
Epileptic Encephalopathies: Etiologies and Mechanisms Kristen Park, MD Assistant Professor of...
26
Epileptic Encephalopathies: Etiologies and Mechanisms Kristen Park, MD Assistant Professor of Pediatrics and Neurology UCHSC
-
Upload
virginia-murphy -
Category
Documents
-
view
216 -
download
1
Transcript of Epileptic Encephalopathies: Etiologies and Mechanisms Kristen Park, MD Assistant Professor of...

Epileptic Encephalopathies: Etiologies and Mechanisms
Kristen Park, MD
Assistant Professor of Pediatrics and Neurology
UCHSC

Outline
• Overview
• Etiology and Mechanisms

Definition
• Age dependent syndrome
• Unique types of frequent seizures
• Abnormal interictal EEG
• Heterogeneous causes
• Pharmacoresistant
• Frequently associated with developmental impairment and/or regression

Epileptic Encephalopathies

Etiology of Encephalopathy
• Epilepsy/epileptogenesis
• Interictal abnormalities
• Underlying pathophysiology

Epilepsy/Epileptogenesis
• Clinical evidence– Longitudinal study of Dravet patients did not correlate
intellectual profile with seizure control• Early appearance of absence seizures associated with worst
developmental outcome• 2 patients with truncation mutations followed and
demonstrated progressive cognitive decline
– Variable timing between onset of encephalopathy and seizures in different syndromes
• Dravet, Doose vs Rett

Epilepsy/Epileptogenesis
• Functional data– MRS in patients with cortical malformations
showed abnormal NAA in the epileptogenic zone• Normal levels when seizures controlled• In patients with ongoing seizures, anatomically
abnormal regions that were not epileptogenic showed normal NAA
• Decrements shown in contralateral hippocampus and thalamus
• Reversible after successful temporal lobectomy

Epilepsy/Epileptogenesis
• Experimental evidence– Mirror foci– Kindling– Animal models– Cellular
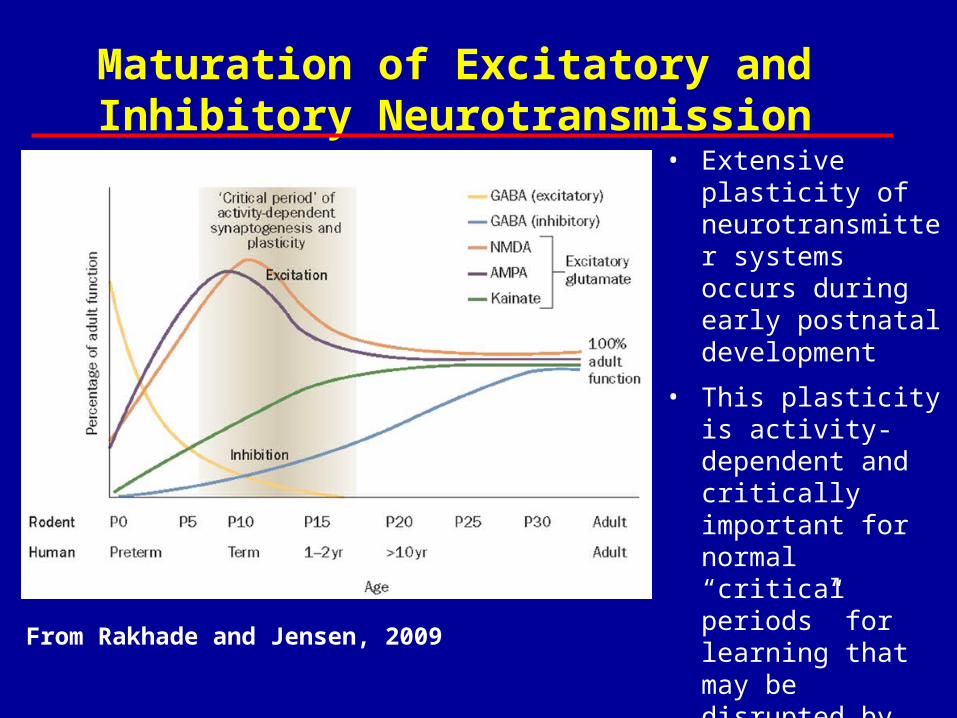
From Rakhade and Jensen, 2009
Maturation of Excitatory and Inhibitory Neurotransmission
• Extensive plasticity of neurotransmitter systems occurs during early postnatal development
• This plasticity is activity-dependent and critically important for normal “critical periods” for learning that may be disrupted by early-life seizure activity

Epileptogenesis: Synaptic Plasticity
• Molecular– Increased Inhibitory Neurotransmission
• Accelerated changes in chloride gradient• Increased postsynaptic GABAA receptor
expression
– Altered Excitatory Neurotransmission• Post-translational changes in GluR1 and GluR2• Decreased dendritic spine density• Reduced AMPA and NMDA receptor expression
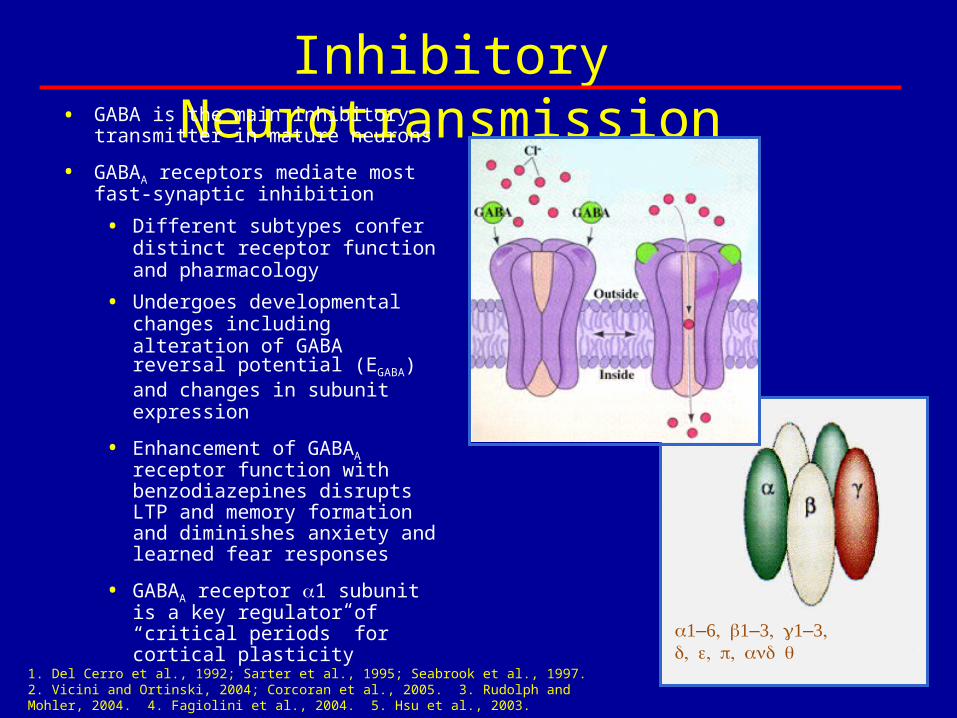
Inhibitory Neurotransmission• GABA is the main inhibitory
transmitter in mature neurons
• GABAA receptors mediate most fast-synaptic inhibition
• Different subtypes confer distinct receptor function and pharmacology
• Undergoes developmental changes including alteration of GABA reversal potential (EGABA) and changes in subunit expression
• Enhancement of GABAA receptor function with benzodiazepines disrupts LTP and memory formation and diminishes anxiety and learned fear responses
• GABAA receptor 1 subunit is a key regulator of “critical periods” for cortical plasticity
–––
1. Del Cerro et al., 1992; Sarter et al., 1995; Seabrook et al., 1997. 2. Vicini and Ortinski, 2004; Corcoran et al., 2005. 3. Rudolph and Mohler, 2004. 4. Fagiolini et al., 2004. 5. Hsu et al., 2003.

Inhibitory Neurotransmission
• Depolarizing GABA currents are critical for Ca++ dependent developmental processes including neuronal proliferation, migration, targeting & synaptogenesis
• Early-life seizures accelerate the switch of EGABA from depolarizing to hyperpolarizing in hippocampal CA1 neurons and are associated with spatial learning deficits
• GABAA receptor changes after prolonged early-life seizures
– Total GABAA receptor and 1 subunit expression is
increased

Excitatory Neurotransmission
• Extensive plasticity of excitatory neurotransmission occurs during
normal postnatal development
• This plasticity is activity-dependent and can be disrupted by early-life
seizure activity
• Excitatory signaling through both the AMPA and NMDA receptors are
critical for different types of LTP and hippocampal learning
• Mutant mice lacking subtypes of AMPA receptors (GluR1 or GluR2
subunits) or NMDA receptors have impaired learning and behavioral
abnormalities.
• Decreased AMPA Receptor GluR2 subunit expression has been shown
after hypoxia-induced seizures, Lithium-Pilocarpine-induced seizures
and febrile seizures at P10
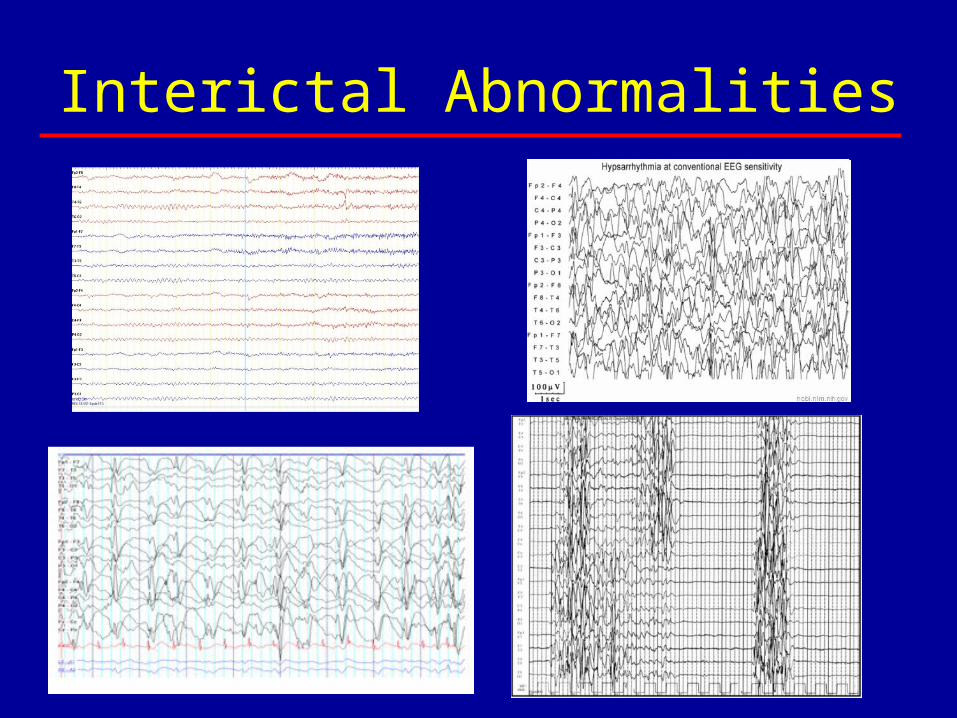
Interictal Abnormalities

Interictal Abnormalities
• Clinical evidence– Resections of focal cortical abnormalities in
West syndrome can lead to resolution and improved development

Interictal Abnormalities
• Functional data– EEG-fMRI studies in West syndrome– EEG in CSWS– fMRI studies in CSWS

Underlying Pathophysiology
• Clinical evidence– Dravet
• Milder phenotypes associated with missense mutations
• More severe phenotype associated with pore mutations
– KCNQ2E – pore domain vs BFNC scattered– Cognitive impairments often related to age at
onset with infantile being more severe

Fragile X Syndrome• Caused by an expanded triplet repeat in the FMR1 gene
that codes for the fragile X mental retardation protein (FMRP), an mRNA-binding protein that binds to and regulates 4% of brain mRNA, including many RNAs important for synaptic plasticity
• FMRP regulates mRNA transport in dendrites and regulates local protein synthesis important for dendritic spine development, synaptic formation and plasticity.
• In the absence of FMRP, excess and dysregulated mRNA translation leads to altered synaptic function and loss of protein synthesis-dependent plasticity
• The hallmark of FXS pathology is the hyperabundance of dendritic spines with a long, thin, and otherwise immature morphology
Comery et al., 1997; Penagarikano et al., 2007; Basel and Warren, 2008; Antar et al., 2004; Feng et al., 1997b; Laggerbauer et al., 2001; Li et al., 2001, Lu et al.,2004; Muddashetty et al., 2007; Zalfa et al., 2003; Gibson et al., 2008
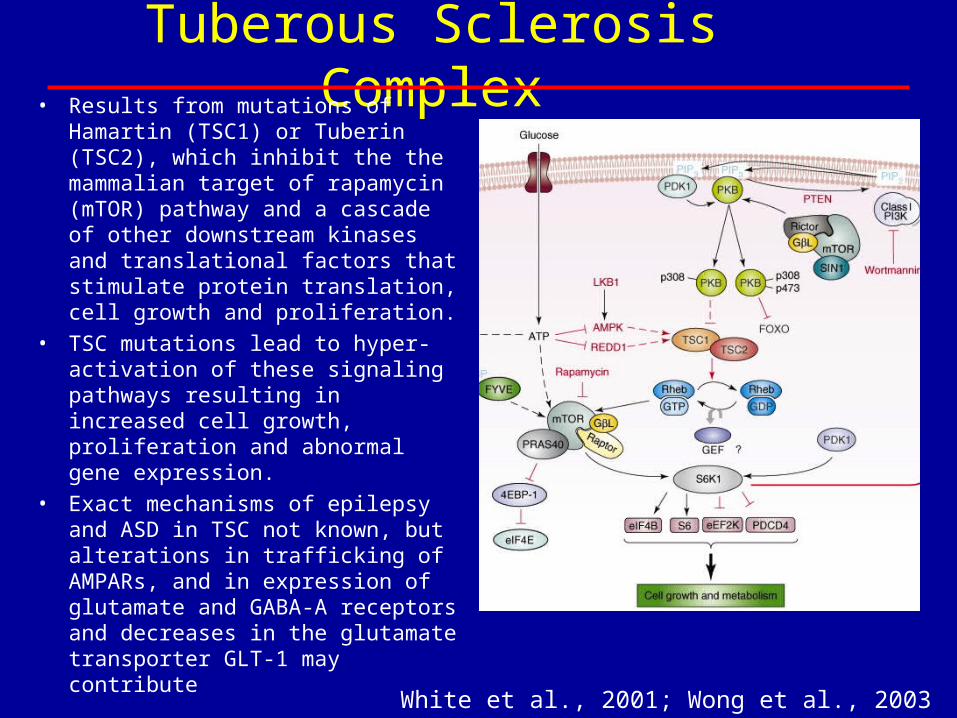
Tuberous Sclerosis Complex• Results from mutations of Hamartin
(TSC1) or Tuberin (TSC2), which inhibit the the mammalian target of rapamycin (mTOR) pathway and a cascade of other downstream kinases and translational factors that stimulate protein translation, cell growth and proliferation.
• TSC mutations lead to hyper-activation of these signaling pathways resulting in increased cell growth, proliferation and abnormal gene expression.
• Exact mechanisms of epilepsy and ASD in TSC not known, but alterations in trafficking of AMPARs, and in expression of glutamate and GABA-A receptors and decreases in the glutamate transporter GLT-1 may contribute
White et al., 2001; Wong et al., 2003

Rett Syndrome• Caused by mutations in the methyl-CpG
binding protein 2 (MeCP2) gene, a transcriptional repressor involved in chromatin remodeling and the modulation of RNA splicing.
• In resting neurons, MeCP2 regulates gene expression by binding to methylated CpG dinucleotides and recruiting HDAC complexes and chromatin remodeling proteins. This leads to chromatin compaction, making the promoter inaccessible to the transcriptional machinery.
• Neuronal activity induces MeCP2 phosphorylation and leads to its release from the promoter region, dissociation of the corepressor complex, and transcription of target genes.
• In Rett, the absence of MeCP2 causes a loss of activity dependent changes in gene expression that may disrupt synaptic plasticity
From Charhour and Zoghbi, Neuron, 2007
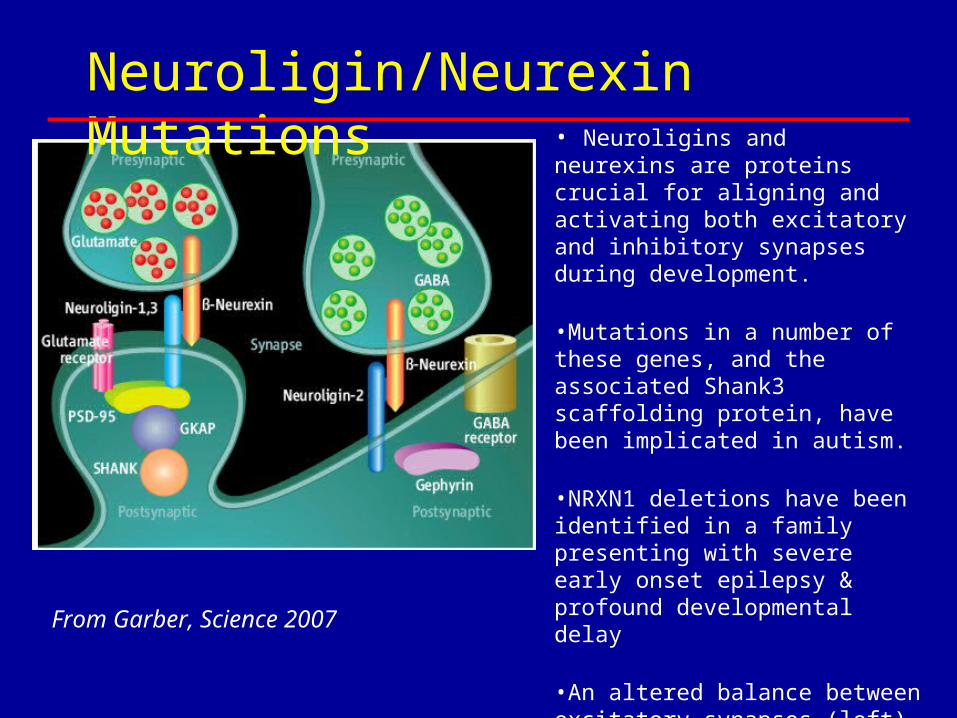
• Neuroligins and neurexins are proteins crucial for aligning and activating both excitatory and inhibitory synapses during development.
•Mutations in a number of these genes, and the associated Shank3 scaffolding protein, have been implicated in autism.
•NRXN1 deletions have been identified in a family presenting with severe early onset epilepsy & profound developmental delay
•An altered balance between excitatory synapses (left) and inhibitory (right) could affect learning and social behavior as well as contribute to epilepsy.
From Garber, Science 2007
Neuroligin/Neurexin Mutations

Interneuronopathies
• Experimental evidence– Critical role of interneurons
• Complex networks coordinate higher functions; excitatory and inhibitory, variable firing patterns
– Developmental abnormalities resulting in reduced numbers of cortical and hippocampal interneuron subtypes have been reported to cause severe early life epilepsies, ID and autism:
• ARX
• NPN2
• Lissencephaly (DCX)
• SCN1A
• TSC1• Cortical dysplasia• PMG• CNTNAP2

Interneuronopathies
• SCN1A– Selective knock-out in basal forebrain– Disruption of learning and memory without
spontaneous seizures– Dysregulation of hippocampal oscillations– Spatial learning deficit
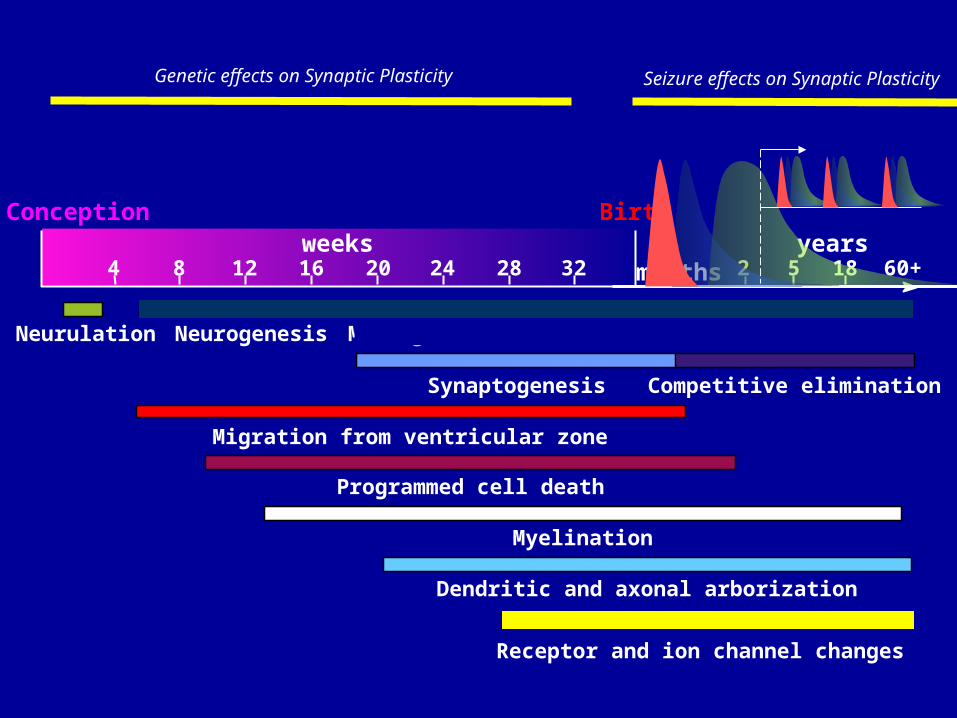
Neurulation Neurogenesis Max. growth
Synaptogenesis Competitive elimination
Migration from ventricular zone
Programmed cell death
Myelination
Dendritic and axonal arborization
Conception Birthweeks
4 8 12 16 20 24 28 324
monthsyears
5 18 60+22
Receptor and ion channel changes
Genetic effects on Synaptic Plasticity Seizure effects on Synaptic Plasticity

Application
• Characterize– Spectrum of clinical presentation– Functional measurements
• EEG• Neuropsychologic profile
• Correlate– Genotype and functional expression– Developmental and neuroanatomic factors

Conclusions


















