibco), 100 units/ mL penicillin (Gibco) and 100 mg/
mL streptomycin (Gibco).
HCV non-structural proteins NS3, NS3-4A and NS4A were amplified from cDNA
clone of HCV genotypes 3a (pS52 strain), accession no GU814263 and cloned in
the pEGFP-C1 and pFLAG-CMV2 vectors. Recombinant vectors containing NS3,
NS3-4A and NS4A genes pEGFP-C1/NS3, pEGFP-C1 /NS3-4A, pEGFP-C1
/NS4A, pFLAG-CMV2/NS3, pFLAG-CMV2 /NS3-4A and pFLAG-CMV2 /NS4A
were transfected using trans-LT1 transfection reagent in Huh7 cells (Mirus 2300,
USA). HCV infection in this study was carried out using cell culture-derived HCV
Jc1 genotype 2a HCVcc (HCV infectious in cell culture) that were maintained in
the presence of 0.4 mg/ mL G418 (Invitrogen). HCVcc infection was carried out at
multiplicity of infection (MOI) of 1.
3.1.2 Chemicals and consumables
All the chemicals were purchased from Sigma St. Louis, USA and Fisher
scientific, USA.
3.1.3 Enzymes
DNA amplification enzyme Platinum® Taq DNA Polymerase High Fidelity and
Proteinase K were purchased from (Life Technologies Invitrogen, USA).

Chapter 3 Materials and methods
38
Restriction endonucleases include (BglII & BamHI), T4 DNA ligase, antarctic
phosphatase was purchased from (New England Biolabs Inc. MA, USA). Rnase
cocktail enzyme mix was obtained from (Life Technologies Ambion, USA).
3.1.4 Antibodies
Antibodies used in present study were purchased from (Cell Signaling Technology,
USA, Sigma, USA, Abcam, USA and Thermo Scientific, USA, Molecular Probe,
USA). Primary antibodies were included the following: rabbit polyclonal anti-
BAX; rabbit monoclonal (Cell Signaling); anti-β-actin (Cell Signaling); rabbit
polyclonal anti-Bcl-xL (Cell Signaling); rabbit polyclonal anti-Caspase 7 (Cell
Signaling); rabbit polyclonal anti-Caspase 9 (Cell Signaling); rabbit polyclonal
anti-Caspase 3 (Cell Signaling); rabbit polyclonal anti-Cytochrome c (Cell
Signaling); rabbit polyclonal anti-PARP (Cell Signaling); rabbit polyclonal anti-
GFP (Cell Signaling); mouse monoclonal anti-FLAG (Sigma); rat monoclonal anti-
KDEL (Abcam); mouse monoclonal anti-HCV core (Thermo Scientific). The
secondary antibodies used for immunofluorescence were Alexa Fluor 350, 488,
568, 594, or 647 anti-donkeys, mouse, rabbit, or goat IgG (Molecular Probe). The
secondary antibodies used for immunoblot analysis were RP-conjugated anti-
mouse IgG (Cell Signaling), HRP-conjugated anti-rabbit IgG (Cell Signaling).

Chapter 3 Materials and methods
39
3.1.5 Antibiotics
Table 3.1: List of antibiotics used in the present study
3.1.6 Molecular weight markers
1 Kb ladder, Mass DNA ladder and 100 bp DNA ladders were obtained from
(Biolabs, USA). Protein molecular weight marker was purchased from MBI
(BioRad, USA).
3.1.7 Reagents
Reagents used in the study were included the following: dNTPS mix (Invitrogen
Life Technologies, USA), Trans-LT1 transfection reagent (Mirus, USA), Super
signal (West Femto Maximum Sensitivity Substrate) (Thermo Scientific, USA),
Immuno Western blot (Chemiluminesent HRP Substrate (Millipore, USA),
MitoTracker CMXRos Red stain (Invtrogen), MitoSOX™ Red mitochondrial
superoxide stain (Invitrogen), Hoechst 33342 stain (Invitrogen), Mounting agent
ProLong® Gold Antifade Reagent with DAPI (Invitrogen).
3.1.8 Media
(A) Bacteria
(a) Luria Bertani (LB) Broth (per liter)
LB (Sigma) 25 g
Reagent Stock solution Final concentration in use
Ampicillin
Kanamycin
IPTG
IPTG
100 mg/ mL
50 mg/ mL
238 mg/mL
1 M
100 µg/ mL
50 µg/ mL
1 µg/mL
1 mM

Chapter 3 Materials and methods
40
Dissolve 25 g LB broth to the 900 mL dH2O. After mixing adjust the volume up to
1 L.
(b) Luria Bertani (LB) Agar (per liter)
LB (Sigma) 37 g
Dissolve 37 g LB agar to the 900 mL dH2O. After mixing adjust the volume up to
1 L.
(B) Media used for cell lines
(a) DMEM (Gibco)
(b) RPMI (Gibco)
(c) Fetal bovine serum (Hyclone)
(d) 1% MEM non-essential amino acids (Gibco)
(e) Penicillin / streptomycin (Gibco)
(f) Geneticin® Selective Antibiotic (G418 Sulfate) (Invitrogen)
3.1.9 Buffers
(a) 1X Tris EDTA (TE) Buffer (500 mL)
Tris HCl 1 M 5 mL
EDTA 0.5 M 1 mL
dH2O - 496 mL
Buffers for Agarose Gel Electrophoresis
(a) Tris Acetate EDTA (TAE) Buffer, pH 8.3 (per Liter)
Tris base 242 g
EDTA (0.5M) 100 mL
Glacial acetic acid 57.1 mL

Chapter 3 Materials and methods
41
Mix all chemicals together in 800 mL dH2O. After mixing adjust to volume up to 1
L.
(b) Ethidium-bromide dye (10 mg/ mL)
Ethidium- bromide 10 mg
Milli Q water 1 mL
(c) 6X loading dye for agarose gel electrophoresis (Biolabs, USA)
Reagents and Buffers for SDS-Page
(a) RIPA buffer Quantity (1mL)
1 M TRIS pH 7.4 50 μL
2.5 M NaCl 60 μL
10 % NP40 100 μL
10 % DOC 25 μL
10 % SDS 10 μL
10 mM PMSF 10 μL / 255 μL
dH2O 74 5μL
(b) Acrylamide (30 %)
Acrylamide 29.2 g
N, N' methylene bisacrylamide 0.8 g
Milli Q water 60 mL

Chapter 3 Materials and methods
42
The solution was stirred to dissolve the acrylamide. The volume was made up to
100 mL and the solution was filtered through Whattman filter paper no. 1 before
use.
(c) Ammonium per sulfate 12.5% (w/v)
(d) TEMED 8.4% (v/v)
(e) Resolving buffer
Tris base 9.08 g
SDS 0.2 g
Dissolve in 60 mL of distilled water and adjust pH to 8.8 with conc. HCl and make
up to 100 mL.
(f) Stacking buffer
Tris base 3.03 g
SDS 0.2 g
Dissolve in 60 mL of distilled water and adjust pH to 6.8 with conc. HCl and make
up to 100 mL.
(g) 10 X Tris-Glycine buffer (per/liter)
Tris base 30.3 g
Glycine 144.1g
SDS 10 g
(h) Coomassie brilliant blue staining solution (per Liter)
Brilliant blue (R250) 1 g
Acetic acid 100 mL

Chapter 3 Materials and methods
43
Methanol 400 mL
Milli Q water 500 mL
(i) Lysis Buffer/ Loading dye
Tris base 1.51 g
SDS 4 g
Glycine 20 g
Bromophenol blue 0.002 g
Dissolve in 60 mL of water and make up to 90 mL. Add 10 mL -mercaptoethanol
just before use.
Reagents for Western blot Analysis
(a) Transfer buffer (per liter)
Tris Base 3 g
Glycine 14.4 g
Methanol 200 mL
(b) Blocking buffer 5 % non-fat milk in TBS
(c) Wash buffer 0.1 % Tween 20 in TBS (TBS-T)
Reagents for cell viability assay
(a) Plaque Staining Solution
Crystal violet 0.2 %

Chapter 3 Materials and methods
44
3.1.10 Instrumentation
Sorvall RT7 plus (Ultra Centrifuge), Eppendorff (5415R) Centrifuge, Premium
Biotech Visible Spectrophotometer Ultra space 1000, Kodak (Digital Science)
image station (440), i cycler (Bio-Rad), Weighing Balance (OHAUS), PH Meter
(Accumet Basic Fisher Scientific), Incubator (Boeuel Scientific), Kodak (M35A-
OMAT Processor), Transilluminator (Fisher Biotech), Olympus X cite (Series
120Q) EXFO, ELx 800 Universal Microplate Reader (Bio-TEK Instruments), New
Bronswick Scientific Excella E25 Incubator Shaker Series, Castel Autoclave
System, Mini-PROTEAN Tetra Handcast System (Bio-Rad), Mini-Sub Cell GT
System (Bio-Rad), Wide Mini-Sub Cell GT System (Bio-Rad).
3.2 Methodology
3.2.1 Primer designing
Primers for amplification of HCV non-structural NS3, NS3-4A and NS4A genes
and primers for the sequencing of positive clones were designed according to
mammalian expression vectors (pFLAG-CMV2 & pEGFP-C1) using online
available oligo calculator and synthesized from (IDT-Integrated DNA
Technologies, USA). Primers sequences are given in the Table 3.2.
3.2.2 Amplification of HCV non-structural NS3, NS3-4A and NS4A genes
Infectious cDNA clone of HCV genotypes 3a (pS52 strain), accession no
GU814263 was used for the amplification of HCV non-structural NS3, NS3-4A &
NS4A genes. These genes were amplified using Platinum® Taq DNA Polymerase
High Fidelity (Invitrogen). The reaction mixture and cycling conditions for PCR
amplification of each gene are given in Table 3.2.

Chapter 3 Materials and methods
45
Table 3.2: Nucleotide sequences of the synthetic primers for sequencing and PCR:
All primers were successfully annealed to their respective targets. The sequences
were designed from reported sequences of HCV genomes (NZL & pS52 of
genotype 3a) available at NCBI. Sequences of the restriction enzymes used for
each primer is italicized while stop codons in reverse primers are underlined.
S. No Primer ID Primer Sequence 5’ to 3’ Restriction site Product Size
PCR PRIMERS
1. NS3-GFP F ATCTAAAAGATCTGGCCGTGAGGTGTTG BglII NS3 (1.9 kb)
NS3-4A (2.1 kb) 2. NS3-FLAG F ATCTAAAAGATCTCGGCCGTGAGGTGTTG BglII
3. NS3 R TTAATTGGATCCTCAGGTGGTTACTTCCAG BamHI
4. NS4A-GFP F ATCTAAAAGATCTGGCCGTGAGGTGTTG BglII NS4A (168 bp)
NS4A (168 bp) 5. NS4A-FLAG F ATCTAAAAGATCTCAGCACCTGGGTGTTG BglII
6. NS4A R TTAATTGGATCCTCAGCACTCCTCCATC BamHI
SEQUENCING PRIMERS
1. pFLAG-CMV2 F ATAACCCCCCGTTG
2. pFLAG-CMV2 R TTAGGACAAGGCTGGTGG
3. pEGFP-C1 F AGCACCCAGTCCGCCCTGAGC
4. pEGFP-C1 R GAAATTTGTGATGCTATTGC

Chapter 3 Materials and methods
46
3.2.3 Amplification of NS3 gene using pFLAG-CMV2 primers
Reaction mixture
10 mM dNTPs--------------------------------------------------------1μL
2 mM MgSO4 (25 mM)----------------------------------------------2 μL
HF Buffer (10X)------------------------------------------------------5 μL
NS3-FLAG F (10 pmole/μL)---------------------------------------1 µL
NS3-FLAG R (10 pmole/μL)---------------------------------------1 μL
Template (DNA 640 ng/ µL)----------------------------------------1.5 μL
HF Taq polymerase (1U)-------------------------------------------0.2 μL
Nuclease free water-------------------------------------------------38.3 μL
Total volume---------------------------------------------------------50 μL
The following conditions were followed for amplification of NS3 gene.
PCR cycling conditions

Chapter 3 Materials and methods
47
3.2.4 Amplification of NS3 gene using pEGFP-C1 primers
Reaction mixture
10 mM dNTPs---------------------------------------------------------1 μL
2 mM MgSO4 (25 mM)----------------------------------------------2 μL
HF Buffer (10 X)------------------------------------------------------5 μL
NS3-FLAG F (10 pmole/ μL)--------------------------------------1 µL
NS3-FLAG R (10 pmole/ μL)--------------------------------------1 μL
Template (DNA 640 ng/ µL)---------------------------------------1.5 μL
HF Taq polymerase (1U)-------------------------------------------0.2 μL
Nuclease free water-------------------------------------------------38.3 μL
Total volume---------------------------------------------------------50 μL
The following conditions were followed for amplification of NS3 gene.
PCR cycling conditions

Chapter 3 Materials and methods
48
3.2.5 Amplification of NS3-4A gene using pFLAG-CMV2 primers
Reaction mixture
10 mM dNTPs--------------------------------------------------------1 μL
2 mM MgSO4 (25 mM)----------------------------------------------2 μL
HF Buffer (10X)------------------------------------------------------5 μL
NS3-FLAG F (10 pmole/ μL)--------------------------------------1 µL
NS3-FLAG R (10 pmole/ μL)---------------------------------------1 μL
Template (DNA 640 ng/ µL)----------------------------------------1.5 μL
HF Taq polymerase (1U)---------------------------------------------0.2 μL
Nuclease free water----------------------------------------------------38.3μL
Total volume------------------------------------------------------------50 μL
The following conditions were followed for amplification of NS3-4A gene
PCR cycling conditions

Chapter 3 Materials and methods
49
3.2.6 Amplification of NS3-4A gene using pEGFP-C1 primers
Reaction mixture
10 mM dNTPs----------------------------------------------------------1 μL
2 mM MgSO4 (25 mM) ----------------------------------------------2 μL
HF Buffer (10 X) -----------------------------------------------------5 μL
NS3-FLAG F (10 pmole/ μL) --------------------------------------1 µL
NS3-FLAG R (10 pmole/ μL) --------------------------------------1 μL
Template (DNA 640 ng/ µL) ---------------------------------------1.5 μL
HF Taq polymerase (1U) --------------------------------------------0.2 μL
Nuclease free water---------------------------------------------------38.3 μL
Total volume----------------------------------------------------------50 μL
The following conditions were followed for amplification of NS3-4A gene.
PCR cycling conditions

Chapter 3 Materials and methods
50
3.2.7 Amplification of NS4A gene using pFLAG-CMV2 primers
Reaction mixture
10 mM dNTPs----------------------------------------------------------1 μL
2 mM MgSO4 (25 mM) -----------------------------------------------2 μL
HF Buffer (10 X) ------------------------------------------------------5 μL
NS3-FLAG F (10 pmole/ μL) ---------------------------------------1 µL
NS3-FLAG R (10 pmole/ μL) ---------------------------------------1 μL
Template (DNA 640 ng/ µL) ----------------------------------------1.5 μL
HF Taq polymerase (1U) ---------------------------------------------0.2 μL
Nuclease free water----------------------------------------------------38.3 μL
Total volume------------------------------------------------------------50 μL
The following conditions were followed for amplification of NS4A gene.
PCR cycling conditions

Chapter 3 Materials and methods
51
3.2.8 Amplification of NS4A gene using pEGFP-C1 primers
Reaction mixture
10 mM dNTPs----------------------------------------------------------1 μL
2 mM MgSO4 (25 mM) -----------------------------------------------2 μL
HF Buffer (10 X) ------------------------------------------------------5 μL
NS3-FLAG F (10 pmole/ μL) --------------------------------------1 µL
NS3-FLAG R (1 0pmole/ μL) --------------------------------------1 μL
Template (DNA 640 ng/ µL) ---------------------------------------1.5 μL
HF Taq polymerase (1U) --------------------------------------------0.2 μL
Nuclease free water---------------------------------------------------38.3 μL
Total volume----------------------------------------------------------50 μL
The following conditions were followed for amplification of NS4A gene.
PCR cycling conditions

Chapter 3 Materials and methods
52
3.2.9 Agarose gel electrophoresis
Agarose gel 1-1.5 % premixed with 10 mg/ mL Ethidium- bromide were used for
the resolving of PCR products when run containing TAE buffer in the gel
electrophoresis chamber. Size of the desired band was determined either by 1 kb or
100 bp ladder, while gel was run at 70 V for 60 minutes and observed in UV light
(Tran illuminator).
3.2.10 Purification of DNA products from agarose gel
Amplified DNA products of the particular genes were eluted from agarose gel
using DNA clean and concentrator kit according to the manufacturer’s instructions.
Briefly, particular DNA sample was run on 1 % agarose gel prepared in TAE
buffer. The required fragment was sliced out under UV light and transferred to 1.5
mL microcentrifuge tube. 2-5 volumes of DNA binding buffer to each volume of
DNA sample was added in the tube, mixed and incubated at 55 oC in water bath for
approximately 10-15 minutes, to dissolve the agarose completely. Then mixture
were transferred to Zymo-spin column, centrifuged for 30 sec at room temperature
and discarded the flow-through. Next, 200 μL wash buffer was added to the
column and centrifuged for 30 sec at room temperature. Washing step was repeated
and then column was air dried, poured with 20-30 µL pre-warmed nuclease free
water, incubated at room temperature for 1 min and spun down for 1 min at 13,000
rpm for the elution of DNA. Concentration of eluted DNA was estimated by
running on 1 % agarose gel, with Mass DNA ladder.

Chapter 3 Materials and methods
53
3.2.11 Construction of pEGFP-C1 recombinant vectors
Respective amplified DNA products of NS3, NS3-4A and NS4A separately and
expression vector pEGFP-C1 were digested with selected restriction enzymes, gel
purified and further ligated using T4 DNA ligase overnight at 4 oC. 2-5 µl of
ligation mixture was mixed with 100 µl to Z-competentTM
E. coli strain competent
cells and incubated on ice for 30 minutes. The tubes were placed at 42 oC in a
water bath for 90 seconds and on ice for 1 minute. After the addition of 400 µl LB
medium, tubes were then incubated at 37 oC for 30 to 60 minutes. After that 150 µl
of transformation culture was plated onto LB agar plate containing ampicillin (100
ug / mL).
3.2.12 Construction of pFLAG-CMV2 recombinant vectors
Respective amplified DNA products of NS3, NS3-4A and NS4A separately and
expression vector pFLAG-CMV2 were digested with selected restriction enzymes,
gel purified and further ligated using T4 DNA ligase overnight at 4 oC. 2-5 µl of
ligation mixture was mixed with 100 µl to Z-competentTM
E. coli strain competent
cells and incubated on ice for 30 minutes. The tubes were placed at 42 oC in a
water bath for 90 seconds and on ice for 1 minute. After the addition of 400 µl LB
medium, tubes were then incubated at 37 oC for 30 to 60 minutes. After that 150 µl
of transformation culture was plated onto LB agar plate containing Kanamycin
(100 ug / mL).

Chapter 3 Materials and methods
54
3.2.13 Selection of positive clones
Positive clones of particular genes were separately screened out first through
restriction digestion (BamHI and BglII) and then by sequencing (Eton Biosciences,
USA). First, plasmids from 32 selected colonies were isolated by “QIAprep®
Miniprep Kit” according to manufacturer’s instruction. Further, were digested
using selected restriction enzymes and was incubated in an eppendorf tube at 37 oC
for 1-2 hrs. The digested DNA was checked by agarose gel electrophoresis.
Selected clones were further confirmed by automated sequencing, which was
performed commercially form Eton Biosciences, USA.
3.2.14 Large scale preparation of plasmid DNA
High concentration of plasmid DNA was prepared by using “Qiagen® plasmid
midi kit” according to manufacturer’s concentration. Shortly, 150 mL LB broth
containing ampicillin (100 μg/ mL) was inoculated with a transformed E. coli
colony and incubated overnight in a shaking incubator at 37 ºC. Further cells were
harvested by centrifuging at 6000 g for 10 min at 4 ºC and bacterial pellet was
resuspended in 4 mL ice chilled buffer P1. 4 mL P2 buffer was poured in the tube
for 5 min to lysed the cells. After 5 min 4 mL ice chilled P3 buffer was poured in
the and immediately inverted 4 - 6 times and incubated on ice for 15 min. Further
tube was centrifuged at 13,000 g for 30 min at 4 ºC and supernatant was placed in a
new centrifugation tube. A QIAGEN-tip column was equilibrated by the addition
of 4 mL buffer QBT and allowed it to empty. Then supernatant was applied to the
column and allowed to drain through by gravity than washed the column by the

Chapter 3 Materials and methods
55
addition of 10 mL QC buffer. The plasmid DNA was eluted by the addition of 5
mL QF buffer and collected in new tube. For cleaning purpose 3.5 mL isopropanol
was added to the eluted DNA and further mixed and centrifuged at 17,000 g for 30
min at 4 ºC. Further, supernatant was discarded and the pellet was washed with 2.5
mL 70 % ethanol and was centrifuged at 17,000 g for 10 min at 4 ºC. Discard the
supernatant and was allowed the pellet to completely dry for 5-10 min and finally
resuspended in 100 μL TE buffer or nuclease free water. DNA concentration was
determined using spectrophotometer at 595nm.
3.2.15 Cell culture and transfection
Huh-7 cells were maintained in Dulbecco’s modified eagle medium (DMEM)
supplemented with 100 μg/mL penicillin; streptomycin, 1 % Non-essential amino
acids (MEM) and 10 % fetal bovine serum (Gibco, USA) at 37 °C with 5 % CO2.
Cells were seeded in 60mm plates and cultured until they became 50-60 %
confluent. Constructed plasmids about 1-2 μg of pEGFP-C1 (non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A, pFLAG-CMV2
(non-expressing control), pFLAG-CMV2/NS3, pFLAG-CMV2/NS3-4A &
pFLAG-CMV2/NS4A were separately transfected in 40 % confluent cells with
trans-LT1 transfection reagent. 6hrs post transfection media (with transfection
reagent and plasmid) was changed.
3.2.16 Preparation of whole cell lysate for SDS page
Whole cell lysate for SDS PAGE was prepared by washing twice with PBS 72hrs
post transfection. Cells were scraped out using scraper and poured in 1.5 mL

Chapter 3 Materials and methods
56
microcentrifuge tube. Further these cells were dissolved in freshly prepared 1 mL
RIPA buffer (1 M TRIS pH 7.4, 2.5M NaCl, 10 %NP40, 10 % DOC, 10 %SDS, 10
mM PMSF and proteinase inhibitor), incubated on ice for 30 mints and sonicated
6-8 times. After being sonication extracts were centrifuged at 12000 rpm for 20
mints at 4 oC, supernatants were poured in new ice chilled 1.5 mL microcentrifuge
tube and stored at -80 oC. Protein Assay kit (Bio-Rad) was used according to
manufacturer’s instructions to measure protein concentration.
3.2.17 SDS polyacrylamide gel electrophoresis
Protein was separated by SDS polyacrylamide gel electrophoresis. 8 and 10 % gels
were used for the separation of protein bands. Following recipe were followed for
the preparation of above mentioned gels (Table 3.3). After addition and mixing of
all ingredients in the tube, TEMED was added in it to allow the gel to polymerize.
Gel was poured between the assembled gel plates up to 2/3 portion.
Small amount of butan-2-ol was added on the top of the resolving gel to allow a
smooth interface on the gel. After polymerization of the gel butan-2-ol was
removed from the top of the gel and washed twice with distilled H2O. Further,
stacking gel was poured on the top of resolving gel and comb was inserted to the
gel for the preparation of wells for loading of protein samples. After
polymerization comb was carefully removed and placed the plates in the
electrophoresis tank having 1X gel running buffer. Protein samples were mixed
with 5X SDS loading buffer and boiled for 5 mints at 100 oC. 50ug of each sample
was loaded in each lane of the gel and precision plus protein standard dual color

Chapter 3 Materials and methods
57
was also loaded in one lane to estimate the size of loaded samples. Finally gel was
electrophoresed at 25 V for 1 hour.
Table 3.3: Composition of different SDS-PAGE gels
3.2.18 Western Blotting
For western blotting proteins bands were transferred to Hybond-C extra
nitrocellulose membrane electrophoretically using tank blotting apparatus. The gel
was removed from the tank and stacking gel discarded from the resolving gel. A
blotting sandwich was made where the gel was placed with a piece of
nitrocellulose membrane, two pieces of 3 MM Whatman paper and two fiber pads.
SDS-PAGE
S. NO Chemical Resolving 8% Resolving 10% Stacking Gel
1. 40 % Acrylamide 2.4 mL 3 mL 0.75 mL
2. 1.5 M Tris-HCl 4 mL 4 mL -
3. 1 M Tris-HCl - - 2.25 mL
4. Water 5.3 mL 4.7 mL 3 mL
5. 10 % SDS 150 μL 150 μL 75 μL
6. 10 % APS 150 μL 150 μL 75 μL
7. TEMED 5 μL 5 μL 5 μL

Chapter 3 Materials and methods
58
The sandwich was fixed in a cassette and was placed into the transblot apparatus
and filled the tank with transfer buffer (Tris glycine buffer). Electrotransfer was
carried out at 100 mA for 1:30 hrs or 25 mA at 4 ºC overnight.
After successfully transferred, blot was blocked with 5 % skimmed milk for 1 hr
at room temperature followed by one washed with TBS-T buffer (0.05 % Tween
20). Further, blots were incubated with appropriate primary antibodies (1:1000)
overnight at 4 oC. After being washing three times with TBS-T buffer blot was
incubated with particular horseradish peroxidase (HRP) labeled secondary
antibodies (1:10000) at room temperature for 1 hr. After being washed three times
with TBS-T buffer, the membrane were incubated with chemiluminescent HRP
substrate for 1 minute at room temperature to visualized the positive bands using
Kodak image station (Digital science, 440) according to the manufacturer’s
instructions.
3.2.19 Immunofluorescence Microscopy
The cells expressing NS3, NS3-4A and NS4A proteins were grown on glass cover
slips. After removal of medium from plates cells were fixed with 4 %
paraformaldehyde for 10 mints at room temperature. After fixation cells were
washed five time using 1 X PBS. MitoTracker CMXRos Red was used to stain
mitochondria in live cells for 10 mints before fixation. Next, cells were blocked
with blocking buffer (3 % BSA, 5 % Goat serum, 0.05 % Triton X-100) for 1 hr at
room temperature. After blocking cells were incubated with particular primary
antibodies (1:1000) overnight at 4 oC. Further, cells were washed thrice with PBS

Chapter 3 Materials and methods
59
and then incubated with particular Alexa flour secondary antibodies (1:10000) in
the dark. After secondary antibody incubation cells were washed thrice with PBS.
Further, nuclei were stained with DAPI. Images were visualized under a 100X oil
objective using an Olympus FluoView 1000 confocal microscope. Quantification
of images was conducted with ImageJ or MBF ImageJ softwares.
3.2.20 Crystal violet cell viability assay
The effect of cell viability was determined by measuring crystal violet dye
absorbance of living cells. Briefly, cells were transfected with pEGFP-C1 (empty
vector/ non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-
C1/NS4A, pFLAG-CMV2 (empty vector/ non-expressing control), pFLAG-CMV2
/NS3, pFLAG-CMV2 /NS3-4A and pFLAG-CMV2 /NS4A recombinant vectors in
three different sets of experiments. 72 hrs post transfections cells were treated with
crystal violet dye, stabilized with 33 % acetic acid and fixed with 4 %
paraformaldehyde. Optical density was measured at wave length 595 nm using
premium biotech visible spectrophotometer ultra-spec 1000.
3.2.21 Subcellular Fractionation
For the isolation and separation of mitochondrial and cytosolic fractions from
experimental cells, Huh-7 cells were transfected with pEGFP-C1 (non-expressing
control/ empty vector), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-
C1/NS4A. 3 days post-transfection, cells were scraped and pure cytosolic and
mitochondrial fractions were isolated using mitochondrial isolation kit for cultured
cells according to the manufacturer’s instructions. Briefly, 800 μL Reagent A were
added in the homogenized cells and incubated on ice for 2 mints. Next, cells were

Chapter 3 Materials and methods
60
dounce homogenized then mixed with 800 μL Reagent C and centrifuged at 700 g
for 10 mints at 4 oC. Supernatant were collected in fresh 1.5 mL microcentrifuge
tube and again centrifuged at 12,000 g for 15 mints at 4 oC. Supernatant were
removed and pellet (mitochondria) were washed with 500 μL Reagent C and
centrifuged at 12,000 g for 5 mints at 4 oC. Supernatant (Cytosolic fraction) were
collected in new tube while pellet (mitochondrial fraction) were resuspended in TE
buffer. Equivalent amounts of protein from each fraction were analyzed by
Western blotting with the indicated primary and secondary antibodies.
3.2.22 Mitochondrial Superoxide Estimation
Huh-7 cells were seeded on glass cover slips and transfected with pEGFP-C1 (non-
expressing control/ empty vector), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and
pEGFP-C1/NS4A for the estimation of mitochondrial superoxide (byproduct of
oxidative phosphorylation of complexes I and III of the electron transport chain).
72 hrs post transfection cell were washed with PBS and stained with MitoSOX™
Red mitochondrial superoxide indicator 10 µM for 10 min at 37 oC. After being
fixed with 4 % paraformaldehyde, superoxide production was estimated at 100X
oil under florescence microscope (Olympus).
3.2.23 Measurement of Mitochondrial Complex I Enzyme Activity
The activity of mitochondrial oxidative phosphorylation respiratory chain complex
I (NADH dehydrogenase) in NS3, NS3-4A and NS4A expressing cells were
measured by using mitochondrial complex I activity assay kit according to the

Chapter 3 Materials and methods
61
manufacturer’s instructions. Briefly, Huh-7 cells were transfected with pFLAG-
CMV2 (non-expressing control/ empty vector), pFLAG-CMV2 /NS3, pFLAG-
CMV2 /NS3-4A and pFLAG-CMV2 /NS4A. Huh-7 cells were used as negative
control (mock) while Huh-7 cells infected with HCVcc used as positive control.
Cells were harvested 72 hrs post transfection and processed for further
experimentation.
3.2.24 Hoechst Staining
Cells were seeded on glass cover slips and transfected with pEGFP-C1 (non-
expressing control/ empty vector), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and
pEGFP-C1/NS4A. At various time points (24hrs, 48hrs and 72hrs) cells were fixed
using 4 % paraformaldehyde, washed five times with PBS and stained with 10mM
Hoechst 33342 at room temperature for 10 min. After incubation cells were gently
washed and mounted. The morphology of the nuclei of the cells was examined at
100 X oil under florescence microscope (Olympus).
3.2.25 Statistical analysis
To analyze the significance of numerical data, Student’s t-test was performed by
using Graph Pad Prism version 5.0 (GraphPad Software, Inc., CA, USA). Data are
presented as mean ± standard error of the mean. p-values < 0.05 were considered
statistically significant (p < 0.05, p < 0.01 and p < 0.001).

Results

Chapter 4 Results
62
RESULTS
4.1 Amplification and construction of recombinant vectors
Genes for HCV non-structural proteins NS3, NS3-4A and NS4A were successfully amplified
from cDNA clone of HCV genotypes 3a (pS52 strain) using two different sets of primers for
each gene i.e. one set of primers for pEGFP-C1 clones and other set of primers for pFLAG-
CMV2 clones. Positive clones were confirmed by restriction digestion analysis. Figure 4.1(A &
B) shows the double digestion of positive clones of pEGFP-C1 /NS3 containing NS3 (1.9 kb),
pEGFP-C1 /NS3-4A containing NS3-4A (2.1kb) and pEGFP-C1/NS4A containing NS4A (165
bp), while figure 4.2(A & B) shows the double digestion of positive clones of pFLAG-CMV2
/NS3 containing NS3 (1.9 kb), pFLAG-CMV2 /NS3-4A containing NS3-4A (2.1 kb) and
pFLAG-CMV2 /NS4A containing NS4A (165 bp).
Maps of above mentioned recombinant vectors are shown in Figure 4.3 (A & B).

Chapter 4 Results
63
Figure 4.1: 1% agarose gel electrophoresis shows the results of double digestion of recombinant
pEGFP-C1 vectors. (A) Lane 1 refer to 1 kb ladder, lane 2 refers to undigested recombinant
vector (pEGFP-C1/NS3), lane 3 refer to digested vector (4.7 kb) releasing NS3 (1.9 kb) gene,
lane 4 refer to undigested recombinant vector (pEGFP-C1/NS3-4A) while, lane 5 refer to
digested vector (4.7 kb) releasing NS3-4A (2.1 kb) gene. (B) Lane 1 refer to undigested
recombinant vector (pEGFP-C1/NS4A), lane 2 refer to digested vector (4.7 kb) releasing NS4A
(165 bp) gene while, lane 3 refer to 1 kb ladder.
(A) (B)

Chapter 4 Results
64
Figure 4.2: 1% agarose gel electrophoresis shows the results of double digestion of recombinant
pFLAG-CMV2 vectors. (A) Lane 1 refer to 1 kb ladder, lane 2 refer to digested vector (4.7 kb)
releasing NS3 (1.9 kb) gene, lane 3 refers to undigested recombinant vector (pFLAG-
CMV2/NS3), lane 4 refer to digested vector (4.7 kb) releasing NS3-4A (2.1 kb) gene while, lane
5 refer to undigested recombinant vector (pFLAG-CMV2/NS3-4A). (B) Lane 1 refer to 1 kb
ladder, lane 2 refer to undigested recombinant vector (pFLAG-CMV2/NS4A), while, lane 3 refer
to digested vector (4.7 kb) releasing NS4A (165 bp) gene.
(A) (B)

Chapter 4 Results
65
Figure 4.3 (A): Maps of recombinant pEGFP-C1 constructs. pEGFP-C1/NS4A vector
containing HCV NS3-4A gene, pEGFP-C1/NS3 vector containing HCV NS3 gene while
pEGFP-C1/NS4A vector containing HCV NS4A gene.
Figure 4.3 (B): Maps of recombinant pFLAG-CMV2 constructs. pFLAF-CMV2/NS3-4A
vector containing HCV NS3-4A gene, PFLAG-CMV2/NS3 vector containing HCV NS3 gene
while pFLAG-CMV2/NS4A vector containing HCV NS4A gene.
(A)
(B)

Chapter 4 Results
66
4.2 Sequencing of GFP-tagged recombinant vectors
Positive clones of pEGFP-C1/NS3, pEGFP-C1/NS3-4A & pEGFP-C1/NS4A were confirmed by
commercial sequencing (Eton Biosciences, USA) from both directions using forward and reverse
primers. Figure 4.4 depicts the representative chromatogram and sequence homology of HCV
non-structural NS3 gene with reported sequences. Figure 4.5 shows the representative
chromatogram and sequence homology of HCV non-structural NS3-4A gene with reported
sequences while; Figure 4.6 is showing the representative chromatogram and sequence
homology of HCV non-structural NS4A gene with reported sequences.

Chapter 4 Results
67
Figure 4.4: Representative chromatograms and sequence homology of HCV non-structural NS3
gene with reported sequences.

Chapter 4 Results
68
Figure 4.5: Representative chromatograms and sequence homology of HCV non-structural NS3-
4A gene with reported sequences.

Chapter 4 Results
69
Figure 4.6: Representative chromatograms and sequence homology of HCV non-structural
NS4A gene with reported sequences.

Chapter 4 Results
70
4.3 Sequencing of FLAG-tagged recombinant vectors
Positive clones of pFLAG-CMV2/NS3, pFLAG-CMV2/NS3-4A, pFLAG-CMV2/NS4A were
confirmed by commercial sequencing from both directions using forward and reverse primers.
Figure 4.7 depicts the representative chromatogram and sequence homology of HCV non-
structural NS3 gene with reported sequences. Figure 4.8 shows the representative chromatogram
and sequence homology of HCV non-structural NS3-4A gene with reported sequences while;
Figure 4.9 is showing the representative chromatogram and sequence homology of HCV non-
structural NS4A gene with reported sequences.

Chapter 4 Results
71
Figure 4.7: Representative chromatograms and sequence homology of HCV non-structural NS3
gene with reported sequences.

Chapter 4 Results
72
Figure 4.8: Representative chromatograms and sequence homology of HCV non-structural NS3-
4A gene with reported sequences.

Chapter 4 Results
73
Figure 4.9: Representative chromatograms and sequence homology of HCV non-structural
NS4A gene with reported sequences.

Chapter 4 Results
74
4.4 Western blot analysis of FLAG-tagged HCV non-structural NS3, NS3-4A and NS4A
proteins
Transient expression of FLAG-tagged HCV non-structural NS3, NS3-4A and NS4A proteins
were analyzed by western blotting. Cell lysates were harvested 72 hrs post transfection using
RIPA buffer and were analyzed with anti-FLAG antibody. Figure 4.10 (A) shows no band (non-
expressing control) in lane 1 while specific band of 7 kDa representing the fusion complex of
FLAG with NS4A protein in lane 2. On the other hand figure 4.10 (B) shows specific band of 72
kDa representing the fusion complex of FLAG with NS3 protein in lane 1 while specific band of
72 kDa representing fusion complex of FLAG with NS3-4A protein in lane 2. Internal loading
control β-actin was used.

Chapter 4 Results
75
Figure 4.10: Western blot analyses of pFLAG-CMV2/NS3, pFLAG-CMV2/NS3-4A,
pFLAG-CMV2/NS4A vectors expressing HCV non-structural NS3, NS3-4A and NS4A
proteins. (A) Lane 2 Shows specific band of 7 kDa representing the fusion complex of FLAG
with NS4A protein. (B) Lane 1 shows specific band of 72 kDa representing the fusion complex
of FLAG with NS3 protein, while lane 2 shows specific band of 72 kDa representing fusion
complex of FLAG with NS3-4A protein. β-actin was used as an internal loading control.
(A) (B)
(A)
Lane 1: Empty vector
Lane 2: NS4A-FLAG
(B)
Lane 1: NS3-FLAG
Lane 2: NS3-4A-FLAG

Chapter 4 Results
76
4.5 Western blot analysis of GFP-tagged HCV non-structural NS3, NS3-4A and NS4A
proteins
Transient expression of GFP-tagged HCV non-structural NS3, NS3-4A and NS4A proteins were
analyzed by western blotting. Cell lysates were harvested 72 hrs post transfection using RIPA
buffer and were analyzed with anti-GFP antibody. Figure 4.11 shows specific band of 27 kDa
representing GFP protein in lane 1, 33 kDa representing the fusion complex of GFP with NS4A
protein in lane 2, 98 kDa representing fusion complex of GFP with NS3 in lane 3 while specific
band of 98k Da representing the fusion complex of GFP with NS3-4A protein in lane 4. β-actin
was used as internal loading control.

Chapter 4 Results
77
Figure 4.11: Western blot analysis of pEGFP-C1/NS3, pEGFP-C1/NS3-4A & pEGFP-
C1/NS4A vectors expressing HCV non-structural NS3, NS3-4A and NS4A proteins. Specific
band of 27 kDa representing GFP protein in lane 1, 33 kDa representing the fusion complex of
GFP with HCV non-structural NS4A protein in lane 2, 98 kDa representing fusion complex of
GFP with HCV non-structural NS3 protein in lane 3 while, specific band of 98 kDa representing
the fusion complex of GFP with HCV non-structural NS3-4A protein in lane 4. β-actin was used
as internal loading control.
Lane 1: Empty vector
Lane 2: NS4A-GFP
Lane 1: NS3-GFP
Lane 2: NS3-4A-GFP

Chapter 4 Results
78
4.6 Immunofluorescence analysis of GFP tagged recombinant vectors
To determine the GFP tagged HCV non-structural NS3, NS3-4A and NS4A protein expression
transiently, cells were fixed 48 hours post transfection on glass coverslips, mounted with DAPI
containing ProLong® Gold Antifade Reagent to stain the nuclei and observed under fluorescence
microscope. Figure 4.12 (A), depicts the transfected GFP tagged pEGFP-C1 (non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing NS3,
NS3-4A and NS4A proteins expressing Huh-7 cells in green, while nuclei counterstained with
DAPI in blue. Figure 4.12 (B) depicts the graphical representation of transfection efficacy of
GFP tagged expressing cells in percentage, pEGFP-C1 (non-expressing control) shows 59 %
transfection, pEGFP-C1/NS3 containing NS3 protein shows 46%, pEGFP-C1/NS3-4A
containing NS3-4A protein shows 45 % while pEGFP-C1/NS4A containing NS4A protein shows
55 % transfection.

Chapter 4 Results
79
Figure 4.12 (A): Immunofluorescence analysis of pEGFP-C1/NS3, pEGFP-C1/NS3-4A &
pEGFP-C1/NS4A vectors expressing HCV non-structural NS3, NS3-4A and NS4A proteins.
Huh-7 cells transiently expressing pEGFP-C1 (non-expressing control), pEGFP-C1/NS3,
pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing HCV non-structural NS3, NS3-4A
and NS4A proteins (left panel) were stained with DAPI (middle panel) to stain nuclei. Overlaid
pictures (right panel) showing transfected and untransfected cells.
(A)

Chapter 4 Results
80
Figure 4.12 (B): Graphical representation of pEGFP-C1/NS3, pEGFP-C1/NS3-4A &
pEGFP-C1/NS4A vectors expressing HCV non-structural NS3, NS3-4A and NS4A proteins.
pEGFP-C1 (non-expressing control) shows 59 % transfection efficiency, NS3/GFP shows 46 %,
NS3-4A/GFP shows 45 % while NS4A/GFP shows 55 % transfection efficiency.
(B)

Chapter 4 Results
81
4.7 Immunofluorescence analysis of FLAG tagged recombinant vectors
FLAG tagged HCV non-structural NS3, NS3-4A and NS4A expression transiently, cells were
fixed 48hours post transfection on glass coverslips, incubated first with anti-FLAG antibody
mouse and second with Alexa Fluor 488 anti-mouse (green) antibody and then mounted with
ProLong® Gold Antifade Reagent with DAPI to stain the nuclei and observed under
fluorescence microscope. Figure 4.13 (A) depicts the transfected FLAG tagged pFLAG-CMV2
(non-expressing control), pFLAG-CMV2 /NS3, pFLAG-CMV2 /NS3-4A, pFLAG-CMV2
/NS4A vectors containing HCV non-structural NS3, NS3-4A and NS4A proteins expressing
Huh-7 cells in green and nuclei counterstained with DAPI in blue. Figure 4.13 (B) depict the
graphical representation of transfection efficacy of FLAG tagged expressing Huh-7 cells in
percentage, pFLAG-CMV2 (non-expressing control) shows 58 % transfection, pFLAG-CMV2
/NS3 containing NS3 protein shows 48 %, pFLAG-CMV2 /NS3-4A containing NS3-4A protein
50 % while pFLAG-CMV2 /NS4A containing NS4A protein shows 49 % transfection.

Chapter 4 Results
82
Figure 4.13 (A): Immunofluorescence analysis of pFLAG-CMV2/NS3, pFLAG-CMV2/NS3-4A,
pFLAG-CMV2/NS4A vectors containing HCV non-structural NS3, NS3-4A and NS4A
proteins. Huh-7 cells transiently expressing pFLAG-CMV2 (non-expressing control), pFLAG-
CMV2 /NS3, pFLAG-CMV2 /NS3-4A and pFLAG-CMV2 /NS4A vectors containing HCV non-
structural NS3, NS3-4A and NS4A proteins (left panel) were incubated first with anti-flag mouse
antibody and second with Alexa Fluor 488 anti-mouse antibody (left panel) and counter stained
with DAPI (middle panel) to stain nuclei. Overlaid pictures (right panel) showing transfected and
untransfected cells.
(A)

Chapter 4 Results
83
Figure 4.13 (B): Graphical representation of pFLAG-CMV2/NS3, pFLAG-CMV2/NS3-4A,
pFLAG-CMV2/NS4A vectors expressing HCV non-structural NS3, NS3-4A and NS4A
proteins. pFLAG-CMV2 (non-expressing control) shows 58 % transfection efficiency,
NS3/FLAG containing NS3 protein shows 48 %, NS3-4A/FLAG containing NS3-4A protein
shows 50 % while NS4A/FLAG containing NS4A protein shows 49 % transfection efficiency.
a)
(B)

Chapter 4 Results
84
4.8 HCV non-structural NS3-4A and NS4A proteins induces cell death
One of the most common application of crystal violet dye use is to determine the maximum cell
viability by checking the absorbance of the dye taken up by the cells (Gillies, Didier et al., 1986;
Shaik, Chatterjee et al., 2004; Thomas, Finnegan et al., 2004). To check the cell viability in
pEGFP-C1 (non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A
vectors containing HCV non-structural NS3, NS3-4A and NS4A proteins. Huh-7 cells
transfected in three sets of experiments were used for each vectors and cell viability was
determined by measuring crystal violet dye absorbance of living cells. pEGFP-C1 (non-
expressing control) shows 100 % cell viability, pEGFP-C1/NS3 containing NS3 protein shows
65 %, pEGFP-C1/NS3-4A containing NS3-4A protein shows 45 % and pEGFP-C1/NS4A
containing NS4A protein shows 46 % cell viability (Fig.4.14 B). These results depict maximum
cell death in NS3-4A and NS4A expressing Huh-7 cells (Fig. 4.14 A).

Chapter 4 Results
85
Figure 4.14: Cell death induced by HCV non-structural NS3-4A and NS4A proteins. Huh-7
cells transiently expressing pEGFP- C1 (empty vector/ non-expressing control), pEGFP-C1/NS3,
pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors containing HCV non-structural NS3, NS3-4A
and NS4A proteins were examined for cell viability by crystal violet cell viability assay. (A)
Huh-7 cells transfected with above mentioned vectors stained with crystal violet dye which
clearly showing the cell death in HCV non-structural NS3-4A and NS4A proteins expressing
(A)
(B)

Chapter 4 Results
86
cells. (B) Graphical representation of cell viability of vectors-harbouring cells. P values were
calculated by using an unpaired Student’s t-test (mean ± SEM; n=03; ***p<0.001, **p<0.01).
While pFLAG-CMV2 (non-expressing control), pFLAG-CMV2 /NS3, pFLAG-CMV2 /NS3-4A
and pFLAG-CMV2 /NS4A vectors containing NS3, NS3-4A and NS4A proteins were also used
to check the cell viability. Huh-7 cells transfected in three sets of experiments were used for each
vectors. pFLAG-CMV2 (non-expressing control) shows 100 % cell viability, pFLAG-CMV2
/NS3 containing NS3 protein shows 62 %, pFLAG-CMV2 /NS3-4A containing NS3-4A protein
shows 45 % and pFLAG-CMV2 /NS4A containing NS4A protein shows only 48 % viability
(Fig.4.15 B). These results depict maximum cell death in HCV non-structural NS3-4A and NS4A
proteins expressing Huh-7 cells (Fig. 4.15 A).

Chapter 4 Results
87
Figure 4.15: Cell death induced by HCV non-structural NS3-4A and NS4A proteins. Huh-7
cells expressing pFLAG-CMV2 (non-expressing control), pFLAG-CMV2 /NS3, pFLAG-CMV2
/NS3-4A, pFLAG-CMV2/NS4A vectors containing HCV non-structural NS3, NS3-4A and
NS4A proteins were examined for cell viability by crystal violet cell viability assay. (A) Huh-7
cells transfected with above mentioned vectors stained with crystal violet dye which clearly
showing the cell death in HCV non-structural NS3-4A and NS4A proteins expressing cells. (B)
(A)
(B)

Chapter 4 Results
88
Graphical representation of cell viability of vectors-harbouring cells. P values were calculated by
using an unpaired Student’s t-test (mean ± SEM; n=03; ***p<0.001, **p<0.01).
4.9 Intracellular localizations of HCV non-structural NS3, NS3-4A and NS4A proteins
We examined the intracellular localization of HCV non-structural NS3, NS3-4A and NS4A
proteins to endoplasmic reticulum and mitochondria. GFP tagged pEGFP-C1 (non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing NS3,
NS3-4A and NS4A proteins were used to observe localization. KDEL antibody was used to stain
Endoplasmic reticulum while MitoTracker CMXRos Red was used to stain mitochondria in live
cells. Confocal immunofluorescence microscopic analysis showed that all HCV non-structural
NS3, NS3-4A and NS4A proteins were localized on endoplasmic reticulum (Fig.4.16). While in
the case of Mitochondria, only HCV non-structural NS3-4A complex and NS4A proteins shown
significance localization to the perinuclear region (Fig.4.17). These analyses suggest that HCV
non-structural NS4A protein alone or in complex with NS3 induces mitochondrial perinuclear
clustering.
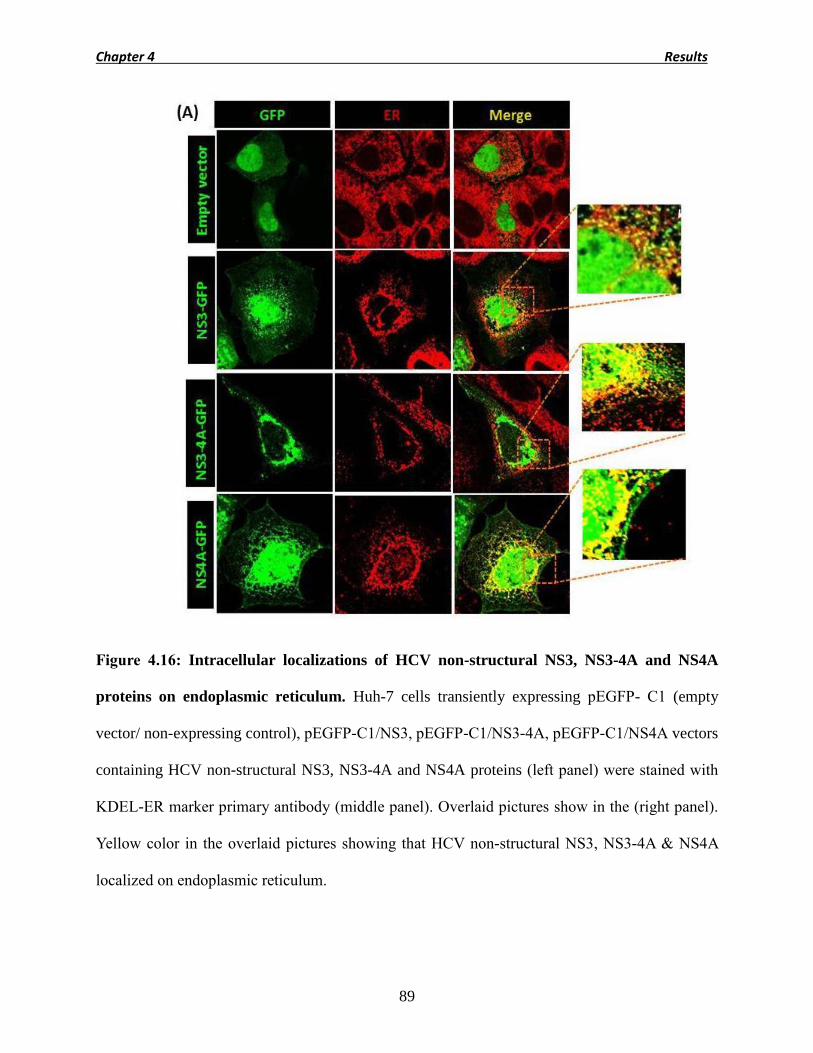
Chapter 4 Results
89
Figure 4.16: Intracellular localizations of HCV non-structural NS3, NS3-4A and NS4A
proteins on endoplasmic reticulum. Huh-7 cells transiently expressing pEGFP- C1 (empty
vector/ non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors
containing HCV non-structural NS3, NS3-4A and NS4A proteins (left panel) were stained with
KDEL-ER marker primary antibody (middle panel). Overlaid pictures show in the (right panel).
Yellow color in the overlaid pictures showing that HCV non-structural NS3, NS3-4A & NS4A
localized on endoplasmic reticulum.

Chapter 4 Results
90
Figure 4.17: Intracellular localizations of HCV non-structural NS3, NS3-4A and NS4A
proteins on mitochondria. Huh-7 cells transiently expressing pEGFP- C1 (empty vector/ non-
expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors containing
HCV non-structural NS3, NS3-4A and NS4A proteins (left panel) were stained with mitotracker
(CMXROS) to stain mitochondria (middle panel). Overlaid pictures are shows in the (right
panel). Yellow color in the overlaid pictures showing that HCV non-structural NS3-4A & NS4A
localized on mitochondria.

Chapter 4 Results
91
4.10 HCV non-structural NS3-4A and NS4A proteins induces mitochondrial
fragmentation/ fission
To examine the induced mitochondrial dynamics changes in HCV non-structural NS3-4A, NS4A
proteins expressing Huh-7 cells, transfected cells were stained with membrane potential
dependent mitotracker-Red. The mitochondria network was severely damaged in pEGFP/NS3-
4A and pEGFP/NS4A expressing cells, while in pEGFP-C1 (non-expressing control) and
pEGFP/NS3 expressing cells shows filamented mitochondria with no damage (Fig. 4.18A).
Graphical representation of percentage of fragmented mitochondria shows almost 82 %
fragmented mitochondria in pEGFP/NS4A containing NSA protein expressing cells, 78 % in
pEGFP/NS3-4A containing NS3-4A protein expressing cells, while 4 % in pEGFP/NS3
containing NS3 protein expressing cells and 1 % in pEGFP-C1 (non-expressing control) (Fig.
4.18 B). Hence these results collectively suggest that HCV non-structural NS4A protein alone
and in complex (NS3-4A) localized into mitochondria and disrupt its morphology.

Chapter 4 Results
92
Figure 4.18 (A): HCV non-structural NS3-4A and NS4A proteins induce mitochondrial
fragmentation. Huh-7 cells transiently expressing pEGFP- C1 (empty vector/ non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors containing HCV non-
structural NS3, NS3-4A and NS4A proteins (middle panel) were stained with mitotracker
(CMXROS) to stain mitochondria (right panel). Overlaid pictures are showing in the (left panel).
HCV non-structural NS3-4A & NS4A proteins expressing cells (right panel) clearly shows the
mitochondrial fragmentation. Areas marked with green boxes were enlarged (zoom-in).

Chapter 4 Results
93
Figure 4.18 (B): Graphical representation of HCV non-structural NS3-4A and NS4A
proteins induces mitochondrial fragmentation. Cells with fragmented mitochondria were
counted randomly. P values were calculated by using an unpaired Student’s t-test (mean ± SEM;
n=03; ***p<0.001, *p<0.05).
(B)

Chapter 4 Results
94
4.11 BAX translocation and Bcl-XL regulation in HCV non-structural NS3-4A and NS4A
proteins expressing Huh-7 cells
BAX is multidomain protein of Bcl-2 family a pro-apoptotic protein and essential for all events
of apoptosis via the intrinsic pathway (Oltvai, Milliman et al., 1993; Wei, Zong et al., 2001).
Normally BAX is expressed in an inactive state in the cytosol and in response to apoptotic
stimuli, conformational alterations regulates its activation and ultimately translocate the BAX
towards mitochondria (Crow, Mani et al., 2004). While Bcl-XL is an anti-apoptotic protein,
mostly localize to the outer membrane of mitochondria and inhibit the cytochrome c release to
the cytosol (Krajewski, Tanaka et al., 1993; Kluck, Bossy-Wetzel et al., 1997).
GFP tagged pEGFP-C1 (non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and
pEGFP-C1/NS4A vectors containing HCV non-structural NS3, NS3-4A and NS4A proteins were
transfected to analyze the BAX translocation. Confocal immunofluorescence analysis shows the
maximum BAX protein translocation towards mitochondria in HCV non-structural NS3-4A and
NS4A expressing cells, while NS3 and pEGFP-C1 (non-expressing control) shows negligible
translocation (Fig. 4.19A). Immunoblotting technique was used to analyze the expression levels
of BAX and Bcl-XL proteins were analyzed using anti-BAX and anti-Bcl-XL antibodies.
Transfection of Huh-7 cells were done with pEGFP-C1 (non-expressing control), pEGFP-
C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing HCV non-structural NS3,
NS3-4A and NS4A proteins, cells treated with staurosporine served as positive control, while
Huh-7 cells serve as mock. BAX protein expression were observed up regulated in positive
control huh-7 cells, HCV non-structural NS3-4A and NS4A proteins expressing cells, while
down regulated in mock cells, pEGFP-C1 (non-expressing control) and in pEGFP-C1/NS3
containing HCV non-structural NS3 protein expressing cells (Fig. 4.19 B). In the case of Bcl-XL

Chapter 4 Results
95
protein expression, down regulated expression were observed in positive control Huh-7 cells,
pEGFP-C1/NS3-4A containing HCV non-structural NS3-4A and in pEGFP-C1/NS4A containing
HCV non-structural NS4A protein expressing cells, while up regulated expression was observed
in mock cells, pEGFP-C1 (non-expressing control) and in pEGFP-C1/NS3 containing HCV non-
structural NS3 protein expressing cells (Fig. 4.19 C). These results overall demonstrate that HCV
non-structural protein NS3-4A, NS4A containing Huh-7 expressing cells and positive control
shown down regulated expression of Bcl-XL protein which is anti-apoptotic in nature while
shown up regulated expression of BAX protein which is pro-apoptotic in nature hence, HCV
non-structural protein NS4A alone and in complex with NS3 (NS3-4A) accumulate on
mitochondria that disturb the mitochondrial dynamics and ultimately up and down regulate the
expression of anti and pro-apoptotic proteins.

Chapter 4 Results
96
Figure 4.19 (A): HCV non-structural NS3-4A and NS4A induces BAX translocation to
mitochondria. Huh-7 cells transiently expressing pEGFP- C1 (empty vector/ non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors containing HCV non-
structural NS3, NS3-4A and NS4A proteins (left panel) were incubate with BAX primary
antibody rabbit (second left panel) and were stained with mitotracker (CMXROS) to stain
mitochondria (second right panel). Yellow color in the overlaid pictures (right panel) showing
that in HCV non-structural NS3-4A & NS4A proteins expressing cells, BAX protein induces
translocation to mitochondria.

Chapter 4 Results
97
Figure 4.19 (B-C): Western blot analysis of pro-apoptotic BAX protein and Bcl-XL protein.
Blot was probed with anti-BAX antibody and up regulated expression was observed in positive
control (Huh-7 cells treated with Staurosporine), pEGFP-C1/NS3-4A vector expressing HCV
non-structural NS3-4A protein & in pEGFP-C1/NS4A vector expressing HCV non-structural
NS4A protein. β-actin was used as an internal loading control. (C) Western blot analysis of anti-
apoptotic Bcl-XL protein. Blot was probed with anti-Bcl-XL antibody and down regulated
expression observed in positive control (huh-7 cells treated with Staurosporine), pEGFP-
C1/NS3-4A vector expressing HCV non-structural NS3-4A protein & in pEGFP-C1/NS4A
vector expressing HCV non-structural NS4A protein. β-actin was used as an internal loading
control.
(B)
Lane 1: + control
Lane 2: Mock
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP
(C)
Lane 1: Mock
Lane 2: + control
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP

Chapter 4 Results
98
4.12 HCV non-structural NS3-4A and NS4A proteins increased mitochondrial superoxide
generation in Huh-7 cells
Oxidative phosphorylation of complexes I and III of electron transport chain produce
mitochondrial superoxide as byproduct (Liu, Fiskum et al., 2002; Kudin, Bimpong-Buta et al.,
2004). In the presence of viral proteins or other environmental stress ROS level become
increases dramatically. For detection of superoxide in the mitochondria of live cells a
MitoSOX™ Red mitochondrial superoxide indicator a fluorogenic dye with highly selective
detection was used. To estimate the mitochondrial superoxide generation in GFP tagged pEGFP-
C1 (non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A
vectors containing NS3, NS3-4A and NS4A proteins expressing cells, 72 hrs post transfection
cells were stained with MitoSOX™ Red dye. Immunofluorescence images show highly
increased superoxide generation in pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors expressing
HCV non-structural NS3-4A and NS4A proteins expressing cells and almost none in pEGFP-C1
(non-expressing control) and pEGFP-C1/NS3 containing HCV non-structural NS3 protein
expressing cells (Fig. 4.20 A). Figure 4.20 B depicts the graphical representation of percentage of
superoxidation in mitochondria, in which pEGFP-C1/NS4A containing HCV non-structural
NS4A proteins expressing cells shows 84 % superoxide generation, pEGFP-C1/NS3-4A
containing HCV non-structural NS3-4A protein shows 79 %, pEGFP-C1/NS3 containing HCV
non-structural NS3 protein shows 4 % and pEGFP-C1 (non-expressing control) shows 3 %
generation.

Chapter 4 Results
99
Figure 4.20 (A): HCV non-structural NS3-4A and NS4A proteins induced the
mitochondrial superoxide generation. Huh-7 cells transiently expressing pEGFP- C1 (empty
vector/ non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors
containing HCV non-structural NS3, NS3-4A and NS4A proteins (left panel) were stained with
MitoSOX™ Red mitochondrial superoxide (right panel). Overlaid pictures show in the (middle)
panel. Right panel pictures showing that HCV non-structural NS3-4A & NS4A proteins
expressing cells generate high amount of superoxide species.

Chapter 4 Results
100
Figure 4.20 (B): Graphical representation of HCV non-structural NS3-4A and NS4A
proteins increased the mitochondrial superoxide generation. Cells with increased level of
superoxide generation were counted randomly. P values were calculated by using an unpaired
Student’s t-test (mean ± SEM; n=03; ***p<0.001).
(B)

Chapter 4 Results
101
4.13 Cytochrome c translocation in HCV non-structural NS3-4A and NS4A proteins
expressing Huh-7 cells
During mitochondrial mediated apoptosis outer membrane permeabilization allowed cytochrome
c release to cytosol (Goldstein, Waterhouse et al., 2000). To assess the cytochrome c
translocation, Huh-7 cells were transfected with pEGFP-C1 (non-expressing control), pEGFP-
C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing HCV non-structural NS3,
NS3-4A and NS4A proteins, cells treated with staurosporine served as positive control, while
Huh-7 cells serve as mock. Immunoblot analysis of mitochondrial fractions shows down
regulated expression in positive control, NS3-4A and NS4A expressing cells and up regulated in
mock, pEGFP-C1 (non-expressing control) and in HCV non-structural NS3 protein expressing
cells (Fig. 4.21 A), while cytosolic fraction shows up regulated expression in positive control,
NS3-4A and NS4A expressing cells and down regulated in mock, pEGFP-C1 (non-expressing
control) and in NS3 expressing cells (Fig.4.21 B). These results collectively represented the
mitochondrial membrane damage and increase in membrane permeability.

Chapter 4 Results
102
Figure 4.21: Cytochrome c translocation in HCV non-structural NS4A and NS3-4A
proteins expressing cells. Huh-7 cells were transfected with pEGFP-C1 (non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing HCV
non-structural NS3, NS3-4A and NS4A proteins. Huh-7 cells treated with staurosporine served as
positive control, while only Huh-7 cells serve as mock. (A) Down regulated expression was
observed in mitochondrial fractions in positive control (treated with Staurosporine), pEGFP-
C1/NS3-4A & pEGFP-C1/NS4A vectors containing HCV non-structural NS3-4A and NS4A
proteins expressing Huh-7 cells. β-actin was used as an internal loading control. (B) Up regulated
expression was observed in cytosolic fractions in positive control (Huh-7 cells treated with
Staurosporine), pEGFP-C1/NS3-4A & pEGFP-C1/NS4A vectors containing HCV non-structural
NS3-4A and NS4A proteins expressing huh-7 cells. β-actin was used as an internal loading
control.
(A & B)
Lane 1: Mock
Lane 2: + control
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP

Chapter 4 Results
103
4.14 Effect of HCV non-structural NS3-4A and NS4A proteins on the mitochondrial
oxidative phosphorylation system
Previous studies show that HCV infection affects the oxidative phosphorylation (Piccoli, Scrima
et al., 2007; Kim, Syed et al., 2013). Hence, we designed an experiment to compare the effects
of OXPHOS Complex I activity between whole HCV genome and individual HCV non-
structural proteins i.e. NS3, NS4A and NS3-4A. Huh-7 cells infected with HCVcc for 3 days
while only Huh-7 cells serve as mock. Graphical representation shows the percentage activity of
OXPHOS Complex I in Transfection of Huh-7 cells was done with pFLAG-CMV2 /NS3,
pFLAG-CMV2 /NS3-4A, pFLAG-CMV2 /NS4A vectors containing HCV non-structural
proteins and pFLAG-CMV2 vector serve as (non-expressing control). 72 hrs post transfection
cells were harvested and mitochondrial I enzyme activity was analyzed as per manufacturer’s
instruction. NS3-4A protein expressing cells shows total 16 % reduction, NS4A protein
expressing cells shows 19 % reduction while NS3 protein expressing cells shows only 15 %
reduction (Fig.4.22 B). Expression levels of HCV non-structural NS3, NS3-4A and NS4A
proteins used in assay were confirmed by immunoblotting (Fig.4.22 C). From these results we
inferred that HCV non-structural NS3, NS3-4A, NS4A proteins individually and HCVcc leads to
overall 15-20 % reduction of OXPHOS Complex I activity as compared to mock.

Chapter 4 Results
104
Figure 4.22 (A): Graphical representation of OXPHOS Complex I activity between whole
HCV genome (HCVcc) and Huh-7 cells (mock). Huh-7 cells infected with HCVcc and
harvested 3 days post infection while only Huh-7 cells used as mock. Activity of mitochondrial I
enzyme was measured according to manufacturer’s instruction. P values were calculated by
using an unpaired Student’s t-test (mean ± SEM; n=03; **p<0.01).
(A)

Chapter 4 Results
105
Figure 4.22 (B): Graphical representation of the effect of HCV individual HCV non-
structural NS3, NS3-4A, NS4A proteins on the reduction of OXPHOS Complex I activity.
Huh-7 cells expressing HCV non-structural NS3, NS3-4A, NS4A proteins and pFLAG-CMV2
vector (non-expressing control) were transfected for 72 hrs and used to analyze the activity of
mitochondrial I enzyme. P values were calculated by using an unpaired Student’s t-test (mean ±
SEM; n=03; **p<0.01, *p<0.05).
(B)

Chapter 4 Results
106
Figure 4.22 (C): Western blot analysis of HCV non-structural NS3, NS3-4A and NS4A proteins
used for the assay reduction of OXPHOS Complex I activity. β-actin was used as an internal
loading control.
Lane 1: Empty vector
Lane 2: NS3-FLAG
Lane 3: NS3-4A-FLAG
Lane 1: empty vector
Lane 2: NS4A-FLAG
Lane 1: Mock
Lane 2: Core protein

Chapter 4 Results
107
4.15 Caspase -3, 7, 9 activation and poly (ADP-ribose) polymerase (PARP) cleavage in HCV
non-structural NS3-4A and NS4A expressing Huh-7 cells
After destruction of mitochondrial functions, next step of intrinsic apoptosis is to activate
procaspases (Crow, Mani et al., 2004). Once cytochrome c released to cytosol it works together
with Apaf-1 (apoptotic protease activating factor 1) to activate procaspase-9 an initiator caspase
(Lawen, 2003). Activation of caspase-9 then activates procaspase-7 and 3 (effector caspases).
Further caspase-3 plays an important role in the cleavage of PARP (poly (ADP-ribose)
polymerase) (Boulares, Yakovlev et al., 1999). Active caspase-3 become capable to cleave
cellular substrate PARP which ultimately induces apoptosis is the cell i.e. blebbing of membrane,
DNA fragmentation and deterioration of cell structure (Coleman, Sahai et al., 2001; Leverrier
and Ridley, 2001; Sebbagh, Renvoize et al., 2001).To confirm the activation of caspase-9, 7, 3
and cleavage of PARP cells were transfected with pEGFP-C1 (non-expressing control), pEGFP-
C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing HCV non-structural NS3,
NS3-4A and NS4A proteins. Cells treated with staurosporine served as positive control, while
Huh-7 cells serve as mock. Blot treated with anti procaspase 9 antibody shown 47 kDa band
representing procaspase-9 in all experimental cell while beside 47 kDa, a polypeptide of 35 kDa
representing the cleavage form of procaspase-9 have also observed in positive control, HCV non-
structural NS3-4A and NS4A proteins expressing Huh-7 cells (Fig.4.23 A). Blot treated with anti
procaspase-7 antibody shown 35 kDa band of procaspase-7 in all experimental cells while beside
35 kDa, a polypeptide of 19 kDa band has also observed in positive control, HCV non-structural
NS3-4A and NS4A protein expressing Huh-7 cells representing the active form of caspase-7
(Fig.4.23 B). For capase-3 activation blot were treated with anti procaspase-3 antibody, all
experimental cells shown procaspase-3 expression with 35 kDa band while positive control,

Chapter 4 Results
108
HCV non-structural NS3-4A and NS4A proteins expressing Huh-7 cells have shown a
polypeptide of 17 kDa band representing the caspase-3 active form (Fig. 4.23C). Consistent with
the caspase-3 activation, western blot analysis of all experimental cells shown the full length
inactive form of PARP protein with 116 kDa band while positive control, HCV non-structural
NS3-4A and NS4A proteins expressing Huh-7 cells shown the cleavage of PARP protein into a
smaller unit of 85 kDa band (Fig. 4.23 D).

Chapter 4 Results
109
Figure 4.23: (A) Western blot analysis of Caspase-9 protein in HCV non-structural NS3-4A
and NS4A proteins expressing Huh-7 cells. Huh-7 cells were transfected with pEGFP-C1
(non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A containing
HCV non-structural NS3, NS3-4A and NS4A proteins. Cells treated with staurosporine served as
positive control, while only Huh-7 cells serve as mock. 72 hrs post transfection cells were
harvested and used for western blot analysis. Blot was probed with anti procaspase-9 antibody
and activation of procaspase-9 was observed only in positive control (treated with
Staurosporine), pEGFP-C1/NS3-4A & pEGFP-C1/NS4A containing HCV non-structural NS3-
4A and NS4A proteins expressing cells. β-actin was used as an internal loading control.
(A)
Lane 1: Mock
Lane 2: + control
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP

Chapter 4 Results
110
Figure 4.23: (B) Western blot analysis of Caspase-7 protein in HCV non-structural NS3-4A
and NS4A proteins expressing Huh-7 cells. Huh-7 cells were transfected with pEGFP-C1
(non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A containing
HCV non-structural NS3, NS3-4A and NS4A proteins. Cells treated with staurosporine served as
positive control, while only Huh-7 cells serve as mock. 72 hrs post transfection cells were
harvested and used for western blot analysis. Blot was probed with anti procaspase-7 antibody
and activation of procaspase-7 was observed only in positive control (treated with
Staurosporine), pEGFP-C1/NS3-4A & pEGFP-C1/NS4A containing HCV non-structural NS3-
4A and NS4A proteins expressing Huh-7 cells. β-actin was used as an internal loading control.
(B)
Lane 1: Mock
Lane 2: + control
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP

Chapter 4 Results
111
Figure 4.23: (C) Western blot analysis of Caspase-3 protein in HCV non-structural NS3-4A
and NS4A proteins expressing Huh-7 cells. Huh-7 cells were transfected with pEGFP-C1 (non-
expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A containing
HCV non-structural NS3, NS3-4A and NS4A proteins. Cells treated with staurosporine served as
positive control, while only Huh-7 cells serve as mock. 72 hrs post transfection cells were
harvested and used for western blot analysis. Blot was probed with anti procaspase-3 antibody
and activation of procaspase-3 was observed only in positive control (treated with
Staurosporine), pEGFP-C1/NS3-4A & pEGFP-C1/NS4A containing HCV non-structural NS3-
4A and NS4A proteins expressing Huh-7 cells. β-actin was used as an internal loading control.
(C)
Lane 1: Mock
Lane 2: + control
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP

Chapter 4 Results
112
Figure 4.23: (D) Western blot analysis of PARP protein in HCV non-structural NS3-4A and
NS4A proteins expressing cells. Huh-7 cells were transfected with pEGFP-C1 (non-expressing
control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-C1/NS4A containing HCV non-
structural NS3, NS3-4A and NS4A proteins. Cells treated with staurosporine served as positive
control, while only Huh-7 cells serve as mock. 72 hrs post transfection cells were harvested and
used for western blot analysis. Blot was probed with anti PARP antibody and cleavage of PARP
was observed only in positive control (treated with Staurosporine), pEGFP-C1/NS3-4A &
pEGFP-C1/NS4A containing HCV non-structural NS3-4A and NS4A proteins expressing Huh-7
cells. β-actin was used as an internal loading control.
(D)
Lane 1: + control
Lane 2: Mock
Lane 3: Empty vector
Lane 4: NS3-GFP
Lane 5: NS3-4A-GFP
Lane 6: NS4A-GFP

Chapter 4 Results
113
4.16 Chromatin condensation in HCV non-structural NS3-4A and NS4A proteins
expressing Huh-7 cells
Apoptosis ends on chromatin condensation which is one of the most important occasion.
Caspase-3, -7 (execution caspases) activates cytoplasmic endonucleases and proteases which
degrades nuclear material and cytoskeletal proteins in the cell (Elmore, 2007). Endonuclease
CAD (Caspase-activated DNAse) degrades chromosomal DNA and cause the chromatin
condensation (Sakahira, Enari et al., 1998). To analyze the condensed nuclei in HCV non-
structural NS4A and NS3-4A proteins expressing Huh 7 cells, cells were transfected with
pEGFP-C1 (non-expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A and pEGFP-
C1/NS4A vectors containing HCV non-structural NS3, NS3-4A and NS4A proteins. Cells were
harvested at different time points (24 hrs, 48 hrs & 72 hrs) and stained with Hoechst 33342 stain
and were observed under the florescence microscope. More condensed nuclei were observed at
72 hrs post transfection in pEGFP-C1/NS3-4A and pEGFP-C1/NS4A vectors containing HCV
non-structural NS3-4A and NS4A proteins while pEGFP-C1 (non-expressing control), pEGFP-
C1/NS3 vector containing HCV non-structural NS3 protein expressing cells shown negligible
amount of condensed nuclei (Fig. 4.24 A). Fig. 4.24 B depicts the graphical representation of
percentage cells with condensed nuclei in which pEGFP-C1/NS3-4A vector containing HCV
non-structural NS3-4A shown 48 % condensed nuclei, pEGFP-C1/NS4A vectors containing
HCV non-structural NS4A shown 50 % while pEGFP-C1 (non-expressing control) and pEGFP-
C1/NS3 vector containing HCV non-structural NS3 shown only 10-15 % condensed nuclei at 72
hrs post transfection.

Chapter 4 Results
114
Figure 4.24 (A): Chromatin condensation in HCV non-structural NS3-4A and NS4A
proteins expressing cells. Huh-7 cells transiently expressing pEGFP- C1 (empty vector/ non-
expressing control), pEGFP-C1/NS3, pEGFP-C1/NS3-4A, pEGFP-C1/NS4A vectors containing
HCV non-structural NS3, NS3-4A and NS4A proteins Huh-7 cells were harvested 24 hrs (left
panel), 48hrs (middle panel) and 72 hrs (right panel) post transfection. Nuclei of the cells were
stained with Hoechst 33342 stain. 72hrs post transfected stained nuclei showing more condensed

Chapter 4 Results
115
nuclei (bright) in pEGFP-C1/NS3-4A & pEGFP-C1/NS4A vectors containing HCV non-
structural NS3-4A and NS4A proteins expressing Huh-7 cells.
Figure 4.24 (B): Graphical representation of chromatin condensation in NS3-4A and NS4A
expressing cells. Cells with condensed nuclei were counted randomly. P values were calculated
by using an unpaired Student’s t-test (mean ± SEM; n=03; ***p<0.001).
(B)

Discussion

Chapter 5 Discussion
116
DISCUSSION
Hepatitis C virus (HCV) is a major cause of viral hepatitis in humans. It is a blood borne
pathogen that targets hepatocytes and moves from acute to chronic phase, favoring
complicated hepatic and extra-hepatic pathologies like apoptosis, fibrosis and cirrhosis,
eventually leads to hepatocellular carcinoma (HCC) (Qadri, et al., 2004; Howe et al., 2004;
Joyce et al., 2009). A strong association is being documented between chronic HCV infection
with genotype 3a and HCC (63.44 % of tested HCC patients) in Pakistani population (Idrees et
al., 2009). HCV shows hepatocyte specific tropism, where it propagates, infects the
neighboring hepatocytes and takes control over the host machinery by perturbing normal
signaling pathways to favor its replication (Clement, et al., 2009; Joyce et al., 2009).
HCV induces several complex pathways leading to generation of reactive oxygen species,
hepatic inflammation, hepatic fibrosis and HCC (Fartoux et al., 2005; Pekow et al., 2007; Bieche
et al., 2005). With the advancement of research in molecular virology study related to HCV
proteomics revealed many underlying pathways involved in HCV associated pathogenesis but
the factors involved in HCV induced pathogenesis are still unclear (Sheikh et al., 2008; Joyce et
al., 2009).
Many viruses including HCV have been shown to induce oxidative stress during replication and/
or protein expression. Hepatitis C infection in human liver as well as different experimental
models is associated with increased hepatic oxidative stress (Liu et al., 2003; Qadri et al., 2006;
Ghaziani et al., 2006). The previous study indicated that HCV and its certain proteins NS4A
and NS4B modulate the cellular signaling pathways in such a way that these proteins

Chapter 5 Discussion
117
accumulates on the mitochondria and ultimately disturb the mitochondrial dynamics which lead
the cells towards the mitochondrial mediated apoptotic pathway.
In the present study we examined the possible effects of HCV non-structural NS4A protein alone
and in complex with NS3 (NS3-4A) in the induction of apoptosis via mitochondrion mediated
pathway. Constructions of intergenotypic and intragenotypic chimeras of strain (JFH-1)
genotype 2a and HCV genotype 1b cloned genome (bicistronic replicons) are working efficiently
to get the advantage of boosted replication (Blight, Kolykhalov et al. 2000; Lindenbach, Evans et
al. 2005). In previous study novel clones of HCV genotype 3a infectious cDNA named as strain
S52 and of genotype 4a known as strain ED43, however robust cell culture model for expression
of individual hepatitis C virus proteins of different genotypes, especially genotype 3a are still
missing. (Gottwein, Scheel et al. 2010) From Pakistan Butt et al. (Butt, Idrees et al. 2011) have
only reported the construction of few mammalian expression plasmids encoding some genes of
HCV genotype 3a.
In the present study, we successfully amplified and cloned the full length NS3, NS3-4A & NS4A
genes of HCV genotype 3a separately in pFLAG-CMV2 and pEGFP-C1 expression vectors.
Positive clones were confirmed by restriction digestion analysis and sequencing from both
directions. Each particular clone was transfected separately to Huh-7 cells and expression was
analyzed using Western blot assays. Expression of transfected cells were also analyzed using
florescence microscopy after having probed with particular florescent antibodies. pFLAG-CMV2
expression vector used in the present study has the ability for intracellular transient expression of
N-terminal Met-FLAG which is fused with desired HCV non-structural NS3, NS3-4A and NS4A
proteins and detected by using anti-FLAG antibody. While pEGFP-C1 expression vector has the

Chapter 5 Discussion
118
red-shifted variant of wild-type GFP protein which is fused with desired HCV non-structural
NS3, NS3-4A and NS4A proteins and can easily detected by using anti GFP antibody.
Fluorescence and confocal microscopy is a very powerful tool for labeling of different organelles
and proteins within a living cell to investigate there interaction with other organelles, protein and
to investigate the transfected proteins expression in the eukaryotic cell. Efficient cell culture
based systems have the ability to express individual HCV non-structural NS3, NS3-4A and
NS4A protein separately in Huh-7 cells.
Apoptosis, programmed/ planned cell death or cell suicide, is a normal physiological process and
occurs normally during development, aging, hemostasis mechanism and defense mechanism in
pathological conditions (Kerr, Winterford et al. 1994; Elmore 2007). Different studies suggest
that apoptosis occurs in the human hepatocytes mostly by mitochondria-mediated apoptosis
pathway but still it is under investigations that whether apoptosis is mediated by T lymphocytes
or by HCV replication itself. (Bantel and Schulze-Osthoff 2003; Deng, Adachi et al. 2008). HCV
infection prompts Bax-induced, mitochondrial mediated, caspase-3 dependent apoptosis when
infected Huh-7.5 cells with chimeric J6/JFH1 strain of HCV genotype 2a was used in their
experiments (Deng, Adachi et al. 2008). Zhu et al. was successful in culturing a human hepatoma
novel cell line (LH86) permitted JFH-1 HCV genotype 2a infection which induced cell apoptosis
which was related to the viral replication in the cell.
Different structural and non-structural proteins of HCV are reported to be involved in the
induction of the mitochondrial-mediated caspase-3 dependent apoptosis pathway when expressed
in different cell culture systems. (Chou, Tsai et al. 2005; Chiou, Hsieh et al. 2006; Nomura-
Takigawa, Nagano-Fujii et al. 2006). HCV Core protein expressing in Huh7 cells activated
TRAIL-mediated apoptosis during the chronological induction of caspase-8 activation by

Chapter 5 Discussion
119
triggering of mitochondrial mediated apoptosis signaling pathway (Chou, Tsai et al. 2005). HCV
E2 protein of HCV have the capability to induce apoptosis because they have the genetic
similarity with the envelope proteins of other viruses of the flaviviridae family i.e. dengue and
Langat virus (Chiou, Hsieh et al. 2006). While Nomura-Takigawa et al. reported that HCV non-
structural NS4A (genotype 1b) is probably involved in the induction of apoptosis through
mitochondrial mediated apoptosis while HCV non-structural NS3-4A complex is more prone to
translocate towards mitochondria as compared to HCV non-structural NS3 protein alone. Our
study clearly shown that HCV non-structural NS4A protein (genotype 3a) alone and in complex
with NS3 (NS3-4A) induces apoptosis via Bax-induced, mitochondrial mediated, caspase -3
dependent pathway.
Our results indicated the significant number of cell death in Huh-7 cells expressing HCV non-
structural NS3-4A and NS4A proteins in contrast to NS3 expressing and vector transfected non-
expressing control cells (Fig. 4.1). Intracellular compartmentalization of HCV protein is
considered as an important aspect and it is clearly understood that HCV structural and non-
structural proteins accumulates on ER for maturation (Moradpour, Penin et al. 2007), while some
of HCV protein were reported to be localized on mitochondria as well. Our results clearly shows
the localization of HCV non-structural NS3, NS3-4A and NS4A proteins to ER (Fig. 4.2A) while
only NS3-4A complex and NS4A shown localization on the mitochondria (Fig. 4.2B) and this
result confirms that both HCV non-structural NS3-4A and NS4A proteins localized on
mitochondria and disturb the mitochondrial dynamics. Furthermore our results clearly showed
the significant mitochondrial fragmentation in HCV NS3-4A and NS4A expressing Huh-7 cells
(Fig. 4.3). Bax proteins and Bcl-2 proteins belong to Bcl-2 family that takes part in prominent
role in mitochondrial mediated apoptotic pathway and in addition to the regulation of

Chapter 5 Discussion
120
cytochrome c release from mitochondria to cytosol (Borner 2003; Qi, Li et al. 2010). A pro-
apoptotic protein Bax, translocates and anchors on the outer mitochondrial membrane (OMM),
while Bcl-2 or Bcl-xL anti-apoptotic proteins inhibit the Bax protein to integrate in the
mitochondria. In the current study we have observed increased accumulation of Bax and
conformational changes on mitochondria in the HCV non-structural NS3-4A and NS4A proteins
expressing Huh-7 cells (Fig 4.4A). The current study also demonstrated the up regulated
expression of Bax protein and down-regulated expression of Bcl-2 protein in HCV non-structural
NS3-4A and NS4A expressing Huh-7 cells (Fig. 4.4B & C). These results overall demonstrate
that HCV non-structural protein NS3-4A, NS4A containing Huh-7 expressing cells and positive
control shown down regulated expression of Bcl-XL protein which is anti-apoptotic in nature
while shown up regulated expression of BAX protein which is pro-apoptotic in nature hence,
HCV non-structural protein NS4A alone and in complex with NS3 (NS3-4A) accumulate on
mitochondria that disturb the mitochondrial dynamics and ultimately up and down regulate the
expression of anti and pro-apoptotic proteins.
I observed increased production of mitochondrial superoxide in HCV non-structural NS3-4A and
NS4A proteins expressing Huh-7 cells (Fig. 4.5) because mitochondrial superoxide is an
important supply of reactive oxygen species (ROS) in the cell and may contribute to prompt the
conformational changes of Bax protein, its dimerization and mitochondrial translocation
(D'Alessio, De Nicola et al. 2005; Nie, Tian et al. 2008). Bax protein ultimately integrates into
the outer mitochondrial membrane (OMM), and from mitochondria cytochrome c releases into
the cytosol. The present research found HCV non-structural NS3-4A and NS4A proteins
expression prompted the release of cytochrome c into the cytosol (Fig. 4.6). Activation of
caspases takes part as dominant character in the completion of apoptosis and in the same manner

Chapter 5 Discussion
121
released cytochrome c from mitochondria activates effector caspases. Normally cytochrome c is
located on the mitochondrial inner membrane although its cofactors Apaf-1 and procaspase-9 are
expressing in the cytosol. Once Cytochrome c is released to the cytosol forms a multimeri Apaf-
1/cytochrome c complex and ultimately activates the procaspase-9. After the activation of
caspase-9, it cleaves and activate the downstream caspase i.e. caspase-7 and -3 (Budihardjo,
Oliver et al. 1999; Denault and Salvesen 2002; Lawen 2003; Gogvadze, Orrenius et al. 2006).
Further active caspase-3 cleaved its substrate PARP into two fragments i.e. p89 and p24 leading
the cell towards apoptosis by DNA fragmentation, Nuclei condensation, membrane blebbing,
nuclear breakdown, apoptotic bodies etc. (Boulares, Yakovlev et al. 1999; Loeffler and Kroemer
2000; Lockshin and Zakeri 2001). Western blot analysis in our study demonstrates the activation
of caspase-9, caspase-7, caspase -3, and cleavage of PARP in HCV non-structural NS3-4A and
NS4A proteins expressing Huh-7 cells (Fig. 4.8 A, B, C & D).
Various research studies clearly indicate that affects of HCV infection on the respiratory chain
complex I enzyme (NADH dehydrogenase) of mitochondria via oxidative phosphorylation which
ultimately promote the destruction of mitochondrial outer membrane (Korenaga, Okuda et al.
2005; Korenaga, Wang et al. 2005; Piccoli, Scrima et al. 2007; Kim, Syed et al. 2013). In the
present study we compare the consequences of OXPHOS Complex I activity of HCV infected
cells (HCVcc) with HCV non-structural NS3, NS3-4A and NS4A proteins expressing Huh-7
cells in which our results shown that there is a 15 % reduction in HCVcc and NS3 expressing
cells (Fig. 4.7A) and 18-20 % reduction in HCV non-structural NS3-4A and NS4A proteins
expressing Huh-7 cells (Fig. 4.7B) as compared mock. These results clearly suggest that HCV
non-structural NS3-4A and NS4A proteins accumulate on mitochondria hence promote the
destruction of mitochondrial outer membrane and induces the mitochondrial-mediated apoptosis.

Chapter 5 Discussion
122
Chromatin condensation which is the terminal stage of apoptosis is the clear evidence of
apoptosis induced cell death. Chromatin condensation has been observed in NS3-4A and NS4A
expressing cells at 72 hrs post transfection (Fig. 4.9). In conclusion, current study clearly
demonstrates the directly involvement of HCV genotype 3a non-structural NS3-4A and NS4A
proteins in the triggering of Bax translocated, mitochondrial-mediated, Caspase-3 dependent
apoptotic pathway.
Future prospects
The role of NS3-4A and NS4A proteins in mitochondrial-mediated apoptosis pathway can also
be confirm by synthesis of the mutants of these particular proteins by producing mutation at
different conserved regions in their genes. Beside the investigation of HCV non-structural NS3,
NS4A and NS3-4A proteins role in the induction of mitochondrial mediated apoptosis pathway,
these transiently expressing cell lines can also be used and facilitate in the development of novel
antiviral strategies to inhibit the replication of this noxious pathogen. These cell culture based
systems can also play a dynamic role to test the novel drugs for its inhibitory action, the
evaluation of vaccine candidates, characterization of humoral immune responses and also in the
evaluation of different signaling pathways.

References

Chapter 6 References
123
REFERENCES
Aaron, S., McMahon, M. J., Danielle, M., Torres, L., Clatts, M., Tortu, S., Mildvan,
D. and Simm, M. (2008). Intranasal transmission of hepatitis C virus:
virological and clinical evidence. Clin. Infect. Dis., 47, 931-934.
Alter, M. J., Holland, P. V., Morrow, A. G., Purcell, R. H., Finestone, S. M. and
Moritsuqu, Y. (1975). Clinical and serological analysis of transfusion-
associated hepatitis. Lancet, 11, 838-841.
Alter, M. J. (2002). Prevention of spread of hepatitis C. Hepatology, 36, S93-S98.
Alter, M. J., Margolis, H. S., Krawczynski, K., Judson, F. N., Mares, A., Alexander,
W. J., Hu, P. Y., Miller, J. K., Gerber, M. A. and Sampliner, R. E. (1992). The
natural history of community-acquired hepatitis C in the United States. The
Sentinel Counties Chronic non-A, non-B Hepatitis Study Team. N. Engl. J.
Med., 327, 1899-1905.
Arababadi, M. K., Nasiri Ahmadabadi, B., Yousefi Daredor, H. and Kennedy, D.
(2012). Epidemiology of occult hepatitis B infection among thalassemic,
hemophilia, and hemodialysis patients. Hepat Mon., 12, 315-319.
Arema, A. P. and Jacobson, I. M. ( 2009). New and experimental therapies for HCV.
Nature Rev. Gastroenterol. Hepatol., 6, 403-411
Arif, F., Fayyaz, J. and Hamid, A. (2008). Awareness among parents of children with
thalassemia major. J. Pak. Med. Assoc., 58, 621-624.

Chapter 6 References
124
Asabe, S. I, Tanji, Y., Satoh, S., Kaneko, T., Kimura, K. and Shimotohno, K. (1997).
The N-terminal region of hepatitis C virus-encoded NS5A is important for
NS4A-dependent phosphorylation. J. Virol., 71, 790-796.
Ashfaq, U. A., Javed, T., Rehman, S., Nawaz, Z. and Riazuddin, S. (2011). An
overview of HCV molecular biology, replication and immune
responses. Virol. J., 8, 161.
Australian Institute of Health and Welfare (2007). Statistics on drug use in Australia
2006 Canberra: Australian Institute of Health and Welfare.
Balayan, M. S., Andjaparidze , A. G. and Savinskaya, S. S. ( 1983). Evidence for a
virus in non-A, non-B hepatitis transmitted via the fecal-oral route.
Intervirology, 20, 23-31.
Bantel, H. and Schulze-Osthoff , K. (2003). Apoptosis in hepatitis C virus infection.
Cell Death Differ., 10, S48-58.
Bartenschlager, R., Laake, L. A., Mous, J. and Jacobsen, H. (1993). Non-structural
protein 3 of the hepatitis C virus encodes a serine-type proteinase required for
cleavage at the NS3/4 and NS4/5 junctions. J. Virol., 67, 3835-3844.
Bartenschlager, R., Lohmann, V., Wilkinson, T. and Koch, J. O. (1995). Complex
formation between the NS3 serine-type proteinase of the hepatitis C virus and
NS4A and its importance for polyprotein maturation. J. Virol., 69, 7519-7528.
Bartenschlager, R. and Lohmann, V. (2000). Replication of hepatitis C virus. J.
Gen. Virol., 81, 1631-1648.

Chapter 6 References
125
Bartenschlager, R., Frese, M. and Pietschmann, T. (2004). Novel insights into
hepatitis С virus replication and persistence. Adv. Virus. Res., 63, 71-180.
Beeson, P. B. (1943). Jaundice occurring one to four months after transfusion of
blood or plasma. Report of seven cases. JAMA., 1, 1332-1334.
Behrens, S. E., Tomei, L. and De, Francesco, R. (1996). Identification and properties
of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO. J., 15,
12-22.
Benali-Furet, NL., Chami, M., Houe, L., De, Giorgi, F., Vernejoul, F., Logorce, D.,
Buscail, L., BratenSchlager, R., Ichas, F., Rizzuto, R. and Paterlini-Brechot, P.
(2005). Hepatitis C virus core triggers apoptosis in liver cells by inducing ER
stress and ER calcium depletion. Oncogene, 24, 4921-4933.
Beran, R. K., Lindenbach, B. D. and Pyle, A. M. (2009). The NS4A protein of
hepatitis C virus promotes RNA-coupled ATP hydrolysis by the NS3 helicase.
J. Virol., 83, 3268-3275.
Bieche, I., Asselah, T., Laurendeau, I., Vidaud, D. and Degot, C. (2005). Molecular
profiling of early stage liver fibrosis in patients with chronic hepatitis C virus
infection. Virology, 332, 130-144
Blight, K. J., Kolykhalov, A. A. and Rice, C. M. (2000). Efficient initiation of HCV
RNA replication in cell culture. Science, 290, 1972-1974.
Bonino, F., Hoyer, B., Shih, J. W., Rizzetto, M., Purcell, R. H. and Gerin, J. L.
(1984). Delta hepatitis agent: structural and antigenic properties of the delta-
associated particle. Infect. Immun., 43, 1000-1005.

Chapter 6 References
126
Borner, C. (2003). The Bcl-2 protein family: sensors and checkpoints for life-or-death
decisions. Mol. Immunol., 39, 615-647.
Boulares, A. H., Yakovlev, A. G., Ivanova, V., Stoica, B. A., Wang, G., Iyer, S. and
Smulson, M. (1999). Role of poly(ADP-ribose) polymerase (PARP) cleavage
in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in
transfected cells. J. Biol. Chem., 274, 22932-22940.
Bracho, M. A., Gosalbes, M. J., Blasco, D., Moya, A. and Gonzalez-Candelas, J.
(2005). Molecular epidemiology of a hepatitis C virus outbreak in a
hemodialysis unit. Clin. Microbiol., 43, 2750-2755.
Branch, A. D., Stump, D. D., Gutierrez, J. A., Eng, F. and Walewski, J. L. (2005). The
hepatitis C virus alternate reading frame (ARF) and its family of novel
products: the alternate reading frame protein/F-protein, the double-frameshift
protein, and others. Semin. Liver Dis., 25, 105-117.
Brass, V., Bieck, E., Montserret, R., Wolk, B., Hellings, J. A., Blum, H. E., Penin, F.
and Moradpour, D. (2002). An amino-terminal amphipathic alpha-helix
mediates membrane association of the hepatitis C virus nonstructural
protein 5A. J. Biol. Chem., 277, 8130-8139.
Brass, V., Moradpour, D. and Blum, H. E. (2006). Molecular virology of hepatitis C
virus (HCV): 2006 update. Int. J. Med. Sci., 3, 29-34.
Budihardjo, I., Oliver, H., Lutter, M., Luo, X., and Wang, X. (1999). Biochemical
pathways of caspase activation during apoptosis. Annu. Rev. Cell. Dev. Biol.,
15, 269-290.

Chapter 6 References
127
Burns, J., Burrow, S., Genovese, E., Pumphrey, M., Sims, E. and Thomson, N.
(2003). The health of Indigenous Australians. South Melbourne, Oxford
University Press, 29, 397-444.
Butcher, S. J., Grimes, J. M., Makeyev, E. V., Bamford, D. H. and Stuart, D. I.
(2001). A mechanism for initiating RNA-dependent RNA polymerization.
Nature, 410, 235-240.
Butt, S., Idrees, M., Rehman, I. U., Ali, L., Hussain, A., Ali, M., Ahmed, N., Saleem,
S. and Fayyaz, M. (2011). Establishment of stable Huh-7 cell lines expressing
various hepatitis C virus genotype 3a protein: an in-vitro testing system for
novel anti-HCV drugs. Genet. Vaccines. Ther., 9, 12.
Carrere-Kremer, S., Montpellier-Pala, C., Cocquerel, L., Wychowski, C., Penin, F.
and Dubuisson, J. (2002). Subcellular localization and topology of the p7
polypeptide of hepatitis C virus. J. Virol., 76, 3720-3730.
Chandra, J., Samali, A. and Orrenius, S. (2000). Triggering and modulation of
apoptosis by oxidative stress. Free Radic. Biol. Med., 29, 323-33.
Chang, T. T., Chang, T. Y., Chen, C. C., Young, K. C., Roan, J. N., Lee, Y. C.,
Cheng, P. N. and Wu, H. L. (2001). Existence of hepatitis C virus in Culex
quinquefasciatus after ingestion of infected blood: experimental approach to
evaluating transmission by mosquitoes. J. Clin. Microbiol., 39, 3353-3355.
Chevaliez, S. and Pawlotsky, J. M. (2006). HCV Genome and Life Cycle. Tan, L. S.
(editor), Chp. 01., Horizon Bioscience, Norfolk, UK.

Chapter 6 References
128
Chevaliez, S., Brillet, R., Lazaro, E., Hezode, C. and Pawlotsky, J. (2007). Analysis
of ribavirin mutagenicity in human hepatitis C virus Infection. J. Virol., 81,
732-7741.
Chiou, H. L., Hsieh, Y. S., Hsieh, M. R. and Chen, T. Y. (2006). HCV E2 may induce
apoptosis of Huh-7 cells via a mitochondrial-related caspase pathway.
Biochem. Biophys. Res. Commun., 345, 453-458.
Choo, Q. L., Kuo, G., Weiner, A. J., Overby, L. R., Bradley, D. W. and Houghton,
M. (1989). Isolation of a cDNA clone derived from a blood-borne non-A,
non-B viral hepatitis genome. Science, 244, 359-362.
Choo, Q. L., Richman, K. H., Han. J. H., Berger, K., Lee, C., Dong, C., Gallegos, C.,
Coit, D., Madina-Selby, R. and Barr, P. J. (1991). Genetic organization and
diversity of the hepatitis C virus. PNAS., 88, 2451-2455.
Chou, A. H., Tsai, H. F., Wu, Y. Y., Hu, C. Y., Hwang, L. H., Hsu, P. I. and Hsu, P.
N. (2005). Hepatitis C virus core protein modulates TRAIL-mediated
apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis
signaling pathway. J. Immunol., 174, 2160-2166.
Clement, S., Pascarella, S. and Negro, F. (2009). Hepatitis C virus infection:
molecular pathways to steatosis, insulin resistance and oxidative stress.
Viruses, 1, 126-143.
Coleman, M. L., Sahai, E. A., Yeo, M., Bosch, M., Dewar, A. and Olson, M. F.
(2001). Membrane blebbing during apoptosis results from caspase-mediated
activation of ROCK I. Nat. Cell. Biol., 3, 339-345.

Chapter 6 References
129
Couto, L. B. and Kolykhalov, A. A. (2006). Animal Models for HCV Study. Tan, L.
S. (editor), Chp. 12., Horizon Bioscience, Norfolk, UK.
Crow, M. T., Mani, K., Nam, Y. J., and Kitsis, R. N. (2004). The mitochondrial death
pathway and cardiac myocyte apoptosis. Circ. Res., 95, 957-970.
D'Alessio, M., De Nicola, M., Coppola, S., Gualandi, G., Pugliese, L., Cerella, C.,
Cristofanon, S., Civitareale, P., Ciriolo, M. R., Bergamaschi, A., Magrini, A.
and Ghibelli, L. (2005). Oxidative Bax dimerization promotes its translocation
to mitochondria independently of apoptosis. FASEB. J., 19, 504-1506.
De Francesco, R. and Migliaccio, G. (2005). Challenges and successes in developing
new therapies for hepatitis C. Nature, 436, 953-960.
Deforges, S., Evlashev, A., Perret, M., Sodoyer, M., Pouzol, S., Scoazec, J. Y.,
Bonnaud, B., Diaz, O., Paranhos-Baccala, G., Lotteau, V. and Andre, P.
(2004). Expression of hepatitis C virus proteins in epithelial intestinal cells in
vivo. J. Gen. Virol., 85, 2515-2523.
Denault, J. B. and Salvesen, G. S. (2002). Caspases: keys in the ignition of cell death.
Chem. Rev., 102, 4489-4500.
Deng, L., Adachi, T., Kitayama, K., Bungyoku, Y., Kitazawa, S., Ishido, S., Shoji, I.,
and Hotta, H. (2008). Hepatitis C virus infection induces apoptosis through a
Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J.
Virol., 82, 10375-10385.
E, Dreschers, S., and Bock, J. (2003). Role of mitochondria in apoptosis. Exp.
Physiol., 88, 85-90.

Chapter 6 References
130
Egger, D., Wolk, B., Gosert, R., Bianchi, L., Blum, H. E., Moradpour, D. and Bienz,
K. (2002). Expression of hepatitis C virus proteins induces distinct membrane
alterations including a candidate viral replication complex. J. Virol., 76, 5974-
5984.
El-Hage, N. and Luo, G. (2003). Replication of hepatitis C virus RNA occurs in a
membrane-bound replication complex containing nonstructural viral proteins
and RNA. J. Gen. Virol., 84, 2761-2769.
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol.,
35, 495-516.
Esteban, J. I., Gomez, J., Martell, M., Cabot, B., Quer, J., Camps, J., Gonzalez, A.,
Otero, T., Moya, A. and Esteban, R. (1996). Transmission of hepatitis C virus
by a cardiac surgeon. N. Eng. J. Med., 334, 555-560
Failla, C., Tommei, L. and Francesco, R. D. (1994). Both NS3 and NS4A are
required for proteolytic processing of hepatitis C virus non-structural
proteins. J. Virol., 68, 3753-3760.
Fan, T. J., Han, L. H., Cong, R. S. and Liang, J. (2005). Caspase family proteases and
apoptosis. Acta. Biochim. Biophys. Sin., 37, 719-27.
Farci, P., Choo, Q. L., Kuo, G., Weiner, A. J., Overby, L. R., Bradely, D. W. and
Houghton, M. (2002). Isolation of a cDNA clone derived from a blood-
borne non-A, non-B viral hepatitis genome. Science, 244, 359-362.
Favaloro, B., Allocati, N., Graziano, V., DiIlio, C. and De Laurenzi, V. (2012). Role
of apoptosis in disease. Aging, 4, 330-349.

Chapter 6 References
131
Feinstone, S. M., Kapikian, A. Z. and Purceli, R. H. (1973). Hepatitis A: detection by
immune electron microscopy of a virus like antigen associated with acute
illness. Science, 182, 1026.
Feinstone, S. M., Kapikian, A. Z., Purcell, R. H., Alter, H. J. and Holland, P. V.
(1975). Transfusion-associated hepatitis not due to viral hepatitis type A or B.
N. Engl. J. Med., 292, 767-770.
Feld, J. J. and Hoofnagle, J. H. (2005). Mechanism of action of interferon and
ribavirin in treatment of hepatitis C. Nature, 436, 967-972.
Fischer, R., Baumert, T. and Blum, H. E. (2007). Hepatitis C virus infection and
apoptosis. World J. Gastroenterol., 13, 4865-4872.
Flint, M., Thomas, J. M., Maidens, C. M., Shotton, C., Levy, S., Barclay, W. S. and
McKeating , J. A. (1999). Functional analysis of cell surface-expressed
hepatitis C virus E2 glycoprotein. J. Virol., 73, 6782-6790.
Foley, Suzanne, B. A., Abou-Saleh. And Mohammed, T. (2009). Risk behaviors and
transmission of hepatitis C in injecting drug users. Addict. Disord. Treat., 8,
13-22.
Forton, D. M., Karayiannis, P., Mahmud, N., Taylor-Robinson, S. D. and Thomas, H.
C. (2004). Identification of unique hepatitis C virus quasispecies in the central
nervous system and comparative analysis of internal translational efficiency of
brain, liver, and serum variants. J. Virol., 78, 5170-5183.

Chapter 6 References
132
Franco, E., Meleleo, C., Serino, L., Sorbara, D. and Zaratti, L. (2012). Hepatitis A:
Epidemiology and prevention in developing countries. World J. Hepatol., 4,
68-73.
Gale, M. J., Jr, Korth, M. J., Tang, N. M., Tan, S. L., Hopkins, D. A., DeverT, E.,
Polyak, S. J., Gretch, D. R. and Katze, M. G. (1997). Evidence that hepatitis
C virus resistance to interferon is mediated through repression of the PKR
protein kinase by the nonstructural 5A protein. Virology, 230, 217-227.
Galle, J., Herzog, C., Schollmeyer, P. and Wanner, C. (1994). Oxygen-derived
radicals stimulate renin release of isolated juxtaglomerular cells. FEBS Lett.,
351, 314-316.
Gallinari, P., Brennan, D., Nardi, C., Brunetti, M., Tomei, L., Steinkuhler, C. and De,
Francesco, R. (1998). Multiple enzymatic activities associated with
recombinant NS3 protein of hepatitis C virus. J. Virol., 72, 6758-6769.
Germi, R., Crance, J. M., Garin, D., Guimet, J., Thelu, M. A., Jouan, A., Zarski, J. P.
and Drouet, E. (2001). Mosquito cells bind and replicate hepatitis C virus. J.
Med. Virol., 64, 6-12.
Ghaziani, T., Shan, Y., Lambrecht, R.W, Donohue, S.E., Pietschmann,T.,
Bartenschlager, R. and Bonkovsky, H. L. (2006). HCV proteins increase
expression of heme oxygenase-1 (HO-1) and decrease expression of Bach1 in
human hepatoma cells. J. Hepatol., 45, 5-12.
Gillies, R. J., Didier, N. and Denton, M. (1986). Determination of cell number in
monolayer cultures. Anal Biochem., 159, 109-113.

Chapter 6 References
133
Gogvadze, V., Orrenius, S. and Zhivotovsky, B. (2006). Multiple pathways of
cytochrome c release from mitochondria in apoptosis. Biochim. Biophys.
Acta., 1757, 639-647.
Goldman, M. (1994). Elda E. Anderson Award. Presented to James G. Tripodes.
Health. Phys., 67, 445-6.
Goldstein, J. C., Waterhouse, N. J., Juin, P., Evan, G. I. and Green, D. R. (2000). The
coordinate release of cytochrome c during apoptosis is rapid, complete and
kinetically invariant. Nat. Cell. Biol., 2, 156-162.
Gosert, R., Egger, D., Lohmann, V., Bartenschlager, R., Blum, H. E., Bienz, K. and
Moradpour, D. (2003). Identification of the hepatitis C virus RNA replication
complex in Huh-7 cells harboring subgenomic replicons. J. Virol., 77, 5487-
5492.
Gottwein, J. M., Scheel, T. K., Callendret, B., Li, Y. P., Eccleston, H. B., Engle, R.
E., Govindarajan, S., Satterfield, W., Purcell, R. H., Walker, C. M. and Bukh,
J. (2010). Novel infectious cDNA clones of hepatitis C virus genotype 3a
(strain S52) and 4a (strain ED43): genetic analyses and in vivo pathogenesis
studies. J. Virol., 84, 5277-5293.
Grakoui, A., McCourt, D. W., Wychowski C., Feinstone, S. M. and Rice, C. M.
(1993). A second hepatitis C virus-encoded proteinase. PNAS., 90, 10583-
10587.

Chapter 6 References
134
Grakoui, A., McCourt, D. W. and Wychowski, C. (1993). Characterization of the
hepatitis C virus-encoded serine proteinase: determination of proteinase-
dependent polyprotein cleavage sites. J . Virol., 67, 2832-2843.
Gretton, S. N., Taylor, A. I. and McLauchlan, J. (2005). Mobility of the hepatitis C
virus NS4B protein on the endoplasmic reticulum membrane and membrane-
associated foci. J. Gen. Virol., 86, 1415-1421.
Griffin, S. D., Harvey, R., Clarke, D. S., Barclay, W. S., Harris, M. and Row, lands,
D. J. (2004). A conserved basic loop in hepatitis C virus p7 protein is required
for amantadine-sensitive ion channel activity in mammalian cells but is
dispensable for localization to mitochondria. J. Gen. Virol., 85, 451-461.
Gulbins, E. (2003). Regulation of death receptor signaling and apoptosis by
ceramide. Pharmacol. Res., 47, 93-399.
Hamid, S., Umar, M., Alam, A., Siddiqui, A. and Qureshi, H. Butt. (2004). PSG
consensus statement on management of hepatitis C virus infection. J. Pak.
Med. Assoc, 54, 146-150.
Hayashi, N. and Takehara, T. (2006). Antiviral therapy for chronic hepatitis C
past,present, and future. J. Gastroenterol., 41, 17-27.
Hellen, C. U. and Sarnow, P. (2001). Internal ribosome entry sites in eukaryotic
mRNA molecules. Genes Dev., 15, 1593-612.
Hockenbery, D. M., Oltvai, Z. N., Yin, X. M., Milliman, C. L. and Korsmeyer, S. J.
(1993). Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell,
75, 241-251.

Chapter 6 References
135
Hoofnagle, J. H. (2002). Course and outcome of hepatitis C. Hepatol., 36, S21-29.
Hope, R. G., Murphy, D. J. and McLauchlan, J. (2002). The domains required to
direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share
common features with plant oleosin proteins. J. Biol. Chem., 277, 4261-4270.
Horner, S. M., Park, H. S. and Gale, M. Jr. (2012). Control of innate immune
signaling and membrane targeting by the Hepatitis C virus NS3/4A protease
are governed by the NS3 helix-α 0 . J . Virol., 86, 3112-3120.
Howard, C., Thomas, Anna, S. F., Lok, Stephen, A., Locarnini, Arie, J. and
Zuckerman, (eds), (2014). Viral hepatitis, ED. 4th, Wiley-Blackwell.
Howe, A.Y.M., Bloom, J., Baldick, C.J., Benetatos, C.A., Cheng, H., Christnsen, J.S.,
Chunduru, S.K., Coburn, G.A., Feld, B., Gopalsamy, A., Gorczyca, W.P.,
Herrmann, S., Johann, S., Jiang, X., Kimberland, M.L., Krisnamurthy, G.,
Olson, M., Orlowski, M., Swanberg, S., Thompson, I., Thorn, M., Vecchio,
A.D., Young, D.C., Zeijl, M.V., Ellingboe, J.W., Upeslacis, J., Collett, M.,
Mansour, T.S. and O’Connell, J.F. (2004). Novel nonnucleoside inhibitor of
hepatitis C virus RNA-dependent RNA polymerase. Antimicrobial agents and
chemotherapy, 48, 4813-4821.
http://www.the-scientist.com/?articles.view/articleNo/24336/title/Culturing-Hepatitis
Idrees, M. and Riazuddin, S. (2008). Frequency distribution of hepatitis C virus
genotypes in different geographical regions of Pakistan and their possible
routes of transmission. BMC Infect. Dis., 8, 69.

Chapter 6 References
136
Imran, M., Waheed, Y., Manzoor, S., Bilal, M., Ashraf, W., Ali, M. and Ashraf, M.
(2012). Interaction of Hepatitis C virus proteins with pattern recognition
receptors.Virol. J., 9, 126.
Ivanov, A. V., Bartosch, B., Smirnova, O. A., Isaguliants, M. G. and Kochetkov, S. N.
(2013). HCV and oxidative stress in the liver. Viruses, 5, 439-469.
Jang, J. Y., Kim, S. J., Cho, E. K., Jeong, S. W., Park, E. J., Lee, W. C., Lee, S. H.,
Kim, S. G., Kim, Y. S., Kim, H. S., Kim, B. S., Lin, W. and Chung, R. T.
(2014). TRAIL Enhances Apoptosis of Human Hepatocellular Carcinoma
Cells Sensitized by Hepatitis C Virus Infection: Therapeutic Implications.
PLoS One, 9, e98171.
Joyce, M. A., Walters, K. A., Lamb, S. E., Yeh, M. M., Zhu, L. F., Kneteman, N.,
Doyle, J. S., Katze, M. G. and Tyrrell, D. L. (2009). HCV Induces Oxidative
and ER Stress, and Sensitizes Infected Cells to Apoptosis in SCID/Alb-uPA
Mice. PLoS Pathog., 5.
Joyce, M. A. and Tyrrell, D. L. (2010). The cell biology of hepatitis C virus. Microbes
Infect., 12, 263-271.
Kato, N., Hijikata, M., Ootsuyama, Y., Nakagawa, M., Ohkoshi, S., Sugimura, T. and
Shimotohno, K. (1990). Molecular cloning of the human hepatitis C virus
genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad
Sci., 87, 9524-9528.
Kaufmann, S. H. and Earnshaw, W. C. (2000). Induction of apoptosis by cancer
chemotherapy. Exp. Cell. Res., 256, 42-9.

Chapter 6 References
137
Kerr, J. F., Winterford, C. M. and Harmon, B. V. (1994). Apoptosis. Its significance
in cancer and cancer therapy. Cancer, 73, 2013-2026.
Khromykh, A. A. and Westaway, E. G. (1997). Subgenomic replicons of the
flavivirus Kunjin: construction and applications. J. Virol., 71, 1497-1505.
Kim, J. L., Morgenstern, K. A., Lin, C., Fox, T. anf Dwyer, M. D., Landro, J. A.,
Chambers, S. P., Markland, W., Lepre, C. A. and O'Malley, E. T.
(1996). Crystal structure of the hepatitis C virus NS3 protease domain
complexed with a synthetic NS4A cofactor peptide. Cell, 87, 343-355.
Kim, S. J., Syed, G. H. and Siddiqui, A. (2013). Hepatitis C virus induces the
mitochondrial translocation of Parkin and subsequent mitophagy. PLoS
Pathog., 9, e1003285.
Kluck, R. M., Bossy-Wetzel, E., Green, D. R. and Newmeyer, D. D. (1997). The
release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation
of apoptosis. Science, 275, 1132-1136.
Kmiec, Z. (2001). Cooperation of liver cells in health and disease. Adv. Anat.
Embryol. Cell. Biol, 161, 1-151.
Kolykhalov, A. A., Mihalik, K., Feinstone, S. M. and Rice, C. M. (2000). Hepatitis C
virus-encoded enzymatic activities and conserved RNA elements in the 3'
nontranslated region are essential for virus replication in vivo. J. Virol., 74,
2046-2051.

Chapter 6 References
138
Korenaga, M., Okuda, M., Otani, K., Wang, T., Li, Y. and Weinman, S. A. (2005).
Mitochondrial dysfunction in hepatitis C. J. Clin. Gastroenterol., 39, S162-
166.
Korenaga, M., Wang, T., Li, Y., Showalter, L. A., Chan, T., Sun, J. and Weinman, S.
A. (2005). Hepatitis C virus core protein inhibits mitochondrial electron
transport and increases reactive oxygen species (ROS) production. J. Biol.
Chem., 280, 37481-37488.
Krajewski, S., Tanaka, S., Takayama, S., Schibler, M. J., Fenton, W. and Reed, J. C.
(1993). Investigation of the subcellular distribution of the bcl-2 oncoprotein:
residence in the nuclear envelope, endoplasmic reticulum, and outer
mitochondrial membranes. Cancer Res., 53, 4701-4714.
Kudin, A. P., Bimpong-Buta, N. Y., Vielhaber, S., Elger, C. E. and Kunz, W. S.
(2004). Characterization of superoxide-producing sites in isolated brain
mitochondria. J. Biol. Chem., 279, 4127-4135.
Kumar, S. (2007). Caspase function in programmed cell death. Cell Death Differ., 14,
32-43.
Kumar, V., Cotran, R.S. and Robins, S.L. (1992). Basic Pathology, ED.5th, W.B.
Saunders Company, 75-77, 305-310.
Lam, A. M. and Frick, D. N. (2006). Hepatitis C virus subgenomic replicon requires
an active NS3 RNA helicase. J. Virol., 80, 404-411.
Lawen, A. (2003). Apoptosis-an introduction. Bioessays, 25, 888-896.

Chapter 6 References
139
Lee, S. H., Kim, Y. K., Kim, C. S., Seol, S. K., Kim, J., Cho, S., Song, Y. L.,
Bartenschlager, R. and Jang, S. K. (2007). Interaction of HCV core protein
with 14-3-3epsilon protein releases Bax to activate apoptosis. Biochem
Biophys. Res. Commun., 352, 756-762.
Lerat, H., Honda, M., Beard, M. R., Loesch, K., Sun, J., Yang, Y., Okuda, M., Gosert,
R., Xiao, S. Y., Weinman, S. A. and Lemon, S. M. (2002). Steatosis and liver
cancer in transgenic mice expressing the structural and nonstructural proteins
of hepatitis C virus. Gastroenterology, 122, 352-365.
Lesburg, C. A., Cable, M. B., Ferrari, E., Hong, Z., Mannarino, A. F. and Weber, P.
C. (1999). Crystal structure of the RNA-dependent RNA polymerase from
hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol., 6, 937-
943.
Leverrier, Y. and Ridley, A. J. (2001). Apoptosis: caspases orchestrate the ROCK 'n'
bleb. Nat. Cell. Biol., 3, 91-93.
Li, J. and Yuan, J. (2008). Caspases in apoptosis and beyond. Oncogene, 27, 6194-
6206.
Lin, C, Wu, J. W., Hsiao, K. and Su, M. S. (1997). The hepatitis C virus NS4A
protein: interactions with the NS4B and NS5A proteins. J. Virol., 71, 6465-
6471.
Lindenbach, B. D., Evans, M. J., Syder, A. J., Wolk, B., Tellinghuisen, T. L., Liu, C.
C., Maruyama, T., Hynes, R. O., Burton, D. R., McKeating, J. A. and Rice, C.

Chapter 6 References
140
M. (2005). Complete replication of hepatitis C virus in cell culture. Science,
309, 623-626.
Liu, Y., Fiskum, G. and Schubert, D. (2002). Generation of reactive oxygen species
by the mitochondrial electron transport chain. J. Neurochem., 80, 780-787.
Liu, C., Gaca, M., Swenson, E. S., Vellucci, V. F., Reiss, M. and Wells, R. G. (2003).
Smad2 and Sn-mad3 are differentially activated by transforming growth factor
beta in quiscent and avctivated hepatic stellate cells in TGF- beta independent.
J. Biol. Chem., 278, 1172-1178.
Lock, G., Dirscherl, M., Obermeier, F., Gelbmann, C. M., Hellerbrand, C., Knoll, A.,
Scholmerich, J. and Jilg, W. (2006). Hepatitis C contamination of
toothbrushes: myth or reality? J. Viral Hepat., 13, 571-573.
Lockshin, R. A. and Zakeri, Z. (2001). Programmed cell death and apoptosis: origins
of the theory. Nat Rev Mol. Cell. Biol., 2, 545-550.
Loeffler, M. and Kroemer, G. (2000). The mitochondrion in cell death control:
certainties and incognita. Exp. Cell. Res., 256, 19-26.
Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L. and Bartenschlager, R.
(1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell
line. Science, 285, 110-113.
Lorenz, I. C., Marcotrigiano, J., Dentzer, T. G. and Rice, C. M. (2006). Structure of
the catalytic domain of the hepatitis C virus NS2-3 protease. Nature, 442,
831-835.

Chapter 6 References
141
Lu, L., Li, C. and Haqedorn, C. H. (2006). Phylogenetic analysis of global
hepatitis E virus sequences: genetic diversity, subtypes and zoonosis. Rev.
Med. Virol., 16, 5-36.
Lu, S. N., Chen, T. M., Lee, C. M., Wang, J. H., Tung, H. D. and Wu, J. C. (2003).
Molecular epidemiological and clinical aspects of hepatitis D virus in a unique
triple hepatitis viruses (B, C, D) endemic community in Taiwan. J. Med.
Virol., 70, 74-80.
Lundin, M., Lindstrom, H., Gronwall, C., Persson, M. A. (2006). Dual topology of the
processed hepatitis C virus protein NS4B is influenced by the NS5A protein.
J. Gen. Virol., 87, 3263-3272.
Lyn, R. K., Kennedy, D. C., Stolow, A., Ridsdale, A. and Pezacki, J. P. (2010).
Dynamics of lipid droplets induced by the hepatitis C virus core protein.
Biochem. Biophys. Res. Commun., 399, 518 -524.
Ma, Y., Yates, J., Liang, Y., Lemon, S. M. and Yi, M. (2008). NS3 helicase domains
involved in infectious intracellular hepatitis C virus particle assembly. J.
Virol., 82, 7624-7639.
MacCallum, F, O. (1947). Homologous serum jaundice. Lancet, 2, 691.
Macdonald, A., Crowder, K., Street, A., McCormick, C. and Harris, M. (2004). The
hepatitis C virus NS5A protein binds to members of the Src family of tyrosine
kinases and regulates kinase activity. J. Gen. Virol., 85, 721-729.

Chapter 6 References
142
MacIntyre, C. R., Burgess, M., Isaacs, D., MacIntyre, P. B., Menzies, R. and Hull, B.
(2007). Epidemiology of severe hepatitis A in Indigenous Australian children.
J. Paed. Child Health., 43, 383-387.
Malarkey, D. E., Johnson, K., Ryan, L., Boorman, G. and Maronpot, R. R. (2005).
New insights into functional aspects of liver morphology. Toxicol. Pathol., 33,
27-34.
Malhi, H. and Gores, G. J. (2008). Cellular and molecular mechanisms of liver injury.
Gastroenterology, 134, 1641-1654.
Malik, R., Selden, C. and Hodgson, H. (2002). The role of non-parenchymal cells in
liver growth. Semin. Cell. Dev. Biol., 13, 425-31.
Marusawa, H., Hijikata, M., Chiba, T. and Shimotohno, K. (1999). Hepatitis C virus
core protein inhibits Fas- and tumor necrosis factor alpha-mediated apoptosis
via NF-kappaB activation. J. Virol., 73, 4713-20.
McCaughan, G. W. and Torzillo, P. J. (2000). Hepatitis A, liver transplants and
Indigenous communities. Med. J. Australia., 172, 6-7.
Mohebbi, S. R. and Nejad, M. R. (2012). Seroepidemiology of hepatitis A and
Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral
hepatitis genome. Science, 244, 359-362.
Monazahian, M., Bohme, I., Bonk, S., Koch, A., Scholz, C., Grethe, S., Thomssen, R.
(1999). Low density lipoprotein receptor as a candidate receptor for hepatitis
C virus. J. Med. Virol., 57, 223-229.

Chapter 6 References
143
Moradpour, D., Penin, F. and Rice, C. M. (2007). Replication of hepatitis C virus.
Nat. Rev. Microbiol., 5, 453-463.
Mordon, S. and Blanchemaison, P. (2008). Re: Histologic evaluation of interstitial
lipolysis comparing a 1064, 1320 and 2100 nm laser in an ex vivo model. by
Khoury JG, Saluja R, Keel D, Detwiler S, Goldman MP. Lasers. Surg. Med.,
40, 402-406.
Mumtaz, K., Hamid, S. S., Adil, S., Afaq, A., Islam, M., Abid, S., Shah, H. A. and
Jafri, W. (2005). Epidemiology and clinical pattern of hepatitis delta virus
infection in Pakistan. J. Gastroenterol. Hepatol., 20, 1503-1507.
Nakamura, M.T., Cheon, Y., Li, Y. and Nara, T.Y. (2004). Mechanisms of regulation
of gene expression by fatty acids. Lipids, 39, 1077-1083.
Neddermann, P., Clementi, A. and De Francesco, R. (1999). Hyperphosphorylation of
the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A,
NS4B, and NS5A encoded on the same polyprotein. J. Virol., 73, 9984-9991.
Nemati, E., Alavian, S. M., Taheri, S., Moradi, M., Pourfarziani, V. and Einollahi, B.
(2009). Hepatitis C virus infection among patients on hemodialysis: A report
from a single center in Iran. Saudi J. Kidney Dis. Transpl., 20, 147-153.
Neumann, A. U., Lam, N. P., Dahari, H. D., Gretch, I., Wiley, T. E., Layden, T. J. and
Perelson, A. S. (1998). HCV dynamics in vivo and the antiviral efficacy of
interferon alfa therapy. Science, 282, 103-107.
Nie, C., Tian, C., Zhao, L., Petit, P. X., Mehrpour, M. and Chen, Q. (2008). Cysteine
62 of Bax is critical for its conformational activation and its proapoptotic

Chapter 6 References
144
activity in response to H2O2-induced apoptosis. J. Biol. Chem., 283, 15359-
15369.
Nomura-Takigawa, Y., Nagano-Fujii, M., Deng, L., Kitazawa, S., Ishido, S., Sada, K.
and Hota, H. (2006). Non-structural protein 4A of Hepatitis C virus
accumulates on mitochondria and renders the cells prone to undergoing
mitochondria-mediated apoptosis. J. Gen. Virol., 87, 1935-1945.
Oltvai, Z. N., Milliman, C. L. and Korsmeyer, S. J. (1993). Bcl-2 heterodimerizes in
vivo with a conserved homolog, Bax, that accelerates programmed cell death.
Cell, 74, 609-619.
Owen, R. A., Molon-Noblot, S., Hubert, M. F., Kindt, M. V., Keenan, K. P. and
Eydelloth, R. S. (1994). The morphology of juxtaglomerular cell hyperplasia
and hypertrophy in normotensive rats and monkeys given an angiotensin II
receptor antagonist. Toxicol. Pathol., 22, 606-19.
Ozben, T. (2007). Oxidative stress and apoptosis: impact on cancer therapy. J.
pharma. Sci., 96, 2181-2196.
Palomares-Jerez, M. F., Nemesio,H. and Villalaín J. (2012). Interaction with
membranes of the full C-terminal domain of protein NS4B from
Hepatitis C virus. Biochim. Biophys. Acta., 1818, 2536-2549.
Park, S. E., Park, C., Kim, S. H., Hossain, M. A., Kim, M. Y., Chung, H. Y., Son, W.
S., Kim, G. Y., Choi, Y. H. and Kim, N. D. (2009). Korean red ginseng extract
induces apoptosis and decreases telomerase activity in human leukemia cells.
J. Ethnopharmacol., 121, 304-312.

Chapter 6 References
145
Pasquier, C., Bujan, L., Daudin, M., Righi, L., Berges, L., Thauvin, L., Berrebi, A.,
Massip, P., Puel, J. and Izopet, J. (2003). Intermittent detection of hepatitis C
virus (HCV) in semen from men with human deficiency virus type 1 (HIV-1)
and HCV. J. Med. Virol., 69, 344-349.
Pekow, J. R., Bhan, A. K., Zheng, H. and Chung, R. T. (2007). Hepatic steatosis is
associated with increased frequency of hepatocellular carcinoma in patients
with hepatitis C-related cirrhosis. Cancer, 109, 2490-2496.
Penin, F., Brass, V., Appel, N., Ramboarina, S., Montserret, R., Ficheux, D., Blum, H.
E., Bartenschlager, R. and Moradpour, D. (2004). Structure and function of
the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J.
Biol. Chem., 279, 40835-40843.
Penin, F. and Dubuisson, J. (2004). Structural biology of hepatitis C virus.
Hepatology, 39, 5-19.
Piccoli, C., Scrima, R., Quarato, G., D'Aprile, A., Ripoli, M., Lecce, L., Boffoli, D.,
Moradpour, D. and Capitanio, N. (2007). Hepatitis C virus protein expression
causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-
oxidative stress. Hepatology, 46, 58-65.
Pileri, P., Uematsu, Y., Campagnoli, S., Galli, G., Falugi, F., Petracca, R., Weiner, A.
J., Houghton, M., Rosa, D., Grandi, G. and Abrignani, S. (1998). Binding of
hepatitis C virus to CD81. Science, 282: 938-941.
Pisarev, A. V., Shirokikh, N. E. and Hellen, U. T. (2005). Translation initiation by
factorindependent binding of eukaryotic ribosomes to internal ribosomal entry
sites. C. R. Biol., 328, 589-605.

Chapter 6 References
146
Pitot, H. C. (1993). The molecular biology of carcinogenesis. Cancer, 72, 962-970.
Pop, C. and Salvesen, G. S. (2009). Human caspases: activation, specificity, and
regulation. J. Biol. Chem., 284, 21777-21781.
Prikhod'ko, E. A., Prikhod'ko, G. G., Sieqel, R. M., Thompson, P., Major, M. E. and
Cohen, J. I. (2004). The NS3 protein of hepatitis C virus induces caspase-8-
mediated apoptosis independent of its protease or helicase activities. Virology,
329, 53-67.
Prince, A. M., Brotman, B., Grady, G. F., Kuhns, W. J., Hazzi, C., Levine, R. W. and
Millian, S. J. (1974). Long-incubation post-transfusion hepatitis without
serological evidence of exposure to hepatitis-B virus. Lancet, 2, 241-246.
Qadri, I., Iwahashi, M., Capasso, J. M., Hopken, M.W., Flores, S., Schaack, J. and
Simon, F.R. (2004). Induced oxidative stress and activated expression of
manganese superoxide dismutase during hepatitis C virus replication: role of
JNK,p38 MAPK and AP-1. Biochem. J., 378, 919-928.
Qadri,I., Iwahashi, M., Kullak-Ublick, G. A. and Simon, F. R. (2006). Hepatocyte
Nuclear Factor (HNF) 1 and HNF4 Mediate Hepatic Multidrug Resistance
Protein 2 Up-Regulation during Hepatitis C Virus Gene Expression. Mol.
Pharmacol., 70, no. 2.
Qi, F., Li, A., Zhao, L., Xu, H., Inagaki, Y., Wang, D., Cui, X., Gao, B., Kokudo, N.,
Nakata, M. and Tang, W. (2010). Cinobufacini, an aqueous extract from Bufo
bufo gargarizans Cantor, induces apoptosis through a mitochondria-mediated
pathway in human hepatocellular carcinoma cells. J. Ethnopharmacol., 128,
654-661.

Chapter 6 References
147
Quarato, G., Scrima, R., Agriesti, F., Moradpour, D., Capitanio, N. and Piccoli, C.
(2013). Targeting mitochondria in the infection strategy of the hepatitis C
virus. Int. J. Biochem. Cell. Biol., 45, 156-166.
Qureshi, S., Batool, U., Iqbal, M., Qureshi, O., Kaleem, R., Aziz, H. and Azhar, M.
(2009). Response rates to standard interferon treatment in HCV genotype 3a.
J. Ayub. Med. Coll. Abbottabad., 21, 10-14.
Raja, N. S. and Janjua, K. A. (2008). Epidemiology of hepatitis C virus infection in
Pakistan. J. Microbiol. Immunol. Infect., 41, 4-8.
Reed, J. C. and Pellecchia, M. (2005). Apoptosis-based therapies for hematologic
malignancies. Blood, 106, 408-418.
Rizzetto, M., Canese, M. G., Aricó, S., Crivelli, O., Trepo, C. and Verme, G. (1977).
Immunofluorescence detection of a new antigen antibody system associated
to the hepatitis B virus in the liver and in the serum of HBsAg carriers. Gut.,
18, 997-1003.
Sakahira, H., Enari, M. and Nagata, S. (1998). Cleavage of CAD inhibitor in CAD
activation and DNA degradation during apoptosis. Nature, 391, 96-99.
Sala, J., Cardena, E., Holgado, M. C., Anez, C., Perez, P., Perinan, R. and Capafons,
A. (2008). The contributions of Ramon y Cajal and other Spanish authors to
hypnosis. Int. J. Clin. Exp. Hypn., 56, 361-72.
Saladin, K. (2012). Just the FACTS101 E-Study Guide For: Anatomy & Physiology:
A Unity of Form and Function. Cram., 101.

Chapter 6 References
148
Scheel, T. K. and Rice, C. M. (2013). Understanding the hepatitis C virus life cycle
paves the way for highly effective therapies. Nat. Med., 19, 837-849.
Schiff, E. R. (2007). Schiff's Diseases of the Liver. 2 Lippincott Williams & Wilkins.
Schreiber, G. B., Busch, M. P., Kleinman, S. H. and Korelitz, J. J. (1996). The risk of
transfusion transmitted viral infections. The retrovirus epidemiology donor
study. N. Engl. J. Med., 334, 1685-1690.
Schultz, R. (2005). Hepatitis A outbreak in Central Australia Northern Territory. Dis.
Control Bulletin., 12, 4-7.
Sebbagh, M., Renvoize, C., Hamelin, J., Riche, N., Bertoglio, J. and Breard, J. (2001).
Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and
apoptotic membrane blebbing. Nat. Cell. Biol., 3, 346-352.
Serebrov, V., Pyle, A. M. (2004). Periodic cycles of RNA unwinding and pausing by
hepatitis C virus NS3 helicase. Nature, 430, 476-480.
Shaik, M. S., Chatterjee, A. and Singh, M. (2004). Effects of monensin liposomes on
the cytotoxicity, apoptosis and expression of multidrug resistance genes in
doxorubicin-resistant human breast tumour (MCF-7/dox) cell-line. J. Pharm.
Pharmacol., 56, 899-907.
Shepard, C. W., Finelli, L. and Alter, M. J. (2005). Global epidemiology of hepatitis
C virus infection. Lancet Infect. Dis., 5, 558-67.

Chapter 6 References
149
Shin, E. C., Shin, J. S., Park, J. H., Kim, J. J., Kim, H. and Kim, S. J. (1998).
Expression of Fas-related genes in human hepatocellular carcinomas. Cancer
Lett., 134, 155-162.
Simmonds, P., Bukh, J., Combet, C., Deleage, G., Enomoto, N., Feinstone, S., Halfon,
P., Inchauspe, G., Kuiken, C., Maertens, G., Mizokami, M., Murphy, D. G.,
Okamoto, H., Pawlotsky, J. M., Penin, F., Sablon, E., Shin-I, T., Stuywer, L.
J., Thiel, H. J., Viazov, S., Weiner, A. J. and Widell, A. (2005). Consensus
proposals for a unified system of nomenclature of hepatitis C virus genotypes.
Hepatology, 42, 962-973.
Strickland, G. T., Mohamed, M. K., Abdel-Hamid, M., Mikhail, N. N., Abdel-Aziz,
F., Mehat, A., Maqder, L. S. and Fix, A. D. (2005). Intrafamilial transmission
of hepatitis C in Egypt. Hepatology, 42, 683-687
Takamizawa, A., Mori, C., Fuke, I., Manabe, S., Murakami, S., Fujita, J., Onishi, E.,
Andoh, T., Yoshida, I. and Okayama, H. (1991). Structure and organization of
the hepatitis C virus genome isolated from human carriers. J. Virol., 65, 1105-
1113.
Tang, H. and Grise, H. (2009). Cellular and molecular biology of HCV infection and
hepatitis. Clin. Sci.,(Lond), 117, 49-65.
Tariq, H., Manzoor, S., Parvaiz, F., Javed, F., Fatima, K. and Qadri, I. (2012). An
overview: in vitro models of HCV replication in different cell cultures. Infect.
Genet. Evol., 12, 13-20.

Chapter 6 References
150
Tatsuo, K., Osamu, Y. and Masao, O. (2013). Hepatitis C Virus and Hepatocellular
Carcinoma. Biology, 2, 304-316.
Tellinghuisen ,T. L. and Rice, C. M. (2002). Interaction between hepatitis C virus
proteins and host cell factors. Curr. Opin. Microbiol., 5, 419-427.
Thomas, M., Finnegan, C. E., Rogers, K. M., Purcell, J. W., Trimble, A., Johnston, P.
G. and Boland, M. P. (2004). STAT1: a modulator of chemotherapy-induced
apoptosis. Cancer Res., 64, 8357-8364.
Timm, J. and Roggendorf, M. (2007). Sequence diversity of hepatitis C virus:
implications for immune control and therapy. World J. Gastroenterol., 13,
4808-4817.
Valva, P., De Matteo, E., Galoppo, M. C., Gismondi, M. I. and Preciado, M. V.
(2010). Apoptosis markers related to pathogenesis of pediatric chronic
hepatitis C virus infection: M30 mirrors the severity of steatosis. J. Med. Virol.
82, 949-957.
Waheed, Y., Shafi, T., Zaman, Safi, S. and Qadri, I. (2009). Hepatitis C virus in
Pakistan: A systematic review of prevalence, genotypes and risk factors.
World J. Gastroenterol., 15, 5647-5653.
Wake, K. (2004). Karl Wilhelm Kupffer And His Contributions To Modern
Hepatology. Comp. Hepatol., 3 Suppl 1, S2.
Wang, A. G., Moon, H. B., Kim, J. M., Hwang, S. B., Yu, D. Y. and Lee, D. S.
(2006). Expression of hepatitis C virus nonstructural 4B in transgenic mice.
Exp. Mol. Med., 38, 241-246.

Chapter 6 References
151
Wang, J., Zhou, W., Yuan, H. and Wang, Y. (2008). Characterization of a novel
fungal chitosanase Csn2 from Gongronella sp. JG. Carbohydr. Res., 343,
2583-2588.
Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A.
J., Roth, K. A., MacGregor, G. R., Thompson, C. B. and Korsmeyer, S. J.
(2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial
dysfunction and death. Science, 292, 727- 735
Williams, R. (2006). Global challenges in liver disease. Hepatology, 44, 521-526.
Wolk, B., Sansonno, D., Krausslich, H. G., Dammacco, F., Rice, C. M., Blum, H. E.
and Moradpour, D. (2000). Subcellular localization, stability, and trans-
cleavage competence of the hepatitis C virus NS3-NS4A complex expressed
in tetracycline-regulated cell lines. J. Virol., 74, 2293-2304.
Yamashita, T., Kaneko, S., Shirota, Y., Qin, W., Nomura, T., Kobayashi, K. and
Murakami, S. (1998). RNA-dependent RNA polymerase activity of the soluble
recombinant hepatitis C virus NS5B protein truncated at the C-terminal
region. J. Biol. hem., 273, 15479-15486.
Zaidi, N., Kanwal, N., Ghazal, A., Fatima, K., Javed, F. and Qadri, I. (2012).
Phylogenetic and structural analysis of HCV nonstructural protein 4A from
Pakistani patients. Virus Genes, 44, 1-7.
Zekri, A. R., Bahnassy, A. A., Hafez, M. M., Hassan, Z. K. and Kamel, M. (2011).
Characterization of chronic HCV infection-induced apoptosis. Comp.
Hepatol., 10, 1476-5926

Chapter 6 References
152
Zenetti, A. R., Tanzi, E. and Newell, M. L. (1999). Mother to infant transmission of
hepatitis C virus. J. Hepatol., 31, 96-100.
Zhao, C. , Ma Z., Harrison TJ., Feng R., Zhang C., Fan J., Ma H., Li M., Song A. and
Wang, Y. (2009). A novel genotype of hepatitis E virus prevalent among
farmed rabbits in China. J. Med. Virol., 81, 1371-1379.
Zhao, P., Han, T., Guo, J. J., Zhu, S. L., Wang, J., Ao, F., Jing, M. Z., She, Y. L., Wu,
Z. H. and Ye, L. B. (2012). HCV NS4B induces apoptosis through the
mitochondrial death pathway. Virus Res., 169, 1-7.
Zhu, H. and Briggs, J. M. (2011). Mechanistic role of NS4A and substrate in the
activation of HCV NS3 protease. Proteins, 79, 2428-2443.
Zhu, H., Dong, H., Eksioqlu, E., Hemminq, A., Cao, M., Crawford, J. M., Nelson, D.
R. and Liu, C. (2007). Hepatitis C virus triggers apoptosis of a newly
developed hepatoma cell line through antiviral defense system.
Gastroenterology, 133, 1649-1659.


















