
Drug Stability - National University Stability...1.1 Drugs susceptible to hydrolytic degradation If...
219
Drug Stability
Transcript of Drug Stability - National University Stability...1.1 Drugs susceptible to hydrolytic degradation If...

Drug Stability

Stability of pharmaceutical products refers to the
capacity of the product or a given drug substance to
remain within established specifications of identity,
potency, and purity during a specified time period.

1. Chemical Stability of Drug
Substances

Many drugs are susceptible to some form of chemical
decomposition when formulated in either liquid or even
solid dosage forms.
Such degradation not only leads to a loss of potency of
the drug but may, in some cases, cause changes in the
physical appearance of the dosage forms, for example,
discoloration following the photochemical
decomposition of the drug.

1. Identification of the product(s) formed provides a
better understanding of the mechanism(s) of these
chemical reactions as well as other valuable information.
2. Other reasons for quantitating drug loss include the
following:
Why study drug stability?

I. The drug may degrade to a toxic substance.
Therefore, it is important to determine not only how
much drug is lost with time but also what are its
degradants. In some cases, the degradants may be
of known toxicity.

II. Degradation of the drug may make the product
esthetically unacceptable. Products are presumed to be
adulterated if significant changes in, for instance, color
or odor have occurred with time.
colorless pink

III. Even though a drug may be stabilized in its intended
formulation, the formulator must show that the drug is
also stable under the pH conditions found in the
gastrointestinal tract, if the drug is intended for oral
use.

Most drug substances are fairly stable at the neutral
pH values found in the small intestine (disregarding
enzymatic degradation) but can be unstable at pH
values found in the stomach. Examples of drugs that
are very acid-labile are various penicillins,
erythromycin and some of its analogs.

Pathways of Chemical
Degradation

Drug substances used as pharmaceuticals have diverse
molecular structures and are, therefore, susceptible to
many and variable degradation pathways.

Possible degradation pathways include:
hydrolysis,
dehydration,
isomerization and racemization,
elimination,
oxidation,
photodegradation,
and complex interactions with excipients and other drugs.

It would be very useful if we could predict the chemical
instability of a drug based on its molecular structure.
This would help both in the design of stability studies
and, at the earliest stages of drug development, in
identifying ways in which problematic drugs could be
formulated to minimize chemical degradation.

1. Hydrolysis

Hydrolysis, in its widest sense, is the breaking of a
chemical bond due to the reaction of water.
Hydrolysis is often the main degradation pathway for
drug substances having ester and amide functional
groups within their structure.

1.1 Drugs susceptible to hydrolytic
degradation
If the drug is a derivative of carboxylic acid or contains
functional groups based on this moiety, for example an
ester, amide, lactone, lactam , imide or carbamate
(Scheme 1) ,then we are dealing with a drug which is
liable to undergo hydrolytic degradation.

Scheme 1:Examples of chemical groups susceptible
to hydrolysis

Drugs that contain ester linkages include acetylsalicylic
acid (aspirin),physostigmine, methyldopate, tetracaine
and procaine.
Ester hydrolysis is usually a bimolecular reaction
involving acyl–oxygen cleavage.

Acid-catalysed hydrolysis
The initial protonation on the carbonyl oxygen produces a
resonance stabilized cation; this increases the
electrophilicity of the carbonyl group, making it susceptible
to attack by the nucleophilic water. Proton transfer from the
water to the alcohol converts the latter into a better leaving
group (G).


Incidentally, this mechanism is the reverse of the
mechanism for formation of an ester from an acid
and an alcohol under acidic conditions (esterification).

Base-catalysed hydrolysis
This reaction is easier to follow; the nucleophile in this
case is the strongly basic OH ion, which attacks the
carbon of the carbonyl group directly.
Note that in base-catalysed hydrolysis the acid formed
by hydrolysis instantaneously reacts with the excess of
base to form the salt of the acid. The free acid may be
obtained, if desired, by acidification of the mixture.


Hydrolysis of the ester group of procaine

The hydrolysis of amides involves the cleavage of the
amide linkage as for example, in the breakdown of the
local anaesthetic cinchocaine

As examples of lactam ring hydrolysis we can consider
the decomposition of nitrazepam and chlordiazepoxide,
Other drugs, apart from the benzodiazepines, which are
susceptible to hydrolysis include the penicillins and
cephalosporins.

Hydrolysis of β-lactam cephalosporins

1.2 Controlling drug hydrolysis in
solution
1.2.1 Optimization of formulation
Hydrolysis is frequently catalyzed by hydrogen ions
(specific acid-catalysis) or hydroxyl ions (specific
base-catalysis) and also by other acidic or basic
species that are commonly encountered as components
of buffers. This latter type of catalysis is referred to as
general acid–base catalysis.

Several methods are available to stabilise a solution of
a drug which is susceptible to acid–base catalyzed
hydrolysis.
1. The usual method is to determine the pH of
maximum stability from kinetic experiments at a
range of pH values and to formulate the product
at this pH.

2. Alteration of the dielectric constant by the addition of
nonaqueous solvents such as alcohol, glycerin or
propylene glycol may in many cases reduce hydrolysis.

3. Since only that portion of the drug which is in
solution will be hydrolyzed, it is possible to suppress
degradation by making the drug less soluble.
The stability of penicillin in procaine–penicillin
suspensions was significantly increased by reducing its
solubility by using additives such as citrates, dextrose,
sorbitol and gluconate.

4. Adding a compound that forms a complex with the
drug can increase stability.
The addition of caffeine to aqueous solutions of
benzocaine, procaine and tetracaine was shown to
decrease the base-catalysed hydrolysis of these local
anaesthetics in this way.

5. In many cases solubilisation of a drug by surfactants
protects against hydrolysis.

1.2.2 Modification of chemical structure of drug
The control of drug stability by modifying chemical
structure using appropriate substituents has been
suggested for drugs for which such a modification does
not reduce therapeutic efficacy.

Azithromycin Clarithromycin
Erythromycin

2. Oxidation

Oxidation processes
Oxidation is a loss of electrons or an increase in the
oxidation state and reduction is a gain in electrons or a
decrease in the oxidation state. Redox reaction is an
electron transfer process:
where the reduced form loses n number of electrons
(e-).

Autoxidation (auto-oxidation) is a complex oxidation
mechanism that proceeds through a free radical chain
process.
It is a common degradation mechanism for
unsaturated fats, but a number of drugs containing
carbon-carbon double bonds also undergo
autoxidation. In general, the free radical chain process
consists of three steps


A free radical is formed in the initiation step, frequently
through the thermal or photochemical hemolytic
cleavage of an R-H bond.
The initiation is catalyzed by metal ions such as Cu 2+ ,
Ni 2+ , and Fe 3+ . Molecular oxygen is added to the free
radical in the propagation step and then in the rate-
determining step (RDS), the peroxyl radical extracts the
hydrogen atom from RH to form another R • radical.

The rate of the RDS depends mainly on the strength of
the C-H bond that is being cleaved. In the termination
step, the chain reaction is broken when two free
radicals react to form nonradical products.
The rancidification of fat, which gives a distinct rancid
smell (due to the formation of volatile aldehydes and
ketones), is an example of autoxidation:


2.1 Drugs susceptible to oxidation
Oxidation mechanisms for drug substances depend
on the chemical structure of the drug and the
presence of reactive oxygen species or other
oxidants.

Catechols such as methyldopa and epinephrine
are readily oxidized to quinones.

Thiols such as 6-mercaptopurine and captopril are
oxidized to disulfides.

Phenothiazines such as promethazine are oxidized via
complex pathways and yield various products
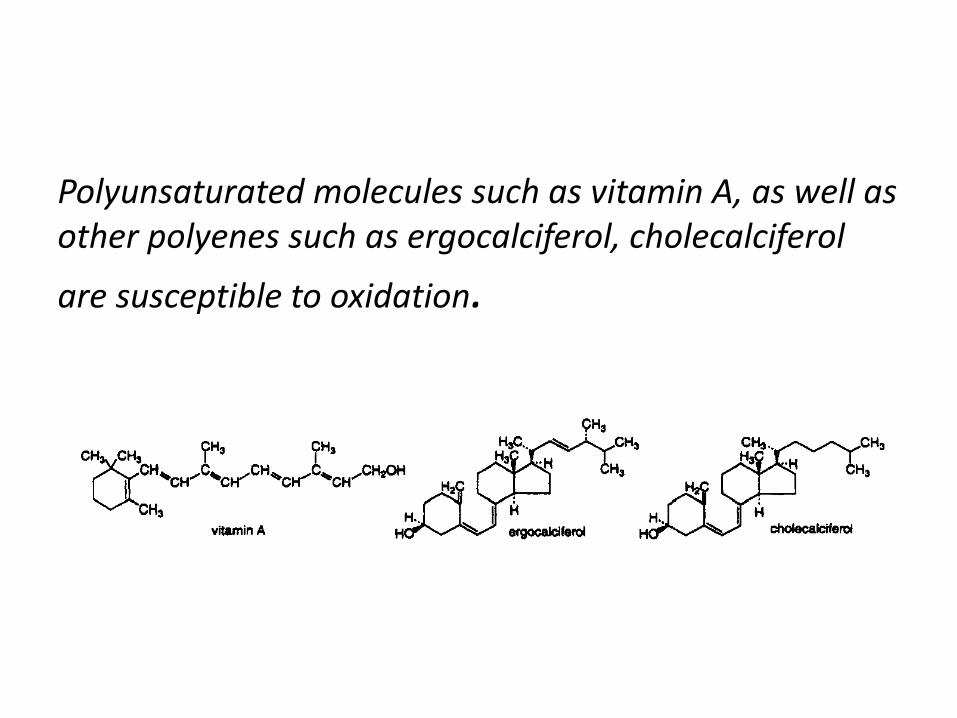
Polyunsaturated molecules such as vitamin A, as well as
other polyenes such as ergocalciferol, cholecalciferol
are susceptible to oxidation.

Polyene antibiotics, such as amphotericin B (II) which
contains seven conjugated double bonds (heptaene
moiety), are subject to attack by peroxyl radicals,
leading to aggregation and loss of activity.


The ether group in drugs such as econazole nitrate (III)
and miconazole nitrate (IV) is susceptible to oxidation.
The process involves removal of hydrogen from the C–H
bonds in the α-position to the oxygen to produce
radicals, which further degrade to α-hydroperoxides
and eventually to aldehydes, ketones,alcohols and
carboxylic acids.


2.2 Stabilization against oxidation
Various precautions should be taken during
manufacture and storage to minimize oxidation.
2.2.1 The oxygen in pharmaceutical containers should
be replaced with nitrogen or carbon dioxide; contact of
the drug with heavy-metal ions such as iron, cobalt or
nickel, which catalyze oxidation, should be avoided; and
storage should be at reduced temperatures.

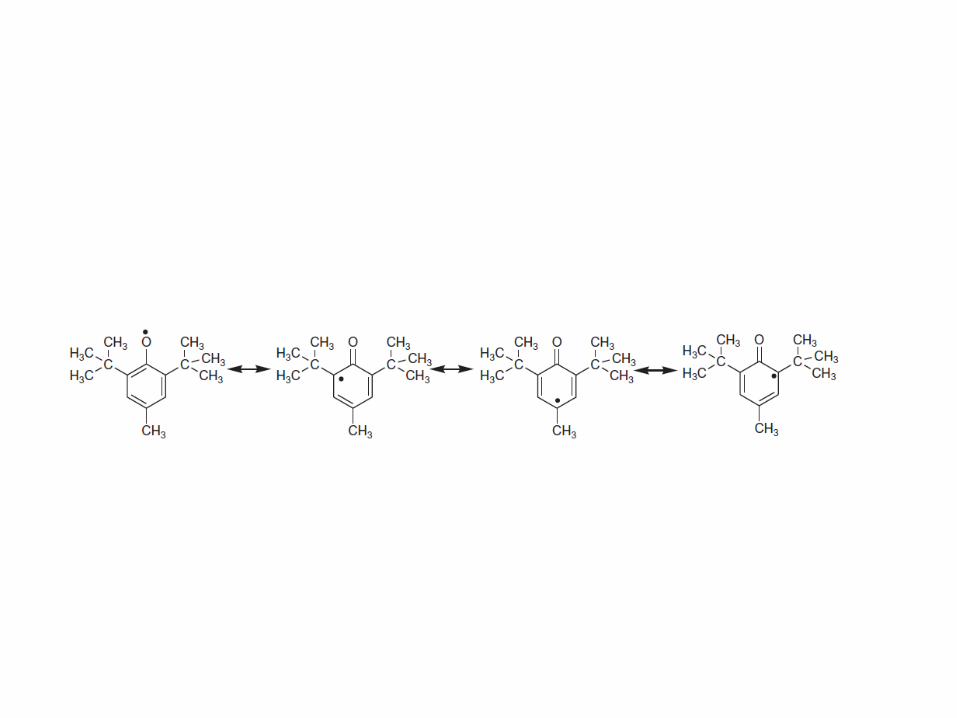
are sterically hindered phenols that react with free
radicals, blocking the chain reaction.
2.2.2 Antioxidants

These hindered phenols are radical-trapping
antioxidants for oxy- and peroxy radicals. The phenoxy
radicals formed with their bulky substituents are
stabilized by steric hindrance and cannot attach drugs
or unsaturated fatty acids to maintain the chain
reaction.

The radical formed is stabilized by delocalization of the
unpaired electron around the phenol ring to form a
stable resonance hybrid (i.e. low-energy radical):

Reducing agents such as sodium metabisulfite
are compounds that have lower redox potentials and,
thus, are more readily oxidized than the drug they are
intended to protect.

2.2.3 Use of chelating agents
Chelating agents, such as disodium edetate, are
used to chelate and remove metal ions.
Chelating agents do not possess antioxidant activity as
such, but enhance the action of phenolic antioxidants
by reacting with catalyzing metal ions to make them
inactive.

2.2.4 Use of amber or coloured glass containers
Amber glass excludes light of wavelengths ‹470 nm and so affords some protection to light-sensitive
compounds.
Special formulations, such as metered dose inhalers
used in the treatment of asthma, also offer protection
from light and oxygen since the drug is dissolved or
suspended in propellant and stored in a sealed
aluminium container.

Isomerisation is the process of conversion of a drug into
its optical or geometric isomers.
Since the various isomers of a drug are frequently of
different activity, such a conversion may be regarded as
a form of degradation, often resulting in a serious loss
of therapeutic activity.
3.Isomerisation and Racemization

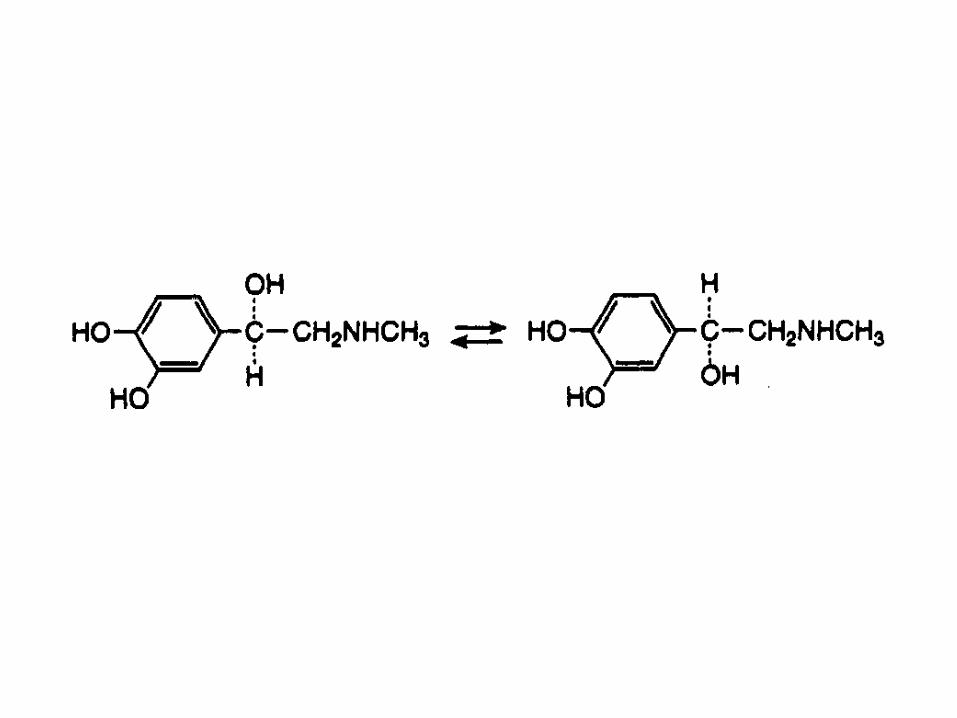
The appreciable loss of activity of solutions of
adrenaline at low pH has been attributed to
racemisation – the conversion of the therapeutically
active form, in this case the levorotary form, into its less
active isomer.

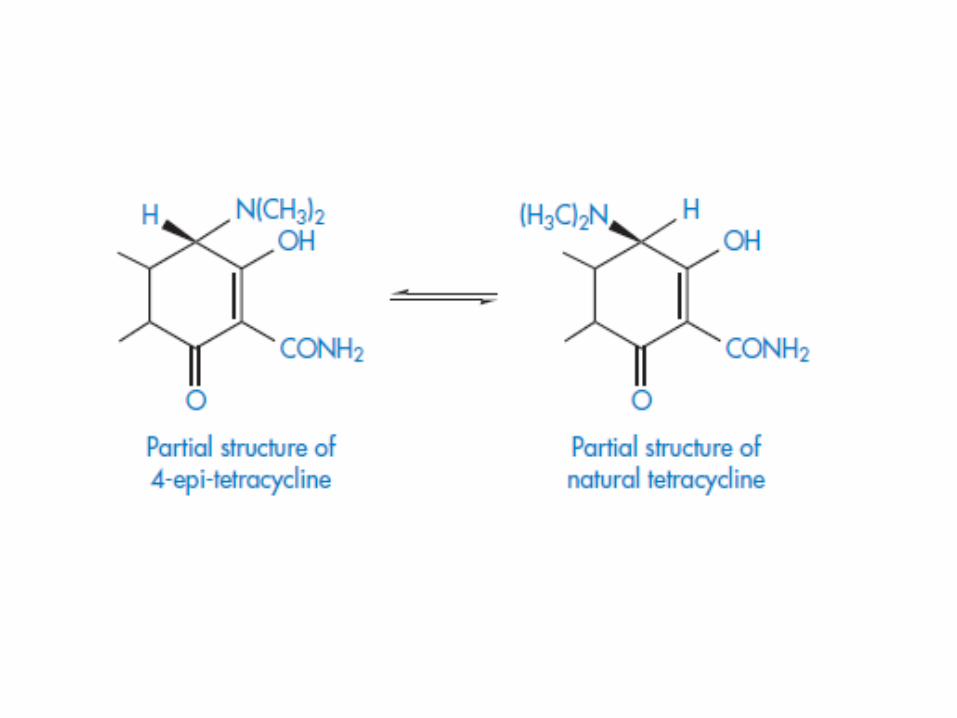
In acidic conditions the tetracyclines undergo
epimerisation at carbon atom 4 to form an equilibrium
mixture of tetracycline and the epimer, 4-epi-
tetracycline. The 4-epi-tetracycline is toxic and its
content in medicines is restricted to not more than 3%.

Cis–trans isomerisation may be a cause of loss
of potency of a drug if the two geometric
isomers have different therapeutic activities.
Vitamin A (all-trans-retinol) is enzymatically
oxidised to the aldehyde and then isomerised
to yield 11-cis-retinal ,which has a decreased
activity compared with the all-trans molecule.


Racemization and epimerization, which are
reversible conversions between optical isomers,
have been reported for many drug substances.
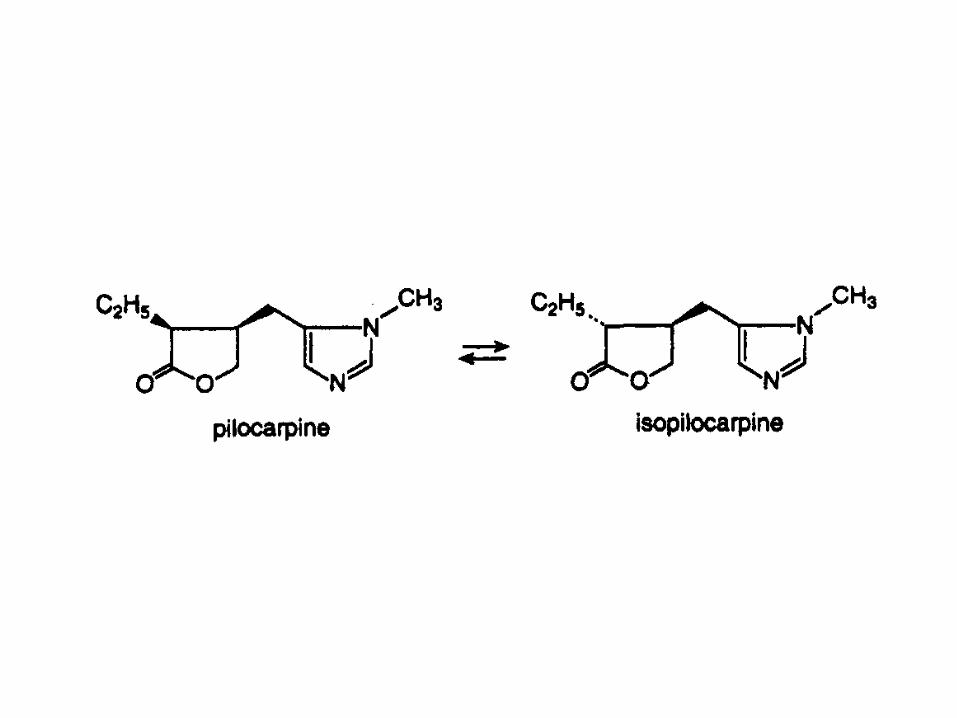
Pilocarpine undergoes epimerization by base
catalysis

Primary photochemical reaction occurs when the
wavelength of the incident light is within the
wavelength range of absorption of the drug
(usually within the ultraviolet range, unless the
drug is coloured), so that the drug molecule
itself absorbs radiation and degrades.
4. Photochemical decomposition

Photodegradation may also occur with drugs
that do not directly absorb the incident
radiation, as a consequence of absorption of
radiation by excipients in the formulation
(photosensitisers) which transfer the
absorbed energy to the drug, causing it to
degrade.

Although it is difficult to predict which drugs
are likely to be prone to photodegradation,
there are certain chemical functions that are
expected to introduce photoreactivity, including
carbonyl, nitroaromatic and N-oxide functions,
aryl halides, alkenes, polyenes and sulfides.

The phenothiazine chlorpromazine (CLP) is
rapidly decomposed under the action of
ultraviolet light, the decomposition being
accompanied by discoloration of the solutions



Stabilization against photochemical
decomposition
1. Pharmaceutical products can be adequately
protected from photo induced decomposition
by the use of coloured glass containers and
storage in the dark. Amber glass excludes light
of wavelength ‹ 470 nm and so affords considerable protection of compounds sensitive
to ultraviolet light.

2. Coating tablets with a polymer film containing
ultraviolet absorbers has been suggested as an
additional method for protection from light.

Polymerisation is the process by which two or more
identical drug molecules combine together to form a
complex molecule.
5. Polymerization

It has been demonstrated that a polymerisation
process occurs during the storage of concentrated
aqueous solutions of aminopenicillins, such as
ampicillin sodium and also formaldehyde.
The reactive β-lactam bond of the ampicillin molecule
is opened by reaction with the side-chain of a second
ampicillin molecule and a dimer is formed.


• Sugars such as glucose and lactose are known to
undergo dehydration to form 5-(hydroxymethyl)
furural.
• Erythromycin is susceptible to acid catalyzed
dehydration,
• Prostaglandins E1 and E2 undergo dehydration
followed by isomerization
6. Dehydration

Dehydration of glucose
Scheme 28

Dehydration of erythromycin
Scheme 29

Dehydration and isomerization of
prostaglandin E2
Scheme 30

Drug substances having a carboxylic acid group
are sometimes susceptible to decarboxylation,
4-Aminosalicylic acid is a good example
7. Decarboxylation and
Elimination


Quite often, reactions can occur between the
drug and one or more additives. Similarly, two
drugs might be formulated in the same product
and react with each other.
8. Drug-Excipient and Drug-
Drug Interactions

8.1 Reactions of Bisulfite, an
Antioxidant
It was reported that epinephrine, a catecholamine,
undergoes displacement of its hydroxy group by
bisulfite, .
Dexamethasone 21-phosphate, an α/β-unsaturated
ketone, is known to undergo addition by bisulfite.


Reducing sugars readily react with primary
amines, including those of amino acids, through
the Maillard reaction. Drug substances with
primary or secondary amine groups undergo this
addition/rearrangement reaction, also called
the browning reaction because of the resulting
discoloration.
8.2 Reaction of Amines with
Reducing Sugars

In the presence of drug substances with hydroxy groups,
aspirin undergoes a reversible transacylation reaction
to form salicylic acid, while acetylating the drug
substance. For example, codeine and sulfadiazine are
acetylated by aspirin.
8.3 Transesterification Reactions


Another example of transesterification is the reaction of
benzocaine with polyvinyl acetate phthalate.

2. Kinetics of chemical
decomposition in solution

Kinetics deals with the study of the rate at which
processes occur and mechanism of chemical reactions
Kinetics Motion or
movement
Velocity, rate or
rate of change

It involves the study of rate of change and the way in
which this rate is influenced by the concentration of
reactants, products, and other chemical species that
may be present, and by factors such as solvents,
pressure, and temperature.

• It gives an in light into the mechanism of changes
involved
• Allows a prediction of the degree of change that will
occur after a given time has elapsed.
WHY DO WE STUDY ABOUT
KINETICS?

RATES
• the speed or velocity of a reaction with which a reactant or reactants undergoes a change.
• It is determined by the change in the concentration of the reactants or products as a
function of time.
• The rate may be determined by the slowest or rate determining step.
RATES AND ORDERS OF
REACTIONS

Before we can predict the shelf-life of a dosage form it
is essential to determine the kinetics of the breakdown
of the drug under carefully controlled conditions.

2.1 REACTION RATE AND HALF-LIFE (t1/2)
For a hypothetical reaction,
aA Product
the rate of reaction of A into product (P) is defined by the
derivative
−dA/dt
where the minus sign means the concentration (or
amount) of A is decreasing with time. For P, the rate of
production is defined by

+dP/dt
Here the concentration (or amount; amount =
concentration × volume) of P always increases
with time. According to the law of mass action, the rate
of chemical reaction is proportional to the product of
the molar concentration of the reactants each carried
to the power equal to the number of moles of the
substance undergoing reaction.

−dA/dt α [A]a
Therefore,
−dA/dt = k[A]a or
dA/dt = −k[A]a
where k is the proportionality constant, also known
as the rate constant, [A] is the molar concentration
of A as a function of time, and a is the number of
moles of A undergoing reaction.

• Half-life (t1/2) is defined as the time required for
half of the initial concentration (or amount)
of reactants to form products.
• Shelf-life is defined as the time for the original
potency (i.e., 100%) of the active drug to be reduced to
95% (t 95 ) or, more frequently, 90% (t 90 ), although
more stringent time limits may apply if the degradation
products are toxic.

2.2 ORDER OF REACTION
• The order of a chemical reaction refers to the
way in which the concentration of the reactant
influences the rate.
• Most commonly, zero-order and first-order reactions
are encountered in pharmacy and will be discussed in
details below. However, the concept of second order will
also be introduced.

2.2.1 Zero-Order Reactions
Again, for a hypothetical reaction,
A → P
The rate equation for a zero-order reaction is
defined as
dA/dt = −k[A]0
Since [A]0= 1, the rate equation can be simply
rewritten as
dA/dt = −k0

The rate equation can be integrated as

and solved to give the integrated rate equation
for zero-order kinetics:
At − A0 = −k0t
or
A0 − At = k0t
where At is the amount of A at any time t, A0 is
the initial amount of A, and k0 is the zero-order
rate constant with the unit of concentration (or
mass)/time.
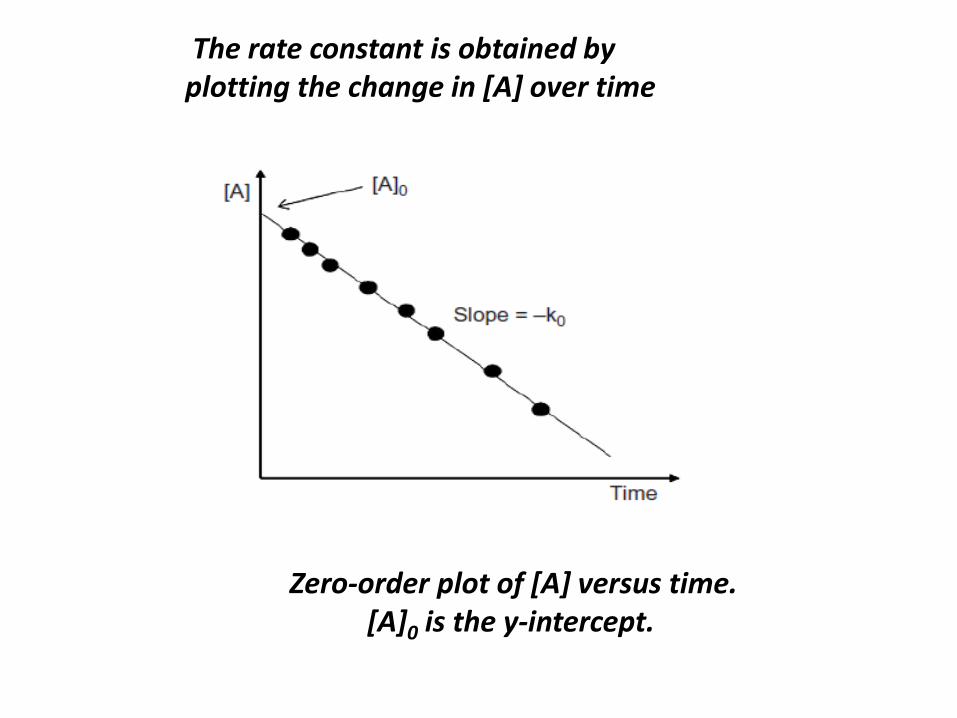
The rate constant is obtained by
plotting the change in [A] over time
Zero-order plot of [A] versus time.
[A]0 is the y-intercept.

The half-life (t1/2) of a zero-order reaction
can be deduced by the fact that at t1/2
At = A0/2
Substituting into the integrated equation for
zero-order reactions will give
A0/2 = A0 − k0 t1/2
or
A0 − A0/2 = k0 t1/2 and A0/2 = k0 t1/2

Therefore, the half-life of zero-order reactions
is defined by
t1/2 = 0.5A0/k0 or A0/2 k0
Therefore, the half-life of zero-order reactions is directly
proportional to the initial concentration of the
reactants.


Example
Drug X degrades by a zero-order process with a
rate constant of 0.05 mg ml-1 year−1 at room
temperature. If a 1% weight/volume (w/v) solution is
prepared and stored at room
temperature:
1. What concentration will remain after 18 months?
2. What is the half-life of the drug?

Answers
A0 = 1% w/v = 10 mg/ml; t =18 months = 1.5 year;
k0 = 0.05 mg ml−1 year −1
1. At = A0 – k0t = 10 – (0.05 × 1.5) = 9.25 mg/ml
2. t1/2 = 0.5 A0 /k0 = (0.5 × 10)/0.05 =100 years

2.2.2 First-Order Reactions
The rate equation for first-order kinetics is
given by
dA/dt = −k1[A] 1
or simply
dA/dt = −k1 [A]

The rate equation can be integrated to give

which is solved to give the integrated rate equation for
first order kinetics:
ln At − ln A0 = −k1t
which can be rewritten as
ln At = ln A0 − k1t or At = A0 e−k
1t
Converting into base 10 log,
log = ln/2.303
log At = log A0 – k1t/2.303

The half-life of a first-order reaction is defined as the
time (t1/2) when At = A0/2.
Substituting this into the integrated equation:
log A0/2 = log A0 – k1t1/2/2.303
which will give
log 2 = k1t1/2/2.303

or
t1/2 = 0.693/k1
and
k1 = 0.693/ t1/2
Note that the half-life of first-order reactions is
independent of the initial concentration of the
reactants.
The units of k1 will be (1/time) or time-1.


Plot of [A] and [P] versus time

First-order plot of ln [A] versus time.
ln [A]0 is the y-intercept.
– /2.303

The characteristic of a first-order reaction is such that
over the same time period, the fraction of unchanged
drug remaining will always be the same as from
At = A0 e−k
1t,
At/A0 = e−k1
t
which is a constant.

Example
Homatropine is an ester that undergoes hydrolysis in
aqueous solutions. Samples were collected and the
homatropine concentration at various time points
calculated:
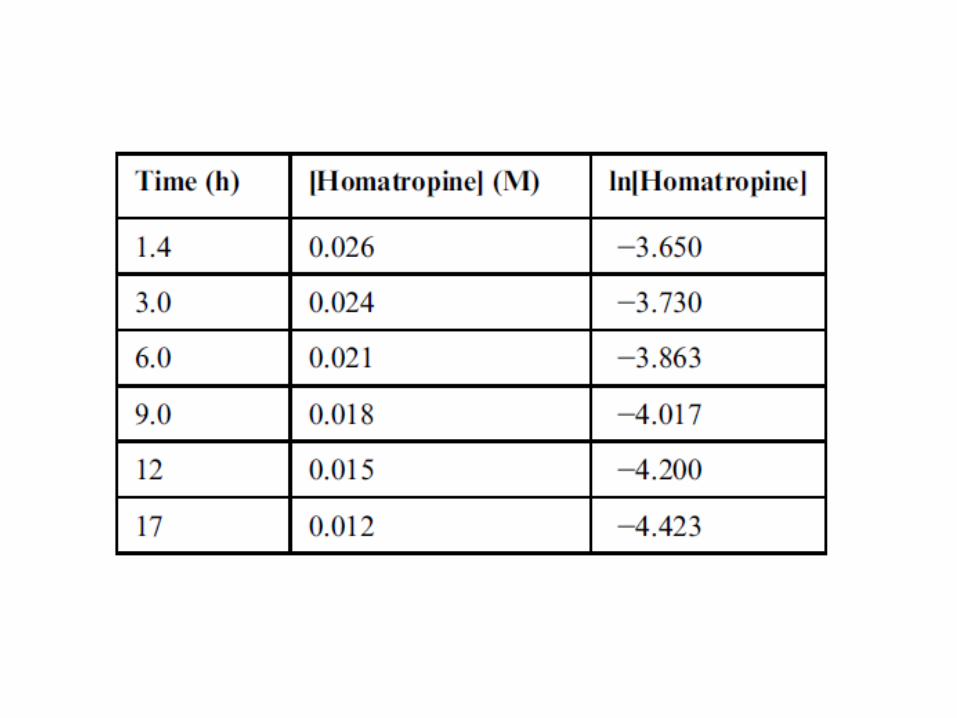



Apparent (or Pseudo) Zero-Order
Reactions for Suspensions
Suspensions (e.g., Amoxil®, Mylanta®, and Maalox®)
are dosage forms in which the concentration of a drug
exceeds the solubility.
Suspension formulations, therefore, have solid
particles suspended in a solution of drug. The
decomposition of drugs in suspensions depends
on the concentration of the drug in solution as
shown below.

Asolid → Asoln → Product
The rate of decomposition of A in solution, therefore, is
given by the first-order expression
dA/dt = −k[A]soln

Since there is excess solid drug present, and it
dissolves continuously to replace the portion of drug
in solution, which is being converted to product, the
concentration of drug in solution (k[A]soln) remains
constant, and we can write
k[A]soln = k′0

which converts the decomposition process an
apparent zero-order process, with k′0 being the
apparent zero-order rate constant, although
the actual decomposition of the drug from the
solution may be first order. The above rate
equation, therefore, can be rewritten as
dA/dt = −k′0

and the integrated form of the apparent zero-order rate
equation is expressed as
At = A0 − k′0 t

Example
An aqueous suspension of drug X contains 200
mg of drug X per teaspoon (5.0 ml). The
solubility of drug X at 25°C is 1 g/350 ml in
water, and the first-order rate constant for
degradation of drug X in solution at 25°C is
3.9 × 10−6 sec−1 .
Calculate (a) the zero-order rate constant and
(b) the shelf life of the liquid preparation.

Answers
(a) From k′0 = k[A]soln =
(3.9 × 10−6 sec−1)(1 g/350 ml) =
1.11 × 10−8 g/ml sec−1
(b) t90 = 0.1[A0]/ k′0 =
(0.1)(0.04 g/ml)/ 1.11 × 10−8 g/ml sec−1=
3.6 × 105 sec = 4.2 days

2.2.3 Second-Order Reactions
The rate of a second-order reaction is proportional to the
concentration of two reactants:
where k2 is the second-order rate constant and [A] and
[B] are the reactant concentrations.

A simpler form of second-order reaction is obtained if
[A]=[B] or if two molecules of A react:


Example
Ethyl acetate undergoes hydrolysis in aqueous alkaline
solutions containing equal concentrations of both the
ester and sodium hydroxide or 0.020 M. Samples were
collected and the ethyl acetate concentration was
determined at two time points:
The initial concentrations of both reactants are the same
and, thus, can be used to calculate the second-order rate
constant.


Since the initial concentrations of both the ester and the
sodium hydroxide are identical, can be used to calculate
the half-life:



EXAMPLE
Calculation of first-order rate constant and
half-life

The following data were obtained for the hydrolysis of
homatropine in 0.226 mol dm-3 HCl at 90°C:Show that the
hydrolysis follows first-order kinetics and calculate (a) the
rate constant, and (b) the half-life.

Answer
(a) The reaction will be first-order if a plot
of the logarithm of the amount of homatropine
remaining against time is linear.

The following figure shows a linear plot with a
slope = -(1.96 - 1.55)/(20 - 2) = -2.278 x 10-2 h-1
Slope = K1/2.303
Therefore,
K1 = 5.25 x 10-2 h-1

b) From this equation
t0.5 = 0.693/K1 = 13.2 h
The half-life of the reaction is 13.2 h.

First-order plot for hydrolysis of homatropine in
hydrochloric acid (0.226 mol dm-3) at 90°C

2.1.3 Determination of the order of
reaction
• The most obvious method of determining the order of
a reaction is to determine the amount of drug
decomposed after various intervals and to substitute
the data into the integrated equations for zero-, first-
and second-order reactions.
• The equation giving the most consistent value of k for
a series of time intervals is that corresponding most
closely to the order of the reaction.


For zero order, At = A0 – k0t.
950 = 1000 − k0(2)
900 = 1000 − k0 (4)
Solving both the equations shows that k0 is the
same (i.e., 25 mg/ml/h). Therefore, this is an
example of zero-order reaction kinetics. The
same conclusion could also be drawn by looking at
the data as the amount lost every 2 hours being
constant

The best way to determine the order of a reaction is to
plot the data according to the equation for zero-order,
first-order ,or second-order reactions:

If a linear zero-order plot is obtained, then the reaction
follows zero-order kinetics. If a nonlinear plot is
observed, then re-plot the data according to a first-
order equation. If a linear plot is observed, then the
reaction is first-order; if not, then try a second-order
plot, and so on.

Fitting data to the standard rate equations may,
however, produce misleading results if a fractional
order of reaction applies.
An alternative method of determining the order of
reaction, which avoids this problem, is based
on the following equations :

where [A]0 is the initial reactant (drug) concentration
and n is the reaction order., t 1/2 is determined at two
different [A]0 values:

The half-life of the reaction is determined for a series of
initial drug concentrations, C0 , and the order, n, is
calculated from the slope of plots of log t0.5 as a
function of log C0 .


Example Determination of the
order of reaction

Example
The kinetics of decomposition of a drug in aqueous
solution were studied using a series of solutions of
different initial drug concentrations, C0 . For each
solution the time taken for half the drug to decompose
(that is, t0.5) was determined with the following results:

Determine the order of reaction and calculate the rate
constant.

Answer Application of the equation requires values for log C0
and log t0.5; thus


A plot of log t0.5 against log C0 is linear with slope (1 - n)
= -1.01. Hence n = 2.01, i.e. the reaction is second-
order.
The intercept of the graph (that is, the value of log t0.5
at log C0 = 0) is 2.60, and
k = 2.51 × 10−3 (mol dm −3) −1 min −1

3.Factors influencing drug
stability
•Liquids
•Semisolids
•Solids

3.1 Liquid dosage forms
1. pH:
Degradation rates of drug substances are generally affected
by pH because most degradation pathways are catalyzed by
hydronium and/or hydroxide ions. Water itself is also a
critical reactant. If the critical path in a reaction involves a
proton transfer or abstraction step, other acids and bases
present in solution (usually buffer species) can affect
the reaction rate.

These reactions will also be pH-dependent because the
fraction of any species present in its acid or base form
will be dependent on its dissociation constant and the
solution pH.
Also, for ionizable drugs, the fraction of drug present in
any particular form will depend on the solution pH.
Therefore, if the reactivity of the drug depends on its
form, its reactivity will be pH-dependent.

When a reaction dependent on hydronium and
hydroxide ion activity is performed at constant pH, it
usually follows pseudo-first-order kinetics, which can be
described by a first-order rate constant kobs. A reaction
in which hydronium ion, hydroxide ion, and water
catalysis are observed can be described by

where kobs is the sum of the specific rate constants
and activities for each parallel pathway, and a H+
and a OH- are the activities of hydronium and
hydroxide ion, respectively. This equation is for the
case when the drug itself is neutral in the pH range
of study, that is, where ionization of the drug does
not have to be taken into account. The pH-rate
profiles for drugs meeting this criterion are relatively
simple, as shown in the following figures

Simple pH-rate profiles for drug
degradation

If the contributions of the first and second terms in the
equation are larger than that of the third term, the pH-rate
profile shown in (panel 1) of the figure is seen. If the second
and third terms are dominant, then the profile illustrated in
(panel 2) is observed. If the first and third terms are
dominant, then a V-type pH-rate profile (panel 3) occurs. If
all terms contribute significantly, the U-shaped pH-rate
profile shown in (panel 4) is observed.

2. Temperature:
• Increase in temperature usually causes a very
pronounced increase in the hydrolysis rate of drugs in
solution. This effect is used as the basis for drug
stability testing.

The equation which describes the effect of temperature
on decomposition is the Arrhenius equation:
log k = log A – Ea /(2.303RT)
and

where Ea is the activation energy (The minimum
amount of energy needed for a reaction to proceed), A
is the frequency factor, R is the gas constant
(8.314 J mol –1 K –1 ) and T is the temperature in kelvins.

The Arrhenius equation predicts that a plot of the log
rate constant, k, against the reciprocal of the
temperature should be linear with a gradient of
– Ea/2.303R.

• Therefore, assuming that there is no change in the
order of reaction with temperature, we can measure
rates of reaction at high temperatures (where the reaction
occurs relatively rapidly) and extrapolate the Arrhenius
plots to estimate the rate constant at room temperature
(where reaction occurs at a very slow rate)
• This method therefore provides a means of speeding up
the measurements of drug stability during preformulation.
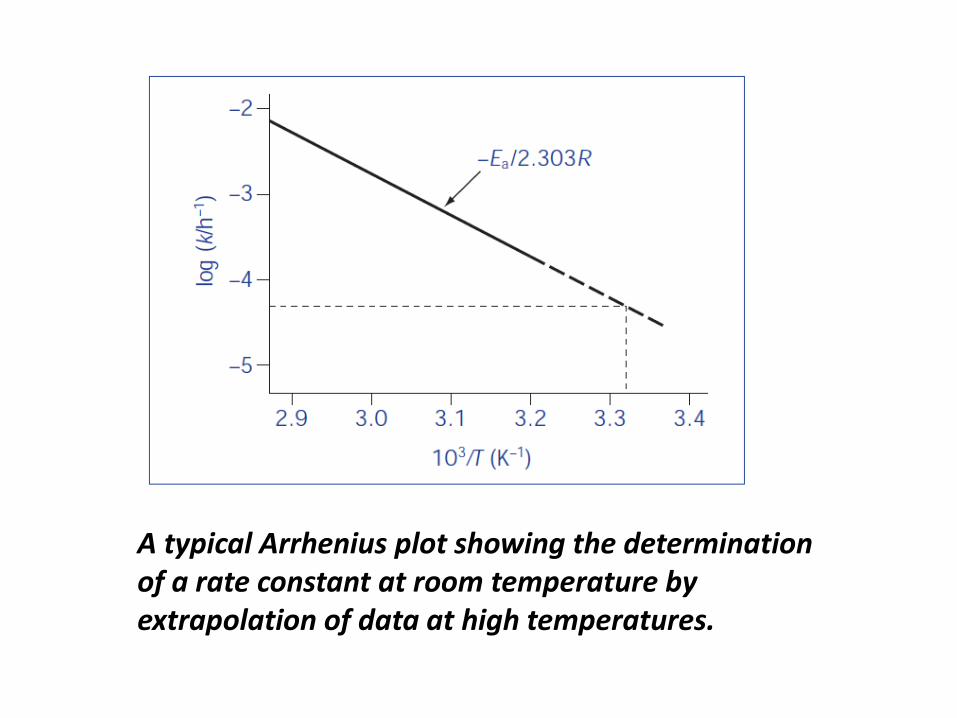
A typical Arrhenius plot showing the determination
of a rate constant at room temperature by
extrapolation of data at high temperatures.

For a given reaction under a given reaction condition
(i.e., reaction media, pH, etc.), the following equations
can be used to calculate k-values at different
temperatures:

Thus, if Ea is known and the rate constant (k1 ) at a
given temperature is T1 , then it is possible to calculate
k 2 at temperature T2

Application of the Arrhenius
Equation

EXAMPLE
The following values were determined for the specific
acid-catalytic constants for an anti-inflammatory drug:

Determine graphically (a) the rate constant for
acid-catalysis at 25°C, (b) the activation energy.

Answer
According to the Arrhenius equation, a plot of
log k against 1/T has a gradient of -Ea/2.303R.
From the graph:
(a) At 1/T = 3.356x 10-3 K-1, log k = -5.85.
Therefore, k at 25°C = 1.41 x10-6 (mol.dm-3)-1 s-1.
(b) Gradient = -5.91x103 K.
Therefore Ea = 113 kJ mol -1.

Calculating the rate constant
The first-order rate constant for the hydrolysis of
sulfacetamide at 120°C is 9x10-6s-1 and the activation
energy is 94 kJ mol-1. Calculate the rate constant at
25°C.

Removing the logarithms,
k120/k25 = 9.55 x103.
Therefore, k25 = 9 x 10-6/9.55 x103
=9.4x10-10 s-1.

Calculation of Ea , t1/2 , and t90
Atropine is an ester that is hydrolyzed in aqueous
solutions. The following data for atropine hydrolysis
was obtained for an aqueous atropine solution:
Using the data in the table, calculate A, Ea , t1/2 , and t90
at room temperature (25 °C).

We start by plotting the data according to
Then we need to convert the data from the table to the
Y (lnk) and X (1/T) values:




Calculating the shelf-life
The initial concentration of active principle in
an aqueous preparation was 5x10-3 g cm-3.
After 20 months the concentration was shown by
analysis to be 4.2x10-3 g cm-3. The drug is known to be
ineffective after it has decomposed to 70% of its original
concentration.
Assuming that decomposition follows first order
kinetics, calculate the expiry date of the drug
preparation.

Substituting into the first-order equation

70% of the initial concentration
= 3.5x10-3 g cm-3

3. Ionic strength:
We often need to add electrolytes to drug solutions, for
example to control their tonicity. Consequently we must
pay particular attention to any effect they may have on
stability.

• The equation which describes the influence of
electrolyte on the rate constant is the Brønsted–
Bjerrum equation:
log k = log k0 + 2AzAzB√μ
where zA and zB are the charge numbers of the two
interacting ions, A is a constant for a given solvent and
temperature and μ is the ionic strength.


• The Brønsted–Bjerrum equation predicts that a plot
of log k against μ1/2 should be linear for a reaction in
the presence of different concentrations of the same
electrolyte with a gradient of 2AzAzB
• The gradient will be positive (i.e. the reaction rate will be
increased by electrolyte addition) when reaction is between
ions of similar charge, for example, the acid-catalysed
hydrolysis of a cationic drug ion.

• The gradient will be negative (i.e. the reaction
rate will be decreased by electrolyte addition) when the
reaction is between ions of opposite charge, for
example, the base-catalysed hydrolysis of positively
charged drug species.
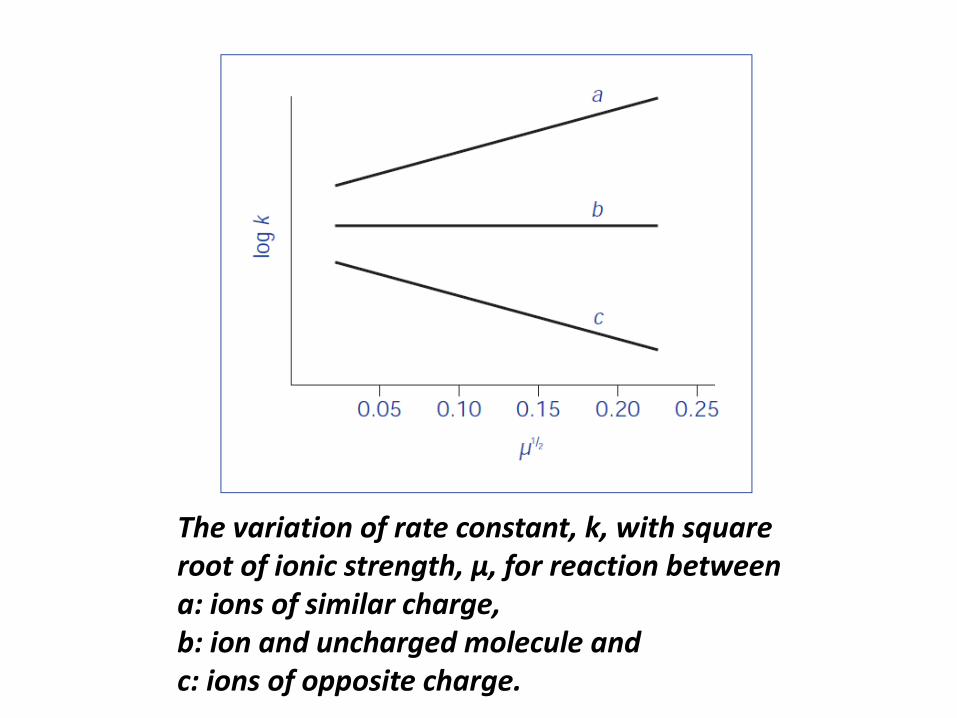
The variation of rate constant, k, with square
root of ionic strength, μ, for reaction between
a: ions of similar charge,
b: ion and uncharged molecule and
c: ions of opposite charge.

4. Oxygen:
Since molecular oxygen is involved in many oxidation
schemes, we could use oxygen as a challenge to find out
whether a particular drug is likely to be affected by
oxidative breakdown. We would do this by storing solutions
of the drug in ampoules purged with oxygen and then
comparing their rate of breakdown with similar solutions
stored under nitrogen.

• Drugs which have a higher rate of decomposition
when exposed to oxygen can be stabilized by replacing
the oxygen in the storage container with nitrogen or
carbon dioxide. These drugs should also be kept out of
contact with heavy metals and should be stabilized with
antioxidants.

5. Solvent effects:
Since we are considering the hydrolysis of drugs it might
seem that an obvious way to reduce the breakdown
would be to replace some or all of the water in the
system with a solvent such as alcohol or propylene
glycol.

The equation that describes the effect of the dielectric
constant, ε , on the rate of hydrolysis is:
log k = log kε=∞ – KzAzB/ε
where K is a constant for a particular reaction at a given
temperature, zA and zB are the charge numbers of the two
interacting ions and kε=∞ is the rate constant in a theoretical
solvent of infinite dielectric constant.

• This equation predicts that a plot of log k against the
reciprocal of the dielectric constant of the solvent
should be linear with a gradient –KzAzB. The intercept
when 1/ε = 0 (i.e. when ε = ∞) is equal to the logarithm of the rate constant, kε=∞ , in a theoretical solvent of
infinite dielectric constant

The variation of rate constant with reciprocal of
dielectric constant for reaction between a , ions
of opposite charge, b, ion and uncharged
molecule and c, ions of similar charge.

• The gradient will be negative when the charges on the
drug ion and the interacting species are the same. This
means that if we replace the water with a solvent of lower
dielectric constant then we will achieve the desired effect of
reducing the reaction rate.
• The gradient will be positive if the drug ion and the
interacting ion are of opposite signs and therefore the
choice of a non-polar solvent will only result in an increase
of decomposition.


6. Light:
Photolabile drugs are usually stored in containers
which exclude ultraviolet light, since exposure to
light in this wavelength range is the most usual
cause of photodegradation . Amber glass is
particularly effective in this respect because it
excludes light of wavelength of less than about 470
nm. As an added precaution, it is always advisable to
store photolabile drugs in the dark.

7.Surfactants:
The presence of surfactants in micellar form
has a modifying effect on the rate of hydrolysis
of drugs. The magnitude of the effect depends
on the difference in the rate constant when the
drug is in aqueous solution and when it is
solubilised within the micelle, and also on the
extent of solubilisation.

4. Stability testing and
prediction of shelf-life

It is clearly most important to be able to ensure
that a particular formulation when packaged in a
specific container will remain within its physical,
chemical, microbiological, therapeutic and
toxicological specifications on storage for a
specified time.

A recently agreed stability-testing requirement for a
Registration Application within the three areas of the
EC, Japan and the USA exemplifies the core stability
data package required for new drug substances and
associated drug products.

Current Stability Testing Concepts
Testing under temperature plus humidity
conditions instead of isothermal conditions
Both drug substance and drug product to be
subjected to similar test conditions

Zone concept for determination
of world-wide storage conditions,
based on calculation of mean
kinetic temperature


Long-term
Zone I
Zone II
Zone III
Zone IV
- 25°C/60%RH
- 30°C/35%RH - 30°C/65%RH
Accelerated For all zones - 40°C/75% RH
Intermediate Zone I and II - 30°C/65%RH

Consideration for product-wise
storage conditions

General Products
Study
Storage Condition
Minimum time
period at
Submission
Long-term Testing
30 ± 2°C/65 ± 5% RH
12 months
Accelerated
Testing
40 ± 2°C/75 ± 5% RH
3 months
Where "significant change" occurs due to accelerated testing, only
long-term testing should be conducted.


Study
Storage condition
Minimum time period
covered by data at
submission
Long term
30 ± 2°C/
40 ± 5% RH
12 months
Accelerated
40 ± 2°C/
not more than 25% RH
6 months
Liquid Products Packed in
Semi-permeable containers

Drug Products Intended for Storage
in a Refrigerator
Study
Storage condition
Minimum time period
covered by data at
submission
Long term
5°C ± 3°C
12 months
Accelerated
If available,
25 ± 2°C/60± 5% RH,
otherwise
30 ± 2°C/65 ± 5% RH
6 months

Drug Products Intended for Storage in a
Freezer
Study
Storage condition
Minimum time
period covered by
data at
submission
Long
term
- 20°C ± 5°C
12 months


















