Eletronic Structure of Molecules
60
2 Electronic Structure of Molecules By G. DOGGETT Department of Chemistry, University of York, Heslington, York YO 1 5DD 1 Introduction The continuing development and improvement of on-line literature retrieval facilities is, by necessity, forcing a change in the nature and form of literature reviews; as Kettle’ has recently commented, the time is rapidly approaching when one must question the usefulness of providing a comprehensive review of the literature, when lists of pertinent papers can be obtained from an efficient information retrieval system. With these thoughts in mind, the main aim of this review is to provide the reader with a perspective of selected recent advances in atomic and molecular electronic structure theory and its applications. By adopting this somewhat pedagogic approach, in which much of the review is written in the form of a commentary, it is hoped that the informed non-specialist may be able to pick his way through the veritable maze of problems, ideas, and models that pervade the field of quantum chemistry. More detailed accounts of selected topics are best found in the Royal Society of Chemistry Specialist Periodical Reports on ‘Theoretical Chemistry’, or in specialist review series such as ‘Advances in Quantum Chemistry’. It is useful, perhaps, to view the development of the theory and its applications in terms of the tree analogy; just like the tree, there are many areas of new growth - in the periphery of the subject (branches), in the main body of the subject (trunk), and in areas of mathematics and physics (roots) that are often obscured from the physical chemist. As with the tree, growth in the peripheral regions is often perceptibly faster than that in the main body of the subject; a situation that arises because it is often easier to apply existing theoretical models to new systems than it is to develop new models. A well known example of this situation is manifested in the literature by the existence of literally hundreds of applications of the Hall-Roothaan form of molecular orbital theory in all its various guises - both ab initio and semi-empirical. Clearly, a considerable amount of useful information and experience has been gained from such calculations; but, of course, it should be remembered that the basic constraints defining the model are often obscured, or even lost, under the weight of an ever increasing number of essentially routine applications, on account of the widespread availability of S. F. A. Kettle, Chem. Br., 1980, 16, 511. Published on 01 January 1980. Downloaded by UNIVERSIDADE FEDERAL DA BAHIA on 25/12/2013 09:19:25. View Article Online / Journal Homepage / Table of Contents for this issue
Transcript of Eletronic Structure of Molecules

2 Electronic Structure of Molecules
By G. DOGGETT
Department of Chemistry, University of York, Heslington, York YO 1 5DD
1 Introduction
The continuing development and improvement of on-line literature retrieval facilities is, by necessity, forcing a change in the nature and form of literature reviews; as Kettle’ has recently commented, the time is rapidly approaching when one must question the usefulness of providing a comprehensive review of the literature, when lists of pertinent papers can be obtained from an efficient information retrieval system. With these thoughts in mind, the main aim of this review is to provide the reader with a perspective of selected recent advances in atomic and molecular electronic structure theory and its applications. By adopting this somewhat pedagogic approach, in which much of the review is written in the form of a commentary, it is hoped that the informed non-specialist may be able to pick his way through the veritable maze of problems, ideas, and models that pervade the field of quantum chemistry. More detailed accounts of selected topics are best found in the Royal Society of Chemistry Specialist Periodical Reports on ‘Theoretical Chemistry’, or in specialist review series such as ‘Advances in Quantum Chemistry’.
It is useful, perhaps, to view the development of the theory and its applications in terms of the tree analogy; just like the tree, there are many areas of new growth - in the periphery of the subject (branches), in the main body of the subject (trunk), and in areas of mathematics and physics (roots) that are often obscured from the physical chemist. As with the tree, growth in the peripheral regions is often perceptibly faster than that in the main body of the subject; a situation that arises because it is often easier to apply existing theoretical models to new systems than it is to develop new models. A well known example of this situation is manifested in the literature by the existence of literally hundreds of applications of the Hall-Roothaan form of molecular orbital theory in all its various guises - both ab initio and semi-empirical. Clearly, a considerable amount of useful information and experience has been gained from such calculations; but, of course, it should be remembered that the basic constraints defining the model are often obscured, or even lost, under the weight of an ever increasing number of essentially routine applications, on account of the widespread availability of
S. F. A. Kettle, Chem. Br., 1980, 16, 511.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online / Journal Homepage / Table of Contents for this issue

4 G. Doggett
high-speed computers. In fact, it has been said that there is a detectable tendency to use computers as a substitute for thinking! Notwithstanding this last comment, there is. and always will be, a place for computational quantum chemistry in the development of the subject: however, there is a case for being more prudent in testing and applying the many theoretical models in current use - primarily because all models contain within them weaknesses that cannot be readily ameliorated. It is perhaps better, therefore, to spend more time developing better models, as well as improving the understanding of existing models, than to go on increasing the routine applications of a given model to larger and larger molecules. The latter temptation is, of course, difficult to resist, particularly as increasing numbers of experiments are being carried out on systems that make themselves amenable to theoretical verification and interpretation - in this respect it is interesting to note that two new journals* devoted to computational quantum chemistry have announced their appearance in 1980.
As indicated above, the basic approach of this review involves taking selected views of the general shape of the subject, insofar as theories of electronic structure calculations are concerned, particular attention being paid to conceptual and model developments that appear, at this juncture, to be growth points - in some cases of existing branches and, in other cases, of new branches.
2 General Considerations
Looking back, the continuing areas of interest and growth have been concerned with the development of improved models for calculating energy minimized geometries and properties of molecular species, as well as with the calculation of potential energy surfaces for simple chemical reactions. The calculation of these surfaces is not an easy task - even for the simplest systems - but there has been growth in their use for calculating cross-sections or rate constants for selected chemical reactions. Now, of course. different models for the electronic structure problem yield surfaces with different characteristics, and it is only just starting to become clear how these differences affect calculated values of observed properties. For this reason it is deemed more useful, for the purposes of the present review, to examine some of the underlying assumptions associated with orbital models of electronic structure before presenting detailed results.
Adiabatic Approximations.-The Born-Oppenheimer approximation, which is used to separate electronic and nuclear motions, forms the foundation (trunk of the tree) of nearly all models currently used to investigate electronic structure problems: furthermore, the concept of molecular shape emerges as a consequence of this approximation in a natural way. This latter point was taken up a few years ago by Woolley3 in a paper in which he sought an answer to the question ‘Must a molecule have shape?’ To many chemists this work must have been very perplexing, because the concepts being questioned by Woolley have a very long and noble history: by raising the question again, Woolley has forced growth in a somewhat unexpected direction. He points out that the concept of shape for an isolated molecule is a classical notion and,
JOurnd of Computationd Chemistry, John Wiley & Sons Inc.: Theochem. Elsevier Scientific Publishing co. R. G. Woo1ley.J. Am. Chem. Soc.. 1978, 100, 1073: Isr. J . Chem.. 1980. 18.30.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 5
because of this, it is difficult to reconcile with a purely quantum mechanical view of matter. The implications of the adiabatic approximation of Born and Oppenheimer (from which the shape concept emerges) have been discussed again this year by W ~ o l l e y , ~ Trir~dle ,~ and Claverie and Diner5 in an important issue of the Israel Journal of Chemistry devoted to the theory of molecular structure and bonding. The general situation is far from clear in this fundamental area of current interest, and Woolley’s original question remains unanswered. Another view of the problem is delineated by Primas6 in a recent review of the (formal) foundations of theoretical chemistry, but, in the section pertaining to molecular shape, he presents a frank view of the unsolved problem: ‘. . . it remains a mystery why the Born-Oppenheimer description should lead to a notion of molecular structure which can be interpreted classically’. Wilson’ has also discussed this problem of defining molecular structure within a quantum mechanical framework, and concludes that, in general, there is no single preferred definition that is without drawbacks. However, for the majority of simple molecules, a definition of molecular structure can be found that reduces to the conventional Born-Oppenheimer equilibrium structure; the extraction of geometrical information about such structures from experimental measurements of spectroscopic frequencies, or from marks on a photographic emulsion, is viewed essentially as a semi-empirical exercise.
It could be said (very loosely), perhaps, that as far as electronic motion is concerned the Bohr and Schrodinger models for the ground state of the hydrogen atom (in the fixed nucleus approximation) display certain similarities to the adiabatic and non-adiabatic approaches for determining the stationary states of a system of electrons and nuclei. In the former, the orbit for the electron is fixed (analogous to fixing the nuclear positions in the Born-Oppenheimer approximation), while in the latter model the radius of the Bohr orbit is found to correspond to the most probable value of the radial co-ordinate, that is, a distribution function is obtained for the radial co-ordinate. This similarity is reflected in the recent work of Bishop and Cheung8 on the calculation of nuclear and electronic distribution functions from the non-adiabatic wave functions for HI+, but, before describing the results of these calculations in detail, it is helpful to establish the notation in the present review by giving a brief resume of the adiabatic approximation (a more recent and detailed review of the vibronic coupling problem is given by Azumi and Matsuzaki9).
In the work of Born and Oppenheimer, the solution of the Schrodinger equation with Hamiltonian
A = fie, + A,, where
m 2M a
H,,=--Z v (I
C. Trindle. Zsr. J. Chem., 1980, 19, 47.
H. Primas in ‘Quantum Dynamics of Molecules’, ed. R. G. Woolley, Plenum Press, New York, 1980. ’ P. Claverie and S . Diner, Isr. J. Chem.. 1980, 19, 54.
’ E. B . Wilson, Int. J . Quantum Chem., S.ymp., 1979, 13, 5. * D. M. Bishop and L. M. Cheung, In!. J. Quantum Chem.. 1979. 15.517.
T. Azumi and K . Matsuzaki, Photochem. Photobiol., 1977,25,3 15.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

6 G. Doggett
is developed in the form
n r n n . m
x,,(R) is the mth nuclear wave function associated with the electronic state Yn. The latter function is obtained as a solution of
He, yn = En(R) y n
in which the nuclei are held in the configuration defined by the vector R in the space of nuclear configurations. So long as the Yfl corresponding to the ground state (n = 1) is non-degenerate, and is well separated in energy from the next electronic state, then the adiabatic approximation [equation (1)l
gives reasonable representations of the vibronic states associated with the ground electronic state LOnrn, Onk are non-interacting under fi, and hence individual vibronic states can be considered; it should be noted, however, that this is a rather over-simplified view of the real situation of a vibrating (rotating) molecule, in which the Hamiltonian has to be specified very carefully - see, for example, the recent review by Sutcliffelo 1.
The approximation of CP by equation (1) leads naturally to the concept of a potential energy curve (diatomic) or hypersurface (polyatomic molecule) in the space of nuclear configurations, because the electronic energy is a function of R and appears as part of the potential energy term associated with the nuclear motion. Very often the accessible parts of the energy hypersurface of interest correspond to a region in the neighbourhood of a minimum, particularly for molecular species in their ground states. In these situations, the excursions of the nuclei involve sampling only a small region of the hypersurface, and a traditional rigid molecule view emerges in which the harmonic approximation is adequate for describing the vibrational motion. However, recent experimental" and theoretical', investigations of systems such as H,O . . . FH, which exhibit low frequency bending and stretching modes, show that the excursions on the energy hypersurface are less confined and the situation is indicative of a non-rigid structure. Not surprisingly, Bouteiller and co-workers12 found that it was necessary to fit the energy hypersurface, for their vibrational analysis, by an expansion that included terms up to fourth order in the deformation co-ordinates.
The whole problem of understanding the nature and categorization of non-rigid molecules is one of continuing interest; a valuable survey of the general situation has been given by Berry.13 Furthermore, the concept of a wreath product has been used by Bala~ubramanian'~ to rationalize the construction of symmetry groups of non-rigid molecules. By investigating further the representation theory of generalized wreath
lo B. T. Sutcliffe in 'Quantum Dynamics of Molecules', ed. R. G . Woolley, Plenum Press, New York, 1980. l 1 J. W. Bevan, Z. Kisiel, A. C. Legon, and D. J . Millen, Proc. R . SOC. London, Ser. A , 1980, 372,441. IZ Y. Bouteiller, M. Allavena, and J . M. Leclercq, J. Chem. Phvs.. 1980, 73, 2851. l 3 R. S. Berry in 'Quantum Dynamics of Molecules', ed. R. G. Woolley, Plenum Press, New York. 1980. l4 K. Balasubramanian, J. Chem. Phys., 1980, 72,665.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 7
product groups, Balasubramanian was able to derive the selection rules for allowed electric dipole transitions, using, as an illustration, the example of triphenyl. Ezra’j has also given a unified approach for elucidating the symmetry properties of a class of such molecules. He takes a dynamical model in which the nuclei execute fast small-amplitude vibrations about an equilibrium configuration that is itself undergoing slow large- amplitude motion.
The cosy familiarity of long-established concepts cannot be taken as a substitute for their veracity; clearly, in developing an answer to Woolley’s question ‘Must a molecule have shape?’ it is necessary to take Cou1son’s16 advice and voyage out into areas that are distinctly mathematical in nature, in order to improve understanding of the root system of the subject. The charting of this area of the subject is somewhat rudimentary, and calculations that avoid explicit use of the Born-Oppenheimer approximation are extremely few in number (for references, see the papers cited in references 3-5). However, an accurate calculation by Bishop and Cheung, already alluded to above, is of especial interest, as these authors have shown how the non-adiabatic wave function for H,+ can be visualized in a simple way.
The wave function is written in the form shown in equation (2), where, for the
rotationless ground state M = 35 and N = 13; {$i}, { x j } form orthonormal sets of electronic and nuclear functions, respectively. New orbitals Yi(r), X,(R) are now defined according to the prescriptions
with the coefficients b,, Umj being chosen so that the expression for Q, can be put in ‘diagonal form’ [equation (3)l. The djj are occupation numbers. By writing Q, in this
N
Q,(r, R) = 1 djj Yj(r) Xj(R) j = 1
(3)
form, a rapidly convergent expansion is obtained: for example, the first four terms yield an energy that agrees to seven significant figures with the energy obtained from the full expansion. The corresponding occupation numbers are 0.9957222, 0.0042443, 0.000033 1, and 0.0000003, and, because of the obvious rapid increase in importance of successive terms in equation (2), it is not surprising to find that the first term alone forms quite a good approximation to Q,. In this case, X , ( R ) is symmetrically peaked about IRI = 2.0 a.u., and contours of Y, ( r ) look very similar to those of the conventional 1 og orbital associated with an internuclear separation of 2.0 a.u. Bishop and Cheung also give plots showing the overall density function averaged over either the nuclear variables or the electronic variables; that is, plots of
p,(r) = J” Q,* OdR and p,(R) = J” Q,* (Ddr
G. S. Ezra,Mol. Phys., 1979, 38,863. l6 C. A. Coulson, Bull. Inst. Math. Appl., 1973, 9, 206.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

8 G. Doggett
In addition, the contour plot of the density @*(r,R) @(r,R) for I R I = 2.0 a.u. is very similar to that obtained from the adiabatic wave function at this geometry. It should be remembered, of course, that a bond length for H,+ is not one of the parameters produced in the calculation; the total energy obtained from either equation ( 2 ) or ( 3 ) represents the energy of the non-rotating molecular species in which vibrational and electronic contributions to the energy are not separated.
Pictorial Models.-It is obviously very difficult to extend the above kinds of calculations to more complicated molecular systems, but the lack of apparent activity in this branch of the subject should not be taken as evidence that all is well with traditional approaches and concepts; an open mind must be kept until a more definitive picture emerges. In fact, it is the lack of a suitable pictorial representation that often blocks the way to general understanding of newer developments. For example, models based on the simple idea of overlapping orbitals have a long pedigree as well as an obvious pictorial and intuitive appeal - so much so that the power of arguments based on such schemes should not be underestimated. Two examples from the 1980 literature will suffice to illustrate this point. First, Schoeller’’ tried to answer the question ‘When is a singlet carbene linear?’, by observing that carbenes, of the form CX,, are linear in their ground states only if the electronegativity of X is less than that of carbon. Schoeller obtained a rationalization of the observed geometries by means of a simple molecular orbital model, in which the carbon lone pair is allowed to delocalize itself over the three centres by invoking the use of empty n orbitals on X. This picture is different from the one preferred by Pauling18 who, in a later communication, presented a pictorial valence bond view in which structures based on the use of the empty carbon 71 orbital are deemed to be important.
The second example arises in the discussion of geometries expected for the van der Waals molecules Ar ... XX’, where XX’ = Cl,, N,, O,, HC1, or CIF. Burdett” suggests that two kinds of orbital interactions are important in determining the observed geometrical arrangement: first, those of ‘type 1’ involving an unoccupied (antibonding) molecular orbital, 4, on XX’ and a (filled) 3 p orbital on Ar (see Figure la); secondly, those of ‘type 2’ (see Figure lb) involving the interaction between a (filled) molecular orbital, #, on XX’ and a (filled) 3 p atomic orbital on Ar. In each case, the particular 3 p orbital used by Ar depends upon the relative orientation of Ar and XX’. Interactions of types 1 and 2 lead to overall stabilization and destabilization, respectively, because in the latter situation the antibonding orbital #a is more destabilized than the bonding orbital #,, is stabilized.
Burdett is able to show, on the basis of a very simple calculation, that the collinear arrangement Ar . . . XX’ is preferred if X’ is more electron attracting than is X (even allowing for electrostatic and dispersion interactions). This result follows from the orbital overlap model because, for the type 1 interaction between 3p, and 20,, greater stabilization is achieved for ArClF than for ArFCl because the antibonding 20, orbital contains a greater weight of the chlorine 3p, atomic orbital. For this latter reason, the destabilization arising from the type 2 interaction is least, because the 20, (occupied) molecular orbital contains the smaller weight of the chlorine 3p, atomic orbital.
W. W. Schoeller,J. Chem. SOC., Chem. Commun., 1980, 124. L. Pauling, J. Chem. SOC., Chem. Commun., 1980, 688.
l9 J. K. Burdett, J. Chem. Phys., 1980, 73, 2825.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 9
I I I I I
\
I I I I
I I 3P.+i\\ I I
‘\wl Ar xx ‘ x x‘ Ar
Figure 1 Outer orbital interaction diagrams for van der Waals molecules. (a) ‘type 1’ interaction; (b) ‘type 2’ interaction
These two examples, taken from the current year’s literature, illustrate the longstanding divergence in views held by different schools of theoretical chemists: on the one hand, as exemplified by the above discussion, there are those (pragmatists) who search for form and structure in the subject, and for whom the appeal of simple pictorial arguments is very strong, while on the other hand there are those (idealists) wedded to the ab initio calculational approach, and any thoughts of semi-empiricism are rejected in their search for form and structure at a deeper level.
The main problem with the calculational approach is that it is very difficult to answer questions posed within a chemical framework. Why, for example, does CO prefer to bond to transition metal via the carbon end of the molecule, and why are complexes containing the isoelectronic N, molecule so much less in evidence? Again, why does insulin form dimers or hexamers so readily? These are pertinent questions to ask within a chemical context, but the point is that, at the present time, the second group of theoretical chemists are asking and answering very different kinds of questions. For example, what is the best choice of basis set for constructing molecular wave functions in order to determine energies and other properties? This is not a spurious question to ask - uninteresting though it may appear at first sight - and a perusal of the recent literature shows evidence of widespread and continuing interest in this problem. Why should this be the case? Again, why is it so difficult to calculate accurate energies for ionization processes and for long-range interactions between closed shell species (Be.. . Be)?
An attempt will be made in the following Sections to answer these and other questions, in order to achieve the original aim of providing perspective views of recent developments in the construction of suitable models for calculating electronic structures from first principles. In the next Section, a resume of orbital model building is given: then follows a detailed discussion of the basis set problem. Finally, the review concludes with Sections devoted to the application of the various orbital models to selected groups of systems of chemical interest, where the calculation of molecular properties is of prime concern.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online
10
3 Orbital Models for the Electronic Wave Function
G. Doggett
Spin Functions.-It is current practice to construct (adiabatic) electronic wave functions, Y(rlR) for N electron systems - written as Y from now on - by allocating the electrons to orbitals di, taking care to couple the spins of individual electrons so that the overall antisymmetric space-spin wave function is an eigenfunction of s2 (the operator corresponding to the square of the total spin angular momentum - spin-orbit coupling effects ignored). Thus for a three electron system, a possible wave function is given by
.Jql (r l ) d2(r2> d 3 ( . , ) 6 = &dl 4 2 4 3 6
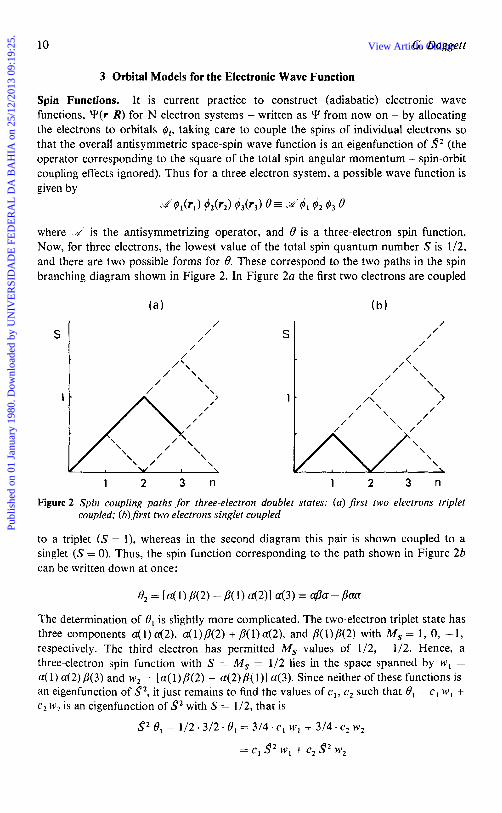
where .w' is the antisymmetrizing operator, and 6 is a three-electron spin function. Now, for three electrons, the lowest value of the total spin quantum number S is 1/2, and there are two possible forms for 8. These correspond to the two paths in the spin branching diagram shown in Figure 2. In Figure 2a the first two electrons are coupled
1 2 3 n 1 2 3 n
Figure 2 Spin coupling paths for three-electron doublet states: (a) jirst two electrons triplet coupled; (b) jirst two electrons singlet coupled
to a triplet ( S = l), whereas in the second diagram this pair is shown coupled to a singlet ( S = 0). Thus, the spin function corresponding to the path shown in Figure 2b can be written down at once:
The determination of 8, is slightly more complicated. The two-electron triplet state has three components 4 1 ) 4 2 ) , 4 l ) p ( 2 ) + p(1) 4 2 ) , and p(1)/3(2) with Ms = 1, 0, -1, respectively. The third electron has permitted M , values of 1/2, -1/2. Hence, a three-electron spin function with S = M , = 1/2 lies in the space spanned by w , =
41) a(2)/3(3) and w 2 = [a( 1)/3(2) + a(2),8( l)] 4 3 ) . Since neither of these functions is an eigenfunction of S2, it just remains to find the values of cl, c2 such that 8, = c, w, + C, w2 is an eigenfunction of S2 with S = 1/2, that is
3 2 el = 1/2.3/2. el = 3/4. cl w, + 314 - c, w,
= c, 3' w, + c, $ 2 w2
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 1 1
which implies c l (s2 - 3/4)w1 + c2(s2 - 3/4)w2 = 0. Taken with the normalization condition c12 + 2c22 = 1, the solution c, = 2, c2 = -1 is obtained.
Since neither spin coupling is the preferred one (at this stage), the overall space-spin wave function is written as
dl'-d$l $2 $3 '1 + d 2 d $ l $2 $3 e2 = &$l $2 $3[dl '1 + d 2 e21
It may well happen, of course, that the orbital coupling chosen here does not correspond to an eigenfunction of the spatial symmetry operators, I? (a necessity since 2, like s2, commutes with I?). Therefore, instead of a single orbital product, it may be necessary to generate a sum of orbital products that parallels the spin situation. For example, the ls2p1 2p-, excited orbital configuration of atomic lithium yields the antisymmetrical functions ,dls2pl 2p-, O1 and d' ls2p1 2p-, 8,, both of which are doublet states, but neither has the correct spatial symmetry characteristics (that is, neither is an eigenfunction of L2). Straightforward analysis of the kind given above for the spin functions, but taking sp2 configurational wave functions with ML = 0, yields the required space-spin eigenfunctions:
where the absence of presence of a bar over the orbital label designates a or p spin, respectively. Also, in the expansions on the right-hand sides of the above equations, an antisymmetrized product vanishes if one orbital is occupied by two electrons with the same spin functions.
Thus, in general, a given orbital configuration yields acceptable wave functions in the form indicated by equation (4) which is a linear combination of antisymmetrized products, a,', of spin orbitals. The dj are variational parameters and the ckj are determined by spatial symmetry requirements.
= 1 a, @,,,I
rn
Now if it happens that different orbital configurations give rise to overall wave functions Y j of the form in equation (4), which possess the same space-spin symmetry characteristics as Yl, then the matrix of I? must be diagonalized to produce improved multi-configuration wave functions of the form given in equation ( 5 )
C xj yj j
Orbital Functions-So far, all orbital models have the above characteristics in common; the differences occur when restrictions are placed on the forms of the $i. In the molecular orbital model, for example, double occupancy is usually forced on the system, and then there is no loss of generality in taking the #i to be orthogonal. In
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

12 G. Doggett
addition, the (Hartree-Fock) orbitals, li, which are, in general, delocalized over the molecular framework, are also eigenfunctions of an effective one-electron operator, h; h#i = e i# , . (for open shells, the definition of h is a little less obvious). The orbitals themselves are usually constructed from a set of basis functions according to the prescription
# i = C c k i X k
Although the xk are often taken to be atomic orbitals centred on the various nuclei in the molecule, this need not be a fundamental assumption of the model; bond-centred functions or bicentric functions are often used. This aspect of the orbital model is discussed in more detail in the next Section.
It is clear that the double-occupancy constraint in single-configuration molecular orbital theory makes the model unsuitable for describing dissociative processes in which there is a change in the number of electron pairs. Thus the model must fail for the process HOH -, H + OH; but a reasonable description is obtained for processes like HeH -+ He + H and HCO -, H + CO. While the double occupancy constraint does lead to difficulties in constructing complete energy hypersurfaces, it is not too severe an approximation in selected regions of the hypersurface, notably around minima corresponding to the presence of well defined molecular species. For systems with n electrons outside a closed shell of rn doubly-occupied orbitals, the number of spin functions needed is reduced fromf2i+n toLs, wheref: = ( 2 s + l ) N ! / [ ( N / 2 + S + 1)!(N/2 - S ) ! ] , since the closed shell electron configuration can be associated with only one path on the spin branching diagram. Thus, for systems with zero or one electron outside a closed shell, f ; m + , = 1 and, subject to spatial symmetry require- ments, the wave function can be represented usually by a single antisymmetrized product of spin orbitals (for n electrons outside a filled shell this result also obtains for the maximum permitted value of S), a situation which is distinctly attractive from the computational point of view.
However, if reliable results are required over more extensive regions of the energy hypersurface then it is necessary to take a multi-configuration expansion for Yl. For such model wave functions, it is usually relatively easy to select configurations required to produce the correct species on bond dissociation - at least for the ground state surface - but it is not so easy to ensure that the complete set of configurations is chosen to produce separated species in appropriate space-spin eigenstates. The two approaches currently in widespread use are the traditional configuration interaction (CI) and multi-configuration self-consistent field (MC SCF) methods. In the former method, the #i obtained as eigenfunctions of h for the single-reference (root) configuration wave function, 'PI, are used to generate the other Yj . The main problem here is associated with the slow convergence of the CI expansion, which results from the use of orbitals that are both unoccupied in Yl, and also too diffuse spatially. This aspect of possible poor convergence characteristics in CI calculations has been examined again recently by Cooper and Pounder.*O They propose that suitable unoccupied orbitals may be determined from a modified one-electron Schrodinger equation of the form
(h + m 2 ) #, = E , $,
*O I. L. Cooper and C. M. Pounder, J. Chem. Phys., 1979, 71,957 (see also ref. 30. p. 103).
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 13
where A. is a (positive) effective charge parameter, P is the electron-nucleus potential energy contribution to fi, and z is the sum of the nuclear charges. This extra term in the effective Hamiltonian ensures that the 4” are spatially more contracted than are the original unoccupied orbitals. Furthermore, the orthogonality of occupied and unoccupied # is maintained by taking 4” = zjcj#j, where #j is a member of the set {#” 1. The cj are determined in the usual way by energy minimization. The method has been applied to HCN, and the resulting CI energies are significantly improved over those obtained from the conventional calculation.
On the other hand, in the MCSCF formulation of the problem, the xj and the occupied #,. in a set of selected configurational wave functions {#,.} are determined iteratively by minimizing the energy of the multi-configuration wave function (the limited number of configurations in such expansions are chosen usually with a view to obtaining the correct description of the dissociation products). This method is now increasingly used in an extended form within the framework of the CI model; here the { Y j 1 form a multi-reference function, and (selected) single and double excitations that results from {‘Pi} are taken together with { Y j } as expansion functions in a CI calculation. Thus, for the MCSCF calculation itself, Y = 2, cK YK, but, in the CI variant
y = 2 (dK ‘J’K + C dKs YKs + C d K D YKD) K S D
where YKs and YKD are wave functions corresponding to singly and doubly excited states, respectively, arising from the reference configurational wave functions Y,. Some of these YKs and YKD states correspond to triply and quadruply excited states relative to the leading configurational wave function, Y in the set of reference states {YK} - this is why their inclusion is helpful in improving the description of electron correlation effects.
The point to notice about the molecular orbital model is that it does not provide a very helpful framework for interpreting the results of a given calculation; this arises because the chemical bond concept, which plays such a fundamental role in chemistry, requires the electron pairs to be localized rather than delocalized over the molecular framework. In fact, localization is not sufficient, because the intrapair correlation effects are also very important, although they are absent in the simpler forms of the molecular orbital model. Thus attempts to transform delocalized orbitals to localized molecular orbitals cannot ameliorate the basic deficiencies in the model. This viewpoint is essentially at the root of the criticism made by Pauling of Schoeller’s work on carbenes, which was discussed earlier.
The MCSCF variant of the molecular orbital model (with or without configuration interaction), is basically an attempt to correct for the lack of correlation between pairs of electrons with opposite spins. The use of this and related models forms an important area of current research, and more complete details are given in later Sections. Before continuing with this model, it is instructive to consider a simple example of bond breaking in order to isolate the reasons for the failure of the molecular orbital theory.
Bond Breaking in Molecular Orbital Theory.-Consider the problem of describing the bonding in the T I ground state of CH. For simplicity, only Is, 2s, and 2p carbon atomic orbitals, and a single 1s hydrogen aiomic orbital (h) are used as basis orbitals
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

14 G. Doggett
in the expansion of the #i, but the general conclusions are unaltered if a more extensive basis set is used.
Three of the four sigma molecular orbitals (ol, 02, and a3) are doubly occupied in the ground state; o4 is unoccupied, and the 2p1 7t orbital on carbon is singly occupied. Thus the overall A = 1, M , = 1/2 component of the wave function is given by
Yl = d o 1 a, 0, a, 0 3 a, 71
Now the o molecular orbitals have the asymptotic forms (R -, 03)
because, in the limit R -+ GO, the molecule is required by the constraints of the molecular orbital model to dissociate into the lowest energy species each containing an integral number of electron pairs - here C+, H- [this is consistent with the limiting forms given by Liu and Verhaegen21 in note (b) of their Table XI]. Thus, as far as electron configurations are concerned
CH(a12 oZ2 oj2 71) -, C+( 1s’ 2s’ 2p1), H-(h2).
With this observation, it is immediately apparent that ground-state neutral-atom dissociation products can be obtained only by including additional wave functions in the CI expansion, which arise from the singly excited configuration aI2 oZ2 o3 CI, n of CH. This latter configuration correlates with Co( ls2 2s2 2p0 2pl), Ho(h).
As shown in the introductory part of this Section, two antisymmetrized wave functions may be constructed from the c ~ ~ ~ o ~ ~ o ~ c ~ ~ n configuration on account of the presence of three singly occupied orbitals; that is
Y2 = &al 6, o2 a 2 [ 0 3 o4 dl1 and Y3 = .dol 6, o2 6 2 [ 0 3 0 , n 6 ~ I .
These two wave functions become more important than Y , as R -+ co. But, in this limit, equations (6a) and (6b) are obtained, and each antisymmetrized spin orbital product
Y, -, d l s ~ 2 s % [ 2 ( h 2 p o Fl) - ( h G o 2p1) - (x2p0 2p,)l
Y3 -, d l s G 2 s 2 s [ ( h 5 , 2p1) - (&2p0 2pl)l
effectively reduces to either Oj‘h or Ok’h, where Oj’, and Ok‘ are antisymmetrized products of carbon atom spin orbitals. This situation obtains because the permutations omitted involve interchanges of electrons between carbon and hydrogen which yield zero contributions to the matrix elements of I?. Thus, as R -+ 03, Y 2 and Y 3 yield the antisymmetric functions d 2 p 0 2 p l , &Go 2p,, d 2 p 0 2p1 and &Go 2p1, d 2 p 0 2p,, respectively, corresponding to the 2p2 configuration of carbon (suppressing the filled s orbitals for notational convenience). Now the ground state of carbon in the 2p2 configuration is 3P, and d 2 p 0 2p, is, in fact, the ML = 1, M , = 1 component wave function corresponding to this term. But the ML = 1 , M , = 0 component wave functions for this configuration are &2pO2p1 + d 2 p 0 2 p 1 (for ’P) amd &2p02p1 - d 2 p 0 2 p 1 (for ‘D). Thus either structure by itself fails to describe the spin decoupling that is necessary to achieve the correct dissociation products; for example, Y 3 yields
21 A. P. D. Liu and G . Verhaegen, J . Chem. Phys., 1970,53,735.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 15
04
T T r
0 3 a 2 - a
. d G o 2 p l and ,cP2p02pl, in which the former is a 50:50 mixture of the 3P and 'D M , = 1, M , = 0 component wave functions, and the latter is the M , = 1, M , = 1 component of 3P.
The key point here is that the M L = 1, M, = 0 component wave functions span a two-dimensional sub-space; this is why it requires both Yz and Y3 to ensure that the sub-space is properly spanned (any two linearly independent combinations of .a/Go2pl and d 2 p 0 q 1 spans the sub-space, and the two represented by Yz and Y3 suffice). In the asymptotic limit, therefore, the energy matrix takes the form
a Ir - 1 I
I I I I I I I I : : 2po A ! I ** I - I - ! 2P, 2P-,
1 - 1 I I h & I :;- I )
I I
! @-2s f : * k l -- j ---:
C+H- COHO
/ x
where the lowest eigenvalue arises from the 2 x 2 block. If all single and double valence excitations from Yl are also considered (the
abbreviation CISD is currently in widespread use for such calculations), then these can be enumerated in a symbolic fashion as shown in Figure 3. The energy matrix in the asymptotic limit now takes the form shown in Figure 4.
The C0(2s2p3), Ho(h) and C0(2s2 2p2), Ho(h) wave functions are non-interacting in the asymptotic limit, as the carbon atom configurations are of different parity. Thus, the somewhat surprising result is obtained that the three-component C I wave function considered originally is sufficient for describing the dissociation process. Furthermore, only the ground state configuration of 2s' 2p2 of carbon is generated, and so the wave function describing the products is not a correlated wave function (for this to be the case, 2p4 configurations of carbon are required and, from Figure 3, it is clear that these can arise only from molecular zll states associated with the triply excited electron configuration oI2 o3 o4 x3. Thus, considering the dissociated species from the 'super- molecule' point of view, the CISD molecular calculation produces a wave function in which there is no configuration interaction in the separated atoms. It appears, therefore, that the configuration interaction in the molecular situation is needed partly to cope with the problem of bond breaking, and partly with improving the description of electron correlation in the molecule. It can be seen from a simple pictorial representation of these results that the accuracy of the calculated dissociation energy is
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online
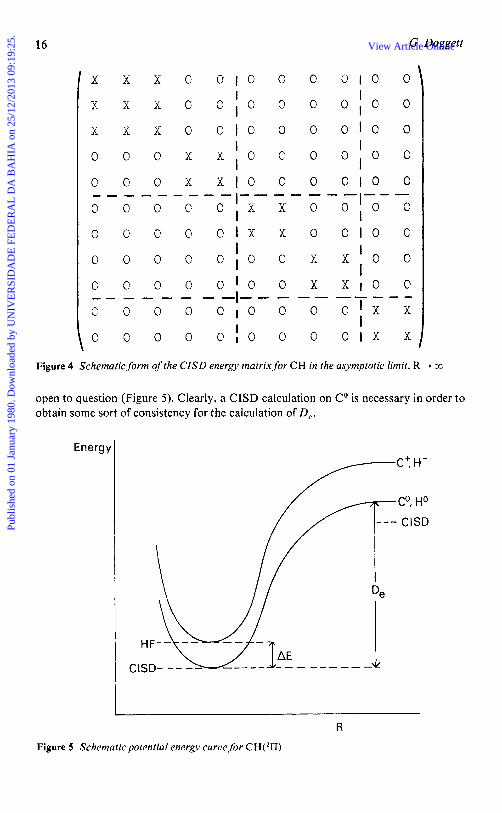
16 G. Doggett
Figure 4 Schematic form of the CISD energy matrix for CH in the asymptotic limit, R + co
open to question (Figure 5) . Clearly, a CISD calculation on Co is necessary in order to obtain some sort of consistency for the calculation of D,.
Energb
Co, Ho
. CISD
R
Figure 5 Schematic potential energy curve for CH('II)
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 17
The imbalance in the description of the correlation effects - which favours the molecular environment - is basically a manifestation of what is called the ‘size-consistency’ problem. It can be overcome onlv bv the tiresome procedure of including wave functions associated with higher order excitations from the reference configuration. When many orbitals are available - as occurs, for example, if either a large basis set is used or the system contains a large number of electrons - then the computational problems become very severe indeed on account of the vast numbers of configurational wave functions that need to be considered. Davidson and Silver22 have, in fact, recently produced an improved version of an earlier empirical formula of the form
AE = AE,,( 1 - cI2)/(2cl2 - 1)
which can be used for estimating the effects of higher order excitations on the calculated energy. In this formula c, is the coefficient of the reference configurational wave function in the CISD calculation and AEsD is the quantity shown in Figure 5; the formula is stated to be most reliable for systems containing between 10 and 30 electrons.
Pople, Seeger, and K r i ~ h n a n , ~ ~ in their investigation of the size-consistency problem suggest the following form for AE
I (rn2 + 2m tan2 28)+ - rn 2(sec 28- 1)
AE = AE,,
Here cos 8 = c, and rn is the number of electrons.
Bond Breaking in Valence Bond Theory.-In contradistinction to the molecular orbital model, the traditional valence bond model places electrons in orbitals that are localized around the various nuclei in the molecule, although, as seen later, the extent of localization can be controlled to a certain degree. In the simpler forms of valence bond theory, which invoke only a covalent description of a bond pair, there is a tendency to over-correlate the motion of electrons of opposite spin; this is why a multi- configuration (multi-structure in valence bond parlance) approach is necessary in order to give greater flexibility to the wave function. However, by avoiding the double occupancy constraint (except, perhaps, for core and lone-pair electrons), it is necessary to include all spin coupling schemes in order to obtain correct dissociative behaviour. Consider, for example, the 211 state of the CH radical in which, for simplicity, the 1s and 2s orbitals are each doubly occupied; the 2p, carbon n-orbital is singly occupied, and a sigma bond is formed between the carbon 2p, and the hydrogen l s (h) orbitals. The two structures arising from this configuration (which dominates the overall wave function at large internuclear separations) are shown schematically in Figure 6, but the filled 1s and 2s orbitals are omitted for simplicity.
In Figure 6(a) the bond pair is singlet coupled, and the appropriate spin coupling scheme for the A = 1, M , = 4 structure wave function is described by
y2 = .d l s i i2sZ[h2po 2p, 8 2 1
22 E. R. Davidson and D. W. Silver, Chem. Phys. Lett., 1977. 52. 403. 23 J. A. Pople. R. Seeger, and R. Krishnan, I n t . J . Quantum Chem., S-vmp., 1977, 11, 149.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online
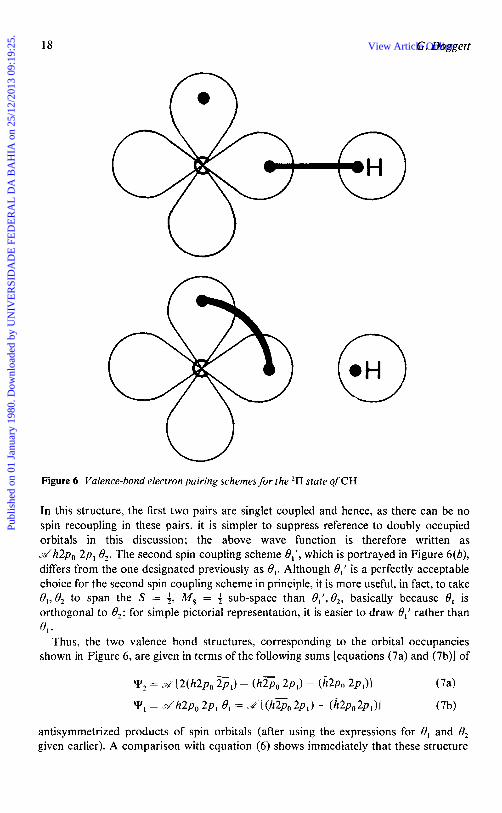
18 G. Doggett
Figure 6 Valence-bond electron pairing schemes for the 21’1 state of CH
In this structure, the first two pairs are singlet coupled and hence, as there can be no spin recoupling in these pairs, it is simpler to suppress reference to doubly occupied orbitals in this discussion; the above wave function is therefore written as ..d‘h2p0 2p, 9,. The second spin coupling scheme el’, which is portrayed in Figure 6(b), differs from the one designated previously as 8,. Although 8,’ is a perfectly acceptable choice for the second spin coupling scheme in principle, it is more useful, in fact, to take B,, 0, to span the S = 4, M , = 4 sub-space than el’, O,, basically because O1 is orthogonal to e2; for simple pictorial representation, it is easier to draw 8,’ rather than
Thus, the two valence bond structures, corresponding to the orbital occupancies shown in Figure 6, are given in terms of the following sums [equations (7a) and (7b)l of
4.
antisymmetrized products of spin orbitals (after using the expressions for 0, and O2 given earlier). A comparison with equation (6) shows immediately that these structure
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 19
wave functions are identical to those obtained in the asymptotic limit for the molecular orbital CI wave function discussed in the previous Section. However, in this variant of the valence bond model, where the forms of the orbitals do not vary with internuclear separation, the wave functions (7a) and (7b) are built from the same components, irrespective of the internuclear separation; it is, in fact the change in structure weights that describes the response of the system to bond formation. It is clear that, as R -+ co, components of term wave functions with definite L,S values can be obtained only if both spin coupling schemes are included in the valence bond model, a result that follows from the earlier observation that it takes two structure wave functions to span the sub-space of M , = 1, M , = 1 component wave functions.
After these preliminary discussions of the basic differences of approach in the molecular orbital and valence bond theories of molecular electronic structure, it is practicable to examine some recent advances in the application of the two models.
Recent Developments in Valence Bond Theory.-The practical problems of implementing the valence bond model in the form described above - that is, by building structures out of orbitals, #i, that are monocentric - stem from the difficulty in structure selection in situations where the set of 4i is such that very many structures can be generated, and from the non-orthogonal nature of the basis orbitals themselves.
The former difficulty arises because, conventionally, large numbers of structures are needed in order to achieve a satisfactory description of the change in electronic structure on molecule formation. Unfortunately, the resulting wave functions are just as unwieldy as their molecular orbital CI counterparts. This suggests that it might be more profitable to adopt a strategy in which attention is focused on improving the quality of the orbitals used in constructing the valence bond structures, thus paralleling the situation in molecular orbital theory where orbitals determined from an MCSCF wave function are generally accepted as yielding more rapidly convergent CI expansions.
Developments of the valence bond model along the above lines are not new. In recent papers, both Gerratt and Raim~ndi*~ and van Lenthe and Balint-Kurti2’ survey and develop two somewhat different reformulations of the valence bond model, each of which is capable of describing the effects of bond making and breaking in a more compact and appealing manner than hitherto. In Gerratt and Raimondi’s approach the number of structures is severely limited initially by the choice of the orbital model. Each electron is ascribed to an orbital, #i, which is expressed as a linear combination of the basis orbitals, xj ; the individual #i are non-orthogonal, and orbital double occupancy is precluded. It is, therefore, necessary to include all spin coupling schemes in order to ensure the correct spin decoupling on bond dissociation. Thus, the simplest such wave function for an N-electron system is written in the form of equation (S), where f = fNs,
and #i = z jd j ix j . In this approach, each di is an eigenfunction of its own effective Hamiltonian operator; hi#i = and the dji ,ck are determined iteratively by minimizing the energy. Clearly, for rn basis functions, there are rn molecular-based
24 J . Gerratt and M. Raimondi, Proc. R . SOC. London, Ser. A , 1980, 371, 525. 25 J . van Lenthe and G. G. Baht-Kurti, Chem. Phys. Lett., 1980,76, 138.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

20 G. Doggett
orbitals that are eigenfunctions of each ii, and only the lowest energy orbital is selected from each such set in constructing equation (8).
Since each orbital, #i, asymptotes to an orbital on a given atom at R -+ co (diatomic assumed), a wave function in the form of equation (8) will yield spin eigenfunctions for the separated atoms. In certain cases, as obtained in BH and BeH, for example, the asymptotic behaviour of a pair of #i - describing the outer valence electrons of either boron or beryllium - is such that they both contain mixtures of valence s and p orbitals; thus, at this level of approximation, the description of the orbital decoupling is not quite right. In fact, the atomic boron or beryllium species is described by a non-variationally determined mixture of valence 2s22pn and 2pn+ configurations. Strangely, this is almost the converse of the situation obtaining in the previously discussed limited CI calculation, but, as already noted and unlike its molecular orbital counterpart, the molecular wave function does describe dissociation into atoms. Gerratt and Raimondi, and also Gerratt and Pyper26 in earlier work, find that the orbital recoupling required can be achieved by admitting configurations in which pairs of valence electrons are excited into 71-orbitals (a double excitation is needed to ensure that a C electronic state arises). The results obtained by Gerratt and Raimondi, using a single-configuration spin-coupled wave function for BeH, are shown in the first line of Table 1. The second line in the Table contains the near Hartree-Fock results of Cade and Huo.
Table 1 Total energies and Re values for BeH*
No. of No. of -E/a.u. R ,/a.u. Ref: basis orbitals configurations 15.1790 2.58 1 130 1 15.1531 2.528 160 1 15.1969 2.579 130,7n 71 15.2324 2.538t 190, 672,46, 24 1039
a b
a C
* Atomic units are used in this and subsequent tables (Energy: 1 a.u. =: 2.62546 MJ mol-'; Length:
t Experimental Geometry. a J. Gerratt and M. Raimondi, Proc. R . SOC. London, Ser. A , 1980,371,525; * P. E. Cade and W. M. Huo,
J. Chern. Phys., 1967,47,614; C . F. Bender and E. R. Davidson, Phvs. Rev., 1969, 183,23.
1 a.u. z 52.92 pm).
Unlike the molecular orbital-based models, the computational problems in extending the spin-coupled form of valence bond theory to large systems are immense, for unless the wave function is constrained by allowing for some core-valence orthogonality or orbital double occupancy (cores, for example), the energy calculation becomes intractable. However, as reported by Gerratt and Raimondi in their investigation of BeH, the spin-coupled scheme has a significant advantage over conventional molecular orbital theory when formulated within a CI framework: in particular, the unoccupied orbitals, arising from the solution of hi#i = E ~ # ~ , are more spatially contracted than are their SCF counterparts, and are hence better suited as correction functions in the description of the electron correlation. The results for a 71 multi-structure valence-bond-based wave function (line 3 of Table 1) compare very
26 J. Gerratt and N. C . Pyper, Proc. R. SOC. London. Ser. A , 1977, 355.407.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 21
well with those obtained by Bender and Davidson in a 1039 configuration CI calcula- tion (line 4, Table 1).
It would seem, therefore, that by the simple expedient of relaxing the double occupancy constraint, it is possible to generate CI-type expansions of much shorter extension than those arising from molecular orbital based theories. There are two reasons for this superiority of the valence-bond-based approach; first, proper dissociative behaviour is built into the model, and hence excessive mixing of configurations is not required in order to yield space-spin eigenfunctions of the separated species, and secondly, as already noted, the use of spatially more contracted unoccupied orbitals improves the convergence characteristics of the multi-con- figuration wave function.
In contradistinction to Gerratt and Raimondi's extension of the valence-bond model, van Lenthe and Balint-Kurti prefer a slightly different formulation of a generalized valence-bond theory, which avoids some of the problems experienced with the spin- coupled multi-configuration approach of the former authors. Basically, Balint-Kurti and van Lenthe adopt the much earlier strategy of Moffitt, and focus attention on the atomic states that may be combined to produce molecular states of appropriate symmetry. Thus, even in zeroth-order, a multi-structure valence-bond wave function is used which differs in detail from all the forms discussed so far. The approach involves building molecular Y i out of antisymmetrized products of appropriate components of L, S eigenfunctions of the combining species: the multi-structure formalism is required, therefore, in order to allow for both covalent and ionic inter- actions. For example, in the case of CH, the reference multi-configuration wave function is built out of wave functions corresponding to the interaction of COHO, C+,H-, and C-,Hf states as follows: Co(3P),Ho(2S), Co( 'D),Ho(2S), C+('D0),H-( ' S ) , C+(2Po),H-( ' S ) , and C-( 2Po),H+. The required molecular symmetry adapted 211 functions, Yi , are obtained by standard procedures; for example, in the case of the fragments C(3P),H(2S), the ML = 1 , M , = 1 , and the M , = 1, M , = 0 atomic space- spin eigenfunctions for carbon have been given earlier. If thedoubly occupied s orbitals are suppressed, then these are M 2p1 2p0 and ;s/ (2p, 2p0 + 2p1 2p0), respectively.
A molecular ' ll(A = 1, M , = 1/2 component) is constructed from aggregate states having ML = 1 , M , = 1/2 (z-axis assumed to lie along the internuclear axis), that is, from 0, = -d2pl 2p0h and Q2 = d (%,2po + 2p1&) h, neither of which is an eigenfunction of $'. The particular linear combination of and @, corresponding to a doublet state (S = 1/2, M , = 1/2) is found by requiring that S2(cl@, + c,@,) = 3/4 (c1@, + c2Q2), that is (7/4 - c , + 2c2) + (cl + 11/4 - c2) = 3/4 (cl + c2) , from which it follows that c, = -2c2. Thus the required component of the 211 state, Yl, say, is given by
Y, = 2 d 2 p 1 2p0h - .S/(G, 2p0 + 2p, Go) h = .d 2p1 2p0 h8,
(a result that should have been obvious from the forms of the spin functions w1 and w2 contained within <D, and 02, respectively). The other combination of a, and <D2 that can be constructed yields the M , = 1/2, A = 1 component of a 417 state.
The second spin-coupling scheme, previously designated by O,, and associated with the 2p, 2p0 h configuration, is obtained from the aggregation of the appropriate components of C('D),H(2S); Y2 = .d 2p1 2p0 h82. Thus, in this model, as R + GO, the
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

22 G. Doggett
two ’ll structures associated with the 2p, 2p, h configuration are just those given by either the CI [equation (6)l or the valence bond [equations (7a) and (7b)l models, when their respective 2 x 2 energy matrices are diagonalized. It is clear, therefore, that van Lenthe and Baht-Kurti’s approach always yields space-spin eigenfunctions after bond breaking, irrespective of the number of structure wave functions used. So far, though, this is a conventional valence-bond approach that starts from the separated atom end of the problem; furthermore, in extending this approach to deal with polarization and other interatomic effects, the conventional treatment involves augmenting the set of Yi with structures arising from more highly excited (and ionic) states of the combining species, thus leading back to an extended form of multi-structure wave function with all its attendant problems of structure selection. van Lenthe and Balint-Kurti avoid these difficulties by taking a limited number of structures based on neutral atom and singly charged ions, as listed above in the case of CH. The resulting valence-bond wave function forms the reference function for a limited form of configuration interaction; for this purpose, the original basis orbitals, xj , are augmented with some unoccupied orbitals so that singly excited configurations can be constructed from the Yi in the reference function. By allowing for the interaction of the reference function with these singly excited states, it is possible to obtain a set of improved orbitals, #i, which are used in place of the original xi to obtain an improved reference function. The whole process is iterated to self-consistency. Thus, at the end of the calculation, a set of molecularly determined orbitals, #i, is obtained for use in constructing a limited multi-structure wave function. The power of the method lies in the options that can be exercised in determining the optimum #i. For example, if the #i are restricted to be monocentric (hybrid orbitals), and orthogonal to other #k on the same centre, then a simple view of the bonding emerges that is consonant with traditional ideas. It is also clear that the imposition of complete orthonormality on the set of #i results in the conventional MCSCF wave function, provided no limitations are placed on the partitioning of the basis orbitals over the #i. The VBSCF wave function therefore contains the MCSCF wave function as a special case.
The results of van Lenthe and Balint-Kurti for OH (’ll), which has the same set of basic structures as CH because of the 2p2,2p4 equivalence, are given in Table 2; the increase in accuracy of the calculated dissociation energy and bond length, when the #i are allowed some flexibility, is immediately apparent. Molecular orbital and CI results
Table 2 Total energies and Re values for OH in the VBSCF model
Basis (6s 12pl ld5 f12s3p3d2f) (5s4p2dl f13s l p ) (9s5p ld,5s l p )
( 1 ls6p2d15s2p)
(10s6p ldl5s l p )
+ [4s2pld/2slpl
-, [4s3p2d/3s2pl
-, [5s3pld/2slpI
No. of spin configs. Method Energy1a.u. D,la.u.
240 1 CI -75.6422 - 1 SCF -75.4208 - 7 VB -75.3633 0.064 7 VBSCF -75.4207 0.121
-lo3 MCSCF -75.5033 0.152
-lo3 CI -75.4852 0.148 Expt. -75.78 0.17
R,la.u. Re$ 1.834‘ a
1.795 2.20 1.87 1.86 C
1.88 1.834
d
“ C . F. Bender and E. R. Davidson, Phys. Rev., 1969, 183, 23; bP. E. Cade and W. M. Huo, J. Chem. Phys., 1967, 47,614; S . P. Walch, T. H. Dunning, R. C. Raffenetti, and F. W. Bobrowicz, J. Chem. Phys., 1980, 72, 406; d R . E. Howard, A. D. McLean, and W. A. Lester, J. Chem. Phys., 1979, 71, 2412;
experimental value assumed
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 23
are also given in Table 2, as are some recent results reported by two other groups of workers whose main interests lie more in the global aspects of the 0 + H2 energy hypersurface. In each case, however, both the basis sets and the numbers of configurations used are more extensive than those selected by van Lenthe and Baht-Kurti in their pilot calculations, and this explains the discrepancy in the quality of the results. Interestingly, the extension of the so called Generalized Valence Bond method used by Walch, Dunning, Raffenetti, and Bobrowicz2’ yields a model lying somewhere between the two models discussed above. As in the Gerratt-Raimondi model, attention is focused more on the molecular situation, but additional orthogonal- ity constraints are included of the kind used by van Lenthe and Baht-Kurti.
In concluding this Section on some aspects of orbital modelling within the valence-bond framework, it should have become clear that a recurring theme in all the approaches relates to the construction of spin eigenfunctions. The methods used in the examples given above are rather laboured,- and, clearly, in dealing with systems containing larger numbers of electrons, it is essential to use more powerful techniques. Fortunately, the recent book by Pauncz28 on the efficient construction of spin eigenfunctions is a very timely addition to the literature, and provides a valuable compendium (and critique) of the various spin coupling schemes; of particular interest is his discussion of the construction and transformation properties of the preferred forms of spin wave functions to be used in the calculation of the matrix elements of H in a systematic manner.
Recent Developments in Molecular Orbital Based CI Theory.-As indicated in the Introduction, the literature continues to be dominated by calculations within the molecular orbital CI formalism. Some of the reasons for this pertain to the special attributes of the molecular orbital model itself. The increased pace of recent applications, however, results directly from the development of very sophisticated techniques that now enable CI calculations to be performed using -104-105 configurational wave functions in the CI expansion. Clearly, for a system containing a large number of basis functions, the limited number of occupied orbitals, and the much greater number of unoccupied orbitals, makes it very difficult to consider the contribution of all configurations in the CI expansion of the total wave function. Even performing a CISD calculation is not good enough, in general, because of the attendant size-consistency problem already discussed at some length. In addition, configurations have to be selected very carefully in order to span the appropriate sub-space of the separated species, and the more configurations there are the more difficult this becomes. Quite frequently, the problem of configuration choice is dealt with by using an energy criterion that is usually based on a perturbation calculation of the contribution of a given wave function; if this contribution lies below some prescribed limit, then the wave function under consideration is precluded from the expansion. However, this procedure is bound to lead to difficulties in constructing both smooth wave functions and energy hypersurfaces; configurations that are important for one set of geometrical parameters may be precluded in other regions of the energy hypersurface, where the geometry of the nuclear framework is different.
The other difficulties posed by very large CI wave functions are conceptual in
*’ S. P. Walch, T. H. Dunning, R. C. Raffenetti, and F. W. Bobrowicz,J. Chem. Phys., 1980, 72,406. R. Pauncz, ‘Spin Eigenfunctions’, Plenum Press, New York, 1979.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

24 G. Doggett
nature; apart from calculating observed properties, some aspects of which are discussed in the following Sections, there remains the problem of using the wave function to provide physical and chemical insight into the system under study. It is extremely unlikely, for example, that even these very extensive multi-configuration wave functions can be used to provide meaningful answers to some of the questions posed towards the end of the Introduction. However, it is still useful to test the basic model to its limits, or at least as far as it is practicable to go.
Now the basic ingredient in a CI calculation is exemplified by the method for evaluating the matrix elements of A:
The constants dg, eg,= are determined by the coefficients in the LCAO expansion of the #k, and by the configurations and space-spin coupling schemes defining Yi, Yj . Thus, sub-sets of the same master set of one- and two-electron integrals are summed with different weights in the evaluation of every matrix element - clearly an inefficient procedure, because the list of integrals requires to be read more than once (there are too many integrals, in general, to store them all in core). A first break-through was made some years ago by Roos, using a so called integral-list driven direct CI method applied to a CISD calculation (for a recent review of methodology see Roos and Siegbahn29); here each integral is taken in turn, and its contributions to the various elements of H are evaluated. Thus, in this method, multiple passes through the list of integrals are avoided, and the matrix elements are built up gradually as the integral list is read.
Ferguson and Handy30 have recently extended the Roos method to allow for the contribution of some triply and quadruply excited configurations - in fact those configurations deemed important for overcoming the size-consistency problem. However, they show that the choice of the forms for the unoccupied orbitals is crucial in the case of calculations on the He, system with an hternuclear separation of 10 a.u. The use of unoccupied orbitals, obtained from the SCF calculation, does not lead to the correct dissociation products, as some multiply excited configurations are missing: these are the ones required to produce two He atoms described by CISD wave functions. Despite this difficulty with the SCF orbitals, Ferguson and Handy are able to show that a choice of (localized) natural orbitals (these orbitals, &, are such that the one-electron density function [equation (9)l is reduced to the form &zkk&*(r)&(r),
p(r) = N I Y*(X,, X2, - - . XN) Y(X1 , X2,. a . XN) dT1.. . dTNdS1 (9)
29 B. 0. Roos and P. E. M. Siegbahn, in 'Modern Theoretical Chemistry', ed. H. F. Schaefer, Plenum Press,
30 W. 1. Ferguson and N. C. Handy, Chem. Phys. Lett., 1980,71,95. New York, Vol. 3, 1977.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 25
where the n k k are occupation numbers) leads to a situation where triply excited configurations make no contribution, and there are sufficient quadruply excited configurations in the 'molecule' to span the CISD space of the separate He atoms. These conclusions follow from an examination of the various energies in Table 3.
Table 3 Energies obtained from CI calculations on He, for r = 10 a.u.
E ( S CF) /a. u . E ( CZSD)/ a. u . E (CISD Q)/a. u . E ( CZSD T )/ a. u . E (CISD TQ )/a .u . 2He -5.723 219 -5.799 410 He, -5.723 219" -5.798 802" -5.798 887" -5.798 803" -5.798 807" He, -5.723 162' -5.798 803' -5.799 412b -5.798 803' -5.799 412'
a SCF orbitals; * localized natural orbitals
A second, more recent, advance in the theory was initiated by Paldus in his application of the unitary group formalism to the CI problem. A later important, and elegant, contribution was made by Shavitt, who developed a graphical method for enumerating configurations and for calculating the matrix elements between con- figurations (full references and details of this and other work are given in the Proceedings of a recent Daresbury Study Weekend on Electron C~rrelation~l). In this approach, the Hamiltonian operator is written in a, perhaps, unfamiliar form involving excitation operators 8,:
H = Aj j + 4 1 gijk-[Eik -@jI - Jj[ gill i j i jkl
where ,??ii(#l#2 . . . #j . . .#N) = . . . #i . , . #N). Because fi has a different representation from usual, it follows, not unexpectedly, that its matrix elements do not involve the 8, as such. A clue to this is already apparent in the mode of operation of the 8,; no spin variables are involved explicitly.
Now, the N-electron orbital product space is closed under unitary transformations on the set of the n orbitals, #k, and the matrices generated by such transformations correspond to (reducible) representations of U(n). It is possible, however, to find linear combinations of these orbital products that transform like irreducible representations of U(n), the linear combinations of interest being selected by their characteristic permutational properties which ensure that they can be associated with states of given total spin, S (that is, when the Hamiltonian operator is independent of spin variables, it is possible to work in a so called spin-free formalism, where all the permutational requirements involved in the use of antisymmetrized products of space-spin functions can be met by working with the group theoretically determined linear combinations of spatial functions alone). Thus, in the unitary group approach, the evaluation of the matrix elements of the Hamiltonian reduces to the evaluation of the matrix elements of 8, and (giki!?jl - 6jkd!?,l) over the symmetry adapted linear combinations of the N- electron orbital products, a procedure that is simplified by using the commutation properties [ f i i j , f i k [ ] = 6jkgil - 6ip!?jk of the Eij , together with the appropriate Shavitt graph.
31 'Electron Correlation', Proc. Daresbury Study Weekend, 17-18 November, 1979, ed. M. F. Guest and S. Wilson, SciencelResearch Council, 1980.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

26 G. Doggett
Brooks and S ~ h a e f e r ~ ~ have recently reported a practical implementation of the Graphical Unitary Group Approach (GUGA) for performing CI calculations. Their ‘loop-driven’ (LD) method is very efficient in comparison with traditional CI methodology, and both single and multi-reference CI wave functions can be accommodated, as can a limited form of configuration selection. In further work, Brooks and co -worke r~~~ demonstrate how the mechanics of the LDGUGA method facilitate the calculation of the two-electron density function from a CI or MCSCF wave function. The role played by the two-electron density function, p,, in the energy expression is easily seen if the Hamiltonian is first written in pair form:
The expected energy is then given by33
E = J Y*(x,, x, . . . xJ RY(x1,x2,. . . X N ) dz
Clearly, this latter expression is valid for any choice of wave function, but Brooks and co-workers show how the form of the CI or MCSCF wave function is especially suited to the calculation of p2, within the framework of the LDGUGA formalism. As an illustration of the power of their technique, a multi-reference root function (containing the SCF reference function; ten singly excited and one hundred and fifteen doubly excited configurations) was used to investigate part of the energy hypersurface for the C3Hz system.
Almost contemporaneously with the above work by Brooks and others, Siegbahn34 has reported, in a series of papers, how the GUGA method can be implemented for performing CISD- and CIS-type calculations. At the CIS level, a test calculation on CH,O,, which is thought to be an intermediate in the ozonolysis of ethene, shows that the lA1 ground state is very nearly degenerate with the 3B, state (for the particular choice of basis set).
Another contribution to this general area has been made by D u c ~ , ~ ~ who used the permutational symmetry properties of the wave function to construct an algorithm for performing a direct CI calculation using general multi-reference expansions. His approach has the advantages of both the loop-driven UGA method of Brooks and S ~ h a e f e r ~ ~ and the (integral-driven) scheme implemented by Siegbahn,34 but no applications of the method are currently available.
Since this Section is concerned more with CI technology, rather than with
32 B. R. Brooks and H. F. Schaefer, J . Chem. Phys., 1979, 70, 5092; B. R. Brooks, W. D. Laidig, P. Saxe,
3 3 R. McWeeny and B. T. Sutcliffe, ‘Methods of Molecular Quantum Mechanics’, Academic Press,
34 P. E. M. Siegbahn, J. Chem. Phvs., 1979, 70,5391; 1980, 72, 1647. 35 W. Duch, Theor. Chim Acta, 1980, 57,299.
and H. F. Schaefer, ibid., 1980, 72, 3837.
London, 1969, Chapter 4.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 21
application, it is pertinent to single out recent contributions concerned with the improvement of energy minimization procedures, particularly with reference to the MCSCF model. In this latter model, it is important to develop quadratically convergent energy minimization procedures, in order to reduce the number of iterations, and hence the number of integral transformations (each time the orbitals, #i, are updated in the iteration scheme, the basic set of integrals also has to be updated in readiness for the variation calculation of the configuration weights).
Roothaan, Detrich, and Hopper,36 Werner and M e ~ e r , ~ ' Polezzo and Fan t~cc i ,~* and also Yeager and Jprrgen~en~~ (and Albertsen) have all proposed improved MCSCF methods that are based on second-order iterative procedures. The latter authors have applied their technique to various states of Be, O,, and C,; where an excited state with the same symmetry as the ground state is of interest, they found it necessary to use a constrained variational procedure that allowed some measure of control over changes in the orbital and configuration spaces. Jmgensen, Albertsen, and Yeager4' have also suggested a useful method for reducing the number of (time-consuming) two-electron integral transformations that occur in a second-order MCSCF procedure. Basically, the full orbital transformation is replaced by an approximate transformation in selectively chosen cycles of the iteration procedure.
The two further basic problems remain: the development of efficient matrix element evaluation routines for non-GUGA calculations, and further development in the use of analytical gradient techniques for locating stationary points on the energy hyper- surface.
As far as the elucidation of the first problem is concerned, Tavan and Schulten4' have reported an efficient algorithm for evaluating general CI matrix elements in calculations that use multi-reference states, but no applications of their technique are currently available. Wormer and P a l d u ~ ~ ~ have also continued with their programme of CI matrix element evaluation within the framework of the unitary and symmetric group approaches. An advance, in a somewhat different direction, is reported by Handy43 who, noting that the majority of two-electron integrals and configurational wave function weights are less than 0.02 in magnitude (a.u. for integrals), suggests that it is practicable to store these quantities as integers in the central memory of the computer. As a result, very large numbers of input/output transactions are eliminated in the evaluation of the matrix element expressions. The success of this novel approach is apparent in Handy's extensive calculations on H,O; 52 210 spin coupled singly and doubly excited configurational wave functions are generated from a multi-reference root function with thirty-seven configurations, the constituent molecular orbitals being expanded in terms of 4 1 basis functions. The calculation yielded an energy of -76.293 65 a.u., compared with the Hartree-Fock value of -76.058 86 a.u. By comparison, a straight CISD calculation, based on the use of a single reference
36 C. C. J . Roothaan. J. Detrich. and D. G. Hopper, Znt. J . Quantum Chem., Symp.. 1979. 13,93. " H.-J. Werner and W. Meyer, J. Chem. Ph-vs.. 1980. 73. 2342. 38 S. Polezzo and P. Fantucci, Mol. Phvs.. 1980, 39, 1527. 39 D. L. Yeager and P. Jsrgensen, Mol. Phvs., 1980, 39, 587: D. L. Yeager, P. Albertsen, and P. Jnrgensen.
40 P. Jmgensen, P. Albertsen, and D. L. Yeager. J. Chem. Phvs., 1980, 72,6466. 4 ' P. Tavan and K. Schu1ten.J. Chem. Phvs., 1980,72,3547. 4 2 P. E. S. Wormer and J. Paldus, Znt. J. Quantum Chem.. 1980. 18,841. " N. C. Handy, Chem. Phvs. Lett., 1980, 74,280.
J. Chem. Phys., 1980,73,28 11.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

28 G. Doggett
function, yielded an energy of -76.289 92 a.u. Although the MRCISD calculation does not yield significantly better results than the CISD calculation for the molecular situation, Handy anticipates that the former method will provide a more useful description of the bond-dissociation process.
The second problem is an important one to resolve simply because calculations at just one point on the energy hypersurface can be very time-consuming (especially if extensive CI is used). It is essential, therefore, to develop efficient methods for locating maxima, minima, and saddle points on the energy hypersurface; a procedure that is accomplished most readily through the use of analytical expressions for the gradient of the energy expression with respect to the nuclear co-ordinates. The main contributors in this area are cited by Krishnan, Schlegel, and P ~ p l e , ~ ~ in their extension of Pulay’s analysis to the CID wave function; application of their methodology to the CISD wave function is straightforward in principle, and is promised to form the subject of a future paper. However, almost simultaneously with the analysis reported by the above authors, Handy4’ and Brooks and c o - ~ o r k e r s ~ ~ demonstrated how analytical gradients could be obtained from the two-electron density function derived from a C I wave function generated from a single reference function (as discussed above, the calculation of the two-electron density function itself is facilitated by working in the LDGUGA formalism). As an illustration of the power of the latter method, Brooks and co-workers calculated the geometry of the transition state for the HNC + HCN isomerization using, for comparison purposes, both the SCF and the CISD model wave functions (the latter calculation involved the use of 12 497 configurations). The calculated geometrical parameters are found to be R,,/a.u. = 2.2572 (2.2179), R,,/a.u. = 2.2063 (2.1824), and RNH/a.u. = 2.6961 (2.7800), where the SCF results are given in parentheses. The barrier height for the isomerization reaction is calculated to be 152 kJ mol-’ (167 kJ mol-’). Somewhat surprisingly, there is not very much difference between the two sets of values for the transition state parameters in this particular example; a result that is known not to persist in general.
At this stage it is convenient to turn to a discussion of CI methodology within the framework of perturbation theory.
Moeller-Plesset Perturbation Theory.-As noted in the previous sub-section, developments of molecular orbital based theories, within the CI framework, have recently undergone a dramatic change in form. This change followed the realization that the unitary group approach provides a systematic and efficient method for evaluating matrix elements of the Hamiltonian. However, all the CI approaches discussed so far are variational in nature.
Perturbation methods provide a strategy for solving Schrodinger’s equation which, in principle, is computationally more advantageous than the variational CI approach. In the Moeller-Plesset form of perturbation theory, for example, the Hamiltonian is first written in the form
A=Ho+ P
44 R. Krishnan, H. B. Schlegel, and J . A. Pople, J . Chem. Phys., 1980, 72,4654. 45 N. C . Handy, in ref. 28. 46 B. R. Brooks, W. D. Laidig, P. Saxe, J. D. Goddard, Y. Yamaguchi, and H. F. Schaefer,J. Chem. Phys.,
1980,72,4652.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 29
where fi0 = c ih^(i), = 1 i < j { l/rij - .I_ #j( 1) l/rij( 1 - pli) $41) dt, 1, pli is the operator that permutes the co-ordinates of electrons 1 and i, h(i) is the usual effective one-electron Hamiltonian operator for electron i, and the antisymmetrized N-fold products of its eigenfunctions, #j, are eigenfunctions of Ho, and P represents the difference between the instantaneous and average potential energies of interaction between pairs of electrons.
In the absence of the perturbation, p, the ground state of the N-electron system is represented by Y , = . 6 # 1 # 2 # 3 . . . #,, which is an eigenfunction of f i 0 with eigenvalue e, + e2 + . . . + E, = E l , say. In the presence of p, the new ground state is described in terms of a linear combination of Yl and the wave functions of higher energy states, in which one, two, three, . . . electrons are promoted from occupied to unoccupied orbitals, the particular weights of which are determined by the equations of perturbation theory. These equations show that the energy of the perturbed ground state may be developed in the form
E = E(O) + E(’ ) + E(’) + . . . , where E(‘) is the perturbation correction of the rth order [this means that E(‘) has a contribution from P in the form of an r-fold product of matrix elements of P, divided by an (r - 1)-fold product of energy factors].
In essence, therefore, the perturbation theoretic approach is concerned with obtaining tractable approximate solutions of the secular determinant for the CI problem, in the basis of the N-electron configurational wave functions, Yj. The main problem arising in this approach stems from the original partitioning of the total Hamiltonian, in order to find a suitable zeroth-order operator, Ho; the assumption being that the resulting perturbation series for the energy converges in a satisfactory manner (not an easy matter to prove). On the other hand, for a given order of perturbation, far fewer matrix elements need to be evaluated in comparison with the variational CI model; in second-order perturbation theory, for example, only matrix elements linking the ground and doubly excited states are required (singly excited states are not involved, as the matrix elements linking such states to Yl are zero if they satisfy the appropriate Hartree-Fock equation, fiqdi = ~ 4 ~ ) .
is given by Yl* PY, dz, and it follows from the defined quantities above that E(O) + E(’) is just the Hartree-Fock energy, EHF, associated with the state Yl. The second-order contribution to the energy has the form given in equation (10) where Y j is a doubly excited configurational wave function
The first order correction to the energy,
- c IS Yl* PYj dZ12/IEj - Ell j # 1
(relative to Y,), and Ej is its energy. On substituting for the individual wave functions in equation (lo), the energy up to second order, EMp2, becomes
occ unocc = E H , - 1 2 Ig’(0, ab)I2/[&a + Eb - &i - &j1
i j ab
where g’(u, ab) = I #:( 1) #;(2). 1/r12- (1 - Pl2) #a( 1) #b(2 ) dt, dz,, and the sum- mations allow for contributions from excited states in which electrons are excited from
also involves only doubly excited configurations. However, as $i, #j to $a, ~ b -
The calculation of
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

30 G. Doggett
recently summarized by Krishnan, Frisch and Pople,,' the evaluation of I?(,) involves the consideration of singly, triply, and quadruply excited configurations in a full treatment, a situation that causes computational problems, primarily because of the large number of multiply excited states that can be constructed (but this depends on the number of unoccupied orbitals available). Krishnan and co-workers use a fairly simple basis set, and report estimates of the relative importance of triply and quadruply excited states on the estimated value of E(,) for some atomic and simple molecular species. The contribution of triply excited states, I?$4), is larger than that of the singly excited states, for all the systems studied. In addition for the ten electron hydrides CH,, NH,, OH,, and HF, as well as for N,, Ei4) is larger than that of individual contributions from singly, doubly, or quadruply excited states. Furthermore, for these species (with the exception of HF) the sum of the magnitudes of contributions from singly, doubly, and quadruply excited states i s less than the magnitude of the contribution from triply excited states. Thus in the case of N,, for example, the accumulative contribution of the various components of the energy to fourth order is as follows (in a.u.):
E H F = -108.943 95 EMP2 = -109.248 19
I?(,) = -0.304 24
E(,) = +0.002 86
= -0.02 1 15
E M p 3 = -109.245 33
EM,, = -109.266 48
These results show clearly that convergence has not been achieved at fourth order. In addition, the total energy is not as good as that obtained by Wilson and Silver48 in their use of the [2/1] Pade approximant to the third-order energy; here, of course, these authors used expansion functions for the #i that differed significantly from those of Krishnan and co-workers.
Guest and Wilson49 have also used a different form of fourth-order perturbation theory in their investigation of the relative importance of triply and quadruply excited states, the [2/ l l Pade approximant being used to estimate the contribution of doubly excited states. As expected, their results parallel those of Krishnan and co-workers, in that the fourth-order contributions by triply and quadruply excited states are negative and positive, respectively, and, for multiply bonded systems - such as CO - the magnitude of the contribution from triples is significantly greater than is that of quadruples. Furthermore, by examining the relative contributions of these multiply excited states as a function of bond length, it becomes apparent that the fourth-order contribution from triply excited states is more sensitive to changes in bond length than is that of quadruples, Eg). Thus, for N, at 2.0741 a.u., the respective contributions are as follows (in a.u.):
I?(') + E( ' ) = -108.976 84 E ( 2 ) + E(3) = -0.3 17 94
EF) = -0.017 60
I?:) = +0.006 28
47 R. Krishnan, M. J. Frisch, and J. A. Pople, J. Chem. Phvs., 1980, 72,4244. 48 S. Wilson and D. M. Silver, J. Chem. Phys., 1980, 72,2159. 49. M. F. Guest and S . Wilson, Chem. Phys. Lett., 1980, 72,49.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 31
while for a bond length of 2.4520 a.u., triples and quadruples are found to contribute -0.034 35 and +0.008 35 to the energy, respectively.
Urban and c o - w ~ r k e r s ~ ~ have also examined the contribution of quadruply excited states to the fourth-order perturbation energy for the ten-electron systems Ne, FH, OH,, and NH,; they find that E$‘) increases in magnitude from Ne to OH,, and then decreases from OH, to NH,.
4 Large Molecules and the Molecular Structure Problem Revisited
Core-Valence Separation.-Although all variants of the CI model discussed above are used with increasing success on small aggregates of nuclei and electrons, the future is still uncertain as far as heavier species or larger aggregates are concerned. In order to make such calculations, even at the SCF level, it becomes increasingly necessary to consider methods for separating core from valence electrons. Formally, this is quite an involved problem as the optimum partition of the N electrons into groups may not always be obvious; but one practical necessity is the imposition of core-valence orthogonality, for otherwise too many contributions to matrix elements arise, and the calculations rapidly become intractable.
The recent paper by Nicolas and DurandS1 contains a discussion of the problems arising in the construction of an effective Hamiltonian for the valence electrons; they also provide detailed references to earlier work in this field. Further analysis of the core-valence separability problem is given by Dixon and Robertson5, in their recent review; these authors are concerned more with a semi-empirical model, in which the effective potential experienced by a valence electron is approximated by a (pseudo)- potential that compensates for the formal exclusion of the core electrons in determining the valence-electron wave functions. A related model is provided by the so called X a method, in which the potential experienced by a given electron is approximated by a suitably chosen local potential, usually taken to be spherically symmetrical within atomic spheres of given radii (parameters of the model), and also outside a larger circumscribing sphere, which touches all the atomic spheres. The potential in the inter-sphere region is usually taken to be constant.
The power of the Xcr method lies in its ease of application to large, chemically interesting, systems, where it is generally agreed that the model provides reasonably reliable estimates for ionization energies. For example, Scheire and c o - ~ o r k e r s ~ ~ have recently used the Xa method for calculating ionization energies and electron distributions for the XeF, systems ( n = 2, 4, or 6). The ambiguities in the values of their calculated molecular properties arise, in part, from the difficulty in deciding whether the atomic spherical regions should be overlapping or non-overlapping.
The Molecular Structure Problem Revisited.-In another application of the X a method to LiH, Cook and KarplusS4 analyse the possibility - originally suggested by Bader, Beddall, and Peslak - that the problem of choosing atomic sphere radii might be
” M. Urban, I. Hubai, V. Kello, and J. Noga, J. Chem. Phvs., 1980, 72, 3378.
5 2 R. N. Dixon and I. L. Robertson in ‘Theoretical Chemistry’, ed. R. N. Dixon and C. Thompson
’’ L. Scheire, P. Phariseau, R. Nuyts, A. E. Foti, and V. H. Smith, Phvsica, 1980. 1OlA. 22. 54 M. Cook and M. Karplus, J. Chem. Phvs., 1980, 72, 7.
C. Nicolas and Ph. Durand,J. Chem. Phys., 1980,72,453.
(Specialist Periodical Reports), The Chemical Society, London, 1978. Vol. 3, p. 100.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

3 2 G . Doggett
related to the more general problem of partitioning space into regions where the ratio of average kinetic energy to potential energy is constant (that is, regions where the virial theorem is satisfied locally). Unfortunately, the results of Cook and Karplus are not conclusive in this matter; nevertheless, it remains an interesting thought that further insight into the X a method might be gained by searching for surfaces with slightly different characteristics than those used hitherto.
Now the more general (zero-flux) surfaces alluded to above, introduced by Bader and his are defined by the condition d(r) - V p ( r ) = 0, for all r contained in the set of points defining the surface; p(r) is the one-electron density function defined in equation (9), which is more fundamentals6 than the wave function itself - primarily because it is amenable to experimental measurements7 - and 6 is the unit vector normal to the surface in question at the point r.
The work of Bader is associated with the investigation of the topological properties of both p(r) and the associated Vp(r) function. The characteristics of the critical points of p(r) [points where V p ( r ) = 01 are of especial interest, and can be used to develop the concept of a molecular graph, which in turn leads naturally to the notion of molecular structure. However, the Bader view of molecular structure is different from the usual geometrical one, in which a point, R, in nuclear configuration space defines the structure in a geometrical sense; in this new approach, the concept of molecular structure is associated with a region of nuclear configuration space surrounding the point R, say. Thus, for two points R,R’ in this region, there is a change in geometry in proceeding from R to R’ , but there is not necessarily a concomitant change in molecular structure. This is basically because the Bader concept of molecular structure is topological in nature, and is therefore not a point property of the system. However, at special ‘catastrophe’ points in nuclear configuration space, discontinuous changes of molecular structure do occur.
It is clear that this generalized view of molecular structure, as formulated by Bader and co-workers, is in conflict with the conclusion of Woolley as set out in the Introduction. dut, as Bader and co-workers3* note, the so called Generator Co-ordinate Method (GCM) also leads to a situation in which the wave function Y(r.R) is no longer associated with a specific molecular geometry, but rather with a molecular structure in the Bader sense, that is, in defining Y(r,R), a region of nuclear configuration space containing R is sampled.
It is evident from the discussion given above that the calculation of p(r), and its topological properties, form an important branch of the subject. However, for the purposes of the present review, this is where the development must be left for the time being, primarily because it is currently possible to calculate a reliable p(r) only from the wave function Y for the system [see equation (9)l. However, Y is itself constructed from orbitals $i which are, in turn, expressed as linear combinations of basis functions, xi. Since the problem underlying all electronic structure calculations is concerned with the choice of xj (for a particular choice of orbital model), attention is now focused on this important branch of the subject.
” R. F. W. Bader. J . Chern. Phys., 1980, 73, 2871; R. F. W. Bader, Y. Tal, S. G. Anderson. and T. T.
’‘ R. G. h r . R. A. DonnellY, M. Levy, and W. E. Palke,J. Chern. Phys., 1978, 68. 3801. Nguyen-Dang, Isr . J . Chern., 1980, 19,8.
Proceedings SAGAMORE V Conference, Phys. Scr., 1977, 15,65.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 33
5 Choice of Basis Functions
N-Electron Expansion Functions.-Up until this point, more emphasis has been placed on the N-electron functions, Yi , which form a basis for describing the multi- configuration expansion of the wave function, than on their constituent #i; in fact, it was not necessary to say very much about the detailed form of the basis functions xi used in expanding the orbitals #i because, in earlier Sections, the discussion was concerned more with method than with calculation.
There are two main aspects of working with a multi-structure or multi-configuration wave function of the form of equation (5). First, the N-electron functions span a sub-space, of a given global symmetry, associated with the Hamiltonian operator, H . This is easily seen because, for a finite number of Y i - p say - the function of BYi does not in general lie in the span of the Yi , that is, flyi cannot be written in the form c1Yl + c2Y, + . . . + cpYp, where c,,c,, . . . cp E a. It is sufficient, however, in calculating expectation values of fi to consider that part of H Y i lying in the span of the {Yi 1. This follows because if Q{(i =p + 1, p + 2, . . .) form the additional N-electron functions required to make the basis complete, then
P a2
HY,= 1 CiYi+ 1 C i @ i i= 1 i = p + 1
Thus, since the Qi are chosen to be orthogonal to the Yi, it follows that IYj* AYldr = x+’= ci I Yj* Y i dr. Hence, H can be diagonalized in the p-dimensional sub-space spanned by the Yi, even though this basis of N-electron functions is incomplete.
The models discussed in the previous Section, which are all rooted in the orbital concept, present different approaches to the problem of finding a suitable set of Yi to span a convenient sub-space of the complete N-electron configuration space. In the single configuration molecular orbital model, for example, p = 1, while in the M configuration CI variant of the molecular orbital model, the states Qi ( i = 2,3 . . . M ) , which are used to augment the single root function Yl, are constructed from the #i
associated with Yl. Furthermore, because the #i are written in terms of a linear combination of xj, it is the number of the xj that determines the number of Qi that can be constructed in this manner; that is, the set of N-electron wave functions is still incomplete. Traditionally, though, the unoccupied orbitals (often called the external orbitals) associated with the SCF calculation on the root function (either Yl or some limited linear combination of Y i for the multi-configuration SCF model) are used to construct the Qi, but, as already seen in the work of Gerratt and Raimondi, some sets of unoccupied orbitals are more useful than others.
Secondly, if M expansion functions, xj, are taken, then M possible orbitals, #j , may be constructed from this orbital basis set; the selection and choice of the Qi can, therefore, be a bewildering matter. It may be better, in fact, to work with fewer - M’ say - and compromise; Yl will then be a poorer single or multi-configuration root wave function, but the ( M - M ’ ) orbitals could then be used to augment the (smaller) set of unoccupied #j , in order to construct a set of Qi. By choosing M large enough, it is also possible to investigate the completeness of the orbital basis set. Such approaches are not common in the literature, but it is interesting that Ladik and Ciiek58 have recently
58 J . Ladik and J . Ciiek, J. Chem. Phvs., 1980, 73. 2357.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

34 G. Doggett
proposed a scheme, similar in principle to the one described above, in which the ( M - M ‘ ) basis functions are centred at one point in the molecule, and these orbitals are used to generate the excited configurations that are required in calculating improved molecular energies that include correlation contributions. Eggarter and Eggarter” have also presented a careful analysis of the completeness of atomic orbital basis sets for atoms; they augmented a standard set of atomic basis functions with a set of functions of the form
For large enough values of nfnax, the results are independent of the choice of A, and hence completeness is achieved. Furthermore, their calculation of second-order perturbation energies, within the Moeller-Plesset formalism, yields from between 80% (for Be) to 99.6% (for F) of the expected correlation energy (the difference between the Hartree-Fock result and the adjusted experimental result). The close proximity in energy of the ls2 2s2, ‘ S and the 1s’ 2p2, ‘ S terms, particularly for Be, results in a slow convergence of the perturbation expansion. In fact in this case higher order perturbation corrections ought to be considered.
The problem of using incomplete basis sets has also been examined by McDowell,60 insofar as Hartree-Fock theory is concerned. He used a diagrammatic perturbation theoretic procedure, which was later reformulated within a differential equation format in order to avoid the computation of awkward integrals. More recently, McDowell has extended his work on the Hartree-Fock model to investigate the consequences of basis set incompleteness in the calculation of perturbative corrections to ground-state energies.
Conventional Basis Functions.-The general situation as far as molecules are concerned is more involved and hazardous. In principle, a complete set of basis functions could be centred on any point in space, but the practicalities of performing calculations dictate that incomplete basis sets (containing a finite number of functions) must be used. The functional form of the basis functions themselves is a matter of choice, and the most complete recent review of the general situation seems to be that given by Burden and Wilson6’ in 1972.
It is usually considered prudent to take sets of xj centred on the various nuclei in the molecule, although quite often the basis set is augmented with bond-centred functions that may or may not be ‘floating’ Slater-type or Gaussian-type (STO or GTO, respectively), which remain the most popular analytical forms for the xi. However, for point properties (electron density on the nucleus), it may be necessary to make modifications to the inner regions of GTOs in order to produce cusped functions (the electron-nuclear cusp property follows from a theoretical requirement on the behaviour of the total wave function when the co-ordinates of an electron coincide with those of a nucleus, and, in order that the total wave function can have the desired property, it is necessary that both the constituent orbitals #i and the xj should have appropriate cusped forms). Steine? has suggested a suitable form for cusped GTOs that can be
J9 E. Eggarter and T. P. Eggarter, J. Ph.vs. B , 1978, 11, 1157; ibid., p. 2069; ibid.. p. 2969. 6o K. McDowell, J. Chem. Phvs., 1979. 70, 3148; Int. J. Quantum Chem., Svmp., 1979, 13, 51: In[. J .
61 F. R. Burden and R. M. Wilson, Adv. Phvs., 1972, 21, 825. b 2 E. Steiner. J. Chem. SOC., Faradav Trans. 2, 1980, 76, 391.
Quantum Chem., 1980, 17,895.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 35
used as basis functions for first row atoms (STOs are cusped in their primitive form, but the cusp condition on the total wave function is unlikely to be satisfied when a small-sized basis set is used).
The different spatial characteristics of STOs and GTOs also has implications as far as the lengths of LCAO expansions of the #i are concerned. Experience suggests that, in general, more primitive GTOs are required than STOs in order to yield molecular basis sets of comparable quality. However, more basis functions means more integrals to evaluate, and, in order to reduce the number of integrals generated over GTOs (such integrals are much easier to evaluate than are their STO counterparts), it has become widespread practice to seek suitable contractions (fixed linear combinations) that have been energy optimized for the separate atoms.
A further difficulty arises in practice, however, because the size of the basis set that can be comfortably accommodated is determined basically by the number of nuclei present in the molecule. In general, for small numbers of nuclei it is possible to use quite extensive basis sets, but, even here, there is room for compromise, because it is often useful to require that a given basis set - for carbon, say - can be carried from molecule to molecule. Thus, for comparison purposes, it is desirable to work with the same basis set for carbon in CH, C2H2, C2H4, C2HZF2, etc., even though it may well be possible to obtain better results for CH using a basis set for carbon that could not be accommodated in the same calculation on C2H2F2. The definition of ‘better’ raises more awkward issues, as the overall objectives of a given calculation should always be apparent; that is, is insight and understanding in a global sense more or less important than the numerical reproduction of a property that can be measured experimentally? In general, it is far too restrictive, therefore, to contemplate widespread use of very extensive basis sets for molecular calculations. This is the motivation lying behind the recent work of Huzinaga and c o - ~ o r k e r s ~ ~ (references to earlier work contained therein), who use an extension of a standard least-squares fitting procedure to find optimum contractions of three GTOs fitted to an STO. The method is different from the one suggested some years ago by Pople and co-workers, in that ns, rather than Is, GTOs are used in expanding the ns STO; there are also other differences of detail. In the present method, the optimum exponents and combining coefficients are determined by an iterative energy minimization procedure. Although the basis set is fairly crude in nature, the authors’ obiectives are to produce acceptable valence atomic orbitals that can be used for reasonably reliable calculations on molecular systems; the results of their calculations on a number of simple species show that geometries and total and orbital energies are closer to the Hartree-Fock results than those obtained using the STO-3G orbitals of Pople and co-workers. The reasons for this poorer behaviour of the latter orbitals are now understood. The least-squares fitting procedure, based as it is on an energy minimization procedure, places too much emphasis on the form of the contracted orbitals in the core region, at the expense of the outer valence region of the atom. This deficiency of the STO-3G orbitals has also been recognized by Binkley, Pople, and Hehre,64 in their design of somewhat different new small GTO basis sets. These basis sets, which are of the split-shell type, are designed for molecular
63 H. Tatewaki and S. Huzinaga, J . Chem. Phys., 1980, 72, 399; A. Tavouktsoglou and S. Huzinaga, J .
64 J. S . Binkley, J. A. Pople, and W. J. Hehre. J. Am. Chem. SOC., 1980, 102, 939. Chem. Phys., 1980, 72, 1385.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

36 G. Doggett
calculations on species containing first-row atoms. First of all a flexible basis set of six GTOs is used to represent the 1s core orbital, then two s-type and twop-type GTOs and another more diffuse s-type and p-type GTO are used to fit the valence-shell orbitals. The exponents are optimized for this 6-21G basis, and the linear coefficients are determined in the usual way. Next, the valence orbitals are left unchanged while the core orbital is fitted using three s-type GTOs. The resulting 3-2 1G basis is expected to yield valence orbitals with acceptable spatial characteristics. Test calculations, using both the 6-21G and the 3-21G basis sets, give rise to geometries, vibrational frequencies, and dipole moments that are consistently in good accord, and yet considerably better overall than the STO-3G basis.
Other small basis sets are provided by floating spherical GTOs (FSGTOs), and the necessary flexibility is obtained by optimizing both the orbital exponent ( k ) and the position of the origin of the GTO. Thus, transferability is a problem, in general, because the bond Gaussian will be located at different points according to the nature of the nearest neighbouring atoms. However, the simplicity of this rather old model (see reference 61) when used within the framework of molecular orbital theory is appealing, and this accounts for its continued use. H ~ b e r ~ ~ has used this model in calcdations on NH,, H,CO, CH,F, etc., in which the core functions are tied to the nuclear positions, and obtained acceptable geometries and dipole moments for a range of ten polyatomic molecules. Pakiari and Linnett66 also reported results for diatomic species, using both open- and closed-shell molecular orbital models (but with the former model there may be difficulties in obtaining dissociation products in appropriate L,S eigenstates). Finally, the FSGTO model has been used by Armstrong, Jamieson, and perk in^,^' in exploratory calculations on the electronic band structure of polymers, and by Spangler and Christoffersen68 in an ongoing investigation of the feasibility of using the molecular fragment concept in building up the electronic structure of large molecules at the minimum basis set level. The former authors are optimistic that the use of the Christoffersen variant of the FSGTO model will, in the near future, enable ab initio calculations to be performed on large polymeric systems.
McLean and Chandler69 have reported several more extensively contracted basis sets for atoms with atomic numbers lying between 11 and 18; detailed references to earlier work (including reviews) are also given, and a useful general discussion is provided of the problems inherent in the choice of basis set. They investigated different ways of contracting twelve s-type and either eight or nine p-type GTOs. The uncontracted basis sets themselves are conventionally designated as (1 2,8) and (1 2,9), respectively. For the former basis set, three contractions were considered: [4,21, [5,31, and [6,41, where the notation [n,m] means that the twelve s-type GTOs are replaced by n linear combinations in which the primitive functions are partitioned over the n new functions in either a disjoint manner (the more usual situation) or with one or more double occurrences. McLean and Chandler’s main objective is to find suitable, yet simple, basis sets of increasingly good quality, in order to investigate the variations in calculated molecular properties with choice of basis set. Basis sets for C1-, P-, and S-
H. Huber, Theor. Chim. Acta, 1980. 55 , 117. 66 A. H. Pakiari and J. W. Linnett, Int. J . Quantum Chem.. 1980, 18,661. ‘’ D. R. Armstrong, J. Jamieson, and P. G. Perkins, Theor. Chim. Acta. 1980, 57.43.
6 9 A. D. McLean and G. S. Chandler,J. Chem. Ph-vs., 1980, 72,5639. D. Spangler and R. E. Christoffersen, Int. J . Quantum Chem.. 1980, 17, 1075.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 37
are also given, as such functions are thought to be more useful for describing the electronic structure of certain molecules containing chlorine, phosphorus, or sulphur. The quality of the various contractions is best seen by comparing the calculated total energies (the uncontracted basis functions must yield the lowest energy, as any constraint on the coefficients, as required in forming contracted orbitals, incurs a loss in energy). Results for neutral phosphorus are given in Table 4. For comparison, a basis of two Is, two 2s, and two 2p STOs yields an energy of -340.715 60 a.u., whereas the Hartree-Fock energy is -340.7 18 69 a.u.
Table 4 Energies for P(4S) using the McLean and Chandler contractions of (1 2,8) and (1 2.9) GTO basis sets
Uncontracted Contracted Disjoint basis basis partitioning ( E + ( 12,8) -
[4,21 Yes [5,31 No [6,41 No
[6,41 No [6,51 No
- (1 239)
340.7079)/a.u. 0.0 0.03624 0.02672 0.00093
-0.0042 5 -0.0028 2 -0.00330
Now these basis sets of McLean and Chandler, which are representative of many other basis sets in current use, possess a certain degree of flexibility for describing the distortion of the atomic orbitals in the molecular environment. Thus, for example, the polarization effects induced to a situation where the (contracted) ‘ls’, ‘2s’, ‘2p’, ‘3s’. and ‘3p’ orbitals are mixed, the basic assumption being that the contracted orbitals are unchanged from their free atom forms (this is the rationale underlying the use of contracted orbitals). However, the primitive GTO exponents and the contraction coefficients, which have been optimized for the free atoms, may no longer be suitable for the molecule (clearly, this problem relates to the use of incomplete basis sets). Computationally, it is very tiresome to reoptimize the non-linear parameters in order to find the molecularly-optimized basis functions; in any event, if the basis set is extensive enough (even in contracted form) this should not be necessary.
Poirier, Daudel, Mezey, and Csizmadia’” have recently explored the possibility of circumventing the difficulties alluded to above in the case of carbon in a molecular environment. They use reasonably compact (uncontracted) basis sets that have inbuilt flexibility for describing the changing chemical environments in which the carbon atom is found. In detail, a simple basis of three GTOs per symmetry type per shell is taken with exponents, a:, obtained from a calculation on the free carbon atom. Similar calculations are performed on the ground state of C+ and C - , in order to produce a set of exponents a,+,ai- appropriate to basis sets of so called uniform quality, a concept associated with the choice oi exponents (see the references cited in Poirier et al. for further details).
All exponents in the molecular environment are now assumed to have a quadratic dependence on a parameter Q; thus
aj(Q) = a j Q2 + bi Q + a:,
’O R. A. Poirier, R. Daudel, P. G. Mezey. and I. G. Csizmadia. Int . J . Quantum Chem., 1980. 18. 715.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

38 G. Doggett
where q(0) = a:. The coefficients ai and bi are determined by requiring that q( I) = ai+ and q(-l) = c q ; Q is then the only variation parameter in the energy minimization procedure. For systems containing one carbon atom, the energy hypersurface, E ( Q , R), is then a function of both Q and the geometrical parameters, R. Furthermore, if R is fixed, minimization of E with respect to Q is particularly easy to achieve.
The parameter Q does not correlate with the formal atomic charge (except for atoms and ions), but its value is strongly influenced by the overall charge on the species, and also on the environment of the carbon atom. For example, in CH,, CH,, CH,-, CH3+, and CH,, the values of Q are 1.9,0.7, -1.0, 2.0 and 0.0, respectively.
In considering C, systems, it is necessary to allow for a different value of Q for each carbon atom (if chemically inequivalent); thus, the minimum in the E ( Q l , Qz ,R) hypersurface is sought. For species containing inequivalent carbon atoms, the two basis sets are found to display an interdependence (as expected).
Non-conventional Basis Functions.-Two other interesting areas in the development of basis sets are of particular note. First, in the case of diatomic species, there is increased interest in the use of bicentric orbitals. These orbitals are obtained from the solutions of the Schrodinger equation for the motion of the electron in the field of two fixed nuclei with selectively chosen charges. The use of these basis sets, which vary with internuclear separation in a natural way, has been investigated by Staryk and K a p y ~ h e v , ~ ~ in their discussion of the He, problem, and also by Vukajlovii: and c o - ~ o r k e r s ~ ~ in non-adiabatic calculations on H,. The latter authors experienced convergence difficulties with the total energy on account of the incompleteness of the basis set used in their calculations. However, the calculated difference in energy between the two lowest vibrational states is in good agreement with the experimentally observed value.
Secondly, Silver and Wilson73 have extended and developed the use of even- tempered atomic orbitals in their search for a universal (that is, atom independent) basis set. Conceptually, this work is very attractive indeed as the use of such a basis means that it becomes possible to compare electronic structures for different (isoelectronic) molecules in a systematic manner: for example, the charge density function [equation (9)l may be written in the form of equation ( l l ) , where the coefficients drs are
determined by the particular choice of model. Normally, the xj for different atoms (and even sometimes the same atom in different molecules) have different functional forms: but, here, with a common set of xi, it becomes profitable to compare the same elements of the d matrix in different molecules. For the single configuration molecular orbital model, it would appear likely that the Mulliken atom and bond populations (computed from the elements of d and the overlap integrals between pairs of basis functions), for example, would provide a sound basis for comparing changes in the electron distributions of isoelectronic species.
V. V. Starykh and V. V. Kapyshev, J. Chem. Phys., 1980,72,2713.
therein). l2 0. A. Mogilevsky, L. I. Ponomerev, and F. R. VukajloviC, J. Phys. B , 1980, 13, 2489 (and references
l 3 D. M. Silver and S. Wilson, J. Chem. Phys., 1978, 69,3787.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 39
The even-tempered orbitals themselves are usually taken to be in either Gaussian or Slater form; ns and np orbitals are expanded in terms of common sets of 1s and 2p primitive functions, respectively. The key feature is that the exponents of the primitive basis orbitals, for a given 1 value, are taken as members of a geometrical progression:
where r = 1, 2, . . . M,, a > 0, p > 1, and M I is the number of primitives of a given 1 value. There are thus only two parameters (a, and P I ) to determine for each atomic orbital, #i, apart from the usual linear coefficients of the respective basis functions, xj. The main advantages in using basis functions in even-tempered form stem from the fact that they span the orbital space uniformly, and also it is easy to generate fairly large basis sets that are free of linear dependence problems. Earlier work by Raffenetti and others (for details see Silver and Wilson73) is concerned with the determination of optimum values of a and p for each orbital type by means of an energy minimization procedure. However, Silver and Wilson suggest that universal basis sets of even-tempered form can be generated by taking arbitrary, but sensible, values for a;, PI , and M,; for example, by taking cq, = 0.5, Po = 1.5, Mo = 9 for ns orbitals, and a1 = 1.0, PI = 1.75, M , = 6 for np orbitals, they found that the results for species as diverse as He, Be, and Ne were very close to those determined by optimizing the values of aand p for each atom separately. The two sets of total energies are compared in Table 5 with those of Clementi and Roetti.
Table 5 Energies for some closed-shell atoms (in a.u.)
Universala Raffenettib Clementi- Atom basis (STO) (STO) Roetti (DZ-ST0)c
He -2.861 68 -2.861 68 (3,O) -2.861 67 Be -14.573 02 -14.573 02 (8,8) -14.572 37 Ne -128.547 01 -128.547 10 (8,6) -128.535 1 1
M , = 9, M , = 6; Tables, 1974, 14, 177.
M,, M , values in parentheses; E. Clementi and C. Roetti, At. Data Nucl. Data
The overall goodness of the universal basis set can be judged by the fact that if the Raffenetti basis set for Ne is used to calculate the energy for Be, then a value of -14.166 02 a.u. is obtained, thus showing the limitations of a basis set tailored to a specific atom. On the other hand, the use of the universal basis set shows consistent good agreement with the results of Raffenetti, irrespective of the choice of atom.
In some earlier work, Silver and N i e u ~ p o o r t ~ ~ used Po = 1.55 and P, = 1.6 and found that the total energies for first-row atoms were very close to those obtained by Clementi and Roetti in all cases; in addition, the energies lie lower than those obtained by Raffenetti from an even-tempered set of GTO functions with Mo = 10 and MI = 6.
As already indicated, the attractiveness of universal even-tempered basis sets stems from their generality, simplicity, and ease of application to the molecular situation. Thus for a fixed geometry it is possible to use one set of integrals to calculate molecular energies for a family of related species. This aspect of the generality of such basis sets has been taken up recently by Wilson and Silver,75 who augmented the basis set of
74 D. M. Silver and W. C. Nieuwpoort, Chem. Phys. Lett., 1978, 57,421. 75 S. Wilson and D. M. Silver, J. Chem. Phys., 1980, 72,2159.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

40 G. Doggett
Silver and Nieuwpoort with a set of three 3d functions with 4 = 1.5, p2 = 1.65, and performed calculations on BF, CO, and N,. Over the range of internuclear separations studied (1.75-2.50 a.u.), the energies are slightly higher than those obtained from conventional SCF calculations using slightly fewer STO basis functions (but, of course, in these latter calculations the number of independent exponents is greater). The results for the universal sp and spd basis sets are given in Table 6, along with some roughly comparable SCF energies.
Table 6 Calculated SCF energies for N,, CO, and BF using universal and conventional basis sets of STOs"
Molecule Un iversa 1 ( sp) Un iv ersa I (spd ) Conventional -108.9055 - 108.992 1 -108.9956 (200,671)
- 1 12.7846 (220, 1271) -124.0514 -124.1077 -124.1090 (220, 1271)
N, - 1 12.7828 co - 1 12.7031
BF
' Energies are in a.u. and the internuclear separation is 2.0 a.u. in all cases. References to the con- ventional SCF calculations are given in S. Wilson and D. M. Silver, J. Chem. Ph-vs., 1980. 72, 2159.
There now follow sub-sections concerned with two important current developments in the selection of basis sets: first, the problem of designing basis sets for post Hartree-Fock calculations (in terms of relative energies) is briefly re-examined and secondly, some calculational aspects of working with incomplete basis sets are looked at anew.
Basis Sets for Correlated Wave Functions.-Nearly all the basis sets discussed so far, with the exception of the even-tempered variety, are designed primarily for use in SCF calculations on molecular species. Calculations concerned with reducing the energy difference between the SCF and the observed energies (adjusted for relativistic corrections, etc.) have also tended to incorporate the same kinds of basis sets that are used for the simpler model calculations.
In the case of BH, for example, the sixteen o orbital basis SCF (single configuration) calculation of Cade and Huo yields an energy of -2.131 37 a.u. at R = 2.336 a.u., an energy that is very close to the basis set limit for the model (the Hartree-Fock energy). However, with a modest basis of free-atom STOs - two per shell per symmetry-type for boron (a double zeta basis) and a free-atom 1s orbital for hydrogen - an energy of -25.134 121 a.u. is achieved from a single configuration spin coupled wave f~nct ion . '~ Extending the basis further to fourteen a-type STOs, and performing limited exponent optimization, yields an energy of -25.169 34 a.u. (experimental value -25.273 a . ~ . ) , ~ ~ a result that recovers 25% of the energy difference between the Hartree-Fock and experimental results, but with a smaller basis than that required by Cade and Huo. Furthermore, the inclusion of a second configuration of the form 0,' oZ2 z', with associated spin couplings, yields 50% of the correlation energy.
From this simple example it is clear that the estimation of the correlation energy depends in an intricate way upon both the choice of basis set and the model. The energy limit for o orbitals is common to both the Hartree-Fock and the spin-coupled models, as the N-electron configuration space is spanned by the same set of antisymmetrized products of basis spin orbitals: { , d x a x b . . . x , , @ k } , where 6, is an
'' G. Doggett and R. J . Russell (unpublished work)
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 41
N-fold product of a and p spin functions, and xa,xb. . . E {xi : i = 1,2, . . . m 1. The choice of model is therefore crucial. As seen earlier, the requirement of double occupancy in the simple molecular orbital model leads to a loss in energy that can be regained only by considerable extra effort. This basic difference between the molecular orbital and valence-bond-based models arises simply because valence-bond models have some correlation effects built into the wave function from the start, and this could be the main reason for the much more rapidly convergent configuration interaction (CI) expansions based on non-orthogonal #i than on their molecular orbital counterparts.
Within the framework of molecular orbital theory there is a growing tendency to look for basis sets that are tailored more to the partial treatment of the correlation problem. Thus, while it is often considered desirable to optimize exponents of basis functions at the Hartree-Fock level, it is unlikely that the values obtained will be appropriate for post-Hartree-Fock calculations (although the need for exponent optimization becomes less important as the size of the basis set increases). Evidence in support of this view is apparent in the slow convergence properties of the conventional CI calculations.
Several different kinds of basis set, which are thought to be more appropriate for recovering part of the correlation energy, have been investigated recently. First, Wilson and Silver75 report the use of universal even-tempered basis sets within the framework of Moeller-Plesset perturbation theory. Their total energies for CO, BF, and N, are obtained from the [2/11 Pade approximant to the third-order perturbation energy, and show significant improvements on the values obtained from the SCF model (cf: Table 6). The respective total energies, at their corresponding equilibrium internuclear separations, are -1 13.2122, -124.5128, and -109.4368 a.u., values which yield 83, 77, and 84%, respectively, of the correlation energy.
Wilson” has also used universal sets of even-tempered GTO primitive functions, of differing extents, within the framework of Moeller-Plesset perturbation theory, to calculate E[2/11 energies (as well as other estimates of the energy including correlation effects) for a series of atomic species containing the ls2 2s2 electron configuration. He took values of M , from 6 in steps of 2 to 20, in order to investigate the completeness property of the basis set. With the exception of Li-, convergence in the energy to four decimal places occurs for M , = 14 or 16. However, for Li-, convergence is not achieved, even for M , = 20; this is indicative of the difficulty encountered in trying to span the orbital space for systems with diffuse orbitals.
In a different direction, but also within the framework of second-order Moeller- Plesset theory, Krishnan, Binkley, Seeger, and P ~ p l e ~ ~ have produced basis sets of contracted GTO split-shell orbitals, in which the exponents are chosen to yield the lowest atomic ground-state energy. As the authors acknowledge, this method is not above criticism, as it is strictly incorrect from the variational point of view (the energy expression does not correspond to an expectation value of the Hamiltonian). However, their rationale in performing the calculations lies in the observation that EM,, tends to a limiting value as the size of the basis set increases; it therefore appears meaningful to think in terms of obtaining a ‘best’ basis set for the calculation of EM,,. Moreover, by determining the orbitals from an expression which includes part of the correlation energy, it is anticipated that ‘better quality’ Oj may emerge for using in CI calculations.
7 7 S. Wilson, Theor. Chim. Acta, 1980. 57, 53; ibid. (in the press). 78 R. Krishnan, J. S. Binkley, R. Seeger, and J. A. Pople,J. Chem. Phvs., 1980, 72,650.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

42 G. Doggett
Dimculties Arising in the Use of Incomplete Basis Sets.-The use of limited (incomplete) sets, of both basis functions and N-electron configurational wave functions, results in further problems of a more subtle nature. These problems usually manifest themselves when calculating energy differences, especially if they are small: for example, when comparing the energies of two conformers, when attempting to calculate the interaction energies of (neutral) closed shell species, or even when calculating dissociation energies.
Now, on the basis of experience, it has been found that careful optimization of the basis set parameters in the molecular environment - a procedure that is of greater significance for small basis sets - yields significantly improved molecular energies. However, this obscures, and may even exacerbate, the problem alluded to above concerning the calculation of energy differences. It is fairly easy to see that in the ‘molecule’ AB there are more basis functions (and hence more Y i ) available to describe the electronic motion, than there are in the separated A and B moieties; this is the origin of the difficulty. In detail, this means that because atomic functions centred on A can be expanded in terms of linear combinations of functions centred on B (and vice versa), the intra-atomic contribution to the correlation energy (within a chosen model) will vary with the separation between A and B. In particular, there are excited configurations involving orbitals of B that arise in the ‘molecular’ situation, but not in the calculation on B alone. For example, in a valence-bond (localized orbital) calculation on Be,, using a basis of Is, 2s, and 2p atomic orbitals on each centre, the separated Be atoms are described optimally by a two configuration wave function built out of ls2 2s’ and ls2 2p2 configurations. But, in the molecule, structures of the kind ,d.2sA 5,[2pA 2pB(@ - /3a)I arise, which involve charge transfer with simultaneous excitation, and give an additional contribution to the intra-atomic correlation energy; for, on expanding 2pA in terms of functions centred on B, it follows that:
i
where JB is a function centred on B, and cj varies with the A-B separation. Thus, structures of the kind just described give rise to additional improvement of the intra-atomic correlation in the ‘molecule’. Since the correlation energies are large in themselves, any attempt to calculate small energy differences will be vitiated by the uncontrolled changes in the intra-atomic contribution to the correlation energy. This problem gives rise to what is usually known as the superposition error, and is an artefact of the incompleteness of the basis set. Clearly, for a complete basis set, the span of { AB } is included in the space spanned by the basis functions, and so no new structures would be generated implicitly (unless the N-electron space was truncated too severely).
As noted originally by Boys and Bernadi,79 the counterpoise (function) method can be used to minimize the error arising from the superposition of the two basis sets in the molecular calculation. In their method the interaction energy is calculated, as usual, by subtracting the energies of A and B from that of AB; but, in evaluating the energy of A, for example, the basis set is now augmented to include the functions centred on B, the position of which is determined by the A-B separation. Thus, as the calculated energies
79 S. F. Boys and F. Bernadi, Mol. Phys., 1970, 19, 553.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 43
are both obtained using a common basis set, the energies of A and B will vary with internuclear separation.
Apart from the superposition error, in which the AB system is described, in effect, by a superior basis set (as far as intra-atomic correlation effects are concerned), the incompleteness also means that basis functions may well be missing that are required for describing intermolecular interactions; these two effects act in opposition, as the former leads to lower energies and the latter to higher energies than the optimum. Clearly, the superposition error can be reduced by working with more extensive basis sets, but even then errors can arise from other sources. Apart from the difficulties just discussed, there is the ongoing problem associated with the lack of size-consistency in the choice N-electron configurational basis functions for AB and A . . .B.
The use of basis orbitals, with exponents optimized for the chosen molecular environment, is often thought to be advantageous insofar as the description of intermolecular interactions is concerned. However, although the AB binding energy is improved by this technique, the use of different orbital basis sets for A,B and A-B again leads to a kind of superposition error, even though the orbitals may improve the description of the interaction between A and B. For example, suppose that a 2s STO possesses exponents k and k + 6 in A and AB, respectively. The orbital in the molecule can be represented in terms of a linear combination of s-type orbitals, with exponent k , by noticing that
2s(k + s) = Nre-(k+S)r = Nre-kr [ I - r + (6r),/2! + . . .1 = 2 4 k ) + C, 3s(k) + C, 4 4 k ) . . .
This latter expression is a contraction of a more extensive set of orbitals on A than is assumed in the free A calculation.
Incomplete basis sets for calculating E(AB) and E(A),E(B) therefore unavoidably give rise to a superposition error, unless the latter is carefully controlled.
McLean and ChandleP9 have recently reiterated the consequences of the super- position (and other) errors: first, calculated dissociation energies for bound molecules will be too large (large energy differences involved) and secondly, the lack of control of the superposition error, in particular, may completely vitiate the calculation of van der Waals energies of interaction or other small energy differences. Insofar as the calculation of dissociation energies is concerned, Bauschlicher" has investigated the size of the superposition error in the calculation of D, for C10 and N,. He used a limited form of CI, and also included bond-centred polarization functions in his basis set. It transpires that the use of such functions leads to an unacceptably large superposition error and, in the case of C10 and N,, it is fortuitous that this error is cancelled by the other errors arising from the use of a finite basis set and limited CI. Since there is no reason to expect cancellation of these errors in general, Bauschlicher cautions against the use of bond-centred functions for describing polarization effects: it is much wiser to centre such functions on one of the nuclei.
Two examples of the latter situation are considered. The first example is taken from the work of Liu and McLean" who report the results of some careful molecular orbital based calculations on the Be, system. The standard Hartree-Fock results
C . W. Bauschlicher, Chem. Ph.vs. Lett.. 1980. 74. 271. " B. Liu and A. D. McLean, J . Chem. Phys., 1980. 72, 34 18
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

44 G. Doggett
predict a repulsive potential energy curve, while earlier CI and more recent perturbation theoretic investigations82 yield discordant results for the position of the minimum (weak) in the potential energy curve. Liu and McLean find a binding energy of about 9.65 kJ mol-I, together with a minimum energy internuclear separation of 4.71 a.u. They obtain these results from a carefully controlled CI calculation, in which the selected configurations are constructed out of localized molecular orbitals, $i, the selection being necessary in order to avoid an unbalanced treatment of the intra-atomic correlation energy (the basis set used consisted of 7s, 5p, 4d, and 2f STOs, and is big enough basis to avoid the complicating features of the superposition error). The second example relates to SCF calculations by Marynick'j on the barriers to inversion in NX, and PX, systems. He finds problems with the two fluoro derivatives; in particular, planar NF, is not bound relative to NF, and F. Marynick concludes that improved calculations need to be performed within the CI framework, in order to resolve the existing uncertainties; however, even at the SCF level, the superposition error ought to be eliminated, as otherwise the uncontrolled change in the intra-atomic correlation energy error, when the geometry changes from the pyramidal to the planar configuration, may play a significant role in influencing the calculated barrier height.
In contradistinction to the problems experienced in using molecular orbital based models to determine the energies of weakly interacting systems, valence-bond models show more stability in their application. In particular, the superposition error is easily avoided by simply omitting charge transfer structures A-B+, in which there is simultaneous excitation of B (and vice versa). This procedural advantage of valence-bond models has been stressed recently by Gerratt and Papadopo~los,'~ in their calculations on the He, system (their paper contains references to earlier work). On the basis of their calculations, these authors conclude that the essential physics of intermolecular interactions is revealed most directly through a very simple valence- bond-based model. Furthermore, they support this view by demonstrating that the He . . . He interaction energy, V, can be represented very accurately by the expression:
5
V = V , + 2 C2nlR2n n = 3
where the repulsive contribution, V,, is obtained from the ground-state wave function, in which only permutations corresponding to zero and single interchanges are used. The coefficients C,, are the usual multipole-multipole coefficients, and R is the intersystem separation.
Another investigation - invalidating earlier work because of the lack of con- sideration of the superposition error - is provided by Meyer, Hariharan, and K~tze ln igg ,~~ in their careful and detailed analysis of the He . . . H, system in the region of the van der Waals minimum (collinear arrangement preferred; cf: the preference for the type 1 interaction of B~rdett '~). They found that the minimum in the previously reported SCF potential energy surface was an artefact resulting from the lack of proper consideration of the basis set superposition error. The counterpoise corrections used in the current work, at both the SCF and CI levels, induces significant changes in the
M. A. Robb and S. Wilson, Mol. Phys., 1980,40, 1333. 83 D. S. Marynick, J. Chem. Phys., 1980, 73, 3939. 84 J. Gerratt and M. Papadopoulos, Mol. Phys., 1980,41, 1071.
W. Meyer, P. C. Hariharan, and W. Kutzelnigg, J. Chem. Phys., 1980, 73, 1880.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 45
earlier results. Intersystem correlation effects are also allowed for by the inclusion of selected triply excited configurations. Despite the apparent success in producing an accurate energy hypersurface, the authors remain apprehensive over the generalization of the method to more complex systems. Clearly, the main difficulties arise from the intricate corrections that have to be made in order to overcome the weaknesses inherent in the chosen orbital model. It remains to be seen whether the alternative approach formulated by Gerratt and Papadopoulos is any more successful with this four-electron system.
A more formal development in the analysis of the wave function used for describing intermolecular interactions has been made by Kutzelnigg.86 He is particularly concerned with the interrelation of the basis sets for the ‘supermolecule’ and fragments, and their respective completeness properties.
In contrast to the developments just discussed, some progress has been made by Sokalski, Hariharan, Kaufman, and Petrongolo8’ in the choice of basis sets at the SCF level for describing intermolecular interactions. Their results for H 2 0 . . . H 2 0 (Table 7) show that it is possible to use small, but prudently chosen, basis sets to obtain interaction energies at the SCF level. The minimal uniform quality basis set (see the
Table 7 Intermolecular interaction energies for (H,O),
Counterpoise Basis Uncorrected E (SCF)/a.u. corrected E (SCF)/a.u. DZ -0.009 778 -0.009 680
Uniform minimal -0.012 875 -0.009 531 STO-3G -0.008 284 -0.002 725
references cited by Sokalski et al. for the definition of uniform quality), which is well balanced and is capable of giving a good representation of the molecular charge distribution, in contrast to the STO-3G basis, which, as noted earlier, cannot provide a really acceptable description of the valence-electron distribution. The STO-3G basis sets therefore seem to be unsuitable (large systems assumed) because, by their very nature, they are unable to yield an adequate representation of the electrostatic contribution to the interaction energy.
Instability Problems.-In the above discussion of basis sets, it is implicitly assumed that only neutral molecules are of interest. However, the choice of basis sets may be crucial in the case of negative ion species for, as originally noted by Ahlrichsg8 (at the SCF level), the occurrence of positive orbital energies leads to an unstable HF wave function.
This instability feature was recently re-examined by England,89 within the context of the SCF and MCSCF models. He finds that, in the MCSCF model, instability occurs if the occupation number of an orbital with positive orbital energy is close to unity. An actual example of the consequences of this instability is available from the work of Cookg0 on 0-. He finds that, although the use of a DZ basis leads to convergence at
86 W. Kutzelnigg, J . Chem. Phys., 1980, 73,343. 87 W. A. Sokalski, P. C. Hariharan, J. J. Kaufman, and C. Petrongolo, Int. J . Quantum Chem., 1980, 18.
88 R. Ahlrichs, Chem. Phys. Lett., 1975, 34, 570. 89 W. B. England,f. Chem. Phys., 1980, 72, 2108. 90 D. B. Cook, J. Chem. SOC., Chem. Commun., 1980,623.
165.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

46 G. Doggett
the SCF level, the system is not stable relative to 0 + e-. Furthermore, Cook shows that this solution is an artefact of the choice of basis set because, if the basis set is augmented with suitably chosen diffuse functions, then the energy of the 1 ~ ~ 2 s * 2 p ~ 3 s ' ( ~ P ) state is lower than that of the ls22s2sp4(3P) (ground) state. He suggests that this failure of the model arises because the electron is trying to occupy an orbital that removes it from the proximity of the oxygen nucleus, that is, the system is trying to lose an electron. Cook notes that other authors have observed this phenomenon for the H- species. He suggests that these anomalies in the HF model can be removed only by allowing each electron to have a separately optimized orbital, independent of the orbitals of the other electrons: but, of course, this is just the fundamental tenet of the spin-coupled approach discussed in an earlier Section. Normally, however, the problem highlighted by Cook is not so apparent for neutral species, but the relaxation of the constraints inherent in the H F model are beneficial, even for these species.
In concluding this Section, which is concerned with an exploration of the branches of the subject close to the trunk, it just remains to reiterate that great care is necessary in calculating experimental observables when using ab initio methods. If, through a poor choice of basis set, the energy hypersurface is inaccurate, then molecular properties associated with critical points on the hypersurface will also be in error. At this stage, therefore. model methodology is put on one side and the remaining Sections are devoted to a discussion of calculations on selected families of molecular species. Given the space available, the species are selected because of their intriniscally interesting electronic structures, and no attempt is made to be exhaustive in this brief survey of orbital model applications.
6 Results for Some Diatomic Molecules
Twelve-electron Species.-The calculation of the nature of the ground states of diatomic isoelectronic species such as LiF, BeO, CN+, and C, poses some very delicate problems, especially for the last two species.
There are eight valence electrons to be accommodated and, in the simplest molecular orbital model, the choice lies between the . . . 17r4 and 17r3 50' configurations, which give rise to 'Et and 311 states, respectively. For CNf these states are nearly degenerate (the energy difference for C, is also small), and it is not an easy matter to demonstrate which one has the lower energy. However, Bruna, Peyerimhoff, and Buenker" have finally resolved this uncertainty in the case of CN+ (the most difficult system), and it now appears that all the species listed above possess a '%+ ground state. This situation does not obtain for diatomic systems containing one or two second-row atoms, since the preferred occupancy of the outer (T orbital leads to competition between 3C-(772 a') and ' I l (n3a ' ) for forming the ground state.91 For example, in SiN+ and Si, a 3C- ground state is predicted, but in CSi and CP+, the 'll state is preferred.
CN'. Detailed calculations within the MCSCF formalism have been presented by Bruna and ~o-workers,~' in which they used several different sets of basis functions. Their results suggest that earlier calculations, which tended to favour a 311 ground
9 1 p. J , Bruna. s. D. Peyerimhoff, and R. J. Buenker. J. Chem. Phvs., 1980, 72, 5437: Chem. Phys. Lett.. 1980. 72. 278.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 47
state, suffered from the effects of inadequate basis sets and poorly convergent CI expansions.
Bruna and co-workers first used a GTO basis of (9s5pld) functions, contracted to a set of [ 4s2p Id1 functions for each atom. Both single reference and multi-reference CISD calculations, the latter with ten reference functions, were then performed with configuration selection based on an energy-threshold criterion. The results in both cases show that the 311 state is the lower of the two possible states, irrespective of the extent of CI; however, in the MR-CISD calculation, the separation is considerably less than that obtained in the corresponding SCF calculations. The best energies of -91.9823 a.u. ('F) and -91.9854 a.u. (311) actually quoted by Bruna and co-workers are obtained from an extrapolation procedure applied to calculations using a number of different configuration selection thresholds; that is, the energies are quoted for an energy threshold of zero a.u.
These authors then showed that the use of a slightly different GTO basis set of (9s5pldl f ) orbitals contracted to [5s3pldlfl, for each atom, led to virtually unchanged SCF energies, yet the energy-based configuration-selected MR-CISD calculations (T = 2.5 x a.u. in the Buenker and Peyerimhoff notation), with 14 reference functions for the 'zc+ and 10 reference functions for the 311 state wave functions, yield energies of -92.0018 a.u. ('C+, R = 2.268 a.u.) and -91.9991 (311, R = 2.362 a.u.), respectively. The 'C+ state is now the ground state, and the 311 state lies 0.0027 a.u. higher in energy. On the other hand, the basic MCSCF calculations themselves yield energies of -91.8169 a.u. (311) and -91.7996 a.u. ('C+), which indicates a 311 ground state. It turns out that the augmented basis set, which includes bond-centred functions, gives rise to a 'C+ ground state when the single reference CISD calculations are extended to include at least five reference configurations in the 'C+ calculation. Further changes in the magnitude of the 1C+-311 separation occurs as the number of reference functions is increased to 14 and 10 in the development of the 'C+ and 311 wave functions, respectively, but the ground state remains non-degenerate. These calculations illustrate very clearly that, apart from the choice of a suitable basis set, it is difficult to ensure that comparable and balanced treatments of correlation effects are maintained as the number of CI reference functions increased.
C,. This species, which is isoelectronic with CN+, has also presented difficulties in the elucidation of its ground state by theoretical methods. It is generally accepted now that the lowest energy state is 'Zgt rather than 311u. On the basis of this assumption, Dupuis and Liu9, have explored the possibility of using the SCF, MCSCF, and CISD models to determine the level of computational effort required in calculating an accurate value for the electron affinity, by subtracting the total energies of the molecule and ion. However, without further analysis, it is difficult to ascertain the magnitude of the error induced by this process, primarily because it is difficult to achieve a balanced treatment of correlation effects in both species. For this reason, the basis set for carbon needs to be sufficiently flexible in order to describe both C, and C,- with comparable accuracy.
The particular basis set chosen by Dupuis and Liu consisted of a (6s4p) set of STOs, augmented with a set of (2s2p3dlf) diffuse functions. This basis set yields ground state energies for C, (. . . 20,' 20,' ln;) and C,- (. . . 20,* 30,' lzrr,4) of -75.405 87 a.u. and -75.565 15 a.u., respectively, that is, the ion is stable with respect to the molecule
92 M. Dupuis and B. Liu, J . Chem. Phvs., 1980. 73. 337
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

48 G. Doggett
by 0.159 28 a.u. (at R = 2.35 a.u.). The CISD calculation at the same geometry yields energies for the molecule and ion of -75.716 66 a.u. and -75.854 18 a.u., respectively (energy difference now 0.137 52 a.u.).
Now, in the case of C,, the configuration . . . 20: 3og2 1x2 gives rise to another 'Cgt wave function, which lies close in energy to the ground state - an observation that led Dupuis and Liu to perform MCSCF CI calculations on the two species, by allowing for all single and double excitations from the two reference configurations. The resulting energies are -75.749 6 2 a.u. and -75.887 02 a.u. (difference 0.137 40 a.u.) for an internuclear separation of 2.35 a.u. However, unlike for the CISD calculation, the minimum energies occur at values for the internuclear separation of less than 2.35 a.u. for the molecule, and greater than 2.35 a.u. for the ion. Thus, although the total energies improve in proceeding from the CISD to the MR-CISD calculations, the estimate for the electron affinity remains virtually unchanged. However, the calculated values of the molecular properties Re, o, and w,xe, for both molecule and ion, show more sensitivity to the choice of model. Interestingly, though, the single reference CISD calculations yield the best overall set of spectroscopic constants, but the calculated electron affinity is greater than the experimental value by 0.0077 a.u. Further improvement is possible only if more extensive basis sets are used, together with the inclusion of higher order excitations in the CI expansion of the wave function. As far as basis functions are concerned, however, calculations on the anion may be particularly susceptible to the lack of an adequate number of diffuse functions (cf: the earlier comments by Cookg0).
The previously cited work of Yeager, Albertsen, and J ~ r g e n s e n ~ ~ contains MCSCF calculations on the 'C,+ excited states of C, . These authors used a (4s3pld) STO basis set, and constructed the MCSCF wave function from the four reference configurations . . . 2oU2 1 n,,", . . .20,' 1 n: 1 ni, . . .20,' 30,' 1 nu3 1 n,', and . . .30: 1 n:. The lowest energy lCg+ excited state was calculated to have an energy of -75.450069 a.u. at R = 2.347 a.u.
Ni+. Despite all the calculations on other members of the isoelectronic series, there has been really very little interest in Ni+. Cobb and c o - ~ o r k e r s ~ ~ have made a contribution in this direction by performing exploratory SCF calculations on the ground (3Cg-) and excited ('C,+, *nu, 311,) states of this species.
As is well known, the dominant configurations for C, and Nit are 10; 10: 20: 2aU2 1 n,," and 10: 10: 20; 2oU2 30: lnU2, respectively, a feature that is a consequence of the increasing 2s-2p orbital energy difference that occurs in progressing from left to right across the periodic table. In order to test the quality of their results, Cobb and co-workers also used the same basis sets for calculating the ground-state energies for N, and N,+. Basis set I was constructed from a set of (10s5p36) GTOs contracted to a set of [7s3p3d] functions; basis set 11, on the other hand, consisted of a set of (1 ls7p3d) uncontracted functions. Not surprisingly, basis I1 gave consistently lower energies, a result that follows, presumably, because the contraction coefficients used in basis I were obtained from a calculation on the free nitrogen atom; with this constraint imposed, it is not possible to generate enough flexibility for describing the highly contracted electron distribution in the dipositive ion. The same phenomenon is apparent in the calculations on the excited states, where
93 M. Cobb, T. F. Moran, R. F. Borkman, and R. Childs, J. Chem. Phys., 1980,72,4463.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 49
there are also marked differences in the predicted excitation energies. At R = 2.4 a.u. (close to the minimum internuclear separation for both basis sets), for example, the differences in energy between the 3Cg-, 'C,+ and 'Cg+, 311u states are 0.0875, 0.045 68 a.u. for basis I, and 0.02 1 98,O.O 19 97 a.u. for basis 11, respectively. Since these energy differences are so sensitive to the choice of basis set, it seems likely that the basis needs augmenting with some additional spatially contracted orbitals in order to improve the description of these doubly ionized states. In fact, it is interesting to compare the results for N, and N2+ with those of Ermler and M ~ L e a n . ~ ~ These latter authors used (4s4p3d) and (6s8p9d2f) STO basis sets in SCF and CISD calculations on the ground state of N, and on the lowest ,Cg+, 211,, and ,CU+ states of N,+. The larger of the two Ermler and McLean basis sets leads to a lower energy for N, than is obtained from the Cobb basis 11; for example, at R = 2.0 a.u. the total energies are -108.995 593 a.u. and -108.990 33 1 a.u., respectively. However, for N2+, the Cobb basis I1 is the better one, yielding an energy of -108.41 1 137 a.u. at R = 2.068 a.u., compared with the Ermler and McLean value of -108.407 526 a.u. at the same internuclear separation.
In comparing the results obtained from these two sets of calculations, it becomes clear that it is very difficult to achieve a consistent level of accuracy with a given basis set, especially if positive ions are of interest. Just because one basis set gives excellent results for a given species, it does not imply that the same basis set spans the orbital space of a related species with the same degree of uniformity. Problems such as these arise if the chosen basis set has inbuilt bias. For example, if the contraction coefficients are obtained from calculations on selected atomic species, the addition of diffuse functions (for anions) or spatially contracted functions (for cations) is unlikely to overcome the bias. This is what makes universal basis sets an attractive proposition in principle, but so far, little experience has been gained in their use.
LiF. The calculations by Stevens,95 on the LiF molecule and its ion, illustrate some of the difficulties attending the extension of traditional methods of calculation to anionic species. Like all species considered in this section, LiF possesses a 'C+ ground state.
Stevens used an STO basis of the form (5s2pld/5s3pld), in which the extra p functions on fluorine are spatially diffuse (exponent optimized for the free F- species), in order to perform SCF and MCSCF calculations on selected states of LiF and its anion. Although the SCF wave function is expected to give a reasonable description of the molecular structure in the vicinity of the equilibrium internuclear separation, its incorrect dissociative behaviour causes problems at large internuclear separations. The energies of the ground states of LiF and LiF- at R = 3.0 a.u. are - 106.979 6 1 a.u. and - 106.990 56 a.u., respectively, values that are close to the respective minimum values at R = 2.965 a.u. and R = 3.097 a.u.
From the calculated values of the spectroscopic constants it transpires that there are five vibrational states for the ground state of LiF-, in which the species is stable relative to LiF + e-. As far as excited states are concerned, Stevens finds that the 22C+ and states are stable with respect to electron loss for R > 6 a.u., and there are no real difficulties in calculating potential energy curves. This is not the situation for smaller internuclear distances, however, as the neutral molecule ground state lies lower in energy than both of the excited ion states; in these circumstances, the variational
94 W. C. Ermler and A. D . McLean, J . Chem. Phys., 1980,73,2297. 95 W . J. Stevens,J. Chern. Phys., 1980, 72, 1536.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

50 G. Doggett
calculation should produce LiF + e- as the lowest energy state. However, because of the limited nature of the basis set used, this cannot be achieved (I$ the earlier observations by Cookg0). SCF calculations on the ionic species are therefore difficult to execute, as the resulting states are in the continuum region for the system consisting of a neutral molecule plus an electron. Despite these difficulties, Stevens makes tentative suggestions as to the origin of the observed electron scattering resonances for alkali-metal anions.
As an aside, it is interesting to note that Olson and Liug6 have also made more extensive CI calculations on the NaH and NaH- systems; here again, the anion yields a ground state that is stable relative to NaH + e-. The calculated adiabatic electron affinity is given as 0.0 132 a.u.
BeO. Some of the problems experienced with other species discussed above are also apparent in calculations on BeO. Bauschlicher and Yark~ny ,~? in their MCSCF calculations on the ground iC+ and excited I l l states of this species at an internuclear separation of 2.5 a.u., focus attention on the calculation of excitation energies. The 'c+ states are expanded in terms of contributions from the four configurations . . . 3a24a2 l#, . . . 3a24a'5a' lf', . . . 3a25a2 lf', and . . . 3a24a2 17r327r1, in which the individual molecular orbitals are constructed from a GTO basis of the form [4s2pld/4s3pldl, the exponent of the additional diffuse oxygen p orbital being optim- ized for 0-.
The 1'C+ state is found to contain only 65% of the first configuration in the list above, and another 25% is contributed by the last configuration. The energy of this state is found to be -89.507 16 a.u., compared with the SCF result of -89.422 82 a.u. The 2'C+ excited state is obtained by optimizing the orbitals in the four configuration MCSCF wave function for the second root of the secular problem. The main improvement over the single configuration SCF calculations results from the better estimate for the magnitude of the energy separation between the two lowest 'C+ states: the difference is now 23 836 cm-', instead of 3551 cm-' at the SCF level (experi- mental value in the region of 2 1 200 cm-').
The calculations on the excited 'I1 states are performed using a three configuration MCSCF wave function, in which the reference configurations are . . . 4a2 50' 1 $, . . . 4 a ' ln4 2n1, and . . . 402 5a2 60' In'. Unlike the calculations on the 'C+ states, the inclusion of the additional configurations leads to very little change in the estimated energy of the lowest 'I1 state: SCF value -89.441 66 a.u., MCSCF value -89.441 89 a.u.
Additional calculations on the ground 'C+ and excited 'I1 states, with three extra configurations added to each list of reference functions, yields energies of -89.5 10 a.u. and -89.4646 a.u., respectively. Thus, as noted by Bauschlicher and Y a r k ~ n y , ~ ? electron correlation effects are more active in lowering the energy of the ground state than that of the first excited state of 'I1 symmetry; furthermore, the smaller sets of reference configurations evidently provide good starting functions for MR-CI calculations, as the small loss in energy resulting from their use should be readily recouped in the CI calculations. The situation does not seem to be so crucial as that obtaining in the case of CN+, for, as seen above, it is not possible to predict the correct
96 R. E. Olson and B. Liu, J . Chem. Phvs., 1980. 73, 2817. '' C. W. Bauschlicher and D. R. Yarkony. J . Chem. Phw.. 1980,72. 1138.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 5 1
ground state for this species uniess CI is admitted in an MCSCF calculation with at least five reference functions in the 'C+ set.
Direct Calculation of Electron Affinities.-For several of the species considered in this Section, the calculated adiabatic or vertical electron affinities are given with variable degrees of confidence, a situation that arises because the usual energy differencing procedure can induce errors of unknown magnitude unless the estimated correlation energies, for molecule and ion, are comparable. Part of the problem arises directly from the use of different sets of orbitals for describing the electron distributions in the molecule and ion; consequently, ASCF or AMCSCF calculations cannot give consistently accurate results for valence-shell ionization or electron attachment energies. The inclusion of CI improves matters (as seen above), but it is still difficult to estimate and isolate errors arising from the use of incomplete basis sets.
On account of these difficulties, in ensuring a balanced treatment of electron correlation effects, some alternative approaches are gaining increasing attention. In the Equations of Motion method (EOM), a compromise position is adopted in which a single set of orbitals is taken for the N and N & 1 electron systems: the key feature of the method lies in the introduction of a transition operator, aAN, which, when applied to the N electron ground-state wave function, produces the Ath state of the N -t 1 electron system. The approach utilizes a different representation of the wave function and Hamiltonian operator than usual (cf. the GUGA method) and, in common with the related propagator method, provides a different route for the calculation of energy differences. Herman, Freed, and Yeager98 have used this technique for calculating the ionization energies of BH, HF, and Ne, and the electron affinities of OH and F: on the other hand, Ortiz and 0hrn9' have applied the related propagator method to estimate the electron affinities of C N and C1,. Pickup, Sabo, and Firsht"' have also calculated the first two ionization energies of H,O using a transition functional scheme in which transition energies are calculated directly.
All these approaches for the direct calculation of transition energies are still dependent upon a prudent choice of basis set, but experience seems to suggest that such approaches lead to a more balanced - and hence more controlled - treatment of correlation effects than is obtained in conventional calculations (in their respective limits, for a given choice of basis set, there is no difference between any of the methods; the problems arise when wave function expansions are truncated to yield a tractable calculational procedure).
7 Some Aspects of Surface Calculations
General Problems.-In comparison with diatomic systems, the construction of ab initio energy hypersurfaces for triatomic (ABC) systems poses several new complicating features: first, as discussed by Davidson,"' the topological properties of the hierarchy of (Born-Oppenheimer determined) surfaces of increasing energy provide subtle and awkward problems, especially where surfaces intersect; secondly. the complete ground-state surface, for example, has to be able to accommodate all
98 M. F. Herman. K. F. Freed. and D. L. Yeager.J. Chem. P h p . . 1980. 72.602: ibid.. P. 61 1 . 99 J. V. Ortiz B. and Y. o h m . J . Chem. Phps.. 1980. 72.5744.
lo ' E. R. Davidson, J. Am. Chem. SOC., 1977. 99. 397. B. T. Pickup. D. W. Sabo. and D. Firsht. Theor. Chim. Acta. 1980.56. 165.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

52 G. Doggett
possible reaction processes consistent with the conservation of spin. Thus, the reaction A + BC + AB + C is just one of the possible processes occurring on the ground-state surface, which also connects with other reactant and product channels. In addition, intermediates ABC, corresponding to minima or saddle points may also be found (for a minimum point, the matrix of second derivatives of E ( R ) with respect to nuclear co-ordinates gives rise to only positive eigenvalues, while for a saddle point of the simplest kind, one negative eigenvalue occurs, but the frequency of the corresponding normal mode is now imaginary). The complicating features of multiple-surface systems - as obtained, for example, with the O(3P, 'D) + H2('Zg+) reactions - are excluded from this short overview of some ABC surfaces, where attention is focused more on those parts of the surface that are of interest either in isomerization reactions, or in the elucidation of the properties of an intermediate.
The reason for taking this somewhat restricted view is quite simple; in calculating the cross sections for reactive processes, for example, the whole surface needs to be sampled, and this is where the problems really loom large. Minimum energy paths from reactants to products can be defined - sometimes in a rather special way (see Kato and Morokumalo2) - and used in approximate calculations: but it is difficult to justify working in such one-dimensional sub-spaces of E ( R ) without further comment.
Apart from the difficulty in finding the right shape of the surface in the 'easy' regions, the presence of points or lines (sub-spaces) where surfaces stick together can cause problems in the study of molecular reaction dynamics. For example, it is not always wise to use the Born-Oppenheimer adiabatic surfaces; instead, depending upon experimental considerations, it is sometimes more appropriate to decouple the surfaces to produce diabatic surfaces (these at least possess continuous derivatives with respect to nuclear co-ordinates).
The ab initio calculation of potential energy hypersurfaces is thus seen as a first step in the elucidation of the dynamical properties of reactive scattering processes. However, this first step is a difficult one to take, as the basis set problem is still not satisfactorily resolved for most systems of chemical interest. Even in the case of simple diatomic systems, for example, it is not easy to span the orbital space (and hence the N-electron space) in a uniform fashion, let alone completely, for different electronic states of the same or related systems. Similarly, on a general surface, the basis set must be flexible enough to describe all the species, both intermediate and asymptotic, with the same degree of accuracy. Thus, in the case of the HeH,+ system considered in detail by Hopper,lo3 it is necessary to ensure that the basis set provides an adequate description of the appropriate ground or excited states of He, HeH, HeH+, He+, H2+, and H,. However, for this system, the ground state surface yields a minimum corresponding to the occurrence of a linear H-H-He+ species, and so the basis set must also be extensive enough to provide a good description of this triatomic intermediate (and, hopefully, without a superposition error).
Clearly, there are severe problems to be overcome in view of the intrinsic limitations of most of the orbital models currently in use. Thus, at the present time, CI calculations with 104-105 expansion functions per point on the energy hypersurface present a viable proposition only if applications are restricted to regions in the vicinity of a critical point. For this reason there has been a growing interest in the development of
lo* S . Kato and K. Morokuma,J. Chem. Phys., 1980. 72, 206; ibid., 73, 3900. Io3 D. G. Hopper, Znf. J . Quantum Chem., Symp., 1978, 12, 305;J. Chem. Phvs., 1980, 73.4528.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 5 3
alternative methods for obtaining surfaces by easier calculational means. On the one hand, Murrell and his have perfected a computationally convenient scheme. in which surfaces are given in terms of a simple algebraic expression, the important feature being that the surfaces are calibrated in such a way that they possess the correct asymptotic behaviour for all reactant and product channels.
On the other hand, the use of simpler orbital models, which incorporate well tried approximation schemes, also provide a route for performing tractable calculations over more extensive regions of the energy hypersurface. The semi-empirical Diatomics in Molecules method (DIM) is regarded as one of the more successful models of this kind: see, for example, the recent review by Tully105 on the nature of the model and the scope of its application. Despite its simplicity, however, the problem of eliminating artefacts of the model remains a constant source of potential difficulty. A timely word of warning in this respect emerges from the work of Schinke and LesterIo6 on the O('D) + H2('Cg+) -, OH('II) + H(,S) reaction: they parameterized an ab initio surface (which includes the stable H,O intermediate) in the spirit of Sorbie and Murrell (SM) and also compared their results with those obtained from the semi-empirical SM surface. It was found that small variations in the energy hypersurface led to significant differences in the calculated cross section for the reaction, especially at very low collision energies. The calculated rate constants are also found to be sensitive to small changes in E ( R ) in the far reactant channel: in fact, changes of 4.2 kJ mol-' in the attractive region of one of the surfaces constructed changed the calculated rate constant at 300 K by a factor of two. On the basis of these observations, Schinke and Lester remain apprehensive over the practicality of obtaining rate constants from calculated energy hypersurfaces.
With these comments in mind, this Section is now concluded with discussions of calculations in which only small regions of the energy hypersurfaces are explored for a number of simple systems.
The Li, System.-This system continues to attract interest as it forms a useful model system for examining the effects of bonding in alkali-metal clusters: also the possibility of forming the anion or cation presents interesting situations worthy of further study. As Gole, Childs, Dixon, and Eades'" note in their recent investigation of the Li, system, molecular beam, e.s.r., and mass spectrometric studies on related alkali-metal clusters provide the impetus for a continuing theoretical study of this, the simplest, alkali-metal cluster.
Gole and co-workers confirm, as a result of their SCF and CISD calculations, that the system Li, is stable with respect to Li, + Li, but the dissociation energy for the process Li3+ + Liz+ + Li is three times greater than that for the neutral triatomic species. In addition, the adiabatic electron affinity and ionization energy for Li, are calculated to be 0.0404 a.u. and 0.1452 a.u., respectively.
As far as the Li, species itself is concerned, the energy hypersurface has several complicating features : first, the molecule is non-rigid, secondly, at the D,, nuclear configuration, the Jahn-Teller effect is operative because the valence-electron
'04 K. S. Sorbie and J. N. Murrell. Mol. Phys.. 1975, 29, 1387; S. Carter and J. N. Murrell, ibid.. 1980. 41.
lo5 J. C. Tully. Adv. Chem. Phys.. 1980, 17. 63. '06 R. S. Schinke and W. A. Lester, J . Chem. Phys.. 1980, 72, 3754. lo' J . L. Gole, R. H. Childs, D. A. Dixon. and R. A. Eades, J . Chem. Phys., 1980. 72. 6368.
567 (and references therein).
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online
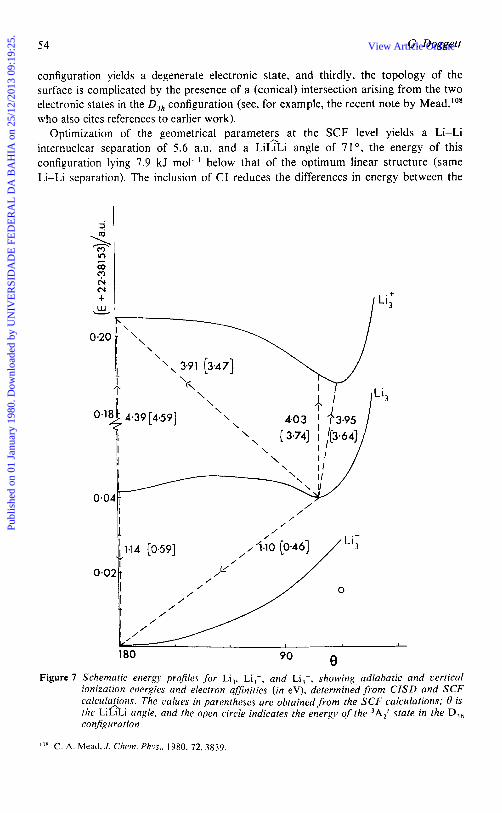
54 G. Doggett
configuration yields a degenerate electronic state, and thirdly, the topology of the surface is complicated by the presence of a (conical) intersection arising from the two electronic states in the D,, configuration (see, for example, the recent note by Mead,lo8 who also cites references to earlier work).
Optimization of the geometrical parameteE at the SCF level yields a Li-Li internuclear separation of 5.6 a.u. and a LiLiLi angle of 71° , the energy of this configuration lying 7.9 kJ mol-' below that of the optimum linear structure (same Li-Li separation). The inclusion of CI reduces the differences in energy between the
(4*59]
'\
4.03 I T3.95 [ 3-74] T ' I ![3*64] I
\ I ; 1 \ . I
\ i '. 0 . 0 4 w
0 0
Figure 7 Schematic energy profiles for Li,, Li,+, and Li3-, showing adiabatic and vertical ionization energies and electron affinities (in ev). determined from CISD and SCF calculalions. The calues in parentheses are obtained from the SCF calculations; 0 is the L i L k angle, and the open circle indicates the energy of the 3A2' state in the D,, co njigu ra t io n
lox C . A. Mead. J . Chern. Phw.. 1980. 72,3839.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 55
two configurations to 3.8 kJ mo1-’, while still favouring the bent structure. However, the behaviour of the energy in the vicinity of the linear confiEration is sensitive to the degree of correlation included in the wave function; as the LiLiLi angle is reduced from 180° to 150°, the SCF energy decreases by 1.3 kJ mol-’, while the CISD energy increases by 7.9 kJ mol-’, thus showing that, at the CI level, the linear structure is also located at a relative minimum on the energy hypersurface.
The calculations on the Li,- species were performed with the Li, basis set augmented with a set of diffuse s and p functions. The SCF calculation yields a lowest energy singlet state corresponding to a collinear arrangement of the nuclei, with a Li-Li internuclear separation of 6.0 a.u., a distance that is reduced to 5.6 a.u. in the CISD calculation. The singlet state energy for the optimum geometry lies lower than the energy of the ,A2’ state at the D,, minimum energy configuration; but, for this latter geometry, the triplet state lies lower than the singlet state - a general feature which is unchanged when correlation effects are included.
In the case of Li,+ there are only two electrons to accommodate in the valence shell, and, not surprisingly, the lowest energy (non-degenerate) state occurs for the D,, nuclear configuration. The difference in energy between the D,, and the D,, structures is 107.5 kJ mol-’ in the SCF calculation, and 67.4 kJ mol-’ in the CISD calculation; the optimum SCF Li-Li separation of 5.78 a.u. is increased by only 0.05 a.u. when CI is admitted.
On the basis of the above discussion, it is clear that the multiple minima on the Li, surface lead to the definition of several adiabatic and vertical electron affinities and ionization energies, as indicated in Figure 7. The SCF values of the electron affinities are approximately 50% of the CI values; for ionization energies, the discrepancy is about 8% at the most. As noted by Gole and co-workers, however, these calculated energy differences are sensitive to the choice of basis set, and comparison with the results obtained by Gerber and Schurna~her , ’~~ who used the CEPA model, suggests that the present basis may be biased towards the linear configuration. Gole and co-workers conclude that further calculations are necessary on all three species, in order to eliminate artefacts arising from the choice of basis set.
The HCO System.-Dunning1l0 has performed SCF CI calculations on those parts of the (HCO) surface that involve reactions of the triatomic moieties HCO and COH:
H C O + H + C O (a)
C O H - + C O + H (b)
COH-HCO (c)
He used a primitive GTO basis set for C, 0, and H of (9s5p/9s5p/4s) orbitals, contracted to [3s2p/3s3p/2s] and then augmented with (3d/3d/2p), the exponents of the latter polarization functions being optimized at the experimental equilibrium geometry for HCO.
The electron configuration for the SCF’A‘ ground state of either triatomic species is (la’)’ . . . (6a’)’(7ar)’( la”)’. As the X-H internuclear separation increases, the 7a’ orbital asymptotes towards a 1s orbital on hydrogen, and hence the SCF wave function
’09 W. H. Gerber and E. Schiimacher,J. Chem. Phvs.. 1978,69, 1692. ‘lo T. H. Dunning, J . Chem. Ph.vs.. 1980. 73, 2304.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online
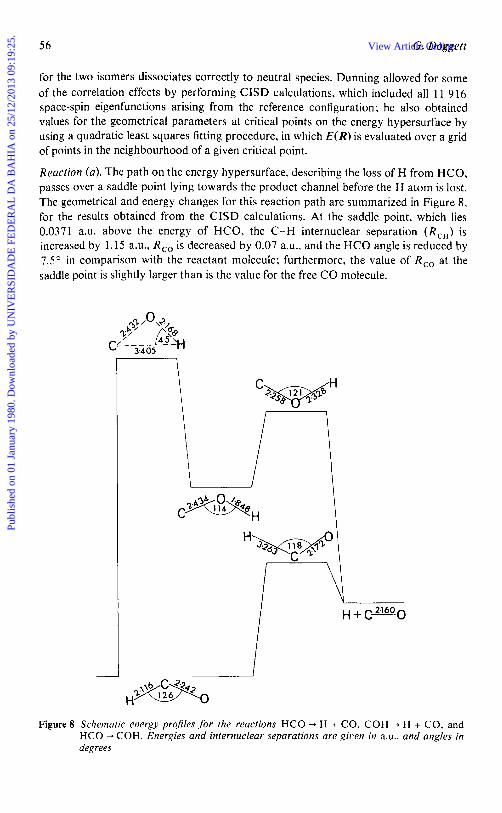
56 G. Doggett
for the two isomers dissociates correctly to neutral species. Dunning allowed for some of the correlation effects by performing CISD calculations, which included all 11 9 16 space-spin eigenfunctions arising from the reference configuration: he also obtained values for the geometrical parameters at critical points on the energy hypersurface by using a quadratic least squares fitting procedure, in which E(R) is evaluated over a grid of points in the neighbourhood of a given critical point.
Reaction (a). The path on the energy hypersurface, describing the loss of H from HCO, passes over a saddle point lying towards the product channel before the H atom is lost. The geometrical and energy changes for this reaction path are summarized in Figure 8, for the results obtained from the CISD calculations. At the saddle point, which lies 0.0371 a.u. above the energy of HCO. the C-H internuclear separation ( R c H ) is increased by 1.15 a.u., R,, is decreased by 0.07 a.u., and the HCO angle is reduced by 7 . 5 O in comparison with the reactant molecule: furthermore, the value of R,, at the saddle point is slightly larger than is the value for the free C O molecule.
Figure 8 Schematic energ?, profiles for the reactions HCO + H + CO, COH -, H + CO, and HCO --t COH. Energies and internuclear separations are given in a.u., and angles in degrees
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 57
Reaction (b). The situation here is different from that arising in reaction (a), because the COH species has an initial energy lying 0.039 1 a.u. above the CO + H dissociation limit. However, the saddle point in the reaction path occurs more towards the reactant channel than is the case with HCO (see Figure 8); at this point, R,, is decreased from the COH valueAbut it is still 0.1 a.u. larger than is the free C O value. Also, the values of R,, and COH at the saddle-point geometry are greater than the corresponding values for the COH molecule.
Reaction (c). It is clear from Figure 8 that HCO is the isomer with the lower energy: thus it is not surprising to find that the minimum energy path for isomerization passes over a saddle point with a barrier height greater than that associated with the dissociation of COH to H + CO. On the basis of this observation, Dunning concludes that the conversion of COH to HCO is most likely to occur through a two-step process, in which dissociation to H + C O is followed by association of the products to form HCO.
The C O , System.-The energy hypersurface for CO, is relatively unexplored, despite the fact that the (collinear) molecule was selected for one of the earliest SCF calculations, a point noted recently by Feller, Katriel, and Davidson ' 1 1 in their search for a cyclic isomer on the surface. The existence of such isomers for systems like NO, and 0, is well established, and this formed the motiva.tion for the current investigation of the CO, system by Feller and co-workers.
In C,, symmetry, the dominant configurations for the ring state are . . . 5a,Z1b,23b221a,26a,2 and . . . 5a,21b,23bzZla,24b,2: and the molecular orbitals themselves are expanded in terms of the 3-21G basis functions of Binkley, Pople, and H e h ~ e ~ ~ in the first instance. Using only C, , preserving distortions, the two- configuration MCSCF energy profile from the collinear arrangement (- 186.5807 a.u.) passes over a saddle point 0.1783 a.u. above the starting configuration, before a shallow minimum is reached 0.003 1 a.u. below the saddle point.Jhe geometrical parameters for t h z e two critical points are R,, = 2.534 a.u., OCO = 9 5 S 0 and R,, = 2.56 a.u., OCO = 81.4O, respectively. Inclusion of two further configurations in the MCSCF treatment, in which an a, outer electron pair is excited into the out-of-plane 2b, molecular orbital, yields a saddle-ring energy difference of 0.0054 a.u. Further CISD calculations, based on the two-configuration reference function, indicate that the saddle point now lies 0.2172 a.u. above the energy of the collinear configuration, and the minimum corresponding to the ring configuration lies 0.01 26 a.u. below the saddle point.
An enlargement of the basis set to include polarization functions yields a similar result. At the MCSCF level, the saddle-ring separation is 0.0008 a.u. - in each case, the ring state lies lower in the energy than the saddle-point configuration. The question of whether the ring isomer will dissociate into CO('C) + O('D) or surmount the barrier over the saddle point to yield the collinear configuration is a sensitive issue. Exploratory calculations suggest that the energy of the dissociation products lies only very slightly above the energy of the transition state. It is clear, therefore, that further calculations need to be carried out on this system in order to obtain a more complete understanding. In the meantime, however, the comparative calculations on the systems
'I1 D. Feller, J . Katriel and E. R. Davidson, J . Chem. Ph-vs., 1980, 73,4517.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

58 G. Doggett
0,, NO,, and CO, show that the ring-saddle barrier at the CISD (3-21G basis) decreases from 0.0601 a.u. for 0, to 0.0031 a.u. for CO,, results indicating that, although a ring isomer for the related BO, system is precluded, such isomers may occur for the NO,-, CO,-, and N,O systems.
XCN Systems.-The study of XCN-CNX isomerization reactions continues to provide a fertile area for testing orbital models. Thee has been a number of recent calculations on these kinds of system, and the relative stabilities of the two identifiable minimum energy isomers is not always as expected.
HCN c* CNH Isomerization. It has been established for a long time now that the HCN form has a lower energy than has CNH on the ground-state energy surface. However, attempts to explore further the ground- and excited-state surfaces have been reported during the last year. As noted earlier, Brooks and c o - ~ o r k e r s ~ ~ used fairly extensive CISD calculation to investigate the geometry and barrier height at the saddle point in the isomerization reaction on the ground state surface. This surface has also been fitted by Carter, Mills, and Murrell,'12 using the semi-empirical method of Sorbie and M~rre1l.l'~ In addition, both Vazquez and Gouyet1l3 and Redmon, Purvis, and Bartlett' l 4 have investigated the ground-state surfaces for several excited states, in an attempt to understand the process responsible for the production of HNC.
Vazquez and Gouyet used a GTO basis set to explore the possibility that the isomerization mechanism involves participation by the 2'A ' excited state. On the ground-state surface, the saddle-point geometry is similar to that found by Brooks and co -w~rke r s ,~~ in that the hydrogen is shifted more towards carbon than nitrogen, but the barrier to the reaction HNC -+ HCN is 70 kJ mol-' greater than the Brooks value. Despite this overestimation of the barrier height, and also of the isomerization energy, it is still possible to obtain useful information about the isomerization scheme, by comparing the characteristics of the two 'A' surfaces. On the 2lA' surface itself, which is calculated with the C-N separation unaltered from the (fixed) ground-state value, there is no barrier to isomerization; in fact, there is a path on the surface along which the energy decreases as hydrogen moves from carbon towards nitrogen, thus vitiating the possibility that HCN ( ,A') can be reformed. The region near the avoided crossing of the two 'A' surfaces occurs on the nitrogen side, where the well on the 2'A' surface coincides with a hill on the ground-state surface, is thought to provide the environment necessary for intersurface transitions to occur. However, in order for the system to gain access to the excited surface, Vazquez and Gouyet suggest it might be necessary to carry out the reaction H(,S) + CN(,II) to yield an excited HCN molecule that is either on the 1 'A " or the 2 'A ' surface; once on the latter surface, the system can undergo intramolecular energy transfer to yield HNC in its ground state.
Redmon and co-workers' l4 have also calculated isomerization energies and barrier heights for the ground-state surface of the (HCN) system. Their main concern, in a perturbation-based investigation (which included some of the effects of singly, doubly, and quadruply excited configurations), was with the sensitivity of the results to the choice of basis set. The largest basis set used, X, consisted of a set of contracted GTOs augmented with d functions on carbon and nitrogen, and p functions on hydrogen - 69
S. Carter, I. M. Mills, and J. N. Murrell, J. Mol. Spedrosc., 1980. 81. 110. G. J. Vazquez and J.-F. Gouyet, Chern. Phys. Lett., in the press. C. T. Redmon, G. D. Purvis, and R. J. Bartlett, J. Chern. Phys., 1980, 72, 986.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Tabl
e 8
Ener
gies
and
bar
rier
hei
ghts for
the
HN
C +
HC
N is
omer
izat
ion
[E(H
CN
)-E
(HN
C)l
Bu
rrie
r he
ight
, M
odel
Ba
sis"
E (H
CN
)/a.
u.
E (H
NC
)/a.
u.
E( T
.S.)/
a.u.
b /k
J m
ol-'
A/kJ
mol
-' SC
F D
ZP
-92.
8890
-9
2.87
35
-92.
81
19
-4 1
16
2 SC
F'
DZ
P -9
2.89
08
-92.
8737
-
-45
SCF
X
-92.
9083
-9
2.89
14
-92.
8318
-4
5 15
6 SD
Q-M
BP
T~
X
-93.
2375
-9
3.21
36
-93.
1603
-6
1
140
-
See
C. T
. Red
mon
, G. D
. Pur
vis,
and
R. J
. Bar
tlett,
J. C
hem
. Phy
s., 1
980,
72,
986
for d
etai
ls;
ener
gy o
f tra
nsiti
on s
tate
; SC
F ca
lcul
atio
n w
ith c
ount
erpo
ise
corr
ectio
n;
pertu
rbat
ion-
base
d m
odel
- se
e '. Pu
blis
hed
on 0
1 Ja
nuar
y 19
80. D
ownl
oade
d by
UN
IVE
RSI
DA
DE
FE
DE
RA
L D
A B
AH
IA o
n 25
/12/
2013
09:
19:2
5.
View Article Online

60 G. Doggett
basis functions in all. The contribution of the superposition error was also assessed with the aid of the counterpoise method, but, for this calculation, a slightly less extensive basis set was used - a so called DZP basis that consisted of the union of the sets of ( ldlldllp) and [4s2p/4s2p/2sl functions. The results given in Table 8 show that the isomerization energy is more sensitive to the choice of model and basis set than is the overall barrier height for the reaction HNC + HCN. The same general trend is also apparent in the work of Haese and W O O ~ S . " ~ They found that the isomerization energy varied from 30.5 (SCFDZ) to 63 kJ mol-' (CI).
Although HCN is the lower energy isomer on the ground-state surface, a reversal of roles occurs in the case of the positive ion species. Murrell and Derzi1l6 have recently examined this situation in the calculation of ionization energies for HCN and HNC. At either the SCF or CISD (with configuration selection) levels of calculation, the *C+ state of HNCf is found to lie below the 211 state of HCN+ (which itself lies below the 2C+ state). All states yield minimum energies for the collinear arrangement, except for the 'II state of HNC+, which is bent. The overall effects of CI are, as usual, fairly marked, and the isomerization energy is sensitive to the inclusion of correlation effects. However, here, unlike the neutral molecular situation, the ground states being compared are of different symmetry types. The SCF ground state energies for HCN+ and HNC+ are -92.4613 a.u. and -92.4839 a.u., respectively, while inclusion of CI lowers these energies to -92.6975 a.u. and -92.7307 a.u., respectively.
BCN - BNC and LiCN * LiNC Isomerizations. Redmon and c o - ~ o r k e r s ~ ~ ~ have also examined these two systems in addition to the (HCN) system discussed above. Here, as in the (HCN)+ system discussed by Murrell and Derzi,l16 the isocyanide is found to have the lower energy of the two isomers (collinear arrangement in both cases), irrespective of whether correlation effects are included or not. However, the correlation energy correction is greater for the cyanide, but it is still not enough to reverse the relative stabilities. Unfortunately, no barriers are given for the isomerization reactions, but the isomerization energy for the Li species is especially small (further detailed comparisons are not possible because there seem to be transposition errors in the published Table of energies).
HeCN+ - HeNC+ Isomerization. Wilson and Green"' have recently calculated the equilibrium geometries for HeCN+ and its isomer HeNC+, both of which are of theoretical and experimental interest (although, as the authors note, in the astrochemical situation destruction of HeCN+ is likely to occur via the exothermic reaction with H2). Despite the problems concerned with the experimental isolation of such systems, the fact that they are isoelectronic with the (HCN) species makes them inherently interesting from the theoretical point of view.
The calculations show that, indeed, the HeCN+ species is linear in its ground state, and of lower energy than is the corresponding isocyanide. In view of the largely ionic character of the bonding, however, it is necessary to augment standard basis sets - say of DZ quality - with a number of suitably chosen polarization functions. Despite the apparent flexibility of such basis sets, the minimum-energy geometry for HeCN+ is found to be sensitive to the choice of basis set functional form; for example, GTO basis
N. N. Haese and R. C . Woods, J. Chem. Phys., 1980,73,4521. J . N. Murrell and A. A1 Derzi, J . Chem. SOC., Faraday Trans. 2 , 1980, 76, 3 19. S. Wilson and S . Green, J. Chem. Phys., 1980, 73 ,4 19.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

Electronic Structure of Molecules 61
functions yield consistently shorter C-N separations than do their STO counterparts. However, the dissociation energy for the process HeCN+ -P He('S) + CN+('C+) is calculated to be 154 and 164 kJ mol-' for the augmented STO and GTO basis sets, respectively. The corresponding calculations on the HeNC+ system, using the augmented STO basis set, yielded a metastable state with R H e N = 1.908 a.u. and RN, = 2.329 a.u. at an energy of 164 kJ mol-' above the ground state (for which the geometrical parameters in the same basis are RHeC = 2.055 a.u. and R C N = 2.188 a-u.), that is, the energy of the HeNC+ lies 10 kJ mol-' above the dissociation products He, CN+, but, like the COH system discussed earlier, there is a barrier of about 40 kJ mol-' to the removal of He. In this particular case, Wilson and Green deduce that HeNC+ is unlikely to be observed.
H,CN+ - HCNH+ f+ CNH, have considered the electronic structures of the species with empirical formula H,CN+ - all isoelectronic with HCN and HeCN+ - in a study of the mechanism for the production of HNC from the reaction between C+ and NH,. They suggest that two routes are possible for the formation of HNC from singlet H,NC+: the first route involves the triplet state of this species as a precursor, and yields HNC exclusively, while the second route proceeds via the linear HCNH+ (singlet) intermediate, and yields HCN and HNC. In both processes, the last step involves electron capture.
The present results on the triplet states of the four geometrical isomers are best considered in the context of the earlier work by Conrad and Schaefer"' on the corresponding singlet states of the various H,CN+ species. The singlet state of the linear HCNH+ species lies lowest in energy and, for all the basis sets used - either at the SCF or CISD levels (GUGA methodology) - the H,-NC+ singlet state has the next lowest energy, followed by singlet H,-CN+, triplet H,NC+, triplet H,-CN+, and so on. Thus, for both singlet and triplet excited states, the hydrogen atoms are bonded preferentially to nitrogen; for the ground state, the system can be considered as HCN, with a proton attached to the nitrogen lone pair.
The path on the energy hypersurface for the isomerization reaction H,-NC+ (singlet) to H-CN-H (singlet) involves passing through a transition state with a barrier height varying between 2 19 (at the DZ SCF level) and 124 kJ mol-' (at the augmented DZ CISD level). In the case of the H,-CN+ (singlet) H-CN-H+ (singlet) isomerization, the barrier is seen only in the SCF and CI calculations using a DZ basis set. It is clear, therefore, that the basis set problem remains a sensitive issue; a more complete treatment of the ground- and excited-state hypersurfaces must include a balanced treatment of all reaction products on the respective surfaces. Thus, it is by no means certain, for example, that the present treatment of the H,CN+ systems is adequate for the species CN+, which occurs in one of the product channels.
H,N-CN - H,N-NC Isomerization. This isomerization reaction has been studied by Vincent and Dykstra120 at selected points on the energy hypersurface. They calculated molecular energies using the SCF and Self Consistent Electron Pair (SCEP) models, for which the orbitals were expanded in terms of either DZ or DZ plus polarization (DZP) basis sets (the SCEP model was not discussed in the section above on orbital
Isomerization. Allen, Goddard and Schaefer
T. L. Allen, J. D. Goddard, and H. F. Schaefer, J . Chem. Phys., 1980, 73, 3255. M. P. Conrad and H. F. Schaefer, Nature (London), 1978, 274,456.
lZo M. A. Vincent and C. E. Dykstra, J. Chem. Phys., 1980,73,3838.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online

6 2 G. Doggett
models, but it yields results of comparable quality to those obtained from the CISD model).
The H,N-CN isomer possesses the lower energy for the two choices of basis set (in either model) and, in contrast to the systems studied earlier, the difference in energy between the equilibrium structures of both isomers is quite large - 176 and 221 kJ mol-’ at the SCF DZ and SCEP DZP levels, respectively. In addition, and as found for the HCN isomerization reaction, the correlation correction to the SCF energy is biased towards the cyanide. The estimates for the barrier height of the H2N- CN -+ H,N-NC rearrangement, which are considerably greater than those found for the other cyanide isomerizations discussed above, are sensitive to both the choice of basis set and the choice of model; thus, for the DZP basis, for example, barriers of 368 and 412 kJ mol-’ are found for the SCF and SCEP calculations, respectively. However, use of a DZ basis for the SCF calculations increases the barrier, from the DZP value of 368 kJ mol-’, to 393 kJ mol-’.
8 Conclusion
Returning to the tree analogy, it is clear that most of this review has been concerned with climbing a relatively small, and yet important number of branches, most of which are close to the trunk. No apologies are made, therefore, for this rather restricted view of the quantum chemical tree; for unless the limitations and range of applicability of the basic models are fully appreciated, much time can be wasted in discussing attributes of molecular properties that are nothing more than artefacts of the choice of basis set - unless, of course, a range of systems is chosen for study, in order to provide a view of relative changes in the property of interest (the experience with the XCN isomerization reactions should act as a warning!). Thus, until molecular energies can be obtained with some degree of reliability, the further use of the wave function itself for calculating other molecular properties is of questionable validity. This follows because the variation theorem ensures that only the energy is improved in a systematic manner with parallel improvement in the basis set, that is, the calculated energy approaches the exact value from above. However, the expectation values of operators other than I? do not have this desirable property, and there are problems in assessing how the error in the calculated molecular property varies with the choice of basis set. Some aspects of these difficulties have been discussed most recently by Ransil.’”
There are many more important branches of the subject to explore: of special mention are those concerned with the construction of momentum space (p) wave functions, the construction and use of r- and p-space density functions, and the development of methods for divulging the physical content of correlated wave functions. However, an assessment of progress in these and other areas is best left to a later report.
Acknowledgement. I would like to thank S. J. Smith for his useful comments on the first draft of the manuscript.
’” B. J. Ransil, Znt. J. Quantum Chem., 1980, 17,467.
Publ
ishe
d on
01
Janu
ary
1980
. Dow
nloa
ded
by U
NIV
ER
SID
AD
E F
ED
ER
AL
DA
BA
HIA
on
25/1
2/20
13 0
9:19
:25.
View Article Online




![C1 - Atoms, Molecules and Stoichiometry [Structure Question]](https://static.fdocuments.net/doc/165x107/544bbbb7b1af9f767d8b48ca/c1-atoms-molecules-and-stoichiometry-structure-question.jpg)