
Electronic-structure investigation of oxidized aluminum...
11
Electronic-structure investigation of oxidized aluminum films with electron-momentum spectroscopy X. Guo, S. Canney, A. S. Kheifets, M. Vos, Z. Fang, S. Utteridge, and I. E. McCarthy Electronic Structure of Materials Centre, The Flinders University of South Australia, Adelaide, South Australia 5001, Australia E. Weigold Research School of Physical Sciences and Engineering, The Australian National University, Canberra, Australian Capital Territory 0200, Australia ~Received 13 May 1996; revised manuscript received 6 September 1996! Electron-momentum spectroscopy ~EMS! or ( e ,2e ) measurements with oxidized aluminum thin films have been performed. Due to the surface sensitive nature of the EMS spectrometer employed, the measured ( e ,2e ) events come from the front oxidized layer as viewed by the electron detectors. The measurements show clearly two major features in the spectral momentum density distribution and they are related to the upper valence band and the lower valence band of aluminum oxide. The first is a ‘‘dual parabola’’ energy-momentum dispersion pattern spanning about 8 eV in the upper valence band. This dual parabola pattern has been qualitatively reproduced by a linear muffin-tin orbital ~LMTO! calculation on spherically averaged a -Al 2 O 3 with nearly the same energy span. In the lower valence band, the LMTO calculation indicates a dispersion spanning about 5 eV, and the measured spectral momentum density plot shows a similar ‘‘bowl’’ shape but with less dispersion. The possible causes that blur the dispersion in the lower valence band are discussed. Other features in the spectral momentum density distribution are also discussed and compared with the LMTO calculation. @S0163-1829~96!08147-7# I. INTRODUCTION Electron momentum-spectroscopy ~EMS! is a spectro- scopic technique that can measure electron-momentum dis- tributions directly and visualize the dispersion relation be- tween energy and momentum of electrons in solids. 1 This was demonstrated by a series of ( e ,2e ) experiments on solid targets 2–10 in the last few years. In these experiments, the ( e ,2e ) spectrometer 11 of The Flinders University of South Australia was used. It has increased ( e ,2e ) coincidence count rates @up to 1000 counts/min ~Ref. 12!# and signifi- cantly improved both energy and momentum resolution. In addition to the energy-resolved momentum distribution of electrons in valence bands 5,8–10 and core levels, 4,9 which can be uniquely measured by EMS, information has also been obtained on the influence of lattice order on the electronic structure, 6 the electronic structure of adsorbates, 7,9 and the annealing effects on the electronic structure of the surface. 3 The targets used in these investigations include carbon ~amorphous carbon, graphite, highly oriented pyrolitic graphite, and diamondlike amorphous carbon!, semiconduc- tors ~amorphous silicon and amorphous germanium!, and a semiconductor compound ~polycrystalline silicon carbide!. In fact EMS is now being applied to the study of a wide range of materials. Among the wide range of materials that can be explored by EMS, metals and metal oxides are a particularly interest- ing group of targets. First of all, EMS on metals and metal oxides may provide an approach to the subject of electron correlation effects, which are an important factor in under- standing electronic structures and properties of solid materi- als from 3 d metals 13 to high-T c superconductors. 14 Second, the interest in metals and metal oxides is not only due to their importance in fundamental aspects in solid-state phys- ics, but also due to their wide application in microelectron- ics. Metal oxides usually have a much more complex elec- tronic structure than metals. This provides a challenge to EMS of whether it can be applied to study materials with complex bulk structures. An interesting example is alumi- num metal and aluminum oxide. Aluminum is a simple case, being a ‘‘free-electron’’ metal with a straightforward face centered cubic ~fcc! crystal structure. A parabolic free elec- tron energy-momentum dispersion pattern should be visual- ized with EMS. As a matter of fact, recent EMS measure- ment on aluminum metal 15 showed excellent agreement with a free-electron model calculation. Aluminum oxide ~Al 2 O 3 ) is an ionic solid and an insulator and has, on the other hand, a much more complex crystal structure. In the a -Al 2 O 3 form, for example, its structure may be described as a hexagonal close packing array of oxygen atoms with two- thirds of the octahedral holes occupied by aluminum atoms. Its peculiar crystal structure and wide-ranging applications have drawn both theoretical and experimental interest. In- deed, the electronic structure of aluminum oxide has been widely studied both theoretically 16–22 and experimentally 23,24 However, there is no general agreement theoretically 17,16,19 and experimentally. 23,25–29 on its band structure details, and particularly few of them have concerned the momentum dis- tribution aspect of its valence-band electrons. 24,30 In this paper we describe our recent investigations of the electronic structure of oxidized aluminum thin films with EMS. The oxidized layers on the aluminum thin film are related to aluminum oxide ~Al 2 O 3 ). To analyze and interpret the experimental results a spherically averaged linear muffin- PHYSICAL REVIEW B 15 DECEMBER 1996-II VOLUME 54, NUMBER 24 54 0163-1829/96/54~24!/17943~11!/$10.00 17 943 © 1996 The American Physical Society
Transcript of Electronic-structure investigation of oxidized aluminum...

ia
PHYSICAL REVIEW B 15 DECEMBER 1996-IIVOLUME 54, NUMBER 24
Electronic-structure investigation of oxidized aluminum filmswith electron-momentum spectroscopy
X. Guo, S. Canney, A. S. Kheifets, M. Vos, Z. Fang, S. Utteridge, and I. E. McCarthyElectronic Structure of Materials Centre, The Flinders University of South Australia, Adelaide, South Australia 5001, Austral
E. WeigoldResearch School of Physical Sciences and Engineering, The Australian National University, Canberra,
Australian Capital Territory 0200, Australia~Received 13 May 1996; revised manuscript received 6 September 1996!
Electron-momentum spectroscopy~EMS! or (e,2e) measurements with oxidized aluminum thin films havebeen performed. Due to the surface sensitive nature of the EMS spectrometer employed, the measured(e,2e) events come from the front oxidized layer as viewed by the electron detectors. The measurements showclearly two major features in the spectral momentum density distribution and they are related to the uppervalence band and the lower valence band of aluminum oxide. The first is a ‘‘dual parabola’’ energy-momentumdispersion pattern spanning about 8 eV in the upper valence band. This dual parabola pattern has beenqualitatively reproduced by a linear muffin-tin orbital~LMTO! calculation on spherically averageda-Al2O3
with nearly the same energy span. In the lower valence band, the LMTO calculation indicates a dispersionspanning about 5 eV, and the measured spectral momentum density plot shows a similar ‘‘bowl’’ shape butwith less dispersion. The possible causes that blur the dispersion in the lower valence band are discussed. Otherfeatures in the spectral momentum density distribution are also discussed and compared with the LMTOcalculation.@S0163-1829~96!08147-7#
de
th
I
eeni
cebotic
e
restatroerte
toys-n-ec-to
ithi-se,e-ual-re-hehetheaso-
ms.nsIn-een
dis-
thehretffin-
I. INTRODUCTION
Electron momentum-spectroscopy~EMS! is a spectro-scopic technique that can measure electron-momentumtributions directly and visualize the dispersion relation btween energy and momentum of electrons in solids.1 Thiswas demonstrated by a series of (e,2e) experiments on solidtargets2–10 in the last few years. In these experiments,(e,2e) spectrometer11 of The Flinders University of SouthAustralia was used. It has increased (e,2e) coincidencecount rates@up to 1000 counts/min~Ref. 12!# and signifi-cantly improved both energy and momentum resolution.addition to theenergy-resolvedmomentum distribution ofelectrons in valence bands5,8–10and core levels,4,9 which canbe uniquely measured by EMS, information has also bobtained on the influence of lattice order on the electrostructure,6 the electronic structure of adsorbates,7,9 and theannealing effects on the electronic structure of the surfa3
The targets used in these investigations include car~amorphous carbon, graphite, highly oriented pyroligraphite, and diamondlike amorphous carbon!, semiconduc-tors ~amorphous silicon and amorphous germanium!, and asemiconductor compound~polycrystalline silicon carbide!.In fact EMS is now being applied to the study of a widrange of materials.
Among the wide range of materials that can be exploby EMS, metals and metal oxides are a particularly intereing group of targets. First of all, EMS on metals and meoxides may provide an approach to the subject of eleccorrelation effects, which are an important factor in undstanding electronic structures and properties of solid maals from 3d metals13 to high-Tc superconductors.
14 Second,
540163-1829/96/54~24!/17943~11!/$10.00
is--
e
n
nc
.n
dt-ln-ri-
the interest in metals and metal oxides is not only duetheir importance in fundamental aspects in solid-state phics, but also due to their wide application in microelectroics. Metal oxides usually have a much more complex eltronic structure than metals. This provides a challengeEMS of whether it can be applied to study materials wcomplex bulk structures. An interesting example is alumnum metal and aluminum oxide. Aluminum is a simple cabeing a ‘‘free-electron’’ metal with a straightforward faccentered cubic~fcc! crystal structure. A parabolic free electron energy-momentum dispersion pattern should be visized with EMS. As a matter of fact, recent EMS measument on aluminum metal15 showed excellent agreement wita free-electron model calculation. Aluminum oxid~Al 2O3) is an ionic solid and an insulator and has, on tother hand, a much more complex crystal structure. Ina-Al2O3 form, for example, its structure may be describeda hexagonal close packing array of oxygen atoms with twthirds of the octahedral holes occupied by aluminum atoIts peculiar crystal structure and wide-ranging applicatiohave drawn both theoretical and experimental interest.deed, the electronic structure of aluminum oxide has bwidely studied both theoretically16–22and experimentally23,24
However, there is no general agreement theoretically17,16,19
and experimentally.23,25–29on its band structure details, anparticularly few of them have concerned the momentum dtribution aspect of its valence-band electrons.24,30
In this paper we describe our recent investigations ofelectronic structure of oxidized aluminum thin films witEMS. The oxidized layers on the aluminum thin film arelated to aluminum oxide~Al 2O3). To analyze and interprethe experimental results a spherically averaged linear mu
17 943 © 1996 The American Physical Society

coum
la-ts
nthothroncss
re
eios
fam-to
uldul--
ewendva
isth
etw
s
f
um
are
icofhisoseingonethedhe
r
of
lec-
l
2
17 944 54X. GUO et al.
tin orbital ~LMTO! calculation ona-Al2O3 has been per-formed. Any of the other forms of Al2O3, although theirband structure in the reduced zone scheme may appearpletely different, are all expected to have similar momentdensities in the real momentum space.
This paper is organized as follows: The LMTO calcution ona-Al2O3 is described in Sec. II. Section III consisof two parts. The EMS technique and the (e,2e) spectrom-eter used in this experiment are described in Sec. III A athe details of the preparation and the characterization ofsamples are given in Sec. III B. In Sec. IV, the spectral mmentum density plots of both the EMS measurement andLMTO calculation are presented. The details of the electenergy-momentum distributions for both the upper valeband and the lower valence band are shown and discualso in Sec. IV. Conclusions are drawn in Sec. V.
II. THEORY
It is known that aluminum oxide has different structuforms such asa-Al2O3 , g-Al2O3, and a-Al2O3. Even so,the broad features of their density of states and plasmoncitations are similar, as indicated by x-ray photoemissspectroscopy23 ~XPS! and electron energy-losspectroscopy.31 The XPS data of Balzarotti and Bianconi23
also suggest that an amorphous aluminum oxide surlayer has similar electronic properties to its crystalline forof a-Al2O3 andg-Al2O3. For that reason it is generally accepted thata-Al2O3 can be taken as a suitable prototypeinterpret experimental spectroscopic results.
We follow this logic and perform anab initio self-consistent calculation of the electronic structure on ba-Al2O3, which we will be referring to in analyzing aninterpreting our experimental data. For the present calction we employ the LMTO method32 in atomic-sphere approximation~ASA! with von Barth and Hedin parametrization for the exchange-correlation potential.33 Although theLMTO method is just one of many computational schemderived within the general density-functional philosophy,find it advantageous in terms of accuracy and computatioefficiency and capable of performing full-scale banstructure calculations on solids with a large number oflence electrons per unit cell.
The atomic arrangement ina-Al2O3 ~corundum structure!is quite complicated. The Bravais lattice of corundumtrigonal with all the primitive lattice vectors of equal lenga ~9.694 a.u.! forming an equal angleb (55°68) betweenany two of them.34 In Cartesian coordinates, the primitivlattice vectors are conveniently expressed in terms of theconstants,s and r ,
S t1t2t3D 5S s•ex 0•ey r •ez
2s/2•ex sA3/2•ey r •ez
s/2•ex 2sA3/2•ey r •ezD , ~1!
wheres5(2a/A3)sin(b/2) ~5.17 a.u.! andr5Aa22s2 ~8.18a.u.!.35 The volume of the elementary cell iV5(3A3/2)s2r .
The unit cell ofa-Al2O3 contains two molecular units oAl 2O3 ~ten atoms altogether!, arranged symmetrically withrespect to the origin as shown in Fig. 1. The four alumin
m-
de-eneed
x-n
ces
k
a-
s
al--
o
atoms are arranged along thez axis at distances60.435r and61.065r .35 The two oxygen groups, each of three atoms,arranged in two planes, which are perpendicular to thez axisand cross it at distances60.750r . The oxygen atoms formequilateral triangles with length of 0.574r .
The essence of the LMTO-ASA method is that the atompolyhedron is filled with a number of atomic spheres, eachwhich represents a nonequivalent atomic position. Tmethod is mostly suitable for close-packed structures whelementary cell can be spanned effectively by touchatomic spheres. ‘‘Open’’ structures such as the diamondcan be treated by adding ‘‘empty’’ atomic spheres atinterstitial sites.36 We follow this scheme for corundum anplace two additional ‘‘empty’’ spheres at the origin and tz axis at a distance of 1.50r . The equivalent position at adistance of21.50r can be reached by a primitive vectotranslation2t12t22t3 and is not included in the muffin-tinbasis. So, we haveN512 atomic spheres in the basis eachthe radius
Rs5S 1N 3
4pV D 1/352.246 a.u.
The valence-band structure ofa-Al2O3 along the twohigh-symmetry directions,GX andGZ, is plotted in Fig. 2together with the Brillouin zone of thea-Al2O3 crystal.There are total of 24 bands, which accommodate 48 e
TABLE I. Valence-band widths ina-Al2O3.
Valence-band width~eV!
Type of calculation Upper Gap Lower Tota
SemiempiricalEvarestovet al. ~Ref. 21! 12.06 10.42 5.45 28.23Ciraci and Batra~Ref. 19! 11.8 6.3 9.5 27.6
First principlesBatra ~Ref. 20! 6 10 3 19Xu and Ching~Ref. 17! 7.39 8.53 3.26 19.18Godin and LaFemina~Ref. 16! 6.5 8.7 3 18.2Present, LMTO 7.31 8.62 3.39 19.3
FIG. 1. The unit cell ofa-Al2O3. Thez axis is indicated.

tu,nc
.he.
Vurre
hd
mi-nd
omy oftheels
ntal
nd
ion
hen-re-jor
sent
sityec-trypa-
bo-he
esmcondikero
54 17 945ELECTRONIC-STRUCTURE INVESTIGATION OF . . .
trons in the elementary cell. The most characteristic feaof the valence band in Al2O3 is its split into two subbandswhich are commonly labeled the lower and upper valebands. The lower valence band~LVB ! comprising six sepa-rate bands originates from the 2s states of six oxygen atomsIn our model the LVB spans approximately 3.4 eV. Tupper valence band~UVB! comprises 18 individual bandsThey are derived from the oxygen 2p orbitals. The UVBspan is approximately 7.3 eV. The interband gap is 8.6 e
It is worthwhile to compare the present band-structcalculation with the earlier theoretical results, which asummarized in Table I. All the calculations ona-Al2O3 re-ported to date can be classified into eitherab initio ~or first-principle calculations!, or semiempirical calculations, whicuse a set of adjustable parameters to fit spectroscopic
FIG. 2. ~a! The valence-band structure ina-Al2O3 along theGX andGZ directions, and~b! the Brillouin zone of thea-Al2O3
crystal.
re
e
.e
ata
available from experiment. As seen from the table, the seempirical models produce a significantly wider valence bathan that from theab initio calculations. This result wasnoted previously by Ciraci and Batra.19 One of the possibleexplanations of this phenomenon is that wider bands frthe spectroscopic experiments are due to the poor qualitthe samples and/or insufficient energy resolution. Byvery nature of the fitting procedure the semiempirical modreproduce these bands.Ab initiomodels are free of any of theadjustable parameters and not affected by any experimeresults.
We should mention that not only are our valence-bawidths in good agreement with otherab initio calculations,but also the intimate details of our band-structure calculatare very close to those reported by other authors~see, forinstance, Fig. 2 of Ref. 20!.
After producing the valence-band structure from tLMTO model we proceed with the spectral momentum desity calculation. This is a quite straightforward procedudescribed elsewhere.37,38 The results of the momentum density calculation are presented in Fig. 3 for the three mahigh-symmetry directionsGX, GY, andGZ, and in Fig. 6 asa spherical average to be compared directly with the preexperimental data.
The two-dimensional band energy and momentum denplots of Fig. 3 allow us to restore the three-dimensional sptral momentum density in the three major high-symmedirections. The LVB disperses along an approximaterabola near the center of the Brillouin zone~BZ!. After itreaches the BZ boundary it either bends down (GZ direction!or remains flat (GX, GY direction!. The momentum densitydecreases fast beyond the first BZ. So only the free paralalike part of the dispersion curve will be observable in texperiment.
The behavior of the UVB is quite peculiar. It dispersdownwards right from the center of the BZ. The momentudensity increases and peaks near the middle of the seBZ. So, experimentally, one would observe a parabolalshape with a minimum in energy shifted away from zemomentum.
m
he
FIG. 3. The band energies and momentudensities for the upper valence band~two toprows! and the lower valence band~two bottomrows! in a-Al2O3. Highlighted are the total mo-mentum density~momentum density plots! andthe band that contributes most significantly to tmomentum density~band energy plots!. The mo-menta are in atomic units.

tive
17 946 54X. GUO et al.
FIG. 4. The schematic representation of the noncoplanar asymmetric geometry used in the (e,2e) measurements.~a! The scatteringgeometry. With this geometry the momenta of the target electrons are detected nominally along they axis. ~b! The incident and outgoingbeams relative to the sample. Shaded area is the exit surface where most detectable (e,2e) events occur. This determines the surface sensinature of the experiment.
itrnllt olo
all
t
ac
i-t
ndth
yicth
oe
es
rfo
t ofre-
e
en-
d
not.en
narhe-hegy.anlowea-ea-
ec-c-
nmh inonmi-fory ofuredomm
ntlytheVmen
silyenticalolar
Since in all the three directions the momentum densand the band energies are quite similar, the same pattethe spectral momentum density will persist after sphericaaveraging over the irreducible wedge of the BZ. The resulthis averaging is shown in Fig. 6 as a linear grey-scale pThe LVB displays itself as a ‘‘bowl’’ while the UVB has apeculiar form of a dual parabolalike shape in the sphericaveraged spectral momentum density plot~Fig. 6, centralpanel!. These characteristic features will be discussed latogether with the experimental data.
III. EXPERIMENTAL DETAILS
A. EMS technique and the spectrometer
Electron-momentum spectroscopy is based on the (e,2e)reaction.39 In an (e,2e) reaction, under suitable conditions,binary collision occurs in which a high-energy incident eletron with energyE0 and momentump0 collides with one ofthe bound electrons in the target and the scattered~the fastone, energyEf , momentumpf) and ejected~the slow one,energyEs , momentumps) electrons are detected in coincdence and analyzed for energy and momentum. Thenbinding energy« and momentumq of the bound electron inthe targetbeforethe collision are determined via energy amomentum conservation neglecting the recoil energy ofion:
E02«5Ef1Es , ~2!
and
p01q5pf1ps , ~3!
whereq is the momentum of the bound electron.It is known1,40 that the (e,2e) cross section at high energ
and high momentum transfer is, in the independent partapproximation, proportional to the modulus square ofbound electron momentum space wave functionuf(«,q)u2,i.e., the electron spectral momentum density. In the casecrystal target the proportionality, ignoring diffraction of thincident and outgoing electrons, may be expressed as40
d5s
dV fdVsdEf5~2p!4N
pfpsp0
f df eeuf~«,q!u2, ~4!
whereN is the number of unit cells in the crystal,f d is adispersion factor, which is nearly 1 in practical caspf(5upf u) andps(5upsu) are fixed when varyingpf andps~angles! to scanq, and f ee is the Mott cross section foelectron-electron scattering, which is very near constant
yofyft.
y
er
-
he
e
lee
f a
,
r
the momentum range of interest. Thus the measuremen(e,2e) cross sections at different kinematic conditions corsponding to different momentaq at different binding ener-gies « via Eqs. ~2! and ~3! is a direct measurement of thspectral momentum densityuf(«,q)u2 of the bound electronsin the target and this is often referred to as electron momtum spectroscopy.
For crystalline solid targets diffraction of the incident anoutgoing electrons in the (e,2e) reaction may occur. Whenthe diffraction happens Eq.~4! may not generally hold. How-ever, for disordered solid targets where long range orderlonger exists, Eq.~4! is applicable. It should be noted thaEMS measures thereal momentum of the bound electrons
The (e,2e) spectrometer used in this experiment has bedescribed in detail elsewhere.11 For clarity a brief descriptionis given here. The spectrometer is set up in a noncoplaasymmetric geometry and in the transmission mode. A scmatic representation of the geometry is shown in Fig. 4. Tincident electron energy is 20 keV plus the binding enerThe thin-film sample is held vertically and positioned atangle of 30° towards the incident beam. The fast and selectrons have energies of 18.8 and 1.2 keV, and are msured in coincidence with two electron analyzers each msuring simultaneously a range of azimuthal angles~out of theplane! and energies at polar angles of 14° and 76°, resptively. With this kinematics the momenta of the target eletrons are detected nominally along they axis within the thinfilm, and the spectrometer is surface sensitive to about 2of the sample on the exit side due to the mean free patthe solid of the 1.2-keV outgoing electrons. The electranalyzer used for measuring the fast electrons is a hespherical analyzer with a pass energy of 100 eV, the onethe slow electrons is a toroidal analyzer with a pass energ200 eV. The ranges of energy and azimuthal angle measby the two analyzers are from 18 790 to 18 810 eV and fr218° to 118° for the hemispherical analyzer, and fro1182 to 1218 eV and fromp26° to p16° for the toroidalanalyzer, respectively. The energy resolution has recebeen improved with a monochromator incorporated withelectron gun.41 An overall measurable energy range of 56 ewith a resolution of 0.9 eV and momentum range fro23.5 to 3.5 a.u. with a resolution of 0.15 a.u. have beachieved.
An in situelectron energy loss measurement can be eaconducted with the spectrometer by adjusting the incidelectron energy to match the setting of the hemispheranalyzer, which views the electrons scattered into the pangle of 14°. Transmission electron diffraction~TED! pat-

nou
rte
donndm
armoo
in
am
se
r
pueeinbw
hi-
s
nsumislu
triotrtdi
d
umatio
thein
edac-heonmpleoftal
the
gara-otherrstor-terwasndwerghin
ofedpleo-
thet-1.5eyer
wo
ter
as
heable
sed.ctraentplecter-onwasec-t 15
54 17 947ELECTRONIC-STRUCTURE INVESTIGATION OF . . .
terns of the samples can be also measuredin situwith a TEDsystem attached to the spectrometer chamber.
B. Preparation and characterization of the samples
Two samples, one a self-supporting aluminum film athe other an evaporated aluminum film on an amorphcarbon substrate, were prepared for this experiment.
The self-supporting sample preparation process stawith nominally 40-nm-thick aluminum films, which wervacuum evaporated on rocksalt~NaCl!. The aluminum filmon rocksalt was cleaved to a 434-mm square and then sliinto an 80% deionized water and 20% methanol solutiThe aluminum film was floated off the rocksalt support athen caught with a molybdenum sample holder. The alunum film covered a few holes~0.7 mm in diameter! of thesample holder. The sample holder was put into an ovenheated slowly to 100 °C for about 1 h toevaporate the watebefore putting it into an ion beam sputtering vacuum chaber for further processing. The 40-nm aluminum film is tthick to be used as a target in our (e,2e) spectrometer. Ionbeam thinning~IBT! was therefore used to prepare the thfilm to a thickness that gives a sufficient (e,2e) coincidencecount rate. This was done in the ion beam sputtering chber with a background pressure in the low 1026-Torr range.A saddle-field ion source was used to produce a well focuAr 1 ion beam. At 5-kV anode voltage a 5-mA Ar1 beamwas produced when pure Ar gas was fed into the ion souto a pressure of 431025 Torr. The sputter thinning tookabout 10 min. During the thinning process, a light wasunderneath the film. The change of color of the film as sfrom an optical microscope indicated when sufficient thning had taken place. After the thinning process the chamwas pumped back to the base pressure and the sampletransferred under vacuum to a preparation chamber, wwas in ultrahigh vacuum~UHV!, and then to the spectrometer chamber under UHV. An (e,2e) coincidence count rateof 35 counts/min was achieved. The estimated thicknesthis self-supporting sample was about 15 nm.
It is known that aluminum oxidizes readily in air to forma saturated oxide layer with a thickness of about 1.5–2~Refs. 42 and 43! that resists further corrosion, known apassivation. Due to the reactive nature of the aluminmetal and the vacuum condition for ion sputtering in thexperiment, an aluminum oxide layer remained on the aminum film after the ion beam thinning. An Auger spectrometer, which has since been upgraded, was available inUHV preparation chamber and was then used to charactethe aluminum film surface, which was viewed by the twanalyzers of the spectrometer. The measured Auger elecspectrum showed a peak at 5462 eV. This peak was thoughto be theL II,IIIVV cross transition between aluminum anoxygen in aluminum oxide, corresponding to a vacancythe L II,III level of aluminum in Al2O3 and cross transitionsfrom oxygen supplying the down~recombination! and up~Auger emission! electrons as explained by Quinto anRobertson.44 The Auger LMM peak of pure aluminumshould be at 70 eV. An oxygen peak at 51862 eV wasclearly observed and most likely related to the aluminoxide layer on the aluminum film. A carbon peak27562 eV was also observed. This carbon contaminat
ds
ed
.
i-
nd
-
-
d
ce
tn-eras
ch
of
m
--heze
on
n
n
might come from the process of vacuum evaporation offilm or the ion beam sputtering due to the modest vacuumthe sputtering chamber, or both.
A TED measurement of this sample was also performat 10, 20, and 30 keV incident electron energies. The diffrtion rings manifested polycrystalline bulk structure of tsample and matched those of aluminum metal. Basedthese measurements, we regarded this self-supporting saas a thin film with aluminum oxide layers of thickness1.5–2 nm sandwiching a polycrystalline aluminum mefilm. The measured (e,2e) events come essentially only fromthe oxide layer viewed by the two analyzers because ofsurface sensitive nature of the spectrometer.
The possibility of contamination of the self-supportinsample due to the modest vacuum condition in the preption process concerned us and we therefore prepared ansample in a different way. This sample was prepared by fievaporating 3 nm of pure aluminum on a 5-nm-thick amphous carbon film, which covered the 1.0-mm-diameholes of a sample holder. The evaporation thicknessmonitored with a crystal thickness monitor. The backgroupressure of the preparation chamber was in the lo10210-Torr range. The pressure went up to the hi1029-Torr range during evaporation. The sample obtainedthis way was then exposed to pure O2 ~99.9%! at 1 atm. for2 min. This produced an oxidized layer with a thicknessabout 1.5 nm. After an EMS measurement, which showboth aluminum and aluminum oxide features, this samwas covered with an additional 1 nm of aluminum by evapration and exposed to pure O2 again at 1 atm. for 2 min. Thereason for the second evaporation and exposure ofsample to O2 is that the mean free path of the 1.2-keV ougoing electrons for aluminum oxide may be larger thannm, which means that (e,2e) events occurring beyond th1.5-nm oxidized layer are still detectable. The oxidized laresists further oxidation to deeper depth.
The sample front surface that would be viewed by the tanalyzers was characterized before and after the O2 expo-sures with our newly installed Auger electron spectrome~PHI Model 3017, Physical Electronics, Inc.!, which has anenergy resolution of 0.6%. The oxidized aluminum layer wclearly identified by the shift of the AugerLMM peak of theevaporated aluminum as shown in Figs. 5~a! and 5~b! whereFig. 5~a! is an enlarged portion of Fig. 5~c! in the kineticenergy range 40–80 eV and Fig. 5~b! is that of Fig. 5~d! inthe same kinetic energy range. The AugerLMM peak of thepure aluminum sample was located at 69 eV@Fig. 5~a!#. Af-ter the oxidation this peak shifted to 57 eV@Fig. 5~b!#, indi-cating the formation of the aluminum oxide layer on tsurface. The carbon and oxygen peaks were still observfor the evaporated aluminum sample@Fig. 5~c!#, but quiteweak. An (e,2e) count rate of 65 counts/min with thicarbon-film-supported aluminum oxide sample was obtain
After the EMS measurement, electron energy loss spewere measured for both of the samples at 18.8-keV incidelectron energy. The spectrum for the self-supporting samshowed a sharp aluminum plasmon peak at 15-eV charaizing the bulk of the sample. The aluminum double plasmpeak could hardly be seen, which indicated the samplevery thin. For the carbon-film-supported sample the sptrum showed both the sharp aluminum plasmon peak a

e
in
.
17 948 54X. GUO et al.
FIG. 5. The Auger electron spectra for thevaporated aluminum sample~a! and~c!, and theoxidized aluminum sample~b! and ~d!. The Au-ger LMM peak of aluminum at 69 eV shifts 12eV down to 57 eV after the oxidation as shown~a! and ~b!. This indicates the formation of thealuminum oxide layer on the aluminum surface
Thethcioineth
sidht
ed
y-eteaeloo
ionh of-i-retral
lec-
ea-nce
ruc-isThes ain
r-eroal-
eV and the broad carbon plasmon feature at 24 eV.electron energy loss measurement with 18.8-keV incidelectron energy is not sensitive to the surface layer ofsamples. The features in the electron energy loss spewere taken into account in the procedure of deconvolutfor the plasmon excitations by the incident and outgoelectrons. The effect of the plasmon excitations on the msured spectral momentum densities will be discussed innext section.
IV. RESULTS AND DISCUSSION
Figure 6 presents the spectral momentum density plotmeasured with the spectrometer for the aluminum oxsamples~left panel for the self-supporting sample, rigpanel for the carbon-film-supported sample! and calculatedusing the LMTO method on a spherically averaga-Al2O3 ~central panel!, respectively. A spectral momentumdensity plot is an intensity distribution in an energmomentum plane. The intensity is represented by the lingrey scale, the darker scale corresponding to a higher insity. The highest density in all these plots has been normized to unity for ease of comparison. The statistical unctainties in the measured data can be gauged from the pshown in Figs. 8–10. The LMTO calculation of spectral m
entetranga-e
ase
arn-l-r-ts-
mentum density of spherically averageda-Al2O3 shown inFig. 6 has been convoluted with the 1-eV energy resolutof the spectrometer. Because of the small mean free patthe slow electron in the solid, the (e,2e) events can be regarded as originating almost conclusively from the alumnum oxide layer. Contributions from the aluminum metal aindeed negligible. This is clear from the measured specmomentum densities~Fig. 6, left and right panels! wherethere is no obvious parabolic dispersion feature of free etrons in aluminum as measured by Canneyet al.15 with thesame EMS spectrometer.
As discussed earlier aluminum oxide has two distinct ftures in its valence-band structure, namely, an upper valeband and a lower valence band. A previous (e,2e)experiment24 on an aluminum-aluminum oxide thin foil withan energy resolution of 4.5 eV reported dispersionless sttures in both the upper and lower valence bands. Thismanifestly not the case in the present measurements.present measured spectral momentum density plot show‘‘dual parabola’’ dispersion pattern spanning about 8 eVthe upper valence band~Fig. 6, left and right panels!. TheLMTO calculation shows a similar ‘‘dual parabola’’ dispesion pattern with nearly the same energy span. Near zmomentum both the EMS measurement and the LMTO c
tslf-
dd
eal-n
FIG. 6. The spectral momentum density ploas measured with the spectrometer for the sesupporting~left panel! and carbon-film-supported~right panel! aluminum oxide samples comparewith a LMTO calculation on spherically averagea-Al2O3 ~central panel!, respectively. The bind-ing energy is relative to the vacuum level. Thhighest density in each panel has been normized to unity. The linear grey scale is shown othe right-hand side.
c
tTncn-nl’dtenhntmu
oni
ro
s
oxy-
e2mxy-at
andceheayes
pec-hemi-ingomthetedrawin
ibu-rgyce-.
en-
ortum
can
y
in-erchonessnheec-Therk.of
ergy
ovlelegeaontruth
ela
54 17 949ELECTRONIC-STRUCTURE INVESTIGATION OF . . .
culation exhibit lower intensity throughout the upper valenband.
There is a gap between the upper valence band andlower valence band. The EMS measurement and the LMcalculation give nearly the same gap. In the lower valeband, the LMTO calculation, after convolution with the eergy resolution of the spectrometer, indicates a dispersioabout 5 eV. The EMS measurement shows a similar ‘‘bowshape in the spectral momentum density, i.e., the full wiof the band becoming narrower with increasing bindingergy in the lower valence band, but with less dispersion. Tpossible causes that blur the dispersion in the lower valeband will be discussed later. Nevertheless, one can seethe measured major features in the valence band of alunum oxide are qualitatively reproduced by the LMTO calclation.
Actually, the two distinct features in the valence bandaluminum oxide can be explained in terms of the electrostructure of its molecular unit Al2O3. Aluminum oxide is anionic solid. Its molecular unit Al2O3 is formed stably wheneach of the two aluminum atoms loses three electrons ftheir outermost orbitals (3s2,3p1) to become cation Al31
and each of the three oxygen atoms gains two electron
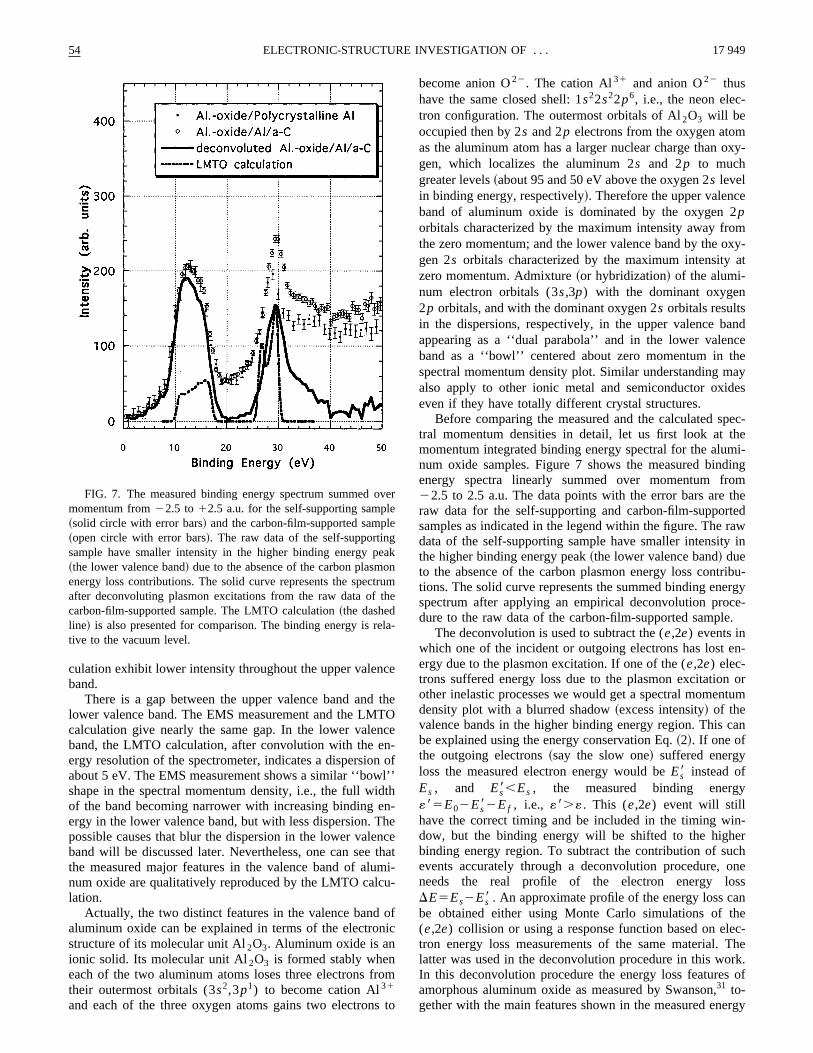
FIG. 7. The measured binding energy spectrum summedmomentum from22.5 to12.5 a.u. for the self-supporting samp~solid circle with error bars! and the carbon-film-supported samp~open circle with error bars!. The raw data of the self-supportinsample have smaller intensity in the higher binding energy p~the lower valence band! due to the absence of the carbon plasmenergy loss contributions. The solid curve represents the specafter deconvoluting plasmon excitations from the raw data ofcarbon-film-supported sample. The LMTO calculation~the dashedline! is also presented for comparison. The binding energy is rtive to the vacuum level.
e
heOe
of’h-ecehati--
fc
m
to
become anion O22. The cation Al31 and anion O22 thushave the same closed shell: 1s22s22p6, i.e., the neon elec-tron configuration. The outermost orbitals of Al2O3 will beoccupied then by 2s and 2p electrons from the oxygen atomas the aluminum atom has a larger nuclear charge thangen, which localizes the aluminum 2s and 2p to muchgreater levels~about 95 and 50 eV above the oxygen 2s levelin binding energy, respectively!. Therefore the upper valencband of aluminum oxide is dominated by the oxygenporbitals characterized by the maximum intensity away frothe zero momentum; and the lower valence band by the ogen 2s orbitals characterized by the maximum intensityzero momentum. Admixture~or hybridization! of the alumi-num electron orbitals (3s,3p) with the dominant oxygen2p orbitals, and with the dominant oxygen 2s orbitals resultsin the dispersions, respectively, in the upper valence bappearing as a ‘‘dual parabola’’ and in the lower valenband as a ‘‘bowl’’ centered about zero momentum in tspectral momentum density plot. Similar understanding malso apply to other ionic metal and semiconductor oxideven if they have totally different crystal structures.
Before comparing the measured and the calculated stral momentum densities in detail, let us first look at tmomentum integrated binding energy spectral for the alunum oxide samples. Figure 7 shows the measured bindenergy spectra linearly summed over momentum fr22.5 to 2.5 a.u. The data points with the error bars areraw data for the self-supporting and carbon-film-supporsamples as indicated in the legend within the figure. Thedata of the self-supporting sample have smaller intensitythe higher binding energy peak~the lower valence band! dueto the absence of the carbon plasmon energy loss contrtions. The solid curve represents the summed binding enespectrum after applying an empirical deconvolution produre to the raw data of the carbon-film-supported sample
The deconvolution is used to subtract the (e,2e) events inwhich one of the incident or outgoing electrons has lostergy due to the plasmon excitation. If one of the (e,2e) elec-trons suffered energy loss due to the plasmon excitationother inelastic processes we would get a spectral momendensity plot with a blurred shadow~excess intensity! of thevalence bands in the higher binding energy region. Thisbe explained using the energy conservation Eq.~2!. If one ofthe outgoing electrons~say the slow one! suffered energyloss the measured electron energy would beEs8 instead ofEs , and Es8,Es , the measured binding energ«85E02Es82Ef , i.e., «8.«. This (e,2e) event will stillhave the correct timing and be included in the timing wdow, but the binding energy will be shifted to the highbinding energy region. To subtract the contribution of suevents accurately through a deconvolution procedure,needs the real profile of the electron energy loDE5Es2Es8 . An approximate profile of the energy loss cabe obtained either using Monte Carlo simulations of t(e,2e) collision or using a response function based on eltron energy loss measurements of the same material.latter was used in the deconvolution procedure in this woIn this deconvolution procedure the energy loss featuresamorphous aluminum oxide as measured by Swanson,31 to-gether with the main features shown in the measured en
er
k
me
-

.0.1
hee
17 950 54X. GUO et al.
FIG. 8. The binding energy spectra from 0to 1.6 a.u. of momentum at equal intervals of 0a.u. The raw data~with the error bars! have beendeconvoluted for plasmon excitations~the fullline!. The dashed curves are results from tLMTO calculation. The binding energy is relativto the vacuum level.
eloc
onfveVon
g
v
ubattheonV
noea-dif-nsi-ltsre-showng
nc-
re-heinseenantria-o-heenin-ity
loss spectrum for the carbon-film-supported sample wtaken into account. The parameters related to the energyfeatures were adjusted in the empirical deconvolution produre to get a reasonable result.
After the deconvolution the intensity between the twmain peaks corresponding to the upper and lower valebands dropped close to zero~Fig. 7!. There is a clear gap o6.5 eV between the upper valence band and the lowerlence band. This compares very well with the gap of 6.6in the LMTO calculation after one convolutes the calculatiwith the energy resolution of the spectrometer.
A LMTO calculation of momentum integrated bindinenergy spectrum ofa-Al2O3 is also shown in Fig. 7. It hasbeen normalized so that the peak heights of the lowerlence bands of the calculation and the measurement~afterdeconvolution! are the same. Compared to the LMTO calclation, the measured upper valence band has a broaderstructure and the measured intensity is much larger thancalculated one with the present arbitrary normalization oftwo. In the lower valence band the measurement showstended tail structure in the 30–35-eV binding energy regiwhereas the LMTO calculation shows a total width of 5 e
resse-
ce
a-
a-
-ndheex-,
consisting of two peaks at around 26.5 and 29 eV andstructure at all beyond 31 eV. To further compare the msurement and the LMTO calculation and discuss theseferences we need to examine the spectral momentum deties in detail. In the following discussion only the resuobtained with the carbon-film-supported sample are psented and discussed as they have better statistics andimproved contrast in comparison with the self-supportialuminum sample.
Figure 8 shows the raw and deconvoluted data as a fution of the binding energy for momentum from 0 to11.6a.u. at equal intervals of 0.1 a.u., together with the corsponding LMTO calculations. Due to symmetry about tmomentum axis, the positive and negative momentum bhave been summed to improve the statistics. It can be sthat the intensity of the upper valence band is fairly constbefore decreasing slowly at quite high momenta. The vation of the intensity of the upper valence band with the mmentum does not agree well with the LMTO calculation. Tcalculation contains a major contribution from the oxyg2p orbital to the upper valence band. This predicts zerotensity at zero momentum with the maximum in the dens

h
tepelcuakndaithmth
en-ctsforen-
ising
sing
s inithistionin-TOuchn inin
ces.nyc-longq.
es
onsen-cesor-
ifi-ngdingAvenro
de-les.r-Thestri-re-enin
nyu-ntso-
ay
n-er
heibu-
eelaT
54 17 951ELECTRONIC-STRUCTURE INVESTIGATION OF . . .
at around 1 a.u. of momentum~see also Fig. 3 and Fig. 6!. Atall momenta the measured intensity is broader and higthan that predicted.
The deconvoluted momentum distributions at selecbinding energies are presented in Fig. 9 for the upvalence-band region. The spherically averaged LMTO calations are also shown in this figure. The calculated pemove symmetrically towards lower momentum as the biing energy increases from 10 to 18 eV, with a second pesymmetric about zero momentum growing in intensity wincreasing binding energy from 12 to 16 eV moving out froclose to zero momentum, and joining the other lobe of
FIG. 9. The momentum distributions at selected binding engies for the upper valence band of aluminum oxide. The experimtal data are the results after deconvolution of the raw data for pmon losses. The dashed curves are results from the LMcalculation.
er
dr-s-k,
e
parabola at 16 eV. This can be seen by taking constantergy cuts through the central panel of Fig. 6; i.e., it reflethe cuts through the ‘‘dual parabola’’ dispersion patternthis upper valence-band structure. In the lower bindingergy region of this band the oxygen 2p orbital character islargely responsible for the momentum distribution, whichpeaked at higher momenta. Moving to increasing bindenergy the aluminum 3p, 3s, and oxygen 2p orbitals hy-bridize and create a considerable dispersion. With increabinding energy, the contribution from aluminum 3p de-creases whereas the contribution from aluminum 3s in-creases in the orbital hybridization. This leads to the peakthe momentum distribution moving to lower momentum wincreasing binding energy as illustrated in Fig. 9. Thisreflected in the edge of the measured momentum distribumoving towards lower momenta as the binding energycreases. However, a quantitative comparison with the LMcalculation shows that the measured spectra have mlarger intensity between the peaks at all energies showFig. 9. Similarly the peaks in Fig. 8 are much broaderenergy than the calculated ones at all momenta.
There are a few possible causes for these differenFirst, it should be caused by multiple elastic scattering of aof the electrons. As mentioned in Sec. III A, the EMS spetrometer measured the momenta of the target electrons athey axis~see Fig. 4!. These momenta are determined by E~3! by accurately measuring the azimuthal angles (fs andf f in Fig. 4! of the outgoing electrons at fixed polar angl(76° cone and 14° cone in Fig. 4!. Occurrence of multipleelastic scattering distorts the incident and outgoing directiof the electrons. As a result, it distorts the measured momtum distribution of the target electrons. This has been notiin (e,2e) measurements of other materials, such as amphous silicon,5 amorphous germanium,9 and the polycrystal-line silicon carbide.10 There also is in these cases a signcant intensity near zero momentum in the low bindienergy region of the valence bands where the corresponLMTO calculations indicate zero or negligible intensity.detailed account of multiple elastic scattering has been giby Vos and Bottema.45 The pronounced difference near zemomentum in the upper valence band~Fig. 9! between thecalculation and the experiment could also be caused byfects that may exist in the near surface region in the sampWe know that only (e,2e) events occurring in the near suface region are detectable with this EMS spectrometer.existence of these defects may affect the momentum dibution in the near zero momentum region due to chargedistribution near the defect site. Similarly, aluminum-oxygbonds at the surface may be quite different from thosebulk.19 Contributions from multiple elastic scattering and alocal disorders are not included in our present LMTO calclation. The quantitative comparison of the measuremewith the theory is hindered by the multiple scattering prcesses. Monte Carlo simulations of (e,2e) collision in solidsin which all possible scattering processes are included mprovide a better comparison.
Now let us turn to the lower valence-band region. In cotrast to the upper valence band, the intensity of the lowvalence band in the binding energy spectra~Fig. 8! dropssignificantly with increasing momentum. To examine tcharacter of the lower valence band, the momentum distr
r-n-s-O

e-etataults
17 952 54X. GUO et al.
FIG. 10. The momentum distributions at slected binding energies for the lower valencband of aluminum oxide. The experimental daare the results after deconvolution of the raw dafor plasmon losses. The dashed curves are resfrom the LMTO calculation.
Odemae
otethbeluec
in
a
lluer
aadonergchi
r
the-eV
in-nd
an
wers ofobleMSumtryhetailght,ex-
ionedin-
mSwithnttedtheperly,a
tions at different binding energies together with the LMTcalculations are presented in Fig. 10. As in Fig. 9 theconvoluted experimental data are used in Fig. 10. Thementum distribution in Fig. 10 peaks at zero momentum29-eV binding energy. This is not surprising since the lowvalence band is dominated by the oxygen 2s orbital. How-ever, due to small aluminum 3p orbital contribution to thebonding combination of aluminum 3s and oxygen 2s orbit-als the LMTO calculation, convoluted with the energy reslution of the spectrometer, indicates a dispersion of aboueV in the lower valence band. As mentioned above, the msured spectral momentum density shows less dispersionthe calculated one. The discrepancy in the dispersiontween the measurement and the LMTO calculation may bpart due to excess oxygen adsorbed on the saturated anum oxide surface. Nevertheless, the ‘‘bowl’’ shaped sptral momentum distribution is clearly seen in Fig. 6~left andright panels! in the lower valence band and is similarshape to that of the LMTO calculation~Fig. 6, central panel!.The features of the adsorbates7 do not completely obliteratethe dispersion features of the aluminum oxide, but msmear it somewhat and consequently make a shallow~lessdispersion! ‘‘bowl’’ in the lower valence band in the spectramomentum density plot. An experiment with controlled aminum oxidation on aluminum may provide a better undstanding of this discrepancy.
One may also notice in Fig. 8 that the measured mpeak in the lower valence band shows a full width at hmaximum~FWHM! of about 4.5 eV at all momenta selectewithin an interval of 0.1 a.u., whereas the LMTO calculatishows the lower valence band with a FWHM of 1.5 eV evafter one convolutes the calculation with the 1-eV eneresolution of the spectrometer. This energy width differenmay be due to the amorphous nature of the sample, whas not been sufficiently described by the calculation.
The intensity of the lower valence band does not appea
-o-tr
-5a-ane-inmi--
y
--
inlf
nyech
to
drop as fast with increasing binding energy as that ofupper valence band. There is a tail structure in the 30–35binding energy region~Fig. 8!. This tail structure cannot beremoved by the deconvolution without creating negativetensity in the region above the upper valence ba(.18-eV binding energy!. Also, the momentum distributionin the 30–35-eV binding energy region~Fig. 10! is moresimilar to the 26–29-eV binding energy region rather ththe 10–17-eV~the upper valence band! region. These argu-ments lead us to associate this tail structure with the lovalence band. Similar structures at higher binding energiethe deepest valence level are observed in the case of ngases, where the satellite structures of valence levels in Ebinding spectra are identified by examining their momentdistributions and allocated to corresponding symmemanifolds.1 Indeed, if one normalizes the theoretical and texperimental lower valence-band features including thestructure in Fig. 8 to equal area rather than equal heiagreement in the upper valence-band region between theperiment and the calculation improves. A clear interpretatof the tail structure in the lower valence band may netheoretical calculations in which electron correlation iscluded.
V. CONCLUSIONS
In conclusion, the visualization of the spectral momentudensity of materials with complex bulk structures using EMhas been demonstrated by performing measurementsoxidized aluminum films that are prepared in two differeways. The oxidized layers on the aluminum films are relato aluminum oxide. The measured two major features inspectral momentum density, which are related to the upand lower valence bands of aluminum oxide, respectiveare represented qualitatively by the LMTO calculation on

Vulanctaoatit
ofaleirksnsofian
54 17 953ELECTRONIC-STRUCTURE INVESTIGATION OF . . .
spherically averageda-Al2O3. In particular, the observedenergy-momentum dispersion pattern spanning about 8 ethe upper valence band is reproduced by the LMTO calction with nearly the same energy span. In the lower valeband, the LMTO calculation indicates a dispersion of aboueV with maximum intensity at zero momentum. The mesurement shows a similar ‘‘bowl’’ shape in the spectral mmentum density plot but with less dispersion and a greband width. Further EMS measurements on aluminum wcontrolled oxidation are in progress.
c-
tss
M
d
d,
ld
nd
E.
d
ld
ld
B. B
y,
rs
s
in-e5--erh
ACKNOWLEDGMENTS
The authors want to thank Dr. Chris J. RossouwCSIRO for providing the aluminum films, and the technicstaff in the Electronic Structure of Materials Centre for thskillful technical support throughout the experiment. Thanalso go to Professor C. E. Brion for meaningful discussioduring his visit to this Centre. The Electronic StructureMaterials Centre is supported by a grant from the AustralResearch Council.
ys.
.
in,
un.
w,
Ut-
1I. E. McCarthy and E. Weigold, Rep. Prog. Phys.54, 789~1991!.2E. Weigold, Y. Q. Cai, S. A. Canney, A. S. Kheifets, I. E. MCarthy, P. Storer, and M. Vos, Aust. J. Phys.49, 543 ~1996!.
3P. Storer, Y. Q. Cai, S. A. Canney, S. A. C. Clark, A. S. KheifeI. E. McCarthy, S. Utteridge, M. Vos, and E. Weigold, J. PhyD 28, 2340~1995!.
4R. S. Caprari, S. A. C. Clark, I. E. McCarthy, P. J. Storer,Vos, and E. Weigold, Phys. Rev. B50, 12 078~1994!.
5M. Vos, P. Storer, Y. Q. Cai, A. S. Kheifets, I. E. McCarthy, anE. Weigold, J. Phys. Condens. Matter7, 279 ~1995!.
6M. Vos, P. Storer, Y. Q. Cai, I. E. McCarthy, and E. WeigolPhys. Rev. B51, 1866~1995!.
7M. Vos, S. A. Canney, P. Storer, I. E. McCarthy, and E. WeigoSurf. Sci.327, 387 ~1995!.
8A. S. Kheifets, J. Lower, K. J. Nygaard, S. Utteridge, M. Vos, aE. Weigold, Phys. Rev. B49, 2113~1994!.
9Y. Q. Cai, P. Storer, A. S. Kheifets, I. E. McCarthy, andWeigold, Surf. Sci.334, 276 ~1995!.
10Y. Q. Cai, M. Vos, P. Storer, A. S. Kheifets, I. E. McCarthy, anE. Weigold, Phys. Rev. B51, 3449~1995!.
11P. Storer, R. S. Caprari, S. A. C. Clark, M. Vos, and E. WeigoRev. Sci. Instrum.65, 2214~1994!.
12M. Vos, R. S. Caprari, P. Storer, I. E. McCarthy, and E. Weigo~unpublished!.
13G. van der Laan, M. Surman, M. A. Hoyland, C. F. J. Flipse,T. Thole, Y. Seino, H. Ogasawara, and A. Kotani, Phys. Rev46, 9336~1992!.
14S. G. Ovchinnikov, Phys. Rev. B49, 9891~1994!.15S. Canney, M. Vos, A. S. Kheifets, N. Clisby, I. E. McCarth
and E. Weigold~unpublished!.16T. J. Godin and John P. LaFemina, Phys. Rev. B49, 7691~1994!.17Y. Xu and W. Y. Ching, Phys. Rev. B43, 4461~1991!.18M. Causa, R. Dovesi, C. Roetti, E. Kotomin, and V. R. Saunde
Chem. Phys. Lett.140, 120 ~1987!.19S. Ciraci and I. P. Batra, Phys. Rev. B28, 982 ~1983!.20I. P. Batra, J. Phys. C15, 5399~1982!.21R. A. Evarestov, A. N. Ermoshkin, and V. A. Lovchenkov, Phy
Status Solidi B99, 387 ~1980!.22M. H. Reily, J. Phys. Chem. Solids31, 1041~1970!.
,.
.
,
,
.
,
.
23A. Balzarotti and A. Bianconi, Phys. Status Solidi B76, 689~1976!.
24P. Hayes, M. A. Bennett, J. Flexman, and J. F. Williams, PhRev. B38, 13 371~1988!.
25G. Drager and J. A. Leiro, Phys. Rev. B41, 12 919~1990!.26C. G. Dodd and G. L. Glen, J. Appl. Phys.39, 5377~1968!.27D. W. Fischer, Adv. X-ray Anal.13, 159 ~1970!.28S. P. Kowalczyk, F. R. McFeely, L. Ley, V. T. Gritsyna, and D
A. Shirley, Solid State Commun.23, 161 ~1977!.29I. A. Brytov and Yu. N. Romashchenko, Fiz. Tverd Tela~Lenin-
grad! 20, 664 ~1978! @Sov. Phys. Solid State20, 384 ~1978!#.30N. M. Persiantseva, N. A. Krasil’nikova, and V. G. Neudach
Zh. Eksp. Teor. Fiz.76, 1047~1979! @Sov. Phys. JETP49, 530~1979!#.
31N. Swanson, Phys. Rev.165, 1067~1968!.32H. L. Skriver, The LMTO Method~Springer-Verlag, Berlin,
1984!.33U. von Barth and L. Hedin, J. Phys. C5, 1629~1972!.34R. Wyckoff, Crystal Structures~Interscience Publishers, New
York, 1963!.35J. C. Slater,Quantum Theory of Molecules and Solids~McGraw-
Hill, New York, 1965!.36D. Glotzel, R. Segall, and O. K. Andersen, Solid State Comm
36, 403 ~1980!.37A. S. Kheifets and Y. Q. Cai, J. Phys. C7, 1821~1995!.38A. S. Kheifets and M. Vos. J. Phys. C7, 3895~1995!.39E. Weigold and I. E. McCarthy, Adv. At. Mol. Phys.14, 127
~1978!.40L. J. Allen, I. E. McCarthy, V. W. Maslen, and C. J. Rossou
Aust. J. Phys.43, 453 ~1990!.41S. Canney, M. J. Brunger, I. E. McCarthy, P. J. Storer, S.
teridge, M. Vos, and E. Weigold~unpublished!.42N. Ishigure, C. Mori, and T. Watanabe, J. Phys. Soc. Jpn.44,
1196 ~1978!.43J. C. Fuggle, L. M. Watson, and D. J. Fabian, Surf. Sci.49, 61
~1975!.44D. T. Quinto and W. D. Robertson, Surf. Sci.27, 645 ~1971!.45M. Vos and M. Bottema, Phys. Rev. B54, 5964~1996!.

















