
Dynorphin A – Interactions with receptors - DiVA - Simple search
53
Dynorphin A– Interactions with receptors and the membrane bilayer Licentiate thesis Johannes Bj ¨ orner ˚ as Under the supervision of profs.Lena M¨ aler and Astrid Gr ¨ aslund Department of Biochemistry and Biophysics, Stockholm University Stockholm, Sweden September 2013
Transcript of Dynorphin A – Interactions with receptors - DiVA - Simple search

Dynorphin A –Interactions with receptorsand the membrane bilayer
Licentiate thesisJohannes Bjorneras
Under the supervisionof profs. LenaMaler andAstridGraslund
Department of Biochemistry and Biophysics, Stockholm University
Stockholm, Sweden September 2013


Abstract
The work presented in this thesis concerns the dynorphin neuropeptides, anddynorphin A (DynA) in particular. DynA belongs to the wider class of typicalopioid peptides that, together with the opioid receptors, a four-membered familyof GPCR membrane proteins, form the opioid system. This biological system isinvolved or implicated in several physiological processes such as analgesia, ad-diction and depression, and effects caused by DynA through this system, mainlythrough interaction with the κ (kappa) subtype of the opioid receptors (KOR), arecalled the opioid effects. In addition to this, non-opioid routes of action for DynAhave been proposed, and earlier studies have shown that direct membrane inter-action is likely to contribute to these non-opioid effects. The results discussed herefall into either of two categories; the interaction between DynA and a fragment ofKOR, and the direct lipid interaction of DynA and two variant peptides.
For the receptor interaction case, DynA most likely causes its physiological effectsthrough binding its N-terminal into a transmembrane site of the receptor protein,while the extracellular regions of the protein, in particular the extracellular loop II(EL2), have been shown to be important for modulating the selectivity of KOR forDynA. Here we have focussed on the EL2, and show the feasibility of transferringthis sequence into a soluble protein scaffold. Studies, predominantly by nuclearmagnetic resonance (NMR) spectroscopy, of EL2 in this new environment showthat the segment has the conformational freedom expected of a disordered loopsequence, while the scaffold keeps its native β-barrel fold. NMR chemical shiftand paramagnetic resonance enhancement experiments show that DynA bindswith high specificity to EL2 with a dissociation constant of approximately 30µm,while binding to the free EL2 peptide is an order of magnitude weaker. Thestrength of these interactions are reasonable for a receptor recognition event. Nobinding to the naked scaffold protein is observed.
In the second project, the molecules of interest were two DynA peptide vari-ants recently found in humans and linked to a neurological disorder. Previouslypublished reports from our group and collaborators pointed at very differentmembrane-perturbing properties for the two variants, and here we present theresults of a follow-up study, where the variants R6W–DynA and L5S–DynA werestudied by NMR and circular dichroism (CD) spectroscopy in solutions of fast-tumbling phospholipid bicelles, and compared with wild type DynA. Our resultsshow that R6W–DynA interacts slightly stronger with lipids compared to wildtype DynA, and much stronger compared to L5S–DynA, in terms of bicelle associa-tion, penetration and structure induction. These results are helpful for explainingthe differences in toxicity, membrane perturbation and relationship to disease,between the studied neuropeptides.

ii
List of publications
I Johannes Björnerås, Martin Kurnik, Mikael Oliveberg, Astrid Gräslund,Lena Mäler and Jens Danielsson; Direct detection of neuropeptide dynorphinA binding to the second extracellular loop of the κ–opioid receptor using asoluble protein scaffold. Submitted manuscript
II Johannes Björnerås, Astrid Gräslund and Lena Mäler; Membrane interactionof disease-related Dynorphin A variants. Biochemistry 52, 4157–4167 (2013)

iii
Abbreviations
DHPC 1,2-dihexanoyl-sn-glycero-3-phosphocholineDMPC 1,2-dimyristoyl-sn-glycero-3-phosphocholineDynA dynorphin ADynB dynorphin BEL2 the extracellular loop II of the κ–opioid receptorKOR κ–opioid receptorHSQC heteronuclear single-quantum coherenceLUV large unilamellar vesicleMTSL 1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl) methanesulfonateNOE nuclear overhauser enhancementOR opioid receptor(s)PC phosphatidylcholinePDYN prodynorphinPRE paramagnetic relaxation enhancementSOD1 human superoxide dismutase 1SUV small unilamellar vesicle

CONTENTS
1 Introduction 11.1 Life, what is it really? . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.1.1 Compartments . . . . . . . . . . . . . . . . . . . . . . . . . . 11.1.2 Signalling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Life, how do we deal with it? . . . . . . . . . . . . . . . . . . . . . . 21.3 Final words (of the introduction) . . . . . . . . . . . . . . . . . . . . 3
2 Biological background 52.1 The opioid system . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
2.1.1 Receptors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52.1.2 Dynorphins . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
3 Research projects 133.1 DynA–KOR interactions . . . . . . . . . . . . . . . . . . . . . . . . . 133.2 Lipid interaction properties of DynA and related peptides . . . . . 13
4 Methods 154.1 Membrane mimetic systems . . . . . . . . . . . . . . . . . . . . . . . 15
4.1.1 Bicelles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154.1.2 Other systems . . . . . . . . . . . . . . . . . . . . . . . . . . . 164.1.3 Scales of length and time . . . . . . . . . . . . . . . . . . . . . 16
4.2 NMR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174.2.1 General theory . . . . . . . . . . . . . . . . . . . . . . . . . . . 174.2.2 Chemical shifts . . . . . . . . . . . . . . . . . . . . . . . . . . 204.2.3 Dynamics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214.2.4 Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 234.2.5 Labelling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4.3 CD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
5 Summary of results 255.1 The construct protein SOD1–EL2 has a β–barrel core, while the
inserted segment is unstructured. (Paper I) . . . . . . . . . . . . . . 255.2 The extracellular loop 2 of the κ opioid receptor, as expressed in the
construct protein SOD1–EL2, interacts weakly with Dynorphin A.(Paper I) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
iv

CONTENTS v
5.3 The two disease-related Dynorphin A variants R6W-DynA and L5S-DynA have markedly different lipid interaction properties. (PaperII) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
6 Conclusions and outlook 29
Acknowledgements 31
BIBLIOGRAPHY 33
APPENDIX 43

vi CONTENTS

1 Introduction
1.1 Life, what is it really?
The questions arising from thinking about the fundamental aspects of life candrive anyone to the brink of insanity. How did life start? Was the origin of life aone-time event, or are there other planets, or even other universes, teeming withweird and wonderful creatures? Is there an ultimate purpose with life in general,and my life in particular? If not, how can I come to terms with this? How shouldmy life best be lived? Such things may, and will occupy the minds of humans– from the curious five-year old to the elder watching the dusk of life deepenaround her. As for other animals, we may, sadly enough, never really know whatthey think.
Even the question of how to define life is debatable (Dawkins et al. 2011, Owen2008), and I will make no attempt to settle it. But despite the disagreements, thereare a few key concepts generally accepted as elements of life. A living organism 1)has some sort of information storage capability – e.g. DNA; 2) is able to reproduceand evolve; 3) is able to convert and use energy. In addition to this, two othercharacteristics of life, significant to the work presented in this thesis, are listedbelow.
1.1.1 Compartments
All known living systems have some way of keeping things in confined spaces.The most obvious example is an overall envelope – like the human skin, the barkof a tree or an outer bacterial membrane – that creates an inside of the organismseparated from the surroundings. Apart from this, the insides of organisms maybe compartmentalised to various degrees of complexity, adding spatial constraintas a parameter for controlling the functions – enzymatic reactions, transport,storage, etc – constituting the life of the organism.
Many physiological responses depend on things happening at these boundaries,for example whether a specific particle (molecule, virus, etc) is able to pass througha cellular membrane. In this thesis, one such system – a signalling peptide of theneural system interacting with neuronal receptors and the cell membrane – isdiscussed.
1

2 Chapter 1 Introduction
1.1.2 Signalling
A second feature characteristic of a living organism, and something that is con-nected with compartmentalisation, is a means of sending and receiving signals(and an ability to process these signals). This can range from rather simple pro-cesses such as sensing the flow from an energy source and move towards it, to theintricate machinery underlying a serious human discussion on, say, contemporaryChinese art [www.youtube.com/watch?v=yA775QobZhg].
At its most general, a biological signalling system involves a ligand – the physi-cal embodiment of the signal, and a receptor – something that ’reads’ the signalthrough interaction with the ligand. But beneath this surface of simplification aremany layers of complexity, a problem that is addressed in one of the projects de-scribed in this thesis by a divide-and-conquer approach where a part of a receptoris removed from the membrane protein and allowed to interact with the ligand ina simplified context.
1.2 Life, how do we deal with it?
Out of the many ways of looking at life, one is from the viewpoint of science. ’Lifescience’ is often used to describe the scientific study of all systems that are in someway associated with living organisms. To state the obvious, this research area isenormous, and encompasses a large number of seemingly very different niches,from data mining to deep sea biology; from proton spin states to prosthetic limbs.
No one, neither Nobel prize laureate nor layman, can deny that our understandingof living systems and our ability to predict and control them, are at a level that wasunimaginable a hundred years ago (as unimaginable as the level of the scientistsin the beginning of the 1900s to their predecessors a century earlier...). But hasthis brought us nearer to an answer to the question of how to deal with life froma personal point of view; i.e. is it easier to live a good, meaningful life todaythan in 1913? Hardly. Human knowledge accumulates, but wisdom arguablydoes not; and conceptions of what is good are often heterogenous, and sometimescontradictory.
But regardless of the incapability of science to perfect human nature, the scientisthas proven to be a key figure in the modern society. Both as a contributor tomaterialistic improvements, and as a producer of knowledge needed to makeinformed decisions. The contributions from life scientists in these two aspects areobvious, from modern blessings such as dental anaesthesia and antibiotics to theinfluence of our increased biological understanding on discussions of racism andsexism.

1.3 Final words (of the introduction) 3
1.3 Final words (of the introduction)
Dear reader, do not see this section as an attempt to write a moral guide to lifeor life science. Or as any other guide for that matter. It is meant merely to serveas a sketch of the world from which this thesis has sprung from a viewpointtoo seldom used in my daily life as a life scientist, and as a reminder that eventhe dullest lab work on a Tuesday afternoon in November is part of the greatmachinery of the world; and that there are reasons to look at our profession withboth humility and pride.

4 Chapter 1 Introduction

2 Biological background
2.1 The opioid system
Opiates are compounds eliciting their effects through the opioid system, andthe drug morphine, a molecule isolated from the opium poppy, is perhaps themost widely known member of the family. Although knowledge of opiates andtheir effects on humans date back hundreds, or even thousands of years (see(Brownstein 1993)), a more detailed model of the cause of their effects is muchyounger.
2.1.1 Receptors
The receptors of the opioid system are G protein-coupled receptors (GPCRs), thelargest class of membrane proteins (Rosenbaum et al. 2009). The research interestin membrane proteins in general has grown steadily over the last decades [REF],and reasons for studying this huge group of biomolecules range from pure basicresearch to pharmaceutical and biotechnological applications. But despite thatmembrane proteins – e.g. receptors, transporters and channels – are predicted toconstitute 20 to 30 % of all proteins (Krogh et al. 2001)), and that they by a roughestimate make up at least half of existing (Drews 2000) and predicted (Bakheetand Doig 2009) drug targets, they remain largely unexplored. A vast body ofbiophysical and biochemical research has greatly expanded the knowledge in thisarea, but for many membrane proteins, characterisation and understanding onan atomic level has been out of reach, due to the difficulties of studying themwith high-resolution spectroscopic techniques such as NMR or X-ray crystal-lography. Numbers that illustrate the effects of these problems may be found indatabases of protein structures, with the database of membrane protein structures(blanco.biomol.uci.edu/mpstruc/) containing approximately 1300 coordinate files,compared to more than 85000 in the Protein Data Bank (PDB, at present the largestdatabase of protein structure information). Much simplified, the problems comedown to the following:
X-ray: A well-defined three-dimensional crystal structure is needed to get awell-resolved diffraction pattern, and since membrane proteins by naturereside in a 2D-like (lipid bilayer) environment, these crystals are not easilyformed.
NMR: The structures of most soluble proteins have been solved with liquid stateNMR, where anisotropy (ideally) averages out intermolecular interactions,and scales down the informational content of the spectra. Since the aver-aging (partly) relies on a rapid overall motion of the protein (much faster
5

6 Chapter 2 Biological background
than the motion of an entire cell or a synthetic vesicle), this approach re-quires that the membrane proteins are either solubilised in some less polarmedium than water, or reconstituted in a membrane-mimicking system –like micelles or bicelles (both of which will be discussed later). The first ofthese approaches gives conditions that poorly mimicks the in-vivo environ-ment, while the second often poses considerable experimental challenges.A related problem is that linewidths in NMR spectra correlate with molec-ular tumbling rates, and since membrane proteins tend to be large, and arelikely to be complexed with lipids (in a membrane mimicking solvent), theline broadening may simply be too severe to get data of sufficient quality toproduce a reasonable structural estimate.
Solid state NMR (ssNMR) on powder-like samples offers the advantageouspossibility of using a much more native-like lipid bilayer as an environ-ment for the membrane protein. However, in the last two decades onlya handful of membrane protein structures have been solved by ssNMR[http://www.drorlist.com/nmr/SPNMR.html], showing that the method isnot yet trivially applicable to protein structure determination, but the ap-proach is in rapid development (see (Hong et al. 2012) for a review), and it islikely that this technique will have a great impact on the field in the comingyears.
The GPCRs are involved in chemical signal transduction, and it is no exaggerationto say that this family of proteins has lately been the superstars of biomolecules,awarding two of the pioneers in the field – Robert Lefkowitz and Brian Kobilka –the Nobel prize in chemistry 2012. In the general agonistic situation, a ligand in-teracts with the receptor, increasing the probability of the receptor to interact withone or several guanine nucleotide binding proteins (G-proteins). The G-proteinsfunction as transducers, and possibly amplifiers, propagating the signal by mod-ulating one or several effector systems (Gudermann et al. 1995, Rosenbaum et al.2009).
As recently as 2005, only one GPCR crystal structure existed, that of bacteri-orhodopsin (Palczewski et al. 2000), but several years of hard work and technicaland methodological improvement has resulted in several new structures in thelast few years, many of which are from the labs of Ray Stevens and Brian Kobilka(see e.g. (Chien et al. 2010, Granier et al. 2012, Manglik et al. 2012, Shimamura et al.2011, Thompson et al. 2012, Warne et al. 2008, Wu et al. 2010; 2012)), and at the timeof this thesis, 20 crystal structures have been produced (http://gpcr.scripps.edu/).Without downplaying any of the remarkable achievements underpinning theexisting GPCR structures, several questions regarding their function remain tobe answered, and several problems remain to be solved. One such aspect isthe scarcity of NMR structures – to date, the CXCR1 receptor is the only GPCRwho has had its structure determined by NMR (Park et al. 2012). In contrast toX-ray crystallography, NMR spectroscopy may be used under near-native condi-tions, and provides valuable information on dynamics on different time scales,

2.1 The opioid system 7
in addition to the molecular structure. Another problem is the lack of structuralinformation on peptides bound to GPCRs – to the author’s knowledge, only onesuch structure exists, that of the CXCR4 chemokine receptor in complex with acyclic peptide (Wu et al. 2010). Being bulkier than small-molecule ligands, theinteraction of peptide agonists or antagonists necessarily involves larger regionsof the GPCRs, e.g. extracellular loops.
Opioid receptors
The existence of the GPCR subfamily opioid receptors (originally called opiatereceptors) was demonstrated by the work of Pert and Snyder (Pert and Snyder1973), Goldstein et al. (Goldstein et al. 1971), the Swede Lars Terenius (Terenius1973a;b, Terenius et al. 1975) and other groups, in the early seventies (for an ex-citing personal account, as well as an historical review of these years, see (Snyderand Pasternak 2003)). Through interactions with various endogenous peptides,these receptors, divided into four classes - κ, μ, δ, and the nociceptin/orphaninFQ peptide receptor – are involved in the modulation of pain ((Przewłocki andPrzewłocka 2001, Wen et al. 1985; 1987)), as well as more complex physiologicalprocesses (Bodnar 2012) such as reward and addiction (Bruchas et al. 2010, Brui-jnzeel 2009, Drews and Zimmer 2009), and different types of mental disorders(Tejeda et al. 2012).
A compelling feature of the opioid receptors (as well as other GPCRs) is theircombination of high similarity – both on a sequence level (60 to 70 %) (Uhl et al.1994), and structurally – and high specificity with respect to various ligands (inparticular peptides or peptide analogues). The opioid receptors all have a GPCRtype A (rhodopsin-like) fold, with seven transmembrane helices interconnectedby intra- and extracellular loops, as is exemplified by the X-ray structure of theKOR shown in Figure 2.1. The recent structure determination of all four opioidreceptors (Granier et al. 2012, Manglik et al. 2012, Thompson et al. 2012, Wu et al.2012) has made it possible to draw some conclusions regarding the similaritiesand differences within this receptor family. For example, in all four receptors thereis a binding pocket in the transmembrane region, a region that is well conserved,with several conserved amino acids having the same position (Filizola and Devi2012). The sequence dissimilarities are mainly located in the termini, and extra-and intracellular loops of the receptors (Uhl et al. 1994); and hence these regionshave been in the focus of interest as conveyors of ligand selectivity (Paterlini 2005).Surprisingly, these regions are also structurally similar (Filizola and Devi 2012),suggesting that only subtle differences modulate the ligand selectivity, and/orthat the existing crystal structures show the proteins in similar states (all fourstructures have an agonist bound), and that this is just one state out of many.Based on the larger conformational freedom of the loop regions, it is reasonable tobelieve that these parts of the receptors show the largest structural differences indifferent receptor states. Chimeric studies have shown that the selectivity patternof one receptor subtype may be transferred to another subtype by interchangingextracellular fragments (Dietrich et al. 1998, Meng et al. 1995, Wang et al. 1994).

8 Chapter 2 Biological background
Figure 2.1: X-ray crystal structure of the κ-opioid receptor where the extracellularloop II has been marked with red. The figure was produced using Pymol, from aPDB structure file, ID 4DJH, at www.pdb.org
The importance of the extracellular loops is further discussed below for the specificcase of the κ-opioid receptor.
Ligands
A wide variety of opioid receptor ligands are known, some of which are isolatedand/or synthesised from natural sources, such as morphine, heroin and salvinorinA (Roth et al. 2002), while others are synthetic. It is beyond the scope of this thesisto give an overview of these ligands, but the topic will be discussed in the specificcontext of the κ-opioid receptor and its interaction with the endogenous peptideligand dynorphin A.
Making the question of receptor–ligand selectivity even more intriguing is thefact that also among the ligands there are many common features, perhaps mostnotably the so called enkephalin sequence: Tyr–Gly–Gly–Phe–Leu/Met, that con-stitutes the N-terminal sequence of all typical opioid peptides (Janecka et al. 2004),including the dynorphin family which is discussed below.

2.1 The opioid system 9
2.1.2 Dynorphins
In the 1970’s, a substance with distinct morphine-like properties was isolatedfrom cow brain (Teschemacher et al. 1975). The substance was later identifiedas an endogenous opioid peptide, named dynorphin after its high potency in anassay using the contractions of guinea pig ileum (Goldstein et al. 1979), and hadits sequence determined (Goldstein et al. 1981). Further research established thatthis powerful peptide exerted its opioid effects mainly through the κ-subtype ofthe opioid receptors (KOR) (Chavkin and Goldstein 1981a;b, Chavkin et al. 1982),and that there are two types of dynorphins, dynorphin A (DynA) and dynorphinB (DynB), emanating from the same precursor protein: prodynorphin (PDYN).
Opioid receptor interactions
During the decades up until present day, the dynorphins, in particular DynA,have been extensively studied and characterised – both for basic research pur-poses, and as a drug template. A large number of research groups have usedtruncations, mutations, chemical modifications and various forms of cyclisationof the peptides, in particular of DynA, to map out the physico-chemical and struc-tural properties needed for potent, selective and tunable opioid receptor agonistand antagonist effects; for examples see (Björnerås et al. 2013) and referencestherein. Many times the ultimate goal with this research has been to developalternatives to morphine, matching its ’gold standard’ analgesic properties whileavoiding the adverse side effects, such as addiction. The results of this researchinclude a number of synthetic non-peptide ligands, one of which was used in thecrystallisation and structure determination of the KOR (Wu et al. 2012).
The many results of this research do not easily condense to a few simple rules,but some aspects are worth highlighting. It is now generally accepted that the N-terminal residues of the opioid peptides (the enkephalin sequence), in particularTyr1 and Phe4 (Turcotte et al. 1984), interact with residues in the transmembranebinding pocket of the receptors to trigger receptor activation. This region of theligand is often called the ’message’, while the C-terminal parts of peptide ligandsare called ’address’ regions; a concept coined by Goldstein et al. in 1981 (Chavkinand Goldstein 1981b). Furthermore it has been shown that the KOR extracellularloop II (EL2) is needed for high affinity OR–dynorphin binding (Wang et al. 1994,Xue et al. 1994). The position of EL2 in the KOR can be seen in 2.1, where EL2 isshown in red. As for the extracellular receptor regions, and the mechanisms forligand selectivity, a few different concepts have been proposed:
Binding/favourable interaction: The message-address model depicts the lig-and as consisting of two (not completely distinct) parts. The N-terminalsequence (message) activates (or blocks) the receptor upon interaction withresidues in the transmembrane binding pocket; while the remaining peptideregion (address) needs to have a favourable interaction with extracellularparts of the receptor for the ligand to reach and bind in the transmembrane

10 Chapter 2 Biological background
site.
Selection through exclusion: Here, selectivity for the interaction is proposedto be impaired through an exclusive mechanism – unfavourable energeticsbetween ligands and extracellular receptor regions keep certain ligands out.See (Metzger and Ferguson 1995).
Conformational mechanisms: In this hypothesis, the extracellular loops con-strain the positions of the transmembrane helices, and, as a consequence,the positions of contact points in binding sites. See (Dietrich et al. 1998).
In the literature there is support for all the proposed mechanisms, and a reason-able (albeit fence-sitting) conclusion is that for any ligand–receptor system, allthree models will be valid, but to different degrees. In the case of DynA–KORinteraction, the abundance of positively charged amino acid residues in DynAand negatively charged residues in the EL2 of KOR, immediately gives rise to theidea that electrostatic interactions are important, or even primarily responsible,for the interaction between the two. This hypothesis has been tested in severalstudies, with mixed results. Schlechtingen and co-workers (Schlechtingen et al.2003), performed binding and activity studies with KOP and various analoguesof DynA, showing that Arg residues, especially Arg6 and Arg7 were crucial for κ-selectivity. Ferguson, on the other hand, reported that replacing charged residues(but not all) in EL2 did not affect DynA affinity and function (Ferguson et al. 2000).Also modelling studies have downplayed the importance of Coulomb forces incomparison with hydrophobic interactions (Paterlini et al. 1997), and experimen-tal studies of the EL2 peptide in micelles lend some support to this (Zhang et al.2002).
Membrane interactions
By blocking the opioid receptors with an antagonist such as naloxone, dynorphinshave been shown to induce a variety of physiological responses, predictably callednon-opiate effects. These effects, most often studied in cellular systems, mice orrats, include paralysis (Bakshi and Faden 1990), changes in the motor system(Walker et al. 1982) and neural cell death (Tan-No et al. 2001). The underlyingsignalling pathways do in some cases involve other receptors, see e.g. (Chen et al.1995, Massardier and Hunt 1989), but direct membrane interactions have alsobeen suggested to play a role. These interactions include translocation into cells(Marinova et al. 2005) and the formation of pores affecting calcium influx into cells(Hugonin et al. 2006). Furthermore, DynA has been suggested to destabilise anddisrupt bilayers by lysis and bilayer fusion, and to induce formation of vesicles inordered bilayers (Naito et al. 2002). Interaction with the membrane may also playa role in the receptor interaction, e.g. by inducing peptide structure, something

2.1 The opioid system 11
that has been observed in model membrane systems (Erne et al. 1985, Hugoninet al. 2008, Lancaster et al. 1991, Lind et al. 2006, Schwyzer 1986). Moreover, themembrane may serve as a surface for peptide adsorption and accumulation priorto receptor binding, as has been suggested by Schwyzer and Sargent (Sargent andSchwyzer 1986, Schwyzer 1986).

12 Chapter 2 Biological background

3 Research projects
3.1 DynA–KOR interactions
A problem of major biological and pharmacological interest is how different lig-ands bind to different members of the opioid receptor family. What distinguishesagonists from antagonists? What are the binding energetics and kinetics? Howhas evolution addressed the issue of receptor–ligand selectivity?
In this project, we look at the interaction between the KOR and its endogenousligand DynA. The main focus of our interest has so far been the interaction betweenone of the extracellular receptor loops and the peptide ligand, since this interactionhas been implicated as crucial for selectivity. The ultimate goal in this project is tostudy the full-length receptor, but the experimental difficulties are considerable,and hence a fragment-based approach has been used. More specifically, the secondextracellular loop (EL2) of KOR was grafted onto a soluble protein scaffold, thecore of human superoxide dismutase 1 (SOD1), resulting in a protein, prosaicallycalled SOD1–EL2. Apart from being an aid in answering the underlying biologicalquestions related to this specific system, the feasibility of using this approach asa general method to study the interaction between membrane protein loops andpeptide ligands was also evaluated.
3.2 Lipid interaction properties of DynA and related pep-tides
The properties of the peptides in the dynorphin family have been investigatedfor a few decades, but many questions remain unanswered. One area of interestis how the peptides interact with lipid bilayers, and for some years researchers inour group have looked at these questions from a biophysical point of view. Whatdistinguishes the dynorphins from other neuropeptides? Are there differences inmembrane interaction properties within this peptide family? What properties ofthe higher-level biological system(s) can be inferred from the biophysical results?Questions like these are at the heart of this project, and form a starting pointfor one of my research projects. More specifically, I have studied the wild-typeDynA, and a few newly discovered members of the dynorphin family, usingfast-tumbling phospholipid bicelles as the (main) membrane mimetic.
These ’new’ dynorphin peptides deserve to have further research devoted tothem, because despite the plethora of synthetically modified DynA peptides andanalogues, up until recently no naturally occurring variants were known. Thenmutations in the gene coding for PDYN were identified and shown to be the causeof a human neurodegenerative disorder (Bakalkin et al. 2010), a discovery that was
13

14 Chapter 3 Research projects
soon followed by the identification of other mutations in this gene (Jezierska et al.2013). Cell toxicity studies, as well as leakage assays showed that the variants hadvery different properties, despite them being linked to the same disorder. Theseinteresting aspects prompted us to begin studying the molecular details of thelipid-interaction properties of these peptides using NMR spectroscopy.

4 Methods
4.1 Membrane mimetic systems
For the life scientist interested in molecules and processes coupled to biologicalmembranes, the complexity and size of a cell envelope effectively prohibit the useof many biophysical and biochemical techniques. This is also true for many othernative membranes, and so the scientist is forced to address the problem by creatinga system sharing the properties of interest with the ’real’ membrane environment,but sufficiently cut down in size and complexity for it to be manageable by atleast some of the methods in the scientist’s toolbox. Such an imitation of a realmembrane is often called a membrane mimetic.
Just as for native biological membranes, the mimetics exploit that lipid moleculeshave certain physicochemical properties that result in rather complicated thermo-dynamical behaviour, with lipids in solution self-assembling into super-molecularcomplexes. A thorough treatment of this is beyond the scope of this thesis, andthe reader is referred to an excellent book by Mouritsen on the subject (Mouritsen2005). For a review of different membrane mimetic systems in the specific contextof NMR spectroscopy, see Warschawski (Warschawski et al. 2011).
4.1.1 Bicelles
In the ideal model, bicelles are patches of bilayer-forming lipids surrounded by arim of short-chain detergent molecules (Andersson and Mäler 2005, Losonczi andPrestegard 1998, Sanders and Schwonek 1992, Vold et al. 1997), as shown in Figure4.1. The bilayer part has very little curvature, similar to a native membrane (moreon this below). By varying the relative amounts of lipids with long and shortchains, as well as parameters such as lipid type, ionic strength, temperature, pHand overall lipid concentration, the morphology of the lipid complexes change,and there is no absolute consensus as to where the boundaries of different bicellemorphologies are in this parameter space (Glover et al. 2001, Lu et al. 2012, Sterninet al. 2001, van Dam et al. 2004). In the projects described in this thesis we haveused bicelles that are so small that their reorientation is fast on an NMR timescale,giving spectra with (reasonably) sharp lines. Such lipid complexes are sometimescalled isotropic bicelles, and are widely used for studies of lipid-interacting proteinsand peptides (Andersson and Mäler 2005; 2006, Prosser et al. 2006).
15

16 Chapter 4 Methods
Figure 4.1: Model of an ideal bicelle. From (Ye 2013), used with permission.
4.1.2 Other systems
An aqueous suspension of only detergent molecules will, above a certain concen-tration, form micelles, which are spherical lipid aggregates with the fatty chainsturned inwards and the polar headgroups facing the water. Micelles have beenused extensively to solubilise membrane-associated peptides and proteins to fa-cilitate biophysical and biochemical characterisation; one arbitrary example outof many is the study of an Arg-rich voltage-sensor domain from a K+ channel(Unnerståle et al. 2009).
Bilayer-forming lipids in solutions without detergents will form various typesof vesicles, i.e. spherical structures with one or several bilayers. From thesecomplexes unilamellar vesicles may be prepared in different sizes, ranging fromsmall unilamellar vesicles (SUVs), with diameters from 20 to 50 nm up to LUVswith diameters of approximately 100 nm.
4.1.3 Scales of length and time
The size differences between different types of membrane mimetics, and to cellularmembranes, are substantial, as shown in Figure 4.2, which gives an approximateoverview of the relative sizes of bicelles, LUVs and an E. coli. bacterial cell. Withnotations from the figure, but in three dimensions, a surface element for a sphereof radius r will be given as S = 2πrh. Here h, the maximum distance (projectedon the radius) between any two points on the surface segment, can be seen asa curvature parameter. This means that for two spheres of different sizes, twosurface elements of equivalent area will have a curvature inversely related to theradii of the spheres. As an example, a 25 nm2 patch on a sphere with r = 100 nm

4.2 NMR 17
will have h ≈ 0.04 nm – less than the length of a chemical bond.
Bicelle, top view
Large unilamellar vesicle
Bacterial celld ~ 10 nm
d ~ 100 nm
d ~ 1000 nm
α
h
r
S
Figure 4.2: Overview of membrane mimetic length scales; d is the diameter of thespherical/disc shaped objects.
4.2 NMR
This is an overview of nuclear magnetic resonance (NMR) spectroscopy, as per-formed on biological samples (in particular polypeptides) in the liquid state. Inaddition to an extremely condensed section on the general theory, which maybe found in several NMR textbooks, such as (Levitt 2008), applications that havebeen exploited in the research treated in this thesis are discussed from a practicalpoint of view.
4.2.1 General theory
Angular momenta
The nucleons making up atomic nuclei all possess both orbital and spin angu-lar momenta, which give a total angular momentum for each nucleon. Thesemomenta then add up to give atomic the nucleus a total angular momentum, orspin, I. The nuclear angular momentum is quantised, and its magnitude takesthe following values:

18 Chapter 4 Methods
|I| = ~√
I(I + 1) (4.1)
where the quantum number I takes an integer or half-integer value. Stable nu-clei are known with I-values between 0 and 15/2. The nuclear angular momen-tum along the x-, y- and z-axis may be represented by operators Ix, Iy and Iz,respectively, and an operator of total angular momentum can be defined as:I2tot = Ix
2+ Iy
2+ Iz
2
Magnetic moment
Associated with the angular momentum is a magnetic momentµI which is parallelor antiparallel to the angular momentum, and with the so-called gyromagnetic(or magnetogyric) ratio γ as the constant of proportionality, according to:
µI = γI (4.2)
Polarisation
In general the spin vectors of the nuclei are isotropically distributed in the absenceof an external magnetic field, yielding no net magnetization vector. If a magneticfield B0 is applied, the combination of magnetic moment and angular momentumwill cause the spin polarisation axis to precess around the applied magnetic field.Since different orientations of the spin polarisation axis with respect to the ap-plied field B0 correspond to different energies, an anisotropic distribution of spinpolarisation directions will be established after some time (if the temperature isfinite). The equilibrium situation is described by Boltzmann statistics, and resultsin a bulk magnetic moment, often expressed as a magnetisation M.
Resonance
In a quantum mechanical treatment a Hamiltonian operator for the interactionenergy between the nuclear magnetic moment and the applied static magneticfield, may be constructed as:
H = −µI · B0 (4.3)
With the magnetic moment proportional to the angular momentum according toEquation 4.2, and the applied field oriented along the z-axis, this simplifies to:
H = −γB0Iz (4.4)
The eigenvalues, and thereby the energy levels, for this Hamiltonian are:

4.2 NMR 19
E = −γ~B0mI, mI = −I,−I + 1, ..., I (4.5)
For a particle with spin 1/2, such as the 1H-nucleus, this corresponds to two energylevels. Transitions between these two energy levels are induced by any interactionwith a frequency corresponding to the energy difference between the levels:
~|ω0| = ∆E = |γ|~B0 (4.6)
A more detailed treatment of the dynamics of superpostition states, correspondingto the direction of precession, gives:
ω0 = −γB0 (4.7)
This so-called Larmor frequency may also be deduced through a classical me-chanical treatment of the system.
Spin ensembles and the density operator
For a macroscopic sample of nuclei, containing on the order of 1019 molecules, theensemble of spins could be treated by adding together all the contributions fromthe single spin states to calculate the macroscopic magnetisation, but this wouldbe rather tedious. Instead an operator which contains the average of all membersin the ensemble is introduced as follows:
σ = |ψ >< ψ| =
(cαcα∗ cαcβ∗cβcα∗ cβcβ∗
)(4.8)
Here cα and cβ are coefficients of a single spin in a superposition state, whilethe overbar denotes averaging over the entire ensemble. The diagonal termsin the density operator matrix represents populations of the two states, whichgives the magnetisation along the z-axis. The off-diagonal terms are the so-calledcoherences between the states, meaning somewhat loosely the degree of alignmentof spin polarisation vectors in the xy-plane.
As an example, a spin-1/2 ensemble in thermal equilibrium will have the followingdensity matrix:
σ =
e−EαkT 0
0 e
−EβkT
(4.9)

20 Chapter 4 Methods
As discussed before, the spins are Boltzmann distributed (Eα =12ω0, Eβ = −
12ω0
in units of ~), and equally distributed in the xy-plane, giving a net magnetisationalong the positive z-axis. Since a perturbation at the Larmor frequency may changethe state of a single spin, the bulk magnetisation vector may be manipulated byapplying a magnetic field oscillating at this frequency, for a given period of time τ– e.g. as a pulse of electromagnetic radiation generated by a current through a coil.The direction of the field is transverse to the static field, in the x-direction, say, andits linear oscillation along the x-axis can be seen as the sum of two fields rotatingin opposite directions in the xy-plane. The field strength Br f is much smallerthan the strengh of static field, typically around four orders of magnitude, butif the frequency of the oscillating field is in (or near) resonance with the Larmorfrequency of the spin, the field will ’join’ the precessing magnetic moment forseveral cycles, letting its effect on the magnetic moment accumulate.
As an example, the bulk magnetisation vector may be ’tipped down’ to the xy-plane where the rotating magnetisation will in turn generate a current in the samecoil used to create the pulse. This response of the system will contain informationon all interactions affecting the energies of single spin states.
4.2.2 Chemical shifts
NMR spectroscopy would be of limited use if every nucleus of a certain type hadthe exact same frequency. Luckily this is not the case, since each nucleus has acertain molecular surrounding, with a specific electron distribution, giving a localmicroscopic magnetic field which will slightly alter the resonance frequency fromthe (theoretical) value of a nucleus only experiencing the applied static field. Thefrequency difference is called the chemical shift, and is usually given as a field-independent offset from the Larmor frequency of the same nucleus in a referencecompound, ωrc, in units of ppm, as follows:
δ =(ω0 − ωrc
ωrc
)(4.10)
Structural information
Although the chemical shift in principle contains all the details of the molecu-lar surroundings, the theoretical framework and modelling capacity required todeduce atomic coordinates directly from the chemical shift values are not yet suf-ficient. With this said, a lot of conformational information may still be deduced,and used together with known theoretical or empirical physicochemical prop-erties of a polypeptide chain, and complementary methods such as molecularmodelling, to estimate protein structure (Cavalli et al. 2007, Robustelli et al. 2010,Wishart and Sykes 1994). For example, atoms belonging to residues in orderedstructural elements will have different chemical shift values than corresponding

4.2 NMR 21
atoms in disordered segments. Hence, a common strategy to estimate structure, orat least structural propensities, is to compare observed chemical shift values withdatabase averages of values from different types of structural elements (Marshet al. 2006).
Ligand binding
If a ligand binds to a receptor, some residues – both on the ligand and the receptor –will necessarily have their chemical environment modified, and nuclei of either thereceptor or ligand, or both, may be studied. Depending on the details of binding(energetics, kinetics, etc), these perturbations will have different magnitudes; acase of ligand-induced structure, e.g., would give large shift differences for thenuclei in the receptor backbone, while for weak binding even nuclei in the vicinityof the binding site may have changes close to the ’chemical shift noise’ level, asseen in Paper I.
Chemical environment
In a situation where a molecule of interest may be in either of two chemicalenvironments the chemical shift values may give information as to the locationof the nuclei even if no ordered structure is present. An example from the thesisat hand is a system where a peptide is dissolved in an aqueous environment andstudied in the absence and presence of a lipid bilayer, as in Paper II.
4.2.3 Dynamics
Relaxation – the return from a non-equilibrium to an equilibrium state – is a richand complex process in general, and this holds true also for the specific case ofatomic nuclei in NMR spectroscopy. For a more complete treatment of the subject,see e.g. (Kowalewski and Mäler 2006) and (Palmer 2007).
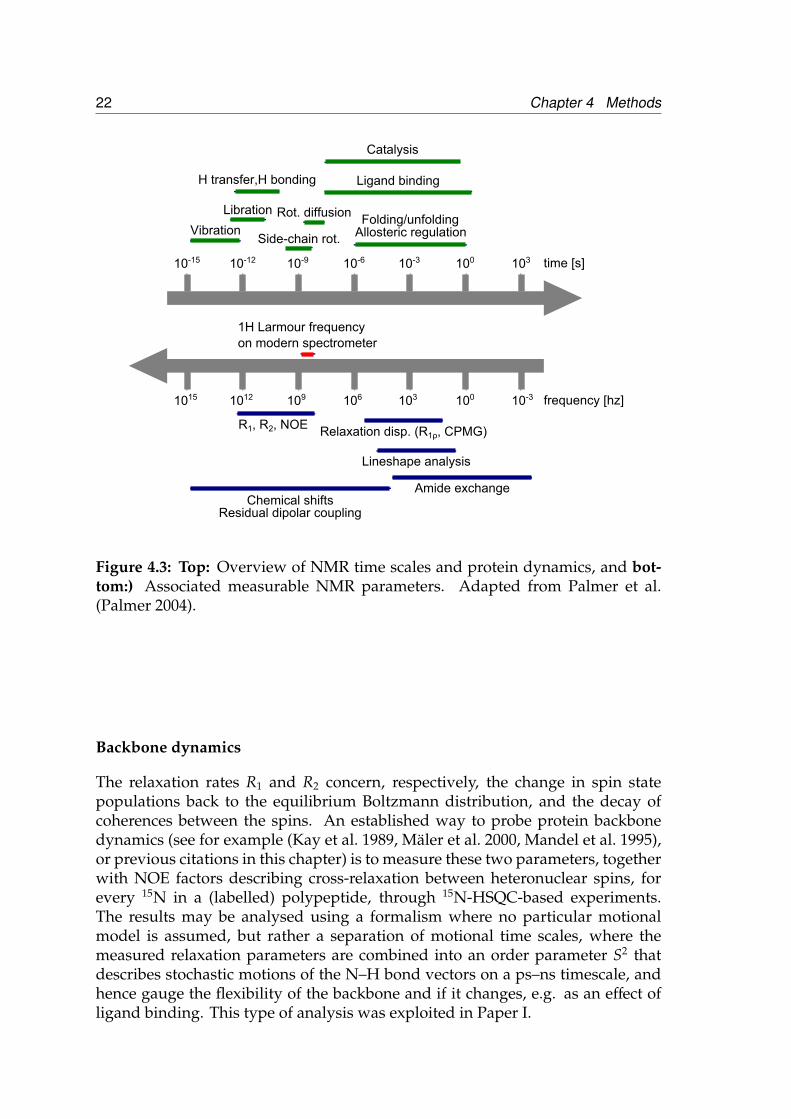
Physically, relaxation in NMR is a consequence of small, local, time-varyingmagnetic field disturbances that, if they contain components at frequencies cor-responding to energy differences between states in the spin system studied, willinduce transitions between these states. Since the transition probabilities willbe (slightly) larger in the direction towards low energy, after some time charac-terised by a relaxation time constant the system will reach equilibrium. The timevariation in the local field at a specific molecular site is generated by varioustypes of molecular motion, and hence the time constants contain information onthese processes. An overview of molecular time scales and the associated NMRobservables is shown in Figure 4.3.
In general, several relaxation mechanisms, some of them correlated, combine togive an effective relaxation from one particular state to another.
22 Chapter 4 Methods
time [s]
frequency [hz]
10-15 10-12 10-9 10-6 10-3 100 103
1015 1012 109 106 103 100 10-3
H transfer,H bonding Ligand binding
Libration
VibrationSide-chain rot.
Rot. diffusion
Catalysis
Folding/unfoldingAllosteric regulation
R1, R2, NOE
Residual dipolar couplingChemical shifts
1H Larmour frequency
on modern spectrometer
Amide exchange
Relaxation disp. (R1ρ, CPMG)
Lineshape analysis
Figure 4.3: Top: Overview of NMR time scales and protein dynamics, and bot-tom:) Associated measurable NMR parameters. Adapted from Palmer et al.(Palmer 2004).
Backbone dynamics
The relaxation rates R1 and R2 concern, respectively, the change in spin statepopulations back to the equilibrium Boltzmann distribution, and the decay ofcoherences between the spins. An established way to probe protein backbonedynamics (see for example (Kay et al. 1989, Mäler et al. 2000, Mandel et al. 1995),or previous citations in this chapter) is to measure these two parameters, togetherwith NOE factors describing cross-relaxation between heteronuclear spins, forevery 15N in a (labelled) polypeptide, through 15N-HSQC-based experiments.The results may be analysed using a formalism where no particular motionalmodel is assumed, but rather a separation of motional time scales, where themeasured relaxation parameters are combined into an order parameter S2 thatdescribes stochastic motions of the N–H bond vectors on a ps–ns timescale, andhence gauge the flexibility of the backbone and if it changes, e.g. as an effect ofligand binding. This type of analysis was exploited in Paper I.

4.2 NMR 23
Paramagnetic relaxation enhancement (PRE)
The (spin) magnetism of an unpaired electron will enhance the relaxation of neigh-bouring nuclei through a dipole–dipole mechanism (Bloembergen and Morgan
1961), with an electron–nucleus distance dependence of1r6 . The dipole–dipole
interaction energy is proportional to the product of the gyromagnetic ratios of thetwo particles, and since the electron gyromagnetic ratio is approximately 650 timeslarger than the nuclear equivalent, the relaxation enhancement may be dramatic(Jahnke 2002).
One way to exploit this phenomenon is to attach a chemical moiety containingan unpaired electron on a ligand, yielding a sensitive probe for ligand–receptorinteraction. This was used to study the interaction between DynA and SOD1–EL2in Paper I.
4.2.4 Diffusion
NMR measurements of molecular diffusion are based on spin echoes – the reemer-gence of induced signal through a refocussing of spin phases – and magnetic fieldgradients. The gradients introduce a position-dependence for the spins, and incombination with spin echoes this can be used to study translational motion ofmolecules in the sample. The interested reader is referred to the book by Morris(Morris 2007).
Here, diffusion measurements were used in Paper II to study the association ofpeptides to bicelles.
4.2.5 Labelling
Although not a formal requisite of NMR spectroscopy, many, if not most, ofpresent-day NMR experiments require isotope labelling of the studied biomolecules.Labelling schemes may range from reasonably simple, e.g. feed bacteria with15N-labelled ammonium chloride during protein production, to targeting spe-cific bacterial pathways with labelled chemicals in order to get certain (parts of)residues labelled. In the projects described here, protein (SOD1–EL2) uniformlylabelled with 15N and/or 13C by growing the over-expressing bacteria (describedin the Experimentals procedure of paper I) in 15N-labelled ammonium chlorideand/or 13C-labelled glucose was used.

24 Chapter 4 Methods
4.3 CD
CD spectroscopy exploits the fact that some materials and molecules absorb cir-cularly polarised light differently depending on the polarisation direction of thelight. The basis of this optical activity is an asymmetry in the molecule, and achemical structure that allows electrons to move in a helical fashion. For biologi-cal molecules, it is usually not the chromophore itself that has this type of opticalactivity, but the electronic environment in which it resides. Hence the method isvery sensitive to structural features of e.g. a polypeptide chain. For a practicalreview of the method, see the article by Kelly (Kelly et al. 2005).
In general, CD spectra provide a good estimate of overall molecular structure –or, more correctly, an ensemble average molecular structure – while more quan-titative information is difficult to extract. By fitting the experimental data to asuperposition of reference spectral profiles, either experimental (Greenfield 2007)or theoretical (Louis-Jeune et al. 2012), one may attempt to obtain the percent-age of different structural elements in the peptide or protein backbone, althoughrecent results suggest a risk of overinterpretation (Lin et al. 2013).
CD spectroscopy was used in both Paper I and II to estimate peptide and proteinoverall secondary structure.

5 Summary of results
5.1 The construct protein SOD1–EL2 has a β–barrelcore, while the inserted segment is unstructured.(Paper I)
In order to probe a purported selectivity mechanism for the KOR – the interactionof the second extracellular loop (EL2) with peptide ligands – while avoiding someof the difficulties of working with the full-length membrane receptor, a constructprotein was engineered. This protein consisted of a soluble β-barrel scaffold ontowhich the EL2 from KOR was grafted. The scaffold was a variant of human su-peroxide dismutase 1 (SOD1) in which the native loops had been truncated (asdescribed in the Paper I), giving the engineered protein the name SOD1–EL2.
Overall, our characterisation of the protein by CD and various types of NMRexperiments paint the picture of a construct where the core part is structurallyvery similar to the ’parent’ scaffold protein SOD1, while the segment taken fromthe KOR behaves as a flexible loop.
5.2 The extracellular loop 2 of the κ opioid receptor,as expressed in the construct protein SOD1–EL2,interacts weakly with Dynorphin A. (Paper I)
The construct discussed in the above section was used to study the interactionbetween the EL2 of KOR and DynA – the primary endogenous ligand of KOR.The effects of (unlabelled) DynA on NMR chemical shifts of (15N-labelled) SOD1–EL2 backbone amides were small and, for many residues, noise-like. A fewresidues, mainly located in the EL2-region or in areas of the scaffold in closeproximity to EL2, showed slightly larger shift differences, differences that alsowere proportional to the DynA concentration.
Also the relaxation rates of amides in the protein backbone [umpublished results]changed very little upon addition of DynA. Fitted order parameters suggested anincrease in backbone rigidity in the presence of ligand, but the quality of fittingwas judged insufficient to use this result as evidence for ligand binding.
Mirroring chemical shift experiments, in which unlabelled protein was titratedonto 15N labelled DynA (L5 and L12) were also performed. In this experimentalapproach SOD1–EL2, EL2 peptide and the naked scaffold protein, SODnoloops,were used. These experiments showed more conclusively an interaction between
25

26 Chapter 5 Summary of results
EL2 (in SOD1–EL2) and DynA. Both the EL2 peptide and SOD1–EL2 caused con-sistent changes in the chemical shifts of DynA, while no chemical shift changeswere observed when the scaffold without the EL2 segment was added, showingthat the EL2-loop is required for DynA interaction. Assuming a one-to-one bind-ing, dissociation constants were estimated from the chemical shift data, and areshown in Table 5.1. The dissociation constants describe a remarkably weak in-teraction, compared with the nanomolar affinities previously reported for DynAbinding to the full-length receptor (Garzón et al. 1983, Kawasaki et al. 1993). Butfrom a biological point of view this makes a lot of sense, since a too strong bindingof the ligand to the extracellular regions of the receptor could impair the bindingof the DynA N-terminal to the KOR transmembrane pocket, or significantly slowdown the release of DynA from the receptor.
Table 5.1: Dissociation constants for two molecules interacting with DynA.
Interacting molecule KD [µm]EL2 peptide 310 ± 25SOD1–EL2 32 ± 5
Further interaction studies were performed with SOD1–EL2, and DynA with aparamagnetic probe (MTSL) attached C-terminally to DynA. As described in theMethods section, these experiments probe the distances between an unpairedelectron (in MTSL) and any nucleus, through the effects of the electron on the re-laxation of the nuclear spin. Using this technique binding was indeed confirmed,with the EL2 loop being the region of the protein most affected by the spin-labelledDynA. Also other areas appear to be ’close enough’ to the probe in DynA to beaffected, but since the location of these regions in the protein are reasonably nearthe inserted EL2 loop, this result is consistent with a selective interaction with theEL2 loop as also shown by the chemical shift analysis.
5.3 The two disease-related Dynorphin A variants R6W-DynA and L5S-DynA have markedly different lipidinteraction properties. (Paper II)
In this project, two recently found (Bakalkin et al. 2010) DynA variants were in-vestigated with CD and NMR spectroscopy. The main focus was on the moleculardetails of peptide–lipid interaction, since a previous study had shown that two ofthe variants had markedly different membrane perturbing properties in a vesicleleakage assay (Madani et al. 2011).
As described in Paper II, a variety of proton NMR techniques were employed,and the results show that one peptide variant – R6W–DynA – has ’aggressive’lipid perturbing properties, with a strong association and comparatively deep

5.3 The two disease-related Dynorphin A variants R6W-DynA and L5S-DynA havemarkedly different lipid interaction properties. (Paper II) 27
N-terminal positioning, while another – the L5S–DynA variant – exhibits a verymild type of interaction. An interesting aspect of this work is the moderate degreeof ordered structure in all the peptides despite evident bilayer penetration. Themore aggressive variant, R6W–DynA, was found to bind bicelles to almost 100 %,while remaining largely unstructured. This has also been observed for wild-typeDynA (Lind et al. 2006) and suggests that the mechanisms by which some ofthese peptides e.g. induce leakage in vesicles are rather complicated, in contrastto strongly channel- or pore-forming peptides such as gramicidin A (Allen et al.2003, Cifu et al. 1992).

28 Chapter 5 Summary of results

6 Conclusions and outlook
All the work in this thesis seek to understand the function of the dynorphin neu-ropeptides, and the fate of these peptides when they come in close contact withthe outer boundary of a cell. Admittedly, the approaches here chosen to study thisbiological system are, like most biophysical approaches, very reductionistic. Theinteraction with the receptor is taken apart, and only a fragment (less than 10 %of the total membrane protein sequence) is studied. Likewise, in the membraneinteraction studies, membrane mimetics involving a maximum of three differentlipid types are used. How valid are these approches, what can we say from ourresults, and what are the possible routes forward?
Starting with the receptor, our approach may be criticised on the ground thata fragment taken out of a protein context will be so far from the features of theoriginal system that any ligand–interaction observed in the model system hasno relevance for the interaction in the native system. In response to this thereare previous studies (Katragadda et al. 2001, Kerman and Ananthanarayanan2005) showing that expressed membrane protein fragments keep the biophysicalproperties, including structure, of the full-size system. Moreover, the segmentwe study has a low structural propensity anyway, so this issue should not be ofcritical importance. A question that is more difficult to address comes from thefact that in the native receptor, the EL2 is most likely additionally constrained bya disulfide bridge between one of its residues and a cysteine in a transmembranehelix. Even though these cysteines are critical for antagonist binding (Ott et al.2004), the importance for the EL2–DynA interaction is difficult to assess, sincethe loss of function may also be a consequence of changes in the transmembraneregion. The conceptual distance from the native system may also be seen as anadvantage; in the native system ligands bind very strongly to a transmembranesite, and a much weaker binding to an extracellular loop would be difficult todisentangle from the strong one.
Our results show that the DynA ligand has a weak, positive interaction withan extracellular loop of its native receptor, KOR. This feeble binding makes bio-logical sense, since a stronger binding could prevent the opioid core (N-terminus)of the peptide from reaching the transmembrane interaction site. Studies with ascrambled DynA peptide [unpublished], suggest that this also interacts weaklywith the loop, thereby precluding strict sequence specificity. Further research onpeptides without potency in activity assays could help answer the question ofwhether there is a positive ligand–receptor loop interaction that ’lets the rightones in’, or a passive obstruction of the transmembrane binding site that keepsthe wrong peptides out. Related to this, fully labelled ligands would be of greathelp, and attempts to produce such peptides are being discussed. Further, con-structs with other opioid receptor loops could be a way to map out the selectivitydifferences between the receptors in this family. And finally, the holy grail of this
29

30 Chapter 6 Conclusions and outlook
work would be studies of the full-length receptor under conditions accessible toliquid-state NMR.
As for the membrane interaction of the disease-related DynA variants, the choiceof lipids in the membrane mimetics may be motivated by the abundance of PClipids in mammalian neural cell membranes. For example PC is the major com-ponent of rat neuronal plasma membranes, constituting almost 30 % of total lipidamount (Calderon et al. 1995) (the second is PE, present at 15 to 20 %). PG is nota major component in neural cells, but its overall negative charge is a substitutefor other charged lipids such as PS and PI which are present to various degreesin biological membranes. It is of course possible that the dynorphin peptidescould interact mainly with membrane regions, in the native cellular environment,enriched in lipids with another headgroup than PC, but the author of this thesisis not aware of any reports of this.
Our work on the dynorphin variants show that small changes of the sequence(single residues) may result in large differences in membrane-interaction proper-ties. The link to a human physiological disorder makes the studied peptides bothrelevant and interesting, and we think that our results may be used in future re-search on the disease. From a biophysical perspective, one way to go forward withthis project would be to make similiar studies of variants with more systematicresidue variations. Another is to investigate if there is any cooperative behaviourinvolved, e.g. accumulation of peptides on/in the membrane, and formation ofintramolecular complexes contributing to membrane destabilisation and possiblydisruption. Here fluorescense spectroscopy techniques would be a complementto the methods described previously.

Acknowledgements
I would like to warmly thank a number of people, whose generous contributionsof time, skill and encouragement have brought this thesis into existence.
First and foremost, main supervisors Lena Mäler and Astrid Gräslund; for yourprofound knowledge, clarity of thought and common sense.
Project collaborators/supervisors, from whom I have learnt immensely: JensDanielsson for his inspiring unwillingness to compromise on what it meansto be a scientist, Martin Kurnik for tolerating the simultaneous presence of hisvirtuosity and my inaptitude in the same confined lab spaces, and Jesper Lindfor scientifical and practical guidance during my first nervous weeks.
Present PhD colleagues: Weihua Ye, Axel Abelein, Scarlett Szpryngiel and JobstLiebau; and past: Anna Wahlström, Sofia Unnerståle and Fatemeh Madani.For being around, for being funny, for helping out. Life is what happens whileyou are busy making experiment plans, and you make that life interesting andcomfortable.
Other colleagues and students, past and present: Jüri Jarvet, Sebastian Wärmlän-der, Göran Eriksson, Luminita Moruz, Viktor Granholm, Erik Sjölund, Chris-tian Seutter von Loetzen, Christina Bösing, Pontus Pettersson, and many others,who are not named, but not forgotten. You deserve more recognition than thesefew lines, and I am confident that you get it.
Senior Research Engineer Torbjörn Astlind, Senior Administrative Officer HaidiAstlind, and Research Engineer Britt-Marie Olsson. Impressive titles, but yourwork is infinitely more impressive. God only knows what we would do withoutyou.
My mentor Andreas Barth: Even if I have been fortunate enough not to need yourservices, I know that you are there to help me.
Last but not least; former supervisors, teachers and evaluators: Mathias Nils-son and Gareth A. Morris of University of Manchester, and Martin Billeter ofGothenburg University. You got me interested in biophysics and NMR, and youhave given solid support at times when I have doubted my professional choicesand competence, something for which I am immensely grateful.
31

32 Chapter 6 Conclusions and outlook

Bibliography
Allen, T. W., O. S. Andersen, and B. Roux. 2003. Structure of gramicidin A ina lipid bilayer environment determined using molecular dynamics simulationsand solid-state NMR data. Journal of the American Chemical Society 125:9868–9877.
Andersson, A., and L. Mäler. 2005. Magnetic resonance investigations of lipidmotion in isotropic bicelles. Langmuir 21:7702–7709.
Andersson, A., and L. Mäler. 2006. Size and shape of fast-tumbling bicelles asdetermined by translational diffusion. Langmuir 22:2447–2449.
Bakalkin, G., H. Watanabe, J. Jezierska, C. Depoorter, C. Verschuuren-Bemelmans,I. Bazov, K. A. Artemenko, T. Yakovleva, D. Dooijes, B. P. C. V. de Warrenburg,R. A. Zubarev, B. Kremer, P. E. Knapp, K. F. Hauser, C. Wijmenga, F. Nyberg,R. J. Sinke, and D. S. Verbeek. 2010. Prodynorphin mutations cause the neurode-generative disorder spinocerebellar ataxia type 23. American Journal of HumanGenetics 87:593–603.
Bakheet, T. M., and A. J. Doig. 2009. Properties and identification of humanprotein drug targets. Bioinformatics 25:451–457.
Bakshi, R., and A. I. Faden. 1990. Competitive and non-competitive NMDAantagonists limit dynorphin A-induced rat hindlimb paralysis. Brain Research507:1–5.
Björnerås, J., A. Gräslund, and L. Mäler. 2013. Membrane interaction of disease-related dynorphin A variants. Biochemistry 52:4157–4167.
Bloembergen, N., and L. O. Morgan. 1961. Proton relaxation times in paramagneticsolutions. effects of electron spin relaxation. The Journal of Chemical Physics 34:842.
Bodnar, R. J. 2012. Endogenous opiates and behavior: 2011. Peptides 38:463–522.
Brownstein, M. J. 1993. A brief history of opiates, opioid peptides, and opioidreceptors. Proceedings of the National Academy of Sciences of the United States ofAmerica 90:5391–5393.
Bruchas, M. R., B. B. Land, and C. Chavkin. 2010. The dynorphin/kappa opioidsystem as a modulator of stress-induced and pro-addictive behaviors. BrainResearch 1314:44–55.
Bruijnzeel, A. W. 2009. kappa-opioid receptor signaling and brain reward func-tion. Brain Research Reviews 62:127–146.
Calderon, R. O., B. Attema, and G. H. DeVries. 1995. Lipid composition ofneuronal cell bodies and neurites from cultured dorsal root ganglia. Journal ofNeurochemistry 64:424–429.
33

34 BIBLIOGRAPHY
Cavalli, A., X. Salvatella, C. M. Dobson, and M. Vendruscolo. 2007. Proteinstructure determination from NMR chemical shifts. Proceedings of the NationalAcademy of Sciences 104:9615–9620.
Chavkin, C., and A. Goldstein. 1981a. Demonstration of a specific dynorphinreceptor in guinea pig ileum myenteric plexus. Nature 291:591–593.
Chavkin, C., and A. Goldstein. 1981b. Specific receptor for the opioid peptidedynorphin: structure–activity relationships. Proceedings of the National Academyof Sciences of the United States of America 78:6543–6547.
Chavkin, C., I. F. James, and A. Goldstein. 1982. Dynorphin is a specific endoge-nous ligand of the kappa opioid receptor. Science 215:413–415.
Chen, L., Y. Gu, and L. Y. Huang. 1995. The mechanism of action for the blockof NMDA receptor channels by the opioid peptide dynorphin. Journal of Neuro-science 15:4602–4611.
Chien, E. Y. T., W. Liu, Q. Zhao, V. Katritch, G. Won Han, M. A. Hanson, L. Shi,A. H. Newman, J. A. Javitch, V. Cherezov, and R. C. Stevens. 2010. Structure ofthe human dopamine D3 receptor in complex with a D2/D3 selective antagonist.Science 330:1091–1095.
Cifu, A., R. K. 2nd, and O. Andersen. 1992. On the supramolecular organizationof gramicidin channels. the elementary conducting unit is a dimer. BiophysicalJournal 61:189 – 203.
Dawkins, R., J. C. Venter, S. Altman, L. Hartwell, P. Davies, C. McKay,L. Krauss, and R. Bingham, 2011. The great debate - what is life? Video,thesciencenetwork.org/programs/the-great-debate-what-is-life.
Dietrich, G., G. Gaibelet, R. Capeyrou, J. L. Butour, F. Pontet, and L. J. Emorine.1998. Implication of the first and third extracellular loops of the mu-opioidreceptor in the formation of the ligand binding site: a study using chimericmu-opioid/angiotensin receptors. Journal of Neurochemistry 70:2106–2111.
Drews, E., and A. Zimmer. 2009. Modulation of alcohol and nicotine responsesthrough the endogenous opioid system. Progress in Neurobiology .
Drews, J. 2000. Drug discovery: A historical perspective. Science 287:1960–1964.
Erne, D., D. F. Sargent, and R. Schwyzer. 1985. Preferred conformation, orienta-tion, and accumulation of dynorphin A-(1-13)-tridecapeptide on the surface ofneutral lipid membranes. Biochemistry 24:4261–4263.
Ferguson, D. M., S. Kramer, T. G. Metzger, P. Y. Law, and P. S. Portoghese. 2000.Isosteric replacement of acidic with neutral residues in extracellular loop-2 of thekappa-opioid receptor does not affect dynorphin A(1–13) affinity and function.Journal of Medicinal Chemistry 43:1251–1252.

BIBLIOGRAPHY 35
Filizola, M., and L. A. Devi. 2012. Structural biology: How opioid drugs bind toreceptors. Nature 485:314–317.
Garzón, J., P. Sánchez-Blázquez, V. Höllt, N. M. Lee, and H. H. Loh. 1983. En-dogenous opioid peptides: comparative evaluation of their receptor affinitiesin the mouse brain. Life Sciences 33 Suppl 1:291–294.
Glover, K. J., J. A. Whiles, G. Wu, N. jun Yu, R. Deems, J. O. Struppe, R. E.Stark, E. A. Komives, and R. R. Vold. 2001. Structural evaluation of phospho-lipid bicelles for solution-state studies of membrane-associated biomolecules.Biophysical Journal 81:2163 – 2171.
Goldstein, A., W. Fischli, L. I. Lowney, M. Hunkapiller, and L. Hood. 1981. Porcinepituitary dynorphin: complete amino acid sequence of the biologically activeheptadecapeptide. Proceedings of the National Academy of Sciences of the UnitedStates of America 78:7219–7223.
Goldstein, A., L. I. Lowney, and B. K. Pal. 1971. Stereospecific and nonspecificinteractions of the morphine congener levorphanol in subcellular fractions ofmouse brain. Proceedings of the National Academy of Sciences of the United States ofAmerica 68:1742–1747.
Goldstein, A., S. Tachibana, L. I. Lowney, M. Hunkapiller, and L. Hood. 1979.Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proceedings of theNational Academy of Sciences of the United States of America 76:6666–6670.
Granier, S., A. Manglik, A. C. Kruse, T. S. Kobilka, F. S. Thian, W. I. Weis, andB. K. Kobilka. 2012. Structure of the delta-opioid receptor bound to naltrindole.Nature 485:400–404.
Greenfield, N. J. 2007. Using circular dichroism spectra to estimate proteinsecondary structure. Nature Protocols 1:2876–2890.
Gudermann, T., B. Nürnberg, and G. Schultz. 1995. Receptors and G proteins asprimary components of transmembrane signal transduction. Journal of MolecularMedicine 73:51–63.
Hong, M., Y. Zhang, and F. Hu. 2012. Membrane protein structure and dynamicsfrom NMR spectroscopy. Annual Review of Physical Chemistry 63:1–24.
Hugonin, L., A. Barth, A. Gräslund, and A. Perálvarez-Marín. 2008. Secondarystructure transitions and aggregation induced in dynorphin neuropeptides bythe detergent sodium dodecyl sulfate. Biochimica et Biophysica Acta 1778:2580–2587.
Hugonin, L., V. Vukojevic, G. Bakalkin, and A. Gräslund. 2006. Membrane leakageinduced by dynorphins. FEBS Letters 580:3201–3205.
Jahnke, W. 2002. Spin labels as a tool to identify and characterize protein-ligandinteractions by NMR spectroscopy. ChemBioChem 3:167–173.

36 BIBLIOGRAPHY
Janecka, A., J. Fichna, and T. Janecki. 2004. Opioid receptors and their ligands.Current Topics in Medicinal Chemistry 4:1–17.
Jezierska, J., G. Stevanin, H. Watanabe, M. R. Fokkens, F. Zagnoli, J. Kok, J.-Y.Goas, P. Bertrand, C. Robin, A. Brice, G. Bakalkin, A. Durr, and D. S. Verbeek.2013. Identification and characterization of novel PDYN mutations in dominantcerebellar ataxia cases. Journal of Neurology .
Katragadda, M., J. L. Alderfer, and P. L. Yeagle. 2001. Assembly of a polytopicmembrane protein structure from the solution structures of overlapping peptidefragments of bacteriorhodopsin. Biophysical Journal 81:1029–1036.
Kawasaki, A. M., R. J. Knapp, A. Walton, W. S. Wire, T. Zalewska, H. I. Yamamura,F. Porreca, T. F. Burks, and V. J. Hruby. 1993. Syntheses, opioid binding affinities,and potencies of dynorphin A analogues substituted in positions, 1, 6, 7, 8 and10. International Journal of Peptide and Protein Research 42:411–419.
Kay, L. E., D. A. Torchia, and A. Bax. 1989. Backbone dynamics of proteins asstudied by 15N inverse detected heteronuclear NMR spectroscopy: applicationto staphylococcal nuclease. Biochemistry 28:8972–8979.
Kelly, S. M., T. J. Jess, and N. C. Price. 2005. How to study proteins by circulardichroism. Biochimica et Biophysica Acta 1751:119–139.
Kerman, A., and V. S. Ananthanarayanan. 2005. Expression and spectroscopiccharacterization of a large fragment of the mu-opioid receptor. Biochimica etBiophysica Acta 1747:133–140.
Kowalewski, J., and L. Mäler. 2006. Nuclear spin relaxation in liquids: Theory,experiments, and applications. CRC Press, Boca Raton, Florida.
Krogh, A., B. Larsson, G. von Heijne, and E. L. Sonnhammer. 2001. Predictingtransmembrane protein topology with a hidden markov model: application tocomplete genomes. Journal of Molecular Biology 305:567–580.
Lancaster, C. R., P. K. Mishra, D. W. Hughes, S. A. St-Pierre, A. A. Bothner-By,and R. M. Epand. 1991. Mimicking the membrane-mediated conformationof dynorphin A-(1-13)-peptide: circular dichroism and nuclear magnetic reso-nance studies in methanolic solution. Biochemistry 30:4715–4726.
Levitt, M. H. 2008. Spin dynamics. 2nd edition. John Wiley & Sons Ltd, Chichester,England.
Lin, K., H. Yang, Z. Gao, F. Li, and S. Yu. 2013. Overestimated accuracy of circu-lar dichroism in determining protein secondary structure. European BiophysicsJournal 42:455–461.
Lind, J., A. Gräslund, and L. Mäler. 2006. Membrane interactions of dynorphins.Biochemistry 45:15931–15940.

BIBLIOGRAPHY 37
Losonczi, J., and J. Prestegard. 1998. Improved dilute bicelle solutions for high-resolution NMR of biological macromolecules. Journal of Biomolecular NMR12:447–451.
Louis-Jeune, C., M. A. Andrade-Navarro, and C. Perez-Iratxeta. 2012. Predic-tion of protein secondary structure from circular dichroism using theoreticallyderived spectra. Proteins: Structure, Function, and Bioinformatics 80:374–381.
Lu, Z., W. D. Van Horn, J. Chen, S. Mathew, R. Zent, and C. R. Sanders. 2012.Bicelles at low concentrations. Molecular Pharmaceutics 9:752–761.
Madani, F., M. M. Taqi, S. K. T. S. Wärmländer, D. S. Verbeek, G. Bakalkin, andA. Gräslund. 2011. Perturbations of model membranes induced by pathogenicdynorphin A mutants causing neurodegeneration in human brain. Biochemicaland Biophysical Research Communications 411:111–114.
Mäler, L., J. Blankenship, M. Rance, and W. J. Chazin. 2000. Site-site communi-cation in the EF-hand Ca2+-binding protein calbindin D9k. Nature StructuralBiology 7:245–250.
Mandel, A. M., M. Akke, and A. G. Palmer. 1995. Backbone dynamics of Es-cherichia coli ribonuclease HI:: correlations with structure and function in anactive enzyme. Journal of Molecular Biology 246:144–163.
Manglik, A., A. C. Kruse, T. S. Kobilka, F. S. Thian, J. M. Mathiesen, R. K. Sunahara,L. Pardo, W. I. Weis, B. K. Kobilka, and S. Granier. 2012. Crystal structure of themu-opioid receptor bound to a morphinan antagonist. Nature .
Marinova, Z., V. Vukojevic, S. Surcheva, T. Yakovleva, G. Cebers, N. Pasikova,I. Usynin, L. Hugonin, W. Fang, M. Hallberg, D. Hirschberg, T. Bergman, U. Lan-gel, K. F. Hauser, A. Pramanik, J. V. Aldrich, A. Gräslund, L. Terenius, andG. Bakalkin. 2005. Translocation of dynorphin neuropeptides across the plasmamembrane. a putative mechanism of signal transmission. Journal of BiologicalChemistry 280:26360–26370.
Marsh, J. A., V. K. Singh, Z. Jia, and J. D. Forman-Kay. 2006. Sensitivity ofsecondary structure propensities to sequence differences between alpha- andgamma-synuclein: implications for fibrillation. Protein Science 15:2795–2804.
Massardier, D., and P. F. Hunt. 1989. A direct non-opiate interaction of dynorphin-(1-13) with the N-methyl-D-aspartate (NMDA) receptor. European Journal ofPharmacology 170:125–126.
Meng, F., M. T. Hoversten, R. C. Thompson, L. Taylor, S. J. Watson, and H. Akil.1995. A chimeric study of the molecular basis of affinity and selectivity of thekappa and the delta opioid receptors. potential role of extracellular domains.Journal of Biological Chemistry 270:12730–12736.
Metzger, T. G., and D. M. Ferguson. 1995. On the role of extracellular loops ofopioid receptors in conferring ligand selectivity. FEBS Letters 375:1–4.

38 BIBLIOGRAPHY
Morris, G. A. 2007. Diffusion-Ordered Spectroscopy. Wiley Blackwell (John Wiley &Sons).
Mouritsen, O. G. 2005. Life - as a matter of fat. Springer-Verlag, Berlin Heidelberg.
Naito, A., T. Nagao, M. Obata, Y. Shindo, M. Okamoto, S. Yokoyama, S. Tuzi,and H. Saitô. 2002. Dynorphin induced magnetic ordering in lipid bilayers asstudied by 31P NMR spectroscopy. Biochimica et Biophysica Acta - Biomembranes1558:34–44.
Ott, D., R. Frischknecht, and A. Plückthun. 2004. Construction and characteriza-tion of a kappa opioid receptor devoid of all free cysteines. Protein Engineering,Design & Selection 17:37–48.
Owen, J. 2008. Virus-infecting virus fuels definition of life debate. National Geo-graphic News http://news.nationalgeographic.com/news/2008/08/080822-giant-virus.html.
Palczewski, K., T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. L.Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto, and M. Miyano.2000. Crystal structure of rhodopsin: A G protein-coupled receptor. Science289:739–745.
Palmer, A. G., 2007. Relaxation and dynamic processes. Pdf E-book.
Palmer, A. G., 3rd. 2004. NMR characterization of the dynamics of biomacro-molecules. Chemical Reviews 104:3623–3640.
Park, S. H., B. B. Das, F. Casagrande, Y. Tian, H. J. Nothnagel, M. Chu, H. Kiefer,K. Maier, A. A. De Angelis, F. M. Marassi, and S. J. Opella. 2012. Structure ofthe chemokine receptor CXCR1 in phospholipid bilayers. Nature 491:779––783.
Paterlini, G., P. S. Portoghese, and D. M. Ferguson. 1997. Molecular simulation ofdynorphin A-(1-10) binding to extracellular loop 2 of the kappa-opioid receptor.a model for receptor activation. Journal of Medicinal Chemistry 40:3254–3262.
Paterlini, M. G. 2005. The function of the extracellular regions in opioid recep-tor binding: insights from computational biology. Current Topics in MedicinalChemistry 5:357–367.
Pert, C. B., and S. H. Snyder. 1973. Opiate receptor: demonstration in nervoustissue. Science 179:1011–1014.
Prosser, R. S., F. Evanics, J. L. Kitevski, and M. S. Al-Abdul-Wahid. 2006. Currentapplications of bicelles in NMR studies of membrane-associated amphiphilesand proteins. Biochemistry 45:8453–8465.
Przewłocki, R., and B. Przewłocka. 2001. Opioids in chronic pain. European Journalof Pharmacology 429:79–91.

BIBLIOGRAPHY 39
Robustelli, P., K. Kohlhoff, A. Cavalli, and M. Vendruscolo. 2010. Using NMRchemical shifts as structural restraints in molecular dynamics simulations ofproteins. Structure 18:923–933.
Rosenbaum, D. M., S. G. F. Rasmussen, and B. K. Kobilka. 2009. The structureand function of G-protein-coupled receptors. Nature 459:356–363.
Roth, B. L., K. Baner, R. Westkaemper, D. Siebert, K. C. Rice, S. Steinberg, P. Erns-berger, and R. B. Rothman. 2002. Salvinorin A: a potent naturally occurring non-nitrogenous kappa opioid selective agonist. Proceedings of the National Academyof Sciences of the United States of America 99:11934–11939.
Sanders, C. R., and J. P. Schwonek. 1992. Characterization of magnetically ori-entable bilayers in mixtures of dihexanoylphosphatidylcholine and dimyris-toylphosphatidylcholine by solid-state NMR. Biochemistry 31:8898–8905.
Sargent, D. F., and R. Schwyzer. 1986. Membrane lipid phase as catalyst forpeptide-receptor interactions. Proceedings of the National Academy of Sciences ofthe United States of America 83:5774–5778.
Schlechtingen, G., R. N. DeHaven, J. D. Daubert, J. A. Cassel, N. N. Chung, P. W.Schiller, J. P. Taulane, and M. Goodman. 2003. Structure-activity relationships ofdynorphin A analogues modified in the address sequence. Journal of MedicinalChemistry 46:2104–2109.
Schwyzer, R. 1986. Estimated conformation, orientation, and accumulation ofdynorphin A-(1-13)-tridecapeptide on the surface of neutral lipid membranes.Biochemistry 25:4281–4286.
Shimamura, T., M. Shiroishi, S. Weyand, H. Tsujimoto, G. Winter, V. Katritch,R. Abagyan, V. Cherezov, W. Liu, G. W. Han, T. Kobayashi, R. C. Stevens, andS. Iwata. 2011. Structure of the human histamine H1 receptor complex withdoxepin. Nature 475:65–70.
Snyder, S. H., and G. W. Pasternak. 2003. Historical review: Opioid receptors.Trends in Pharmacological Sciences 24:198–205.
Sternin, E., D. Nizza, and K. Gawrisch. 2001. Temperature dependence ofDMPC/DHPC mixing in a bicellar solution and its structural implications. Lang-muir 17:2610–2616.
Tan-No, K., G. Cebers, T. Yakovleva, B. H. Goh, I. Gileva, K. Reznikov, M. Aguilar-Santelises, K. F. Hauser, L. Terenius, and G. Bakalkin. 2001. Cytotoxic effectsof dynorphins through nonopioid intracellular mechanisms. Experimental CellResearch 269:54–63.
Tejeda, H. A., T. S. Shippenberg, and R. Henriksson. 2012. The dynorphin/kappa-opioid receptor system and its role in psychiatric disorders. Cellular and Molec-ular Life Sciences 69:857–896.

40 BIBLIOGRAPHY
Terenius, L. 1973a. Characteristics of the "receptor" for narcotic analgesics insynaptic plasma membrane fraction from rat brain. Acta Pharmacol Toxicol(Copenh) 33:377–384.
Terenius, L. 1973b. Stereospecific interaction between narcotic analgesics and asynaptic plasm a membrane fraction of rat cerebral cortex. Acta Pharmacol Toxicol(Copenh) 32:317–320.
Terenius, L., W. H. Gispen, and D. De Wied. 1975. ACTH-like peptides andopiate receptors in the rat brain: Structure-activity studies. European Journal ofPharmacology 33:395–399.
Teschemacher, H., K. E. Opheim, B. M. Cox, and A. Goldstein. 1975. A peptide-like substance from pituitary that acts like morphine. i. isolation. Life Sciences16:1771–1775.
Thompson, A. A., W. Liu, E. Chun, V. Katritch, H. Wu, E. Vardy, X.-P. Huang,C. Trapella, R. Guerrini, G. Calo, B. L. Roth, V. Cherezov, and R. C. Stevens. 2012.Structure of the nociceptin/orphanin FQ receptor in complex with a peptidemimetic. Nature 485:395–399.
Turcotte, A., J.-M. Lalonde, S. St-Pierre, and S. Lemaire. 1984. Dynorphin-(1–13). i.structure-function relationships of Ala-containing analogs. International Journalof Peptide and Protein Research 23:361–367.
Uhl, G. R., S. Childers, and G. Pasternak. 1994. An opiate-receptor gene familyreunion. Trends in Neurosciences 17:89 – 93.
Unnerståle, S., J. Lind, E. Papadopoulos, and L. Mäler. 2009. Solution structure ofthe HsapBK K+ channel voltage-sensor paddle sequence. Biochemistry 48:5813–5821.
van Dam, L., G. Karlsson, and K. Edwards. 2004. Direct observation and charac-terization of DMPC/DHPC aggregates under conditions relevant for biologicalsolution {NMR}. Biochimica et Biophysica Acta (BBA) - Biomembranes 1664:241 –256.
Vold, R., R. Prosser, and A. Deese. 1997. Isotropic solutions of phospholipid bi-celles: A new membrane mimetic for high-resolution NMR studies of polypep-tides. Journal of Biomolecular NMR 9:329–335.
Walker, J. M., H. C. Moises, D. H. Coy, G. Baldrighi, and H. Akil. 1982. Nonopiateeffects of dynorphin and des-Tyr-dynorphin. Science 218:1136–1138.
Wang, J. B., P. S. Johnson, J. M. Wu, W. F. Wang, and G. R. Uhl. 1994. Humankappa opiate receptor second extracellular loop elevates dynorphin’s affinity forhuman mu/kappa chimeras. The Journal Of Biological Chemistry 269:25966–25969.
Warne, T., M. J. Serrano-Vega, J. G. Baker, R. Moukhametzianov, P. C. Edwards,R. Henderson, A. G. W. Leslie, C. G. Tate, and G. F. X. Schertler. 2008. Structureof a [bgr]1-adrenergic G-protein-coupled receptor. Nature 454:486–491.

BIBLIOGRAPHY 41
Warschawski, D. E., A. A. Arnold, M. Beaugrand, A. Gravel, Étienne Chartrand,and I. Marcotte. 2011. Choosing membrane mimetics for NMR structural stud-ies of transmembrane proteins. Biochimica et Biophysica Acta - Biomembranes1808:1957 – 1974.
Wen, H., Z. Mehal, B. Ong, W. Ho, and D. Wen. 1985. Intrathecal administrationof beta-endorphin and dynorphin-(1-13) for the treatment of intractable pain.Life Sciences 37:1213 – 1220.
Wen, H. L., Z. D. Mehal, B. H. Ong, and W. K. Ho. 1987. Treatment of pain incancer patients by intrathecal administration of dynorphin. Peptides 8:191–193.
Wishart, D. S., and B. D. Sykes. 1994. Chemical shifts as a tool for structuredetermination. Methods in Enzymology 239:363–392.
Wu, B., E. Y. T. Chien, C. D. Mol, G. Fenalti, W. Liu, V. Katritch, R. Abagyan,A. Brooun, P. Wells, F. C. Bi, D. J. Hamel, P. Kuhn, T. M. Handel, V. Cherezov,and R. C. Stevens. 2010. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071.
Wu, H., D. Wacker, M. Mileni, V. Katritch, G. W. Han, E. Vardy, W. Liu, A. A.Thompson, X.-P. Huang, F. I. Carroll, S. W. Mascarella, R. B. Westkaemper, P. D.Mosier, B. L. Roth, V. Cherezov, and R. C. Stevens. 2012. Structure of the humankappa-opioid receptor in complex with JDTic. Nature 485:327–332.
Xue, J. C., C. Chen, J. Zhu, S. Kunapuli, J. K. DeRiel, L. Yu, and L. Y. Liu-Chen.1994. Differential binding domains of peptide and non-peptide ligands in thecloned rat kappa opioid receptor. Journal of Biological Chemistry 269:30195–30199.
Ye, W., 2013. Biophysical studies of membrane associated biomolecular Interac-tions. Licentiate thesis, Department of biochemistry and biophysics, StockholmUniversity.
Zhang, L., R. DeHaven, and M. Goodman. 2002. NMR and modeling studies ofa synthetic extracellular loop ii of the kappa opioid receptor in a DPC micelle.Biochemistry 41:61–68.

42 BIBLIOGRAPHY

Paper I


Paper II


















