
Electronic Supplementary Information Programmed ... · Electronic Supplementary Information...
21
S1 Electronic Supplementary Information Programmed stereoselective assembly of DNA-binding helical metallopeptides Ilaria Gamba, Gustavo Rama, Elizabeth Ortega-Carrasco, Jean-Didier Maréchal, José Martínez-Costas, M. Eugenio Vázquez* and Miguel Vázquez López* I. Gamba, G. Rama, Prof. M. Vázquez López. Departamento de Química Inorgánica and Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS). Universidade de Santiago de Compostela, 15782 Santiago de Compostela. Spain. E-mail: [email protected] E. Ortega-Carrasco, Dr. J.-D. Maréchal. Departament de Química. Universitat Autònoma de Barcelona, 08193 Cerdanyola. Spain. Prof. J. Martínez-Costas. Departamento de Bioquímica y Biología Molecular, and Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS). Universidade de Santiago de Compostela, 15782 Santiago de Compostela. Spain. Prof. M. E. Vázquez. Departamento de Química Orgánica and Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS). Universidade de Santiago de Compostela, 15782 Santiago de Compostela. Spain. E-mail: [email protected] Electronic Supplementary Material (ESI) for ChemComm. This journal is © The Royal Society of Chemistry 2014
Transcript of Electronic Supplementary Information Programmed ... · Electronic Supplementary Information...

S1
Electronic Supplementary Information
Programmed stereoselective assembly of DNA-binding helical metallopeptides Ilaria Gamba, Gustavo Rama, Elizabeth Ortega-Carrasco, Jean-Didier Maréchal, José Martínez-Costas, M. Eugenio Vázquez* and Miguel Vázquez López*
I. Gamba, G. Rama, Prof. M. Vázquez López. Departamento de Química Inorgánica and Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS). Universidade de Santiago de Compostela, 15782 Santiago de Compostela. Spain. E-mail: [email protected]
E. Ortega-Carrasco, Dr. J.-D. Maréchal. Departament de Química. Universitat Autònoma de Barcelona, 08193 Cerdanyola. Spain.
Prof. J. Martínez-Costas. Departamento de Bioquímica y Biología Molecular, and Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS). Universidade de Santiago de Compostela, 15782 Santiago de Compostela. Spain.
Prof. M. E. Vázquez. Departamento de Química Orgánica and Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS). Universidade de Santiago de Compostela, 15782 Santiago de Compostela. Spain. E-mail: [email protected]
Electronic Supplementary Material (ESI) for ChemComm.This journal is © The Royal Society of Chemistry 2014

S2
1. General All reagents were acquired from commercial sources: Dimethylformamide (DMF) and Trifluoroacetic acid (TFA) were purchased from Scharlau, CH2Cl2 from Panreac, CH3CN from Merck, HBTU (O-Benzotriazole-N,N,N',N'-tetramethyl-uronium-hexafluorophosphate) and HATU (2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate methanaminium) from GL Biochem (Shanghai) Ltd. All other chemicals were purchased from Sigma-Aldrich or Fluka. All solvents were dry and synthesis grade, unless specifically noted. (NH4)Fe2(SO4)2.6H2O salt from Sigma-Aldrich was used as Fe(II) ion source.
Reactions were followed by analytical RP-HPLC with an Agilent 1100 series LC/MS using a Luna C18 (250 x 4.60 mm) analytical column from Phenomenex. Standard conditions for analytical RP-HPLC consisted on a linear gradient from 15% to 95% of solvent B for 30 min at a flow rate of 1 mL/min (A: water with 0.1% TFA, B: acetonitrile with 0.1% TFA). Compounds were detected by UV absorption at 222, 254 and 310 nm. High-Performance Liquid Chromatography (HPLC) was performed using an Agilent 1100 series Liquid Chromatograph Mass Spectrometer system. Analytical HPLC was run using a Luna C18 (250 x 4.60 mm) reverse phase analytical column; compounds were detected by UV absorption at 222, 254 and 310 nm. The purification of the peptides was performed on a Luna C18 (250 x 10 mm) semi-preparative reverse phase column from Phenomenex. The standard gradient used for analytical and semi-preparative HPLC was 90:10 to 50:50 over 30 min (water/acetonitrile, 0.1% TFA). Electrospray Ionization Mass Spectrometry (ESI/MS) was performed with an Agilent 1100 Series LC/MSD VL model in positive scan mode using direct injection of the purified peptide solution into the MS. Peptides purification was performed by semi-preparative RP-HPLC with an Agilent 1100 series Liquid Chromatograph using a Luna 5u C18(2) 100A (5 µm, 10 × 250 mm) reverse-phase column from Phenomenex. Standard conditions for analytical and semi-preparative RP-HPLC consisted on an isocratic regime during the first 5 min, followed by a linear gradient from 10% to 50% of solvent B for 30 min (A: water 0.1% TFA, B: acetonitrile 0.1% TFA). Compounds were detected by UV absorption (222 nm) and by ESI+−MS. The fractions containing the products were freeze-dried, and their identity was confirmed by ESI+−MS and MALDI-TOF.
Electrospray Ionization Mass Spectrometry (ESI/MS) was performed with an Agilent 1100 Series LC/MS model in positive scan mode using direct injection of the purified peptide solution into the MS.
Matrix-assisted laser desorption/ionization mass spectrometry (MALDI/MS) was performed with a Bruker Autoflex MALDI/TOF model in positive scan mode by direct irradiation of the matrix-absorbed peptide.
UV measurements were made in a Jasco V-630 spectrophotometer coupled to a Jasco ETC-717 temperature controller, using a standard Hellma semi-micro cuvette (140.002-QS). Measurements were made at 20 ºC.
Luminescence experiments were made with a Jobin-Yvon Fluoromax-3 fluorescence spectrometers (DataMax 2.20), coupled to a Wavelength Electronics LFI−3751 temperature controller. All measurements were made with a Hellma semi-micro cuvette (114F-QS) at 20 ºC.
Circular dichroism measurements were made with a Jasco J-715 coupled to a Neslab RTE-111 termostated water bath, using a Hellma 100-QS cuvette (2 mm light pass). Measurements were made at 20 ºC. Samples contained (300 µL) 10 mM PBS buffer (pH = 7.2), 50 μM of peptides; the successive additions of Fe(II) aliquots ((NH4)2Fe(SO4)2·6H2O, ammonium iron(II) sulfate (Mohr's salt), 3 µL, 10 mM aqueous solution) complete the complex formation (Fe(II), 100 µM). The spectra are the average of 2 scans and were processed using the program Sigmaplot 11.0 (Systat Software Inc).

S3
2. Synthesis of the unnatural coordinating residue Fmoc-O1PenBpy-OH (1) The bipyridine residue was obtained following the route previously reported by us,1 and which is based on a method reported by Newkome et al.2, starting from 5,5'-diethyl-2,2'-bipyridine, which was obtained from 5,5-dimethil-2,2-bipyridine using the method reported by Francis H. Case3 and Whittle et al.4
Scheme S1. Synthesis of the coordinating amino acid 1.
NNH N
CO2H
N NMe Me
1. KMnO4 / H2O2. EtOH / H2SO43. H2N-NH2 / EtOH toluene, 80 ºC
N NCO2Et
1. HCl / NaNO22. reflux, EtOH/Xylene
N NNH
CO2EtEtO
OEtOH / NaOH
a) SOCl2
b) 2, DIEA CH2Cl2
N NH2N CO2H
2
NH
O
H2N
FmocHN OO
OHO
O
NHFmoc
Fmoc-O1PenBpy-OH (1)Fmoc-O1Pen-OH

S4
3. Peptide synthesis All peptide synthesis reagents and Fmoc amino acid derivatives were purchased from GL Biochem (Shanghai) Ltd. Novabiochem. Fmoc-O1Pen-OH was purchased from IRIS Biotech (Cat. #: FAA1565).
C-terminal amide peptides were synthesized following standard peptide synthesis protocols (Fmoc/tBu strategy) on a 0.05 mmol scale using a 0.48 mmol/g loading Fmoc-PAL-PEG-PS resin from Life technologies with a PS-3 automatic peptide synthesizer from Protein Tecnologies. The amino acids were coupled in 5-fold excess using HBTU as activating agent, except for the synthetic Fmoc-O1PenBpy-OH coordinating residue, which was coupled using HATU as activating agent.
Each amino acid was activated for 30 seconds in DMF before being added onto the resin and couplings were conducted for 30 min. Deprotection of the temporal Fmoc protecting group was performed by treating the resin with 20% piperidine in DMF for 10 min.
General procedure for cleavage-deprotection: A resin-bound peptide dried overnight (0.025 mmol) was placed in a 50 mL falcon tube, to which 3 mL of the cleavage cocktail (50 µL CH2Cl2, 25 µL of H2O, 25 µL of TIS (Triisopropylsilane) and 940 TFA µL) were added. The resulting suspension was shaken for 3 h. The resin was filtered, and the TFA filtrate was concentrated with a nitrogen current to an approximate volume of 1 mL, which was added to ice-cold diethyl ether (10 mL). After 10 min, the precipitate was centrifuged and washed again with 15 mL of ice-cold ether. The solid residue was dried under argon and redissolved in acetonitrile/water 1:1 (1 mL) and purified by semi-preparative reverse-phase HPLC. The collected fractions were lyophilized and stored at −20 °C. Their identity was confirmed by ESI+-MS and MALDI-TOF.
LL-H:
ESI-MS (m/z) [M+H]+ calc for C108H113N31O24 = 2227.8; found = 1114.6, calc. for [M+2H]2+ = 1114.9; found = 743.2, calc. for [M+3H]3+ = 746.6.
UV (H20) λmax, nm (ε): 308 (169700) M−1cm−1
Yield: 52%
DD-H:
ESI-MS (m/z) [M+H]+ calc for C108H113N31O24 = 2227.8; found = 1114.6, calc. for [M+2H]2+ = 1114.9; found = 743.2, calc. for [M+3H]3+ = 746.6.
UV (H20) λmax, nm (ε): 308 (169700) M−1cm−1
Yield: 43%
Synthesis of the fluorescent peptides
The coupling of the peptide with the rhodamine fluorescent probe was performed directly on the solid support: 5-(and-6)-carboxy-X-rhodamine (6.7 mg, 9.6 µmol), activated as succinimidyl ester, was dissolved

S5
in 1.5 mL of DIEA solution in DMF (0.19 M, 0.29 mmol). The mixture was sonicated during 2 minutes and then loaded in a SPPS reactor, containing the Rink Amide resin (40 mg, 9.5 µmol) where the LL-H or DD-H peptides were previously grown, as described before. The mixture was gently shaken during 2 hours at rt. The resin was extensively washed and dried. The desired peptide was cleaved from the solid support, using the standard acidic cocktail (5 mL, 2.5 hrs), and purified by HPLC.
LL-RhH:
ESI-MS (m/z) [M+H]+ calc for C141H142N33O28 = 2745,07; found = 1374.1, calc. for [M+2H]2+ = 1373,5; found = 915.8, calc. for [M+3H]3+ = 916.0. UV (H20) λmax, nm (ε): 575 (81000) M−1cm−1.
Yield: 54%
DD-RhH:
ESI-MS (m/z) [M+H]+ calc for C141H142N33O28 = 2745,07; found = 1374.1, calc. for [M+2H]2+ = 1373,5; found = 915.8, calc. for [M+3H]3+ = 916.0. UV (H20) λmax, nm (ε): 575 (81,000) M−1cm−1.
Yield: 48%
The above described protocol was also followed to synthetize the corresponding fluorescein derivatives, using fluorescein isothiocyanate (FITC) as fluorescein source (Scheme S1).
Scheme S2. Structure of the peptide ligands functionalized with a fluorescein in the N-terminal end of their peptidic sequences (LL-FluH and DD-FluH).

S6
LL-FluH:
ESI-MS (m/z) [M+H]+ calc for C129H126N32O29S = 2618,91; found = 1310.5, calc. for [M+2H]2+ = 1310,0; found = 873,6, calc. for [M+3H]3+ = 873.9.
Yield: 57%
DD-FluH:
ESI-MS (m/z) [M+H]+ calc for C129H126N32O29S = 2618,91; found = 1310.5, calc. for [M+2H]2+ = 1310,0; found = 873,6, calc. for [M+3H]3+ = 873.9.
Yield: 53%

S7
4. Peptide helicates synthesis The peptide helicates were synthetized by addition of aliquots of an aqueous solution containing Fe(II) metal ions [Mohr’s salt; the total amount of Fe(II) added was 2 equivalents] to a mother solution of the peptide ligand in PBS buffer (pH = 7.4, 298 K). All the coordination reactions were quantitative. The peptide helicates derived from the ligands LL-FluH and DD-FluH were poorly soluble in water.

S8
5. MALDI-TOF mass spectrometry of the peptide helicates ΔΔ-[Fe2(DD-H)]4+: MALDI-TOF (m/z) [M]+ calc for C108Fe2H113N31O24 = 2339.7, found = 2337.9
ΛΛ-[Fe2(LL-H)]4+ : MALDI-TOF (m/z) [M]+ calc for C108Fe2H113N31O24 = 2339.7, found = 2337.9
ΔΔ-[Fe2(DD-RhH)]4+: MALDI-TOF (m/z) [M]+ calc for C141H142Fe2N33O28 = 2856,94, found = 2851.9
ΛΛ-[Fe2(LL-RhH)]4+ : MALDI-TOF (m/z) [M]+ calc for C141H142Fe2N33O28 = 2856,94, found = 2854.8
ΔΔ-[Fe2(DD-FluH)]4+: MALDI-TOF (m/z) [M]+ calc for C129H123Fe2N32O29S = 2727,78, found = 2727,5
ΛΛ-[Fe2(LL-FluH)]4+ : MALDI-TOF (m/z) [M]+ calc for C129H123Fe2N32O29S = 2727,78, found = 2727,5

S9
6. Computational Details Molecular Dynamics The optimization of the loop was performed on an initial model of the ΛΛ isomer where the metal centers satisfies the chiral induction. The simulation were performed under the UCSF chimera environment5 using the MMTK Molecular Dynamics driver.6 We used the built-in interface of UCSF Chimera for AmberTools,7 to generate the GAFF force field of the system.8 Its charges were generated through the Gasteiger method.9 Considering that the first coordination sphere of the metal would not impact drastically on the dynamical properties of the loop, the simulations were performed in absence of metal and with the bipyridine rings fixed. A MD run of 50000 steps with an integration time of 1fs and 10000 of heating steps. The entire simulation was post-processed using the NMRCLUST on the production part of the simulation.10 The 4 most populated clustered were selected for further calculations.
QM/MM refinement Quantum Mechanics/Molecular Mechanics geometry optimizations were performed using the ONIOM procedure as implemented in Gaussian09,11,12 and combining DFT(B3LYP),13,14 and AMBER force field.7 For the total 279 atoms, the Quantum Mechanical part which includes the six bipyridine groups and the two Fe ions, is composed by 110 atoms. Two basis sets were used: 6-311G* 15,16 for the main group atoms and aug-cc-pVTZ for the Fe centres.17 All calculations have been carried out using implicit solvent (water in this case) with the CPCM model,18,19 and electronic embedding with the Fe atoms at the oxidation state +II and low spin.20
Figure S1. Molecular representation of the most stable ΛΛ–[Fe2(LL-H)]4+ complex highlighting the interaction of the ammonium group of the N-terminal end with the C-terminal loop
A total of 4 geometries were obtained that mainly differ from geometries of the loops and the cis/trans conformation of the amide groups in the loops and the spacers. The potential energy difference between the four geometries does not exceed 20 kJ/mol, which is consistent with a certain amount of flexibility of the system. For clarity, only the more stable complex was further analyzed.
The lowest energy model presents a helicoidal geometry (Figure S1) with both metal ions in octahedral configurations and the six bipyridine rings perfectly ordered around the metal centres.

S10
The average distance between the Fe(II) ions and their coordinating nitrogen is 2.03 Å and the bipyridine groups present none to very little out-of-plane displacement (average value of 0.1 Å).
All the amino acid residues of the peptide ligand, including the two proline residues, present trans configuration. Despite their proximity, no hydrogen bonds are identified between the amide groups of the different spacers. The entire helicate is not perfectly symmetric, and both loops are oriented towards the same side of the helicate with an approximate angle of 75º with respect to the helicate axis. The two proline residues are at the most external part of the loops with atoms reaching up to 9 Å out of the helicate axis. Interestingly, the C-terminal proline (P1, Scheme 1 in the main body of the manuscript) is less displaced from the helicate axis, and the entire C-terminal loop presents a more compact geometry as a result of the interaction of three carbonyl groups of the peptide backbone (G1, P1, G2, Scheme 1) with the N-terminal amine, which is expected to be protonated at neutral pH (Figure S1). Altogether, it appears that a certain amount of molecular complexity and organization have been reached in this metallopeptide, since remote regions of the peptide chain are interacting with one another and influencing the folding of the system
S11
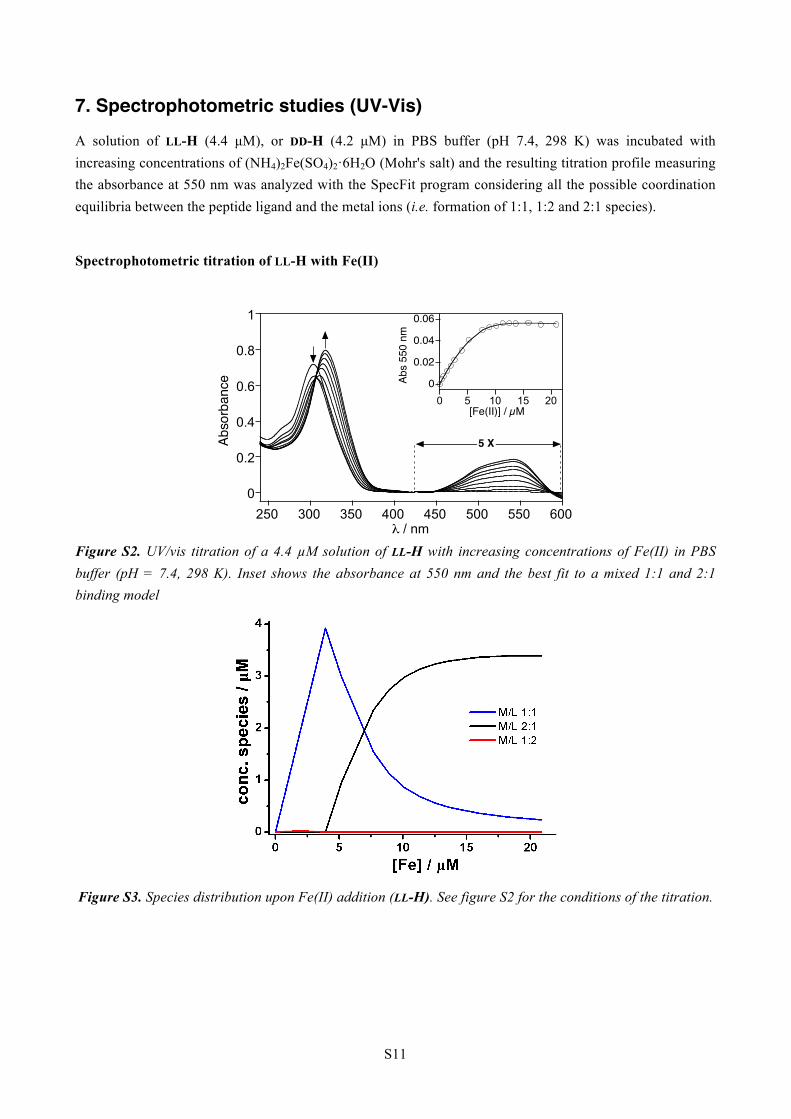
7. Spectrophotometric studies (UV-Vis)
A solution of LL-H (4.4 µM), or DD-H (4.2 µM) in PBS buffer (pH 7.4, 298 K) was incubated with increasing concentrations of (NH4)2Fe(SO4)2·6H2O (Mohr's salt) and the resulting titration profile measuring the absorbance at 550 nm was analyzed with the SpecFit program considering all the possible coordination equilibria between the peptide ligand and the metal ions (i.e. formation of 1:1, 1:2 and 2:1 species).
Spectrophotometric titration of LL-H with Fe(II)
Figure S2. UV/vis titration of a 4.4 µM solution of LL-H with increasing concentrations of Fe(II) in PBS buffer (pH = 7.4, 298 K). Inset shows the absorbance at 550 nm and the best fit to a mixed 1:1 and 2:1 binding model
Figure S3. Species distribution upon Fe(II) addition (LL-H). See figure S2 for the conditions of the titration.
0
0.2
0.4
0.6
0.8
1
250 300 350 400 450 500 550 600
Abso
rban
ce
λ / nm
5 X
0
0.02
0.04
0.06
0 5 10 15 20Ab
s 55
0 nm
[Fe(II)] / µM

S12
Spectrophotometric titration of DD-H with Fe(II)
a. b. c. Figure S4. a. Uv-vis spectra recorded upon titration of a 4.2 µM solution of DD-H with increasing concentrations of Fe(II) ions (PBS buffer, pH = 7.4, 298 K); b. plot Abs at 550 nm vs. [Fe]; c. species distribution upon Fe(II) addition.

S13
8. Circular dichroism Circular dichroism measurements were made in a Jasco J-715 CD spectrophotometer coupled to a Neslab RTE-111 thermostated water bath, using a Hellma 100-QS cuvette (2 mm light pass). Measurements were made at 20ºC. Samples (300 µL, 10 mM PBS buffer, pH = 7.4) contained 50 µM of LL-H or DD-H peptides. The successive additions of Fe(II) aliquots [10 mM aqueous solution of (NH4)2Fe(SO4)2·6H2O (Mohr's salt), aliquots of 3 µL] complete the formation of the complex (final [Fe(II)] = 100 µM). The spectra showed are the average of 2 scans and were processed using the program Sigmaplot 11.0 (Systat Software Inc).

S14
9. Fluorescence anisotropy titrations with three-way DNA junctions A 1.0 mL solution of the peptidic helicate (ΔΔ-[Fe2(DD-RhH)]4+or ΛΛ-[Fe2(LL-RhH)]4+,1 µM) in 10 mM PBS buffer (10 mM NaCl, pH 7.3) was titrated with increasing concentrations of the Y1Y2Y3 DNA junction. The fluorescence anisotropy of the rhodamine fluorophore, measured at 611 nm, was recorded after each addition, and the resulting profile was analyzed using the program DynaFit, considering a 1:1 binding mode and the contribution of non-specific electrostatic complexes to the overall signal. The experiments were performed at 20 ºC in a 1 cm cuvette (excitation slit: 3 nm; emission slit: 10 nm; integration time 5 s).
S15
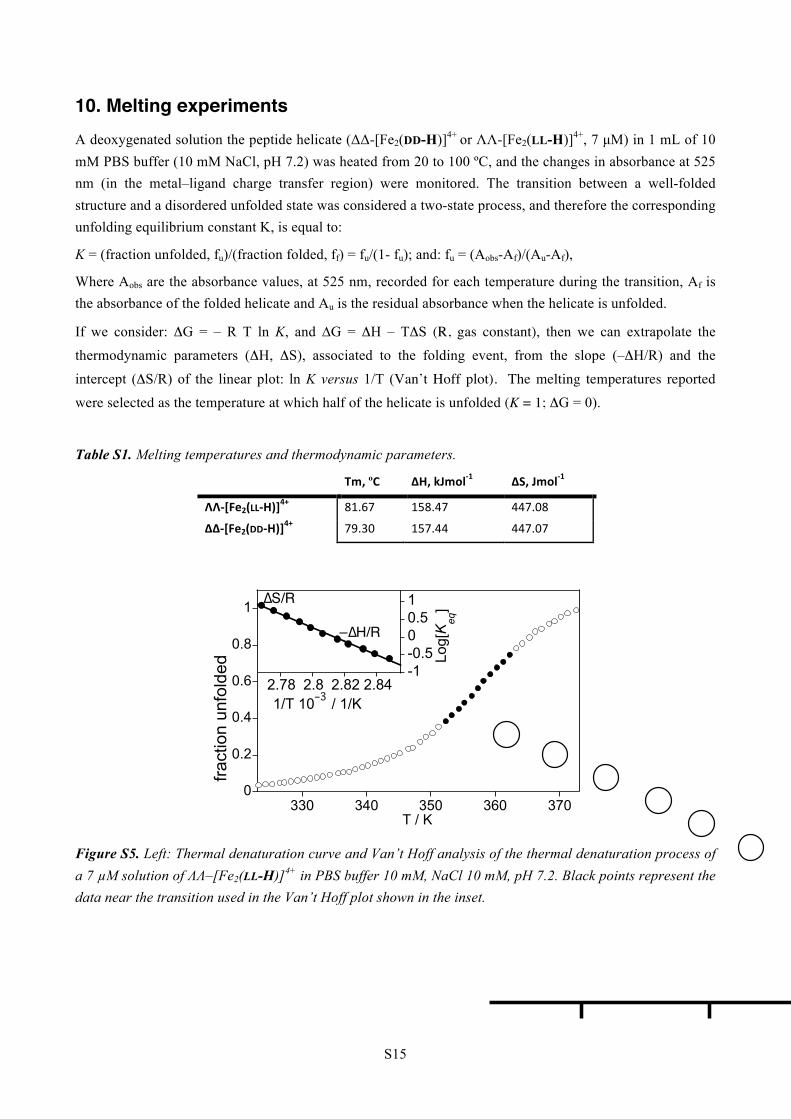
10. Melting experiments A deoxygenated solution the peptide helicate (ΔΔ-[Fe2(DD-H)]4+ or ΛΛ-[Fe2(LL-H)]4+, 7 µM) in 1 mL of 10 mM PBS buffer (10 mM NaCl, pH 7.2) was heated from 20 to 100 ºC, and the changes in absorbance at 525 nm (in the metal–ligand charge transfer region) were monitored. The transition between a well-folded structure and a disordered unfolded state was considered a two-state process, and therefore the corresponding unfolding equilibrium constant K, is equal to:
K = (fraction unfolded, fu)/(fraction folded, ff) = fu/(1- fu); and: fu = (Aobs-Af)/(Au-Af),
Where Aobs are the absorbance values, at 525 nm, recorded for each temperature during the transition, Af is the absorbance of the folded helicate and Au is the residual absorbance when the helicate is unfolded.
If we consider: ΔG = – R T ln K, and ∆G = ΔH – TΔS (R, gas constant), then we can extrapolate the
thermodynamic parameters (ΔH, ΔS), associated to the folding event, from the slope (–ΔH/R) and the
intercept (ΔS/R) of the linear plot: ln K versus 1/T (Van’t Hoff plot). The melting temperatures reported were selected as the temperature at which half of the helicate is unfolded (K = 1; ΔG = 0).
Table S1. Melting temperatures and thermodynamic parameters.
Tm, ºC ΔH, kJmol-‐1 ΔS, Jmol-‐1
ΛΛ-‐[Fe2(LL-‐H)]4+ 81.67 158.47 447.08
ΔΔ-‐[Fe2(DD-‐H)]4+ 79.30 157.44 447.07
Figure S5. Left: Thermal denaturation curve and Van’t Hoff analysis of the thermal denaturation process of a 7 µM solution of ΛΛ–[Fe2(LL-H)]4+ in PBS buffer 10 mM, NaCl 10 mM, pH 7.2. Black points represent the data near the transition used in the Van’t Hoff plot shown in the inset.
2.78 2.8 2.82 2.84-1-0.500.51
Log[Keq
]
1/T 10–3 / 1/K
0
0.2
0.4
0.6
0.8
1
330 340 350 360 370
fract
ion
unfo
lded
T / K
∆S/R
–∆H/R

S16
Figure S6. ΛΛ-[Fe2(LL-H)]4+: Absorbance melting profile at 525 nm
a. b. c.
Figure S7. ΔΔ-[Fe2(DD-H)]4+: a. Absorbance melting profile at 525 nm; b. Fraction unfolded variation versus temperature; c. Van’t Hoff plot.

S17
11. Fluorescence anisotropy titrations with duplex DNA A 1 µM solution of the ΛΛ-[Fe2(LL-RhH)]4+ helicate in PBS buffer (100 mM / NaCl 100 mM, pH= 7.3) was prepared. Aliquots of a stock DNA solution (34 µM) were sequentially added over the helicate solution, gradually increasing the concentration of DNA in the mixture. The corresponding anisotropy value at 611 nm was recorded after each addition, and the resulting titration profile was analyzed with the DynaFit program using a 1:1 binding model including the contribution of higher association complexes.21 The experiments were performed at 20 ºC in a 1 cm cuvette (excitation slit: 3 nm; emission slit: 10 nm; integration time 5 s).
Figure S8. Anisotropy titration of 1 µM ΛΛ-[Fe2(LL-RhH)]4+ helicate in PBS buffer. Experimental points are the mean of three independent experiments, and the curve represents the best fit a 1:1 binding model including the contribution of higher association complexes. The resulting dissociation constant for an A/T-rich DNA hairpin is of approximately ≈ 59 µM, which is even weaker than the worse binding constant measured for the mismatched ∆∆ isomer for the three-way junction, thus indicating that the regular B-DNA structure is not a high-affinity target for these type of structures.
0.08
0.09
0.1
0.11
0.12
0.13
0.14
0.15
0 0.4 0.8 1.2 1.6 2 2.4 2.8 3.2
Anis
otro
py
[DNA] / µM

S18
12. Binding to Three-way DNA Junctions Despite the significant chemical differences, the structure of the described peptide helicate is in many ways similar to that of dinuclear supramolecular helicates previously described by Hannon et. al;22 a plausible binding model is based on the reported X-ray structure of the complex between the tetracationic triple helical supramolecular cylinder [Fe2(C25H20N4)3]Cl4 and a three-way DNA junction, as deposited in the PDB database (PDB ID# 2ET0). The minimized structure of the peptide helicate was superimposed with the original metallosupramolecular helicate in the X-ray (specifically, the 2 and 2’ carbon atoms in all the coordinating bipyridines were selected for superposition). Inspection of the resulting composite structure does not reveal any major incompatibilities, and in fact shows that the strands of both the organic and the peptide ligands can adopt a similar arrangement inside the three-way junction cavity. Structure superposition was made with the “Pair Fitting Wizard” as impolemented in MacPyMOL (PyMOL Molecular Graphics System, v1.6.0.0, Shrödinger LLC).
Figure S9. Result of first-order molecular modeling in which the metallopeptide (orange) is superimposed with the structure of the metallohelicate (blue) in complex with the DNA three-way junction (light blue surface). X-ray data taken from ref. 22.
S19
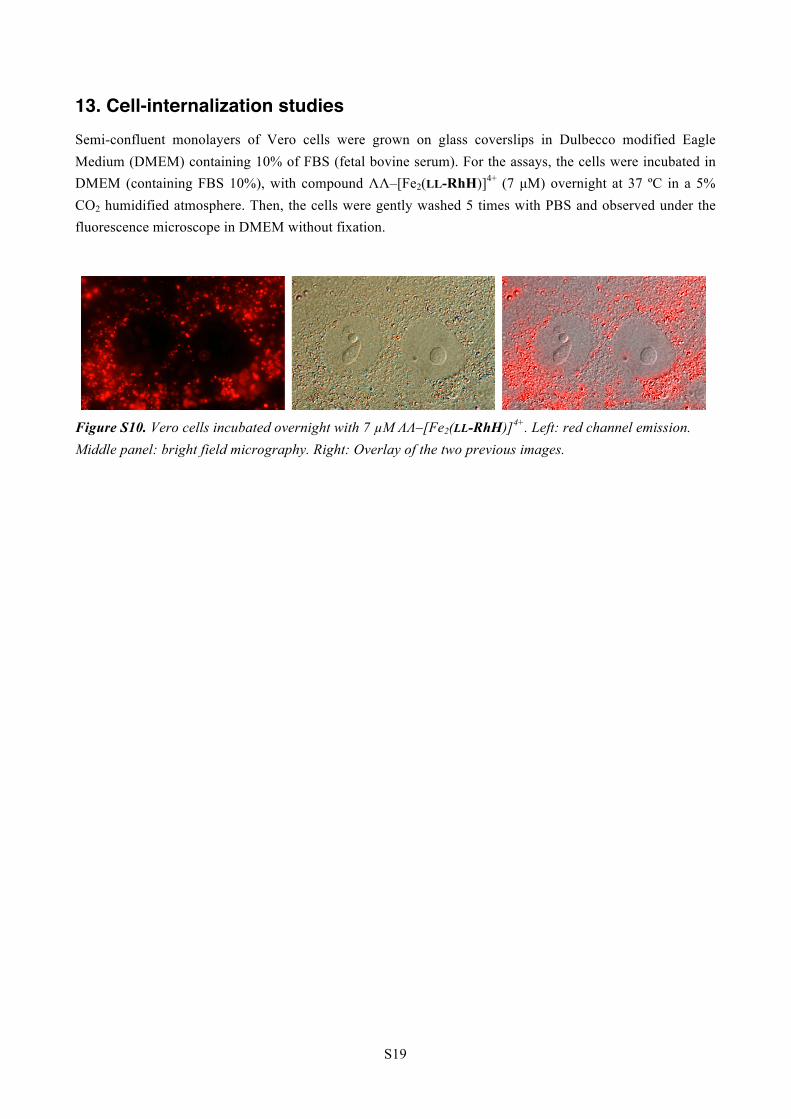
13. Cell-internalization studies Semi-confluent monolayers of Vero cells were grown on glass coverslips in Dulbecco modified Eagle Medium (DMEM) containing 10% of FBS (fetal bovine serum). For the assays, the cells were incubated in DMEM (containing FBS 10%), with compound ΛΛ–[Fe2(LL-RhH)]4+ (7 µM) overnight at 37 ºC in a 5% CO2 humidified atmosphere. Then, the cells were gently washed 5 times with PBS and observed under the fluorescence microscope in DMEM without fixation.
Figure S10. Vero cells incubated overnight with 7 µM ΛΛ–[Fe2(LL-RhH)]4+. Left: red channel emission. Middle panel: bright field micrography. Right: Overlay of the two previous images.

S20
References
1 G. Rama, A. Ardá, J.-D. Maréchal, I. Gamba, H. Ishida, J. Jiménez-Barbero, M. E. Vázquez, M. Vázquez López, Chem. Eur. J. 2012, 18, 7030.
2 G. R. Newkome, J. Gross, A. K. Patri, J. Org. Chem. 1997, 62, 3013.
3 F. H. Case, J. Am. Chem. Soc. 1946, 68, 2574
4 C. P. Whittle, J. Heterocyclic Chem. 1977, 14, 191.
5 E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, T. E. J Comput. Chem. 2004, 13, 1605.
6 K. Hinsen J. Comp. Chem. 2000, 21, 79.
7 D. A. Case., T. A. Darden., T.E. Cheatham III, C.L. J. Simmerling, R. E. Wang, R. Duke, R. C. Luo, Walker, W. Zhang, K.M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A.W. Goetz, I. Kolossváry, K.F. Wong, F. Paesani, J. Vanicek, R.M. Wolf, J. Liu, X. Wu, S.R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D.R. Roe, D.H. Mathews, M.G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, P.A. Kollman (2012), AMBER 12, University of California, San Francisco.
8 J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman, D. A. Case J. Comput. Chem. 2004, 25, 1157.
9 J. Gasteiger M. Marsili Tetrahedron 1980, 36, 3219.
10 L. A. Kelley, S. P. Gardner M. J. Sutcliffe. Protein Eng. 1997, 11, 1063.
11 S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma, M. J. Frisch J. Mol. Struct. (Theochem) 1999, 462, 1.
12 Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
13 A. D. Becke J. Chem. Phys. 1993, 98, 5648.
14 C. Lee, W. Yang, R. G. Parr, Phys. Rev. B 1988, 37, 785.
15 A. D. McLean G. S. Chandler, J. Chem. Phys. 1980, 72, 5639.
16 K. Raghavachari, J. S. Binkley, R. Seeger, J. A. Pople J. Chem. Phys. 1980, 72, 650.
17 N. B. Balabanov, K. A. Peterson J. Chem. Phys. 2005, 123, 64107.

S21
18 V. Barone M. Cossi J. Phys. Chem. A 1998, 102, 1995.
19 M. Cossi, N. Rega, G. Scalmani, V. Barone J. Comp. Chem. 2003, 24, 669.
20 T. Vreven, B. Mennucci, C. O. da Silva, K. Morokuma, J. Tomasi J. Chem. Phys. 2001, 115, 62.
21 P. Kuzmic, Anal. Biochem., 1996, 237, 260.
22 A. Oleksi, A. G. Blanco, R. Boer, I. Usón, J. Aymamí, A. Rodger, M. J. Hannon, M. Coll, Angew. Chem. Int. Ed., 2006, 45, 1227.


















