
Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
368
D r. L en gye l B él a • D r. C sá k vári B éla Általános és szervetlen kémi ai prakt ikum
Transcript of Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 1/366
Dr. Lengyel Béla • Dr. Csákvári Béla
Általános és szervetlen
kémiai praktikum
T a z d t ö n y r l t i a d ó

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 2/366
ÁLTALÁNOS ÉS SZERVETLEN
KÉMIAI PRAKTIKUM
M Á S O D I K K Ö N Y V
P R A K T I K U M H A L A D Ó K S Z Á M Á R A
S Z E R K E S Z T E T T E :
DR. L E N G Y E L B É L A
ÉS
D R. C S Á K V Á R I B É L A
Második , bővített és átdolgozott kiadás
T A N K Ö N Y V K I A D Ó , B U D A P E S T
19 6 7

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 3/366
E G Y E T E M I T A N K Ö N Y V
I R T A :
DR. L E N G Y E L B É L A
KOSSUTH-DÍJAS EGYETEMI TANÁR,
A MTA LE VE LE ZŐ TA GJA
D R. C S Á K V Á R I B É L A
e g y e t e m i d o c e n s , a k é m i a i
TUDOMÁNYOK KANDIDÁTUSA
DR . K O V Á T S Z O L T Á N
KÖNYVTÁRIGAZGATÓ
K i a dását a m velődésügyi m i n i sz ter r en de l t e el

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 4/366
Tartalomjegyzék
Előszó a II. könyvhöz.......................................................................................................... 13
Előszó a második kiadáshoz.......................................................................................................... 14
Általános rész
42. A laboratóriumi munkában használatos szerkezeti anyagok 17
42.1. Üveg ................................................................................................................................ 1742.2. Tűzálló (kerámiai) anyagok.......................................................................................... 21
42.2.1. Alumínium-szilikát alapú anyagok 2142.2.2. Oxidkerámiai anyagok ................................................................................. 21
42.2.3. Szén- (grafit-)tartalmú anyagok 2142.3. Fém ek.................................................................................................................................. 2542.4. Műanyagok ............................................................ 3042.5. Kitt-tömítések................................................................................................................... 31
42.5.1. Irreverzíbilis kittek 3142.5.2. Reverzibilis kittek 32
42.6. Zsírok ............................................................................... 3342.7. Parafa, papír és gyapot 34
43. Alacsony hőmérséklet előállítása 36
43.1. Hőszigetelők 3543.2. Hűtőanyagok..................................................................................................................... 35
43.2.1. Hűtőkeverékek 3543.2.2. Szénsavhó ............................................................................................................ 3643.2.3. Folyékony levegő 37
44. Magas hőmérséklet előállítása 39
44.1. Gáztüzelésű kemencék 3944.2. Elektromos kemencék 40
44.2.1. Ellenállás-kemencék 40
44.2.2. ívfénykemencék.......................................................................................... 4144.2.3. Nagyfrekvenciás indukciós kemencék........................................................ 42
46. Hőmérsékletmérés és hőszabályozás 43
45.1. Gáz- és folyadékhőmérők................................................. 43
45.1.1. Gázhőmérők .................... : ................................................................................. 4345.1.2. Folyadékhőmérők 43

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 5/366
4 TARTALOMJEGYZÉK
45.2. Elektromos elven működő hőmérők 44
46.2.1. Ellenállás-hőmérők 4445.2.2. Termisztorok.............................. 4445.2.3. Termoelemek 47
45.3. Optikai pirométerek 4845.4. Termokolor festékek............................................................ 4845.5. Seger-gúla ............................................................................ 4945.6. Hőmérséklet-szabályozás, térmosztátok 49
45.6.1. TermoBztátok töltő- (hőközlő) anyagai .................. 5045.6.2. Önműködő hőmérséklet-szabályozók (regulátorok) 6045.6.3. Termosztálás fázisegyensúlyok felhasználásával 52
46. Vákuumtechnika 54
46.1. Általános tudnivalók 5446.2. Szivattyúk 56
46.2.1. Vízlégszivattyúk............................................... 6646.2.2. Forgó olajszivattyúk (rotációs szivattyúk) 5646.2.3. Gőzsugárszivattyú 5846.2*.4. Diffúziós szivattyú .................. 59
46.3. Vákuummérés 60
46.3.1. Vákuumindikálás elektromos kisüléssel ................................................... 6046.3.2. Higanyos manométerek, a közlekedőedények elvén működő vákuum-
mérő berendezések ................................................................................ 61
46.3.3. Kompresszióé vákuummérő .......................................................................... 6246.3.4. Radiométer típusú nyomásm érők......... ..................................................... 6346.3.5. Gázok belső súrlódásának nyomásfüggésén alapuló vákuummérők 6346.3.6. A hővezető-képesség nyomásfüggésén alapuló vákuummérők 63
'46.3.7. Ionizációs vákuummérők 66
46.4. A vákuumberendezée tartozékai 67
46.4.1. Kifagyasztócsapdák, terelőlemezek 6746.4.2. Csővezetékek, csapok, illesztés....................... 67
46.5. Gázadszorpció üveg- és fémfelületen, kigázosítás . . . . ...................................... 7346.6. A vákuumberendezés felépítése, üzemeltetése; hibahelykeresés 75
47. Munka nyomás alatt 78
47.1. Bombacső.................................................................. 7847.2. Autoklávok ............................... 8047.3. Nagynyomású manométerek............................................. 82
48. Az anyagok tisztítása 84
48.1. Halmazállapot-változás nélkül végbemenő elválasztás 84
48.1.1. Szilárd fázisok szétválasztása....................................................................... 8448.1.2. Szilárd —folyékony fázisok elválasztása ............................ 8648.1.3. Folyékony halmazállapotú anyagkeverék komponenseinek elválasz
tása ......................................................................... 8948.1.4. Géz elválasztása szilárd vagy folyékony anyagtól 9148.1.5. Gázelegyek komponenseinek elválasztása 92
48.2. Anyagok tisztítása halmazállapot-változás útján 93
48.2.1. Kristályosítás 9348.2.2. Desztillálás 9648.2.3. Szúblimálás ................................................................. 10248.2.4. Kondenzáció, kifagyasztés 104

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 6/366
TARTALOMJEGYZÉK 5
48.2.5. Kiolvasztás ...........................................................................................................48.2.6. Kioldás, extrakció
48.3. Adszorpció, deszorpció, kromatográfia, ioncsere
48.3.1. Adszorpció, deszorpció ....................................................................................48.3.2. Gázkromatográfia, ioncserélő kromatográfia48.3.3. Molekuláris szűrők
48.4. Tisztaságvizsgálat
49. Gázok
49.1. Gázfejlesztés ................................................................49.2. Gázok tisztítása, szárítása
49.2.1. Általános módszerek ..............................49.2.2. Hidrogénszennyezés eltávolítása49.2.3. Oxigén eltávolítása .........................
49.2.4. Szén-dioxid eltávolítása.......................
49.2.5. Szén-monoxid eltávolítása . . . . .49.2.6. Illékony szénvegyületek eltávolítása49.2.7. Nitrogén eltávolítása .........................................................49.2.8. Egyéb szennyezések eltávolítása
49.3. Szárítóanyagok .................................................................................................................49.4. A laboratóriumi gyakorlatban sűrűbben előforduló gázok szokásos szeny-
nyezései ...............................................................................................
49.5. Áramlássebesség- és nyomásszabályozás, tárolás49.5.1. Gázok áramoltatása . . . . ....................
49.5.2. Az áramlássebesség mérése49.5.3. Áramló gázok keverése49.5.4. Gázok tárolása
.50. Oldószerek
50.1. Halogén elemek ....................................................
50.2. Hidrogén-halogenidek50.3. Hidrogén-cianid
50.4. Kén-hidrogén50.5. Kén-dioxid50.6. Ammónia ...........................................................
50.7. Szénhidrogének50.8. Kloroform ....................................................................................................................50.9. Szén-tetraklorid.............................................................................
50.10. Szilícium- és ón-tetraklorid50.11. Alkoholok50.12. Dietiléter50.13. Aceton ...............................................................................................................................50.14. Szén-diszulfid50.15. Piridin
50.16. Víz
51. Az elektrolízis alkalmazása preparatív célokra
51.1. Bevezetés ..................................................
51.2. Gázok előállítása51.3. Fémek leválasztása......................................................................................................51.4. Katódos redukció fémkiválasztás nélkül................................................................51.5. Anódos oxidáció ........................................................... .................................................
61.6. Olvadékelektrolízis
104105
106
106107110
113
115
115117
117121
122
124124125125125
126
129
131131132133134
138
138139139
139140140140140140141141141142142142
143
144
144145146146147147

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 7/366
6 T A R T A L O M J E Q Y Z É K
62. Félmikro- és mikropreparatív munkamódszerek 150
52.1. Laboratóriumi alapműveletek 15052.2. Desztillálás, szublimálás ................................................................... 15552.3. Műveletek oxigénre érzékeny anyagokkal 157
Részletes rész
53. Nemfémes elemek 165
53.1. Hidrogén 16553.2. Fluor 16553.3. Klór 16653.4. Bróm 166
53.5. Jód .................................................................................................................. 16753.6. Oxigén 16753.7. Kén 16763.8. Szelén 16753.9. Tellúr .............................................................................................. 16853.10. Nitrogén 16853.11. Sáígaarzén 16953.12. Antimon 16953.13. Sz én .......................................................... 170
53.13.1. Aktív szén .............................. 17053.13.2. Grafitbevonat 170
54. Fémek 171
54.1. Fémek és néhány nemfémes elem előállítása fémtermiés reakcióval 171
54.1.1. Szilícium 17354.1.2. Bór ............................. 17354.1.3. Mangán 17354.1.4.. Molibdén 17454.1.5. Vanádium ............................................................................................ 17454.1.6. Alkálifémek előállítása cirkóniummal történő redukcióval 17454.1.7. Titán ................................................................. 17554.1.8. Cirkónium 176
54.2. Fémek előállítása hidrogénes redukcióval . . . . 176
54.2.1. Vas . 17754.2.2. Kobalt 17754.2.3. Nikkel ............................... 17754.2.4. Molibdén 17854.2.5. Volfrám ....................... 17854.2.6. Germánium 178
54.3. Fémek előállítása elektrolízissel 178
54.3.1. Kadmium 179
54.3.2. Lítium 17954.3.3. Kalciu m .................. 17054.3.4. Magnézium 181
54.4. Fémek előállítása termikusbontással 182
54.4.1. Titán ................................; ..................................................................... 18254.4.2. Cirkónium 18454.4.3. Hafnium 18454.4.4. Tórium ............... 18454.4.5. Alkálifémek előállítása azidok elbontásával 185

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 8/366
TARTALOMJEGYZÉK 7
54.5. Alkálifémek tisztítása és eltartása 186
54.5.1. Alkálifémek tisztítása vákuumdesztillációval ......................................... 18654.5.2. Az alkálifémek eltartása és használatra való előkészítése 18654.5.3. Finom eloszlású alkálifémek előállítása 187
54.6. Platinafémek 187
'54.6.1. Platinaszivacs ......................................................................................; ........... 18754.6.2. Platina- és palládiumazbeszt 18754.6.3. Platinakorom 18854.6.4. Palládiumkorom 18854.6.5. Platinázóoldat 188
•55. Hidrogénvegyületek 189
55.1. Nemfémek hidrogénvegyületei 18955.1.1. Hidrogén-fluorid........................................ 18955.1.2. Hidrogón-klorid ............................................................................................... 19055.1.3. Hidrogén-bromid 19055.1.4. Hidrogén-jodid 19255.1.5. Kén-hidrogén ........................................................................................ .......... 19355.1.6. Hidrogén-poliszulfid 19455.1.7. Szelén-hidrogén.................................... : .......................................................... 19555.1.8. Tellúr-hidrogén 19555.1.9. Ammónia ......... 19655.1.10. Arzén-hidrogén ............................................................................................... 19655.1.11. Antimon-hidrogén 197
55.1.12. Szilánok 19755.2. Fémek hidridjei .............................................................................................(................... 198
55.2.1. Lítiumrhidrid ........................ 19855.2.2. Lítium-aíumínium-hidrid 10955.2.3. Alkálifém-hidridek . . , .................................................................. 20055.2.4. Alkáliföldfém-hidridek 20155.2.5. Titán-hidrogén 20155.2.6. Réz-hidrogén 202
56. Vízmentes halogenidek 203
56.1. Vízmentes halogenidek előállítása elemi szintézissel 20356.1.1. Vas(III)-klorid 20456.1.2. Króip(III)-klorid........... 20556.1.3. Volfrám( VI)-klorid 20556.1.4. Ón(IV)-klorid .................................................................................................... 20656.1.5. Antimon-pentaklorid 20656.1.6. Foszfor-triklorid . . 20756.1.7. Foszfor-pentaklorid 20856.1.8. Szilícium-tetraklorid .................................................................... 20856.1.9. Diszilícium-hexaklorid és homológjai 20956.1.10. Dikén-diklorid ____ 21056.1.11. Szelén-tetraklorid 21056.1.12. Bór-trikíorid ...................................................................................................... 21056.1.13. Alumínium-bromid 21156.1.14. Ón(IV)-bromid ................................................ 21156.1.15. Bizmut-tribromid 21256.1.16. Bór-tribromid ................................................................................................. 212
,56.1.17. Foszfor-tribrom id............................................................................................. 21356.1.18. Szilícium-tetrabromid és króin(III)-bromid .................................. 21356.1.19. Cirkónium-, titán-, szilícium-tetrajodid és alumínium-jodid 21456.1.20. Ón(IV)-jodid, alumínium-jodid és foszfor-trijodid . . . 21456.1.21. Alumínium-jodid, difoszfor-tetrajodid és bizmut-jodid 215

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 9/366
8 TARTALOMJEGYZÉK
56.2. Vízmentes halogenidek előállítása oxidok halogénezósével 215
56.2.1. Titán-tetraklorid, szilícium-tetraklorid és bór-triklorid 21556.2.2. Cirkónium- és tórium-tetrabromid 216
56.3. Vízmentes halogenidek előállítása halogénátvivő segítségével 216
56.3.1. Titán-tetraklorid ......................................................................................... 21756.3.2. Lantán (Ill)-klorid 21756.3.3. Bór-triklorid ...................................................................................................... 21756.3.4. Cirkónium-tetraklorid, rénium-, nióbium- és tantál-pentaklorid 21 &56.3.5. Bór-tribromid .................. 22056.3.6. Volfrám(VI)-bromid 22056.3.7. Szén-tetrabromid 220'56.3.8. Szén-tetrajodid 221
56.4. Vízmentes halogenidek előállítása halogén-hidrogénnel 22156.4.1. Alumínium-klorid 22156.4.2. Bór-trifluorid . . . . 22256.4.3. Arzén-trifluorid 222
57. Keverékhalogenidek........................................... 222
57.1. Szén-diklorid-dibromid . . . ................................................................. 22357.2. Szilícium-diklorid-dijodid 22357.3. Alumínium-klorid-dijodid 224
58. Alacsonyabb vegyértékű halogenidek 225
58.1. Antimon-triklorid 22558.2. Réz(I)-klorid ...................................... 22658.3. Titán-triklorid 22658.4. Vas(II)-klorid............................................................................................................. 227
. 58.5. Króm(II)-klorid 22858.6. Urán(IV)-klorid.................... 22858.7. Vanádium(III)-klorid 22858.8. Vanádium(II)-klorid 228
59. Kristályvíztartalmú halogenidek 230
59.1. [Hexakvo-króm(HI)]-klorid ...................................... 23059.2. [Diklór-tetrakvo-króm(III)]-klorid-dihidrát 231
59.3. Vanádium(III)-klorid-hexahidrét 23159.4. Berillium(II)-klorid-tetrahidrát 232
60. Interhalogenidek és pszeudohalogenidek 233
60.1. Jód-monoklorid 23360.2. Jód-triklorid ............................................................................ 23360.3. Jód-monobromid 23460.4. Dicián ...................................................... 23460.5. Rubeán-hidrogénsav 235
61. Sav-halogenidek 236
61.1. Tionil-klorid és foszfor-oxid-klorid 23661.2. Tionil-bromid ....................... 23761.3. Foszfor-tioklorid 23861.4. Kloro-kénsav ......................... 23961.5. Piroszulfuril-klorid 24061.6. Szelenil-klorid 24061.7. Nitrozil-klorid 24161.8. Kromil-klorid ......................................................................................................... 24261.9. Vanádium-oxid-triklorid 24361.10. Nióbium-oxid-klorid 243

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 10/366
TARTALOMJEGYZÉK 9
62. Oxidok 244
62.1. Difluor-monoxid 244
62.2. Klór-dioxid 24562.3. Jód-pentoxid ...................................................................................... 246'62.4. Jód(III)-oxid-jodát 24662.5. Kén-trioxidok 24762.6. Dikén-trioxid 24762.7. Szelén-dioxid 24862.8. Tellúr-dioxid . .................................. 24962.9. Dinitrogén-oxid ............................................................................................................. 25062.10. Molibdén(VI)-oxid 25062.11. Volfrám(VI)-oxid 25162.12. Urán(VI)-oxid .............. 25162.13. Rénium(VII)-oxid 251
63. Alacsonyabb vegyértékű fém oxidok 253
63.1. Titán-trioxid ................................ 25363.2. Vanádium(IH)-oxid 25463.3. Króm(III)-oxid ........ 25463.4. Molibdén(IV)-oxid 25463.5. Volfrám(IV)-oxíd 25463.6. Réz(I)-oxid 255
64. Oxosavak és sóik 256
64.1. Kálium-klorát................ 25664.2. Magnézium-perklorát 25764.3. Kólium-bromát 25864.4. Jódsav ......................... 25864.5. Nátrium-perjodátok 25864.6. Kálium-perjodát 25964.7. Bárium-perjodát 25964.8. Per jódsav ...........................................................................................................; ........... 26064.9. Ammónium-vanádium-timsó .................................................................................. 26064.10. Ammónium-vanódium(II)-szulfát-hexahidrát 26164.11. Kobalt(III)-szulfát-oktadekahidrét 26264.12. Antimort(III)-szulfát 263
64.13. Higany(II)-szulfát 26364.14. Foszforsav 26364.15. Foszforossav 26464.16. Hipofoszforossav............................................................................ 26564.17. Bárium-hipofoszfit-monohidrát 26564.18. Nátrium-pirofoszfát 26564.19. Nátrium-metafoszfát .................................................................................................... 26664.20. Vas(II)-ortofoszfát-oktahidrát 26664.21. Kálium-manganát(VI)......................... 26664.22. Nátrium-hipomanganát (V)-hidrát 26764.23. Kalcium-volframát 26764.24. Ólom-tetraacetát.................... 26764.25. Szilícium-tetraaeetát 26864.26. Bór-triacetát 26864.27. Szilikagél 268
65. Peroxidok, peroxisavak 270
65.1. Nátrium-peroxid 27065.2. Hidrogén-peroxid .......................................................................................................... 27065.3. Kálium-tetraperoxo-kromát(V) ............*............................................................... 27165.4. Ammónium-peroxi-diszulfát és kálium-peroxi-szulfát 271

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 11/366
10 T ARTALOMJEGYZÉK
66. Szulfidok és tiovegvületek 273
66.1. Higany(II)-szulfid 27366.2. Króm(III)-szulfid ............................................................................... 27366.3. Foazfor-pentaszulfid 27466.4. Molibdén(IV)-szulfid 27466.5. Bárium-tiokarbonát 27466.6. Arzén-orfcotiofoszfát 27566.7. Nátrium-tiokromit ....................................................................................................... 27566.8. Bárium-ditionát-dihidrát 27566.9. Kálium-vas(III)-azulfid .................................................... 27666.10. Kálium-bizmut(III)-Bzulfid 27666.11. Alumínium-szelenid 276
67. Nitridek, foszfidok, arzenidek, antimonidok, bizmutidok, karbidok, azilieidek
éa bori dók 27767.1. Lítium-nitrid 27767.2. Bór-nitrid...................................................................... 27767.3. Magnézium-nitrid 27867.4. Kalcium-nitrid 27967.5. Titén-nitrid ....................................................................................... 27967.6. Alkálifémekfoszfidjai, arzenidjei, antimonidjai éa bizmutidjai......................... 279
67.6.1. Lítium-foszfid, lítium-arzenid, lítium-antimonid és lítium-bizmutid 28067.6.2. Nátrium-foazfid, nátrium-arzenid, nátrium-antimonid éa nátrium-
-bizmutid 28067.7. Kalcium -foszfid ....................................................................................................... 281
67.8. Alumínium-foszfid 28167.9. Kalcium-karbid 28167.10. Vas-karbid ................ 28267.11. Titán-karbid 28267.12. Szilicidek 283
67.12.1. Magnézium-szilicid 28367.12.2. Kalcium-azilicid . . . 28467.12.3. Kalcium-diszilicid 28467.12.4. Titán(IV)-szilicid 284
67.13. Boridok 284
67.13.1. Titán-borid 285
68. Ammono- és egyéb nitrogénvegyületek 286
68.1. Alkálifém-amidok 286
68.1.1. Nátrium-amid.................................................. 28668. V.2. Lítium-amid éa kálium-amid 288
68.2. Nátrium-azid 28868.3. Hidrogén-azid 28968.4. Ólom-azid............................................................................................................................ 28968.5. Hidroxil-ammónium-klorid 28968.6. Hidroxil-ammónium-foszfát 29068.7. Hidroxil-amin ..................................................................................................... 29068.8. Foszfor-nitrid-klorid 291
69. Komplex vegyületek 292
69.1. Acidokomplexek 292
69.1.1. Hidrogén-[tetrafluoro-borát(III)] 29269.1.2. Kálium-[tetrafluoro-borát(IÍI)] ................................................................. 29269.1.3. Ammónium-[hexakloro-plumbát(IV)] 29369.1.4. Kálium-[hexakloro-plumbát(IV)] 29369.1.5. Kálium-[trijodo-plumbit(II)] 294

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 12/366
TARTALOMJEGYZÉK 1 1
69.1.6. Hidrogén-[hexaciano-ferrát(H)] 294
69.1.7. Káli^m-[trioxa]áto-ferrát(HI)]..................................................................... 29569.1.8. Ammónium-[hexakloro-platinát(IV)] 295
69.2. Hidroxokomplcxck 295
69.2.1. Nátrium-[hexahidroxo-sztannát(IV)].......................................................... 29669.2.2. Nátrium-[hexahidroxo-pl umbát(IV)] 29669.2.3. Nátrium-[tetrahidroxo-kuprát(II)] ............................................................296
69.3. Kelátkomplexek ............................................... 297
69.3.1. [Tetraacetil-acetonát-cirkónium(IV)] 29769.3.2. [Triacetil-acetonát-króm(III)]............................... 29769.3.3. [Diglikokollát-réz(II)]........... 297
70. Karbonilok és nitrozilok 29870.1. [Tetrakarbonil-nikkel(O)] . . . . 29870.2. [Tetrakarbonil-dijodid-vas(II)] 29970.3. [Pentakarbonil-vas(O)] ................................................................... 29970.4. Dihidrogcn-[tetrakarbonil-ferrát(II)] . . . 30070.5. Hidrogén-[tetrakarbonil-kobaltát( —1)] 30070.6. Kobalt-karbonilok .................................... 30170.7. [Pentakarbonil-szulfid-dikobalt(I)] 30270.8. [Dinitrozil-dikarbonil-ferrát(O)]................................................. 30270.9. Dinátrium-[pentaciano-nitrozil-ferrát(III}] 303
71. Izo-polisavak, heteropolisavak és sóik 304
71.1. Nátrium-izo-polivanadátok 304
71.1.1. Nábrium-pirovanadát 30471.1.2. Nátrium-metavanadát ......................... 30471.1.3. A pentavanadinsav nátriumsója 304
71.2. Izo-polimolibdátok 305
71.2.1. Nátrium-paramolibdát 30571.2.2. Nátrium-metamolibdát 30571.2.3. Ammónium-molibdát 305
71.3. Heteropolisavak és sóik 305
71.3.1. Bór-volfrámsav 30671.3.2. Sziliko-volfrámsav ......... 303
71.3.3. Kálium-sziliko-volframát 307
72. Ritkaföldfémek és vegyületeik 308
72.1. Lantán 30&72.2. Cérium ............................. 30^72.3. Lantán(III)-klorid 30972.4. Lantán(III)-szulfát 30972.5. Cérium(TV)-oxid . . . . 30972.6. Cérium(IV)-szulfát 31072.7. Cérium{III)-oxid ........................................................................................... 31072.8. A ritkaföldfémek szétválasztása ioncserélő gyanta alkalmazásával 312
73. Amalgámok 312
73.1. Nátriumamalgám 312
73.1.1. Folyékony atnalgám, kb. 1% Na-tartalommal 31273.1.2. Szilárd amalgám, 2 —3% Na-tartalommal 313
73.2. Kálóiumamalgám 31373.3. Báriumamalgám 314
74. Fémorganikus vegyületek 314
74.1. Szerves lítium vegyületek 314
74.1.1. Fenil-lítium 315

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 13/366
TAB.TA LOM JEG YZÉK 12
74.2. Szerves higanyvcgyületek 317
74.2.1. Fenil-higany (II)-acetát 31774.2.2. Fenil-higany(II)-klorid 31774.2.3. Di(izo-propil)-higany(II) 318
74.3. Szerves bórvcgyületek 319
74.3.1. Trifenil-bór ................................................................... 32074.3.2. Nátrium-[trifenil-eiano-borót(HI)] 32274.3.3. Metil-bór-diklorid 322
74.4. Szerves alumíniumvegyületek 323
74.4.1. Metil-alumínium-kloridok ................................................. 32374.4.2. Di(izo-propoxi)-alumfnium-klorid 325
74.5. Szerves szilíciumvegyületek 325
74.5.1. Tetraetoxi-szilén................................. 32774.5.2. Metil-klór-szilánok 32874.5.3. Trimotil-fenil-szilén 33174.5.4. Fenil-trietnxi-szilán 33274.5.5. Trimetil-p-tolil-szilán . . ............................................................................ 33374.5.6. Tetrametil-szilán ............................................... 33374.5.7. Tetrametil-disziloxán-J,3-diol 33474.5.8. Fenil-triklór-szilán 335
74.6. Szerves ónvegyületek 337
74.6.1. Tetraetil-ón 33774.6.2. Tetíabutil-ón . . . . 33874.6.3. Dietil-ón-diklorid .33974.6.4. Dietil-ón-oxid . . . 33974.6.5. Dietil-ón-diacetát . 33974.6.6. Dibenzil-ón-diklorid 34074.6.7. Tribenzil-ón-klorid 340
74.7. Szerves ólomvegyületek 341
74.7.1. Tetraetil-ólom................................................... 34174.7.2. Trietil-ólom-p-toluol-szulfonát 341
74.8. Szerves titánvegyületek 342
74.8.1. Tetraétoxi-titán .............................................................. 34274.8.2. Trietoxi-titán-klorid 343
75. Baleseti veszély és óvatossági rendszabályok 344
75.1. Mérgezések ......... 34475.2. Robbanás, tűz 347
75.2.1. Gázpalackok kezelésével kapcsolatos előírások ............................. 35075.2.2. Nyomás alatti berendezésekkel kapcsolatos előírások 35175.2.3. Tűzvédelmi tudnivalók és előírások 351
75.3. Áramütés 35275.4. Sebesülések .................................................................... 35375.5. Radioaktív sugárzás okozta fertőzések 353
Az elemek periódusos rendszere és atomsúlya 354
összefoglaló irodalom a részletes részhez 355
Képletmutató 357
Tárgymutató 362

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 14/366
Előszó a II. könyvhöz
Az Általános és szervetlen kémiai praktikum most útjára induló II. könyve kettős
feladatnak kíván megfelelni. Szolgálni óhajtja egyrészt — az I. könyvhöz csatlakozva, de magasabb szinten — a szervetlen kémia egyetemi oktatásának ügyét, segít
séget nyújtva a vegyészeknek a felsőbb évfolyamon előírt szervetlen kémiaigyakorlatok korszerű, elmélyedő elvégzéséhez. Ennek érdekében a klasszikus értelemben vett szervetlen kémiai preparatív munka területét igyekszik kiterjesztenia különleges körülmények között, mint nagy vákuumban, nagy nyomáson, ala
csony és magas hőmérsékleten, továbbá nemvizes1közegben lejátszódó folyamatoktanulmányozására is.
A szerzők azonban másrészt figyelemmel voltak arra, hogy a magyar nyelvű
szakirodalomban hiányzik olyan mű, amely az önálló szervetlen kémiai munkához
nélkülözhetetlen alapismereteket, mint a szerkezeti anyagokat, a berendezéseképítését és használatát, a tekintetbe jövő kiindulási anyagok minősítését és kiválasztását áttekintően és összefoglalóan tárgyalná. Ezt a hiányt főként a könyv általános része hivatott pótolni.
Ilyenformán ez a munka az egyetemi oktatásban betöltött szerepe mellett azzal az igénnyel is fellép, hogy mondanivalóját tanulmányaikat már befejezett szak
emberek is felhasználhassák, akik a legkülönfélébb tudományos-műszaki területenkerültek kapcsolatba a szervetlen kémia gyakorlatával.
Ennek a nagy anyagnak az összegyűjtésében és a /vázolt szempontok szerinti
feldolgozásában a munka dandárját szerzőtársaim, dr. Csáhoári Béla és dr. Kováts Zoltán vállalták, akik a Bzóban forgó területen folytatott oktató tevékenységük sokéves tapasztalatára is támaszkodhattak a témák kiválasztásában és a tárgyalás
színvonalának kialakításában.Törekvésünk az volt, hogy csak olyan feladatokat vegyünk fel az anyagba,
melyeket gyakorlatilag kipróbáltunk. Jóllehet e tekintetben több évre visszamenő,szisztematikus munkára hivatkozhatunk, melyben a tanszék többi munkatársai is
hathatósan támogattak bennünket, e célkitűzést mégsem sikerült maradéktalanul
megvalósítanunk. Az e téren esetleg jelentkező hiányosságokat bőséges irodalmi hivatkozással igyekeztünk kiküszöbölni, vagy legalábbis enyhíteni.
Végül köszönettel tartozunk lektorainknak, Dr. Szabó Zoltán és Dr. Szarvas Pál egyetemi tanároknak, akik fáradtságok nem kímélve nézték át a kéziratot. Meg
jegyzéseiket, észrevételeiket eredményesen használtuk fel a könyv végleges elkészítése során. Köszönjük továbbá Arató Gyula grafikusnak az ábrák elkészítésében vég
zett kitűnő munkáját.
Budapest, 1961. június. Dr. Lengyel Béla

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 15/366
14 ELŐSZÓ
Előszó a második kiadáshoz
A Praktikum II. kötetének új kiadása az első kiadáshoz viszonyítva számottevő mértékben bővült. Ez lehetővé tette számos olyan, a gyakorlatban egyre inkábbhasználatos módszer ismertetését, mint a mikro- és félmikrotechnika vagy az ion
cserélők alkalmazása, amelyek tárgyalására az előző kiadásban a korlátozott terjesdelem miatt nem kerülhetett sör.Bemutatjuk a preparatfv munkamódszerek újabb fejlődési irányait, így a
molekuláris szűrők, a gázkromatográfia, termisztorok, egyes újabb műanyagokstb. alkalmazását.
A Praktikum „Részletes rész” -e újabb, a szerzők által reprodukált előállításimódszerekkel is bővült. Célunk az volt, hogy a vegyészhallgatók az elemek és ve-gyületek minél szélesebb körét ismerjék meg gyakorlati munkájuk során. E didaktikai szempontok érvényesítése mindinkább döntő befolyásoló tényezőjévé válik azegyetemi kémiaoktatásnak, miután az oktatási reform szellemében az előadások
keretében egyre kevesebb tárgyi ismeretanyag foglal helyet. A „Eémorganikus vegyületek” -ről szóló fejezet tárgyalásmódja eltér az eddig
szokásostól. Behatóan foglalkozunk e vegyületek tulajdonságaival, valamint elméleti vonatkozásaival is. Ezt nemcsak- ezek gyakran mérgező vagy öngyulladó voltaindokolja, hanem az a tény is, hogy a vegyész-, ill. vegyészmérnök-képzés kereténbelül a fémorganikus vegyületekkel — melyek ipari szempontból is mind nagyobb
jelentőségűek — az alapelőadások csak érintőlegesen foglalkoznak. Az új kiadás szerkesztésének és bővítésének munkáját túlnyomó íészben
Dr. Csákvári Béla docens vállalta magára, aki e téren másfél évtizedes oktatói tevé
kenységének tapasztalataira támaszkodhatott.őszinte köszönetét mondunk e mű lektorának, Dr. Szarvas Pál egyetemi tanárnak, akinek javaslatait és megjegyzéseit eredményesen használtuk fel. Külön köszönjük Dr. Szarvas Pál-nak az első kiadásról megjelent könyvbírálatát, mely hasznos útmutatást nyújtott, s egyben ösztönzésül is szolgált az újabb kiadás kidolgozásához.
Budapest, 1967. Dr. Lengyel Béla

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 16/366
ÁLTALÁNOS RÉSZ

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 17/366
42 . A laboratóriumi munkában használatos szerkezeti anyagok
42.1. Üveg
A laboratóriumi gyakorlatban legáltalánosabban használt szerkezeti anyag az
üveg. Legfőbb előnye átlátszósága és kémiai ellenálló-képessége. Hátránya törékenysége és a hirtelen hőmérsékleti változások iránti érzékenysége.
A korszerű technika a gyakorlatban használatos üvegekkel szemben igen különböző igényeket támaszt. Ennek megfelelően az ipar igen sok üvegfajtát állít elő,
amelyek mindegyike bizonyos tulajdonságában egy-egy speciális követelményt optimálisan kielégít. A felhasználhatóság szempontjából legfontosabb tulajdonságok:
a megmunkálhatóság, a hőmérsékleti változások iránti érzékenység, a különbözőegyéb szerkezeti anyagokkal (porcelán, fémek stb.) való összeolvaszthatóság, a me
chanikaiszilárdság, az optikai és elektromos sajátságok, valamint a kémiai ellenálló-
-képesség és végül a kereskedelmi ár. A megmunkálhatóság szempontjából legelőnyösebbek az ún. „hosszú” üvegek,
amelyeknek a viszkozitása a hőmérséklet függvényében aránylag kismértékben vál
tozik. Ilyen üvegek a szintén alacsony lágyuláspontú, de „rövid” ablaküveggelszemben az ún. thüringiai üvegek, valamint a jénai normál üvegek, melyek igen al
kalmasak bonyolult berendezések készítésére. A hőmérsékleti változásokkal szemben mutatott ellenállás igényét a thüringiai
és jénai normál, ún. lágy üvegeknél lényegesen jobban kielégíti a magasabb lágyu
láspontú, keményebb jénai „Oeráte 20-as” , Rasotherm, Durán, Pyrex, Ergon,
Supremax és kvarcüveg. Ezek kémiai ellenálló-képessége is nagyobb. Az üveg lágyu
láspontja (kb. 107 poise viszkozitásnak megfelelő hőmérséklet) csak abban az esetben jelenti az alkalmazhatóság felső határát, ha az üveg nincs hosszabb ideig tartó,
nagyobb mechanikai igénybevételnek kitéve (pl. vákuum.)Gyakorlati szempontból fontos adat az ún. transzformációs pont, a 1013 poise
viszkozitásnak megfelelő hőmérséklet. Ha az üveget hosszabb ideig ezen a hőmérsékleten tartjuk, feszültsége belső szerkezeti átrendeződés folytán kiegyenlítődik.
A kvarcüveg kb. 1350 C°-ig használható. Legelőnyösebb tulajdonsága az, hogy afelsoroltak közt legkisebb a hőtágulási együtthatója, és ezért a gyors hőmérséklet-
-változást jól bírja. Alkalmazásakor tekintetbe kell venni, hogy 1000—1100 C° fölött
számottevő a kristályosodási sebessége, ezért huzamosabb ideig ezen a hőmérsékleten nem alkalmazható. A kristályosodott, üvegtelenedett kvarcüvegtárgy mikro
kristályos szövetűvé és porózus szerkezetűvé válik. A kvarcüveg még magas hőmérsékleten is igen jó elektromos szigetelő (ellenállása 1000C°-on 10®í? cm-1). A 42.1.1.táblázatban látható a leggyakrabban használatos üvegfajták összetétele és néhányfontosabb fizikai tulajdonsága.
Az üvegek egymással való összeforrasztása szempontjából is fontos adat a hőtá-
2 Ált. és szervetlen kémiai prakt. — 4284/H.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 18/366
N é h á n y g y a k o r l
a t b a n h a s z n á l a t o s ü v e g ö s s z e t é t e l e é s t u l a j d o n s á g a i
18 S Z E R K E ZE T I A N Y A G O K
ss-o3
43_, -H o o| 3
£ öohco3B
©1
O 6 9 4
CDCD
1oIC 7 0 0
7 7 6
<£<+ j©o© 8
5 0
7 7 6
0
- 7 3
0
7 9 5
1 5 0 0
M*0 © CD <N A
IC O © ©eo o 00 ©
Owo . IC o IC IC © © © © © r- ©1 eo 1 1 © eo © eo 1 ÍN 2
ifi Oo O © © o © t> © © © « 2c< .5 o © ÍN ©uE- 2 © IC IC
o ot— IC© © Tfl © „-©o <N 00 eo eo eo © -2hí 00 © Tfl 00 00 co 00 © o
h £ ' 8 00 1 pH
K §?©* 00 CD© 1<M ©
«o 1eo pH pH pH o3* 1 <N©~ 1 i ©* ©“ ©‘ i o <N* 1 1JÍ o 1
flN
t-o pH U3 00 © ©_ *c_ ©____
' M 1 © 00 (N CD © •T 1PJ
o pH
n
o IC LC 00 t- H eo t- H ©M CO I IC © © <N eo l4
D o p © © © © ASÖ©"»
bes I ©‘ 00 ©~ 1 öo 1 1 1 1
•Öa> O lO eo © ICéti ©* eo 00* 00 1ffl
4>
o o © © 00 pH (N IC+10> éti
O 00 IC © ""r-H TjT ©" © 1 1N
CD o © ff* © © p H ©W 1
09 1 eo* ©* © © © o 1 1
«O o 00 CD t- »q .éti i •oT 00 CD © © ©“ 4T 1& <N p H
lOM r* © eo 00 © H © ©
i <N <M IC CD © CD <N* Ti oco IC t- t- r- © 00 r- t- ©CD H

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 19/366
A Z Ü V E G 19
gulási együttható (a). Jól olvaszthatok egymással azok az üvegek, amelyeknél ac értéke 10 %-nál nem sokkal nagyobb mértékben különbözik, így pl. a különböző thü-
ringiai üvegek. Ha az összeforrasztandó üvegfajták tágulási együtthatója a megengedettnél nagyobb mértékben különbözik egymástól, akkor az áthidalás a különbség nagyságától függően több részletben végezhető el ún. „összekötő” üvegek segítrségével. Ezek megfelelő számú, fokozatosan változó a-jú, de kétoldali szomszédjukkal jól forrasztható üvegből készült gyűrűből állnak. Pl. a ihüringiai és &jénai „Gerate 20”-as üveg 4-tagú, &jénai „Geráte 20” -as és a kvarcüveg 7-tagú „összekötővel”forrasztható össze. Hasonló összekötők segítségével oldható meg az üveg—porcelánés üveg—fém forrasztása is. Készülnek speciális üvegek meghatározott fémek, illetve különleges fémötvözetek hőtágulásához hasonló a-val, meghatározott üvegtípus-ba való beforrasztás céljából.
Az üvegcsövek illesztésének az összeforrasztásnál egyszerűbb, de kevésbé tökéletes módja a gumi- vagy egyéb műanyagcsővel való összekötés. Az így illesztett vezetékben az áramló anyag kisebb-nagyobb mértékben szennyeződhet, mert az üvegnél sokkal kevésbé ellenálló gumicső alkotórészeivel kölcsönhatásba léphet. Ezt aveszélyt küszöböli ki a csiszolatos illesztés. Ma. már olyan tökéletes kivitelben készülnek csiszolatos illesztések, hogy szívás alatt sem hatol át rajtuk a higany, még zsíro-zatlan állapotban sem. Csövek illesztésére legelterjedtebb a kúpos csiszolat. A tengelyre merőleges irányú elmozdulások lehetősége esetén, a kúpos csiszolat törésénekveszélye miatt újabban gömbcsiszolatot használnak. Ez kétségtelenül jobb, de
drágább megoldás. A régebben készült kúpos cBiszölatok átmérője és palástjának a tengellyel bezárt szöge műhelyenként, sőt ezen belül is változott. Ezeknek az ún. egyedi csiszolatoknak a tokjai, illetve csapjai — kölcsönös összecsiszolás eredményeként — csak
Normálcsiszolatok méretei 42.1.2. táblázat
Jelölés Normál DIN 12 242
Rövid DIN 12 248
Hosszú DIN 12 243
régi
új A csiszolat méretei mm-ben
legnagyobb0
legkisebb0 hossza
legkisebb0 hossza
legkisebb0 hossza
NS 100 94 6078/10 NS 85 79,5 60 —
— — —
64/10 NS 70 66 50 — — — —
56/10 NS 60 55,4 43 — — — —
45/10 NS 50 — — 47,5 25 —
40/10 NS 45 41 36 — — 40 5030/10 NS 34,6 31 30 — —, — —
25/10 NS 29 25,8 26 26,9 21 24,8 4220/10 NS 24 21,1 23 22,2 18 — —
16/10 NS 19 16,4 21 17,8 12 15,2 3811/10 NS 14,5 12,2 18 13,3 12 11 359/10 NS 12,6 10,4 16 — — 9,25 32,57/10 NS 10 8,1 15 — — — —
6/10 NS 7,5 6,9 10 - - —
3/10 NS 5 3,7 10 — —
2

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 20/366
20 S Z E R K E Z E T I A N Y A G O K
a párjukkal voltak illeszthetők. Lényegesen olcsóbbá tette a csiszolatok alkalmazását a méretnorma bevezetése. Az NS (Normalschliff) jelzésű, ún. normálcsiszólatok
nemzetközi megegyezés szerint meghatározott méretekben készülnek (lásd a 42.1.2.táblázatot). A palást hajlásszöge is mindenütt azonos: 2° 52', a tűrés ± 1#. Az NSmegjelölésére használt szám a csiszolat legnagyobb átmérőjét jelenti.
Nagyobb átmérőjű alkatrészek illesztésére síkcsiszolatokat használunk (pl.exszikkátor és fedele).
Keverők tömítésére, pipetták, dugattyúk készítésére hengercsiszolatok ké
szülnek. Ezek az ún. KPG-csövek geometriailag pontos keresztmetszetűek. Tűrésük0,01 mm vagy még kevesebb. Készülnek ezenkívül még ovális, négyszög, illetve hatszög keresztmetszetű precíziós csövek is.
Bár az üvegnek igen előnyös tulajdonsága kémiai ellenálló-képessége, bizonyos
körülmények között mégis intenzív kémiai reakciókra képes. Rendkívül fontos ezérta laboratóriumi gyakorlatban, hogy az üveg kémiai ellenálló-képességének határait
jól ismerjük.Gázokkal szemben — kivéve a fluortartalmúakat — az üveg igen ellenálló, de
magasabb hőmérsékleten számolni kell a korróziójával. így pl. a H2-gáz 500 C°-nálmagasabb hőmérsékleten, főleg az PbO-, Sb20 3- és As20 3-tartalmú üvegekből ki
választja az illető elemet, és az üveg megfeketedik. Alkálifémgőzök szintén redukáló hatásúak, az üveg bámulását idézik elő való
színűleg Si-kópződés * miatt. A kvarcüveg alkálifémgőzöknek 1300 C°-ig ellenáll,
Cl2- és HCl-gázzal 1200 C°-ig nem reagál, de alkálifém-oxid-tartalmú üvegek eseténkb. 400 C°-on (pl. thüringiai üveg) a két utóbbi reakció nem hanyagolható el.
Legkevésbé ellenálló az üveg a fluortartalmú gázokkal szemben. Legagresszí-
vebb a CIF3, amely üveggyapottal tűzjelenség közben reagál. A HF-gáz szobahő
mérsékleten is megtámadja az üveg felületét. A tiszta, száraz F2-gáz kevésbé reakcióképes, pl. kvarcedénybén szobahőmérsékleten' eltartható. Tiszta BF3-, SiF4-,
PF5-dal végzendő műveletek üvegkészülékben végrehajthatók.
Olvadékokkal szemben az üveg általában már kevésbé ellenálló. Oxidmentes fé
mek elegendő magas lágyuláspontú üvegedényben (kvarcüveg) megolvaszthatok
ugyan, de a Mg és az A1 olvadéka már 700—800 C°-on megtámadja az üveget, sőtaz alkálifémek már alacsonyabb hőmérsékleten is (pl. a Li 250 C°-on)reagálnak;1050 C° felett már szénnel is megindul a reakció.
Az alacsony olvadáspontú alkáli-kloridok 1000 C° körüli hőmérsékleten illékonySiCl4képződése közben megtámadják a kvarcüveget. Olyan olvadékokkal szemben,
amelyek termikus bomlása közben bázisos oxidok képződnek (pl. Na2C03, K N 03,Ba(N03)2 stb.), a kvarcüveg csak néhány száz fokig ellenálló. Fontos megjegyezni,
hogy a gyakran használt P20 5 300 C° fölötti hőmérsékleten ugyancsak megtámadjaaz üveget.
A lúgok, valamint a savak közül a foszforsav főleg melegen, a hidrogén-fluorid
hidegen is jelentős mértékben oldja az üveget. Savak és víz hatására is az alkáliféméé alkáliföldfém-ionok H+-ionokkal cserélődnek ki, és az üveg felületén hidratált ko
vasavas védőréteg képződik. Az üvegfelület alkálileadását az előbbi folyamat meggyorsítása útján, forró vízgőz tartós bevezetésével lehet csökkenteni.
Teljesen alkálimentes közeget kvarcüvegben vagy műanyag-, illetve fémedényben lehet előállítani.
Az átlátszó kvarcüveggel teljesen azonos kémiai tulajdonságai vannak a finom
levegőzárványok miatt selymes fényű, át nem látszó Eotosil- vagy Vitreosil-kvarc

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 21/366
T Ű Z Á L L Ó A N Y A G O K 21
tárgyaknak is. Az átlátszatlanság az alacsonyabb hőmérsékleten történt megmunkálás következménye. A Vitreosil-kvarc alkalmazási területének felső határa akristálykvarcénál alacsonyabb lágyuláspontja miatt néhány száz fokkal alacsonyabb. Speciális, HF-nak ellenálló üvegfajta is készülalumínium-foszfátból, Fluor ex néven; ezt a NaOH oldja.
\
42.2. Tűzálló (kerámiai) anyagok
Azok a szerkezeti anyagok, melyek magasabb hőmérsékleten használhatók,
mint az üveg, három nagy csoportba oszthatók (42.2.1. táblázat).
42.2.1. Alumínium-szilikát alapú anyagok
Az ide tartozó anyagok fontosabb fizikai tulajdonságait a 42.2.1. táblázat foglalja össze. Általában porózus szerkezetűek, azért a tiszta kvarcüvegnél kevésbégázzárók. Alkalmazhatóságuk felső hőmérsékleti határa, valamint kémiai ellenálló-képességük az Al20 3-tartalom növekedésével no.
Legkevésbé ellenállóak a magasabb hőmérsékleten ható alkálikus vagy erősenredukáló reagensekkel (nemnemes fémekkel) szemben. Érdekes megjegyezni, hogya laboratóriumi'technikában gyakran használt HC1- és Cl2-gázzal szemben a porcelán eszközök csak 800, illetve 1000 C°-ig ellenállók, szén jelenlétében ennél lényegesen alacsonyabb hőmérsékleten is korrodeálódnak.
42.2.2. Oxidkerámiai anyagok
Magas olvadáspontú tiszta oxidokból is készülnek laboratóriumi eszközök(zsugorítással). Ezeknek a zsugorított oxideszközöknek igen kiváló kémiai ellenálló--képeöségük van, de alkalmazásukkor figyelembe kell venni specifikus érzékenységü
ket egyes reagensekkel szemben. A „szinterkorund” a fluor kivételével minden gázzal szemben 1700 C°-ig indifferens, a F2 azonban 500 C°-on felül megtámadja. Fluorral magasabb hőmérsékleten CaF2-ból készült eszközökben dolgozhatunk (1000C°-ig). A zsugorított oxidokból készült eszközökről tudnunk kell, hogy nedves helyen raktározva vizet vesznek fel. Ezt 200—250 C°-on való gondos szárítással el kelltávolítani a magasabb hőmérsékleten való igénybevétel előtt, mert különben megrepedhetnek. Ellenálló-képességüket a gyakrabban használatos fémek, oxidok, hid-roxidok és sók olvadékával, valamint a tömény savakkal és lúgokkal szemben a42.2.2.1., 42.2.2.2. és 42.2.2.3. táblázat foglalja össze.
42.2.3. Szén- (grafit-) tartalmú anyagok
A szén (grafit) indifferens, illetve redukáló atmoszférában a legmagasabb hőmérsékleten alkalmazható szerkezeti anyag. Használhatóságának határát akarbid-képződés hőmérséklete szabja meg. Mechanikai sajátságainak javítása céljából különböző kötőanyagokat (pl. agyagot) alkalmaznak. Ennek következtében azonbanhóallósága csökken.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 22/366
T ű z á l l ó a n y a g o k
t u l a j d o
n s á g a i
4 2 .
2 .
1 .
t á b
l á z a t
22 S Z E R K E Z E TI A N Y A G O K
Pcd&>
•O.S « 2 “
Sg;®s c 8
i f M *1 1 1 2 ”'O d NM kjjj 00 mffi
9 jí| ,§
h t
^d Ed ® M © © .g;®2 3>1^O Ö-O 5fc
g •9 tí j £ ^ :I i ‘g l l - 8 .
S f l ^ s »
I á i S& §s ® S § » S2 bow S'S> « a d r ! -S *P-P
© ö' B 6 ^
•a ®ff|jsS s | g g
S tíyOJ e9V I éU ^8 | « 2 e
bo
© N:0> J4 S bO
kJT CD*»I ^©•
‘S $h
_3 <yn OS©5 io
^ IN
-P
á l1 s í «S g § 8 £ c
CB-S.&S m' o '8-íÍS •a 8 | 8i>£ g '2
s - . g SS e
c £P .Í 2 g i
a ®£ IqS8~ 0=3-02 gtS * £
S'O ®& ^ m a rá
"3 S C QD ®bp© © 08 nj«J«Í^ >'©
M
'S^T-o ® ^g 6 ® |
§3 bój ||g8l l| ö ,:3 :0 O
H ő i n g a -
d o z á s o k k a l
s z e m b e n i
e l l e n á l l á s
<sbí>S.d ©
O♦ r^bo <> c©
£boÍ2© bo '©
bű
ór -oo~*o Gs ©® .SP
s«oEh
.bp
o‘ •©ü —O g S.§>
be
OoO s j00 -í->
I*.*0 GoJoO► P.o
AEw o
Ü A
o o o o © o00 LO U3 © (N kpCD 00 00 00 00 ©
i-H (N
•o-cd
S hÍ "
•gae®4 ! 5
w s a
oo
oo
©
oo
•g^O •<ÖlO 3
Ő't'Tí > S ‘T ^ ’3o I|O .13 a >!• § ö © « oa Ő3 io O 3geo^í p n ©
obo be d d -g >>J8
a3 -£P J*a s§ s
o& T3 u tí p 3IPS 3*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 23/366
T Ű ZÁ L LÓ A N Y A G O K 23
<0 tí
'3 “ g
3 _
w „ q | *|>a'? ö za ,®J§ 'g 3
'P'o ©3 ©g,*&m 8, 3 3 »o 3a g — g>2- Sí ^ tí fc S mÜ> tí © r T fi ® Öd bO
cc
gJ3s
S Msí!- ° fi® >© © *« 3-0s 3 g % 5 scc
— -o© "T i
á g 1 3a g
ig « 01
-s.*©
1 1 - 5 1 1s© 2 ^ 0 § | s
“III-g 0,:fi :0 O
•3 M T3 ® _»g S .§ -g s
u 5 |§ 3 8 ,1 1§g?II| ÉV3 :0 O
•3^T3 ®$ B' ®Jd> .© ©,
üt!a °*:3!0
'S^l'O ®
-3 ^, T3 ~
58S®JdSfiS®^S
o •§-g x $-g2'5 8,-0 2 § 3 8)-o 2íL fi ® S £ & c ® § $5P5 > £ > 5P5 > £ £o ÍVfi :0 O a asfi JO O
3 Jd <ó-o ® . ©+a
IJfSJilfI l i i ! Hlf l l l l f ' f s
U •‘í'B
k ö z e p e s
j ó
r o s s z
e l é g j ó
j ó
k i t ű n ő
i g e n j ó
m9 s
•O >0 0 | <• "fi fi — "* «§ "* j a sJd A
© 3fi NN "O
•8 IS2. 3 ft
2 7 0 0
2 6 0 0
3 0 0 0
2 7 0 0
2 1 3 5
g y a k o r
l a t i l a g
n e m o
l -
v v a s z t -
h a t ó t n e g
1 5 0 0 C °
f e l e t t
b o m l i k
2 6
0 0
2 2 0 0
2 8
0 0
2
2 0 0
1 9
0 0
3 0
0 0
f e l e t t
1 5 0 0
Z s u g o r í t o t t
c i r k ó n i u m -
- o x i d
Z s u g o r í t o t t
b e r i l l i u m -
- o x i d
Z s u g o r í t o t t
t ó r i u m - o x i d
Z s u g o r í t o t t
m a g n é z i u m -
- o x i d
Z s u g o r í t o t t
s p i n e l l
S z ó n ( g r a f i t )
S z i l í c i u m -
- k a r b i d
M Z, ©O - t4-^ 3 X 0 . a
Z 2 Bn ö c o h o ö © áS *
°L%Í°”ó o 35t<§ © 3 © © V wPQ H fi
t i ,'ON
CG
2 .
3 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 24/366
24 S Z E R K E Z E T I A N Y A G O K
42.2.2.1. táblázat
Fémolvadékok hatása kerámiai oxidokra
Fémek c- AljOj Zr02 BeO MgO
Li (Hg-atm.-ban) 700 + + __ + + + +
Na (H2) 700 - — -
K (H2) 800 — - - —
Be (H2) 1500 + + —Mg (H.) 800 — — + + —
Ca (H2) 1000 — + + + + +
A í (H2)..................................
1000 — —
+ + + +S i(H 2) .........................v - . . . . 1600 — — + + + +
Sb 800 — —
Bi 600 — — +
Cu .......................- .................1200 — — +
Ti (H2) 1800 + + + +
fcr (H 2) 1700 + + - — -
C r .............................1900 + + + +
Cr (H2) 1900 - - -
Mn .............................................1600 + + + + + +
Mn (H2) 1600 - — +
Fe ........................................... 1600 + + + + + + +
Fe (Hs)-----
1700 - —
-Ni, Co, Pt 1600 — — —
Pb 600
- gyakorlatilag nem támadja meg-(- kismértékben megtámadja
-f + megtámadja
42.2.2.2. táblázat
Oxid- hidroxid- és sóolvadékok hatása kerámiai oxidokra
c° A1203 Zr02 BeO MgO
500 _, _
NaOH, KOH 500 •— — — -
Li2C 03 1000 - —
Na2C 03 1000 - — — —
k 2c o 3 1000 - + + —
B20 3 .............................1250 — + + — 4- 4-
SiOj 1780 + — + + + 4-
PbO 900 + + + + —
Cr2Oa ...........1900 + + + + + + 4- +
MiL jO j , Mn30 4 1600 + + + + + + + 4-FeO, Fe20 3 1600 + + 4- + + 4- H —b
N a F ......................................... 1200 + + - + +
Na2B40 - 1200 — - 4 + +
k h s o 4 500 - — —
KCN 800 +
—gyakorlatilag nem támadja meg+ kismértékben megtámadja
+ + megtámadja

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 25/366
F É M E K 25
42.2.2.S. táblázat
Tömény savak és lúgok hatása kerámiai oxidokra
C° AI2O3 Z r0 2 BeO MgO
cc. H2S 04 338 ._
+ + + + +
cc. HC1 110 + — + + ' H" +cc. H N 03 122 + + H—hCC. H 3 P O 4 160 — - + + + +
cc. H F ........... .................... 120 — — + + + +
20%-os NaOH 103 — — — —
- gyakorlatilag nem támadja meg + kismértékben megtámadja
+ + megtámadja
42.3. Fémek
Az üvegnél és porcelánnál nagyobb mechanikai követelményeknek megfelelőlaboratóriumi eszközök fémből készülnek. Jó elektromosság- és hő vezető-képességüksok esetben magas olvadásponttal és nagymértékű kémiai ellenálló-képességgel párosul. Egészen speciális tulajdonságú ötvözetek is készülnek különleges összetétellel
és rendeltetéssel. A nemesfémek sokféle felhasználásának alapja ezek ismert kémiai ellenálló-ké
pessége. Leggyakrabban az ezüstöt és a platinát alkalmazzák, azonban többnyirenem kémiailag tiszta állapotban, hanem ötvözet alakjában.
A legfontosabb laboratóriumi fémszerkezeti anyag a platina. A kereskedelmiforgalomban kapható ún. platina eszközök általában néhány tized százalék irídiumot is tartalmazó ötvözetek. Az irídiumot a színfém mechanikai szilárdságának éskémiai ellenálló-képességének növelése céljából alkalmazzák. Nagyobb szilárdságikövetelményeket magasabb hőmérsékleten is kielégítő platina eszközök 5-—30 %
iridiumtartalmú ötvözetből készülnek. A 35 %-nál nagyobb iridiumtartalmú ötvözetek törékenységük miatt már nem megmunkálhatok. Az iridiumtartalommal nőa platinaötvözetek izzítási vesztesége.
Noha a gyakorlatban használatos platinafémek tenziója és párolgási sebességenem különbözik számottevő mértékben egymástól, az oxidképződést és az oxidoktenzióját is figyelembe véve, az egyes fémeszközök izzítási vesztesége a szokásosigénybevétel hőmérsékletén Rh, Pt, Pd, ír esetében úgy viszonylik egymáshoz, mint1 :2 :6 :60. A Ru és Os még a felsoroltaknál is lényegesen illékonyabb. A platina tenziója ródiummal való ötvözéssel csökkenthető. A Pt—Rh-ötvözetek ezért fűtőszálak készítésére alkalmasak. A platinaötvözetből készült eszközök legfeljebb 1600C°-ig, az iridiumtégelyek 2000 C° féletti hőmérsékletig használhatók. Utóbbiaknálazonban ügyelnünk kell arra, hogy a 750—1140 C° hőmérsékleti közön gyorsan haladjunk át, mert az irídium itt oxidálódik.
Bár a platina savakkal (a királyvizet kivéve) és egyéb vegyszerekkel szembenigen ellenálló, használata közben néhány fontos óvatossági rendszabályt figyelembe kell vennünk. így pl. nem szabad világító, kormozó lángban hevíteni a kárbidkép-ződés veszélye miatt. Az ötvöződés és a képződött ötvözet alacsonyabb olvadáspont
ja következtében veszély áll fenn akkor, ha a hevített platinával könnyen ötvöződő

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 26/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 27/366
FÉMEK 27
42.3.2. táblázat
Korrózióálló ötvözetek
Kereskedelminév összetétel (súly %) Tulajdonság
Speciálisfelhasználás
Acidur Si-tartalmú vasötvözet savállóContracid 68-61 NI, 12-19,5 Fe, 15
Cr, 2 - 2 Mn (Mo, W , Co, Be) saválló orvosi műszerek
Ferroterm 0, 15 -2 C, 6 - 3 0 Cr,a maradék Fe hő- és korrózióálló öntvények
Illium 60 Ni, 21 Cr, 6 -Cu, 5 Mo,
2 W, 1 - 1 Mn (Si, Al, Fe) saválló kalöriméter-bombákIronac 13,2 Si, 1,1 C, 0,8 Mn,
0,05 S, a maradék Fe saválló öntvényekKaroni 13,5 Cr, 0,5 Ni, 0,5 Mn,
0,3- I S i , 0,15 C, a maradékFe
hőálló acél (800 C°-ig)
K-monelfém 63 Ni, 30 Cu, 3,5 Al,1,5 Fe, 0,2 C 6 Mn, Si, stb. korrózióálló műszerek nem-
mágneses részeiMangánbronz 94 - 95 Cu, 6 - 5 Mn vízzel és vizes sóol-
dattal szemben kor- termosztátok al-rózióálló katrészei
Monelfém 6 6 -7 5 Ni, 2 5 - 30 Cu, amaradék: Fe, Mn, Si, C, vízgőznek ellenáll kondenzátorcsö-S, P vek
NCT-acól 0,16 C, 1 5 -6 0 Ni, 1 5 - 2 5 hőálló (1200 C°-ig) öntvényekCr, a maradék Fe és korrózióálló
Újezüst 4 6 - 6 6 Cu, 1 9 - 31 Zn, korrózióálló evőeszközök13-36 Ni
Nicorros 67 Ni, 33 Cu vizes sóoldatokkalszemben rendkívül korrózióálló
hajóépítés
Nicrosilal 2 - 4 C, 18 Ni, 6 Si, 2 Cr, a maradék tecbn. Fe
hőálló, nem mágne-ses öntvények
Niresist 2 - 4 C, 1 3 - 1 6 Ni, 6 - 8 Cu,2 —6 Cr, a maradók techn. saválló, nem mágne-.Fe ses öntvények
Remanite Rozsdamentes acólötvöze-tek.pl. 16 Cr, 1 Ni, rozsdamentesa maradék Fe korrózióálló alkatrészek
V2A-acól 0,1 C, 18 Cr, 8 Ni,a maradók Fe korrózióálló acél autokláv
VM-aoél 0 , 1 - 0 , 5 C, 10 -1 5 Cr, 1 -—3 Ni, a maradék Fe korrózióálló acél szelepek

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 28/366
28 S Z E R K E Z E T I A N Y A G O K
A volfrám, mólibdén és tantál kis tenziójú és hőtágulási együtthatójú (keményüvegekbe jól beforrasztható) fémek, melyek kitűnnek nagy szilárdságukkal, és főként a vákuumtechnikában használatosak. A volfrám és a mólibdén fűtőellenállás
céljára is alkalmas (W- és Mo-keraencék), magas hőmérsékleten azonban csak védőgáz-, illetve redukáló atmoszférában (H2 vagy H2-|- N2) használható.
A fémek kémiai ellenálló-képességét leginkább a halogénekkel szemben mutatottviselkedésük jellemzi. Ismeretes azonban, hogy a halogének hatását jelentős mérték
ben'befolyásolj a azok nedvességtartalma. Teljesen száraz klór pl. szobahőfokon soknemnemes fémet sem támad meg. Nedves klórgáz viszont pl. az aranyat vagy ezüstöt szobahőfokon is megtámadja. A platina szobahőmérsékleten nedves klórgázzalszemben is indifferens, de 250 C°-on már észlelhető korrozív hatás jelentkezik, éséOO C° felett a korrózió számottevő. Az arany folyékony klórban annak forráspont
ján gyorsan oldódik. Klórral szemben a fémek között a legellenállóbb a platina-iri-dium-ötvözet, amely 400 C° alatt teljesen indifferens.Fluorral, illetve fluor-hidrogénnel végzendő műveletekhez platinán kívül vö
rösréz, magnézium, illetve monelfém (30 % Cu, 70 % Ni) használható szerkezetianyagként. A réz fluorral szemben 350 C°-ig ellenáll, a hidrogén-fluorid viszont1200 C°-ig nem támadja meg. A magnézium felületén védőréteg alakul ki, amely afluor további agresszív hatását 280 C°-ig megakadályozza. A platina fluor hatására300 C°-ig nem korrodeálódik számottevő mértékben. Az alumínium, nikkel és krómfluorral szemben csak szobahőfokon stabilis. Száraz fluorgáz szobahőfokon ezüstbőlkészült edényben is eltartható. A keményforrasz fluorral szemben eléggé ellenálló,
a lágyforrasz viszont nem. Ezért fluorgázzal érintkezésbe kerülő forrasztások csakaz előbbi felhasználásával készíthetők.
A nemesfémek ellenálló-képességéről tömény ásványi savakkal szemben a42.3.3. táblázat ad felvilágosítást.
42.3.3. táblázat
A nemesfémek ellenálló-képessége savakkal szemben (80—100 C°-on)
j A g A u P l Rh ír P d
40%-os HF "H- 37%-osHCl + — - — - -
65%-os HN03 + + — - — - 4- 4 -
96%-os H gSü4 + + — + — — 4 - 4 -
k i r á l y v í z + + + + 4 - + + — 4 " +
- gyakorlatilag nem támadja meg+ kismértékben megtámadja+ + oldja
A laboratóriumban használt fémeszközök gyakran kerülhetnek érintkezésbe
fémhigannyal, illetve higanysókkal. Mindkét esetben fennáll az amalgámképződésveszélye, különösen magasabb hőmérsékleten. A higannyal szemben teljesen közömbösek. a Fe, W, Ta, ír, Be. A fontosabb fémek olvadáspontját, tepziój át és
hőtágulási együtthatóját a, 42.3.4. táblázat tartalmazza. 1 A gázoknak a fémekben való oldhatóságára, illetve a fémek gázáteresztő
képességére szintén tekintettel kell lennünk a fémek felhasználása során. Néhány fém különböző gázokkal többé-kevésbé stabilis vegyületeket, pl. hidride

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 29/366
F É M E K 29
42.3.4. táblázat
Néhány fém fizikai tulajdonságai
OlvadáspontC°
10- ö Hgmm gőznyomásnak megfelelő hőmérsék
let, C°
Hőtágulási együttható (25 C°-on) a-107
ír 2454 1993 66Rh 1966 1681 9ÖPt 1773,5 1606 89Pd 1555 1156 106 Au 1063 767 142 Ag 960,5 1083 187W 3380 2554 44
Mo 2622 1923 53Ta 3030 24Ö7 65Zr 1860 1627 143Cu .1084 946 165Ni 1453 1157 125Fe 1535 1094 116
A1 658 724 231
két, oxidokat, nitrideket képez. Ezek a. fémek az illető gázokat nagyobb
mennyiségben kötik meg majd kisebb parciális nyomáson vagy magasabb hőmérsékleten újra leadják. így viselkedik aTi, Pd és a Ta hidrogénnel, vagy, a Cú és Auoxigénnel szemben.
A 42.3.5. táblázatban tájékoztatásulközöljük néhány fém gázáteresztő képességét 400 C°-on (1 dm2 fémfelületen, 1 mm fal-vastagságú fémlapon áthaladt hidrogénmennyisége ml/órában kifejezve abban azesetben, ha a fémlap két oldalán a gáznyo
más 0, illetve 760 Hgmm).Fémeszközök részeinek illesztése he
gesztéssel vagy forrasztással történik. Az alkalmazott fémek közül igen könnyen hegeszthető a Pt, jól hegeszthető még fújtatólángban is az Pb, Ni, Sn, Cu, Ag, Au, Fe. A W, Mo, Ta legkönnyebben elektromosponthegesztéssel egyesíthető. ^
A hegesztésen kívül gyakran előforduló fémkötési eljárás a forrasztás. Erre acélra különböző mennyiségű ólmot tartalmazó ónötvözetet használunk. A forrasztandó fémfelületeket előzetesen ZnCl2 + NH4Cl-tartalmú forrasztóvfzzel vagy gyan
tás „forrasztózsírral” gondosan meg kell tisztítani. Az említett forrasZtóötvözetekolvadáspontja 180-250 C° körül van, azért ezeket lágyforrasznak nevezzük.Magasabb hőmérsékleten olvadó forrasztások készülnek ezüstöt és rezet tartal
mazó keményforrasz-szál. Ezek használatához a felület megtisztítására bóraxolvadékszolgál.
Fémeknek üveg-, illetve porcelán felülethez való forrasztása különösen a vákuumtechnikában nagy jelentőségű. A gyakorlatban ezt úgy végzik, hogy az üveg
42.3.5. táblázat
Fémek gázáteresztő képessége400 C°-on
Fém Gáz Áteresztőképesség
Pd h 2 18 000Fe h 2 14Ni h 2 6
Pt h 2 0,13Cu ' H 2 0,041 Ag 0 2 0,015

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 30/366
30 S Z E R K E Z E T I A N Y A G O K
vagy porcelán felületére grafitot visznek fel vagy platinát égetnek rá, amelyre né
hány tized mm vastagságú rezet vagy ezüstöt galvanizálnak. Ehhez.a legtöbb fémegyszerű lágyforrasszal köthető.
42.4. Műanyagok
A korszerű laboratóriumi gyakorlatban egyre nagyobb jelentőséget kapnak aműanyagok. Azonfelül, hogy kémiai ellenálló-képességük kiváló, általában jól megmunkálhatok és hegeszthetők. Különböző laboratóriumi edényeket, ventillátoro
kat, szűrőket, csöveket, tömítőgyűrűket, kesztyűket stb. készítenek belőlük.
Poli(vinil-klorid) /PVC). Lágyító nélkül mint kemény, lágyítókkal pedig mintlágy PVC-t alkalmazzák. Az egyéb klórtartalmú, nagymolekulájú polimerekhez hasonlóan fény és hosszabb ideig tartó melegítés (200 C°) hatására bomlik. Ez a tulaj
donsága megszabja feldolgozásának és alkalmazásának körülményeit. Lágyulási
hőmérséklete 60 —80 C° között van. Mechanikai igénybevételre ennél magasabb hő
mérsékleten nem alkalmas. 180 —200 C°-os levegőáram bán hegeszthető. A PVC neméghető, oxidáló tömény savaknak: krómkénsavnak, klórgáznak, ózonnak, számos
szerves oldószernek (pl. benzinnek) ellenálló anyag, vízfelvevő-képessége 0,05 %-nál
kisebb. A lágyított PVC kémiai ellenálló-képessége általában kisebb, és a lágyítóSzer
minőségétől is jelentős mértékben függ.1
Polisztirol (trolitul). Kiváló elektromos szigetelő, átlátszó, fényálló, 85 C°-onlágyul, benzolban jól oldható, tehát ragasztható. A belőle készült edények jól eva-kuálhatók, és folyékony levegővel hűthetők. Salaknak — HF-nak is —, továbbá
lúgoknak is ellenálló. Polietilén. 120 C°-ig használható, konc. savaknak és lúgoknak ellenáll. A halo
gének és a füstölgő kénsav megtámadja. Poli(tetrafluor-etilén) (teflon)..Kb. 320 C°-ig mutatja a műanyagok kiváló ké
miai tulajdonságait. Valamennyi savnak — így HF-nak és király víznek is — még
forráspontjukon is ellenáll. A kénsav 300 C°-on felül csak enyhe duzzadást és súly-növekedést idéz elő rajta. K 2S20 7-olvadéknak vagy KOH —NaOH-olvadéknak
350 C°-ig ellenáll. Csupán az alkálifémek és a Na20 2-olvadék roncsolja el magasabb
hőmérsékleten.Szilikonokból is készül kaucsukhoz hasonló anyag (pl. „Silatic ” , Siloprén” stb.)
amely rugalmasságát —90 C°-tól -f 200 C°-ig; megtartja, kémiai ellenálló-képessége
ugyancsak kiváló. Az utóbbi' időben sikerült a szilikongumi és a teflon előnyös tulajdonságait
egyesítő, sőt e komponensek hőállóságát meghaladó stabilitású kombinációs anya
got előállítani.Szerves polikarbonátok. Kiváló mechanikai tulajdonságaik vannak; 300 C°-ighasználhatók. Híg ásványi savakkal szemben rendkívül ellenállók, viszont lúgok
(6 %-os KOH, NH4OH) és szerves oldószerek (alkohol kivételével) megtámadják.Gyakorlati szempontból igen előnyös tulajdonságuk, hogy átlátszóak. Alkalmazá
suk az utóbbi időben terjedt el. Polimetákrilál (plexiüveg). Igen jó elektromos szigetelőanyag (fajlagos vezető-
1 Carlowitz, B .: Kunststoff-Tabellen. Schiftmann, Bensberg-Frankenforst, 1963.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 31/366
M Ű A N Y A G O K — K I T T E K 31
42.4.1. táblázat
A vulkanizált gumi relatív gázáteresztő képessége és a gázok oldha
tóságára vonatkozó adatok (20 C°-on)
képessége < 10“ 12 ljQ cm). Átlátszósága és az üvegnél nagyságrendekkel nagyobbütési és hajlítási szilárdsága miatt kiválóan alkalmas védőfalak, manipulációs szek
rények stb. készítésére. 30 %-os H2S04, 20 %-osH N03, 10'%-os HC1, 75 %-os HF,valamint 50 %-os KOH nem támadja meg. Szén-tetrakloriddal, alkoholokkal,benzollal szemben viszont nem ellenálló.Folyékony klór gyorsan ronc.sölja.
Gumi (kaucsuk). Régebben sokkal nagyobb jelentősége volt a laboratóriumi technikában. Rugalmassága és viszonylag jó kémiai ellenálló-képessége miatt főként csövek,dugók, tömítések készülnek belőle. Bár a
korszerű műanyagok a gumi valamennyi tulajdonságát túlszárnyalják, a laboratóriumimunkában a vulkanizált gumit ma is széleskörűen alkalmazzuk. Rugalmassága a hőmérséklet függvényében erősen változik,gyakorlatilag —20 és + 150 C° között használható. Levegő és ultraibolya fény, illetveózonnyomok hatására törékennyé válik.Hosszabb ideig sötét, hűvös helyen ammónia-atmoBzférában vagy glicerines víz alatt tart
ható el. Lúgok, halogének, oxidáló savak,apoláros oldószerek megtámadják. Olajok és zsírok hatására duzzad. Speciálisfajtái, mint pl. a neoprén, perbunán, stb. olajokkal szemben is ellenálló.
A gázokat a vulkanizált gumi észrevehető mértékben oldja, de az egyensúlyiállapot beállásához hosszabb idő, szobahőfokon kb. 1 nap szükséges. Természetesenmindazon gázok, illetve folyadékok, amelyek a gumiban oldódnak, bizonyos mértékig áthatolnak azon. A 42.4.1. táblázatban különböző gázok gumiban való oldhatóságára és a vulkanizált gumi relatív gázáteresztő képességére vonatkozó tájékoztató adatok találhatók. Megjegyzendő, hogy a P2Os felett kiszárított gumi porozi-
tása jelentős mértékben nagyobb. Különböző tömítő anyagokkal viszont a vulkanizált gumi gázáteresztő képessége nagymértékben csökkenthető.
Gáz
Relatívgáz
áteresztőképesség
Gázok oldhatóságavulkanizált gumi
ban (téri. %-ban,normálállapotravonatkoztatva)
N H , 50 930co2 18 100c h 4 2,2 25
O* 2,8 7,3co 1,3 6,2n 2 1,0 3,5
h 2 6,2 1,0He 4,0 1,0
42.5. Kitt-tömítések
A gázokkal végzett laboratóriumi munkában és a vákuumtechnikában használatos ragasztások.
42.5.1. Irreverzibilis kittek
Nagy mechanikai szilárdságuk, tűzáUóságuk és specifikus kémiai ellenálló-képességük miatt előnyösek.
A vízüvegből készült kittek nem vízállók, de kitűnően tapadnak az üveghez ésmég vörösizzáson is használhatók.
Ha finoman porított talkumból vagy azbesztből, és esetleg ZnO-ból vagy MgO-ból vízüveggel' sűrű pépet készítünk, a kapott kitt néhány órán belül megdermed.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 32/366
32 S Z E R K E Z E T I A N Y A G O K
Őlom-oxid — glicerin. 40 g sárga ólom-oxidot rövid ideig 200 C°-ra melegítünk.Lehűlés után 10 ml igen tiszta glicerinnel sűrű péppé dörzsöljük. Teljesen tiszta, zsír
talanított üveg, porcelán, fém- és fafelületekhez tapad. Célszerű a ragasztandó felületeket tiszta glicerinnel megnédvesíteni. Kb. %óra alatt köt. 260 C°-ig használható,lúgokkal leoldható.
Cink-oxid — cink-ldorid. 60 %-os ZnCl2-oldathoz kevert karbonátmentes (frissen kihevített) ZnO néhány perc alatt alaktartó és kémény, polírozható felületű tömeggé szilárdul.
Tűzálló kitt készíthető 90 % kaolin és 10 % bórax keverékéből kevés lenolaj vagyvíz hozzáadásával. A kittel összetapasztott helyet lassú szárítás után gyenge vörös-izzásig kell hevíteni.
Műanyag kittek. Különböző műgyanták, mint a Hostacol, Araldit stb. megolvasztás előtt néhány tized százalék keményedést gyorsító katalizátorral keverve,fémhez és üveghez igen jól tapadó kittet szolgáltatnak.
42.5.2. Reverzibilis kittek
, Az ide sorolható kittek mechanikai szilárdsága csekély. Általában enyhe melegítésre megolvadnak, lehűtve rideggé és törékennyé válnak. Kémiai ellenálló-képességük is gyenge. Savakkal, lúgokkal, organikus oldószerekkel, sőt még meleg vízzelszemben sem teljesen ellenállók. A ragasztott felületről megfelelő oldószerrel köny-nyen eltávolíthatók. Legfontosabb felhasználási területük a vákuumtechnika.
Picein. Legfontosabb szerepe olyan kötések létrehozásában van, amelyék forrasztással nem egyesíthetők. Fekete színű, kaucsuktaftalmú anyag. Kb. 50 C°-onlágyul, de szobahőmérsékleten is kimutathatóan plasztikus. 80, illetve 105 C°-on olvadó minőségben is készül. Magasabb hőmérsékleten szénkiválás közben bomlik. Tenziója 20 C°-on 3 •10“ 4 Hgmm. Vízben, alkoholban nem, benzolban, szén-tet-rakloridban jól oldható.
Színtelen pecsétviasz. Szobahőmérsékleten szilárdabb, de ridegebb is, plint apicéin. Tulajdonságai és alkalmazása egyébként a piceinhez hasonló.
Apiezon-viasz. Alacsony tenziója miatt (20 C°-on 5 •10-7 Hgmm) a vákuum-technika számára fontos. Szobahőmérsékleten is képlékeny. Oldószere a xilol. Bekhotinsky-cement. Üvegre és fémekre a picémnél erősebben köt. Tenziója
20 C°-on 2* 10-3 Hgmm. 60—80 C°-on lágyul. Melegítve fenolra emlékeztető szagú. Kaucsukaszfalt (Marineleim). 2 rész aszfaltot és 1 rész nyerskaucsukot petróle
umban oldva és (olajfürdőben) 140 C°-on melegítve, klórnak, savaknak ellenálló kittet kapunk.
AgCl-kitt. Megolvasztott AgCl (op. 455 C°) jól tapad üveg-, kvarc- és fémfelületekhez (Ag, Pt, Mo, W), azonban nem megbízhatóan vákuumtartó. Vízben, savakban, organikus oldószerekben nem oldódik, tioszulfátban, cianidban és ammóniábanigen. 71,5 % TaCl-dal keverve, alacsonyabb olvadáspontú elegyet (op. 210 C°) ad,mely lehűlve a tiszta AgCl-kittnél elasztikusabb.
Üvegalapú kitt. Tulajdonképpen alaesony (350 C°) olvadáspontú, nagy ólom--oxid-tartalmú üveg. 5 g finom eloszlású Si02, 16 g mínium és 4 g vízmentes bóraxelegyét porcelán tégelyben hígan folyó olvadékká ömlesztve homogenizáljuk. Az olvadékot V alakúra hajlított fémlemezre öntve, óvatosan lehűtjük.,.Btt»sett-lángbankönnyen elfölyósítható. Jól tapad üveg, vas, réz és sárgaréz felületeken, alumíniumon és kvarcon nem.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 33/366
Z S Í R O K 33
42.6. Zsírok
A laboratóriumi technikában használatos kenőanyagok. Alkalmazásuk céljakettős: egyrészt — különösen a vákuumtechnikában — a gázok diffúzióját.akadályozzák meg a csiszolatok mentén, tehát tömítésül szolgálnak, másrészt viszontaz üvegcsiszolatok korrózióját, illetve^azok „beragadásának” veszélyét csökkentik.
A vákuumtechnikai célokra használatos csapzsír legfontosabb tulajdonsága akis tenzió. Ezzel a megfelelő fejezetben részletesen foglalkozunk (lásd 46.4.2).
A laboratóriumi gyakorlatban alkalmazható zsírok kiválasztásánál tekintettelkell lenni azok ellenálló-képességére. A közhasználatú Ramsay-zBÍi (összetételétlásd. 46.4.2.), valamint a szilikoncsapzsír nem megfelelőek abban az esetben, haszabad halogének vagy más agresszív gázok, pl. HBr, BC13, BF3, COCl2, B2H6 stb.
hatásának lehetnek kitéve.Ha halogének vagy nitrogén-oxidok jelenlétére szállíthatunk, akkor a következő kenőanyag készítését javasoljuk. Sztearin és paraffin 3:2 arányú keverékét150 C°-on klórozzuk, majd NOC1 hatásának tesszük ki, végül vákuumban gáztala-nítjuk.
Meglehetősen ellenálló halogénekkel szemben a tiszta vazelin, vagy a szilárdparaffinból és paraffinolajból készült zsír is. Utóbbiak fluor jelenlétében is használhatók.
Amennyiben csekély mennyiségű vízgőz nem okoz zavart, agresszív gázok jelenlétében szirupsűrűségű foszforsav vagy metafoszforsav is jól használható.
Metafoszforsav alapú kenőanyag készítésére alkalmas eljárás: 10 g HP03-atés 2 g H3B 03-at 100 ml vízben intenzív keverés közben oldunk, majd az oldatot25 ml-re bepároljuk. Ezután 1 ml 85 %-os H3P 0 4-at adunk hozzá, és keverés közbena bepárlást addig folytatjuk, míg az oldat forráspontja el nem éri a 122 C°-ot. Azátlátszó, kissé higroszkópos, viszkózus kenőanyag kristályosodás veszélye nélkülhónapokig eltartható.
A tömény kénsav is használatos kenőanyagként, különösen agresszív gázok jelenlétében, bár kis viszkozitása nem előnyös.
Szerves anyagokkal dolgozva, amelyek általában a közhasználatú zsírokat jól oldják, zsírmentes ke nőanyagokat alkalmaznak. Éterrel vagy benzollal történő lúunka esetén Kapsenberg nyomán áz alábbi kenőanyag használata célszerű:25-35 g dextrint 35 ml glicerinnel porcelán tálban jól összekeverünk, majd az elegyetintenzív keverés közben felmelegítjük. A szirupsűrűségű anyagot a habzás megindulásáig kétszer egymás után felhevítjük, majd még melegen vattán átszűrjük. Az így kapott kenőanyag valamivel ridegebb, mint a vazelin, és higroszkópos.Megjegyzendő, hogy alkohol, aceton, piridin, ecetsav és anilin esetében a fenti kenőanyag nem használható.
Zsírmentes kenőanyagként általában jól alkalmazható a nagy diszperzitásfokú
grafit, illetőleg annak megfelelően választott oldószerben való szuszpenziója.Szükség esetén kenőanyagok alkalmazását higanyzáras csapok és csiszolatok
használatával kerüljük ki.Magasabb hőmérsékleten alkalmazható kenőanyagok: 200 C°-ig a szilikonzsír,
e hőmérséklet felett pedig olvasztott sók (kevés kaolinporral keverve), mint pl.KSCN 175-320 C° között, vagy KNOa 340 — 360 C° között.
3 Ált. és szervetlen kémiai prakt. — 4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 34/366
S Z E R K E Z E T I A N Y A G O K 34
42.7. Parafa, papír és gyapot•
A laboratóriumi munkában gyakran használt nyersanyagok közé tartozik aparafa (dugó), a papír (szűrő) és a gyapot. A parafának sok szerves anyaggal szemben nagyobb a kémiai ellenálló-képessége, mint a vulkanizált guminak, azonban te
kintettel kell lennünk arra, hogy a terpentinolaj, valamint az alkoholgőzök a parafát megtámadják. Hasonlóan hatnak a tömény kénsav, tömény salétromsav, a halogének és a tömény lúgok is. Kémiai ellenálló-képességének növelését és a gázát
eresztő képességének csökkentését 120 C°-os paraffinban történő áztatással, majdsztaniolpapírba való csomagolással biztosíthatjuk.
A különböző szűrőpapírjajták kémiai ellenálló-képessége és alkalmazhatóságiterülete nagymértékben függ azok nyersanyagától és készítési módjától. Töménysavak és lúgok, különösen melegen, csökkentik a szűrőpapír mechanikai szilárdsá
gát és annak szétmálását idézik elő. A szűrőpapír meglehetősen higroszkópos, lég-száraz állapotban 5-6 súly % adszorbeált vizet tartalmaz, amire alkalmazása során
tekintettel kell lenni. A gyapot (vatta) szűrésre vagy hordozóanyagként (pl. szárítóanyagok esetében
a tömítés elkerülésére) használatos. Tömény savak, sőt melegen híg savak is gélszerű anyaggá roncsolják szét. 5%-osnál töményebb lúgok hidegen is megtámadják.
Irodalom
Fr i ed r i chs , F . : Das Glas im chemischen Laboratórium, Jakobs, Rheydt, 1951.Mo r e y , O. W . : The properties of glass. Reinhold Publ. Corp., Ne w York , 1938.
Kovács L.\ Műanyagzsebkönyv. Hűszaki Könyvkiadó, Budapest, 1959.J l ener oe, B . A . : PeaHHOBhxe TexHHMecKHe M3geJinji. rocxMMK3flaT, MocKBa, 1959.Sa lmang, H . : Die physikalisohen und chemischen Grundlagen dér Keramik. Springer,
Berlin, 1961.Micksch , K . : Taschenbuoh dér Kitté und Klebstoffe. W iss. Verlag., Stuttgart. 1952. Saechbli ng —Zebr owsk i : Kunststoff-Taschenbuch. Hanser, München, 1959.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 35/366
43 . Alacsony hőm érséklet előállítása
43.1. Hőszigetelők
A 0 C°-nál alacsonyabb hőmérsékleten végzendő műveleteknél elsősorban ahőszigetelés kérdését kell megoldanunk. Erre a célra leginkább a rossz hővezető-
képeBségű, porózus állapotú anyagok alkalmasak. Ilyenek pl. a parafaliszt, fűrész-por, gyapot, üvegvatta, azbeszt, különböző műanyag habok, pl. Iporka stb. Ezek
hővezető képessége kb. 0,028 kcal/m*óra*fok. Ennél is kb. 30 %-kal kisebb a szárazlevegő hővezető-képessége. Viszonylag jó hőszigetelő hatást lehet elérni tehát légköpennyel. Valamennyi felsorolt anyagnál jobban szigetel azonban a vákuumkö
peny. Az ilyen ún. Dewar-iéke kettősfalú edény hőmérséklet-változást jól bíró
üvegből készül, és a két fala közötti térben a maradéknyomás (vákuum) kb. 10-5Hgmm. Ezen a nyomáson a levegő vezetőképessége elhanyagolható, hővezetés
csupán a külső és belső üvegfal összeforrasztásának helyén, az üvegnyakon át lép
fel. A Detüar-edények nyakát éppen ezért le szokták szűkíteni, hogy a vezetőkeresztmetszet kisebb legyen. A hőszigetelő hatást tovább növelni a hősugárzás
csökkentésével lehet. Ebből a célból a vákuumköpeny belső felületét ezüst- vagyréztükörrel vonják be.
A légköpenyes edény hőszigetelő képessége az evakuálás hatására kb. háromszorosára, ezüstözéssel pedig további tízszeresére nő.
Ha a hűtött tér belsejének megfigyelésére van szükség, akkor az ezüstözésben
néhány milliméter széles átlátszó csíkot hagynak. Az edény hűtőközeggel valómegtöltésekor ajánlatos a védőszemüveg használata, mert az evakuált tér impló-
ziója következhet be. Az összeforrasztás helyén megmaradt üvegfeszültségek miatt
az edény nyakát óvni kell a hirtelen hő változástól. A Dewar-e dényeket fém- vagyfatokkal szokták ütődés, illetve karcolódás ellen is védeni.
Liternyi térfogatú gázok rövidebb ideig tartó szállítására fémből készült kettős falú edényeket használnak.
43.2. Hűtőanyagok
A hűtőanyag megválasztását az elérendő hőmérséklet szabja meg.
43.2.1. Hűtőkeverékek
0 C°-tól kb. —50 C°-ig terjedő határok között jeget, illetve különböző víztartalmú hűtőkeverékeket alkalmazhatunk.
Kriokidrátok. Ha a rendszer hőmérsékletét a jég olvadáspontja alá, de nempontosan definiált hőmérsékletre akarjuk csökkenteni, akkor úgy járhatunk el,
hogy az apróra tört jeget valamely sónak az elérendő hőmérséklettől függő mennyiségével egyszerűen összekeverjük. Az ily módon elvileg elérhető legalacsonyabb
3*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 36/366
36 A L A C S O N Y H Ő M É R S É K L E T E L Ő Á L L ÍT Á S A
hőmérséklet a kriohidrát olvadáspontja. Amennyiben a kriohidrát olvadáspontját
a hőmérséklet stabilizálására is fel akarjuk használni, akkor a megfelelő összetételű oldatot más hűtőeljárással meg kell fagyasztani. A leggyakrabban használt kriohidrátok:Jég—konyhasó. 33 rész finomra porított techn. nátrium-kloridot 100 rész hóval
vagy apróra tört jéggel összekeverünk. Az eutektikum összetétele: NaCl-2 H20,op. —21,2 C°.
Jég—MgCl2. 84 rész MgCl2*6 H20 - f 100 rész jég egyszerű összekeverésével—27 C° hőmérsékletet érhetünk el. Az eutektikum összetétele: MgCl2*12 HaO.
Jég—CaCl2. Elérhető hőmérséklet —40 C°. Keverési arány: 143 rész CaCl2*•6 H20 + 100 rész jég. Az eutektikum olvadáspontja —55 C°.
Tömény savas hűtőkeverékek. Hatásuk azon alapszik, hogy hígítéshőjükugyan exoterm, de lényegesen kisebb, mint a jég endoterm olvadáshője. Ilyenpl. a 2 rész jég, 1 rész cc. HN03 elegye. Elérhető hőmérséklet —56 C°.
Jég nélkül előállítható hűtőfürdók. A jelentékeny endoterm oldáshőjű sók közülazok, amelyek nagymértékben oldhatók is, alkalmasak a környezet hőmérsékletének leszállítására. így pl. 10 C°-os víz hőmérséklete azonos súlyú NH4N03-tal—20 C°-ra szállítható le, vagy 2 rész NH4SCN és 1 rész +10 C°-os víz —25 C°-osoldatot ad.
A 43.2.1.1 táblázatban szerepel még néhány használatos hűtőfürdő és azáltaluk elérhető hőmérsékleteséB.
43.2.1.1. táblázatHűtőfürdők
összetétel Hőmérsékletesés, C°
100 g víz, 90 g CH ,CO ONa3 H ,0 15,4100 g víz, 30 g NH 4C1 18,4100 g víz, 65,2 g NaNOa .................. 18,5100 g víz, 250 g CaClj-6 H20 .....................100 g víz, 38 g NajSOj-10 HtO, 23,5 g NaNOj, 23,5 g N H 4C1
23,2
25100 g víz, 60 g NaNOa ..................................
100 g víz, 100 g NH 4NO a27,230
100 g víz, 200 g N H 4SCN 35
43.2.2. Szénsav hő
A —50 CP-ná\ alacsonyabb bőmérsékletek előállítására (kb. —100 C°-ig) leggyakrabban a szilárd szén-dioxidot alkalmazzák. Előnye, hogy igen olcsó és szükségesetén a laboratóriumban használatos cseppfolyósított szén-djoxidból is előállítható,továbbá, hogy maradéktalanul elillan.
Lefelé fordított acélpalackból a folyékony szén-dioxidot vastag, lyukacsos falúzacskóba fejtjük. A nagy felületen bekövetkező gyors elpárolgás következtébena COa visszamaradó része megfagy. Az így előállított szénsavhó finom eloszlású,gyorsan szublimál. A kereskedelmi forgalomban kapható szárazjég ugyancsak szilárd szénsav, de nagy nyomással kemény tömbökké préselik, hogy gyorsabb párolgását a felület csökkentésével megakadályozzák. Az ilyen szénsavtömbök papírbacsomagolva napokig, Dewar-edényben pedig akár hetekig is eltarthatók. A szilárdCOa a bőrfelületre jutva néhány perc alatt égési sebhez hasonló sérülést okoz.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 37/366
HŐTÖAHYAGOK 37
43.2.2.1. táblázat
Szilárd szén-dioxidos hűtŐkeverékek
összetételHőmérséklet
C°
Szil. CO, — alkohol................ - 72-Szil. C 02 — kloroform - 7 7Szil. COj — éter ..................... - 7 7Szil. COj — folyékony SOs . . - 8 2Szil. C 02 — aceton - 8 5
Hűtőberendezésben való alkalmazásakor a hőátadás növelése érdekében kémiailag indifferens folyadékkal hűtőkeverékeket készítünk. Az elporított szénsav
jeget alacsony fagyáspontú folyadékkal híg péppé keverjük. Az alkoholos, éteres,acetonos stb. hűtőkeverékkel elérhető hőmérsékletek felsorolását lásd a 43.2.2.1.táblázatban. A C02 szublimációs pontja 1 atm nyomáson —78,5 C°. A parciálisnyomás csökkentésével ez a hőmérséklet csökkenthető, pl. 20Hgmm-en—116,7 C°.
43.2.3. Folyékony levegő
—100 C° alatti hőmérsékletek megvalósítására (kb. —200 C°-ig) folyékonylevegőt, illetve nitrogént alkalmazunk. A cseppfolyós levegő forráspontja párolgása
43.2.3.1. táblázat
Oxigén—nitrogén-elegyek forráspontja
Mól % ForráspontC®
Mól % ForráspontC«
0 , N* O, N*
0 100 -19 5 ,8 0 50 60 -19 1 ,895 96 - 195,57 55 45 -191,29
10 90 -195 ,28 60 40 -190 ,6715 85 - 194,95 65 35 -1 89 ,9320 80 - 194,59 70 30 -189 ,1726 76 - 194,22 75 26 -188 ,3030 70 -193,82 80 20 -1 87 ,4336 65 -193 ,39 86 15 - 186,4640 60 - 192,92 90 10 -185 ,3545 66 - 192,40 96 6 -184,21
100 0 - 182,97
közben állandóan változik, tekintve, hogy a nitrogén forráspontja (—196 C°) azoxigénénél (—183 C°) alacsonyabb (43.2.3.1. táblázat). így a Dewar-edényben visz-szamaradó folyékony fázis idővel oxigénben egyre inkább feldúsul, ami nagy fajlagos felületű éghető anyagokkal való érintkezésnél robbanást okozhat. (Lásd:folyékony levegőt tartalmazó robbanóanyagok, pl. oxilikvit.) A tiszta N2 forráspontja alatti hőmérséklet a parciális nyomás csökkentésével érhető el. így pl.H2-gáz gyors átbuborékoltatásával a hőmérséklet —200 C°-ra szállítható le.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 38/366
38 A L A C S O N Y H Ő M É R S É K L E T E L Ő Á L L ÍT Á S A
A folyékony levegő felhasználása közben fokozottan vigyázni kell a hőpalackok érzékeny Össze-forrasztási helyére. Nem ajánlatos a hideg folyadéknak a lényegesen magasabb hőmérsékletű edénynyakán át való kiöntése. Az áttöltésre a 43.2.3.1.
ábra szerinti adagoló berendezés használható.
43.2.3.1. ábra.Folyékony levegő éttöltésére alkalmas készülék
Irodalom
van Lammeren, J. A.: Technik dér tiefen Temperaturen. Springer, Berlin, 1941.Z)’ Ans, J és Lax, E.: Tasohenbuch für Chemiker und Physiker. Springer, Berlin, 1949. nepnAtMüH , B. H. : KpaTKHü cnpaBOMHHKxhmhk3. rocxHMH3aaT, MocKBa, 1954.
Nikolski, B. P .: Handbuchdes Chemikers, Bd. I, II, III. VEB Verlag Technik, Berlin, 1966-1959.
Preisich M .: Vegyészek zsebkönyve. Műszaki Könyvkiadó, Budapest, 1959. öarlowitz, B .: Kunststoff-Tabellen. Schiftmann, Bensberg- —Frankenforst, 1963.
Plath, E., Plath, L.: Tasohenbuch dér Kitté und Klebstoffe. Wissensohaftliche V. Stuttgart, 1963.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 39/366
44. Magas hőmérséklet előállítása
A kísérleti technikában szükséges magasabb hőmérsékletek előállítására általában az exoterm gázreakciók hőjét vagy az elektromos energiát használják fel. A különböző kemencetípusokban a munkatér hőmérsékletét a megfelelő fű
tésen kívül alkalmas hőszigeteléssel kell biztosítani.
44.1. Gáztüzelésű kemencék
Az elérhető hőmérséklet nagymértékben függ a tüzelésre felhasznált gáz égés
hőjétől. A leggyakrabban használt világítógázt levegőben elégetve kb. 1300 C°,
oxigénnel keverve 1800 C° érhető el. Durranógáz-kemencében 2000 C°, acetilén(Dissous-gáz) —oxigén-keverékkel 3200 C° hőmérséklet valósítható meg.
A legprimitívebb hőszigetelő berendezés az egyszerűsamottköpeny (44.1.1. ábra). Lényegesen jobb a 44.1.2. áb
rán vázolt oldaltüzelésű kis tégelykemence hőszigetelése.Utóbbi termolit téglából házilag is kifaragható. Fújtatóláng-gal(világítógázzal) fűtve 1650 C° is elérhető vele.
Magasabb kalóriájú fűtőanyaggal hasonló berendezésben2000 C°-on felüli hőmérséklet is megvalósítható, de ilyen eset
ben a tűzteret megfelelőmagas olvadáspontú
anyaggal (MgO, Zr02)kell kibólelni.
Használatosak mégkülönböző rendszerűpetróleumtüzelésű labo
ratóriumi kemencék. Az
elérhető legmagasabbhőmérséklet oxigén
áramban 2600 C°.Ha az elvégzendő
műveletet mégha táro-
44.1.1. ábra. Samott- köpeny
44.1.2. ábra. Tégely kemence termolit téglából
zott gázatmoszférában kell végrehajtani, akkor célszerűen különböző átmérőjűüveg- vagy porcelán csöveket használunk. Kvarcot hidrogéntartalmú anyaggal
fűtött csőkemencében nem alkalmazhatunk, mert a kvarc magasabb hőmérsékleten hidrogénre áteresztővé válik.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 40/366
40 M A G A S H Ő M É R S É K L E T E L Ő Á L L Í T Á S A
44.2. Elektromos kemencék
Fő előnyük a gáztüzelésű kemencékkel szemben, hogy könnyebben szabályozhatók és korrodeáló hatású égéstermékeket nem fejlesztenek.
44.2.1. Ellenállás-kemencék
A különböző hőmérsékleti tartományokban célszerűen alkalmazható fűtőhuzalokat, ill. pálcákat és a velük megvalósítható maximális hőmérsékleteket a44.2.1.1. táblázatban foglaljuk össze.
44.2.1.1. táblázat
Fűtőtestek ellenállás-kemencéhez
A fűtőtest (huzal) minőségeMax. üzemi
hőmérsékletC°
Konstantin (64% Cu, 46% Ni) 600Króm-nikkel (20% Or, 8 0 % .Ni) ................ 1150Kantél (30% Or, 65% Fe, 5% Al) 1300
Platina ................................................................ 1300Platina-ródium (70% Pt, 30% Rh) 1700
Szilit 1450
G rafi t.................................................................... 2500Molibdén (redukáló atmoszférában vagy
vákuumban) ....................................................
Volfrám (redukáló atmoszférában vagy2000
vákuumban).......................%.......................... 3000
A vezeték (pálcák) megfelelő elhelyezésével különböző alakú munkaterek (té-
gelykemence, tokos kemence, csőkemence stb.) fűtése oldható meg.Ellenállás-kemencék készítése házilag is könnyen megoldható. Az alábbiakbanegy példán bemutatjuk az igen gyakran használt csőkemencék tervezését és készítését.
A célnak megfelelő méretű csőre tekercseljük a fűtőhuzalt. Fűtőcsőként üveg,alumínium (500 C°-ig), vas- (700 C°-ig), kvarc- (1500 C°-ig), ill. 1600 C°-nál magasabb hőmérsékleten porcelán, szillimanit, szinterkorund csövek használhatók. A szigetelőanyagból készült csövekre a fűtőhuzalt közvetlenül rá tekercselhetjük. A sima felületű üveg- vagy kvarccsövekre célszerű a cső hosszában, egymástólnéhány cm-riyi távolságra helyezett azbesztzsinórra tekercselni a fűtőhuzalt, amivel
megakadályozhatjuk a fűtőhuzal elcsúszását és érintkezését (lásd 74.5.2.2. ábra).Közepes hőizoláciő esetén, ami a fűtőcsőnél egy-két cm-rel nagyobb átmérőjű porcelán vágy üvegcsőbe helyezéssel, vagy három-négyszeres azbesztzsinóros tekercseléssel érhető el, a kívánt hőmérséklettől függően az alábbi teljesítményű fűtőszálakalkalmazása célszerű:
300 C°-ig dm2-enként 20 W, minden további 100 C°-ra egészen 700 C°-ig további20 W dm2-enként, 700—1000 C° között 100 C°-onként újabb 30 W/dm2, végül1100-1300C0 között 100 C°-onként már további 40 W/dm2 teljesítmény számolható.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 41/366
k e m e n c é k 41
A rendelkezésre álló áramforrás feszültsége és a szükséges teljesítmény ismeretében kiszámítható a megfelelő áramerősség. Biztonság kedvéért a számításoksorán a névleges feszültségnél 10 %-kal kisebb feszültséget vegyünk figyelembe,
így 220 V esetén 200 V-tal kalkuláljunk. Példaként válasszuk egy 2 cm átmérőjűés 30 cm hosszú munkatérrel rendelkező, 1000 C°-ra fűthető laboratóriumi csőke
mence készítését. A fűtendő felület 2 mm = 2>1-3,14.3 = 2 dm2. 1000 C°-ra valófelfűtéshez az előbbiekben vázolt empirikus szabály értelmében 180 W/dm2, tehátösszesen 360 W szükséges. így az alkalmazható áramerősség 360/200= 1,8 A, és a
fűtőhuzal ellenállása R = V j I = 200/1,8=110 Ohm. A közhasználatú fűtőhuzalok
maximális terhelhetősége átlagosan 0,6 A/0,1 mm átmérő (1 mm átmérőjű huzalesetén 6 A) . 1 m hosszú fűtőhuzalok ellenállása kb. 1,3 Ohm /mm2. így pl. 0,4 mm
átmérőjű huzal ellenállása: l,3/0,22:t = 1,3/0,125= 10,5 Ohm/1 m huzalhossz.Következésképpen az adott huzalból 10,2 m szükséges, mivel annak ellenállása a
számított 110 Ohm. A huzal átmérőjének kiválasztása során tekintettel kell lennünk a munkatér méreteire is. Célszerű kb. 2 mm-es tekercselési távolságot alkal
mazni, s mivel a példában szereplő (2 cm 0 -jű) fűtőcső kerülete 6,5 cm, és amenetszám 150 (a munkatér hossza 30 cm), a szükséges huzal hossza kb. 10 m-re
tehető. A számítások alapján tehát megállapítható, hogy a 0,4 mm 0 -jű huzal —amennyiben 2 mm-es tekercselési távolságot kívánunk elérni — a megadott spe
cifikáció szerinti csőkemence készítésére alkalmas.
44.2.2. Ivfénykemencék
Ha a megfelelő intenzitású elektromos ív környezetét kellően magas olvadás
pontú szigetelővel vesszük körül, akkor 3 —4000 C° közötti hőmérsékletű munkatéris előállítható. Házilag is elkészíthető a 44.2.2.1. ábrán látható egyszerű berende
zés, amelynek lényege egy elektródul is szolgáló grafittégely és a vele szemben elhelyezett süllyeszthető
szénelektród. Szükség esetén a grafittégelyt vi-rágcserépbeillesztett szénrúd is helyettesítheti. Ez a megoldás azonban célszerűen csak akkor alkalmazható, ha a termék
kvarcablak
44.2.2.1. ábra. í vfénykemence

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 42/366
42 M A G A S H Ő M É R S É K L E T E L Ő Á L L Í T Á S A
tisztasága iránt nincsenek nagyobb követelmények. Egyenáram alkalmazásávalaz anédon egyenletesebb működés és magasabb hőmérséklet érhető el. A Mois- scm-típusú kemencében az elektromos ív nem éri a munkatérben elhelyezettanyagot, hanem ebben az esetben a fölötte elhelyezkedő ívből csak a sugárzó hőthasznosítjuk.
44.2.3, Nagyfrekvenciás indukciós kemencék
Igen alkalmasak elektromosan vezető anyagok, elsősorban fémek megolvasztására, szublimálására, főleg, ha a fűtött tér evakuálása is szükséges. Bosszul vezetőanyagok megfelelő olvadáspontú fém- vagy grafittégelyben ugyancsak felhevíthetőkbenne. A berendezést költséges volta miatt a szokványos laboratóriumi munkábanegyelőre ritkábban alkalmazzák, viszont vákuumtechnikai célokra manapság ez alegelterjedtebb (44.2.3.1. ábra).
Irodalom
Lebeau, P.: Les hautes températures. Masson, Paris, 1950. Hessenbruch, W.: Metalle und Legierungen für hohe Temperaturen. Springer, Berlin,
1940.Wartenberg, H.: Handbuch dér Experimentalphysik Bd. I X . Harms, Wien, 1929.
Pirani, M.: Elektrothermie. Springer, Berlin, 1930. Angerer, E.: Technische Kunstgriffe bei physikalischen Untersuchungen 8. Aufl. Wieweg,
Braunschweig, 1952.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 43/366
45 . Hőm érsékletmérés és hőszabályozás
A hőmérséklet mérésére elvben bármely, a hőmérséklettel arányosan változó fizikai tulajdonság felhasználható. A gyakorlatban legtöbbször a térfogatváltozást használják fel hőmérsékletmérésre, de emellett a nyomás, elektromos ellenállás,termoelektromotoros erő vagy a sugárzó energia stb. változása is mértéke lehet ahőmérséklet-változásnak. A hőmérséklet-különbség egysége a C°, amely a víz 1 atmnyomáson mért fagyás- és forráspontja közötti különbség 1/100 része.
45.1. Gáz- és folyadékhőmérők
A hőmérséklet változásakor legnagyobb mértékben a gáznemű anyagok térfogata változik, a legérzékenyebb méréseket tehát gázhőmérővel végezhetjük. Egyszerűbb kezelhetőségük miatt mégis a folyadékhőmérők az elterjedtebbek.
45.1.1. Gázhőmérők
A gázhőmérők elve az ideális gázok hőkiterjedésének törvényszerűségén alapszik. Éppen ezért gázhőmérők töltésére az ideális gáz tulajdonságait legjobbanmegközelítő hidrogén és hélium alkalmas. A hidrogén- és héliumtöltésű gázhőmérők igen alacsony hőmérsékletek mérésére használatosak, magasabb hőmérsékleten a nitrogén is megfelelő.
45.1.2. Folyadékhőmérők
Töltésükre leggyakrabban a higanyt használják (fagyáspontja —38,9 C°, forráspontja 357 C°). Hőmérsékletmérésre jóval a higany forráspontja felett is megfelelők, ha a kapillárist indifferens gázzal töltik meg. Ilyen gáztöltésű, kvarcüvegbőlkészült higanyos hőmérők 750 C°-ig is használhatók. 8,5 % talliumtartalmú amal-gámmal a higanyos hőmérő alsó méréshatára —60 C°-ig tolható ki. Igen előnyöstulajdonsága a higanynak, hogy az üvegfelülethez nem tapad, viszkozitása is csak
kismértékben változik, ezért egyéb hőmérőfolyadékok használatánál jelentkezőutánfolyásból eredő hibától mentes.
A magasabb hőmérsékletek mérésére készült folyadékhőmérőket újabban gal- ímmfémmel (op. 29,5 C°, fp. 2064 C°) töltik. Az ilyen hőmérő kapillárisa kvarcbólkészül. Huzamosabban 1000 C°-ig, rövid időre 1400 C°-ig használható. Ha a folyadékhőmérők kapillárisának hőmérséklete különbözik a tartály hőmérsékletétől,fonalkorrekciót kell alkalmazni.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 44/366
44 H Ő M É R S É K L u E T M É R É S É S H Ő S Z A B Á L Y O Z Á S
Alacsony hőmérsékletek mérésére alacsony olvadáspontú anyaggal töltötthőmérők alkalmasak. Ilyen anyag pl. az izo-pentán (alkalmazhatóságának alsóhatára —195 C°), normál-pentán (—130 C°), amilalkohol ( —110 C°), toluol (—90 C°)Hátránya ezeknek a folyadékoknak, hogy a kapilláris falát nedvesítik, azonkívülviszkozitásuk a fagyáspont közelében jelentékeny mértékben megnő. Ezért mérésközben a higanyos hőmérőkkel ellentétben csak a folyadékgömbjüket szabad amérendő alacsony hőmérsékletű térbe helyezni, és mérés közben csak függőlegeshelyzetben, kellő utánfolyási idő elteltével olvasható le róluk a kb. ±0 ,5 C° pontosságú hőmérsékleti érték.
45.2. Elektromos elven működő hőmérők
A folyadékhőmérőkhöz hasonlóan könnyen kezelhetők, hőkapacitásuk sokkalkisebb, tehát a mérendő tér hőmérsékletét sokkal hamarabb átveszik, ezért gyorshőmérséklet-változások követésére a folyadékhőmérőknél alkalmasabbak.
46.2.1. Ellenállás-hőmérők
A gyakorlatban csak a platinának van ezen a téren jelentősége. A berendezéshőérzékelő szerve vékony kvarcüveg csőben elhelyezett vagy kvarcrudacskára spirális alakjában ráforrasztott platinahuzal, amelynek ellenállását általában úgyválasztják meg, hogy 0 C°-on éppen 100 ü legyen. Ez az ellenállás + 500 C°-on már280Q. Az ellenállás mérésére általában a kompenzációs eljárást használják. Az ellenállás-hőmérővel elérhető maximális pontosság 0,001 C°. A platina ellenállás-hőmérő—200 C°-tól kb. 1100 C°-ig alkalmas hőmérséklet mérésére. A —200 C° alattihőmérsékletek mérésére ólom ellenállás-hőmérőt használnak.
46.2.2. Termisztorok
A termiszlor a modern kapcsolástechnika legegyszerűbb felépítésű és igen sokoldalúan használható kapcsolási eleme. A termisztorok tulajdonképpen különbözőfém-oxidókból álló, negatív hőmérsékleti együtthatójú félvezető ellenállások. Ismeretes, hogy a fémek ellenállása a hőmérséklet emelésével nő, a félvezetőké viszontugyanakkor csökken. Míg azonban például a platina fajlagos ellenállása —100 C°és +400 C° között tízszeresre nő, a termisztor ellenállása 10_7-szeresére csökken(45.2.2.1. ábra).
A termisztorok sokrétű alkalmazása éppen e nagy hőmérséklet-érzékenységbőlkövetkezik. Tulajdonságaik jellemzésére az alábbi adatok szolgálnak:
a) hőmérséklet—ellenállás-karakterisztika;b) áramerősség —feszültség-karakterisztika;c) idő —áramerősség-karakterisztika.
A termisztor ellenállását hőmérsékleti hatással kétféleképpen befolyásolhat juk: egyrészt a környezet hőmérsékletének változtatásával, másrészt a termisz-torba betáplált teljesítmény, tehát a termisztoron áthaladó áram növelésével. A környezet hőmérsékletének mérése során célszerű a termisztor ellenállását kisáramerősségekkel meghatározni. Ilyenkor a termisztor nem melegszik fel, tehát

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 45/366
T K R M I S Z T O B O K 45
úgynevezett hidegellenállást kapunk. A 45.2.2.1. ábra a termisztorokra jel
lemző ellenállás—hőmérséklet-görbétmutatja be, amelyet jó közelítéssel az
ohm
R t . — R ib (4 í - t )
1
*8
10r
101
XT
101
vrmis r ior
pn
100 0 100 200 300 400 C
h m ér sék let
exponenciális egyenlettel írhatunk le,
a h o l J ^és B ^a T ltilletve a T2 hőmérsékleten mért hidegellenállás, B pedig
anyagi állandó.
A termisztorok ellenállásának jellemzésére a legalább két hőmérsékleten (pl. 20 és 80 C°-on) mért ellenál
lást és az adott hőmérsékletre vonat
kozó százalékos hőmérsékleti koefficienst, vagy a B értékét alkalmazzák.
(Bővebben lásd: Dobos D.\ Elektronikus kémiai mérőkészülékek. MűszakiKönyvkiadó Budapest, 1962. 61. old.
és Egyesült Izzólámpa és Villamossági
RT.: Tungsram Termisztorok.) A termisztorok B-értéke 3 000 —4 000 K° között
mozog, s ennek C°-onként szobahőmérsékleten —3,6 %, illetve —4,5 % ellen
állás-változás felel meg.
A különféle felhasználási területek eltérő követelményeinek kielégítésére a
45.2.2.2. ábrán vázolt, különböző alakú (rúd, gyöngy, tárcsa stb.) és tulajdonságú
termisztorok kerülnek forgalomba, melyeknek 20 C°-on mért ellenállása nagymértékben különbözik.
Ha a termisztoron fokozatosan nö
vekvő áramot bocsátunk át, és közben
45.2.2.1. ábra. Termisztor ellenállás —hőmérséklet-jelleggörbéje
folyamatosan mérjük a termisztor sarkain
( —o— -*) I1
(£> Pm
45.2.2.2. ábra. Termisztorok. a) rúd alakú termisztor; b) gyöngytermisztor; c) tárcsa-
termisztor
45.2.2.3. ábra. Termisztor feszültség —
■ — áramerősség-jelleggörbéje

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 46/366
46 H Ő M É R S É K L E T M É R É S É S H Ő S Z A B Á L Y O Z Á S
fellépő feszültséget, tapasztalhatjuk, hogy kis áramerősségek esetén a feszültség
arányos az áramerősséggel, tehát érvényes az Oáwi-törvény. Az áramerősségettovább növelve, a feszültség növekedése csökkenő tendenciát mutat, sőt bizonyos áramerősség felett a feszültség csökken (45.2.2.3. ábra). A feszültség maximuma olyan áramerősség mellett lép fel, 'ahol a termisztoron átfolyó áramnakmegfelelő teljesítmény már olyan nagy, hogy a J ou l e -fél e hő a termisztort a környezet hőmérséklete fölé tudja melegíteni. A feszültség—áramerősség-görbe megközelítően vízszintes szakasza lehetővé teszi a termisztoroknak feszültségstabilizálásra történő felhasználását.
A feszültség maximuma a 20 C°-on azonos ellenállású, de különböző alakúés méretű termisztorok esetén különböző áramerősségnél jelentkezik. De nemcsaka termisztor nagysága és hőkapacitása, hanem a környezet is nagymértékben befolyásolja annak melegedését, illetve a feszültség maximális értékét. Annak jellemzésére, hogy valamely adott termisztor meghatározott körülmények között milyenteljesítménnyel melegíthető 1 C°-kal a környezet hőmérséklete fölé, a disszipációs állandó mérvadó, melynek értékét mW/C°-ban szokás kifejezni. Egyazon termisz-
torra vonatkozóan a disszipációs állándó értéke természetesen erősen változik atermisztor környezetének hővezető-képessége szerint. A termisztor egyensúlyi hőmérséklete nemcsak a termisztort körülvevő közeg minőségétől, hanem annakáramlási sebességétől, sőt gázok esetében azok nyomásától is függ. A fent tárgyaltaklehetővé teszik a folyadéknlvó (lásd 48.1.2.), az áramlási sebesség (lásd 49.5.2.)és a gáznyomás (lásd 46.3.6.) termisztorral történő mérését.
A termisztor idő —áramerősség-karakterisztikájából (45.2.2.4.ábra) megállapítható, hogy a termisztort bizonyos feszültségre kapcsolva, a rajta átfolyó áram
s ezzel együtt a termisztor ellenállása is az
idő meghatározott függvénye. A termisztortezen tulajdonsága jelfogók időzítésére teszialkalmassá.
A termisztoros hőmérő elvileg bármilyenellenállás-hőmérő kapcsolásának megfelelőenhasználható. Rendkívüli hőmérséklet-érzékenységük folytán ezekkel a hőmérőkkel egyszerű WheaJtstone-hiőőaX, 2 • 10“ 10 érzékenységű galvanométerrel 0,001 C° hőmér-
ter m isztor
cseng
45.2.2.5. ábra. Termisztoros hőmérsékletjelző

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 47/366
T E R M O E L E M E K 47
séklet-külonbség is kimutatható. A termisztoros hőmérőket rendkívül kis hőkapa
citásuk folytán különösen a mikropreparatív technikában, illetve kis mennyiségűanyagok tisztaságvizsgálata és azonosítása (forráspont-, fagyáspont-, tenziómé-rés, stb.) terén alkalmazzák.
Gyakori a termisztoroknak hőmérséklet]’elzŐként történő felhasználása. A 45.2.2.5. ábrán egy termisztoros hőmérséklet]elző kapcsolási elve látható. A berendezés különös előnye annak egyszerűsége, kis térigénye, csekély áramfogyasztása és alacsony ára.
45.2.3. Termoelemek
A termoelemek 1000 C° feletti hőmérséklet mérésére is alkalmasak. A 45.2.3.1.táblázatban közöljük a különböző összetételű termoelem-párok alkalmazhatóságának hőmérsékleti határait.
45.2.3.1. táblázat
Termoelemek fcszültségkülönbsége (mV-ban)
Hőmérséklet
C°
Konstantán/
réz *
Konstantán/
vas
Konstantán /
ezüst
Nikkel/króm
nikkel
Pt/Pt +10%
Rh.
Volfrám/
molibdén
- 2 0 0 - 8 ,2 7 - 6 , 5 4- 1 8 0 - 7 ,7 5 - 5 ,2 0 — — — —
- 1 0 0 - 4 , 8 2 - 3 , 3 5 — — — —
0 0,0 0,0 0,0 0,0 0,0 —
100 5,40 4,28 4,12 3,85 0,64 —
200 10,99 9,29 8,84 8,02 1,42 - - 0 ,7 0300 16,56 14,86 14,10 11,97 2,29 —
400 22,07 20,87 19,77 15,26 3,21 - 1 ,3 4500 27,58 — 25,79 18,42 4,17 —
600 33,27 — 32,15 21,74 5,18 - 1 , 5 4700 39,30 — — 25,32 6,23 —
800 46,72 — — 28,86 7,31 - 1 ,4 0900 52,29 — — 32,47 8,43 —
1000 58,22 — — 36,04 9,56 - 1 , 01100 — — — 39,73 10,72 —
1200 — — — — 11,89 + 0 ,2 "1300 — — — — 13,07 + 0 ,31400 — — — — 14,26 + 0 ,81500 — — — — 15,45 + 1 , 5
1600 — — — — 16,63 + 2,11700 — — -r — — + 2 ,86
1800 — — — — + 3,Q
A termoelemek elektromos feszültségkülönbségének (1. 45.2.3.1. táblázat) meghatározására a kívánt pontosságnak megfelelő mérőeljárást választják ki. Napjainkban a kereskedelmi forgalomban néhány gyakrabban használatos termoelemhez ka-ibrált hőfokleolvasó műszerek (mV-mérők) is kaphatók. A 45.2.3.1. ábra a különböző platinafém-tartalmú termoelemek termofeszültségét tünteti fel a hőmérsékletfüggvényében.
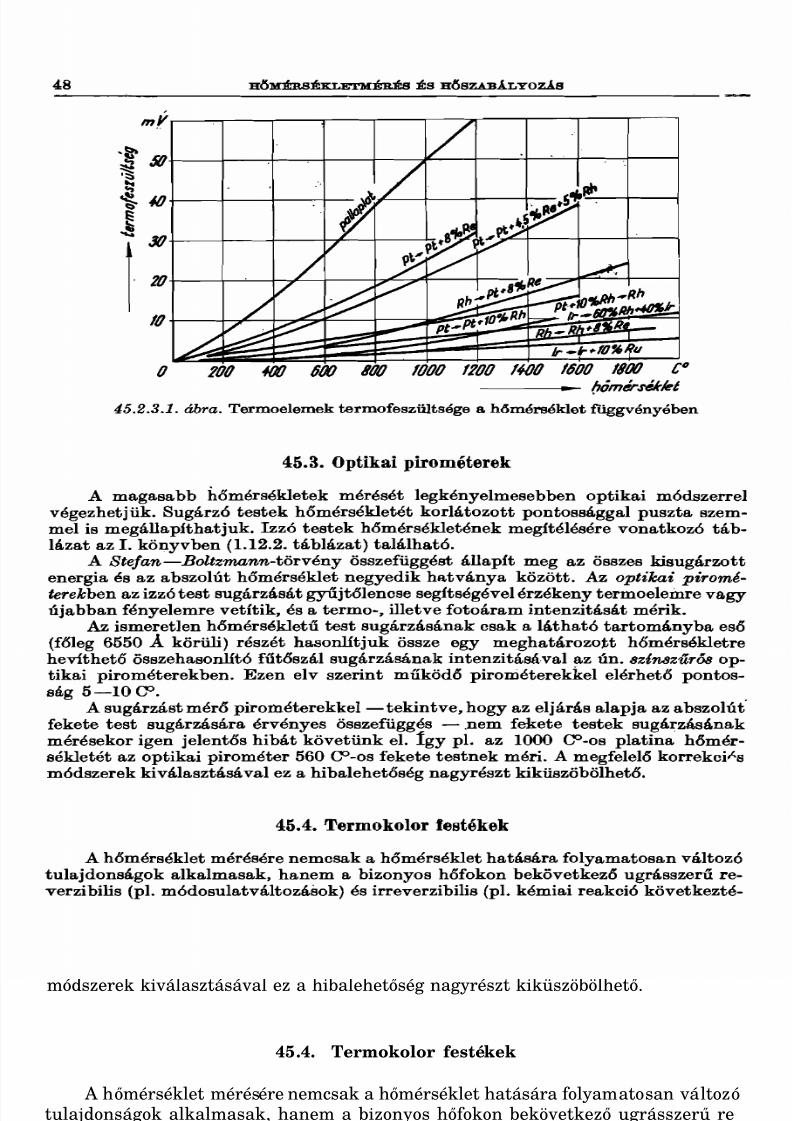
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 48/366
48 H Ö M É R S É K X E T M É B É S É S H Ő S Z A B ÁL Y O Z Á S
mV
£Q
1^ 40
l 30
20
to
f i
$
-J í trVSm Rh
H*40%t
200 400 600- h mérséklet
45.2.3.1. ábra. Termoelemek termofeszültsége a hőmérséklet függvényében
45.3. Optikai pirométerek
A magasabb hőmérsékletek mérését legkényelmesebben optikai módszerrelvégezhetjük. Sugárzó testek hőmérsékletét korlátozott pontossággal puszta szemmel is megállapíthatjuk. Izzó testek hőmérsékletének megítélésére vonatkozó táblázat az I. könyvben (1.12.2. táblázat) található.
A Stefan— Boltzmann-tÖTvény összefüggést állapít meg az összes kisugárzottenergia és az abszolút hőmérséklet negyedik hatványa között. Az optikai piromé- terekben az izzó test sugárzását gyűjtőlencse segítségével érzékeny termoelemre vagy
újabban fényelemre vetítik, és a termo-, illetve fotoáram intenzitását mérik. Az ismeretlen hőmérsékletű test sugárzásának csak a látható tartományba eső(főleg 6550 Á körüli) részét hasonlítjuk össze egy meghatározott hőmérsékletrehevíthető összehasonlító fűtőszál sugárzásának intenzitásával az ún. szinszűrős optikai pirométerekben. Ezen elv szerint működő pirométerekkel elérhető pontosság 5—10 C°.
A sugárzást mérő pirométerekkel —tekintve, hogy az eljárás alapja az abszolútfekete test sugárzására érvényes összefüggés — nem fekete testek sugárzásánakmérésekor igen jelentős hibát követünk el. így pl. az 1000 C°-os platina hőmérsékletét az optikai pirométer 560 C°-os fekete testnek méri. A megfelelő korrekciós
módszerek kiválasztásával ez a hibalehetőség nagyrészt kiküszöbölhető.
45.4. Termokolor festékek
A hőmérséklet mérésére nemcsak a hőmérséklet hatására folyamatosan változótulajdonságok alkalmasak, hanem a bizonyos hőfokon bekövetkező ugrásszerű reverzibilis (pl. módosulatváltozások) és irreverzibilis (pl. kémiai reakció következté-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 49/366
HŐMÉRSÉKLETMÉRÉS 49
ben fellépő) színváltozások is felhasználhatók. Ilyen színváltozás észlelhető pl. reverzibilis módosulatváltozás közben a Hgl2, valamint a réz(I)- és ezüst-[jodo-mer-
kurát(II)] esetén (I. könyv 39.6.5. és 40.14.4.).Bizonyos körülmények között, meghatározott hőmérsékleten egyes Cu-, Co-,Ni-, Mo-, Mn-, U-, V- és Cr-vegyületek gyors, irreverzibilis színváltozással járó bomlást szenvednek^ Ezek közül egyesek fokozatosan bomlanak el többszörös színváltozás közben (45.4.1. táblázat).
45.4.1. táblázat
Néhány termokolor festékként használatos vegyület átcsapási hőmérséklete
NiCl2 •2 C6H 18N4 •10 H ,0 halvány- 60° sárga 100° ibolyazöld
(N H4)sH 6[Fe(Mo04)fl]-7 H 20 fehér 80° sárga 220° fekete
[Cu/piridin/J (SCN)2 zöld 135° sárga 220° fekete
CoNH4P04.H 20 vörös 140° kék 500° halvány szürkéskék
[Cr(NH3)6]4(P 20 7)s sárga 145° ibolya 226° világoskék280® barna
Speciális célokra több komponenBŰ keverékek is készíthetők, amelyek ± 5 C°pontossággal az elért maximális hőmérsékletnek megfelelő színt veszik fel. A színnekmegfelelő hőmérséklet összehasonlító táblázatról olvasható le. Az ilyen festékele-gyek nagy jelentőségűek és igen elterjedtek egyes gépalkatrészek üzemi hőmérsékletének jelzésére.
45.5. Seger-gúla
Eredetileg a kerámiai iparban használták az égetőkemeneék hőmérsékleténekmérésére a különböző szilikátáaványok keverékéből készített háromszög alapú pira
misokat. Ezek meghatározott lágyuláspontján bekövetkező alakváltozás jelzi azelért hőmérsékletet. 600 —2000 C° között 64 tagból álló precíziós sorozat segítségével kb. + 20 C° pontossággal meghatározható a munkatér hőmérséklete.
45.6. Hőmérséklet-szabályozás, termosztatok
A laboratóriumi munkában gyakran felmerülő feladat a munkatér hőmérsékletének állandósítása. Erre a célra szolgálnak a különböző rendszerű termosztátok. A
környezet ingadozásaitól független, meghatározott pontosságú hőmérséklet megvalósítása annál nehezebb, minél jobban különbözik a kívánt hőmérséklet a környezetétől.
Igen alacsony és igen magas hőmérsékletek esetén fokozott jelentősége van ahőszigetelésnek. Alacsony hőmérsékleten ennek szokásos és legeredményesebb mód
ja a Dewar-edény, magas hőmérsékleten pedig a különböző tűzálló, magas olvadáspontú porózus anyagok alkalmazása.
4 Ált. és szervetlen kémiai prakt. —4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 50/366
50 HŐMÉRSÉKLETTMÉRÉS £IS HŐSZABÁLYOZÁS
45.6.1. Termosztátok töltő (hőközlő) anyagai
Meghatározott hőmérséklet stabilizálására a termosztátokat használjuk. Igenkedvezően befolyásolja a termoBztátok stabilitását a nagy hőkapacitás. Ezért a termosztát töltésére lehetőleg nagy mennyiségű, jelentős hőkapacitású hőközlő anyagot alkalmaznak.
Folyadéktermosztátokb&n állandó és egyenletes hőmérséklet biztosítása csak intenzív ke veréssel valósítható meg. A stabilizálandó hőmérséklettől függően a következő töltőfolyadékok használhatók:
Alacsony hőmérsékleten —150 C°-tól petroléter, —80 C°-tól alkohol, 0 és 95 C°között víz, 100—200 C° között paraffinolaj, 300 C°-ig kénsav vagy turbinaolaj, tar-tósabb igénybevételre 300 C° felett (350 C°-ig) pedig a szilikonolaj alkalmas. Magasabb hőmérsékleten (350—600 C°-on) főleg K N 03- és NaN03- tartalmú sóolvadékokvagy alacsony olvadáspontú fémek (Wood-íém, Pb, Sn stb.) használatosak. Mégmagasabb hőmérsékleten (600—1400 C°-ig) 7 sr. BaCl2+ 2 sr. NaCl olvadéka használható vastag falú samott-tégelyben.
Hőközlőként szilárd fázisú anyagok is tekintetbe jöhetnek. Ilyenek a jó hővezető képességű fémekből készült hűtő-, illetve fűtőblokkok. így pl. —180 C°-ig ter
jedő hőmérsékletet folyékony levegőver vagy nitrogénnel hűtött, Dewar-edénnyelhőszigetelt alumínium blokkban állandósíthatunk. A hőelvonás sebességét a fém-b’lokk folyékony levegőbe merülő részének süllyesztésével vagy emelésével változ
tathatjuk. A légtermosztátok tulajdonképpen jól szigetelt falú, ajtóval ellátott, zárhatószekrények, amelyekben a kívánt hőmérséklet stabilizálásáról megfelelő automatikus működésű szerkezet gondoskodik. Ide sorolhatók a különböző jégszekrények,szárítószekrények és végeredményben a szabályozható hőmérsékletű kemencék is. A légtermosztátok hőmérséklet-ingadozása az egyéb, nagy hőkapacitású termosztátokénál jóval nagyobb.
45.6.2. Önműködő hőmérséklet-szabályozók (regulátorok)
Az elektromos fűtés a régebben használatos gázfűtést már csaknem teljesen kiszorította. Kisebb teljesítményű termosztátokban fűtőellenállásként nagyon jólhasználhatók a kereskedelemben kapható merülő vízforralók. Nagyobb teljesítményre megfelelően méretezett ellenállásokat kell készíteni. A keramikus szigetelésű ellenálláshuzalt célszerű jói hővezetőből (pl. alumíniumból vagy vörösrézből) készültcsőbe helyezni. A fűtőáram kapcsolására különböző rendszerű regulátorok használatosak. Különleges berendezésekkel (ultratermosztát) ±0,005 C° pontosság is elérhető.
Gázfűtésű termosztátok szabályozására használható pl. a toluolos regulátor. A
termosztát felmelegedése következtében a toluol kiterjed, és a kapillárisban levő higany elzárja a fűtőgázáram útját. Hogy az égő ebben az esetben se aludjon el teljesen, néhány mm-es gyújtóláng táplálására mellékutat kell a gáz számára biztosítanunk. A toluolos regulátorral (45.6.2.1. ábra) a kívánt hőmérsékletet a benne levőhigany mennyiségének megfelelő megválasztásával állíthatjuk be.
Ha a toluolos regulatort elektromos fűtésű termosztát szabályozására használ juk, akkor a higanyszint emelkedése nem a gázáram útját, hanem az elektromos fűtést kikapcsoló relé áramkörét zárja.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 51/366
H Ő M É R S É K L E T - S Z A B Á L Y O Z Ó K 51
Lényegében azonos elv alapján mőködik az ún. kontakt hőmérő (45.6.2.2. ábra).Ennek higanyszála ugyancsak az elektromos fűtést kapcsoló relé áramkörét zárja.
A hőmérő kapillárisába beépített felső platina kontaktus magassága, s ezzel a relékapcsolásának hőmérséklete változtatható.
45.6.2.1. ábra. Toluolos regulátor
45.6.2.2. ábra. Kontakt hőmérő
45.6.2.3. ábra. Bimetall hőmérő
A bimetall hőérzékélőt két különböző kiterjedési együtthatójú fémből készült lemez alkotja. Ezek lapjukon összeforrasztva úgy helyezendők el a szabályozandó hőmérsékletű térben, hogy a kettős lemez egyik vége rögzítve, a másik vége egy kontaktuscsavar közelében szabadon legyen. A hőmérséklet-csökkenés folytán bekövetkező elmozdulás a bimetall hőmérő (45.6.2.3. ábra) szabad
végén olyan irányú legyen, hogy a fűtőáramkört zárja. A bimetall hőmérő előnye, hogy magasabb hőmérsékleten használható, mint a kontakt hőmérő, és mérsékelterősségű fűtőáramot közvetlenül is kapcsolhat.
A termosztátfűtés áramkörének kapcsolására különböző rendszerű reléket használhatunk,.amilyen pl. az elektromágneses, tirátronos és gáznyomásrelé.
Az utóbbi időben igen elterjedt a termtsztoros hőszabályozás. Ennek legegyszerűbb kapcsolása a 45.6.2.4. 'ábránlátható. Nagy disszipációs állandójú (lásd 45.2.2.) termisz-torral a vázolt kapcsolásban 30 C° körül elérhető érzékenység: a jelfogó 0,3 C°-oshőmérséklet-emelkedésre húz be.
Nagyobb pontosságú mérésekhez folyamatos hőszábályozóksX alkalmazhatunk.Termisztorral aránylag egyszerű elektronikus berendezéssel készíthető a folyamatos hőszabályozó.1
45.6.2.4. ábra. Termisz- toros hőszabályozás
kapcsolási vázlata
1 Dobos, D.: Elektronikus kémiai mérőkészülékek. Műszaki Könyvkiadó, Budapest, 1962, 61. old.
4*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 52/366
52 HŐMÉRSÉKLETMÉBÉS é s HŐSZABÁI j YOZÁS
45.6.3. Termosztálás fázisegyensúlyok felhasználásával
A szilárd—cseppfolyós fázisegyensúlyt termosztálásra annak az elvnek azalapján használhatjuk fel, hogy mindaddig, amíg a két fázis egyidejűleg jelen van, állandó nyomáson a hőmérséklet sem változik, és a környezette] szemben jelentkezőhőkicserélődés csupán a fázisok mennyiségének arányát változtatja meg. Ilyenrendszereket egy anyagból, vagy több anyagból (pl. az eutektikus ponton) állíthatunk össze. Termosztáló fürdők készítésére alkalmasak a kriohidrátok (45.6.3.1.táblázat) és az alacsony olvadáspontú anyagok (45.6.3.2. táblázat).
45.6.3.2. táblázat
45.6.3.1 táblázat Alacsony hőmérsékletű termosztálóKriohidrátok fürdők készítésére használatos anyagok
Anyag minőségeGramm
anyag/100 g jég
Eutektikushőmérséklet
C°
H j B 0 8 .................... 1,0 - 0,7NaaS04 *10 H20 . 10,2 - 1,25Na.CO.10 H.O 19,0 - 2,1KNOa .................. 12,2 - 2,9BaClj-2 B 20 36 - 7,8K C 1 ....................... 24,6 -1 0 ,7NH*C1.................. 22,9 - 1 5 ,8NaCl (2 H20 ) 30,7 -2 1 ,2CaCl2*6 H20 143 - 5 5HC1 (3 HjO) 33 - 8 6
Anyag minősége*
OlvadáspontC°
Anilin ......................... - 6,6Benzaldehid........... - 14,0S zén-tetraklorid - 2 2,9Bróm-benzol - 30,6Etilén-klorid - 35,6Klór-benzol - 45,2Kloroform — 63,5 Aceton .............. - 95Metilalkohol........... - 97,8Szén-diszulfid - 1 1 1 , 6Dietiléter -■ 116 ,2Etil-klorid,.............. - 1 3 8 , 7Tetrafluor-metán - 1 8 3 , 6
Különböző anyagok forráspontja az előbb ismertetett elvek alapján ugyancsak felhasználható a hőmérséklet állandósítására. A 45.6.3.3. táblázat az alacsonyhőmérsékletek stabilizálására alkalmas anyagokat foglalja magában.
45.6.3.1. ábra. Gőztermosztát alacsonyhőmérsékletre

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 53/366
T E R M O S Z T Á L Á S 53
45.6.3.3. táblázat
Alacsony hőmérsékletű gőztcrmosz-t&tok üzemeltetésére használatos
anyagok
A 45.6.3.1. ábrán vázolt berendezés az alkalmazott anyag atmoszféra nyomásonmórt forráspontjának hőmérsékletét állandósítja. Lényege folyékony levegővel vagy
száraz jéggel működtetett visszafolyós hűtő,mely az átlaghőmérsékletnél jóval alacsonyabb forráspontú termosztáló folyadék (pl.NH3, CH4 stb.) kondenzátumának a hűtőtérbe való visszajuttatását szolgálja. Nyomáscsökkentéssel a rendszer hőmérsékletecsökkenthető. A környezetnél magasabbhőmérsékletek állandósítására különbözőanyagokkal töltött gőztermosztátok alkal
masak (45.6.3.2. ábra). A gyakrabban használt munkaanyagok a 45.6.3.4. táblázatbanláthatók.
A hőmérsékletmérésről és hőszabályozásról szóló fejezetben csupán a megfelelőmódszer kiválasztásához kívántunk elviszempontokat nyújtani. A gyakorlati megvalósításhoz szükséges terjedelmes műszaki részletekre vonatkozóan azkészülékek használati utasítására, illetve a bővebb szakirodalomra utalunk.
A n y a g m in őség eF o r r á s p o n t
e°
Kén-dioxid - 10,02 Ammónia - 33,5Propán - 44,5Etán - 88,5Etilén -103,9Metán -164
egyes
45.6.3.4. táblázatGŐztermoszt&tban használatos anyagok
A n y a g m in ő ség eF o r r á s p o n t
C° A n y a g m in ős ég eF o r r á s p o n t
C °
Etil-klorid 13,1 Jégecet ..................... 118,1Dietiléter ................ 34,60
46,2Klór-benzol 132
Szén-diszulfid Bróm-benzol 165,6 Aceton 56,3
61,21 Anilin 184,4
Kloroform .............. Tetral in .................. 207,3
Metilalkohol......... 64,7 Nitro-benzol 210,9Szén-tetraklorid 76,7 Naftalin 217,90Etilalkohol 78,3 Difenil 264,9Benzol ..................... 80,12 Benzofenon 305,9Etilén-klorid
Víz83,7
100,00Kén 444,6
Irodalom
Eder, F.: Moderné Messmethoden dér Physik. VEB. Deutscher Verlag d. Wiss., Berlin,
1956. Henning, F.: Temperaturmessung. J. A. Barth Verlag, Leipzig, 1935. Erdey-Grúz T. —Proszt J .: Fizikai-kémiai praktikum I-II. Tankönyvkiadó, Budapest,
1965.Hdb. d. technischen Betriebskontrolle Bd. III. Phyaikalische Messmethoden. Akad.
Verlaggesellschaft, Leipzig, 1951. Archív für teohnisohes Messen. Hengsteriberg, J., Sturm, B., Winkler, O.: Messen und Regein in dér ehemischen Technik.
Springer, Berlin—Göttingen—Heidelberg, 1967. Dobos D.: Elektronikus kémiai mérőkészülékek. Műszaki Könyvkiadó, Budapest, 1962.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 54/366
46. Vákuumtechnika
A kémiai laboratóriumban és az üzemi gyakorlatban gyakran szükséges, hogyvalamely műveletet levegőtől és idegen gáztól mentes, vagy definiált nyomású és ismert összetételű gáztérben valósítsunk meg. Ilyen kísérleti körülmények megvalósításához a rendszerben csökkentett nyomást, vákuumot kell létesítenünk. Az alábbiakban röviden, a teljességre való törekvés nélkül ismertetjük a vákuum előállítására és mérésére szolgáló berendezéseket és a szivattyúrendszerek felépítését.
46.1. Általános tudnivalók
Égy rendszer, vákuumállapotának jellemzésére az abban uralkodó nyomás és a jelenlevő gáz minősége szolgál. A gyakorlatban használt nyomásegységekről és azokátszámításáról a 46.1.1. táblázat ad felvilágosítást.
A vákuumszivattyú teljesítőképességét a vég vákuummal, a kipufogó oldalonmérhető nyomással és a szívássebességgel jellemezhetjük.
Végvákuumon (P0) á szivattyú szívócsonkján mérhető legkisebb nyomást értjük. A kipufogó oldal nyomása (elővákuum) az a nyomás, amellyel szemben a szivattyúműködtethető. Ez szivattyúfajtánként és típusonként különböző lehet, értéke1 atm-tól 10-2 Hgmm-ig változik. A szívássebesség (8 ) az időegység alatt eltávolított
gázmennyiség liter/sec vagy m3/óra egységben kifejezve egy meghatározott nyomá-
Nyomá Hegységek
Hgmm (torr) din /cma milibar
1 Hgm m (torr) = 1 mm higany- .. oszlop nyomása ................................ 1 1,333 22-10® 1,333 22
10"®1 din/cm2 = 1 mikrobar ( fib) ...........
1 millibar (mb) = 10® d in /c m * .........
0,760 06-10“ ® 10,760 06 10® 1
1 bar (b) — 10® din/cm* .............. 760,06 10® 10®1 kg/m* ss 1 mm vízoszlopnyomás . 0,735 66 10 -* 0,980 665-10* 0,980 665 -10“ *1 kg/cm* = 1 a t ..................................... 735,56 0,980 665-10® 0,980 665-10®1 atm — 760 Hgmm ........................... 760 1,013 25-10® 1,013 25-10®1 mikron = 1 *10“ ®H g m m ................ 10"® 1,333 22 1,333 22-10“ ®
% vákuum = 760 100;

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 55/366
S Z Í V Á S S E B E S S É G 66
són. Mechanikus szivattyúk szívásBebességét általában 1 atm nyomású levegőre
(volumetrikus szívássebesség), a gőzsugár- és diffúziós szivattyú esetén 10-3 vagy10” 4 Hgmm nyomásra vonatkoztatva szokták megadni. A vákuumszivattyú szívássebessége az alábbi összefüggés szerint függ a nyo
mástól :
E = s ( 1 —
ahol E a pillanatnyi szívássebesség, P a recipiensben uralkodó nyomás, P0 a végvákuum. Következésképpen a nyomás csökkenésével E állandóan csökken, és P = P0nyomáson zérussá válik. Bármely rendszerben a mért szívássebesség nemcsak a szivattyú belső szívósebességének (S) és a végvákuumnak, hanem a rendszert a szivattyúval összekötő csővezetékek, csapok stb. áramlási ellenállásának is függvénye.Egy F vezetőképességű (1 jF ellenállású) csővezetékkel összekötött, S szívássebességű szivattyú eredő szívássebességét az
1 ISe = l j S + l j F
összefüggéssel számíthatjuk, ahol Se a mért valódi szívássebésség a recipiensben. F értékét azon nyomástartományban ( ~ 760 Hgmm-től ~ 10-2 Hgmm-ig), ahol agázmolekulák közepes Bzabad úthossza kisebb, mint a cső átmérője (A <k 2r) és
P -1-P P 1—P2 « —---- -, a Hagen— Poiseuille-törvény adja meg, amely szerint
1 103P — = W = -------
F r4
8 M
n 1333 (P j-f P 2)[sec/1],
ahol L a cső hossza, r a cső sugara, P l és P2 a nyomás a cső két végén, M a gáz molekulasúlya.
Kis nyomásokon (P si lO -3 ), ahol A » 2 r, vagyis az ún. molekuláris áramlás
területén a csővezeték ellenállását a Knudsen által levezetett összefüggés alapján
átszámítása 46.1.1. táblázat
barkg/m*
(mm-vízoszlop) kg/cm* (at)atm (760Hgmm) mikron (n)
1,333 22-10~3 13,595 1 1,359 51-10-a 1,315 79-10-* 10*io-« 1,019 72-10-2 1,019 72.10-« 0,986 92-10-« 0,760 06
10~8 10,019 72 1,019 72.10-3 0,986 92-10-3 0,750 06-10»1 1,019 72-104 1,019 72 0,986 92 0,750 06-10»0,980 665-10-4 1 10-* 0,967 84-10-* 0,735 56-10*0,980 665 10* 1 0,967 84 0,735 66-10«1,013 25 1,033 23-104 1,033 23 1 0,760-10«1,333 22 10-« 1,359 51-10-* 1,359 51-10-4 1,315 79-10-4 1
Hgmm = 760(-% vákuum
100

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 56/366
66 V Á K U U M T E C H N IK A
számíthatjuk. Az alábbiakban a gyakorlat számára fontos esetekre megadjuk a vezetékellenállás számítására alkalmas összefüggéseket.
Hosszú, kör keresztmetszetű cső (L » r ) esetén, 20 C°-on, levegő jelenlétében
W = — •— [sec/1].r3 100
Rövid, kör keresztmetszetű cső (L^2r) esetén,-a fenti körülmények között
W =
8 L
3 7+1 '
3,63 - 10 r2[sec/1].
^ Végül kör keresztmetszetű kiömlőnyílásnál (L r), 20C°-on, levegő jelenlétében:
1W =
3,63 •10 r2[sec/1].
46.2. Szivattyúk1
A preparatív laboratóriumi munkában felhasználandó szivattyútípust a szükséges szívássebesség, a végvákuum, a munkatérben fejlődő korrozív gázok, stb.figyelembevételével kell megválasztani.
46.2.1. Vízlégszivattyúk
A laboratóriumban közhasználatú vízlégszivattyúk (lásd 1.14.) üvegből vagyfémből készülnek. Szívássebességük egyedenként is különböző, kb. 0,15 1/sec, megfelelőenméretezett fémszivattyúkra 0,5 l/sec is lehet. 1,5—3 átm víznyomás eseténa víz hőmérsékletétől függően 10—15 Hgmm végvákuum is elérhető velük.
46.2.2. Forgó olajsziyattyúk (rotációs szivattyúk)
A forgószivattyúk működési elve: térfogatnövelés a szívóoldalon, komprimálása nyomóoldalon. A forgó olajsziyattyúk különböző mechanikai megoldásúak. AGaede-féle forgónyelves olajszivattyú elvi rajzát a 46.2.2.1. ábra mutatja, amelynekalapján működése megérthető. Az A henger alakú furatban forog a B excentrikushelyzetű dob, melynek középsíkjában, a rajta áthaladó horonyban két nyelv helyezkedik el. Ezeket a közöttük feszülő nyomórugó szorítja a ház falához úgy, hogy forgás közben azon csúsznak végig. Amikor a dob a nyíllal jelzett irányban forog, a C tér növekszik, ennek következtében a D nyíláson át levegőt szív. Ugyanakkor amár bezárt E térben levő levegőt az F nyelv összenyomja, és végül a G visszacsapó-szelepen keresztül kinyomja.
A zárószelep felett olajnívó van, és az egész szívóhenger maga is olajjal töltöttházban helyezkedik el. Ennek egyrészt az a célja, hogy megakadályozza a külsőlevegő bejutását a kompressziós térbe, másrészt a kompresszió végén a szelep alászivárgó olaj a „káros teret” kitöltve, biztosítja az összenyomott levegő teljes kiürítését, valamint a mozgó részek kenését is.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 57/366
S Z I V A T T Y Ú K 57
A Oaede-féle szivattyún kívül a gyakorlatban leginkább a Kinney-Tendszerü szivattyú terjedt el. Ennek működése a 46.2.2.2. ábra alapján követhető. A Kinney- rendszerű szivattyú forgó excenterdobját az A gyűrű veszi körül, amely forgásakorvégighalad a szivattyúház belső, hengeres fala mentén. A gyűrű a széles, lapos B tolattyúban folytatódik. Ez az excenterdob forgása közben a C csap vezetékében
fel-alá mozog, és azzal együtt lengő mozgást végez. A tolattyú üreges? A dob for
gása közben ezen az üregen keresztül jut a levegő a szívótérbe, ugyanakkor a dobmásik oldalán az előzőleg beszívott levegő összenyomódik, és a D rugóval terheltnyomószelepen át kilökődik az olajjal töltött F térbe. Ez a levegő olaj cseppeketvisz magával, amelyek a szivattyúházhoz kapcsolt olaj szeparátorban ütközés folytán le választódnak. A külső atmoszférába csak a tiszta levegő jut. Az excenterdobforgása közben a tolattyú alsó E része bizonyos helyzetben elzárja a beeresztőnyí-lást, és a teljes fordulat ideje alatt a beszívott levegőt elválasztja az evakuálandótértől. Az így beszívott levegő kinyomása egy újabb fordulatot vesz igénybe. A tel jes periódus tehát két fordulat alatt megy végbe.
A nagyobb teljesítményű szivattyúkat hűtés cél jából vízköpennyel veszik körül. A megfelelő végvákuum elérése nagymérték
ben függ a használt olaj minőségétől és mennyiségétől. A minőséget illetően fontos az alacsonytenzió és az üzemi hőmérsékleten megfelelő viszkozitás. Mechanikai szennyezéseket nem tartalmazhat az olaj. Az eg^es szivattyútípusokhozszükséges olaj minőségét és mennyiségét a gyártóvállalatok megadják. Ennek betartása rendkívül
fontos a szivattyú helyes működése szempontjából. A fent leírt rotációs szivattyúk végvákuumaáltalában 5• 10“ 3—10~3Hgmm, szívássebességük 0,5 m3/órától 750 m3/óráig terjed.
Az eddig leírt olaj szivattyúk legfeljebb 10-3 Hgmm-es végvákuumot szolgáltatnak. Lényegesen nagyobb végvákuumot adnak az ún. kétfokozatú rotációsszivattyúk. A kétfokozatú vákuumszivattyú, melynek sémáját a 46.2.2.3. ábramutatja, lényegében két sorba kapcsolt egyfokozatú szivattyú, ahol az 1 pumpa a
2 előszivattyújaként szolgál. A kétfokozatú szivattyúkkal elérhető végvákuum
46.2.2.3. ábra. Kétfokozatú rotációs szivattyú

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 58/366
58 V Á K U U M T E C H N IK A
10“ 5 Hgmm körül van, és szívássebességük lényegesen nagyobb a kisebb nyomások
felé, mint az egyfokozatú szivattyúké. A forgó olajszivattyú működése közben előbb komprimálja az eltávolítandógázt, s ezalatt a benne levő gőzök kondenzálódhatnak a hengerben. Az olajba kerültkondenzátum nagymértékben lerontja a szivattyú teljesítőképességét, mert a vákuumtérrel érintkező szennyezett olaj gázt ad le, csökkentve ezzel a recipiens vákuumát. A beszívott gőzök kondenzálódását megakadályozhatjuk a Gaede általalkalmazott ún. gázballaszttal. Ha a szivattyúház megfelelő részén (a kompressziósoldalon) szelep segítségével atmoszféra nyomású levegőt engedünk be, a külső száraz levegő a szivattyúban levő gőzt mintegy felhígítja, s ezzel parciális nyomását akondenzációhoz szükséges nyomásnál kisebb értékre csökkenti. A szükséges bal
lasztlevegő mennyisége annál nagyobb, minél nagyobb a szivattyú szívássebessége,és minél nagyobb hányadát alkotja a kondenzálható gőz a szivattyú által szállítottgázmennyiségnek. A* ballasztlevegő mennyiségét a beengedőnyílás szelepének állításával változtathatjuk. Ha a szelep zárva van, a szivattyú mint közönséges olajszivattyú működik. Gázballasztszivattyúk ugyancsak készülnek egy- és kétfokozatú kivitelben. Végvákuumuk mindig kisebb, mint a közönséges rotációs szivaty-tyúké. Alkalmazási területek a kémiai laboratóriumi és üzemi technikában van,különösen a vákuumdesztillációk, vákuumszárítás stb. területén.
%48.2.8. Gőzsugárszivattyú
A gőzsugárszivattyú különböző fajtáiban (46.2.3.1. ábra) víz, organikus folyadék gőze vagy higanygőz áramlik nagy sebességgel divergens fúvókán keresztül
(XavaZ-fúvóka). A gőz kiterjed, miközben nyomásenergiájasebességenergiává alakul, és a nagy sebességgel áramló gáztömege a fúvókából a légtartályon keresztül irányítottáramlással a konvergens — divergens-diffuzorba jut. A lég-kamrán átmenő gőz bizonyos mennyiségű gázt ragad magával a leszívandó térből. A befogott gáztömegnek átadja
sebessége egy részét, miközben saját sebessége csökken. A két tömeg így az ún. eredő sebességgel jut a diffijzorba,ahol sebességi energiájának legnagyobb része nyomássáalakul vissza. Ez lehetővé teszi, hogy a szivattyú az eredőtömeget a leszívandó térben uralkodó nyomásnál lényegesen magasabb nyomáson távolítsa el. A befogott gáz tehátkis abszolút nyomásról valamilyen nagyobb abszolút nyomásra komprimálódik.
A gőzsugárszivattyút leginkább ipari berendezésekbenalkalmazzák, ahol nagy mennyiségű gőzt kell leszívni nem
túlságosan kis nyomáson. Üvegből készült berendezés,korrozív gőzök és gázok elszívására különösen alkalmas. Vízgőzzel működtetett ilyen rendszerű szivattyúval 1 Hgmm
körüli végvákuum érhető el. Higany vagy organikus folyadékok gőzével működtetve, a végvákuum 10~3 Hgmm lehet, a hozzá szükséges elővákuum pedig 20—1Hgmm körüli. Organikus folyadékok gőzével működtetett szivattyúk szívássebességének maximuma 10-2 H|mm körüli nyomáson van, és a méretektől függően10 m3/órától 1000 m3/óráig terjed.
46.2.3.1. ábra. Gőzsugárszivattyú

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 59/366
D I F F Ú Z IÓ S S Z I V A T T Y Ú 59
46.2.4. Diffúziós szivattyú
A diffúziós szivattyúban (46.2.4.1. ábra) pl. higanygőz áramlik az A , gázzal vagyelektromosan fűtött kazánból felfelé, és a B fúvókákpn át a C térbe jut. A higanygőz, amikor impulzusának egy részét már átadta a jelenlevő gázmolekuláknak, ahűtött D felületen gyorsan kondenzálódik. A leszívandó térből bediffundált gázmolekulák a higanygőzökkel való ütközés következtében olyan sebességkomponensre tesznek szert, mely őket a fúvóka közfeléből elviszi, és ezáltal szívást hoz létre. A higanyatomoknak a gázmolekulákkal ellentétes irányú diffúzióját megfelelőhűtéssel akadályozzuk meg (kifagyasztás). A lefeléáramló gázmolekulákat rotációs elő vákuum-szivattyútávolítja el.
A diffúziós szivattyúk működésének feltétele az,hogy a recipiensből levegőmolekulák diffundáljanaka szivattyú felé. Megfelelő hatásfokkal ez csak akkorkövetkezik be, ha a gázmolekulák közepes szabadúthossza elég nagy (kb. 10-1 Hgmm nyomás alatt). Az ilyen típusú szivattyú működtetéséhez tehát elő-vákuum szükséges, annál is inkább, mert atmoszférikus nyomáson a higany erősen oxidálódna.
A használatos diffúziós szivattyúk két vagy há
rom fokozatúak, ezek egyike többnyire gőzsugárfo-kozat. Az üvegből készült diffúziós szivattyúk szívás-sebessége 1 —30 1 /sec, fémszivattyúké 20—7000 1 /sec(10-3 Hgmm nyomáson mérve). Végvákuumuk 10“ 6Hgmm körüli értékű lehet, ha a higanygőzök visszatartásáról megfelelő kifagyasztócsapda segítségévelgondoskodunk. A higany gőznyomása 20 C°-on1,7* 10-3 Hgmm.
Az utóbbi időben igen jelentőssé vált az a típus,amelyben higany helyett organikus folyadékot, „ola
jokat” használnak. Ennek előnye a .higanydiffúziósszivattyúval szemben a nagyobb szívósebesség. A használt olajok tenziÓja ugyanis lényegesen kisebb, mint a higanyé, és általuk feleslegessé válik a kifagyasztócsapda alkalmazása. Alacsony tenziójú olajokkal folyékony levegős kifagyasztócsapda alkalmazása nélkül is elérhető olyan vákuum, amilyen higanyszivattyúval csak folyékony levegős kifagyasztócsapda segítségévelvalósítható meg.
Az ún. frakcionáló szivattyúnak olyan a szerkezeti felépítése, hogy folytonosfrakcionálást végez, vagyis benne az olaj különböző forráspontú összetevőire válik
szét. így azonos olajat használva is jobb vákuumot ad, mint a többi diffúziós szivattyú. Az olajdiffúziós szivattyúkkal elérhető végvákuum 10~6—10"8 Hgmm, atípustól és a használt olaj minőségétől függően. A 46.2.4.1. táblázat a gyakorlatbanhasznált diffúziós olajok kereskedelmi elnevezését és gőznyomását tartalmazza.
A használatban levő olajok — a szilikonolaj kivételével — igen érzékenyekoxidációval szemben, ezért az olajdiffúziós szivattyúk a higanyos szivattyúknál isnagyobb elővákuumot igényelnek (általában 5* 10“ 2 Hgmm-t). Az olaj használatának hátránya, hogy benne — ellentétben a higannyal — mindenféle gáz és gőz jól
46.2.4.1. ábra. Diffúziós szivattyú

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 60/366
60 V Á K U U M T E C H N IK A
46.2.4.1. táblázat
Diffúziós (kondenzációs) szivattyúkban használatos olajok gőznyomása
Kereskedelmi elnevezés
Z'
összetétel PMC°, Hgmm
Butil-ftalát di-n-butil-ftalát 3,3 • 10-®Oktil-ftalát di-n-óktil-ftalát 4,0 • 10“8Octoil ......................... di-3-etil-hexil-f tálát 3,3 •1 0 "7 —3,3 •10-*Narcoil 40 di-( 3,5,5)-trimetil-hexil-ftalát 6,0* 10“8
Amoil ......................... di-i-amil-ftolát 1,3 • 10-® Amoil S di-i-amil-szebacát 1,0 •10~6
Octoil S di-3-etil-hexil-szebacát 2,0 • 1 0 "8m-Cr tri-m-krezil-foszfát 9,0 10~8p-Cr ............................. tri-p-krezil-foszfát 2,0 •lO"8
Ápiezon A petróleumdesztillációs termék 2,0 • 10“ 6 Apiezon B petróleumdesztillációs termék 4’0 •10~7 Apiezon C petróleumdesztillációs termék 1,0 10“ 8 Arochlor , .................. klórozott szénhidrogén 8.0 •10~6Szilikon ÜC metil-fenil-szilikonolaj 6,0 •10-®
oldódik. A frissen töltött vagy lelevegőzött olajdiffúziós szivattyú maximális végvákuumát csak 8—10 órai üzemeltetés után éri el.
A diffúziós szivattyúk működtetésének általános szempontjai a következők: A higany- és az olajdiffúziós szivattyúk használatánál is ügyelni kell a kazán megfelelő fűtő teljesítm ényé re. Az optimálisnál nagyobb vagy kisebb fűtésnél ugyanis aszívássebesség-jelentős mértékben csökken. A kereskedelmi forgalomban levő szivattyútípusok optimális fűtőteljesítményét a gyártó vállalat pontosan megadja.Ügyelnünk kell még az elővák uvm értékének megtartására. Minden diffúziós szivaty-tyútípushoz tartozik egy olyan elővákuum-érték (az un. letörési nyomás), amelyalatt a szivattyú nem működik, felette pedig szívássebessége független az elővá-kuumtól. Diffúziós szivattyúknál, különösen a higannyal működőknél, lényeges a
szivattyú megfelelő hűtése. Elégtelen hűtés a szívássebesség rohamos csökkenését ésa higanynak vagy olajnak a leszívandó térbe való bejutását eredményezi. Olajdiffúziós szivattyúk a hűtésre kevésbé érzékenyek, készülnek léghűtéses kivitelben is.Ezeknél többnyire elég vízzel hűtött olajcsapdát használni, de 10~7 Hgmm-nél
jobb vákuum elérésére itt is célszerű a folyékony levegős kifagyasztócsapda alkalmazása. Higanydiffúziós szivattyúk esetén, ha 10-3 Hgmm-nél jobb végvákuumotakarunk elérni, mindenkor cseppfolyós levegővel hűtött kifagyasztócsapdát kellalkalmazni.
46.3. Vákuum mérés
Kis nyomások mérésére a vákuumtechnikai irodalom igen sokféle módszert ír le. Az alábbiakban a kémiai gyakorlat szempontjából jelentős mérési módszereket foglaljuk össze.
46.3.1. Vákuummáik álás elektromos kisüléssel
Elektromos kisüléssel észlelni tudjuk a vákuum beállását 3 •10“ 1és 10~3 Hgmmhatárok között, és legalább a nagyságrendet jól meg tudjuk becsülni. Ha a leszívotttérhez kisülési csövet kapcsolunk, és elektródjait nagyfeszültségű áramforrással

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 61/366
VÁ K T JU M M É BÉ S 61
kötjük össze, kb. 30 Hgmm alatt gázkisülést kapunk, melynek képe a nyomás csökkenésével változik. 10-1 Hgmm alatt az üvegfal is fluoreszkál, 10-2 Hgmm-nélkisebb nyomásokon már csak a fluoreszcencia marad meg, és 10-3 Hgmm alatt ezis fokozatosan megszűnik. A kisülés színe a gáz anyagától függ.
46.3>2. Higanyos manométerek, a közlekedőedények elvén mfiködővákuummérő berendezések
Legegyszerűbb változatuk azún. nyitott differencidlmanométer. Ez csökkentettnyomások mérésére 760 mm-nél magasabb szárú, U alakúra hajlított cső, melyben
a külső levegőt a vákuumtér gázaitól kb. 80 cm hosszú higanyszál választja el. Akét végére ható nyomás különbségét a higanyfelszínek magasságkülönbsége mutat ja. Az atmoszféra nyomástól csak kismértékben eltérő nyomások mérésére a higanyigen alkalmas manométer-zárófolyadék, mert tenziója szobahőmérsékleten kicsi,ezenkívül a felületével érintkező gázokatnem oldja. Hátránya nagy felületi feszültsége folytán a csővezetékben fellépő kapilláris depresszió. Tiszta higany és csőfelület esetén ez 8 mm belső átmérőjű csőbencsak ± 0,07 mm, 16 mm átmérőjű csőbenlegfeljebb ±0,01 mm pontossággal reprodukálható. A két meniszkusz különbségével mérő nyitott manométer esetén a depressziós hiba kiesik ugyan, de csak akkor,ha mindkét felület azonos összetételű ésnyomású gázzal érintkezik. A gyakorlatban, a kémiai laboratóriumban fennállómérési körülmények között a higany felülete kémiai szennyeződésnek van kitéve, ezért a mérések reprodukálhatósága az
előbbinél lényegesen rosszabb. Az 1 —200 Hgmm közötti nyomások mérésére általában a rövidített manométer használatos. A fent említett hibalehetőségek elkerülése céljából a‘gondosan tisztított és szárított manóm étercsövet nagyvákuumban desztillált higannyal töltjükmeg. A 46.3.2.1. ábrán rövidített manométerek készítésére alkalmas készüléketmutatunk be. Az A csonkhoz csatlakozó szivattyúval evakuált edényt és higanytleforrasztás előtt gáztalanítjuk. Ezután a manométercsövek megtöltését egyszerűátöntéssel végezhetjük. A rövidített manométérrel való mérés pontosságát legnagyobb mértékben a Torricdli-ű .rbe esetleg bejutó gázok befolyásolják (nyomásuk aszintkülönbséget csökkenti). A nyitott szárban levő meniszkusz szennyeződéséből
származó hibát jelentősen csökkenti a megfelelő helyen beépített ún.^Bzmsew-csiúcs(46.3.2.2. ábra), mely a rajta átáramló higany felületi szennyeződéseit visszatartja. A rövidített manométerek érzékenysége, ha azokat szilíkonolajjal töltjük, az alacsony nyomások felé egy nagyságrenddel növelhető. Ha a higanymanométerrelnagyon kis nyomásokat akarunk mérni, akkor a szintkülönbség mérését katetomé-terrel kell végezni. A depressziós hiba a manométercső átmérőjének növelésévelküszöbölhető ki.
46.3.2.1. ábra. Rövidített manométerek készítése
46.3.2.2. ábra. Bunsen-CBÚcs

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 62/366
62 VÁ K.UTJ MTE C H N IK A
46.8.3. Kompresszióé vókuummérő
A MacLeod-féle manométer az ideális gázok nyomása és térfogata közötti összefüggést kifejező Boyle—Mariotte-törvény alapján működik (46.3.3.1. ábra). A mérést úgy végezzük, hogy a higany szintjét az A segédkapillárisban a B mérőkapilláris csúcsáig emeljük. A mérőkapillárisban a higanyszint k mm-rel alacsonyabb lesz,mert ott a kapillárist és a hozzá csatlakozó G térfogatot kezdetben betöltő gáz kom-
primálódott. A q keresztmetszetű mérőkapillárisban akompresszió előtti p nyomás p'-re változik. Ezek ismeretében felírhatjuk a következő összefüggést:
p' [h-q) = p-C.
A komprimált gáz p' nyomása h Hgmm-rel nagyobb azeredeti p -nél, tehát
p' = p + h.
A két egyenletet összesítve:
P'h-q + h2-q = p-G ,
amelyből megengedhető közelítéssel:leveg
el vakuum
p = l - h 2. C
— a műszerre jellemző konstans érték; a skála kvadrati-
46.3.3.2.^ó6m.^f^£.eo(i-^ ]jUSkalibrálása a konstans meghatározásával történik. A műszer annál érzékenyebb, minél kisebb p-t jelez az
egységnyi h szintkülönbség, vagyis minél kisebb — számértéke. Ennek gyakorlatiGhatára részint a nagy tömegű higany felemelésének nehézkessége (maximálisan300—500 ml), a keskeny kapillárisban pedig a higany tapadása (minimális átmérő0,5—0,7 mm). A tapadást többnyire a higany oxidációs termékei és egyéb fém-oxi-dok okozzák, ezért gondosan tisztított, vákuumban desztillált higanyt célszerűhasználni. A MacLeod-manométer alsó mérési határa általában 10-5 Hgmm, különleges berendezések méréshatára 10~7 Hgmm is lehet. A mérési határ nagyobbnyomások felé kétszeres (alul bővebb) mérőkapilláris alkalmazásúval terjeszthetőki. Ennek megfelelően egy bővebb és egy szűkebb segédkapillárisra van szükség.
A MaeLeod-m&nométeT használatát kényelmesebbé tehetjük, ha a higanyszintemelését/nívóedény helyett a külső légköri nyomással végeztetjük. Az ábrán látható csapot a külső levegő felé fordítva, a higanyszint emelkedik, elővákuumraállítva pedig a higany leszívható, és a műszer újabb mérésre kész. Előnye, hogyegyszerű kalibrálással, q és C meghatározása után abszolút értékben adja anyomást. Hátránya, hogy a könnyen kondenzálható gőzök nyomását nem mutatjamegbízhatóan, mivel ezek a kapillárisban a kompresszió következtében cseppfolyó-sodhatnak. Ez a mérendő gázban levő gőzök gőznyomásától és a hőmérséklettől

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 63/366
VÁKTTUM MÉRÖK 63
függően következhet be. Kifagyasztócsapda alkalmazásával ez a hiba kiküszöbölhető, de ez esetben a ifocLeod-vákuummérő csak a permanens gázok parciális nyomását méri, éB nem a rendszer teljes nyomását. A Jfocieod-manométer működésielvéből következik, hogy a mérés szakaszos, tehát folyamatos mérésre vagy gyorsváltozások észlelésére nem alkalmas.
46.11.4. Radiométer típusú nyomásmérők
Kis nyomású gáztérben két különböző hőmérsékleten tartott felület közötterő lép fel. Ez annak tulajdonítható, hogy a melegebb felületbe ütköző molekuláknagyobb, a hideg felületbe ütközők kisebb átlagos kinetikus energiával verődnek
vissza. Ezt az elvet használják fel a Knudsen-féle vá- kuummérőben, melynek vázlata a 46.3.4.1. ábrán látható. A fűtött B —B felületről jövő molekulák .kinetikus energiája nagyobb, mint az A lemez B —B-vel ellentétesoldalába ütköző molekulák energiája. Ez az A —A lemez elfordulását eredményezi. Kimutatható, hogy a fellépő erő a 10-4—10-8 Hgmm tartományban a gáznyomás és a fennálló hőmérséklet függvénye, és nem függ a
jelenlevő gáz anyagi minőségétől. Mivel az elfordulást anyomás függvényében leíró Összefüggés minden adata
közvetlenül mérhető, a Knud&en-féle vákuummérőabszolút műszernek tekinthető és más vákuummérőműszerek kalibrálására használható. A kereskedelmiorgalomban levő Kmidsen-féle vákuummérők mérésha-ára 5» 10-4—10-7Hgmm-ig terjed, pontosságuk 1—2 %.
46.3.4.1 . áb r a .A K n u d sen - -féle nyomásmérő vázlata
46.3.5. Gázok belső súrlódásának nyomásfüggésén alapuló vákuummérők
A vákuumtérben lengésbe hozott kvarcszál lengési amplitúdójának felére
csökkenéséhez szükséges időből (fi/2) a gázok közepes molekulasúlyának (M^) ismeretében kiszámíthatjuk a nyomást a
p -YMh = — — ahl2
összefüggés alapján ahol p a nyomás, a és b a kvarcszál méreteitől függő állandó.Mérési határa 10-1 —10-4 Hgmm között van, össznyomást mérő műszer. Mindengázra kalibrálni kell. Alkalmazása akkor indokolt, ha rendkívül korrozív gázokvagy gőzök jelenlétében kell vákuummérést végezni.
46.3.6. A hővezető-képesség nyomásfüggésén alapuló vákuummérők
Ismeretes, hogy kis nyomásokon, ahol a hideg és a meleg felület közötti távolság Összemérhető, vagy kisebb, mint a gázmolekulák közepes szabad úthossza, agáz hővezető képessége függ a nyomástól. Adott (Tt—T0s 100 C°) hőmérséklet-- különbségnél és 1—10~4 Hgmm nyomáson, ahol a sugárzási és egyéb veszteségekelhanyagolhatók, a gáz hővezetőképesség-változásából származó veszteség ará-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 64/366
64 V Á K U U M T E C H N IK A
nyosan változik a nyomással. A gáz hővezető képességének változása alapjánműködő nyomásmérő műszerek bemenő energiáját állandó értéken tartjuk. Ennekkövetkeztében a felület hőmérséklete növekvő nyomással csökken.
A hővezetőképesség-változás alapján működő, különböző típusú műszer eseténkülönböző módszereket, használunk a hőmérséklet-változás mérésére. A termő-elemes típusban a hőmérsékletet a fűtött fémszál közepére hegesztett termoelem*mel (termokereszt) mérjük. A termoelemes nyomásmérő csövet és a hozzá tartozóelektromos áramkört a 46.3.6.1. ábra mutatja.
vákuum mér fej r & ly , -
vákuum
fútöszót
termokereszt
a I ó\b b
46.3.6.1. ábra. Termoelemes nyomásmérő
vákuum
46.3.6.2. ábra. APiram-manométer elektromos kapcsolása
A Piráni- féle vagy ellenállásmérő típusban a nyomást fűtött fémszál (Pt, Nivagy W) ellenállás-változásából határozzuk meg. A Pirani-féle nyomásmérőt éselektromos kapcsolását a 46.3.6.2. ábra mutatja. A mérőhíd egyik ágába egy igenkis nyomásra leszívott, a mérőcsővel azonos felépítésű leforrasztott csövet kapcsolunk a külső hő mérséklet-változás kompenzálása céljából.
A term isztoros vákuum m érő elve lényégében azonos a Pirani- fé le nyomásmérőelvével. A disszipációé körülmények és ezzel együtt a termisztor feszültség—áramerősség-karakterisztikája különböző nyomások esetén nagymértékben változik(lásd 45.2.2.) A mérés érzékenysége különösen a nagyobb nyomáshatárok felé bővül(10 Hgmm-ig). 10-3 Hgmm nyomás alatt a termisztor érzéketlen, mert a gázrészecskék közepes szabad úthossza itt már igen nagy, a hőelvezetés csekély, a termisztormajdnem kizárólag sugárzás útján adja le teljesítményét. A termisztornak a Piran i -
manométerrel szemben az az előnye, hogy mérete lényegesen kisebb, s ugyanakkor
szennyezésekkel szemben kevésbé érzékeny. A mérőberendezés kapcsolási elve,amit a 49.5.2.3. ábrán szemléltetünk, lényegében azonos az áramlásméréshez alkalmazott kapcsolásokkal.
A hővezető-képesség nyomásfüggésén alapuló műszerek össznyomást mérnek,méréshatáruk 10—10-4 Hgmm-ig terjed, egyes különleges berendezésekben ennéllényegesen kisebb vákuumok is mérhetők. Érzékenységük függ a jelenlevő gázminőségétől, tehát minden gázra vagy gázkeverékre kalibrálni kell.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 65/366
VÁKUtTBOCÉltŐK 65
46.S.7. Ionizációs vákuummérök
Mérési elvük: ha gáztérben adott sebességű elektronok haladnak, ezek a gáz-molekulákkal ütközni fognak. Amennyiben az elektronok kinetikus energiája egybizonyos értéket meghalad, az ütközés következtében a gáz ionizálódik, pozitív ionés egy elektron keletkezik. Ugyanazon gáz és elektron-kinetikus energia eseténaz egységnyi úthosszon keletkező ionok száma, az ún. differenciális ionizáció csakaz ütközések valószínűségétől, vagyis a nyomástól függ. Az ionizációs manométe-rek méréshatárát a nagyobb nyomások felé az ion szabad úthossza, illetve rekombi-nálódása határozza meg, továbbá az, hogy a gáznyomásnak olyan kicsinek kelllennie, amin az izzókatód (mely az elektronokat szolgáltatja)még nem oxidálódik,illetve mérgeződik.
Az ionáram és elektronáram közötti viszony a nyomásra jellemző, és a következőképpen fejezhető ki:
ahol 8 a mérőcső érzékenységi állandója, mely függ a cső geometriai méreteitőlés a jelenlevő gáz minőségétől. Az ionizációs vákuummérő cső lényegében három-elektródás elektroncső, trióda, mely a mérendő nyomású rendszerhez csatlakozik.Elektronforrás a cső katódja, mely általában volfrám, néha emittáló réteggel be
vont platina. A trióda rácsa az elektronkollektor szerepét tölti be és-(-150-1— 180Vfeszültségen van, míg anódja az ionkollektor, mely a katódhoz viszonyítva —20—25 V feszültségű. Az ionizációs vákuummérőkelvi kapcsolását a 46.3.7.1. ábra szemlélteti.
A gyakorlatban nem az ionáram/elek -tronáram értéket, hanem az állandó elektronáram, továbbá elektronkollektor- és ion-kollektor-feszültség mellett jelentkező ion-áramot mérjük. Az elektronáram és a kollek-
torfeszültségek állandó értéken tartását aziparilag gyártott készülékekben elektronikusstabilizátor, illetve szabályozórendszer biztosítja. Az ionizációs manométer céljárahasznált mérőcsövek alkatrészeinek könnyenés jól gáztalaníthatóknak kell lenniük, mertkis nyomások mérésénél a cső belső felületén megkötött gázok csak lassan de-szorbeálódnak és jelentősen meghamisítják a mért nyomásértéket. Az izzókatódosionizációs vákuummérök méréshatára 10-3 —10-8 Hgmm-ig terjed, ennél alacsonyabb nyomások közönséges ionizációs vákuummérő csövekkel nem mérhetők,mert a csőnek a katódból az elektronkollektorba ütköző elektronok hatására keletkező lágy Röntgensugárzás folytán maradékárama van, mely független a nyomástól. Az általánosan használt ionizációs vákuummérőknek csak 10“ 8 Hgmmfeletti nyomásokon van olyan ionáramuk, mely észrevehetően nagyobb a maradékáramnál. EgyeB speciális ionizációs vákuummérő csövekkel 10“ 11 Hgmm nyomásig lehet mérni.
Az ionizációs vákuummérő össznyomást mérő műszer, érzékenysége függ a5 Ált. és szervetlen kémiai prakt. —4284 f i i .
46.3.7.1. ábra. Ionizációs manométer kapcsolási vázlata

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 66/366
66 V Á K U U M T E C H N I K A
gáz minőségétől. Irodalmi adatok szerint argonra vonatkoztatott érzékenységi állan
dójának és egy adott gázra vonatkoztatott állandójának hányadosa ( — = r J.. .. .. „ ....................... \Qx J kuionbozo geometnajú csövek es eltérő üzemadatok esetében sem nagyon tér elegymástól. E hányadosok ismeretében elég a csövet egy gázra kalibrálni (argon),ebben az esetben g az r-értékek ismeretében más gázokra kiszámítható.
46.3.7.2. ábra. A Penning-féle manométer elektromos kapcsolása
Izzókatódos vákuummérők működésük közben gázt fogyasztanak (sajátszívás).Ezt a hibát a mérésnél figyelembe kell venni, illetve a mérőcsövet úgy kell összekötni a mérendő térrel, hogy a vezeték ellenállása kicsi legyen, mert ellenkező esetbennyomáskülönbség lép fel a manométercső és a mérendő tér között, és a vákuummérő
a valódinál kisebb nyomást mér. Az ionizációs vákuum-mérő szívássebessége 0,1—1 1/perc érték között van.
Az ionizációs vákuummérőknek még két fajtájaterjedt el a gyakorlatban: a Penning-ié\e hidegkatódosionizációs vákuummérő és az Alfatron vákuummérő.
A hidegkatódos vagy Penning-féle vákuummérö csőben az ionizáló elektronok cirkónium- vagy vaskatódbólnagy feszültség (2000 V) hatására lépnek ki. Az elektronok úthosszának és evvel az ionizáció hatásfokának megnövelésére Penning a kisülési teret egy kb. 500 Gauss erősségű mágneses térbe helyezte. A mágneses térremerőlegesen haladó elektronok a mágneses tér hatásáracsavarvonalú pályára kényszerülnek, és így a két elektróda között megtett út az eredetinek sokszorosára nő. A jelenlevő gázok nyomását a teljes kisülési áram jelzi. A Penning által eredetileg készített cső 5* 10“ 3—10-5Hgmm nyomástartományban mért. A később Penning és Nienhuis által módosított cső méréshatára
5*10-3—10-7 Hgmm-ig terjed. A Penning-féle cső és a hozzá szükséges elektromos rendszer kapcsolási rajzát a 46.3.7.2. ábra szemlélteti. A hidegkatódos ionizációs vákuummérőt igen elterjedtén használják a gyakorlatban egyszerűsége éslevegőbetörésre való érzéketlensége miatt. Ossznyomást mérő műszer, érzékenysége függ a mérendő gáz minőségétől. Minden gázra kalibrálni kell.
3 0 -4 0 V
Er sít ,
0leolvasó műszer
rács
fémköpeny
u sa------ °k t '. v
™ | készítmény
vákuum
46.3.7.3. ábra. Alfatron

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 67/366
A V Á K Ü T T M B E R E N D E ZÉ S T A R T O Z É K A I 67
Az Alfatron vákuummérőben az ionizációt a mérőfejben elhelyezett radioaktívpreparátumból származó alfa-részecskék végzik. A 46.3.7.3. ábra az Alfatron vá
kuummérő felépítését és kapcsolását mutatja be. A műszer hátránya, hogy az ion-áram erőssége az izzókatódos vagy a Penning-féle vákuummérőkkel szemben sokkalkisebb, továbbá, hogy használatánál radioaktív sugárzás miatt sugárzásvédelemrőlkell gondoskodni. Méréshatára 10-2—10-5 Hgmm-ig terjed, össznyomást mér,érzékenysége függ a jelenlevő gáz minőségétől.
46.4. A vákuumberendezés tartozékai
46.4.1. Kifagyasztócsapdák, terelőlemezek
hddct n
A higannyal működő szivattyúknál 10-3 Hgmm-nél jobb vákuum elérésérekifagyasztócsapdákat kell használni. Kifagyasztás céljából a vezeték egy szakaszát C02-os hűtőkeverékkel vagy folyékony levegővel hűtjük, és a kifagyasztottgőzöket összegyűjtjük. Ezeket a gŐz-csapdákat úgy kell megszerkeszteni, hogynagy legyen a hűtőfelületük, de az áramlási ellenállást ne növeljék meg túlságosan.Előnyös, ha a kondenzált anyag könnyen
eltávolítható. Néhány típust a 46.4.1.1.ábrán mutatunk be. A szárazjég-keverékek hőmérsékletén
(—79 C°-on) a higany tenziója 3*10-9Hgmm, a higany tehát gyakorlatilag tel
jesen távol tartható. A higany szivattyúkkal szemben előnyös az olyan olaj szivattyúk használata,
amelyek szobahőmérsékleten csak igen kis tenziójú (10—5Hgmm) olajjal működnek. A csapdákban fellépő hátrányos ellenállás-növekedés miatt az újabb, olajdiffúziósszivattyúval működő vákuumkészülékekben jóval kisebb ellenállású, vízzel hűtöttterelőket vagy olaj csapdákat alkalmaznak.
W
46.4.1.1. ábra. Fagyasztó- és szárítócsapdák
46.4.2. Csővezetékek, csapok, illesztés
A vákuumtechnika szerkezeti anyagai között kezdettől fogva szerepelnek az üvegés a kvarc mellett a különböző fémek. A laboratóriumi berendezések ma is főkéntüvegből és kvarcból készülnek, a nagyobb méretek esetén fennálló fokozott szilárdsági és biztonsági igények miatt azonban újabban egyre nagyobb mértékben alkalmaznak fémalkatrészeket.
A felhasználásra kerülő vezetékek geometriai méreteitől nagymértékben függa rendszer áramlási ellenállása (lásd 46.1.). Minél kisebb ellenállás biztosítása céljából a vezeték a lehető legrövidebb legyen, belső átmérője pedig 15—20 mm-nél nelegyen kisebb. Elkerülendők a meredek hajlítások és a szűkületek. A csapok minimális furatátmérője 10 mm. Az alkalmazott csapok összeillő felületeit finomankell csiszolni, és a biztos zárás érdekében megfelelő nagyságúra kell méretezni, A furatokat nem célszerű egymással szemben elhelyezni, hanem az ún .ferde furaté csapok alkalmazása előnyös (46.4.2.1. ábra). Ezek elforgatásakor is keletkeznek
5*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 68/366
68 V Á K U U M T E C H N IK A
ugyan a csapzsír-rétegben vájátok, de rajtuk keresztül nem léphet fel zárt helyzet
ben szivárgás, mivel különböző magasságban képződnek és egymás között nemközlekedhetnek.Nem ajánlatos a csapok testét tömör üvegből készíteni, mert ezek melegítésre
sokkal érzékenyebbek, mint az üregesek. Az üreges testű csapokat viszont célszerűúgy szerkeszteni, hogy belsejük kapcsolatban álljon az evakuált térrel (46.4.2.2.ábra). Ilyenkor ugyanis a külső nyomás nagy erővel nyomja a csapot a tokba, éselősegíti a biztos zárást.
A csapzsírban képződő légvezető váj átok kiküszöbölhetők higanyzáras csapok alkalmazásával. Ennek két típusát mutatja be a 46.4.2.3. ábra.
46.4.2.1. ábra. 46.4.2.2. ábra. 46.4.2.3. ábra. Higanyzáras vákuumcsapok Vákuumcsapok Vákuumcsap
Ha a vezetéket bizonyos helyen rendszeresen szét kell szedni, a csatlakozástcsiszolattal lehet megoldani. Ha a tengely körüli elforgás veszélye áll fenn, akkor kúpos csiszolatot, egyidejű hajlítás lehetőségénél gömbcsiszolatot, kisebb párhuzamoselmozdulások biztosítására pedig síkcsiszolatot alkalmazunk.
A kúpos csiszolatok közül a nemzetközi szabvány által meghatározott hajlásszögű, egymás között cserélhető ún. normálcsiszolatokat javasoljuk.
A csapok és csiszolatok összeillő felületei a leggondosabb megmunkálás ellenére
sem akadályozzák meg a levegő beszivárgását a vákuumrendszerbe. A mozgat-hatóan illesztett felületeket ezért csapzsírral, a ritkábban szétszedett csiszolatokatpedig reverzibilis kittekkel szokás tömíteni. A vákuumtechnikai célokra használatos csapzsír legfontosabb tulajdonsága az alacsony tenzió. Kisebb igények esetén(10-4 Hgmm-ig) a Bamsay-zsírt használjuk. Tenziója 20 C°-on 10-4—10-5 Hgmm.Paraffinból, vazelinből és kaucsukból készül 1 : 3 : 7 — 1: 8 : 16 közötti súlyarányban, egyszerű összeolvasztással.
A különböző típusú Apiezon-zsírok (L, M, N) tenziója 10-5 Hgmm és ennélkisebb. Megfelelő gáztalanítással tenziójuk mérhetetlenül kicsivé tehető. Típusuktól függően 20—80 C°-ig használhatók. Kitűnő minőségű vákuumcsapzsír készülszilikonokból is. A szilikonzsírok 200 C°-ig is használhatók.
Ha a berendezésben a csapzsírt megtámadó vagy oldó gőzök képződnek, azsírok helyettesítésére szükség esetén 10"2—10-4 Hgmm nyomásig P20 5 vagyH2S04 használható.
Az említett szükséganyagok használatát elkerülhetjük kenést nem igénylő szelepek alkalmázásával. Ilyenek a különböző fémmembrán- (Cu, Ag, Pt), tányér-,tű-, üveg- és higanyos szelepek. Ezek közül a korrozív gáz tulajdonságainak ismeretében ki tudjuk választani a megfelelőt.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 69/366
A V Á K U T T M B E R E N D E ZÉ S T A R T O Z É K A I 69
A csiszolatok és csapok tömítése akkor jó , ha a zsír vékony és egyenletesrétegben vonja be az illeszkedő felületeket. Ilyenkor a csapzsír teljesen átlátszó,
benne semmiféle légbuborék vagy egyéb zárvány nincs, sem áramlási vonalak,nem láthatók. Ezt úgy érhetjük el, ha a csapot előzőleg gondosan megtisztítjuk(szén-tetrakloriddal, benzollal), és a csapdugóra visszük fel a szükséges mennyiségűzsírt. Ezt egy-egy gyűrű alakú rétegben a furat alatt és fölött helyezzük el. Ezutána nyitott helyzetnek megfelelően gyengén belenyomjuk a dugót az előzőén enyhénmelegített tokba, és a kenőanyag egyenletes eloszlatását olyan kis'szögben végzettforgató mozgással végezzük, hogy közben a csap sohase záródjék teljesen. A csapotcsak ezután forgassuk körbe. A csapok forgatását mindig olyan lassan végezzük,hogy közben túl ne lépjük a csapzsír folyási sebességét, mert ez a kenőréteg szakadás
sát eredményezi és tömítetlenséget okoz. Az elforgatás sebességhatárának túllépését a forgási ellenállás megnövekedése jelzi. A vékony csapzsírréteg néhány hónapalatt többnyire megkeményedik, ezért időnként meg kell újítani.
A ritkán mozgatott csiszolatok összeragasztásán kívül a reverzibilis kitteket használják fel a különböző anyagból készült (tehát különböző tágulási együtthatójú) alkatrészek illesztésének tömítésére is. így pl. a fémdiffúziós szivattyúkszívócsonkját a rendszer üvegből készült részéhez piceinnel ragasztott csiszolattalszokás csatlakoztatni. A picein réteg csak akkor zár megfelelően, hateljesen száraz és zsírtalanított felü
letre tud tapadni. Az üveget ezértenyhén előmelegítjük, és a meglágyított picein ráhelyezése után melegítését a kitt elfolyósodásáig folytatjuk. Vigyázzunk arra, hogy közben a picein lángra ne lobbanjon,sem pedig bomlást, illetve kokszo-sodást ne szenvedjen. A másik,ugyancsak előmelegített felületet ilyenkor illesztjük a piceinre. Enyhe melegítés
közben lassú forgatással gondoskodunk az egyenletes eloszlásról és a hézagtalantapadásról.Csővezetékek csiszolat és forrasztás nélküli csatlakoztatását különböző kittek
segítségével a 46.4.2.4. ábra szerint végezzük. A vákuumtechnikában használatos reverzibilis kittek összetételét, tenzióját
és egyéb tulajdonságait a 42.5.2. pontban már tárgyaltuk. Az irreverzibilis kittek(ólom-oxid—glicerin, vízüveg—talkum, kaolin—bórax) nem elég vákuumzárók,csak reverzibilis kittel bevonva használhatók.
A vákuumberendezés üveg- és fémalkatrészeinek összekötését forrasztással ismegoldhatjuk. Erre vonatkozóan lásd 42.3.
Újabban egyre nagyobb mértékben terjed a fémek alkalmazása a vákuum-technikában. A fémből készült nagy teljesítményű szivattyúkhoz nagyméretű
fémcsapok és szelepek, rugalmas és hajlítható fémvezetékek készülnek. Előnyükaz üvegnél nagyobb mechanikai szilárdság, kisebb törési és repedési veszély, valamint a méretek és a teljesítmény egyszerűbb növelési lehetősége, egyszóval a jobbkezelhetőség, aminek főleg az ipari alkalmazás esetén van nagy jelentősége. A fémalkatrészek egymáshoz illesztése finoman megmunkált és zsírozott síkcsiszolatokkal,vagy a szemben fekvő részek között elhelyezett, néhány milliméter átmérőjű, gyűrű
46.4.2.4. ábra. Csővezetékek csatlakoztatása

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 70/366
70 V Á K U U M T E C H N IK A
alakú vájatba helyezett gumikarikával lehetséges. Rugalmas, hajlítható fémvezetékek a cső tengelyére merőlegesen hullámosított tombaklemezből készülnek(46.4.2.5. ábra).
Nagy vákuumban használható, megfelelő kis áramlási ellenállású hajlékonyvezetékek csakis fémből készíthetők. Az ún. vákuumgumicsövek legfeljebb 10-3Hgmm-ig jöhetnek tekintetbe. Ugyanis nagy falvastagságuk ellenére sem eléggégázzárók, és a vízgőzt is jelentékeny mértékben áteresztik.
Az előbbiekben tárgyalt fémvezetékekbencsapok helyett a nagy keresztmetszettel nyíló
46.4.2.5. ábra. Hajlítható fémcső síkcsiszolatos csatlakoztatással
46.4.2.6. ábra. Tányérszelep nyitott és zárt helyzetben
mikrométer- csavar
f i
tányérszelepeket alkalmazzák (46.4.2.6. ábra). Előnyük a csapokkal és tűszelepekkel szemben az, hogy lényegesen kisebb az áramlási ellenállásuk.
A gyakorlatban egyéb területeken is használatos más szerkezetű tű-, memb
rán-, redukciós stb. szelepekkel az első könyvben foglalkoztunk. Agresszív, fémeket korro-
deáló gázokkal végzendő munkákhoz, valamint olyan esetekben, amikor csapzsír vagy egyébkenőanyagok nem használhatók, j ó szolgálatot tesz a telj esen üvegből készült Bodenstein-féle szelep (46.4.2.7. ábra). Ebben a fém-
membránt üvegből készülő ru;galmas test helyettesíti, mely atengelye irányában kb. 0,5—1,0mm össznyomást tud elviselni.
A beforrasztott kapilláris nyílását vele gondosan összecsiszolt üveggömb zárja. A két zsírozatlan üvegfelület pontosösszenyomását mikrométercsavarral végezzük. A szelep nyomáscsökkentőként ishasználható, és vákuumbiztosan zár.
Üvegkészülékekben, ha nem kell tartani a higany korróziójától, kényelmesenhasználhatók a zsírozatlanul működő Stoők-féle szelepek (46.4.2.8. ábra). Az első
üvégharm on ika gömbcsiszola t
46.4.2.7. ábra. Bodenstein-féle üvegszelep

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 71/366
A V Á K U U M B E R E N D E Z É S T A R T O Z É K A I 71
típusban a zárást a higany felületén úszó csiszolt gömbfelületű szeleptest végzi. A másik működésének az alapja az, hogy a fritt (zsugorított üvegszűrő, porózusüveglemez) a gázok számára átjárható, higanyra nézve nem. Az utóbbinak hátránya
a fritten fellépő nagy áramlási ellenállás.Preparatív feladatok megoldása közben, ha csak egyszeri nyitásra van szükség,a drága Bodenstein-ezelep helyett törőszelepet használunk. A finom végű kapillárisban vagy vékony falú gömbben végződő tartályt hozzáforrasztjuk a csővezetékhez, amelybe előzetesen beleforrasztott vasmagot tartalmazó kis üvegcsövet, „kalapácsot” teszünk (46.4.2.9. ábra). (A kalapács két végébe a fém és üveg közécsipetnyi azbesztvattát tegyünk, hogy a kalapács el ne törjön.) Függőleges helyzetben mág-nes segítségével kb. 10—15 cm magasra emelve
vákuum
i múnka- ■j-i térhez
46.4.2.8. ábra. Stock-féle szelepek 46.4.2.9. ábra. Mágneses törőszelep
a kalapácsot, ráejtjük a törőszelepre. A nyíláson át az anyag bejuthat a munkatérbe,amit azután a csővezeték leforrasztásával újra elzárhatunk. Mivel nagyobb átmérőjűcsöveket vákuumban nehéz leforrasztani, célszerű előre leszűkített és megvastagított falú szakaszokat képezni a törőszelep használata után újra lezárandó csővezetéken. A leforrasztandó szűkületet igen óvatosan és egyenletesen többször meglágyítjuk, végül a legmelegebb fúvólángban megolvasztjuk úgy, hogy a cső kihúzás
és falának elvékonyodása nélkül záródjék. Ha az egjfk oldalán erősebben meglágyult üveget a vákuum beszívja, a homorú résznél könnyen bekövetkezhet a repedés, törés. Az előzőleg elzáródott üvegcsövet szabad csak kihúzással megszakítani. A képződött üvegszálat kis gömbbé olvasztjuk össze és lassan lehűtjük.
A 46.4.2.10. ábrán bemutatott két törőszelep működtetéséhez nem szükségesmágnes. Közülük az egyik (a) szilárd, a másik (b) folyékony anyagnak az evakuálttérbe juttatását oldja meg.
A törőszelepek alkalmazási területéhez hasonló feladatok megoldására jól felhasználható a vákuumvezetéknek egy csaknem kapillárissá szűkített csőszakasza,
mely könnyen olvadó Wood-íémme\ zárható el.Ha a reakciót vagy mérést meghatározott gáz adott nyomásán akarjuk végrehajtani, előzőleg evakuáljuk és kigázosítjuk a berendezést, majd meghatározottsebességű gázáramot engedünk a készülékbe, amelyben a szívássebességtől függővégvákuum fog beállni. A nyomásszabályozás tehát a gázbevezetés sebességénekszabályozásával történik. Ez általában korlátozott pontosságú módszer, és főleg1 Hgmm-nél nagyobb nyomásokon alkalmazzák.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 72/366
72 V Á K U U M T E C H N IK A
A 46.4.2.11. ábrán levő berendezés tűszelepszerűen működik. Az áramlásiellenállást, illetve a gázáram sebességét a kapillárisban elmozdítható drót benyú-lásának mértéke szabja meg.
A Wóhl-fÜe csap (46.4.2.12. ábra) úgy szabályozza a gázáramot, hogy az elve
zetőcső állandó keresztmetszetű nyílása előtt a csapdugó ék alakú nyílása az elforgatás mértékétől függő nagyságú rést hagyszabadon. A gázáram ilyen módon jólreprodukálhatóan beállítható. Zsíroznicsak a rés alatt és fölött szabad a csapot.
munkatér
46.4.2.10. ábra. Törőszelepek 46.4.2.11. ábra. TŰBzelepes gázáram-szabályozó
Újabban 10“ 6 Hgmm-ig használható, önműködően szabályozható, tűszelepes,fémből készült, vákuummérővel egybeépített gázáramszabályozók kerültek forgalomba.
Közepes nyomásokon a gázáram szabályozása automatikussá tehető az ún.manosztátohkal. Ezek egyik ismert formája a Cártesius-féle manosztát (46.4.2.13.
ábra). A benne elhelyezett úszó tetején levő membrán zárjavagy nyitja az elszívó vezetéket. Üzembe helyezése úgy törté
nik, hogy az úszó belsejével összekötött teret a készüléktöbbi részével együtt a kívánt nyomásértékre szívjuk le. A tartályrészt azután csappal elzárjuk. Ha a nyomás az úszó fölöttitérben a megengedettnél alacsonyabb értékre csökken, az úszófelemelkedik és elzárja a szívócsonkot.
A Prytz-féle manosztát (46.4.2.14. ábra) tulajdonképpenhiganyos differenciálmanométer. Egyik szárába a kívánt nyomáskülönbségnek megfelelő magasságban kúposán kiképzettüvegfrittet vagy porcelán szűrőt építünk be. A nyomáskülönb
ség a nívóedény segítségével tetszés szerint beállítható. Ha aszívó oldalon a nyomás csökken, a higanyszintek különbségenövekszik, a kúpnak nagyobb felülete válik szabaddá, meggyorsul rajta a gázáramlás. A gyorsabb gázbeáramlás csökkenti a nyomáskülönbséget, a felemelkedő higanyszint kisebb kúpfelszínen engedi csak át a gázt. Ezzel a módszerrel kb.i 2—3 Hgmm pontosságú szabályozás lehetséges.
46.4.2.12. ábra.
Wohl-íéle csap

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 73/366
O Á Z A D S Z O B P C I Ó Ü V E G - É S F É M T E L Ü L E T E N 73
Az eddig tárgyalt közvetlen nyomásszabályozókon kívül közvetett manosz-tátok is használatosak. A többnyire elektromágnessel működtetett szelepet kontakt manométer vezérli. Kontakt manométerként — megfelelő módon átalakítva — bármely típus használható, pl. közönséges barométer, rövidített manométer (46.4.2.15.ábra) vagy nagy manométer.
A kontakt manométerek érzékenységét a zárófolyadék fajsúlyának csökkentésével emelhetjük. Erre a célra legjobban a koncentrált
szívás kéneav felel meg. Hátránya, hogy a kontaktus zárásakorfellépő elektrolízis elkerülése végett váltóáramú relét kellhasználni.
46.4.2.13. ábra. Cartesius- 46.4.2.14. ábra. Prytz-féle manosztét -féle önműködő nyomás-
szabályozó
46.4.2.15. ábra. Kontakt
manométer
Az ismertetett manosztátok nemcsak 1—760 Hgmm közötti nyomáson, hanem kb. 1 atm túlnyomásig használhatók. A szabályozás pontossága kedvező
esetben ± 0,2 Hgmm-t is elérhet.
46.5. Gázadszorpció üveg* és fémfelületeD, kigázosítás
Régen megfigyelték már, hogy teljesen lezárt üvegedényben a vákuum előszörrohamosan, majd exponenciálisan csökkenő mértékben romlik. Ennek oka az üvegfelületén adszorbeált gázok felszabadulása. Lágyabb üvegek az adszorbeált gázokatkb. 200 C°-on, keményebb üvegek magasabb, kb. 300 C° hőmérsékleten adják le. Az abszorpció útján megkötött gázok csak ennél is magasabb hőmérsékleten, alágyuláspont közelében kezdenek felszabadulni. A felszabadult gázmennyiség legnagyobb része vízgőz, C02, és csak viszonylag kis részét (mintegy 1 %-át) teszik ki atöbbi gázok (N2 stb.). Langmuir számítása szerint az 500 C°-ra hevített lámpaburából deszorbeálódott vízgőz mennyisége a monomolekulás rétegvastagsághoz szükségesnek 56-szöröse, a C02 pedig 4,8-szerese.
A nagyobb alkálitartalmú, lágyabb üvegek hőkezelésük alatt nagyobb mennyiségű gázt adnak le, mint a keményebb üvegek. A kifűtésükhöz szükséges magasabb hőmérséklet ellenére a keményebb üvegek alkalmasabbak vákuumberen-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 74/366
'74 V Á K U U M T E C H N IK A
46 .5.1. ábra. Szárítóedény
<iezések építésére. Az üvegfelület tisztítására felhasznált szerves oldószerek közülkülönösen nagy az alkohol adszorpciója. Legkönnyebben deszorbeálódik a CC14.
A vízgőz és C02 mellett jelentéktelen mennyiségben előforduló egyéb/gázoknemcsak felületi adszorpció, hanem részben az üveg bizonyos mértékű áteresztő-képessége következtében jutnak az evakuált térbe. Ismeretesek arra vonatkozóirodalmi adatok, hogy vékonyabb falú üvegedények — különösen magasabb hőmérsékleten — a He-ot, H2-t, N2-t és egyéb gázokat bizonyos mértékben átengedik.
Az üvegedények falán adszorbeált gázokat, különösen a vízgőzt, legcélszerűbben megfelelő méretű és alakú elektromos kályhával 3—400 C°-ra való fel-
Jűtéssel távolíthatjuk el. Ennek hiányában a kifűtést az üvegre tekert hőszigeteltellenálláshuzallal vagy gázlánggal is végezhetjük, de ilyenkor a legnagyobb óva
tossággal kell eljárni, hogy a szívatás alatt levő készülék meg ne repedjen és ennekkövetkeztében szét ne robbanjon (védőszemüveg!). A deszorpció következtében felszabaduló Vízgőzt a szi
vattyú is eltávolítja ugyan, de igen meggyorsíthatjuk ezt a folyamatot kifagyasztással vagy szárítással. A kifagyasztócsapda hűtésére ebben az esetben azonban nem elegendő a szénsavhó-keve-rék, mert a víznek —80 C°-on még jelentős tenziója van (5* 10-4Hgmm). A folyékony levegő hőmérsékletén ez a gőznyomás már
jelentéktelen. Vákuumberendezés nedvességének megkötésére fagyasz
tócsapdákon kívül szárítóanyagokat is alkalmazhatunk. Legalkalmasabb erre a célra a megfelelő edényben elhelyezett
~P20 5. A 46.5.1. ábrán bemutatott szárítóedényben néhány darab üveggolyó isvan, hogy az esetleg elfolyósodott felületet a csap körüli elforgatással meg lehessenújítani. A P20 5-dal egyensúlyban levő levegőben 2 -1 0 '5 mg vízgőz van literenként.
Másik ilyen elterjedt szárítóanyag a vákuumtechnikában a Mg(C104)2. A visz-szamaradó vízgőz 10"3 mg/liter.
A maradékgázok eltávolítására különböző nagy felületű adszorbenseket vagy
gettereket alkalmazhatunk. Adszorpcióval pl. az aktív szén a folyékony nitrogénhőmérsékletén 02-t, N2-t, C02-ot, kisebb mértékben H2-t és nemesgázokat tudmegkötni. Az ugyancsak használatos szilikagél adszorpciós tulajdonságai az aktívszénnél kevésbé előnyösek. Egyes fémek: Ta, Nb, Ti, Zr, Th, A1 finom eloszlásbanszintén igen alkalmasak különböző gázok megkötésére. Ebben az adszorpción kívülaz oldódásnak és a kemoszorpciónak is szerepe van. Nyomáscsökkentés és hőmérséklet-növelés hatására a megkötött gázokat a fémek részben leadhatják, ezértcélszerű a vákuumberendezés fémalkatrészeit és még fokozottabb mértékben azadott esetben a munkatérben felhasználásra kerülő fémporokat (pl. Zr-, A1-, Ti-
jodidok elemi szintézisénél) előzetesen kigázosítani. Az evakuált térben jelenlevő
fémek kigázosítása legcélszerűbben kívülről történő nagyfrekvenciás kifűtésseloldható meg. A berendezésnek a kérdéses fémet tartalmazó részét nagyfrekvenciásáramot vivő tekercs terébe helyezzük. A keletkező örvényáramok a fémet felhevítik.Szokásos a fémek előzetes kiizzítása vákuumkályhában. A nagyfrekvenciás áramelőállítására szikraközös vagy elektroncsöves adót alkalmazunk. Az elsőt 100 kHz-ig, a másodikat 0,5—1 MHz-ig használjuk.
A szivattyúzás után visszamaradó, illetve felszabaduló gázok megkötésére-alkalmazott másik eljárás a getterezés^Getterezésre alkálifémek, illetve alkáliföld-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 75/366
A V Á K U U M B E R E N D E Z É S Ü Z E M E L T E T É S E 75
fémek és ezek Ötvözetei használatosak, amelyek az evakuált térben elpárologtatva,kémiai reakcióba lépnek a visszamaradt gázokkal, és nemillékony vegyületek kép
ződése közben megkötik azokat. A báriumnak különös előnye getterezés szempont jából, hogy lecsapódás után, hidegen még a folyékony levegő hőmérsékletén isaktív.
46.6. A vákuumberendezés felépítése, üzemeltetése; hibahelykeresés
A vákuumberendezés munkatere a legmesszebbmenően alkalmazkodik az elvégzendő feladathoz, tehát mindig egészen speciális. A rendszer többi része, avákuumot létesítő szivattyúk, a különböző mérőműszerek, kifagyasztócsapdák,
nyomáskiegyenlítő pufferedények stb. sokkal kevésbé speciális szempontok szerintkészülnek, és a munkatér megfelelő változtatásával többféle célra is alkalmasak. A 46.6.1. ábrán közölt szerkezeti vázlat egy preparatív kémiai célokra megfelelő,max. 10-6 Hgmm végvákuum létesítésére alkalmas berendezést mutat.
46.6.1. ábra. Vákuumberendezés preparatív kémiai célokra
A készülékben rotációs olaj szivattyú szolgáltatja az elő vákuumot. A hozzácsatlakozó egyik (kb. 5 literes) pufferedény a hirtelen nyomásváltozás kiegyenlítését, a másik a MacLeod-manométer működtetését szolgálja. A végvákuumothárom fokozatú, fémből készült higanydiffúziós pumpával állítjuk elő. A berendezés többi része olcsón készíthető üvegből. Ennek előnye, hogy az alkatrészekkönnyen megtisztíthatók, és teljes zsírtalanítás (krómkénsav, szén-tetraklorid),majd szárítás után összeforraszthatók. A diffúziós szivattyú fém elszívócsonkjáhozcsatlakozó üvegtokot piceinnel tömítjük.
Használatbavétel előtt a berendezést gondosan ki kell próbálni, vákuumbankigázosítahi, és üzemszünetben is meg kell védeni a külső levegőtől és annaknedvességtartalmától. Ezért a szivattyú és a munkatér közötti szakaszt lehetőleg

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 76/366
76 V Á K U U M T E C H N IK A
állandóan vákuum alatt tartjuk. A készülék üzemeltetését az elővákuum és adiffúziós szivattyú hűtővizének megindításával kezdjük. A higanydiffúziós szivattyú elektromos fűtését csak az 5»10-2 Hgmm elérése után kapcsoljuk be.Ugyanakkor üzembe helyezzük a kifagyasztócsapdákat, és óvatosan megnyitjuk amunkateret a szivattyúval összekötő csapokat. A higanydiffúziós szivattyú a fűtésmegkezdése után kb. egy órával kezd csak szívni. Addig is és utána folyamatosanellenőrizni kell a hűtővíz hőmérsékletét, amely legfeljebb 1 —2 C°-kal emelkedhet abeömlő víz hőmérséklete fölé.
Az összeállított vákuumberendezés hibalehetőségei közül meg kell említenia tömítetlenséget, melynek következtében a külső levegő behatol a leszívott térbe,és rontja az előállított vákuumot. Tömítetlenség a kapilláris méreteknél is több
nyire jóval kisebb méretű üveghiba, lyuk, repedés stb. helyén szokott fellépni. Megszüntetése
általában igen egyszerű, megkeresése azonbanannál nehezebb feladat. A lyukkereséshez nagyon hasznos segédeszköz az ún. teslázó, amellyelnagyfrekvenciájú nagyfeszültséget hozunk létre. A kisülési fény intenzitása és színe a lyuk közelében más, így ez felismerhető. Ha a gya
nús hely fémalkatrészen van, más módszertkell alkalmaznunk. így pl. a recipienBen belülkisülést hozunk létre, és megfigyeljük, változik-e a színe, ha a feltételezett hibahelyre olyangázt, pl. világítógázt engedünk, amely a lyukon át bejutva, változást okozhat a kisülésszínében.
A hibahelykeresés további egyszerű mód jára az ad lehetőséget, hogy a hővezetéses vákuummérőknél az érzékenység függ a gáz minő
ségétől. Ha a hibára gyanús helyre hidrogéngáztfúvatunk, és ott beszívás van, a hővezetéses vákuummérő ezt ugrásszerű kitéréssel jelzi. Hibakeresés céljára a Piráni-féle vákuummérővel célszerű kiegyenlítetthídban dolgozni, és műszerét érzékeny galvanométerrel helyettesíteni. Ha a mérő-csövet a diffúziós szivattyú elővákuum oldalánál helyezzük el, a módszer érzékenysége fokozódik, és még 10"4—10“ 5 Hgmm nyomás környezetében is jól használható.
Hibakeresésre nagyvákuumú rendszerekben rendkívül sok módszer ismeretes,melyek leírása túlmegy könyvünk keretein. A különböző módszerekkel kapcsolat
ban a megfelelő szakirodalomra utalunk.Igen előnyösek az utóbbi időben forgalomba került, teljesen fémből készültnagy vákuum-berendezések. A 46.6.2. ábrán látható egy ilyen, vertikális recipienscsatlakozású készülék.
A 46. fejezet csak rövid áttekintést ad a vákuumtechnikai módszerekről,amelyekről bővebb tájékozódást az alábbiakban felsorolt irodalomjegyzék alapjánkaphatunk.
46.6.2. ábra. Fémből készült nagyvákuumszivattyú vázlata

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 77/366
I R O D A L O M A V Á K U U M T E C H N IK Á H O Z 77
Irodalom
Jaeckel , R . : Kleinste Drucke, ihre Messung und Erzeugung. Springer Verlag, Berlin, 1950.
Duahman S . : A vákuumtechnika tudományos alapjai. Akadémiai Kiadó, Budapest, 1959.
Mönch , O. 0.: Hochvakuumtechnik. Kudolf A . Láng Verlag, Pössneck, 1950.Ghithrie, A.: Vacuum Equipment and Techniques. McG-raw—Hill Book Company Inc., New York, 1949.
Diela, K .—Jaeckel , R . : Leybold Vakuum-Taschenbuch für Laboratórium und Betrieb. Springer Verlag, Berlin, 1958.
L ec k ,J .H .: Pressure Measurement in Vacuum Systems. Institute of Physics, London, 1957.
Hei nze, W.: Einführungin die Vakuumtechnik. VEB Verlag Technik, Berlin, 1955. Lapor te, H . : Vakuummessungen. VEB Verlag Technik, Berlin, 1955.Winter E. és munkatársai: Vákuumtechnika I —H —IIT. kötet. Nehézipari Könyvkiadó,
Budapest, 1954.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 78/366
47. Munka nyomás alatt
Olyan kémiai reakciók végrehajtásához, amelyek valamelyik komponensénektenziója az adott hőmérsékleten az 1 atm-t meghaladja, bombacsövet vagy auto-klávot használunk.
47.1 Bombacső
Kisebb anyagmennyiségek esetén célszerűen az ún. bombacső alkalmazható. A bombacsőhöz nagyméretű és mindenképpen hibátlan üvegcső szükséges. Lég-buborék vagy karcolás nagymértékben csökkenti a nyomásállóságot, kellő finomságú csiszolás, felületi megmunkálás ellenben nem. A cső belsejében ható és aszakítási szilárdságot igénybevevő nyomásnak az üveg mintegy négyszer kevésbé
tud ellenállni, mint a kívülről ható nyomásnak.. így pl. 11 mm átmérőjű, 1 mmfalvastagságú cső 20 C°-on 104 atm belső nyomást képes elviselni, ugyanakkor460 atm külső nyomásnak áll ellen. A falvastagság növelésével nem lehet korlátlanul kiterjeszteni a nyomáshatárt. 1—3 mm-nél nagyobb falvastagságnálugyanis az egyenlőtlen felmelegedés, a belső feszülések folytán nagyobb a törésiveszély. Az átmérő növelése természetesen csökkenti a cső szilárdságát. A leggyakrabban alkalmazott méret 10—25 mm 0 . Ha a befogadóképesség növelése szükséges, inkább a cső hosszát kell növelnünk, nem pedig az átmérőjét.
A legnagyobb megengedhető nyomás igen nagymértékben függ az alkalmazott
hőmérséklettől. A transzformációs hőmérséklet közelében a 20 C°-on elviseltmaximális nyomás 50 %-a mát 3—4 hét alatt szétrepesztheti a csövet. Viszont40 —50 C°-kal a transzformációs hőmérséklet alatt a cső már tartósan elviseli a20 C°-on mért maximális nyomás 4/5 részét.
Néhány fontosabb üvegfajta nyomásállósága a 47.1.1. táblázatban látható.
Üvegek nyomásáUósága 47.1.1. táblázat
Ü v e g f a j t a T h ü rin g ia i Jénát G - 2 0 D u r á n S u p r e m a x
Maximális nyomás 20 C°-on, atm 480 450 650 500
Transzformációs hőmérséklet, C° 520 570 540 720

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 79/366
B O M B A C S Ő 79'
Ha vizes oldatokat bombacsőben kezelünk, tekintetbe kell vennünk a legtöbb használatos üvegféleség szokatlan érzékenységét a nagynyomású, forró vízgőzzel szemben. Kb. 300 C° hőmérsékleten a víz nemcsak az ún. lágy üvegeket,hanem az egyébként igen kiváló kémiai ellenállást tanúsító eszközüvegeket (pl. aPyrexet) is néhány óra alatt teljesen elbontja. Bizonyos mértékig ellenáll enneka hatásnak a jénai égetőcső anyaga, valamint néhány, igen sok PbO-ot tartalmazóoptikai üvegfajta. A jénai égetőcső falát a víz pl. 500 C°-on is csak kb. 1 mmmélységig alakítja át laza, kristályos szerkezetűvé.
A bombacsövek méretének megválasztásakor nemcsak a kiindulási anyagokmennyiségére kell tekintettel lennünk, hanem a várható termékekre is. A csöveketlegfeljebb félig, vagy inkább csak 1/3 részben szabad megtölteni. Ha gázfejlődés
várható, a töltet még kevesebb legyen. Töltés előtt — a könnyebb leforraszthatóságérdekében — a cső nyitott végét 15—20 cm-es darabon kb. 6 mm belső átmérőreszűkítjük le. Az anyagot megfelelő szárvastagságú tölcséren át töltjük a csőbe,és a leforrasztandó csőszakasz külső és belső felületét a leggondosabban tisztántartjuk.
Ha a reagálandó anyagoknak csak a leforrasztás után szabad elegyedniük,elhelyezhetjük az egyik komponenst a 47.1.1. ábra szerinti hosszú szárú kehelyben,vagy vékony falú leforrasztott ampullában, amelyet majd erélyes rázással összetudunk törni.
Bizonyos esetekben szükséges lehet a reakciótér oxigénmentesítése. Ezt külön
böző teljesítőképességű szivattyúk segítségével szokták végrehajtani. A leforrasz-tást fújtatólánggal, lehetőleg gáz—oxigén-eleggyel végezzük. Vigyázzunk arra,hogy a forrasztás helyén a falvastagság egyenletes maradjon, és az üveget fokozatosan, lassan hűtsük le. A 6 mm átmérőjű csővégződést is célszerű először mintegy1/2 mm-re szűkíteni, és csak teljes kihűlés után fejezni be a mintegy 10—15 mm,hosszú kapilláris leömlesztésével a bombacső leforrasztását.
Jó minőségű üvegből készült hibátlan bombacsőben vizes oldatok különösveszély nélkül felmelegíthetők 200 C°-ra. Ha nincs jelentős gázfejlődés, ez kb.20 atm belső nyomásnak felel meg. Ha még nagyobb nyomás fellépésével szá
molhatunk, az üvegből készült bombacsövet acél védőcsőbe helyezzük. A papírbavagy azbesztlemezbe csomagolt bombacsövet úgy helyezzük el a védőcsőben, hogykapillárissá húzott vége 1—2 cm-re kiálljon. Az acél- és üvegfal közötti hézagottöbbnyire kiizzított kovafölddel töltik ki. Homok erre a célra semmiképpen semalkalmas, mert egyetlen éles karcolásnak a bombacső felületén könnyen robbanáslehet a következménye.
A bombacső 50 atm-nál nagyobb nyomást is elvisel, ha kívülrőlmegfelelő ellennyomással kompenzáljuk. Ilyenkor az üvegből készültbombacsövet légmentesen zárható acél védőcsőbe helyezzük, és a köztitérbe olyan anyagot viszünk, amelynek nagyobb a tenziója az alkalma
zott hőmérsékleten a cső belsejében várható nyomásnál. Víz erre a célraaz említett korróziós hatás miatt nem megfelelő. A kb. 10 mm falvastagságú acélcső nyílása menetes fedéllel zárható el. Tömítésére 3 mm vastaglágy vörösréz lemezt szokás használni, amely az acélcső peremébe vá-
47.1.1. ábra. Bombacső a nyomás alatt reagáltatandó anyagokelkülönítését szolgáló ampullával

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 80/366
80 M U N K A N Y O M Á S A L A T T
gott három, kb. 1 mm mély, koncentrikus vájatba nyomódva, tökéletes zárástbiztosít. Hy módon megoldható az üvegcső tartalmának kb. 400 C°-ra való heví
tése — ha víz nincs jelen — törés veszélye nélkül. A bombacsövek melegítésére különböző folyadékfürdőket (víz, olaj, paraffin),
csőkemencéket vagy alumínium blokkokat alkalmazunk. Cél a minél egyenletesebbhőátadás biztosítása. Nyitott végű acélcsőben vagy anélkül melegített bombacsöveket rongyba csavartan célszerű elhelyezni a folyadékfürdőben, és robbanásukesetére megfelelő szilánkvédelemről (drótháló és vastag üvegfal) ajánlatos gondoskodni. Ha ismeretlen kimenetelű kísérlettel állnak szemben, az említett biztonságiintézkedéseken kívül a ferdén felfüggesztett bombacsőnek először csak a folyékonyfázist nem tartalmazd gőzterét melegítik, és csak fokozatosan terjesztik ki a
hevítést. Végül külön óvatosságot igényel a bombacsövek felnyitása. Csak akkor nyúl
junk a berendezéshez, ha az teljesen kihűlt / / Kezünket vastag bőrkesztyűvel, szemün ket és arcunkat drót védőálarccal biztosítsuk ! A szokásos módon üvegvágóval csakakkor nyissuk fel a kapilláris végét, ha biztosak vagyunk abban, hogy a belsőnyomás nem lehet több 1 atm-nál. A belső nyomás csökkentését fokozhatjuk abombacső gömbölyű végének mélyhűtésével is. Ha még mindig fenáll a belsőtúlnyomás veszélye, helyezzük el drótháló és üvegfal mögött a felnyitandó bombacsövet, és a kapilláris vége alá tegyünk megfelelő méretű hegyes lángú Bunsen-égőt.így várjuk meg, amíg a megolvadt üveget a belső nyomás kifújja.
Az előbbiekben leírt módon, vastag falú kvarccsőben állítható elő pl. a titán (II)--klorid titán(IV)-kloridból és fémtitán forgácsból úgy, hogy a fémet a ferde helyzetben rögzített bombacső felső, 800—900 C°-ra Hevített részében, a TiCl4-ot pediga cső alsó, alacsonyabb hőmérsékletű részében helyezzük el.1 Ha a reakció szemmelláthatóan nagyrészt végbement, az egész osövet néhány napig 600—700 C°-on tartva,az anyagot homogenizáljuk.
Az SiHF3 sziliko-kloroformból állítható elő TiF4 vagy SnF4 segítségével, ha akeveréket száraz bombacsőben hosszabb ideig 100—120 (P-on tartjuk.2
47.2. Autoklávok
Autoklávokat akkor használunk, ha nagyobb anyagmennyiséggel dolgozunknyomás alatt. Az autokláv többnyire benyúló hőmérővel, nyomásmérő műszerrel,esetleg ke verő vei ellátott fűthető edény, amelynek csavarosán rögzíthető, jól zárófedele van. Kisebb nyomást (15 atm) üvegből készült autoklávok is elviselnek,a több száz atmoszférára méretezett készülékek többnyire megfelelő szakítószilárdságú, rozsdamentes acélból készülnek.
Az<üveg-„autoklávok” legegyszerűbb alakja az ún. nyomáspalack. Ez vastagDurán-üvegből készült edény, amelynek síkcsiszolatú Bzáját hozzácsiszolt vastagüveglappal szorítókengyel segítségével lehet lezárni (47.2.1. ábra). Más szerkezetűüvegautoklávok is használatosak. Valamennyinek közös előnye az átlátszóság, amia folyamatok megfigyelését lehetővé teszi. Az egyéb szerkezeti anyagból készültautoklávökön e célból megfelelő módon tömített, kis felületű, vastag falú ablakokat lehet alkalmazni.
1 Klemm W.,-Grimm L.: Z. anorg. alig. Chem. 249, 198 (1942). * Bújj O., Albert C.: Bér. dtsch. chem. Ges. 38, 56 (1905).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 81/366
A U T O K L Á V O K 81
A néhány atmoszféránál nagyobb nyomású autoklávok fedelének tömítéséreólom- vagy lágy vörösréz lemezt használnak; 400 atm nyomásig és 350 C° hőmér-
sékletig lágyvas, monelfém vagy ezüst is szóba kerülhet. A még magasabb nyomáson és hőmérsékleten használt autoklávok fedele kónuszos illesztéssel záródik. A kúposán kialakított fedelet a tompább szögben csiszolt autokláv nyílásába csavarszorítja be. Különleges esetekben (pl. redukció alkálifémekkel) alkalmazzák azautoklávok hegesztéssel történő zárását (lásd 54.1.5.).
Az autokláv belsejét az acél védelme érdekében a korróziót okozó anyag kémiaireakcióképességének megfelelően megválasztott bevonattal látják el. Ismeretes pl.
nikkel-, króm-, ezüst- vagy zománc-, újabban műanyag bevonat is. Üveg- vagy porcelán betétek szintén használatosak.
d A
U
47.2.1. ábra. Üvegautokláv s zorítókengyellel
47.2.2. ábra. Forgatható autokláv
Az újabb konstrukciójú autoklávokról lehetőleg elhagyják a kívülről benyúlókeverőtengelyt, mert tömítése nehézkesen oldható meg.
A keverést egyes típusoknál egyszerűen az autokláv forgatásával eszközölhetjük (47.2.2. ábra). Az autoklávban történő keverés zárt térben elhelyezettmotorral, vagy ami még célravezetőbb, mágneses keverő segítségével is megoldható.
Az utóbbi megoldás a hazai gyártmányú autoklávokon is használatos.3
Az autoklávok fűtése gázzal vagy elektromos úton történik. Egyes esetekbena felmelegedés egyenletesebbé tételére olaj- vagy fémfürdőt (ólom- vagy Pb —Sb--ötvözetet) használnak. Magasabb hőmérsékletek elérésére az autoklávot belsőfűtéssel kell ellátni. Ismeretesek olyan autoklávok is, amelyekben beépített szűrősegítségével nyomás alatt végrehajtható a szűrés, illetve más szerkezetű berendezésben pl. az extrakció.
A nagynyomású autoklávok alkalmazása során különösen nagy figyelmet kellfordítani a biztonsági rendszabályok betartására. így az autoklávokat könnyű,ún. repülőtetővel ellátott helyiségben kell üzemeltetni; az,autoklávok fűtésének,
keverésének szabályozását másik helyiségből kell megoldani; az autokláv-helyiség-ben csak abban az esetben tartózkodhatunk, ha az autokláv nincs fűtés, illetvenyomás alatt.
Nagynyomású gázokkal végzendő műveletek közben előforduló feladat azautokláv légtelenítése, amit többszöri evakuálással és a használandó gázzal való
8 Lampart gyártmányú vertikális autokláv mágneses keverővei. Ty pe: LÁM— 1(1, 2, 3, 4 literes kapacitással).
6 Alt. és szervetlen kémiai prakt. —4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 82/366
82 M U N K A N Y O M Á S A L A T T
átöblítéssel érhetünk el. A megfelelő nyomás elérése érdekében elegendő olykora gázpalack csatlakoztatása az autoklávhoz. A kereskedelmi forgalomban nemkapható gázok megfelelő nyomáson való alkalmazásához kompresszor vagy elegendő mértékben hűthető, nyomásálló kondenzációs edény szükséges a laboratóriumban előállított gáz cseppfolyósítására és összegyűjtésére.
Újabban kísérleti célokra extrém magas nyomásokat is előállítottak (100 000atm), s itt nagyszámú érdekes polimorf módosulatváltozást észleltek. A szerkezetianyagok megválasztása ilyen körülmények között még fokozottabb jelentőségű.Példaként említhetjük meg, hogy a vas 9000 atm nyomáson hidrogénnel közvetlenérintkezésben szobahőmérsékleten is másodpercek alatt törékennyé válik. Az üveg-és kvarcablakok pl. olajban és glicerinben sokszorosan nagyobb nyomást viselnek
el, mint vízben vagy éterben.
47.3. Nagynyomású manométerek
Az 1 atm-nál jelentékenyen nagyobb nyomások mérésére kb. 40 atm-ig memb-ránmanométereket, azon felül kb. 4000 atm-ig csőrugós manóm étereket használnak. 10 000 atm nagyságrenden felül a nyomás mérésére az észlelhető vezetőképesség-változást használják fel. így pl. a manganin huzal ellenállása — állandóhőmérsékleten — 10 000 atm hatására kb. 2—3 %-kal változik. Lökésszerű nagy
nyomások (robbanások) mérésére a piezoelektromos hatás ad lehetőséget. A zárt rendszerben várható nyomás hozzávetőleges becsléséhez közöljük néhány
anyag tenziójának hőmérsékletfüggését a kritikus pontig (47.3.1. ábra).
47.3.1. ábra. Néhány anyag tenziógörbéje

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 83/366
I RODALOM 83
Irodalom
Weisberger A.'. Technique ofOrganic Chemistry, Vol. H. Interscience Publisbers, New
Y ork—London, 1948. Handbuch dér Experimentalphyaik, Bd. V U L, Harms, Wien, 1929. Jánecke E.\ Die Welt. dér chemischen Körper bei hohen und tiefen Temperaturen und
Drucken. Verlag Chemie, Weinheim, 1960.Gomings E. W .: Nagynyomású technológia. Műszaki Könyvkiadó, Budapest, 1959. Archív für Techniaches Mesaen. Korndorf B. A.: Hochdrucktechnik in dér Chemie. VE B Verlag Technik, Berlin, 1966.
6'

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 84/366
48. Az anyagok tisztítása
A szokásos fizikai tisztítási műveleteket elvileg két csoportra oszthatjuk. Az egyikbe soroljuk azokat, amelyekben az elválasztás során a komponensekhalmazállapota nem változik meg, a másikba azokat, amelyekben az elválasztáshalmazállapot-változással jár. A fizikai műveleteken kívül természetesen igen nagy
jelentősége van az anyagok kémiai úton való tisztításának is. Az erre vonatkozóadatok a részletes részben találhatók.
48.1. Halmazállapot-változás nélkül végbemenő elválasztás
48.1.1. Szilárd fázisok szétválasztása Az iparban gyakran, a laboratóriumi technikában ritkábban előforduló műve
let. Az elválasztandó komponensek keverékét először el kell porítani. Meg kell jegyezni, hogy még a legfinomabban elporított állapotban sem lesz tökéletes atisztítás. A szilárd halmazállapotú anyagok aprítására vonatkozóan lásd 1.13.
Aprítás közben az anyagnak gyakrannemcsak a szemcsemérete változik, hanembizonyos fokú szennyeződésen kívül egyébfizikai és kémiai tulajdonságai is megváltoznak (pl. kristályvízvesztés vagy vízfelvétel, színváltozás, fluoreszkáló képesség megszűnése, oxidálódás stb.).Higrosz-kópos vagy oxidációra hajlamos anyagok
48.1.1.1. ábra. Vákuum-golyósmalom aprítását éppen ezért védőgáz-atmoszfé-
rában vagy vákuumban szokás végezni.
Vákuumban történő aprításhoz vastag falú üvegből készült kis golyósmalomhasználatos (48.1.1.1. ábra), amelyet megfelelő biztonsági rendszabályok betartása
mellett (védőfal, drótháló) rázógépre szerelhetünk. Az aprítás célja meghatározott szemcseméret elérése. Ennek ellenőrzése meg
felelő szövetszámú (lyukméretű) szitával történik. A különböző szövetszámú szitákból szabványsorozat van forgalomban. A 48.1.1.1. táblázatban a sziták szabványszerinti (DIN 1171) szövetszámát, a szitaszem szélességét, a szita készítéséhezalkalmazott drót átmérőjét és a szitaszemek cm2-enkénti számát tüntettük fel.
Az aprítás folyamán a szemcseméret-eloszlás változik, és nagymértékben függhet a keverék komponenseinek anyagi minőségétől. Az egyes szemcseméretek kiválasztása útján egyes alkatrészek bizonyos mértékben feldúsíthatók.
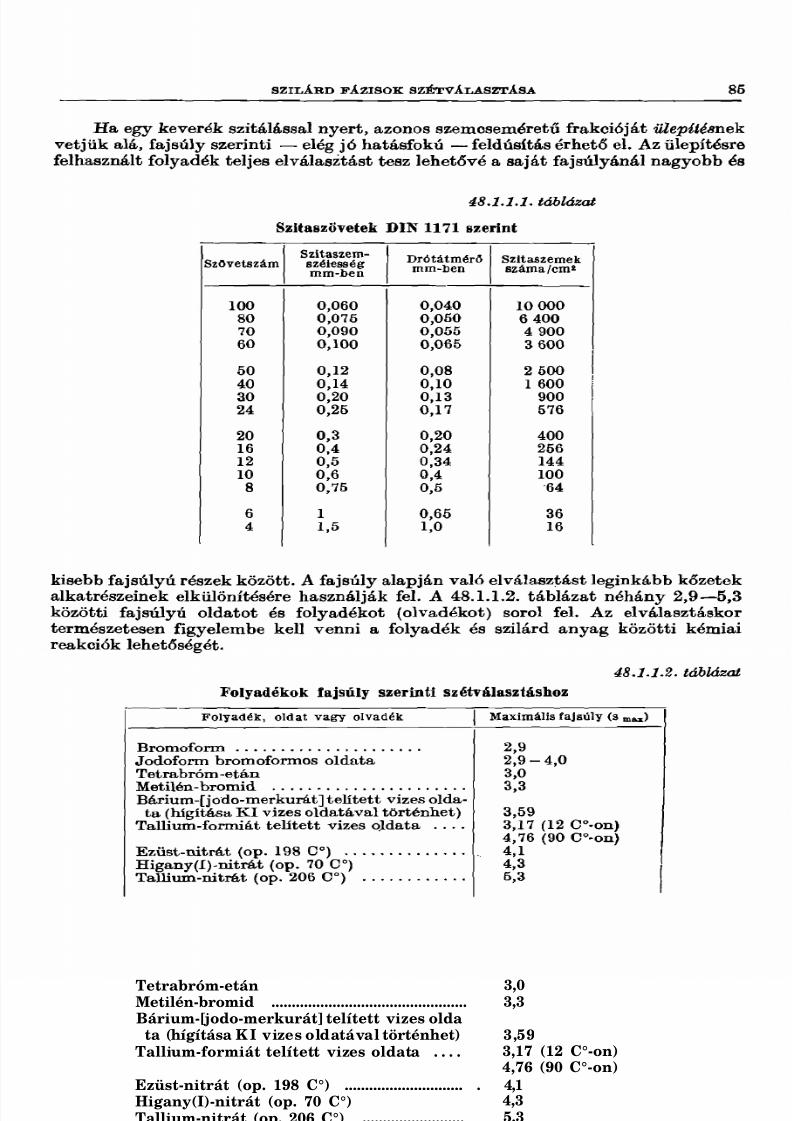
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 85/366
S Z I L Á R D F Á Z I S O K S Z É T V Á L A S Z T Á S A 86
Ha egy keverék szitálással nyert, azonos szemcseméretű frakcióját útépítésnek vetjük alá, fajsúly szerinti — elég jó hatásfokú — feldúsítás érhető el. Az ülepítésrefelhasznált folyadék teljes elválasztást tesz lehetővé a saját fajsúlyánál nagyobb és
48.1.1.1- táblázat
SzltaszÖvetek DIN 1171 szerint
SzövetszámSzitaszem-széiességmm-ben
Drótátmérőmm-ben
Szltaszemekszáma/cm*
100 0,060 0,040 10 00080 0,075 0,050 6 40070 0,090 0,055 4 90060 0,100 0,065 3 600
50 0,12 0,08 2 50040 0,14 0,10 1 60030 0,20 0,13 90024 0,25 0,17 576
20 0,3 0,20 40016 0,4 0,24 256
12 0,5 0,34 14410 0,6 0,4 100
8 0,75 0,5 64
6 1 0,65 364 1,5 1,0 16
kisebb fajsúlyú részek között. A fajsúly alapján való elválasztást leginkább kőzetekalkatrészeinek elkülönítésére használják fel. A 48.1.1.2. táblázat néhány 2,9—5,3
közötti fajsúlyú oldatot és folyadékot (olvadékot) sorol fel. Az elválasztáskortermészetesen figyelembe kell venni a folyadék és szilárd anyag közötti kémiaireakciók lehetőségét.
48.1.1.2. táblázat
Folyadékok fajsúly szerinti szétválasztáshoz
F o l y a d é k , o l d a t v a g y o l v a d é k Maximális fajsúly (s mai)
Bromoform.............................................. 2,9Jodoform bromoformos oldata 2 , 9 - 4 , 0
Tetrabróm-etán 3,0Metilén-bromid ................................................ 3,3Bárium-[jodo-merkurát] telített vizes oldata (hígítása K I vizes oldatával történhet) 3,59
Tallium-formiát telített vizes oldata . . . . 3,17 (12 C°-on)
Ezüst-nitrát (op. 198 C°) .............................
4,76 (90 C°-on) . 4,1
Higany(I)-nitrát (op. 70 C°) 4,3
Tallium-nitrát (op. 206 C°) ......................... 5,3

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 86/366
86 A Z A N Y A G O K T IS Z T ÍT Á S A
Elsősorban a felületi tulajdonságok felhasználásán alapuló elválasztási módszer az ún. flotálás, melynek különösen az iparban van nagy jelentősége.
48.1.2. Szilárd—folyékony fázisok, elválasztása
Ez a feladat dekantálással, szűréssel, eentrifugálással oldható meg. Az egyszerűbb eljárásokra vonatkozóan lásd 1.14. Az ott tárgyalt vákuumszűrés folyamána folyadékfelszínre nehezedő külső légköri nyomással szemben a szívópalack belse jének nyomását megközelítően nullára csökkentve maximálisan 1 atm nyomás-különbség érhető el. Kolloid dimenziójú részecskék visszatartására olyan finompórusméretű szűrőréteg szükséges, amellyel a szűrősebesség 1 atm nyomáskülönb-
48.1.2.2. ábra. Kapsenberg~£éle automatikus szűrő
ség esetén még igen lassú. A gyorsabb szűréshez szükséges nagyobb nyomáskülönbség csak a folyadékfelszínre ható nyomás növelésével valósítható meg. Ezen azelven működnek a különböző ultraszűrők.
A túlnyomás létesítésére indifferens gázokat, általában nitrogént használnak.Megjegyezzük, hogy a kereskedelmi forgalomban kapható ultraszűrőket a maximális nyomásra specifikálják. Az üvegfalú ultraszűrőket általában max. 5 atm-ig,
az acélból készült szűrőket pedig 100 atm-ig lehet alkalmazni. A szűrés sorángondoskodni kell a folyadékot tartalmazó szűrőkhöz vezető csatlakozások megfelelő tömítéséről.
A laboratóriumi munka során gyakran merül fel nagyobb mennyiségű oldatszűrésének szükségessége. A 48.1.2.1. ábrán látható automatikus szűrő lehetővéteszi a folyamatos szűrést és az állandó utántöltés kiküszöbölését. Működése akövetkező: a hajlított kivezetőcsövet kis gumidugóval eltömjük, a csapot kinyit juk, és a tölcséren keresztül megtöltjük az edényt a szűrendő oldattal. Ezután a

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 87/366
S Z Ű U É S 87
tölcsért gumidugóval zárjuk, -és a csapon át vízlégszivattyúval addig szívatunk,amíg a tölcsér szárából a levegő nem kezd az oldaton át buborékolni. Ekkor a
belső térben a nyomás annyival kisebb a külső légnyomásnál, mint amekkora aszintkülönbségnek megfelelő hidrosztatikai nyomás. A csapot ezután zárjuk, és
a kivezetőcsőről a gumidugót eltávolítjuk. Miután a tölcsért a kivezetőcső alá helyeztük, az edényből az oldatot a csap óvatos elforgatásával — jelzett magasságig —a tölcsérbe folyatjuk, majd a csapot újra zárjuk, és a dugót a beöntőtölcsérről eltávolítjuk. A tölcsérben a folyadéknívó magassága szűrés alatt állandó marad.
A Kapsenberg-féle automatikus szűrő (48.1.2.2. ábra) működési elve a következő : a beépített üvegszűrő fölött levő folyadéknívó magassága a szűrőberendezésben létesített megfelelő csökkentett nyomás segítségével biztosítható.
A levegővel (oxigénnel) szemben érzékeny anyagok szűrését C02-atmoszférá-ban végezhetjük. A 48.1.2.3. ábra ennek igen egyszerűen végrehajtható megoldásátszemlélteti. Oxigénnel vagy nedvességgel szemben nagyon érzékeny anyagokszűrését és előállítását (kicsapását) ugyanazon berendezésben hajtjuk végre. A 74.1.1. ábrán bemutatott berendezés A edényében indifferens atmoszférában állít
juk elő a terméket, majd az oldatot a készülék megfordításával C üvegszűrőn át a B edénybe szűrjük át.
Gyakran felmerül annak szükségessége — az előbbiekben tárgyalt automatikusszűrésen kívül is —, hogy a folyadékszint állandóságát biztosítsuk. Ezt könnyenmegoldhatjuk a 48.1.2.4. ábrán bemutatott egyszerű berendezéssel, mely lényegében a 48.1.2.1. ábrán vázolt automatikus szűrőkészülékkel azonos elv alapjánműködik.
A folyadékok transzportálása, automatikus szűrés vagy cseppfolyós gázokbanmint oldószerben végrehajtott reakciók során igen hasznos a folyadéknívó mérésérealkalmas folyadékszint-jelző használata. Ilyen termisztoros folyadékszint-jelzőműködési elve a disszipációs állandó változásán alapszik (lásd 45.2.2.). Levegőbena hőelvezetés rosszabb, a hőegyensúly sokkal magasabb hőmérsékleten jön létre,

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 88/366
88 A Z A N Y A G O K T IS Z T ÍT Á S A
mint, pl. vízben vagy folyékony ammóniában. A 48.1.2.5. ábrán vázolt kapcsolásesetén az alkalmazott tárcsatermisztor (lásd 45.2.2.2. ábra) levegőben felmelegszik,ellenállása lecsökken, ezáltal a kör árama megnövekszik és a skálaizzó világít. Ha
viszont a termisztor vízbe merül, az egyensúlyihőmérséklet sokkal alacsonyabb lesz, ezáltalmegnő a termisztor ellenállása, és a kör áramának csökkenése következtében a skálaizzó kialszik.
20 V
48,1.2.4. ábra. Folyadékszint állandó
ságát biztosító berendezés
48.1.2.5. ábra. Termisztoros folyadékszint jelző
kapcsolási vázlata
Az ülepítéssel végzett szétválasztás hatásfoka nagymértékben fokozhatócentrifugálással. A centrifugában ható nyomás a kerületi sebességgel arányosannövekszik (kerületi sebesség: V = 2r-rt'n, ahol r — sugár, n — percenkénti fordu
latszám). Az adott kerületi sebességhez tartozó nyomásértékeket a48.1.2.1. táblázat tartalmazza.
A legegyszerűbb kézi centrifu
gával kb. 1000 fordulat/perc érhetőel. Az elektromos meghajtású, golyóscsapágyon futó laboratóriumicentrifugák fordulatszáma általában 2000 —4000 között van, nohavannak ennél lényegesen gyorsabbak is. Különleges csapágyazássalés többnyire turbinameghajtássalkészülnek az ún. ultracentrifugák,
amelyek percenként 20000—200 000fordulatra is képesek. A fordulatszám növekedésével nő a tengely igénybevétele. Ilyenkor fokozot
tan szükség van az egymással szemben levő centrifugaedények pontos kiegyensúlyozására. Az esetleges tengely- vagy edénytörés következményei ellen kb. 1000fordulaton felül acél védőköpeny használata szükséges. Az ultracentrifugákathasonló ok miatt betongödörben helyezik el.
48.1.2.1 táblázat
Nyomás a centrifugában
V m/sec
n (10 cm sugarúcentrifugában)
Nyomásatm
10 955 0,5114 1 335 1,020 1 910 2,0440 3 820 8,1080 7 640 32,6
140 13 350 100

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 89/366
K I R Á Z Á S 89
48.1.3. Folyékony halmazállapotú anyagkeverék komponenseinek elválasztása
Erre több lehetőség kínálkozik. Emulziók esetén pl. kisózással vagy centri-fugálással el lehet különíteni a diszperz részt. A fázisok különválása után a nagyobbfajsúlyú alsó fázist a választótölcsér alján levő csapon át leengedjük, vagy a felső,könnyebb komponenst leszivornyázzuk.
Az egymással nem elegyedő folyékony fázisok egyszerű, mechanikus elkülönítésével a következő laboratóriumi feladatok oldhatók meg:
a) Folyékony anyag oldott szennyezésektől való megtisztítása, pl. az éter megtisztítása alkohol szennyezéstől vízzel vagy vizes CaCl2-oldattal való kirázással;
b) Több komponensű rendszer szelektív extrakciója. így pl. az analitikában gya
kori feladat több kation közös vizes oldatából a szerves oldószerben oldhatókomplex kirázása. Ugyanezt az elvet nagyobb méretekben nemcsak preparatív,hanem ipari célokra is alkalmazzák.
Az említett esetekben az oldott anyag egymással nem elegyedő oldószerekközötti megoszlási egyensúlyát használjuk fel. A csapadékok kimosásához hasonlóan a folyadékok extrahálásánál is nagyobb mennyiségű oldószer helyett sokkalcélszerűbb több kisebb részlet alkalmazása.
Az n számú kirázás után a visszamaradó anyag koncentrációja (Cn) a következő képlettel számítható:
L y L-\-ccl I
ahol C0 a kezdeti koncentráció, L a kirázandóoldat térfogata, l az oldószer térfogata, a pedig az adott hőmérsékleten érvényes megoszlásihányados.
A szakaszosan ismétlődő extrahálást automatizálja a laboratóriumban használatos ún.
perforátor készülék (48.1.3.1. ábra), amely működési elvében a 8oxhlet-íé\e extraktorhoz hasonló. A készülék csak abban az esetben alkalmazható, ha az extraháló folyadék fajsúlya nagyobb az oldaténál. Az extrahálandó oldatot akészülék A tartályszerű részében helyezzük el. A fölötte levő B üvegszűrőn át a kondenzálóhűtő felületéről lecsöpögő extrahálószer finompermet alakjában jut az oldat felszínére, és nagyobb fajsúlya következtében a tartály aljára
süllyed. Speciális kivitelű készülékekben a tartály oldalát olyan alakúra képezik ki, hogy azextrahálószer lefelé tartó cseppjeit a benyúlóterelőlapokon minél hosszabb út megtételérekényszerítse. Az extraháló folyadék a C túlfolyón át az alsó, D lombikba jut. A készülékcsak abban az esetben működik folyamatosan, ha a két folyadék forráspontja között
48 .1.3 .1. ábra. 48 .1.3 .2. ábra. Perforátor Perforátor

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 90/366
30 A Z A N Y A G O K T IS Z T ÍT Á S A
elegendő különbség van, azonkívül nem elegyednek egymással korlátlanul és nemképeznek stabil emulziót.
Abban az esetben, ha az oldaténál kisebb fajsúlyú extrahálószer alkalmazásaszükséges, a hűtőben kondenzált fázist a folyadékoszlop alján fordított állásúüvegszűrő alá vezetjük (48.1.3.2. ábra).
Több komponensű oldatok valamennyi alkotórészének szétválasztására aperforátor nem alkalmas, helyette a szakaszos ellenáramú kirázás elvén működő,Craig által szerkesztett készülék használható.
A 48.1.3.3. ábrán látható a készüléknek néhány (üvegből készült) „megoszlásicellája” . Ezeknek nagyszámú (200 cellából álló) sorozatával 24 óra alatt 150 000 kirázás is megvalósítható. A módszer teljesítőképes
ségéről fogalmat alkothatunk magunknak, ha a megoszlási egyensúly tárgyalásakor ismertetett képlet szerint kiszámítjuk a visszamaradó anyag Cn koncentrációját pl. 200 kirázás után.
48.1.3.3. ábra. Craig-készülék a) vízszintes állásban; b) függőleges állásban
A készülék a következőképpen működik: Az elválasztandó elegynek megfelelő oldószerpárt mindenekelőtt kölcsönösen te
lítik egymással, majd a készülék valamennyi egységét megtöltik a nehezebb oldószer akkora mennyiségével, mely a csövet függőleges helyzetben, annak alsó ki-úgazásáig tölti meg. Ez alkotja a rendszer stacionárius fázisát. Ezután vízszinteshelyzetben az A csövön keresztül betöltik az oldószerpár kisebbik fajsúlyú tagját.
A sorozat elején viszont 2—3 cellában a szétválasztandó anyagok keverékét tartalmazó oldatot helyezik el. A szétválasztandó anyagok keverékét az oldószerpárnak akár a nagyobb, akár a kisebbik fajsúlyú tagja tartalmazhatja.
Ha az elegyet a nehezebb fázis tartalmazza, azt az első néhány cella aljábana tiszta oldószer helyére juttatjuk, és megfelelő mennyiségű könnyebb oldószert isrétegezünk föléje. A megoszlási egyensúlyt a cellák vízszintes helyzete körüli billeg-tetésével érhetjük el, majd meredekebb állásban megvárjuk a két fázis elkülönülését. Ezután a cellát a rajz szerinti függőleges helyzetbe állítva, a kisebb fajsúlyúoldószer a B tartályból a C csövön keresztül a következő sorszámú cella D tartály-részébe jut. Újra vízszintes helyzetbe hozva a készüléket, a mozgó fázis az E csövönát most már a következő cella stacionárius fázisával kerül érintkezésbe. Minden

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 91/366
K I R Á Z Á S 91
egyes kirázás után az első cellából tovább vitt mozgó fázist megfelelő mennyiségűtiszta oldószerrel (a könnyebb fajsúlyúval) pótolni kell.
Mindegyik komponens a választott oldószerpárban fennálló megoszlási hányadosának megfelelő sorszámú cellában fog feldúsulni. A kirázások számának elegendő növelésével az elválasztás gyakorlatilag teljessé tehető. Az elválasztandómennyiségnek határt szab a készülék mérete. Ennek ellenére a szervetlen prepara-tív munkában nagy teljesítőképessége miatt speciális feladatok megoldására, pl.Zr3+—Hf3+ elválasztására használják benzol—tiofenil-trifluor-aceton oldószerpársegítségével.
Nagyobb oldatmennyiségek extrahálására az eddig ismertetett készülékekegyike sem alkalmas. A 48.1.3.4. ábrán vázolt berendezés a folyamatos ellenáramú
kirázásé Ivén működik.1 Ebben a két folyadékfázis keveredése egy álló és egy forgó henger palástjai közöttkialakult néhány milliméteres résben megy végbe.
Szervetlen sóknak vizes oldatokból történő ex-trakciójánál nehézséget okozhat az akvokomplex-képződés. Jól oldódó elektrolitok (HC1, LiCl,NH4SCN, NH4N03) nagy mennyiségének hozzáadásával vízelvonó hatást érhetünk el. Ugyanakkormegfelelő anion vagy komplexképző kiválasztásávala szerves oldószerben való oldhatóságot növelhetjük.
Az említett extrakciós módszerek alkalmazásával olyan tisztítási és elválasztási feladatok is megoldhatók, amelyek sem lecsapással, sem frakcionáltkristályosítással stb. nem hajthatók végre. így pl.Sc-vegyületeknek lantanidáktól való tökéletes pre-paratív elválasztása tömény vizes NH4SCN-oldatbóléteres kioldással valósítható meg.
A folyadékfázisok közt végbemenő frakcionáltextrakció speciális formája az ún. megoszlási kroma-
tográfia. Ezt az eljárást biológiai anyagok vizsgálatára dolgozták ki, majd az analitikában tett szertnagyobb jelentőségre.
Kolloid oldatok elektrolitszennyezéseit féligáteresztő hártya segítségével is kivonhatjuk tiszta oldószerrel. A dialízisnek nevezetteljárás ismertetését lásd 14.10.
48.1.4. Gáz elválasztása szilárd vágy folyékony anyagtól
Ebben az esetben az egyik feladat a gáz megtisztítása a lebegő szilárd vagy
folyékony részecskéktől, a másik a szilárd fázis megtisztítása a felületén adszor-beálódott gázoktól, végül a harmadik az oldott gázok eltávolítása a folyékonyanyagból.
Gázok portalanítása vagy kodtelenítése legegyszerűbben szűréssel végezhető el. A lebegő részek méretétől függően egyszerű gyapot- vagy üveggyapot réteget,nagyobb diszperzitásfok esetén különböző pórusméretű zsugorított üvegszőrőt
1 Short J. F., Twigg G. H.: Ind. Engng. Cheín. 43, 2932 (1951).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 92/366
92 A Z A N Y A G O K T IS Z T ÍT Á S A
(G3—G4) használhatunk. Ez az eljárás csak a 10~4—10“ 5 cm-nél nagyobb 0 -jűrészek eltávolítására alkalmas.
A visszamaradó finomabb szennyezések a Tyndall-jelenség alapján mutat*hatók ki. Az ilyen kolloid dimenziójú részecskék eltávolítását illetően lásd 49.2.1.
A kolloid részecskék kondenzációs gócképző hatását használja fel az a módszer, melynek során az aeroszolt forrásban levő vízrétegen (vagy egyéb folyadékon)buborékol tatjuk át. A forralóedényhez csatlakozó hűtőben a kondenzáció a lebegőrészecskéken indul meg, amelyek azután a rárakódott folyadékeseppel együtt amosófolyadékba kerülnek.
Szilárd anyagok felületén adszorbeált gázok eltávolítását a hőmérséklet növelésével és a gáz parciális nyomásának'ösökkentésé vei végezhetjük. A parciális
nyomás csökkentését evakuálással vagy az adszorpció szempontjából indifferensgázárammal érhetjük el. így pl. klórozás után a termékből és a készülékből a felületen megkötött klórt N2- vagy C02-árammal szoríthatjuk ki.
A folyadékban oldott (abszorbeált) gázok koncentrációja azonos hőmérsékleten azok parciális nyomásával arányos (Henry-törvény). Eltávolításukra éppenezért a szilárd fázisok felületén adszorptíve kötött gázoknál alkalmazott eljárások
használhatók, így pl. a vízben oldott oxigén eltávolíthatóforralással és nitrogén- vagy szén-dióxid-átbuborékoltatásközben való hűtéssel.
48.1.5. Gázelegyek komponenseinek elválasztása
A legelterjedtebb módszer erre a célra a szelektív adszorpció. Részletesebb tárgyalását lásd 48.3. Ennek azelvnek az alkalmazásán alapszik a gázkromatográfiás el
járás. Eredményesen használták pl. szénhidrogének, illetve klór-szilán-homológok elegyének vizsgálatára. Újabban preparatív célokra is alkalmazzák.
Lényegesen nagyobb méretekben is megvalósítható
gázelválasztási eljárás a frakcionált diffúzió. így végeztékpl. kezdetben nagyüzemi berendezésben az U235 és U238-izotóp hexafluoridjának az elválasztását. Porózus agyagfalon 800-szor kellett megismételni a diffúziót ahhoz,hogy a végén az U235-ot gyakorlatilag tisztán megkap-
U W U . 1 ' 1
matos termodiffúziós nassaK.elválasztás sémája Az előzőnél is érzékenyebb módszer — amellyel izotó
pok elválasztása is megvalósítható — a termodiffúziós el járás. Elve a 48.1.5.1. sematikus ábrán'látható. Pl. sósavgáz termodiffúziójávalsikerült a 35C1- és 37Cl-izotópot 99,5 %-os tisztaságban előállítani. Sikerrel
alkalmazható a termodiífúzió nemesgázok gyors és kvantitatív szétválasztására is.2
Izotópok elválasztására az előbbi módszereken kívül alkalmazzák még többekközött az ultracentrifugát és a tömegspektrométert is.
könn y kom ponens
v gázkeverék a d ago lása
Tn ehéz komponens
ÁR7 A 7 / t 7h*/V A írtlTTn
2 Clusius K., Dickel O.: Z. physik. Chem. 44y 397 és 451 (1939).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 93/366
K R I S T Á L Y O S Í T Á S 93
48.2. Anyagok tisztítása halmazállapot-változás útján
A laboratóriumban leggyakrabban használt anyagelválasztási, tisztítási módszerek — talán a szűrés kivételével — ebbe a kategóriába tartoznak.
Szilárd anyagkeverékek komponenseinek szétválasztásakor a folyékony halmazállapotba (kiolvasztás, oldás) és a gázállapotba való átvitel (szublimálás) egyaránt előfordul. Szilárd és folyékony fázisok keverékét szűrésen kívül desztillálássalválaszthatjuk el egymástól.
Folyékony elegyek szó választására leggyakrabban alkalmazott eljárás a desztillációs bizonyos esetekben az egyik komponens kifagyasztása is célravezető.
Gázelegyek alkatrészei eltérő kondenzációs pontjuk vagy fagyáspontjuk alap
ján is elkülöníthetők. A felsorolt elválasztások nagy része néhány alapműveletre vezethető vissza(oldás—kristályosítás, desztillálás, illetve szublimálás), ezért az elválasztást ezekkel a műveletekkel kapcsolatban tárgyaljuk.
48.2.1. Kristályosítás
Az aprítás, oldás, keverés műveleteit az 1.13., a bepárlást az 1.18. pontbantárgyaltuk. Az 1.15.-ben ismertettük a kristályosítás alapműveleteit, ezért továbbiakban csak a speciális célokat szolgáló kristályosító eljárásokkal foglalkozunk.
Az átkristályosítással való tisztítás közben fellépő anyagveszteség általában jelentékeny. Értékesebb anyagok vagy igen rossz termelési hányad esetén (pl.izomorf anyagok elválasztásakor) a veszteség csökkentése érdekében frakcionált kristályosítást alkalmazunk. A művelet lefolyásának vázlatát a 48.2.1.1. ábra tüntetifel. Ezen a szilárd kristályos anyag útját kihúzott vonalak, az oldott anyagét(anyalúg) szaggatottak jelzik.
48.2.1.1. ábra. Frakcionált kristályosítás menete

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 94/366
94 A Z A N Y A G O K T IS Z T ÍT Á S A
Készítsünk a szétválasztandó anyagból forrón telített oldatot, és lehűtés,illetve bepárlás közben bontsuk fel a szükséges számú (a példában A — E-ig) frak
cióra. Ezek közül az elsőt célszerűen a többi egyenlő rész kétszeres súlyára méretezzük. A továbbiakban az egyeB frakciókat annyi tiszta oldószerben oldjuk, hogylehűlés után a szilárd anyagnak kb. a fele kristályosodjék ki. Az 1 A frakció kiválásaután visszamaradó anyalúgot az 1 B szilárd termékének oldására használjuk fel, saz ábra szerinti elv alapján dolgozzuk fel a többi frakciót is. A nehezebben oldódóanyag ily módon kristályos alakban összegyűjthető, a jobban oldódó rész pedig azanyalúgban dúsul fel.
Az eljárás hátránya, hogy igen lassú, időtrabló; ennek ellenére használjákkémiai reakciók útján nehezen végrehajtható elválasztásokra, pl. Th-, Ce- és La-tól
megtisztított ceritföldfémsók szétválasztására. A kikristályosított só az illetőelemeknek a magnéziummal képezett kettős nitrátja. — Káliummentes rubídiumelőállítása a megfelelő vastimsók frakcionált kristályosításával történhet. — Háf-nium és cirkónium elválasztására is alkalmas az eljárás ammónium-hexafluoro-komplexeik alakjában.
Kristálynövesztés. A kristályos anyagok általában optikai, elektromos stb.fizikai tulajdonságaik tekintetében anizotropok, és az anizotróp sajátságok akémiai reakcióképességben is megmutatkoznak. Az egyes tulajdonságok egyértelmű, reprodukálható meghatározása csak jól fejlett, morfológiailag hibátlan példányokban lehetséges.
Az ún. egykristályok előállítása különleges technológiával történik. Az ismertebb módszerek a következők: kristályosítás oldatból vagy olvadékból, szublimáció,elektrolízis. Valamennyi eljárás közös alapja az a követelmény, hogy a gócképződés sebességét minimálisra csökkentve, biztosítsuk a kristályképződés feltételeit.
Leggyakoribb feladat a kristálynövesztés oldatból. A kristályosodást vékonyfonálon függő, lehetőleg szabályosan kialakult apró kristállyal indítjuk meg. Túltelített oldatba mártott fonálon képződő sok kis kristály közül nagyítóval kiválasztjuk a legszebb példányt, közvetlenül alatta ollóval levágjuk a fonalat,fölötte pedig kézzel lemorzsoljuk a többi kristályt, és a kiválasztott kristályt
használjuk fel az anyalúg beoltására. A beoltott, melegen telített oldatot óvjuk a gyors lehűléstől és a vele járó túl-telítődéstől, mert az meggyorsítja a kristálygócképződést. Éppen ezért a kristályosító edényt jól szigetelt termosztát ládában helyezzük el, forró vízzel töltött,nagy hőkapacitású, kettős falú fémtartály belsejében.
Termosztát hiányában úgy is megóvhatjuk a rendszert a gyors lehűléstől, hogyamikor a lehűlő oldat hőmérséklete a szobahőmérsékletnél már csak alig valamivelmagasabb, leöntjük az addig kivált kristályokról, és beoltással ezt használjuk felegykristály növesztésére. Hátránya ennek az eljárásnak, hogy viszonylag nagytérfogatú oldatot igényel.
Csökkenthető az oldat térfogata akkor, ha a kiválott anyagmennyiség folyamatos pótlásáról gondoskodunk. Ezt az elyet valósítja meg a 48.2.1.2. ábrán látható készülék. Ebben a berendezésben az anyalúg két egymástól alig különböző(Ti és T2) hőmérsékletű, jól termosztált edény között cirkulál. A melegebb edényben a szilárd anyag oldódása következtében az oldat folyamatosan telítődik. Azalig hidegebb kristályosító edényben ily módon minimális mértékű, de állandó túltelítettség lép fel. Ilyen körülmények között a folyadék áramlása nem okoz zavarta kristály fejlődésében.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 95/366
K R I S T Á L Y O S Í T ÁS 95
Különböző szervetlen anyagok kristályosítására — a leggyakrabban használtvizen kívül — oldószerként egyéb szervetlen folyadékok (NH3, S02, halogén-hidro
gének, H2S, halogének, CS2 stb.) és szerves oldószerek is alkalmasak. A kristályosításhoz szükséges, megfelelő mértékű túltelítettség nemcsak a hő
mérséklet lassú változtatásával érhető el, hanem az oldószerenként változó oldhatóság felhasználásával is. így pl. a vízben igen jól oldódó [Cu(NH3)4]S04 szépenkristályosítható, ha a vizes oldatra óvatosan tömény alkoholt rétegezünk. A diffúziókövetkeztében előálló alkoholos közegben a só oldhatósága fokozatosan csökken(lásd 39.6.3.).
Különösen magasabb olvadáspontú oxidok kristályosítására oldószerként fel-használhatók olyan sóolvadékok, amelyek az illető anyaggal nem képeznek szilárd
oldatot. Szép MgO kristályok állíthatók elő például MgCl2-olvadékból 1000 C° körüli hőmérsékletenközönséges porcelán tégelyben. A gázlángban képződő vízgőz hatására a klorid hidrolizál, és a képződőMgO a MgCl2 fölöslegében oldódva, szépen fejlett kristályok alakjában válik ki a tégely falán.
A Si02 (tridimit és krisztobalit) kristályosításáraennek Na2W 04-, kevés P20 5-, V20 5-, illetve As^g-tar-talmú olvadéka alkalmas. Az olvadéknak nemcsakmint oldószernek van szerepe, hanem az egyébként
igen lassan végbemenő allotrop átalakulást is meggyorsítja.
Az előzőekben ismertetett eljárások közös vonása az volt, hogy a kristályt az anyagnak valamelyfolyadékkal (olvadékkal) készült oldatából állítottukelő. Az alábbi módszerek szerint a kristályt olvadékának szabályozott megszilárdításával hozzuk létre.
Az olvadékból történő egykristály-élőállítás következő módjai ismertebbek:
A Tammann-eljárás, melynek lényege, hogy az olvadékot tégelyben (Pt, grafit»porcelán) vagy leforrasztott (üveg, kvarc, egyéb) csőben elhelyezve nagyon lassanáthúzzuk az olvadáspont környezetében fekvő igen meredek hőmérsékleti gradiensen. így pl. LiF-egykristály előállítása céljából két stabilizált hőmérsékletű teretlétesítünk, amelyek egyikét a só olvadáspontja felett, a másikat az olvadáspontjaalatt kb. 60 C°-ra szabályozzuk be. Az így kialakuló 120 C° hőmérséklet-különbségnek kb. 0,1 mm-en belül kell fennállnia, hogy az eljárás sikeréhez szükséges, mintegy 1000 C°/mm gradiens létrejöjjön. Az olvadéknak a gradiensen való áthaladásakb. 1 mm/óra sebességű legyen.
Ebben az esetben is fontos, hogy az olvadékban csak egy kristálygóc növekedésére legyen lehetőség. Az ólomegykristály előállításakor ezt pl. úgy érhetjük el,hogy a vákuumban megolvasztott, legtisztább kiindulási anyagot tartalmazóüvegcső alját előzetesen vékony kapillárissá húzzuk ki. Az ebben képződő kristály-gócok közül csak az tud majd növekedni, amelynek a leggyorsabban növekedőkristálylapja a cső tengelyében helyezkedik el.
Az előzőnél sokkal egyszerűbb eszközökkel valósítható meg az egykristály előállítása, főleg könnyen olvadó fémekből és sókból, az ún. húzóéljárással. Az olvadáspontnál nem sokkal magasabb hőmérsékleten tartott olvadék felszínéhez előmele-
48.2.1.2. ábra. Berendezés oldatból való kristály
növesztésre

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 96/366
96 A Z A N Y A G O K T IS Z T ÍT Á S A
gített platina, üveg vagy porcelán csövet érintünk. A csővéghez tapadó kristálytóraműves szerkezettel lassan emelve, hosszú egykristályokat nyerhetünk, amelyekkeresztmetszete a húzás sebességétől függ. Ezen az elven alapszik a nagy gyakorlati jelentőségű Si- és Ge-tranzisztorok gyártása, azzal az eltéréssel, hogy a spektroszkópiai tisztaságú kristályok előállítása nagy Vákuumban történik. Az egykristályelőállítására vonatkozó tudnivalók annyira speciálisak, hogy a gyakorlati munkához szükséges részletek tekintetében az irodalomra utalunk.3
48.2.2. Desztillálás
A korlátlanul elegyedő folyadékok komponensekre való bontását az teszilehetővé, hogy egyensúlyi állapotban a folyadék és a gőzfázis összetétele egymástól
különbözik. A hőmérséklet—összetétel-diagramokon (48.2.2.1. ábra) ezért az egyensúlyi helyzetet két görbével ábrázolhatjuk: az egyik a folyadék összetételét kifejező
forráspontgörbe (alsó), a másik a gőzfázisét mutatóharmatpontgörbe (felső). Az egyensúlyi diagramoknagymértékben függnek a külső nyomástól, es általában az 1 atm-n fennálló állapotot tüntetik fel.
Az egyszerű desztillálás csak abban az esetbenalkalmas anyagkeverékek (szilárd—folyadék, folyadék—folyadék) komponenseinek egymástól való el
választására, ha tenzióik olyan mértékben különböznek, hogy a folyadékkal egyensúlyban levő gőz gyakorlatilag csak az illékony komponenst tartalmazza(pl. olajsav—etiléter). A valóságban ez csak ritkánfordul elő, ezért az elválasztás hatásfokának növelésére visszafolyós hűtővel, ún. deflegmátorral elősegítjük a gőz nehezebben illó komponensének akondenzálását, és gondoskodunk a folyékony fázisbavaló visszavezetéséről.
A deflegmálás hatását valamely folyadékelegy harmatpont- és forráspont-
diagramja alapján vizsgálhatjuk (48.2.2.1. ábra). A desztilláló üstben levő X mösszetételű folyadékból B hőmérsékleten Yb összetételű gőz távozik. A deflegmátor-ban ez a gőz G hőmérsékletre hűl le. Mivel az 7 b összetételű gőz a C hőmérsékletennem lehet egyensúlyban az X m összetételű folyadékkal, két fázisra fog bomlani,amelyek közül a kondenzált folyadék összetétele a D pontnak megfelelő X it a gázfázisé pedig az E pontnak megfelelő Yd (valóságban az egyensúly sohasem áll beteljesen). Az elválasztás annál tökéletesebb, minél nagyobb hányadát vezetjükvissza az elpárolgó gőznek, vagyis minél nagyobb az ún. reflux.
Azonban a refluxarány s ezzel a desztillátum tisztasága nem növelhető tetsző
legesen. Ha nem helyezünk súlyt a desztillátum időegység alatt elérhető mennyiségére, hanem az elérhető maximális tisztaságra törekszünk, vagyis az ábrán az F pontot tetszőlegesen megközelítjük, akkor a desztillátum maximálisan elérhetőtisztaságát a G pont adja .meg.
3 Mark H., Polányi M., Schmid E .: Z. Physik 12, 60 (1923); Kyropoulos S .: Z. Physik 63, 850 (1930); Morgenstern H .: Kristallogr. A. 100, 223 (1938); Hannay N. B .: Semiconductors, Reinhold P. C., New York, 1959.
--------^ X,Y
48.2.2.1. ábra. Harmatpont- és forráspontdiagram

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 97/366
D E S Z T I L L Á L Á S 97
A tárgyalt deflegmáció csak egyik különleges és nem is nagyon hatékonyalkalmazása egy általánosabb elvnek, a rektifikálás ebének. Ennek lényege akövetkező: valamely gőzáram ellenáramban érintkezik a folyadékárammal, amely -lyel nincs egyensúlyban. Előnyös a minél nagyobb érintkezési felület. Az érintkezéskövetkeztében a gőzben az illékonyabb, a folyadékban pedig a nehezebben illőkomponens dúsul fel. Hatásosabbá tehető a rektifikáció azáltal, hogy a visszafolyófolyadékot lapos edényekben, „tányérokon” fogjuk fel. Ezeken buborékol át a felszálló gőz, a kondenzált folyadék pedig tányérról tányérra lefelé csurog. Ilyen módona rektifikálás egyesíthető a frakcionált desztillációval, mivel a folyadék mindenegyes tányérról újra desztillál.
Az elérhető szétválasztás annál jobb, minél több tányér van az oszlopban. A rektifikáló oszlopok hatásfokának jellemzésére az elméleti tányérszám használa
tos, amely megadja, hogy az oszlop teljesítménye hány ideálisanműködő tányérral egyenértékű.4 Megfelelően méretezett rektifikáló oszlopon gyakorlatilag teljes elválasztás érhető el. Meg kellazonban jegyezni, hogy az adott nyomáson állandó forráspontú,ún. azeotróp elegyek a szétválasztás szempontjából külön komponensnek tekintendők, összetevőikre rektifikálással sem bonthatók szét.
A frakcionáló lombikból és kondenzáló hűtőből álló legegyszerűbb laboratóriumi desztilláló berendezés adólombikjának
nyakában is végbemegy bizonyos fokú deflegmálás. Ennek hatásfoka különböző hosszúságú és rendszerű deflegmáló hűtővelnövelhető, amellyel mintegy megnyújtjuk a lombik nyakát.Deflegmátor helyett a rektifikálás elvét is hasznosító frakcionálófeltéteket, illetve kolonnákat alkalmaznak nagyobb követelményeket támasztó feladatok megoldására. Ezek legkezdetlege- ^sebb formája felületnövelő anyagokkal töltött szélesebb cső Frakcionáló f e l t é t
{48.2.2.2. ábra.) A gyakorlatban igen sokféle megoldású kolonna használatos. Egy részükben
tányérszerűen kiképzett emeletek vannak, amelyeken a folyadék összegyűlik, és a
felfelétörő gőz ezeken buborékol keresztül. Gyakoribb kolonnatípus a tányérokatnem tartalmazó rektifikáló oszlop. A felületet megnövelő töltőanyag lehet változatos méretű, golyó vagy gyűrű alakú üveg, illetve kerámiai anyag, fémből készültspirális, lánc vagy egyéb. Ennek felületén jön létre a folyadék és gőz közötti ki-cserélődés.
Az egyszerű, üres csőnek is van rektifikáló hatása. Az 1 cm 0 -jű, 1 m hosszúcső elméleti tányérszáma kb. 1. Ugyanez a cső megfelelően töltve 20—22 elméletitányérnak felel meg. Tájékoztatásul szolgáljon, hogy a benzol és toluol elegye 12 elméleti tányérszámú oszlopon gyakorlatilag teljesen szétválasztható (benzol fp. 80,1
0°, toluol fp. 110,6 C°).Irodalmi adatok5 szerint a kolonna fémbetétjének gyors (kb. 1000 ford./perc)forgatásával a hatásfok mintegy négyszeresére növelhető. 10,7 m magas, kb. 500 el-
4 Az elméleti tányérszám meghatározását lásd E r d ey-Gr úzT . — Pr oszt J .: Fizikai-kémiai praktikum, I.-II. Tankönyvkiadó, Budapest, 1965.
5 Lesesne, S. D., Lochter, H. L .: Ind. Engng. Chem. Analyt. Edit. 10, 450 (1938).
7 Ált. és szervetlen kémiai prakt. —4284/ 1 1 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 98/366
98 A Z A N Y A G O K T IS Z T ÍT Á S A
méleti tányérszámú kolonnában a vízgőz l80-izotóp tartalmú frakciója jól feldúsítható.6
Kolonnák építésekor nagy gondot kell fordítani az alapos hőszigetelésre. Enél-kül a gőz teljes mennyisége kondenzál, és visszajut az adólombikba (100%-osreflux). A környezettel való hőkicserélés kikapcsolása biztosíthatja csak a frakcionálás egyesfázisaiban az egyensúly minél jobb megközelítését. A gyakorlatban ezt a kolonnaazbesztzsinórral való körültekerésével, külön szabályozható elektromos fűtéssel
vagy evakuált és ezüstözött köpennyel (Dewar) valósítják meg. Ha a kolonna tányérszámát azoszlop magasságának növelésével szaporítjuk,megnövekszik az ún. „holdup”, amely a desztillá
landó anyagnak a működő oszlopot megtöltőmennyiségét jelenti. Ezzel együtt hátrányosanmegnő az elválasztáshoz szükséges legkisebbanyagmennyiség is.
Ha a desztillált anyag a forráspont hőmérsékletén reakcióba léphet a levegő alkatrészeivel,védőgáz-atmoszférát alkalmazunk. Eljárhatunkúgy is, hogy csökkentett nyomáson végezzük a
desztillálást, amikor is nemcsak a hőmérséklet csökkentése szorítja vissza a zavaróreakciók sebességét, hanem a reagáló komponensek parciális nyomásának csökken
tése is ebben az irányban hat.Vákuumdesztillációt alkalmazunk akkor is, amikor az atmoszferikus desztil-
láció hőmérsékletén az anyag termikus bomlása következnék be. A nyomáscsökken -tés némely esetben az elválasztandó komponensek forráspontkülönbségének növekedésétis előidézheti, vagyis megkönnyíti az elválasztást. Vákuumdesztilláció esetén a nyomáscsökkentés mértékét a célnak megfelelően határozzuk meg. Vízlégszivattyúval 10—20 Hgmm-ig
terjedő nyomáscsökkentést hajthatunk végre,rotációs olaj szivattyúval kb. 0,1 Hgmm-ig, többfokozatú olaj- vagy higanydiffúziós szivattyúval pedig 10-3 Hgmm alá csökkenthetjük anyomást. A szivattyúkra vonatkozó részletektekintetében a vákuumtechnikai részre utalunk.
Az alkalmazott berendezés a légköri nyomáson végzett desztillációra felhasználttól any-nyiban is különbözik, hogy finoman kihúzottkapillárison keresztül levegőt vagy indifferens
gázt vezetünk a desztilláló lombik aljára. Ennek az a célja, hogy a folyadékot állandó mozgásban tartsa, és megakadályozza a túlhevü-lést. Hogy a kapilláris mellett a forráspont ellenőrzésére szolgáló hőmérőt is el tudjuk helyezni, 48.2.2.4. ábra. Négyujjú szedőfeltét
a Pegram, G. B ., Üreg, H. C., Huffman, J. R.: Physic. Rév. 49, 883 (1936); Ind. Engng. Chem. 29, 631 (1937).
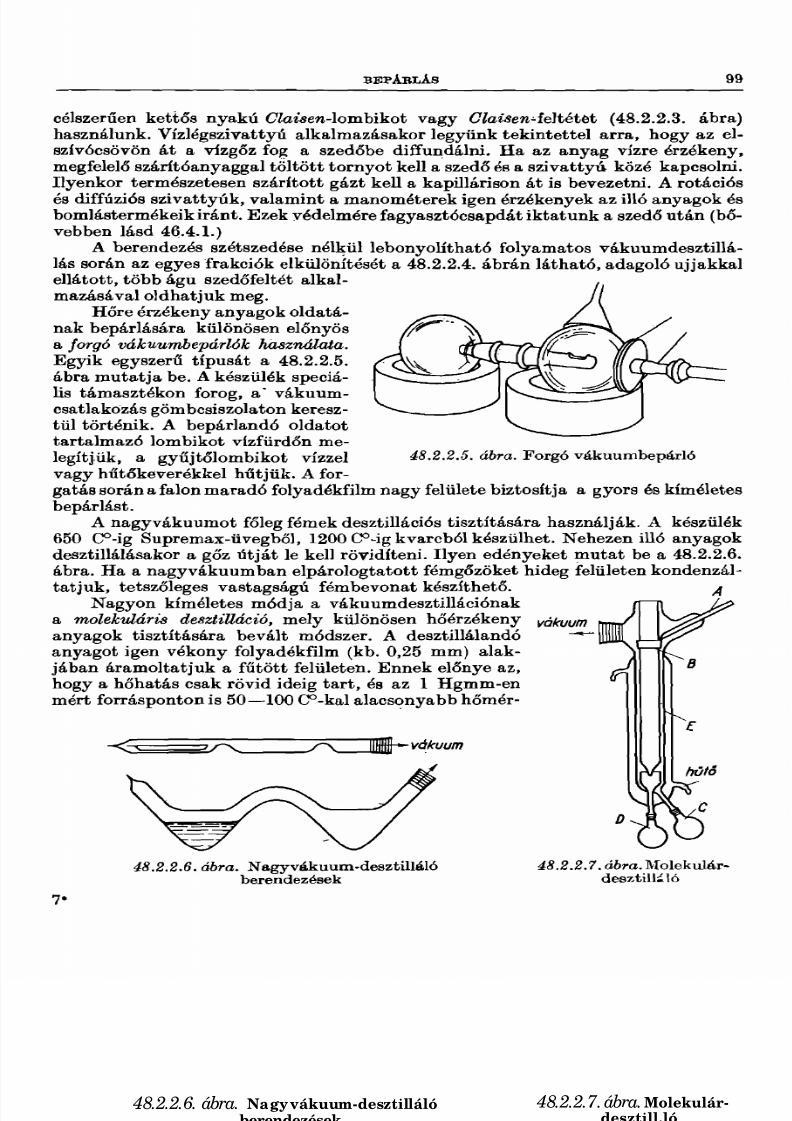
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 99/366
b e p A b l A s 99
célszerűen kettős nyakú CWaew-lombikot vagy Claisen-feltétét (48.2.2.3. ábra)használunk. Vízlégszivattyú alkalmazásakor legyünk tekintettel arra, hogy az el-
szívócsövön át a vízgőz fog a szedőbe diffundálni. Ha az anyag vízre érzékeny,megfelelő szárítóanyaggal töltött tornyot kell a szedő és a szivattyú közé kapcsolni.Ilyenkor természetesen szárított gázt kell a kapillárison át is bevezetni. A rotációsés diffúziós szivattyúk, valamint a manométerek igen érzékenyek az illó anyagok ésbomlástermékeik iránt. Ezek védelmére fagyasztócsapdát iktatunk a szedő után (bővebben lásd 46.4.1.)
A berendezés szétszedése nélkül lebonyolítható folyamatos vákuumdesztillá-lás során az egyes frakciók elkülönítését a 48.2.2.4. ábrán látható, adagoló ujjakkalellátott, több ágú szedőfeltét alkalmazásával oldhatjuk meg.
Hőre érzékeny anyagok oldatának bepárlására különösen előnyösa forgó vákuumbepárlók használata.
Egyik egyszerű típusát a 48.2.2.5.ábra mutatja be. A készülék speciális támasztékon forog, a' vákuum-csatlakozás gÖmbcsiszolaton keresztül történik. A bepárlandó oldatottartalmazó lombikot vízfürdőn me
legítjük, a gyűjtőlombikot vízzelvagy hűtőkeverékkel hűtjük. A for-gatás során a falon maradó folyadékfilm nagy felülete biztosítja a gyors és kíméletesbepárlást.
A nagy vákuumot főleg fémek desztillációs tisztítására használják. A készülék650 C°-ig Supremax-üvegből, 1200 C°-ig kvarcból készülhet. Nehezen illó anyagokdesztillálásakor a gőz útját le kell rövidíteni. Ilyen edényeket mutat be a 48.2.2.6.ábra. Ha a nagyvákuumban elpárologtatott fémgőzöket hideg felületen kondenzáltat juk, tetszőleges vastagságú fémbevonat készíthető.
Nagyon kíméletes módja a vákuumdesztillációnak
a molekuláris desztilláció, mely különösen hőérzékeny vákuumanyagok tisztítására bevált módszer. A desztillálandóanyagot igen vékony folyadékfilm (kb. 0,25 mm) alak
jában áramoltatjuk a fűtött felületen. Ennek előnye az,hogy a hőhatás csak rövid ideig tart, és az 1 Hgmm-enmért forrásponton is 50—100 C°-kal alacsonyabb hőmér-
48.2.2.5. ábra. Forgó vákuumbepárló
s~\ \ \§T vákuM
48.2.2.6. ábra. Nagy vákuum-desztilláló berendezések
48.2.2.7. ábra. Molekulár- desztill.ló

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 100/366
100 A Z A N Y A G O K T IS Z T ÍT Á S A
sékleten hajtható végre az elválasztás. A 48.2.2.7. ábrán látható készülék a követ
kezőképpen működik. Az A csövön át folyamatosan táplálják be az előzetesen gázmentesített oldatot. A B fűtött felületen osztógyűrű segítségével képződik a lefeléáramló folyadékfilm. Ettől kb. 1—1,5 cm távolságban van az E hűtőköpeny fala.Mivel az itt használt, 10-3 —10-4 Hgmm körüli nyomáson a molekulák közepes sza
bad úthossza 2—5 cm, biztosítva van annak a lehetősége, hogy a folyadékrétegből kilépő molekulák krak-kolódást okozó ütközések nélkül jussanak a hűtöttkondenzáló felületre. A maradékot a D, a desztillátu-mot pedig a C csövön át vezetjük el.
Az egyéb molekulárdesztillációs berendezések
lényegében szintén a fenti elv alapján működnek,így pl. az ún. vákuum-gyorsbepárlók7nagy folyadék-térfogatok alacsony hőmérsékleten végrehajthatóelpárologtatását teszik lehetővé. Nagy jelentősége
-=\ deszfillátum van ennek az eljárásnak pl. kolloid oldatok kíméleteskoncentrálásában.
A molekuláris dcsztilláció általában csak anyagoktisztítására, pontosabban a nem illékony komponenstől való elválasztására használható fel. Frakcionálásraazzal a módosítással alkalmazható, hogy a párolgásisebességet a párologtatási hőmérséklet leszállításávalnagymértékben korlátozzuk. Ennek az lesz az ered-
hutövíz-tüt fo lyó
48.2.2.8. ábra. Automatikus vízdesztilláló
és H2S,
ménye, hogy a készüléken átfutó folyadék illékony komponensének is csakkis százaléka jut el egy-egy alkalommala kondenzáló felületre. A kitermeléstúgy lehet javítani, hogy a desztillációsmaradékot többször is átvezetjük aberendezésen. A módszer anorganikus
prepáratív alkalmazására példa a H2S5elválasztása.8 A desztilláció automatizálása az at
moszferikus és a vákuumeljárások esetében aránylag egyszerű eszközökkelmegvalósítható. Minden esetben a desztilláló lombikban levő folyadék folyamatos pótlását kell megoldanunk.
A kémiai laboratóriumokban közismert vízdesztilláló tulajdonképpen at
moszferikus nyomáson működő automatikus berendezés (48.2.2.8. ábra). Az elpárolgott vízmennyiséget a kondenzáló gőzöktől felmelegedett hűtő-
vákuum
Hg nívó-
edény
48.2.2.9. ábra. Higanydesztilláló
7 Jantzen, E., Schmalfuss,‘H Chem. Fabr. 1, 373, 390 és 701 (1928); » Prasnitz, H.: Koll. Z. 86, 63 (1939).
Fehér, F ., Bandler, M.:Z. anorg. alig. Chem. 258, 140 (1949).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 101/366
D E S Z T I L L Á L Á S 101
vízből pótolják. A kazánban a víz szintjét változtatható magasságú túlfolyó biztosítja.
Automatizált vákuumdesztilláló berendezést használunk pl. a higany tisztítására (48.2.2.9. ábra). Ennél az állandó folyadékszintet a külső légköri nyomás biztosítja. A forralás megkezdése előtt a rendszerből a levegőt higanydifflíziós szivattyúval le kell szívni. Ennek következtében, a 76 cm magas fenékcsövön át a higany alombikba nyomul. Ilyen körülmények között a forráspont kb. 180 C°. A gőzök főtömege a speciálisan kiképzett lombik körbefutó csatornájában, a maradék pedig afüggőlegesen lefelé vezető ágban kondenzál. Ennek keskenyebb alsó része ugyan
csak kb. 76 cm hosszú, és túlfolyóban végződik. Néhány órai üzemeltetés után a külső elszívás megszüntethető; a továbbiakban akészülék önmaga biztosítja a vákuumot. Akészülék üvegalkatrószeit a higany nagy faj-súlya miatt különlegesen erősre kell méretezni. A szabályozott hőközlés alumínium blokkal történhet. Az elektromos fűtés automatikus kikapcsolása a higanytartályba megfelelőmagasságban beépített két kontaktus segít
ségével biztosítható. Ha a higany szintjeennél alacsonyabbra süllyed, az áramkörmegszakad.
Vízgőzdesztillációval olyan folyadékoktisztítása célszerű, amelyek hőérzékenyek,vízben nem oldódnak, gőznyomásuk kicsi, demolekulasúlyuk elég nagy (illóolajok). Ilyen folyadékok vizes keverékét forralva, adesztillátum a komponenseket az alábbi egyenlet által megszabott arányban tartalmazza :
9 i P i ' M y
48.2.2.10. ábra. Vízgőzdesztilláló
92 P r M 2
ahol g a tömeget, P a parciális nyomást, M a molekulasúlyt jelenti. Mivel az elegyforrása akkor következik be, amikor két komponens parciális nyomásának összegeeléri a külső légnyomást, a kisebb tenziójú anyag lényegesen a forráspontja alattihőmérsékleten desztillálható. Ebből a szempontból a vízgőzdesztilláció a vákuum-desztillációhoz hasonló hatású, bár lényegesen egyszerűbb eszközökkel valósíthatómeg. A vízgőzdesztillációt — mely a szerves preparatív munkában nagy jelentőségű — a szervetlen laboratóriumi gyakorlatban ritkán alkalmazzák.
A 48.2.2.10. ábrán látható berendezés lombikjában helyezzük el a tisztítandóanyag keverékét, és forráspontjáig melegítjük. Ezután a kazánból, amely szokásszerint vörösrézből készül, folyamatosan vízgőzt vezetünk át a lombikon. A gőz-fejlesztőn biztonsági célokat szolgál a vízszintjelző és a túlnyomáscső (Steig-rohr). A desztilláció a lombik és a kazán közé iktatott háromágú csappal bármikor megszakítható.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 102/366
102 A Z A N Y A G O K T IS Z T ÍT Á S A
48.2.3* Szublimélás
Szublimálásaal olyan szilárd anyagok tisztíthatok, amelyek tenziója eléri vagymegközelíti a rendszerben uralkodó nyomást, vagyis amelyek megolvadás nélkül isgyorsan elpárologtathatok. A szublimálható anyagok közismert példája a jód, mely
nek gőznyomása olvadáspontja alatt is számottevő,tehát vákuum nélkül is gyorsan szublimálható. Az ecélra szolgáló legegyszerűbb berendezést főzőpohárból vagy bepárlócsószóből és hideg vízzel telt lombikból állíthatjuk össze. Az egyenletesen melegítettedényből a jódgőzök a ráhelyezett lombik hűtött al
jára csapódnak ki.
48.2.3.1. ábra. SbCl3 szublimá- 48.2.3.2. ábra. Sorozatos szublimációra használhatólása légnedvesség kizárásával tekesorozat
Ha az atmoszferikus nyomáson szublimálható anyag a légnedvességre érzékény, a vízgőz távoltartását a 48.2.3.1. ábra szerinti készülékkel oldhatjuk meg
Kb. 200 g SbCl3 tisztítását elvégezhetjük úgy, hogy 500 ml-es frakcionáló lombikaqhelyezzük, melynek oldalcsövét előzőleg P20 5-dal töltött szárítócsővel kapcsoltukössze. Nyakára széles szájú gömblombikot illesztünk jól záró parafadugó segítségével, és az ábra szerinti helyzetben vízfürdőre tesszük. A lombik felső részére szűrőpapírt borítunk, és csepegő vízzel állandóan nedvesen tarjuk. így az említett meny-nyiség kb. egy nap alatt szépen fejlett kristályok alakjában lerakódik a hűtött lombikfalon. A maradék megdermedése után a kristályok gyenge kopogtatással leválaszthatók a lombik faláról, és a szedőbe rázhatók.
Azok az anyagok, amelyeknek jelentős tenziójuk van, a vízgőzdesztilláció elvé
hez hasonlóan vivőgázáramban sokkal gyorsabban szublimálhatok, mint anélkül. Példaerre a halogenidek, pl. a FeCl3 szublimálásaCl2 segítségével.
Közömbös gázatmoszférát alkalmazunkakkor, ha az anyagot óvni kell pl. a levegőoxigénjétől. A 48.2.3.2. ábrán látható készülék előnye, hogy benne a szublimálást közömbös atmoszférában vagy ugyanilyen gázáramban egymás után többször is megismé
telhetjük, és a legtisztább frakciót az utolsótekében le is forraszthatjuk. A tekesorozatotcsiszolat vagy tűzálló kitt segítségével közvetlenül csatlakoztathatjuk a szublimálandóanyagot előállító készülékhez is. Mivel aszilárd anyagok hővezető-képessége (a fémekkivételével) általában rossz, a szublimálandóanyagot mindig vékony rétegben kiterítve
48.2.3.3. ábra. Vákuumszublimáló készülék

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 103/366
S Z U B L I M Á L Á S 103
(finoman elporított állapotban és gondosan szárítva) vetjük alá a szublimálásnak,
hogy a helyi túlmelegedéseket elkerüljük.Ha mégis fennáll a termikus bomlás veszélye, akkor a szublimálás hőmérséklete a nyomás csökkentésével jelentős mértékben leszállítható.
A 48.2.3.3. ábrán egy egyszerűen kivitelezhető vákuumszublimáló készüléketmutatunk be.
Abban az esetben, ha a hűtött felületet igen közel visszük a szublimálandóanyaghoz, a szilárd fázisból kilépett molekuláknak sokkal nagyobb hányada jut eladott hőmérsékleten a hűtőre. Ezzel a főként analitikai célokra használt mikroszvb- limálási módszerrel lehetővé válik olyan anyagok szublimálása légköri nyomáson,amelyek hőérzékenységük miatt egyébként
csak nagyvákuumban lennének szublímál-hatók.
VákuumszublimálásTSi a nehezen illóanyagok nagy vákuumban való desztillálá-sához ajánlott készülékek is alkalmazhatók.Hátrányuk, hogy a szublimátümot nehézbelőlük kiszedni. Ha az anyagot nem lehetkiolvasztani, a készüléket szét kell vágni. A48.2.3.4. ábrán bemutatott készülék ezt a
nehézséget küszöböli ki. Az evakuálható csiszolatos csövet az elszublimáló anyagfelfogására szánt béléscsővel látjuk el, ebben helyezzük el az anyagot tartalmazófiolát. Ha az anyag az üveget megtámadja, béléscsőként egyik végén zárt platinaosövet alkalmazunk, melynek nyitott fele a hideg térbe nyúlik.
Ha a szublimálandó anyag gőznyomását célszerű alacsony értéken tartani,akkor a 48.2.3.5. ábra szerinti megoldás alkalmas arra, hogy a hűtött tér a szublimáló felület közelébe kerüljön.
A 48.2.3.6. ábra alacsony hőmérsékleten végzendő vákuumszublimálásra alkalmas berendezést ábrázol.9 A készülék függőleges helyzetben működtethető. Aljatermosztáló fürdőbe merül, a belsejébe nyúló edény pedig szénsavhóval vagy más
hűtőkeverékkel tölthető.Szublimálással egykristályt is állíthatunk elő10 az oldatoknál tárgyalt elvek
alapján. A polikristályos anyagot evakuált csőben helyezzük el. Ennek két végétegymástól csak kismértékben különböző hőmérsékletreállítjuk be. A szabályosan fejlett oltókristályt a hidegebboldalon, az anyag többi részét pedig a melegebb oldalonhelyezzük el. Az áramló oldatból történő egykristály-
48.2.3.5. ábra. Vákuumszublimáló berendezés 48.2.3.6. ábra. Mélyhűtőé vákuum-
szublimáló berendezés
növesztéshez hasonlóan, az elszublimált anyagnak a kristálygóchoz való továbbítására közömbös gázáramot alkalmazhatunk.
» Marberg, G. M .: J. Am. Chem. Soo. 60, 1509 (1938).10 Stranski, I. N .: österr. Chemiker Ztg. 45, 145 (1942).
Ál-blokk
o D
48.2.3.4. ábra. Vákuumszublimáló berendezés

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 104/366
104 A Z A N Y A G O K T IS Z T ÍT Á S A
48.2.4. Kondenzáció, kifagyasztás
Gázkeverékek elválasztására, illetve gázok tisztítására eredményesen alkalmazható a részlegesen vagy fokozatosan végrehajtott kondenzálás, illetve kifagyasztás.
Tisztítás céljából vezessük át a gázt lassú áramban ún. fagyasztócsapdán,amelynek hőmérsékletét úgy választjuk meg, hogy azon a kivált szennyezésnek márszámottevő tenziója ne legyen. így pl. folyékony levegővel hűtött csapdában aH2, He, Ne, Na, 0 2, F2>Ar, CH4, CO, NO kivételével minden anyag kifagyasztható,vagyis a felsorolt gázok egymáson kivül minden egyéb szennyezéstől megtisztíthatók.
Ha különböző hőmérsékletre hűtött csapdasoron vezetjük át a gázelegyet, azegyes komponensek a kondenzálási pontjuknak megfelelő hőmérsékletű csapdábanfognak feldúsulni. Célszerű a csapdákban a hideg felület növelése érdekében, nagyfajlagos felületű adszorbenseket elhelyezni.
Két, egymáshoz nagymértékben hasonló tenziójú szilárd anyag szublimálásútján való elválasztása gyakorlatilag kivihetetlen. Ennek legfőbb oka a szilárd testek rossz hővezető-képessége (a fémek kivételével). A fűtött felületen többnyire helyi túlhevülések lépnek fel, és itt mindkét anyag maradéktalan elpárolgása következik be.
Ilyen esetekben könnyebben célt érünk a frakcionált kifagyasztással. Ezt úgyhajtjuk végre, hogy vákuumban, alacsony hőmérsékleten elpárologtatjuk a teljes
anyagmennyiséget lehetőleg úgy, hogy a komponensek parciális nyomása kb. 1—2Hgmm legyen. A képződött gőzöket fokozatosan egyre alacsonyabb hőmérsékletrehűtött U-cső sorozaton vezetjük át. Az egyes frakciók a különböző hőmérsékletűszedőkben gyűlnek össze.> Az utolsó szedőt a vákuumberendezés védelme érdekében folyékony levegővelhűtjük. Mivel a fenti esetben a kondenzált fázis szilárd, vigyázni kell arra, hogy azU-csövet valamelyik frakció el ne tömje. A frakcionált kifagyasztás igen hosszadalmas, lassú művelet. Igen kis, néhány grammos mennyiségek feldolgozására is 24—36óra szükséges.
Folyékony halmazállapotú anyagkeverékek komponenseinek szétválasztása is
végezhető kifagyasztással. Példaként megemlítjük a tengervíz sótartalmának koncentrálását az oldószer fő tömegének kifagyasztása útján, vagy a paraffinolaj paraffintartalmának kinyerését. További, nagy jelentőségű alkalmazási területe a fentieljárásnak a kohászatban van. Nem ötvöződő fémek közös olvadékából kifagyasztható a magasabb olvadáspontú elem.
Oldott szennyezéseket tartalmazó anyagok tisztítására is felhasználható ez amódszer, amennyiben az oldószernek tekinthető tisztítandó anyag fő tömegét tisztaállapotban kifagyaszthatjuk, miközben a szennyezés a megmaradó folyékony fázisban koncentrálódik. így állítható elő pl. a kristálybenzol.
48.2.5. Kiolvasztás
Szilárd halmazállapotú anyagkeverékek alkatrészei, ha olvadáspontjuk közöttelég nagy különbség van, egyszerűen elválaszthatók egymástól az alacsonyabb olvadáspontú rész megolvasztása és kicsurgatása útján. Tipikus példa erre a Frask-fé- le kénbányászati eljárás.
Ugyancsak kiolvasztással tisztítható meg a fémh'tium a felületén levegőn képződő osidrétegtől, ha a műveletet megfelelő készülékben vákuumban végezzük.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 105/366
E X T R A K C I Ó 105-
48.2.6. Kioldás, extrakció
Ennél az eljárásnál a szilárd anyagkeverék alkatrészeinek elválasztására az.alkalmazott oldószerben mutatott különböző oldékonyságukat használjuk fel. Ennek egyszerű példáját tárgyaltuk a 2.1. pontban (CaCÓ3—NaCl).
A preparátumok, illetve csapadékok kimosása a preparatív és az analitikaikémiában egyaránt a leggyakrabban előforduló műveletek egyike. Ha lehetőség:
48.2.6.1. ábra. Sor klet -extrakto r
szerint tökéletes elválasztást akarunk elérni, vagy a kioldás folyamata hosszadalmas, automatikusan működő készülékeket alkalmazhatunk. Ezek legismertebb típusa az ún. Soxhlet-féle extralctor (48.2.6.1. ábra). Szerkezete és működése lényegében azonos a 48.1.3. pontban ismertetett perforátorokéval. A forralólombikból eldesztillált folyadék a készülék felső részében levő hűtőből visszacsöpögve, állandóanfriss oldószere az áteresztő falú hüvelyben elhelyezett anyagnak. A túlfolyó szintjének elérése után az extraktumot tartalmazó oldat lefolyik a lombikba, ahol az oldó
szert újra elpárologtatjuk.Előfordul, hogy valamely preparátum tisztítása folyamán alacsony hőmérsék
leten cseppfolyósodó gázokkal történő extrakciót kell alkalmaznunk. így pl. alkáli-fém-amidok tisztításához folyékony-ammóniás extrakció használatos.11
Cseppfolyós gázokkal (folyékony NH3, folyékony SOa, stb.) való extrakcióhoz.a 48.2.6.2. ábrán vázolt készülék alkalmazható.
11 Juza, R., Fasold, K., Haeberle, C.: Z. anorg. alig. Chem. 234, 75 (1937).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 106/366
106 A Z A N Y A G O K T IS Z T ÍT Á S A
Az extrahálandó anyagot az A edényben levő B üvegszűrőre helyezzük. A C csövön át gondosan szárított (fémnátriumról ledesztillált) ammóniát vezetünk be, és a
szárazjeges hűtőkeverékkel hűtött A edényben kondenzáltat]uk. Miután az ammóniában oldható komponenseket kioldottuk, a csapok megfelelő állításával és a D edény hűtésével a folyékony ammóniát az A edényben fellépő túlnyomással a D edénybe nyomatjuk át. Szükség esetén a hűtés megcserélésével az ammónia ismétaz A edénybe desztillálható át, és az extrakció tetszés szerint megismételhető.
48.3. Adszorpció, deszorpció, kromatográfia, ioncsere
Az anyagtisztítás módszerei közül ebben a csoportban azokat tárgyaljuk,
amelyek folyamán egyrészt nem mindig dönthető el, hogy bekövetkezik-e a fázis-átalakulás vagy sem (pl. gázadszorpció, kapillárkondenzáció), másrészt, amely eljárásokban bizonyos fokig kémiai reakcióknak is szerepe van (ioncsere).
48.3.1. Adszorpció, deszorpció
Az adszorpció egyik alkalmazási módja a laboratóriumi gyakorlatban és iparitechnológiában egyaránt gyakran használt derítés. Részleteire vonatkozóan lásd1.15. és 14.1.1.
Gázok tisztítása, illetve a gázkeverékek szétválasztása hatékonyabbá tehető,
ha a kondenzációs módszereket adszorpcióval kombináljuk. Preparatív célokra azadszorpció főleg akkor alkalmazható, ha a gázelegy egyes alkotórészeinek adszorpcióképessége — az adott adszorbensen — egymástői jelentős mértékben különbözik.
A tisztítandó gáznál sokkal nehezebben illó szennyezések (csapzsírgőzök, vízgőz,C02 stb.) pl. mélyhűtött szilikagélen vagy aktív szénen történő adszorpcióval távolíthatók el. A hűtés hőmérsékletét úgy kell megválasztani, hogy elkerüljük a tisztítandó gáz kapillárkondenzációját. — A módszert a szennyezés fő tömegének pl.kémiai módszerrel vagy kifagyasztással végzett eltávolítása után az utolsó nyomokkiküszöbölésére célszerű használni.
Aktívszén adszorbensek hűtésére a folyékony levegőt igen ajánlatos cseppfo
lyós nitrogénnel helyettesíteni, mivel az edény esetleges cltörése következtében azaktív szénnel keveredő folyékony levegő robbanó keveréket alkot (oxilikvit). Ugyanezen okból nem szabad gázok esetleges oxigénszennyezését (folyékony nitrogénnelhűtött) aktív szénen adszorbeáltatni.
A deszorpcióval kombinált adszorpció segítségével gázkeverékek kevésbé illókomponense is tisztán előállítható. így pl. folyékony hidrogénnel hűtött szilikagél-lel a ncongáz héliumtartalma ezen a hőmérsékleten az adszorbensről nyomás csökkentésével teljesen eltávolítható, azután pedig a hőmérséklet emelésével a tisztaneon deszorpciója is végrehajtható.12
Miként az adszorpció a frakcionált kondenzációval hasonlítható össze, a frak-cionált deszorpció megfelelőjének a frakcionált desztillációt tekinthetjük. A deszorp-ciót szükségszerűen megelőző adszorpció körülményeinek megfelelő megválasztásanagymértékben fokozhatja a deszorpcióval elérhető elválasztás hatásosságát.
Az említett adszorbenseket (aktív szén, szilikagél, A1203) alkalmazás előtt vákuumban, 300 C° körüli hőmérsékleten több órán át tartó kigázosításnak kell alé-
12 Edse, R., Harteck, P.: Angew. Chem. 53, 210 (1940).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 107/366
K R O M A T O G R Á F I A 107
vetni az adszorbeáló edényben. A szilikagélt nem szabad 350 C°-nál nagyobb hőmérsékletre hevíteni, mert adszorbeáló képessége irreverzibilis módon csökken.
Az aktív szén bizonyos anyagok (02, F2, Cl2, S) minimális mennyiségeit irreverzibilis módon megköti. Ezek csak magasabb (600 C° feletti) hőmérsékleten, vákuumban távolíthatók el, és akkor CO, C02, CF4, CC14, CS2 alakjában kapjuk visszaazokat.
A deszorpció hőmérsékletének beállításakor számolni kell az aktív szén igenrossz hővezető képességével. A termosztát hőmérsékletét a szén — különösenvákuumban — igen nehezen veszi fel. A szilikagélnek ilyen szempontból valamivelkedvezőbb tulajdonságai vannak. A frakcionált deszorpció hatásfoka nagyvákuum-ban igen előnyösen növelhető.
Az adszorbeált gázok deszorpciójának megkezdése előtt a berendezést alaposan evakuáljuk (10-4 Hgmm-re). Ez után a termosztáló fürdővel beállítjuk a legkönnyebben illó komponens deszorpciós hőmérsékletét. A deszorpció megindulásáta vákuum csökkenése jelzi. Ha a rendszer nyomása az alkalmazott szívássebességmellett elérte az 1*10-2 —5-10“ 2 Hgmm közötti értéket, a hőmérséklet továbbinövelését megszüntetjük, és a frakció teljes leszívatását ezen a hőmérsékletenvégezzük el. Amikor a vákuum elérte az eredeti értéket, akkor befejeződött az elsőkomponens deszorpciója, és a hőmérséklet emelésével a fenti módon elkezdhetjüka következő részlet leszívatását.
Az alkalmazott adszorbens mennyisége elsősorban az adszorbeálandó anyagok
mennyiségétől függ. 3—50 ml normál gázkeverék deszorpciós szétválasztására általában a szokásos adszorbenseknek 1 grammja szükséges.
A módszer főleg gázanalitikai szempontból jelentős, de a preparatív kémiábanis alkalmazzák gázelegyek szétválasztására, pl. az orto- és parahidrogén, vagy H2és D2 elválasztására.13
A frakcionált deszorpció végrehajtható vákuum és melegítés helyett gyakorlatilag nem adszorbeálódó, indifferens gázáram segítségével is. Ezt az elvet alkalmazza a gázkromatográfiás eljárás.
48.3.2. Gázkromatográfia, ioncserélő kromatográfia
Kromatográfiának nevezzük azokat az elválasztási módszereket, amelyekbenmozgó gáz- vagy folyadékfázis nagy felületű álló fázissal érintkezve, többé-kevésbéegyensúlyra vezető folyamatok (adszorpció, deszorpció, megoszlás, kioldás, diffúzió, ioncsere stb.) eredményeképpen az elegyfázisban levő anyagok elkülönülhetnekegymástól. A mozgó fázis lehet légnemű (gázkromatográfia) vagy cseppfolyós(adszorpciós, megoszlási vagy ioncserélő kromatográfia).
Az álló fázis többnyire szilárd, vagy szilárd fázison megkötött folyadékréteg. Az elválasztást eredményező folyamatok a fázisok érintkezési felületén mennek
végbe, azért célszerű álló fázisként nagy fajlagos felületű szilárd anyagot alkalmazni.
Speciális metodikája miatt a többi eljárásoktól elkülöníthető a gázkromatográfia. A gázkromatográfia egyre nagyobb szerepet kap a preparatív, különösen a mikro-preparatív laboratóriumi munkában. Ez a gázkromatográfia nagy szelektivitásávalmagyarázható, amely egyedülálló lehetőségeket biztosít a legnehezebb szétválasz-
19 Edse, R., Harteck, P.: Angew. Chem. 53, 210 (1940).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 108/366
108 A Z A N Y A G O K T IS Z T ÍT Á S A
tási feladatok megoldására is, mint pl. azeotrópképző vagy hőérzékeny anyagokszétválasztása, vagy spektroszkópiai tisztaságú anyagok előállítása.
A preparatív gázkromatográfiai eljárásokban a szokásos analitikai gázkromatográfiai módszerekhez viszonyítva nagyobb feldolgozandó anyagmennyiségeknekmegfelelően növelni kell az oszlopok átmérőjét. E téren problémát jelent az akörülmény, hogy az oszlopok hatásossága az átmérő növelésével általában romlik.Ez a hátrány nagyrészt kiküszöbölhető szűk szemcsefrakciójú töltetek és belső gázterelőlapok alkalmazásával. Ily módon 10 cm 0-ig terjedő oszlopokkal 200—500 ganyag betáplálása esetén igen hatásos szétválasztás érhető el. A kapacitás növelésének másik módja több kisebb átmérőjű oszlop párhuzamos bekötése.
A preparatív gázkromatográfia vonalán jelentős fejlődést hozott az automa
tikus mintaadagolás és frakciószedés megoldása, aminek segítségével a tulajdonképpen szakaszos művelet ismétlésével napi néhány kilogramm tiszta anyag iselőállítható.14
A gázkromatográfiás oszlopok tölteteként általában diatomaföld alapú hordozókat alkalmaznak, melyeket speciális előkészítéssel viszonylag homogén felületűvéalakítanak. A hordozók felületét különböző utókezeléssel, pl. savas vagy lúgosmosással, a felület szilanizálásával, hőkezeléssel stb. módosítják. Erősen polárosanyagok szétválasztása esetén az adszorpciós hatás kiküszöbölésére 0,1—0,2 %magas forráspontú poláros anyaggal, ún. korróziós inhibitorokkal is borítják a felületet. Speciális esetekben töltetként üveg-, teflon- vagy termolittégla-port használ
nak. A töltet nedvesítésére alkalmazott folyadékok választéka igen nagy. 150—200
C°-ig általában különböző polaritású poliglikolok és poliészterek használatosak.Magasabb hőmérsékleten (400 C°-ig) Apiezon-zsírok, szilikonpolimerek és aszfaltének alkalmasak. 400 C° feletti tartományban szervetlen só-eutektikumok a nedvesítő folyadékok.
A gyakrabban alkalmazott detektálási módszerek mellett, amilyenek pl. a hő-vezetőképesség-mérés, ionizációs detektálás, az utóbbi időben számos specifikusdetektortípust is használnak (elektronbefogási, lángemissziós, fotoionizációs detek
tor, tömegspektrométer, infravörös spektrométer, stb.). \ A gázkromatográfia elsősorban szerves anyagok analízisére és szétválasztásárahasználatos. Űjabban azonban a fémorganikus, valamint szervetlen vegyületek, pl.illékony halogenidek, magasabb hőmérsékleten pedig fém-halogenidek és -karbonátok elemzésére és szétválasztására is alkalmazzák.
A kromatográfia különböző változatai közül a szervetlen preparatív kémiábanlegnagyobb jelentősége az ioncserélő oszlopkromatográfiának van. Alkalmazásátidegen ionoktól mentes víz előállítására már a 14.3.-ban tárgyaltuk. Ezenkívülegymáshoz igen hasonló kémiai viselkedésű anyagok elválasztására is jól alkalmazható módszer. A ritkaföldfémek 5,0—5,5 pH-jú, 0,8 %-os ammónium-citrát-puffer
segítségével külön-külön leoldhatók az adszorbensről. Adszorpcióképességük ilyenkörülmények között a lantántól a lutécium felé haladva a rendszám növekedésévelfolyamatosan csökken.15 Irodalmi adatok szerint16 az eljárás tiszta cirkónium előállítására, illetve Zr —Hf elválasztására is alkalmas.
14 Best, Y. D. N Gas Chromatography. Universities Press, Belfast, 1958. 288.old.18 Spedding, F. H. és munkatársai: J. Am. Chem. Soc. 70, 1671 (1948); 75, 2529 (1953).18 Ayrea, J. A.: J. Am. Chem. Soc. 69, 2879 (1947);
Liater, B . A. J.: J. Chem. Soc. [London] 3123 (1951).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 109/366
I O N C S E R É L Ő K 109
Az ioncserélőket két csoportba oszthatjuk: szervetlen ioncserélőkre és gyantákra. A szervetlen ioncserélőkhöz az alumínium-szilikátok és különböző oxid alapú
ioncserélők sorolhatók. Az alumínium-szilikátok az első szintetikus úton előállítottioncserélők, melyek alumínium-szulfát- és nátrium-szilikát-oldatok elegyítésévelkészíthetők. A kapott kemény gélt elporítjuk, mossuk, majd kiszárítjuk. A gélbenlevő nátriumionok kicserélhetők kalcium-, ammónium- vagy egyéb ionokra. Azalumínium-szilikátok ioncsere-kapacitása számottevő, azonban hátrányos tulajdonságuk, hogy savak és lúgok hatására egyaránt bomlanak, ami jelentős mértékben korlátozza alkalmazhatóságukat. Az oxid alapú ioncserélők, pl., a Zr02, Th02,TiOaés Sn02 az oldatok pH-jától függően kation- és egyúttal anioncserélő tulajdon-ságúak lehetnek. A cirkónium-dioxid erősen lúgos közegben kationcserélő, viszont
pH-2-nél anioncserélőként működik (kapacitása 1 milliekvivalens ion/g ioncserélő).Különösen jó kationcserélő tulajdonságúak a négyértékű fémek foszfátjai,molibdátjai és vanadátjai. A cirkónium-foszfát pl. dihidrogén-foszfát funkcionáliscsoportokat tartalmazó lineáris polimernek tekinthető: [ —Zr(H2P 04)2—0 —]n, és12 -es pH-nál 13 milliekvivalens/g elméleti kapacitása van. Az oxid alapú ioncserélők finom por vagy különböző méretű granulált gél alakjában készíthetők.Preparatív célokra az utóbbiak használatosak, míg a por alakú ioncserélők analitikaicélokat szolgálnak.
A szervetlen ioncserélők a hőmérséklet-változással szemben a gyantáknálkevésbé érzékenyek, és szervetlen ionokra nagyobb szelektivitásúak..
Az ioncserélő gyantákat lényegesen szélesebb körben alkalmazzák, mint aszervetlen ioncserélőket. Ezek általában sztirol és divinil-benzol kopolimepízációjarévén előállított műgyanták, amelyek funkcionális csoportokat — pl. erősensavanyú jellegű S03H- vagy gyengén savas C03H-, esetleg bázikus nitrogéntartalmú csoportot — is tartalmaznak. E savanyú vagy bázikus funkcionális csoportokat tartalmazó gyanták vizes oldatokban reverzibilis ioncsere-reakcióba lépnek:
(R~S03)H + Na+ ^ (R~S03)N a+ H +,
(R-NR3)0H + Cl" ^ (R-NR3)C1 + OH".
Az ioncserélő gyanták specifikus fajtája a hidrokinon, fenol és formaldehidkondenzációjából előállítható redoxitulajdonságú gyanta, amely egyenlő számúkínon- és hidrokinoncsoportot tartalmaz, így redoxipotenciálja közelítőleg azonosa kínon—hidrokinon-oldatokéval. Amennyiben e gyantából készült oszloponnagyobb redoxipotenciálú vizes elektrolitoldatot csurgatunk át, redukció következik be: pl. a Fe3+-oldat Fe2+-oldattá redukálódik. Az ilyen típusú ún. elektron
cser élők különösen analitikai szempontból jelentősek. A kereskedelmi forgalomban levő ioncserélő műgyanták szemcsemérete duz
zasztott állapotban 20^-tól 2 mm 0-ig térj ed. Rendszerint megfelelő csomószámúsziták közötti frakció kapható, tehát közelítőleg homodiszperz gyanta. Minél kisebba szemcseméret, annál rövidebb idő szükséges a környező oldattal való egyensúlybeállásához, vagyis annál kisebb az „elméleti tányérszám”-nak megfelelő oszlopmagassága. A diszperzitásfok növelése azonban nagymértékben emeli az oszlopáramlási ellenállását, és ezért preparatív célokra általában 0,25—0,40 mm-esszemcseátmérőjű ioncserélő gyanta használatos. A kereskedelmi forgalomban levőgyanták kapacitása 10—100 milliekvivalens/g gyanta, a gyakorlatban azonban azioncserélő oszlopokban csak e kapacitás néhány százalékát használjuk ki.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 110/366
110 A Z A N Y A G O K T IS Z T ÍT Á S A
A preparatív munkára alkalmas ioncserélő oszlop előállítása igen egyszerű.
A kísérleti berendezés egy áteresztő csappal és beépítettG
l-es üvegszűrővel ellátott — szükség szerint 6—20 mm 0 -jű és 60—200 cm hosszú — üvegcső (48.3.2.1.ábra). Szükség esetén üvegszűrő helyett PFaíf-lemez és üveggyapot is alkalmazható. Az ioncserélő megfelelő csomószámú sziták közötti frakcióját használjuk. Azoszlopot félig vízzel töltjük, majd a gyantát fokozatosan adagoljuk, és néhányórán át duzzadni hagyjuk. Kationcserélő gyanta esetében az oszlopot 5 n sósavval(kb 10 percig) alaposan átmossuk, hogy az esetleg benne levő kationokat hidrogén
ionra cseréljük ki. Ezt követően az-oszlopot desztillált vízzelkloridmentességig mossuk. Az ioncserére kerülő oldatot óvatosanrétegezzük az oszlopra. Amennyiben egy szabad sav preparatív
előállítására törekszünk, elegendő, hogyha 10—15 perces állásután az oldatot desztillált vízzel kimossuk az oszlopból. Az oszlop ioncserélő kapacitásának meghatározásához a
megfelelően előkészített oszlopba 5 g NaClot tartalmazó 50 mloldatot rétegezünk, és az ioncserélő oszlopon áthaladt oldat savtartalmát (anioncserélő gyanta esetében lúgtartalmát) megtitrál- juk.
Amennyiben többféle kation elválasztását kívánjuk elérni,úgy az oszlop eluálását (kimosását) híg savval vagy még inkábbolyan specifikus oldatokkal végezzük, amelyek egyes kationok
kal komplexeket képeznek. Specifikus elválasztások esetén természetesen a megfelelő irodalmi adatokra célszerű támaszkodni.
48.3.3. Molekuláris szűrők
A molekuláris szűrők a szilikát alapú adszorbensek speciáliscsoportját alkotják, és tulajdonságaikat illetően lényegesen különböznek az egyéb adszorbensektől. A molekuláris szűrők ter
mészetes és mesterséges zeolitók, azaz nagy mennyiségű zeolitosvizet tartalmazó alkáli- és alkáliföldfém-alumínium-hidro-szili-kátok, amelyek hőkezelés hatására kristály vázuk megtartásamellett víztartalmukat elvesztik. Az eltávozó zeolitos víz helyénmolekuláris nagyságrendű, egyenletes eloszlású és azonos mé
retű pórusok rendszere marad vissza, amelyek térfogata bizonyos esetekben a kristály térfogatának 50 %-át is elérheti. A pórusméreteket a kristályszerkezet és azeolitos víz eltávozásának mértéke határozza meg. A hőkezeléssel aktivált zeolit-kristály a pórus méreténél kisebb átmérőjű molekulákat képes adszorbeálni. Innenered a molekuláris szűrő elnevezés is.
A természetes zeolitok inhomogenitása és lelőhelyeinek ritkasága szükségessétette szintetikus zeolitok előállítását. E kiváló tulajdonságú molekuláris szűrőkneknégy fő típusát gyártják: 4 A , 5 A, 10 X és 13 X. Az elnevezésben szereplő számoka pórusok „ablakai ” -nak közelítő méretét adják meg (Á-ben), az Aéaa,X betűkpedig a megfelelő kristály típust jelölik. A molekuláris szűrők szabályos kristály-rendszerű zeolitok. A 4 A típusú zeolitkristály összetételét az alábbi képlet fejezi ki:
48.3.2.1. ábra. Ioncserélő oszlop preparatív
célokra
2)12(^ ^ 2)121 *27 H20.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 111/366
M O L E K U L Á R I S S Z Ű K Ö K 111
nagy pórus ablaka
nagy pórus
A rácsállandó: 12,32 Á, tércsoport: 0£Pm3m. A kristályt A104- és S i04- tetraéderekből álló térháló alkotja, amelyben a tetraédereket oxigénhidak kötik össze. Az AlOg-ionok töltését elemi cellánként 12 Na+-ion kompenzálja. Utóbbiakkönnyen kicserélhetők egyéb egy- vagy kétértékű kationra. Az elemi cella (110) síkban készített metszetét a 48.3.3.1. ábra vázolja. Az alumínium-szilikát-váz 8 dboxigénion által alkotott gyűrűből áll, amelyek 4,2 Á nagyságú „ablak ” -kal rendelkező pórust zárnak körül. Az elemi cella középpontjában levő nagy pórushoz akocka lapjai irányában további 6azonos méretű pórus csatlakozik.
Az elemi cellát magában foglalókocka csúcsai irányában további 8
kis pórus foglal helyet, amelyek átmérője 2,6 A. Az X típusú zeolit szintén
szabályos kristályszerkezetű és atermészetes faujasitéhoz hasonló.
A molekuláris szűrőknek kristályszerkezetükből kifolyólag azalábbi adszorpciós tulajdonságaikvannak:
A zeolitváz pórusai szigorúanazonos méretűek, úgyszintén apórusok ablakai is. Ezért csupánazok a molekulák adszorbeálódnak,amelyek be tudnak hatolni e pórusokba, tehát az egyes zeolittípu-sokra jellemző kritikus méretnélkisebbek. Egy adott molekulárisszűrő—adszorbátűm anyagpár eseténaz adszorpció bekövetkezése tehát
mindenekelőtt az „ablak” és az adszorbátum méretétől függ. Az adszorbátum e kritikus mérete gömb alakú molekulák esetén azok átmérő jével azonos, láncmolekulák esetében viszont azok hosszanti tengelyére merőlegeslegnagyobb keresztmetszet átmérőjével egyenlő. így valamennyi normál-szénhidrogén kritikus mérete azonosnak tekinthető, ezért ezek a n-tetradekánig bezárólagbehatolnak az 5 A típusú zeolit pórusaiba (48.3.3.1. táblázat). Tekintettel arra,hogy növekedő szénatomszámmal a diffúzió sebessége számottevően, csökken, anagyobb szénatomszámú normál-szénhidrogének esetén az adszorpció hosszú időtigényel. A 48.3.3.1. táblázat növekvő „ablak”-méret sorrendjében tünteti fel akülönbözó' molekuláris szűrőtípusok alkalmazásának lehetőségeit17.
Tekintettel arra, hogy a zeolitváz pórusai molekuláris méretűek (4—20 A),azok fajlagos felülete — következésképpen adszorpciós kapacitásuk is — rendkívülnagy. Az adszorpciós kapacitás — a szihkátváz erősen poláros jellege miatt — függa cserélhető kation minőségétől (méret, töltés, polarizáló erő), valamint az adszorbátum polaritásától és polarizálhatóságától is. így a poláros vagy könnyen polarizálható adszorbátumok adszorpciós kapacitása növekszik. A zeolit déhidratáció-
48.3.3.1. ábra. A 4 A típusú zeolit kristályszerkezeti metszete az (1 1 0 ) síkban (a szaggatott vonal egy elemi cellát határol, a nagyobb körök oxigén-, a kisebbek nátriumionokat jelképeznek).
Méretezés Á-ben
17 Barrer, R. M.: Brit. Chem. Engineering, May, 1959.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 112/366
A Z A N Y A G O K T IS Z T ÍT Á S A 112
48.3.3.1. táblázat
A molekuláris szűrők osztályozása, valamint az azokkal adszorbeálható
fontosabb anyagok
M o le k u las z ű rő -- t ípus
A d szo rb á tu m
4 A He, Ne, Ar, CO, H 2, 0 2, N2, NH 3, H 20 , Kr, X e, CH4, C2H 6,
CH3OH, CH3N H 2, CHgCl, CH3.Br, co2, c 2h 2, c s 2
5 A C3H8, n-C4H1(), n-C7H16, C2H 5C1, C2H 5Br, C2H 5OH,
C2H 5N H 2, CH2C12, CH2Br2, CHF2C1, CHFa, (CH3)2NH,
c h 3i , b 2h 6, c f 4, c 2f 6, c f 2o i2, c f 3c i , CHFC12
10 X SF6, í -C4H 10, í -C5H 12, C8H 18, CHC13, CHBr3, CHI3,
(CH3)2CHOH, (CH3)2CHC1, n-C3F8, n-C4F10, n-C7Fj6, B5H 9,
(CH3)3N, (C2H 5)3N, C(CH3)4, C(CH3)3C1, C(CHs)3Br,
C(CH3)3OH, CC14, CBr4, C2F2C14, C6H 6, C8H 5CH3, C6H4(CH3)2, ciklohexán, tiofén, furán, piridin, dioxán,
B4H 14, naftalin, kinolin, 6-decil-l,2,3,4-tetrahidro-nafta- lin, 2-butil-l-hexil-indán, C6FnCF3
13X 1 ,3,5-trietil-benzol
Nem adszorbeálja a (n-C4F9)3N-t vagy annál nagyobb molekulákat
48.3.3.2. táblázat
Egy cserélőiont tartalmazó A és X típusú zeolitok adszorpciós kapacitása18
Adszorbátum Hőmérséklet
C°
Egyensúlyinyomás
Hgmm
Egyensúlyi adszorpciós kapacitássúly %
1
típusú zeolitNa Ca Na Ca
h 2o 25 0,02 12,5 13,7 22,9
0,10 21,0 20,3 - —
4,5 25,3 27,3 29,3 33,3
c o 2 25 1,6 5,9 5,9 — —
80,0 15,0 19,5 — —
730,0 18,9 24,4 — -
CO 0 25 - — — 2,5
750 — 7,0 — 5,1
HgS 25 0,5 8,5 12,5 —
11,0 26,4 21,0 — —
n-Hexán 25 20,0 — — 19,2 -
s o 2 szoba- 700,0 26,8 26,8 — —
Metanol szoba- 90,0 15,9 18,8 — —
n-Bután szoba- 700,0 0,019 11,55 ' — —
i-Bután szoba- 400,0 0,6 0,5
is T ó th K . M a g y . K é m . L a p j a 1 6 , 440 (1961) .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 113/366
T I S Z T A S Á G V I Z S G Á L A T 113
jának mértéke, vagy az előzetesen adszorbeált anyagok minősége és mennyisége is jelentős mértékben befolyásolja annak kapacitását.
A molekuláris szűrők adszorpciós kapacitása az adszorbátum csekély parciálisnyomása esetén is igen nagy, különösen poláros jellegű adszorbátumok esetében(pl. vízgőz, szén-dioxid stb). A 48.3.3.2. táblázat ismerteti az A és az X típusúzeolitok adszorpciós kapacitását az egyensúlyi nyomás függvényében néhány fontosabb adszorbátumra vonatkozóan.
A molekuláris szűrők adszorpciós kapacitása magasabb hőmérsékleten isszámottevő. Míg az ismert legjobb adszorbensek 100 C° körül már minimáliskapacitásúak (pl. a szilikagél, az alumínium-oxid), a molekuláris szűrők viszontmeleg gázelegyek szétválasztására vagy meleg jgázok szárítására is alkalmasak(lásd 49.3.).
A molekuláris szűrők felhasználási területe az utóbbi időben rohamosan növekszik. Igen finom (1—5/z részecskeméretű) kristálypor alakjában vagy kevés kötőanyaggal néhány milliméter átmérőjű szemcsékké alakítva hozzák forgalomba.
Elegyek szétválasztására — laboratóriumi méretekben — sztatikus módszeris alkalmazható, amelynek lényege, hogy az előzőleg vákuumban vagy gázárambankihevített molekuláris szűrő nyugalmi állapotban érintkezik a szétválasztandóeleggyel. Sokkal célravezetőbb — főleg nagyobb mennyiségek esetén — a dinamikusmódszer, amikor is a gáz- vagy folyadékelegyet a molekuláris szűrővel töltöttoszlopon vezetjük át.
Gyakorlatban a molekuláris szűrők kapacitását maximálisan 50 %-osan használjuk ki. A szűrő regenerálása evakuálással és melegítéssel végezhető el. A regenerálás folyamán a molekuláris szűrőt lehetőleg kevéssé adszorbeálódó gázzal történőöblítés kpzben hűtjük le. Több párhuzamosan működtethető adszorbenssel a folyamatos üzemeltetés is egyszerűen megoldható.
A molekuláris szűrők közé sorolhatók az előzőekben tárgyalt zeolitokon kívülegyéb térhálós szerkezetű anyagok is, pl. a keményítő és más poliszacharidók,melyek azonban gyakorlati szempontból kisebb jelentőségűek.
48.4. Tisztaságyizsgálat
A tisztítási eljárások eredményességéről a preparatív munka végén az anyagolvadáspontja, forráspontja, illetve tenziójának mérése alapján tájékozódhatunk.Ezek az adatok a szokásos analitikai vizsgálatok eredményeitől eltérően, a szennyezés minőségére való tekintet nélkül csupán annak távollétére vagy hozzávetőlegesmennyiségére utalnak.
Raovlt törvénye értelmében a tiszta anyag gőznyomása más anyaggal valószennyeződés következtében az idegen anyag móltörtjével arányos mértékben
csökken; az oldatok fagyáspontcsökkenése és forráspont-emelkedése szintén a móltört függvénye.így az anyagok mért fizikai állandóinak az irodalomban található értékétől
való eltérés a szennyezettség fokmérője. Az olvadáspont- és forráspont-meghatározások kivitelét lásd 2.3. és 2.8. A tenziómérések különböző módjait a fizikaikémiai praktikumok tárgyalják.
8 Ált. és szervetlen kémiai prakt. — 4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 114/366
A Z A N Y A G O K T IS Z T ÍT Á S A 114
Irodalom
Weissberger, A.: Technique of Organic Chemistry. Vol. ITT, és IV.Interscience Publishers, New York— London, 1950.
Weissberger, A.: Technique of Organic Chemistry. Vol. V. Interscience Publishers, New York— London, 1951. Erdey-Grúz T., Proszt J. : Fizikai-kémiai praktikum. I —II. kötet IX . kiadás. Tankönyv-
kiadó, Budapest, 1965. Lux, H.: Anorganisch-chemische Experimentierkunst. Johann Ambrosius Barth Verlag,
Leipzig, 1954. Kufferatk, A.: Filtration und Filter. Bodenbendér, Berlin, 1952. Lederer, E. és M . : Chromatography. Elsevier P. C., Amsterdam— London— New York—
Princeton, 1957. Prausnitz, P. H., Reitstölter, J .: Elektrophorese, Elektroosmose, Elektrodialyse in
Flüssigkeiten. Dresden, 1931.Vámos E.: Kromatográfia. Műszaki Könyvkiadó, Budapest, 1959.
Grubitsch, H .: Anorganisch-práparative Chemie. Springer, Wien, 1950. Kroll, E .: Handbuch dér Laboratoriumsdestillation. VEB Deutscher Verlag d. Wiss.,
Berlin, 1958. Mikes J.: Ioncserélő műgyanták és alkalmazási technológiáink. Műszaki Könyvkiadó,
Budapest, 1958. Nyúl Gy.: Lepárlás. Műszaki Könyvkiadó, Budapest, 1955. Brenner, N., Gálién, J., Weis, M .: Gas Chromatography. Academic Press Inc., New
York— London, 1962. Heftmann, E .: Chromatography. Reinold Publ. Corp. New York, 1963.yjnyToe,K. B.: Hccn egO BaH He CBOHCTBa HOHOoöMeHHbix MaTepHajiOB. H3^aTejibCTB 0 H a yK a ,
M o c K B a , 1964.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 115/366
49. Gázok
Fejlett vegyiparral rendelkező országokban igen sokféle gáz készül nagyipari
méretben, és ezek legtöbbje nyomásálló acélpalackba töltve a kereskedelmi forgalomban is kapható. A gázpalack használatát illetően lásd 1.19. Ha a palackozottgáz tisztasági foka nem megfelelő, esetleg a gáz speciális, nehezen eltávolíthatószennyezést tartalmaz, vagy csak igen kis mennyiségű gázra van szükség, akkorezeket laboratóriumban állítják elő. A gyakran használatos, továbbá a ritkábban
szükséges, készen nem kapható gázok előállítását is a részletes részben tárgyaljuk.Itt csak az előállítás technikai megvalósításának általános tudnivalóit foglaljukröviden össze.
49.1. Gkzfejlesztés A gázfejlesztés leggyakoribb és legismertebb módja a szilárd anyagból folyadék
kal történő előállítás (pl. C02 fejlesztése CaC03-ból HCl-val).Ennek a feladatnak a technikai megoldása igen egyszerű. A gázfejlesztő
készülék frakcionáló lombikból és csapos tölcsérből is összeállítható (lásd 1.19). Az így előállított gáz azonban levegővel szennyezett, még akkor is,
ha a lombik légterét már átöblítettük a fejlődő gázzal. Nagyobbtisztasági igények esetén a gázfejlesztésre felhasznált folyadék (többnyire vizes oldat) lcvegőtartalmát is ki kell űzni. Erre a célra leg
alkalmasabb maga az előállított gáz. A megfelelő készülék (49.1.1.ábra) működése a következő. Először a csepegtető tölcsért levesszüka gázfejlesztő lombikról, és vízlégszivattyú segítségével teleszívat
juk a fejlesztésre felhasználandó folyadékkal, majd a 2 és 4 csapmegnyitása után az i-en át leengedett folyadékkal megindítjuk a
gázfejlesztést. A kezdetben levegővel kevert gáz átbuborékol a csepegtető tölcsérben levő, oldott levegőt tartalmazó folyadékon. Egy
idő múlva a fejlődő gáz egyre inkább légmentessé válik, és kiűziaz egyéb oldott gázokat. Miután már a fejlesztőbe jutó folyadék
sem tartalmaz oldott levegőt, a 4 csapot elzárhatjuk, és a 3 csap
megnyitásával megkezdhetjük a tiszta gáz elvezetését.Szilárd anyag és folyadék kölcsönhatása alapján működő automatikus gáz-
fejlesztőket ( Kipp , Devüle stb.) az 1.19.-ben ismertettünk.Bizonyos esetekben a szilárd fázisra csepegő folyadék igen heves reakciót,
veszélyes helyi túlmelegedést okozhat, vagy nagy térfogatú iszap és hab képződésével jár, esetleg a költséges szilárd komponensnek rossz a kihasználhatósága.
8*
49.1.1. ábra. Gázfejlesztő készülék
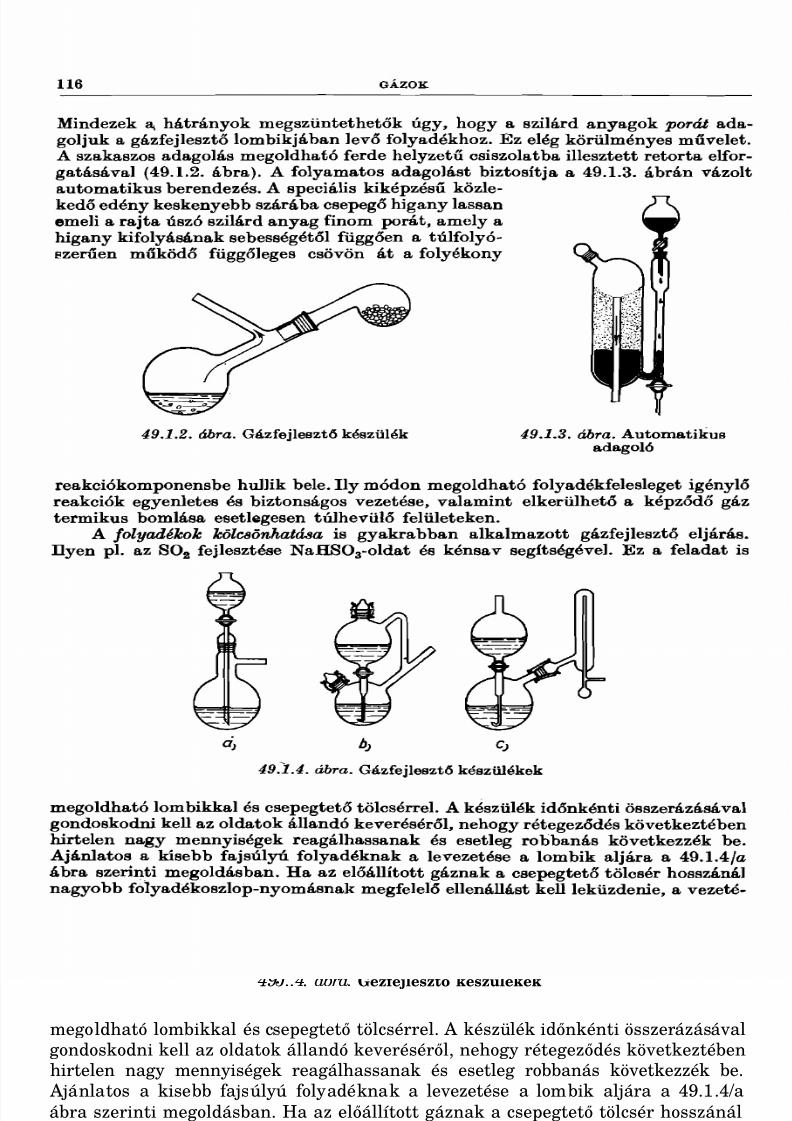
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 116/366
116 G Á Z O K
Mindezek a, hátrányok megszüntethetek úgy, hogy a szilárd anyagok porát adagoljuk a gázfejlesztő lombikjában levő folyadékhoz. Ez elég körülményes művelet.
A szakaszos adagolás megoldható ferde helyzetű csiszolatba illesztett retorta elforgatásával (49.1.2. ábra). A folyamatos adagolást biztosítja a 49.1.3. ábrán vázoltautomatikus berendezés. A speciális kiképzésű közlekedő edény keskenyebb szárába csepegő higany lassanemeli a rajta úszó szilárd anyag finom porát, amely ahigany kifolyásának sebességétől függően a túlfolyó-szérűén működő függőleges csövön át a folyékony
49.1.2. ábra. Gézfejlesztő készülék 49.1.3. ábra. Automatikus adagoló
reakciókomponensbe hullik bele. Ily módon megoldható folyadékfelesleget igénylőreakciók egyenletes és biztonságos vezetése, valamint elkerülhető a képződő gáztermikus bomlása esetlegesen túlhevülő felületeken.
A folyadékok kölcsönhatása is gyakrabban alkalmazott gázfejlesztő eljárás.Ilyen pl. az S02 fejlesztése NaHS03-oldat és kénsav segítségével. Ez a feladat is
49J..4. ábra. Gézfejlesztő készülékek
megoldható lombikkal és csepegtető tölcsérrel. A készülék időnkénti összerázásávalgondoskodni kell az oldatok állandó keveréséről, nehogy rétegeződés következtébenhirtelen nagy mennyiségek reagálhassanak és esetleg robbanás következzék be. Ajánlatos a kisebb fajsúlyú folyadéknak a levezetése a lombik aljára a 49.1.4/aábra szerinti megoldásban. Ha az előállított gáznak a csepegtető tölcsér hosszánálnagyobb folyadékoszlop-nyomásnak megfelelő ellenállást kell leküzdenie, a vezeté-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 117/366
O Á Z F E J L E S Z T É S 117
két bekötjük a zárt adagoló tölcsér gázterébe (49.1.4/b ábra). A 49.1.4/c ábrán bemutatott készülék akkor tesz jó szolgálatot, ha a reakcióelegy habzik.
A 49.1.5. ábrán vázolt készülék előnye, hogy folyamatosan működtethető.Folyadékok kölcsönhatása alapján működő automatikus gázfejlesztő készüléketmutat be a 49.1.6. ábra.
Szilárd komponensek keverékének reakciója útján is előállítható gáz pl. a CH4:
CH3COONa + Ba(OH)2 = CH4+ BaC03 + NaOH.
Az ilyen természetű műveleteket célszerű retortában vagy vízszintes' helyzetű,egyik végén zárt és kb. a keresztmetszet feléig töltött üvegcsőben végrehajtani. A szükséges hőmérsékletet megfelelően szabályozható csőkemencével vagy termosztáló fürdővel biztosítjuk. A gázáram ilyenkor mindig
jelentős mennyiségű port visz magával, amelynek eltávolítását a tisztítással foglalkozó fejezetben (49.2) tárgyaljuk.
A szilárd anyag termikus disszociációja út ján való gázfejlesztésre példa az igen tiszta Cl2
előállítása PtCl4, A uC13, illetve AuCl-ból. A feladat technikai megoldása az előzővel teljesenazonos.
Szilárd anyag és gáz kölcsönhatására példa a
HgO + 2 CL, = C120 + HgCl2.
reakció. Az előállítás módja hasonló az előzőkettőhöz. A vízszintesen fekvő, félig töltött
csőben a szilárd anyag felett vezetjük el a gáztmeghatározott hőmérsékleten. Két gáz kölcsönhatásával is előállítható egy
harmadik gáz. Pl. a S02C12 előállítása:
S02 + Cl2 = S02C12.
Az első részfeladat rendszerint megfelelő arányú gázkeverék készítése. Erre vonatkozóan lásd a gázok áramlásának szabályozásával foglalkozó részt. A továbbiakbanleginkább katalizátorral töltött oszlopon vezetik át a gázelegyet, melynek megfelelőhűtésével a képződéshő elvezethető. A terméket hűtött szedőben kondenzáltatjuk.
Oldott gázt tartalmazó folyadékok forralásával is előállíthatók egyes gázok. Pl. azNH3 vagy HC1 felszabadítható tömény vizes oldataik forralásával. Az oldószer jelentős részét a forralólombikhoz csatlakozó visszafolyós hűtővel kondenzáltatjuk.
Olvadék vagy (vizes) oldat elektrolízisével is többféle gáz, pl. F2, H2, 0 2 stb.fejleszthető. Az elektrolízis gyakorlati megoldásával külön fejezet (51.) foglalkozik.
19.1.5. ábra. Folyamatos gázfejlesztő
49.1.6. ábra. Automatikus gázfejlesztő

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 118/366
118 G Á Z O K
49.2. Gázok tisztítása, szárítása
49.2.1. Általános módszerek
A különböző módszerekkel fejlesztett, illetve nagynyomású acélpalackból.redukciós szelepen át lefejtett gáz az esetek nagy részében nem használható felközvetlenül, tisztítás nélkül. Tisztítás szempontjából más eljárást kell alkalmaznunk a gáznemű, és megint mást a szilárd vagy cseppfolyós állapotú szennyezésekeltávolítására. Ha a szennyezés szilárd vagy cseppfolyós, a durvább részt, így a portés folyadékcseppeket kb. 10 cm-es tömött vattarétegen, szűrőpapíron vagy G3 —G4-es zsugorított üvegszűrőn lehet visszatartani. A hatásfok ellenőrzésére felhasználhatjuk a Tyndáll- jelenséget. A 10-5 cm-nél kisebb méretű részecskéket
tartalmazó kolloid füst, illetve köd kiszűrése jóval nehezebb feladat, és az előbbimódon megoldhatatlan.
A legfinomabb aeroszolok megszüntetésére a következő eljárások használhatók :
Cottrell— Möller-módszer, vagyis a részecskék elektromos feltöltése és nagy-feszültségű kondenzátorlemezen való lecsapása. A nagyiparban gyakran alkalmazott eljárás laboratóriumban is megvalósítható. Fémfóliával borított kvarcvagy üvegcső tengelyében feszítsünk ki fémhuzalt. A két fegyverzettel párhuzamosan kapcsoljunk nagy kapacitású kondenzátort (leydeni palackot), és kössük
össze dörzselektromos géppel. Ha van nagyfeszültséget adó (10 0Ő0 V) elektron-csöves egyenirányítónk, kapcsoljuk a huzalra a negatív pólust, és földeljük a külsőfegyverzetet. A cső tengelyében kifeszített elektródon ionizálódó gázt a kolloidrészecskék adszorbeálják, és az elektrosztatikus vonzás következtében megkötődnek a cső falán.
Termodiffúziós eljárás. A berendezés az előbbihez hasonló. Hosszú Liebig-hntő tengelyében feszítsünk ki ellenálláshuzalt, amfelynek üzemi hőmérséklete kb. 600 C°. A magas hőmérsékleten intenzív mozgást végző kolloid részecskék a hideg falfelületen lecsapódnak. Az eljárás csak akkor alkalmazható, ha a gáz bomlás nélkülelviseli a 600 C°-ot.
A részecskéje méretének növelése. Könnyen megvalósítható, de nem mindigeredményes eljárás a következő: a tisztítandó gázt forró vizen vezetjük át, ilymódon vízgőzzel telítjük. A vezeték következő, hűtött szakaszán a vízgőz konden-zálása elsősorban a gócként szereplő kolloid részecskéken indul meg, és ha a cseppelég nagyra tud nőni, lecsapódik és magával rántja a szennyező részeket.
49.2.1.1. ábra. Drechsel-féle mosópalack
49.2.1.2. ábra. Muencke-íéle mosópalack
49.2.1.3. ábra. Spirélceöves mosópalack
49.2.1.4. ábra. Csavarfelületű mosópalack

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 119/366
G Á Z M O S Ó K 119
A kolloid rendszer megszüntetése elpárologtatás útján. Ha a diszperz részecskékanyagának a tenziója elég nagy, akkor ajánlható az elpárologtatásuk és a gőzökkémiai úton vagy adszorptive történő' megkötése.
A gázalakú szennyezések eltávolítására ugyancsak használhatunk fizikaivagy kémiai módszereket. Nagy forráspontkülönbség esetén sokszor igencélszerű a gázelegy frakcionált desztillálása (lásd a gáz—gáz elválasztására vonatkozó részt). Általánosabban alkalmazott módszer a gáz mosása különböző reagensekkel, illetve tisztítása szilárd anyagokkal végbemenő reakció (kemoszorpció, illetve adszorpció) útján. A folyékony reagensek hatásossága a gázzal való érintkezés intenzitásától és időtartamától függ. Az intenzitást a buborékméret csökkentésével, az időtartamot pedig az úthossz növelésével lehet fokozni. Az eljá
rás különböző rendszerű mosópalackokb&n hajtható végre. Ezek mindegyike egy--egy speciális igény kielégítésére alkalmas. így pl. a DrechseUTendszerű csiszolatosmosópalack (49.2.1.1. ábra) a vezeték megbontása nélkül a mosófolyadék gyors
49.2.1.1 táblázat
Gázelegy: 99% N2és 1% C02, mosófolyadék térfogata 100 ml
C02-abszorpció %-osan
Mosófolyadék Áramlási sebesség Egyszerű( Drechsel-féle) mosópalack
Csavarfelületűmosópalack
4 % -os KOH-oldat 3,8 liter/óra 87,5 99,94%-os KOH-oldat 11,5 liter/óra 83,4 99,4
15%-os KOH-oldat 3,8 liter/óra 96,3 99,9
kicserélését teszi lehetővé, a Muencke-iéle pedig (49.2.1.2. ábra) a gázáram megfordulása esetén sem enged mosófolyadékot a csővezetékbe.
A mosás hatásosságát a buborék úthosszának növelésével fokozzák a spirálcsöves és csavarfelületű mosópalackok (49.2.1.3. és 49.2.1.4. ábra). Meg kell említeni a gázt finoman eloszlató, beépített-üvegszűrős ún. intenzív mosópalackot(49.2.1.5. ábra). A különböző típusú mosópalackok hatásosságának összehasonlítására szolgáljon a 49.2.1.1 és 49.2.1.2. táblázat.
Az üvegszűrők jelzését, pórusméretét es buboréknyomását vízben, illetve szén--tetrakloridban a 49.2.1.3. táblázat foglalja össze. A G3-nál finomabb pórusú üveg-
49.2.1.2. táblázat
Gázelegy: 93% IV2 és 7% 02, mosófolyadék:5%-os CrCl2-oldat, felhasznált mosófolyadék-
-magasság: 70 mm
Mosópalack típusaMaximális áramlásisebesség, amely mel
lett móg teljes abszorpció következik be
Egyszerű (Drechsel-féle)Csavarfelületü ................
Üvegszűrős (Gl-es) Üvegszűrős (G2-es)
66 liter /óra 112 liter/óra 204 liter /óra 49.2.1.5. ábra. Üvegszűrős
mosópalackok

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 120/366
120 G Á Z O K
szűrőket nagy ellenállásuk miatt mosópalackokban ritkán alkalmazzák. A bubo-.rékméret további csökkentése érdekében figyelembe vesszük a felületi hatásokat is,
pl. a lúgos mosófolyadékokban porcelán szűrővel finomabb eloszlású gázlioszoltkapunk, mint ugyanolyan pórusméretű üvegszűrővel. Savanyú közegben fordította hetyzet.
49.2.1.3. táblázat
Üvegszűrők fontosabb adatai a BS. 1752/1952szabvány alapján
Üvegsz űrő jele
Pórusátmérő f*
Buboréknyomás vízben
Hgmm
Buborék-nyomásCCI4-ban
Hgmm
G00 ........... 2 5 0 -5 0 0 4 - 9GO .............. 150 -250 9 - 1 5G1 9 0 -1 5 0 1 5 -2 5G2 4 0 - 9 0 2 5 - 5 5G3 1 5 -4 0 6 5 -1 4 5G4 5 - 1 5 145 - 435 5 5-1 60G6 <2 >1000 >400
Kis mennyiségű mosófolyadék számára konstruált eszközt mutat a 49.2.1.6.ábra.
A mosófolyadék regenerálását is elvégzi a 49.2.1.7. ábrán látható berendezés,melyben az átbuborékoló gáz a mosófolyadékot körforgásban tartja. Előnyösenalkalmazható pl. ez a készülék kis mennyiségű oxigénszennyezés folyamatos eltávolítására, amennyiben a Cr2+-tartalmú savanyú mosófolyadék az amalgamált Zn--tölteten átáramolva, állandóan regenerálódik.
Lassan lefolyó reakcióban végbemenő tisztító eljárásra ajánlható az üveggyönggyel töltött mosó, amely nemcsak a buborékméret csökkentésével, hanem az
érintkezés időtartamának meghosszabbítása útján is fokozza a hatást. Pl. lúgos
49.2.1.6. ábra. 49.2.1.7. ábra.Speciális Speciális gázmosó
mosópalack
49.2.1.8. ábra. Üveg- gyöngyös gázmosó
49.2.1.9. ábra. Mosótorony

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 121/366
G Á Z O K T I S Z T Í T Á S A 121
Na2S20 4-tal való 0 2-mentesítésre célszerű a 49.2.1.8. ábrán vázolt mosó. Az üvegtöltet átlagos átmérője azonban legfeljebb 0,3—0,5 mm legyen.
Az eddig tárgyalt mosópalackok többnyire jelentékeny áramlási ellenállástokoznak, ezért nem minden esetben, főleg nem tetszőleges számban alkalmazhatók.Gyakorlatilag alig jelent ellenállást a gáz útjában az ún. mosótorony, amelyben areagenssel nedvesített felületű szilárd töltet közötti hézagokon át áramlik a gáz. Az iparban ezt a módszert kiterjedten alkalmazzák gázok tisztítására, laboratóriumban ritkábban. Laboratóriumi használatra alkalmas automatikus működésűmosótorony vázlatát a 49.2.1.9. ábra mutatja, melyben a mosófolyadékot a nyílirányából beáramló gáz folyadékliftszerűen viszi fel a torony tetejére.
Szilárd hatóanyagot első
sorban a gázok legtöbbjébenszennyeződésként előfordulóvíz és vízgőz eltávolításáraszoktak használni. A leggyakrabban alkalmazott eszköz
szárítótorony néven ismeretes.Nincsen számottevő -áramlásiellenállása. Függőleges helyzetben célszerű alkalmazni,nehogy a gáz az esetleg víz
szintes helyzetű csőben lazánelhelyezett adszorbens fölöttváltozatlanul haladhasson át.
A szárítótornyok szilárd töltetéből a túlságosan aprórészeket és finom port elkell távolítani, egyrészt, hogya torony teljes keresztmetszete el ne tömődjék, másrészt, hogy a port a gázáram magával ne vigye. A gáz
áramba került porrészecskék visszatartására a szárítótorony töltetét megfelelő vastagságú vatta- vagy üveggyapot réteg közé szokás helyezni. A szárítótorony alsó,kiöblösödő része (49.2.1.10. ábra) az esetleg elfolyósodó szárítóanyag cseppjeinek.összegyűjtésére szolgál. — Más rendszerű szárítóberendezésben, pl. U-csőben az.előzőekben említett elfolyósodás a gáz útjának elzáródására vezethet.
b) c)
49.2.1.10. ábra. Gázok szárítására szolgáló eszközök. a) CaCl2-os cső; ó^U-csője) szárítótorony
49.2.2. Hidrogénszennyezés eltávolítása
Csaknem valamennyi illékony hidrogénvegyülethez — előállításának körülményeitől függően — több-kevesebb hidrogéngáz is elegyedik. Ugyancsak mindig
tartalmaznak hidrogént az elektrolitikus úton előállított gázok. Amíg azonban aH2S, PH3, SíH4 stb.-hez elegyedett nagyobb mennyiségű (1—10 %) hidrogén'semzavarja különösebben az említett gázok szokásos felhasználását, oxigénben, szén--monoxidban vagy klórban sokkal kisebb koncentrációja is kellemetlen lehet.
Eltávolítására jól használható a legtöbb gázénál sokkal kisebb adszorpció-képessége és nagyobb illékonysága. Ennek következtében a hozzá e tekintetbenhasonló héliumon és neonon kívül valamennyi más gáztól elválasztható frakcionáltlepárlással. A gyakorlatban HBr, Hl, H2S, PH3, CO és N2 hidrogénmentesítését szók-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 122/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 123/366
G Á Z O K T I S Z T Í T Á S A 123
Az aktív töltet készítése a következő: 120 g bázisos réz(II)-karbonátot 2 1 cc.ammónium-hidroxidban feloldunk. Az oldatot 420 g tisztított infuzóriafölddel itat
juk fel, és porcelán tálban vízfürdőn bepároljuk. A még kissé nedves masszát mintegy i/2 cm vastag rétegben üveglapra kenjük, és késsel kb. % cm élhosszúságú kockákká daraboljuk, majd 150—180 C°-os szárítószekrényben teljesen megszárítjuk. Atörmeléktől és finomabb portól megtisztított töltetet az alján üveggyapottal elzárt
•csőbe helyezzük, beleállítjuk a hőmérőt, és megindítjuk a H2-áramot. Amikor ahőmérséklet eléri a 200 C°-ot, megindul a karbonátos és hidroxidos rézvegyületekbomlása, és a finom eloszlású fémréz ibolya színe jelzi a redukció megtörténtét. A képződő víz a cső aljához csatlakozó tartályban gyűlik össze. Ha a vízképződés megszűnt, a berendezés
használatra kész. A maradék 0 2 10-5—10~á%
nagyságrendű. A mangán(II)-karbonát hevítésével előállított mangán(II)--oxid is kiválóan alkalmas egyes gázokban (N2, H2) jelenlevőoxigénnyomok eltávolítására.1 Oxigénabszorpcióra használatos még a semleges vagy gyengén savanyú CrCl2-oldat 0,2—2,0mólos koncentrációban. Előállítható sósavas CrCl3-oldatbólamalgamált Zn-darával. Ha nem H2 tisztítására használjuk,figyelembe kell venni, hogy melegen és nagyon savanyú közegben, valamint bizonyos fémszennyezések jelenlétében állandóan hidrogént fejleszt. A CrSO, hasonló körülmények között
H2S-t ad le. 'Igen hatásos oxigénelnyelő lehet a 300 g rézdróttal és a
következő összetételű folyadékkal töltött mosó: 25 ml telített(N H^CO^ 25 ml cc. NH4OH, 50 ml H20 és 10 g NH4C1.Kicsiny ón darabot is tegyünk bele. A mosófolyadék a képződő [Cu (NH3)4]2+ komplex ionoktól az elnyelt oxigénnel arányos mértékben megkékül. A szén-monoxidot is megköti. 4-reakció kvalitatív oxigénkimutatásra is felhasználható.
Gyakran alkalmazott abszorpciós oldat a lúgos pirogallol. Összetétele: 1 súly-
rész 25 %-os vizes pirogallololdat és 9 súlyrész 60 %-os KOH. Gyengébben lúgosoldatban a tisztítandó gázt állandóan szén-monoxiddal szennyezi. A lúgos oxi--hidrokinon-oldat ugyancsak jó oxigénabszorbens, és CO-ot sem ad le.
Klórgáz oxigénszennyezésének eltávolítására a többször megismételt vákuum-desztilláció a legmegfelelőbb. Cseppfolyós klóron (—78 C°-on) tiszta hidrogénnapokig tartó átbuborékoltatása is eredményes lehet. Vörösen izzó szén felettelvezetve szintén oxigénmentesíthető a klór. A képződő szén-monoxid utólag frak-cionált desztillációval távolítható el.
A fluor 20—30 Hgmm nyomáson, folyékony levegővel hűtött kvarc kisülési csőben tisztítható meg az oxigéntől. A képződő F20 2 adott hőmérsékleten nem illékony.
A szén-monoxid 7—900 O-os szénrétegen átvezetve oxigénmentesíthető. Aképződő szén-dioxidot már könnyű eltávolítani.
Ózon jelentékenyebb mennyiségben csak a fluor-hidrogén előállításakor képződik. Nyomokban előfordul még az elektrolízissel és a KMn04-ból előállított oxigénben. Ez azonban egyszerűen eltávolítható a gázelegy 300 C°-ra való hevítésévelvagy higanyrétegen való átbuborékoltatással.
49.2.3.1. ábra.
Oxigénmentesítő készülék
1 Schulek , E . Pungor E .: Magy. K ém . Foly . 56, 213 (1950).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 124/366
124 G Á Z O K
49.2.4. Szén-dioxid eltávolítása
A szén-dioxid gyakori szennyezése a palackozott, valamint a laboratóriumban előállított gázoknak. Eltávolítása aránylag egyszerű feladat: erősen lúgosreagensek segítségével megköthető. Leggyorsabban a 40 %-os KOH-oldat abszorbeálja, a NaOH jóval lassabban. Nyomnyi mennyiségű C02 eltávolítására telítettBa(OH)2-oldat vagy nedves felületű szilárd KOH alkalmasabb. Száraz KOH nemmegfelelő abszorbens. Nagyobb mennyiségű szénsav megkötésére nátronmeszet’szokás használni ugyancsak nedves állapotban. A nátronmész tömény NaOH-dalmegoltott CaO.
Az ammónia COa-mentesítésére elsősorban szilárd KOH vagy nátronmészalkalmas, mivel vízben az ammónia igen jól oldódik.
A nitrogén-monoxid nátrium-hidroxiddal, a karbonil-szulfid pedig 30 %-oskálium-hidroxiddal a szénsavtól eredményesen megtisztítható.
Nem használható fel szén-dioxid-mentesítésre — a fellépő kémiai reakciókmiatt — a KOH a következő gázok esetében: CS2, COCJ2, N02, Cl2, HCN, H2S stb.Sokszor segít a szén-dioxid kifagyasztása, de pl. H2S és HC1 esetében ez az elválasztás is igen rossz hatásfokú a tisztítandó gáz és a szennyezés nagyon közelállótenziója miatt.
A C02 kvalitatív kimutatására a Ba(OH)2-oldatból kiváló BaC03-csapadékszolgál.
49.2.5. Szén-monoxld eltávolítása
Szén-monoxid fordul elő kis mennyiségben az elektrolitikusan előállítottklórgázban, azonkívül az acetilénben, karbonil-szulfidban és cián-hidrogénben. Valamennyi esetben alkalmazható a cseppfolyósítás és az azt követő frakcionálás.
Kémiai módszerekkel is megoldható a CO eltávolítása gázelegybol. A szén--monoxidnak az analitikai gyakorlatból ismert abszorbense a réz(I)-klorid-oldatsemleges, illetőleg ammóniás vagy sósavas közegben. A semleges oldat összetétele:125 g CuCl, 265 g NH4C1 és 750 ml nitrogénáramban kiforralt víz. Az oxidáció
megakadályozására egy darab rézdróthálót is teszünk bele. A szén-monoxidot areagens CuCl*C0?2H20 összetételű vegyület alakjában köti meg. Ennek disszo-ciációs nyomása azonban jelentékeny, úgyhogy egyensúly esetén a gáztérben azeredeti CO-koncentrációnak mintegy 3 %-a változatlanul megmarad. A CuCl-olda-toknál sokkal jobban abszorbeálja a CO-ot a K4[Ni2(CN)6] oldata,2 amelyben areakciótermék összetétele: K2rNi(CN)3CO]. Ez Ca(OH)2-oldattal forralva könnyenregenerálható. Teljesnek mondható a CO abszorpciója 15 % réz(I)-oxidofctartalmazócc. kénsavas szuszpenzióban, ha a vele töltött mosópalackok közül kettőt 60 C°-on,egyet pedig szobahőmérsékleten működtetünk.
Indifferens gázok CO-tartalma 300 C°-on CuO felületén szén-dioxiddá oxidálódik. A katalitikus reakció részben már 0 C°-on is végbemegy, 40 % CuO-tartalmúMn02-keveréken (Hopkalit). A I20 5 160—170 C°-on, ugyanez kénsavas oldatban0 C°-on is oxidálja a szén-monoxidot.
A szén-monoxid kimutatásának legérzékenyebb módja a kénsavas ammónium--molibdát-oldattal és palládium-kloriddal impregnált szilikagélen történő eljárásmely már 10-3 % CO kimutatására alkalmas.
* D. R. P. 801 635 (1951).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 125/366
G Á Z O K T I S Z T Í T Á S A 125
49.2.6. Illékony szénvegyületek eltávolítása
Szénhidrogének nyomai csaknem valamennyi komprimált gázban megtalálhatók. Forrásuk a kompresszor kenőolaja, amely többnyire finom permet alakjában szennyezi a gázt. Fém-karbidok savas elbontásakor is képződnek különbözőmolekulasúlyú szénhidrogének. Ezek előfordulnak az acetilénben és metánban,valamint foszfor- és szilíciumhidrogénben is.
Az olaj cseppek mechanikusan, vagy kifagyasztással és aktívszenes adszorpcióval eltávolíthatók a gázelegyből, a kisebb molekulasúlyú szénhidrogének, különösena metán azonban csak igen nehezen. A legtöbb szerves vegyület oxigénmentesatmoszférában is, 500 C°-os CuO felületén szén-dioxiddá és vízzé ég el. A metán
itt is nehezebben reagál. Acetiléntől a gázelegy 0,5 % Ag2S04- és kevés NiS04-tartalmú cc. kénsavval ismegtisztítható- A metil -és etilalkoholt ugyancsak megköti a cc. kénsav, de a cc.KOH és CaCl2 is; a CaO viszont nem. Étergőzt cc. kénsavval, acetont NaHS03-öldattal lehet abszorbeálni.
49.2.7. Nitrogén eltávolítása
A nitrogén igen gyakran előforduló, de többnyire ártalmatlan szennyezés.Nitrogénmentes gázok előállításakor biztosítani kell a levegő mint nitrogénforrás
kizárását. Nehezen kerülhető el a N2 jelenléte a nemesgázokban, dinitrogén-oxid-ban és nitrogén-monoxidban. A felsoroltak közül frakcionálással csak a dinitrogén--oxidtól választható el.
Nemesgázok nitrogénmentesítése Li, Mg, Ca vagy Ti segítségével végezhető.Bár a Li már szobahőmérsékleten is megköt bizonyos mennyiségű nitrogént, mégislegalkalmasabb erre a Ca, különösen kb. Y2 % lítiummal vagy nátriummal aktiválva. Az így aktivált kalciumforgács nemcsak nitrogént, hanem 440 C°-on oxigént,metánt és egyéb szénhidrogéneket is megköthet. Titánpor kvarccsőben, 850 C°-onugyancsak reagál a nitrogénen kívül szén-monoxiddal, szén-dioxiddal, valamint
oxigénnel is. Hidrogénnek nitrogéntől való tisztítására nátriummal aktivált magnéziumforgács alkalmas, 600 C°-on,Nitrogénnyomok kimutatására spektroszkópos vizsgálatot ajánl az irodalom.
49.2.8. Egyéb szennyezések eltávolítása
Ammónia. Igen ritkán előforduló szennyezés. Vizes kénsavoldatban maradéktalanul elnyelődik.
Nitrogén-dioxid. Igen kis mennyiségben nitrogénben fordulhat elő, s ebből kifagyasztással könnyen eltávolítható. Koncentrált H2S04 és 20 %-os KOH is jólabszorbeálja.
Kén-hidrogén. Gyakran kimutatható szennyezés a következő gázokban: C2H2,COS, CH4, SíH4, p h 3 és HC1. Híg kénsavból nascens hidrogén hatására is keletkezik bizonyos mennyiségben. Katalizátorméreg, a gázelegyből gondosan eltá-volítandó. Tömény lúgokban jól oldódik, ezekkel megköthető, száraz KOH nemköti meg. Sósavtól való elkülönítésére megfelelő módszert nem ismerünk. Kismennyiségű (10“ 5 %) H2S is kimutatható ammóniás ólom-acetát-oldattal. (A PH3és A sH3 ugyancsak adja a reakciót.)

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 126/366
126 G Á Z O K
Foszfor-hidrogén. Igen kellemetlen, mérgező komponense sok technikai minőségű gáznak, pl. a H2S, C2H2, CH4, HBr, H l stb. gázoknak. A szárításukra felhasznált foszfor-pentoxid esetleges P20 3-tartalma vízzel PH3 képződése közben reagál.Tömény CuS04- vagy telített KMn04-oldattal a gázból kimosható.
Arzén-hidrogén. Főként a laboratóriumban fejlesztett hidrogént szennyezi,különösen, ha a kiindulási anyagok valamelyike arzéntartalmú. Ezért nem szabada Kipp-készülék hidrogénjét tartósan a laboratórium levegőjébe engedni. Azelőző kettőhöz hasonlóan katalizátor- és sejtméreg. Megköthetjük 5—10 %-os AgN03- vagy telített KMnÖ4-oldattal.
Sósavgáz. A HCl kisebb mennyiségben minden sósav segítségével előállítottgázban előfordul. Vízben igen jól oldódik. Klórból kalcium-oxiddal távolítható el.
Olyan gázoktól, amelyek nedvesség hatására elbomlanak, kondenzálással és frak-cionálással, vagy horzsakőre felvitt vízmentes réz(II)-szulfáttal lehet elválasztani. Az utóbbi esetben képződő addíciós vegyület, a CuS04*2 HCl elég stabilis. Kimutatása ezüst-nitráttal lehetséges.
Halogének. Ezek a megfelelő halogén-hidrogénben és egyéb halogénvegyületek-ben is előfordulhatnak. Megkötésükre legalkalmasabb a higany. Mivel a reakciócsak a felületen következik be, a higany finom eloszlatásáról, esetleg elpárologtatósáról kell gondoskodni. Egyes esetekben elegendő, ha a tisztítandó anyagothigannyal összerázzuk. Vizes abszorpciós folyadékként használható még a lúgosarzenit vagy maga a lúg. Az egyes halogének más halogénszennyezéstől mentes állapotban való előállítása külön probléma. Ezt a speciális részben tárgyaljuk.
49.3. Szárítóanyagok
A víz mint szennyezés valamennyi gázban előfordulhat. Eltávolítása, azaz agázok szárítása sokféle módon, különböző igényeknek megfelelő mértékben oldható
meg‘ ,
A víz cseppfolyós részecskéit a már tárgyalt mechanikus eljárásokkal küszö
bölhetjük ki. Kifagyasztása és a gázelegy frakcionálása ugyancsak hatásos módszer.Leggyakrabban mégis az adszorpciót és abszorpciót, valamint az ezekkel kapcsolatos kémiai folyamatokat alkalmazzák a víznek és gőzének különböző gázelegyek-ből való eltávolítására.
A laboratóriumi gyakorlatban használatos ismertebb szárítóanyagok: cc.H2S04, szilikagél, CaCl2, Mg(C104)2, P20 5, KOH, BaO, CaO, CaS04, CuS04.
Tömény kénsav. Nem alkalmas sem bázisos jellegű, sem könnyen oxidálódógázok, pl. NH3, Hl, HBr, H2S, AsH3, PH3, C2H2, HCN, (CN)2, N02 szárítására.Nem ajánlható még H2, NO és HF szárítására sem.
Tiszta cc.kénsavval nagyon jól szárítható a HCl, Cl2, Br2, CO, N20 és CH4. A CO
és a H2 (a nascens H még inkább) magasabb hőmérsékleten Hg- vagy Se-katalizá-tor jelenlétében redukáló hatású, és a kénsavból kén-dioxidot szabadít fel, ami aszárítandó gázt szennyezi.
Előnye a kénsavnak mint szárítóanyagnak, hogy igen gyors és erélyes hatású,továbbá vízmegkötő kapacitása is nagy. Ezek a tulajdonságai a hőmérséklet emelésével sem csökkennek jelentősebb mértékben. így pl. 5 % vizet tartalmazó kénsavfölött a vízgőztenzió még 100 C°-on sem több, mint vízmentes CaCl2 fölött szoba-hőmérsékleten.
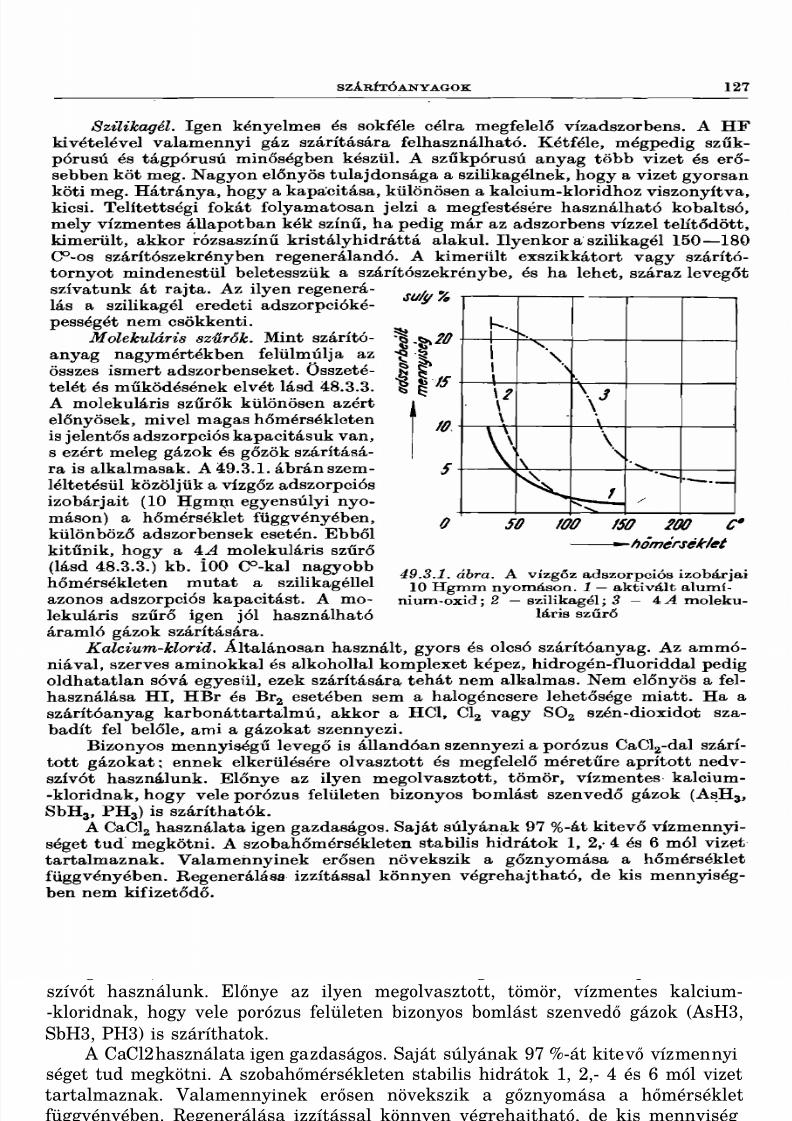
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 127/366
S Z ÁR ÍT Ó A N Y A G O K 127
Szilikonéi. Igen kényelmes és sokféle célra megfelelő vízadszorbens. A HFkivételével valamennyi gáz szárítására felhasználható. Kétféle, mégpedig szűk-pórusú és tágpórusú minőségben készül. A szűkpórusú anyag több vizet és erősebben köt meg. Nagyon előnyös tulajdonsága a szilikagélnek, hogy a vizet gyorsanköti meg. Hátránya, hogy a kapacitása, különösen a kalcium-kloridhoz viszonyítva,kicsi. Telítettségi fokát folyamatosan jelzi a megfestésére használható kobaltsó,mely vízmentes állapotban kék színű, ha pedig már az adszorbens vízzel telítődött,kimerült, akkor rózsaszínű kristályhidráttá alakul. Ilyenkor a szilikagél 150—180C°-os szárítószekrényben regenerálandó. A kimerült exszikkátort vagy szárító-tornyot mindenestül beletesszük a szárítószekrénybe, és ha lehet, száraz levegőtszívatunk át rajta. Az ilyen regenerá
lás a szilikagél eredeti adszorpcióképességét nem csökkenti. Molekuláris szűrők. Mint szárító
anyag nagymértékben felülmúlja azösszes ismert adszorbenseket. összetételét és működésének elvét lásd 48.3.3. A molekuláris szűrők különösen azértelőnyösek, mivel magas hőmérsékletenis jelentős adszorpciós kapacitásuk van,s ezért meleg gázok és gőzök szárítására is alkalmasak. A 49.3.1. ábrán szemléltetésül közöljük a vízgőz adszorpciósizobárjait (10 Hgmpi egyensúlyi nyomáson) a hőmérséklet függvényében,különböző adszorbensek esetén. Ebbőlkitűnik, hogy a 4 A molekuláris szűrő(lásd 48.3.3.) kb. Í00 C°-kal nagyobbhőmérsékleten mutat a szilikagéllelazonos adszorpciós kapacitást. A mo
lekuláris szűrő igen jól használhatóáramló gázok szárítására. Kalcium-klorid. Általánosan használt, gyors és olcsó szárítóanyag. Az ammó
niával, szerves aminokkal és alkohollal komplexet képez, hidrogén-fluoriddal pedigoldhatatlan sóvá egyesül, ezek szárítására tehát nem alkalmas. Nem előnyös a fel-használása Hl, HBr és Br2 esetében sem a halogéncsere lehetősége miatt. Ha aszárítóanyag karbonáttartalmú, akkor a HC1, Cl2 vagy S02 szén-dioxidot szabadít fel belőle, ami a gázokat szennyezi.
Bizonyos mennyiségű levegő is állandóan szennyezi a porózus CaCl2-dal szárított gázokat; ennek elkerülésére olvasztott és megfelelő méretűre aprított nedvszívót használunk. Előnye az ilyen megolvasztott, tömör, vízmentes kalcium--kloridnak, hogy vele porózus felületen bizonyos bomlást szenvedő gázok (AsH3,SbH3, PH3) is száríthatok.
A CaCl2 használata igen gazdaságos. Saját súlyának 97 %-át kitevő vízmennyiséget tud megkötni. A szobahőmérsékleten stabilis hidrátok 1, 2,- 4 és 6 mól vizettartalmaznak. Valamennyinek erősen növekszik a gőznyomása a hőmérsékletfüggvényében. Regenerálása izzítással könnyen végrehajtható, de kis mennyiségben nem kifizetődő.
49.3.1. ábra. A vízgőz adszorpciós izobárjai 10 H gm m nyomáson. 1 — ak tivá lt alum í-
nium-oxid; 2 — szilika gél ; 3 — 4 A moleku
láris szűrő

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 128/366
128 G Á Z O K
Magnézium-per klórát. A Mg(C104)2 ugyancsak semleges só, az előbbinél erélyesebb reagens. Az ammóniát nem köti meg [a Ba(C104)2 igen]. Hidrátjai 2, 4, és
6 mól vizet tartalmaznak. A két, kevesebb kristályvizet tartalmazó anyag valószínűleg kettős sót képez, mert ismeretes Mg(C104)2*3 HaO bruttó összetételű só is,amely még mindig jól köti meg a vizet. A vízmentes vegyületen (Anhydron) kívüla trihidrát (Dehydrit) is a kereskedelmi forgalomban kapható szárítóanyag. Hevítés
sel regenerálható, de a dihidrát csak135 C° fölött és 5 Hgmm alatti vákuumban adja le utolsó kristályvizét. A vízmentes b ó ezért még 135C°-on is erélyes vízelvonószer. A
szerves elegyrészeket (szénhidrogéneket vagy oldószergőzöket stb.,éghető anyagot) tartalmazó gázokszárítása után a regenerálás a fennálló robbanásveszély miatt igen kockázatos.
Foszfor-pentoxid. A P20 5 valamennyi ismert vízelvonószer közötta leghatásosabb. Kénsavból is vizetvon el. Vigyázzunk ezért, hogy a fo
kozatos szárítás közben kénsavcseppék ne jussanak a víztelenítő sorvégére kapcsolt P20 5-os toronyba,mert a szárított gáz kén-trioxiddalszennyeződik.
A foszfor-pentoxid nem használható NH3, HF, HC1 és HBr, kevéssé alkalmas a H2S, Cl2 és Br2 szárítására. A teljesen száraz ammó
niával ugyan a szintén száraz P20 5nem reagál, de nedvességnyomokhatására megindulhat a hőfejlődéssel járó gyors és heves reakció. Halogén-hidrogének ugyancsak reak
cióba lépnek a foszfor-pentoxiddal foszfor-oxid-halogenidek képződése közben. A kén--hidrogén csak oxigénmentes gázelegyben szárítható, a Cl2 és Br2 csak megfelelőelőzetes víztelenítés után.
Mellékreakció veszélye nélkül felhasználható a foszfor-pentoxid a következőgázok szárítására: 0 2, N02, NO, N20 , CO, C2H2, PH3, AsH3. A N02-dal szemben
azonban 250 C° fölött már nem indifferens, és az olefin-szénhidrogéneket is megköti.
49.3.2. ábra. Kü lönböző szárítóanyagok fölött kialakuló vízgőztenzió egyensúlyi helyzetben
A foszfor-pentoxidot nagyiparilag a sárgafoszfor levegőn való elégetésévelállítják elő. Közben elkerülhetetlen az alacsonyabb vegyértékű oxidok képződése,amelyek vízzel foszforossavat, majd foszforsavat és foszfor-hidrogént is szolgáltathatnak. Különösen a katalitikus folyamatokhoz előkészített gázok PH3-szennye-zése veszélyes, mert nemcsak a katalizátort teszi tönkre, hanem közben a reakció
sebességet is megváltoztatva, helytelen kinetikai következtetésekre adhat alapot.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 129/366
G Á Z O K S ZO K Á S OS S Z E N N Y E Z É S E I 129
Még az analitikai készítmények is kb. 1 % ilyen alacsonyabb oxidot tartalmaznak. A tisztítást száraz oxigénáramban szublimálással végzik 270—300 C°-on (kiterme
lés kb. 10 %). A tiszta készítménynek vízfelvétel után enyhén melegítve nem szabadmegsárgulnia, sem PH3-szagú nem lehet. Erélyes hatása ellenére a P20 5 fölöttlassan alakul ki a vízgőzegyensúly. Az utolsó nyomok megkötése napokig tart.
Egyéb szárítóanyagok. A KOH, CaO és BaO bázisos jellegű vagy egyébként in-differens gázok, pl. NH3, PH3 és N20 szárítására alkalmas, egyben azonban savastulajdonságú szennyezések megkötésére is. Fel kell hívni a figyelmet arra, hogy ateljesen száraz KOH nem reagál szén-dioxiddal. Szén-monoxiddal csak 100 C°fölött, klórral és brómmal pedig csak vörösizzáson indul meg a reakció. A vízmentes KOH egyébként erélyes vízelvonószer, hatása megközelíti a kénsavét.
A CaS04 és Á120 3 előnye semleges kémhatása és az adszorpció gyors bekövet
kezése. Vízzel cserebomlásba lépő anyagok is felhasználhatók gázok szárítására, ha
a keletkezett termékek egyike azonos a szárítandó gázzal, a többi pedig nem illékony vagy könnyen eltávolítható. Hidrogén víztelenítésére a legerélyesebb eljárásaz olvasztott alkálifém vagy alkáliföldfém fölött való elvezetés. A nátrium vagykálium felületén tapadó petróleumot rendkívül gondosan el kell előbb távolítani.
Néhány szárítóanyag hatását a 49.3.2. ábra alapján hasonlíthatjuk össze.
49.4. A laboratóriumi gyakorlatban sűrűbben előforduló gázok szokásos szennyezései
Nitrogén. A legtöbb célra megfelelő minőségű a nagynyomású acélpalackbanforgalomba kerülő nagyipari termék.
40 literes, 5 m3 normálállapotú gázt tartalmazó, 150 atm nyomású tartálybanszállítják. Jelzőszíne: zöld csík. Csatlakozó csonkja jobbmenetes.
N2-tartalma minimum 99,5 %, megengedett szennyezései: 0 2 0,5 %, azonkívülnemesgázok (MSZ 1605—52 szabvány).
Oxigén. 40 literes, 5 m3 normálállapotú gázt tartalmazó, maximum 150 atm
töltőnyomású palackban kapható. Jelzése kék csík, csatlakozása jobbmenetes. 0 2-tartalma minimum 98,8 %, megengedett szennyezése: N2, H2, nemesgázok (MSZ1604—52).
Fontos balesetvédelmi rendszabály: a nagynyomású oxigén útjába semmiféle éghető anyag nem kerülhet! Robbanásveszély! Redukciós szelep zsírozása, éghető tömítések használata szigorúan tilos !
Hidrogén. 40 literes, 5 m3 gázt befogadó, 20 C°-on 125 atm nyomásra töltöttacélpalackban kerül forgalomba. Jelzése vörös csík, a redukciós szelep és vezetékcsatlakozása balmenetes. A H2-tartalom minimum 99,5 %, a megengedett szennye
zés: 0 2 maximum 0,5 % (MSZ 3294—52).Levegővel és oxigénnel igen tág térfogatarányban robbanó elegyet alkot. Biztonsági rendszabályok: meggyújtás előtt az oxigénmentességet kémcsőnyi meny-
nyiségű próbával ellenőrizzük!
Szén-dioxid. Többnyire 10 literes palackokban tárolják. Kritikus hőmérséklete31,4 C°, kritikus nyomása 73 atm, 20 C°-on cseppfolyós, nyomása 57 atm, 50 C°-ongáz, nyomása 187 atm. Jelzése szürke szín, csatlakozása jobbmenetes. C02-tarta-lom minimum 97 térfogat %, 0 2 maximum 0,1 %, szénhidrogén maximum 2 %, N2
9 Alt. és szervetlen kémiai'prakt. —4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 130/366
130 G Á Z O K
maximum 1 %, olaj maximum 0,005 súly %, víz maximum 0,05 súly % (MSZ
20 915-55). Biztonsági intézkedések: a palackot óvni kell a felmelegedéstől! Kritikus pontjafölött a gáz nyomása hirtelen megnő.
Klór. Különböző alakú és méretű tartályokba töltik. 20 C°-on folyadék, nyomása 6,6 atm. A tartály 30 atm, a szelepet 35 atm nyomással kell ellenőrizni. Jelzése szürke szín, csatlakoztatás jobb menettel. Cl2-tartalom minimum 99,5 súly %.Megengedett szennyezés: víz maximum 0,1% (MSZ 20 917-55). Előfordul mégbenne: 02, N2, CO, C02, HC1, C0C12 stb.
Ammónia. A palackok mérete nem szabványosított. 20 C°-on folyadék, nyomása 8,5 atm. Jelölése szürke, csatlakoztatása jobbmenetes.
NH3-tartalma minimum 97 %, gáznemű szennyezés maximum 3 térfogat %.Cseppfolyós szennyezés a folyékony fázisban 0,2—0,5 térfogat %. 105 CP-on nemilló szennyezés: 10 mg/100 ml kondenzátum (MSZ 20 916-55). A szintetikus KHSszennyezése főleg C02 és víz. A gázgyári NH3 szennyezései: víz, piridin, metil-aminstb.
Biztonsági rendszabályok: amrrióniával levegő jelenlétében rezet tartalmazó szer
kezeti anyagok nem érintkezhetnek. Szelepek és vezetékek vasból készülnek. Argon. 40 literes, 5 m3 befogadóképességű, 20 C°-on 150 atm nyomásra töltött
palackokban kerül forgalomba. Kétféle minőségben gyártják: a H jelzésű a hegesz
tésre használatos argon, az N jelzésűt az izzólámpagyártásban alkalmazzák. Szabvány szerinti (MSZ 6271-T-58) összetétele a 49.4.1. táblázatban látható.
49.4.1. táblázat
A kereskedelmi Ar összetétele (térfogat %-ban)
H - A r N - A r
A rg o n ......................... >99,7% >90,0%
Nitrogén....................
< 0,3% < 10
,0
%Oxigén < 0 , 1 % < 0,2 %Hidrogén •................ nincs előírás < 0, 1 %Szénhidrogének nincs előírás < 0,0 0 1%Szén-dioxid .............. nincs előírás ' < 0,02%Összes megengedett szennyezés < 0,3% < 10 ,0 %
Hélium. Szennyezései: hidrogén, nitrogén. Dissous-gáz. 40 literes tartályokba töltik. Porózus anyaggal felitatott aceton-
ban nyomás alatt oldott acetiléngáz. Szabvány (MSZ 1602-53) szerint megengedettszennyezései: levegő, H2, egyéb vízben oldódó gáz, összesen 2 térfogat %, H2S maximum 0,02 %, PH3 maximum 0,05 %, nedvesség maximum 7 g/m3.
Propán—bután-gáz. Különböző nagyságú alumínium tartályokban kerül fel-használásra. 20 C°-on folyékony, gőznyomása kb. 4 atm. Csatlakozó csonkjabalmenetes.
Kén-dioxid. 20 C°-on folyékony, nyomása 3,2 atm. A rezet tartalmazó szerkezeti anyagokat erősen korrodálja. Minőségét nem szabványosították. Előfordulószennyezései: C02, 0 2, N2 és víz.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 131/366
G Á Z O K Á R A M O L T A T Á S A 131
Fo8zgén. Nem kerülhet réztartalmú szerkezeti anyagokkal érintkezésbe akorrózió veszély miatt. Rendkívül mérgező.
Várható szennyezései: C02, HC1, 02 és N2.Külföldön ugyancsak palackozott formában kapható még számos laboratóriumban használatos gáz, pl.: H2S, HC1, N20, HCN, HF, F2, BF3, valamint különböző nemesgázok.
49.5. Áramlássebesség- és nyomásszabályozás, tárolás
A gázok laboratóriumi alkalmazása folyamán a gázokat különböző tisztító-berendezéseken át a felhasználás helyére kell juttatni. Ehhez a művelethez az áramol
tatás, nyomásszabályozás és tárolás problémáit kell megoldani.
49.5.1. Gázok áramoltatása
Az áramlást a megfelelő mértékűre csökkentett túlnyomás biztosítja. Enneknagysága a gázvezeték áramlási ellenállásától és az időegységenként felhasználandógáz mennyiségétől függ. A reakcióba lépő gáz nyomását a vezeték végén, a reakció-tér előtt közvetlenül elhelyezett nyomásszabályozóval állíthatjuk a kívánt értékre(lásd 1.19.).
A laboratóriumi gázfejlesztőkkel előállított vagy néhány atmoszféra nyomásnál nem nagyobb mértékben komprimált gázok több fokozatú tisztítására összeállított berendezés és a vezeték áramlási ellenállásának leküzdése a rendszerben külön áramoltatóegységbeiktatását teheti szükségessé. Hasonló a probléma,amikor a zárt rendszerben kell ugyanazt a gázmennyiséget többször egymás utón, pl. valamely adszorbensoszlopon átvezetni.
Igen egyszerű a 49. 5.1.1. ábrán vázolt szerkezetűdugattyús készülék működése. A megfelelő szerkezeti
anyagból (pl. üvegből vagy a higannyal nem amalga-málódó fémből) készült közlekedőedény egyik szárában a higanyszintet mechanikusan (pl. elektromotorés excenter segítségével) meghajtott dugattyú változtatja. A másik szárához csatlakozó vezetékbenezért a benne elhelyezett visszacsapószelepek működése következtében szakaszos áramlás lép fel. Vízlégszivattyú segítségével működtethető aűtomatikus hi* 49.5.1.1. ábra. Higanyosganypumpa látható a 49.5.1.2. ábrán. Ha a vízlég- gáztovábbító
szivattyú az A térbe szívta a B edényke tartalmát a0 kapillárison át, .4-ba levegő jut, megszűnik a vákuum, és a higanyszintek kiegyenlítődnek. Ezzel a D-ben levő gáz a vezetékbe nyomódik, és a B edény is újramegtelik higannyal, elzárva a kapilláris alját és az A tér nyílását. A vízlégszivattyúmost újra felszívhatja a függőleges csőben a higanyt, és ezzel megtölti a D edénytgázzal. A C kapilláris áramlási ellenállását, illetve a B tartály térfogatát úgy kellméretezni, hogy addig ne jusson levegő az A térbe, míg a higanyszint meg nem közelítette a maximális magasságot. A készülék lökésszerű gázáramot ad.
9*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 132/366
132 G Á Z O K
Használatosak még különböző köpüs- és membránszivattyúk. Csak üvegalkat
részekkel érintkezik a gáz az ún. elektromágneses szivattyúban. Lényege függőlegeshelyzetű, 12mm0-jű KPG-cső (lásd. 42.1.), belefor-rasztott vasmagot tartalmazó dugattyúval. A csövet kétvízzel hűthető elektromágneses tekercs veszi körül, és eztartja a dugattyút kis amplitúdójú, nagy rezgésszámúmozgásban. Az ilyen rendszerű szivattyú kb. 100 Hgmmellenállás leküzdésére képes, és 1 atmoszféra nyomásonkb. 5 1/perc teljesítményű.
Kis nyomáson (kb. 25 Hgmm elővákuummal) üzemeltethető a higany-gőzsugárszivattyú. Végvákuuma kb.
10-3 Hgmm. Zárt rendszerben kisnyomású gázok áramoltatására is alkalmas.Egészen kis ellenállású rendszerben elegendő a ve
zeték egyik függőleges szakaszában fűtést alkalmazni,indifferens gázok esetén pl. platina ellenálláshuzallal. Afelszálló meleg gáz helyére alulról hidegebb áramlik. Ezaz ún. „termoszifon ” elve.
49.5.2. Az áramlássebessé? mérése 49.5.1.2. ábra. Automatikus
gáztovábbító E célra többféle szerkezetű gázóra alkalmas. A turbinakerekes vagy folyadékzáras gázóra lapátkerekének
fordulatszáma mutatja a bizonyos idő alatt átáramlott gáz egész mennyiségét,de a pillanatnyi sebességváltozásokról nem ad felvilágosítást.
Folyamatos sebességmérésre és nagyobb sebességeknél a rotaméterek (49.5.2.1.ábra) alkalmazása célszerű. Lényeges része a rotaméternek egy pontosan függőleges helyzetű, kissé kónikusüvegcső, amelyben megfelelőméretű és súlyú, kúp alakú
dugó van. A dugó felületébevésett, meredek csavarvája-tokban fölfelé áramló gáz a sebességének megfelelő forgásrakényszeríti a kúpot, és a csőben is meghatározott magasságra emeli. A lebegő kúp helyzete a gáz viszkozitásától, tehát hőmérsékletétől is nagymértékben függ. A hőmérsék
let stabilizálása tehát fontos.Legegyszerűbb gázfajtánkéntkülön kalibrálnia műszer skáláját, de a relatív viszkozitások ismeretében az azonos 49.5.2.1. ábra. Rotaméter 49.5.2.2. ábra. Reométer
skálaértékhez tartozó, különböző sebességértékek számítással is meghatározhatók. A gázt minden por- vagyfolyadékcsepptől előzetesen meg kell tisztítani.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 133/366
Á Ü A M L Á S S E B E S S É G 133
ÁramlássebeBség-mérésekre elég tág határokon belül a reométer alkalmas(49.5.2.2. ábra), néhány ml/óra — mintegy 20 1/óra sebességig. Pontossága kb. 2 %.
Az áramló gázt megfelelő keresztmetszetű kapillárison vezetjük át, ésmérjük a két végén képződő nyomáskülönbséget. A műszer méréshatárát a kapilláris keresztmetszetével és a manó méterfolyadék faj-súlyának változtatásával szabályozhatjuk. A nyomáskülönbség ugyancsak függvénye a gáz viszkozitásának és hőmérsékletének. Mindengázra külön kalibrálandó.
Újabban egyre nagyobb szerepet kap a termisztoros ámmMssebes- ség-mÁrés. A mérési elv lényege az,hogy a termisztort körülvevő közegáramlási sebességének függvényében a disszipációs körülményekmegváltoznak. Az áramlási sebességnövekedésével a termisztor lehűl,
illetve disszipációs állandója megnő(lásd 45.2.2.). A változás meghatározásával lehetővé válik a gázok és folyadékok áramlási sebességének mérése.
A 49.5.2.3. ábrán egy termisztoros gázáramlásmérő berendezés elvi kapcsolásivázlatát mutatjuk be. A kompenzáló termisztor ellenállása /R(TX)/ közelítőlegegyenlő a mérotermisztoréval /R{T ) /, és a környezete ugyanolyan minőségű, de'szta-tikus állapotban levő közeg.
49.5.2.3. ábra. Termisztoros áramlásmérő kapcsolási vázlata. .É?-fűtŐtelep, Px és P2 a fűtést szabályozó, illetve a terinisztorok ellenállás-ki- egyenlítését szolgáló potenciométerek, R(TX) és
R(TS) termisztorok, Rt és R2 híd- ill. összehasonlító ellenállások, K xés K 2kapcsolók, M indi
káló műszer.
49.5.3. Áramló gázok keverése
Meghatározott arányú keveréshez mindenekelőtt külön-külön kell ismernünka komponensek áramlási sebességét, majd mindkettőt meghatározott sebességűreszabályozva, keverőedényen át engedjük a vezetékbe. Ha a keverékarány számértéke nagyon eltér az 1-től, a keverést több fokozatban hajtjuk végre.
A 49.5.3.1. ábrán vázolt készülék igen alkalmas nagy hígítású gázelegyek előállítására.
A laboratóriumi munkában gyakran szükségünk van arra, hogy gázokat vagylevegőt adott hőmérsékleten különböző folyadékok (leggyakrabban víz) gőzéveltelítsünk. E célra jól alkalmazhatók az üveggyöngyökkel vagy üveggyapottal töl
tött mosótornyok. A torony tetején permetezzük be az adott folyadékot, és a telítendő gázt a torony alján vezetjük be. Előfordul, hogy a gázhoz a telítettségi parciális nyomásnál kisebb mennyiségű gőzt kívánunk adagolni. Ilyenkor úgy járhatunk el, hogy a gázáram egy részét a folyadék gőzével telítjük, majd ismert meny-nyiségű tiszta gázzal keverjük. Lényegesen egyszerűbb azonban a gázt megfelelőparciális nyomású tömény sóoldatokkal permetezett tornyokon átvezetni. Néhánytelített sóoldat parciális nyomásának változásáról a hőmérséklet függvényében a49.5.3.2. ábra ad tájékoztatást.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 134/366
134 g á z o k :
vivogaz,
k is mennyit __
hozzákever i gáz
leveg ,viv gáz
n )
49.5.3.1. ábra. Csekély gázmeny- nyiség hozzákeverésére alkalmas
készülék
parciális nyomása a hőmérséklet függvényében
40.5.4. Gázok tárolása
Az előállítás és áramoltatás problémáin kívül különleges feladatot jelent éskülön felszerelést igényel a gázalakú anyagok összegyűjtése és tárólása/is. Ha nyílásával lefelé zárófolyadékba merített és azzal töltött edénybe gázt vezetünk, a faj-súlykülönbség alapján a gáz a folyadékréteg fölött gyűlik össze. A jó zárófolyadéknak nem szabad a gáztér komponenseivel reagálnia, sem azokat oldania.
Ezeknek a követelményeknek legjobban a higany felel meg, amelyben eddigegyetlen gáznak az oldott nyomait sem tudták kimutatni, és amely a legtöbb ag
resszív gáz (N20, NO, PH3, S02, BF3, Sí F4, C2H2, COCl2, HC1) kémiai hatásának is ellenáll. Hátránya, hogy drága és az egészségre ártalmas anyag, valamint az,
hogy néhány gáz, illetve kondenzált fázisa mégis reakcióba léphet vele. így pl. ahalogének, a HBr különösen napfény hatására, a Hl, SeH2, TeH2 sötétben is megtámadják. A tiszta, száraz H2S gázállapotban nem reagál vele, cseppfolyósán azonban igen! Az AsH3 főleg nedves higanyfelületen elbomlik. A BC13 és SiBr4 melegensem reagál higannyal, CHC13 és CC14 kb. 100 C°-on már megtámadja.
A gázok vízben való oldhatóságát (49.5.4.1. táblázat) különféle elektrolitokkalhatásosan lehet csökkenteni. Leginkább alkalmazott vizes zárófolyadék a közel telített NaCl-oldat vagy kénsavas Na2S04-oldat. Az utóbbi úgy készül, hogy 200 g víz-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 135/366
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 136/366
136 g á z o k ;
végű kapillárist a gáztérbe nyújtjuk, és az áteresztőcsap megnyitásával az alsótartályt megtöltjük gázzal. Még mielőtt egészen megtelne, *újra belemerítjükhiganyba a kapilláris végét, és azt higannyal teleszíva, az edénybe jutott gáztkét higanyfelület közé zárjuk. Az anyag felhasználása a vízlégszivattyú felső
csatlakozásának megszüntetése után a csapon át leeresztett higany segítségével
lehetséges.
49.5.4.2. ábra. Higanyos gáztartály
49.5.4.3. ábra. Higanyos géz- büretta
Hasonló elv szerint működik a 49.5.4.3. ábrán vázolt higanyos gázbüretta,
amely különösen kisebb gázmennyiségek áttöltésére vagy a gáz analíziséhezmintavételre alkalmas.
Egyéb zárófolyadék nélküli megoldások is szükségesek akkor, ha pl. a gáznedvességgel is, higannyal is reagál. Tiszta gázok eltartása — főleg hosszabb időn
át — üvegcsőbe beforrasztva lehetséges.Gázok tárolására használnak különféle úszószelepekkel elzárt tartályokat is.
Ezeknek többféle típusát mutatja be szerkezeti vázlatban a 49.5.4.4. ábra. Valamennyihez manométer és folyékony levegővel hűthető kondenzációs tér is tartozik,
melynek térfogatmérő skáláján cseppfolyósítás után az edényben tárolt gáz meny-nyisége leolvasható.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 137/366
G Á Z O K T Á R O L Á S A 137
Kompresszor vágy más költséges berendezés nélkül is nagynyomású acélpalackba tölthető bármely (pl. folyékony levegővel) kondenzálható gáz a követ
kező egyszerű laboratóriumi összeállítással. A gyűjtött gázt a folyékony levegővel hűtött, 49.5.4.5. ábrán látható alakú
kisebb acélpalackban kondenzáljuk. Bizonyos idő elteltével a fejlesztő felőli tűszelepet zárjuk, majd az előzőleg evakuált gázpalack sarokszelepét megnyitjuk, és
49.5.4.4. ábra. Szelepes gáztartályok 49.5.4.5. ábra. Kondenzálható gáz áttöltéseacélpalackba
a hűtést megszüntetjük. A kondenzátum elpárolog, és a nyomás a kis térfogatú
kondenzáló- és a nagyobb tartályedény között kiegyenlítődik. Most elzárjuk akettő közötti sarokszelepet, és újra lehűtjük a kondenzálóbombát. Ha a bennemaradt gáz már cseppfolyósodott, megnyitjuk a fejlesztőhöz csatlakozó vezetéket,és folytatjuk a kondenzálást.
Irodalom
Klemenc, A.: Die Behandlung und Keindarstellung von Gasen. 2 . Aufl. Springer, Wien, 1948.
Lux, H.\ Anorganisch-chemische Experimentierkunst. J. A. Barth Veri., Leipzig, 1954.
Bayer, F . : Gasanalyse. 2 . Aufl. Enke, Stuttgart, 1941. Hargittay E.: Gázelemző műszerek. Műszaki Könyvkiadó, Budapest, 1959.Stáhler, A.: Handbuch dér Arbeitsmethoden in dér anorgánischen Chemie. Bd. IV,
Gruyter Co., Berlin —Leipzig, 1926.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 138/366
50. Oldószerek
Az oldás folyamatának a laboratóriumi technikában alapvető fontossága van.
A szilárd testnek az oldószerben való oldhatósága a két anyag fizikai és kémiaitulajdonságaitól egyaránt elég bonyolult módon függ. Nagy fontosságú ebből aszempontból a szilárd anyag kristályainak rácsenergiája, valamint az oldószermolekuláival végbemenő kapcsolódás, szolvatálódás energiamérlege. A szolvatá-ciós hő többek között nagymértékben függ a folyadék dielektromos állandójától,móltérfogatától stb. Még további bonyodalmakat jelent az oldás folyamatában azelektrolitos disszociáció, komplexképzés, asszociáció, szolvolízis stb.
Mindezek alapján könnyen belátható, hogy az egyes, nem túlságos alacsonyhőmérsékleten kondenzálható gázok, illetve egyes, közönséges hőmérsékleten szilárd
testek megolvasztása útján nyert folyékony fázisok oldószerként egészen máskéntviselkednek, mint a legközönségesebben használt víz és a szerves oldószerek.
Az oldószerként felhasználandó gázokat célszerű még kondenzálás előtt minden szennyezéstől, főleg a víztől megtisztítani.
Néhány nemvizes oldószerként használható, nem túlságosan körülményesenelőállítható, alacsony és magas hőmérsékleten cseppfolyós szervetlen anyag, valamint a víz és néhány szerves oldószer fontosabb fizikai tulajdonságait, szárítását,illetve tisztítását a következőkben tekintjük át.
50.1. Halogén elemek
Ezek dözül a fluor igen alacsony forráspontja (—188 C°) és nagy reakció-képessége miatt nem szerepel a nem vizes oldószerek között.
A klór olvadáspontja —100,5 C°, forráspontja —33,95 C°, dielektromosállandója e — 1,97 (0 C°-on), fajlagos vezetőképessége x = l-10” 18f2-1 *cm-1.Oldja a C, Si, Ti, Sn és Pb tetrakloridját, az AsCla-ot, POCl3-ot és S2Cl2-ot. PCl3-daloldhatatlan PCl5-dá egyesül.
Bróm. Op. —7,3 C°, fp. 58 C°, e = 32 (23 C°-on), « = 1,33.10-“ (17 C°-on).Oldja a C, Al, Fe, Sb, Sn, és Ti brómidjait, a ICl3-ot és POCl3-ot. Vízmentesítése
konc. kénsavról való ledesztillálással történhet. Jód. Op. 113,6 C°, fp. 183 C°, e = 11,1 (118 C°-on), x = kb. l«10-s. A nyomás
alatt megolvasztott jód számos jodidnak jó oldószere, az alkáliföldfémek és néhánynehézfém jodidja azonban nem oldódik benne. A K, Ag és Hg cianidjával ICN és
I~, a HgCl2-dal IC1 és 1“ képződése közben reagál. Az interhalogenidek közül a BrF3 és a IF3 viselkedik oldószerként.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 139/366
N E M V I Z E S - O L D Ó S Z E R E K 139
50.2. Hidrogén-halogenidek
Közülük vízmentes oldószerként jól ismert a hidrogén-fluorid. Op. —83,1 C°,fp. 19,5 C°, e = 83,6 (0 C°-on), « = 1,4* 10-5 . Noha dielektromos állandója avízéhez igen hasonló, tulajdonságai sokban eltérnek attól. Az alkálifém- és alkáli-földfém-fluoridok jól oldódnak a hidrogén-fluoridban. Az AgF és TIF szintén jóloldódik, egyéb fém-fluoridok azonban nem. A hidrogén-fluorid a többi halogén--hidrogén legtöbb sójából fluoridképződés közben felszabadítja a savat. A szabadhalogén-hidrogének azonban rosszul oldódnak benne. Az oldódó fluoridok elektromosan jól vezetővé teszik az oldószert. A hidrogén-fluoridban a disszociáció és aszolvolízis egyaránt nagymértékű. Az oxidok közül az Sn02 és HgO indiferens, de abázisosabbak közül a Ba, Ca, Mg, Pb és Agoxidja hevesen reagál HF-dal, a hidro-xidok szintén.
Sósav. öp. —114,2 C°, fp. —85,0 C°, e = 4,6 (+ 27 C°-on), * = 2-10 -’ . A hidrogén-fluoridhoz viszonyítva rossz oldószer. Az ón- és foszfor-kloridokonkívül alig oldható benne szervetlen anyag. A I2 sósavban sötétvörös színnel oldódik.Cseppfolyós C02-dal és H2S-nel a folyékony HC1 minden arányban elegyedik.
Hidrogén-bromid. Op. —86,9 C°, fp. —66,8 C°, e = 3,82 ( + 24 C°-on). A H2S,P0C13, PBrs oldódik benne, Br2 és I2 alig, sók egyáltalában nem.
Hidrogén-jodid. Op. —50,7 C°, fp. —35,4 C°. Csak a jódot és a foszfor-oxid--kloridot oldja.
50.3. Hidrogén-cianid
Op. —13,4 C°, fp. 25,0 C°, e = 152 (0 C°-on), x = 3,3-10"6 (0 C°-on). Jóoldószer, de használatát és vizsgálatát igen mérgező volta nehezíti. Valamennyiismert oldószer között legnagyobb a dielektromos állandója. Ennek ellenére nemoldja különösebben jól az egyes sókat, és az elektrolitos disszociációt sem növeli avárakozásnak megfelelő mértékben. Az alkálisók többsége korlátoltan oldódikbenne, legjobban néhány alkálifém-jodid és -rodanid, valamint a kálium-hidrogén--szulfát. Ezek az oldatok jól vezetik az elektromosságot. Molekularácsos vegyületek
általában ugyancsak jól oldódnak, de az oldatok elektromos vezetőképességecsekély. A HCN a szerves vegyületeket is többnyire jól oldja, kivéve a telített szén-hidrogéneket. Jól oldódik benne pl. a benzol, kloroform, metil- és etilalkohol, éter,aceton és piridin.
Érdekes, hogy alkálifém-cianidok hatására a HCN először megbámul, majdelgyantásodik.
50.4. Kén-hidrogén
Op. —85,5 C°, fp. —60,4 C°, e = 10,2 (—60 C°-on), x = 3,7-10"11. Az egyszerű ionrácsos vegyületeket általában nem oldja. A molekularácsos halogenide-ket inkább oldja, de ezek közül sokkal nehezen oldható szulfidokat képezve reagál(pl. a Cu, Ag, Hg, Bi, Sb, As halogenidjeivel). A CC14 és SiCl4 korlátlanul elegyíthető H2S-nel. Halogén-hidrogének is jól oldódnak benne, valamint a kénsav és azecetsav is. A halogén elemek ugyancsak jól oldódnak kén-hidrogénben, de a I2kivételével reakcióba is lépnek vele. A cseppfolyós H2S jó oldószere a legtöbb szerves anyagnak, még a szénhidrogéneknek is.
A kén halogénvegyiileteinek (S2C12, S0C12, S02C12, valamint SeF4 és SeOCl2)oldószerként való használhatósága ugyancsak ismeretes.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 140/366
140 O L D Ó S Z E R E K
50.5. Kén-dioxid
Op. —75,7 C°, fpv —10,0 C°, e = 13,8 (+ 15 C°-on), x = M C r7 (0 C°-on).Oldó hatása korlátolt. Általában csak az alkáli-jodidokat és -rodanidokat oldja jól,és ezek oldatai vezetik is az elektromosságot. Az oldott halogenidek kisebb-nagyobbsebességgel reakcióba is lépnek az oldószerrel, aminek végtermékeként szabadhalogén, elemi kén és szulfát képződik. A több értékű elemek halogenidjei közül jóloldódnak az Al, Fe, Sb, C, Si, Ge és P kloridjai. Jól oldódik azonkívül folyékonykén-dioxidban sok szerves vegyület is. Közülük a telített alifás szénhidrogénekoldhatósága igen rossz, a telítetleneké és aromásoké viszont jó.
50.6. Ammónia
Op. —77,8 CP, fp. —33,5 C°, e = 22 (- 3 4 C°-on), x = 3.10"8 ( -3 7 C°-on). Az ammónia a vízhez sok tekintetben hasonló tulajdonságú oldószer, viselkedésétsokoldalúan és részletesen tanulmányozták. Nevezetes tulajdonsága, hogy azalkálifémeket és alkáliföldfémeket kék színnel fizikailag oldja. Valamennyi ammó-niás fémoldat jól vezeti az elektromos áramot. Igen jól oldódnak az alkálifém--halogenidek. Áz oldhatóság azonos anion esetében az NH4+, Li+, Na+, K + sorrendben csökken, azonos kation esetében pedig a F - -tól a I~-ig nő. Az ezüst--halogenidek oldhatósága ugyanígy változik, tehát ellenkező irányban, mintha víz
az oldószer: az AgF nem oldódik, az AgI igen. Jól oldódnak még a nitrátok. Azoxidok, szulfátok, karbonátok kivétel nélkül csaknem oldhatatlanok. A I2, Hgl2,Cu (N0 3)2 és Pb(N 03)2 oldódás közben kémiai reakcióba lép az ammóniával. Szervesvegyületek általában jobban oldódnak benne, mint alkoholban, éterben vagy vízben.
Néhány egyéb nitrogéntartalmú vegyület, pl. a N2H4, NHaOH, HN03 ésNOC1, valamint a PC13, P0C13, SbCl3 ugyancsak ismert oldószerek.
50.7. Szénhidrogének
A benzin és petroléter forráspontja és olvadáspontja igen változó. A benzololvadáspontja 5,49 C°, forráspontja 80,12 C°, atoluolé —95,0 C°, illetve 110,8 C°.
Az említett szénhidrogének a legtöbb szervetlen anyagot nem oldják; az Snl4, AlBr3, SbCl3, SbBr3 benzolban és toluolban oldódik. Feltűnő még az AgC104 jelentékeny oldódása toluolban. Tisztításuk desztillációval, szárításuk foszfor-pentoxiddalvagy nátriumdróttal (dróttá préselt fémnátriummal), illetve folyékony kálium—— nátrium-ötvözettel történik.
50.8. Kloroform
A CHC13 (op. —63,5 C°, fp. 61,2 C°) jó oldószere a brómnak és a jódnak,
valamint a zsíroknak és egyéb szerves anyagoknak. Szárítására felhasználható aCaCl2, P20 5, valamint CaS04 és Na2S04. Szigorúan tilos azonban a fémnátriumvagy nátrium—kálium-ötvözet használata robbanásveszély miatt! A szárítóanyagon napokig állni hagyjuk, azután megszűrjük és végül desztilláljuk.
50.9. Szén-tetraklorid
A CC14 (op. —33,8 C°, fp. 76,6 C°) az előbbihez hasonló módon tisztítható.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 141/366
N E M V I ZE 8 O L D Ó S Z E R EK 141
50.10. Szilícium- és ón-tetraklorid
A SiCl4 (op. —67,7 C°, fp. 56,7 C°) és az SnCl4 (op. —33,0 C°, fp. 113 C°)víznyomok hatására is hidrolizál. Gondosan óvni kell a légnedvességtől is. Szárításuk problémája a hidrolízistermékektől (oxidok és sósav) való elválasztássalkapcsolatos.
50.11. Alkoholok
A CH3OH olvadáspontja —97,1 C°, forráspontja 64,7. C°, az C2H5OH olvadáspontja —114,2 C°, forráspontja 78,3 C°. Noha a legtöbb szervetlen vegyületet
a víznél jóval kevésbé oldják, ez az oldó hatás mégis sok esetben jelentős és gyakorlati szempontból fontos. Az alkálifém-halogenidek oldhatósága a kation atomsúlyának csökkenésével, valamint az anionénak a növekedésével nő, tehát legjobbanoldódik a Lil. Az NH3 kivételével a legtöbb gáz jobban oldódik alkoholban, mintvízben, így a HC1, N2, H2, S02, C02 stb. Az említett alkoholok vízzel korlátlanulelegyednek.
Az etilalkohol leggyakoribb szennyezése az acetaldehid. Eltávolítható, ha kb.5 % KOH-dal 8—10 órán át visszafolyatás mellett főzzük, majd a vörösbarnaszínű kondenzációs termékről ledesztilláljuk.
Vízmentesítése egyszerű desztillációval nem oldható meg. A nagyiparban
azeotrop desztillációval készülő, ún. abszolút alkohol még mindig kb. 4 %-nyivizet tartalmaz. Ennek a készítménynek a további vízmentesítése kalcium-oxiddalvagy vízmentes kalcium-szulfáttal néhány napon át végzett rázogatással, végüldesztillációval oldható meg. A kalcium-klorid nem felel meg erre a célra, mertalkoholt köt meg, a kénsav és a foszfor-pentoxid pedig reakcióba lép vele. Azutolsó víznyomokat jóddal vagy etil-bromiddal aktivált fémmagnéziumról valódesztillációval távolítják el. Még aktiválás előtt el kell azonban bontani az esetleges magnézium-nitrid nyomokat, mert különben az absz. alkohol ammóniávalszennyeződik.
Még erélyesebb módja az alkohol szárításának a fém-Na és a magas forráspontú etilészterek (pl. borostyánkősav-dietilészter, fp. 216,5 C° vagy ftálsav-die-tilészter, fp. 298 C°) együttes alkalmazása. 1 liter etilalkoholhoz mintegy 7 g Na és25 g észter szükséges. Vízzel a nátrium hidroxidot képez, a lúg elhidrolizálja azésztert, és etilalkoholon kívül a sav nátriumsója lesz a reakció terméke. A tisztaalkohol az észter fölöslegétől jelentős forráspontkülönbségük alapján desztillációval elválasztható. Az ilyen alkohol nedvszívó, a légnedvességtől is gondosan védenikell.
A metilalkohol—víz-elválasztás ismételt desztillációval is eredményesen megoldható.
50.12. Dietiléter
A dietiléter (op. —116,3 C°, fp. 34,6 C°) igen elterjedten alkalmazott oldószer. Alacsony forráspontja miatt könnyen párolog, gőze gyúlékony, sőt oxigénnel (levegővel) elegyedve robban is. Gyulladási hőmérséklete alacsony, kb. 180 C°, tehátnagyobb izzólámpa felületén, vagy kapcsoló, villany csengő, elektromotor villamosszikrájának hatására is bekövetkezhet a robbanás. Az étergőz nehezebb a levegőnél,

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 142/366
142 O L D Ó S Z E R E K
zárt helyen jelentékeny távolságra „elfolyhat”. A tűzrendészeit szabályok követke
zetes betartása kötelező, különben súlyos szerencsétlenséget okozhat! A tűzveszélynél is kellemetlenebb az éter peroxidtartalma, amely desztillálás
vagy lepárlás közben rendkívül veszélyes robbanást idézhet elő! A peroxidrobbanásveszélye mindenféle éter, sőt dioxán és tetrahidrofurán esetén is fenáll. Ezek fel-használása előtt ajánlatos elvégezni a következő vizsgálatot: 5 ml hidegen telítettvizes benzidinoldathoz 5 ml telített NaCl-oldatot, majd néhány csepp híg FeS04-oldatot (kb. gombostűfejnyi kristály 5 ml vízben) adunk. Kevés peroxidtaxtalmúéter néhány cseppjétől az oldat 1—2 perc alatt megkékül. Azonnali kékülés sokperoxidot jelent. A savanyú kálium-jodidos próba kevésbé érzékeny.
A peroxid eltávolítása a következő módon végezhető. Először savanyú Fe2+-oldattal kimossuk az éterből, és elbontjuk. A mosófolyadék összetétele: 600 gFeS04«7 H20, 60 ml cc. H2S04 és 1100 ml víz, ez kb. négyszeres térfogatú étertisztítására elegendő. Alapos összerázás után választótölcsérrel elkülönítjük a kétfázist, majd az étert szilárd nátrium-hidroxidról ledesztilláljuk. A képződött acet-aldehidet Cr03-oldattal oxidáljuk, majd az étert vízmentes nátrium-karbonátrólújra ledesztilláljuk, végül Na-dróttal szárítjuk. Harmadszori desztillálás után Na-drót fölött °/10-ig töltött barna üvegben hosszabb ideig peroxidmentesen eltartható.Egyébként aldehid jelenlétében, levegő és fény hatására már néhány óra alatt jólkimutatható mennyiségű peroxid képződik benne.
Ha nincs szükségünk vízmentes éterre, FeS04-oldattal érintkezésben peroxidmentesen tárolhatjuk. A peroxid cc. sósavval bepárolva is elbontható.
50.13. Aceton
Az aceton (op. —95,0 C°, fp. 56,3 C°) a sók legtöbbjét nehezen vagy egyáltalánnem oldja. Vízzel korlátlanul elegyíthető, víztartalma természetesen erősen befolyásolja az egyes anyagok oldhatóságát. Tisztítása, vízmentesítése desztillálóval
kezdődhet. A maradék víznyomok eltávolítására erősen lúgos szárítóanyag nemalkalmas, mert gyakran kondenzációt okoz. Vízmentes K2C03 és egyéb részlegesvízelvonószerek használhatók erre a célra.
50.14. Szén-diszulfid
A CS2 (op. —112,1 C°, fp. 45,2 C°) fontos oldószere a foszfornak és sok szervesvegyületnek. Szárítására CaCl2 és P20 5 ajánlható.
50.15. Piridin
Op. —42 C°, fp. 115,5 C°. Bázikus jellegű, nitrogéntartalmú szerves oldószer.Jól oldja a következő sókat: LiCl, LiN03, AgN03, BeBr2, Znl2 és Hgl2. Vízzelminden arányban elegyíthető. Vízmaradványok eltávolítására lúgos tulajdonságúszárítóanyagok alkalmazhatók: KOH, BaO, CaO, K2C03 stb.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 143/366
víz 143
50.16. Tíz
Op. 0,00 C°, fp. 100 C°, e= 81,1 (18 C°), «=4,4 *10"2 (18 C°). A legtöbb laboratóriumi vegyszer tisztasági fokának megfelelő minőségű víz általában a felhasználás helyén készül desztillálás vagy ioncserélő berendezés segítségével. Az ilyen
víznek leglényegesebb szennyezései illékony komponensek, különféle szerves anya
gok, ezenkívül főleg C02 és NH3. További tisztítása újabb desztillációval végez
hető csiszolatos üvegkészülékben, lúgos permanganátról (kb. 3 g NaOH és 0,5 gKMn04/liter). Bőséges elő- és utópárlatot hagyva a víz szénsavmentes lesz, és aszerves anyagok zöme is eloxidálódik. Az ammóniától csak a harmadik desztillálásárán szabadulhatunk
meg. Az adólombikba3 g kálium-hidrogén-
-szulfátot vagy 5 ml, kevés kálium-permanga-
náttal elegyített 20 %-osfoszforsavat teszünk li
terenként. Az ilyen tisz
taságú vizet úgy véd jük az átjutó elektrolitok ellen, hogy a gőz
teret a hűtővel össze- 50.16.1. ábra. Kvarcüvegből készült desztilláló vezetőképes-kötő, függőleges csősza- ségi víz előállításához N
kaszt elektromos fűtés
sel vagy alumínium blokk segítségével kb. 150 C°-ra hevítjük. Az üvegből kioldott
vagy a cseppekkel átvitt végső elektrolitnyomoktól a vizet kvarcból készült berendezésben tisztíthatjuk meg teljesen (50.16.1. ábra). A készülék részei forrasztássalcsatlakoznak egymáshoz. A lombik nyaka egyúttal a túlhevítő. A leszálló Liébig-hű
tőhöz U alakú, vezetőképesség mérésére kialakított túlfolyó illeszkedik. A víz el
tartására csiszolatos burával ellátott kvarclombik szükséges. Speciális célokra pla
tina vagy műanyag (polietilén) edényzetet. használnak. Rövid ideig tartó tárolásra jól kigőzölt jénai üveg is megfelel.
Az ilyen víz már csak kevés oldott C02-ot tartalmaz, s teljesen tiszta, indiffe-rens gáz átvezetésével ettől is viszonylag rövid idő alatt megszabadítható.
Ha különösen kis vezetőképességű vízre van szükségünk, a 80—90 C°-os vízbe20—30 órán át gondosan'szűrt és C02-mentesített levegőt vezetünk, majd az előbbiek szerint kezelt levegő igen lassú átbuborékoltatása közben teljesen kvarcból
készült berendezésből újra ledesztilláljuk egyenesen abba. az edénybe, amelyben
majd felhasználjuk.
r
Irodalom
Weisaberger, A .: Technique of Organic Chemistry, Vol. VIII, Interscience Publishers, New Y ork —London, 1955.
Jan der , G. : Die Chemie in wasseráhnlichen Lösungsmitteln. Springer Veri., Berlin —Göttingen —Heidelberg, 1949.
Audrieth, F . L., Kleinberg, J .: Non-Aqueous Solvents. John Willey, New York, 1953. Nikolski, B. P .: Handbuch des Chemikers, Bd. I., II., III., VEB Veri. Technik, Berlin,
1956-1959.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 144/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 145/366
G Á Z O K E L Ő Á L L Í T Á S A 145
Az előállítandó terméket tartalmazó rendszer redoxipotenciáljának ismeretében a megfelelő elektródok megválasztásával kényelmesen lehet közbenső oxi
dációsfokú termékeket is tisztán előállítani. így pl. Pt-katód alkalmazásával a4- 4 vagy 4- 5 oxidációs állapotú vanádiumsók -f- 3 állapotúvá redukálhatók, mígPb-katódon a redukció tovább megy a + 2 oxidációszámig. A különböző oxidációszámú vanádiumionokat tartalmazó erősen savanyú oldatok (pH= 0) redoxipo-tenciálja:
V0+ + 2 H+ 4- e ^ V 02+ + H20 e0= 1,0 V,
V 02+ + 2 H + + e V3+ + H20 e0= 0,34 V,
V3+ -f- e st V2+ e0= — 0,20 V.
Az alábbiak néhány rövid utalást tartalmaznak az elektrolízis gyakorlati alrkalmazására a szervetlen preparatív munkában.
A részletes részben ezenfelül számos elektrolízises preparatív előállítási mód-,szer kimerítő leírását adjuk.
51.2. Gázok előállítása
Hidrogén (H2). Nagy tisztaságú hidrogén előállítása céljából 30 %-os KOH-oldatot elektrolizálunk megfelelő berendezésben.1
Oxigén (02). íiszta oxigén ugyancsak elektrolízis útján készíthető a hidrogénelőállítására felhasznált berendezésben.2
ózon (03). 12,5 %-os 0 3-tartalmú oxigén 1,08 fajsúlyú kénsavnak nagy anód-áram-sűrűséggel történő elektrolízisével állítható elő (—10 C° alatti hőmérsékleten). Lényegesen jobb kitermelést szolgáltat azonban 40 %-os perklórsavnak —56C°-on végrehajtott elektrolízise.3
Klór (Cl2). Vizes sósavoldat közepes áramsűrűséggel történő elektrolízisével4tiszta, gyakorlatilag oxigénmentes klór állítható elő, ha az oldat sósavtartalma23 %-nál nagyobb.
Fluor (P2), Szabad fluor előállítása KP és HP keverékének elektrolíziséveloldható meg.5Germánium-hidrogén (GeHJ. Gérmánium-dioxid-tartalmú, jéggel hűtött kén-
savas oldat elektrolízisével állítható elő ólomkatód alkalmazásával.6Ón-hidrogén (SnH4). Előállítható igen kis mennyiségben ólom- vagy ónelekt
ródok alkalmazásával katódos redukcióval,7 ha a redukálandó oldat Sn2+-tar-talma csekély.
1 Paneth, F., Peters , K .: Z. physik. Chem. 134 , 365 (1928); Brauer, G.: Z. anorg. alig. Chem. 255, 105 (1947).
2
Paneth, F., Peters, K.: Z. physik. Chem.
134, 365 (1928).
M ole s, E ., Gonzales, F . : J. Chim. physique 19, 310 (1921). L u x , H .: Z. Elektrochem. 48, 213 (1942).
3 Lash , E . I . , Horn beck, R . D ., F u tná m , G. L ., Boé lter, F .: J. Elecirochem. Soc. 98, 134 (1951).
4 Haber , F ., Grinberg, S .: Z. anorg. alig. Chem. 16, 213 (1898).* Gady, G. H .: J. Am. Chem. Soc. 56 , 1431 (1934).
Schumacher , H . J ,: Z. anorg. alig. Chem. 245, 221 (1940).6 Paneth, F ., Rabinovitsch, É .: Bér. dfcsch. chem.'Ges.' 58; 1141 (1925).7 Paneth, F ., Rabinovitsch, E .: Bér. dtsch. chem. Ges. 57 , 1889, 1891 (1924).
10 Ált. és szervetlen kémiai prakt. - 4284/11

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 146/366
146 A Z E L E K T R O L ÍZ IS A L K A L M A Z Á S A P R E P A R A T ÍV C É L O K R A
Antimon-hidrogén (SbH3), 8 g antimon- és 80 g borkősav-tartalmú, 1,7 liter4n H2S04-oldat 0 C°-on végzett elektrolízise közben platina-iridium-katódon képződik.8
Ólom(lV) -hidrogén (PbH4) és bizmut-hidrogén (BiH3). Kisebb koncentrációban ólom-, illetve bizmutkatódon képződik kénsavas oldatok elektrolízise során®.
51.3. Fémek leválasztása
Sóoldatokból katódos redukcióval előállított fémek legtöbbje igen tiszta, különösen C-, S-, P- és N-szennyezéstől mentes.
A nemesebb fémek kiválasztása nem okoz nehézséget. Zn, Cd, Ga, In, TI, Pb, Sb,Ni, Co, Fe, Mn és Cr is leválasztható katódosan vizes oldatból bizonyos körülmények között. Vizes oitrátoldatból Mo, W, Ta és Nb elektrolitikusan előállítható.10
Felhasználva a hidrogén túlfeszültségét higanykatódon, valamint a higanynakaz alkáli- és alkáliföldfémekkel szemben tanúsított jó amalgámképző hajlamát, vizes oldatból történő elektrolízissel az alkálifémek és -földfémek — az amalgámonát — kényelmesen előállíthatók. Más elemeknek, pl. a Si, Al, Ti, Zr, V, W, Th, U-nák vagy a ritkaföldfémeknek az amalgámképzésre való hajlama csekély. Ezérthiganykatódon csak igen rossz kitermeléssel választhatók ki.
Nemvizes oldószerek alkalmazása ritkábban vezet gyakorlatban felhasználható eredményre, de pl. a Li ilyen körülmények között preparatíve előállítható: vízmentes, piridines lítium-klorid-oldat elektrolízisével teljesen tiszta fémlítium nyerhető.11
Egyes nemnemes-férnek, pl. a La, Nd vagy Ce sóik abszolút etilalkoholos oldatából higanykatódra kiválaszthatók. A kapott amalgámból a higany ledesztillálásávala tiszta fémhez juthatunk.12
Alumínium is előállítható fémorganikus vegyületek xilolos oldatából katódosredukcióval.13
Az elektrolízis körülményeinek megfelelő megválasztásával kompakt fémré
teg, illetve por alakú termék előállítása mellett megoldották a fémegykristályokelőállítását is.14
51.4. Katódos redukció fémkiválasztás nélkül
Katódos redukcióhoz többnyire azért alkalmaznak sima felületet, mert azonnagyobb a hidrogén túlfeszültsége. Erre a célra felhasználható pl. a Hg, Pb, TI, Zn,Cd és Sn, illetve amalgámjuk. A redukált oldatok részleges oxidációjának megakadályozása végett a katód- és anódteret célszerű egymástól elválasztani. Erre a célra
8 Reismann, A., Berkenblit, M., Hass, E. C., Oaines, A.: J. Electrochem. Soc. 101, 387 (1954).9Paneth, F,, Nöring, O.: Bér. dtsch. Chem. Ges. 53,1700 (1920); Z. Elektroehem. 26,
452 (1920).10 Yntema, L. F.: J. Am . Chem. Soc. 54, 3775 (1932).11 Müller, R., Hölzl, F., Pontont, F., Wintersteiner, O.: Mh. Chem. 43, 419 (1922). 18 Meints, R. E., Hopkins, B. S., Audrieth, L. F.: Z. anorg. alig. Chem. 211, 237 (1933). 18 Mernél, W Z. anorg. alig. Chem. 269, 52 (1952).14 Erdey-Orúz, T., Kardos, R. F.: Z. phyBik. Chem., Abt. A 178, 255 és 256 (1937);
172, 157 (1935).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 147/366
O X ID Á CIÓ - R E D U K C I Ó 147
többnyire porózus agyagdiafragmát alkalmaznak. Ilyenkor azonban rendszerintkis mennyiségű vas kéri# a diafragmából az oldatba, aminek következtében nem
csak a termék szennyeződhet, hanem a katód szennyeződése folytán a túlfeszültségis csökkenhet. Porózus üvegdiafragma alkalmazásával az említett veszély lényegesen csökkenthető.
A katódos redukció nemcsak hidridek előállítására, illetve szerves vegyüle-tek redukciójára alkalmas, hanem alacsonyabb oxidációfokú fémsók előállítására is, amilyen pl. az NH4V(S04)a*12 HaO (lásd 64.9.) és az (NH4)a(S04)2* 6H jO(lásd 64. 10.).
Katódos redukcióval állítják elő a ritkaföldfémek háromértékű, oldható szulfátjainak keverékéből pl. az EuS04-ot és az YbS04-ot a kétértékű szulfátok kis oldhatósága alapján. A redukciót kénsavas közegben higanykatód alkalmazásával
végzik.15
51.5. Anődos oxidáció
Amennyiben nem magát az anód anyagát akarjuk oxidálni, anódként sima platinát vagy platina-irídiumot alkalmazhatunk, mely az oxidáló hatásnak viszonylag jól ellenáll, illetve amelyén az oxigén jelentős túlfeszültséggel fejlődik. Kénsavas közegben ólomanód is használható. Grafiton kicsi az oxigén túlfeszültsége, kénsavasoldatban anódként gyorsan korrodeálódik, halogén-hidrogénekben azonban alkalmazható.
Anódos oxidációt használunk a részletes részben leírt, alábbiakban felsorolt ve-gyületek előállítására is: Cu20, (NH4)2S20 8, KC103, KC104, Co2(S04)3*18 HaO.
51.6. Olvadékelektrolízis
Az olvadékelektrolízis módszere a nagyiparban gyakran használatos, laborató"riumi méretekben azonban általában kényelmetlen és a kedvezőtlen kitermelés miatt nem szívesen használt eljárás. A Na,'K, Be, Mg, Ca, A1 előállítása olvadékélekt-rolízis útján csak ritkán alkalmazott a laboratóriumi technikában, s csupán a mód
szer megismertetése céljából szerepel a legtöbb praktikumban. Nagy gyakorlati jelentősége van ezzel szemben a tiszta Li, Ta, Th, U, La és más ritkaföldfémek laboratóriumi előállításának.
Elektrolitként általában halogenidolvadék használatos, különösen némelyfluoridol vadék, amelyben a legtöbb oxid igen jól oldódik. A kloridolvadékok elektrolízisét lehetőleg alacsony hőmérsékleten célszerű végrehajtani, illékonyságukmiatt. Ez a hőmérséklet rendszerint kisebb az előállítandó fém olvadáspontjánál,aminek az a hátrányos következménye, hogy a termék nem regulusként gyűlik ösz-sze az előállítás során.
A kitermelést gyakran befolyásolja a fémek oldhatósága a sóolvadékban, így
pl. a BaCJa elektrolízise közben termelődő fémbárium jelentékeny része komplexetképezve újra feloldódik az olvadékban. Megjegyzendő, hogy alkáli-halogenid hozzáadásával a legtöbb fém oldhatósága jelentős mértékban csökkenthető. A rossz kitermelés egyik oka lehet az, hogy a katódon leváló fém részben az olvadékba kerül,
16 Kapfenberger, W.: Z. anal. Chem. 105,' 199 (1936); Z. anorg. alig. Chem. 238> 2n5 (1938);
Brukl, A.: Angew. Chem. 50, 25 (1937).
10

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 148/366
148 A Z E L E K T R O L ÍZ IS Á É K A 'l M A Z Á S A P R E P A R A T Í V C É L O K RA
diffúzió és keveredés folytán az anódhoz jut és újra oxidálódik. Ennek megakadá
lyozására a katód- és anódtéret célszerű alkalmas módon elkülöníteni, amit U alakúkorundedény vagy elektródvédőcső (lásd 54.3.2.) alkalmazásával érhetünk el. Mivelaz oldhatóság és a diffúzió sebessége a hőmérséklettel emelkedik, a hatásfok annálkisqbb, minél magasabb a hőmérséklet. Az áramsűrűség csökkenésével szintén
csökken a hatásfok.Sóelegyek elektrolízisének hatásfokát áz is csökkenti, ha kétféle fém válik le a
katódon, és az egyik gyorsan oxidálódik. így ph az Al elektrolitikus előállítása köz
ben az A120 3 és Na3[AlE6] elegyének olvadékából bizonyos körülményék között akatódon A.1 mellett Na is kiválik, mely azonban a levegőn oxidálódik. A katódon levált fémnek az oxidációját azzal is csökkenthetjük, hogy az anódot szintén védőcső
vel látjuk el, amelyen ét gyakran a mérgező ánódgázokat is (pl. Cl2, CO vagy fluorve-gyületek) leszívatjuk. Kívánatos, hogy az előállítandó fém olvadáspontja feletti, deminél alacsonyabb hőmérsékleten elektrolizáljunk, ezért olyan sóelegyet válasszunk,
melynek alacsony az olvadáspontja, és az előállított fémet is csak kismértékben
oldja. A laboratóriumi méretekben végrehajtott olvadékelektrolízishez általában por
celán, üveg-, korund- vagy grafittégely használatos. A sókeveréket megfelelő tégely
ben megolvasztjuk, majd réz- vagy alumínium lapra öntjük. Kihűlés után durván
elporítjuk, és az így kapott anyagot, használjuk fel az elektrolízishez. Ennek az azelőnye, hogy az elektrolízis megindítása előtt az eutektikus hőmérsékletet nem kell
sokkal meghaladni, és ily módon csökken a tégely korróziós veszélye. Sókeverékek
alkalmazásánál figyelembe kell venni, hogy a különböző sók bomlási feszültsége kü
lönböző mértékben változik a hőmérséklettel, és ennek következtében a hőmérséklet változtatásával megváltozhat a sók bomlási feszültségének sorrendje is. Arány
lag alacsony hőmérsékleten pl. a lítium-halogenidek bomlási feszültsége kisebb,-mint a kálium-halogenideké, magasabb hőmérsékleten viszont fordított a sorrend.
Ezt a lítium előállításakor figyelembe kell venni. A nagyobb áramsűrűséggel való elektrolízisnek határt szab az ún. anódeffektus.
Ha a fejlődő gáz nem tud elég gyorsan eltávozni az anódról, hanem majdnem telje
sén összefüggő rétegben hártyaszerűen veszi körül azt, az elektrolit csak egyes pon--tökon érintkezik :az anóddal. Ilyenkor ezeken a pontokon az áramsűrűség igen nagyra nő, és az áramátadás nagyfeszültségű ív alakjában következik be. Az anódeffek-
tús bonyolult jelenség, melyben szerepe van az anód és az elektrolit felületi feszültségének, az elektrolit viszkozitásának stb. Ezért a kritikus áramsűrűség, amelyen
az anódeffektus létrejön, nagymértékben függ az elektród és elektrolit minőségétől.Legnagyobb a kritikus áramsűrűség; az alkálirhidroxid-oldatokban (20 amp/cm2nagyságrendű), legkisebb a fluoridoldatokban (grafitanódon kb. 0,5 amp/cm2).
Az anódeffektus hátrányos, mert gátolja a nagyobb áramsűrűséggel való elektrolí
zist, ezért a gyakorlatban lehetőleg olyan adalékokkal készítik az olvadékot, amelyek megnehezítik az anódeffektus kialakulását; ilyenek pl. a különböző fém-oxi-
áok. , A fémlítium, fémkalcium és'fémmagnézium olvadék-elektrolízis útján történő
, előállításának leírása a részletes részben található.
A fémurán előállítására olyan olvadék használatos, amely 1-1 rész NaCl-ból ésrC}aCl2rból áll, és kb, 6 % KjTJF0]-ot tartalmaz.16 75Q C°-on molibdénkatódon a fém-
16 Driggs. F~ H., Lilliendahl, W. G.: Ind. Engng. Chem. 22, 516 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 149/366
O L V A B É K E L E K T R O L ÍZ i a 149
urán szürke, szivacsos maöszíaként válik le. A gráfittégely anódként ázoígál. A nyers
terméket vízzel, ecetsavval, majd alkohollal mosva, tiszta fémporhoz jutunk, amelymár szinterezésre felhasználható. Fém-Gd és -La előállítására KC1, LiCl és GdCl3
(illetve LaCl3) keverékéből készült olvadékot használhatunk, katódként pedig meg
olvasztott kadmiumot.17 A kapott, kb. 6 %-os ötvözetből a kadmium nagyvákuumban desztillálással eltávolítható.
Irodalom
MüUer , E . , Rev i her , H . : Elektrochemisches Praktikum. Th. Steinkopf, Dresden —Leipzig, 1960.
MüUer , R . : Elektroohemie nichtmetallischer Stoffe. Springer Veri., Wien, 1937.Bár t fa i B . : Galvanizálók zsebkönyve. Műflzak^KönyVkiadój-Budapest, 1968.
Erdey-Grúz T., Prosti, J .; Fizikai -kémiai praktikum I. —H. TankönyVkia dó, Budapest, 1965. <.
Krause, H.: Galvanotechnik. 13. Aufl. Jönecke, Leipzig, 1952.
í.
A -jv Kf" ’
17 Trombe, F.: Bull. Soc. Chim. Francé 2, 660 (1935); Ann. Chimie 6, 443 (1936).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 150/366
62. Félm ikro- és mikropreparatív munkam ódszerek
A preparatív feladatok megoldásában a kis anyagmennyiségek használata egyre inkább tért hódit. A félmikro- és mikropreparatív munkamódszerek bevezetését
és általános gyakorlatát számos tényező tette indokolttá. Lényeges és általános
szempont a jelentős idő-, anyag- és munkahely-megtakarítás. E módszer használa
tát különösen előnyösen értékesíthetjük a több lépésből álló szintetikus munkák
nál, mivel az előzetes tájékoztató kísérleteket mikrodimenzióban végezve, nem koc
káztatjuk az esetleg hosszadalmas munkával előállított és a szintézis közben úgy
is egyre csökkenő mennyiségű köztitermékeket. Az izotóp elemek felhasználásávalvégzett preparatív munka során — tekintettel az anyagok magas árára, radioaktív
izotópok esetén az esetleges sugárveszélyre is — úgyszólván nélkülözhetetlen a kis
anyagmennyiségekkel való munka. A mikropreparatív munka nevelő hatása isemlítést érdemel. A vegyész, ha a szokásos anyagmennyiségekkel elérhető ered
ményt mikrodimenzióban is biztosítani kívánja, kénytelen lényegesen gondosabban
és körültekintőbben dolgozni. Ez a pontosság bizonyos idő múlva megszokottá vá
lik, ami az általános preparatív munkában is érezteti a hatását. A preparatív kémiában használatos makro, félmikro és mikro megjelölés
mennyiségi tartalma eltér az analitikában használatostól. A preparatív munkában
1 grammnál több reakcióba vitt anyagmennyiség esetén makro-, 0,1 —1 g anyagfelhasználás esetén félmikro-, 0,01 — 0,1 g anyagfelhasználáskor mikroméretről
beszélünk. A mikropreparatív technika úttörője F. Emich volt, aki 1911-ben „Lehrbuch
dér Mikrochemie” című, könyvében összefoglalta és továbbfejlesztette az addigi
ismereteket, és meghatározta a módszer elvi alapjait. Napjainkban már számos,
főleg újabb kiadású kézikönyv foglalkozik evvel a területtel; ezeknek egy részét efejezet irodalmában soroljuk fel. A preparatív mikro- és félmikrotechnika sokféle
képpen és különböző készülékekkel alkalmazható, de eredményes és széleskörű be
vezetése csak egyszerű és különösebb anyagi befektetés nélkül realizálható mód
szer esetén remélhető. Az e fejezetben tárgyalt technika ennek a gyakorlati elvnek
a megvalósítására irányul.
52.1. Laboratóriumi alapműveletek
, A kémiai preparatív gyakorlatban a legegyszerűbb laboratóriumi műveletek
pl. az'anyagok porítása, oldása, bepárlása, kristályosítása mikro- és félmikro-di-menzióban nemcsak az alkalmazott eszközök mérete szempontjából különbözik a
szokásostól, hanem gyakran elvileg is.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 151/366
LÁB ORATÓRIUMI ALAPMŰ VELETEK 151
Az oldás sorén a mikrotechnikában úgy járunk el, hogy tiszta oldószerrelszobahőmérsékleten telítetlen oldatot készítünk. Szükség esetén szűrünk, és az ol
datot ezt követően a melegen való telítettségig bepároljuk. 'Erre elsősorban a kisfolyadékmennyiségek csekély hőkapacitása miatt van szükség, aminek következ
tében a forrón telített oldatokban idő előtti, esetleg szűrésközben bekövetkező kristályosodás állhat elő. Az oldáshoz az
52.1.1. ábrán jelölt méretezésű rövid, de viszonylag széles
kémcsöveket vagy centrifugacsöveket alkalmazunk. A poritást
15 7,5 ÍO
LVméretek mm-ben
52.1.1. ábra. Kémcsövek és centrifuga- csövek méretezése
10cm
L ű.
52.1.2. ábra. Melegítés visszafolyós
hűtővel
lehetőség szerint abban az edényben végezzük, amelyben az oldást vagy más, soronkövetkező műveletet akarunk végrehajtani; ezáltal az anyagveszteség a minimumra csökkenthető. A porításhoz leolvasztott vagy az edény alakjához formált végű
üvegbotot alkalmazunk. * Az oldatok melegítése vízfürdőben, vagy — kellő óvatossággal — mikroláng
felett történhet. Vízfürdőként Erlenmeyer- -lombik (lásd 52.1.2. ábra) használható,amennyiben azonban több kémcsőben vagy
esetleg hoszabb ideig kívánunk melegíteni,úgy az 52.1.3. ábrán vázolt kristályosítócsé
széből álló, tartóálvánnyal és túlfolyóval el
látott egyszerű berendezést alkalmazunk.Szükség esetén a kémcsövet parafadugós
vagy normálcsiszolatos csatlakozással gömbös kiképzésű visszafolyós hűtővel látjuk el(52.1.2. ábra). Alacsonyabb forráspontú oldó
szerek (90 C° alatt) alkalmazása során a visz-
szafolyós hűtőt megnedvesített szűrőpapírral tekercseljük be. Ha egyes anyagok lassú
oldódása miatt az oldatot huzamosabb ideig 52.1.3. ábra. Túlfolyó egyszerű megkell forralnunk, akkor — különösen a na- oldása

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 152/366
152 FÉLMI BÍRÓ- ÉS MIKRO FREPA RATÍV MUNKAMÓDSZEREK
gyobb méretű kémcsövek használata esetén — eredményesen alkalmazhatjuk az52.1.4. ábrán bemutatott merülő hűtőt.
Az oldatok szűrését csak indokolt esetben végezzük el, , tehát csak akkor, ha azoldat szemmel láthatóan szilárd makroszkópos vagy kolloid méretű szennyeződéseket tartalmaz. Oldatok és folyadékok transzport(Uására,va,gyÍB egyik edényből amásikba történő kvantitatív átvitelére igen gyakran van szükség. E művelet egyiklegfontosabb alapeszköze a mikrotechnikábah a szívógömb, amelynek készítése igenegyszerű, némi gyakorlatot és egy üvegtechnikai fúvólángot igényel. Az 5—10 mmátmérőjű, normál falvastagságú, alacsony olvadáspontú üvegcsövet széles
' fuvólángban kb. 1 mm átmérőjű kapillárissá
52.1.4. ábra.Merülő hűtő 52il.5. ábra. Szívógömb készítése
méretének megfelelően a cső leszűkített részétől 10—40 mm távolságban hasonlóméretű másik kapillárist húzunk le (52.1.5. ábra). Ezután mindkét kapillárist10 —15 cm hosszúságúra levágjuk, az egyik kapillárist leforrasztjuk, majd a cső lenem szűkített középső részét erősebb fúvólángban meglágyítjuk, és forgatás köz
ben gömb alakúvá formáljuk (a leforrasztott végű kapillárist szükség esetén lerövidítjük). A megfelelő falvastagságú szívógömb 30—40 cm magasságból falapraejtve nem törik össze. A szívógömb méretezését a transzportálásra kerülő oldatmennyiségéhez szabjuk (térfogata az oldat térfogatának kb. kétszerese legyen).Használatának elve az, hogy az egyik végén leforrasztott szívógömb felmelegítésével, majd azt követő lehűtésével csökkentett nyomást állítunk elő, s ennek segítségével a gömbbe folyadékot szívhatunk fel.
Gyakori feladat pl. besűrített oldatok viszonylag csekély maradékának kvantitatív összegyűjtése és átvitele egy másik edénybe vagy kémcsőbe. Ilyenkor azedénybe 1—2 ml oldószert viszünk be, és azt vízfürdővel vagy mikrolánggal nagyrészt elgőzölögtetjük; a gőzök a lombik falán kondenzálva, az odatapadt anyagotaz edény aljára mossák. A műveletet néhányszor megismételve elérhetjük, hogy azanyag az edény legmélyebb pontján az oldószerben összegyűljék. Ezt követően azoldószer mennyiségéhez méretezett szívógömböt — miután a rövidebb kapilláristleforrasztottuk — kissé felmelegítjük, és lehűlés közben a tiszta oldószerből keveseta nyitott kapillárison át a gömbbe szívatunk. Ezután a szívógömbben levő oldószert enyhe melegítéssel ismét elgőzölögtetjük — miáltal a levegő nagy részét kiszorítjuk a gömbből —, majd a szabad kapilláris végét az. edény legmélyebb pont-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 153/366
L A B O R A T Ó R I U M I A L A P M Ű V E L E T E K 153
jára, a folyadékfelszín alá helyezzük. A gömbben lehűlő gőzök kondenzálódnak, akeletkező vákuum következtében az oldat felszívódik. Szükség esetén a szívógömb
be történő átszívatás közben az edényt kevés oldószerrel mossuk. A megtöltöttszívógömb tartalmát enyhe melegítéssel lehet átfolyatni a kívánt edénybe. Azedény és a szívógömb kimosása végett a műveletet kevés oldószerrel néhányszormegismételjük. Ezáltal biztosíthatjuk, hogy az áttöltés kvantitatív legyen.
Amennyiben az anyagot magában a szívógömbben kívánjuk tárolni, célszerűszakaszos felszívatást alkalmazni. Ilyenkor a részben megtelt szívógömböt nyitott
52.1.6. ábra. Bepárlás
52.1.7. ábra. Bepárlás inertgáz-atmoBzfórában,
illetve nedvesség kizárása mellett
végével felfelé fordítjuk, megvárjuk, míg a felső kapillárisból az oldat teljesen lefolyik, a gömbben levő oldatot óvatosan felmelegítjük, és az edényt hirtelen megfordítva a visszamaradt oldatot vagy a mosófolyadékot kiszívjuk. Szükség eseténa műveletet többször megismételjük. Alacsony forráspontú oldószerrel ez a szakaszos felszívatás nagy gyakorlatot igényel. Az átmosás befejezése után a szívókapillárist is leforrasztjuk, így az oldatot tulajdonképpen ampullázva tároljuk.
A mikropreparatív munka során az anyagok oldásakor általában szobahőmérsékleten telítetlen oldatokat készítünk. Ezért gyakran van szükség megfelelő töménységre történő hepárlásra. A lepárlásnak egyszerűen kivitelezhető módja, ha akémcsőben az oldat felett levő gőzöket az 52.1.6. ábrán bemutatott berendezéssegítségével elszívatjuk. Az elszívóberendezés kapillárisának végét kb. 1 cm-re azoldat felszíne felett helyezzük el, az egyenletes forrást forráskönnyítő kapillárissalbiztosítjuk.
Olyan anyagok bepárlását, amelyeknek a levegő nedvesség-, szén-dioxid- vagyoxigéntartalma árthat, az 52.1.7. ábrán vázolt egyszerű berendezéssel, megfelelőkapacitású szárítórendszer eléje iktatásával vagy nyomásbiztosítással stabilizáltindifferens gázáramban oldhatjuk meg.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 154/366
154 F É L M I K R O - Í S M I K B O P B E P A B A T Í V M t TN K A M Ó D SZ E RE K
A mikropreparatív technikában a kristályok elkülönítésének módja a kristályos anyag minőségétől és mennyiségétől függ. A kis anyagmennyiségekből eredő
viszonylagosan nagy műveleti anyagveszteségek elkerülése érdekében azt a módszert érdemes előnyben részesíteni, amelynek során a kristály—anyalúg-szuszpen-ziót nem szükséges az eredeti edényből átönteni. Ez viszonylag könnyen megoldható az ún. fordított szűrés alkalmazásával, amit többféleképpen végezhetünk el.Jól fejlett, gyorsan ülepedő kristályok esetén igen jól használható a szívógömb-
52.1.8. ábra. Fordított szűrés Emich- -féle szűrő bottal
technika. Ilyenkor a szívógömb kapillárisának végét finomabbra húzzuk ki, és azanyalúgot a kristályok közül az előzőekben ismertetett módon felszívjuk. Ameny-nyiben szűrés közben a kristályok elzárnák a kapilláris végét, úgy azokat a szívógömb óvatos forgatásával eltávolítjuk a kapilláris végéről, ügyelve arra, hogy aművelet közben levegő ne jusson a gömb belsejébe. Az anyalúg kiszívatása útánnéhány csepp oldószerrel lemossuk a kristályokat, és a mosófolyadékot is felszívatjuk.
A fordított szűrés végrehajtásához jól használhatók az Emich-íé\e szűrőbotokis, amelyek 2—5 mm szárátmérővel és 3—6—9 mm korongátmérővel, zsugorítottüvegszűrő betéttel (G2, G3) készülnek. Az 52.1.8. ábrán vázolt egyszerű berendezéssel az anyalúg (mosófolyadék) összegyűjtése is könnyen lebonyolítható. A szívatás sebessége az ábrán feltüntetett csappal szabályozható.
Kis mennyiségű anyagok esetén jól alkalmazható a mázatlan porcdán lapon való szűrés. A kristályokat az anyalúggal együtt cseppentővei a porcelán lapra visz-szük, amely az anyalúgot magába szívja. A szilárd maradékot mikrospatulával alap még eddig nem használt részére kaparjuk, ott kinyomkodjuk, majd egy másikhelyen minimális oldószerrel megint lemossuk. A porcelán lapnak természetesen

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 155/366
D E S Z T I L L Á L Á S — S Z U B L I M Á L Á S 155
megfelelő porozitásúnak és keménységűnek kell lennie. Amennyiben a kristályokon kívül az anyalúgban oldott anyagra is szükségünk van, úgy az a porcelán lap
megfelelő részének oldószeres extrahálásával megoldható. A félmikro-preparatív munkában jól használhatók az analitikában bevált
préaszűrők is. A présszűrő (52.1.9. ábra) összeszűkülő végébe kevés vattát rakunk,fémspatulával vagy vékony üvegbottal megtömködjük. A szűrendő folyadékot azígy előkészített présszűrőbe öntjük. Ha az oldat lassan szűrődik, a körtefecskendővel átpréseljük. A préselés végén a pumpát kezünkkel Összeszorítva tartjuk, éscsak akkor engedjük el, amikor kiemeltük a présszűrőből. Ellenkező esetben a fellépő szívóhatás következtébena gumifecskendő felszippantja
a csapadékos vattát. Ameny-nyiben a vattán levő kristályos anyagot átkristályosításvagy továbbfeldolgozás céljából fel kívánjuk oldani, ’a prés-szűrőt megfelelő mennyiségűoldószert tartalmazó kémcsőbe helyezzük, és a kémcsövetmikrolánggal vagy vízfürdőnmelegítjük.
Félmikro dimenziókbanalkalmazhatjuk az 52.1.10.ábrán vázolt szívókémcsövetis. A 10—15 mm átmérőjű tölcsér szárába vékony, a felsővégén lapított üvegbotocskát helyezünk, amelynek fejére 7—8 mm átmérőjű köralakú szűrőpapír lapot teszünk. A kristályos anyagot tartalmazó anyalúgot cseppen-tős kapillárissal vagy szívógömbbel fokozatosan visszük a szűrőbe oly módon, hogy akapillárist a kör alakú szűrőpapír lap közepéhez érintjük. Egyidejűleg az oldal-
csövön át az edénykét enyhén megszívatjuk. A kristályokat át is moshatjuk úgy,hogy a mosófolyadékot cseppenként a szűrőbe visszük, majd leszívatjuk. Az anyag szárítását kémcsövekben, szilikagél felett egyszerű, dugóval zárható
edényben végezhetjük. Vákuumszárításra normálcsiszolatos, csappal ellátott nagyobb kémcsövet alkalmazhatunk (52.1.11. ábra).
52.2. Desztillálás, szublimálás
A mikro- és félmikroméretekben végrehajtott desztillálások során a kis folyadékmennyiségek csekély hőkapacitása következtében általában nincs szükség víz
hűtéses kondenzálásra. Az 52.2.1. ábrán egy desztiUációs hűtőcső méretezése látható. Az 52.2.2. ábrán
vázolt egyszerű berendezés szobahőmérséklet körül dermedő anyagok desztillá-lására alkalmazható, míg magasabb dermedéspontú anyagok esetében előnyösenhasználható az 52.2.3. ábrán vázolt — csiszolatos hőmérővel ellátott — mikrodesztilláló.
Viszonylag alacsonyabb forráspontú (4- 80 C° alatti) folyadékok desztillálásáraaz 52.2.4. ábrán bemutatott, hűtőujjal ellátott desztilláló berendezést ajánljuk,
gumicso-
SZ.1.11. ábra. Vákuum- exszikkétor

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 156/366
156 F É L M I K R O - É S M I K B O F B Q F A B A T Í V M U N K A M Ó D S Z E R E K
52.2.3. ábra. Mikrodésztilláló magas forráspontú anyagokhoz
52.2.4. ábra. Mikrodesztilláló alacsony forráspontú anyagokhoz

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 157/366
M Ű V E L E T E K O X I G ÉN R E É R Z É K E N Y A N Y A G O K K A L 157
amely egyben csökkentett nyomáson végrehajtott defíztillációra is alkalmas. Kis-mennyiségű anyagok frakcionált desztillációjához az Ellis—TFet/ crod-féle1 mikro-
frakcionáló berendezés használható (52.2.5. ábra). Utóbbi lehetővé teszi a konden-zátumnak úgyszólván cseppenkénti szétválasztását az alkalmazott forgatható csiszolatos szedő segítségével. A hűtőujj bemerülési mélységének szabályozásával
ÓH.2.5. ábra. EUis- Weygand-féle mikro- frakcionáló
52.2.6. ábra. Mikro- szublimáló
a hűtési effektus kényelmesén változtatható. A berendezés vákuumdesztillációra,valamint indifferens gázáram alkalmazásával végrehajtott desztillációra is használható.
A ntikroszublimáló berendezések főként méretezés szempontjából különböznek a
makrodimenziójú szublimálóktól. Az 52.2.6. ábrán igen egyszerű kivitelű mikro-szublimálót mutatunk be, amely vákuumban és inertgáz-atmoszférában történőszubliinálásra egyaránt alkalmas. Az edény alján helyezzük el a szublimálandóanyagot, majd föléje üveggyapotot; a szublimátum az edény középső részén rakódik le. Szükség esetén az edény falát nedves szűrőpapír csíkkal hűtjük.
52.3. Műveletek oxigénre érzékeny anyagokkal
A szervetlen kémiai preparatív laboratóriumi munka területén számos oxigénnel, vízgőzzel szemben érzékeny anyaggal találkozunk. Ilyen pl. a hidrideknagy része (alkálifémek, bór, alumínium stb. hidridjei), a kloridok, bromidok és
jodidok többsége, valamint számos fémorganikus vegyidet". Az oxigénre érzékeny anyagokkal végzétt műveletek során — tekintette] arra,
hogy azok sok esetben szobahőmérsékleten is piroforos tulajdonságnak — a veszély
1 Weygand, C.: Organisch-chemieche Experimentierkunst. Leipzig, J. A. Barth, 1938, 112. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 158/366
158 F É L M I K R O - É S M I K R O P R E P A R A T ÍV M U N K A M Ó D S Z E R E K
csökkentése érdekében célszerű a nagyobb anyagmennyiségek használatát kerülni. A fokozott szennyeződési veszély miatt nem ajánlatos mikromennyiségek alkal
mazása sem. Fentiek miatt az oxigénre érzékeny anyagokkal végzendő műveleteksorán legmegfelelőbb félmikro- (0,1—1,0 g) mennyiségeket alkalmazni. A félmikro-technika gazdaságossági szempontból is kívánatos, tekintettel a nagy tisztaságú
inért gázok magas árára. Az oxigénre érzékeny anyagokkal
folytatott kísérleti preparatív munkasorán gyakran használatosak a mani
pulációs szekrénykék. Ezek aránylagegyszerűen házilag is elkészíthetők.
Az 52.3.1. ábrán két darab csapos kivezetőcsővel és beépített gumikesztyűkkel ellátott, felül nyitható, részbenüvegfalú fémdoboz látható, amelybena műveleteket kézzel elvégezhetjük. A manipulációs szekrénykék előnye,hogy azokban a műveletek — inertgáz--atmoszférában — a szokásos berendezések segítségével hajthatók végre.
Hátránya viszont, hogy a manipuláció eléggé nehézkes, és figyelembevéve a kis
anyagmennyiségekkel kapcsolatos munka során megkívánt precizitást, a műveletekhez nagy gyakorlat és figyelem szükséges.
Az oxigénre érzékeny anyagokkal kapcsolatos preparatív munka végrehajtásának másik lehetősége, hogy a műveletekhez alkalmazott berendezéseket — azinertgáz-atmoszféra igényének megfelelően — módosítjuk. Ez esetben az oxigénre érzékeny anyagok áttöltését is meg kell oldani.
Az oxigénre érzékeny anyagokkal végzett munkában az elsődleges feladat azinertgáz-atmoszféra biztosítása. Inertgáz használata esetén gyakran célszerű inert-
gáz-vákuumrend8zer alkalmazása, melynek sematikus vázlata az 52.3.2. ábrán lát-
52.3.1. ábra. Manipulációs szekrény
52.3.2. ábra. Inertgáz-vákuumrendszer

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 159/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 160/366
160 F É L M I K R O - É S M I K R O P R E P A R A T Í V M U N K A M Ó D S Z E R EK
tű oxigénmentesítése az 52.3.5. ábrán feltüntetett, két végén gumisapkával zártüvegcsövecskével kényelmesen megoldható. Az A jelű tűn keresztül a csövecskétinertgázáram-rendszerbe kötjük. Amennyiben az injekciós edény KPG-csiszolatanem vákuumzáró, úgy enyhe szilikonzsíros kenés alkalmazható. Az injekciós tűnek
52.3.5. ábra. Injekciós tű és edény étöblítése inért gázzal
és tartálynak nitrogénnel történő alapos átoblítése után az A jelű tűcsatlakozás
kihúzható. A szintén gumisapkával zárt edényből a kívánt mennyiségű anyagot az52.3.6. ábrán feltüntetett módon felszívjuk, majd a mintavevő injekciós tű végétújra a nitrogénnel töltött gumisapkákkal zárt csövecskébe húzzuk vissza. Az anyagígy szennyeződés veszélye nélkül juttatható a gumisapkával zárt munkatérbe.
Nagyobb mennyiségű, oxigénre érzékeny folyadék transzportálása inertgáz--atmoszférájú adagolóbürettából az 52.3.7. ábrán látható feltéten át történhet.
52.3.6. ábra. Folyadék áttöltése injekciós módszerrel
52.3.7. ábra. Folyadékok áttöltésére alkalmas inertgóz-atmoszférét biztosító feltét
A büretta gumidugós tömítéssel csatlakozik a csapos feltéthez, utóbbi pedig nor-málcsiszolattal a reagensedényhez. A feltét inertgáz-vákuumrendszerrel van össze
kötve. A piroforos tulajdonságú anyagok, pl; az alumínium-alkilek raktározása is
megköveteli aZ> óvatossági rendszabályok betartását. így pl. nagyobb mennyiségtárolása esetén az inertgáz-atmoszférájú, vastag falú üvegedényeket homokba kellbeágyazni. Piroforos tulajdonságú anyagokat már 100 grammnyi mennyiségekesetén inertgáz-atmoszférájú edényben tároljuk, és az edényt szintén oxigénmentes, nitrogénnel töltött PVC-zsákba zárjuk, majd homokot tartalmazó fa- vagyfémládába helyezzük.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 161/366
MŰVELETEK OX IGÉNKE ÉRZÉKENY ANY AGOKKAL 161
Az oxigénre érzékeny anyagokkal kapcsolatos preparatív munkához használható reaktoredényeket és egyéb berendezéseket természetesen az egyes feladatok
speeiális'jellege szabja meg. Félmikro- és mikrodimenziókban a reaktoredényekbentörténőkeverést általában mágneses keverő segítségével oldjuk meg, amint az az52.3.8. ábrán látható. E reaktorok inertgáz-atmoszférában végzendő munkára isalkalmasak.
52.3.8. ábra. Mágneses keverővei ellátott mikroreaktor-edények
Megjegyezzük, hogy e rövid fejezetben a félmikro- és mikrotechnikának csupánnéhány általánosan használatos alapműveletére tértünk ki, és a speciális problémák részleteire vonatkozóan utalunk a megfelelő szakirodalomra.
i
Irodalom
E m ich, F . : Lehrbuch dér Mikrochemie. Bergmann, München, 1911. E ric h, F., Schneider, F .: Microchemical Laboratory Manual. John Wiley, New York, 1932. Lieb, H., Schöniger, W.: Anleitung zűr Darstellung organischer Praparate mit kleinen
Substanzmengen, Springer, Wien, 1950.MaAH poeK. J l . : KaMecTBeHHuümhkpoxhmHhcckhíí aHajina. H3flaTejibCTB0MOCKOBCKoroyHH-
v B ep cu re T a, MocKBa, 1951.Koff ler , L ., K off le r, A.: Thermo-Mikromethoden zűr Kennzeichnung organischer Stoffe.
Verlag Chemie, Berlin, 1954.Cheronis, N . D . : Micro and Semimicro Methode. (A. Weissberger: Technique of Organic
Chemistry-sorozat VI. kötet) Interscience Publishers, New York, 1954.Kovács ö . : Szerves preparatív munka kis anyagmennyiségekkel. Magyar Kémikusok Lap
ja 13, 61 és 258 (1958).de Liefde Mejjer, H. J ., Janssen , M . «/., van dér Kerk, G. J . M . : Studies in the Organic
Chemistry of Vanadium. Institute fór Organic Chemistry, Utrecht, 1963.
11 Ált. és szervetlen kémiai prakt. —4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 162/366
RÉSZLETES RÉSZ

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 163/366
53. Nemfémes elemek
53.1. Hidrogén (H2)
A hidrogén laboratóriumban kényelmesen előállítható hígított sósav (1:1)és cink reakciójával ifípp-készülékben. Megfelelő tisztaságú vegyszerekkel tisztahidrogént kapunk. A hidrogénfejlődés gyorsítása végett a sósavhoz kevés réz
vagy kobaltsót adunk. Ha a cink nem teljesen tiszta (szulfid-, arzén-, foszfid-
vagy karbidtartalmú), akkor a hidrogén a következő anyagokkal szennyeződhet:H2S, A sH3, foszfor-hidrogének, szénhidrogének, esetleg Na-, 0 2-, C02-nyomok.Ilyenkor a hidrogént kémiai tisztításnak vetjük alá. A gázt tömény (kb. 50 súly %-os) kálium-hidroxid-oldattal töltött mosón, majd semleges, telített KMn04-olda-
ton vezetjük át. Az oxigénnyomokat 300 C°-on palládiumazbeszt katalizátorsegítségével köthetjük meg. Végül a gázt szilárd KOH vagy P205 töltetű tornyok
ban szárítjuk. Szárítóanyagként tömény kénsavat nem célszerű használni, merta hidrogén a tömény kénsavat kismértékben redukálhatja, ami a gáz szennye
ződéséhez vezet. Igen tiszta hidrogén állítható elő 15 %-os kálium-hidroxid (vagy
telített bárium-hidroxidj elektrolízisével, platina- vagy nikkelkatódon.Kis mennyiségű, rendkívüli tisztaságú hidrogén palládium szivacs alkalma
zásával nyerhető. Ilyenkor a palládiumszivacsot nagyvákuumban 300 C° felettgáztalanítjuk, majd 80 és 100 C° között tiszta hidrogéngázt vezetünk az evakuálttérbe. A szivacsot 1 atm nyomáson lehűtjük. Vákuumban 300 C°-on enyhe szívás
közben egyenletes hidrogéngázáramot kaphatunk, melynek nitrogéntartalmamaximálisan 5 •10
-4 %. A palládiumszivacs előállítását lásd 54.6.
Ismeretes, hogy a hidrogén levegővel vagy oxigénnel robbanó elegyet alkot,ezért hidrogénfejlesztéskor ellenőrizni kell, hogy a készülék teljesen levegőmentes-e
(lásd 15.1.1.). Op. —259,2 C°, fp. —252,8 C°.
53.2. Fluor (F.*),
Megolvasztott KF.3 HF elektrolízisével állítható elő. Anódként 3 mm átmé
rőjű nikkeldrótot alkalmazunk. Az elektrolízist kb. 100 C°-on hajtjuk végre. Az
53.2.1. ábrán látható elektrolizálóedény1 — amely egyben kátédként is szerepel —rézből vagy magnéziumból készíthető. Az anód szigetelésére CaF2-ot használunk.Ebből készült dugóba ragasztjuk ólom-oxid—glicerin-kittel az anódul szolgáló
vastag nikkeldrótot. A fedelet az edény peremén körbefutó vályúban elhelyezetthárom fluoritdarabra állítjuk, és keményítővel kevert fluöritporral tömködjük
1 Wartenberg, H.: Z. anorg. alig. Chem. 193, 409 (1930) és 244, 337 (1940).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 164/366
166 N E M F É M E S E L E M E K
lazán körül. Hogy az anódon képződő fluort ne szennyezze az edény alján fejlődőhidrogén, az anódvédőcső alját rézlemezzel kell elzárni úgy, hogy a cső pereme és a
fenéklemez között néhány mm-es rés maradjon az elekro-lit áramlásának biztosítására. A cellát 1,2 kg tiszta,gondosan szárított KHF2 és 300 g frissen desztillált,vízmentes HF (készítését lásd 55.1.1.)keverékével tölt
jük. A berendezést elektromosan fűtjük. A kísérletmegindítása előtt a keveréket 70 C°-ra melegítjük fel. Az elektrolízist 10—20 V feszültséggel és 4—5 amperáramerősséggel végezzük. A melegítést elektrolízis közben megszüntetjük, és a fűtőtestet tulajdonképpen csak
az üzeméltetés szünetében használjuk. Az olvadék súlyveszteségének meghatározása lehetővé teszi az elhasználódott HF időnkénti pótlását. Száraz reakcióelegy használata esetén a víznyomok az elektrolízis első óráibaneltűnnek, az addig képződő oxigénnyomok a fluorgáztólnem választhatók el. A- fluor fejlődését úgy ellenőrizhetjük, hogy a kiáramló gáz útjába világítógázt vezetünk, amely a fluortól azonnal meggyullad.
A száraz fluorgáz az üveget nem támadja meg.Magasabb hőmérsékleten (600 C°-ig) platina, nikkel
vagy szinterkorund csövekben reagáltatható. A rezet350 C°-ig gyakorlatilag nem támadja meg. Op. —223C°, fp. —187 C°.
53.3. Klór (Cl2)
Az alkáli-klorid-elektrolízis útján nyert, kereskedelmi forgalomban kaphatócseppfolyós klór nem tiszta, ezért a 49.4.-ben leírt tisztítási eljárásnak célszerűalávetni.
Laboratóriumban kényelmesen előállítható a következő reakció alapján:
Mn02 + 4 HC1 = MnCljj + 2 H20 + Cl*.
A gázfejlesztő lombikba helyezett mangán-dioxid-hidrátra tömény, levegőmentessósavoldatot csepegtetünk. A gázfejlődés melegítéssel is szabályozható. Ilyenkoris célszerű a kapott klórt tisztításnak alávetni.
A klór a gumit, parafát, csapzsírt, higanyt megtámadja,; üvegedényekbenco. kénsav felett, valamint cseppfolyós alakban acélpalackban eltartható. Op.
— 101,0 C°, fp. —34,0 C°.
53.2.1. ábra. Elektrolizáló edény fluor előállításához
53.4. Bróm (Br2)
A kereskedelmi forgalomban kapható legtisztább bróm is kb. 0,05 % klórtés nyomokban jódot tartalmaz, amire különleges tisztaság igénye esetén figyelemmel kell lenni. Nagyobb mennyiségű klórszennyezés eltávolítására finomanporított kálium-bromidot adunk a tisztítandó brómhoz. A brómot nagyvákuumban

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 165/366
HA LOGÉ NE K —OXIGÉ NC SOPOR T 167
szárazjeges—alkoholos hűtés közben ledesztilláljuk. Op. —7,3 C°, fp. +58 C°, s == 3,14. Oldhatóság vízben 20 C°-on: 3,63 g Br2/100 g HaO.
Teljesen tiszta halogénmentes bróm előállítására a Hónigschmid2 vagy Baxter 3
által ajánlott módszer alkalmazható.
53.5. Jód (I2)
A kereskedelmi jódnak kálium-jodiddal történő halogénmentesítése nemszolgáltat teljesen tiszta terméket a KI szennyezései (Cl- , Br- , alkáli-szulfát stb.)miatt. Mindazáltal, ha nincsenek különleges tisztasági igények, a kereskedelmi
jódot Plotnyilcov* ajánlatára kálium-jodidos, majd bárium-óxidos keverékből szublimálva tisztíthatjuk, és exszikkátorban P20 5 felett tartjuk el.Teljesen tiszta jód előállítására Hőnigsckmid5 módszerét alkalmazhatjuk.
Op. 114 C°, fp. 183 C°, s = 4,93. Oldhatóság (20 C°-on) 0,029 g/100 ml H20.
53.6. Oxigén (0 2)
Acélbombákban tárolt ipari oxigén tisztításával az általános részben (49.2.)foglalkoztunk. Tiszta oxigén előállítása céljából 30 %-os KOH-oldatot elektro-lizálunk nikkelelektródok alkalmazásával. A hidrogénnyomokat palládiumazbesztkatalizátor segítségével távolítjuk el, majd a gáz szárítását cc. kénsavval végezzük.
Igen tiszta oxigén állítható elő hidrogén-peroxid katalitikus bontásával.®
53.7. Kén (S)
A kereskedelemben kapható kén tisztítását CS2-oldatból való többszöri átkris-tályosítással végezhetjük. 70 g p.a. CS2-ben 31,5 g ként oldunk szobahőfokon.
Szűrés után az oldatot zárt ^r/e«.weyer-lombikban hosszabb időn át jeges vízzelhűtjük. Az átkristályosítás többszöri megismétlése után a tiszta terméket finomanelporítjuk, és 90 C° alatt szárítjuk.
^ 53.8. Szelén (Se)
A szelén általában kén-, továbbá kis mennyiségű tellúr- és vasszennyezésttartalmaz. A szennyezésektől való megtisztítása végett Se02-dá oxidáljuk, amelyet többszörös szublimációval tisztítunk meg. A tiszta Se02-szublimátumot vízbenfeloldjuk, és 10 %-os hidrazónium-hidroxid-oldattal redukáljuk. A szelén ilyenkorvörös por alakjában csapódik ki, amely meleg fürdőben szürke csapadékká
* Hőnigsckmid, O., Zintl, E.: Liebigs Ann. Chem. 433, 216 (1923).8 Baxter, Q. P., Moore, Q. J., Boylstónr A . G.: J. Am. Chem. Soc. 34, 260 (1912).4 Plotnikow, W. A., Robotján, W. E.: Z. physik. Chem. 84, 365 (1913).1 Hónigachmid, O., Striébel, W Z. phyaik. Qhem. (A) 166 (Bodenstein-Festband)
286 (1931).8Wartenherg, H. V Z. anorg. alig. Chem. 238, 297 (1938).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 166/366
168 NEMFÉMES ELEMEK
áll össze. Kerüljük a redukálószer fölöslegét, mert hatására a Se poliszelenidekképződése közben oldódik. A kész terméket szűrjük és forró vízzel dekantálva
mossuk, amíg a szűrlet KI-os keményítőoldattal már nem reagál. A tisztítottszelén 170 C°-on szárítható, a víznyomok nitrogénáramban történő desztillálássaltávolíthatók el.7Op. 220 C°, fp. 685 C°, s = 4,26.
53.9. Tellúr (Te)
A kereskedelmi forgalomban levő tellúr rendszerint Te02 mellett kevés Se-,S- és nehézfém-szennyezést tartalmaz. Tisztítás céljából a fémet kvarccsőben f a
áramban desztilláljuk* miután előzőleg achátmozsárban elporítva kvarccsónakbahelyeztük. A tellúr a cső hidegebb részein apró golyók alakjában kondenzál. Ezutána csövet H2-áramban lehűtjük, a regulusokat platina csipesszel kiszedjük, majdcc. HCl-ban kevés HN0 3 hozzáadásával oldjuk. A HN0 3 feleslegét tartós melegítéssel elbontjuk, és az elegyet vízzel annyira hígítjuk, hogy hidrolízis éppen ne következzék be. Szűrés után az oldatot frissen desztillált hidrazónium-hidroxid-oldattalredukáljuk.
A kivált tellúrt vízzel és alkohollal mossuk, és cc. H2S04felett szárítjuk. Az ilymódon kapott finom eloszlású elemet ezután HN03-ban (s = 1,25) 70 C°-on feloldjuk. (Magasabb hőmérsékleten nehezen oldható Te02 csapódik ki.) Bepárlással
jól kristályosítható bázisos só, a Te20 3(0H)N0 3 válik le, amelyet HN03-as oldatbólújra átkristályosítunk, majd porcelán tégelyben elektromos kemencében Te02-dáizzítunk ki. Ezt HCl-ban (s = 1,12) oldjuk, és hidrazónium-hidroxid-oldattal redukáljuk elemi teliúrrá. Mivel a finom eloszlású Te a levegő oxigénjének hatásáraoxidálódhat, kvarccsónakban H2-gázáramban olvasztjuk meg (op. 452 C°, fp.1390 C°).
Különleges tisztasági igény esetén kvarcberendezésben nagyvákuumban alacsony hőmérsékleten desztilláljuk8.
53.10. Nitrogén (N2)
A nitrogén laboratóriumi előállításáraH
53.10.1. altra. Nitrogén előállítása azidokból
csak különleges esetekben kerül sor. Az indifferens gázként gyakranhasználatos, acélbombában kaphatókereskedelmi N2 a 49.2.-ben leírtmódon tisztítható. Igen tiszta N2
állítható elő alkáli-azidok elbontásaútján. Erre a célra az 53.10.1. áb
rán bemutatott berendezés alkalmazható.9 A NaN 3 bomlási hőmérséklete 280 C°, a KN3-é 360 C°. Aberendezést higanydiffúziós sziva,ty-tyúval evakuáljuk és kifűtéssel szá-
7 H ónig schm id , O., K apfenberger, W .: Z. anorg. alig. Chem. 212, 198 (1933).8 H ónig schm id , O., B audrexle r , H .: Z. anorg. alig. Chemie 223, 91 (1935).9 J u s l i , E .: Ann. Physik [5] 10, 895 (1931).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 167/366
169NITBOGÉNCSOPOBT
rítjuk. Ilyenkor az azidot tartalmazó oldalcsövet is melegítjük, azonban ügyelnikell rá, hogy ez a rész a bomlási hőmérséklet alatt maradjon. Ezután a H csapot
elzárjuk, és az A csövet világító lánggal melegítjük. A nyomásnövekedést a G manométeren észleljük. A finom eloszlású Na-por a C edényben ülepszik le; amelegítés megszüntetése után az F csapot megnyitjuk, és a nitrogént az E edénybe eresztjük. A folyamatot addig ismételjük, amíg csak elegendő mennyiségű nitrogén nem gyűlik össze. Op. —210 C°, fp. —195,8 C°.
53.11. Sárgaarzén (As)
A sárgaarzént az arzéngőzök hirtelen lehűtésével állítjuk elő. Az 53.11.1.ábrán látható készülék segítségével az arzéngőzöket közvetlenül az oldószerkéntszolgáló CS2-be vezetjük.10Mivel a sárga módosulat rendkívül fényérzékeny, sötét
ben, pl. az ábrán is látható dobozban hajtjuk végre a reakciót. Az arzént a bevezetőalumínium oldalcsőben helyezzük el. A melegített csőből a gőz tömény H2S04-valszárított C02-gáz vivőárammal kérülaCS2-ot tartalmazó edénybe, amelyet jéggelhűtünk. A beáramló meleg gázkeveréket —20 C°-os C02-árammal hűtjük. A CS2 a beáramló arzéngőzöket oldja. Az oldatot szűrjük, és (sötétben) vízfürdőn kb.felére pároljuk, végül aceton—szárazjég-keverékkél —70 C°-ra hűtjük. Ekkor asárgaarzén kicsapódik. Célszerű több szén-kéneges elnyelőedényt sorbakapcsolni.
Az első két edényből érdemes az arzént kinyerni.
53.12. Antimon (Sb)
Igen tiszta antimon antimon(III)-oxidnak kálium-cianiddal történő redukció jával állítható elő. A komponenseket ekvivalens mennyiségben tartalmazó keveréket porcelán tégelyben megolvasztjuk, és a hőmérsékletet az antimon olvadáspontja
10 Erdmann, H., Unruh, M.: Z. anorg. alig. Chem. 32, 439 (1902).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 168/366
170 N E M F É M E S E L E M E K
fölé emeljük. Lehűlés után a regulust vízben való főzéssel megtisztítjuk a szennyezésektől, és átolvasztjuk. Op. 630 C°, fp. 1635 C°.
53.13. Szén (G)
Az alábbiakban néhány, a laboratóriumi gyakorlatban használatos speciáliskészítmény előállítását tárgyaljuk.
53.13.1. Aktív szén
Előállításakor a lehetőleg tiszta szenet igen óvatos oxidációval fellazítjuk. Eztúgy végezzük, hogy C02- vagy vízgőzáramban 950 C°-on addig hevítjük, amíg a
szén eredeti súlya kb. felére csökken. Az adszorbeált C02 és HaO eltávolítása atermék 300 C°-on nagyvákuumban történő hevítésével érhető el.11
A nagy adszorbeáló képességű aktív szén pl. szobahőmérsékleten CCl4-dalfélig telített gőztérből eredeti súlyával egyenlő mennyiségű CCl4-ot tud megkötni.
53.13*2. Grafitbevonat
Gyakori feladat üveg, kvarc vagy kerámiai anyagok felületének bevonásavékony grafitréteggel, amelyhez felhígított szénhidrogéngőzök termikus bontásá
val juthatunk.12
Ha pl. a kvarccső belBŐ felületét kívánjuk grafitozni, a csövet 850 C°-ra hevít jük, és olyan N2-gázáramot vezetünk át rajta, amelyet előzőleg szobahőfokon víz-és benzingőzökkel telítettünk oly módon, hogy a gázt vízzel, illetve benzinnel töltött intenzív mosópalackokon buborékoltattuk át. A használt benzin forráspontja100 C° körüli legyen. A nitrogén—benzin-áram csekély vízgőztartalma tükörsimafelület képződését segíti elő („Glanzkohle”). A kívánt vastagságú grafitréteg képződése után a csövet nitrogénáramban lehűtjük. Amennyiben grafitkristályok előállítása volt a célunk, úgy a reakciót hosszabb időn (kb. 4 órán) át folytatjuk. A kapott terméket lehűlés után a kvarccső faláról lekaparjuk.
Nitrogén—benzin-keverék helyett propángázt is alkalmazhatunk. Ilyenkor akvarccsövet vízlégszivattyúval megszívatjuk, és a propángáz—vízgőz-elegy (a gőza szivattyúból diffundál vissza) nyomását kb. 20 Hgmm-re állítjuk be.
A tárgyalt eljárásokat alkalmazzák szénellenállások előállítására is.
11 Ruff, O., Rössner, O.: Bér. dtsch. chem. Ges. 60, 411 (1927); Hoffmann, U. és munkatársai: Z. anorg. alig. Chem. 255, 196 (1947).
11 Ruess, O.: Z. anorg. alig. Chem. 255, 263 (1947).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 169/366
54. Fémek
54.1. Fémek és néhány nemfémes elem előállításafémtermiás reakcióval
A fém-oxidok, fém-szulfidok, illetve fém-halogenidek számos fémmel (Al, Mg,Ca, Na, K stb.) erősen exoterm módon reagálnak. Ezeket a reakciókat előnyösen
használhatjuk fel magas olvadáspontú fémek előállítására. Ezekben az ún. termit- reakciókbán a reakció fenntartásához szükséges hőmennyiséget maga a reakcióhő
szolgáltatja. A reakció megindításához rendszerint kisebb mennyiségű gyújtó
keverék elegendő. A fém és a salak általában folyékony állapotban képződik, és
többé-kevésbé szétválik. A termitreakcióban redukálószerként leginkább az alumíniumot alkalmazzák. A hőfejlődés szabályozását indifferens anyagok (pl. A120 3)
hozzáadásával vagy a keverék oxigéntartalmának változtatásával (pl. Ba02,Na2Oa stb.) végezhetjük el. A képződött termék alumíniummal való ötvöződésének
megakadályozására célszerű a redukálandó oxidból kis felesleget alkalmazni.Ennek ellenére a termék rendszerint kb. 1 % alumíniumot is tartalmaz.
Az aluminotermiás eljárást általában akkor alkalmazzuk, amikor a fémetkompakt regulus alakjában kívánjuk előállítani. Könnyen redukálható fémeket,
amilyen pl. a Fe, Mn, Cr, úgyszólván elméleti kitermelésben kaphatunk, hacsaknem dolgozunk túl kis méretekben. Legalkalmasabb a reakciót néhány kilogram
mos anyagmennyiséggel végezni. Olyan fémek előállítására, amelyeknek viszonylag
nagy a tenziójuk (ilyen pl. az Pb, Zn, Ti, U, Be, Mg stb.), az aluminotermiás előállítás hem alkalmazható.
A reakciót közönséges agyagedényben, pl. virágcserépben is végezhetjük;ilyenkor a virágcserepet nagyobb, homokkal töltött cserépbe helyezzük. Reaktor
edényként célszerűbb azonban agyagcserép helyett a hőmérséklet-változást jobbanelviselő samott-tégelyt alkalmazni. Mindkét esetben fennáll azonban a termékszilíciummal való szennyeződésének a veszélye. Ennek kiküszöbölésére a tégelytmagnézium-oxid béléssel lehet ellátni, esetleg magnézia tégelyt alkalmazni.
A leggyakrabban használatos gyújtókeverék összetétele: 10 súlyrész Al-por,
40 súlyrész Ba02, 7 súlyrész KC103. A komponensek porítását külön-külön, és
összekeverésüket igen óvatosan (pl. tollal) végezzük. A gyújtókeverékből kevésóteres kollódiumoldattal kb. 1 g súlyú golyót formálunk, amelybe 10—15 cm hosszú
magnézium szalagot szúrunk. A meggyújtáskor igen óvatosnak kell lennünk, nehogy a Mg-szalag égő vége letörjön, ób a vártnál korábban gyújtva meg a keveréket,sérülést okozzon. (A Mg meggyújtásához ezért védőszemüveget és azbeszt kesztyűt
használjunk.) A magnézium szalagot célszerű kis forrasztólámpával meggyújtani. A reakciót szabadban végezzük!
Aluminotermiás úton állíthatók elő pl. a következő elemek: Cr, Mn, Mo, V ,B és
Si. A fémtermikus reakciókban alkalmazott redukálószerek összehasonlítására az

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 170/366
172 F É M E K
egy oxigénatomra vonatkozó képződéshőket szokás figyelembe venni (lásd az 54.1.1.táblázatot). Ez a módszer tévedésekre vezethet, mert a képződéshő nem a tény
leges reakcióhőmérsékletre vonatkozik, és mert a legtöbb termikus reakcióban azegyensúly egyes komponensek illékonysága következtében egyik vagy másikirányba eltolódhat. A reakció irányát gyakran befolyásolhatja ötvözetképződés is. A termitreakciók lefolyásának pontos megállapítására a reakció hőmérsékletéheztartozó szabadentalpiák és a tenziók ismerete szükséges.
54.1.1. táblázat
Egy oxigénatomra vonatkoztatott képződéshők
Anyag Q (kcal) Anyag Q (kcal)
CaO -151 ,7 7 , T i0 2 - 1 0 9b 2 T h 0 2 -146 ,6 V* S i 0 2 - 10 2
MgO -1 4 3 ,7 Na20 - 99,5BeO -143,1 ZnO - 83,5
V 3 ba20 3 -142 ,9 7 3 Fe20 3 - 66,27 3 a i 20 3 - 133 ,4 h 2o - 57,8
V. ZrOz - 1 2 9 7 , c o 2 - 47,2
Az 54.1.1. táblázat szerint a, fémkalcium igen erélyes redukálószer, mivel azonban magasabb hőmérsékleten illékony, az alumíniumnál vagy a szilíciumnál csakabban az esetben redukál jobban, ha a párolgását megakadályozzuk. Ezt úgyvalósíthatjuk meg, hogy a reakciót acélbombában hajtjuk végre. Az anyagkeveréket acélbombába helyezve behegesztjük, azután — a reakció megindulásáig —kívülről melegítjük. Katalizátorként célszerű fémnátriumot is tenni a keverékbe. Az elért hőmérséklettől, valamint az előállított fém olvadáspontjától függően areakcióterméket por vagy kisebb regulusok alakjában kapjuk. Oxidokból, kloridok-ból, esetleg fluoridokból fémkalciummal számos olyan fémet sikerült előállítani,
amely aluminotermiás úton nehezen vagy nem nyerhető; ilyenek pl. a Ti, Zr, Hf,Th, U, V, Ta, m>.Tiszta fémek előállítására a kalcium alkalmazása azért hátrányos, mert tiszta
állapotban rendszerint nem áll rendelkezésre, és felhasználása előtt többszörösvákuumdesztillációval kell tisztítani. Könnyen redukálható fémek, pl. a Mo, V, Cr,Mn előállíthatók kloridjaik cinkkel történő redukciója útján. A szén nem alkalmasredukálószer tiszta fémek készítésére. A szilícium sok esetben szilicidet alkot, mégisalkalmazzák igen nehezen redukálható fémek, pl. Nb vagy Ta előállítására. Ilyenkor azonban a reakciót vákuumban hajtják végre, és úgy szabályozzák, hogy aSiillékony SiO alakjában a rendszerből eltávozzék. m
Az igen kis tenziójú cirkónium alkálifémek előállítására alkalmas. Céziumotpl. úgy állíthatunk elő, hogy finom eloszlású Zr és Cs2Cr20 7keverékét vákuumbanmelegítjük. Ilyenkor az egyensúlyban levő komponensek közül a legillékonyabb —esetünkben a cézium — átdesztillál.
Más reakciókörülmények között viszont az alkálifémek használatosak pl. a Ti,Zr, U vagy V előállítására. A megfelelő kloridókat vagy fluoridokat fémnátriummalzárt acélbombában reagáltatjuk. A nem teljesen veszélytelen reakció mérséklésérecélszerű alkáli-kloridot is adagolni a reagensekhez.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 171/366
ELEMEK ELŐÁLLÍTÁSA FÉMTERMIÁS REAKCIÓVAL 173
54.1.1. Szilícium (Si)
90 g szárított és szitált tengeri homokot, 100 g alumínium darát és 120 g kénvirágot összekeverünk, s a homokos edénybe helyezett samott-tégelyt (esetlegagyagcserepet) félig megtöltjük vele. A reakciót gyújtókeverékkel indítjuk meg.Lehűlés után a tégelyt összetörjük, és a durván elporított terméket vízbe tesszük(lehetőleg szabadban vagy jól húzó fülkében, mert a képződött alumínium-szulfidhidrolízise következtében nagy mennyiségű H2S-gáz fejlődik.) A vízzel kilúgozott,illetve kimosott termékből dekantálás után a Si regulusait mechanikusan elkülönítjük a salaktól, majd többször megismételt meleg sósavas kezelésnek vetjük alá,míg az alumínium szennyezés teljesen fel nem oldódik. Könnyebb szennyezésekülepítéssel is elválaszthatók a kristályos szilíciumtól.
A több napon át tartó tömény sósavas kezelést dekantálás után hidrogén--fluoridos tisztítás követi, amelyet platina tégelyben szoktunk végezni. A digerálástvízfürdőn kb. 1 órán át folytatjuk, majd a terméket vízzel többször kimossuk, ésa leszűrt kristályokat szárítószekrényben (100 C°-on) megszárítjuk. Az eljárássalkb. 20 g szürke, kristályos szilíciumot állíthatunk elő.
Az előbbinél lényegesen jobb a kitermelés, ha nagyobb mennyiségű anyagkeverékből indulunk ki. Ez esetben 10—12 kg anyagot, amely 4 súlyrész alumínium darából, 5 súlyrész kénporból és 3,6 súlyrész tengeri homokból áll, megfelelőméretű tégelybe helyezünk oly módon, hogy annak 4/5 részét megtöltse. A keveré
ket célszerű ilyenkor több helyen begyújtani. A reakció közben a tégelybe újabb6—8 kg keveréket adagolhatunk. A nyerstermék 85—90 %-os Si, amelyet a fentiekben leírt tisztítási eljárásnak vetünk alá.
54.1.2. Bőr (B)
' 50g vízmentes B20 3-ból, 75 g kén virágból és 100 g Al-darából keveréket készítünk. A reakciót és a termék tisztítását a Si aluminotermiás előállításához hasonlóanvégezzük. A kapott kristályos bór szürkés, fémfényű. Súlya kb. 7,5 g, összetétele AlBl2-nek felel meg.
Ha a redukciót Mg-mal végezzük, amorf, kb. 86 % bórt tartalmazó termékhez jutunk.
54.1.3. Mangán (Mn)
A Mn02 és az A1 közötti reakció robbanásszerű, ezért a mangán aluminotermiás előállítására a viszonylag kevesebb oxigént tartalmazó Mn 30 4-ot használjuk,amelyet finoman elporított Mn02-nak 800—900 C°-on végzett hevítésével kapunk.450 g Mn 30 4-ból és 150 g alumínium darából keveréket készítünk. A keveréknek
kb. 1/10részét gyújtókeverékkel begyújtjuk, majd a többit kisebb részletekben, delehetőleg gyorsan hozzáadjuk. Az adagolást célszerű azbesztkesztyűben és védő-szemüveggel végezni. Lehűlés után az agyagtégelyt összetörjük, és a mangángolyókat a salaktól mechanikusan elkülönítjük. A kiindulási mennyiség csökkentése esetén a termelés rohamosan romlik. A kapott terméket vákuumdesztillációvaltisztíthatjuk.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 172/366
174 F É M E K
64.1.4. Molibdén (Mo)
A MoOa illékonysága miatt a fémmolibdén aluminotermiás előállításáhozMoOj-ból1 indulunk ki, amelyet Mo03-nak vörösizzáson H2-nel történő redukció jával állíthatunk elő. A reakcióhoz 80 g MoOa és 21 g Al-dara vagy -por keverékéthasználjuk fel. A kitermelés kb. 90 %-os, a regulus molibdéntartalma mintegy98 %. A szennyezés: Si, Fe és Al.
54.1.6. Yanádlum (Y)
A redukciót kalciummal végezzük, melynek illékonysága miatt a reakciót zárt
acélbombában hajtjuk végre.2 175 g Va0 3, 300 g aprított Ca éB 300 g előzőleg 450C°-on hevített CaClj keverékét acélbombába helyezzük, és légmentesen hegesztjük(lásd az 54.1.5.1. ábrát). A keverékhez kevés nátriumot vagy káliumot adunk, hogya bombában levő vízgőzt és oxigént megkösse. Az oxigéngáz a bombából úgy is el
távolítható, hogy azt előzetesen evakuáljuk, majd argonnaltöltjük. A bomba lezárása után annak hőmérsékletét 900—950 C°-ra emeljük, és egy órán át ezen a hőmérsékleten tart
juk. Teljes lehűlés után felnyitjuk és az anyagot kikapar-hegesztés juk, majd nagyobb mennyiségű (kb. 20 1) vízzel elkeverjük,
hogy elkerüljük a lokális felmelegedéseket. A tömör anyag
szétmálik. Vízzel, majd 2n HCl-val többször mossuk.
54.1.6. Alkálifémek előállítása 'cirkónium mal történő redukcióval(Cs, Rb, K, Ka, Li)
Tiszta alkálifémek előállítása úgy valósítható meg, ha a54.1.5.1. ábra. képződő fém az előállítás közben gázokkal nem érintkezik, és
Vanádium előállítása a rendszerben levő egyéb komponensek illékonysága rendkívül kicsi. Ezek a feltételek biztosíthatók az alkáli-kromátok-
nak (-molibdátoknak, -volframátoknak) Zr-porral történő redukciója útján.3 Kismennyiségű Cs, Rb vagy K úgy állítható elő, hogy egy súlyrész CsaCr04 (illetveI^CrC^) és négy súlyrész finom Zr-por keverékét kis rudakká préseljük, előzetesengáztalanított kvarccsőben nagyvákuumban leforrasztjuk, és óvatosan hevítjük. A reakciók 725 C°-on lassan indulnak, a hőmérsékletet 1000 C°-ig emeljük. A képződött alkálifém szépen csillogó bevonatként a cső hidegebb részén csapódik le.
A termék oxidmentes. A kitermelés Cs-ra 90—96 %, Rb-ra gyakorlatilag kvantitatív, K-ra 80 %.
A lítium előállítására LiaCr04-ot 8 súlyrész Zr-mal keverünk. A redukciótvákuumban 450—600 C° között végezzük. A kitermelés igen rossz.
Ha a Cs, Rb és K kromátjából való előállításakor a szükséges hőmérsékletapparatív okokból nem biztosítható, a következő összetételű dikromátkeverékethasználhatjuk: 1 súlyrész CsaCraC^ (Rb2Cra0 7, illetve KaCra0 7) és 10 súlyrész Zr. A redukció már 380 C° körül lassan megindul. A kitermelés 80—90 %-os. A Rb,
1 Fűnk, H.: Darstellung dér Metalle im Laboratórium. Stuttgart, 1938, 69. old. * Maráén, J. W., Rentschler, H. G.: Ind. Engng. Chem. 19, 97 (1927).8 de Boer, J. ff., Broos, J., Eminens, H .: Z. anorg. alig. Chem. 191, 113 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 173/366
A L K Á L IF É M E K — T IT Á N 175
valamint a K oxidmentes, a Cs kevés oxidot tartalmaz. Ha azonban egy súlyrész
Cs2Cr20 7-ra 20 súlyrész Zr jut, akkor a Cs-ot is oxidmentesen kapjuk meg.Kis mennyiségű tiszta nátrium előállítására egy súlyréBZ Na2Mo04 vagyNa2W04 és 4 súlyrész Zr-por keverékét használjuk. A nátrium képződése kb. 550,illetve 450 C°-on indul meg. A kapott termék oxidmentes, a kitermelés 100, illetvekb. 80 %-os.
Az 54.1.6.1. táblázatban összefoglaljuk az alkálifémek fontosabb tulajdonságát.
54.1.6.1. táblázat
Az alkálifémek néhány fontosabb fizikai adata
Alkálifém OlvadáspontC°
Forr áspontCe ®18°
Keménység(Mohs)
Li 179 1336 0,534 0,6
Na 97,8 883 0,97 0,4
K 63,5 776 1,47 0,6
Rb 39,0 696 1,521,89
0,3
Cs 28,6 670 0,2
54.1.7. Titán (TI)
A titánnak oxigénhez, nitrogénhez és szénhez való nagy affinitása miatt a-tiszta fém előállítása nehéz. így a Ti02és Ca reakciójával még a legkörültekintőbbmunkával is legfeljebb 98 %-os titán nyerhető. A tiszta fém, amely a törékenyszennyezett termékkel szemben szobahőfokon is képlékeny, fémtermikus reakcióvalcsak halogenidekből állítható elő. A legnagyobb tisztaságú fém (szennyezések:0,013 % szén és kb. 0,06 % nitrogén) a titán-jodid termikus bontásával készíthető(lásd 54.4.1.).
a) Dolgozhatunk az alábbi reakció Bzerint:Ti02 + 2 Ca = Ti + 2 CaO.
25 g szilloiummentes Ti02, 40 g vákuumban desztillált Ca és 20 g Na keverékét V2A-acélból készült, hegesztéssel zárt bombában 20 percig 1000 C° körüli hőmérsékleten hevítjük. Lehűlés és a bombacső nyitása után a borsónagyságú reguluso-kat (kb. 13 g nyers Ti-t) alkohollal, vízzel, majd növekvő töménységű sósavvalkezeljük, azután kloridmentesre mossuk, alkohollal víztelenítjük és 110 C°-onszárítjuk.4
b) Kiindulhatunk nátrium-[hexafluoro-titanát(IV)]-ből:5
NaJTiFj] + 4 Na = Ti + 6 NaF.
Az előállításhoz szükséges Na^TiFJ úgy készül, hogy tiszta Ti0 2-ot töményHF-oldatban melegen feloldunk, majd számított mennyiségű NaOH-ot adunk hoz-
4 Kroll, W.: Z. anorg. alig. Chem. 234, 42 (1937).* de Boer, J. H., Fost, J. D.: Z. anorg. alig. Chem. 187, 182 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 174/366
176 F É M E K
zá. Az oldatot 60 C° alatti hőmérsékleten bepároljuk, a kivált termék vízből valótöbbszöri átkristályosításával (alkoholos kicsapás segítségével) 99,9 %-os tisztaságú Na2[TiF6]-ot kaphatunk.
A redukcióhoz 10 % nátriumfelesleget használunk. A nátriumot apróra daraboljuk, és összekeverjük a nátrium-[hexafluoro-titanát]-tal. A reakciót V 2 A-bom-bában 1000 C°-on végeztük. Ha a használt Na2[TiF6] nem teljesén száraz, robbanáskövetkezhet be. A nyers terméket forró vízzel kifőzzük, és a folyadék fluoridmentes-ségéig dekantálunk. A maradékot NaOH-oldattal ismételten kifőzzük, majd hideg,hígított sósavval Öblítjük és vízzel kimossuk.
c) A titán-tetrakloridot nátriummal redukáljuk:6
TiCl4 -j- 4 Na - Ti + 4 NaCl.
A redukcióhoz sztöchiometriai mennyiségű nátriumot használunk. Amennyibena TiCl4 mennyisége 100 g-nál nagyobb, a reakciót hegesztéssel zárható acélbombában hajtjuk végre. A TiCl4nyomását a bombában kétféle úton lehet alacsonyabbszinten tartani: KC10 3—Na-pasztilla hozzáadásával elérhetjük, hogy a reakcióalacsonyabb hőmérsékleten meginduljon, vagy a bombacsövet úgy szerkesztjük,hogy abban jelentős hőmérsékletesés legyen, de a nátrium hőmérséklete a reakcióhoz szükséges kb. 7—800 C°-on maradjon. A termék egy része félig megolvadtregulusokból áll, más része igen finom por, amely könnyen oxidálódik.
d) Fémtitán előállítása magnéziumos redukcióval.7 A magnézium alkalmazásának előnye a nátriuméval szemben, hogy igen tiszta állapotban állítható elő, éskülönösebb rendszabályok nélkül levegőn eltartható. Mivel a reakció hőmérsékletén (~ 1100 C°) a titán vassal reagál, a reakciót molibdén bélésű tégelyben végezzük.
£4.1.8. Cirkónium (Zr)
A fémcirkon előállítása a titánnál leírt nehézségekbe ütközik; a fémtermikus
■ előállítási módszerek a titánnál tárgyaltakhoz hasonlóak. így Zr02-nak Ca-mal,valamint K 2[ZrFfl]-nak és ZrCl4-nak fémnátriummal történő redukciója egyaránthasználatos.8
54.2. Fémek előállítása hidrogénes redukcióval
Fém-oxidok termikus bontása csak ritkán alkalmazható a fémek előállítására,a Hg és az Ag kinyerésére azonban ez a gyakorlatban is jól bevált módszer. Azoxidok redukciója fémmé sok esetben hidrogéngázzal kényelmesen megvalósítható.Ezt a módszert a következő elemek előállítására alkalmazhatjuk: B, Cr, Zn, Fe, Ni,Co, Cd, Ge, As, Sb, Bi, Ga ,In, TI, Re, Mo, W, Se, Te. Egyes esetekben, pl. a Ga, Inós TI hidrogénezéssel történő előállításakor először alacsonyabb vegyértékű oxidok
9 Fást, J. D .: Z. anorg. alig. Chem. 241, 42 (1930).
7 Króll, W .: Trans. Elektrochem. Soc. 78, 161 (1940).
Oillemot, L Magyar Tud. Akadém ia V I . Oszt . Közi . 14, 303 (1954); Ac ta Techn. A c a d . Sci . H u n g. 10, 221 (1955) ; Neue H ütte 2, [2 /3] 85 (1957).
® de Boer, J. H., Fást, J. D.: Z. anorg. alig. Chem. 187, 177 és 198 (19 30 );
Kroll, ÍV.: Z. anorg. alig. Chem. 234, 42 (1937).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 175/366
F É M E S E L Ő Á L L ÍT Á S A H X D H O O É N E S R E D U K C I Ó V A L 177
képződnek, amelyeknek további redukciója az előzőnél lényegesen lassúbb folyamat. A halogenidek (fluoridok kivételével) vagy a szulfidok hidrogénes redukciója
általában nehezebben folyik le, mivel a hidrogénnek klórhoz, brómhoz, jódhoz,illetve kénhez való affinitása kisebb, mint az oxigénhez vagy fluorhoz való. Bár afluoridok és egyes esetekben a kloridok hidrogénes redukciója gyakorlatban is jólvégrehajtható, mégis csupán olyan esetekben szokás alkalmazni ezt a módszert,amikor a megfelelő oxidok teljes redukcióját oxidos keverékfázisok képződése gátolja. így pl. a VCla, VC14vagy NbCl5 hidrogénnel por alakú tiszta fémmé redukálható.
A hidrogén igen tiszta redukálószer és viszonylag könnyű tisztán előállítani.Előnye még, hogy leszívatással vagy hevítéssel a felesleget, illetve az adszorbeáltgázt könnyen el lehet távolítani. Különösen akkor alkalmazzuk, ha drága és ritkafémek tiszta állapotban való előállítására van szükségünk (pl. Ge, Mo, W, Pt stb.).
Mivel a hidrogénes redukció gyakran elég alacsony hőmérsékleten is végrehajtható, lehetővé válik igen finom eloszlású fémek előállítása. Ha fennáll az a veszély,hogy a kapott termék zsugorodás következtében még teljesen át nem alakult részeket tartalmaz, célszerű a nyers terméket finoman megőrölve újabb hidrogénes redukciónak alávetni.
A hidrogén helyett redukálószerként egyes esetekben NH 3 is alkalmazható. AzNH 3 a fém-oxidok redukciójára csak abban az esetben — pl. Co, Ni és Bi előállításakor— használható, ha a nitridek képződésének veszélye nem áll fenn.
64.2.1. VaB (Fe)
Piroforos vas (ferrum reductum) előállítására legegyszerűbb tiszta Fe(OH)a-bólkiindulni, amelyet Fe(N03)3-oldatból NH4OH-dal kaphatunk. Ezt a csapadékot kb.65 C°-on szárítjuk, majd finoman elporítjuk, és korundcsónakban (porcelán vagykvarccsőben), növekvő hőmérsékleten H2*gázzal redukáljuk. Ha a hőmérsékletetcsak 550 C°-ig emeljük, a kapott termék piroforos tulajdonságú. Ha nem piroforosvasra van szükségünk, a hőmérsékletet kb. 40 perc alatt 400 C°-ról 700 C°-ra emeljük,azután kb. 20 percig 700 C°-on tartjuk (20 g Fe^Oj kiindulási mennyiség esetén).
Ha kevésbé reakcióképes kiindulási anyagot alkalmazunk (pl. 110—120 C°-on szárított Fe(OH) 3-ot), akkor a redukció lényegesen magasabb hőmérsékleten (kb.1100 C°-on) megy végbe és hosszabb ideig (kb. 60 óra) tart.
64.2.2. Kobalt (Co)
Kobalt-Oxidok (CoO, Co 30 4) vagy 120 C°-on szárított kobalt-oxalát hidrogénesredukciójával állíthatók elő. A 250 CP körüli hőmérsékleten előállított termék piroforos. Ha a redukciót 5—6 órán át kb. 500 C°-on hajtjuk végre, por alakú termékhez
jutunk, amely száraz levegőn állandó. Az így kapott fém köbös rácsú, míg az alacsony hőmérsékleten előállított fém hexagonális szerkezetű.
64.2.3. Nikkel (Ni)
Tiszta, oxigénmentes, P20 5-dal szárított Hj-gázt 15 órán át 300 —400 C°-on aNi(N03)a*6 HaO termikus bontásával nyert NiO felett vezetünk el. A termékethidrogénáramban hűtjük le, és a berendezéshez tartozó ampullába forrasztjuk.
12 Ált. és szervetlen kémiai prakt. — 4284/II.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 176/366
F É M E K 178
A piroforos tulajdonságú fémpor alkoholban is eltartható. Hidrogénezéshez katalizátornak alkalmazzák.
54.2.4. Molibdén (Mo)
Az ammónium-molibdát/(NH4)5H(Mo60 2i) *3 H20/hevítésével előállított Mo03-ot 500 CP-on hidrogénáramban az alacsonyabb vegyértékű Mo kevésbé illékonyoxidjává redukáljuk. Ez után a redukciót 1000 C°-on folytatjuk. A terméket hidrogénáramban hűtjük le. A finom eloszlású molibdén a levegőn fokozatosan oxidálódik, ezért célszerű védőgázban vagy vákuumban leforrasztani.
54.2.5. Volfrám ( W)Tiszta, kihevített W 03-ot mázatlan porcelán csőbe helyezett porcelán vagy
nikkelcsónakban H2-gázzal redukálunk.9 A reakciót egy ideig 800 C°-on, majd1000—1200 C°-on végezzük. A redukció elég gyors, végét a vízgőzképződés elmaradása jelzi. A terméket hidrogénáramban hűtjük. A redukciót 800 C°-on is elvégezhetjük, ilyenkor azonban hosszabb időt igényel (8 órát).
A kapott termék diBzperzitásfoka nem a kiindulási termék (W 03) diszperzitás-fokának függvénye, hanem elsősorban a reakció hőmérsékletétől, a hevítés időtartamától és a hidrogénáram sebességétől függ. Nagyobb szemcsenagyságú terméket
akkor kapunk, ha a redukciót nedves H^-gázzal 1500 C°-felett végezzük.
54.2.6. Germánium (Ge)
Mázatlan porcelán csónakot Ge02-dal töltünk meg, és porcelán vagy kvarc-csőbe helyezünk. A redukciót erős hidrogénáramban 600 C°-on végezzük. A csőbőlkiáramló gázt célszerű lefelé hajlított vezetékszakaszon át kivezetni, hogy a le-osapódó vízgőz ne folyjon vissza az izzó reaktorba. 40 g Ge02-nál kisebb mennyiségből indulva ki, a redukció 3 —4 órát igényel. Ügyeljünk arra, hogy a reakció hőmérséklete ne haladja.meg az említett értéket, mert zsugorodás, illetve a GeO párolgása
következhet be.10 Az anyagot hidrogénáramban hűtjük le. A kapott termék szürkés-fekete por, ebből a tömör fém összeolvasztással állítható elő. Az olvasztást mázatlanporcelán tégelyben, átfúrt fedő alatt fitose-tégely), H2-atmoszférában végezzük. A germánium olvadáspontját (959 C°) meghaladó hőmérsékletet oxigénnel tápláltgázlánggal érhetünk el. A tégely rendszerint megreped, mivel a germánium dermedéskor kitágul.
A germánium összeolvasztása konyhasó védőréteg alatt is elvégezhető.
54.3. Fémek előállítása elektrolízissel
A fémek elektrolitikus úton történő redukciójának elméleti kérdéseivel és agyakorlatban felmerülő problémákkal az általános részben foglalkoztunk (51. fe
jezet). Fémek előállítására a vizes oldatban, nem vizes oldószerben és olvadékbanvégrehajtott elektrolízis egyaránt használatos.
9 Fűnk, H .: Darstellung dér Metalle im Laborató rium. En ke, Stuttga rt, 1938, 72 old.
10 Schwarz, R., Elstner, G.: Z. anorg. alig. Chem. 217, 289 (1934).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 177/366
F É M E K E L Ő Á L L Í T ÁS A E L E K T R O L ÍZ IS S E L 17 9
54.3.1. Kadmium (Cd)
A J oneá-reduktorként használatos kadmíumpor tömény CdS04-oldat H2S04-
-val megsavanyított vizes oldatának platinaelektródok között végzett elektrolízisévelállítható elő.11 Áramsűrűség 0,1—0^3 A/cm2. Az elektrolízist henger alakú üvegedényben végezzük. Az egymás alatt 5 cm távolságra elhelyezett lemez alakú elektródok közül az alsót használjuk katódként. Az elektrölizáló edény elég gyorsantelik meg ezüstösen csillogó kristályporral. A voluminózus kristályport üvegbottalidőnként lenyomkodj uky-nefiogy az anóddal érintkezve rövidzárlatot okozzon. Amikor az elektrolit Cd-koncentrációjának csökkenése folytán a katódon hidrogén-fejlődés lép fel, a fürdőhöz CdS04-ot adagolunk, ellenkező esetben a Cd szivacsosanválik le (ez túl nagy áramsűrűség esetén is bekövetkezhet).
54.3.2. Lítium (Li)
a) Előállítható a lítium-klorid telített piridines oldatának elektrolízisével.12 Azelektrolízist szobahőmérsékleten végezzük diafragma nélkül. Anódként grafítlemez,katódként sima vas- vagy platinalemez használható. 14 V' feszültség és 0,2—0,3
A/100 cm2 áramsűrűség esetén jól tapadó ezüstfehér lítiumbevonat képződik. Azelektrolízist nedvesség távoltartásával célszerű végezni, mert a katódos polarizációa cella ellenállását jelentősen megnöveli. Ezérta piridint kálium-hidroxiddal víztelenítjük és
frissen desztilláljuk, a lítium-kloridot pedigfelhasználás előtt kihevítjük.
b) Tiszta lítiumot lítium-bromid és -kloridolvadékelektrolízisével is kaphatunk. Erre acélra az 54.3.2.1. ábrán vázolt, Muthmann-ié\e, rézből készült elektrölizáló edényt alkalmazzuk.13 Az anód grafitrúd, katódként pedig kétvasdrótot használunk. Az elektrolizálóedényfelső része vízzel hűthető. A 10—15 % lítium-
-kloridot tartalmazó lítium-bromidot ívfénnyelmegolvasztjuk (op. 520 C°). Az elektrolízist550 C°-on 10 V kapocsfeszültséggel és 10 A áramintenzitással végezzük. A kivált fémet időnkéntvaskanállal kiemeljük, és a megdermedt olvadéktól kőlapra öntve különítjük el. A sótartalmú nyers fémet 180—200 C°-on megolvasztva,a só és a fém faj súlykülönbségének felhasználásával tisztítjuk meg.
c) Káliummal szennyezett lítium állítható elő LiGl és KC1 egyenlő súlyarányúolvadékának eletrolízisével. Az előállításhoz az 54.3.2.2. ábrán látható berendezésthasználjuk. A kb. 5 cm átmérőjű porcelán tégelyt kevés azbesztpapírral vagy
54.3.2.1. ábra. Lítium elektrolitikus előállítása Muthmann-fé\e cellában
11 Treadwell, F . P .: Lehrbuch d. analyt. Chem ., B d. 2, W ien 1949, 624 old. 18 Kahleriberg, L .: J. PhyB. Chem. 3, 602 (1899).
Pattén, H. B., Mott, W. B .: J. Phys. Chem. 12, 49 (1908).
18 Buff, O., Johansen, O.: Z. Elektrochem. 12, 186 (1906).
12

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 178/366
180 f é m e k
azbesztzsinórral kibélelt vastégelybe helyezzük. Az olvadékot tartalmazó tégelytgázlánggal fűthető samott kemencébe tesszük. Az elektrolízist 450—500 C°-on
végezzük. Az anód 8 mm átmérőjű grafitrúd, amelyre üvegharangot húzunk aképződött klór leszívatására. Katódként 3 mm átmérőjű vasdrótot használunk,
amelyet kvarcból vagy keményüvegből készült védőcsőbe helyezünk olyan módon,
hogy a vasdrót elmozdulását, illetve a védőcsővel való érintkezését a benne el
helyezett dugóval megakadályozzuk. A védőcsőtörése így is gyakran bekövetkezik, ezért tarta
lékról kell gondoskodnunk. A katód védőcsö
vén használt dugón kis furatot készítünk, hogy
az elektrolízis megindulásakor képződő kis
mennyiségű hidrogén el tudjon távozni. Az olvadék hőmérsékletét Pt/Ptj0Lh termoelemmel
mérhetjük. Amennyiben má<! termoelemet használunk (pl. vas/konstantáúJPagy nikkel/króm-- nikkel), akkor azt nem helyezhetjük közvetle
nül az olvadékba, hanem ketnényüvegből készült védőcsővel látjuk el. Az elektródokat kb.
1 cn4 mélyen mártjuk be az olvadékba. Az elek
trolízist 8—10 A áramintezitással és kb. 10 Vfeszültséggel végezzük. Túl nagy feszültségnél
anódeffektus lép fel. Az elektrolízist kb. fél óránát folytatjuk.
A képződött fémlítium az elektródok kieme
lése után nagyobb golyó alakjában az olvadék
felületén úszik. A regulust előmelegített vaskanállal emelhetjük ki. A lítiumot széles nyakú,
gondosan vízmentesített paraffinolajjal töltöttlombikba tesszük. Az elektrolit lítiumtartalmának pótlására az olvadékhoz
kevés megolvasztott LiCl-ot adunk, és az elektrolízist tovább folytatjuk. Az áram-
kihasználás a KC1—LiCl elektrolízisekor kb. 70 %. A sóolvadék maradékától valómegtisztításra a lítiumot kb. 200 C°-on paraffin fürdőben (a paraffint előzőleg
Na-dróttal szárítjuk) megolvasztjuk, majd vízmentes benzinnel mossuk. A lítium ezüstfehér, csillogó fém. Olvadáspontja hőmérsékletén az üveg vagy
kvarc szilikátvázát redukálja. Ampullázását az 54.5.2.1. ábra szerint nagyváku
umban végezzük.
Az előbbi eljárással kapott lítium 2—5 % káliumszennyezést tartalmaz. Ennek
a terméknek hidrogénáramban 700—800 C°-on LiH-dé való alakítása és termikusbontása útján kapjuk a tiszta lítiumot.14
54*8.3. Kalcium (Ca)
Fémkalciumot legegyszerűbben olvadékelektrolízissel állíthatunk elő.15 100
súlyrész kalcium-klorid és 16,5 súlyrész kalcium-fluorid keverékét porcelán tégely
ben megolvasztjuk, majd vastálba öntjük. Lehűlés után az anyagot durván el-
efaklrnn^- — elektród- szén
í f '/ sSF'~!' vm , termoelem
üv eg
védöbu ra
po rcelán tégely
va stégely
54.3.2.2. ábra. L ítium előállítása
egyszerű olvadékelektrolízissel
M Quntz, A . , Broniewski, W .: J. chim. phis. 7, 468 (1909).18 Lengyel B .: Matematikai és Természettudományi Értesítő X I V , 42 és 249 (1898).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 179/366
F É M E K E L Ő Á L L Í TÁ S A E L E K T R O L ÍZ IS S E L 181
porítjuk. Az így előkészített sókeveréket használjuk fel az elektrolízisre. Az elektronlízist korundtégelyben végezzük 800 C°-on, grafitanód (0 kb. 14 mm) és vaskatód
(0 2 mm) alkalmazásával. A katódot úgy helyezzük el, hogy az olvadék felületévelmég éppen érintkezzék. Az elektrolízist 8 V feszültséggel és 8 A intenzitással
végezzük. A kiváló fémkalcium a katódon golyóvá olvad össze; ha már elég naggyáfejlődött, akkor a katódot kihúzzuk az olvadékból. Ilyenkor a kalciumgolyó azolvadék felületén úszik, ahonnan előmelegített vaskanállal kiemelhető. Az így elő
állított fém a kereskedelmj. forgalomban kapható kalciumhoz hasonlóan kis mennyiségű nitrid- és oxidszennyezést tartalmaz. Tisztításáravákuumdesztillációt alkalmazunk.16
A fémkalcium vákuumdesztillációjáboz az 54.3.3.1.ábrán látható berendezést használjuk. 40g nyers kalciumot 1 mm falvastagságú elektrohtvasból vagy V 2 A-acél-
ból készült tégelybe helyezünk. A tégely alatt porce
lán lemezke van, mely megakadályozza, hogy az acél
tégely a külső kvarccsővel érintkezzék. A kvarccsőbefémből (rézből, nikkelből vagy acélból) készült hűtőnyúlik. A csiszolatot ólom csőkígyóval hűtjük.
A desztilláció első fázisában, 10_ 1—10-2 Hgmm-en,
700 C°-on nagyobbrészt az alkálifémek (főleg Na) és ke
vés Ca rakódik le a belső hűtőcsőre. Az oldott gázok le
adása folytán a megfelelő vákuum csak akkor biztosítható, ha a berendezést diffúziós szivattyúval (folyékony levegővel való kifagyasztás közben) folytonosan
szívatjuk. Lehűlés után tisztított argongázzal vagy szén--dioxiddal töltjük, és a hűtőcsövet kicseréljük. Ilyenkor
az alkálifém rendszerint a levegőn meggyullad. A fém-
nátrium kidesztillálását követő fő desztilláció 10“3 Hgmm-nél kisebb nyomáson és kb. 850 C°-on megy végbe. A fémkalcium ilyenkor a belső, hűtött csövön ezüst
színű, fénylő, hosszú krisztallitok alakjában válik ki.
Amennyiben 100—150 C°-kal magasabb hőmérsékleten desztillálunk, a fém tömörebb szerkezetű lesz. A fém diszperzitásfoka a hűtőcső (hűtőujj) hőmérsékle
tétől is függ, ugyanis minél erősebb a hűtés, annál finomabb eloszlású a kiválófém. Lehűtés után a berendezést argonnal vagy C02-dal töltjük meg, és a hűtő-
csőről a fémet megfelelő spatulával leszedjük. Ha a desztillációt többször megismételjük, 99,7% tisztaságú kalciumot kaphatunk (század százalékot kitevő szeny-
nyezések: O, N, Cl, Fe, S, Mg).
54.3.4. Magnézium (Mg)
A magnézium olvadékelektrolízissel való előállítására főleg a fém kloridjaíhasználhatók. A magnézium-klorid azonban kristályvizet tartalmaz (MgCl2*6H20),
és 180 C°-ra hevítve 4 H20-t veszít. Magasabb hőmérsékleten sem adja le fennmaradó kristályvizét anélkül, hogy hidrolízissel részben bázikus sóvá vagy oxiddáne alakulna át. A hidrolízis csökkentésére kristályos magnézium-klorid és ammóni-
\hűtővíz
54.3.3.1. ábra. Kalcium
desztillálása
ie Antropow, A ., Germán, E .: Z. physik. Chem. 127, 209 (1928).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 180/366
182 F É M E K
um-klorid keverékét hevítjük. Ha kiindulási termékként karnallitot (KCl-MgCl2*6
H20) használunk, 160C°-on, 1 atm nyomáson KC1 és MgCl2«2H20 keveréket kapunk, amely további hevítéssel leadja a vizet anélkül, hogy számottevő hidrolíziskövetkezne be. Az elektrolízishez az 54.3.4.1. ábrán vázolt berendezést alkalmazzuk.17
Célszerű frissen víztelenített, még megolvadt anyagot használni. A 10 cm0 -jű és ugyanilyen magasságú vastégelyben levő olvadékot tokos kemencében
gyenge vörösizzásig (700—750 C°) hevítjük fel. A vastégely kátédként is szolgál.
A kontaktust a tégely felső részéhez illeszkedő rézgyűrű biztosítja. Az anód kb. 25mm-es grafitrúd. Kezdetben 10 amperrel elek-
trolizálunk. Az elektrolit felületén finom hab
képződik, mivel a még nyomokban jelenlevő
víz elbomlik. Kb. 1/4óra múlva az áramerősséget 12—13 A-re emeljük (kapocsfeszültség 8 V),
1000 g olvadékra összesen 180—200 amperórát
használunk fel. A képződött magnézium egy
része az olvadék felületén, másik része a tégelyalján gyűlik össze, mivel a fém és az elektrolit
sűrűsége nem különbözik számottevően. Azelektrolízis befejezésekor a felületen levő mag
nézium golyókat előmelegített vaskanállalemeljük ki, majd a tégely egész tartalmát vas
tálba öntjük. Lehűlés után az anyagot durvánelporítjuk, és a magnézium golyókat kiválogat
juk. A kapott magnéziumot vízzel vagy alkohollal mossuk, és megszárítjuk. Az áramkihasználás kb. 50 %-os. A nyersfém kevés
Fe-, A1-, Si-, illetve nitrogénszennyezés mellett nagyobb mennyiségű, kb. 3%
kloridot tartalmaz. A nyerstermék átolvasztással, majd vákuumszublimációvaltisztítható.
~ 1° )
samott-
tok
54.3.4.1.- ábra. Berendezés magn ézium előállítására
54.4. Fémek előállítása termikus bontással
Ez az eljárás kis mennyiségű, de igen tiszta fém előállítására használatos. A jodidok elbontása nagy vákuumban van Arkel—de Boer módszerével végezhető.
Azidok elbontásával, szintén nagyvákuumban, Suhrmann és Clusius rendkívüli
tisztaságú, gázmentes alkálifémeket állított elő.
54.4.1. Titán (Ti)
Kis mennyiségű fémtitánt (20—30 g) jodidjának (Til4) termikus bontásával
készíthetünk.18 Ilyenkor azonban kiindulási anyagként nem a higroszkópos tulaj
donságú jodidját használjuk, hanem nyerstitánból és jódból indulunk ki. Ezek
17 Mutter, E., Reuther, ff . : Elektrochemisches Praktikum, Neunte Auflage, Theodor
Őteinkopff, Dresden — Leipzig, 1953, 355 old.18 van Arkel, A. E., de Boer, J. f f . : Z. anorg. alig. Chem. 148, 345 (1952); Fást, J. D.: Z. anorg. alig. Chem. 241, 42 (1939).Campbell, J. E., Jaffe, R. / . , Blocher, J. M., Gurland, J., Gonser, B. W.: Trans.
Electrochem. Soc. 93, 271 (1948).Gonser, B. W.: Metall Progress 55, 193 (1949);
Litton, F. B., Gonser, B. W.: Metall Progress 55, 346 (1949).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 181/366
F É M E K E L Ő Á L L Í TÁ S A TE R M IK U S B O N T ÁS S A L 183
reakciójában képződő Til4csak átmeneti termék. Az*oxid, nitrid és karbid a jóddalnem reagál, így a nyerstitán nemfémes szennyezései nem kerülnek a termékbe.
Az eljárás hátránya viszont, hogy számos fém, pl. a Zr, Hf, Th, V, azonkívül a B ésSi, valamint a hasonlóan viselkedő A1 és Fe is átkerül a késztermékbe. Ezért, ha a
felsorolt fémektől mentes titánt akarunk előállítani, a felhasznált nyerstitánnak azelőbbiektől mentesnek kell lennie.
A termikus bontást kemépyüvégből készült, az 54.4.1.1. ábrán vázolt edényben
hajtjuk végre. A háromszög alakban elhelye
zett A, B és G jelzésű, 6 mm 0 -jű volfrám-
botok be vannak forrasztva az üvegbe. Atitán a 400 mm hosszú és 0,04 mm 0 -jű D4,
D2volfrámhuzalon válik le. A G edénybe 40 g nyerstitánt, a H jel
zésű, törőszeleppel (L) zárt csőbe pedig 12 g
jódot forrasztunk be. A berendezést 10-3
Hgmm-nél kisebb nyomásra leszívatjuk, és
a fémet gáztalanítás céljából kb. 500 C°-ra
hevítjük. A gázleadás megszűnése után avolfrámszálakat a szükséges áramerősséggel(kb. A) 1400 C°-ra felfűtjük. A szál hő
mérsékletét optikai pirométerrel ellenőrizhet jük. Ekkor a berendezést lehűlés után J-nél
leforrasztjuk, és elektromágnes segítségével
az L válaszfalat az N acélgolyóval széttör jük. Amennyiben a H csőben levő jódot nem
különítjük el szeleppel, akkor a készülék gáztalanítása alatt a H csövet folyékony
levegővel hűtjük. A következő lépésként a jódot átszublimáljuk a G edénybe. Ilyenkor előfordul, hogy a jód titánnal való reakciója (200 C° körüli hőmérsékleten a
reakciósebesség már igen nagy) fénytüneményt okoz. Ezután az edényt K -nál leforrasztjuk, és kemencében 550 C°-ra hevítjük, miközben az izzószálakat kb.
1400 C°-ra fűtjük. A szál hőmérsékletét a kísérlet folyamán optikai pirométerrelfolyamatosan ellenőrizzük, mivel a titán leválása folytán vastagodó szál hőmérsékletének fenntartása céljából a fűtőáram folyamatos növelése szükséges.
A berendezésben lefolyó egyensúlyi folyamatok jelentős mértékben függnék a
nyersfém hőmérsékletétől. így 250 C° alatt Til4képződik, amely a magas hőmérsékletű szálon bomlást szenved. Magasabb hőmérsékleten viszont a Til4 reagál anyerstitánnal Til2képződése közben, amely jelentős mértékben kisebb gőznyomású, A Til4bomlása csak 500 C° feletti hőmérsékleten válik olyan mértékűvé, hogy az
izzószálon észrevehető legyen a titánleválás. A titán leválása folyamán a rendszerben levő kis mennyiségű levegő követ
keztében a termék kevés nitriddel szennyeződhet. Ennek elkerülésére úgy járunk
el, hogy először csak a Dl fűtőszálat fűtjük fel, majd bizonyos idő után az előbbi
szál fűtését megszüntetjük, és a Z>2szál fűtésével folytatjuk a kísérletet, amikor is atovábbiakban nitridmentes titán válik ki. Nagyobb mennyiségű titán leválása (5 mm
szálátmérő elérése) után a fűtőáramot 200 amperig kell növelni. A kísérlet folyamán a kemence fűtését teljesen beszüntetjük, mivel a meg
vastagodott Ti-huzal elegendő hőt termel ahhoz, hogy az egész edényt megfelelőhőmérsékleten tartsa.
A B C

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 182/366
184 FÉMEK
Kellő tisztaságú titánból 'kiindulva, ezzel a módszerrel gyakorlatilag teljesen
tiszta titánrúd készíthető, melynek volfrámtartalma 0,01 % alatt van. A kapottfém tömör és hidegen is jól kovácsolható. Amennyiben por alakú titánra van szükségünk, a tömör fémet kb. 600 C°-on
H2-áramban kezeljük. A képződött hidrid merev és jól porítható. Vákuumban1000 C°-ra hevítve a hidrogént leadja.
Újabban ezt a módszert kidolgozták nagyobb mennyiségű (700 g) titán előállítására is.
54.4.2. Cirkóniui Zr)
Jodidjának termikus bontásával állítjuk elő.19Keményüvegből készült edénybe (54.4.2.1. ábra) két, 2—6 mm 0 -jű volfrámdrótot forrasztunk be. A két volfrám-
drót között 0,04 mm 0 -jű volfrámszál van. A bal oldalonlevő kisebb edénybe kevés foszfor-pentoxid felett szárított jódot teszünk. 1 g jód nagyobb mennyiségű cirkónium előállítására is elegendő, mivel a reakció folyamána jód nem használódik el. A berendezést nagy vákuumban evakuáljuk, és az edénybe vitt cirkóniumot 550 C°-on gáztalanítjuk, mialatt a toldalékban levő jódot fo
lyékony levegővel hűtjük. Gáztalanítás után a volfrám-szálat 1800 C°-ra fűtjük fel, végül az evakuált edénytszívatás közben leforrasztjuk.
A berendezést elektromos kemencében 600 - 650C°-ra hevítjük, és a fűtőszál hőmérsékletét kb. 1800 C°-ra állítjuk be. A jód a cirkóniummal tűztünemény közben egyesül Zrl4-dá, amelynek zöldessárga gőze azizzó volfrámszálon termikus bomlást szenved. A fűtőszál hőmérsékletét a kísérlet közben pirométerrel ellen
őrizzük, és az áramerősség változtatásával a szükségeshőfokon tartjuk. Eközben az áramerősséget fokozatosan 0,25 A-ről 200 A-re növeljük. A kb. 10 mm hosszú és 5 mm 0 -jű cirkóniumrúd volfrámtartalma 0,01 %körül van.
54.4.8. Hafnium (Hf)
van Arkel—de Boer módszere a hafnium előállítására lényegében hasonló acirkónium előállításához. A fűtőszál hőmérsékletét kb. 1600 C°-on tartjuk20.
54.4.4. Tórium (Th)
Igen tiszta és oxigénmentes tórium nyerhető van Arkel—de Boer módszereszerint.21 A berendezés és a munkamenet lényegében hasonló a titán előállítására
54.4.2.1. ábra. Cirkónium előállítása
19 van ArTcel, A. E., de Boer, J. H.: Z. anorg. alig. Chem. 148, 345 (1925). *° van Arkel, A. E.: Metallwirtschaft 13, 405 és 511 (1934).
21 van Arkel, A. E., de Boer, J. H .: Z. anorg. alig. Chem. 148, 345 (1925).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 183/366
A L K Á L IF É M E K E L Ő Á L L ÍT Á S A A Z 1 D O K E L B O N T Á S Á V A L 185
alkalmazott eljáráshoz. A fűtőszál hőmérsékletét 1700 C°-on tartjuk. Nyersanyagként bármely nyerstórium használható, feltéve, hogy nem tartalmaz szennyezéskéntolyan fémet, amelynek jodidjai szintén termikus bomlást szenvednek.
54.4.5. Alkálifémek előállítása azidok elbontásával
Az azidok elbontására használható berendezést (54.4.5.1. ábra) magas olvadáspontú üvegből készítjük. A jénai „Oerate” üveget a reakció hőmérsékletén a Na, K,Rb és Cs nem támadja meg, mivel azonban a Li reakcióba lép az üveggel, ez a módszer lítium előállítására nem alkalmazható.
Amennyiben abszolút tiszta alkálifémet kívánunk előállítani, a berendezést
csiszolatok és csapok nélkül célszerű megoldani. Az azidokat achátmozsárban finoman eíporítjuk, és az E edénybe helyezzük(az alkalmazott mennyiség kb. 10 g NaN3, illetve egyéb alkáli-azidokból kb. 6 g).
A RbN3-ot és a CsN3-ot nem közvetlenül az edénybe, hanemegyik oldalán nyitott kvarctokbahelyezzük, s ez után az E edényta berendezés többi részéhez forrasztjuk. A berendezést nagyszívássebességű elővákuum-szi-
vattyúval (pl. higanygőz-szivaty-tyúval) evakuáljuk és kifűtj ük. A B1 és B2 kifagyasztócsapdákat az egész kísérlet folyamánfolyékony levegővel hűtjük. Az E edényt elektromos kemencében12 órán át 200 C°-on tartjuk, állandó szívatás közben. Csakisilyen óvatos és hosszadalmas ki
fűtés után emeljük az edény hőmérsékletét az azidok bomlásáig.
A hőmérséklet emelését óvatosan kell végezni, hogy a túlságosan heves bomláskövetkeztében képződő N2 ne ragadja magával az azid egy részét. Az egyes azidokbomlási hőmérséklete a következő: NaN3 275 C°, KN3 355 C°, RbN3 396 C°, CsN3390 C°. A NaN3 olvadáspontjának elérése előtt bomlik, a KNa 343 C°-on olvad(a RbN3 olvadáspontja 321 C°, a CsN3-é 326 C°). A bomlás megindulását a nyomásnövekedése jelzi. A hőmérsékletet úgy szabályozzuk, hogy p nyomás 0,1 Hgmmfölé ne emelkedjék. A hirtelen nyomásnövekedés megakadályozására nagyobb (kb.
8 literes) leszívatott pufferedényt csatlakoztatunk a berendezéshez. A bomlásihőmérséklet elérése esetén (különösen a KN3 előállításakor) a bomlás gyakran csak3—4 óra múlva indul meg. Ennek ellenére óvakodjunk az azidok túlhevítésétől,mivel robbanásszerű bomlás következtében a hirtelen nyomásváltozás a berendezéstszétvetheti. A bomlás befejeződése után a nyomás újra lecsökken, és a képződöttalkálifém az E edényből az szedőbe desztillál át. Ezt követően D-nél és C-nél azedényt leforrasztjuk. Utána üzembe helyezzük a vákuumszivattyút, és az A higanyszelepet megnyitjuk. Ha a nyomás 10"6 Hgmm alá csökken, az alkálifém már enyhe
54.4.5.1. ábra. Alkálifémek előállítása azidokelbontásával

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 184/366
186 F É M E K
melegítés hatására átdesztillál az F2 szedőbe. Ezt követően a készterméket nagyvákuumban leforrasztjuk.
Az eljárás hosszadalmas, legalább 3 napig tart. A kitermelés NaN3-ra 100 %, KN3-ra 80 %, RbN3-ra 60 %, CsN3-ra pedig 90 %.22
54.5. Alkálifémek tisztítása és eltartása
54.5.1. Alkálifémek tisztítása YákuumdesztilláciÓYal
A 54.5.1.1. ábrán látható az alkálifémek desztillációjára alkalmas berendezésvázlata. A készüléket magas olvadáspontú keményüvegből készítjük. Az edényt
A-ná.\ leforrasztjuk, és F-en keresztül higanydiffúziós szivattyúhoz csatlakoztatjuk. Állandó szívatás közben 400 —450 C°-on több órán át gáztalanítunk, majda berendezést F-en keresztül szárazN2-gázzal töltjük meg. Ezután .4-nálmegnyitjuk az edényt, és nitrogén-el-lenáramban a B csőszakaszba könnyű-benzinnel mosott és alkálifémet tartalmazó fedett csónakot helyezünk. Akönnyűbenzint először nitrogén-ellen-
áramban az A nyíláson keresztül elpárologtatjuk, majd a B edényt újra
beforrasztjuk, és 10“ 4 Hgmm nyomásra leszívatjuk. A B edényt és a C szűkületetelektromos kemencében felmelegítjük, és az alkálifémet az ampullákat tartalmazó D szedőbe desztilláljuk át. Egyidejűleg az E hűtőcsapdát folyékony levegővel vagyszén-dioxid—aceton hűtőkeverékkel hű tjük. Lehűlés után azedényt újra száraz nitrogénnel töltjük, és az ampullákat leforrasztjuk. ^
Á lítium desztillációjához célszerűen az alkáliföldfémek
desztillációjánál tárgyalt berendezést (54.3.3.1. ábra) használjuk.
54.5.2. Az alkálifémek eltartása és használatra való előkészítése
A lítiumot petroléter alatt, a nátriumot leginkább petróleumalatt tartjuk el. A nátriumot és a lítiumot használat előtt szűrőpapírral megszárítjuk, és abszolút alkohollal, majd petroléterrelmossuk. A káliumot kevés alkoholt tartalmazó éterben a barnaszínű felületi réteg eltávolításáig forgatjuk, majd petroléter benmossuk. A céziumot és a rubídiumot paraffinolaj alatt tartjukel. Az olajat használat előtt benzollal vagy petroléterrel mossuk le.
Ampulláknak vagy üveggolyóknak a megtöltését oxidmen-tes alkálifémekkel az 54.5,2.1. ábrán vázolt módszerrel végezzük. Az edényt és a benne levő kapillárist vákuumban kifűtjük. Lehűlés és száraz nitrogénárammal való töltés után az ábrán megjelölthelyre oxidmentes, desztillált, tömör nátriumdarabot teszünk.
vákuum
54.5.2.1. ábra. Üvegedénykék
töltése oxidmentes alkálifémmel
** Suhrmann, R., Clausius, K .: Z. anorg. alig. Chem. 152, 52 (1926).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 185/366
P L A T I N A F É M E K 187
Az edényt újra evakuáljuk és újra felmelegltjük. A fém megolvad, és az edényalsó részébe folyik le. A folyékony fémet kevés száraz nitrogén beáramoltatásá-val a golyóba nyomjuk, amíg az félig meg nem telik. Ezután a nitrogént újra leszívatjuk. Ilyenkor a kapillárisból kifolyik a megolvasztott fém, de a golyó alsófelében megmarad. Lehűlés után a berendezést száraz nitrogéngázzal telítjük. A csapot levéve, az edény alsó részét az olajfürdőbe tesszük, a fém megolvadásaután a golyót kiemeljük és leforrasztjuk. A fel nem használt nátriumot toluolvagyxilol alatt összeolvasztjuk. Kis mennyiségű nátrium megsemmisítésére, illetve azedény kimosására alkoholt használunk.
54.5.3. Finom eloszlású alkálifémek előállítása
Vastag falú, visszafolyós hűtővel ellátott 1 literes lombikban kb. 5 g előzetesenmegtisztított káliumot 200 ml vízmentes toluolba (Na-hoz xilolt, Li-hoz petróleumot használunk) teszünk, majd forráspontig melegítjük. Ezután a főzőlombikotzárjuk, és ronggyal körültekerve erősen rázogatjuk (védőszemüveg, azbesztkesztyű). A rázogatás idejétől és mértékétől függően a fém egészen por alakúvá is diszpergál-ható. (Részletesebb leírás 74.5.5. és 74.6.2.)
Lényegesen finomabb eloszlású alkálifémhez úgy juthatunk, ha azt folyékonyammóniában oldjuk, és az ammónia elpárologtatósa után visszamaradó ammin-komplexet az ammónia leszívatásával (hidegen, hogy az amidképződést megakadályozzuk) elbontjuk. Az így előállított fémpor rendkívül aktív, és a levegőn magától meggyullad.
54.6. Platinafémek
54.6.1. Platinaszivacs
A platinaszivas előállítása céljából (NH4)2[PtCl6]-ot platina edényben vörös-izzásra (kb. 600 C°-ra )hevítünk. A kiizzított szivacsot híg sósavval, majd desztillált vízzel kifőzzük, végül az izzítást megismételjük.
54.6.2. Platina- és palládiumazlbeszt
Csekély Pt-tartalmú (0,1—1 %) azbeszt előállítására a következő eljárás alkalmas. Az azbesztet megfelelő mennyiségű H2[PtCl6] •6 H20 alkoholos oldatávalátitatjuk. A szükséges platinasó-mennyiség felvétele után az alkoholos azbesztetmeggyújtjuk. Kiégetés után platina bottal összekeverjük.
Több (5—10 %) platinát tartalmazó azbesztet úgy készíthetünk, hogy az azbesztet H2[PtCl6]*6 H20 megfelelő koncentrációjú vizes oldatába mártjuk. Az ol
datot ezután híg lúggal gyengén meglúgosítjuk, és kemencében, 300—400 C°-on aplatinát nátrium-formiáttal kiredukáljuk. Végül az azbesztet hideg vízzel alkáli-mentesre mossuk, és néhányszáz fokos kemencében kiszárítjuk.
A megfelelő palládiumazbeszt a platinaazbeszthez hasonló módon állíthatóelő.23
23 Brauer, G.: Handbuch dér práparativen anorganischen Chemie. Stuttgart, 1954, 1169. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 186/366
188 F É M E K
54.6.3. Platinakorom
H2[PtCl6] 5 %-os vizes oldatát forralás közben NaaC03-tal semlegesítjük, ésforró nátrium-formiát-oldatba öntjük.24 A kapott fekete csapadékot forró vízzeldekántálva mossuk, szűrőpapír közt nyomkodva és Pa0 5 fölött exszikkátorbanszárítjuk. A termék hidrogénnel szemben igen aktív.
54.6.4. Palládiumkorom
Palládium(II)-klorid vizes oldatából nátrium-formiáttal redukálva képződik.25Szobahőmérsékleten a folyamat lassú, de 50 C°-on már pillanatszerű.
54.6.5. Plátlnázőoldat
Üvegfelületeknek platina réteggel való bevonására használható platinázó oldata következőképpen készíthető.
A felsorolás sorrendjében összekeverünk1 g H J P t C y-ot,
3 ml abszolút alkoholt,10 ml telített alkoholos bórsavoldatot.20 ml, terpentinből és levendulaolajból álló elegyet, olyan összetételben, hogy
a platinázóoldat viszkozitása ne legyen túlságosan kicsi. Az oldatot az üvegfelületre ecsettel rákenjük, majd negyedórán át 500 C° körüli hőmérsékleten hevítjük.
24Otdbier, A., Maisch, O.: Bér. dtsch. Chem. Ges. 52, 1370 (1919).28 Böttger, H.: Jahresber. d. phya. Vereins, Frankfurt a/M ., 11. old. (1872— 73).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 187/366
55. Hidrogén? egy öletek
55.1. Nemfémek hidrogénvegy ületei
55.1.1. Hidrogé n-fluorld (HF)
Vízmentes hidrogén-fluoridot vízmentes kálium-hidrogén-fluoridból 500 C°-on
állíthatunk elő.1 A desztilláoiót rézből készült berendezésben végezzük. A KHF2-ot
felhasználás előtt gondosan ki kell szárítani. Ennek érdekében a szárítandó anyagot
az 55.1.1.1. ábra szerinti, rézből készült retortában száraz levegőáramban 150 C°-rahevítjük. Ez a művelet több napot igényel. A lehűlt sót golyósmalomban elporítjuk.
A finom eloszlású porral csak gázálarcot használva ajánlatos dolgozni.
A hidrogén-fluorid fejlesztése végett az A edényt megtöltjük KHFa-dal. A fémkónuszos csatlakozásokat grafit —
-paraffinolaj-szuszpenzióval kenjük.
A terméket konyhasó - jég-hűtéssel
ellátott csapdában gyűjtjük össze. Az ehhez csatlakozó második szá
razjég —acetonos kifagyasztó egész
ségvédelmi célokat szolgál. A hevítést lassan fokozzuk olyan módon,
hogy kb. 3/4 óra alatt érjük el az
500 C°-ot. A desztilláció kezdeténelőször víztartalmú hidrogén-fluo
rid kondenzál a hűtő végén elhe
lyezett kis platina edényben. Az itt összegyűlő, kb. 5—10 ml-es próbákon ellenőrizzük a desztillátum víztartalmát a következőképpen: ha a mintával átitatott
szűrőpapír darabka azonnal zselatinosodik, majd elszenesedik, akkor a desztillátum már gyakorlatilag vízmentes. Ezt elérve, a platina tégelyben újabb 10 ml-esadagot fogunk fel, és azután csatlakoztatjuk csak a hűtőcsapdát oly módon, hogy
a fűtést ilyenkor átmenetileg megszakítjuk, s a hűtő végén levő savcseppet szűrőpapírral leitatjuk. Ezt követően illesztjük rá a hűtőcsapda kónuszos csatlakozását,
majd rácsavarjuk a második, biztonsági hűtőcsapdát, amely kettős célt szolgál:egyrészt megakadályozza, hogy a HF desztilláció közben szennyezze a levegőt, más
részt kifagyasztja a levegőben levő vízgőzt. Kb. 3 órás desztilláció után a HF-fej-
lődés lecsökken, a fűtést megszüntetjük, s a felfogóedényt rézkónuszokkal elzárjuk. A retortában visszamaradó fluoridot bifluorid előállítására használhatjuk fel.
A nyersterméket a kevés KF-szennyezéstől ismételt desztillációval tisztíthat juk meg (op. —85 C°, fp. 19,5 CP).
55.1,1.1. ábra. Hidrogén-fluorid desztillálása
1 Wartenberg, H., Klinkotl, Q.: Z. anorg. alig. Chem. 193, 409 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 188/366
190 H I D R O G É N V E G Y Ü L E T E K
A hidrogén-fluorid vizes oldatának laboratóriumi előállítására igen ritkán vanszükség. 35 %-os oldata műanyag vagy paraffin edényekben kapható a kereskedel
mi forgalomban.
55.1.2. Hidrogén-klorid (HC1)
Jól szabályozható sósavgázáram könnyen előállítható tömény sósavnak tömény kénsavba való adagolásával. Az 55.1.2.1. ábrán látható berendezés lényegesrésze a kapilláris cső, amelyet használat előtt tömény sósavval megtöltünk, hogy a
könnyebb sósav a nehezebb tömény kénsavat tartalmazó edényaljára csurogjon. Az A csapos edénybe 200 ml tömény kénsavat
töltünk, és a B csapos tölcséren keresztül adagoljuk az 1,18 fajsúlyú sósavat. A kénsavval egyenlő térfogatú sósav hozzáadásaután célszerű a kénsavat megújítani. A képződött gázt cc.H2S04-as mosóval szárítjuk (P20 5 nem alkalmazható, mert illófoszforvegyületek képződhetnek) és folyékony-levegős csapdában kifagyasztjuk.
Igen tiszta vizes HCl-oldatot a kereskedelmi tömény sósavból a következőképpen állíthatunk elő. Az oldatot 20 %-osrahígítjuk, kevés KMn04-ot adunk hozzá; a bróm- és a jódszennye-zés eltávolítására ismételten kifőzzük az oldatot, majd kvarchű
tőt használva ledesztilláljuk.
55.1.3. Hidrogén-bromid (HBr)
55.1.2.1. ábra. Vízmentes HBr előállítására kényelmesen alkalmazható aHC1 fejlesztése tetralin és a Br2 reakciója:
C10H12 + 4 Br2 = C10H8Br4 + 4 HBr
A tetralint felhasználás előtt Na2S04-tal szárítjuk és desztilláljuk (fp. 207 C°) *
A reakciót gázfejlesztő készülékben hajtjuk végre oly módon, hogy a tetralinho2— amelybe kevés vasreszeléket teszünk — csepegtető tölcséren át adagoljuk abrómot. A gázfejlesztő lombikot 30—40 C°-os vízfürdőbe helyezzük, és a gázt kismennyiségű brómszennyezésének eltávolítása céljából tetralinnal töltött mosópalackon vezetjük át. Nedvességnyomok eltávolítására —60 C°-os hűtőcsapdáthasználunk. Ha az előállított HBr-ot további tisztításnak kívánjuk alávetni, akkora folyékony levegővel kifagyasztott HBr-ot szublimáljuk. A középső frakció igentiszta HBr (fp. —67 C°). Az eljárás hátránya, hogy a brómnak csak a fele-hasznosítható.
Elemi szintézissel a HBr az 55.1.3.1. ábrán vázolt berendezésben állítható elő. A hidrogéngázt az A kénsavas mosón át a Br2-mal töltött B edény aljára vezetjük. A brómgőzökkel kevert hidrogéngázt ezután a kb. 50 cm hosszú, 2—4 cm 0 -jű ,keményüvegből készült, elektromosan fűthető D reaktorcsőbe vezetjük. A reaktorcsövet üveggyapottal kevert platinaazbeszttel vagy platinázott szilikagéllel tölt
jük meg. A függőleges helyzetű F cső Raschig-gyűrűre vagy üvegtörmelékre szórtvörösfoszfort tartalmaz. A továbbiakban csatlakozó G mosóban kevés desztillált vízvan, hogy a gázból a képződő foszforvegyületeket kimoshassuk. A HBr —H2 gázkeveréket a CaCl2-dal vagy CaBr2-dal (az utóbbi ajánlatosabb) töltött H szárító-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 189/366
H A L O G É N - H I D R O G É N E K 191
csövön átvezetve szárítjuk, és végül a J hűtőcsapdában folyékony levegővel kifagyasztjuk belőle a HBr-ot.
A reakció megindítása előtt hidrogénárammal kiűzzük a berendezésből a levegőt, majd az E elektromos kemencét 350 C°-ra fűtjük fel. A B edénybe kb. 50 mlbrómot teszünk. Az elreagált bróm pótlása a C csapos tölcséren át történik (a nyomáskiegyenlítődést azzal biztosítjuk, hogy a csapos tölcsér edényét csatlakoztat
juk az A és B edényt összekötő csőhöz). Ügyelnünk kell arra, hogy a gázkeverékmindig jelentős mennyiségű H2-felesleget tartalmazzon. Ezt a hidrogéngázárammegfelelő beállításával (melynek változását a konc. H2S04-as A mosópalackban
észlelhetjük), valamint a folyékony bróm enyhe melegítésével vagy hűtésével szabályozhatjuk. Figyelnünk kell arra is, hogy a katalizátor esetleges zsugorodásának
eredményeképpen a D edényben nem képződött-e olyan csatorna, amelyben a H2 —Br2 gázkeverék a katalizátorral nem érintkezve változatlanul halad át.HBr-oldatot egyszerűen úgy állíthatunk elő, hogy a HBr-gázt tetralinnal töl
tött mosópalackon átvezetve, jég —konyhasó-keverékkel hűtött vizes elnyeletŐbenfeloldjuk. Mivel a HBr vízben igen hevesen oldódik, a bevezetőcsőnek célszerűen avízszint fölött néhány mm-rel kell végződnie. A vizes HBr-oldat közvetlenül iselőállítható a következő reakció alapján:
H2S04 -I- KBr = KHS04 + HBr.
Hígított kénsavat alkalmazzunk, mivel a tömény kénsav a HBr-ot Br2-máoxidálná. A reakciót úgy végezzük, hogy 120 g elporított KBr-hoz 200 ml vizetadunk, és lassan, hűtés közben, 90 ml tömény kénsavat csurgatunk hozzá. Ügyelnikell arra, hogy az oldat hőmérséklete ne emelkedjék 75 C° fölé. A teljes kénsav-mennyiség hozzáadása után az oldatot szobahőfokra hűtjük le, és a kivált KHS04-ot leszűrjük. A szűrletről a vizet ledesztilláljuk, és — ha állandó forráspontú, azeo-tróp HBr-oldatra van szükségünk — desztilláció közben a megfelelő frakciót felfogjuk (az azeotróp elegy forráspontja 126 C°). A kitermelés 85 %-os. Az így kapotttermék rendszerint kevés (0,01 %) H2S04-szennyezést tartalmaz.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 190/366
192 H I D BOGÉNVEGYÜ I jE T EK
66.1.4. HWrogén-Jodid (H l)
Vízmentes H l előállítására legalkalmasabb az elemi szintézis, melyet az55.1.4.1. ábrán vázolt berendezésben hajtunk végre. A Ha-gázt a jódot tartalmazó A edényen keresztül vezetjük, amelyből a gáz
keverék a magas olvadáspontú B csőbe kerül. Ennek a csőnek azt a részét, amelyeta Pt-azbeszt tölt ki, 500 C°-ra felfűtött kemence veszi körül. A B csőhöz először aCaI2-ot tartalmazó C jelzésű U-cső (a H l szárítására), azután a Kl-ot tartalmazóZ>-vel jelzett U-cső (a jódnyomok megkötésére), majd az E, —78 C°-ra hűtött kifagyasztócsapda csatlakozik. Utána a levegő nedvességének visszatartására P20 5-os F szárítót alkalmazunk. A berendezést célszerű úgy összeállítani, hogy a csapok meg
felelő állításával a .B csőben Összegyűlt, el nem reagált jód a hidrogénáram megfordításával az A edénybe visszaszublimálható legyen.
Az alkalmazott IÍ2-gázt gondosan oxigénmentesítjük (Pd-kontakton) és szárít juk. A jódot előzőleg megtisztítjuk a Cl2- és Br2-szennyezéstől (lásd 53.5.), és vákuumban P20 5 felett gondosan kiszárítjuk.
Miután a jódot az A edénybe helyeztük, a berendezésből a levegőt száraz Ingázzál kiszorítjuk, s azután kezdjük el a H2 bevezetését. A katalizátor felfűtéseután az A edényt olyan mértékben melegítjük, hogy a B cső hideg részébe csakigen kevés jódgőz kerüljön. Ügyelnünk kell arra is, hogy az A edény és a katalizátor között kondenzált jód ne okozzon eltömődést, ezért időnként Bunsen-égővelóvatosan elszublimáljuk onnan a jódot. A képződött Hl-ot a fagyasztócsapdábangyűjtjük össze. A nyersterméket többszörös desztillációval tisztíthatjuk (op.-0 ,9 C°, fp. - 35,4 C°).
Amennyiben tömény vizes Hl-oldatunk van, akkor a vízmentes Hl-ot előállíthatjuk úgy is, hogy az oldatot P205-dal víztelenítjük. Ilyenkor a gázfejlesztőlombikot P20 5-dal töltjük meg, és egyidejű hűtés közben a csapos tölcséren átcseppenként adagoljuk a Hl-oldatot. A fejlődő Hl-gázt P2Os-dal szárítjuk.
A vizes Hl-oldatot úgy állíthatjuk elő, hogy a szintézis útján előállított HI-gázt hűtés közben vízben elnyeletjük.
Azeotróp Hl-oldathoz az alábbi egyszerű módon juthatunk:500 ml-es lombikban 120 g jódot 150 ml vízben szuszpendálunk, és a lombikot
gázbevezető-, valamint kivezetőcsővel és ke verő vei látjuk el. Erős keverés közbenkén-hidrogén-gázt vezetünk be. A H2S-áramot úgy szabályozzuk, hogy a gáz

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 191/366
K É N - H I D R O G É N 193
állandó feleslegben legyen. A kivezetőcsövön kiáramló H2S-gázt NaOH oldat fölé Vezetve nyeletjük el. Kb. egy óra alatt a jód színe eltűnik. A kiváló kéntől az olda
tot dekantálással és szűréssel tisztítjuk meg, majd á H2S-nyomok eltávolítására
rövid ideig forraljuk.Ezután a szulfidreakciót már nem mutató oldatot desztilláljuk, és a 125—127
C° közötti frakciót felfogjuk. A azeotrop elegy forráspontja 126 C°, Hl-tartalma57 %, s = 1,70. Levegőn erősen füstölgő oldat, leforrasztott vagy leparaffinozottsötétbarna üvegben célszerű eltartani.
55.1.5. Kén-hidrogén (H2S)
A technikai FeS-ból hígított sósavval (1 :1) vagy kénsavval (1 : 6) Kipp-
készülékben fejlesztett H2S szennyezésként savgőzöket, illetve az alábbi gázokattartalmazza: H2, C02, AsH3, N2 és 0 2. A HCLot desztillált vízzel kimoshatjuk. Az AsH3-től való tisztítás céljából a CaC^-dal megszárított gázt száraz (üveggyapoton eloszlatott) jóddal töltött U-csövön vezetjük át. Az Asl3 kicsapódik, azegyidejűleg képződött H l pedig desztillált vízzel kimosható. A permanens gázok,mint H2, N2 és 0 2 olyan módon távolíthatók el, hogy a gázkeverék P2Oi-os szárításaután szárazjeges hűtőkeverékkel a H2S-t kondenzáljuk, majd desztilláljuk.
Tiszta H2S kalcium-szulfiddal és magnézium-kloriddal állítható elő a következő reakciók alapján:
MgCl2 + 2 H20 = Mg(OH)2 + 2HC1,CaS + 2HC1 = CaCl2 + H2fe.
A gázfejlesztő lombikba egy súlyrész CaS és két súlyrész MgCl2 •6 H20 keverékét tesszük, és annyi kiforralt desztillált vizet adunk hozzá, hogy híg kásakeletkezzék. A keverékből enyhe melegítésre a H2S egyenletesen fejlődik. Előszördesztillált vizes mosást alkalmazunk, a gáz szárítását CaCl2-dal és P20 5-dal végezzük. Tömény kénsav használata nem ajánlatos, mert redukálódik. Többszörikondenzálással és leszívatással teljesen oxigén- és nitrogénmentes gázt kaphatunk.
Igen nagy tisztaságú H2S elemi szintézis útján nyerhető.2 Az erre alkalmasberendezés vázlata az 55.1.5.1. ábrán látható.
1 Klemenc, A., Bankowski, O.: Z. a n o r g . a l i g . Chem. 208, 348 (1932).
13 Á l t . és s zerv etle n kém iai p ra k t . — 4 28 4 /1 1 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 192/366
194 H I D R O G É N V E G Y Ü L E T E K
A gömblombikot 250 g gondosan megtisztított kénnel töltjük meg. A hozzácsatlakozó ferde állású reaktorcső (átmérője 2,5 cm, hossza kb. 150 cm) felét borsónagyságú kaviccsal vagy üveggyönggyel töltjük. A berendezést magas olvadáspontú üvegből készítjük, mivel a reaktorcsövet 600 C°-ra kell felmelegíteni. A reaktor felső végére ólom hűtőkígyót helyezünk. A cső végéhez mosók (desztillált vizesmosás), majd többszöri kondenzációra alkalmas berendezés csatlakozik.
A reakció megindítása előtt a berendezést tiszta N2-gázzal öblítjük át. A levegőteljes kiszorítása után H2-t vezetünk be 8—9 1/óra áramlási sebességgel. Egyidejűleg a reaktorcsövet fűteni kezdjük. Ha elértük a. 600 C°-ot, a csapdákat megfelelő hűtőkeverékkel, illetve folyékony levegővel lehűtjük, és a gömblombikbanlevő ként forrásig melegítjük. A kifagyasztócsapdában laza, csillogó H2S-kristályok
válnak ki. A hűtés csökkentésével célszerű a kristályokat időnként megolvasztani,hogy a berendezés esetleges eltömődését megelőzzük. E reakció befejeztével le-forraszt juk a hűtőcsapdát, és amennyiben szükséges, a desztillációt megismételjük. Á kondenzáláshoz ilyenkor szárazjeges hűtőkeveréket használunk. Az első és azutolsó frakció nem elég tiszta, a fő frakció összes szennyezése 0,1 % alatt van(op. —85 C°, fp. —60 C°).
55.1.6. Hidrogén-poliszulfid (H 2S,)
Nátrium-poliszulfidnak sósavval végzett elbontása útján kaphatjuk:
. Na2Sx + 2 HC1 = H2Sx + 2 NaCl.
A kiindulási anyagként használt Na2S5 a következőképpen állítható elő. 2literes hosszúnyakú gömblombikban 500 g Na2S*9 H20 és 250 g S keverékét erőteljes rázás közben kb. 3 órán át vízfürdőn melegítjük. A szulfidkristály vizébenmegolvad, és a kén legnagyobb részét oldja. Lehűtés után a kapott sötét vörös-barna olvadékot 400 ml vízzel hígítjuk, azután szűrjük, és a szűrletet vízzel kétszeresére hígítjuk. Jól elzárva, sötétben az oldat néhány napig eltartható.
A savas elbontást 5 literes konyhasó — jég-keverékkel hűtött edényben végezzük. 2 kg darált jeget és 2 liter tömény sósavat összekeverünk, s amikor —15-—20 C°-ra lehűlt, erős keverés közben csepegtetjük hozzá a Na2Sx-oldatot. A képződött H2SX sárga, olajszerű folyadékként gyűlik össze az edény alján, míg felettetej szerű, vizes kénemulzió foglal helyet. Az adagolás sebességét úgy állítsuk be,hogy a hőmérséklet semmiképpen ne emelkedjék —5 C° fölé. Az egész Na2Sx-oldathozzáadása után a terméket néhány percig szobahőmérsékleten tartjuk, és a kétfázist választótölcsérben elkülönítjük. A víznyomokat a poliszulfidból úgy távolít-
juk el, hogy P20 5-ot tartalmazó üveggyapoton szűrjük át. Eltartására előzőlegforró tömény sósavval kifőzött, majd desztillált vízzel kiöblített és gondosan ki
szárított edény szükséges. A friss H2SXbenzolban színtelenül oldódik. A kitermelésa felhasznált kénre vonatkoztatva 87 %. A keverék analitikai összetétele H2S5-nekfelel meg. Olívaolajhoz hasonló, sárga folyadék. Nincs definiált fagyáspontja, afolyékony levegő hőmérsékletén üvegszerű anyaggá fagy meg, amely felmelegedéskor hosszabb hőmérséklet-tartományban lágyul. Melegítésre alacsonyabb kéntartalmú hidrogén-poliszulfidokká (H2S2, H2S3) bomlik. Krakkolása 15 Hgmmnyomáson, 110 C°-on végezhető. A H2S2-ot vákuumdésztillációval tisztán is előállíthatjuk (fp. 70,7 C°).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 193/366
A Z O X IG É N C S O P O R T E L E M E IN E K H ID K O G É N V E G Y Ü L E T E I 195
A H2S 3 melegítve H^S-j-ra, H2S-re és S-re bomlik. A H2S2-ot és H2S3-ot tartalmazó nyerstermékből a vákuumdesztillációval kinyerhető, maradékából pedig
a Ü2S3 molekuláris desztillációval különíthető el.3 A H2S4-ot a nyers H2S5-ból szintén molekuláris desztillációval kaphatjuk meg.
55.1.7. Szelén-hidrogén (H2Se)
A kén-hidrogénhez hasonlóan elemi szintézissel is előállítható. A reakció aH2S-nél leírt (55.1.5.) berendezésben hajtható végre.4 Oxigénmentes H2 és szeléngőzök keverékét égetőcsőben kb 400 C°-ra hevítjük. Kifagyasztása, valaminttisztítása a H2S előállításánál leírt módon történik. A H2Se színtelen, kellemetlen,rothadt retekre emlékeztető szagú gáz. A H2S-nél is sokkal mérgezőbb, az orr és a
szem nyálkahártyáját megtámadja, ún. szelénnáthát okoz. Előállításakor ezértigen gondosan zárt készülékeket és jól húzó fülkét kell használni. Op. ■—65 C°,fp. 42 C°.
55.1.8. Tellúr-hidrogén (H2Te)
Előállítható a tellúr elektrolitikus redukciójával,5amelyhez az 55.1.8.1. ábránvázolt berendezést használjuk.
Az üvegből készült A elektrolizáló cella Zn-lemezből álló B edényben helyez
kedik el. Hőszigetelés céljából a berendezést vastagfalú C fatokba helyezzük. Azelektrolizáló edénybe alulról a katód nyúlik be. A kató-dot a következőképpen készít jük: vékony falú, végéri .1^-forrasztott üvegcsőben megolvasztunk néhány grammtellúrt, és megdermedése előttrézdrótot nyomunk bele. Lehűlés után az üvegcsövet
gipsszel töltjük még. Felhasználás előtt az üvegcső leforrasztott végét lerepesztjük, ésaz edénybe helyezzük. Azanód ólomlemez. Az edénytaz anód felső széléig 50 %-oskénsavval töltjük meg, és kívülről szárazjegéé hűtőkeverékkel hűtjük. A kísérletetelsötétített helyiségben kell végezni, mert a H2Te a fény hatására gyorsanbomlik. Az elektrolízist 4,6 amperrel, 75—110 volt feszültséggel végezzük. Vigyáz
zunk arra, hogy az eközbén fejlődő hő és a külső hűtés hatására beálló hőmérséklet 0 C° körül legyen. A leszívott gázkeverék H2 mellett maximálisan 45 % H2Te-ot tartalmaz.
Szárítását CaCl2-dal és P205-dal végezzük. Gumicsatlakozások nem használhatók.
55.1.8.1. ábra. H 2Te előállítása katódos redukcióval
8 Fehér, F., Baudler, M.: Z. anorg. .alig. chem. 258, 147 (1949).4 Backer, H. J.: Recueil Trav. Chim. Pays Bas .62, 580 (1943).® Klemenc, A.: Die Behandlung und Reindarstellung von_Gasen. Leipzig, 1938. 186. old.
13*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 194/366
196 H I D R O G É N V E G Y Ü L E T E K
A tellúr-hidrogént folyékony-levegős csapdában kondenzáljak, amelyet a légnedvességtől P20 5-os toronnyal védünk. A H2Te csak sötétben tartható el, alacsony
hőmérsékleten kondenzált állapotban vagy szobahőfokon gázállapotban, leforrasztott üvegedényben. Zárófolyadékként higany nem használható.. Színtelen, kellemetlen, AsH3-re emlékeztető szagú gáz, mérgező, vízgőznyomoktól bomlik, parafával tellúrkiválás közben reagál. A teljesen tiszta H2Te fényérzékenységét illetőenaz irodalmi adatok ellentmondók.
55.L9. Ammónia (NH3)
Laboratóriumban gyakorlatilag nem állítják elő, mióta tiszta szintetikus NH 3
kereskedelmi forgalomban kapható. Az igen kis mennyiségű szennyezés (olajgőzök,szén-dioxid stb.) eltávolítására elegendő a gázt frissen izzított aktív szénen átvezetni. Szárítására Na2C0 3-tal, KOH-dal, BaO-dal vagy Na-dróttal töltött csőhasználatos. Végül P2Os-on vezetjük át, mivel a teljesen száraz NH 3 a P20 5-dalnem reagál. A reakció elmaradása bizonyítéka a sikeres szárításnak. Eljárhatunkúgy is, hogy az NH3-gázt kondenzáljuk, és kevés nátriumot oldunk benne. A kékszínű oldatról elpárolgó NH 3 tökéletesen száraz.
Divers-folyadék. A cseppfolyós NH 3 tenzióját a benne oldott NH4N0 3 olyannagy mértékben csökkenti, hogy a —33 C° forráspontú tiszta folyadék helyetttelített NH4N 03-os oldata 0 C°-on is eltartható. Enyhe melegítésre bármikor tiszta,
száraz NH, szabadítható fel belőle. 1 mól NH.NO, kb.\ NH 3 2 mól ammóniát tart megkötve.
Ügy állítjuk elő, hogy a teljesen száraz ammóni-um-nitrátot lombikban 0 C° alá hűtjük le, és állandóhűtés közben ugyancsak száraz ammóniát vezetünkbele. A só rövid idő alatt elfolyósodik, feloldódik a megkötött ammóniában.
65.1.10. Arzén-hidrogén (AsHa)
Előállítható az alábbi reakció szerint :6
Na3As + 3NH4Br = 3NaBr + 3NH 3 + A s H3.
A reakciót folyékony-ammóniás közegben végezzükaz 55.1.10.1. ábrán látható berendezésben. A száraz
jeges hűtőkeverékkel hűtött kondenzálóedénybe előszörkevés nátriumot teszünk, és erre kondenzáljuk a gondosan szárított ammóniát (lásd 55.1.9.). Ezután kiemel
jük az NH 3 bevezetésére használt csövet és A rögzítő-
rúdját, beletesszük a bemért arzénport, és az edény száját dugóval elzárjuk. Utánaa B adagolót elforgatva, hozzáadjuk a száraz NH4Br-ot. A fejlődő gázkeveréket vízzel mossuk, és P205-os szárítón átvezetve, folyékony levegővel kondenzál juk (op. —“113,5 C°, fp. —54,8 C°>.
55.1.10.1. ábra. A bH 3
előállítása
* Durrant, A. A., Parson, Th. Q., Robertson, D. L.: J. Chem. Soc. (London) 1934, 731.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 195/366
S Z I L Á N O K 197
56.1.11. Antimon-hl drogén (SbH3)
Előállítható antimon elektrolitikus redukciójával.7 Agyagdiafragmát három-furatú gumidugóval zárunk el. Az egyik furatba antimon rúd kerül, ez a katód, amásikba hőmérő, a harmadikba pedig gázkivezető cső. A diafragmát nagyobbüvegedénybe helyezzük, amelybe anódként ólomlemezt teszünk, A diafragmamindkét oldalára 4 n H2S04-oldatot öntünk, és az egész cellát jéggel hűtjük. Az áramerősség 5 A/dm2 katódfelület. A katódon képződő gáz SbH3-tartalmú H2,amelyből P20 6-os szárítás után az SbH 3 folyékony levegővel kondenzálható.
Hőfejlődéssel bomló gáz, ezért könnyen robban, op. —91 C°, fp. —18 C°.Forráspontja felett már elemeire bomlik. Igen mérgező.
55.1.12. Szilánok
A szilánok magnézium-szilicidből híg sósav hatására, vagy — lényegesen jobbkitermeléssel — folyékony-ammóniás közegben NH4Br-dal állíthatók elő.8 A kiindulási anyagként használatos Mg2Si a 67.12.1.-ben leírt módon állítható elő. A szilánoknak a
Mg2Si + 4 NH4Br = SiH4+ 2 MgBr2+ 4 NH 3
reakció alapján történő előállítására az 55.1.12.1. ábrán vázolt berendezés használ
ható.
Az A bontóedényben — amely azonos az 55.1.10.1. ábra szerintivel — száraz jeges hűtőkeverékkel hűtve kondenzáljuk az edény aljáig érő csövön át bevezetettNH 3-gázt. Az A edényben előre elhelyezett NH4Br 'oldásához a folyékony ammóniát
7 Paneth, F .: Z. Elektrochem. 26, 463 (1920).8 Kraus, Gh. A., Brown, G. L.: J. Am. Chem. Soc. 52, 4031, (1930)
Kraus, Gh. A., Gamey, E. S.: J. Am. Chem. Soc. 56, 765 (1934)

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 196/366
198 H I D K O G É N V E G Y Ü L E T E K
50 %-os feleslegben alkalmazzuk. A reakciót —33 C°-ra állított hűtőfürdőbenvégezzük, forrásban levő folyékony ammóniával. Kísérlet közben az oldatot NH3-
gáz bevezetésével keverjük. A ferde oldalcsövön át a B csiszolatos edényke elforgatásával adagolhatjuk a Mg2Si-et. A képződő gázok a gázfejlesztő edényt kb.250 Hgmm túlnyomással hagyják el, amit a C nívóedény segítségével ellensúlyozhatunk. A gázokat a D abszorpciós edényben igen híg, kiforralt sósavval mossuk. A mosófolyadékot gyakran kell cserélni, mert az elnyelt ammóniától lúgossá váltoldat a szilánokat gyorsan bontja. A H2-t és a szilánokat tartalmazó gázkeveréketa mosóból az E P20 5-os szárítócsövön vezetjük át, és szilántartalmát folyékonynitrogénnel kondenzáljuk.
A kapott keverék tisztítását frakcionált kondenzációval végezhetjük. A frak-
cionálásra 20 Hgmm nyomáson —120 C°-os, —150 C°-os és folyékony nitrogénnelhűtött csapda szükséges. A monoszilán főleg a folyékony nitrogén hőmérsékleténkondenzál, a magasabb mólsúlyú szilánok már a kevésbé hideg hűtőben is.
SiH4 op. —185 C°, fp. -1120°
Si2H6 op.—132 C°, fp. -1 5C °
Mindkét frakció további tisztításra szorul. A nyers szilánra vonatkozó kitermelés 70—80 %. A monoszilán színtelen, mérgező gáz, szobahőfokon állandó. Nagyobb mennyiségben levegőn állva robbanásszerűen bomlik. A diszilán még kis
mennyiségben sem tartható el szobahőmérsékletű levegőn.
55,2. Fémek hidridjei
55.2.1. Lítium-hidrid (LiH)
A lítium-hidrid, mely az alkáli-hidridek közül a legstabilisabb, lítiumnak -a tmoszférában történő hevítésekor képződik. Célszerű a reakció sebességét lehetőlegcsökkenteni, amit úgy érünk el, hogy a lítium hőmérsékletét csökkentett nyomású
H2-atmoszférában lassan emeljük. Nehezen kiküszöbölhető akadályt jelent az,hogy a lítium (op. 180 C°) folyékony állapotban, kis ionátmérője következtében,magasabb hőmérsékleten az edény anyagát képező vason vagy nikkelen keresztülhatol. Egyéb anyagok nem jöhetnek számításba, mert a kvarcüveget erősen megtámadja, a legtöbb más fémmel pedig ötvöződik. Ezért úgy járunk el, hogy a lítiumotelektrolitvasból készült mély csónakba tesszük, amelyet másik nagyobb vascsónakba helyezünk.
A LiH nem tud a vason áthatolni, a nyersterméket nagyobbrészt a másodikcsónakban találjuk meg. (Nagyobb mennyiségek feldolgozásához tégely alakú edényeket használnak.) A két egymásba illő csónakot nikkellemezzel bélelt porcelán
csőben helyezzük el. Hogy a vas és a nikkel teljesen oxidm'entes legyen, a berendezést előzőleg tiszta, száraz hidrogéngázáramban (elektrolitikus hidrogént palládiumazbeszttel 300 C°-on oxigénmentesítünk, és CaCl2-dal, valamint P20 5-dal megszárítjuk) 800 C°-on hevítjük. Lehűlés után a vízmentes éterrel lemosott lítiumotgyorsan a kisebb elektrolitvas csónakba helyezzük. A reakcióteret evakuáljuk (vízlégszivattyúval, mely elé CaCl2-oá és P20 5-os szárítótornyokat kapcsolunk), és 200 C°-ra melegítjük, hogy az oldószer maradéka elillanjon. Azután hidrogéngázt vezetünk át a csövön, és folytatjuk a melegítést. A reakció 440 C° körül megindul, 600 C°

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 197/366
L Í H U M - H I D B I D — L ÍT I U M - A L U M Í N IU M -H l D K I D 199
felett igen hevessé válik. Ha elérjük ezt a hőmérsékletet, a hidrogén nyomását400 —500 Hgmm-re csökkentjük és a hőmérsékletet gyorsan 700C°-ra növeljük. A
700 C°-ot nem ajánlatos jelentős mértékben túllépni, mert a LiH elszublimál. A reakció 10—20 perc alatt befejeződik. A terméket hidrogénáramban hűtjük le szobahőmérsékletűre, majd a hidrogént száraz levegővel kiszorítjuk. Az anyagot a csónakból éles spatulával kaparjuk ki, száraz levegő- vagy nitrogénáramban ampullába tesszük, és leforrasztjuk.
A lítium-hidrid fehér anyag, mely vízzel hidrogént fejleszt, miközben LiOHképződik. Fény hatására színeződik (rózsaszín, szürkésbarna vagy kék lesz). Ultraibolya fény hatására ez a színeződés szinte pillanatok alatt bekövetkezik. (A színeződés Li-nak kolloid oldata LiH-ben.) A lítium-hidrid alkáli-klorid-olvadékbail
bomlás nélkül oldható, egyéb oldószerei nem ismeretesek.
55.2.2. Lítium-alumínium-hidrid (LiAlH4)
Előállításának reakcióegyenlete:
4 LiH + A1C1 3 = Lí A1H4 + 3 LiCl.
Visszafolyatásra állított hűtővel, csapos tölcsérrel és higanyzáras keverőveiellátott háromnyakú lombikban 3,05 g (0,08 mól) LiAlH4-et (a szükséges kis meny-
nyiségű kiindulási LiAlH4előállítására a későbbiekben visszatérünk) 30 ml éterbenfeloldunk, majd 23,5 g LiH-et hozzáadva, az oldatot rövid ideig keverjük. További200 ml éter hozzáadása után az oldathoz 71,2 g A1C1 3 300 ml éterrel készült oldatátcsepegtetjük olyan sebességgel, hogy a folyadék forrásban maradjon. Ezután méga reakció befejezéséig keverünk, és az elegyet egy ideig állni hagyjuk. A feleslegbenlevő LiH-et, valamint a képződő LiCl-ot nitrogénatmoszférában üvegszűrővel leszűrjük, és a szűrletet atmoszféra nyomáson szirupsűrűvé pároljuk be. Az étermaradékot vákuumban 70 C°-on leszívatjuk. Kitermelés: 85 %.
Az iniciátorként használt kis mennyiségű LiAlH4-et a következő módszerekkel
állíthatjuk elő.a) 7,0 g AlCl3-ot 4 g finoman elporított LiH-del nitrogénatmoszférában csiszolatos lombikban összekeverünk. Az edényt ezután evakuáljuk, és folyékony-nitro-génes külső hűtés közben kb. 15 ml száraz és peroxidmentes étert kondenzálunk bele. Ha a hűtőkeverékből a lombikot kivesszük, a reakció gyors felmelegedés közbenmegy végbe. A reakciót úgy szabályozhatjuk', hogy az edényt időről időre újra lehűtjük. 5 perc alatt a reakció teljesen befejeződik. Ez a módszer nagyobb mennyiségű Lí A1H4előállítására veszélyessége miatt nem használható.
b) 7,0 g finoman elporított LiH-et és 15,96 g AlCl3-ot nitrogénatmoszférában500 ml-es főzőlombikban összekeverünk, és 150 ml dioxánt adunk hozzá. A hőmér
séklet + 50 C°-ra emelkedik. A keveréket visszafolyatás mellett kb. fél óráig főzzük, lehűtjük, 135 ml éterrel hígítjuk, és további három órán át — ugyancsak visz-szafolyatás mellett — főzzük.
A további feldolgozás mindkét esetben azonos a fentebb leírtakkal. Abban az esetben, ha az előállításnál nem alkalmazzuk a LiAlH4 íniciátort, a
reakció vagy egyáltalán nem játszódik le, vagy pedig bizonyos idő után robbanás-szerű hevességgel indul meg. Szén-dioxid jelenlétében bepárláskor szintén robbanáskövetkezhet be, ezért C02-mentes étert kell alkalmazni. A berendezést gondosan

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 198/366
200 H I D H O G É N V E G Y Ü L E T E K
meg kell szárítani, mert a rendszer vízzel igen hevesen reagál. Célszerű C02-mentesnitrogénatmoszférában dolgozni. A kitermelés szempontjából igen jelentős, hogy
az alkalmazott éter peroxidmentes és teljesen száraz legyen. Szárítását NaH-del vagyCaH2-del kell elvégezni. A LiAlH4éteres oldata, ha a nedvességtől és a C02-tól elzárjuk, hosszú ideig
eltartható. A Lí A1H4újabban nagy jelentőségre tett szert mint redukálószer. Fém-haloge-
nidek és fém-alkilek LiAlH4-del hidrideket, illetve keverékhidrideket adnak. BF3-ból vagy BC]3-ból LiAlH4-del a boránok könnyen előállíthatók. A LiAlH4-del a reakciót általában úgy végezzük, hogy éteres oldatát a redukálandó anyaghoz visz-szafolyatásra állított hűtő alkalmazása és nedvesség kizárása mellett lassan hozzácsepegtetjük .
55.2.3. Alkálifém-hidridek (NaH, S H , RbH és CsH)
Az NaH, KH, RbH és CsH előállításához az 55.2.3.1. ábrán vázolt kvarc- vagySupremax-üvegből készült hidrogénező berendezés használható.9 A reaktorcsövetaz alkálifémgőzöktől acélból készült A béléscsővel óvjuk. Az alkálifémet vasból készült B csónakba, majd egyik oldalán zárt C vascsőbe helyezzük. A fémfelületek oxid-mentesítésére a berendezést hosszabb ideig szívatjuk, miközben a D csapon oxi-.génmentes hidrogént vezetünk be (amelyet szívás közben az E4 csiszolaton veze
tünk ki).
A hidrogénáram átvezetése közben elektromos kemencével a reaktorcső hőmérsékletét gyenge vörösizzásig emeljük. Az alkalmazott elektrolitikus hidrogéntigen gondosan kell oxigén- és vízmentesíteni.
A felhasználásra kerülő nátriumot és káliumot xilol alatt többször megolvaszt juk, és lehetőleg oxidmentesen juttatjuk a vascsónakba. A rubídium és a cézium magában a berendezésben állítandó elő oly módon, hogy Rb2C0 3 (vagy CsjjCC ) és mag-
Zintl, E., Harder, A.: Z. physik. Chem. (B) 14, 265 (1931).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 199/366
A L K Á L IF É M - É S A L K Á L IF Ö L D F É M -H ID R ID E K 201
néziumpor 1 : 3 mólarányú, előzőleg 150 C°-on vákuumban kiszárított keverékét helyezzük a vascsónakba, majd a berendezést leszívatjuk, és az anyagot lassan 620C°-ra hevítjük. (Amennyiben a módszert Na és K hidridjének előállítására használnánk, úgy az anyagkeveréket 300—350 C°-ra melegítjük fel.)
Az alkálifémek az acélcsőben az A helyen kondenzálnak. Lehűlés után a G csövet a benne levő csónakkal együtt H2-áramban E$-n át kiemeljük. Ez után a G ma-nométerszelepet visszahelyezve, a berendezésbe került levegőt Ha-gázzal kiszorít
juk, és az alkálifémet 1 atmoszféra nyomású álló (nem áramló!) hidrogéngázban lassan 300 —400 C°-ra hevítjük. Az alkálifémgőzök a H2-nel reagálva hidridet képeznek, mely a kemence két oldalán a csőben színtelen, finom kristályos tűk alakjábanrakódik le. Ha a fémet túl gyorsan párologtatjuk, a hidrid kondenzált fémmel szeny-
nyeződhet. Reakció közben időnként a berendezést hidrogénnel egy atmoszféra nyomásra feltöltjük. Ha a manométerszelep további nyomáscsökkenést már nem jelez,a berendezést lehűtjük, H2-áramban platina kanállal a hidridet az F csőbe kotorjuk,és az E3 csiszolatot zárjuk. Az Ei csiszolaton át a terméket H2-áramban más edénybe tölthetjük át.
Az alkáli-hidridek színtelen, nedvességre bomló anyagok, reakcióképességükoxigénnel szemben a LiH-től a CsH felé haladva erősen nő, a LiH csak vörösizzáson reagál, a NaH oxigénatmoszférában kb. 230 C°-on gyullad meg, a KH, RbH ésCsH már szobahőfokon is.
A lítium-hidrid gőznyomása 23,5 C°-on 0,023 Hgmm, 640 C°-on kb. 70 Hgmm,
a nátrium-hidridé 300 C°-on 8 Hgmm, a kálium-hidridé 300 C°-on 7,3 Hgmm, $rubídium-hidridé 370 C°-on kb. 100 Hgmm, végül a cézium-hidridé 300 C°-on27,8 Hgmm.
55.2.4. Alkállföldfém-hidridek (CaU2, SrH2, BaH2)
A vákuumban desztillált fémet argonatmoszférában a felületén levő oxidtólmegtisztítjuk, és az alkáli-hidridek előállításánál leírt berendezésbe helyezzük. Ahidrogénezésre gondosan tisztított és szárított H2-gázt használunk. A Ca és Sr ese
tében a reakció 400 —500 C° között, a Ba-nál 200—300 C°-on indul meg. A termékaprókristályos, fehér anyag. A nyerstermék hidrogénáramban való desztillálássaltisztítható. A hidridek 1000 C°-ra hevítve disszöciálnak, a fém elpárolog, de a berendezés hidegebb részén a komponensekből újra hidrid képződik. A csónakbanvisszamarad a főleg karbidokból és oxidokból álló szennyeződés. Désztillálássalmakroszkopikus, kb. 1 mm hosszú kristályokból álló terméket kaphatunk. Ahidridek stabilitása a CaH2 -*■ BaHa irányban csökken.
55.2.5. Titán-hidrogén (Ti/H)
A fémtitán jelentős mennyiségű hidrogént oldhat, e mellett hidridet is képezTiH-TiH2közötti összetétellel. A TiH2-nek megfelelő felső határ csak igen tiszta Tiés H2alkalmazása esetén érhető el.
Legkönnyebben a titánszivacs hidrogénezhető. Ti-lemezen a Hidrogénabszorpció 300 C°-on indul meg, 400 C°-on már gyorsan bekövetkezik a reakció. A részbenhidrogénezett titán már szobahőfokon is reagál a H2-nel. Nagy vákuumban 400 CPfelett a hidrogént újra leadja.
Ha adott mennyiségű hidrogént tartalmazó hidridet kívánunk előállítani, a kö-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 200/366
202 HID'ROCrÉTJVÉCJYÜXiETEK
vetkezőképpen járhatunk el.10 A fémtitánt korund vagy rozsdamentes acéltégelybe tesszük, azt pedig kvárccsőbe helyezzük. A kvárccső égjük végéhez vákuumszi
vattyú, a másik végéhez gázbürettán át (nívóedénnyel és higanytartállyal) H2-fej-lesztŐ csatlakozik. Először meghatározzuk a kvarccső térfogatát,- azután a fémet550 C°-ra hevítve, a berendezést leszívatjuk. Az abszorpció hőmérsékletének és abevezetett hidrogéngáz mennyiségének megfelelő megválasztásával a kívánt meny-nyiségű hidrogént tartalmazó ti tán-hidrogén állítható elő.
55.2.6. Réz-hidrogén (CuH)
Előállítható rézsóoldatoknak hipofoszforossavval történő redukciójával.11
21 g H 3P02-at 300 ml vízben oldunk, és hozzáöntjük az előmelegített (65 C°-os) savanyú rézsóoldatot (25 g CuS04*5 H20 100 ml víz és 20 ml 2 n H2S04keverékébenoldva). Egy napi állás után a csapadékot szűrjük, majd vízzel, alkohollal és éterrelmossuk. Kis mennyiségű vas- vagy halogénion zavarja a hidrid leválását. A CuHfrissen leválasztva vörösbarna-csokoládébarna színű csapadék. Száraz állapotbanigen robbanékony, étertől nedvesen sem teljesen veszélytelen. Levegőn oxidálódik,hevítés hatására a szabaddá váló hidrogén meggyullad.
10 Sieverts, A. és munkatársai: Z. physik. Chem. 145, 227 (1929); Z. anorg. alig. Chem. 153, 289 (1926); 172, 1 (1928); 187, 15Ő (1930); 199, 384 (1930);
Gibb, T. R. P., Kruschnitz, H. W J. Am. Chem. Soc. 72, 5305 (1950).11 Nennhoeffer, O., Nerdel, F.: J. prakt. Chem. 144, 63 (1935).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 201/366
56 . Vízmentes halogenidek
A halogenidekben található kötés típusát tekintve lehet ionos vagy kovalens jellegű, attól függően, hogy mekkora az illető központi elem elektronegativitása.
A kis elektronegativitású elemek halogenidjei sószerű vegyületek, ezek az plektro-negativitások közötti nagy különbség folytán ionrácsot alkotnak, melynek vi
szonylag nagy rácsenergia, magas olvadás- és forráspont a következménye. Olvadékuk jól vezeti az elektromos áramot. Az elektrónegativitások közötti különbség
csökkenésével a kötés jellege a kovalens kötés irányában tolódik el, ami az inter-
molekuláris erők csökkenését és viszonylag alacsony olvadás- és forráspontot
eredményez. A közepes elektronegativitású elemek halogenidjeinek nagy része vízzel érint
kezve hidrolízist szenved, ami gyakran irreverzíbilis folyamat. A hidrolízisre való
hajlam a halogén elektronegativitásának csökkenésével nő.
56.1. Vízmentes halogenidek előállítása elemi szintézissel
Ismeretes, hogy a halogének a legtöbb elemmel közvetlenül egyesülnek. Bár a
fluor a legaktívabb köztük, reakciója a legtöbb fémmel csak lassan megy végbe. Ez
annak a következménye, hogy a fém felületén képződött sónak igen magas a szub
limációs pontja (forráspontja). Vízmentes fluoridok előállítására az elemi szintézismódszerének a laboratóriumi gyakorlatban nincs különösebb jelentősége. Annál ki-
terjedtebb azonban a többi halogénnel végzett elemi szintézis alkalmazása. A brómo-
zás és a jódozás a klórozásnál kevésbé gyorsan végbemenő folyamat, és kisebbhőfejlődéssel jár, mivel a képződéshő a fluoridoktól a jodidok felé haladva csökken.
Há az elemi szintézis körülményei között többféle összetételű halogénvegyületképződésére van lehetőség, a végtermék mindig a reakció hőmérsékletén stabilis
vegyület. így pl. az antimon és foszfor klórozása alkalmával nem penta-, hanem
trikloridok keletkeznek, mert a pentakloridok az adott hőmérsékleten trikloridraés klórra bomlanak. A gyakorlatban a szintézist nem tiszta fémekkel, hanem sok
szor a lényegesen könnyebben előállítható és olcsóbb ötvözetekkel hajtják végre.
Az ilyenkor képződött termék az ötvözetet alkotó fémek halogenidjeinek keveréke.
Tenziójuk különbözősége alapján ezek egymástól többnyire frakcionálással elvá
laszthatók. Az 56.1.1. ábrán láthatjuk több fontos klorid tenziójának változását ahőmérséklet függvényében. Ezek alapján elbírálható, hogy milyen esetekben vezet
eredményre az egyszerű desztilláció, illetve szublimáció. így pl. a SiCl4előállításárarendszerint ferroszilíciumot használnak, melyszennyezésként sokszor alumíniumot
és mangánt is tartalmaz. A klórozás folyamán ezért SiCl^, FeCl3, A1C1 3 és MnCl2 isképződik. Az 57,0 C° forrásponté SiCl4könnyen elválasztható a lényegesen kisebbtenziójú egyéb kloridoktól.
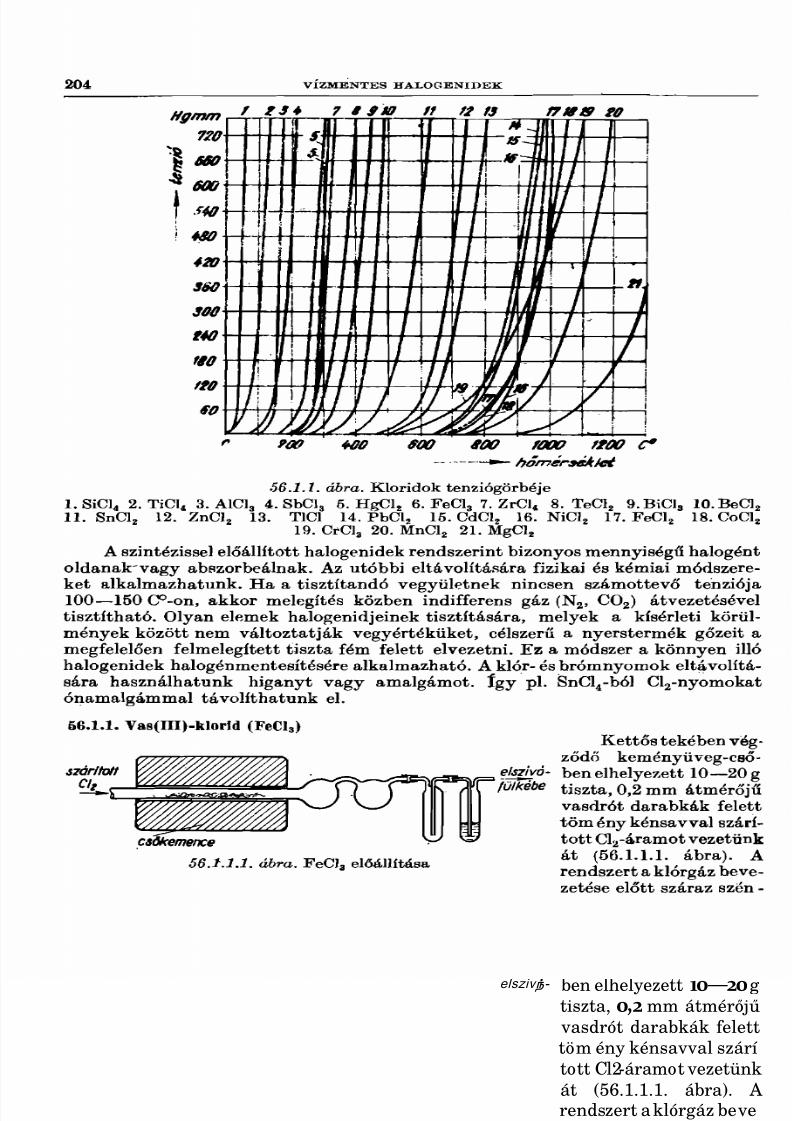
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 202/366
204 V ÍZ M E N T E S H A L O G E N ID E K
56.1.1. ábra. Kloridok tenziógörbéje
I . SiCl4 2. TiCl4 3. A1C13 4. SbCl3 5. HgCl2 6. FeCl3 7. ZrCl4 8 . TeCI2 9.BiCl3 10.BeCl2II. SnCl2 12. ZnCl2 13. T1C1 14. PbCl2 15. CdCl2 16. NiCl2 17. FeCl2 18. CoCl2
19. CrCl3 20. MnCl2 21. MgCl2
A szintézissel előállított halogenidek rendszerint bizonyos mennyiségű halogéntoldanak vagy abszorbeálnak. Az utóbbi eltávolítására fizikai és kémiai módszereket alkalmazhatunk. Ha a tisztítandó vegyületnek nincsen számottevő tenziója100—150 C°-on, akkor melegítés közben indifferens gáz (N2, C02) átvezetésével
tisztítható. Olyan elemek halogenidjeinek tisztítására, melyek a kísérleti körülmények között nem változtatják vegyértéküket, célszerű a nyerstermék gőzeit amegfelelően felmelegített tiszta fém felett elvezetni. Ez a módszer a könnyen illóhalogenidek halogénmentesítésére alkalmazható. A klór- és brómnyomok eltávolítására használhatunk higanyt vagy amalgámot. így pl. SnCl4-ból Cl2-nyomokatónamalgámmal távolíthatunk el.
56.1.1. Va8(III)-klorid (FeCls)
elszivó-
Kettős tekében végződő keményüveg-cső-
ben elhelyezett 10—20 gtiszta, 0,2 mm átmérőjűvasdrót darabkák feletttöm ény kénsavval szárított Cl2-áramot vezetünkát (56.1.1.1. ábra). Arendszert a klórgáz bevezetése előtt száraz szén -

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 203/366
V ÍZ M E N T E S K L Ó R I D Ó K E L Ő Á L L ÍT Á S A E L E M I S Z IN T É Z IS S E L 205
-dioxiddal gondosan kiszárítjuk. A reakció 250—400 C° között megy végbe, ezért acsövet elektromos csőkemencében helyezzük el úgy, hogy a teke a kemencén kívül
maradva léghűtést kapjon és szedőként szolgáljon. A klór áramlási sebességét azelvezetőcsőhöz kapcsolt kénsavas mosópalackban látható buborékolással ellenőrizhetjük. A szedőbe való visszaszívódás ellen fordítva kapcsolt mosópalackkal védekezünk.
A képződött terméket klóráramban 220—300 C° között átszublimáljuk a második tekébe, lehűlés után szárított N2-gázzal kiszorítjuk a klórt, majd a tekét leforrasztjuk. A FeCl 3 fémes csillogású, zöldes színű lemezkékből áll. Olvadáspontjaklóratmoszférában 280 C°. Vákuumban szublimálva részben bomlik. (s25o = 2,90.)
56.1.2. Króm(III)-klorid (CrCl3)
Durva porrá tört krómot (kb. 20 g-ot) légnedvességtől védett rendszerbenvörösizzáson száraz klóráram hatásának teszünk ki. Mivel a keletkező CrCl 3 nehezenilló, a cső hidegebb részein lerakódik. Ezért célszerűen elég nagynak (3 cm) választ
juk a kvarcból vagy porcelánból készült reaktorcső átmérőjét. A cső mégis előforduló eltömődésének kellemetlen következményei ellen védekezésül a mosópalackelé T-csöves kénsavas biztosító-manométert iktatunk. A reakció végeztével a klórtC02-dal kiűzzük, a kihűlt csőből a terméket mechanikusan eltávolítjuk. Tisztításaazon a tulajdonságán alapszik, hogy a tiszta CrCl2ásványi savakban és vízben rend
kívül kis sebességgel oldódik. Kevés CrCl2 jelenlétében az oldódás igen nagymértékben meggyorsul. A HCl-ban kifőzött terméket desztillált vízzel klórmentesre mossuk, és 200 C° körüli hőmérsékleten szárítjuk. Fénylő, vörösesibolya színű pikkelyek. Op. kb. 1150 C°, klóráramban szublimálható. (sl5o = 2,76.)
56.1.3. Volfrám(VI)-klorÍd (\VC16)
A volfrám(VI)-klorid előállítására szűkületekkel több (4—5) részre osztottSupremax-csövet használunk.1 Az első, hosszabb részen kvarc- vagy porcelán
csónakban kb. 5 g volfrámport helyezünk el. A felület oxidmentesítése céljából1—2 órán keresztül 700—1000 C° közti hőmérsékleten hidrogént vezetünk be.Hidrogéngázban történt lehűtés után oxigénmentesített (lásd a klór tisztítását)klórgázt hajtunk át a készüléken, és a csónakot magában foglaló részt 600 C°-ramelegítjük. A redukció ellenére kezdetben mégis képződhet vörös színű volf-rám(VI)-oxid-klorid. A klórozás ideje alatt gondoskodni kell a szűkületek melegen-(350—400 C°-on) tartásáról, hogy a szublimáló termék el ne tömje azokat. A reakció végeztével először az illékonyabb WOCl4-ot (fp. 232 C°) melegítéssel elűzzük acső végére, majd a volfrám(VI)-kloridot klóráramban többszörösen megismételtszublimálással tisztítva, összegyűjtjük az utolsó, szűkületekkel határolt csőszakaszban-. Ekkor— változatlanul klóratmoszférában — megolvasztjuk a terméket.Lehűlés közben az olvadék apró darabokra törik. Ez után a klórt tiszta, száraz nitrogénnel kiszorítjuk, és a készítményt leforrasztjuk. Kékesfekete, nedvességre érzékeny kristályok (op. 275 C°, fp. 347 C°). Exszikkátorban cc. H 2S04fölött sötétben jól eltartható. Szerves oldószerekben jól oldódik. (s?5» = 3,52.)
Hónigachmid, O., Merni, W.: Z. anorg. alig. Chem. 229, 581 (1936).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 204/366
206 V ÍZ M E N T E S H A E O G E N ID E K
66.1.4. Ón(IV)-kIorid (SnCl4)
Cl,
Előállításához az 56.1.4.1. ábrán látható berendezést használjuk. A klórgázt akénsav visszaszívása ellen biztosítékul szolgáló fordított mosópalackon és cc. kénsavas manométeren keresztül a cc. kénsavas mosóba vezetjük. Innen a gáz a gra
nulált ónt (kb. 25 g-ot) tartal- elszivó 1 mazó nagy kémcsőbe jut, ahol fülkébe * már szobahőmérsékleten megin
dul a reakció. A hőfejlődés miatta fémón megolvad. A klóráramotcélszerű úgy szabályozni, hogycsak ritkán észleljünk szikrázásta kémcsőben. A szedőlombikba,melyet só—jég-keverékkel hűtünk, kevés ónfóliát vagy ón-amalgámot helyezünk. Mivel aklór a parafát és a gumit megtámadja, célszerű PVC-dugót és-csövet használni. A reakció megindítása előtt a berendezés szárítását és a klórozás befejeztével a
klórfelesleg eltávolítását N2-árammal végezzük. A nyersterméket atmoszféra nyomáson desztillálással tisztítjuk (op. 30 C°, fp. 124 C°).
Színtelen, légnedvességtől hidrolizáló folyadék. (s2o° = 2,23.)
66.1.5. Antimon-pentaklorid (SbCl5)
A por alakú törmeléktől elválasztott antimon borsó nagyságú darabjait (kb.50 g) az 56.1.5.1. ábrán látható üvegcsőben helyezzük el. Ennek szűkített és lehajlovége a szedőül szolgáló frakcionáló lombik gömbj ériek közepéig ér, és a lombik
nyakában klórálló anyagból készült dugóval (PVC) csatlakozik. A frakcionáló lombik oldalcsövén át bevezetett száraz klór felfelé haladva a reaktorként használtcsőben melegítés nélkül is reakcióba lép az antimonnal. Az exoterm reakció során ahőmérséklet izzásig fokozódhat. Ezen a hőmérsékleten az SbCl 3 a stabilis termék,

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 205/366
V ÍZ M E N T E S K L O B ID O K E L Ő Á L L ÍT Á S A E L E M I S Z IN T É Z IS S E L 207
amely megolvadt állapotban a klórárammal szemben végigfölyik a megdöntöttcsövön, és a klóratmoszférát tartalmazó szedőbe csöpög. Az itt uralkodó alacsonyabb hőmérsékleten részben SbCl5-dá alakul. Az így készült triklorid —pentakloridkeverék SbCl5-dá dolgozható fel. Elvben ugyanaz az eljárás, ha tiszta SbCl3-bólindulunk ki. A megolvasztott SbCl3-on száraz klóráramot vezetünk át. Ügyeljünkarra, hogy az olvadék hőmérséklete ne emelkedjék és az SbClg termikus disszociációja be ne következzék. A klórf^lesleget száraz levegő vagy N2 átbuborékoltatásá-val űzzük ki. A további tisztítás vákuumdesztillációyal történhet (fp. 14 Hgmm-en
68 C°). A berendezés vázlata a következő (56.1.5.2. ábra): A terméket megkell védeni a vízlégszivattyú felől érkező vízgőztől, a higanymanométert pedig a desztillálóból származó klórgáztól. A kettős feladat megoldására legalkalmasabb a szilárdKOH-dal töltött szárítótorony, amely a vízgőzt is és a klórgázt is megköti.
Színtelen, erősen hidrolizáló folyadék. Op. 2,8 C°, fp. 140 C°, de ezen a hőmérsékleten már erősen- bomlik. (á20->= 2,35.)
56.1.6. Foszfor-triklorid (PC13)
Szintézise a szokványos desztillálóberendezés frakcionáló lombikjában hajtható végre. A lombik nyakába gázbevezetőcsövet és hőmérőt illesztünk. A klóralkalmazása miatt célszerű erre csiszolatos edényt vagy PVC-dugót használni.Kiindulási anyagként akár a sárga-, akár a vörösfoszfor megfelel teljesen szárazállapotban. Ennek érdekében a sárga-, illetve vörösfoszfort hosszabb ideig P205 fölött exszikkátorban tartjuk. Az ugyancsak jól megszárított klórgázzal a sárgafoszfor hevesen, a vörösfoszfor csak szublimációs pontjáig (280 C°) hevítve lép reakcióba. A klóráram megindítása előtt a levegőt C02-dal ki kell szorítani a készülékből. Ha nagy a klórfelesleg, PC15képződik, mely az edény falán mint fehér verődékismerhető fel. Ellenkező esetben, vagy ha túl erős a melegítés, sárga foszforverődékmutatkozik a lombik hidegebb részein. Ilyenkor a megfelelő körülményeket többklór adagolásával vagy a hevítés csökkentésével biztosítjuk. A képződött PC1 3 folyamatosan' a szárítófeltéttel védett szedőbe gyűlik össze, és mindig tartalmaz

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 206/366
208 V ÍZ M E N T E S H A L O G É N !D E K
bizonyos mennyiségű PCl5-ot, valamint foszfor-oxid-kloridot. Tisztítását vörös-foszforról való desztillálóval végezzük.
Színtelen, erősen hidrolizáló folyadék (fp. 76 C°; s = 2", 11).
66.1.7. Foszfor-pentaklorid (PC15)
Foszfor-trikloridból és klórból állítható elő:
PC1 3 + Cla = PC15.
A folyadék halmazállapotú PCl3-tól eltérően szilárd. Ha a klóráramot egyszerűen a folyadékfázison vezetnénk át, a képződött pentaklorid sok átalakulatlan tri-klorid zárványt tartalmazna. Ennek kiküszöbölése érdekében a folyékony PCl3-otcsöpögtetjük lassan a Cl2-gázzal töltött edénybe. Hogy a szilárd termék könnyeneltávolítható legyen, széles szájú lombikot használunk 3-furatú PVC- vagy pa-raffinozott parafa dugóval. A furatok közül kettő a klór ki- és bevezetésére, a harmadik a csepegtető tölcsér számára való. A PC1 3 adagolója a P20s-dal töltött szárítófeltéten keresztül érintkezik a külső levegővel. Az ugyancsak erősen hidrolizálóterméket a klórelvezető csőhöz csatlakozó szárítótorony védi a nedvességtől. A készPClg-ot leforrasztva vagy leparaffinozott csiszolatos üvegben tarthatjuk el.
Fehér, kristályos anyag, op. 149 C°, (nyomás alatt) 100 C°-on szublimálható.< 8 ,5 . = 2 , 1 1 . )
66.1.8i Szilícium-tetraklorid (SiCl4)
A klórozás kiindulási anyagául az aluminotermiás úton előállíható nyers-Si,-vagy a kohászati termékként kapott különféle Si-ötvözetek szolgálhatnak:
Si + 2 Cl2 = SiCl4.
56.1.8.1. ábra. SiCl4 előállítása

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 207/366
V ÍZ M E N T E S K L O B ID O K E L Ő Á L L ÍT Á S A E L E M I S Z IN T É Z IS S E L 209
Leggyakrabban nagy szilíciumtartalmú ferroszilíciumot alkalmaznak. Az apróratört fémötvözetet porcelán- vagy kvarccsőben elhelyezve (56.1.8.1. ábra) 300—400
C° hőmérsékleten klórozzuk. A klór teljesen száraz és oxigénmentes legyen. Ezért areakció megindítása előtt klórral kell kiszorítani a készülékből a levegőt. Ha magnéziummal vagy alumíniummal redukált nyers szilíciumot klórozunk, 300—310 C°felett a
Mgo + vc i 2 = M g c i2 + y 2o 2
egyenlet szerint a szennyezésként jelenlevő MgO-ból oxigén képződhet. Ebben azesetben is, mint levegő jelenlétében, az elemi szilícium felületén oxidréteg képződik, mely a SiCl4-termelést erősen lerontja, esetleg teljesen meggátolja. A hőmérsékletet ilyenkor nem szabad 300 C° fölé emelni.
A készüléket úgy állítjuk össze, hogy a reaktorcsőhöz nagyobb átmérőjű, .kaviccsal lazán töltött szakasz csatlakozzék, amelynek rendeltetése a SiCl4-dal együttképződő, de annál jóval kevésbé illékony FeClg és A1C1 3 megkötése. A továbbjutóSiCl4 Liebii7-hűtőben kondenzálva kerül a konyhasó—jég-keverékkel hűtött és szá-rítófeltéttel védett szedőbe. Az így nyert anyag jelentékeny mennyiségű klórt, valamint nagyobb mólsúlyú sziliéium-kloridokat (SinCl2n+2, ahol n = 2—6) tartalmaz.Tisztítása frakcionálással végezhető. Előpárlatként a klór zöme kondenzálás nélküleltávolítható, a főpárlat az 57,5 C° forráspontú SiCl4. A homológ sor két következőtagjától jól elválasztható: a SiaClfl forráspontja 145 C°, a Si 3ClBforráspontja pedig
216 C°. A SiCl4maradék klórtartalma fémhigannyal távolítható el. Kb. 1 napig kevéshigannyal állni hagyjuk és időnként összerázzuk. A SiCl4színtelen, igen erősen hid-rolizáló folyadék. (s20» = 1,48.)
Leforrasztott ampullában tartjuk el.
56.1.9. Diszilfclum-hexaklorid és homológjai (SlnCl2n+2)
Keletkezési reakciójuk:
CaSi2 -f- Cl, -► CaCl2 -f- SinCl2n+2 (n = 1—6).
Kb. 30 % kalciumot tartalmazó szilíciumötvözetből kiindulva jelentős mennyiségűmagasabb szilícium-klorid-homológ (kb. 30 % SigCl , 4 % Si 3Cl8, 1 % Si4_6) állíthatóelő aránylag alacsony hőmérsékleten (max. 250 C°-on) végrehajtott klórozással. A SiCl4előállítására felhasznált készülék itt is alkalmazható. 250 g Si—Ca-Ötvözet-ből kiindulva kb. 14—15 napig tart a reakció a szükséges lassú, legfeljebb mp-en-ként 2-buborék-sebességű klóráramban. A nyerstermékről atmoszféra nyomáson akb. 65 %-nyi SiCl4ledesztillálható, A magasabb homológok azonban a bomlás ve
szélye nélkül csak vákuumdesztillációval különíthetők el egymástól.2 A Si2Cla fp.-ja 20 Hgmm-en 50 C°,.a Si 3Cl8-é 17 Hgmm-en 100 C°. A 4. és 5. tagaz előzőkhöz hasonlóan szobahőmérsékleten ugyancsak folyékony, a 6. szilárd,fehér anyag. Valamennyi a nedvesség nyomai iránt is érzékeny. Levegővel elegyedett gőzeik gyúlékonyak.
» M artin, <?.: J. Chem. Soc. (London) 105, 2Ö36 (1914).
14 Ált. és szervetlen kémiai prakt.--42B 4/II.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 208/366
210 V ÍZ M E N T E S H A L O G E N ID E K
56.1.10. Bikén-diklorid (S2C1,)
A kén kloridjai közül a legegyszerűbben a szobahőmérsékleten állandó S2C12
állítható elő szintézissel. A klórozást frakcionáló lombikban végezhetjük, amelyhez Liébig-hűtő és szedő csatlakozik. A ként megolvasztjuk, és a kb. 130 C°-on tartottlombikba száraz klóráramot vezetünk. Nagy klórfelesleg esetén SC12 is képződik.Célszerű a reakciót a kén teljes reagálása előtt befejezni. Ha a termék mégis SCl2-dalszennyeződött volna, atni vöröses színéről felismerhető, elemi kén jelenlétébenvégzett rektifikációval tisztítható. A tisztított termék halványsárga színű, erősenhidrolizáló anyag, olvadáspontja —77 C°, forráspontja + 138 C°, s20° = 1,68.
56.1.11. Szelén-tetraklorid (SeCl*)
Az elemi szelén klórgázzal szobahőmérsékleten is hevesen reagál. Kényelmesebben irányítható a reakció oly módon, ha a szelént CCl4-ban szuszpendált állapotban klórozzuk.3 Először Se2Cl2 képződik, amely a szén-tetrakloridban oldható.További klórozásra fehér por alakjában SeCl4 válik ki. Nedvesség kizárásávalüvegszűrőn szűrjük, CCl4-dal mossiik, és vákuumexszikkátorban szilikagél felettszárítjuk. Szublimálási pontja 196 C°, op. (nyomás alatt) 305 C°, s20<>=3,80.
56.1.12. Bór-triklorid (BCla)
Elemi szintézissel való előállítására kiindulási anyagként leggyakrabban azaluminotermiás úton előállított, jelentős mennyiségű alumíniumot tartalmazó kristályos bórt használják. A szintézis porcelán vagy kvarccsőben végezhető 500—600C° körüli hőmérsékleten. A klórozáB előtt a levegő- és nedvésségnyomok eltávolításacéljából száraz, meleg N2- vagy C02-áramot vezétünk át a berendezésen. Az oxigént azért célszerű klórozás előtt eltávolítani, mert a részleges oxidáció nagymértékben csökkenti a kloridképződés sebességét. A készülék szárítását és a terméknedvességtől való védelmét igen gondosan kell végezni, mert a hidrolízis folyamán
használt csapzsírokkal is reagál, a csiszolatokat kénsavval kenjük. Célszerűbbazonban a csiszolatokat forrasztással helyettesíteni. A folyamatot magasabb hőmérsékleten azért nem ajánlatos végrehajtani, mert a csőből a klór hatására szilícium redukálódhat ki, és így a termék SiCl4-dal szennyeződhet.
A reaktorcső végéhez üveggyapottal töltött, lefelé hajló csövet csatlakoztatunk,amely A1C13- és FeCl3-csapdaként működik. A szedőhöz — amelyet szárazjég ésdenaturált szesz keverékével hűtünk — P205-os torony, utóbbihoz pedig a kürtőbevezető cső csatlakozik.
A gondosan szárított Cl2-gázt lassan áramoltatjuk, hogy a szedőbe lehetőlegkevés klór kerüljön. A nyerstermékben levő fölös klórt leforrasztott bombacsőben
higannyal rázogatva lehet megkötni. (Védőfal mögött! Robbanásveszély !) A leforrasztott ampullákat csak száraz jeges hűtés utánra megfelelő óvatossági rendszabályokbetartása mellett szabad kinyitni (lásd 47.1.). A BC1 3 desztillálása során az adólombikot infralámpával melegítjük. A BC1 3 víztiszta, levegőn füstölgő folyadék.Op. —107 C°, fp. + 12,5 C°, s2o= 1,43.
* Lenher, V.: J. Am. Chem. Soc. 42, 2498 (1920).
Bell, H C., Gibson, G. S.: J. Chem. Soc. (London) 127', 1877 (1925).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 209/366
V ÍZ M E N T E S B R O M ID O K E L Ő Á L L ÍT Á S A E L E M I S Z IN T É Z IS S E L 211
56.1.13. Alumínium-bromid (AlBr3)
Alumínium és bróm reakciója útján állítható elő. A lombik alját üveggyapottal borítjuk úgy, hogy a csepegtető tölcsér vége a gyapot alá érjen, a gyapotraCCl4-dal zsírmentesített alumínium drótot teszünk célszerűen spirális alakra hajlított 20—30 cm-es darabokban (56.1.13.1. ábra). Az előzetesen cc. HjSOa-róldesztillált brómot csapos tölcsérből adagoljuk. A tölcsércsiszolatát elfolyósított P20 5-dal kenjük. Ha a folyékony 'u { ^ bróm érintkezésbe jut az alumínium dróttal, fénytüne-mény és erős hőfejlődés kíséretében reagál. Ezért vezetjüka gyapot alá, így a fém csak a bróm gőzeivel reagálhat. A
csatlakozásokhoz gumidugót is használhatunk, ha a dugótgondosan alumínium fóliába csavarjuk; de célszerűbb acsiszolatos csatlakozás. A szedő oldalcsövéhez CaCl2-oscsövet kötünk.
A reakciót úgy indítjuk meg, hogy a reakcióteret nyíltlánggal 100 C° körüli hőmérsékletre melegítjük, majd aláng eltávolítása után óvatosan csepegtetve brómot adunkhozzá. A reakció megindulását ködképződés jelzi. Ha eznem következnék be (a reakció sokszor nehezen indul),igyekszünk telített brómgőzteret előállítani, majd olyan
helyen hűtjük nedves szűrőpapírral a lombik falát, aholvalamelyik alumínium spirál hozzáér. A kondenzáló brómmegindítja a reakciót. A termék a lombik alján gyűlik össze. A bróm a ké >zŐdött AlBr3-ban oldódik, ez a reakció lelassulását eredményezi. Az Al-felesleg ellenére a teljes bróm-mennyiség csak visszafolyatás mellett végzett főzéssel vihető reakcióba. A kész AlBr3-ot N2-atmoszférában leforrasztható szedőampullába desztilláljuk át (op. 97,5 C°, fp. 255 C°), mert magasabb hőmérsékleten 0 2 jelenlétében Br2 és Al^C^ képződése közben bomlik.
Színtelen, csillogó, kristályos anyag, vízzel robbanásszerűen reagál, levegőnfüstölög, könnyen oldható CS2-ban,acetonban, benzolban, toluolban.
56.1.14. Ón(rVJ-bromid (SnBr4)
A reakció folyékony halmazállapotú bróm és ón között szobahőmérsékleten is hevesen megy végbe.Ezért a brómot fokozatosan, finomra kihúzott végű csepegtető töl
csérből adagoljuk úgy, hogy a fejlődő reakcióhő ellenére a brómnakminél kisebb része párologjon él. A reakcióedényül szolgáló frakció-náló lombikot ferdén befogva, annak- kivezetőcsövétj visszafolyatás-ra állított hűtőként használjuk(56.1.14.1. ábra). Célszerű olyan
56.1.13.1. ábra. AlBra
előállítása
14*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 210/366
212 V ÍZ M E N T E S H A L O G E N ID E K
frakcionáló lombikot választani, amelynek hosszú nyaka van és az oldalcsöve minélmélyebbről indul. A Br2 adagolását befejezzük, mielőtt még az ón teljesen kifogyott
volna, majd az elegyet forráspontjáig hevítve elősegítjük a megmaradt Bra elrea-gálását, illetve előpárlatként eltávolítjuk azt (op. 33 C°, fp. 201 C°, s35. = 3,35).
A tiszta SnBr4színtelen, kristályos anyag. Higroszkópos, hidrolízisre hajlamos.
56.1.16. Bizmut-tribromid (BiBr8)
A bizmut és a folyékony bróm közönséges hőmérsékleten mérsékelt sebességgelreagál. A visszafolyatásra állított hűtővel ellátott frakcionáló lombikban elhelyezett bizmutporra a sztöchiometrikusnál kisebb mennyiségű, kénsavról desztilláltbrómot öntünk. A keveréket a brómgőzök eltűnéséig főzzük, majd a terméketvákuumdesztillációval tisztítjuk. Előállíthatjuk úgy is, hogy nehezen olvadó üvegcsőben N2- vagy COa-vivőgázzal továbbított Br2-gőzöket 700 C°-on bizmutolva-dékkal reagálhatunk.4
Sárgás színű, higroszkópos, hidrolizáló anyag (op. 218 G°, fp. 441 C°, s^* = 5,70).>
56.1.16. BÖr-tribromid (BBr8)
KvarccBŐben 200—250 C°-ra hevített, aluminotermiásan készült kristályosbór felett kénsavról desztillált Br2-gőzt áramoltatunk H2-vivőgázzal.5 A redukáló
atmoszférára azért van szükség, hogy a bór felületét megvédjük a magasabb hőmérsékleten fennálló oxidáció veszélyétől. Az 56.1.16.1. ábrán vázolt berendezéslehetővé teszi, hogy a Br2-gőz koncentrációját a reakciótérben a hidrogénárammegszakítása nélkül tetszés szerint változtassuk.
A szállított bróm mennyiségét úgy szabályozzuk, hogy a szedőbe brómfölös-
leg ne kerüljön. A szedőt só—jég-keverékkel hűtjük és szárítófeltéttel védjük alégnedvességtől. A berendezés részeit célszerű kénsavval vagy elfolyósított P20 5-dal kent csiszolatokkal, ennek hiányában pedig PVC-dugóval és csővel csatlakoztatni.
4 Hőnigachmidt, O., Birckenbach, L.: Z. Elektrochem. 26,'403 (1920)} Meyer, F .: Liebigs Ann. Chem. 264, 122 (1891).5 Meyer, F., Zappner, R.: Bér. dtsch. chem. Ges. 54, 551 (1921).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 211/366
V ÍZ M E N T E S B B O M ID O K E L Ő Á L L ÍT Á S A E L E M I S Z IN T É Z IS S E L 213
A brómszennyezés eltávolítása céljából a nyersterméket fémhigany jelenlétében visszafolyatás közben elszíntelenedésig főzzük. A BBra-gőzöket a desztillálássorán a forráspontjukon végbemenő oxidációtól közömbösgáz-atmoszférával védjük(op. —46 C°, fp. 90,8 C°). Színtelen, igen könnyen hidrolizáló folyadék. (s0„== 2,65.)
56.1.17. Foszfor-tribromid (PBrs)
A vörösfoszfor a brómmal hevesen reagál. A reakciósebesség csökkentésecéljából a szintézist indifferens oldószerben hajtjuk végre.6 Az oldószer lehet-szén-tetraklorid, tiofén-
mentes benzol, vagy foszfor-tribromid, esetleg fosz-for-triklorid.
A reakciót olyan Clai- sen-lombikban végezzük,amely a hűtő átfordításával visszafolyatásra, illetvedesztillálásra használható(56.1.17.1. ábra). A csiszo-latokat elfolyósított P20 5-dal vagy cc. H2S04-valkenjük.
50 g (P205 felett szárított) vörösfoszfort kb. 200 ml CCl4-ban szuszpendá-lunk. Felforralás után acsepegtető tölcsérből adagoljuk hozzá a brómotolyan ütemben, hogy a re-
akcióhŐ az elegyet forrásban tartsa. Brómfeleslegesetén PBr5 képződik, ezért a sztöchiometriai mennyiségnél kevesebb brómot kellhasználni. Az összes bróm hozzáadagolása után az elegyet 15 percig viBSzafolya-tás közben forraljuk, majd ledesztilláljuk először a CCl4-ot, azután vízhűtés nélkülaPBr3-ot (op. — 41,5C°,fp. 172,9 C°). Színtelen, levegőn füstölgő, hidrolizáló folyadék. (s18«=2,85.)
56.1.18. Szilícium-tetrabromid és króm(Ill)-bromid (SlBr4 és CrBrs)
Kvarccsőben elhelyezett elemi szilíciumot (ill. krómot) a vörösizzás hőmérsékletén Br2-gőzökkel reagáltatva SiBr4 (ill. CrBra) képződik. A laboratóriumi méretben aluminotermiásan is előállítható szilíciumot porcelán csónakban helyezzükel a cső belsejében. A Bra-gŐzöket változtatható hőmérsékletű vízfürdőbe helyezett,vízmentes brómmal töltött mosópalack segítségével állítjuk elő úgy, hogy az ugyan-
6 Fernelius, W. <7.:Inorganic Syntheses, Vol. EL McGraw— HillB ookC ., New York— London, 1946, 147. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 212/366
214 V ÍZ M E N T E S H A L O O E N T D E K
csak szárított indifferens vivőgázt (nitrogént vagy argont) átbuborékoltatjuka mosópalackon. A reakoió megindulásához szükséges hőmérséklet (kb. 650 C°)elérése előtt először a száraz és oxigénmentes gázzal kiszárítjuk a berendezést, éskiszorítjuk belőle a levegőt. Csak azután hevítjük vörösizzásra a csónakban levőszilíciumot (ül. krómot). A képződő illékony SiBr4-ot csiszolattal csatlakozó, só —
jég-keverékkel hűtött szedőbe vezetjük, melynek a fülkébe vezető nyílását foszfor-pentoxidos toronnyal védjük a légnedvességtől. A gázáram brómtartalmát úgyszabályozzuk, hogy lehetőleg teljesen elreagáljon, s a szedőbe minél kevesebb brómkerüljön. A nyersterméket desztiUációval tisztítjuk meg a brómszennyeződés zömétől, annak utolsó nyomait pedig rézporral távolítjuk el.
A tiszta SiBr4 átlátszó, színtelen folyadék. Op. 5 C°, fp. 153 C°lt s25o= 2,81.
A CrBr 3 sötét vörösbama, zöldesen áttetsző kristályokat alkot, vízben oldhatatlan (825° = 4,25).
56*1.19. Cirkónium-, titán-,szílícíum-tetrajodid és alumínium-jodid (Z rl4, T il 4, S il4, A lls)msss-
Noha a halogének közül a jód a legkevésbé reakcióképes, mégis sok elemmelközvetlenül egyesül. A reakciót célszerűen vákuumban végezzük. Az alkalmazottkészülék lényege magas olvadáspontú üvegből készült kettős teke, amelynek össze
kötőcsövét a leforrasztás megkönnyítésére előre elszűkítettük. Mindegyik tekének külön töltőnyílása,
azonkívül egyiknek még a szivattyúhoz csatlakozócsiszolata, a másiknak a jód lehűtésére szolgáló nyúlványa is van (56.1.19.1. ábra). Az egyik tekébe a fém-port, a másikba a P20 5 fölött megszárított jódot tesz-szük, majd a töltőnyílásokat leforrasztjuk. A higany-diffúziós szivattyút ajánlatos folyékony nitrogénnelhűtött aktívszenes csapdával védeni a jódgozöktől.Ezután a berendezést evakuáljuk, a fémport kigázo-
56.1.19.1. ábra. Zrl4előállítása sítjuk és vákuumban leforrasztjuk. Ki gázosítás közben csak a Zr-tartalmú tekét fűtjük, a I2-ot ugyan
akkor folyékony levegővel hűtjük. A jól leszívott kettős tekét 400 —500 C°-os kemencébe tesszük. A cirkónium tűztünemény közben egyesül a jóddal. A komponensek közül a fémet mindig kis feleslegben alkalmazzuk. A reakció végeztével a terméket tisztítás céljából visszaszublimáljuk a kiürült jódos tekébe, és a gömböt akét tekét összekötő szűkületnél is leforrasztjuk. Az anyag igen higroszkópos, hidrolízisre hajlamos.
A többi felsorolt fém-jodidot teljesen hasonló módon állítjuk elő.
Zrl4 op. 499 C° (6,3 atm), szubl. p. 331 C° s = 2,80Til
4op. 150 c ° ,
fp.365 C°
s2s=«= 4,40Sil4 op. 120,í)C°, fp. 287,5 C° s = 4,02a i i 3 op. 191 C°, fp. 382 c ° , an« = 3,95
56.1.20. ón (V)-jodid , alumínium-jodid és toszfpr-trijodld (Snl4, A1IS, PI8)
Szintézisük indifferens oldószerrel (pl. CSa) készült jódoldattal is elvégezhető. A reakciósebességet a komponensek koncentrációján kívül a hőmérséklet szabjameg. Mivel a hőmérséklet a CS2 alacsony forráspontja miatt alig variálható, és a szi-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 213/366
V ÍZ M ENTES HALOG ENID EK ELŐÁLLÍT ÁSA O X ID O K HALOGÉNEZÉSÉVEL 216
lárd anyagként is jelenlevő reakciókomponens koncentrációja is adott, egyedül a jódkoncentrációjának változtatására van lehetőség.
Az Snl4előállítására pl. 1 súlyrész ónra öntsünk 6 súlyrész oldószert, és ebbenoldjunk fel 4 súlyrész jódot. A reakciót csiszolatos, visszafolyatásra állított hűtővelellátott gömblombikban végezzük. A hűtő hosszú legyen, és intenzív hűtésről gondoskodjunk, hogy az igen gyúlékony és mérgező oldószer gőze a laboratórium levegőjébe ne jusson. Nagyobb mennyiség esetén a reakció meggyorsulhat, úgyhogy alombik hűtésére is fel kell készülni.
A reakció végeztével a sárga színű oldatot a fémmaradékról leöntjük, és azanyag vízmentességét biztosítva, bepároljuk. Az Snl4 vörös színű kristálytömegalakjában marad vissza.
Az A1I 3 előállításakor a túlmelegedés veszélye nem áll fenn, a reakcióelegyet —a megfelelő tűzbiztonsági rendszabályokat betartva — inkább melegíteni kell. A PI 3 készítése7az előbbiektől annyiban különbözik, hogy mivel az oldószer a
foszfort is oldja, a jódoldatöt nem egyszerre adjuk hozzá. A lombik feltétjére tehátnemcsak visszafolyatásra állított hűtőt, hanem csepegtető tölcsért is helyezünk.
Snl4op. 143,5 C°, fp. 340 C°; PI 3 op. 61 C°.
66.1.21. Alumínium-jodid, difoszfor-tetrajodid és bizmut-jodid (A1I3, P2I4, és B il3)
Előállíthatók a megfelelően aprított szilárd komponensek melegítése és össze
olvasztása útján. így pl. A1I 3 220 C°-on bombacsőben is szintetizálható. 2 g Al-for-gácsot és 20 g I2-ot kb. 20 mm átmérőjű, 50 cm hosszú bombacsőbe forrasztunk, ésa csövet három óra hosszat 220 C°-os olaj fürdőben vagy alumínium blokkban tart juk. A képződött sötétszürke nyersterméket még desztillációval tisztítani kell.
A P2I4és Bil- szintéziséhez nyomásra nincs szükség. A sztöchiometrikus arányúelegyet megfelelő tűzálló edényzetben egyszerűen összeolvasztjuk. A BiI3-ot szubli-málással tisztít j uk.
P2I4op. 124,5 C°; Bil 3 op. 408 C°.
56.2. Vízmentes halogenidek előállítása oxidok halogénezésével
A halogenidek elemi szintéziséhez szükséges egyes elemek előállítása bizonyosesetekben elég körülményes. Gyakran az oxid könnyebben hozzáférhető anyag,ezért gyakorlati szempontból igen jelentős a fém-oxidok klórozásának megoldása.Sok fém-oxiddal a klór magasabb hőmérsékleten is csak olyan kismértékben reagál,hogy erre preparatív módszert alapítani nem lehet. Szén jelenlétében azonban elégmagas hőmérsékleten megindul a halogénképződés, noha általában ezek a fém-oxidok szénnel önmagukban nem redukálhatok. A szilárd elegyrészeket megfelelő fi
nom eloszlásban kell egymással elkeverni.
66.2.1. Titán-tetraklorid, szilícium-tetraklorid és bór-triklorid (TiCl4, SiCl4és BC13)
A titán-tetraklorid készítésére kiindulási anyagul technikai Ti02-ot használhatunk. Ennek 100 g-ját 25 g korommal és kevés keményítőcsirizzel vagy telített cukoroldattal sűrű masszává gyúrjuk. Ebből kb. 1 /2 cm átmérőjű golyókat formálunk,
7 Ger mann , F . E. E . , Tr ax ler , R. N - : J. Am . Chem. Soc. 49, 307 (1927).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 214/366
216 VÍZ MENTES HALOGENID EK
vagy ilyen rétegvastagságban üveglapra kikenve, késsel 1/2 cm élhosszúságú kockákká aprítjuk. Szárítószekrényben 150—200 C°-on teljes vízmentességig szárítjuk,
végül tégelyben, kevés korommal befedve fújtatólánggal enyhén ki is izzítjuk. Azígy előkészített anyagot megfelelő méretű tűzálló (porcelán, kvarc- vagy Supre-max) csőben helyezzük el, és száraz C02-dal a nedvesség utolsó nyomait is eltávolít
juk a kihevített anyagból és készülékből. A berendezéshez (lásd az 56.1.8.1. ábrát)a gáz szárítására szolgáló kénsavas mosópalackon és ugyancsak kénsavas túlnyomásszelepen kívül az izzítócső másik végén csatlakozó leszálló hűtő és szedő tartozik. A hűtő eleje széles, kb. 4—5 cm átmérőjű, 30 cm hosszú cső, amelyet a Ti02 vasszennyeződéséből képződő FeCl 3 megkötésére kvarckaviccsal töltünk meg.Ennek folytatása a minél hatásosabb kondenzáló hűtő, amely a jéggel hűtött szedőbe
torkollik. A szedő másik nyílása a gázfölösleg elvezetésére szolgál, és a légnedvességbehatolásának megakadályozására P205-os toronnyal áll kapcsolatban.Ha a berendezés és az anyagok vízmentesek, élénk klóráramot vezetünk át a
készülékben, és az izzítócső töltetét lassan vörösizzásig melegítjük. A vasszennyezéstől megszabadított TiCl4átlátszó, színtelen, nagy fénytörésű, levegőn füstölgő,könnyen hidrolizáló folyadék. További tisztítása desztillációval történik.
A szzllcium-tetraJdorid ugyanilyen készülékben állítható elő, de alacsonyabbforráspontja miatt fokozottan kell gondoskodni a kondenzálásról és a szedő hűtéséről. Ha van rá lehetőség, a hűtővizet is só—jég-keverékbe ágyazott vörösréz csőkígyón átvezetve hűtjük.
Legalacsonyabb az eddig tárgyalt vegyületek közül a BC1 3 forráspontja. Ennekszedőjét és kondenzáló csőspirálját alkoholos szénsavhóval kell hűteni.
b c i 3 op. —107 CP, fp. 12,5 C°, Sq o — l i f 3 ,
SiCl4 op. — 70 C°, fp. 57,0 C°, S jjo — 1,48,
TiCl4 op. - 25 C°, fp. 136,4 C°, ®2o° =
56.2.2. Cirkónium- és tórium-tetrabromid (ZrBr4, ThBr4)
Előállításuk8
a klórozáshoz teljesen hasonló módon, ugyanolyan elvek alapjánösszeállított készülékben végezhető. A folyékony brómot vízfürdővel melegíthetőmosópalackon átbuborékoltatott száraz CÖ2-vivőgázzal juttatjuk el a vörösizzásrahevített reakciópartnerhez.
56.3. Vízmentes halogenidek előállítása halogénátvivő segítségével
A különböző vízmentes halogenidek előállítására felhasznált halogénátvivőketkétféle elv szerint alkalmazhatjuk: sztatikus és dinamikus eljárásban. Az első típus
hoz tartozó módszernél pl. leforrasztott bombacsövet használunk, a másikban a klórozószer gőzét a csőkemencében izzított szilárd komponens felett vezetjük át.
A sztatikus eljárás előnye, hógv kevesebb az anyagveszteség, és folyékony halmazállapotú klórozószer használata esetén a termék szépen kikristályosodhat. Akomponensek mólsúlyviszonyának megfelelő változtatásával különböző összetételűtermékek előállítására nyílik lehetőség. A bombacsöves módszer hátránya, hogy
8 Femél i u s, W . G .: Inorganic Syntheses, Vol. II . N ew Yo rk— London 1946, 114. old

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 215/366
VÍZMENTES K LO RID O K ELŐÁLLÍT ÁSA HALOGÉNÁTVIV Ő SEGÍT SÉGÉVEL 217
csak viszonylag kis mennyiségekre alkalmazható, nagyobb mennyiségek feldolgozására pedig drága autokláv szükséges. A közben fellépő nagy nyomás veszélye ishátránya a zárt rendszerben végzett reakcióknak.
A dinamikus eljárás előnye a folyamatosabb munkamenet, a szennyezésekazonnali eltávolításának lehetősége. A dinamikus eljárásban felhasználható berendezés részben az előző pontban leírt, részben a bromidok elemi szintézisére javasolt összeállításhoz hasonló.
56.3.1. Titán-tetraklorid (TiCl4)
A Ti02-ot porcelán csónakban kb. 500 C°-ra hevíthető üveg- vagy kvarc
csőben helyezzük el, melyet elektromos- vagy gázfűtésű kemencével melegítünk. A halogénátvivő CC14 gőzeit indifferens, száraz gáz (C02) szállítja. A klórozószerkoncentrációját a szén-tetrakloridos mosópalack vízfürdőjének hőmérsékletévelszabályozzuk. A reakció megindítása előtt a rendszert gondosan kiszárítjuk, és aterméket P20 5-os toronnyal szintén gondosan óvjuk a légnedvességtől. A
Ti02 + 2 CC14= TiCl4+ 2 COCl2
reakcióban képződő COCl2-ot jól húzó fülke kéményébe vezetjük.
56.3.2. Lantán(IQ)-klorid (LaCl3)Fém-oxidok klórozására előnyösen felhasználható az NH4C1 is. Ebben az eset
ben valószínűleg a termikus disszociációban keletkező NH 3 redukálja az oxidot.10 g lantán-oxidot vagy egyéb ritkaföldfém-oxidot 20 g NH4Cl-dal mozsárban
finoman elporítunk és összekeverünk, majd porcelán tálba téve, Bunsen-ég6n óvatosan, állandó keverés közben izzítjuk. A reakció végét az jelzi, hogy a kivettpróba vízben maradéktalanul oldható. A maradék NH4Cl-tól vákuumszublimációval tisztítjuk meg az anyagot. A vákuumberendezés munkatere kb. 30 cmhosszú, vízszintes üvegcső. Ennek végében elhelyezett anyagot elektromos cső
kemencében lassan 300 C°-ra hevítjük, és néhány órán át ezen a hőmérsékleten tart juk. Ezalatt az NH4C1 a cső hidegebb részében gyűlik össze. Vákuumban hűtjükki, és a vákuumot vízmentes körülmények között szüntetjük meg. A kapott LaCl 3 igen higroszkópos, fehér por, NH^-iont és La20 3-ot nem tartalmaz, vízben maradéktalanul oldódik (op. 872 C°, 8250= 3,84).
Az eljárás egyéb ritkaföldfém-kloridok előállítására szintén alkalmas.
56.3.3. Bór-triklorid (BC1S)
A BC1 3
elemi szintézisénél jobb kitermelést ad az a módszer,amelynek alapjaa BF3, illetve KBF4-nak AlCl3-dal végbemenő cserebomlása.9
a) Az egyik változat szerint a KBF4-ot AlCl3-dal elkeverve speciális frak-cionáló lombikban, olajfürdőn óvatosan 150—170 C°-ra kell felmelegíteni. A képződő KF és A1F 3 a lombikban marad, az illékony BC1 3 pedig a szénsavhóval vagyfolyékony levegővel hűtött szedőben gyűlik össze. A kitermelés kb. 40— 50 %.
9 Gambl e, E . L Inorganic Syntheaes Vol. H L New York— London, 1950, 27. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 216/366
218 VÍZ MENTES K ALOQ ENIDEK
b) Még kedvezőbb termelést érhetünk el a fenti eljárás másik változatával,melynek egyenlete:
BF 3 + A1C1 3 = BC1 3 + a i f 3.
A műveletet az 56.3.3.1. ábrán közölt rajz szerinti normálcsiszolatos berendezésben hajtjuk végre.
A készülék 1 literes lombikjába kb. 0,2 kg AlCl3-ot töltünk, és homokfürdőn180—200 C°-ra hevítjük. A BF 3 bevezetésére szolgáló cső a lombiknak kb. a közepéig nyúljon be, és az AlCl3-töltet fölött mintegy két ujjnyira végződjék. A szedőtcsiszolattal kapcsoljuk a lombikhoz. A mérgező hatású BF3-felesleget a légned
vesség távoltartására PVC-csővei csatlakozó P20 5-os tornyon át kivezetjük akürtőbe. A szedőt intenzíven kell hűteni. Legalkalmasabb erre az alkoholos szén-savhó. A folyékony levegő felesleges, sőt káros, mert hatására kondenzál a BCl3-banamúgy is jól oldódó BF3, melynek jelenléte megnehezíti a termék kezelését.
A BCI 3 előállítására felhasznált BF3-ot KBF4-ból állítjuk elő :10
6 KBF4+ B20 3 + 6 H2S04 = 8 BF 3 + 6 KHS04 + 3 H20. A folyamat üvegből készült gázfejlesztő lombikban lejátszatható. Az ábra
szerinti bevezetőcsőhöz kénsavas mosó közbeiktatásával, T-cső segítségével kétgázfejlesztőt kapcsolunk. A kb. Y2 literes gázfejlesztőt esetenként 80 g KBF4és 16 gB20 3 finoman elporított keverékével, majd 240 ml cc. H2S04-val töltjük meg. A BF3-fejlődés hidegen nem megy végbe, ezért a két gázfejlesztőt homokfürdőn melegít-
10 Booth , M . / . , Wil son, K . J . A m . Chem. Soc. 57, 2273, (1935).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 217/366
VÍZ MENTES K LO BID O K ELŐÁLLÍT ÁSA HAL OGÉN ÁT VT VŐ SEGÍT SÉGÉVEL 219
jük. A csatlakozásokhoz PVC-csövet használjunk. A szükséges KBF4-hoz a 69.1.2szerinti módszerrel juthatunk.
Ezzel az eljárással nyert BC1 3 nyersen sárgás színű, és többszörös tisztításnak vetendő alá. Desztillálásához kizárólag üvegből készült csiszolatos berendezésthasználunk. Az előpárlat többnyire elég jelentős mennyiségű, és zömét az oldottBF 3 teszi ki. A desztillálást infralámpával gyorsíthatjuk. A desztillátumban visszamaradt szennyezéseket finom eloszlású higannyal való rázogatással, majd újra-desztillálással lehet eltávolítani. Ezt a tisztítást viszont az anyag forráspontjafeletti hőmérsékleten, 25 C°-on kell végezni oly módon, hogy a szedőbe kevéshiganyt töltünk, majd a szedőt leforrasztjuk, és az anyag felmelegedése után kb.fél óráig rázatjuk. Ez a folyamat a tisztítás legveszélyesebb lépése, amelyet csak
kellő biztonsági intézkedések mellett (fülke, védőszemüveg, drótháló, bombacső)szabad elvégezni. A leírt higanyos tisztítást és az azt követő desztillálást mindaddig ismétel
jük, amíg a desztillátum teljesen színtelen nem lesz (op. —107 C°, fp. 12,5 C°,s0O= 1,43).
56.3.4. Cirkónium-tetraklorid, rénium-, nióbium- és tantál-pentaklorid (ZrCl4, ReCla,NbCla, TaClj)
Vízmentes fém-halogenidek mikro- és félmikroméretekben történő előállítá
sára alkalmas eljárás alapja a megfelelő oxidok szén-tetraklóriddal való klórozása.11M2On + n CC14 2 MCln + n COCl2.
SchüUng-iivegből készült, vastag falú, 0,5—2 cmbelső átmérőjű és 5—10 cm hosszú bombacsőbe 0,1 —0,5 g fém-oxidot töltünk, majd kb. ötszörös feleslegbenszén-tetrakloridót adunk hozzá: (A bombacső méreteitcélszerűen úgy választjuk meg, hogy a betöltött anyagaz össztérfogat 1/5-ét töltse ki.) Jeges hűtés mellett abombacsövet leforrasztjuk és autoklávba helyezzük.
Az autoklávba 10—20 ml CCl4-ot is töltünk és lezárjuk.Ez után az autoklávot 400 C°-ra fűtjük fel, aminek hatására a nyomás kb. 100 atmoszférára növekszik. Bár eművelet során robbanási veszély nem áll fenn, a nagynyomású autokláv alkalmazásakor előírt rendszabályokat (külön helyiség, más helyiségből vezérelhető fűtés)be kell tartani (lásd 47.2.). A reakció 3—4 óra alatt lezajlik ; az autokláv lehűlése után a szelepet a ventillációbekapcsolása mellett megnyitjuk, majd az autokláv
fedelét levesszük. A bombacsövet jeges hűtés után amegfelelő óvatossági rendszabályok betartása mellett(lásd 47.1.), fülkeajtó védelme mögött (védőszemüveg!)nyitjuk meg, és gyorsan egy előkészített, csappal ellátott csiszolatos edénybe helyezzük (56.3.4.1. ábra).Utóbbit vákuumgumicsővel -szárazjegéé csapdán és biztosító pufferedényen keresztül vízlégszivattyúhoz csatlakoztatjuk. A szén-tetrakloridot enyhe forralás közben
56 .3.4 .1* ábra. Mikro- -vákuumbepárló
11 Kl ein ber g , J . : Inorganic Syntheses Vo l. VI I. N ew Yo rk— London, 1963, 163. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 218/366
220 VÍZMENTES HALO OEN1DEK
(esetleg infralámpás melegítéssel) a fele mennyiségre bepároljuk, amivel a C0C12 iseltávolítható. A bepárlás befejezésekor a csapot zárjuk és az edényt szárítóoszloponátlevegőztetjük, majd P20 5-tartalmú exszikkátorba helyezzük. A kivált kristályokmosását (absz. CCl4-dal) és a szűrést szívógömbtechnikával végezzük (52 1.1.),ennek során azonban tekintettel kell lennünk a termék rendkívül higroszkópos voltára.
ZrCl4színtelen kristályok, op. 437 C° (nyomás alatt), szubl. p. 331 C°, s=2,80;ReCl5zöld kristályok, op. alatt diszproporcionálva bomlik, vákuumban 200 C°-
on szublimálható;NbClg citromsárga, tűs kristályok, op. 194 C°, fp. 240,5 C3, s = 2,75;TaCl5 színtelen kristályok, op. 221, fp. 241,6 C°, s = 3,68.
56.3.5. Bór-tribromid (BBr3)
Készítésére az 56.3.3.1. ábra szerinti berendezést használhatjuk. A lejátszódófolyamat:
BF 3 + AlBr 3 = BBr 3 + A1F3.
A feladat annyival egyszerűbb, hogy a BBra forráspontja jóval magasabb akloridénál, ezért szénsavhó helyett só—jég-keverék is megfelelő hűtőanyag. A bróm-fölösleg eltávolítása céljából a nyersterméket — visszafolyós hűtőt alkalmazva— fémhiganyon főzzük. Desztillálni csak száraz C02-atmoszférában lehet, merta nedvesség és az oxigénnyomok is brómképződés közben végbemenő bomlástokoznak. Tiszta állapotban színtelen folyadék. (Op. —46 C!°, fp. 90,8 C°, s0O= 2,65.)
56.3.6. Vottrám(VI)-bromid (WBre)
Bombacsőben 200 C°-on kényelmesen előállítható W 03-ból és CBr4-ból. A folyamatban kétféle mechanizmussal számolhatunk:
2 WOj + 6 CBr4 = 2 WBr9+ 6 COBr2,
2 W 0 3 + 3 CBr4 = 2 WBr6+ 3 C02.
Ha a teljesen Bzáraz W 03-ot és a CBr4-ot 1: 3 mólarányban forrasztjuk be,az egyenlet szerint COBr2-t kellene kapnunk. A valóságban azonban mindig képződik C02 is, ami a bombacsőben jelentős nyomásnövekedést okoz. Ezért célszerűa gáznemű termékeket a cső felnyitása előtt folyékony levegővel hűtve kondenzálni. A termék sötét színű, kristályos tömeg, erősen higroszkópos. Az adszorbeált bróm-tól és bróm-föszgéntől vákuumban tisztítjuk meg. A CBr4 felesleget CCl4-dal iskimoshatjuk. Szublimációval nem tisztítható, mert elég bomlékony (s = 6,9).
56.3.7. Szén-tetrajiromid (CBr4)
Az AlBr 3 a CCl4-dal 100 C°-on bombacsőben cserebomlásra képes. Igen fontos,hogy az AlBr 3 teljesen vízmentes legyen, különben igen heves reakciók következhetnek be. Az alumínium-bromidot ezért — az ekvivalens mennyiségű CCl4hozzáadása előtt — a bombacsőben megolvasztva is víztelenítjük. A reakció 100 C°-onnéhány óra alatt végbemegy. Utána az elegyet sok vízbe dobjuk. A képződött

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 219/366
VÍZM EN TES HAL OGÉN ED EK ELŐÁLLÍT ÁSA H ALOGÉN-H IDROGÉNNEL 221
AICI 3 feloldódik, a CBr4 leülepszik az edény aljára. Szűrőre gyűjtjük, vízzel több- -szőr kimossuk és Pa0 5 fölött szárítjuk. Vákuumszublimálással vagy alkoholból
való átkristályosltással tisztítható. Fehér, vízben rosszul oldódó kristályos anyag.200 C°-on C02-ra és HBr-ra hidrolizál. (Op. 93,7 C°, fp. 189,5 C°, s = 3,4.)
66.8.8. Szén-tetrajodid (CI4)
Előállítható A1 Cl3-katalizátorral a következő egyenlet szerint:
CC14+ 4 C2H5I = CI4+ 4 C2H6C1.
A reakció szobahőmérsékleten is gyorsan megindul. 15 g CCl4-ot 60 g etil-
-jodiddal 500 ml-es gömblombikban elegyítünk. Mindkét komponens és az edényis száraz legyen. Katalizátorként hozzáadunk még 1 g AlCl 3-ot, majd az edénytCaCl2-os vagy P»05-os szárítófeltéttel elzárjuk. Az oldat csakhamar felmelegszikés megvörösödik annak jeléül, hogy a reakció megindult. A képződő etil-klorid(fp. 13,1 C°) a szárítófeltéten át elpárolog. Kb. 1/2 óra múlva kiválik az oldatból avörös kristályos CI4. Dekantáljuk, majd a katalizátort kb. 50 ml jeges vízzel elhidro-lizáljuk, és a terméket üvegszűrőre gyűjtjük. A felületén kivált I2-ot alkohollallemossuk, végül vákuumexszikkátorban P20 5 vagy szilikagél fölött szárítjuk (op.171 C°).
A CI4 élénkvörös színű kristályos anyag, vízben és alkoholban oldhatatlan,
de hosszabb idő alatt hatásukra elbomlik. Hő- és fényérzékeny, (s = 4,3.)
56.4. Vízmentes halogenidek előállítása halogén-hidrogénnel
66.4.1. Alumínlum-klorld (A1C1S)
Fém-alumínium sósavgázzal 200 C° hőmérsékleten alumínium-klorid és hidrogén képződése közben reagál,
A berendezés lényeges része szabályozható csőkemencével fűthető, tűzállóüvegcső, melynek alakját az 56.4.1.1. ábra mutatja. Ebben helyezzük el lazán atiszta, zsírtalanított felületű alumínium forgácsot. A képződő A1C1 3 a csőből ahozzá csatlakozó szobahőmérsékletű tekébe szublimál. Innen-Bunsen-égővel tovább szublimálhat
juk abba a széles szájú porüvegbe, amelyben Cleparaffinozva el is tartható. A tekéből a porüvegbe átvezető CSŐ vége azért szélesedik ki, mert 56.4.1.1. ábra. Reaktorcső A1C1,ezt a végét a porüveg dugójába ágyazzuk bele, előállításáratehát nem melegíthető. Ezzel megakadályozzuk,hogy a megszilárduló szublimátum eltömje a csövet. A képződő hidrogént egykivezetőcsövön át tovább vezetjük a fülkébe, de útjába P2Os-os tornyot iktatunka hidrolízisre hajlamos termék légnedvesség elleni védelme érdekében. Hogy afolyamat sebességét hozzávetőleg ellenőrizhessük, paraffinolajjal töltött buborék-számlálón is átvezetjük a hidrogénáramot.
A felhasznált HCl-ot NH4Cl-ból kénsavval állítjuk elő. A gázfejlesztő lombikjában elhelyezett szilárd sóra csepegtetjük meghatározott sebességgel a savat.Ha az elegy erősen habzana, kevés absz. alkohollal csökkentjük a felületi feszült-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 220/366
222 VÍZ M ENTES HALOOENID EK
ségét. A sósavgázt két kénsavas mosóban víztelenítjük, és az esetleg bekövetkeződugulás jelzésére kénsavba merülő T-alakú túlnyomásszelepen at vezetjük a reak
ciótérbe. A tiszta A1C1 3 fehér, kristályos, igen higroszkópos, 177,8 C°-on szublimálható anyag. Leforrasztva vagy paraffinozott dugójú porüvegbén tartható el.
56.4.2. Bór-tritluorld (BFS)
Az alábbi reakcióban képződik:
6 KBF4+ 6 HaS04+ B20 3 = 8 BF 3 f 6 KHS04 + 3 H20.
45 g KBF4-ot 7,5 g finoman elporított B20 3-dal jól összekeverünk, és csiszo
latos gázfejlesztőben 100 ml cc. H2S04-at öntünk hozzá. A reakció 150—240 C°közötti hőmérsékleten megy végbe; ezt a hőmérsékletet Bunsen-égŐvel melegítetthomokfürdővel biztosítjuk.
A BF 3 igen stabilis vegyidet. Az üveget nedvesen sem támadja meg. Nemhidrolizál, hanem mono- vagy dihidrát alakban megköti a vizet. Forráspontjaigen alacsony (—101 C°), ezért kondenzálására folyékony levegő szükséges. Vízmentes BF3-ot úgy állítunk elő, hogy kondenzálás előtt cc. kénsavas mosóval vagyszénsavhóval hűtött ldfagyasztócsapdával megszárítjuk. Kényelmesen eltarthatóa BF 3 anizolban oldva, amelyből melegítéssel visszanyerhető. Fejlett szerves
kémiai iparral rendelkező államokban acélpalackokban komprimálva is kapható.
56.4.3. Arzén-trifluorid (AsF ,)
A bór-trifluoridhoz hasonló elv alapján állítható elő arzén-trioxidból és aCaF2 -HFLjSC^ reakcióban képződő HF-ból.12 Kevésbé stabilis, mint a BFa. Levegőnhidrolizál, nedvesen lassan megtámadja az üveget. Kondenzálására és eltartásáraaz üvegnél alkalmasabb szerkezeti anyag az ólom vagy platina.
200 ml-es frakcionáló lombikba 25 g As20 3 és ugyanannyi CaF2 keverékéttesszük, majd 50 ml cc. kénsavat öntünk rá. A melegítés hatására fejlődő gőzöket
só—jég-keverékkel hűtött ólomspirálisban kondenzáljuk, és a kondenzátumot lehetőleg ólom- vagy platina szedőbe gyűjtjük. Az AsF 3 gőze rendkívül mérgező, ezértcsak jó huzatú fülkében készíthető. A kondenzátum két fázisból áll; a kisebb fajsúlyú vizet tartalmaz. Az alsó fázist választótölcsérben elválasztjuk és kisebb lombikból még egyszer ledesztilláljuk (op. —8,5 C°, fp. 63 C°). Az AsF 3 színtelen, füstölgő, mérgező folyadék (s = 2,67).
57. Kcverékhalogenidek
Több értékű elemek egyidejűleg különböző halogénatomokkal is kapcsolódhatnak. Ezek kőiül a halogénvegyületek közül aránylag stabilisak a szén, valaminta szilícium vegyes halogenidjei. Közülük a CF2C12 freon néven a hűtőgépiparbannagy gyakorlati jelentőségre tett szert.
12 Brauer , G.: Handbuch dér Práparativen Anorganischen Chemie, Enke, Stuttgart, 1954, 159. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 221/366
KEVEKÉKHJ lLOGENIDEK 223
57.1. Szén-diklorid-dibromid (CCl2Br2)
A vegyes szén-halogenidek legrégibb előállításmódja pl. a parciálisán halogénezett metán maradék hidrogénjének más halogénatommal való kicserélése
bombacsőben, magasabb hőmérsékleten. A keletkezett termék sohasem'egységes,mindig képződik bizonyos mennyiség valamennyi lehetséges származékból.
Kb. 2 g CH2Cl2-ot 8 g Br2-mal 2 cm 0 -jű, 50 cm hosszú bombacsőbe forrasz
tunk, és acél védőköpenyben olajfürdő segítségével kb. 150 C°-ra felmelegítjük.
Főleg a következő reakció végbemenetelére számíthatunk:
CH2C12 + 2 Br2 = C a2Br2 + 2 HBr,
A képződő termék op.-ja 38 C°, fp.-ja pedig 150 C°.
57.2. Szilícium-diklorid-dijodid (SiCl2I2)
Előállításának többféle lehetősége közül legérdekesebb a szilícium és az inter-
halogenidek között magasabb hőmérsékleten végbemenő reakció,1 melyet az elemi
szintézis módszerével hajtunk végre. A ICl-ot' közvetlenül a felhasználás előtt
állíthatjuk elő úgy, hogy kisebb, erősen hűtött frakcionáló lombikban elhelyezett,P20 5 fölött szárított jódra áramoltatjuk az ugyancsak gondosan szárított klórt.
A képződő IC1 (op. 27,2 C°, fp. 97,4 C°) elpárolgását forró vízfürdővel segíthetjük
elő. A IC1 hatására képződő termék azonban nem tiszta SiCLjI^ hanem a könnyen
végbemenő halogéncsere lehetősége miatt SiClI3-ot és SiCl 3I-ot is tartalmaz. A kép
ződött nyersterméket vákuumdesztillációval lehet
komponenseire bontani. Az egyes vegyes halogeni-dek fizikai tulajdonságai fokozatosan változnak a két
határeset között. A szilicium-jodid-kloridok előállítására legalkal
masabb a SiCl4 és H l közötti halogéncsere:2
SiCl4+ H l = SiCl3I + HC1,
SiCl4+ 2 Hl = Sia2I2 + 2 HC1,'
S ia4+ 3 H l = SiCH 3 + 3 HC1.
A megfelelő mólarányban összekevert vízmentes H l és SiCl4 elegyét vörösizzásra hevített porce
lán vagy kvarccsövön vezetjük át. A reaktorból agázelegyet kondenzáló spirálhűtővel ellátott, száraz
jég—aceton-hűtőkeverékkel hűtött szedőbe vezet
jük, és kondenzáltatjuk. (A —112 C° forráspontúHCl-ot a rendszerből elvezetjük.) Tekintettel arra,
hogy a konverzió kis hatásfokú (kb. 10—20 %), a képződött szilícium-klorid-
-jodidokról lehajtott H l és SiCl4 elegyét célszerű a reaktorba visszajuttatni.
1 Hech t , H . : Práparative Anorganische Chemie. Springer, Berlin, 1951, 110. old.
2 Besson , M . A . : Comptes Rendus, 1891, 611.
57 .2.1. ábra. Szilíoium-klorid-
-jodidok forráspontja

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 222/366
224 VÍZ MENTES ÚALO GEN ID EK .
Az 57.2.1. ábra a szilícium-klorid-jodidok forráspontjának a SiCl4és Sil4forráspontja közötti lineáris változását mutatja.
57.3. Alumínium-klorid-dijodid (A1C1I2)
CC14 és A1I 3 halogéncseréjének eredményeképpen jöhet létre:
CC14+ A1IS = A1C1I2 + CC1 3I.
A komponensek szén-diszulfidos oldatát hosszabb ideig 0 C°-on taríjuk, majd azoldószert és a maradék CCl4-ot ledesztillálva, azt tapasztaljuk, hogy a hőmérséklethirtelen 142 C°-ra emelkedik. Ez a szén-triklorid-jodid (CC1 3I) forráspontja. A desz-tillációt ezután vákuumban folytatjuk. A szén-triklorid-jodid ledesztillálása utánújabb jelentékeny forráspont-emelkedés jelzi az A1CH2 megjelenését. A fő frakcióátdesztillálása után a lombikban e terméknél is magasabb forráspontú A1I 3 bizo
nyos hányada maradhat vissza.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 223/366
58. Alacsonyabb vegyértékű halogenidek
A különböző fémek alacsonyabb vegyértékű halogenidjei általában bázikusabb
sajátságúak, mint a magasabb oxidációfokúak. A kötéstípus is kevésbé apoláros jellegű, mert a központi fémiont körülvevő halogenidek kisebb száma miatt azokárnyékoló hatása is kisebb. Ennek következtében a „ + ” töltésű fémion hatásabizonyos mértékben a szomszédos molekula „ —” töltésű részei felé is érvényesül.
Előállításuk legegyszerűbb módja a termikus bontás a következő egyenletalapján:
2 MeXn = 2 MeXn-x + X 2.
Az egyensúly a hőmérséklet-emelés következtében jobbra tolódik. Egyes esetekben (pl. PtCl4 esetén) a fokozatos disszociáció a hőmérséklet növelésével egészen
az elemi fémkiválásig folytatódik. Alacsonyabb oxidációfokú halogenidek alacsonyabb hőmérsékleten diszpro-
porcionálódásra hajlamosak:
2 TiCl 3 = TiCl2 + TiCl4,
4 TiCl 3 = 3 TiCl4+ Ti.
Más lehetőség az alacsonyabb vegyértékű halogenidek előállítására a magasabb oxidációfokú halogenidek redukciója.
58.1. Antimon-triklorid (SbCl3)
Magát a halogenidet képező elemi állapotú fémet használjuk fel redukcióraaz SbCl 3 előállításakor:
3 SbCl5+ 2 Sb = 5SbCl3.
Az SbCl5 előállítására megadott módszer (lásd 56.1.5.) segítségével termelttriklorid—pentaklorid-keveréket tartalmazó nyerstermékhez darabos antimont
adva, frakcionáló lombikban forraljuk mindaddig, amíg a fenti reakció végbe nemmegy. Csak az exoterm reakció végén tanácsos finomabb eloszlású antimon-porral siettetni a teljes átalakulást, mert kezdetben, mikor az SbCl5 móltörtje nagy,a reakciósebesség robbanásszerűvé fokozódhat. A fémmaradványoktól az SbCl 3 atmoszférikus desztillációval elválasztható (op. 73 C°, fp. 223 C°).
Igen tiszta SbCl 3 állítható elő a desztillált SbCl 3 nedvességnyomoktól védetttérben végrehajtott szublimálásával. A műveletet nagyobb frakcionáló lombik-
15 Ált. és szervetlen kémiai prakt. — 4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 224/366
226 ALACSONYABB VEG YÉRTÉKŰ HALOGENID EK ------------------------- -----------------------------------------------------------------------------------------h
bán végezhetjük, melynek nyakára a 48.2.3.1. ábra szerinti módon illesztjük atermék eltartására szolgáló széles szájú lombikot vagy porüveget. A szublimálás-
kor fellépő nyomáskülönbség a frakcionáló lombik oldalcsövén át egyenlítődhetki, melyet ezért P20 5-dal töltött szárítófeltéttel látunk el. A lombik tetejét folyamatosan nedvesített szűrőpapírral vagy ronggyal hűtjük, és a desztillált SbCl3-otvízfürdőn óvatosan melegítjük. Az anyag a hűtött felületen szépen fejlett, tű alakúkristályokban válik ki, melyeket az edény kihűlése után belerázhatunk a szedőbe. A készüléket szétszedjük, sa tisztított terméket tartalmazó üveg dugóját leparaffi-nozzuk.
Az SbCl3 színtelen, kristályos, higroszkópos és hidrolizáló anyag. (s25„— 3,14.)
58.2. Réz(I)-klorid (CnCl)
Vízmentes állapotban képződik vizes sósavoldatban is Cu(II)sók redukciójaútján. A redukálószer lehet SnCl2, S02, N2H4, NH2OH*HCl és fémréz is.
Készítsünk % literes Erlenmeyer-lombikb&n 125 ml cc. HC1 segítségével50 g CuS04 •5 H2Ó-ból és 25 g NaCl-ból oldatot. Tegyünk bele 20 g zsírtalanítottfelületű finom vörösrézport. Melegítsük vízfürdőn az elegyet elszíntelenedésig. Az oldat a réz(I)-klorid acidokomplexe, amely hígításra nagymértékben disszociál,és a nehezen oldható CuCl ennek következtében kicsapódik. Közben vigyáznunk
kell arra, hogy oxigén és fém együttes hatására a nedves CuCl ne oxidálódjék. A feladatot úgy oldhatjuk meg, hogy a fenti szuszpenzió tisztáját tartós forralássaloxigénmentesített és kevés S02-ot tartalmazó kb. 1 liter vízbe öntjük. A fehérCuCl-ot ugyancsak oxigénmentes, S02-tartalmú vízzel többs*zör dekantáljuk, üvegszűrőn mossuk, végül alkohollal, majd éterrel vízmentesítve, C02-áramban 100 C°-on megszárítjuk (op. 422 C°). Levegőn bázisos sók képződése miatt megzöldül. A tömény NH4OH amminkomplex, a cc. HC1 sósavas komplex alakjában oldja. A szén-monoxidot mindkét oldat megköti.
58.3. Titán-triklorid (TiCl3)
A titán-tetraklorid hidrogénes redukciójával állítható elő.1 A folyamat 1000C° körüli hőmérsékleten megy végbe, és a képződött TiCl3 diszproporcionálódásagyors lehűtéssel akadályozható meg. A feladatot ún. hideg-meleg-csőben (Saint- -Clair— Deville) oldhatjuk meg. Ezt 60x1,8 cm kvarccsőből készíthetjük, melynek belsejébe vékonyabb porcelán csőre 10 cm hosszan kantái- vagy platinafűtőellenállást tekerünk, és az egészet hűtővel körülvett 6 cm 0-jű Rasotherm--üvegcsőbe toljuk. A porcelán cső belsejében elfér még a vékony huzalból össze-hegesztett termoelem. A fűtőellenállást a túlzott keresztmetszet-növekedéB elkerülése céljából huzal helyett 2 mm széles, 0,1 mm vastag szalagból készítjük(58.3.Íja ábra). A teljes berendezést az 58.3.1/5 ábra szemlélteti. A vázlat szerintihelyen csatlakoztatjuk a száraz és oxigénmentes H2-áramot TiCl4-gőzzel telítőmosópalackokat. A négyutas váltócsap segítségével a H2-áramot először balról jobbfelé irányítjuk, és a levegőt a készülékből kiszorítjuk (durranógázpróba). Ezután a
1 Klemm, W., Krose, E.: Z. anorg. alig. Chem. 253, 209 (1947).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 225/366
V A S ( l l ) -K L O B I D 227
kvarccsövet 1000—1200 C°-ra fűtjük fel, és a csepegtető tölcséren ét a 60 C°-osvízfürdővel felmelegített mosópalackot megtöltjük TiCl4-dal. A cső meleg részein
végbemegy a TiCl4 redukciója, és a gázkeverékből a vízzel hűtött üvegköpenyrekiválnak a TiCl 3 iboly^színű kristálylemezei. A maradék TiCl4a másik hűtött mosópalackban só—jég-keverékkel kondenzálható. A gázáram sebességét úgy szabályozzuk, hogy a jegelt edényben éppen csak észrevehető legyen a TÍCI4kondenzé-lása. A hidrogén—sósav-keveréket P20 5-os tornyon át a fülke kéményébe vezetjük.Ha az első palack TiCl4-töltete elpárolgott, a termosztáló fürdőket felcseréljük,^ovébbá a gázáram útját a váltócsap segítségével megfordítva, folytatjuk a reduk-
h t
58.3.1. ábra. TiCl3 előállítása
ciót az anyag teljes felhasználásáig. A készüléket hidrogénnel töltve hűtjük le, majdhidegen nitrogénnel öblítjük át. A szén-dioxid azért nem alkalmas erre a célra-mert a cső szétszedésekor és a termék kiszedése közben—nagy fajsúlya miatt — ki‘folyik a készülékből, és a helyére nyomuló nedves levegőben a TiCl 3 elhidrolizál. A kristályokat szénsavval töltött ampullákba tesszük, és oxigént, vízgőzt távoltart-
va leforrasztjuk. A TiCl 3 432 C°-on szublimál.
58.4. Vas(II)-klorid (FeCl2)
A vas vízmentes halogenidjei közül klórgáz hatására a vasból FeCl3, szárazHCl-dal hasonló körülmények között FeCl2képződik:
Fe + 2 HC1 = FeClí + H2.
1000 C°-ra fűthető (elektromos) csőkemence 3 cm 0 -jű porcelán béléscsövébe15 g lazán elhelyezett, zsírtalanított vékony vasdrótot teszünk. A csövön szárazés óxigénmentes HCl-gázt vezetünk át, aminek eredményeképpen a cső hidegebbrészén szürkésfehér, higroszkópos kristályok alakjában FeCl2 rakódik le (szublimál kb. 670 C°-on; s = 2,98).
15

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 226/366
228 A L A C S O N Y A B B V E G Y É R T É K Ű H A L O G E N T D E K
58.5. Króm(II)-klorid (CrCl2)
A FeCl2-hoz teljesen hasonló módon készül. Apróra tört krómdarabokra szárazHCl-áramot vezetünk.2 A kiindulási anyagot porcelán csónakban porcelán vagykvarccsőbe helyezzük. A csövet fehérizzásig kell hevíteni; a képződő CrCl2 még ezena hőmérsékleten sem illékony, hanem szálas szerkezetű kristályaival megtölti acsónakot. A kristályok többnyire el nem reagált fémkróm zárványokat tartalmaznak. Színük tiszta állapotban fehér, a szennyezetteké azonban szürke. A HCl-banlehűtött anyagot C02-dal töltött ampullákba forrasztjuk, mégpedig csónakostól,hogy a levegő oxigénjével és nedvességével való érintkezés és az ennek következtében lehetséges oxidáció veszélyét csökkentsük, (s = 2,75.)
58.6. Urán(IY)-klorid (UC14)
Az U 02halogénezését enyhébb oxidációs hatású klórozóanyaggal (CC14, SOC^)végezzük.3 A tionil-klorid részben így is redukálódik S2Cl2-dá, aminek következtében bizonyos mennyiségben magasabb vegyértékű halogenidek is képződnek.
2—>3 g U 02-ot fölös mennyiségű SOCl2-dal bombacsőbe forrasztunk, és két napon át 200 C°-os olajfürdőben tartjuk. A végbemenő reakció:
U 02 + 2 SOCl2 = UC14+ 2 S02. A bombacső felnyitását óvatosan végezzük, előtte a képződő S02-ot szénsav
hóval kondenzáljuk. A termék zöld színű kristályos anyag. Szublimálással tisztítható (618 C°-on szublimál), igen higroszkópos. (s20° = 4,73.)
58.7. Yanádmm(III)-klorid (YC13)
Előállítható a vanádium(IH)-oxidnak tionil-kloriddal bombacsőben történő
klórozásával. 2,5 g por alakú V 20 3-ot 8,5 mól tionil-kloriddal mintegy 1,5 cm 0 -jűbombacsőbe forrasztunk, és 24 órán át 200C°-on tartjuk. Száraz jeges —alkoholoshűtés után (S02 kondenzálása) a bombacsövet felnyitjuk. A keletkezett VC1 3 halvány rózsaszínű kristálylemezek alakjában válik ki a tionil-klorid fölöslegéből. A kén-dioxid, tionil-klorid és egyéb gáznemű anyagok elpárolgása után lassú nitrogénáramban enyhe melegítéssel (100 —200 C°-on) elpárologtatjuk a magasabbvanádium-kloridokat is, amelyek mindegyike illékonyabb a VCl 3-nál. 800 C° körül VCl2-ra és VCl4-ra történő diszproporció közben bomlik (s20o = 3,00).
58.8. Vanádmm(II)-klorid (VC12)
A TiCl3-hoz hasonlóan magasabb oxidációfokú kloridjából, a halvány rózsaszín VCl3-ból állítható elő hidrogénes redukcióval, 430 C°-on. Vigyázzunk árra, hogy redukció közben a hőmérséklet ne emelkedjék 500 C°-ra, mert akkor fémvanádium
* Heckt, H .: Prftparative Anorganische Chemie. Springer, Berlin, 1951, 80. old. 8 Heckt, H., Jander, G., Schlapmann, H.: Z. anorg. alig. Chem. 254, 255 (1947).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 227/366
v a n á d i i t m ( i i ) -k l o r i d 229
képződik a diklorid helyett. A kitermelés közel 90 %-os. Előáhítása közben legnehezebb feladat az oxigén és a nedvességnyomok minél teljesebb távoltartása,mert ezek jelenlétében igen könnyen oxidálódik. Az almazöld színű kristályos anyagrendkívül nedvszívó és hidrolízisre hajlamos. Vízben oldva hidrogénfejlődés közbenoxidálódik. Éterben és alkoholban is oldható. Szublimálással szokták tisztítani.
Op. kb. 1350 C°, s = 3,09.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 228/366
59 . Kristályvíztartalmú halogenidek
Rácsszerkezetükbe is beépített vízmolekulát tartalmazó kristályokat a halo-
génvegyületek közül főleg egyes sószerű, poláros vegyületek alkotnak. Ezek vizesoldatból egyszerűen kikristályosíthatók. Preparatív nehézséget legfeljebb a meghatározott víztartalmú hidrát izolálása okoz.
Az apoláros jellegű, molekularácsos halogénvegyületek közül kristályvíztartalmú módosulatban csak kevés állítható elő, az is csupán különleges kísérletikörülmények között. A legnagyobb nehézség az ilyen anyagok víz jelenlétébenbekövetkező hidrolízise, amit egyszerűbb esetekben a közeg megfelelő halogenid-koncentrációjának biztosításával lehet visszaszorítani.
Egyes fémek különböző halogenidjei többféle kristályhidrátot is képeznek.
Ezek közül némelyek — eltérő tulajdonságaik ellenére — azonos összetételűek. Az analitikailag azonosnak talált kristályvízmennyiséget nyilván többféle módonkötheti meg az anyag. A vízmolekulák helyzete, kötésmódja ezekben a vegyület-típusokban igen hasonló más semleges molekuláknak (pl. NH3, különböző szervesvegyületek, stb.) a komplex vegyületekben elfoglalt helyzetéhez.
59.1. [Kexakvo-krdm(IlI)]'klorid ([Cr(H20 )9]Cl3)
Kb. 50 g zöld színű nyers króm(IÜ)-klorid-dihidrátot 40 ml vízben feloldunk,
és az oldatot felforraljuk. Az oldat szabadsósav-tartalmát króm(II)-oxid-hidráttalközömbösítjük mindaddig, amíg a gőzben indikátorpapírral sav már nem mutatható ki. (Az oxid-hidrátot híg meleg CrCl3-oldatból NH4OH-dal csapjuk le, ésforró vízzel mossuk.)
A semleges oldatot gyorsan mintegy 50 ml-re bepároljuk, majd só—jég-keverékkel 0 C° alá hűtjük, és utána hidegen sósavgázzal telítjük. A sósavval a
[Cr(H20 )9]Cl 3 ^ [Cr(HaO)fl]3+ + 3 Cl-
disszociációt szorítjuk vissza. Vigyázzunk arra, hogy a hőmérséklet 0 C° fölé ne
emelkedjék, mert a magasabb hőmérséklet az izomerizációs egyensúlynak a zöldszínű komplex felé való eltolódását segíti elő. Néhány óra múlva az anyag apró,ibolya színű kristályok alakjában kiválik. Üvegszűrőre gyűjtjük, hideg cc. HO-ol-dattal, majd acetonnal mossuk.
Kiindulhatunk a [Cr(H20)fl](N0 3) 3.H20 ugyancsak ibolyaszínű kristályaiból is. Az ibolyaszín itt is a hexakvokomplex jelenlétét mutatja. A sónak kb. 40 g-ját40 ml hideg cc. HCl-ban feloldjuk, és a [hexakvo-króm(III)]-kloridot 0 C° alatti
t

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 229/366
K R I ST Á X Y V ÍZ T A K T A L M Ú H A L O G E N I D E K 231
hőmérsékleten sósavgázzal telítve kristályosíthatjuk ki. A nyersterméket hidegvízből átkristályosíthatjuk. Telített vizes oldatában 0 C° alatti hőmérsékleten is
mert módon HCl-gázt oldunk. Ennek hatására az anyag szép nagy kristályokbancsaknem kvantitatíve kiválik. Acetonnal mossuk, és kénsavas exszikkátorban szárítjuk (op. 95 C°, 8=2,76).
59.2. [Dikloro-tetrakvo-krÓm(III)]-klorid-dihidrát
([ Cr( H20 )4C12]C1 •2H20 )
Visszafolyatásra állított hűtővel felszerelt literes gömblombikban 100 g
króm (Ill)-oxidot és 400 g cc. sósavat 2 —3 órán át főzünk. Ezalatt a vörös színűCrOjC^-gőzök távozása és a klórfejlődés megszűnik, s az oldat teljesen zöld színű lesz.Ezután porcelán tálban addig pároljuk az oldatot, amíg a CrCl 3 •6 H20-nak megfelelő elméleti 266 g súlyt el nem éri, és megvárjuk, amíg kihűl. A sűrű kristálypépet mázatlan agyagtányéron, CaO-dal töltött exszikkátorban szárítjuk.
Vizes oldatból átkristályosítva tisztítjuk. 50 g nyerstérméket kb. 40 ml vízbenoldunk. Az oldatot szűrés után jégbe hűtve sósavgázzal telítjük. A néhány óraalatt átkristályosodó tömeget szűrőre gyűjtjük, mosás nélkül 2 napig ex'szikkátor-ban szárítjuk, majd sósavas éterrel és acetonnal addig mossuk, míg a szűrlet
színtelen nem lesz. Vákuumexszikkátorban 80 %-os kénsav felett szárítjuk (op.83 C°,s=2,76).
59.3. Vanádium(III)-klorid-hexahidrát (VC13-6H20)
A vanádium-pentoxid sósavban oldva vanádium(IV)-oxid-kloriddá redukálódik. A redukció elősegítésére kevés oxálsavat teszünk az oldatba. A vanádium (III)-má való redukciót platinakatódon elektrolitikusan végezzük1, majd a triklorid--hexahidrátot hidegen kikristályosítjuk.
Kb. 25 g V 20 3-ot 250 ml cc. HCl-ban oldunk. A VOO2 további redukcióját
literes főzőpohárban végezzük. A kb. 5—7 cm 0 -jű agyagdiafragmában helyezzükel a redukálandó anyagot. Katódul — ha nincs elég nagy platina lemezünk — platina tégelyt merítünk az oldatba. Anódfolyadékul sósavat használunk, melybőlannyit töltünk a pohárba, hogy szintje azonos magasságban álljon a katódfolyadékdiafragmán belüli szintjével. A diafragmán kívüli sósavoldatba párhuzamos kapcsolásban három szénelektródot merítünk. Akkumulátortöltő egyenirányítóról vett12 V feszültséggel és 4 A áramintenzitással (kb. 0,1 A/cm2 áramsűrűséggel elekt-rolizálunk.
A vanádium háromértékű állapotát zöld színéről ismerhetjük fel. Hosszú időn
át tartó redukció hatására a katódfolyadék kékesibolya színűre változhat, ami avanádium(II) jelenlétének bizonyítéka. Ez azonban levegőátszívatással könnyenvanádium(III)-má oxidálható.
A zöld színű vanádium(IH)-tartalmú katódfolyadékot porcelán csészében aszükséges mértékben bepároljuk, majd jéghűtés közben HCl-gázzal telítjük. A képződött zöld színű kristályhidrátot üvegszűrőre gyűjtjük és erősen leszívatjuk. Vízzel nem szabad mosni, mert hidrolizál.
1 Piccini, A., Brizzi, N Z. anorg. alig. Chera. 19, 394 (1899).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 230/366
232 K H I S T Á L Y V Í Z T A K T A X M Ú H A L O G E N I D E K
59.4. Berillium(II)-klorid-tetrahidrát (BeCl2.4H 20)
Berillium-oxidot porcelén tálban,vízfürdőn cc. HCl-ban oldunk. A maradékotmindaddig újabb sósavmennyiséggel főzzük, míg az egész anyag üledék nélkültisztán nem oldódik. A tiszta oldatot töményre pároljuk, majd be- és kivezető
csővel ellátott lombikban hűtés közben sósavgázzal telítjük. A kivált sárgás kristályokat üvegszűrőn, sósav atmoszférában jól leszívatjuk.Tisztítása céljából sósavas telített oldatát kénsavas exszikkátorban víztele
nítjük mindaddig, míg a tetrahidrát jól fejlett kristályok alakjában ki nem válik.Levegőn gyorsan elfolyósodó, hidrolízis következtében sósavszagú, kristá
lyos anyag. A berillium bromidjának hasonló tetrahidrátja ismeretes, a jodidé nem.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 231/366
60 . Interhalogenidek és pszeudohalogenidek
60.1. Jód-monoklorid (IC1)
Képződik elemeiből szobahőmérsékleten gázállapotú klór felhasználásával, és
—35 C° alatti hőmérsékleten cseppfolyós klórban is. Az előbbi esetben megfelelőméretű frakcionáló lombikban elhelyezett, finoman elporított száraz elemi jódraugyancsak szárított klórgázt vezetünk. A reakció, folyamán a jód elfolyósodását
észleljük. A klórbevezetést akkor szüntetjük meg, amikor a sötét vörösbama IC1-folyadékban megjelennek a klórfölösleg hatására képződő ICl 3-kristályok. A nyerstermék desztilláció útján nem tisztítható meg eléggé, mert az atmoszférikus nyomá
son elért forráshőmérsékleten jelentékeny mértékben bomlik.Tisztább IC1 állítható elő a következő módon.1 Ismert súlyú edénybe száraz
jég—alkoholos-keverékben való hűtés közben klórt kondenzálunk. A becsléssel
megállapított térfogatból számított klórmennyiséggel ekvivalens jódnak kb. a felétpontosan lemérjük, és finoman elporítva a folyékony klórba szórjuk. 80 g jódhozkb. 30 ml klór szükséges. Mivel a képződő anyagok olvadáspontja magasabb,az elegy megszilárdul. Ezután megszüntetjük a hűtést, és lehetővé tesszük a klór-
felesleg elpárolgását szobahőmérsékleten. A klórgázt fülke kéményébe vezetjük.
Ennek végeztével megmérjük a lombikot, kiszámítjuk a klórfelesleget és a veleekvivalens jód mennyiségét. Ezt a hiányzó jódot most pótoljuk, és legalább egy
napot vétünk, amíg a ICI 3-I-I2 = 3IC1 reakció szobahőmérsékleten végbemegy.
A lombik száját ezalatt üvegdugóval lazán elzárva tartjuk. A képződött nyersterméket úgy lehet tovább tisztítani, hogy lassan lehűtjük,
mindaddig, amíg az anyagoknak kb. 80 %-a meg nem szilárdul, azután a folyékony
fázist leöntjük róla. Az eljárást többször megismételjük.
Vörösbama, mérgező és maró folyadék (op. a-ICl 13,9, /3-IC1 27,2 C°, fp.
101 C°, s 29. = 3,10).
60.2. Jód-triklorid (IC13)
Fölöslegben alkalmazott folyékony klórból és jódból állítható elő.2 A felhasz
nálandó jódmennyiséget már előre a klór kondenzálására alkalmazott hosszú nyakú,
csiszolatos gömblombikban helyezhetjük el. A reakció kondenzálás közben folyamatosán megy végbe, s a klórfelesleget a sárga színű, szabad Cl2-eseppek megjelenése jelzi. A gázbevezetést nemsokára megszüntetjük, de az elegyet a reakció
teljessé tétele céljából még néhány órán át a klór forráspontja alatti hőmérsékleten
Booth, H. I .: Inorganic Syntheses, Vol. I, New York— London 1939, 166. old.1 Birk, E.: Angew. Chem. 41, 751 (1928); Z. anorg. alig. Chem. 172, 399 (1928).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 232/366
234 I N T E R H A X O G E N I D E K É S P S ZE U D O H A L O G E N 1 D E K
tartjuk. Végül a hűtést megszüntetve a klórfelesleget szobahőmérsékleten elpárologtatjuk. Ha nagyobb mennyiséget állítanak elő, gazdaságossági szempontból atávozó klórt újra kondenzálják, és a következő gyártásfolyamatban felhasználják.
A kitermelés ily módon csaknem kvantitatív.Szúrós szagú, maró hatású, narancsszínű kristályos anyag. Szublimációs pontja
64 C°, 77 C°-on már teljesen ICl-|-Cl2-ra disszociál, s_40„=3,2.
60.3. Jód-monobromid (IBr)
Szintézise szobahőmérsékleten elemeinek egyszerű elegyítésével végrehajtható.Csiszolatos frakcionáló lombikba tegyünk ismert mennyiségű elporított jódot,
és csapos tölcsérből csepegtessünk rá az ekvivalensnél valamivel több brómot. A reakció hőfejlődés közben megy végbe, aminek látható jele a szilárd fázis elfo-lyósodása. A hőmérsékletet 30—50 C° között tartjuk. Ennél magasabb hőmérsékleten brómveszteség léphet fel. A reakció végeztével kihűtjük az elegyet, amelykemény, sötét masszává szilárdul. Ezután gázbevezető csövet teszünk a csapostölcsér helyére, és a szabad brómot C02-áramban 50 C°-on ledesztilláljuk. A visszamaradó igen kemény IBr-massza színe a jódéhoz hasonló. (Op. 40 C°, fp. 116 C,0s=4,42.)
60.4. 'Dicián (CN)2
Többféle módon állítható elő, legegyszerűbben a Cu(CN)2 diszproporcionáló-dásának felhasználásával. Igen mérgező anyag. Előállításánál fülke és gázálarchasználata szükséges.
200 g CuS04*5 H20-t 400 ml oxigénmentesített, kiforralt vízben oldunk, ésliteres gázfejlesztő lombikba töltjük. A feltét csapos tölcsérén át a réz-szulfáthoz
tömény KCN-oldatot csepegtetünk, miután előzőleg vízlégszivattyúval evakuáltuk a lombik légterét. A reakció egyenletes diciángázáramot szolgáltat. Mielőttaz oldatban a Cu2+ teljesen elfogyna, meleg vízfürdőbe állítjuk a lombikot, mirea gázfejlődés megélénkül. A (CN)2 szennyezésképpen HCN-ot, C02-ot és vízgőzttartalmaz. A cián-hidrogéntől AgN03-oldattal átitatott vattaszűrő segítségéveltisztítjuk meg, utána a nedvességtartalmát kötjük meg CaCLj-os szárítótoronyban. A C02-tól a dicián frakcionált kondenzációval különíthető el. Az előbbi módontisztított diciánt célszerűen vastag falú, szénsavhóval hűtött bombacsőben kondenzáljuk és tartjuk el. A gázbevezetőcső elég nagy keresztmetszetű legyen, hogya dicián bele ne fagyjon, és el ne tömje (op. —34,4 C°, fp. —20,7 C°).
A maradék CuCN-ot is felhasználhatjuk diciántermelésre a következő reakcióalapján:
CuCN + FeCl 3 = Cud + FeCl2 + 1/2 (CN)2.
A lombikban maradt CuCN-csapadékot dekantáljuk, és a gázfejlesztőfeltét csapostölcséréből kb. 30 %-os (s=l,26) FeCl3-oldatot adunk hozzá.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 233/366
B U B E Á 2 Í - H I D R 0 G É N S A V 235
60.5. Rubeán-hidrogénsav (H4C2N2S2)
Diciánból és kén-hidrogénből szintetizálható.3 Fontos mikrokémiai reagens. A diciánt Cu(CN)2-ból állítjuk elő ammóniás közegben, amelynek lúgos pH-ja
megakadályozza a savas természetű mérgező (CN)2-nak a légtérbe jutását. A műveletet biztonsági szempontból mégis fülkében végezzük.
Erlenmeyer-lombikbskn tömény vizes CuS04-oldatot készítünk, és addig öntünkhozzá cc. NH4OH-ot, amíg a csapadék éppen feloldódik. A sötétkék [Cu(NH3)4]2+-ttartalmazó oldathoz ezután annyi tömény KCN-oldatot adunk, amennyi az oldatotéppen elszínteleníti. A színváltozás a K 3[Cu(CN)4]-komplex és a (CN)2-képzŐdésmegtörténtét jelzi. Az oldatot most megszűrjük az esetleges szilárd szennyezésektől,
majd élénk ütemben H2S-t vezetünk bele. A kén-hidrogéntől az oldat először megsárgul, azután narancssárga kristályok alakjában kiválik belőle a rubeán-hidrogén-sav. A kén-hidrogén oldódását és a csapadékki válást jeges hűtéssel segítjük elő. A képződött terméket gyorsan nuccsra gyűjtjük, hideg vízzel mossuk, és alkoholbólvagy jégecetből átkristályosítjuk.
* Formanek, Q.: Bér. dtsch. chem. Ges. 22, 2655 (1899).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 234/366
61. Sav-halogenidek
Ha az oxisavak jellemző hidroxidgyökét klórral helyettesítjük, a sav-kloridok jellegzetes vegyületcsoportját kapjuk. Jellemzőjük a kovalens kötéstípus, az illé-konyság és a hidrolízisre való hajlam. Előállításuk metodikáját is túlnyomóan ezeka tulajdonságok szabják meg. Általában szükséges a vízmentes közeg és a légnedvesség távoltartása. Gyakran alkalmazható az elemi szintézis, illetve a megfelelőmolekulák egyesítése (pl. CO+.CLj = CO02! S02+C12 = S02C12, stb.) többnyirekatalizátor jelenlétében. Némely nemvizes oldószer ún. szolvolízises reakciója issav-halogenidek előállítására ad lehetőséget. Erre vonatkozó példák:
502 -f PC15 = POCI 3 + SOCl2,
50 3 + CC14 = S02Cl2 + COCl2.
Sav-halogenidek részleges hidrolízisével vagy az oxisav hidroxidjainak csupán parciális klórszubsztitúciója útján olyan vegyülettípushoz jutunk, amely az oxisavakra
jellemző hidroxidcsoporttal és a sav-halogenidekre jellemző módon kötött halogénatommal is rendelkezik. Ezeket megfelelő vízelvonószerrel (pl. P20 5-dal) kezelve,pirosa v-halogenideket kapunk:
2 C1S020H = C12S20 5 + H20.
61.1. Tionil-klorid és foszfor-oxid-klorid (S0C12 és P0C13)
Egyszerre képződnek a PC15 ésSOareakciójában. Visszafolyós hűtővel és gázbevezetőcsővel ellátott gömblombikot kb. 1 /3-ig PCl5-dal töltünk meg. Légnedvességtől az egész rendszert gondosan óvni kell. A visszafolyós hűtőt a fülkekéménnyelösszekötő vezetékbe ezért P2Os-os szárítótornyot kell iktatni, és valamennyi alkatrészt összeszerelés előtt jól ki kell szárítani. Ha nincs csiszolatos készülékünk, paraffinnal átitatott parafa dugót használjunk, ennek felületéről azonban még melegen
jól törüljük le a felesleges paraffint. A lombik két furatának egyikébe kerül a hűtő,
a másikba a gázbevezetőcső. A reakcióban felhasználandó S02-ot is alaposan meg kell szárítani. Akár
NaHS03-ból fejlesztjük, akár acélpalackból vesszük a kén-dioxidot, célszerűen kétkénsavas mosópalackon, utána a folyadékcseppektől való megtisztítás céljából vattadugón vezetjük át, mielőtt a lombiknak csaknem a fenekéig érő csövön át a PC15--hoz vezetnénk. A reakció haladására a PC15 elfolyósodásából lehet következtetni,annak teljes végbemenetelét enyhe melegítéssel segítjük elő.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 235/366
T I O N I L - B R O M I D 237
A termék a két sav-klorid elegye, mely egymástól csak többszörös desztillálás-sal választható el. A tionil-klorid fp.-ja 78,8 C°, a foszfor-oxid-kloridé 105,3 C°.
Az elválasztást kb. 40 cm magas, Raschig-gyűrűvel töltött frakcionáló feltét segítségével végezzük. Legjobb erre a célra is összeforrasztott vagy csiszolatos készüléket használni. A légnedvesség teljes távoltartásával végrehajtott desztillálás ter
mékét négy részletben fogjuk fel: elsőként a 82 C° alatti frakciót, majd a 82 —92 C°
közöttit, azután a 92 —102 C° közötti és végül a 102 —112 C° közötti részletet. A második és harmadik frakciót újra desztilláljuk, s az előbbihez hasonlóan ugyanazt a
négy frakcióját különítjük el és egyesítjük az előző desztillálás megfelelő frakcióival.
Ily módon a két középső frakció mennyisége egyre csökken, a szélsőké pedig nő. Amikor eléggé lecsökkent a 82 —102 (P közötti forráspontú elegyek mennyisége,
a főfrakciókat tiszta és száraz feltéttel újra ledesztilláljuk. A tionil-kloridot először óvatosan, visszafolyatásra állított hűtő használata
mellett kb. 1/2 órán át főzzük, hogy az el nem reagált S02-ot kiforraljuk belőle.Utána kezdjük meg a frakcionálást, és a 78 —79 C° körüli, állandó forráspontú főfrakciót elkülönítve fogjuk fel.
A POClg-ot ismételten tisztított edényzetből, vízmentes körülmények között
desztilláljuk. A fő frakció 105 —106 C° között szedhető. Az ismételt frakcionálás ellenére a két sav-klorid kölcsönösen szennyezi egy
mást. A tionil-klorid szennyezettségének mértékét a hidrolíziskor képződő foszfát
molibdátos reakciója alapján ítélhetjük meg. A POCl 3 kéntartalmát szulfát alakban
mutatjuk ki a hidrolizált és oxidált mintában.Mindkét anyag színtelen, nagy fénytörésű, szúrós szagú, hidrolizáló folyadék.
SOCl2: op. -1 0 5 C 0, fp. 78,8 C°, s = 1,66;
P0C13: op. 2 C°, fp. 105,3 C°, s = 1,68.
61.2. Tionil-bromid (S0Br2)
Foszfor-pentabromidnak folyékony kén-dioxidban végrehajtott szolvolízisé-vel állítjuk elő. A reakció egyenlete:
PBrs + S02 = POBr 3 4- SOBr2.
A szobahőmérsékleten végrehajtott folyamat igen lassú, még 30 — 40 C°-Qn is eltart2 —3 napig. Ezen a hőmérsékleten pedig a kén-dioxid egyensúlyi nyomása 5 —8 atm.
Kisebb mennyiséget teljes biztonsággal beforraszthatunk a szokásos méretű (2 cm
0 -jű) bombacsőbe, mert ezek 2 mm falvastagság esetén 100 atm nyomást is elvisel
nek. A megengedhető nyomás azonban a cső átmérőjének növekedésével rohamosan
csökken. Ha a reakciót nagyobb anyagmennyiséggel akarjuk végrehajtani, a csőnekinkább a hosszát növeljük, a keresztmetszetét csak a legszükségesebb mértékben.
A falvastagság 2 —3 mm-en túli növelése nem jelent arányos szilárdságnövekedést. A fenti szempontok alapján méretezett ún. Volhard-csőbe akár 50 g foszfor-penta-
bromidot is bemérhetünk. Erre —20 C°-os hőmérsékletre hűtött állapotban 100 ml
cseppfolyósított kén-dioxidot öntünk. Mivel a S02 —10 C°-on forr, a cső zárt végétleforrasztás közben is hűtőkeverékben tartjuk, és a felhevített csőrészt leforrasztás
után lassan hűtjük le szobahőmérsékletre.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 236/366
238 S A V - H A L O G E N I D E K
A csövet ezután kivesszük a hűtőberendezésből. A folyamat megindulását csakhosszabb idő elteltével észlelhetjük a PBrfi-kristályok mennyiségének csökkenésén
és az oldat vörösbarna színeződésén. A hőmérsékletet legfeljebb 40 C°-ra emeljük. A folyamat végét felismerhetjük a szilárd PBrs elfogyásáról. A cső felnyitásakor acső alja ugyancsak hűtőkeverékbe ágyazandó. A kapilláris végét erős hegyes lángban megolvasztjuk, hogy a belső nyomás a meglágyult üveget kifújhassa. Ezután azoldószert szobahőmérsékleten elpárologtatjuk, és a tionil-bromid a bombacsőbenvisszamarad. Mivel forráspontján már bomlik, vákuumban desztillálva tisztítjuk.
Bombacső nélkül is kényelmesen előállítható a tionil-bromid a foszfor vegyeshalogenidjeinek (PCl 3Br2) kén-dioxidos cserebomlása útján:
PCl 3Br2 + S02 = SOBr2 + POCl3.
A reakciót gázbevezetőcsővel (illetve csepegtető tölcsérrel) és visszafolyatásra állított hűtővel ellátott CZatsen-lombikbaii végezhetjük (lásd 56.1.17.1. ábrát). A megfelelő szögben hajlított végű hűtőt függőleges helyzetből elforgatva, desztilláláskorkondenzáló hűtőként használhatjuk.
A gondosan kiszárított készüléket összeállítása után a csapos tölcséren át először száraz (és oxigénmentes) N2-nel töltjük meg, azután a lombikjába 138 g PCl3-otteszünk. így készítjük —5 C°-on végzett brómaddícióval a SOBr2-elŐállításárahasználatos vegyes halogenidet. A mágneses keverő behelyezése után a lombikotsó —jég-keverékben lehűtjük, és a csapos tölcséren át a PCl3-ba állandó keverés közben 160 g (kb. 50 ml) brómot csepegtetünk. Ennek végeztével a PCl 3Br2-ba ekvimo-láris mennyiségű kén-dioxidot vezetünk. A folyamat befejeződését a hőfejlődés megszűnéséről állapíthatjuk meg. Az elegy ben oldódott S 02-felesleget ezután szobahőmérsékleten hagyjuk elpárologni, illetve a berendezést vákuumdesztillációra átállítva, enyhe szívatással eltávolítjuk.
A nyerstermék vörösbarna színű, foszfor-oxid-kloriddal, brómmal és S2Br2-dalszennyezett. Tisztítása vákuumdesztillációval történik. A berendezés alkatrészeita szedő után következő szilárd KOH-os toronyig csiszolattal illesztjük, mivel aSOBr2 és a Br2 a PVC-t is megtámadja. A KOH-os torony nemcsak a gumivezeté
ket és a manométert védi a korróziótól, hanem a terméket is a vízlégszivattyú felőldiffundáló vízgőztől. 20 Hgmm nyomáson végezve a desztillációt, a 46 —48 C° alattihőmérsékleten felfogott sötét színű előpárlat után külön szedőbe gyűjtjük a narancs-vörös színű SOBr2-ot, amely még mindig jelentős mennyiségű foszfor-trikloriddallehet szennyezve. A teljes elválasztás csak nagy tányérszámú vákuumkolonnábanvégezhető el. ASOBr2 raktározás közben is lassan bomlik, (s = 2,6.)
61.3. Foszfor-tioklorid (PSC13)
Nehézfém-kloridok (SbCl3, PbCl2) és foszfor-péntaszulfid keverékének összeolvasztásakor képződik a tetratio-ortofoszforsavas só mellett a következő reakcióban:
SbCJ 3 + P2S5 = SbPS4+ PSC13.
Szintetizálható PCl3-ból és elemi kénből is AlCl3-kátalizátor alkalmazásával.1 A folyamatot légnedvességtől elzárt térben kell végrehajtani. A berendezést gömb-
1 Knotz, F .: öeterr. Chemiker Z. 50, 128 (1949).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 237/366
K L O R O - K É N S A V 239
lombikból CaCl2-°s vagy P20 5-os csővel elzárt végű, visszafolyatásra állított hűtőbőlés adagoló feltétből állíthatjuk össze. Alkalmas feltét látható a 68.1.1.3. ábrán.
A lombikba 24 g kénport és 100 g PCl 3-ot, az adagolóba pedig 5 g vízmentes AlClj-ot teszünk. Ennek hatására a kén elég heves reakcióban feloldódik. A reakcióannyira heves lehet, hogy néha a lombikot kissé*hűteni is kell. Kb. 10 perc alatt végbemegy az átalakulás, az oldat narancssárga színű lesz.
Ezután lehűtjük a folyadékot, és nagy választótölcsérben, sok vízzel összerázzuk. Ennek célja az A1C1 3 és egyéb oldható szennyezések eltávolítása. Az elegy el-színtelenedik, és a PSC1 3 nehezebb fázisként a tölcsér alján gyűlik össze. Elválasz
tása után CaCl2-dal szárítjuk, majd ledesztilláljuk (op. —35 C°, fp. 125 C°). A PSC1 3 színtelen folyadék, (s = 1,67.)
61.4. Kloro-kénsav (C1S03H)
Előállítását a következő reakció alapján végezzük:
H2S04+ PC15 = POCl 3 + HC1 + C1S0 3H.
Literes gömblombikba 200 g 98 %-os (s15o = 1,84) kénsavat öntünk, és apránként150 g PCl5-ot adunk hozzá. Közben melegítés hatására sósav szabadul fel, amelyetaz elszívónyílásba vezetünk. A PCl5-adagolás befejeztével a maradék sósavat kifor-raljuk, azután a gömblombikot desztillálófeltéttel látjuk el, és a 165 C°-ig átdesztilláló frakciót vízhűtés nélkül kondenzáljuk. Hűtőként egyszerű, vízköpeny nélküli,40 —50cm hosszú és kb. 1 cm 0 - jű tűzálló üvegcsövet használunk. A magas forráspontú anyag léghűtéssel is tökéletesen kondenzálható. Áramló vizet nem is szabad hűtésre alkalmazni, mert a legtöbb hűtő üveganyaga nem viseli el törés nélkül a külsőés belső felület között fellépő, kb. 150 C° hőmérséklet-különbséget. A nyers kloro-
-kénsavat csiszolatos készülékből újra desztilláljuk, és a 152 C°-on átmenő frakciótfogjuk fel. A kloro-kénsav előállításának másik módja a kén-trioxid és sósavgáz egyesíté
se. Nagyobb mosópalackba enyhe melegítéssel megolvasztott, 80 % szabad S0 3-tar-talmú óleumot (kb. 200 g) öntünk. Ezen jól megszárított sósavgázt vezetünk át,amikor is már szobahőmérsékleten megindul az
S0 3 + HC1 = H0S02C1
reakció. A folyamat exoterm, ezért a mosópalackot hűteni kell. Ha a sósavgáz már
nem reagál a füstölgő kénsavval, maradékát kiforraljuk az elegyből, a kénsavból pedig az előző leírás szerint csiszolatos készülékben ledesztilláljuk a 152 C° forráspontú kloro-kénsavat (op. —80 C°, fp. 152 C°).
A kloro-kénsav színtelen, szúrós szagú folyadék. Vízelvonószer, pl. S03, P205
jelenlétében piroszulfuril-klorid képződik belőle. Vízzel erős hőfejlődés közben kénsavra és sósavra hidrolizál (s = 1,79).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 238/366
240 S A V - H A L O G E N I D E K
61.5. Piroszulfuril-klorid (S20 5C12)
Ha a kloro-kénsavat foszfor-pentoxidról desztilláljuk le, piroszulfuril-kloridképződik.^ Magas forráspontja miatt vízhűtés nélküli csőben kondenzálható. Az
esetleg el nem reagált kloro-kénsavtól újabb P20 5-mennyiségről való desztillálássaltisztítjuk meg (op. —37 C°, fp. 152—153 C°). Hosszabb ideig leforrasztott ampullában tartjuk el. (s20<>= 1,84.)
61.6. Szelenil-klorid (SeOCl2)
SeOC^ képződik az alábbi reakcióban:
SeCl4.+ Se02 = 2SeOCl2.
Mindkét kiindulási komponens szilárd anyag. A reakciót a szelén-tetraklorid
CCl4-os szuszpenziójában hajtjuk végre. A terméket a 0C14oldja, s attól desztillá-
cióval különíthető el.
A kiindulási anyagként felhasznált SeCl4-szuszpenziót a reakcióedényben állít juk elő úgy, hogy a szén-tetrakloridos közegben vörösszelént szuszpendálunk, és
száraz klórgázt vezetünk bele. A berendezést (61.6.1. ábra) csiszolatos frakcionáló-lombikhoz csatlakozó IAebig-hűtöhől állítjuk össze. A lombik rövid szára felfelé irá
nyuljon, és a hozzá kapcsolt hűtő csöve még a köpeny előtt olyan szögben legyen
2 Prandtl, W., Borinski, P.: Z. anorg. alig. Chem. 62, 24 (1909); Sanger, Gh. R., Riegel, E. R.: Z. anorg. alig. Chem. 76, 79 (1912).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 239/366
N I T R O Z I I / - K L O B I D 241
meghajlítva, hogy egyrészt függőleges helyzetben visszafolyós hűtőként, másrésztlefordítva mint leszálló hűtő működjék. A klórt a lombik nyakán át a fenekéig érőgázbevezetőcsövön át juttatjuk a szuszpenzióba. Az első lépésben SegCLj képződik,
amely jó oldószere p-z elemi Se-nek, ezért a reakció rövidesen meggyorsul. A követ
kező fázisban a klórfelesleg hatására SeCl4képződik, amit fehér csapadék megjelenéséről ismerhetünk fel, mivel a közeg nem oldja. Most hozzáadjuk a számítottmennyiségű Se02-ot. Célszerű a szuszpenziót mágneses keverő segítségével keverni.
A reakció végét a szilárd fázisok eltűnése jelzi.
Az oldott SeOC^ a CC14 ledesztillálása után a lombikban marad vissza. Halványsárga, maró folyadék. Higroszkópos, a levegőn füstölög. Az anyag forráspontjafeletti hőmérsékleten bömlékony, tehát ha tiszta SeOCl2-ra van szükségünk, vá--
kuumban desztilláljuk (op. +10,8 C°, fp. 176,4 C°, s22„ = 2,42).
61.7. Nitrozil-klorid (N0C1)
NO
40—50 C° hőmérsékletű aktívszén-katalizátor felületén, száraz NO-ból és Cl2-ból képződik. A készülék (61.7.1.-ábra) a szárított gázkomponensek keverésére szol
gáló tér után alkalmazott függőleges helyzetű golyós hűtő, amelyet előzőleg vízgőzzel aktivált és 150 —200 C°-on vákuumban
szárított és kihűtött, borsónyi szemcsézetű,
pormentes, üveggyapotra ágyazott aktívszénnel töltöttünk meg.
A felhasználásra kerülő NO többfélemódon állítható elő. Igen egyszerű a kb.
30 %-os salétromsavból és vörösréz hulladék
ból történő gázfejlesztés, amelyet JST/pp-készü-lékben végezhetünk. Az így fejlesztett NO
azonban igen szennyezett, kb. 15 % N20-otés mintegy 10 % N2-t tartalmaz.
Kisebb mennyiségű, tisztább NO-ot állíthatunk elő salétromsav-tartalmú kénsav ésfémhigany reakciója alapján:
2 HN0 3 + 3 H2S04+ 6 Hg =
= 2 NO + 3 Hg 2S04 -|- 4 H20.
Csapos tölcsérrel és gázbevezetőcsővel ellátott lapos fenekű gázfejlesztő lombikot néhány milliliter híján teljesen megtöltünk kb.
2 súly % NaNOg-ot tartalmazó cc kénsavval. A csapos tölcséren át annyi higanyt enge
dünk a kénsavba, hogy a lombik alját éppen
ellepje. Egyenletes, több órán át tartó gázáramot kapunk. Magasabb oxidációfokú pitrogén-oxidoktól a gázt 2 n NaOH-dal
mosva tisztítjuk meg. A lúg nem tartalmazhat szerves szennyezéseket, mert akkora NO dinitrogén-oxiddá redukálódik. Szárítását kénsavas mosóval vagy foszfor--pentoxidos toronnyal végezzük.
16 Alt. és szervetlen kémiai prakt. —4284/II.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 240/366
242 S A V - H A I ^ O G E I Í I D E K
NOCl-ot állíthatunk elő nitrogén-dioxidból és alkálifém- vagy alkáliföldfém--kloridból is:
Cl" + N204 = N03- + NOC1.
Kb. 60 cm hosszú, 2 cm 0 -jű, függőlegesen elhelyezett üvegcsövet megtöltünk2,5 % nedvességtartalmú KCl-dal. Ha ebbe a csőbe nitrogén-dioxidot (vagy dimer-
jét, N204-ot) vezetünk be, a kristályok felületén végbemegy az egyenletben leírtreakció. A reakciózóna határozottan felismerhető. Éles határvonal válaszja el egymástól a fehér kristályok alkotta háttérben a barna N 02-dal töltött teret a sárgászöld NOCl-gőztértől. Ha a nitrogén-dioxid áramlását megfelelő módon szabályozzuk, a barna fázishatár nem halad túl a KCl-oszlopon, és N02-tól mentes NOCl-otkészíthetünk.
A NOC1 előállításához szükséges N 02-ot 35 —40 %-os NaNOa-oldatból 20 %-oskénsavval állítjuk elő úgy, hogy a képződött N20 3-ot oxigénnel N20 4-dá oxidáljuk. A gázfejlesztő lombikból távozó gázt a folyadékcseppektől üveggyapottal töltöttU-csövön átvezetve tisztítjuk meg, majd só —jég-keverékkel hűtött szedőben kondenzáljuk. A kapott zöldes színű folyadék különböző nitrogén-oxidok elegye. Mérsékelt jeges hűtés közben ezen a folyadékon tiszta és száraz oxigént buborékolta-tunk át. Az oxidáció végét a folyadék megbarnulásáról ismerhetjük fel. EgyenletesN 02-áramot a 21,2 C° forráspontú (op. —11 C°) folyadék lassú elpárologtatásávalkaphatunk.
A NOC1 előállítására további lehetőséget nyújt a2 HC1 + N20 3 = 2 NOC1 + H jjO
reakció. Kondenzált nitrogén-trioxidba vízelvonószer jelenlétében száraz HCl-otvezetünk.
A N20 3 előállításának egyik egyszerű módját a 24.1.5.-ben ismertettük. A zöldszínű kondenzátumot kb. —40 C°-ra hűtött edénybe helyezzük, és óvatosan P2Os-otteszünk bele a további reakció során képződő víz megkötésére. Ezután száraz sósavgázt vezetünk be. A reakció —20 C°-on megy végbe jó kitermeléssel. A kép
ződő NOCl-ot só —jég-keverékkel hűtött szedőbe vezetjük. Ampullákba forrasztvatartható el. Az ampullák leforrasztásakor vigyázzunk arra, hogy a forrasztólángégéstermékei, főleg vízgőz ne jusson az anyaghoz. Sárgásvörös folyadék (op. 1,5 C°,fp. 6,5 C°). Vízzel salétromsavvá és sósavvá hidrolizál.
61.8. Kromil-klorid (Cr02Cl2)
Krómsav és sósav elegyéből képződik vízelvonószerek (pl cc. kénsav) hatására. A kénsav vízelvonó hatásán kívül egyúttal sójából fel is szabadítja a két kiindulási,
anyagot. A reakció bruttó egyenlete:K 2Cr20 7 + 4 NaCl 4- 3 H ^ = 2 Cr02Cl2 + K £ 0 4+ 2 Na2S04+ 3 HaO.
A felhasználható anyagok árát is figyelembe véve a legolcsóbb a K 2Cr20 7ésNaCl keveréke. 300 g K 2Gr20 7és 240 g NaGl jól homogenizált elegyét lehetőleg alacsony hőmérsékleten összeolvasztjuk, és az olvadékot vasbádogra öntve kihűlnihagyjuk. Durva darabokra törve csiszolatos desztilláló berendezés adólombikjábarakjuk, és a lombik nyakába helyezett csepegtetőtölcsérből apránként óvatosan

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 241/366
F É M - O X I D - I L L O W D O K 243
300 g kénsavat adagolunk hozzá. A reakció különösen eleinte igen heves, de ha mármérsékló'dött, a 118 C° forráspontú barna színű kromil-kloridot az elegyből ledesz
tilláljuk. A nyerstermék tisztítása ismételt desztillációval történik (op. —96,5 C°, fp.118 C°). ACr02Cl2 tiszta állapotban rézvörös színű, hidrolizáló folyadék (s = 1,91).Leforrasztva, fénymentes helyen tartható el.
61.9. Vanádium-oxid-triklorid (V0C13)
V 20 5-ból SOCl2-dal állítható elő 100 C°-on. Visszafolyatásra állított hűtővelellátott csiszolatos gömblombikba 25 g V 20 5-ot és 30 ml SOCl2-ot mérünk be, majdvízfürdőn 6—8 óra hosszat forraljuk. A légköri nedvességet P2Os-os szárítófeltéttelgondosan távoltartjuk. Használatbavétel előtt természetesen az anyagokat és azeszközöket is jól kiszárítjuk. A reakció végeztével a visszafolyatásra állított hűtőtleszálló hűtővel cseréljük fel, és az anyagot ledesztilláljuk (op. —77 C°, fp. 127 C°)Ha nem alkalmaztunk SOC^-felesleget, tiszta VOCl3-ot foghatunk fel a szedőben. Világossárga, hidrolizáló folyadék (s = 1,83).
61.10. Nióbium-oxid-triklorid (NbOCl3)
A NbOCl3-ot bombacsőben 200 C°-on állítjuk elő 3 a következő egyenlet alap ján:
Nb20 5 -f 3 SOCl2 = NbOCl 3 + 3 S02 .
Kb. 1 g-nyi Nb20 6-hoz pontosan bemért, a fenti egyenletnek megfelelő sztöohio-metriai mennyiségű SOCl2-ot adagolunk a kb. 2 cm átmérőjű bombacsőbe. Ennekbeforrasztását úgy végezzük, hogy lehetőleg hosszú, vastag falú kapillárissal zár
juk, mely a felnyitás után lehetővé teszi az újból való beforrasztást. Erre azért van
szükség, hogy az acél védőcső alkalmazásával alumínium blokkban, csőkemencében vagy olajfürdőben 3 órán át 200 C°-on tartott, bombacsőben képződött S02-otszobahőmérsékleten elpárologtatva, a reakció egyensúlyát a képződés irányába tol
juk el. A bombacsövet természetesen megfelelő biztonsági intézkedések megtételeután nyitjuk ki (védőszemüveg, a gáznemű termékek kondenzálása szénsavas — alkoholos hűtőkeverékkel stb.). Újból való leforrasztás után további melegítéssel tel
jessé tesszük a reakciót. A NbOCl 3 selyemfényű, fehér, kristályos anyag, 400 C°-onszublimálható.
3 Hecht, H., Jander, G., Schlapmann, H.: Z. anorg. alig. Chem. 254, 260 (1947).
16

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 242/366
62 . Ox idő F
62.1. Diíluor-monoxid (F20 )
Elemi fluor és híg NaOH reakciójával képződik szobahőmérsékleten i1
2 Fa + 2 NaOH = FaO + 2 NaF + BLjO .
A difluor-monoxid igen illékony, a cseppfolyós levegő hőmérsékletén sárgásbarna
folyadék. Hőmérsékleti, fény- és elektromos hatásokkal szemben aránylag stabilis,
a ClaO-nál jóval kevésbé reakcióképes anyag. A száraz üveg- vagy kvarcfelületet
nem támadja meg, vízben csak kismértékben oldódik. Mérgező hatású anyag, ezért
minden vele való műveletet jól húzó fülkében kell végezni.Képződési reakciójával párhuzamosan NaOH-ban a bomlása is végbemegy,
ezért a komponensek egymásrahatásának időtartamát, koncentrációját, stb. a ki
termelés optimumának elérése^ p . céljából pontosan megszabott
CS értéken kell tartani.
A különböző módszerek
kel előállítható fluorgázt 2 mmbelső 0 -jű platina csövönkeresztül, 2 1 /óra sebességgel,2 %-os NaOH-oldaton bubo-rékoltatjuk át. A 62.1.1. ábrán
látható túlfolyós edényben aNaOH-oldatot is folyamatosan cseréljük kb. 1 1 /óra sebességgel. A platina cső 2 cm-re
merüljön a folyadékszint alá. A képződő F20-ot vizes mosópaláckban fluormen
tesítjük, azután folyékony oxigénnel hűtött edényben kondenzáljuk. A vizes mo
són is túljutó szennyezése főleg a bomlástermékként keletkező oxigén. Ezt a cseppfolyós FaO jól oldja. Forráspontjuk különbsége lehetővé teszi a két komponenselválasztását vákuumdesztillációval. Az oxigén forráspontján tartott elegyből az
oxigén 20 Hgmm nyomáson előpárlatként távozik, a magasabb forráspontú F20-ot
a második, cseppfolyós oxigénnel hűtött szedőben aránylag tisztán (kb.89 %) foghatjuk fel (op. —223,8 C°, fp. —144,8 C°). Hogy a levegőbe ne kerüljön mérgezőF20, a szivattyú elé KI-os oldatot tartalmazó elnyelőedényt kapcsolunk.
2 % .
N q O h \ •M
l y
í 1
í l n i! ■?
h 2 o '-113 -183
fülkébe
62.1.1. ábra. F.O előállítása
1 Lebeau, P., Damiem, A.: C. B . Hebd. Séancea Acad. Sci. 188, 1263 (1928); Puff, O., Menzel, W.: Z. anorg. alig. Chem. 190, 267 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 243/366
K L Ó R - D I O X I D 245
62.2. Klór-dioxid (Ci02)
Kálium-klorátból cc. H2S04 hatására képződik. Veszélyes, könnyen robbanóanyag, ezért fontos, hogy egyszerre csak kis mennyiséget (2 — 3 g-ot) készítsünkés tároljunk belőle. Fényérzékeny, bomlik.
20 g KC103-ot 60 g oxidálható szennyezéstől mentes, mosott és kiizzított kvarc-homokkal keverünk össze. A homogenizáláshoz nem szabad dörzscsészét használni,mert dörzsölésre a KC10 3 robban. Biztonságosan úgy végezhető el a művelet, hogypapírlapon többször ide-oda öntögetjük az elegyet, amíg elég egyenletesen elkeveredik. Ezt a keveréket olyan csiszolatos gázfejlesztőlombikba töltjük, melynek csepegtető tölcsérén — a szerves kenő
anyag kiküszöbölése érdekében — nem forgócsap van, hanem a62.2.1. ábra szerinti kézzel működtethető zárócsiszolat.2 A kénsavatis, meg a klorátkeveréket is jeges vízzel lehűtjük, majd a lombikhűtését állandóan fenntartva, lassan belecsepegtetjük a kénsavat. A képződő C102-ot kevés klór szennyezi. A gázokat Pa0 5-os toronyban szárítjuk, majd szénsavhóval vagy folyékony levegővel hűtöttcsapdában kifagyasztjuk. A gázfejlesztés és kondenzálás egész folyamatát csökkentett nyomáson, kb. 20 Hgmm-en végezzük. Haerre a célra vízlégszivattyút használunk, akkor közéje és a kondenzálóedény közé is tegyünk szárítótornyot vagy fagyasztócsap
dát. A klór-dioxid sárga színű gáz vagy vörösbarna színű folyadék
(op. —59 C°, fp. 9,9 C°). Ha a legalább —80 C°-ra hűtött anyag gőzterét jól evakuáljuk ( — 80 C°-on tenziója gyakorlatilag nulla), a klór-dioxidampullába forrasztható a robbanás veszélye nélkül.
Az előbb ismertetett eljárásnál a csökkentett nyomás és az éghető anyagoktávoltartása (forgócsapok kiküszöbölése) ellenére fennáll a robbanás bizonyos valószínűsége. Hatásosabban csökkenthető a robbanásveszély, ha a d 0 2 koncentrációját indifferens gázzal hígítva csökkentjük.3
Az alábbi reakcióban a C02 egyidejű fejlődésén kívül a KC10 3 teljes kihasználása is biztosítva van:
62.2.1. ábr a.
CIO, előállítása
2 KC10 3 + (COOH)2 .2 HaO + 2 H2S04 =
= 2 d 0 2 + 2 C02 + 4 H20 + 2 KHS04 .
Kisebb csiszolatos gázfejlesztő lombikban 20 g KC10 3 és 16 g (COOH)a. 2 H^Okeverékére 70 ml víz és 10 ml kénsav jégbehűtött oldatát öntjük. Vízfürdővel enyhén melegítve, egyenletes gázfejlődés jelzi a meginduló reakciót. A klór-dioxid —— szénsav-keveréket, az előbbihez hasonló módon, foszfor-pentoxidos toronyban
szárítjuk, és szénsavhóval hűtött edényben kifagyasztjuk.Tisztítása vákuumdesztillációval történik. A jól lehűtött elegyből elszublimáljuk a szénsavat, majd a klór-dioxidot is vákuumban tiszta szénsavhóval hűtöttszedőbe desztilláljuk. Ha a tisztasági követelmény nagy, csak a középső frakcióthasználjuk fel.
* Bodenstei n , M ., H ar t eck , P .t Pádéi t , E .: Z. anorg. alig. Chem. 147, 233 (1925). 9 B ray , W . : Z. physik. Chem. 54, 569 (1906);
Schumácher , H . / . , Sti eger , O.: Z . physik. Chem. (B) 7, 364 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 244/366
246 O X I D O K
Ha a Idór-dioxid vizes oldatára van szükségünk, az előbbi módon fejlesztettgázelegyet só—jég-keverékkel hűtött, vizet tartalmazó elnyelőedénybe vezetjük.
Egy térfogat víz 4 C°-on 20 térfogat C102-ot old. Oldat készítésére nagyobb gáz-fejlesztőt és nagyobb mennyiségű kiindulási anyagot is használhatunk (kb. 120 gKC10a-ot és arányosan többet az egyéb komponensekből, másfél literes lombikban).
62.3. Jód-pentoxid (I2Os)
Laboratóriumi előállítása jódsavból történik vízelvonással, 250 C° alatti hőmérsékleten. A vízelvonás két fokozatban megy végbe.4Először H I0 3 *I20 5 képző
dik 70 — 200 C° közötti hőmérsékleten: 3 H K )3 = HI0 3-I20 5+ H20.Gyors hőközlés esetén a jódsav 110 C°-on megolvad, és így kristályvizénekegy részét elveszti. A vízvesztés második részfolyamata csak 200 C° feletti hőmérsékleten indul meg:
2 H I 0 3-I20 5 = 2 I20 5 + H20 .
A vízelvonást száraz és igen tiszta — redukáló szennyezésektől mentes — levegőáramban végezzük. A jódsavat porcelán csónakban kiterítve elég tágas üvegcsőben helyezzük el, melynek hőmérsékletét jól szabályozható elektromos csőkemencével először hosszabb ideig 100 C°-on tartjuk. A vízvesztés első fázisa ugyanis
jóval lassúbb folyamat, mint a második. Bizonyos mennyiségű I20 5 hozzáelegyí-tése a H I03-hoz igen kedvezően befolyásolja az átalakulás sebességét. A teljes vízvesztés azután 240 C°-on 1 óra alatt végbemegy.
A felhasznált levegőáram szárítását és tisztítását igen gondosan kell végezni. A kénsavas mosás egyes szerzők szerint elkerülendő.5 A portalanítás, redukálóanyagok eltávolítása, stb. inkább lúgos KMn04-tal, a szárítás pedig szilárd KOH-dalv$gy P2Os-dal történik. A kiindulásra felhasznált jódsavat célszerű a jód HC103-asoxidációja útján és nem a salétromsavas, illetve hidrogén-peroxidos módszerrelelőállítani. A tapasztalat szerint az így kapott jód-pentoxid kevéssé lesz rózsaszín
árnyalatú. A IaOs fehér, kristályos anyag, mely a redukció következtében esetleg képző
dött, elemi jódtól halvány rózsaszínű lesz. Higroszkópos, hő hatására bomlik. AI2-ra és 0 2-re való bomlás 275 C°-on indul meg és 350 C°-on válik teljessé (s20<>== 4,8).
62.4. Jód(HI)-oxid-jodát (I20 4)
Előállítása úgy történik, hogy jódsavat tömény kénsavval elegyítve exszikká-torban állni hagyunk.6 A reakció a következő két részfolyamatból tevődik össze:
2 HIOs + H2S04 = ( I0 )2S04•HaO + Oa + HaO,
(I0)2S04•HaO + 2 H I0 3 = 2 120 4+ H2S04 + HaO.
4 Lamb, A . B ., Bra'tf; W. G., Geldard, W. J .: J. Am. Chem. Soc. 42, 1644 (1920). * Móle8, E., Perez-Vitoria, A .: Z. physik. Chem. A 156a, 583 (1931).
8 Baki, B. K ., Pertington, J. R .: J. Chem. Soc. (London) 1935, 1258;
Kappeler, H .: Bér. dtsch, chem. Ges. 44, 3496 (1911).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 245/366
A K É N O X ID J A I 247
A 0,5—10 g H I03-at néhány ml cc. H2S04-val egy félmikro-méretű kémcsőben(52.1.1. ábra) elkeverjük, és néhány napra cc. H2S04-aB exszikkátorba helyezzük.
A folyamat akkor fejeződött be, ha az elegy kis részletét vízzel összerázva nemtapasztalunk lényeges jódkiválást. Ha igen, még néhány napot várunk, és megismételjük a próbát. Ha jódkiválás már nem következik be, úgy szívógömb alkalmazásával szulfátmentesre mossuk. Az anyag nem higroszkópos és vízben nehezenoldódik. Vizes mosás után absz. alkohollal, majd éterrel mossuk, végül CaCl2-osexszikkátorban szárítjuk. 130 C°-on I2-dá és I2Og-dá bomlik. (s2o<» = 4,2.)
62.5. Kén-trioxid (S03)
Nagyiparilag a kén-dioxid katalitikus oxidációja útján állítják elő. Laboratóriumi célra sokkal egyszerűbb módszer a füstölgő kénsav (óleum) oldott kén-trioxid--tartalmának kiforralása és Összegyűjtése. A kén-trioxid igen higroszkópos anyag. A munkakörülményektől függő mértékben több-kevesebb vizet szokott kénsavalakban tartalmazni. Már 10-6 mól víz jelenlétében megváltozik a szilárd fázisszerkezete. A vízmentes S0 3 megszilárdulásakor képződő jégszerű (y) módosulatop.-ja 16,8 C°, fp.-ja 44,6 C°. A víz (kénsav) jelenlétében képződő rostos (/?) módosulat op.-ja 32,5 CP, az a-módosulaté pedig 62,3 C°.
62.6. Dikén-trioxid (S20 3)
Heves reakcióban képződik, ha cseppfolyós S03-ban elemi ként oldunk.7 Akén-trioxidot füstölgő kénsavból desztilláljuk ki. Előállítására a 62.6.1. ábrán bé-mutatott csiszolatos berendezést használjuk. A három, kb. 300 ml-es összekapcsoltírakcionáló lombik a kén-trioxid desztillálását és tiszta állapotban való kondenzá-lását szolgálja.
A következő, szélesebb kémcsőszerű edényben hajtjuk végre a reakciót, és ahozzá csatlakozó U-cső a légnedvesség távoltartására szolgáló P
20 5-ot tartalmazza.
7 Vogel , J ., Per k i ngt on, J . B .: J. Chem. Soc. (London) 127, 1514 (1925); Wöh l er , L ., Weyn i t z, O .: Z. anorg. alig. Chem. 213, 129 (1933).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 246/366
248 O X I D O K
A reagenscBŐbe a tetején levő széles csiszolaton 4t 0,5—1 g igen gondosan megtisztított kénport szórunk, az első lombikba pedig 200 ml 65 %-os óleumot öntünk.
A második lombikot jeges vízzel, a harmadikat só—jég-keverékkel hűtjük, és azelső lombik tartalmát megfelelő folyadékfiirdővel (pl. cc. kénsavval) lassan melegít jük. Ha már enyhe melegítésre (max. 50 C°) több S0 3 nem desztillál át, a kénsav-tartalmú adólombikot lekapcsoljuk, és a szedőül szolgáló második lombik nyílásátazonnal elzárjuk, hogy a légnedvesség behatolását megakadályozzuk. A hűtést ezután egy lépcsővel jobbra toljuk, a só—jég-keverékbe a reakcióedényt állítjuk, és aharmadik lombiknak adjuk az enyhébb jegesvizes hűtést. Ezután a második lombikban levő S03-nak kb. a 3/4 részét átdesztilláljuk a harmadikba. Ha a termékmég mindig nem elég tiszta (op. + 16 C°), az időközben kimosott és kiszárított elsőés második lombik felhasználásával ismételten ledesztilláljuk. A reakcióban felhasználandó 15 ml kén-trioxidot megolvasztás után a harmadik lombik elforgatásával az oldalcsövön át öntjük rá az elemi kénre (a fülkeajtó védelme mellett). Azelegy pillanatszerűen megkékül, majd kb. 30 mp múlva heves reakció, fehér füst-képződés indul meg — célszerű ilyenkor néhány percre levenni a foszfor-pentoxidosU-csövet, mert a kén-trioxidnak úgyis elég nagy a nyomása az edényben ahhoz,hogy a nedves levegő beáramlását megakadályozza.
A reakciótér hőmérsékletét tanácsos ez alatt minél pontosabban +16 C°-ontartani, mert egyrészt ennél magasabb hőmérsékleten jelentőssé válik az S20 3 bomlása, másrészt alacsonyabb hőmérsékleten megszilárdul a S0 3-fölösleg, mely azután
csak erősebben felmelegítve, a késztermék egy részének elbomlása közben desztillálható le. A heves reakció kb. két perc alatt megszűnik, a foszfor-pentoxidos szárítófeltétet visszahelyezzük, és az elegyet néhányszor alaposan összerázzuk. Mintegy 5perc alatt leülepszik a folyékony S03-fázis aljára a kékeszöld kristályokból állóS20 3, melyről a S0 3 a berendezés megfelelő elfordításával dekantálható. A harmadiklombikba visszaöntött színtelen S0 3 újabb S20 3-mennyiség termelésére használható fel.
A kristályok felületén tapadó kén-trioxidot kb. 40 C°-on száraz C02-árambanpárologtatjuk el. Célszerű ezt az 52.1.7. ábrán vázolt módszerrel (csiszolatos meg
oldásban) végezni. Időközönként a csiszolatot levéve, üvegbottal óvatosan szétnyomjuk a kék kristálytömeget, hogy a belsejében tapadó kén-trioxid-fölösleget iskiszellőztessük. Ha az anyag felszíne helyenként kezd barnára változni, akkor lehűtjük és maximum + 10 C°-on még mintegy fél órán át folytatjuk a C02-átveze-tést. Ennek végeztével az anyagot gyakorlatilag tisztának tekinthetjük.
Rendkívül higroszkópos, kék színű, kristályos anyag. +15 C° feletti hőmérsékleten már bomlik.
62.7. Szelén-dioxid (Se02)
Legegyszerűbben az elemi szelén oxigénáramban (kevés N02 jelenlétében)való elégetésével állíthatjuk elő,8 Az anyag mérgező és elég higroszkópos. Ha tel
jesen vízmentes állapotban akarjuk előállítani, akkor az oxigénáramot (miután cc.HN03-on átbuborékoltatva elegendő N02-ot vett fel) P20 5-dal szárítjuk.
Az égetést elég nagy 0 -jű (3—4 cm-es), kb. 70 cm hosszú, nehezen olvadóüvegcsőben végezzük. 50—70 g-nyi száraz vörösszelént nagyobb porcelán csónak-
8 M eyer , J . : Bér. dtsch. chem. Ges. 55, 2082 (1922).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 247/366
T E L L Ú R - D I O X I D 249
bán az üvegcső kezdeti szakaszába helyezünk, és élénk oxigénáramot vezetve át,nagyobb gázégővel hevíteni kezdjük az anyagot. Egy idő múlva a szelén meggyul
lad, és kék lánggal elég. Az égéstermékként keletkező Se02a cső hidegebb részénlerakódik, de hogy el ne tömje a gázáram útját, egy második égő segítségével tovább szublimáljuk.
A nyers termék az esetleg el nem oxidálódott szelénrészecskéktől elszíneződött,fehér kristályos anyag. Ha kellően meg nem tisztított oxigént vagy levegőáramothasználnánk, a redukáló szennyezések, porrészecskék a tiszta Se02-ot is redukálva,hasonló színesedést okoznának. Az oxigénáramban megismételt szublimálás közbenmegvan a lehetősége az ilyen szelénmaradványok oxidálásának. A termék a csővégéhez csatlakoztatott, nagyobb térfogatú, kétnyílásos üveggömbben egészen
tiszta minőségben, hófehér, kristályos alakban összegyűjthető.Ha teljesen vízmentes Se02-ra van szükségünk, akkor a szedőként alkalmazottkétnyílású üveggömb kivezetőnyílását szárítófeltéttel látjuk el. A Se02 szublimációs pontja 350 C°. (s 15 o = 3,95.)
62.8. Tellúr-dioxid (TeOa)
Elemi teliúrból cc. salétromsavas oxidációval állíthatjuk elő.9Egy literes lombikban kb. 20 g Te-port 200 ml vízben szuszpendálunk. Ehhez apránként 95 mlcc. HN03-at adunk, és keverés, rázogatás közben feloldjuk benne a tellúrt. A kiindulási anyagban jelenlevő esetleges szelenid-, illetve telluridszennyezések némelyike nem oldódik. Ezektől az oldatot zsugorított üvegszűrő segítségével gondosanmegtisztítjuk. A tiszta szűrlethez további 65 ml cc. salétromsavat adunk, és anitrózus gőzök távozásának megszűnéséig forraljuk.
Az eközben képződő fehér színű csapadék legtöbbször a bizmut- és antimon-szennyezés (bázisos nitrát). Űjabb üvegfritten való átszívatás után az oldatot vízfürdőn kb. 100 ml végtérfogatra pároljuk. A bepárlócsészét ezalatt óvjuk a levegőporszennyeződésétől úgy, hogy nagyobb átmérőjű tölcsért borítunk föléje, amelyet
száránál fogva tartunk megfelelő magasságban. A gőz egy része így a tölcsér száránát távozhat, megakadályozva a porszemcsék belehullását, más része a tölcsér falánkondenzálva nem csepeg vissza a bepárlócsészébe.
A bepárlás eredményeképpen kiváló fehér kristályos csapadék összetétele:Te20 3(0H)N03. A csapadékot üvegszűrőre gyűjtjük, többször kevés hideg vízzelmossuk, és mázatlan agyagtányérra terítve, pormentes helyen szárítjuk. Ha a termék tisztasága, színe nem lenne megfelelő, salétromsavból (s= 1,25) átkristályosítjuk. A légszáraz anyagot porcelán tálban vagy tégelyben 2 órán át 400—430 C°-on hevítjük, miközben szintén nagyon vigyázunk arra, hogy por bele ne hulljon,mert akkor az anyag redukciója és elszíneződése következik be. A hőkezelést homokfürdőn vagy elektromos kemencében végezhetjük.
A Te02 egyszerű módon előállítható a tellúrsav (H8Te06) termikus bontásávalis. Atellúrsavat először porcelán tálban 150—200 C°-os homokfürdőn melegítjük,vagy hosszabb időn át szárítószekrényben tartjuk. Utána 600 C°-os kemencébentovább izzítjuk mindaddig, amíg kihűlés után teljesen fehér nem lesz. Az esetleges
Inorganic Syntheses, Vol. II I. N ew Yo rk— London, 1950, 143. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 248/366
250 O X I D O K
sárga színeződést a még el nem bomlott Te0 3 okozza. Ilyenkor az izzítást továbbfolytatjuk.
Fehér, mérgező, nem higroszkópos anyag. (Op. 733 C°, s=6,02.)
62.9. Dmitrogén-oxid (N20 )
Az NH4NO 3 termikus bontásával állíthatjuk elő. Az ammónium-nitrát szárítása, megolvasztása és aprítása óvatosságot igényel, mert néha látszólag ok nélkülrobban, ezért tanácsos apróbb részletekben alkalmazni.
Jó minőségű, tiszta NH4N 03-ot 160—170 C°-os szárítószekrényben teljesenmegszárítunk, foszfor-pentoxidos exszikkátorban való lehűtése után pedig a meg
dermedt olvadékot nagyjábólfelaprítjuk. A gázfejlesztő berendezés kifagyasztócsapdávalösszeforrasztott frakcionálólom-bik, amelybe az olvadék hőmérsékletének ellenőrzésére higanyoshőmérő illeszthető. A képződővíz zömének kondenzálását szolgáló, alul kiszélesedő U-cső alakúcsapda után a gázvezetékbe hi
ganyos biztonsági túlnyomásszelepet (egyszerű, lefelé nyitottT-cső, amelynek vége változtatható mélységig higanyos edényfolyadékszintje alá nyúlik) ésáteresztőcsapot iktatunk (62.9.1.ábra). A lombikba kisebb meny-nyiségű, kb. 100 g szárított am-
mónium-nitrátot teszünk, és azbesztes dróthálón át lassan melegítjük. Az exoterm
reakció 170 C°-on indul meg. Vigyázzunk, hogy az olvadék hőmérséklete ne haladjameg a 250 C°-ot, mivel akkor már jelentékeny mennyiségű N2 és NO is képződik. A lombik nyakát célszerű lazán rátekert ellenálláshuzallal 100 C° fölötti hőmérsékleten tartani, hogy a reakcióban képződő vízgőz vissza ne kondenzáljon az olvadékba. A fejlődő N20 vagy közvetlenül gázalakban használható fel, vagy folyékony levegővel hűtött szedőedényben kondenzálható. Tisztítása frakcionált desztillálássaltörténhet (op. —90,6 C°, fp. —88,5 C°).
A közvetlenül gázalakban felhasználásra kerülő N20-ot 4 n KOH-dal mossukés lúgos ditionitoldattal oxigénmentesítjük. A ditionitoldat összetétele: 25 gNa2S204, 125 ml víz és 20 ml 70 %-os KOH.
62.10. Molibdén(VI)-oxid (Mo03)
Ügy állíthatjuk elő, hogy arnmónium-molibdátból cc. salétromsavval molib-dénsavat (H2Mo04) választunk le, majd ezt víztelenítjük.
A felhasznált (NH4)2Mo04-ot többször átkristályosítjuk, majd forró töményoldatához ugyancsak forró cc. HN03-at öntünk, aminek következtében sárga csapadék válik ki az oldatból. Néhány órán át hűlni hagyjuk, majd hidegen üvegszű-
62 .9 .1. ábr a. N »0 előállítása

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 249/366
Y 01i F B Á M (V l ) -0 X I D — U R Á N (v i )- O X I D 251
rőre gyűjtjük, és több részletben hideg vízzel mossuk. Víztartalmát kb. 2 nap alatt150C°-os szárítószekrényben elveszíti, és Mo03-dá alakul. 780 C°-on szublimálással
tisztítható (op. 795 C°, fp. 1155 C°). Melegen sárga, szobahőmérsékleten fehérpor. (s=4,70.)
62.11. Vollrám(VI)-oxid (W 0 3)
Volframátok oldatából savfölösleg hatására különböző mennyiségű hidrát-vizet tartalmazó alakban W 0 3 válik le.
Forrásban tartott cc. sósavba cseppenként ugyancsak meleg, telített nátrium--volframát-oldatot csepegtetünk. A sósav térfogata a felhasználandó volframát-oldatnak kb. 2—3-szorosa legyen. A lecsapás befejeztével még mintegy y2—1%
órán át forró vízfürdőn tartjuk az elegyet, azután szűrjük, és a csapadékot 5 %-osNH4N03 -oldattal kloridmentesre mossuk. Szárítását 120 C°-on kezdjük, végül600 C°-on fejezzük be (op. 1470 C°, fp. 1700 C°). Citromsárga, kristályos anyag.(s= 7,20.)
62.12. Urán(VX)-oxid (U 03)
Egyszerű és aránylag tiszta terméket szolgáltató előállítási módja az urán--peroxid-hidrát (U04»2H20) 350 C°-on végbemenő termikus bontása.10 Az urán--peroxid-hidrát előállítása sem kíván különösebb felszerelést. Forró, 10 %-os ura-nil-nitrát-oldatba cseppenként addig adunk 30 C°-os H202-ot, amíg csapadék
képződést észlelünk. A képződött finom fehér port igen szűk pórusú szűrőn szűrjük,és forró vízzel mossuk. A terméket mázatlan agyagtányéron először légszárazzáalakítjuk, azután 100 C°-on, P2Os felett vákuumexszikkátorban szárítjuk. Az ígyelőkészített U 04*2 H20 5—10 g-ját akár egyszerű üvegtálkában, akár porcelántégelyben elektromos tégelykemencébe tesszük. Fedélnyílásán át élénk oxigénáramot vezetünk bele, és az urán-peroxidot így létrehozott oxigénatmoszférában3—5 órán át 350 C°-on tartjuk, majd utána még kb. 1 óra hosszat 400 C°-ra melegítjük.
Ha nem fontos, hogy az urán(VI)-oxid teljesen vízmentes legyen, úgy is eljár
hatunk, hogy közvetlenül az uranil-nitrátot bontjuk el ugyancsak oxigénatmosz-férában.11 Az oxigén parciális nyomásának növelése mindkét eljárás esetén a
3 U 0 3 = U 30 8+ !/ 20 2
reakció szerint végbemenő urán(VT)-oxid bomlásának visszaszorítását szolgálja.Narancssárga, igen higroszkópos anyag. Szobahőmérsékleten, vízgőz jelenlété
ben egy nap alatt monohidráttá alakul. (s= 7,29.)
62.13. Rénium(VII)-oxid (Re20 7)
A Re20 7 a legtöbb réniumvegyület (alacsonyabb oxidációs számú oxidok, ha-logenidek, valamint a tiszta fém) előállításának kiindulási anyaga. KRe04-bőlelőször a fémet kell előállítani, amely azután oxigénáramban elégethető.12
10 B iltz , W ., MiU ler, H .: Z. anorg. alig. Chem. 163, 258 (1927).
11 H üttig , O. F ., Sch röder , E .: Z. anorg. alig. Chem. 121, 250 (1922).
12 B rauer, Q . : H andbuch dér Práparativen Anorganischen Chemie, H . Bd . Enke Verlag, Stuttgart, 1960, 1293. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 250/366
262 O X I D O K
A színtelen K R e04-ot kvarccsónakban kvarcégetőcsőbe-helyezzük, és 400 C°-os elektromos kemencében tiszta hidrogénáramban redukáljuk (a hidrogénéramot
még hidegen megindítjuk, a fűtést azonban csak akkor kapcsoljuk be, ha a durranógázpróba negatív):
2 K Re04+ 7 Ha = 2 Re + 2 KOH + 6 HaO.
A redukció végét arról ismerjük fel, hogy a vízképződés megszűnik.
A fém mellett képződő KOH-ot vízzel kilúgozzuk az elegyből. Az egyidejűlegképződő alacsonyabb vegyértékű rénium-oxidok a heptoxid-előállítás további menetét nem zavarják. A kimosás után nitrogénben megszárított nyersfémet ezután aredukcióra felhasznált készülékben száraz oxigénáramban felhevítjük (kb. 300 C°-
on). A Re20 7sárga szublimátum alakjában a cső hidegebb részén rakódik le. Enyhe
melegítéssel alkalmas szedőbe szublimálva egyben tisztítását is elvégeztük. (Op.296 C°, fp. 363 C°, s= 8,2.)

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 251/366
63. Alacsonyabb Yegyértékű fém -oxidok
A magasabb vegyértékű fém-oxidokból legegyszerűbben hidrogéneB redukcióval
nyerhetők. Ritkábban a redukciót NH3-val vagy az oxidnak elemi fémmel való
összeolvasztásával, illetve zsugorításával hajtjuk végre. Egyes esetekben fémekből
kiindulva, azok részleges oxidációja útján is előállít hatók.
63.1. Titán-trioxid (Ti20 3)
I
A következő egyenlet szerint képződik1:
3 Ti02 + Ti = 2 Ti20 3.
A fémtitánt por alakban vagy legalábbis minél finomabban felaprítva elke
verjük a dioxiddal. Az elegyet evakuált alumínium-oxid csőben felizzítjuk. Mivelaz Al^O^cső porózus, a szivattyút a hőkezelés közben is működtetjük, a vákuumennek ellenére sem lesz 1 Hgmm-nél jobb. A szükséges magas hőmérsékletet elvi
selő — egyik végén zárt — A^CVcsőbe
kisebb átmérőjű hasonló csövet teszünk,és ennek nyílását ugyanilyen másik csővel
zárjuk el. A belső csőben helyezzük el —
még kisebb átmérőjű tégelyben — az izzítandó keveréket (63.1.1. ábra).
Ha a redukálásra felhasznált fém nem
volt elég finom eloszlású, akkor a nyerstermék kihűlés után nem homogén, hanem felismerhető benne az aprított fém
darabkák alakja. A fém azonban ilyenkor már elveszíti eredeti szívósságát, és
rideg, könnyen porítható lesz. Homogenizálás és újabb felhevítés után egységesterméket kapunk.
A titán-trioxid titán-dioxidból 1300—1500 C° körüli hőmérsékleten hidrogénes
redukcióval is előállítható.2
Az előállítást az alkalmazott magas hőmérséklet miattkvarc- vagy alundum csőben végezzük. Az eljárás teljesen hasonló a V, Cr, Mo
oxidjaira vonatkozó, részletesen leírt eljárásokhoz.
Sötét ibolya színű, kristályos anyag (op. 1900 C°, s=4,49).
63 .1.1. ábr a. T i,O a előállítása
* Ehr l i ch , P . : Z. Elektrochem. 45, 362 (1939).
2 BeJiHKOBa, H., KoMap, A., MnxaHJiOB B.: MeTanjiypmcT 14, 23 (1939).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 252/366
254 A L A C S O N Y A B B V E G Y É R T É K Ű F É M -O X J D O K
63.2. Vanádmm(IIl)-oxid (V 20 3)
Vanádium-pentoxidból hidrogénes redukcióval állítjuk elő. A kiindulási anyagot finom por alakjában égetőcsónakban elég tágas kvarc-
vagy porcelán csőbe helyezzük. A redukcióra használt hidrogént gondosan meg kelltisztítani. A levegő kiszorítása után a csövet elektromos kemencében először kb.600 C°-ra hevítjük. Vigyázzunk arra, hogy a hőmérséklet meg ne haladja a V 20 5 olvadáspontját, a 658 C°-ot. Ha már a reakció nagyobbrészt végbement (kb. 2 óra múlva), a hőmérsékletet 900—1000 C°-ra emeljük, és a hidrogénezést további6 órán át folytatjuk. A hűtést ugyancsak H2-atmoszférában végezzük.
Fénytelen, fekete por (op. 1970 C°, s=4,87).
63.3. Króm(III)-oxid (Cr20 3)
Ammónium-dikromátból igen egyszerűen állítható elő. A teljesen száraz, el-porított (NH4)2Cr20 7-nak kb. 5 g-ját porcelán tálba helyezzük, és egyik pontjánizzó vasdróttal megérintjük. Az érintés helyén megindul a reakció, s gyorsan továbbterjedve, az anyag egész tömege felizzik. A kihűlő termék Cr203.
Zöld színű por (op. 1990 C°, s=5,21.)
63.4. Molibdén(IV)-oxid (M o02)
Molibdén (VI)-oxidot hidrogénáramban 450 C°-on redukálva MoOa-ot nyerünk.3 A kiindulási anyagot porcelán csónakban helyezzük el, amelyet tűzálló üvegcsőbeteszünk. Ha már kiszorítottuk a csőből a levegőt (ellenőrizzük durranógázpróbával!), bekapcsolhatjuk a kemence fűtését. A redukció kb. 6 óra alatt fő tömegébenvégbemegy. A maradék Mo0 3 sósavgázáramban vörösizzáson H2(Mo0 3Cl2)-vé alakul és elszublimál. Az így tisztított Mo02-ot a sósavas kezelés után hidrogénáram
ban hűtjük le.Ibolyásbarna, kristályos por. Vízben oldhatatlan. (s25o=4,7.)
63.5. Volfrám(I V) - oxid (W 0 2)
A W 02 900 C°-on, kb. 1/2—1/2 atm parciális nyomású H2- és vízgőzatmoszférában állandó.4
A redukálandó volfrám(VI)-oxidot 900 C°-ra fűthető elektromos csőkemencében elhelyezett kvarc- vagy porcelán csőbe tesszük. Célszerű a port égetőcsónakbana cső közepére helyezni. A kívánt összetételű H2—vízgőz-elegyet úgy állítjuk elő,hogy a (száraz) hidrogénáramot megfelelő tenziójú vizet tartalmazó edényen bubo-rékoltatjuk át. Erre a célra 85 C°-ra temperált, vízzel töltött üvegszűrős mosópalack a legalkalmasabb. Hogy a gázkeverék összetétele a vízgőz kondenzálása
K j h o mh h o b, H. B.: PyKOBOACTBo no HeopraminecKOMy cnHTe3y. rocxHMH3fl;aT, MoCKBa, 1953 CTp. 134.Gl emser , O., Sau er , H .: Z. anorg. alig. Chem. 252, 151 (1943).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 253/366
BÉZ(l)-OXID 255
folytán ne változzék meg, a vezetékét a rácsavart ellenálláshuzallal 100 C°-ra fűtjük
fel. A redukció kb. 2 óra alatt végbemegy, az egyensúly beáll. A készítményt
oxigénmentes N2-áramban (lásd a gáztisztításra vonatkozó részt) hűtjük le. A nitrogénvezetéket a melegvizes mosópalack utón kapcsoljuk a rendszerbe, háromágú
csap segítségével.
63.6. Réz(I)-oxid (Cu20)
Kétféle módosulata ismeretes; a sárga színű 0 C° körüli, a vörös színű 50 C°fölötti hőmérsékleten képződik. Kényelmesen előállítható elektrolitikus oxidációútján. Semleges vagy KOH-dal gyengén meglúgosított telített KCl-oldatban réz-
anódon képződik alacsony (0,06 A/cm2) áramsűrűséggel végrehajtott elektrolízis
alkalmával. Az elektrolízist főzőpohárban végezzük, katódtérnek célszerű a pohárba
helyezett diafragma belsejét választani. Az anód megfelelő tisztaságú (elektrolit-)rézből készült henger vagy spirális alakjában veszi körül a diafragmát. Katódként
platina lemez vagy tű használható. A kívánt módosulatnak megfelelően az előállításhőmérsékletén telített KCl-oldatot vagy 70 C°-ra melegítjük, vagy jeges fürdővelés merülő hűtőspirólissal hűtjük. Az elektrolitot tiszta N2-gáz bevezetésével kever
jük. A képződött csapadékot gyorsan leszívatjuk, majd desztillált vízzel és kevés
alkohollal mossuk, és CaG2-dal töltött vákuumexszikkátorban szárítjuk. Hosszabbidő tartamára ampullába-forrasztva tartjuk el.
*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 254/366
64. Oxosavak és sóik
64.1. Kálium-klorát (KC103)
Előállítására forró KOH-oldatba feleslegben klórgázt vezetünk (lásd 29.10.).
Előállítható továbbá a KC1 vizes oldatának elektrolitikus oxidációjával is a következő megfontolások alapján kialakított módszerrel. Az alkáli-klorid-elektrolízis
eredményeképpen a katódon H2, az anódon Cl2 fejlődik, és az oldat KOH képződé
se folytán lúgossá válik. Az anódfolyamat:
12 Cl" = 6C12+ 12 e~
Az anódtérben keletkező klórgáz a lúgos közegben Cl- - és C10“ -ionok képződése
közben oldódik:
6 Cl2 +1 2 OH“ = 6 CIO" + 6 Cl- + 6 H20.
Amint a CIO “ -koncentráció bizonyos küszöbértéket elért, megindul az anódon a
kloráttá való oxidáció a következő egyenletek alapján:
6 CIO" = 6 CIO + 6 e“ ,
6 CIO + 3 H20 = 2 HQ10 3 + 4 HC1 + V/2 0 2,
vagyis az anódon 12 egyenérték klorát képződése közben 6 egyenérték oxigéngáz
szabadul fel. Minden elfogyasztott 18 F árammennyiségből tehát csak 12 F használható fel klorátelőállításra, az áramkihasználás ezért 12/18 = 66,6 %-os.
Javíthatjuk ezt az értéket a kémiai klorátképződés feltételeinek egyidejű biz
tosításával, amennyiben a KCIO-oldatot gyengén megsavanyítva, a felszabadítotthipoklórossav disszociációját visszaszorítjuk. Ennek következtében nemcsak az
anódtérben, hanem az oldat egész térfogatában végbemehet a következő reakció:
2 KCIO + 2 HC1 = 2 HCIO + 2 KC1, j
KCIO + 2 HCIO = KC10 3 + 2 HC1.
Az iparban ily módon — az oxigénfejlődés megindulásakor az oldatot a hipo-
kloritionok elektrolitikus oxidációjának megakadályozása céljából megsavanyít
va — csaknem 100 %-os áramkihasználást sikerül megvalósítani. A jó áramkihasz
nálás további feltétele, hogy'megakadályozzuk a katódon a klorát redukcióját. Ezt
néhány tized % K 2Cr04 hozzáadásával valósítjuk meg. Az elektrolízist 70—80
C°-on végezzük, alacsonyabb (30—40 C°) hőmérsékleten KC104is képződik.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 255/366
KXOKÁTOK 267
A fentiek alapján a következő kísérleti berendezést állítjuk össze (64.1.1. ábra):400 ml-es, magas hengerpohárban anódként 25 x 40 mm-es sima platinaelektródot
és ettől kétoldalt kb. 2 cm távolságban azonos kerületű, 0,5 mm 0 -jű platina drótból készült keretkatódot helyezünk el. Az elektródokat speciális, 3 pólusú elektród-tartóban vagy szükség esetén megfelelő méretű parafa dugón fúrt lyukakban erősítjük meg. Az elektródokon kívül hőmérő és szén-dioxiddal működő gázkeverő rögzítéséről is gondoskodunk. Az áramforrás kb. 4 A ésmintegy 12 V teljesítésére képes egyenirányító vagyakkutöltő lehet. A megadott méretű elektródok esetén ezaz anódon (mindkét oldalát számítva) 0,2 A/cm2áramsűrűséget jelent. Az elektrolizáló feszültséget megfelelőenméretezett és potenciometrikus kapcsolásban alkalma
zott ellenállással szabályozhatjuk. Az áramintenzitás jelzésére ampermérőt, a fogyasztott árammennyiség mérésére pedig coulombmétert kapcsolunk sorba az elektródokkal. A feszültségmérőt párhuzamosan kapcsoljuka cellával, de csak időnként ellenőrizzük a kapocsfeszültséget.
Ezután megtöltjük a poharat az elektrolittal, amelyúgy készül, hogy 33 g KCl-ot és 0,2 g K 2Cr04-ot 100ml vízben feloldunk. Az áram bekapcsolása után kb.
34 órával megindul az anódon az oxigénfejlődés. Ekkorpipettával 1 ml cc. sósavat adunk az oldathoz, lassúáramban megindítjuk a keverést biztosító szénsavat, ésfelmelegítjük az elektrolitot a termelés szempontjábóloptimális 70—80 C°-ra. A további elektrolízis során időnként ellenőrizzük a pH-t, és szükség esetén néhány tized ml cc. sósavval visszaállítjuk a gyengén savanyú közeget. Ha az áramkihasználáson kívül az energiafogyasztást is mérjük, akkor egyidejűleg a voltmérő állását is feljegyezzük. Mintegy33 amperóra felhasználása után befejezzük az elektrolízist. Az oldatot só—jég--keverékkel lehűtjük, az anyalúgot üvegszűrőn leszívatjuk, és a csapadékot aprórészletekben alkalmazott jéghideg mosóvízzel kloridmentesre mossuk.
Színtelen, táblás kristályok (op. 356 C°, bomlik). Éghető anyagokkal kevervedörzsölésre, ütésre robban. Gyorsan hevítve bomlása magában is robbanásszerű,Mn02-dal keverve lassú, egyenletes, (s j. = 3,34.)
64 .1.1. ábr a . KClO, elő
állítása
64.2. Magnézium-perklorát /Mg(C104)2 /
Vizes oldatból 6 mól kristályvízzel kristályosodik. Gyakorlati jelentősége a kris
tályvízmentes és 1—3 molekula kristályvíztartalmú anyagnak van, amelyet gázokszárítására, a vákuumtechnikában vízgőz megkötésére használunk.Előállítása a kristályvíztartalmú sónak vákuumban (0,1—10 Hgmm), 120—
250 C° közötti hőmérsékleten végrehajtott víztelenítésével történik.1 A hexahidrá-tot viszont úgy állítjuk elő, hogy a kereskedelmi forgalomban kapható 70 %-os pér-
1 Smi t h , A . F . , Koch , E . O.: Z . a n o r g . a l i g . Chem. 223, 18 (1936).
17 Á lt . és s zerv etlen kém ia i p r a k t . — 4 2 8 4 /1 1 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 256/366
258 0 X 0 S A VA K ÉS SÓ IK
klórsavat MgO-dal semlegesítjük, az oxidfelesleget kiszűrjük, majd az anyagot a bepárolt oldatból kikristályosítjuk. Hasonló módon képződik a Mg(C104)2 a perklór-sav és MgCl2 vagy Mg(N03)2 közötti cserebomlásban is.
64.3. Kálium-bromát (KBr03)
Az elemi bróm lúgos oldatában a Tdoráthoz hasonlóan diszproporcionálódásútján képződik. Gramm-molekulasúlynyi KOH-ból 50 %-os oldatot készítünk, ésa grammatomsúlynyi brómnál (25,5 ml) mintegy 10 %-kal kevesebbet fülke alattóvatosan hozzácsepegtetünk. Az oldat végül tartósan sárgára színeződik és erősen
felmelegszik. Még melegen megindul a bromátkristályok kiválása. Lehűlés után acsapadékot szűrőre gyűjtjük, és a nyers terméket 120 ml forró vízből átkristályosít juk. Tisztaságvizsgálat céljából a bromátminta vizes oldatát kénsavval megsavanyítjuk. Ha maradt még benne Br_ion, az a Br03~ion hatásárabrómmá oxidálódik.
Színtelen, kristályos anyag. (Op 434 C°,s = 3,25.)
64.4.Jódsav (HI03)
Elemi jódnak cc. salétromsavas oxidációjával állítható elő:
3 12 + 10 HN03 = 6 H I03 + 10 NO + 2 H20.
Visszafolyós hűtővel ellátott csiszolatos gömblombikban 50 g reszublimált jó-dot fölös mennyiségű (250 g) cc. salétromsavban 70—80 C°-on melegítünk, amíg azoldat halványsárga színűvé válik. A képződő nitrózus gőzöket a hűtőn át a fülke elszívó nyílásába vezetjük.
A reakció befejeztével az elegyet vízfürdőn szárazra pároljuk, majd kevés vízzel felvéve ezt a műveletet megismételjük. A maradékot lehetőleg kevés tömény salétromsavban oldjuk, és jeges hűtőkeverékkel hirtelen lehűtjük. A kivált kristályos jódsavat üvegszűrőre gyűjtjük és exszikkátorban KOH felett szárítjuk.
Színtelen, kristályos anyag. Hevítve vizet veszít és bomlik. (Op. 110 C°,s = 4,63.)
64.5. Nátrium-perj odátok (Na3H2I06,NaI04)
Előállításukra kiindulhatunk akár elemi jódból, akár nátrium-jódátból. 100 g jódból kiindulva első lépésként NaI03-ot állítunk elő (lásd 1. kötet 20.2.6.), amelyetki sem kell kristályosítanunk, hanem 140 g szilárd NaOH-dal meglúgosítva (vagy
156, lgN a I03 500 ml-es vizes oldatából kiindulva), 100 — 200 ml vízzel kiegészítve 5literes lombikban felforralunk, és nagy átmérőjű (legalább 10 mm-eB) üvegcső vönélénk klóráramot vezetünk át rajta:
NaI03 + 4 NaOH + Cl2 = Na3H2I0 6 + 2 NaCl + Ha.O.
A gázbevezetőcsövet azért kell elég bőnek választanunk, hogy a képződő per- jodát a nyílását el ne tömje. Mellette a kétfuratú dugóba elvezetőcsövet is illesztünk, és azt a fülke elszívónyílásáig PVC-cső vei meghosszabbítjuk. Melegítés közben

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 257/366
PEBJODÁTOK 259
fogjuk be állványba az edényt, mert a csapadékkiválás megindulása után az anyalúg igen hajlamos a túlhevülésre, és az oldat nagyokat lökve forr. A reakció végezté
vel az oldatot kevés NaOH-dal meglúgosítjuk, hogy a jobban oldódó Na^alC^-ota kevésbé oldható Na 3H2I06-tá alakítsuk át. Lehűlése után a csapadékot szűrőregyűjtjük és hideg vízzel mossuk. A terméket 110 C°-on száríthatjuk. A NaI0 3 oxidációját teljesen hasonló módon K 2S20 8-dal is elvégezhetjük ((NH4)2S208használata esetén rossz a kitermelés), de ilyenkor a terméket mindig bizonyos mennyiségűszulfát szennyezi.
Elemi jódból vagy jodidból is készíthető NaI04forró, erősen lúgos közegben,klóros vagy brómos oxidációval. Utóbbi esetben is a termék a nehezen oldódónátrium-hidrogén-perjodát (Na 3H2I0 6). Ha NaI04-ot akarunk előállítani, akkora Na 3H2I06salétromsavas oldatából kell átkristályosítani:
Na 3H2IO« + 2 HN0 3 = NaI04+ 2 NaN0 3 + 2 H20.
100g Na 3H2I 0 6-ot 200 ml víz és 55 ml cc. H N0 3 elegyében oldunk. Ha az oldatnem teljesen átlátszó, üvegszűrőn megszűrjük. Ezután mindaddig pároljuk, amígmeg nem indul a kristályosodás, majd az oldatot 20 C°-ra lehűtjük. A hőmérsékletnek ezt az alsó határát szigorúan meg kell tartani, mert 20 C° alatti hőmérsékleten a meta-perjodát helyett annak trihidrátja válik ki. A csapadékot szűrőregyűjtjük, hideg vízzel mossuk és 110 C°-on szárítjuk. Az oldatban maradt elég
jelentős mennyiségű Na-perjodátot a nehezebben oldódó K I04alakjában KN03-talválaszthatjuk le. A NaI04fehér kristályos anyag. {Op. 300 C°, bomlik, slfl0=3,87.)
64.6. Kálium-perjodát (K I0 4)
A K I04 teljesen a nátrium-perjodáthoz hasonló módon készíthető. A lúgosoldatban itt is először a jobban oldódó K 4I209 képződik, es csak semleges vagygyengén savanyú közegben válik ki a képletnek megfelelő, alig oldható K I04.
Fehér, kristályos anyag (s15o= 3,62).
64.7. Bárium-perjódát /Ba3H4(I0 fl)2/
Az alkáli-perjodátok oldatából Ba(N03)2-tal csapható le. Célszerű viszonylag jól oldódó Na 3H2IOfl-ból (lásd 64.5.) kiindulni. A 100 g jódból előállítható mennyiséget (kb. 225 g Na 3H2IOfl) egy liter víz és 10 ml cc. HN0 3 elegyében oldjuk ésfelforraljuk. Intenzív keverés közben 325 g Ba(N0 3)2 telített forró vizes oldatát
öntjük hozzá, és további állandó keverés közben még kb. 2 órán át forraljuk. Végül az oldatot Ba(OH)2-dal közömbösítjük és kihűlni hagyjuk. A képződő fehérkristályos osapadékot dekantáljuk, forró vízzel mossuk és leszívatás után szárítjuk.
17

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 258/366
260 O X O S A V A K É S S Ó I K
64.8. Perjódsav (H5IOa)
Előállításának alapja az, hogy cc. salétromsavban a Ba(N0 3)2rosszul oldódik,a perjódsav viszont jól.2
Ba 3H4(IOe)2+ 6 HN0 3 = 2 H5IO0+ 3 Ba(N03)2.
75 ml vízzel megnedvesített 100 g Ba 3H4(I06)2-ra 200 ml cc. HN03-at öntünk. A salétromsav teljesen színtelen legyen, ne tartalmazzon vörösbarna nitrózus gőzöket, mert azok a perjódsavat redukálják. Ilyen salétromsavat úgy célszerű előállítani, hogy csiszolatos desztilláltvizes üvegben (64.8.1. ábra) vízlégszivattyúval
addig szívatunk át rajta tiszta, pormen-
mlégszivóttyu tes levegőt, amíg teljesen színtelen nemlesz. Az így tisztított salétromsavat felhasználva az elegyet kb. egy órán át keverés közben 60—70 C°-ra melegítjük, azután 30—40 C°-ra lehűtjük, és kiszűrjükbelőle a képződött Ba(N0 3)2-csapadékot. A csapadékon tapadó perjódsavat cc.HN03-mal kimossuk és a szűrlethez önt
jük. Az egyesített cc. salétromsavas per
jódsav anyalúg-frakciókat 60—70 C°-osvízfürdőn vízlégszivattyúval létesített vákuumban bepároljuk. Közben finom kristályliszt alakjában újra Ba(N0 3 -csapadék képződhet, amely jól megkülönböztethető a csillogó perjódsavkristályok-tól. Ez esetben a szűrést megismételjük,majd folytatjuk a bepárlást a perjódsavkezdődő kiválásáig. Másnap szűrünk, ésa kristályokat 50 C°-on célszerűen levegő-
áramban szárítjuk. Az anyalúg bepár-kitermelés ez esetben kb. 90 %-os.
A HsIOa előállítása során kerüljük a gumidugók vagy gumicsövek alkalmazását, mert az a HNOa-val reagálva redukáló nitrózus gőzöket idézhet elő.
Színtelen, higroszkópos, kristályos anyag, olvadáspontján (130 C°-on) I20 5 képződése közben bomlik.
64.9. Ammómum-vanádiuni-timsó / NH4Y(S04)2*12 H20/
Az NH4 V 0 3 kénsavas közegben véghezvitt redukciója útján készíthető. Az öt-értékű vanádiumot először kén-dioxiddal négyértékűvé redukáljuk. A timsóképzés-hez szükséges háromértékű alakot elektrolitikus redukcióval hozzuk létre a négyértékű vanádiumból.
300 ml-es Erlenmeyer-lombikban 25 g NH4 V 03-ot 100 ml 50 %-os kénsavbanoldunk. Ha nem járunk el elég körültekintően, a képződő vanádiumsav dehidratáló-
64.8.1. ábra. H N 0 3 nitrogén-oxid- - mentesítése
lásával további frakció nyerhető. A
* Booth, H. S.: Inorg. Syntheaes Vol. I ,. New York— London, 1939, 172. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 259/366
V A N Á D IU M S Ó K 261
dik, és Va0 5-gél képződik, amely igen nehezen redukálható. Ezt legegyszerűbbenúgy kerülhetjük el, hogy meleg kénsavval, kisebb részletekben, a kén-dioxid beve
zetése közben, a redukció folyamatát megszakítva végezzük az oldást. A gázbevezetést, illetve a felesleg elvezetését kétfuratú dugóba illesztett üvegcsövek segítségévelvégezzük jól húzó fülke alatt. Ha az oldat az utolsó részlet vanadát feloldása utánis megkékült már, a S0a feleslegét C02 bevezetésével űzzük ki. S0a helyett használhatunk redukálószerként oxálsavat is. Ilyenkor a kénsavas oldathoz részletekbenadagoljuk az ammónium-metavanadátot és az oxálsavat állandó melegítés és keverés közben. Az oxálsav feleslege forralásra elbomlik.
Ezután következik a négyértékű vanádium elektrolitikus redukciója három-értékűvé, ólomelektródok alkalmazásával. A katód 2 y2 X 10 cm, azaz kb. 50 cm2
felületű legyen, az alkalmazott áramsűrűség pedig 0,1 A/cm2 vagy inkább valamivelkevesebb (0,08 A/cm2). Feszültség kb. 14 V. Az anód lehet kisebb felületű ólomlemez is, amelyet a katódtértől mázatlan agyaghenger fala választ el. Az elekt-rolizáló edény magas hengerüveg, amelybe valamivel kisebb agyagdiafragmáthelyezünk. A vanadil-szuliát (katolit) keverését COa bevezetésével oldjuk meg. Anódfolyadékul 33 %-os kénsavat használunk. Az elektródokat megfelelő teljesítményű egyenirányítóval vagy akkutöltővel kapcsoljuk össze, és az áramkörbefeszültségszabályozó ellenállást és ampermérőt iktatunk, valamint a kapocsfeszültség ellenőrzésére szolgáló voltmérőt is beépítünk. A redukciót mindaddig folytatjuk,amíg az oldat meg nem zöldül. A folyamat az adott körülmények között a kétértékű
vanádium képződéséig is végbemehet. A redukció fokát KMn04-os titrálással ellenőrizhetjük a következőképpen:
tetszőleges mennyiségű (néhány tized ml-nyi) mintát veszünk a katódfolyadékból,és kénsavas közegben 0,1 n KMn04-tal megtitráljuk. A végpontot csak megfelelőhígítású vanádiumsó-oldatban látjuk jól; ennek koncentrációját tapasztalati útonállapíthatjuk meg. A permanganáttól ötértékűvé oxidálódott vanádiumot S02-dalismét négyértékűvé redukáljuk, és a kén-dioxid -felesleget COa bevezetésével ésmelegítéssel gondosan kiűzzük, majd újra megtitráljuk az előbbi 0,1 n KMn04-tal.Ha a két fogyás viszonya 2 : 1, akkor a katódfolyadékban a vanádium Ijvantitatíve
három értékűvé redukálódott. Mivel mindkét titrálást ugyanazon a kivett folyadék-mintán végezzük, nem szükséges annak sem pontos mennyiségét, sem a KMn04faktorát ismernünk. A katódtér tartalmát a redukció végeztével jéggel hűtött pohárba öntjük, és valamely izomorf timsóval beoltva, az NH4 V(S04)a*12 HaO-tkikristályosítjuk.
64.10. Ammónium-vanádium(n)-szulfát-hexahidrát
/(N H4) 2 V(S04)2 6H20 /
A vanádiumtimsóhoz (64.9.) hasonló módon állítható elő. A módosítások mind
az oxidáló hatásokra érzékenyebb kétértékű vanádiumvegyület levegőtől valóvédelmét szolgálják. Az ammónium-vanadát oldását és redukcióját a négyértékűállapotig ugyanúgy végezzük, mint az előző leírásban. Az elektrolitikus redukciótis hasonló módon ólomelektródok között, COa-védőgáz-atmoszférában hajtjukvégre, de katódtérnek most célszerűbb az intenzív keverhetoség és jobb gázzárásmiatt a diafragmán belüli teret választani. Ebbe az elektródon kívül mechanikuskeverést és szénsavatmoszférát biztosító gázbevezető- és kivezetőcső nyúlik jólzáró gumidugóba illesztve. A kivezetőcsőhöz a levegő bediffundálásának megaka-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 260/366
262 0 X 0 S A VA K Í S S Ó IK
dályózására kevés vízzel töltött mosópalackot csatlakoztatunk. Az anódfolyadék33 %-os kénsav. Az elektrolizálő áramsűrűség kb. 0,1 Aj cm1 legyen a katód mindkét
oldalát figyelembe véve. A redukció előrehaladását ugyancsak KMn04-os titrá-lással ellenőrizzük. A folyamatot akkor tekintjük befejezettnek, ha a kén-dioxidos
redukció előtti fogyás háromszorosa amásodiknak. Ez után kis feleslegben szi-
vókuum lárd (NH4)2S04-ot teszünk a katolitba, ésaz elektrolízist még rövid ideig folytatjuk.
Az anyag kikristályosítását is szénsavatmoszférában végezzük. Erre a célraa kikristályosító edényt vákuumexszik-
kátorba helyezzük, és fedélnyílását olyankétfuratú dugóval zárjuk el, amelynekegyikén a kristályosító csésze aljáig érőüvegcső, a másikon csapos elszívócsonknyúlik az exszikkátorba. Az exszikkátortszénsavval telítjük. A hosszabb üvegcsővégét gumicsővel a redukált oldatba nyúlógázbevezetőcsőhöz csatlakoztatjuk: ilymódon megoldható az oldatnak a kristályosító csészébe juttatása anélkül, hogy
levegővel érintkeznék (64.10.1. ábra). Az átszívatás elősegítésére és az oxigénmentes atmoszféra biztosítására az exszik-kátornak vízlégszivattyúval való megszí-vatásával egyidejűleg a katódtérben cse
kély C02-túlnyomást biztosítunk. A CC^-atmoszférában végzendő szűréshez a48.1.2.3. ábra szerinti egyszerű megoldást is választhatjuk. Az ammónium-vaná-dium(II)-szulfát-hexahidrát apró, piros kristályok alakjában válik ki. A kristályokat ugyancsak oxigénmentes atmoszférában szárítjuk.
64.10.1. ábra. Vanádium(II)-oldat átszívatáaa kristályosítócsészébe
64.11. Kobalt(III)-szuUát-oktadekahidrát /Co2(S04)3* 18H20 /
A CoS04kénsavas oldatában elektrolitikus oxidációval Pt-anódon 3 képződik.Előállítására az ammónium-vanádium-timsó leírásában (lásd 64.9.) ismerte
tett összeállítás alkalmas, de Pb- helyett Pt-anód és Cu-katód használatos. Anód-térnek a diafragma belsejét választjuk. A benne elhelyezett elektród kb. 4 x 12 cm,vagyis mintegy 100 cm2 felületű, henger alakúra hajtott sima platina lemez legyen.
A porózus agyaghengert kívülről körülvevő rézelektródot még nagyobbra, kb. két
szeres méretűre készítsük. A katódfolyadék 8 n kénsav, amelynek szintjét az anódfolyadék nívójával azonosra állítjuk. Az anódtérbe kerül 24 g CoS04*7 H20 75 ml8 n kénsavval készült meleg oldata.
Mielőtt az elektrolízist megindítanánk, az elektrolizáló edényt jeges hűtő-fürdőbe állítjuk, és megvárjuk, amíg az anódtér hőmérséklete 30 C°-ra hűl. Az oldat
8 Müller, E.t Benther, H .: Elektrochemisches Praktikum, Steinkopf, Dresden— Leipzig, 1963, 322. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 261/366
SZULFÁT OK — FOSZFORSAV 263
ezen a hőmérsékleten válik CoS04•7 H20-ra telítetté. Á csapadékkiválás mégsem
indul meg, mert az anódos oxidáció következtében a Co(II)-koncentrációja egyrecsökken. A szabályozó ellenállással a cellán át folyó áram intenzitását kb. 1 A-reállítjuk be. Mintegy 12 óra elteltével az agyag diafragmában sűrű zöldeskék kristálymassza alakjában kiválik a Co2(S04)3*18 H20. Gyorsan üvegszűrőre borítjuk éserősen leszívatjuk. Mázatlan agyagtányéron szárítható. Néhány hét alatt oxigén-fejlődés közben CoS04-tá redukálódik. A három értékű állapot tartósan fenntarthatócéziumtimsó alakjában: CsCo(S04)2*12 H20.
64.12. A ntimon(lll)-szulfát /Sb2(S04)3/
Finom eloszlású antimonnak vagy antimon-oxidnak cc. kénBavban való oldásával készül.
100 ml forró tömény kénsavba apránként finoman elporított antimont vagyantimon(III)-oxidot adagolunk. Állandó kevergetés és melegítés közben — a kénsavforráspontjához közeli hőmérsékleten — az anyag lassan feloldódik. Lehűlés utánaz oldatból csillogó tűs kristályok alakjában kiválik az antimon-szulfát. Ezután azanyalúgot 80 ml vízmentes jégecettel hígítjuk, és — kihűlés után — üvegszűrőnleszívatjuk.
Jégecettel, majd absz. éterrel mossuk, és néhány napon át kénsavas vákuum-exszikkátorban szárítjuk. (s=3,63.)
64.13. Higany(II)-szulfát (HgS04)
Előállítható fémhiganynak forró tömény kénsavas oldásával.10 g higanyt 15 g cc. H2S04-val porcelán tálban — fülke alatt — addig mele
gítünk, míg a fém fel nem oldódik. Azután a savfölösleget elfüstölve a visszamaradó
fehér sótömeghez — lehűlése után — annyi, kis víztartalmú kénsavat adunk,amennyiben az éppen feloldható. Az anyag kikristályosításához szükséges töménységet úgy érjük el, hogy a .vízfölösleget cc. kénsav hozzáadásával elvonjuk. AHgS04színtelen, négyszögletes kristálylemezek alakjában válik ki (s = 6,47).
64.14. Foszforsav (H3P04)
Vízmentes, kristályos állapotban a kereskedelmi forgalomban kapható koncentrált vizes oldatból vákuumban4 állítható elő.
Csiszolatos vákuumbepárló készülékben (64.14.1. ábra) 500 ml cc. foszforsav-oldaton levegőáramot szívatunk keresztül. A levegőt előzőleg kiszárítjuk: előszörCaCl2-dal töltött szárítótomyon, azután cc. H2S04-as mosópalackon, végül P20 5-osedényen vezetjük át. A bevezetőnyílást kapillárissá szűkítjük, hogy a rendszerbebeáramló levegő mennyiségét megfelelő módon csökkentve, a vízlégszivattyú segítségével a foszforsav fölött vákuumot hozhassunk létre. A vízgőznyomás növelése
4K j h o h h k k o b , H . I \ : P yK o B o fl C T B o n o H e o p ra H f la e cK O M y cn H T e s y . r o c x f t M i í3« a T , M o c K B a ,
1953, ctp. 221,

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 262/366
264 0 X 0 S A V A K É S S Ó IK
érdekében a lombik tartalmát vízfürdőn 35 —38 C°-ra melegítjük. Az anyag kb.2 óra alatt víztartalmának nagyobb részét elveszíti, ezért a továbbiakban erélyesebben kell evakuálnunk, és a lombikot kb. 1 —2 Hgmm végvákuumot létesítő rotációs szivattyúhoz kapcsoljuk. Természetesen megfelelő szárító- vagy kifagyasz
tócsapda (P20 5, Mg(C104)2, illetőleg szénsavhó ——alkoholos hűtőkeverék) segítségével meg kellakadályoznuk, hogy az elszívott vízgőz a szivattyúolajba jusson. A víztelenítendő foszfor-savat most is az előbb megadott hőmérsékleten tartjuk, mert a H 3P04*V 2 H2O most már
30 C° alatt kikristályosodhat, 42,4 C°-on pediga H 3P 04megolvad, és megindulhat a H4P20 7-képződés. Kb. 2 —3 órás újabb vákuumbepár-lás után várható az ortofoszforsav kikristályosodása. Ha ez nehezen indulna meg, az olvadékot beoltjuk. Az oltáshoz szükséges foszforsav -kristályt úgy készítjük, hogy a vízmentesítettfoszforsav egy kis részét kémcsőben erélyesenlehűtve megszilárdulásra késztetjük, s ezt visz-szadobjuk a lombikba. Szükség esetén a vele
izomorf arzénsav kristálykáját is felhasználhatjuk.
« Ha az anyagnak már kb. a fele kikristályosodott, leöntjük róla a még víznyomokattartalmazó folyadékot, és porcelán spatulával jól záródó csiszoltdugós üvegbe tesszük. Hosz-Bzabb megőrzés céljából le is paraffin ózzuk.
Az így előállított foszforsav a felületén tapadó folyadékfázis víztartalma miatt még min
dig nem teljesen vízmentes. Teljes szárítása— a fenti hőmérséklet- és nyomásviszonyokközött — napokig tartó folyamat. (Op. 42,4 C°,8= 1,88.)
64.14.1. ábra. Csiszolatos vákuum- bepárló készülék
64.15. Foszíorossav (H3P03)
A PC1 3 hidrolízise útján állítható elő:
PC13 + 3 H20 = H 3P03 + 3 Ha.
Három, sorba kapcsolt mosópalack közül az elsőbe töltjük a P a 3-ot, a következő kettőbe pedig vizet. A foszfor-trildoridot kb. 60 C°-os vízfürdővel melegítjük,a vizet pedig jéggel hűtjük. A vízlégszivattyú segítségével átáramoltatott levegőmagával viszi a P a 3 gőzt, melynek hidrolízisterméke csakhamar színtelen kris-tálypépi alakjában összegyűlik az első vizes mosóban. Üvegszűrőn leszívatjuk, jegesvízzel mossuk és vákuumban szilikagél vagy szilárd KOH felett szárítjuk.
Elfolyósodó színtelen kristályok. (Op. 73,6 C°, s= 1,64.)

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 263/366
H I P O F O S Z F O B O S S A V É S S ÓI 265
64.16. Hipofoszforossav (H3P02)
A Ba(H2P 02)2*H20-ból kénsavval szabadítható fel:
Ba(H2P02)2.H20 + H2S04= 2 H 3P 02 + BaS04+ H20.
A bárium-hipofoszfithoz ekvimoláris mennyiségben 30 %-os H2S04-at Óntünk. A leülepedő BaS04-ról az oldatot leöntjük, bepároljuk, végül 0 C° alá hűtve kikristályosítjuk. Az olaj szerűvé sűrűsödő hipofoszforossav-oldat bepárlását célszerűen vákuumban fejezzük be. A termék fehér, leveles kristályokat alkotó anyag.(Op.26,5 C°, s17o—1,49.)
64.17. Bárium-hipofoszfit-monohidrát / Ba(H2P02)2 *H20 /
Bárium-hidroxid-oldatból és sárgafoszforból képződik:
8 P + 3 Ba(OH)2*8 H20 + H20 = 3 Ba(H2P 02)2.H20 + 2 PH3.
Csiszolatos gázmosófeltéttel ellátott 1/2 literes gömblombikban 15 g Ba(OH)2*•8 H20-ra 200 ml vizet öntve, feloldjuk, és 5 g (víz alatt feldarabolt) sárgafoszfort
dobunk bele. A gázelvezetőt megfelelően hajlított üvegcsővel meghosszabbítvavízzel telt főzőpohárba, a vízfelszín alá vezetjük. A bevezetőcsövön át nitrogénárammal kiszorítjuk az oxigént a berendezés légteréből, azután felforraljuk azoldatot. A meginduló reakció során képződő foszfor-hidrogéneket a lassú nitrogén-áram folyamatosan eltávolítja a lombikból, s azok a vízen átbuborékolva a szabadlevegőn meg is gyulladnak (ha difoszfint vagy magasabb homológot tartalmaznak). A foszfor-hidrogén igen mérgező anyag, a berendezést ezért jól húzó fülkében kellelhelyezni. Ha a foszfor már feloldódott, a nitrogén helyett szén-dioxidot vezetünkát a lombikon, hogy a bárium-hidroxid fölöslegét semlegesítsük. Ezután a bárium-
-karbonátot kiszűrjük, és a szűrletet kristály képződésig töményítjük, majd lehűtvea selyemfényű tűs kristályok alakjában képződő bárium-hipofoszfitot kikristályosítjuk. Tisztítás céljából forró vízből átkristályosítható. (s=2,90.)
64.18. Nátrium-pirofoszf át (Na4P20 7)
A megolvasztott Na2HP04« 12 H20 víztelenítésével készül. A szekunder foszfátot platina tégelyben megolvasztjuk, és 3—4 órán át 700 C° hőmérsékleten tart
juk, noha az átalakulás már jóval alacsonyabb hőmérsékleten (250 C° körül) megindul. Az izzítást addig folytatjuk, míg az olvadék kivett próbája AgN03-tal piro-foszfátra jellemző fehér csapadékot nem ad (az ortofoszfátra jellemző sárga csapadék helyett). Ha a vízmentes pirofoszfátot kristályos állapotban akarjuk előállítani, olvadáspontja fölötti hőmérsékletre hevítjük (880 C°), és az olvadékot lassanlehűtjük, (s = 2,53.)
Ha a pirofoszfát dekahidrátj ára van szükségünk, a vízmentes só finom porátminimális mennyiségű forró vízben oldjuk. A telített oldatból kiválik a Na4P20 7-•10 HzO, amely 79,5 C°-on megolvad, és saját kristályvizében oldódik.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 264/366
266 0 X 0 S A V A K É 9 SÓIK
64.19. Nátrium-metafoszfát /(NaP03)n/
NaH2P 04vagy inkább a NaNH4H P04hevítése útján állítható elő. A nátrium--ammónium-hidrogén-foszfátot platina edényben megolvasztva addig melegítjük,amíg az olvadék viszkózussá nem válik. A vízgőz- és ammóniafejlődés miatt a sűrűanyag erősen habzik, ezért kevergetni kell. A habzás megszűnte után a hőmérsékletet fokozatosan 700—800 C°-ra emeljük, majd az olvadékot platina vagy nikkel-lemezre öntjük. Ha kihűlése nem elég gyors, különböző polimetafoszfátok elegyeképződik.
A primer foszfátok víztelenítését, illetve a NaNH4H P04kihevítését vákuumban is végezhetjük.
A NaNH4H P04-ot használva kiindulási anyagul, az olvadékot porcelán, esetleg platina csészében lassan 200—250 C°-ra melegítjük. A megkeményedő tömegetlehűtjük, és finom porrá törve hosszúnyakú, vastagfalú gömblombikba töltjük,amit kétfuratú gumidugóval zárunk el. Az egyik furatba hőmérő, a másikba elszívóüvegcső kerül, amelyet 1—2 Hgmm végvákuumot teljesítő rotációs szivattyúhozkapcsolunk (fülke!). A hőmérő higanygömbje közvetlenül érintkezzék a hevítendőanyaggal. A lombikot homokfürdőre helyezve, a benne levő foszfátot 3—4 óraalatt vákuumban 320 C°-ra melegítjük. A képződő riátrium-metafoszfát higroszkó-pos fehér por. (Op. 619 C°, s = 2,48.)
64.20. Vas(II)-ortofoszfát-oktahidrát /Fe3(P04)2 •8H2Ö/
FeS04 vagy (NHá)2Fe(S04)2 telített oldatából csapható le híg Na2HP04-tal.Mivel a termék levegőn könnyen oxidálódik, célszerű az oldatokat kiforralt
vízzel készíteni, és a reakció végrehajtására is C02-dal töltött edényt használni. Csiszoltdugós folyadéküvegben vagy lombikban 12 sr. Mohr-só, ill. 8 sr. FeS04*•7 H20 telített oldatához hozzáöntjük 10 sr. Na2HP04*12 HaO és 2 sr. CH 3COONa*•3 H20 150 sr. vízzel készült oldatát. A szén-dioxiddal telt bedugaszolt üveget rázó
gépen 2—3 órán át rázzuk, majd egy nap múlva C02-atmoszférában (48.1.2.3. ábra)a képződött világoszöld kristályokat leszívatjuk. Vizes alkohollal mossuk és szűrőpapír között megszárítjuk. Az eltartására szánt jól záró üveget is célszerű szén--dioxiddal tölteni, hogy az oxidációtól minél hosszabb időre megvédjük. (s = 2,58.)
64.21. Kálium-manganát(VI) (K2Mn04)
Előállítása céljából a Mn02-ot KC103-tal lúgos olvadékban manganáttá oxidáljuk :
Mn02+ KC10 3 + 2 KOH = K 2Mn04+ KC1 + H20 + 0 2 .
Megfelelő méretű vastégelyben Mn02, KOH és KC10 3 (2 : 2 : 1 súlyarányú)elegyét megolvasztjuk. Az olvadékot üvegbottal kevergetve rövidesen észre vesz-szük, hogy annak viszkozitása erősen nő. Amikor már nem lehet kevergetni, kivakarjuk a tégelyből, és porcelán tálban szárítószekrénybe helyezzük. Ha teljesenmegszáradt, finoman elporítjuk, és a vastégelybe visszahelyezve 3 órán át 500 C°-rafelfűtött elektromos kemencében izzítjuk. Ha nincs elektromos kemencénk, gon-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 265/366
0 X 0 S A V A K SÓ I 267
doskodni kell a redukáló hatású gázok távoltartásáról. Kihűlés után újra elporitjukaz anyagot, és minimális mennyiségű vízben feloldjuk. Üvegszűrőn megszűrjük és
porcelán tálba bepároljuk. A kiváló K 2Mn04 sötétzöld, csaknem fekete anyag. Akristálypépet mázatlan agyagtányérra kenve szabadítjuk meg az anyalúgtól. Op.200 C° (bomlik).
64.22. Nátrium-hipomanganát(Y)-hidrát (Na3Mn04-xH20)
A Mn(V) csak tömény lúgos közegben stabilis. Előállítható Mn(VII)-bőlNa2S03-os redukcióval.5
1 súlyrész NaOH és 2,5 súlyrész víz felhasználásával készült nátronlúg 50 miében 2 g igen finomra elporított KMn04-ot oldunk, majd 0 C°-ra hűtjük és apránként ugyancsak finom porrá dörzsölt Na2S03*7 H^O-ot keverünk bele; kb. 10 percalatt a permanganát jellemző ibolyaszíne világoskékre változik, jelezve a mán-gán(VTI) vegyértékváltozását mangán(V)-té. A kristályos csapadékot üvegszűrőn jól leszívhatjuk és a fenti töménységű NaOH-oldattal mossuk. Aránylag rövid időalatt bombk.
64.23. Kalcium-volframát (CaW04)
Yolfrámsavanhidridből és kalcium-karbonátból képződik magasabb hőmérsékleten. A komponensek gondosan kiszárított, finoman elporított és alaposan homoge
nizált elegyét 2—3 órán át 800 C°-on izzítjuk. Kihűlése után újra jól elporitjuk, ésmost már — kb. 2 óra időtartamra — 1100 C°-ra hevítjük.
A termék mindkét esetben fehér, levegőn állandó anyag. A reakció teljes vég-bemeneteléről úgy győződünk meg, hogy a vizsgálandó minta porát 1 ; 1 hígításúsósavval öntjük le. Ha még észrevehető mértékben szén-dioxidot fejleszt, a hőkezelést porítás után újra megismételjük. (S = 6,06.)
64.24. Ólom-tetraacetát /Pb(CH3C00)4/
Míniumnak jégecetben való oldásával készíthető®. A Pb02 alkalmazása csakakkor jár az elméletileg várható jobb kihasználással, ha az ólom(IV)-oxid egészenfrissen készült; különben a jégecetben nem oldódik.
500 ml-es széles szájú Erlenmeyer-lombikot keverő vei és hőmérővel ellátva 60 C°-os vízfürdőre helyezünk. Beleöntünk 150 ml jégecetet, és több részletben 60—65 gPb 30 4-ot oldunk benne. A jégecetbe szórt mínium finom eloszlású, laza por legyen,
a durvább résztől és csomóktól szitálással gondosan megszabadítjuk. Megvárjuk,amíg minden részlet teljesen feloldódik, csak azután szórjuk bele a következőt. Ateljesen színtelen oldatot melegen megszűrjük. Lehűlés közben az ólom-tetraacetátszíntelen, tűs kristályok alakjában kiválik. Nuccson szűrjük, majd 20 ml 50 C°-os
jégecetből átkristályosítva tisztítjuk. A szerves preparatív kémiában használatosoxidálószer. (Op. 175 C°, s17®= 2,23.)
* Lux, H.: Z. Naturforschung 1, 281 (1946).• Inorganic Syntheses, Vol. I . New York— London, 1948, 138. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 266/366
268 O X O S A V A K É S S Ó I K
64.25. Szilícium-tetraacetát /Si(CH3C00)4/
Ecetsavanhidrides oldatban SiCl4és CH3COOH reakciójában képződik. Yisszafolyós hűtővel ellátott csiszolatos gömblombikot egyharmadáig jégece
tet tartalmazó ecetsavanhidriddel töltünk meg, és beleadagolunk a számítottnálvalamivel kevesebb szilícium-tetrakloridot. Vízfürdőn addig főzzük az oldatot,amíg a sósavfejlődés meg nem szűnik. Az oldatból kihűlés közben néhány óra alattfehér kristályos szilícium-tetraacetát válik ki. Az anyalúgot dekantáljuk, a csapadékot absz. éterrel mossuk és vákuumban (5—6 Hgmm) 148 C°-on desztilláljuk.
Nedvességre érzékeny anyag, levegőn is hidrolizál, desztillációját is szárító-feltét védelme alatt végezzük. Op. 110 C°, fp. 160—170 C° (bomlik).
64.26. Bór-triacetát /B(CH3C00)3/
Bórsavból ecetsavanhidrid hatására képződik:
B(OH)3 + 3 (CH 3C0)20 = B(CH 3COO)3 + 3 CH 3COOH.
Visszafolyatásra állított hűtővel felszerelt csiszolatos gömblombikba 1 : 5 súlyarányban bórsavat és ecetsavanhidridet teszünk. Az elegyet lassan vízfürdőn melegítjük. 60 C° hőmérsékleten a reakció hirtelen, hevesen megindul, a jégecet felforr,és a bórsav acetát alakjában feloldódik. A bór-triacetát lehűlés közben csaknemmaradéktalanul kikristályosodik. Igen könnyen hidrolizáló anyag. Tisztítás céljából ecetsavanhidridből kristályosíthatjuk át, s a csapadékot absz. éterrel mossuk,Desztillálás közben vákuumban is bomlik. Fehér tűs kristályok (op. 147—148 C°).
64.27. Szilikagél (nSi02«xH20)
A vizes alkáli-szilikát-oldatból savanyításra leváló polikovasavat szárítószekrényben víztelenítjük. A kapott nagy felületű porózus anyag adszorpciós tulajdonságai igen nagy mértékben függnek a reakció és aggregáció folyamatának körülményeitől.
A következőkben leírt eljárással nagy felületű (kb. 500 m2 /g), kromatográfiaés szárítás céljára egyaránt alkalmas szilikagélfajtához juthatunk.
Jó minőségű (pl. Merck-gyártmányú) 1,38—1,40 fajsúlyú nátrium-szilikát-ol-
dat 3,4 literét 1 liter desztillált vízzel hígítva, intenzív keverés közben, lassan annyicc. sósavval elegyítjük, hogy a kivett próbát a timolkék éppen savasnak indikálja.Kb. 2 órai keverés után a csapadékot leszívatjuk, és desztillált vízzel addig mossuk,míg a mosóvíz savanyú reakciót már nem ad. A gélt ezután porcelán tálban kiterítve, 12 óra tartamára 200 C°-os szárítószekrénybe tesszük, majd tetszőlegesszemcseméretre aprítjuk, végül kloridmentesre mossuk. Az így készült anyagot250 C°-on súly állandóságig szárítjuk.
Ha teljesen elektrolitmentes készítményre van szükségünk, más eljárást kellválasztanunk, mert az előző, savanyítással lecsapott szilikagélt soha nem lehet tel-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 267/366
S Z 1 L I K A G É L 269
jesen tisztára mosni. Az idégen ionoktól mentes szilikagólt ortoko vasa vas etilészterből (lásd 74.5.1.) hidrolízissel készítjük:7
Si(OC2H5)4+ 4 H20 = H4Si04+ 4 C2H5OH.
A szilikátészter 2 térfogatát 1 térf. etilalkohollal és 100 térf. vízzel elegyítjük.Mivel a keveredés nem megy végbe azonnal és tökéletesen, jól zárható folyadéküvegben (célszerűen polietilén edényben) rázógépen addig rázzuk, amíg a kívántegyenletes elegyedés be nem következik. Ezután az oldatot lapos (szintén műanyag)edénybe öntjük, és kb. 15—17 C°-os hőmérsékletű helyiségben helyezzük el. Néhánynap múlva az oldat megzavarosodik, majd viszkózus lesz, végül néhány hét elteltével megkocsonyásodik. A gélből az alkohol és a víz szűréssel, ill. szárítással a fentimódon eltávolítható.
7 Thieaaen, P. A., Koerner, P.: Z. anorg. alig. Chem. 189-y 168 (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 268/366
65 . Peroxidok, peroxisavak
65.1. Nátrium-peroxid (Na20 2)
Nátrium levegőáramban történő elégetésekor képződik. Az égetőcsövön át
szívott levegőt egy 30 %-os NaOH-ot és két cc. H2S04-at tartalmazó mosó segítségével gondosan megtisztítjuk a szén-dioxid- és nedvességnyomoktól. Az így elő
készített levegő kb. 30 cm hosszú, tágas Supremax-csőbe kerül. Ebben helyezzük el
15 cm hosszú, elég magas falú, vékony alumínium csónakban az elégetésre szánt
2—3 g nátriumot. Az égetőcső elszűkített vége ugyancsak 30 cm hosszú, kb. 3 cmátmérőjű üvegcsőbe torkollik, amelyet a képződő nátrium-peroxid-por összegyűj
tése céljából laza üveggyapot töltéssel látunk el. A terméket a vízlégszivattyú felől
diffundáló nedvességtől szilárd KOH-töltetű toronnyal védjük. A szivattyú és aszedő közé még vizes mosót is iktatunk.
A levegőáramoltatás megindítása után a fémnátriumot égősorral melegíteni
kezdjük. Kb. 300 C°-on megindul az exoterm reakció, és az előbbinél jóval kisebbhőközlés hatására is folytatódik. Az égetőcsövet célszerű a légáram belépőnyílása
felőli végén kissé magasabbra emelni, mert a nátriumolvadék hajlamos a légáram
lással szembeni folyásra, és vízszintesen elhelyezett csőben a csónakból is kiléphet.
A vízlégszivattyúval létesített élénk levegőáramot mindaddig fenntartjuk, míg acsónak tartalmának izzása alább nem hagy.
A termék sárga színű por, mely nedvesség és szénsav hatására bomlik. (Op.460 C°, s= 2,81.)
65.2. Hidrogén-peroxid (H20 2)
Speciális készülékben végrehajtott vákuumdesztillációval a kereskedelembenkapható p.a. minőségű 30 százalékos oldatát kb. 85—90 % H2Oa-tartalmúvá tudjuk
töményíteni.1 A további vízmentesítés a hidrogén-peroxid kifagyasztásával törté
nik. A H20 2-kristályokról az anyalúgot lecentrifugáljuk, s az eljárást többszörmegismételjük.
A vákuumdesztilláció céljára olyan frakcionáló lombikot használunk, amelynek nyakán normálcsiszolatos dugó van. A csiszolatnak ilyen fordított alkalmazásátaz teszi szükségessé, hogy a lombikban visszamaradó 85 —90 %-os termék kiön
tésekor a csiszolat érdes felületén elbomlana, ha szokás szerint a csiszolat hüvelyétforrasztottuk volna a lombik nyakához. A kb. 500 ml-es frakcionáló lombikhoz természetesen csiszolatos hűtő és szedő
tartozik. A lombikba kb. 150—200 ml 30 %-os hidrogén-peroxidot teszünk, és16—20 Hgmm-es vákuumban 4o—50 C°-os vízfürdőn az oldatnak kb. 90 %-át
1 Staedél, W.: Z. angew. Chem. 15, 642 (1902).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 269/366
P E R O X l S A V A K S Ó I 271
lepároljuk. A hőmérséklet ne haladja meg az 52 C°-ot, mert akkor a termék megsárgul és használhatatlanná válik. A visszamaradó, kb. 15 ml oldat 85—90 %
koncentrációjú, és (védó'álarc alkalmazása mellett) tiszta, paraffinozott felületűvagy polietilén edénybe önthető.
Az így kapott tömény H20 2-oldatot apoláros belső felületű edényben —35 C°-os hűtőfürdőbe tesszük, és oltással kikristályosítjuk belőle a hidrogén-peroxidot.2
Az oldat kis részletének folyékony levegővel való fagyasztása útján oltó-kristályhoz juthatunk. Ezt a lehűtött oldatba dobva, azonnal megindul a tűszerűkristályok növekedése (op. 1,7 C°). Néhány perc múlva a terméket perforált fenekű,paraffinozott falú szűrőküvettábanlecentrifugáljuk. (Újabban műanyag centrifuga-küvetták is kaphatók.) A műveletet gyorsan végezzük, nem szükséges hozzá nagy
fordulatszámú centrifuga.
65.3. Kálmm-tetraperoxo-kromát(V) (K3Cr08)
10 ml 30 %-os H20 2-ot 50 ml vízzel hígítva 5 ml 50 %-os KOH-dal meglúgo-sítunk, és 100 ml-es Z?rZe7meyer-lombikban só—jég-keverékkel intenzíven lehűtünk. A lombik tartalma a kiváló peroxid-hidráttól rövidesen megkásásodik. Ez után5 g porrá tört K 2Cr04-ot adunk hozzá, összerázzuk, majd további 2 óra tartamáravisszatesszük a hűtőkeverékbe. Ez alatt a kásás tömeg helyett apró vörösbarnakristályok alakjában kiválik a K 3Cr08. Üvegszűrőn leszívatjuk, vízzel, alkohollal,
majd éterrel mossuk és exszikkátorban szárítjuk.Ha a lúgos közegben előállított peroxi-kromátot vízben oldjuk és kénsavval
megsavanyítjuk, az oldat a képződő bomlékony peroxi-dikromáttól átmenetilegmegkékül. Az anyag éterrel kirázható,
65.4. Ammónium-peroxi-diszulfát és kálium-peroxi-diszulíát /(NH4)2S20 8, k 2s2o 8/
Telített,[semleges (NH4)2S20 4-oldatban nagy anódáram-sűrűséggel végrehajtott
elektrolitikus oxidáció útján S20|_ képződik. A műveletet diafragma nélküli berendezésben is végrehajthatjuk, ha biztosítjuk a semleges pH-t, és 1 % K 2Cr04-adalékkalmegakadályozzuk a képződött termék redukcióját a kátédon.3 A savanyú és alúgos közeg egyaránt mellékreakciókra és rosszabb áramkihasználásra vezet. Savanyú közegben a peroxi-dikénsav hidrolízise folytán Caro-sav és hidrogén-peroxidképződik, lúgosán pedig az ammónia oxidációja megy végbe. Elektrolízis közbenezért folyamatosan gondoskodunk a katód- és anódfolyamatokat Összevontan kifejező
2 (NH4)2S04+ 2 F = (NH4)2S20 8+ 2 NH 3 + H2
egyenlet szerint képződő ammónia H2S04-val való semlegesítéséről. Mivel a hőmérséklet emelkedése ugyancsak a peroxi-diszulfát elbontásának kedvez, külső és belsőhűtéssel egyaránt biztosítani kell a 20 C° alatti hőmérsékletet.
Nagyobb akkumulátoredénybe 5 x 5 cm-es sima platina lemezt függesztünkkatódul, melléje kétoldalt pedig 1—1 V 2 mm keresztmetszetű platina drótból ké-
* M oss, O., H esber g , O. W .: J. Am. Chem. Soc. 42, 2569 (1920).8 Mül l e r , E ., Reuther , H . : Elektrochemisches Praktikum. Steinkopf, Dresden— Leipzig,
1953, 305 old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 270/366
272 P E R O X I D O K , P E R O X I SA V A K
szült keretelektródot helyezünk, amelyek mindegyiké a katód kerületét követő
.alakban 20—20 cm hosszú huzalból készült.
A cellába 2 liter, a hűtővíz hőmérsékletén telített és 2 g K 2Cr04-ot tartalmazóEe- és Mn-mentes (NH4)2S04-oldatot öntünk. (1 liter 10 C°-os víz 763 g ammó-
nium-szulfátot old.) A három elektródot üvegből hajlított hűtőkígyó veszi körül,melynek mindkét végével kívül kell érnie a cella falán, mert
különben a csatlakozó gumivezetéket a fejlődő ózonnyomok
igen hamar tönkreteszik. Az elektrolitot tartalmazó edényben
még hőmérő, keverőlapát és a semleges pH biztosítására szolgáló kénsavas adagolótölcsér kap helyet. Ennek a speciális
kénsavadagolónak a rajzát a 65.4.1. ábra mutatja. A meg
adott méretű elektródokra 14 V kapocsfeszültségű egyenirá
nyítóról 10 A áramot adunk a szükséges anódáram-sűrűség
biztosítására.Elektrolízis közben kb. % óránként indikátorpapírral el
lenőrizzük az elektrolit kémhatását, és megfelelőképpen szabá
lyozzuk a kénsavadagolás sebességét. Az oldat inkább vala
mivel savanyúbb legyen, mint lúgos. Ezt egyébként az1 % K 2Cr04 színe folyamatosan jelzi, amennyiben a savanyúközegben képződő Cr20?“ -ionok vöröses színe lúgOBodáskor
teljesen visszasárgul.
Egy ideig a keletkezett (NH4)2S20 8 oldódik, és a kris
tályképződés csak az elektrolit telítődése után, néhány óra
elteltével indul meg.
Az anyalúgból kiszűrt kristályokat kevés hideg vízzel
mossuk. A szulfáttartalmú anyalúg-zárványoktól úgy tisztít juk meg a terméket, hogy 40 C°-os vízből átkristályosítjuk. Melegebb oldatbana S2Og_-ionok bomlása már számottevő. A nedves kristályfrakciót hidegen, má-
zatlan agyagtányéron vagy szűrőpapír között szárítjuk, mert csak teljesen szárazállapotban raktározható (nedvesen HS04, 0 2- és 03-ra bomlik).
A 15 C°-os elektrolit literenként még 748 g (NH4)2S20 8-ot tartalmaz oldott
állapotban, amelyhez közepes töménységű K 2S04-ot csepegtetve, kicsapódik a sokkal kevésbé oldható K 2S20 8. Az anyalúgot 0 C°-ra hűtve, abban literenként csak
17 g kálium-peroxi-diszulfát marad.
65.4.1. ábra. H 2SOá-adagoló

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 271/366
66. Szulfidok és tiovegyülctek
66.1. Higany(II)-szulfid (HgS)
Kétféle módosulatban ismeretes: a vizes Hg2+-oldatokból H2S hatására kép
ződő fekete HgS és a természetben előforduló élénk vörös HgS (cinna barit) alakjában. Az utóbbi a nagyobb stabilitású, de az átalakulás sebessége közönséges hőmérsékleten igen kicsi. Kísérleti előállítására leginkább azt a tényt használják fel,hogy az alkáli-szulfidokkal képezett komplexéből (NH4)2S-ban nem képződik,a kevésbé oldódó vörös módosulata válik ki. Az eljárás elve tehát a következő:a fekete higany(II)-szulfidot minél nagyobb térfogatú, elég tömény K 2S-oldatbanszuszpendáljuk. A képződő komplex (K 2[HgS2]) telített oldatából a nehezebbenoldható vörös módosulat alakjában válik ki a HgS, amely a jobban oldódó feketemódosulat jelenlevő szilárd fázisából folyamatosan pótlódik.
Nagyobb méretű dörzscsészében 10 g fémhiganyt és 4 g kénport kevés (NH4)2S-
oldattal alaposan ósszedörzsölünk. A képződő fekete HgS-ot a kiindulási anyagokmaradványával együtt 12 g 20 %-os KOH-óldattal öntjük le, és kb. egy héten átmintegy 50 C° hőmérsékletű helyen tartjuk. A közben elpárolgó vizet napontatöbbször is pótoljuk, s ekkor egyúttal az elegyet jól meg is kevergetjük. Néhánynap múlva észrevehetővé válik a csapadék színének megváltozása, ami egy hétalatt gyakorlatilag teljesen meg is vörösödik. A kénfölöslegtől és az át nem alakult(valamivel kisebb fajsúlyú) fekete HgS-tól sokszoros vizes dekantálással tisztítjukmeg. A még ezután is benne maradó ként NajjSC^-oldattal főzve Na2S20 3-tá alakítjuk, és újabb forró vizes mosással távolítjuk el. A terméket 100-—110 C° körüli
hőmérsékleten szárítjuk. A képződött cinóbervörös por 583,5 C°-on szublimálható. (s=8,10.)
66.2. Króm(III)-szulfid (Cr2S3)
Közismert, hogy a króm(III)-szulfid hidrolízisre való hajlandósága miatt vizesoldatból nem állítható elő. A magasabb hőmérsékleten más módszerrel előállítotttermék viszont vízben és nem oxidáló savakban nem oldódik.
A Cr2S 3-o t , CrCl3-ból vörösizzáson H2S-nel állítjuk elő. A felhasznált kén-
-hidrogént szükség esetén mossuk (vízzel), majd CaCl2-os és P20 5-os toronybanmegszárítjuk. A gázáramot kvarc- vagy Supremax-csövön vezetjük át, melyetgáz- vagy elektromos fűtésű csőkemencével 500—600 C°-ra fűthetünk. Ebbenhelyezzük el a vízmentes CrCl3-ot tartalmazó porcelán csónakot. Néhány óra alattvégbemegy a reakció. A terméket kén-hidrogén-áramban hűtjük le. A Cr2S 3 fekete,grafitszerű kristályokat alkot. (s20°= 3,78.)
18 Ált. és szervetlen kémiai prakt. —4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 272/366
274 SZUXiFIDOK ÉS TIOVEG YÜLE TEK
66.3. Foszfor-pentaszulfid (P2S5)
A foszfor a kénnel többféle mólviszonyban is közvetlenül egyesülhet. Az exo-term reakciót elegendő az elegy egyetlen pontjának felhevítésével megindítani.Reakció közben a levegőt (oxigént) távol kell tartani, mert különben a komponensek egyesülése helyett mindkettő oxidálódhat.
A kiindulási anyagokat előzetesen gondosan meg kell szárítani. A vörösfoszfort ajánlatos néhány százalékos NaOH-dal mosni, majd a lúgot forró vízzel kimosva, alkohollal vízteleníteni, végül vákuumexszikkátorban szárítani. Az így előkészített foszfort azonos súlyú kénporral keverjük össze. A reakciót függőlegeshelyzetben befogott hosszú nyakú tűzálló gömblombikban fülke alatt végezzük.
A levegőt szén-dioxiddal kiszorítjuk belőle, és porcelán kanállal keveset beledobunka keverékből, azután gázlánggal óvatosan hevítjük az elegy egyik szélét, és megindítjuk a reakciót. A lángot most elvesszük, és folyamatosan adagolva az elegyet,fenntartjuk a folyamatot. Ha a lombik mégis kihűlne, újra megmelegítjük.
Célszerű a lombikot állványostól vastálcára tenni, és oltóhomokot is készenlétbe helyezni, mert előfordulhat, hogy a lombik eltörik és az égő olvadék szétfolyik. Az adagolókanálban is meggyulladhat a keverék, és attól az egész előkészítettanyagmennyiség. A reakció végeztével a kihűlő olvadék beleszilárdul a lombikba.Legegyszerűbben úgy vehetjük ki, hogy összetörjük a lombikot. A nyers termékkissé higroszkópos, szürke tömeg.
Tisztítás céljából durván felaprítjuk, és széles szájú (egynyílású) retortábólszáraz lombikba desztilláljuk (op. 280 C°, fp. 514 C°). Az előpárlatot nem használjuk fel. A termék fő része világossága* egy tömegben megszilárduló olvadék.
Igen tiszta, kristályos P2S5 úgy állítható elő, hogy a fenti nyersterméket jólelporítva zsugorított üvegszűrőbe helyezzük, és azt a SoxMet-extTaktOT feltétjébenhatszoros mennyiségű CS2-dal extraháljuk. Igaz, hogy az anyag oldhatósága csakkb. Yi %> de a sokszor megismételt kioldás eredményeképpen a foszfor-pentaszulfid•szép sárga kristályok alakjában gyűlik össze az extrakciós lombikban. Gondoljunk a CS2 tűzveszélyes voltára és alacsony gyúláshőmérsékletére (236 C°)! Az extraktort
csak elektromos belső fűtésű vízfürdővel üzemeltessük! (s=2,09.)
66.4. Molibáén(IY)-szulfid (MoS2)
Az oldható tiomolibdátokból savanyításra először MoS 3 csapadék képződik.Ha ezt a sötétbarna port oxigénmentes atmoszférában hevítjük, kénkiválás közbengrafitszerű, fémfényű MoS2-dá alakul. Előállítható elemi szintézissel is finomeloszlású keverék hevítésével, vascsőben.1
66.5. Bárium-tiokarbonát (BaCS3)
Melegen telített BaS-oldatból CS2-dal rázogatva állítjuk elő. 32 g Ba(OH)2*•8 H20-t 100 ml forró vízben oldunk, majd a karbonát csapadéktól azbesztrétegenátszűrve megtisztítjuk. Az oldat Telét 60—70 C° hőmérsékleten H2S-nel telítjük.Fontos, hogy a Ba(OH)2 teljes mennyisége Ba(SH)2-dá alakuljon át. Ezután az
1 van Arkel, A. E.: Recueil Trav. Chim. Pays Bas 45, 442 (1926).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 273/366
T I O S Ó K 276
oldat félretett másik-felével melegen közömbösítjük, és újabb szűrés után 8 gC&j-dal elegyítve rázógépbe tesszük. Az oldatból sárga, porszerű kristályos alakbanBaGS 3 válik ki. Üvegszűrőn leszívatjuk, vizes, majd tömény alkohollal mossuk,
végül vákuumexszikkátorban szárítjuk. Az anyalúgból alkohollal újabb frakciócsapható ki. A kitermelés az elméletinek kb. 50—60 %-a.
Az66.6. Arzén-ortotiofoszfát (AsPS4)
As2S 3 + P2S5 = 2 AsPS4reakcióban képződik.2
A foszfor-pentaszulfidból bizonyos fölösleget alkalmazunk. A szárított komponenseket elporítjuk, majd az As2S3-ból 26,2 g-ot ésaP2S5-ból 47,4g-ot jól összekever
ve, magas olvadáspontú üvegedényben, pl. retortábanmegolvasztjuk, majd a foszfor-pentaszulfid fölöslegét eldesztilláljuk. Ennek maradéktalan eltávolítása bizonyosmennyiségű As2S3-veszteséggel is jár. Az olvadék kihűlve zöldessárga tömeggé szilárdul. Kikristályosítani úgylehet az anyagot, ha homokfürdőbe ágyazott porcelán tégelyben megolvasztjuk, s igen lassan hütjük le. Vákuumdesztillációval kardszárú lombikban (66.6.1.ábra) vagy szublimálással is tisztítható, de magas forráspontja miatt csak kvarcedényben. Sárgászöld színű kris
tályok.
66.6.1. ábra. Kardszárú
lombik
66.7. Nátrium-tiokromit (NaCrS2)
Kromát vagy dikromát elemi kénnel végrehajtott redukciója során képződikalkáli -karbonátos olvadékfázisban.3
1 g K 2Cr04-ot vagy megfelelő mennyiségű K 2Cr20 7-ot 20—30 g vízmentesNa2C03-tal és kb. ugyanannyi kénporral összekeverve, megfelelő méretű ezüstvagy vastégelyben megolvasztunk. Kb. fél óra alatt végbemegy a reakció. Az olvadékot lassan hűtjük ki, és utána a jól oldható karbonátot kilúgozzuk belőle. A nát-
rium-tiokromit vékony, fénylő, vörös színű kristályok alakjában marad vissza. Mégszebben fejlett kristályok képződnek^ ha a Na2C0 3 olvadáspontját kb. azonos súlyúK 2C03-tal leszállítjuk, és ebben a sókeverékben végezzük a redukciót és kristályosítást. Á káliumtartalmú olvadékból is nátrium-tiokromit kristályosodik ki, valószínűleg azért, mert oldhatatlanabb a megfelelő káliumsónál.
66.8. Bárium-ditionát-dihidrát (BaS20 6-2H20 )
Szulfitok óvatos oxidációjának eredményeképpen állítható elő a következőegyenlet alapján:
2 SO2- = S20 2- + 2 c - .
Az oxidálószer Mn(IV) vagy Fe(III) lehet.
2 Glatzel, E.: Z. anorg. alig. Chem. 4, 186 (1893); Klement, R .; Z. anorg. alig. Chem. 253, 247 (1947).
3 Schneider, F.: J. prakt. Chem. [2] 56, 415 (1897); Rudolph, W., Stegemann, K .: Z. anorg. alig. Chem. 251, 376 (1943).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 274/366
276 s z ü l f i d o k é s t i o v e q y ü l e t e k
50 g finoman elporított Mn02-ot 250 ml vízben özuszpendálva só—jég-keverékben lehűtünk, és megindítjuk a S02-be vezetést. Kb. 2 óra alatt az Mn02 csaknem
teljesen feloldódik és redukálódik. Ezután az oldatot 1,5 literre hígítjuk, felforral juk, és a MnS206-ból a Mn(OH)2-ot 200 g Ba(OH)2*8 H20 forrón telített oldatávalfelszabadítjuk. A bárium-hidroxidot kis feleslegben alkalmazzuk, és az oldatot megszűrjük. A szűrlet Mn-mentességét (NH4)2S-dal ellenőrizzük. Ezután C02-ot vezetünk a meleg BaS2Oe-oldatba a szabad Ba(0H)2 becsapása céljából. Űjabb szűrésután az oldatot 80 C°-os vízfürdőn kristályképződésig bepároljuk, és a kihűlés közben kikristályosodó bárium-ditionátot leszívatva, szűrőpapírral megszárítjuk.
66.9. Kálium-vas(III)-szulfid (KFeS2)
Karbonátos olvadékban képződik fém vasból és elemi kénből:
6 Fe + 4 K 2C0 3 + 13 S = 6 KFeS2 + K ^ + 4 C02.
150 g K 2C0 3 és 30 g Na2C0 3 keverékéből megfelelő olvadáspontú elegyet készítünk. Belekeverünk még 30 g vasport, 180 g kénport, és elegendő nagy agyagtégelyben megolvasztva kb. egy órán át olvadékállapotban tartjuk. Minél lassabban hagy
juk kihűlni az anyagot, annál szebben fejlett kristályokat kapunk. Az alkáli-karbonátokat és -szulfidokat kioldva a kálium-vas(III)-szulfid sötét, fémfényű kristályok alakjában marad vissza. A vízzel és alkohollal mosott kristályokat szárítószekrényben száríthatjuk.
66.10. Kálium-bizmut(III)-szulfid (KjE^SJ
A KFeS2-hoz hasonló módon készül (lásd 66.9.). A karbonátolvadék is azonossúlyarányú lehet (5:1).
3 g finom elosztású bizmutport 18 g kénporral és azonos súlyú karbonátelegy-gyel alaposan összekeverve agyagtégelyben összeolvasztunk. 1 —2 óra múlva lassan
lehűtjük, és utána kilúgozzuk. A kálium-bizmut-szulfid élénk fényű, acélszürke, szabályos oktaéderekben képződik.
66.11. Alumínimn-szelenid (Al2Se3)
Elemi szintézis útján állítható elő.4 A szeléngőz mérgező hatású, a reakciót feltétlenül jól húzó fülkében kell végezni.
15 g alumínium port 25 g finoman elporított szelénnel egyenletesen összekeverünk, megfelelő méretű samott-tégelybe töltjük, és középen átfúrt samottfedővellefedjük. Az elegybe a fedő nyílásán át magnézium szalagot szúrunk és meggyújt
juk. (A reakciót szabadban kell végezni és védőszemüveget kell használni!) Különgyújtókeverék nem szükséges.
Kihűlés után a tégelyt széttörjük, és az Al2Se3-et jól záró üvegbe tesszük. A termék kissé porózus szerkezetű, higroszkópos tömeg, amely bizonyos mennyiségű Al20 3-szennyezést tartalmaz, de ennek ellenére SeH2 fejlesztésére teljes mértékbenmegfelel.
* Ferneliiis, W . G.: Inorganic Syntheses, Vol. II . New York— London. 1946,184. old

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 275/366
67. Nitridek, foszfid ok ^rz en ide k, antimonidok, bizmutidok,
karbidok, szilicidek és boridok
E vegyületek egyrészt szerkezetük, másrészt előállításuk szempontjából sok
hasonlóságot mutatnak.
67.1. Lítium-nitrid (Li3N)
Az elemi szintézist a 67.1.1. ábrán vázolt berendezésben hajtjuk végre.1 A lítiumot — melynek felületét előzőleg argonatmoszférában megtisztítjuk — megol
vasztott lítium-fluoriddal (op. 840 C°) bevont Zr02-tégelyben azotáljuk. Ennek a té
gelynek az az előnye a legtöbb kerámiai- és fémtégellyel szemben, hogy folyékony
lítiumnak 800 C°-ig tökéletesen ellenáll, és ezáltal lehetővé válik tiszta lítium vegyü
letek előállítása fémlítiumból. A tégelyt vas védőtégelybehelyezzük, amelyet Pythagoras-anyagból készült, alsó végén
zárt csőbe állítunk. A Pythagoras-csőhöz csiszolatos üvegfeltétet csatlakoztatunk piceines vagy pecsétviaszos tömítéssel.
A kittezett részt áramló-vizes ólomspirálissal hűtjük. Nagyobb nitridkristályok előállításakor az azotálást 400 C°-on
kezdjük, s a hőmérsékletet lassanként 800 C°-ra emeljük. A
gondosan tisztított és szárított nitrogént kb. 20 térfogat % ar- ^
gonnal hígítjuk. A Li3N rubin vörös, áttetsző, kristályos anyag,amely nedvesség hatására NH3-képződés közben bomlik.
67.2. Bór-nitrid (BN)
Bór-trioxid és ammónia reakciója útján állítható elő : y
h t víz
B20 3 + 2 NHa = 2 BN + 3 H20.67.1.1. ábra.
LigN előállításaEgy rész finoman elporított B20 3 és két rész Ca 3(P04)2 ke
verékét hevítéssel víztelenítjük. A hordozóanyag alkalmazásával elkerülhetjük a B20 3 összeolvadását. A kapott termék porózus, anyag. A he
vítést mázatlan agyagtégelyben vagy korund tégelyben végezzük. A tégelyt átfúrtés bevezetőcsővel ellátott fedővel zárjuk le. A tégelyfedél tömítésére vízüvegből és
1 Zintl, E., Brauer, O.: Z. Elektrochem. 41, 102 (1935); Zintl, E., WoUersdorf, O.: Z. Elektrochem, 41, 86.7 (1935).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 276/366
278 NITR IDE K
JA tH 3
Al20 3-ból készült kittet használhatunk. A fedőn átvezetett kettős falú cső az ammónia bevezetésére és a képződött vízgőz kivezetésére alkalmas (67.2.1. ábra). A he
vítést és a reakciót gázfűtésű, termolit téglából épült kis kemencében végezhetjük(44.1.2. ábra). Lehűlés után a tégely tartalmát kevés vízzel felkavarjuk, és nagyobb főzőpohárba töltjük át. A szuszpenzióhoz az alkalmazott foszfátnak megfelelő mennyiségűsósavat adunk, vízzel hígítjuk és forrásig melegítjük. Lehűlés után a visszamaradó BN-et híg sósavval dekantáljuk, ésaddig mossuk, amíg kalcium- és foszfátionok már nem mutathatók ki az oldatban. Szűrés és vízzel való mosás után aterméket vákuumexszikkátorban szárítjuk. A kitermelés kb.90 %-os a felhasznált B203-ra vonatkoztatva. A fenti eljárással készült BN kismértékben szennyezett.
'Amennyiben igen nagy tisztaságú anyagra van szükségünk, az alábbi előállítást alkalmazhatjuk:
BBr 3 + NH 3 = BN + 3 HBr.67.2.1. ábra.
BN előállítása A BBr3-ot H2-gázáramban folyékony ammóniába csepegtetjük.2 Átmeneti termékként először bór-amid képző
dik, amely bór-imiddé alakul át. Az ammóniafelesleg elpárologtatása után a mara
dékot száraz ammónia átvezetése közben 750 C°-ra hevítve, az imid bór-nitriddébomlik. A kitermelés csaknem 100 %-os. A BN fehér, könnyű, amorf por, amelyvízben nehezen oldódik, főzés hatására bomlik.
67.3. Magnézium-nitrid (Mg3N2)
A finom eloszlású magnézium magasabb hőmérsékleten nitrogénnel reagál, haazonban a N2nem teljes tiszta, nem kapunk oxidmentes nitridet. Nitrid előállításáraezért inkább a száraz NH3-gázban történő hevítés használatos.
A magnézium forgácsot porcelán vagy szintermagnézia csónakban porceláncsőbe helyezzük, mely elé háromágú csappal száraz NH3-gáz és N2-gáz bevezetéséreszolgáló vezeték kapcsolódik. A porcelán cső kivezető végéhez egy-egy CaO-dal ésKOH-dal töltött U-cső, majd az NH3-gáz feleslegének elnyelését szolgáló két hígkénsavas mosópalack csatlakozik, amelyek közül az elsőnek a bevezetőcsöve rö-videbb és nem ér bele az oldatba. A porcelán csőből N2-nel kiszorítjuk a levegőt, ésezután kezdjük el a CaO-dal és KOH-dal szárított, bombából nyert NH3-gáz áramoltatását. A reakció 800—850 C°-on (elektromos fűtésű csőkemencében) kb. 4 ó-rán át tart. A nitridképződést a H2-gáz képződése jelzi. A reakció elején gyors NH3-
gázáram szükséges, hogy. a reakciótérben biztosítva legyen a megfelelő nyomás, ésne következzék be az abszorbeáló oldat visszaszívása. Á kész preparátumot az ad-szorbeált NH 3 eltávolítására a reakcióval azonos hőmérsékleten 1 % órán keresztülN2-árammal öblítjük át. A lehűtést N2-áramban végezzük, és a levegő nedvességével szemben igen érzékeny terméket erős N2-áramban vesszük ki a porcelán csőből. Az Mg 3N2 zöldessárga ill. narancssárga színű laza por.
2 Stock, A., Holle, W.: Éter. dtech. Chem. Ges. 54, 2096 (1908).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 277/366
NITR IDE K 279
67.4. Kalcium-nitrid (CaaN2)
A desztillált kalciumot nikkelcsónakban, tisztított N2-árammal 3—4 órán át450 C°-on reagáltatjuk. Ezen a hőmérsékleten az azotálás különösen gyorsan megyvégbe, mivel módosulat változás következtében a fémrács fellazul. A kalcium felületére szublimált igen kis mennyiségű nátrium aktiválja a reakciót, mivel megakadályozza összefüggő nitridhártya képződését.
Ha az alkalmazott kalcium nem eléggé tiszta, vagy a nitrogén oxigénnyomokattartalmaz, a reakció csak kb. 800 C° felett következik be. 800 C°-on NHa-gázzalCa-hidriddel szennyezett nitridet kapunk.
67.5. Titán-nitrid (TiN)
Az alábbi reakció alapján állíthatjuk elő:
2 Ti02 + 4C + N2 = 2 TiN + 4CO.
Az egyenlet szerinti mólarányban készült TiÚ2és 1200 C°-on gáztalanított acetilénkorom keverékét molibdén- vagy korundcsónakban 3 órán át kb. 1250 C°-onN2-áramban hevítjük. Ez a technikai előállítás nem szolgáltat teljesen tiszta terméket, kb. 2 % oxidszennyezést tartalmaz.
Igen tiszta termék nyerhető a
2 TiCl4 + N2 + 4 H2 = 2 TiN + 8 HC1
reakció alapján.3 A készülék lényeges része a gazométer, amelyet egyenlő térfogatrész H2-nel és N2-nel töltünk meg. Ezt a gázkeveréket TiCl4-dal töltött, kb. 36 C°-ramelegített mosón buborékolta tjükát. Az áramlási sebesség elég kicsilegyen ahhoz, hogy a TiCl4-dal valótelítődés bekövetkezhessék. A tulaj
donképpeni reaktoredény kemény-üvegből készült gömblombik, amelynek mindkét oldalára egy-egy, agázkeverék be- és kivezetésére szolgáló cső van forrasztva (67.5.1.ábra).
A lombik nyakához csiszolattalcsatlakoztatjuk azt a dugót, amelybe a volfrámelektródokat tartó vastagabb volfrámbevezetéseket forrasztottuk. Azizzószál átmérője kb. 0,2 mm, és rövid nikkelhídon át csatlakozik a vastagabb volf-rámrúdhoz. Az izzószálakat a kísérlet alatt kb. 1450 C°-ra hevítjük. Mivel a leválóTiN szintén jó vezető, kísérlet közben az áramerősséget 40 perc alatt lOÁ-rol 22 A-reemeljük. Nehézséget okoz a hőmérséklet ellenőrzése, mivel a TiCl4bomlása következtében képződő TiCl 3 az edény falán kicsapódik, és ezért a hőmérsékletet a reakció
vák u u m
3 van Ar k el , A . E . , de Boer ,J . H .: Z. anorg. alig. Chem. 148, 345 (1925);Pot ta r d, F . H Wo o d w a r d , P .: J. Chem. Soc. (London) 1948 , 1709; Trans. Faraday
Soc. 46, 190 (1960).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 278/366
280 FOSZFIDOK
megindítása után optikai pirométerrel már nem mérhetjük, csak becsülhetjük. Areakcióra legalkalmasabb nyomás 30—40 Hgmm, amit manó mérettel ellenőrizhe
tünk. (A higany felületét vékony butil-ftalát hártyával védjük.) A nitrid finomkristályos bevonatot képez az izzószálon; színe rézvörös—aranysárga.
67.6. Alkálifémek foszfidjai, arzemdjei,tmtimonidjai és bizmutidjai
Ezek a vegyületek elemi szintézissel állíthatók elő.4Kiindulási anyagok: köny-nyűbenzin alatt kéregtelenített és a benzintől nagyvákuumban megtisztított alkálifémek, frissen desztillált vagy teljesen száraz sárgafoszfornak 275 C°-on hosszabb
ideig történt hevítésével nyert vörösfoszfor, N2-áramban szublimált arzén, igen
tiszta antimon és bizmut.
67.6.1. Lítium-foszfid (Li3P), lítium-arzenid (li3As), lítium-antimonid (Li3Sb),és Iítium-bizmutid (Li Ei)
Lítium-fluoriddal bevont cirkónium-dioxid tégelybe helyezzük a megfelelő
mennyiségű Li- és P-, illetve Li- és As-keveréket, amelyet légmentesen záró vastégelyben 680, illetve 800 C°-ra hevítve megolvasztunk. A képződő vörösbarna Li 3P,
illetve sötétbarna Li3As teljesen tiszta. A terméket a tégelyből védőgáz-atmoszférá
ban csiszolatos üvegedénybe tesszük át, és argon alatt tartjuk. A Li3As és Li3Sb előállítható oly módon is, hogy frissen elporított As, illetve Sb
vízmentes, kondenzált NH3-val készült szuszpenziójához rázogatás közben részle
tekben adagoljuk a Li folyékony-ammóniás oldatát. A reakció végét csekély lítium
felesleg által okozott kék színeződés jelzi. Az NH3-t ezután kiforraljuk, utolsó nyo
mait evakuálással távolítjuk fel. A visszamaradó barna színű Li 3 As, illetve szürkéskék Li3Sb levegőre rendkívül érzékeny, de N2-, illetve Ar-atmoszférában eltartható.
A Li3Bi lítium és bizmut összeolvasztásával állítható elő Ar-atmoszférában.
67.6.2. Nátrium-foszfid (NaaP), nátrium-arzenid (Na3As), nátrium-antimonid (Na3Sb)és nátrium-bizmutid (Na3Bi)
A Na3P számított mennyiségű nátrium és vörösfoszfor összeolvasztásával nyerhető. A műveletet gázzáró vastégelybe helyezett korundtégelyben, Ar-atmoszférá
ban végezzük. A Na3As előállítására a Na-gőzöket felhevített arzén felett vezetjük el. Magas
olvadáspontú, evakuált üvegcsőben vascsónakba helyezzük a nátriumot, és mellette
korundcsónakba (tégelybe) az arzénport. A nátriumos csónakot mozgatható kis elek
tromos eeőkemencével 350—400 C°-ra melegítjük, és a gőzöket a kb. 200 C°-ra melegí
tett As fölé vezetjük. Ha már a teljes As-mennyiség barnásibolya Na 3 As-dé alakultát, a Na feleslegét 450 C°-on magas vákuumban kiszívatjuk a csőből.
A Na3Sb és Na3Bi Ar-atmoszférában való összeolvasztással komponenseiből
állítható elő.
* Z i n t l , E . ,Brauer , G .: Z. Elektrochem. 41, 297 (1935).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 279/366
F 0 S Z F I D 0 K 281
67.7. Kalcium-foszfid (Ca3P2)
Desztillált kalcium és vörösfoszfor keverékét jól evakuált bombacsőben óvato
san hevítjük, míg az egyesülés erős fénytünemény kíséretében be nem következik. A reakció hevességét csökkenthetjük, ha a foszfort lassan a gyenge vörösizzásra hevített, korundcsónakba helyezett kalciumra desztilláljuk. Lehűlés után a csövet
felnyitjuk, és C02-dal töltjük meg. Ha fémmentes terméket akarunk, akkor a nyersterméket foszforral összekeverve, evakuált és leforrasztott csőben hosszabb időn át600 C°-on tartjuk.
PH 3 előállítására alkalmas Ca 3P2-ot készíthetünk aluminotermiás úton is. Hátránya ennek a módszernek az, hogy a kapott foszfid az ALj03-tól nem választható el.
232 g por alakú száraz kalcium-foszfátot és 180 g alumínium darát összekeverünk, és gyújtókeverékkel megindítjuk a reakciót. Célszerű, ha a begyújtást megelőzően a tégelyt 500 C°-ra előmelegítjük.
67.8. Alumínium-foszfid (A1P)
Elemi szintézissel állítható elő.51,8 g finom eloszlású — lehetőleg oxidmentes — alumíniumport 2,9 g tisztított
és szárított vörösfoszforral mozsárban elkeverünk. A reaktor keményüyegből ké
szült, 20 mm átmérőjű, két végén 6 mm-re leszűkített cső. A keveréket a cső közepébe töltjük. Egy vörösfoszfort tartalmazó frakcionáló lombikot összekötünk a rea
genscsővel, és gondosan tisztított H2-gázt vezetünk át a készüléken. A reaktor-csövet az anyagokkal 1/2—1 órán át H2-áramban 80—100 C°-on szárítjuk. A szárítás közben lehetséges foszforveszteség elkerülése végett a frakcionáló lombikot areaktorcsővel azonos hőmérsékleten tartjuk. A kiáramló foszfortartalmú H2-gáztüveggyapottal töltött tornyon vezetjük át, majd a toronyhoz kapcsolt kihúzott
üvegcső végén meggyújtjuk (durranógázpróba). Lehűlés után változatlanul -á ramoltatás közben a frakcionáló lombikot továbbra is melegen tartjuk, hogy a hideg
A1—P-keverékhez kevés foszfor kondenzáljon. Ezután kis fujtatólánggal a keveréket felhevítjük. Rövid, heves reakció után a cső felmelegítésével a foszforfelesleget
eltávolítjuk. A csövet a reakció helyén szétvágjuk. Az AlP-ot H2-atmoszférábantarthatjuk el. A kapott termék kb. 93 %-os A1P, a maradék A1 és A120 3.
Szürke színű, kb. 1000 C°-on (olvadáspont alatt) bomló kristályok- Vízzel fosz-finképződés közben reagál.
67.9. Kalcium-karbid (CaC2)
A kalcium-karbidot tiszta CaO és szén elektromos ívfényben történő összeolvasztásával állíthatjuk elő az alábbi reakció alapján:
CaO + 3 C = CaC2 + CO.
Ez laboratóriumban a következőképpen végezhető e l:
Nagyobb porcelán tégelyt (magassága 6 cm, felső átmérője kb. 8 cm) nem túlsá
gosan finomra porított CaO és faszén egyenlő súlyarányú keverékével töltünk meg
9 Whi te, W . E ., Bu sh ey , A . H .: J. Am. Chem. Soc. 66, 1666 (1944).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 280/366
282 KARBIDOK
a tégely felső peremétől-kb. 1 cm magasságig. Két, kb. 15 mm 0 -jű és 20 cm hosszúgrafitrudat felülről ferdén úgy helyezünk el, hogy azok kihegyezett alsó vége közötti
távolság szabályozható legyen. Az elektródok felső végén rézgyűrűből készült csatlakozás van. Mindkét elektródot azbeszttel izoláljuk, és parafamentes fogó segítsé
gével úgy állítjuk be, hogy kihegyezett végük a tégely közepén kb. 10 mm távolság
ra legyen egymástól. A keveréket a tégely közepén púpozottan felhalmozzuk, és az
besztpapírral befedjük. Az elektródokat 16 mm2keresztmetszetű szigetelt rézdróttal ampermérőn és reguláló ellenálláson keresztül egyenáramú áramforráshoz csat
lakoztatjuk (az egyenáramú ív sokkal csendesebb, mint a váltóáramú). Az elektródok közötti kapocsfeszültséget voltmérővel mérjük.
A reguláló ellenállás teljes bekapcsolása (6 ohm, 220 V feszültség mellett) után
megindítjuk az áramot. Rövid idő alatt az ívfény begyullad. Ezután az áramerősséget 30—40 A-re állítjuk be. A voltmérőn a feszültség 50—70 V között legyen. Hasokkal nagyobb, úgy ez annak a következménye, hogy az elektródok túl távol van
nak egymástól, ha pedig ennél kisebb, akkor túl közel. Helyes üzemeltetéskor a tégelyből távozó szén-monoxid hosszú lánggal ég. 5—10 perc múlva a feszültséget ki
kapcsoljuk, és a rendszert lehűlni hagyjuk. A grafitcsúcsok alatt található az össze
olvadt vagy zsugorított néhány gramm kalcium-karbid. A CaC2 előállítására előnyösen alkalmazható a 44.2.2.1. ábrán vázolt ívfényke
mence is, ahol az elektródok közötti távolság jól szabályozható.Teljesen tiszta állapotban a CaC2 színtelen, kristályos anyag. A tárgyalt mód
szerrel nyert termék azonban szénfelvétel következtében sötét árnyalatú, bronz•vagy kék színű futtatásokkal. Vízzel acetilént fejleszt; ennek meghatározása alap
ján megmérhetjük a nyerstermék karbidtartalmát.
67.10. Vas-karbid (Fe3C)
Elektrolitvas lemez felületét benzolt égetve bekormozzuk, hosszabb ideig vákuumban 700 C°-on hevítjük, s lassan lehűlni hagyjuk. Ha a lemezt anódként kap
csolva semleges FeCl2-oldatot elektrolizálunk kis áramerősséggel6, a képződő Fe3Ckülönválasztható. A Fe 3C. igen tiszta alakban gyűlik össze az edény alján. Szürke
por, amelyet híg ecetsavval, vízzel, alkohollal vagy éterrel mosunk, és vákuumban.szárítunk.
Az Fe3C 1050 C° felett összetevőire bomlik.
67.11. Titán-karbid (TiC)
Technikai előállítása titán(IV)-oxid redukálásával történik a
Ti02 + 3 C = TiC + 2CO
egyenlet szerint, és oxiddal szennyezett terméket ad.Igen tiszta TiC-ot kaphatunk von Arkel—de Boer eljárása szerint. Ennek lénye
ge, hogy a titán valamelyik illékony halogénvegyületének és valamely szénvegyületnek (pl. toluolnak) a keverékét H2-atmoszférában hevítjük.
6Naes& r , G.: Miit. KWI für Eisenforschung 16, 211 (1934.)

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 281/366
SZILICIDEK 283
A reaktoredény hasonló a TiN előállításánál tárgyalthoz (67.5.1. ábra). A teljesen N2- és 0 2-mentes elektrolithidrogén-gázáramot kettéosztjuk. Egyik ágát TiCl4-dal töltött szobahőfokú mosópalackon, másik ágát pedig 15 C° hőmérsékletű to-luolos mosón vezetjük át. A gázkeveréket egyesítés után a reagenstérbe vezetjük. Az izzószálat (W) kb. 1600 C°-on tartjuk, ehhez kísérlet közben emelni kfell az átmenő áram erősségét. A hidrogén jelenléte jelentős mértékben megkönnyíti a reakciótaz izzószálon, és a halogénvegyületek bomlási hőmérsékletét leszállítva, szükségtelenné teszi a csökkentett nyomás alkalmazását.7
67.12. Szilicidek
A szilicidek klasszikus előállítási módja' a komponensek összeolvasztása útjánvégzett elemi szintézis. Eközben általában olyan magas hőmérsékletet kell alkalmaznunk, amelyen az olvadék a szilikáttartalmú szerkezeti anyagokat is megtámadja. A kitermelés és a termék tisztasága is csekély.
Egyes esetekben, mint pl. a Ti, Zr, V, Ta, Mo, W stb. szilicidjeinek előállításáhozelegendő, ha a poralakú keveréket viszonylag alacsony hőmérsékleten (1500 C°alatt), vákuumban vagy argonatmoszférában zsugorítjuk. Ilyenkor a termék tisztaságát a kiindulási anyagok tisztasága szabja meg.
A fém-oxidok redukciója Si-mal, SiC-dal, vagy Si02-dal és szénnel általában
igen magas hőmérsékletet igényel, és olyan megolvasztott terméket szolgáltat,amelyből a megfelelő szilicid elkülönítése igen nehéz.
A szilicideknek aluminotermiás vagy magnéziumos előállítása fém-oxidokbólés Si02-ból csak abban az esetben szolgáltat tiszta terméket, ha a képződött szilici-det a redukálószer (pl. alumínium) feleslegébe beolvadva kapjuk. Ezekben a fémre-gulusokban a termék elkülönül a salakanyagoktól. Kémiai úton, lúgos vagy híg savas kezeléssel a szilicid kristálypor alakjában kinyerhető.
Némely szilicid olyan módon is előállítható, hogy a megfelelő fém és a szilíciumkeverékét oldószerként szolgáló fémolvadékban (pl. alumíniumban vagy rézben)
összeolvasztjuk. Egyes esetekben megfelelő Al-ötvözetek összeolvasztásával is célhoz érünk. így pl. Th—Al- és Si—Al-ötvözetekből szintetizálható a ThSi2.
67.12.1. Magnézium-szilicid *(Mg2Si) /
Elporított Si és Mg-forgács 1 : 3 súlyarányú keverékét zsugorított magnezitcsónakba töltjük, és nagyvákuümban hevítjük.8 A reakció 450 C°-on indul meg,és néhány perc alatt befejeződik. A kapott termék Mg-felesleget tartalmaz, amely700 C°-on ledesztillálható, vagy porítás után a nyerstermékből etil-jodiddal vízmentes éteres közegben kioldható. Éteres mosás után 300 C°-on szárítjuk. A kapott
Mg2Si kékes árnyalatú, kristályos anyag. A szilánok előállítására használatos nagyobb mennyiségű Mg2Si magnézium
ból és Si02-ból termitreakcióval készül:
Si02 + 4 Mg -f- Mg2Si = 2 MgO,
7 Agt e, G., M oer s, K .: Z. anorg. alig. Chem. 198, 233 (1933).8Gir c, G .: C. R. Hebd. Sóances Acad. Sci. 206, 1405 (1933).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 282/366
284 SZIU C IDE K, B OR I DÓK
Foszfor- és kénmentes precipitált kovasavat több órás gyenge vörösizzásontörténő hevítéssel víztelenítünk. A gondosan elporított Si02-ot kétszeres súlyú Mg-porral összekeverjük, és a keverék 100 g-ját kb. egy literes, hideg vízbe állított vas-tégelyben gyújtókeverékkel reakcióba hozzuk. Közvetlenül a reakció megindulásátkövetően gázbeyezetőcsővel ellátott fedőt helyezünk a tégelyre, és a csövön át erőshidrogénáramot vezetünk bele. Lehűlés után a kapott termék igen kemény éstörékeny, kékes színű kristályokból áll, lúgokkal szemben ellenálló, savak szilánokés hidrogén képződése közben elbontják.
67*12.2. Kalcium-szilicid (CaSi)
Ca-forgács és tiszta Si (15 % felesleg) keverékét mázatlan porcelán csónakbatöltjük, és a cső szabad végén át gyors mozdulattal C02-áramban felfűtött kvarccső1000 C° hőmérsékletű terébe toljuk. Néhány másodperc múlva bekövetkezik-aheves reakció, és az anyag megolvad. A csónak kivétele után a terméket azonnallehűtjük; fémes csillogású szürkés CaSi kristályokat kapunk.
Híg sósav hatására öngyulladó szilánok képződése közben gyorsan bomlik.
67.12.3. Kalcium-diszilicid (CaSi2)
Nagy tisztaságú CaSi2 nem állítható elő az elemek közvetlen összeolvasztásaútján. Ehelyett CaSi és számított mennyiségű elemi Si keverékét Ni-csónakban,hidrogénáramban 1000 C°-on hevítjük.9 A reakció a végén igen lelassul, ezért15 órai hevítés szükséges.
Sósavban sárga színű sziloxán kiválása közben oldódik.
67.12.4. Titán(IV)-szüleid (TiSi2)
Előállítható poralakú elemi komponenseinek sztöchiometrikus összetételű keverékéből összeolvasztással vagy szintereléssel, agyagtégelyben, argon védőgáz at
moszférában.10 A reakció 1500 C° körüli hőmérsékleten indul meg, s mivel rendkívülexoterm, csak kis mennyiségű anyagokkal célszerű végrehajtani.Fémtitán helyett elporított és vákuumban kb. 500 C°-on kihevített titán-
-hidrogént is használhatunk. Ez a szilíciummal H2-atmoszférában hevítve már900 C°-on szintereit terméket szolgáltat.
A TiSi2 szürkésfehér, csillogó kristályai jól vezetik az elektromos áramot. Ásványi savakban a H2F2-ot kivéve nem oldódik.
67.13. Boridok
A komponenseikből való összeolvasztás igen magas hőmérsékletet igényel,amelyet egyszerű laboratóriumban nehéz megvalósítani. Azonfelül ez a módszernagymértékben szennyezett terméket szolgáltat. Viszont tiszta bórból és fémpor-
• Brauer, O.: Handbuch dér Práparativen Anorganischen Chemie. Stuttgart; 1964,717. old.
10 Alexander, P. P.< Metals and Alloys 9 , 179 (1938).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 283/366
BORIDOK 285
ból evakuált kvarocsőben, volfrámcsónakban vákuumban, vagy Mo-tégelyben Ar-
atmoszférában zsugorítással tiszta boridok nyerhetők. így kaphatjuk pl. a Ti, Zr,
Cr, Mo és W boridjait. Az A1-, Mg- vagy Si-mal történő redukciós előállítás az ősidőkből indul ki,
és ez lehetővé teszi a tiszta bór előállításának megkerülését. Ezzel szemben azeljárás kellemetlen velejárója, hogy a képződött boridkristályokat a kísérőanyagok
tól kémiai úton kell elválasztani.
67.13.1. Titán-borid (TiBz)
Egyes esetekben, pl. a titán-borid előállításakor a megfelelő fémporból, bór-
-karbidból (B4C) és oxidból lehet kiindulni. A folyamatot az alábbi reakciók mutatják:
7 Ti + 3 B4C + B20 3 = 7 TíB2+ 3 CO
vagy3 Ti + 2 B4C + T i02 = 4 TiB2 + 2 CO.
Az egyenletek szerinti összetételű keverékhez kis oxidfelesleget kell adni.
Az anyagkeverékből pasztillákat préselünk, amelyeket alkalmas kemencében11 H2-atmoszférában W-tálcán 2D00 C°-ra hevítünk.
11 K i effer , R ., Beneaovsky, F ., H ón ak , E. R.: Z. anorg. alig. Chem. 268, 191 (1952).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 284/366
68. A m m on o- és egyéb nitrogénvegyületek
68.1. Alkálifém-amidok
Az alkálifém-amidok előállítására két módszer használatos. Az egyiket a megolvasztott fémfelesleg és ammóniagáz reakciója, a másikat az alkálifémnek kondenzált ammóniafázisban való oldása jellemzi. Az utóbbi reakció ugyan a nátrium
esetében lassan megy végbe (a káliumnál aránylag gyorsabban), de megfelelő katalizátorral meggyorsítható. A kálium-amid lényegesen jobban oldódik folyékony am
móniában, mint a nátrium-amid. Az oldékonyságuk közötti különbséget is fel
használhatjuk nátrium-amid előállítására úgy, hogy azt a kálium-amid folyékony-
ammóniás oldatából nátrium-jodiddal kicsapjuk.
Ili j n
68.1.1. Nátrium-amid (NaNH2)
Nátrium és ammónia kölcsönhatása útján kaphatjuk:
Na + NH 3 = NaNH2+ i / 2H2.
a) Fedővel zárható vasedénybe (68.1.1.1. ábra) kisebb nikkelcsészét állítunk
és ebbe 10—100 g lehámozott és gondosan megtisztított nátriumot teszünk.
A vasedény fedelén levő furatok egyikébe a nikkelcsésze aljáig érő gázbevezetőt, a másikba rövid gázelvezetőcsövet illesztünk, mindkettőt ugyancsak vasból.
A fedél és a gázcsövek tömítését azbesztzsinórral és tűz
álló vízüveges kittel végezzük. A gázpalack és a készülék közé szilárd KOH-os szárítótomyot és paraffin-olajba merülő biztonsági szelepet iktatunk, a kivezetőcsövet pedig a fülke elszívónyílásáig meghosszabbítjuk.
A fém megolvasztása előtt indítjuk meg a gázáramot,
hogy a levegőt a készülékből kiszorítva a robbanásve
szélyt elkerüljük, majd a vasedényt kb. 300 C°-ra he
vítjük fel. 100 g Na-ból kiindulva kb. 6 órás reakcióután a csövet kihúzzuk az olvadékból, és a reakció befe
jeződését a következőképpen ellenőrizzük: a készülékből
távozó gázokat higany felett fogjuk fel, majd vízzelösszerázzuk. Ha az NH 3 abszorpciója után nem marad
vissza H2-gázmaradék,' a reakció teljes. Ameniiyiben az egész Na elreagált, aterméket NH3-gázáramban lehűtjük. Az így készült nátrium-amid peroXid- ésvasmentes. A nagyipar is ezt a módszert alkalmazza.
b) A nátrium-amid előállítására a folyékony ammónia és fémnátrium között
vaskatalizátor jelenlétében lefolyó reakciót is alkalmazhatjuk. Hatásosságának
r1
r A
68.1.1.1 . v ábr a. NaNH,
előállítása

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 285/366
NÁTRIUM-AMID 287
fokozása végett magában a folyékony ammóniában állítják elő a katalizátort. A reakció végén az ammóniafelesleget elpárologtatjuk. A következő berendezést
használjuk (68.1.1.2. ábra): A gondosan megszárított ammóniát a 3-nyakú csiszolatos lombikba vezetjük. A lombikot alkoholos száraz jéggel hűtjük. Az ammó-
petróteum
azbeszt zsin ór
KOH
petróleumot buborékszámláló
68 .1.1.2 . ábra. NaNH2előállítása folyékony ammóniával
niaveszteség csökkentése végett szárazjég— alkohol-eleggyel hűtött csőkígyót-alkalmazunk kondenzálóhűtőként. A gáz áramlását figyelemmel kísérhetjük, haa kálium-hidroxidos szárítótornyok elé és a hűtőkígyó után petróleummal töltött
gázmosót iktatunk a készülékbe. A csiszolatos lombikba higanyzáras vagy KPG-keverőt szerelünk.Ha a lombikban kb. 200—250 ml folyékony ammónia
gyűlt össze, az ammóniabevezetést megszüntetjük.'^ Az ammónia cseppfolyósítása közben lOg fémnátriumot
készítünk elő. Apró darabkákra vágjuk, a petróleumotpetroléterrel lemossuk, gondosan megszárítjuk és nitrogénnel töltött gyűjtőlombikba dobjuk.
A katalizátor készítésére kiindulási anyagként kristályos vas(III)-nitrátot alkalmazunk. Egy mól nátriumra
0,2—0,3 g durván elporított Fe(N03)3*9 H20-t veszünk,és a Na feloldása előtt adjuk a folyékony ammóniához agázbevezetőcső helyett az A csiszolathoz csatlakozó 68.1.1.3. 68 .1.1.3 . ábra.
ábra szerinti adagolófeltéten át. Az oldatot erőteljesen Adagolófeltét8—10 percig keverjük, hogy minél finomabban eloszlassuk.Ez után a vassó súlyának két-háromszorosát kitevő tiszta fémnátriumot adagolunkaz oldathoz. A fémnátrium a vas(III)-nitrátot fém vassá redukálja, miközben részben nátrium-hidroxiddá, részben nátrium-nitráttá alakul. A reakció folyamán a

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 286/366
288 A M M O N O - É S E G Y É B N IT R O G É N V E G Y Ü L E T E K
nátrium feleslege az ammóniával lép reakcióba nátrium-amid képződése és hidrogénfejlődése közben. A finom eloszlású vas az oldatot sötétre színezi.
A nátrium-amid képződésére katalitikus hatást fejt ki a nátrium-peroxid is. A katalizátor elkészítése után — az előbb leírt módon — kis részletekben
fémnátriumot adagolunk az elegyhez. A fémnátrium adagolásának sebességét úgyszabályozzuk, hogy a hűtőkígyó a reakcióhő folytán keletkező gázalakú ammóniátmég cseppfolyósítani tudja. Ha az oldatba juttatott nátriumadag elreagált, ahidrogéngáz fejlődése megszűnik. Ezt a készülék után kapcsolt petróleumos gázmosón figyelhetjük meg. A reakció közben a keverést lassú ütemben folytatjuk,csak az utolsó nátriumdarab beadagolása után gyorsítjuk meg, hogy az esetlegaz edény falára tapadó nátriumdarabkákat is belemossuk az oldatba. A nátrium-
-amid rosszul oldódik folyékony ammóniában, és porszerű csapadék alakjában kiválik. Á csapadékról dekantálással eltávolítjuk a folyékony ammóniát. Egyszerűmegoldás, ha a reakcióelegyet lassan felmelegedni engedjük, amikor is az oldószerelpárolog. Az adszorbeált ammónia eltávolítása végett a terméket fél óra hosszatvákuumban tartjuk, majd nitrogénatmoszférában tároljuk.
A nátrium-amid fehér, kristályos anyag* 210 C°-on olvad, vákuumban szublimálható. Sötétvörös izzás hőmérsékletén elemeire bomlik. A levegőtől és a nedvességtől óvni kell. Az oxidált termék hevítése rpbbanást okozhat. A szerves kémiaiiparban és a laboratóriumi technikában redukálószerként alkalmazzák.
68.1.2. Lítium-amid (LiNH2) és kálium-amid (KNH2)
Előállításuk és tulajdonságaik a nátrium-amidéhoz hasonlók. Kis mennyiségűLiNH2-ot úgy állíthatunk elő, hogy bombacsőben kevés lítiumot folyékony ammóniában oldunk és leforrasztjuk. Vas védőcsőben néhány órán át 60 C°-on tartjuk,így a reakció katalizátor nélkül is végbemegy.
Nagyobb mennyiség előállítására ezüstcsónakban levő lítium felett 400 C°-onammóniát vezetünk át.
A KNH2-ot folyékony ammóniával történő reakcióval kényelmesen előállít
hatjuk. Mivel a reakció gyorsabb, mint a nátrium-amid esetén, katalizátor nélkülis elkészíthetjük, ha vasmentes anyagra van szükségünk.
68.2. Nátrium-azid (NaN3)
Nátrium-amid és dinitrogén-oxid reakciója útján kaphatjuk:
2 NaNH2 + N20 = NaN3 + NaOH + NH3.
A nátrium-azid előállításához ugyanazt a készüléket használjuk, mint a nát-Tium-amid előállításához (68.1.1.1. ábra). A NaNH2 előállítása után az ammónia-fejleBztő helyére N20-fejlesztőt kapcsolunk. A N20-ot a 24.1.1.-ben leírt módonNH4N03 termikus bontásával nyerjük. A reakcióedény elé higanyos biztosítószelepet iktatunk, és a N20-ot Na2C03-tal, illetve NaOH-dal töltött toronyban szárítjuk. A bevezetőcsőnek nem szabad a NaNH2 -olvadékba érnie, mivel a NaN3 a reakcióhőmérsékletén szilárd halmazállapotú, és dugulás következhet be. 25 g NaNH2-bólkiindulva kb. 5 óra alatt fejeződik be a reakció. A nyersterméket, amennyiben nemhasználják fel egyéb azidok vagy HN3 előállítására, forró vízből való átkristályo-sftással tisztítjuk. A színtelen, kristályos NaN3 hevítéskor elemeire bomlik.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 287/366
H I D R O G É N -A Z I D — Ó L O M -A Z ID 289
68.3. Hidrogén-azid (HNa)
Nátrium-azidnak kénsavval végzett elbontása útján állíthatjuk elő:NaN3 + H2S04 = HN3 + NaHS04.
A NaNg vizes oldatát 2 : 1 hígítású kénsavval 60 C°-on megsavanyítjuk, ésaz oldatot desztillálásnak vetjük alá. Többszörös frakcionálás és CaCl2-ról valódesztillálás eredményeképpen vízmentes HN3-ot kaphatunk. Az eljárás hátránya,hogy az előállítás folyamán gyakran heves robbanás lép fel. Ez kiküszöbölhetőGünther és Meyer szerint, ha kénsav helyett sztearinsavat alkalmazunk.1 A tisztaNaNg-ot —40 C°-on sztearinsavval keverjük össze. Evakuálás után a keverékmelegítésével képződő HN3-ot a fejlesztőlombikkal összeforrasztott, folyékony levegővel hűtött csapdában fogjuk fel. A nyersterméket —50 és —80 C° között vákuum-desztillálással tisztíthatjuk.
A HN3 színtelen folyadék, kellemetlen szagú, gőzei is mérgezők (op. —80 C°,fp. 37 C°). Rendkívüli robbanékonysága miatt célszerű az oldatát alkalmazni,amely viszonylag veszélytelen. Vizes oldatát a következőképpen készíthetjük:250 ml-es gázfejlesztőlombikban 15 g NaN3-ot és 5 g NaOH-ot 150 ml vízbenoldunk. A lombikhoz hűtő közbeiktatásával 100 ml vizet tartalmazó 250 ml-esmosópalackot csatlakoztatunk. A lombik tartalmát forrásig melegítjük, és 90 ml40 %-os kénsavat csepegtetünk hozzá. A desztillációt addig folytatjuk, amíg a
lombikban kb. 50 ml desztillációs maradék marad vissza. A kapott oldat kb. 3 %HNg-ot tartalmaz. A HNg-ot vizes oldatából éterrel kirázhatjuk.
68.4. Ólom-azid /Pb(N3)2/
Előállítására vízben való csekély oldhatóságát használjuk fel. A reakció egyenlete:
Pb(N03)2 + 2 NaNg = Pb(N3)2 + 2NaNOa.
NaNg vizes oldatához erős keverés közben az egyenlet szerinti mennyiségűPb(N03)2 oldatát adagoljuk. Az erőteljes keverés megakadályozza a nagyobbkristályok képződését, amelyek később porításkor robbannának. Nuccsolás utána terméket vízzel kimossuk, és exszikkátorban megszárítjuk. A finomkristályosfehér por iniciálgyújtásra használatos.
68.5. Hidroxil-ammónium-klorid /(NH30H)C1/
Előállításának lényege a HN03 elektrolitikus redukciója, melyet a 68.5.1.ábrán vázolt készülékben hajtunk végre.2 Kb. 10 cm 0 -jű és ugyanolyan magasságú, katódként is szolgáló ólomedénybe — miután azt drótkefével gondosanmegtisztítottuk, majd Hg(N03)2-oldattal és higannyal beamalgámoztuk — agyag-diafragmát állítunk, amelynek belseje anódtérként szolgál. Az anód ólomból
1Günther, P., Meyer, R.: Z. Elektrochem. 41, 541 (1935).2 Tafel, E .: Z. anorg. alig. Chem. 31, 332 (1902).
19 Á lt . és szervetle n k ém ia i p ra k t . — 4 2 8 4 /1 1 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 288/366
290 A M M O N O - É S E G Y É B N IT R O G É N V E G Y Ü L E T E K
készült hűtőspirál. Az ólomedényt jeges hűtőkeverékbe helyezzük. Ugyanebbeüvegkígyót is ágyazunk, amelyet az anódul kapcsolt ólomcsőtekerccsel összekötve,
az anód hűtővizének előhűtésére használunk. Az agyagdiafragma köré mechanikuskörkeverőt helyezünk. Először az anódteret töltjükmeg kb. 50 %-os kénsavval,majd amikor az agyagdiafragma átnedvesedett, akatódtérbe azonos töménységű savat töltünk, és akeverés megindítása után24 amperes árammal elkezdjük az elektrolízist.Kapilláris csőben végződőtölcsérből kb. 2 óra alatt20 g 30 ml vízzel hígítottsalétromsavat csepegtetünk a katódfolyadékba. A hőmérséklet közben neemelkedjék 15 C° fölé. Kb.1 órával a HN03 hozzáadá
sa után a kivett próbában(FeS04-os éstöményHjjSO^as reakcióval) ellenőrizzük,
hogy a katódfolyadék tartalmaz-e nitrátot. Befejezésül a katódfolyadékot kiöntjük az edényből, egyenlő mennyiségű vízzel hígítjuk, és a szulfátot hűtés közben BaCl2-oldattal kicsapjuk. A BaS04-ot leszűrjük, kimossuk, és a szűrletet vákuumban vízfürdőn beszárítjuk. A száraz maradékot vízből átkristályosítjuk. A(NH3OH)Cl színtelen kristályokból áll. Hevítésre bomlik. Erős redukálószer.
68.6. Hidroxil-ammónium-foszfát /(NH30H )P 04/
Előállítható az alábbi reakció szerint:
[Na3P 04 + 3 (NH3OH)Cl = (NH30H)3P 04 + 3 NaCl.
1 liter forró vízben 500 g Na3P 04-12 H20-t , illetve 600 ml vízben szinténmelegen 273 g (NH3OH)Cl-ot oldunk. A megszűrt oldatokat összeöntjük, és lehűlésután szűrjük. Kb. 200 g hidroxil-ammónium-foszfátot kapunk. Az anyalúg bepárlá-sával 90 %-os kitermelést érhetünk el.
Fehér, kristályos anyag, amely hevítésre hidroxil-amint ad le.
68.7. Hidroxil-amin (NH2OH)
Az alábbi reakció alapján állíthatjuk elő:
(NH30H)3P 04 = H3P 04 + 3 NH2OH.
100 ml-es vákuumdesztilláló berendezés lombikjába 20 g lehetőleg durvakristályos hidroxil-ammónium-foszfátot teszünk. 13 Hgmm nyomáson a lombikot
ölomcsö
68.5.1. ábra. (NHsOH)01 előállítása

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 289/366
F O S Z F O R -N I T B I D - K X O B I D 291
150—170 C°-ra melegítjük, ügyelve arra, hogy a tenzió 30—40 Hgmm fölé neemelkedjék. A hőmérő — amelyet úgy állítunk be, hogy majdnem a lombik aljáig
érjen — ilyenkor 135—137 C°-ot mutat. Desztilláláskor védőszemüveget használunk, mert a hőmérséklet kismértékű túllépése is robbanást okozhat. A kapott
kb. 5 g csekély víztartalmú terméket abszolút alkoholból átkristályosíthatjuk, és
vákuumexszikkátorban tömény kénsav felett megBzáríthatjuk. (A hidroxil-amin
illékonysága miatt az evakuálást rövid ideig végezzük.) Igen higroszkópos, 33 C°-onolvadó, szagtalan anyag.
Előállítható hidroxil-ammónium-kloridból is Na-alkoholáttal 3 a következőreakció alapján:
NH 3OHCI + C2H5ONa = NH2OH + NaCl + C2H5OH
abszolút alkoholos közegben. A kiváló NaCl-ot leszűrjük, és a szabad hidroxil--amint —18 C°-on kifagyasztjuk.
68.8. Foszfor-nitrid-klorid /(PNCl2)n/
A különböző polimerizációfokú (PNCl2)n (n = 3—7) foszfor-pentaklorid és
ammónium-klorid kölcsönhatása folyamán képződik magasabb hőmérsékleten:4
PC15 + NH4d = PNC12+ 4 HC1,
Kb. 50 g foszfor-pentakloridot azonos súlyú vagy még több ammónium-klo-riddal alaposan összekeverünk, majd gömblombikba tesszük, és újabb NH4Cl-meny-
nyiséggel 3—5 cm vastagon befedjük. 150 C°-os (±1 0 C°) olajfürdőn 4—6 óra
alatt végbemegy a reakció, egyidejűleg a polimerizáció is megindul. A lombik
hidegebb részén lerakódik az elszublimált trimer, az alján elasztikus, színtelen,átlátszó massza alakjában főleg magasabb homológok elegye marad vissza. A trimer
további részleteit ebből a masszából vagy vízgozdesztillációval, vagy meleg
petroléteres extrakcióval nyerhetjük ki. Tisztítása vákuumdesztillációval, esetleg
vízmentes jégecetből való átkristályosítással történhet [op. 114 C°, fp. 127 C°(13 Hgmm-en), s = 1,98].
Aromás szagú, gőze mérgező, ellenszere híg ammónia belélegzése.
8 Lechner , H . , H of jm an n , J . : Bér. dtsch. chem. Ges. 55, 916 (1922).4 Schenck , R R m e r , G . : Bér., dtsch. chem. Ges. 57, 1345 (1924);Stein man n, R ., Sch i r mer , F . B . Audr ieth , L . F . : J. Am. Chem. Soc. 64, 2377 (1942); Audr i eth , L . F . , Stei n mann , R ., Toy , A . D . F . : Chem. Reviews 32, 109 (1943).
19*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 290/366
69. Komplex v egy öletek
Az ammóniakátokhoz tartozó néhány kobalt-, nikkel- és krómkomplex elő
állítását az első kötetben tárgyaltuk. Ebben a fejezetben több acido-, hidroxo,illetve kelátkomlex előállítását írjuk le.
69.1. Acidokomplexek
69.1.1. Hidrogén-[tetrafluoro-borát(m)] (H[BF4])
Bórsavból és hidrogén-fluoridból kapjuk:
H 3B 0 3 + 4 HF = H[BF4] + 3 H20.
Vasedényben, jéghűtés közben 70—90 %-os H2F2-oldathoz részletekbenh 3b o 3-at adunk kis feleslegben. A reakció hőfejlődés közben magától megy
végbe. Befejezésekor a H 3B 03-felesleget leülepítjük, és a tiszta H[BF4]-oldatotdekantáljuk. Ez színtelen folyadék, az üveget szobahőfokon nem támadja meg.
69.1.2. Kálium-[tetrafluoro-borát(III)] (K [BF4] )
4—5 literes, keverővei felszerelt gömblombikban 1 liter tömény (32 %-os)
sósavhoz keverés közben 175 g bórsavat adunk kisebb részletekben. Negyedórás
keverés után — további erélyes keverés közben — részletekben 450 g NaF-ot adagolunk a szuszpenzióhoz. A reakció következtében az oldat 50—70 C°-ra melegszik fel. Az anyagot egy éjjelen át állni hagyjuk, 1,25 liter vízzel hígítjuk és 2
órás erős keverés közben feloldjuk. A visszamaradt kis oldhatatlan rész főleg
K 2[SiF6]-ból áll (a felhasznált NaF káliummal szennyezett), amelyet nuccsal
leszűrünk. A 155 g KOH-ot 0,5 liter vízben oldjuk, és keverés közben a szűrlet-
hez öntjük. További lúg hozzáadása, illetve a semlegesítés nem célszerű, mert
a kitermelést csak kismértékben javítaná, viszont a vas(III)-hidroxid kiválásafolytán a termék szennyeződését okozná. Az oldatot 0 C°-ra hű tjük, hidegen nuccsol-
juk, a csapadékot hideg vízzel savmentesre mossuk és szárítószekrényben megszá
rítjuk. A kitermelés kb. 80 %-os. Az eljárás előnye, hogy nem igényel különleges edényzetet.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 291/366
PLUMBÁTOK 293
69.1.3. Ammónium-[hexakloro-plumbát(IV)] /(NH«)2[PbCl0]/
Az elektrokémiai úton előállított H2[PbCla]-oldatból nyerjük a rosszul oldódóammóniumsót NH4Cl-oldattal kicsapva. A H2[PbCla]-oldatot sósavoldat elektrolízisével állítjuk elő ólomkatód és két
különböző anód alkalmazásával, amelyek közül az egyik oldódó ólomanód, amelyaz ólomionokat szolgáltatja, a másik pedig indifferens grafitanód, amelyen anégyértékű ólommá való oxidáció következik be.
Akkumulátoredény közepére agyagdiafragmát (69.1.3.1. ábra), ennek belsejébe7 cm hosszú, 3,5 cm széles ólomlemezt helyezünk. Az edény aljára grafitlemeztteszünk, amely anódként szolgál. Ebbe 1,5 cm0 -jű grafitrudat illesztünk, amelyre valamivel na
gyobb belső átmérőjű üvegcsövet húzunk. Azanódtérbe oldódó anódként két, egyenként 27 cmhosszú és 5 cm széles hullámos ólomlemezt is állítunk. Valamennyi ólomelektródot a kísérlet megkezdése előtt drótkefével jól megtisztítjuk. . Agrafit- és az ólomanódokat párhuzamosan köt
jük, és mindkét párhuzamos áramkörbe szabályozó ellenállást és ampermérőt is kapcsolunk.
Az anódtérbe 1200 ml sósavat (fajsúly 1,18), a ka-
tódtérbe 225 ml sósavat (fajsúly 1,10) öntünk. Azelektrolízist 12—14 V feszültséggel, mindkét áramkörben 2 A áramerősséggel folytatjuk, amikor isaz ólomanódokon az áramsűrűség 0,05 A/cm2, agrafitanódon pedig 0,03 A/cm2.
A kísérlet időtartamát úgy állítjuk be, hogy1 liter anódfolyadókra 20—25 amperóra jusson.
Az áramkihasználás 70—80 %-os. A kapott narancssárga anódfolyadékot főzőpohárba öntjükát, és jeges hűtés közben 10 %-os NH4Cl-oldatot adunk hozzá feleslegben. Sárga
színű (NH4)2[PbCl6] csapadék válik ki. Jéggel hűtött szűrőn szűrjük, a csapadékot jéggel hűtött abszolút alkohollal és éterrel addig mossuk, amíg a szűrletHC1- és Cl2-mentessé nem válik. Agyagtányéron, exszikkátorban szárítjuk. A kitermelés kb. 65 g tiszta (NH4)2[PbCl6 j. Vízzel Pb02 kiválása közben hidrolizál,20 %-os sósavban oldódik.
69.1.4. Kálium- [hexakloro-plumbát(lV)] (K2[PbCl6])
Előállításának alapját1 az alábbi egyenletek fejezik ki:
PbCl2 + Cl2 + 2 HC1 = H2[PbCIB],H2[PbCl6] + 2 KC1 = K 2[PbCle] + 2 HG.
Először a H2[PbClfl]-oldatot álhtjuk elő. 30 g PbCÜ2-ot 600 ml tömény sósavbanszuszpendálunk és szobahőfokon klórral oxidálunk. Miután az oldatot klórral
69.1.3.1. ábra. H^PbClgj-oldat előállítása
1 Brauer, O.: Handbuch dér Práparativen Anorganischen Chemie. Enke, Verlag, Stuttgart, 1954, 568. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 292/366
294 K O M P L E X V E G Y Ü L E T E K
telítettük, az át nem alákult PbCl2-ról dekantáljuk. és jéggel lehűtjük. 500 ml
tiszta H2[PbCl6]-oldathoz szintén 0 C°-ra hűtött, 15 g KCl-ot tartalmazó 200 mlvizet adunk, és állandó hűtés közben az oldatba tiszta HCl-gázt vezetünk. Rövididő alatt megindul a citromsárga K 2[PbCl6] kiválása. A HC1 bevezetését telítődésigfolytatjuk. Jéggel hűtött üvegszűrőn szűrünk, a csapadékot kevés hideg töménysósavval mossuk és agyaglemezek között nyomkodva szárítjuk. A termék tisztasága szempontjából fontos, hogy a H2[PbCl6]-oldatot annak előállítása utánnyomban feldolgozzuk, mert az állás közben esetleg képződő PbC^ jelenlétébensötét narancsszínű, ismeretlen szerkezetű termék válik ki. A K 2[PbCl6] vízzelhidrolizál, 20 %-os sósavban oldódik.
69.1.6. Kálium- [trijodo-plum bit(II)] (K [PbI8] -2 H20 )
Ólom(II)-nitrátból és kálium-jodidból állítjuk elő:
Pb(N0 3)2+ 3 KI = K[PbI3] + 2 KN03.
Előállításához 4 g Pb(N03)2-ot 15 ml meleg desztillált vízben oldunk, és keverés közben 15 g Kl-nak 15 ml vízben készült meleg oldatát adjuk hozzá. Előszörsárga Bzínű Pbl2 válik ki, amely lehűléskor halványsárga K[PbI3] •2 H20 -tá alakulát. Ismételt felmelegítéskor a komplex disszociációja következtében újra a sárga
színű Pbl2keletkezik. A sót hidegen, üvegszűrőn nuccsoljuk, és szűrőpapír közöttnyomkodva vagy agyagtányéron szárítjuk. A vízmentes vegyület a dihidrátból vákuumexszikkátorban tömény kénsavas
szárítással, vagy a dihidrát acetonos oldásával és kétszeres térfogatú éterrel történőkicsapásával állítható elő.2
A vízmentes K[PbI3] víz kimutatására szolgáló, igen érzékeny reagens. Vízzelnem a dihidrát visszaalakulásával reagál, hanem Pbl2-kiválással, tehát sárga színeződést okoz. Reagensként a szilárd anyagot vagy acetonos oldatát használjuk.
A K[PbI3]-2 H20 vizes oldatban nagyobb Ki-felesleg jelenlétében állandóvegyület.
69.1.6. Hidrogén-[hexaciano-ferrát(II)] (H4[Fe(CN)6J)
Főzőpohárban 42 g K 4[Fe(CN)fl]*3 H20-t 350 ml vízben oldunk, és 100 mltömény sósavat adunk hozzá. Az esetleg kiváló KCl-ot kevés víz hozzáadásávalfeloldjuk, teljes lehűlés után kb. 50 ml étert adunk hozzá, néhány órán át állnihagyjuk, amíg az éteres komplex színtelen kristályok alakjában kiválik:
K 4[Fe(CN)6] •3 H20 + 4 HC1 + 2 (C2H5)20 =
= H4[Fe(CN)6] . 2 (C2H5)20 + 4 KC1 + 3 H20.
Lenuccsoljuk, kevés étertartalmú híg sósavval mossuk, majd 50 g alkoholbanújra feloldjuk. Miután a fel nem oldódott KCl-tól szűréssel elkülönítettük, az étereskomplexet 50 g éter hozzáadásával újra kicsapjuk, szűrjük és éterrel mossuk. Az éteres komplexet 40—50 C°-on vízlégszivattyúval csökkentett nyomáson elbontjuk, és megkapjuk a H4[Fe(CN)fl]-ot.3 Ez tiszta állapotban hófehér, teljesen
1 Biüz, W.: Bér. dtsch. chem. Ges. 40, 2182 (1907). • Biüz, W., Z. anorg. alig. Chem. 170, 161 (1928).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 293/366
A C ID O - É S H lD R O X O K O M P L E X E K 295
száraz állapotban állandó, nedves levegő jelenlétében kék színűvé válik. Citrom
sárga vizes oldata melegítésre vagy fény hatására bomlik.
69.1.7. Kálium-[trloxaláto-lerrát(III)l (Kg[Fe(C204)3] •3H20)
35 g kristályos vas(II)-szulfátot 100 ml vízben feloldunk, és forralás közben kisadagokban 10—15 ml tömény salétromsavat adunk hozzá. Az oxidáció folyamánnitrogén-oxidok fejlődnek, ezért célszerű fülke alatt dolgozni. Az oxidáció befejeződésekor (ellenőrzés K 3[Fe(CN)6]-tal) az oldatot 2 literre hígítjuk, NH4OH-dal gyengén meglúgosítjuk, és a kicsapódó vas(III)-hidroxidot öt-hatszor vízzel dekantálvemossuk. Ezután üvegszűrőn szűrjük és kevés forró vízzel mossuk, amíg a mosófo
lyadék szulfátmentessé válik. A nedves hidroxidot részletekben 44 g savanyúkálium-oxalát 100 ml 40 C°-os vízzel készült oldatához adagolva oldjuk. Ezt aműveletet, valamint a következő munkálatokat sötét üvegből készült edényekben,vagy vörös fénnyel megvilágított helyiségben célszerű végrehajtani, mivel a tri-oxalát fényérzékeny. Az oldatot szűrjük és a szűrletet kezdődő kristályki válásigbepároljuk. Lehűtésre a zöld színű trioxalátkristályok kiválnak. Vízzel, majd éterrelmossuk, exszikkátorban szárítjuk. A terméket sötét üvegből készült vagy feketepapírba csomagolt edényben tároljuk.
69.1.8. Amm6nium-[hexakloro-platinát(rV)l /(NH4)t[PtCl,)l/
H2[PtCl6] (előállítását lásd 38.3.) híg, gyengén sósavas oldatához először kevésH202-ot, majd az utóbbinak a kifőzése után feleslegben NH4Cl-ot (egy súlyrészplatinára legalább 3 súlyrész NH4C1) adunk. Az oldatot ezután vízfürdőn lassanbepároljuk. A maradékot üvegbottal többször fellazítjuk, és vízfürdőn addig melegítjük, míg a sósav szaga már nem érezhető. A száraz sómaradékot kevés desztilláltvízzel megnedvesítjük és hidegen telített NH4Cl-oldatban szuszpendáljuk. Szűréskor először hideg NH4Cl-oldattal, majd alkohollal mosunk. Az anyalúgnak teljesenszíntelennek kell lennie és platinát csak nyomokban tartalmazhat. (Kimutatása
H2S-neI és SnCl2-dal.) Amennyiben a kapott termék nem citromsárga kristályokbóláll, hanem vöröses árnyalatot is mutat, ez egyéb platinafémekkel (Pt, ír vagy Rh)való szennyezettségére utal. Az (NH4)2[PtCle] vízben kevéssé, NH4Cl-tartalmúvizes oldatokban igen rosszul oldódik.
69.2. Hidroxokomplexek
A hidroxosók közé azokat a komplex vegyületeket soroljuk, amelyek komplex
anionjában a központi fématom körül hidroxidionok helyezkednek el. mint ligan-dumok. A hidroxosók kationjai alkálifém- vagy alkáliföldfém-ionok. Egyes esetekben nehézfémek hidroxokomplexei is eloállíthatók.
A komplex hidroxoanionok képződése az oldatban az
Me(OH)x + y OH" ^ [Me(OH)I+y7 '
egyensúly következtében erős bázisok hatására megy végbe nehezen oldódó fém--hidroxidokból. Az alkáli-hidroxosók többsége csak tömény lúgos oldatok üledéke-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 294/366
296 K O M P L E X V E G Y Ü L E T E K
ként állandó. A hőmérséklet emelkedése a fenti egyensúlyt balra tolja el. Csak azSn(IV), Pb(IV), Sb(V) hexahidroxosója oldható szobahőmérsékleten bomlás nélkül
vízben.
09.2.1. Nátrium-[bexabiaroxo-sztaiinftt(IV)] (Na2\Sn(UH)8])
SnCl4 híg sósavas oldatát karbonátmentes NaOH-dal metiloranzsra semlege
sítjük. A lenuccsolt és vízzel kloridmentesre mosott Sn02»aq-t részletekben fölös,tömény, kb. 100 C°-os NaOH-ba adagoljuk, amelyben gyorsan és tisztán oldódik.Rövid idő múlva kiválik a színtelen, kristályos hexahidroxosó. A kristálykásátC02-mentes atmoszférában szűrjük, 30 %-os NaOH-dal, majd alkohollal és éterrelmossuk. A termék C02-dal szemben igen érzékeny.
69.2.2. Nátrium-[hexahidroxo-plumbát(IV)] (Na2[Pb(0H)6])
A nátrium-[trihidroxo-plumbit(II)] anódos oxidációja útján kaphatjuk4:
Na[Pb(OH)s] + NaOH + 2 OH" = Na2[Pb(OH)6] + 2 e".
300 ml 13 n NaOH-ban forralás közben 18,5 g sárga PbO-ot oldunk. Az oldatotüvegszűrőn megszűrjük, és C02 kizárása mellett lehűlni hagyjuk. Mivel az ígykapott Na[Pb(OH)3]-oldat instabilis, ezért szobahőmérsékletre való lehűlés utáncélszerűen nyomban felhasználjuk az elektrolitikus oxidációra. Ellenkező esetben
az esetleg kivált PbO-ról dekantáljuk az oldatot.Elektrolizáló cellaként 300 ml-es gumilemezzel zárható üvegedényt haszná
lunk. Megfelelő furatokban gázkivezetőcsövet, hőmérőt, higanyzáras keverőt,üvegcsőbe kittezett anódhoz csatlakozó vezetéket és katódtérként szolgáló kisebbagyagdiafragmát helyezünk bele. Sima, 5 x 5 cm felületű platinaelektródokat használunk. Szobahőfokon elektrolizálunk 0,02—0,03 amper/cm2 áramsűrűséggel. Azanódtérben levő erősen lúgos Na[Pb(OH)3]-oldatot elektrolízis közben erőteljesenkeverjük. A katódtérbe tömény lúgot teszünk. A Na2[Pb(OH)fl] fehér kristálypermet alakjában válik ki. Leülepítjük, a tiszta oldatot leszívatjuk, a kristálypépet
abszolút alkohollal leöntjük, és az egészet átöntve egy kisebb edénybe, addig dekantáljuk abszolút alkohollal, amíg a mosófolyadék már nem ad lúgos reakciót. A fehér kristályok vákuuínexszikkátorban történő szárításkor kissé megsárgulnak.
69.2.3. Nátrium-[tetrahidroxo-kuprát(II)] (Na2 [Cu(0H )J] )
Réz(II)-oxidból állítjuk elő:
CuO + HzO + 2 NaOH = Na2[Cu(OH)4].
500 g NaOH-ból és 330 ml HaO-ből készült karbonátmentes
lúgban 15 g tiszta CuO-ot visszafolyatásra állított hűtővel ellátott lombikban forralva oldunk. A 110 C°-ra lehűlt sötétkékoldatot a visszafolyós hűtőn át óvatosan 140 ml vízzel hígítjuk,és kb. 100 C°-on a fel nem oldódott CuO-ról dekantálva, elő-
69.2.3.1. ábra. melegített ezüstedénybe öntjük. Az edényt 50 %-os KOH-ot Péligot-cs6 tartalmazó Peligot-csővel (69.2.3.1. ábra) zárjuk, és elektromos
4 Orube, Q.: Z. Elektrochem. 28, 273 (1922); Simon, A.: Z. anorg. alig. Chem. 177, 109 (1929).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 295/366
K E L Á T K O M P I / E X E K 297
szárítószekrényben kb. 6 napon át 75 C°-on tartjuk. A sötét színű kristályokatC02 kizárása mellett nuccsoljuk. Kevés, 50-, majd 45 %-os, szobahőmérsékletűNaOH-dal mossuk, és agyaglemezen H2S04 felett szárítjuk. Kitermelés: 13 g.
69.3. Kelátkomplexek
69.3.1. [Tetraacetll-acetonát-cirkónium(IV) [Zr(C5H70 2)4]
5,8 g ZrOCl2*8 H20 50 ml-es vizes oldatát 15 C°-ra lehűtjük, és hozzáöntjük10 g acetil-aceton 50 ml 10 %-os Na2C03-ban keverés közben előállított és jégbehűtött oldatát. A képződött kristályokat kb. 1 óra múlva leszívatjuk, és hidegvízzel kimossuk. A nyersterméket, amely a komplex dekahidrát ja, 25 ml benzolbanoldjuk, és kiszűrjük belőle az oldhatatlan cirkónium-oxidot. A benzolos oldatból
petroléteres hígításkor a cirkónium-acetil-acetonát vízmentes alakban kristályosodik ki.5
69.3.2. [Triacetil-acetonát-króm (lll)] [Cr(C5H70 2)3]
Alkoholos oldatban a vízmentes Cr(N0 3)3 közvetlenül egyesül a hozzáadottacetil-acetonnal.
20 g Cr(N03)3-ot alkoholban oldva visszafolyós hűtővel ellátott csiszolatoslombikba öntünk, és háromszoros mólsúlynyi (15 g) acetil-acetonnal enyhén mele
gítjük. A kelátkomplex kialakulását az oldat intenzív ibolyaszínűvé válása jelzi. Az oldószert ledesztillálva, csillogó, vörösesibolya kristályok alakjában kapjuk akróm-acetil-acetonát komplexet. Tisztítás céljából benzol —kloroform-elegybőikristályosíthatjuk át, vagy vákuumban szublimáljuk. (Op. 216 C°, valamivelelőbb már szublimál.)
69.3.3. [Diglikokollát-réz(II)] [Cu(C2H40 2N)2]
Előállítása céljából réz(II)-hidroxidot glikokollban oldunk.12,5 g CuS04*5H20-t szükséges mennyiségű vízben oldva az oldathoz kis
feleslegben híg KOH-ot adunk. A levált Cu(OH)2-ot vízzel jól kimossuk, és 7,5 gglikokoll vizes oldatában melegen oldjuk. A [diglikokollát-réz(II)]-monohidrát kristályai kékesibolya színű tűk.
* Femélius, W. G.: Inorganio Syntlieses, Vol. II . New York— London, 1946. 121. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 296/366
70. Karbonilok és nitrozilok
A karbonilok klasszikus előállítási módja a CO és különböző fémek (Mo, Fe,Co, Ni) közvetlen reakcióján alapszik, ami azonban csak magasabb hőmérsékleten(100—200 C°) és általában nagy nyomáson (150—200 atm) megy végbe. A reakciókvégrehajtására csak különleges, szén-monoxidnak ellenálló anyaggal "(pl- Cu—Ag-Ötvözettel) bélelt autoklávök használhatók. Ezért a fent említett módszert a gyakorlatban igen ritkán alkalmazzák.
Az alábbiakban a karbonilok néhány olyan előállítási módját tárgyaljuk,amelyek a szokásos laboratóriumi felszerelés segítségével is végrehajthatók.
70.1 . [Tetrakarbonil-nikkel(O)] [Ni(C0)4]
Előállítására szén-monoxidnak és NiS04-ból, NH 3-ából, valamint Na2S20 4-bólkönnyen előállítható nikkel-szulfoxilát-ammóniakát |(NH 3)xNiS02|vizes oldatánakreakciója használható:1
NiS04+ Na2S20 4+ (s + 2) NH 3 + HaO = (NH 3)xNiS02 + Na2S04+ (NH4) j£ 0 39
(NH 3)xNiS02 + H20 + 4CO = [Ni(CO)4] + (NH4)2S0 3 + (x—2) NH3.
A reakció végrehajtására a 70.1.1. ábrán látható berendezést használjuk. Azüvegszűrős ún. intenzív mosó (A) felső nyílásába csapos rázóedényt (B) illesztünk,elvezetőcsövéhez pedig CaCl2-ot és P2Os-ot tartalmazó U-csövet (C), majd 3 dbkondenzáló szedőt csatlakoztatunk.
A mosópalackba 14,0 g NiS04*7 H20-nak 400 ml vízzel és 60 ml 25 %-osNH4OH-dal készült oldatát öntjük. A rázótölcsérből a levegőt kiszorítjuk, és 12,5 gNa2S20 4-ot teszünk bele, amelyet N2-ellenáramban hozzáöntött 30 ml cc. NH4OH-nak 80 ml vízzel készült oldatában rázogatással feloldunk. Az ammóniás ditionitottartalmazó csapos tölcsért gumidugóval a mosópalack felső nyílásába illesztjük.Ezután megindítjuk az ammóniás NiS04-oldatot tartalmazó mosópalackon át a
tiszta CO-gázáramot, és néhány perc múlva lassan ammóniás ditionitoldatot csepegtetünk bele. A képződő [Ni(CO)4] a száraz jéggel hűtött szedőkben kondenzál.
Az el nem reagált CO a berendezésből kiáramolva elégethető. A reakció végeztévelaz eredetileg kék színű nikkelsóoldat elszíntelenedik. A berendezésben a csatlakozások gumicsővel is megoldhatók, de ha tiszta anyagot kívánunk előállítani, akkor
1 Hieber, W., Fischer, E. O., Böckly E.: Z. anorg. alig. Chem. 269, 308 (1952).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 297/366
VAS-KARBONILOK 299
a nyersterméket CO-áramban üvegcsiszolattal csatlakozó edénybe desztilláljuk át. A kitermelés kb. 85 %-os.
Színtelen folyadék (op. —25 C°, fp. -1-40 C°). Levegőn hevesen oxidálódik,
gőze igen mérgező.
70.2. [Tetrakarboml-dijodid-vas(n)] ([Fe(C0)4I2])
Szintézis útján vízmentes Fel2-ból és CO-ból 6,3 atm nyomás felett közönségeslaboratóriumi autoklávban állítható elő.2 Abszolút éteres oldatban dolgozunk és1 g FeI2-hoz 50 ml étert használunk. Az autoklávot evakuálással és CO-átöblítésselelőzőleg légraentesftjük. A CO-ot közvetlenül bombából vezetjük az autoklávba.
A kitermelés majdnem elméleti, bár az előállítás napokig is eltarthat. A kapott
termék fényérzékeny.
70.3. [Pentakarbonil-vas(O)] ([Fe(C0)5])
A [Fe(CO)5] ipari termék, amelyet a klasszikus karbonilszintézis szerint CO
és finom eloszlású vas reakciójával állítanak elő. Preparatív munkákhoz a [Fe(CO)5]
általában kiindulási anyagként használatos. Szintézise a laboratóriumi normál
autoklávokban nem oldható meg, mert CO-nak ellenálló különleges bélést igényel
(pl. Cu —Ag-öt vözetet).
A [Fe(CO)5] laboratóriumi előállítására az alábbi reakció alkalmas:3
5 [Fe(CO)4I2] + 10 Cu = 10 Cul + 4 [Fe(CO)5] + Fe.
A [Fe(CO)4I2] és a finom eloszlású rézpor 1: 1 súlyarányú keverékét CO-áramban40 C°-ra melegítjük, majd a hőrúérsékletet lassan 55 C°-ra emeljük. —50 C°-os
szedőben a képződő [Fe(CO)5] felfogható. Kitermelés kb. 30 %-os. Ha a reakciót
* Hieber, W., Lagálly, H.: Z. anorg. alig. Chem. 245, 305 (1940). 8 Hieber, W., Lagálly, H.: Z. anorg. alig. Chem. 245, 295 (1940).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 298/366
300 K A B B O N I L O K É S N I T R Q Z I L O K
autoklávban 10 atm CQ-nyomáson hajtjuk végre, akkor a kitermelés gyakorlatilagkvantitatív.
A [Fe(CO)5] szobahőfokon sárga, olajszerű folyadék (op. —21 C°, fp. 4- 102,6 C°).Fényérzékeny, levegőn öngyulladó. (s20°= 1,46.)
70.4. Dihidrogén-[tetrakarbonil-ferrát(II)] (H2[Fe(C0)4])
A H2[Fe(CO)4] alkáli-, ammónium- és alkáliföldfémsóinak oldata a [Fe(CO)s]és a megfelelő hidroxidok oldatának vagy szuszpenziójának oxigénmentes térbentörténő rázásával állítható elő.4Sav hozzáadására a hidrid felszabadítható.
[[Fe(CO)s] + 3 NaOH = [Fe(CO)JHNa + Na2C0 3 + H20,
[Fe(CO)4]HNa -f H2S04 = H2[Fe(CO)4] + NaHS04.
A reakciót a [Ni(CO)4] előállítására használt berendezésben (70.1.1. ábra) hajt juk végre. A rázóedénybe evakuálás után 7 ml [Fe(CO)5]-t, majd 50 ml ki forraltvízben oldott 25 g NaOH-ot öntünk, és az edényt teljesen megtöltjük oxigénmentes nitrogénnel vagy szén-monoxiddal. 5 órás rázás után a H2[Fe(CO)4] nátriumsó
jának homogén, sárga színű oldatát kapjuk. Ebből a hidridet kénsavval tesszük
szabaddá. Az intenzív mosóba 50 ml 1: 1 hígítású H2S04-at öntünk. A rázótölcsért a70.1.1. ábrán látható módon felülről csatlakoztatjuk a mosóhoz, az egész berendezést levegőmentes CO-dal átöblítjük, és a [Fe(CO)4]HNa-oldatot cseppenként hozzáadagoljuk. A képződő karbonil-hidrid a CO-árammal a szárazjeges hűtőkeverékkel hűtött szedőkbe kerül és kondenzál. A kapott nyerstermék nagyvákuumbantörténő desztillálással tisztítható.
A H2[Fe(CO)4] a folyékony levegő hőmérsékletén színtelen, kristályos anyag(op. —70 C°). —10 C°-on a színtelen folyadék enyhe vörösödés közben bomlásnakindul, és magasabb hőmérsékleten teljesen elbomlik.
70.5. Hidrogén-[tetrakarbonil-kobaltát(-I)] (H[Co(CO)4])
Lúgos, kevés alkáli-cianid-tartalmú oldatban szuszpendált Co(OH)2 a szén--monoxidot megköti a H[Co(CO)J alkálisójának képződése mellett, amelyből a hidrid savval szabaddá tehető.5
2 Co (N 0 3)2 + 11 CO + 12 KOH = 2 K[Co(CO)J + 4 KN0 3 + 3 K 2C0 3 + 6 HaO,
K[Co (CO)4] + H2S04= H[Co (CO)4] + KHS04.
A reakciót a [Ni(CO)J előállításához használt berendezésben (70.1.1. ábra)hajthatjuk végre. Az evakuált rázóedénybe ( B ) egymás után 30 ml vízben oldott14,6 g Co (N 03)2«6 H20-t, 10 ml vizet, 22 ml vízben oldott 22,4 g KOH-ot, 10 mlvizet, majd 6 ml vízben oldott 3,2 g KCN-ot és végül újra 10 ml vizet folyatunk.
4 Hieber, W., Vetter, H.: Z. anorg. alig. Chem. 212, 146 (1933).5 Hieber, W ., Schulten, H.: Z. anorg. alig. Chem. 232, 29 (1937); Blanchard, A. A., Oilmont, P.: J. Am. Chem. Soc. 62, 1192 (1940).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 299/366
K O B A L T - K A K B O N I L O K 301
Ez után az egyik csapon át lúgos pirogallololdatot tartalmazó intenzív mosó közbeiktatásával 6 1 CO-ot tartalmazó gazométerrel kötjük össze, és rázógépben erőtel jesen rázatjuk. A CO abszorpciója az intenzív mosó segítségével észlelhető. A reakció folyamán először a sárga oldatban barna színű szuszpenzió képződik, ezután ez
további CO felvételével majdnem szuszpenziómentes K[Co(CO)4]-oldattá alakul. A hidridet kénsavval szabadítjuk fel. Az intenzív mosóba 60 ml 1 : 1 hígítású
H^SO^at teszünk, a rázótölcsért a 70.1.1. ábrán látható módon felülről csatlakoztatjuk a mosóhoz, és az egész berendezést levegőmentes CO-dal átöblítjük. A kiáramló CO-felesleget elégetjük. Miután az intenzív mosó tartalmát konyhasó—jég
hűtőkeverékkel —10 C° alá, a kondenzáló szedőket (szárazjég —aceton keverékkel)—80 C°-ra hűtöttük, az alkálikus karboniloldatot cseppenként a kénsavba enged
jük. A szabaddá váló hidrid a CO-árammal a szedőbe kerül. A K[Co(CO)4]-oldathozzáadásának befejeztével a CO bevezetését még kb. egy órán át folytatjuk,
ameddig a szedők előtti csapon át kiáramló CO-gáz meggyújtva már nem mutatvilágító, karakterisztikus „karbonil” -lángot. A hidridet végül —30 C°-on nagy vá
kuumban alacsonyabb hőmérsékletű szedőbe szublimáljuk át. A tiszta hidrid szilárd állapotban színtelen, kristályos anyag, amely —26,2
C°-on világossárga folyadékká olvad, és valamivel magasabb hőmérsékleten bomlás és [Co2(CO)8] képződése következtében narancssárga színűvé válik. A H[Co(CO)4gőzei rendkívül mérgezők.
70.6. Kobalt-karbonilok /([Co(CO)8], [Co(CO)3]4/
Az [óktakarbonil-dikobalt(O)] a H[Co(CO)4] termikus bomlásával képződik:6
2 H[Co(CO)4] = [Co2(CO)8] + H2 .
A 70.5. szerint frissen előállított és a 70.1.1. ábra alapján, (szárazjég — acetonos
hűtőkeverékkel) —80 C°-ra hűtött szedőbe szublimált H[Co(CO)4]-ot néhány óránát —30 (P-os hűtőben tartjuk, majd a hűtés megszüntetésével lassú CO-árambanforrni hagyjuk. (A H[Co(CO)4] forráspontja —26,2 C°.) A forráspontnál valamivelmagasabb hőfokon a bomlás is bekövetkezik. A maradék kristályos narancssárgaszínű [Co2(CO)8], vízben nem oldódik, alkoholban, éterben és más szerves oldószerben viszont igen. Levegőn ibolya színű bázisos kobalt-karbonáttá bomlik. Olvadáspontja 51 C°, 53C°-on inért gáz-atmoszférában is észrevehetően bomlik, és 60 C°-on
néhány nap alatt kvantitatíve trikarbonillá alakul át:
2 [Co2(CO)8] = [Co(CO)3]4+ 4 CO.
Az így képződő [Co(CO)3]4fekete, kristályos anyag, mely meleg benzolból átkristályosítva tisztítható.
8 Hiéber, W., Mühlbauer, F., Ehmann, E. A.: Bér. dtsch. Chem. Ges. 65, 1090 (1932),

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 300/366
302 KARBONILOK ÉS NITROZILOK
70.7. [Pentakarbonil-szulfid-dikobalt(I)] [Co2S(CO)5]
[Oktakarbonil-dikobalt(0)]-ból n-heptánban oldott kénnel állítható elő.7 A 70.7.1. ábrán vázolt mágneses keverővei, temperáló köpennyel vagy fürdővel
ellátott kb. 100 ml-es reaktoredényben ( A ) 0,10 g ként (sulphur praecipitatum)30 ml n-heptánban oldunk, majd a légteret CO-dal telítjük. A B jelű üvegkosárkába—levegő kizárása mellett — 0,7 g (3,13 mmól) kristályos [Co2(CO)8]-ot mérünk be,és a Ofjelű dugó hosszabbításán levő kampóra akasztjuk (manipulációs szekrényben).Ezt követően a berendezést újra átöblítjük szén-monoxiddal. A termosztálást 25C°-ra állítjuk be, és megindítjuk a mágneses keverőt. A hőmérséklet beállása után
70.7.1. ábra. [Co2S(CO)6) előállítása
a G dugó elforgatásával a [Co2(CO)8]-ot a kéntartalmú heptános oldatba juttatjuk.Gyors oldódás után már néhány perc múlva szén-monoxid-fejlődés közben megindul a fémfényű, fekete kristályok kiválása. Később finom kristályos csapadék válikki. Mintegy 3 óra alatt 9,8 mmól CO szabadul fel, vagyis 1 mól felhasznált [Co2(CO)8]-ra 3,12 mól. A kivált [Co2S(CO)5]-ot CO-atmoszférában szűrjük (célszerűen a48.1.2.4. ábrán vázolt megoldással), és n-heptánnal mossuk. Ezután 200 ml melegbenzollal oldjuk, és szűrés után 24 órán át állni hagyjuk az oldatot. A [Co2S(CO)5]fénylő fekete kristályok alakjában válik ki. Szobahőfokon levegőn is eltartható,vízben nem oldódik. A kitermelés kb. 30 %-os.
7 Markó,-L., Bor, Gy., Almásy, Q.: Chem. Bér. 94, 847 (1961).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 301/366
VAS-K AB BO N IL -N IT B O ZIL O K 303
70.8. [Dinitrozil-dikarboml-ferrát(O)] [Fe(N0)2(C0)2]
A nitrozil-karbonilok a megfelelő karbonil-hidrid alkálisóinak oldatábólekvivalens mennyiségű nitrit, majd sav hatására képződnek:8
[Fe(CO)4]HNa + 2 NaN02 + 3 CH3COOH =
= [Fe(NO)2(CO)2] -f- 2 CO + 3 CH3COONa + 2 H20.
A karbonil-hidrid alkálisójának előállítását a 70.4.-ben tárgyalt módon végezzük
el. A nátrium-nitrit-oldat beszívatás útján történő hozzáadását közvetlenül követősavas elbontást a karbonil-hidridek előállításával kapcsolatban leírt kiindulási
mennyiségek esetén 50 ml jégecettel végezzük. A nyerstermék tisztítását nagyvákuumban és alacsony hőfokon végzett desztillálással vagy szublimálással hajtjuk
végre. Ebben az esetben a terméket célszerűen leforrasztható szedőbe kondenzáljuk.
A [Fe(NO)2(CO)2] sötétvörös, kristályos anyag, op. 18,5 C°, bomlási hőmérsék
lete 50 C°. Levegőn gyorsan oxidálódik.
70.9. Dinátrium-[pentaciano-mtrozil-ferrát(III) ] -hidrát
(Na2 [ Fe(CN) 5N0 ] •H20 )
500 ml-es főzőpohárban 4 g elporított káhum-[hexacianoferrát(II)]-t /K 4[Fe(CN)6]*2 H20 / 60 ml desztillált vízben oldunk. Keverés közben az oldathoz64 ml 38 %-os salétromsavat adunk, és vízfürdőn digeráljuk a reakció befejezéséig,
amit oly módon ellenőrizhetünk, hogy a kivett próba egy cseppj éhez egy csepp
vas(II)-szulfát-oldatot adunk. Ha a csapadék már nem kék, hanem zöld, a folya
mat befejeződött. Néhány napos állás után állandó keverés közben Na2C0 3-oldat-tal óvatosan semlegesítünk: a lúgfelesleget gondosan kerüljük. A semlegesített olda
tot forrásig melegítjük, forrón szűrjük és gyorsan 40 —50 ml térfogatra bepároljuk.
Lehűlés után egyenlő térfogatú alkoholt adunk az oldathoz, amire a képződöttKN 0 3 nagyobb része kiválik (a nitroprusszid-nátrium alkoholban jól oldódik). Azoldatot újra bepároljuk. A sötétvörös oldatból állás közben kiválnak a rubinvörös
kristályok. Az anyalúg további bepárlásával újabb kristályfrakciókhoz jutunk. Akapott kristályfrakciókat egyesítjük, és kevés vízből átkristályosítjuk.
• Hieber, W., Anderson, J. St.: Z. anorg. alig. Chem. 208, 288 (1932); 211, 132 (1938).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 302/366
71. Izo-polisavak, heteropolisavak és sóik
A szabad izo-polisavak nem izolálhatók, mivel hidrogénion-feleBleg esetén folyamatos aggregáció következik be egészen a nagymolekulájú oldhatatlan oxid-hid-rátok képződéséig. Ez alól kivétel az ún. metavolfrámsav (Ha0*4 W 0 3*aq), amely
kémiai és krisztallográfiai szempontból a heteropolisavakhoz sorolható. Az izo-poli
savak sói bizonyos feltételek megtartása esetén kristályos állapotban előállíthatok. A szabad heteropolisa vak és azok sói egyaránt állandók, egyszerűen előállíthatók.
71.1. Nátrium-izo-polivanadátok
71.1.1. Nátrium-pirovanadát (2Na20 -V 20 c-aq) Az alábbi reakció alapján kaphatjuk:1
2 Na 3 V 0 4*aq -f 2 HC104= 2 Na20 ’ V 20 5-aq + 2 NaC104.
100 ml 1,1 mólos Na 3 V 04-oldathoz (amelyet úgy állítunk elő, hogy 10,0 g V 20 5-ot és 13,2 g karbonátmentes NaOH-ot 100 ml vízben feloldunk) cseppenként,keverés közben 24,9 ml 4,44 n HC104-at adunk. Ezután rövid ideig vízfürdőn melegítjük, amíg a narancsszínű folyadék el nem színtelenedik, majd vákuumban 30 C°alatt bepároljuk. A kiváló színtelen kristályokat leszűrjük és kevés vízzel mossuk.
71.1.2. Nátrium-metavanadót (Na20 -V 2Os aq)
Előállítható az alábbi reakció szerint :2
2Na 3 V 04-aq + 4 HC104= Naa0.V 20 6-aq + 4NaC104.
100 ml 0,812 mólos Na 3 V 04-oldathoz (literenként 73,9 g V 206és 90 g NaOH)cseppenként, keverés közben 57,5 ml 2,54 n HC104-at adunk, vízfürdőn az oldat el
színtelenedéséig melegítjük, majd 30 C°-on vákuumban bepároljuk. A színtelenkristályokat leszűrjük és kevés vízzel kimossuk.
71.1.3. A pentavanadinsav nátrium sója (3 Na20 -5 V20 6 aq)
A következő reakció szerint állítjuk elő :3
10 Na 3 V 04.aq + 24 HC104= 3 Na20-5 V 20 5.aq + 24 NaC104
1 2 3 Jander, O., Jahr, K. F . : Z. anorg. alig. Chem. 211, 53 (1933); Kolloid Beih. 41 35 (1935).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 303/366
I Z O - P O I íI M O L I B d A T O K — H E T E B O P O U S A V A X 305
150 ml 0,812 mólos NajVO*-oldathoz (lásd 71.1.2.) cseppenként, keverés közben 381 ml 0,8 n HC104-oldatot adunk, és kb. 14 napig zárt edényben állni hagyjuk.
A halvány narancsszínű oldatot 26—30 C° között vákuumban bepároljuk, a kiválónarancsszínű kristályokat kevés vízzel mossuk.ÖJu.
71.2. Izo-polimolibdátok
71.2.1. Nátrium-paramollbdát (6N a,0 *12M o03 aq)
8 g NaOH-ot tartalmazó vizes oldatban melegítés közben feloldunk 28,79 gMo03-ot, és bepárlás után'a terméket hidegen kristályosítjuk. Fényes prizmákban
kristályosodó anyag.
71.2.2. Nátrlum-metamolibdát (Na^O-áMoOs’ aq)
Az alábbi reakció útján állíthatjuk elő :4
4 Na2Mo04*aq + 6 HC1 = NaaO*4 Mo03*aq + 6 NaCl.
9,3 g NajjMoC^-ot 8 ml forró vízben oldunk, és a még meleg oldathoz bürettábólcseppenként 11 ml 5,5 n HCl-at adunk. Az átmenetileg képződő csapadék újra fel
oldódik, és az oldat sárgás színűvé válik. Bedugaszolt ^VZemreet/er-lombikban hideghelyen több napon át állni hagyjuk. Szűrés után a kristályokat hideg vízzel mossukés a levegőn szárítjuk. Kitermelés 6 g.
71.2.3. Ammónium-molibdát /6 (NH4)20 •12 M o03 •7 H20 /
Mo0 3 vizes —ammóniás oldatát 60 C° hőmérsékleten bepároljuk. Ezalatt az am-móniafelesleg elpárolog, és a fenti összetételű só kiválik. Kristályai nagy, színtelen,hexagonális prizmák. Vízben közepesen oldódik, főzésre hidrolizál.
71.3. Heteropolisavak és sóik
A heteropolisavak képzésére fémek és nemfémek gyenge oxisavai, pl. volfrám-sav, molibdénsav, vanádiumsav, illetve bórsav, foszforsav, kovasav, arzénsav stb.hajlamosak.
A szabad heteropolisavak előállítására Drechsél5.a következő módszert ajánlja: A heteropolisavak nátriumsójának a lehető legtöményebb, gyakran szirupsűrű
ségű oldatához választótölcsérben */3 térfogatnyi étert adunk. Erélyes összerázással
az oldatot éterrel telítjük, azután jéggel hűtött, vasmentes tömény sósavat vagy33 %-os fLjS04-at adunk hozzá kis részletekben, és minden részlet után jól összerázzuk. A folyadéknak eközben nem szabad felmelegednie, ezért ha szükséges, a választótölcsért hűtjük. A felszabadított sav az éterrel nyomban addíciós vegyidettéegyesül, és nehéz, olajszerű cseppek alakjában lesüllyed, megtisztulása után elkülö-
4 Wempe, O.: Z. anorg. alig. Chem. 78, 302 (1912).® Drechael, E.: Bér. dtsch. chem. Ges. 20, 1452 (1887).
20 Á lt . és szerv etle n k ém ia i p ra k t . — 428 4 /1 1 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 304/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 305/366
S Z I U K O - V O L F R A M Á T O K 307
próba szűrletébŐl híg sósav hatására már nem válik ki volfrámsav, a reakció lezajlott. Az Si02-feleslegtől szűréssel megtisztított oldatot éterrel és tömény sósavval
rázzuk ki. A további feldolgozás részleteit illetően lásd 71.3.Színtelen, 23 C°-on olvadó kristályok.
71.3.3. K&linm-sziliko-volframát (2 K20-S i0 a-12 W 03-aq)
A szabad sav vizes oldatához (egy súlyrész sav és három-négy súlyrész víz)számított mennyiségű szilárd K 2C03-ot adunk (2 mól K 2C03: 1 mól sav) és melegít
jük. A tiszta oldatot, amely még savanyú kémhatású, vízfürdőn kb. 1/3 térfogatrabepároljuk. Lehűtés után kiválik a káliumsó, amely meleg vízből átkristályosítható.7
7 Rosenheim, A., Jaenicke, J Z. anorg. alig. Chem. 101, 243 (1917).
20*

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 306/366
72 . Ritkaföldfémek és vegyületeik
72.1. Lantán (La)
A kompakt fém előállítása LaCl3-ot tartalmazó olvadék elektrolízisével történik.1 Kiindulási anyagként leggyakrabban La^Og áll rendelkezésre, amelyet szükséges mennyiségű tömény sósavban feloldunk, és kb. 1 y2-szeres mennyiségűNHdCl-ot adunk hozzá. Az oldatot erős keverés közben lehetőleg szárazra pároljuk,
lehűlés alatt a keverést folytatjuk, és ezzelaz anyagot elporítjuk. A kapott port azNH4C1 nagyobb részének eltávolítására platina tálban, keverés közben, lassan emelkedőhőmérsékleten erős ködképződésig hevítjük,majd exszikkátorban lehűtjük. A kapott
LaCl 3 osekély NH4Cl-tartalma hasznos, mertaz oxid-klorid képződését gátolja.
Az olvadékelektrolízishez használhatóberendezés a 72.1.1. ábrán látható. Elektromos fűtésű kemencébe magas, vastag falúgrafittégelyt helyezünk, amely az olvadékottartalmazza és egyben anódként szerepel.
Az áram hozzá vezetésére a tégely alsó részénlevő vasgyűrű szolgál. A katód 10 mm
0 -jű, 100 mm hosszú Mo-rúd, amely felülszinterkorund csőbe helyezett - vascsőhözvan erősítve. A katód forgatható, ami azolvadék keverését teszi lehetővé. Az áramhozzá vezetését szénkefe-kontaktus léteBÍti.
A leváló fémlantán összegyűjtésére a katódalá kisebb korundtégelyt helyezünk, amely egyben a grafiton anódosan fejlődőklórtól is megóvja a terméket. Üzembehelyezéskor a kemencét 1000 C°-ra fűtjük,és a tégelyt a következő összetételű sókeverékkel töltjük meg: 27,4 % LaCl3, 68 %KC1 és 4,6 % CaF2.
Célszerű először a LaCl 3 teljes mennyiségét kevés folyósitóanyaggal összekeverve betölteni, az NH4C1 esetleges elpárolgását megvárni és csak ezután adnihozzá a többi folyósítóanyagot. A katód 1—2 forgást végezzen másodpercenként.
A kísérlet befejezésekor az áramot megszakítjuk, néhány percig még keverünk, scsak azután vesszük ki a tégelyt. Jó áramkihasználás érhető el 25 amperórás
C_
s z én k e f e
ej ' / / + ~rowbo'—vascs
koründcs
Mo rúd gra f i t - tégely
korund- tégely
72.1.1. ábra. La nté n előállítása
1 Weibke, F., Sieber, </..* Z. Elektrochem. 45, 518 (1939).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 307/366
L A í í T Á N ' É S O É B I X m a Ó K 300
elektrolízissel 7 A/cm2áramsűrűsóg mellett, 1000 C°-on. 980 C°-on mér nem kapunkösszeolvadt regulust, hanem legfeljebb 5 mm
0-jű golyócskákat. 1000 C° felett vi
szont az áramkihasználés csökken. A kapott termék 99 %-os, fémfényű, kovácsolható. (Op. 885 C°.) Nedves levegő megtámadja.
72.2. Cérium (Ce)
A kompakt fém CeCl3, KC1 és NaCl-ból álló olvadék elektrolízisével állíthatóelő. Az elektrolízist a Muthmann-féle elektrolizáló edényben (54.3.2.1. ábra) hajtjukvégre. Az edény rézből készül, elektródként grafitot használunk. A tégelyben 200 g
CeCl 3 és 15—20 g 1: 1 mólarányú K.C1—Nad-o t tartalmazó sókeveréket olvasztunkmeg. 30—40 A-es, 12—16 V feszültségű árammal elektrolizélunk. Az elektrolízist700—750 C° között végezzük. Az olvadék megdermedése után a regulusokat kiszedjük, és KC1—NaCl-olvadék alatt korundtégelyben átolvasztjuk.
72.3. Lantán(IH)-klorid (LaCl3)
A vízmentes só LaCl 3«aq-ból vízmentesítéssel nyerhető úgy, hogy a víztartalmú anyaghoz 5—6-szoros mennyiségű NH4Cl-ot keverünk, és addig hevítjük,
amíg az NH4Cl-gőzök távozása meg nem szűnik.Előállíthatjuk La20 3-nak NH4Cl-dal történő klórozésával is. Az utóbbi eljá
rással az 56.3.2.-ben is foglalkoztunk.Nagyobb mennyiségű LaCl 3 előállítása céljából 60 g La203-ot és kétszeres
súlyú NH4Cl-ot olyan 250 ml-es kvarclombikba teszünk, amelynek a nyakáhozcsiszolattal 25 cm hosszú, 3 cm 0-jű kvarccső csatlakozik. Az edényt 250 CF-os lég-fürdŐn tartjuk. Kb. 8 óra elteltével az NH3-fejlődés megszűnik, a kvarccsövetcsiszolatos dugóval elzárjuk és hűlni hagyjuk.
A maradék NH4Cl-ot az edényből olajszivattyúhoz kapcsolva, 300—400 C°
közötti hőmérsékleten távolítjuk el.2
72.4. Lantán(III)-szuUát /La2(S 04)3/
La203-ot platina tégelyben tömény sósavban oldunk, és ezt az oldatot töménykénsavval 360 C°-on elfüstöljük. A képződött savanyú szulfátot 480 C°-on súlyállandóságig hevítjük, aminek hatására semleges só, La2(S04)3 képződik.3
72.5. Cérmm(IV)-oxid (Ce02)
A kereskedelmi készítmény tisztítására az alábbi módszer ajánlatos: 10súlyrész anyagot 7 súlyrész hidrokinont tartalmazó tömény sósavban forralás közben oldunk. A hidrokinon oxidációs termékeitől való elválasztásra a oériumot
* Brukl, A .: Angew. Chem. 52, 152 (1939).
3 BocJe, R.: Angew. Chem. 62, 375 (1950) és Z. anorg. alig. Chem. 263, 146 (1960).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 308/366
310 R íT K A F Ö L D F É M E K É S V E G Y Ü L E T E IK
oxálsavval kicsapjuk, majd az oxalátot tömény salétromsavban feloldjuk. Az oldatot ezután majdnem szárazra pároljuk, és a felhasznált CeOa-daI egyenlő súlyú
NH4N03-tal összekeverve, tömény salétromsavval többször bepároljuk. A narancsszínű (NH4)2Ce(N03)ö kettős nitrátot üvegszűrőn leszűrjük, tömény salétromsavbólátkristályosítjuk, és nitrogén-oxid-mentes 6 n HN03-ban oldjuk, végül peroxid-mentes éterrel kioldjuk. A salétromsavás oxidációt és az éteres kioldást a maradékkal megismételjük. Az éteres kivonatokat egyesítjük, víz hozzáadása után az étertledesztilláljuk és szűrjük. Az így kapott cérium(IV)-nitrát-oldatot kb. 2 n HN03-val megsavanyítva forró, tömény oxálsavoldatba csurgatjuk. Az aprókristályoscsapadékot kiszűrjük, sok vízzel mossuk, szárítjuk és oxiddá izzítjuk. A nitrátoldatának bepárlásával és termikus bontásával a Ce02 szintén előállítható.
72.6. Cérium(IY)-szulfát /Ce(S 04)2/
Előállítható Ce02 cc. kénsavas oldásával. A szükséges oxidot viszont az(NH4)2Ce(N0 3)6 termikus bontásával, izzításával készíthetjük el (lásd 72.5.). ACe02 tiszta állapotban sárgás színű anyag. Sötétebb árnyalata prazeodimszennye-zésre vall. Finoman elporítva cc. H2S04-ban szuszpendáljuk, majd melegítjük.Észrevehető oldódás nélkül néhány óra alatt sötétebb sárga, kristályos Ce(SÓ4)2-táalakul át. Kihűlés után a cc. kénsavat leöntjük róla, majd vízmentes jégecettel
többször dekantáljuk, végül üvegszűrore gyűjtve szulfátmentesre mossuk. KOHfelett vákuumexszikkátorban szárítjuk, (s =. 3,91.)
72.7. Cérium(III)-oxid (Ce20 3)
Korundcsónakba helyezett Ce02-ot oxigénmentes és szárított H2-nel redukálunk.4 A tiszta Ce02 lényegesen lassabban redukálódik, mint a kereskedelembenkapható termék. így pl. a 72.5.-ben leírt módon tisztított 3 g Ce02 redukciójához
1000 C°-on 80 óra szükséges, 1100 C°-on pedig kb. 45 óra. A redukció befejeződésétaz átmeneti termékek kékesfekete színének eltűnése jelzi. A Ce20 3 aranysárgaszínű, levegőn lassan oxidálódik, (s = 6,86.)
72.8. A ritkaföldfémek szétválasztása ioncserélő gyanta alkalmazásával5
Technikai Ce(N03)a híg HN03-as oldatához (125 g Ce(N03)a-ot 0,5 1 In HN03-ban oldunk) addig adagolunk forralás közben NaaCOa-oldatot, amíg az oldat
2—3 lesz. Ekkor a cériumtartalom egy része cérium(IV)-oxid-hidrát alakjában kicsapódik. Az elegyhez KMn04-oldatot adunk, míg az oldat maradandóan rózsaszínűvé nem válik. Ez után forralás közben KMn04- és Na2C03-tartalmú oldat(mólarány 1 : 4) adagolásával a pH-t 5—6-ra állítjuk be, majd 10 percig intenzívkeverés (keverőmotorral) közben forraljuk. Melegen szűrünk és meleg vízzel mos-
4 Bemer, G., Holtschmidt, U .: Z. anorg. alig. Chem. 265, 105 (1951). s N*achod, F . G., Schubert, J .: Ion Exchange Technology. N ew Yo rk, 1956 , 365. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 309/366
A R IT K A F Ö L D F É M E K S Z É T V Á L A S Z T Á S A IO N C S E R É L Ő V E L 311
suk a csapadékot, amely a technikai cériumsó teljes cériummennyiségét cérium(IV)--karbonátok alakjában tartalmazza. Az oldatban találhatók az egyéb ritkaföldfémionok (La3+, Pr3+, Nd3+, stb.), amelyek elválasztásához az oldat közvetlenül felhasználható.
Az elválasztás a 48.3. fejezetben tárgyalt elvek alapján történik.
250 ml kationcserélőt (0,2—0,4 mm 0 -jű Dowex-50 vagy Wofatit KPS-200)az előbbiek szerint előkészített ritkaföldfémion-tartalmú oldatban áztatva telítünk. Az így előkészített ioncserélőt 4 cm 0-jű, részben vízzel telt oszlopba töltjük(lásd 48.3.).
Utóbbihoz hasonló átmérőjű, 350 ml Zn2+-nal telített kationcserélőt tartalmazó oszlop csatlakozik.
Az eluáló oldat — amely mindkét egymást követő oszlopon folyamatosan átfolyik — 2 %-os nitrilo-triecetsav (NH3-val 7 pH-ra pufferőit) vizes oldata. Ha azeluálás sebessége nagyobb, mint 0,5 mól/sec, úgy csapadékkiválás az oszlopbannem lép fel. Az eluátumban a lantanidák a növekvő ionrádiusz sorrendjében jelent
keznek, és külön-külön felfoghatók. Az oszlopban visszamaradt La3+ 4 % nitrilo--triecetsavat és 2,4 % NH4Cl-ot tartalmazó (9 pH-jú) oldattal gyorsan eluálható. Az egyes frakciókból a ritkaföldfémek forrás közben oxálsavval csaphatók ki.
Az oldatot 20 percig 80 C°-on állni hagyjuk, majd szűrünk, és a csapadékot oxiddáizzítjuk. Az első frakciók kevés Zn2+-nal lehetnek szennyezve.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 310/366
73. Amalgámok
Alacsonyabb hőmérsékleten (1000 C° alatt) olvadó fémek higanyötvözeteinek
előállítása — ámennyiben egyáltalán amalgámokat képeznek — könnyen véghezvihető a komponenseknek zárt vastégelyben vagy üvegbombában történő egyesítésével. Egyes esetekben — pl. alkálifémamalgámok előállítása esetén — a szoba
hőfokon is túlságosan heves reakció sebességének csökkentését úgy biztosítjuk,
hogy az egyes komponenseket indifferens oldószerben szuszpendálva reagáltatjuk. A megfelelő fémsóoldatok higanykatódon történő redukciója is használható
módszer amalgámok előállítására.
A laboratóriumi gyakorlatban gyakran használatosak a nemnemes fémekfolyékony, illetve félig szilárd amalgámjai, főleg redukálószerként.
73.1. Nátriumamalgám
73.1.1. Folyékony amalgám, kb. 1% Na-tartalommal
250 ml-es í/rZenmeyer-lombikban 3,5 g nátriumot kb. 15 ml toluol-védőréteg
alatt főzőlapon melegítve megolvasztunk, és rázás közben cseppenként 25 ml
higanyt adunk hozzá. Az eleinte igen heves reakció később lelassul. A toluol közbenállandóan forr (visszafolyós hűtő). Befejezésül a toluolt dekantáljuk.
73.1.2. Szilárd amalgám, kb. 2— 3% Na-tartalommal
250 ml-es háromnyakú gömblombikba 6,9 g tiszta, feldarabolt nátriumot (2 %-
os amalgám esetén, vagy 10 g Na-ot 3 %-os amalgám előállításakor) teszünk. Alombik egyik nyakán az öblítő nitrogéngáz bevezetésére szolgáló cső, másik nyakán
a kivezetőcső és a középsőn a higany adagolására szolgáló csapos tölcsér van.
Az edényt nitrogénnel gondosan átöblítjük, és folyamatos nitrogénáramban
10 ml higanyt adunk a nátriumhoz, azután szabad lánggal óvatosan hevítjük a
reakció megindulásáig. Ha a reakció mérséklődött, lassanként hozzáadunk további
15 ml higanyt. A reakció végefelé időnként melegíteni kell az elegyet. A folyamat
befejeztével a megolvadt amalgámot tiszta lapra öntjük, s még meleg és törékeny
állapotban felaprítjuk.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 311/366
A L K Á L IF Ö L D F É M -A M A L Q Á M O K 313
73.2. Kalciuruamalgám
Csavarmenettel zárható, gáznyomással mozgatható D dugattyúval, Rossignol- szeleppel és manométerrel ellátott kis acélbombába (73.2.1. ábra), először 35 mlhiganyt és 7—10 g felaprított kalciumot teszünk. A dugattyú behelyezése utáp a fedőt rácsavarjuk, ésa szelepen át nitrogént vezetünk be, amíg a bombában el nem érjük a 60—70 atm nyomást. A megindulóreakciót, amely 16 perc alatt be is fejeződik, a bombaalsó részének melegedése jelzi.1 Lehűlés után a szelepet nyitjuk, a fedőt lecsavarjuk, ,és az amalgámot
gyorsan áttesszük egy száraz, jól záró üvegdugósporüvegbe.Higannyal hígítva az amalgám jelentős mér
tékben felmelegszik, összerázás után lehűtjük.
73.3. Báriumamalgám
nagynyomású N i palackból
73.2.1. ábra. Acé lbomba du
gattyúval , Rossignol-szelep- pel és manométerrel
250 [ml-es főzőpohárba 18 ml (250 g) higanytteszünk, amelyet katódul használunk. A higannyal
a kontaktust üvegcsőbe forrasztott platina dróttalhozzuk létre. Az anód vízszintesen elhelyezett, 5—10cm2 felületű platina lemez. A 100 ml telített BaCl2--oldatot 6—7 V feszültséggel, 1,75—2,5 A-es árammal 2—2,5 órán át elektrolizál-
juk. Elektrolízis közben a higany felületét keverjük. Végül az oldatot leöntjük,az amalgámot desztillált vízzel, majd alkohollal és éterrel mossuk. Levegőtől elzárva tartjuk el. Az amalgám kb. 3 % báriumot tartalmaz.2
1 Brukl, A .: Angew. Chem. 52, 151 (1939).
* Booth, H. S .: Inorganic Syntheses, Vo l . I . N ew York— London, 1939, 11. old.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 312/366
74. Fémorganikus vegyületek
A fémorganikus vegyületek — elméleti és preparatív szempontból egyaránt —átmenetet képeznek a szervetlen és a szerves vegyületek között. Nagy számuk és
igen különböző'tulajdonságaik következtében célszerűbb ezeket a szervetlen vegyületek között tárgyalni annak ellenére, hogy preparatív előállításukat illetőenegy részük a szerves vegyületek csoportjához áll közelebb. Ebben a fejezetben aszerves fémvegyületek nagy területéről csak az általánosan ismert, gyakorlati ésmódszertani szempontból fontosabb vegyületek előállítását tárgyaljuk.
74.1. Szerves lítium vegyületek
A szerves lítiumvegyületek előállítását kétféleképpen csoportosítják: egyrészt
a lítiumforrás, másrészt a kiindulásul felhasznált szénvegyületek szerint.1 A közvetlen eljárás fémlítiumból indul ki, a közvetett eljárás pedig lítiumvegyületekből. A kiindulásul alkalmazott szénvegyület lehet szerves halogenid (direkt eljárás),aktívabb hidrogént tartalmazó szénhidrogén (hidrogén—lítium-csere: Sorigin-reakció) vagy más szerves fémvegyület (fémorganikus szintézis), stb.
A szerves lítiumvegyületek rendkívül reakcióképesek. Előállításuk és reagál-tatásuk ezért nagy körültekintést és elővigyázatosságot igényel.
A szerves lítiumvegyületek igen érzékenyek a nedvességre és az aktív hidrogénre egyaránt. Ezért előállításuk aktív hidrogént (pl-, a —COOH, —OH stb.hidrogénje) és nedvességnyomokat nem tartalmazó vízmentes, inért oldószerbentörténik. Ilyen oldószer pl. a benzin, petroléter (alifás szénhidrogén), aromás szén-hidrogén vagy éter. Természetesen ezeket az oldószereket is pl. fémnátriumrólvaló desztillálással gondosan vízmentesítem kell.
Általában legaktívabbak a tercier szénatomra kapcsolódó lítiumot tartalmazóvegyületek. Aktivitásban utána a szekunder, majd a primer és végül az aromáslítiumvegyületek következnek.
A tercier és a szekunder szénatomra kapcsolódó lítium annyira aktív, hogymég az aromás benzollal is reakcióba léphet, különösen forraláskor. Ezért az ilyenlítiumvegyületek készítésére oldószerként csak alifás szénhidrogén (benzin, petrol
éter) alkalmazható. Ezzel szemben a primer szénatomra kapcsolódó lítiumot tartalmazó vegyületek éteres közegben is előállíthatók.Nedvességnyomokat a reagensedény légtere sem tartalmazhat, tehát feltétle
nül vízmentes atmoszférát kell biztosítani. A szerves lítiumvegyületek sokszor nem-
1 Kocséskov,K. A ., Talalajeva, T. V.: Szintetikus mód szerek a szerves L i, N a, K , R b és Cs- vegyületek köréből. Akad émiai Kia dó , B udapest, 1954.
Földesi, M TA Kém . Tud. Oszt. Közi . 25, 159 (196 6).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 313/366
S Z E R V E S I / Í T I U M V E Q Y Ü T iE T E K 316
csak a nedvességre érzékenyek, hanem a levegő oxigénjével is tűztünemény közbenreagálnak. Ezért indifferens ún. inért atmoszférára van szükség. Ilyen inért atmoszféraként gondosan oxigénmentesített nitrogén, nemesgázok (hélium, argon),etán, bután stb. alkalmas.
Az inért gázok vízmentesítését P20 5-dal vagy szilikagéllel végezhetjük. Álevegő (oxigén) beáramlását a készülékbe úgy akadályozhatjuk meg, hogy állandó,lassú inért gázáramban dolgozunk. A gáz beáramlását és távozását két paraffinolajos zár beiktatásával ellenőrizhetjük.
A szerves lítiumvegyületeket rendszerint további fémorganikus szintézisek átmeneti termékeként állít juk elő, s ilyenkor általában a lítium vegyületet ugyanazonedényben készítjük el, amelyben reagáltatásukat is végrehajtjuk.
Célszerűbb azonban olyan berendezéseket használni, melyek egyikrészében a szerves lítiumvegyület előállítását végezzük, másikrészében pedig azok reagáltatását hajtjuk végre. Az előállításhoz a legtöbb esetben a fémlítiumot feleslegben alkalmazzuk, ésez sok esetben zavarná a szerves lítium vegyülettel tervezett reakciót (pl. egyéb melléktermékek is keletkezhetnek). Ezért a szerveslítiumvegyülettől a feleslegben visszamaradt fémlítiumot el kellválasztanunk. Mivel a szerves lítiumvegyületek nagy részeszerves oldószerben jól oldódik, azok oldatából a lítium szűrésútján eltávolítható. Ilyen készüléket mutat be a 74.1.1. ábra.
Az A jelű lombikban készült szerves lítium vegyület oldatát— a berendezésnek 180°-kal,történő elfordítása után — a közbeiktatott szűrőn (G) a B jelű lombikba szűrjük át. A szerves lítium-vegyület reagáltatása ugyanebben a lombikban történik. Néhány szerves lítiumvegyület szerves oldószerekben kevésbé oldódik. Ezek a szerves oldószerben feliszapolhatók, és a, 74.1.1.ábrán bemutatott — ez esetben szűrőbetét (C) nélkül használt— berendezés A jelű lombikjában készült szerves lítiumvegyület feliszapolása után a feleslegben levő fémlítiumtól dekantá-
lással és a B jelű lombikba történő átöntéssel elválaszthatók. A fémlítiumot alkalmazhatjuk darabolt formában, reszelék vagy préselt drótalakjában. Amennyiben még aktívabb lítiumra van szükség, úgy viszonylag nagyobb diszperzitásfokú ún. lítiumhomokot készítünk.
A lítiumhomolc előállítása úgy történik, hogy a fémlítiumot paraffin alatt250 C°-on megolvasztjuk, és erőteljes keverés vagy rázás mellett lehűlni hagyjuk.Inért gázként argon, bután vagy etán alkalmazható, nitrogén azonban nem, miutánlítium-nitrid képződhet.
74.1.1. ábra. Szűrőbetótes
készülék szerves lítiumvegyületek
előállítására
74.1.1. Fenil-lítium (C6H«Li)
Közvetlen eljárással állíthatjuk elő:2
C0H5Br + 2 Li = C0H5Li + LiBr.
A 74.1.1.1. ábrán bemutatott készülék 250 ml-es lombikjába (A) bemérünk 60 mlvízmentes benzolt, majd hozzáadagolunk 3,3 g késsel meghámozott és felaprított
2 Gilman, H., Ingham, R. K., Schmidt, S. G.: J. org. chem. 18, 1743 (1953).
Földesi, 1., Gömöry, P .: A cta Chim . H u n g. 45, 231 (1965).
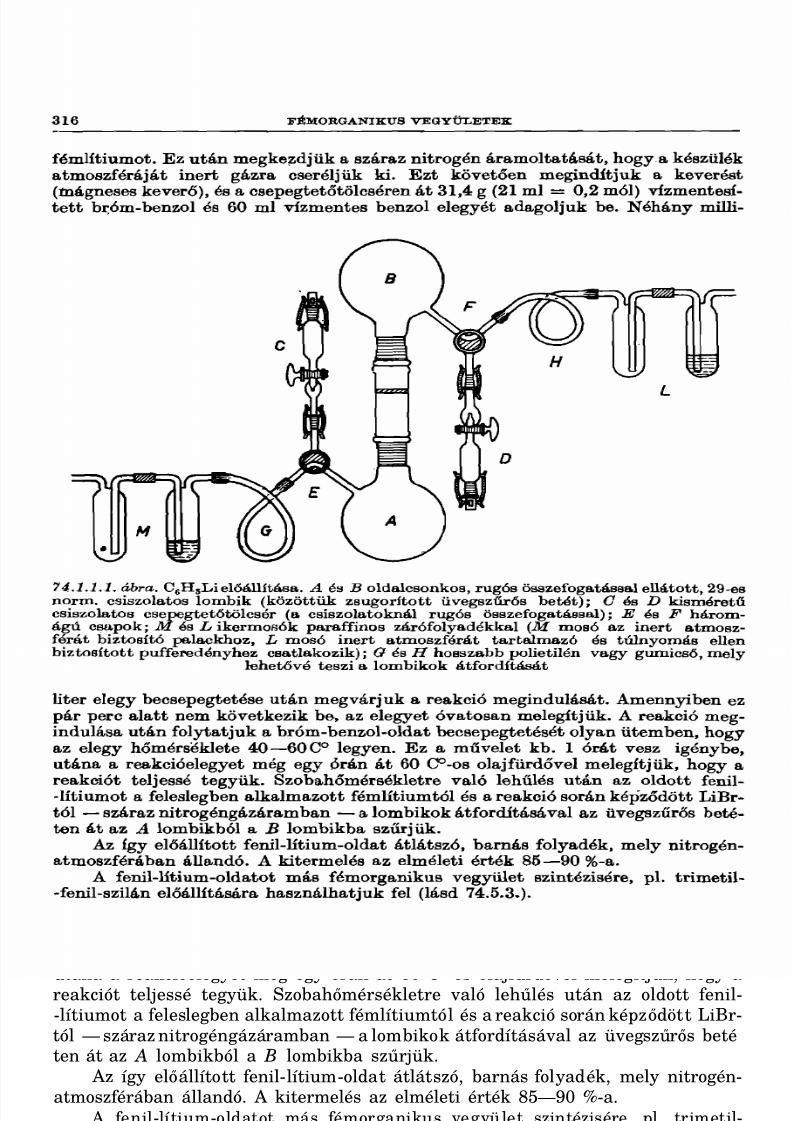
7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 314/366
316 F É M O R G A N I K U S V E G Y Ü X E T E K
fémlítiumot. Ez után megkezdjük a száraz nitrogén áramoltatását, hogy a készülékatmoszféráját inért gázra cseréljük ki. Ezt követően megindítjuk a keverést(mágneses keverő), és a csepegtetőtöleséren át 31,4 g (21 ml = 0,2 mól) vízmentesített bróm-benzol és 60 ml vízmentes benzol elegyét adagoljuk be. Néhány milli-
74.1.1.1. ábra. C6H 5L i előállítása. A és B oldalcsonkoa, rugós összefogatással ellátott, 29-es norm . csiszolatos lom bik (kö zöttü k zsugor ított üvegszűrős be tét ); <7 és D kisméretű
csiszolatos csepegtetŐtölcsór (a csiszolatoknál rugós összefogatésBal); E és F háromágú csapok; M és L ikerm osók p araffinos zárófolyad ékkal (M mosó az inért atmoszférát biztosító palackhoz, L mosó inért atmoszférát tartalm azó és túlnyomás ellen bizto sított pufferedényhez csa tlakozik ) ; G és H hosszabb polietilén vagy gum icső, m ely
lehetővé teszi a lombikok átfordítását
liter elegy becsepegtetése után megvárjuk a reakció megindulását. Amennyiben ezpár perc alatt nem következik be, az elegyet óvatosan melegítjük. A reakció megindulása után folytatjuk a bróm-benzol-oldat becsepegtetését olyan ütemben, hogyaz elegy hőmérséklete 40—60 C° legyen. Ez a művelet kb. 1 órát vesz igénybe,
utána a reakcióelegyet még egy órán át 60 C°-os olajfürdővel melegítjük, hogy areakciót teljessé tegyük. Szobahőmérsékletre való lehűlés után az oldott fenil--lítiumot a feleslegben alkalmazott fémlítiumtól és a reakció során képződött LiBr-tól — száraz nitrogéngázáramban — a lombikok átfordításával az üvegszűrős betéten át az A lombikból a B lombikba szűrjük.
Az így előállított fenil-lítium-oldat átlátszó, barnás folyadék, mely nitrogén-atmoszférában állandó. A kitermelés az elméleti érték 85—90 %-a.
A fenil-lítium-oldatot más fémorganikus vegyület szintézisére, pl. trimetil--fenil-szilán előállítására használhatjuk fel (lásd 74.5.30-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 315/366
S Z E R V E S H I G A Ü Y V E G Y Ü I jE T E K 3 17
74.2. Szerves higanyvegyületek
A szerves higanyvegyületek a legstabilisabb fémorganikus vegyületek közé tar
toznak; sem vízzel, sem oxigénnel szemben nem érzékenyek. Ezért előállításuksorán nincs szükség vízmentes oldószerek vagy inért atmoszféra alkalmazására.Mivel a szerves higanyvegyületek általában alacsony forráspontú és magas
gőztenziójú anyagok, előállításuk és alkalmazásuk higanymórgezés veszélyét rejtimagában. A szerves higanyvegyületekkel ezért igen nagy körültekintéssel és óvatossággal kell dolgozni. Egy részük fényérzékeny és higanykiválás közben bomlik,ezért tárolásukra célszerűen sötét üveget használunk
74.8.1. Fenil-higany(II)-acetát (CGH6HgOOCCH3)
Előállítható a benzol merkurálása útján:
CeHe + Hg(OOCCH 3)2 = C6H5HgOOCCH 3 + CH 3COOH.
VisBzafolyós hűtővel ellátott, 250 ml-es gömblombikban 80 ml (lehetőleg tio-fénmentes) benzolt, 15 g higany-acetátot és. 20 ml 95 %-os etilalkoholt adagolunkbe. Az azonnal kiváló sárga csapadékot néhány milliliter jégecet ho/záadásávaloldatba visszük, majd az elegyet 5 órán át forraljuk. Énnek hatására 50—70 %-banlejátszódik a reakció. Ha az 5 órás forralás után még 20 ml alkoholt adagolunk
az elegyhez, és további 40—50 órán át folytatjuk a forralást, úgy a reakció kb.80 %-ban végbemegy. Lehűlés után a reagálatlanul visszamaradt kevés (kb. 1 g)higany-acetátról a fenil-higany(II)-acetát oldatát leszűrjük, és a csapadékot kevésbenzollal mossuk. A szűrlet szárazra párolásával kinyerjük a fenil-higany(H)-ace-tátot, amelyet 95 %-os etilalkoholban átkristályosítva tisztíthatunk. (Op. 149 C°.)
74.8.2. Fenti-higany(Il)-klorld (C6H5HgCl)
aj A fenil-higany(II)-acetátot — ha. szükséges —könnyen fenil-higany(II)-klo-
riddá alakíthatjuk. A 74.2.1. szerint előállított benzolos oldatát negyedére párol juk, és a még meleg oldathoz CaCl2alkoholos oldatát keverjük fölös mennyiségben:
2 C6H5HgOOCCH 3 f CaCl2 = 2 C6H5HgCl + Ca(OOCCH3)2.
A fenil-higany(II)-klorid jól szűrhető csapadék alakjában válik ki. A szűréssorán a csapadékot meleg vízzel mossuk, és szívatás közben szárítjuk. Tisztításaalkoholból való átkristályosítással történhet. (Op. 252 C°.)
A szubsztituált benzolszármazékok merkurálása általában még könnyebbenelvégezhető.3 Ilyenkor — elsőosztályú szubsztituensek esetén — orto- és para-
sZubsztituált higanyvegyületek keletkeznek. Különösen könnyen merkurálhatóka fenolok4és az aromás aminok5.
8 Coffey, S .: J. Chem. Soo. 127, 1029 (1925).
* Whümore, F. G., Hamon, E. R.: Org. Synth. Coll. Vol. 1, 161 (1961). 6 Dimroth, 0 .: Bér. 35, 2032, 2035 (1903).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 316/366
318 F É M O R G A N IK U S V E G Y Ü L E T E K
b) Fenil-higany(II)-klorid előállítható diazotálással6:
C6H5- N H 2 C6H5—N2C1
C6Hs —N2C1 + HgCl2 = C6H5—N2C1. HgCl2,
C6H5- N 2d •HgCl2 + 2 Cu aceton , C6H5 —HgCl + 2 CuCl + N2.
500 ml-es főzőpohárban 18,6 g (0,2 mól) amiint 60 ml tömény sósavban— esetleg melegítés közben —oldunk. A lehűlt oldathoz 60 g darabos jeget adunk, és+ 5 C° alatti hőmérsékleten erélyes keverés közben 14 g (0,2 mól) NaN02 töményvizes oldatát csepegtetve hozzá, diazotálünk. A nátrium-nitrit-oldat adagolása kb.
20 percet vesz igénybe, és a reakció az adagolás után kb. 20 perc alatt befejeződik.Közben egy literes főzőpohárban 54 g (0,2 mól) higany(II)-kloridot 54 ml
tömény sósavban oldunk. Lehűlés után 60 g apróra darált jeget adunk hozzá,és mechanikus keverés mellett a fentiek szerint előállított diazotált oldatot egyszerre beleöntjük. Az elegyítés után közvetlenül kiválik a diazónium kettős só.20 perces keverés után nuccson szűrjük, vízzel, alkohollal és éterrel mossuk, majdszívatással szárítjuk. Kitermelés 80 —90%.
A kettős só bontásához cementált rézport készítünk: 25 g réz-szulfát (CuS04*•5 H20) 150 —200 ml-es vizes oldatához 6 g cinkport adunk, éskb. 20 percig rázogat-
juk. Az oldat lehűlése a cementálás végét jelzi. A cementált rezet liuccson szűrjük,vízzel, alkohollal majd éterrel mossuk, és szívatással szárítjuk. A megfelelően kiszárított réz világos színű.
Mechanikus keverővei és csepegtetőtölcsérrel ellátott 500 ml-es főzőpohárba80 ml acetont és 6 g cementált, kiszárított rézport teszünk. Keverés közben a szuszpenziót külső só—jég-hűtéssel —10 C° alá hűtjük, majd az előző módon elkészítettszáraz diazónium kettős sóból 20 g-ot részletekben, a —10 C° alatti hőmérsékletbetartása mellett, beadagolunk. További félórás keverés után a szuszpenziót desztilláló készülékbe öntjük, és lehajtjuk róla az acetont. A száraz maradékból Soxhlet- készülékben acetonnal extrahálva nyerjük ki a fenil-higany(II)-kloridot (az extra
háló aceton lehajtása után). A terméket acetonból vagy kloroformból átkristályosítva tisztíthatjuk. Kitermelés: 75 %-os. (Op. 258 C°.)
Amennyiben a diazónium kettős só bontását a rézpor nagy feleslege mellett ésammóniával történő forralás közben végezzük, úgy bisz(organo)-higany-származé-kot kapunk.7
74.2.3. Di(izo-propil)-higany(II), /[(C H 3)2CH]2H g /
Egyszerűbb alifás ketonok higanykatódos elektrolitikus redukciója megfelelő
körülmények között szerves higanyvegyületeket eredményez8:
2 £ > 0 - 0 + Hg + 6H+ + 6e- = ( ^ ^ C h W + 2 H ,0 .CH«' \CH*/ /
6 Nesmejanow, A . N .: Bér. 62, 1010 (1929).
7 Nesmejanov, A . N., Kohn E. D .: Org. Synth. Coll. Vol. 2, 381 (1950) « Tafel, J>: Bér. 39, 3626 (1906).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 317/366
S Z E R V ES H I G A N Y - É S B Ó R V E G Y ÍT L E T E K 319
Az elektrolízist a 74.2.3.1. ábra szerinti készülékben hajtjuk végre. A katódtérkb. 11,5 cm átmérőjű, 16 cm magas főzőpohár, melynek aljára 1 cm vastagságú ré
tegben higanyt öntünk. A higanyba üvegcsőbe forrasztott platina tűelektródot helyezünk. Az anódtér mintegy 7 cm átmérőjű, henger alakú porózus diafragma, amelyet megfelelő módon befogva a katódtér be függesztünk úgy, hogy a tégely alja ahigany felszínétől 2—2,5 cm-re legyen. A diafragmát célszerű paraffinnal átitatott fafedővel ellátni. Anódvezetékként a diafragma belse jébe ólomlemezt erősítünk. Az anódtér melegedését hűtőspirálisban cir
kuláltatott hűtővízzel mérsékelhet jük. Az anódfolyadék 200 g 20 %-oskénsav. A katódfolyadék 300 g30 %-os kénsav, melyhez 9,4 g (11,4ml = 0,166 mól) acetont keverünk. A katódtér hőmérsékletének csökkentésére 40 C°-os vízfürdőt alkalmazunk. A reakció optimális hőfoka45 —50 C°. Az optimális feszültségkülönbség 7,8—9,4 V. 10 A áram
erősség mellett az optimális reakcióidő 1 óra '20 perc, amely égyben a legjobb áramkihasználást (kb. 60 %) is jelenti. Más feszültség és áramerősség (I) mellett az elektrolízis idejét ki lehet számítani. (A szükséges idő: 96 500/2-7 mp.) Az áram bekapcsolása után néhány perccelborostyánkősárga cseppekben megindul a diizó-propil-higany kiválása. A keletkezett diizo-propil-higanyt választótölcsérrel elválasztjuk, a kitermelés 11—15 g. Azelektrolízist az elválasztott higannyal és új katódfolyadékkal megismételhetjük. Adiizo-propil-higanyt 2 g Na2S04-on szárítjuk, és szűrés után vákuumdesztillálássaltisztítjuk. Fp. 80 C°/16 Hgmm.
-'imrwTnmnnnnrff
h t m
/TlOt__ r __
edény
h t víz
SOX-asÁénsav* oceéon
higany
74.2.3.1. ábra. Di-(izo-propil)-higany(II) előállítása Hg-katódos elektrolízissel
74.3. Szerves bórvegyületek
A három vegyértékű bornak háromféle szeryes származéka ismeretes :9
KBXj B^BX RjB
(R alkil- vagy arilgyököt, X halogénatomot jelöl).
A szerves bórvegyületek igen hajlamosak elektrondonor-vegyületekkel valókomplexképzésre:
R3B + NR3 -> R3B - NR3,R3B + RNa -* [R4B]"Na+,
ami a szerves bórvegyületek számát nagymértékben megnöveli. A komplex szervesbórvegyületek sem a levegő oxigénjére, sem aktív hidrogénre, így nedvességre semérzékenyek, ezért ezekkel levegő jelenlétében vagy vizes oldatban is lehet dolgozni.
• Qerard, W.: The Organic Chemistry of Boron. Academyc Press, London, 1961.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 318/366
320 FÉMORGAJSUKTTS VE G YÜ LE TE K
A nemkomplex kötésű szerves bórvegyületek, különösen a trialkil-bór azonban,ba vízre nem is, de a levegő oxigénjére igen érzékeny, azzal gyakran robbanásszerű
hevességgel lángra lobban. Ezért előállításukhoz feltétlenül inért atmoszféra (Na)szükséges.
74.3.1. Trifenil-Mr/[B(C6H6)a] /
A bór^avészterek a GW^nard-reagenssel általában mono-organo-bórsav-származékot |[RB(OR2j||eredményeznek, abór-halogenidek azonban triorgano-bórt. Mivela bór-halogenidek általában alacsony forráspontúak (BF3 fp. —101 C°, BC13 fp.13 C°) és szobahőmérsékleten gázok, célszerű ezek helyett bór-trifluorid-éterátotalkalmazni.
Bór-trifluorid-dietiléter előállítása:
BF3 + OíCaHs), - BF3.(C2H5)20.
A szükséges bór-trifluoridot KBF4-ból nyerjük a következő reakció alapján:
6 KBF4 + Ba03 + 6 H2S04 = 8 BF3 + 6 KHS04 + 3 H20.
500 ml-es normálcsiszolatos gázfejlesztő lombikban 104 g (0,82 mól) KBF4-ot,16 g (0,23 mól) B20 3-ot és 240 ml tömény kénsavat elegyítünk. A bór-trifluoridotkénsavas mosón átvezetve 58 g (0,8 mól) fémnátriumról desztillált vízmentes éterben nyeletjük el. A bevezetőcső néhány milliméterre merüljön az éter felszíne alá,
és a visszaszívás elleni biztosítás cél jából a bevezetőcsövön néhánygolyós bővületet képezzünk ki(74.3.1.1. ábra). Melegítésre megindul a BF3 fejlődése {lásd 56.3.3). Az éternek bór-trifluoriddal valótelítődését a BF3-gáz távozása jelzi. A keletkező éterát-komplex stabi
lis és desztillálással tisztítható (fp.123-126 C°), a kitermelés közel kvantitatív. A Grignard-reakció:10
C6H5Br + Mg - CflH5MgBr,
3C flH5MgBr + BF3-
-* [(CflH5)]3B + 3MgBrF.
Ke verővel, gázbevezetőcsővel, cse
pegtetőtölcsérrel és visszafolyásraállított hűtővel ellátott 700 ml-esnógynyakú lombikban (74.3.1.2.ábra) 250 ml (fémnátriumról desz
tillált) vízmentes éterben 12 g (0,5 g-atom) magnézium forgácsot és pár szemjódotszuszpendálunk, majd 78 g (0,5 mól) bróm-benzol kis részletének hozzáadásával
10 Krause, E., Nitache, R.: Bér. 54, 2784 (1921).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 319/366
T R I F E N I L - B Ó R 321
74.3.1.2. ábra. Trifenil-bórelőállításaTJWymzrci-reakeióval
<(esetleg melegítéssel) megindítjuk a Qrignard-reagens képződését. Ezután az éterenyhe forrása mellett kb 1 óra alatt becsepegtetjük a maradék bróm-benzolt.
A beadagolás befejeztével az moior■ oldatot még fél érán át forral- ^ CJ3 juk, hogy a Qrignard-reagensképződését teljessé tegyük.Ezt követően az oldathoz hűtés közben 17,7 g (0,125 mól)bór-trifluorid-éterátot csepegtetünk. A reakcióhő hatásáraaz éter felforr, ezért az éterátbecsepegtetését az éter forrásának ütemében folytatjuk,majd befejezésül még egy óránát forralunk visszafolyatásmellett, majd áramló nitrogénben átszereljük a készülé-ket (74.3.1.3. ábra), és az étertvízfürdőn lehajtjuk. Az utóbbi befejeztével — nitrogén-áramban—szedőlombikot cserélünk, és a csepegtetőtölcsér helyére kapillárist helye
zünk; a N2-bevezetőcső helyére dugót iktatunk, majd a nitrogént a kapillárishozcsatlakoztatva a készüléketvákuumra kapcsoljuk. A tri-fenil-bórt 13-15 Hgmm-esvákuumban 245—250 C°-osolajfürdő alkalmazásával akb. 250 ml-es szedőbe desztilláljuk. A szedőt kormozó lánggal enyhén melegítjük, hogy atrifenil-bór megdermedését
megakadályozzuk. A desztil-láció befejeztével a trifenil--bór a szedőben hamarosanmegdermed. Op. 136 C°, fp.203 C° (15 Hgmm). A kitermelés kb. 50%-os. A készüléketnitrogénáramban szedj ük szét.
A szedő lekapcsolása és bedu-gaszolása után a reaktorlom
bik tartalmát alkohollal, majdvízzel, végül 10%-os sósavval
'74.3.1.3. ábra. Oldószer lehajlására és átszereléssel kódju k.vákuumdesztülációra alkalmas készülék Ügyeljünk a trifeml-bort
tartalmazó szedőre, nehogy alevegő oxigénje érje. Nitrogén alatt eltartható. A trifenil-bór éterben gyengén,benzolban és toluolban jól oldódik. Alkohollal borát keletkezése közben reagál.
21 Alt. és «2ervetlen kémiai prakt. —4284/11.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 320/366
322 F É M O B G A N I K U S V E G Y Ü X E T E K
74.3.2. Nátrium-[triíenil-ciano-borát(III)] /Na[(C6H5)3CNB] /
A szedőben N2-atmoszférában felfogott trifenil-bórhoz számított NaCN-tartalmú (kb. 5 %-os)_, híg vizes oldatot adva a nátrium- [trifenil-ciano-borát] vizes oldatakeletkezik, amely Kasignost -Te&gvnskéTi t ismeretes, és alkálifémionok, aminok ésaminszármazékok analitikai kimutatására alkalmas.11
74.3.3. Metil-bór-diklorid (CH3BC12)
Metil-alumínium-dikloridból állítható elő az alábbi reakció alapján12:
CH3A1C12 + BF3 CH3BC12 + A1F3.
A reakció gázfázisban folyik le, és az A1F3 csekély tenziója következtében a reakció a felső nyíl irányában teljessé válik. A (CH3A1C12)2 helyett ekvivalens mennyiségű (CH j ^AL jC^ és A1C13 keverékét használjuk.
ő4
74.3.3.1. ábra. CH3BC12 előállítása
A reakció végrehajtására a 74.3.3.1. ábrán bemutatott készüléket szereljükössze. Az egyenként 500 ml-es gázfejlesztő készülékhez (Ax és A2) kénsavas mosó(B), majd a 2 1-es, kettős tekéjű reaktorlombik (C) csatlakozik. Utóbbihoz mélyhűtött szedőt (D), pufferedényt (E), majd a légnedvesség visszatartására P20 5-dal töltött szárít ótornyot (F) kapcsolunk. A reaktoredénybe bemérünk 79 g (0,6mól) szublimált AlCl3-ot, majd a légteret száraz nitrogéngázzal kicserélve hozzányomatunk 121 g (0,6 mól) trimetil-alumínium-szeszkvikloridot /(CH3)3A12C13/.
A gázfejlesztő készüléket BÍT3 előállítására használjuk (részletesebben lásd 56.3.3). A BF3-fejlődés melegítésre indul meg. A reakció beindításakor a reaktorlombiktartalmát kezdődő forrásig melegítjük (óvatosan, homokfürdő alkalmazásával), további melegítésre azonban a reakció exoterm volta miatt nincsen szükség. A reakcióidőtartama kb 4 ó ra. A kitermelés BF3-ra vonatkoztatva kb. 90 %.
11 Wittig, O., Buff, B.: LiebigB Ann. 573, 195 (1951).12 Lengyel, B., Csdkvári, B,: Z. anorg. alig, Chem. 322, 103 (1963).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 321/366
SZEBVES AIiUMÍNIUMVEGYÜI/ETEK 323
A nyerstermék nitrogénatmoszférában végzett desztillálással tisztítható, és nit-rogénatmoszférájú ampullában leforrasztva hidegen eltartható. Színtelen folyadék.
(Fp. 11,5 C°.)74.4. Szerves alumíniumvegyűletek
A szerves alumíniumvegyűletek a levegő' oxigénjének jelenlétében öngyulladók.Különösen nagy az öngyulladás veszélye a trimetil-alumínium esetében. A széniánohosszának növelése vagy a szerves csoportok egy részének helyettesítése halogénnel némileg csökkenti az öngyulladásra való hajlamot, de nem szünteti meg azt. Amíg az A1—C-kötés fennáll, az öngyulladó jelleg is megmarad. A nedvességnyomok katalizálják az öngyulladást. Víz vagy légnedvesség hatására a szerves alumí-niumvegyületek gyorsan hidrolizálnak. Vízbe öntve robbanásszerűen bomlanak,ezért a velük való műveleteket vízgőzmentes, inért atmoszférában kell végezni.Legalkalmasabb erre a célra a száraz és oxigénmentes nitrogén vagy argon.
A nagyfokú tűzveszély miatt fokozott óvatosságra van szükség. Alumínium-organikus vegyületekkel végzett reakciók vagy azok előállítása során a készüléketfelügyelet nélkül működtetni nem szabad! A szerves alumíniumvegyelületek koncentrálódási helyét, pl. a szedőlombikot vastepsiben, száraz homokba célszerű állítani. A felhasználásig történő tárolás laza üvegdugóval bédugott (nem légmentesenlezárt), száraz homokba ágyazott lombikban történhet. A munka során különös
gondot kell fordítani a visszaszívódás megakadályozására. így pl. a tömény kénsavas mosókat fordított állású üres gázmosóval biztosítjuk. Ha valamilyen okbóla már elindított reakciót le kívánjuk állítani, akkor várjuk meg, míg a készülékN2-áramban teljesen lehűlt. Szerves alumíniumvegyületeket oldószer nélkül is előlehet állítani. Szükség esetén természetesen vízmentes és reaktív csoportot nem tartalmazó oldószert kell alkalmaznunk. Az oldószer olefinkötést sem tartalmazhat,mivel szerves alumíniumvegyűletek jelenlétében ezek polimerizálódhatnak.
74.4.1. Metil-alumínium-kloridok /(CH3A1C12)2és (CH3)2(A1C1)2/
Az R 3 AIX 3 típusú vegyületek előállításának legrégebbi és az alábbiakban részletesen leírt módja — amelyet ma is a legkönnyebben kivitelezhetőnek tartanak —a következő egyenlettel fejezhető ki:
6 RX + 4 A1 = (R2 A1X )2 + (RA1X 2)2
(R alkil- vagy arilgyök, X valamelyik halogén).Meg kell jegyeznünk, hogy a reakció során ekvivalens mennyiségben keletkező
mono- és dialkil-alumínium-kloridok dimer vegyületet, ún. metil-alumínium-szeszkvikloridot képeznek: (CH 3) 3 Al2Cl 3.
Hasonlóan a Orignard- vegyületek előállításához, Iegkönyebben jodidokkal, legnehezebben kloridokkal megy végbe a reakció. Arra az esetre, ha R X gázhatalmazállapotú, egyes szerzők autokláv használatát ajánlják, azonban az átalakulás akkoris bekövetkezik, ha megfelelő hőfokon tartott alumínium forgács felett vezetjük elaz RX-gázt. A reakciót meti-kloriddal végezve az optimális hőmérséklet 150 C°.Magasabb hőmérséklet a (CH 3 AICI2)2képződésének kedvez, sőt kátrányosodás is bekövetkezhet.
21

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 322/366
32 4 FÉMOÍttiANIKUS VEGYÜLETEK
Tiszta alumínium helyett célszerű réztartalmú Al-ötvözet alkalmazása. Azalábbi művelethez a kereskedelmi, 5 % rezet tartalmazó Al-ötvözetet és a kereske
delmi metil-kloridot használjuk. Az előállításhoz alkalmas készülék vázlata a74.4.1.1. ábrán látható. A reaktor kb. 150 cm hosszú, 40 mm átmérőjű üvegcső.Ha a lehajló rész függőleges helyzetben van, az üvegcső lejtése kb. 10°. A reak
torhoz csiszolattal vagy gumidugóval 11-es szedőlombik csatlakozik. A gömblombikon oldalt 4 cm hosszú kivezetőcső van a szárítócsapdák csatlakoztatására és a gázfelesleg elvezetésére. A reaktort elektromos fűtőtest, azbeszt szigetelés veszi körül, mely
nek segítségével a hőmérséklet 150 C°-ig emelhető. A fűtőtest a reaktor felső háromnegyedét borítja, alsó egynegyed része a léghűtés biztosítására szabadon van. A reaktorban a gázt gumidugóba illesztett üvegcsövön keresztül vezetjük be. Előtte aCH 3Cl-ot tömény H2S04-val szárítjuk. A szedőlombik kivezetőcsövéhez ugyancsakkénsavas mosópalackok csatlakoznak (biztosítás fordított mosópalackkal). A gázbevezetőcső előtti mosók és a CH3C1-os palack közé higanyos manométert kapcsolunk.
Az alumíniumot finoman forgácsolva szorosan a fűtőtesttel körülvett reaktor-részbe töltjük. A cső elején közvetlenül az Al-forgácsra kb. 5 g vízmentes AlCl3-portszórunk, amely katalizálja a reakció megindulását. Kedvező esetben a reakció2—3 óra múlva megindul, amit a manométer nyomáscsökkenése mutat. Ezután agázáramot úgy állítjuk be, hogy a készülék semmi esetre se szívjon vissza, és minélkevesebb gáz távozzék. A távozó gázokat jól húzó kéménybe vezetjük. Az egész folyamat alatt ügyeljünk a gáz kezelésére, mert a metil-klorid erősen mérgező anyag! A műveletet száraz nitrogénatmoszférában kell végezni. A szerves alumíniumve-gyületet a levegő oxigéntartalmától és a nedvességtől egyaránt védeni kell. A szedőlombikot vastepsiben levő homokfürdőbe állítjuk. A termék nagyfokú tűzveszélyessége miatt körültekintő óvatosságra van szükség. A készülék felügyelet nélkül nem
működtethető. Leálláskor a készüléket minden esetben N2-áramban teljesen lehűlni hagyjuk. Az alumíniumorganikus vegyület magas forráspontú 70—100 C° 10 Hgmm-
en), tiszta állapotban színtelen anyag, amely a léghűtéses csőszakaszban csepp-folyósodik és a szedőlombikba csepeg. Amennyiben az alumínium elég tiszta és ahőmérséklet a 110 C°-ot nem haladja meg, akkor csak cseppfolyós termék keletkezik. Ha a hőmérséklet túl magas, a termék részben kátrányosodik, részben kristályos AlCl 3-dá, illetve (CH 3 AlCl2)2-dá alakul, amely a reaktort el is tömheti. Ha a tér-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 323/366
S Z E R V E S A L U M Í N I U M - É S S Z I L ÍC I U M V E G Y Ü L E T E K 325
mék narancssárga színű folyadék, akkor az gyakorlatilag (CH 3) 3 Al Cl3-nak tekinthető, mely a mono- és dimetil-alumínium-kloridok dimer vegyülete.
Az előállított alumíniumorganikus vegyületet pl. szilánszintézisre használhat juk fel.
74.4.2. Di(izo-propoxi)-alumínium-klorid
Az Al-O-C kötésű vegyületek levegő oxigénjére ugyan nem érzékenyek, de vízgőz, illetve víz hatására hidrolizálnak. Ezért ezek előállítása során inért gáz nemszükséges, de a levegő nedvességétől a reaktort és a terméket óvni kell.
Csepegtetőtölcsérrel, keverővei és visszafolyatásra állított hűtővel felszerelt
2 literes háromnyakú gömblombikba 27 g^l g-atomsúly) száraz és nem oxidálódott Al-fóliát mérüpk be, majd 350 ml vízmentes (Na-ról desztillált) izo-propil-alkoholtöntünk rá. Ez után hozzáadunk 1 g HgCl2-ot. A hűtő felső végét a levegő nedvességétől CaCl2-os csővel óvjuk meg. Zárt elektromos főzőlapon az izo-propil-alkoholtforrásig melegítjük, majd a csepegtetőtölcséren át 85 g (0,55 mól) vízmentesített(előpárlat vétele mellett ledesztillált) szén-tetrakloridot csepegtetünk hozzá. A reakció megindulásakor a melegítést megszüntetjük, és az esetleg túlhevessé váló reakció szükség szerinti mérséklésére jeges vizes fürdőt helyezünk készenlétbe. A reakció megindulása után az elegyet a szén-tetraklorid megfelelő ütemben történőadagolásával tartjuk forrásban. A beadagolás ideje kb. 1 óra. Az elegyet utána még
áz A1 eltűnéséig melegítjük (kb. 1 —2 órán át). Az elegy színe szürkésfehér. Rövidállás után a főleg higanytartalmú fekete csapadék leülepszik; a tiszta, átlátszó oldatot desztilláló lombikba dekantáljuk, majd lehajtunk róla kb. 200 ml benzolt.
A visszamaradt oldatból már melegen kezd kiválni a fehér kristályos di-izo-propo-xi-alumínium-klorid, melyet az oldat lehűlése után száraz üvegszűrőn leszívatunkés kevés száraz izo-propil-alkohollal, majd száraz petroléterrel mosunk. A szárításkénsavat és paraffint tartalmazó vákuumexszikkátorban történik. A kitermelés72—75 %-os.13
74.5. Szerves szilíciumvegyületek
A szén —szilícium-kötés sem oxigénre, sem nedvességre nem különösebben érzékeny. Annál érzékenyebb nedvességre a szilícium —halogén-kötés.
Az alkilezettségi fokot tekintve a következő szerves szilíciumvegyületek készíthetők :
R4Si R 3SíC1 R2SíC12 RS íC13.
Ezek sorrendben non-, mono-, di- és trifunkciós klórszilánok. Rövid jelölésük N, M, D, illetve T (a SiCl4-ot röviden Q-val szokás jelölni). Hidrolizálva a mo-
noklór-szilán disziloxánná, a diklór-szilán láncpolimerré és a triklór-szilán térhálóspolimerré alakul.
A szerves szilíciumvegyületek előállításában egyaránt fontos szerepe van a di-rekt szintézisnek és a fémorganikus szintézisnek. A direkt szintézis a metil-klór-szi-lánok előállítása során főtermékként dimetil-diklór-szilánt, melléktermékként me-
18 Földest, I .: Acta. Chim. Hung. 37, 329 (1963).Gál, Qy., Tokár, G., Simonyi,!.: Magy. Kém. Foly. 61, 268 (1955).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 324/366
326 FÉMORQANIKUH VEOYÜLETEK
til-triklór-szilánt, továbbá trimetil-klór-szilánt eredményez. A közel álló forráspontok miatt (74.6.1. táblázat) a metil-klór-szilánokelegye csak kolonnás desztillálóval
választható szét. Nagy problémát okoz azonban a direkt szintézis, valamint a SiCl4Grignard-reagenssel való alkilezése során a reakció közben képződő vagy változatlanul visszamaradt SiCl4-nak a trimetil-klór-szilántól való szétválasztása, mivelutóbbiak azeotrop elegyet képeznek.
74.5.1. táblázat
A metil-klór-szilánok fizikai állandói
0 p .
c°F p . / 7 6 0 H g m m
C°
Sűrűség,Sjo
T ö ré s m u t at ó ,n ÍO
(CH3)4Si íoc= 102 , 12 26 0,6464 1,3648 (10 C°)1/3=101,7 i »
(CH3)3SiCl - 5 6 57,3 0,8581 1,3884(CH.bSiCla -7 6 ,5 70 1,06CHgSíC^ -7 7 ,5 66,2
A metil-klór-szilán-veszteséget okozó azeotropképződés elkerülhető a szervesalumíniumvegyülettel történő alkilezési eljárással14, mely főleg dimetil-diklór-szi-lánt eredményez.
2 (CH 3) 3 A12C1 3 + 3 SiCl4-v 3 (CH 3)2SiCl2 + 2 A12C16.
A reakció azonban nem ilyen egyszerű, mivel a szerves alumíniumvegyület amár alkilezett szilícium-halogenidet könnyebben — kisebb energiabefektetéssel —alkilezi tovább. Ezért először tetrametil-szilán keletkezik?
4 (CH 3) 3 A12C1 3 + 3 SiCl4 = 3 (CH3)4Si + 4 A12C16.
A reakció során keletkező alumínium-klorid azonban katalizálja a (CH3)4Si ésa SiCl4 közötti egyensúlyi reakciót, melynek eredménye a dimetil-diklór-szilánképződése:
(CH3)4Si + SiCl4
^ 2 (CH 3
)2SiCl2.
Az eljárással kapott termék SiCl4-ot és (CH 3) 3SiCl-ot egyidejűleg nem tartalmaz, és így az azeotróp képződése ki van zárva.
A desztillációs elválasztással járó nehézséget kiküszöbölhetjük, ha tetraetoxi--szilánt (xrogríiard-reagenssel alkilezünk :
Si(OC2H5)4+■ 2 CH3MgX - (CH 3)2Si(OC2H5)2 + MgX 2 + Mg(OC2H5)2.
Természetesen a dimetil-dietoxi-szilán mellett más alkilezettségi fokú etoxi--szilán-származékok is keletkeznek. Ezek azonban számottevően eltérő forráspont
juk (lásd 74.5.2. táblázat) következtében desztillációval jól szétfrakcionálhatók.Utóbbi eljárás előnyös azért is, mert lehetővé teszi az üzemi szempontból nemkívánatos éter mellőzését, vagyis az oldószer nélküli Grignard-reakció megvalósítását.15 Természetesen víz hatására az alkil-etoxi-szilánok és a tetra-etoxi-szilánetoxicsoportja is lehidrolizál, ezért az etoxi-szilánok előállítását is vízmenteskörülmények között kell végezni.
14 Lengyel, B., Székely, T.: Magy. Tud. Akad. Kém. Oszt. Közi. 8, 427 (1957). 18 Nagy, J.: Periodica Polytech. 2, 241 (1958).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 325/366
S Z E R V E S S Z I L Í C I U M V E G Y Ü L E T E K 327
74.5.2. táblázat
A metil-etoxi-szilánok fizikai állandói
F p . / 7 6 0 H g m m
C°
Sűrűség ,
S8JT ö ré s m u t at ó ,
n í0
(CH3)3SiOC2H 5 74 0,7573 1,3443(CH3)2Si(OC2H 5)2 113 0,8401 1,3814CH3Si(OC2H 5)3 141 0,8947 1,3831Si(ÓC2H 5)4 160,5 0,9346 1,3830
A szilícium-halogenid alkilezése elvégezhető más fémorganikus vegyület alkalmazásával is. Felhasználhatjuk pl. a 74.1. szerint előállított szerves lítium vegyüle-tet is alkilezőszer gyanánt:
(CH3)3SiCl + LiR = (CH3)3SiR + LiCl.
Tetraalkil- vagy aril-szubsztituált szilánok előállítására a Wurtz-reakció is alkalmazható, melyre a következőkben szintén találunk példát.
A szilícium —halogén (és természetesen a szilícium—alkoholát)-kötés, mintmár említettük, víz hatására könnyen hidrolizál:
^Si—C l -h H — 0 — H - ^>Si— OH + HC1.
A kialakuló -^Si—OH kötésből savas vagy lúgos közegben vízvesztéssel szil-
oxánkötés keletkezik:
4 S i— OH -(- HO—Si— -> ^ S i—0 —Si^ + HoO.
Monofunkciós szilánból disziloxán, difunkciós vegyületből láncpolimer (esetenként 3 vagy 4 szilíciumatomot tartalmazó ciklo-trí-, illetve -tetrasziloxán, rövid
jelöléssel D3, ill. D4) jön létre, trifunkciós vegyületből pedig térhálós polimer keletkezik. A kondenzációt már az Si —Cl-kötés hidrolízise során keletkező HC1 is előtudja idézni, sőt hő hatására, azaz melegítéskor is bekövetkezik. Ezért, ha hid-roxidkötést is tartalmazó vegyületet kívánunk előállítani, úgy ezt csak hűtésmellett és semleges kémhatású oldatokban (esetleg pufferolással) lehet végrehajtani.Ilyen példával találkozunk e fejezet során a tetrametil-disziloxán-diol előáUításávalkapcsolatban.
74.5.1. Tetraetoxi-szil&n /Si(OC2H5)4/ Szilícium-tetrakloridból absz, alkohollal állítható elő16:
4 CaHsOH + SiCl4= (C2H50 )4Si + 4 HC1.
16Thisaen, P. A., Koemer, 0 .: Z. anorg. alig. Chem. 189, (1930).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 326/366
328 F É M O R G A N I K U S V E G Y Ü L E T E K
Csepegtető tölcsérrel, KPG-keverővel és visszafolyatásra állított hűtővel ellátott 1 literes háromnyakú gömblombikba (74.5.1.1. ábra) 202 g (254 ml = 4,4 mól)
teljesen vízmentes etilalkoholt(lásd 50.11.) mérünk be, majda csepegtető tölcséren át —hűtőfürdő alkalmazása nélkül —hozzácsepegtetünk 170 g (114ml = 1 mól) SiCl4-ot. Erős sósavfejlődés mellett megindula reakció. Az adagolás befe jeztével még egy órán át for
rásban tartjuk az oldatot,hogy a felszabaduló HCl-otkiforraljuk, és az a hűtőn, valamint a Cad2-os csövön át akürtőbe távozhasson. Utána
jeges vízben lehűtjük az oldatot, és a maradék sósavatNa-alkoholáttal (kongóvörösindikátor jelenlétében) semle-
74.5.1.1. ábra. (Cj,H50)4Si előállítása gesítjük. A kicsapódó NaCl-tól száraz üvegszűrőn történő
szűréssel elválasztjuk (a csapadékot kevés vízmentes alkohollal mossuk). A szűrletet desztilláló készülékbe öntjük, és atmoszférikus nyomáson lehajtjukróla a fölös alkoholt, majd a maradékot frakciónáljuk. A tetraetoxi-szilán (ortoko-vasavas etilészter) forráspontja 168 C°. A lombikban magasabb forráspontú meta-és polikovasavas észterek maradnak vissza.
74.5.2. Metil-klór-szllánok /(CH3)3SiCl, (CH3)3SiCl2, CH3SiCl3 /
A metil-klór-szilán- elegyek előállításmódjai:a) Si és CH 3CI direkt reakciója:17
Si + 2 CH 3C1 -> (CH 3)2SiCl2+ /(CHgjgSiCl, CH 3SiCl3/.
A reakciót a réz katalizálja. A rezet tartalmazó szilíciumot, mely alkalmas arra,hogy vele a CH 3C1 elfogadható sebességgel reagáljon, kontaktmasszának szokásnevezni. A reakcióhoz legmegfelelőbb az 5—25 % rezet tartalmazó kontaktmassza,mely úgy készül, hogy a réz és a szilícium megfelelő arányú finomra őrölt porát-1060 C°-on H2- vagy N2-atmoszférábán hevítjük. De készíthető a kontaktmasszaoly módon is, hogy a CuCl és a ferroszilícium jól megőrölt porát reaktorcsőbenösszekeverjük, és N2-áramban lassan 300 C°-ra melegítjük. A réz kiválik a szilíciumra, a keletkező SiCl4pedig eltávozik a reaktorcsőből.
A direkt szintézist a 74.5.2.1. ábrán bemutatott csigakeverős reaktorban hajt juk végre.18 A reaktorcső ( A ) keményüvegből készült, 30 mm 0 -jű, 800 mm hosszú
17 Rochow, E. G.: U. S. Pat. 2, 380 995 (1941),• J. Am. Chem. Soc. 67, 963 (1945).18 Lengyel, B., Csákvári, B.,: Acta Chim. Hung. 39, 27 (1963).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 327/366
n-H
CaC/2 H2S04
74.5.2.1. ábra. Metil-klór-szilánok előállítása direkt szintézissel. A reaktor, B fütŐhuzal, (J köpeny, D termoelem, E keverő, F léghűtő, G vízhűtés, H szárazjeges —alkoholos hűtésr
J reométer, K pufferedény, M motor
üvegcső, amelyet szintén keményüvegből készült, 50 mm 0 -jű csőbe helyezünk.
Utóbbira tekercseljük a fűtőszálat (B) a cső hosszában
elhelyezett néhány szál azbesztzsinórra csavarva, hogyannak elcsúszását kiküszöböljük (74.5.2.2. ábra). Ezt afűtőcsövet hőszigetelés céljából újabb 60 mm 0 -jű üvegcsőbe helyezzük. A fűtést ejtőkengyeles hőfokszabályozóval szabályozzuk, a termoelemet (D) a fűtőcső és a reaktor-
cső közé helyezzük. A rézből készült csigakeverő tengelyegumidugón keresztül jut a reaktorba. Az alkalmazott reaktor előnye, hogy a reakciótér látható, ami lehetővé teszi akeverés szabályozását úgy, hogy a kontaktmassza a reaktorban a kívánt magasságig emelkedjék. A metil-kloridotpalackból vesszük, az áramlás sebességét reduktorszelep-
pel szabályozzuk, és reométerrel (J) ellenőrizzük. A terméket lég-, vízhűtéssel, majd szárazjeges—alkoholos hűtés
sel kondenzáljuk. A CH 3Cl-gáz feleslegét a kürtőbe vezet-
ízbesztzmór
74.5.2.2. ábra. A fűtőhuzal tekercselése

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 328/366
330 F É M O B Q A .N I K U S V E G Y Ü L E T E K
jük ki. A termék összetételének meghatározását legcélszerűbben gázkromatográ
fiás analízissel végezhetjük el.1® Amennyiben a direkt szintézist CHaCl és HCI gázeleggyel végezzük, úgy a főtermék CH 3SiCI3. A könnyen reakcióba vihető aktív hidrogént (Si—H-kötést)tartalmazó metil-klór-szilánok (pl. CH 3SiHCl2) mennyiségét a termékben úgy növelhetjük, hogy a CH 3C1 és HCI gázelegyhez kevés (1 % alatti mennyiségű) BC13-ot is keverünk.20
b) SiCJ4alkilezése metil-alumínium-szeszkvikloriddal. A SiCl4és a metil-alumínium-kloridok reakciójának eredményeképpen a követ
kező termékek képződnek:
(CH 3) 38iCl, (CH 3)2SíC12, CH 3SíCJ 3 és A1C13,tehát mono-, di-, illetve trifunkciós szubsztituált szilánok keverékét kapjuk. A reakció atmoszferikus nyomáson nem következik be, mivel a reakciósebesség gyakorlatilag csak 200 C° felett kielégítő. 'Tekintettel arra, hogy a kiindulási anyagoknak ezen a hőmérsékleten mintegy 30 atm tenziója van, a reakciót autoklávbankell végrehajtani.
Gondosan kiszárított 1 literes talpas lombikba kétfuratú gumidugóval a desztillált vizes üvegéhez hasonló feltétet helyezünk, melyet ugyancsak gondosan ki-«
szárítunk. A lombik fenekéig érő csö
vet nitrogén palackkal köt j ük össze, ésa lombikot nitrogénnel töltjük meg. A 74.1.-ben foglaltak szerint előállítottalumíniumorganikus vegyületet — pl.
metil-alumínium-szeszkvikloridot —nitrogénnel óvatosan az előzőleg N2-nelmegtöltött talpas lombikba nyomat
juk (74.5.2.3. ábra.). Az alumíniumorga-nikus vegyület levegőn azonnal meg
gyullad, ezért a műveletet igen nagyóvatossággal kell végrehajtani. Az átnyomatott alumíniumorganikus vegyület mennyisége kb. 200 g legyen. A bemérést oly módon végezzük, hogy a
lombikot nitrogénnel megtöltött és gumidugóval lezárt állapotban üresen lemérjük,majd az alumíniumorganikus vegyület benyomatása után dugóval azonnal lezárva,ismét lemérjük.
Az alumíniumorganikus Vegyülettel való műveletek során elkerülhetetlen,hogy kis mennyiségű anyag szabad levegőre ne kerüljön. így pl. a 74.5.2.3. ábrán
vázolt nyomatócső, amelynek segítségével az alumíniumorganikus vegyületet akeverőlombikba juttatjuk, az alumíniumorganikus vegyülettől nedvesen kerül■ érintkezésbe a levegővel. Ez a kis mennyiségű anyag a levegőn meggyullad és elég
74.5.2.3. ábra. Az alumíniumorganikus vegyü- letek átnyomatása
1® Garzó, T:, Till, F ., Till, I.,: Magy. Kém. Foly. 68, 328 (1962). Lengyel, B., Garzó, G., Székely, T ,: Acta Chim. Hong. 37, 37 (1963).
20 Csákvári, B ., Garzó, G., fenei, S.: Acta Chim. Hung. 39, 33 (1963).Caákvári, B., Jenei, S„ Knausz, D., Télegdi, L .: Acta Chim. Hung. 45, 31 (1965).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 329/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 330/366
332 F É M O R G A N I K U S V E G Y Ü L E T E K
csapot az inért gáz áramoltatásának megfelelően állítjuk be, majd az A és B lombikot kezdeti állásba hozzuk, (az A lombik alul helyezkedik el), és a G csepegtető
tölcséren át 50—50 ml absz. benzol és absz. alkohol elegyét adagoljuk az A lombikba. A lítium zömének elreagáltatása után az alkoholos folyadékkal mindkétlombikot átöblítjük. A készüléket csak a fenti műveletek elvégzése után szedhet
jük szét. A mosófolyadékként használt elegyet az előzőleg kinyomatott termékkelegyesítjük. A keletkezett LiCl és LiBr oldására még 50—100 ml vizet adunk hozzá. Az elegyből a felső réteget választótölcsérben elválasztjuk, és az alsó vizes rétegetmég két alkalommal 30—30 ml benzollal vagy éterrel rázzuk össze. A szerves frakciókat egyesítjük, 10 g vízmentes Na2S04-on szárítjuk, és redős szűrőn 250 ml-esgömblombikba szűrjük át. Előbb az oldószert desztilláljuk le, majd a trimetil-
-fenil-szilánt, mely vízsugárvákuumban (80 Hgmm) 98—99 C°-on forr. Kitermelés17—18 g (az elméletinek 68—77 %-a).
74.5.4. Fenil-trietoxi'Szilán /CeHsSi(OCjH6)3/
Tetra-etoxi-szilánból GWgward-reakcióval állítjuk elő:23
Si(OC2H5), + C„H5MgBr CeHsSi(OC,Hs)3 + Mg(OC2H5)Br.
Visszafolyós hűtővel, KPG-keverővel, csepegtetőtölcsérrel és folyadékba érőhőmérővel ellátott; 2 literes, négynyakú gömblombikba bemérünk 80 g (3,3 g-atom)magnézium forgácsot, 6,24 g (672 ml = 3 mól) frissen desztillált tetraetoxi-szilánt,1,5—2 g jódot, 10 ml vízmentés étert és katalizátorként 4 g vízmentes réz(I)-klori-dot. Ezután zárt elektromos melegítővel vagy merülőforralóval melegített vízfürdőalkalmazásával 70—75 C° belső hőmérsékletre melegítjük az elegyet. A csepegtetőtölcséren át 354 g (233 ml = 2,25 mól) bróm-benzolból pár millilitert becsepegtetünk,és megvárjuk, míg a Grignard-reagens képzése megindul. A melegítést elvesszük,és a bróm-benzol becsepegtetésének ütemével tartjuk 70—80 C°-on a reakcióelegyhőmérsékletét. Becsepegtetés után külső melegítéssel 5—6 órán át 80—100 C° közötti hőfokon tartjuk az elegyet. A magnézium a reakció során oldatba megy, akivált csapadéktól az oldatot üvegszűrőn megszűrjük, 2 literes lombikba öntjük,
Claisen-feltétet teszünk rá, melyhez vízhűtéses szedőlombik csatlakozik. Kevésváltozatlanul megmaradt tetraetoxi-szilán távozása után főpárlatként a fenil-triet-oxi-szilán, majd kevés utópárlatként a difenil-dietoxi-szilán desztillál át. (vö. 74.5.3.táblázat). A fenil-trietoxi-szilánra számított kitermelés kb. 60 %-os. Szükségesetén a terméket ismételt frakcionált desztillációval tisztítjuk.
74.5.3. táblázat
A fenil-etoxi-szilánok fizikai állandói
Fp./760Hgmm
C°
Op. (C°) Sűrűség,s20
Törésmutató,TI20
Si(OC2H 5)4 165,5 0,9284 1,3830c'Gff5s i ( 0 c 2lI5)3 235 0,9961 1,4632(C6H 5)2Si(OC2H 5)2 296 1,0200 1,5236(C6H s)3SiOC2H 5 34 4 65(C6H 5)4Si 428
i
23 4
23 Nagy, J., Gábor, T.: Periodica Polytech. 6, (3) 149 (1962).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 331/366
M E T I L - S Z I L Á N O K 333
74.5.5. Trimetil-p-íolil-szilán /p-CHaCaH4Si(CH3)a/
A Wurtz-TCBkoió útján állítható elő:24(CH3)3SiCl + p-CH 3C6H4Cl + 2 Na — ► p-CH 3CBH4Si(CH 3)3 + 2 NaCl.
Visszafolyós hűtővel (tetején CaCI2-os csővel) ellátott egy literes, egynyakú,vastagfalú gömblombikban 200 ml nátriumról ledesztillált vízmentes xilol alatt29 g (1,2 g-atomsúly+kb. 5 % felesleg) fémnátriumot olvasztunk meg célszerűenN2-atmoszférában. Ez után a hűtőt levesszük, és a lombikot
jól bedugaszoljuk. Védőszemüveg, illetve védőálarc és azbesztkesztyű használata mellett, száraz homokfürdő felett,
a lombik intenzív rázogatása közben az elegyet lehűlni hagy juk. A megolvadt nátrium finom szemcsék alakjában mintnátriumhomok dermed meg. A xilolt dekantálás után leöntjük,s maradékát vízmentes éterrel (50—50 ml) dekantálva mossuk le. Ezután a nátriumhomokra 300 inl vízmentes étertöntünk, a lombikot feltéttel látjuk el, amelynek egyik ágábavisszafolyós hűtőt, a másikba csepegtetőtölcsért iktatunk(74.5.5.1. ábra). A visszafolyásra állított hűtő felső végét alevegő nedvességének távoltartására CaCl2-os csővel dugjukbe. Az Y-feltét egyenes ágába állított csepegtetőtölcséren át
76,2 g (71 ml = 0,6 mól) vízmentes p-tolil-klorid és 65,2 g(76,2 ml = 0,6 mól) trimetil-klór-szilán, összekevert elegyébőlnéhány millilitert becsepegtetünk. (A vízmentes p-tolil-klori-dot az anyag előpárlat vétele mellett történő desztillálásávalkapjuk, az előpárlat ugyanis azeotróposan elveszi a csekélyvíztartalmat.) Zárt elektromos melegítővel forralva beindít
juk a reakciót, majd a melegítést megszüntetve az elegyet abecsspegtetés ütemének szabályozásával enyhe forrásban tart
juk. Az adagolás befejeztével kb. egy órás forralással teljessé
tesszük a reakciót. Lehűlés után az elegyhez a feleslegben levőnátrium elbontására 100 ml vízmentes etilalkoholt csepegtetünk. Fél órás állás után óvatosan hozzáadjuk a kivált NaCloldásához szükséges vizet. A jó szétválás elérése végett a lúgos oldatot savval semlegesítjük, és választótölcsér segítségével elválasztjuk. Az alsó vizes részt 50^-50 ml-éterrel néhányszor kirázzuk. Az éteres rétegeket egyesítjük, és 10 gvízmentes Na2S04-on szárítjuk. Szűrés után desztilláló készülékben lehajtjuk az étert, és a maradékot vízsugárszi-vattyú-vákuumban desztilláljuk le. A termék 64—82 g, ami 65—83 %-os kiter
melésnek felel meg. Fp. 109—110 C°/30 Hgmm.
74.5.5.1. ábra. Wurtz- reakció Na-homokkal
74.5.6. Tetrametil-szilán /(CH3)4Si/
A szilícium-tetraklorid (CH 3) 3 Al2Cl3-dal történő alkilczésének kinetikai vizsgálata során bebizonyosodott, hogy a gyökcserés folyamatokban a Sí—Cl-kötés
M Földesi, Acta Chim. Hung. 45, 237 (1965).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 332/366
334 F É M O R G A N I K U S V E G Y Ü L E T E K
74.5.6.1. ábra. (CH3)4Si előállítása
annál könnyebben támadható, minél több alkilkötés vanugyanazon a klór-szilán-molekulán.25 Következésképpen a metil-klór-szilánok között legkönnyebben a(C ^ ^ iC l alkilezhetŐ:
(CH 3) 3 AJ2C1 3 + 3 (CH3)3SiCl = 3 (CH3)4Si + A12C1,.
A reakciót viszafolyós hűtó'vei, nitrogénbevezető-csővel, csepegtetőtölcsérrel, valamint mágneses keverő-vei ellátott 500 ml-es gömblombikban végezzük (74.5.6.1.ábra). A visszafolyásra állított hűtő felső végét csiszo-laton át csatlakoztatjuk egy mélyhűtött szedőedényhez.
A lombikot vízmentes nitrogénnel töltjük meg, ésnitrogénnel 85,0 g (0,41 mól) metil-alumínium-szesz-kvikloridot nyomatunk a lombikba, majd óvatosan,homokfürdőn 70 C°-ra melegítjük, hogy a reakciópillanatszérűen lejátszódjék. Ezzel elkerüljük a trimetil--klór-szilán felhalmozódását, miáltal a robbanásveszélycsökkenthető. Az előmelegített reagenshez mágneseskeverővei való állandó keverés közben 88 g (0,81 mól)(CH 3) 3SiCl-t csepegtetünk. A reakció előrehaladásávalaz elegy hőmérséklete az alacsony forráspontú tetrame-
til-szilán keletkezése folytán fokozatosan csökken. A keverés viszont a viszkozitás növekedése miatt egyre nehezebbé válik. A reakció lezajlása után — erős vissza-folyás mellett — a keletkezett tetrametil-szilánt a mélyhűtött szedőbe desztilláljuk. A nyert termék 58 g, a(CH 3) 3SiCl-ra vonatkoztatott kitermelés 82 %-os. A termék gázkromatográfiai úton kimutatható szennyeződéstnem tartalmaz.26
74.5.7. Tetrametil-disziloxán-l,3-dÍoI /H 0(C H,)2Sl-0-Si(CH3)20 H /
A dimetil-diklór-szilán hideg, semleges pH-jú oldatban végrehajtott hidrolízlsével állítható elő.27
CHaX / C1Si
CH3/ ^C 1 Hi0
c h 3 / c i
Si
c h / \ c i
c h 3X x o h
Si
CH3/ \ O H
CH^ ^O H
Si
CH/ \ O H /
CH3X /OH
Si
CH
CH,
/ \O
Si
C H , / /-O H
2# Lengyel, B., Székely, T.: Z. anorg. alig. Chem. 287, 273 (1956). Lengyel, B., Székely, T.,Jenei,S., Oarzó, G.: Z. anorg. alig. Chem. 323, 66 (1963).
*6 Székely, T., Gsákvári, B., Foldesi, / . , Újszászi, A..* Annales Univ. Sói. Budapestiensis Sect. Chim. 6, 103 (1964).
27 Lucas, Q. E., Martin, R. W.: J. Am. Chem. Soc. 74, 6225 (1962).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 333/366
F E N I L - T R I K L Ó R - S Z I L Á N 335
Folyadékba érő hőmérővel és gázbevezetőcsővel, valamint csepegtetőtölcsérrel ellátott 2 literes, négynyakú gömblombikba 1140 ml desztillált vizet mérünk be.(A negyedik tubust, mely levegőztetésre szolgál, szabadon hagyjuk.)A műveletetcélszerű fülkében végezni. Indikátorként brómtimolkék és fenolftalein keverékéthasználjuk, amelynek az átcsapási £>H-ja 7 körül van. (Savas közegben kék, lúgosközegben piros). A lombikot hűtőfürdőben (só—jég) + 2 C°-ra hűtjük le, és a csepegtetőtölcséren át megkezdjük a dimetil-diklór-szilán (100 g = 94,4 ml = 0,62 mól)becsepegtetését, és ezzel egyidőben az NH3-gáz bevezetését. A gázbevezetés sebességét úgy szabályozzuk, hogy a pH-érték 6,5—8,5 között legyen (az alkalmazottindikátor átcsapási intervallumának megfelelően), és az elegy hőfoka mindvégig0 és + 2 C° között maradjon. A beadagolás üteme érthetően nagymértékben függ
a hűtés intenzitásától, és kb. 2—4 órát vesz igénybe. Ennek befejeztével kb. 1 óránát folytatjuk az ammóniagáz — szükség szerinti — bevezetését, hogy az esetlegutólag bekövetkező megsavanyodást elkerüljük. Ezután az elegyhez adagolunk380 g jól elporftottNaCl-ot, s ezt keverés közben feloldjuk. Az oldatot egy éjszakánát -f-10 C° alatti hőmérsékleten állni hagyjuk, és a kristályosán kivált tetrametil--disziloxán-l,3-diolt szűrés után petroléterből átkristályosítva tisztítjuk. Op. 67—68C°. Gondos és pontos munka esetén a kitermelés 40—50 %-os.
74.5.8, Fenil-triklór-szilán (C6H5SiCla)
Az Si—H-kötésű vegyületek alkil- vagy aril-halogenidekkel ún. magas hőfokú kondenzációra is képesek, ami preparatív célra gyakran jól hasznosítható.28 így afenil-tfiklór-szilánt a triklór-szilán és a klór-benzol reakciójával állítjuk elő:
C„H5C1 + HSiCl 3 jL5Q- 75°g!_^ C6H5SiCl 3 + HC1.
A reakciót a 74.5.8.1. ábra szerinti berendezésben hajtjuk végre. A tulajdonképpeni reaktor egy kb. 20 mm 0 -jű és 1 m hosszú kvarccső, mely ejtőkengyelesműszerrel szabályozott elektromos fűtéssel van ellátva. A reagálandó komponenseket — megfelelő arányban való összekeverés után — szervomotorral hajtott
dugattyú adagolja. Az adagolópipettában paraffinolaj és higany közvetítőfolyadékot használunk. A szabályozható szervomotor (M ) forgását nagy keresztmetszetű fogaskerékkel át tételezzük, és így nyomatjuk le a dugattyút, mely olaj-,illetve higanydugattyú közvetítésével az adagoló (Aj terében levő elegyet az elpárologtatóba nyomja. Az elpárologtatóba adagolt elegy gőzzé alakul, s így jut a reaktorcsőbe. (A B jelű gázbevezetőcső azt a célt szolgálja, hogy szükség esetén a re-akcióelegyhez valamilyen gázt is adagoljunk.) A reaktorból, szárazjég—aceton--hűtőkeverékkel hűtött, leeresztőcsappal is ellátott szedőedényekbe (C) kerül akondenzációs reakcióval keletkezett termék (a változatlanul maradt kiindulási
anyagokkal együtt). A reakció során képződött HC1 a szedőkhöz csatlakozó vissza-folyós hűtőn (D), majd P20 5-os szárítón át a kürtőbe távozik. Az adagolót, kiürüléseesetén, az anyagtartályban levő reakcióelegyből a háromágú csapon (E) át tölthetjük fel.
28 IleTpoB, A. fl., Mhpohob, B. d>.. IIoHOMapeHKO, B. A., HepHtmieB, E. A.: CiiHTea KpeM- HHflopraHHHecKHx MOHOMepoB. MasaTejibCTBo AKafleMHH Hayn CCCP, MocKaa, 1961, CTp. 149 h 326. Knauez, D., Oarzó, O., Gömöry, P., Telegdi, L.: Magy. Kém. Foly. 70, 119 (1961

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 334/366
336 r É M O B Ü A N I K ü S V E G Y Ü L E T E K
A fenil-triklór-szilán készítésének megfelelő optimális körülmények: a reaktorhőmérséklete 680 C°, kontakt idő: 20 mp. A C6H5C1: HSiCl 3 = 2 : 1 összetételmellett a SiHCl3-ra vonatkoztatott konverzió, tehát a kitermelés kb. 50 %-os.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 335/366
S Z E R V E S Ó N V E G Y Ü L E T E K 337
74.6. Szerves ónvegyületek
A szerves ónvegyületek sem oxigénre, sem vízre nem érzékenyek. 9
Több eljárás is ismeretes, melyek során szerves ónvegyületek készítésére vagy átalakításáravizes közeget alkalmazunk. %
A szerves ónvegyületek kellemetlenül átható szagúak. Egyes irodalmi adatokkarcinogén hatásukról is beszámolnak, mások viszont vitatják ezt. Toxikus hatásuklényegesen kisebb, mint a szerves ólom-, vagy higanyvegyületeké. Az illékonyabbvegyületek toxicitása természetesen nagyobb, mint a kevésbé illékonyaké. Mindenesetre megállapíthatjuk, hogy a szerves ónvegyületekkel való műveleteket ajánlatos kellő óvatossággal végezni. Ha bőrfelületre cseppen, célszerű bő szappanosvízzel lemosni.
74.6.1. Tetraetil-ón /(C 2H5)4 Sn/
Gngrnard-reakcióval állítjuk elő.30
C2H5Br + Mg = C2H5MgBr,
4 C2H5MgBr + SnCl4= (C2H5)4Sn + 4 MgBrCl.
Visszafolyós hűtővel (tetején a levegő nedvességének távoltartására szolgálóCaCl2-os csővel), KPG-keverővel és csepegtetőtölosérrel felszerelt egy literes háromnyakú lombikban (74.6.1.1. ábra) 30 g
(1,24 g-atomsúly) Mg-reszelék, 150 ml fém-nátriumról desztillált vízmentes éter és párszem jód elegyébe 200 ml vízmentes toluol-ban oldott 150 g (105,5 ml = 1,39 mól) etil--bromidból pár ml-t csepegtetünk. Közbenzárt elektromos fűtőtesttel vagy vízfürdőnaddig melegítjük, míg a Grignard-re&gensképződése megindul, majd a melegítést megszüntetjük. Az esetleges heves reakció szükség szerinti mérséklésére jegesvizes fürdőt
helyezünk készenlétbe. A maradék toluolosetil-bromidot olyan ütemben csepegtetjükbe, hogy az éter enyhe forrásban maradjon. A csepegtetés kb. egy—másfélórát vesz igénybe, s befejezésül az elegyet még félórán átforraljuk. Ez után hűtés (só—jég) és intenzív keverés közben becsepegtetünk 50 g (22,3ml = 0,193 mól) vízmentes SnCl4-o£, majd100 ml vízmentes toluolt. A heves reakciólezajlása után a visszafolyásra állított hűtőtleszálló állásba helyezzük. (74.6.1.2. ábra),és a fölös étert kidesztilláljuk (míg a párlat
29 van dér Kerk, G. J. M., Luijten, J. G. A., Noltes, J. G.: Angew. Chem. 70, 298 (1968). Ingham, R. K., Rosenberg' S. D., Gilman, M.: Chem. Rév. 60, 459 (1961).
30 Luijten, J. G. A., van dér Kerk, G. J. M.: Investigation in the Field of Organotin Chemistry. Utrecht, 1959.
Luijten, J. F. A., van dér Kerk, G. J. M.: Org. Synth. 36, 86 (1956).
22 Ált. és szervetlen kémiai prakt. —4248/11.
74.6.1.1. ábra. GViffnard-reakció vissza- folyatásra állított hűtővel

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 336/366
338 F É M O B G A N I K U S V E G Y Ü L E T E K
hőfoka a Í00C°-ot el nem éri). Két órán át 100 C°-on melegítve tesszük teljessé azalkilezést. Lehűlés után a Grignard-vegyület bontására 150 ml vizet, majd 250 ml10%-os sósavat csepegtetünk be jéghűtés közben. A készüléket ezután szétszed
jük, a szerves részeket elválasztjuk, és a vizes részt néhányszor 50—50 ml toluollal
motor
CJ3
rázzuk ki. Az egyesített szerves frakciókat 50 ml 10 %-os sósavval, majd újra vízzelrázzuk ki, és 10 g vízmentes CaCl2-on történő szárítás után redős szűrőn szűrjük.Desztillálókészülékben először lehajtjuk az oldószert, majd 12 Hgmm-en, 63—65 C°között szedjük a főpárlatot. A kitermelés kb. 90%-os.31
74.6.2. Tetrabutil-ón /(C4H„)4Sn/
PFwríz-reakcióval készítjük:32
4 C4H9C1 + SnCl4+ 8 N a = ( C4He)4Sn + 8 NaCl.
Erős falú, egynyakú, kb. egy literes gömblombikba 400 ml vízmentesített silóit és 57,5 g (2,5 g-atomsúly) hámozott fémnátriumot mérünk be. A lombiknyakára CaCl2-os csövet tartalmazó visszafolyós hűtőt helyezünk, majd a fémnátriumot elektromos melegítővel megolvasztjuk. IJzután a hűtőt levesszük, és a lombikot vasdugóval bedugaszoljuk. Védőszemüveget és azbeszt kesztyűt veszünk fel. A
lombikot száraz homokfürdő fölött erőteljesen rázogatva hűtjük. A fémnátrium finom homok alakjában szilárdul meg. A xilolt a nátriumról dekantáljuk, és helyére40— 60 C° forráspontú vízmentesített petrolétert rétegezünk. A lombikra feltétet(74.5.5.1. ábra), annak egyik szárába csepegtetőtölcsért, másik ágába pedig vissza-folyásra állított hűtőt helyezünk. Elektromos fűtőtesttel forraljuk, és a szuszpen-
31 Földesi, Acta Ohim. Hung. 45, 237 (1965).32 van dér Kerk, G. J. M.; Luijten, J. G. A .: J. Appl. Chem. 4, 301 (1954).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 337/366
S Z E RV E S Ó N V E G Y Ü L E T E K 339
zióhoz csepegtetőtölcséren át 81,5 g (36,5 ml = 0,314 mól) vízmentes SnCI4és 115 g(130 ml = 1,25 mól) vízmentesített butil-klorid elegyéből néhány millilitert cse
pegtetünk. (A butil-klorid vízmentesítése desztillációval történik; az előpárlat aze-otróposan elviszi a víznyomokat, a főpárlat vízmentes butil-klorid. Fp. 78 C°,s = 0,888). A melegítést néhány .percig, illetve a reakció megindulásáig folytatjuk.Ez után az elegy beesepegtetésének ütemével szabályozzuk a forralást. Az adagoláskb. másfél órát vesz igénybe, befejezésül az elegyet még két órán át forraljuk, majdlehűlés után a visszamaradt kevés fém-Na reagáltatására 50 ml alkoholt csepegtetünk hozzá. A Na reagáltatása (esetleg melegítés) után a NaCl oldására rázogatásközben kb. 200 ml vizet csepegtetünk. A készüléket szétszedjük, az elegyet választótölcsérben elválasztjuk. Az alsó vizes réteget 50—50 ml petroléterrel újra kirázzuk, és az egyesített petroléteres frakciókat kb. 10 g vízmentes Na2S04-on szárítjuk.Szűrés után az oldószert desztilláló készülékben lehajtjuk, a maradékot 4 Hgmm-esvákuumban desztilláljuk. A tetrabutil-ónt tartalmazó főpárlatot 130—134 C°-onvesszük le. A kitermelés 64 —72 g, tehát 66 —75 %-osrrak felel meg.
74.6.3. Dietil-6n-diklorid /(C2H5)2SnCl2/
Ekvilibrálással állítjuk elő.33
(C2H5)4Sn + SnCl4^ 2 (C2H5)2SnCl2.
250 ml-es száraz gömblombikban jéghűtés közben 58,75 g (0,25 mól) tetraetil-
-ónt 65,1 g (0,25 mól) SnCl4-dal elegyítünk. A lombikra visszafolyós hűtőt helyezünk,melynek felső végéhez a levegő nedvességének távoltartására CaCl2-os csővet csatlakoztatunk. Az elegyet kb. 2 órán át 220 C°-os olajfürdőben melegítjük, majd"-Rascfogr-gyűrűs feltét alkalmazásával szétfrakcionáljuk. 8 Hgmm nyomáson 94—95C°-on, illetve 14 Hgmm-en 108—109 C°-on kapjuk a dietil-ón-dikloridot, mely aszedőben megdermed (op. 84 C°). Ezért célszerű a szedőedényt közvetlenül a desztilláló feltéthez kapcsolni. A kitermelés kb. 90 %-os.
74.6.4. Dietil-ón-oxid /(CaHsLSnO/n
Dietil-ón-diklorid hidrolízisével állítjuk elő.34
(C2H5)2SnCl2 -f- 2 KOH = 1 /n [(C2H5)2SnO]n + 2 KC1 + H20.
Visszafolyós hűtővel, keverővei, csepegtetőtölcsérrel felszerelt egy literes, háromnyakú lombikba 18 g (0,3 mól) KOH-ot, 0,3 g zsíralkohol-szulfonátot (mikepon,mavepon) és 380 ml vizet mérünk be. Az oldatot kb. 90 C°-ra felmelegítjük, és keverés közben a csepegtetőtölcséren át kb. 1/2 óra alatt 36,5 g (0,148 mól) dietil-ón--diklorid 40 —50ml-es alkoholos oldatát csepegtetjük hozzá. A tölcsért 10 ml alkohollal átmossuk, és a mosófolyadékot szintén hozzácsepegtetjük. Ezután még kb. 1/2órán keresztül melegítjük az elegyet, mire a hidrolízis teljessé válik. (Utóbbi úgy állapítható meg, hogy az alsó olajos dietil-ón-dikloridos réteg eltűnik.) A dietil-ón--oxid-polimert fehér csapadék alakjában kapjuk meg. Lehűlés után nuccson szűr
jük (a szűrést könnyebben végezhetjük, ha a gyengén lúgos oldatot előbb C02-gázbevezetésével semlegesítjük), és szívatás közben vízzel kloridmentességig mossuk. Végül kb. 50 C°-on vákuum-szárítószekrényben súlyállandóságig szárítjuk. A kitermelés gyakorlatilag kvantitatív.
33 Kocheehkov, K. A.: Bér. 66, 1661 (1939).34 Földesi, I., Strdner, Qy.: Acta Chim. Hung. 45, 313 (1965).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 338/366
340 F É M O R G A N I K U S V E G Y Ü L E T E K
74.6.5. Dietil-ón-diacetát /(C2H5)2Sn(OOCCH3)2/
Az előállítás módját az alábbi egyenlet fejezi ki:l/n[(C2H5)2SnO]n + (CH 3C0)20 - (C2H5)2Sn(OOCCH3)2.
100 ml-es desztilláló lombikba 38,5 g (0,2 mól) dietil-ón-oxidot és 24 g (23,6ml = 0,235 mól) ecetsavanhidridet mérünk. (Ecetsavanhidrid helyett megfelelőmennyiségű ecetsav is alkalmazható.) 100 C°-on melegítve vagy jól összerázva azelegy homogén oldattá alakul. A fölös ecetsavanhidridet 100—120 C°-os olaj fürdőhőmérsékletén hajtjuk le, és a maradékot vákuumban (rotációs szivattyú!) desztillálva tisztítjuk. (Sav alkalmazása esetén a reakció során víz képződik, melyet toluolhozzáadásával azeotróposan desztillálunk le, és ezután végezzük el a vákuumdesz-
tillációt.) 1,5 Hgmm nyomáson a fp. 84—85 C°. A szedőbe bedermedő dietil-ón-diacetát (op. 89 C°) petroléterből átkristályosítva tisztítható.
74.6.6. D ibcn zil-ón-d iklorid /(C6H 5CH2)2SnCl2/
Direkt szintézissel állítjuk elő:35
2 C6H6CH2C1 + Sn = (C6H5CH2)2SnCl2
Ónpor tisztítása: A kereskedelmi ónport kétszeres súlyú 10 %-os vizes NaOH-
-oldattál 10 percen át rázogatjuk, hogy a felületre tapadt ón(II)-oxidot és zsírsavat(melyet a porítás során adnak hozzá) eltávolítsuk. Ezután vízzel semlegesre mossuk, és nedves állapotban frissen használjuk fel.
20 g — a fenti tisztításnak alávetett — nedves ónport 300 ml-es háromnyakúlombikba teszünk, és 150 ml toluolt adunk hozzá. A lombikot keverővei, visszafo-lyós hűtővel és csepegtetőtölcsérrel szereljük fel. Elektromos melegítővel erélyeskeverés közben felforraljuk az oldatot. 5 perces forralás után 20 g benzil-kloridotcsepegtetünk hozzá, és a keverést, valamint a forralást 5 órán át folytatjuk. A maradék, feleslegben levő óntól az oldatot még melegen szűrjük, az ónport acetonnalmossuk. A szerves oldószert desztilláló készülékben lehajtjuk, és a maradékot etil-
-acetátból átkristályosítva tisztíthatjuk. Op. 161—163 C°. A kitermelés 65—70 %-os.
74.6.7. T ribenzil- ón-k lorld /(C6H 5CH2)3SnC l/
Közvetlen reakcióval nyerhető:
3 C6H5CH2C1 + 2 Sn = (C6H5CH2)3SnCl + SnCl2.
A 74.6.6. alatt leírtakhoz hasonló módon felszerelt 300 ml-es lombikba 20 gtisztított ónport (lásd 74.6.6.) és 150 ml vizet adunk, és forraljuk. Keverés közben
5 perc alatt 20 g benzil-kloridot csepegtetünk hozzá. A forralást és a keverést 3—5órán át folytatjuk, mire az ón nagy része reakcióba lép. A termék a forró vízben ola jos szuszpenziót képez, mely lehűléskor megdermed. Lehűlés után nuccson leszívat juk, az ónport is tartalmazó csapadékot $oa;Meí-készülékben acetonnal extraháljuk. A tribenzil-ón-klorid az aceton lepárlása után kristályosán bedermed. Etil-acetátbólátkristályosítva tisztítjuk. (Op. 142—144 C°). Kitermelés 70—75 %-os.
35 Sishido, K., Tákeda, Y., Kinugawa, Z.: J. Ám. Chem. Soc. 83, 538 (19 61).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 339/366
• S Z E R V E S Ó L O M V E O Y Ü L E T E E 341
74 .7. Szerves ólomvegyületek
Oxigénre, vízre különösebben nem érzékenyek. Toxicitásuk viszont igen nagy,ezért a velük való műveleteknél a legnagyobb körültekintéssel és óvatossággal kelleljárni. Az illékonyság növekedésével emelkedik az ólommérgezés veszélye, ezértilyen vegyületekkel fülkében, vagy — célszerűbben — zárt készülékben dolgozzunk.36
74.7.1. Tetraetii-óiom /(C 2H5)4Pb/
Ólomötvözet felhasználásával az alábbi reakció alapján állítható elő:37
4 Pb-Na + 4 C2H5Br - (C2H5)4Pb + 4 NaBr + 3 Pb.ólomötvözet készítése: fafedővel, erős vaske verő vei ellátott öntöttvas edénybe
200 g fémólmot mérünk. 325 C°-on megolvasztjuk, 100 g gépolajat vagy szilárdparaffint adunk hozzá, és ömledék állapotban tartva, intenzíven kavargatva, aprórészletekben 18 g fémnátriumot és 3 g fémkáliumot adagolunk az elegybe (védő-szemüveget és azbeszt kesztyűt használunk!). Erős kevergetés közben hagyjuk lehűlni, mire az ötvözet szemcsés alakban dermed meg. Teljes lehűlés után dekan-táljuk róla az olajat, majd petroléterrel lemossuk, és felhasználásig petroléteralatt tartjuk el.
Visszafolyós hűtővel és csepegtetőtölcsérrel ellátott 1 literes kétnyakú lombikba visszük át az ötvözetet, és kb. 200 ml 80—100 C° forráspontú petroléterrelfedjük. Rázogatás és melegítés közben 109 g etil-bromidnak kb. egy tized részétadagoljuk hozzá. A melegítést addig folytatjuk, míg a reakció megindul, ez után amelegítés mellőzésével tovább csepegtetjük bele az etil-bromidot úgy, ahogy azexoterm reakció ezt megengedi. A beadagolás befejezése után az oldatot kb. 2 óránát forraljuk. A reakció végét az jelzi, hogy az ötvözet Na- és K-tartalmának reagálása folytán az ötvözet ólomporrá esik szét. Még biztosabban mutatja a végpontotaz, hogy víz becsepegtetésére már nincs pezsgés.
A terméket vízgőzzel ledesztilláljuk (lásd 48.2.2.) majd a felső petroléteresólom-etil-tartalmú réteget elválasztjuk. A vizes fázist 50—50 ml petroléterrel néhányszor kirázzuk. Az egyesített petroléteres oldatokból desztilláló készülékbenlehajtjuk az oldószert, és a maradékot váküumdesztillálással tisztítjuk. A tetra-etil-ólom 13—14 Hgmm-en 80—85 C° között forr. A kitermelés 70—80%-os.
A szerves ólomvegyületekkel kapcsolatos minden műveletet jól húzó fülkébenés lezárt készülékben kell végezni. A fülke gőztelenítése brómos vagy klóros kloroformmal történik. Vigyázzunk, hogy ólom-tetraetil-gőzöket semmiképpen ne lélegezzünk be és ólom-tetraetil ne cseppenjen a bőrfelületre.
74.7.2. Trietü-61om-p-toluól-sziilíohát /(C2H5)3PI)0S02C8H4CB3/
Az előző eljárás alapján készült, 65,5 g desztillált tetraetil-ólomhoz a szedőben24,5 g p-toluol-szulfonsavat és pár szem szilikagélt adunk, majd az anyagot vízfürdőn visszafolyásra állított hűtő alkalmazásával 20—30 percig forraljuk. Bcn-
36 Willem8ens,L.G.: Organolead Chemistry. Intem . Lead. Zinc. Rés. Org.. New York, 1964.87Shapiro, H.: Adván. Chem. Ser. 23, 290 (1959).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 340/366
342 F É M O R G A N I K U S V É G Y Ö L E T E K
zol bán oldjuk, a benzolt célszerű már a reakció elején hozzáadagolni (kb. 50 ml). A kicsapást a benzolos oldatból petroléter hozzáadásával végezzük. (Op. 167 —
-168 C°.)
74.8. Szerves titánvegyületek
A T f—C-kötésű szerves titánvegyületek csak igen alacsony hőmérsékletenállandók, és előállításukra csak a legutóbbi időben került sor. Annál gyakoribbaka titán-alkoxidok, azaz a titánészterek. Utóbbiak oxigénre nem, vízre azonbanigen nagy mértékben érzékenyek, ezért előállításuk és reagáltatásuk során vízmentes oldószerek alkalmazása és a légnedvesség távoltartása szükséges.
74.8.1. Tetraetoxi-titán /Ti(OC2H5)4/
Legkényelmesebben a titán-amin-klorid addíciós vegyületen át állíthatóelő :38
TiCl4 + 6 NH 3 = t :í c i4.6 n h 3,
TíC14*6 NH 3 + 4 C2H5OH = Ti(OC2H5)4 + 4 NH4C1 + 2 NH3.
Csepegtetőtölcsérrel, KPG-keverővel, gázbevezetőcsővel és visszafolyásra állított hűtővel (melynek felső végére a levegő nedvességének távoltartására P20 5-dal
töltött szárítót iktatunk) felszerelt négynyakú lombikba 35 g (0,184 mól) TiCl4-otmérünk. A keverést megindítjuk, és mivel a reakció erősen exoterm, a lombikot
jeges fürdőbe állítjuk. Ezután megindítjuk a (két KOH-tornyon átvezetve szárított) NH3-gáz bevezetését a TiCl4-oldatba, amit addig folytatunk, míg a titán--tetraklorid-hexamminnak megfelelő súlyszaporulat (18,9 g) nem következik be.
A titán-tetraklorid-hexammin barnássárga, por alakú vegyület, mely levegőtőlelzártan nedvességmentes térben bomlás nélkül eltartható. A levegő nedvességétőlazonban hidrolizál, és fokozatos kifehéredés (T i02-képződés) közben ammóniát ad le.
Az amminkomplex előállítása után a gázbevezetőcső helyére dugót teszünk.
A csepegtetőtölcséren át a titán-tetraklorid-hexamminhoz apró részletekben 100 ——150 ml vízmentes alkoholt csepegtetünk, majd 2—5 órán át vízfürdőn forralunk. Az alkoholízis közben NH4Cl-csapadék, NH 3-gáz, valamint a folyékony halmaz-állapotú tetraetoxi-titán keletkezik. A kivált NH4C1 csapadékot melegen szűrjük,s a csapadékot 50—100 ml vízmentes benzollal szívatás közben mossuk. (Az NH4C1-csapadék a bemért TiCl40,5—2,5%-át tartalmazza.)
Az elegyet 12 órán át állni hagyjuk, amire kevés csapadék még kiválik. Újraszűrünk, majd az oldószert atmoszferikus nyomáson ledesztilláljuk. Ezután a titán-észtert vákuumban desztilláljuk le. (Fp. 132 C°/3 Hgmm., 134 C°/4 Hgmm,150 C°/ll Hgmm, 168 C°/35 Hgmm, 180 C°/85 Hgmm, 235 ,C°/760 Hgmm.)
A kitermelés 65%-os. A desztillációs maradék barnás színű, titánt is tartalmazó,gyantás anyag. A nyers desztillátum kb. 0,2% NH4Cl-szennyeződést tartalmaz,színe világossárga. Újradesztillálással színtelen termékhez jutunk. A tetraetoxi-titán gyengén gyümölcsszagú, higroszkópos folyadék. A levegő nedvességétől elzárva bomlás nélkül eltartható.
A fenti eljárás egyéb titán-alkoxidok előállítására is alkalmas.
38 Lengyel B., Garat T.: Magy. Kém. Foly. 59, 343 (1955).

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 341/366
S Z E R V E S T I T Á N V E G Y Ü X E T E K 343
74.8.2. Trietoxi-titán-klorid /(C2H.,0)3TiCl/
Az alábbi reakció alapján állítható elő39:TiCl4+ 3 HOC2H5 4- 3 C5H5N = (C2H50)3TiCl + 3C6H5N.HC1.
Keverővei, csepegtetőtölcsérrel és visszafolyós hűtővel felszerelt, egy litereslombikba 92 g (2 mól) absz. etilalkoholt 150 g (1,9 mól)KOH-on szárított absz.piridint és 300 ml vízmentes benzolt mérünk. A visszafolyatásra állított hűtő felsővégén a levegő nedvességétől való óvás céljából CaCl2-os csővel csatlakoztatunk.
A lombikot az exoterm reakció miatt jegesvizes fürdőbe merítjük. Megindítjuk akeverőt, és az oldat lehűlése után becsepegtetünk 95 g (0,5 mól) TiCl4-ot. Ezután
az elegyet a reakció teljessé tétele végett 2 órán át vízfürdőn forraljuk, végülhűtőkeverékkel (só—jég) lehűtjük, és az oldatot a kivált piridin-hidrogén--klorid csapadékról leszívatjuk, (a csapadékot kevés vízmentes benzollal még mossuk). A fölös alkoholt, piridint és benzolt ledesztilláljuk, s a maradékot vákuum-desztillálással tisztítjuk. A trietoxi-titán-klorid 18 Hgmm nyomáson 174—176 C°-on forr. Igen higroszkópos folyadék. A kitermelés kb. 70%-os.
^ Horima O . B.: C h h t c 3 opraHHH ecKHx coef lHHeHHH. CÖopHHK, M ocK Ba , 1952, c i p . 144'.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 342/366
7 5. Baleseti veszély és óvato ssági rendszabályok
A laboratóriumi munkában a legnagyobb veszélyt a váratlanul bekövetkezőfejlemények jelentik. Megfelelő felszereléssel és rendszabályokkal gyakorlatilagminden előrelátott jelenség ártalmas volta nagymértékben csökkenthető, sőt esetlegteljesen kiküszöbölhető. Ezért a laboratóriumi balesetek elkerülése céljából a legfontosabb a veszély felismerése, megelőzése és a megfelelő lélekjelenlét.
E fejezetben a laboratóriumi munka során előforduló mérgezésekkel, tűz- ésegyéb baleseti veszélyekkel, azok megelőzésével, valamint a balesetek bekövetkezésekor követendő eljárásokkal, illetve sególynyújtással foglalkozunk. Megjegyezzük, hogy az itt ismertetett egyes preparátumok előállításáról szóló leírásokban
az ezekkel kapcsolatos munkák közben fellépő veszélyekre szintén felhívtuk afigyelmet.
75.1. Mérgezések
Az egyes — laboratóriumokban gyakran használt — vegyszerek, főleg oldószerek és gázok mérgező hatása közismert. Az ilyen gázok és gőzök belélegzéseellen általánosan elterjedt védekezési mód a jól húzó fülke használata, illetve,ahol ez nem lehetséges vagy nem elég hatásos, megfelelő töltetű gázálarc viselése.
Halált idézhet elő olyan helyiségben való egészen rövid ideig l}/2—1 órán át)
tartó tartózkodás is, melynek légterében pl. csak 0,000 05 térf.% Hg vagy 0,000 76térf.% Br2 van. Az illékony nehézfémvegyiileteknek /[Ni(CO)4], Hg(CH3)2, stb./,
valamint fémgőzöknek (Cd-, Be-por) egészen rövidideig tartó belélegzése is halálos lehet, az Os04füstje viszont vakságot okoz. A CS2-gőzök tartÓBbelélegzésének súlyos mérgezés lehet a következménye. Ugyancsak erősen mérgező a 75.1.1. táblázatban felsorolt anyagok gőze a megadottnálnagyobb koncentrációban.
A táblázat szerinti — és még egyéb fel nemsorolt — anyagok tartós behatás esetén a megadottnál kisebb koncentrációban is okozhatnakidült mérgezést. Rendkívül alattomos és többnyirekevéssé ismert egyes szerves oldószereknek tartós használatakor fellépő foglalkozási ártalom.Ilyen oldószerek pl. a benzol és homológjai, valamint ezek Származékai. Igen mérgezőek továbbá
75.1.1. táblázat
Néhány gáz erős mérgező hatást kifejtő koncentrációja
(térfogatszázalékban)
C12 0,0017
p h 3 0,0036HCN 0 ,0 1 1
s o 2 0,015 Amiin 0,018H jjS 0,043HC1 0,12
CO 0,26N H , 0,50c o 2 4,0

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 343/366
M É R G E Z É S E K 345
a telítetlen szénhidrogének, ezenkívül valamennyi metilszármazék (metil-klorid,dimetil-szulfát, metilészterek, metilalkohol), tetraklór-etán, etilén-klórhidrin, eti-
lénglikol, etilén-oxid, etilén-diamin, dioxán és sok más vegyidet is.Gázálarc használata esetén természetesen figyelemmel kell lenni a megfelelő
szűrőbetétek alkalmazására. A 75.1.2. táblázat a gázálarc-szűrőbetétek jelzéséről,színéről és védő hatásáról ad tájékoztatást.
75.1.2. táblázat
Gázálarc-szűrőbetétek jelzései és védő hatása
Jelzés Szín Védő hatás
A barna szerves anyagok gőzeiB szürke oldószerek, halogének, haloid-
savak, nitrózus gázokC szürke + sárga kolloid porD kék cián-hidrogénE sárga kénsavI kék + barna ciklonK zöld ammóniaL sárga + piros kén-hidrogénM sárga + kék kén-hidrogén -f ammóniaO sárga + zöld foszfor- és arzén-hidrogén
R sárga + barna kén-hidrogén és csekély meny- nyisé^ü szerves oldószergőz
Pb fehér + barna ólomgózCO szürke szén-monoxidTu piros égési gázok és pernyeCl zöld + sárga klórF piros füst
Teljesen ártalmatlan oldószernek’ csak a vizet lehet tekinteni. Megfelelő körül
mények között használva viszonylag kevéssé ártalmasak a következő oldószerek:tiszta benzin, dekalin, etilalkohol, dietiléter, aceton, ecetsavas etilészter, metilén--klorid, vinil-klorid, tetraklór-etilén.
Körültekintést és óvatosságot igényel a mérgező anyagok pipettázása. A száj-üregbe kerülő mérgező gőzök a nyálkahártyán át felszívódva halált is okozhatnak.Ilyen veszélyek kiküszöbölésére valók a különböző méregpipetták. Legmegfelelőbbek az olyan konstrukciók, amelyekben a pipettához csatlakozó evakuált csaposedény megnyitásával szívható fel a mérgező folyadék.
A mérgezésnek a laboratóriumban nemcsak a tüdőn és gyomron át van lehetősége, hanem a bőrön keresztül is. Évekig tartó fájdalmas fekélyeket okoz a bőröna tömény HF-oldat vagy annak gőzei. A pusztulás idővel a. csontokra is átterdje.Irodalomból ismeretes baleset szenvedő szereplője több ujjpercét vésztette annakkövetkeztében, hogy kalcium-fluoriddal és kénsavval végzett üvegmaratás közbenkezét kb. 2 percen át érte a tömény fluor-hidrogén-gáz. Hasonló esetben azonnalibő vizes öblítés és magnézium-oxidból glicerinnel készült kenőcs alkalmazása segíthet.
Az ép bőrön keresztül is felszívódó mérgező anyagok közé sorolhatók még:a HgCl2, a nehézfémek zsírsavas sói, stb.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 344/366
346 B A L E S E T I V E S Z É L Y É S Ó V AT O S SÁ G I R E N D S Z A B Á L Y O K
Igen súlyos bőrsérüléseket okoz — különösen melegen — a fröccsenő töménylúg, azonkívül a konc. kénsav és salétromsav, a folyékony bróm és egyéb anyagok.Nagyobb üvegből való áttöltésük esetén védőszemüveg, gumikesztyű és gumikötény használata kötelező. Bróm- és tömény hidrogén-fluorid-oldat bármily csekély mennyiségével történő művelet során kötelező a gumikesztyű használata.
Különlegesen erős mérgeket (lásd 75.1.3. táblázat), illetve ezek oldatait tartalmazó edényeket „Méreg” jelzéssel kell ellátni, és lezárt ún. méregszekrényekbentárolni.
75.1.3. táblázat
A laboratóriumi munka során használatos különlegesen
erős mérgek
Anilin Ani lin-klór-hidrát Antimon vízben oldódó sói Arzén és vegyületei Bárium és vegyületei (BaS04kivételével)
Brucin és vegyületei Cián és alkáli-cianidok Dietil-anilin Dimetil-anilin Dimetil-szulfát Diklór-anilin-szulfát Fenolok nitroszármazékai Fluor-ecetsavFluor-hidrogén és vegyületei Fluor-foszfátok F oszfor-halogenidek Higany sók Kálium-klorát Kálium-kromát
Klór-anilinKrómsavMetilalkoholMetil-halogenidek
Monoetil-anilinMonoklór-ecetsavNátrium-kromátNátrium-klorátNikotin és sóiNitro-anilinNitro-benzolÓlom-tetraetilOzmiumsavOxálsav és oldható sóiPikrinsavSzelén-hidrogén és szelenidek Sztrichnin és vegyületei Tallium és vegyületei Xilil-bromid
A mérgezések közül elsősegélynyújtásra heveny mérgezések esetén kerül sor,minthogy az idült mérgezések már foglalkozási ártalmaknak számítanak, és azokorvosi kezelést igényelnek.
Heveny mérgezés esetén az élet megmentésében a helyes elsősegélynyújtásszerepe döntő lehet. Legfontosabb a gyorsaság! Éppen ezért az elsősegélynyújtásmódjai közül mindig azt alkalmazzuk először, ami azonnal rendelkezésünkre áll,még abban az esetben is, ha az kisebb értékű, mint egy másik eljárás, amelynekalkalmazására csak időveszteség árán kerülhet sor. A hatásosabb eljárást ilyenkorkésőbb alkalmazzuk.
A beteget a mérgező légkörből azonnal el kell távolítani a mérgező anyagnaka szervezetbe történő további bejutása és behatásainak megakadályozása érdekében.
A gyomor- és bélrendszerbe jutott mérget hánytatással, hashajtással távolítjukel, illetve aktivált szénnel (Carbo activatus, Carbo medicinalis) hatástalanítjuk. A leghatásosabb egyszerű hánytatási mód az aktívszenes hánytatás. Egy liter langyos vízben 2—3 evőkanálnyi aktív szenet elkeverünk, és ebből 1/2 liternyit megitatunk, majd azonnal garatingerlést alkalmazunk. Ha a mérgezett a szuszpen

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 345/366
M É R G E Z É S E K — R O B B A N Á S , T Ű Z 347
ziót kihányta, úgy a hánytatást újabb adaggal néhányszor megismételjük. Ha ahányás alatt a mérgezett fuldokolni kezd, arca elkékül, úgy valószínűleg hányadék
jutott a légutakba. Ilyenkor a mérgezett felsőtestét előrehajtjuk, hátát a lapockákközött erélyesen, de nem durván ütöget jük. Ha a darabos hányadék a garatban akadtmeg, úgy azt zsebkendőbe csavart ujjal eltávolítjuk.
Tilos hánytatást alkalmazni az alábbi esetekben:— eszméletlen vagy görcsös állapotban, mert ilyenkor fulladást okozhat;— erősen legyengült állapotban;— maró anyagokkal való mérgezés esetén (ilyenkor a mérgezettel vizet vagy
célszerűbben tejet itatunk);— ha a mérgezett véreset vagy vért hány.
A már egészen vagy részben felszívódott méreg hatástalanítása elsősorban
orvosi feladat. Egyes esetekben, így oxálsavval vagy szervetlen fluorvegyületekkelvaló mérgezés esetén a mérgezettnek az orvosi beavatkozásig szájon át valamilyenkalciumsó oldatát (kalcium-klorid. kalcium-glukonát) adjuk.
A bőrre került maróanyagot (lúg vagy sav) előbb ruhával vagy szűrőpapírralitatjuk fel, s ezután a bőrt bő vízzel mossuk, majd a gyulladt bőrfelületet vazelinesvagy olajos kötéssel borítjuk. Ha törlőruha vagy szűrőpapiros nincs kéznél, úgybőséges vízzel, éspedig egyszerre nagyobb mennyiséggel mossuk le.
A szemek kimosását mindig tiszta, lehetőleg langyos vízzel végezzük, és savatvagy lúgot, akkor sem használunk, ha a szembe lúg vagy sav került. Ilyenkor a követendő eljárás a következő: a fejet mérsékelten oldalt fordítjuk, hüvelyk és mutatóujjunkkal az alsó és felső szemhéjjat széthúzzuk és a folyadékot 10 —15 cm magasságból a belső (az orr felé eső) szemzug felé irányítva, a szembe csurgatjuk.
Ha a szembe oltatlan mész került, úgy azt mindig 5%-os cukoroldattal mossuk ki. ,
Ha gáz vagy gőz belélegzése okozott mérgezést, úgy a mérgezettet friss levegőre, vagy tiszta levegőjű, más (nyitott ablakú) helyiségbe visszük, és ott fektetjük. A felsőruhákat a törzsről eltávolítjuk, és a mérgezettet könnyű takarókkal betakarjuk. Eszméletlenség esetén a mérgezettet lefektetjük, fejét balra fordítjuk.
Légzési zavarok esetén ammóniát szagoltatunk. Ismeretlen gázzal történt
mérgezésnél mesterséges lélegeztetést csak végső szükségben szabad alkalmazni,mivel számos, pl. klór-, kén-dioxid, arzén-hidrogén, bróm, nitrózus gőzök, stb.-gázmérgezés esetén a mesterséges lélegeztetés tilos! Szén-dioxid-, szén-monoxid-,kén-hidrogén-mérgezéskor friss levegőn mesterséges légzést alkalmazunk.
75.2. Robbanás, tűz
Robbanások bekövetkezhetnek bármely, nyomáskülönbségnek kitett edényfal mechanikus sérülése esetén. Evakuált edények berobbanása (implóziója)'éppúgy okozhat szilánksérülést, mint az explózió. Új, használatlan vákuumexszikká-torokat és egyéb vákuumberendezéseket célszerű első alkalommal drótháló vagytakaró alatt leszívatni, hogy az esetleges robbanáskor szétrepülő éles üvegszilánkok sérülést ne okozzanak. Igen veszélyes robbanást okozhat nagynyomásúgázpalackok feldőlése az egyébként jelentéktelen mértékű korróziós sérülés helyénmeginduló repedés következtében. H2-, 0 2-, N2- stb. gázpalackot ezért csak bizto-

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 346/366
348 B A L E S E T I V E S Z É L Y É S Ó V A T O SS Á G I R E N D S Z A B Á L Y O K
san rögzített helyzetben szabad használni. Olyan edényeket, ampullákat, amelyek
ben túlnyomás tételezhető fel, védőszemüveggel, kesztyűben, esetleg további, méghatásosabb védőberendezések alkalmazásával szabad csak felnyitni. Egyszerűbbesetekben az ampulla rongyba való csavarásával is jelentősen csökkenthetjük aszilánksérülés veszélyét.
Éghető gázok levegővel vagy oxigénnel keveredve veszélyes robbanó elegyeketalkothatnak. A 75.2.1. táblázat néhány robbanó elegy alsó és felső koncentráció-határát, lobbanáspontját (lásd 75.2.3.) és öngyulladási hőmérsékletét szemlélteti.
75.2.1. táblázat
Néhány szervetlen és szerves anyag levegővel alkotott elegyének robbanási határkoncentrá-
ciója és öngyulladási hőmérséklete (folyadékok esetében azok lobbanáspontja),valamint az alkalmazható oltóanyagok
A n y a g
Robbanási határ-koncentráció
Lobbanáspontöngyulla
dási hőmérséklet
C°
Alkalmazható oltóanyagokalsó j felső
(térf. %)
C°
1. 2. 3. 4. 5. 6.
Hidrogén 4,1 74,2 580
Ammónia 16 27,0 — 780 C 02, poroltóSzén-monoxid 12,5 74,2 — 651 víz, C 02, poroltóSzón-diszulfid 1,0 50 - 3 0 124 víz, C 02, poroltóMetán 6,3 13,9 — 537 —
Etán 3,12 15,0 — 510 —
Acetilén 2,5 80 - 335 —
Propán 2,37 9,7 — 466 —
Bután 1,6 85 - 6 0 430 —
Pentán 1,4 8,0 - 4 0 309 haboltó, C 02, poroltóBenzol 1,4 8 - 1 1 580 haboltó, C 02, poroltóToluol 1,27 7,0 4 619 haboltó, C 02, poroltóBenzin 1 ,2 6,0 - 6 . .. + 8 230 haboltó, C 02, poroltó
Petroléter 1,4 5,9 -4 6 246 C 02, poroltóMetilalkohol 6,0 36,5 1 2 ' 400 C 02, poroltóEtilalkohol 3,28 19 13 426 víz, C 02, poroltó
Aceton 2,15 13,0 - 1 8 604 víz, C 02, poroltóDietiléter 1,7 48 -2 9 186 C 02, poroltó
Megfelelő körültekintést, óvatosságot igényel ezért minden olyan művelet,ahol durranógáz képződhet: vizes oldatok elektrolízise, akkumulátortöltés stb.Éghető gázokat fejlesztő készülékekben (H2,H2S, C2H2 stb.) a kezdetben jelenlevő,levegőt tartalmazó keverék mindig robbanásveszélyes. Kémcsőben felfogott kisebbmennyiségű mintán felhasználás előtt mindig győződjünk meg a gáz oxigénmentességéről. Fújtatólángok légvezetékének csapját használat után el kell zárni, merta pufferedénybe világítógáz szivároghat át — még akkor is, ha a gáz kis lánggalég —, és meghatározott összetétel kialakulásakor veszélyes robbanás fordulhat elő.
Gázelegy gyulladását elektromotor vagy kapcsoló, csengő vagy telefon szikrája is előidézheti. A CS2— levegő-elegy gyulladási hőmérséklete igen alacsony(lásd 75.2.1. táblázat), úgyhogy teljesen szikramentes kapcsolók alkalmazása esetén is előfordulhat a gyulladás, pl. égő villanykörte felületén.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 347/366

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 348/366
350 B A L E S E T I V E S Z É L Y É S Ó V A T O SS Á G I R E N D S Z A B Á L Y O K
75.2.1. Gázpalackok kezelésével kapcsolatos előírások
A gázpalackok szállításával és tárolásával kapcsolatban az alábbiakra kellfigyelemmel lenni:
— Csak zárókupakkal és szelepvédő sapkával ellátott palack szállítható vagytárolható.
— A gázpalackok csak az arra kijelölt, a föld színénél magasabban fekvő raktárban tárolhatók, ahol egyéb anyagok tárolása tilos!
— Éghető és neméghető gázokat tartalmazó palackok, valamint üres gázpalackok külön tárolandók! Az üres gázpalackok krétával feltűnően megjelölendők!
— Tilos gázpalackokat fűtőtest közelében vagy szabadban, napfénynek kitett
helyen tárolni.— A gázpalackokat feldőlés ellen lánckikötéssel vagy bilinccsel kell biztosítani.
— Gázpalackot egy személy egyedül nem szállíthat, kivéve a targoncás palack-szállítást.
A gázpalackok üzemeltetésével kapcsolatos tudnivalók:— Használatbavétel előtt meg kell győződni a sarokszelep zárásáról, valamint
arról, hogy a biztosító sasszeg a helyén ván-e. Tilos bármilyen javítást végezni agázpalackon és annak szelepén, erre csak a töltőállomás illetékes. Hibás szelepűgázpalack használata tilos!
— Munkakezdés előtt a nyomásszabályozó tömítését meg kell vizsgálni.— Tilos javításokat végezni a nyomásszabályozón, vezetékeken mindaddig,
amíg a gázpalack nincs teljesen elzárva.— Folyékony gázzal töltött gázpalackokat használat közben 45°-nál nagyobb
mértékben megdönteni nem szabad.— Acetilént, ammóniát tartalmazó gázpalackhoz rézcsatlakozás nem hasz
nálható.— Oxigénpalackokat tilos olajos kézzel vagy ronggyal megfogni, a nyomás-
csökkentő szelepét zsírozni.— Nyomásszabályozó alkalmazása nélkül tilos a palackból való gázhasználat!— A gázpalackokat használat után el kell zárni.
A gázpalackok színjelzéseit a 75.2.1.1. táblázatban tüntettük fel.
75.2.1.1. táblázat
A gázpalackok színjelzése és a nyomásszabályozó csatlakozása
(ráz Színjelzés Csatlakozás
Ammónia szürke
jobbmenetes vasszelep Argon sárga jobbmenetDisszugáz sárga kengyeles vasszelepHidrogén piros balmenetKén-dioxid szürke jobbmenetKlór szürke klóros szelep, jobbmenetesNitrogén zöld jobbmenetOxigén kék jobbmenetSzén-dioxid szürke jobbmenet

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 349/366
T Ű Z V É D E L M I E L Ő Í B Á S O K 351
75.2.2. Nyomás- és vákuum alatti berendezésekkel kapcsolatos előírások
Nyomás alatt használatos berendezéseket, pl. autoklávokat vagy gázpalacko
kat előzetes nyomáspróbának kell alávetni. E nyomáspróbák gyakoriságát szabványok írják elő. Minden berendezésen fel kell tüntetni a nyomáspróba, a maximális üzemnyomás és hőmérséklet adatait, valamint a nyomáspróba időpontját.
Az autoklávokon levő feszmérő, biztonsági szelep és hőmérő működését üzemeltetés előtt ellenőrizni kell.
Nagynyomású autoklávok alkalmazása során a biztonsági előírások betartására különösen nagy figyelmet fordítsunk! Ezeket az autoklávokat könnyebb, ún.repülőtetővel ellátott helyiségben üzemeltetjük. Az autoklávok fűtésének, keverésének szabályozását más helyiségből végezzük,.
Vákuum alatt működő berendezésekkel végzett műbeletek során, — amilyen
pl. a vákuumdesztilláció, szublimáció — védőfülke vagy védőháló, továbbá védőálarc vagy védőszemüveg használata kötelező!
Vákuum vagy nyomás alatt végzett műveletekkel részletesen a 46. ill. 47.fejezet foglalkozik, melyek alapos áttanulmányozására külön felhívjuk a figyelmet.
75.2.3. Tűzvédelmi tudnivalók és előírások
A tűzveszélyes anyagúkat lobbanáspontjuk alapján osztályozzák. Lobbanáspont az a hőmérséklet, amelyen az adott folyadék felett levő gőzök a folyadék fölé
tartott gyújtólángtól fellobbannak anélkül, hogy a folyadék maga meggyulladna. A tűzveszélyes anyagokat és folyadékokat lobbanáspontjuk alapján öt veszélyességifokozatba sorolják(I—V). Minél alacsonyabb valamely folyadék lobbanáspontja,annál tűzveszélyesebb. A 75.2.3.1. táblázat néhány, a laboratóriumi munka sorángyakrabban használatos folyadék lobbanáspontját és veszélyességi fokát szemlélteti.
75.2.3.1. táblázat
Tűzveszélyes folyadékok osztályozása
Folyadék Lobbanáspont
C°
Veszélyes
ségi fokFolyadék Lobbanáspont
C°
Veszélyes
ségi fok
Acetaldehid + 28 II Etiléter -2 9 I Aceton -1 8 I Izo-bután + 60 I Amil-acetát + 21 - +34 II Izo-batil-ac^tát + 18 I Anilin + 71 III Izo-butilalkohol + 22 IIBenzin, könnyű + 58 - + 10 I Izo-propil-aoetát + 6 IBenzin, közepes 0 - + 1 0 I Izo-propil-éter + 28 II
Benzin, lakk + 23 II Ligroin + 8 IBenzol - 1 1 I Metil-acetát + 13 I
Bróm-benzol + 65 IIIMetilalkohol - 1 - + 3 2 II
Butil-aoetát + 22 II Metiléter + 41 IIButilalkoholok + 22 - +44 1 1 Monoklór-benzol + 29 IIDenat. szesz + 12 I n-Heptán + 4 IDimetiléter -4 0 I n-Pentán - 10 IDioxán + 1 2 - +18 I n-Oktán + 13 IEtil-acetát - 3 I Petróleum + 2 1 IIEtil-benzin + 5 I Petroléter -4 6 I

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 350/366
352 BALESETI VESZÉLY ÉS ÓVATOSSÁGI RENDSZABÁLYOK
Az I. veszélyességi fokú anyagok különálló raktárban, csak a tűzrendészeti ha
tóság által megengedett mennyiségben és edényben tárolhatók. Előkészítő helyiségekben és a felhasználó laboratóriumokban szintén csak a tűzrendészet általengedélyezett mennyiségű napi szükséglet — általában 1 —1 kg — tárolható.
Tűz esetén — a tűz keletkezésekor jelenlevő — minden személynek törvénybenelőírt kötelessége a tűz megfékezésében, eloltásában résztvenni. A tűzoltás megkezdése előtt a helyiséget áramtalanítani kell, ha ez nem lehetséges, úgy kizárólagporoltót és szén-dioxidos oltót szabad csak használni. A keletkezett tüzet lokalizálni kell, közeléből mindennemű éghető és gyúlékony anyag eltávolítandó!
A kézi tűzoltó készülékek közül általánosan használható a szén-dioxidos oltó
készülék ; ezt azonban pincékben, rosszul szellőztethető helyiségekben ne alkalmazzuk! Laboratóriumokban szintén általánosan használatos a poroltó készülék, amelyszén-dioxid mellett igen finom eloszlású karbonátport is tartalmaz. Emiatt gépek ésműszerek közelében lehetőleg ne alkalmazzuk! Vizzel nem reagáló folyadékokathaboltóval oltsunk. A 75.2.1. táblázat néhány általánosan használt vegyszer égésekor alkalmazható oltóanyagról ad tájékoztatást. Felhívjuk a figyelmet arra,hogy nem áramtalanított helyiségben vagy elektromos zárlat okozta tűzeseteknéltilos és életveszélyes vízzel vagy"haboltóval oltani!
Ha valakinek a ruhája meggyulladt, pokrócot vagy köpenyt borítunk az illetőre, és azzal igyekszünk a tüzet elfojtani. Ha még komolyabb égési sérüléseket
nem szenvedett, úgy célszerű tus alá állítani, azonban égési sebeket vízzel mosnitilos!
75.3. Áramütés
Az emberi testen végigfutó 2—8 mA intenzitású áram már fájdalmat okoz.12 mA izomgörcsöt vált ki, aminek az lehet a következménye, hogy az áramütöttnem tud szabadulni az áramforrástól, és szívgörcs vagy égés következtében elpusztul. A test átlagos ellenállása — kézen át a lábig mérve — kb. 1000 Q. Ha az
Ohm-törvény alapján számítva a 12 V nem is, de kb. 40 V-nál nagyobb feszültségkedvezőtlen körülmények között már halálos lehet. Gyakorlott villanyszerelő száraz kézzel és szigetelt talpazaton ennek többszörösét is károsodás nélkül elviselheti. — A tapasztalat szerint a váltakozó feszültség súlyosabb élettani hatású,mint az egyenfeszültség.
Az alábbiakban az elektromos berendezésekkel kapcsolatos munkák, szerelésekelőírásait foglaljuk össze:
Szigorúan tilos előzetesen nem feszültségmentesített elektromos berendezésenszerelőmunkát vagy javítást végezni! Ugyancsak tilos elektromos szerelést ideiglenes biztosíték alkalmazásával, pl. talpalással megoldani!
Védőföldeléssel minden 110 vagy 220 volttal működő készüléket el kell látni!Kemencék vagy egyéb melegítők használata előtt azokat ellenőrizni kell, hogynem zárlatosak-e.
Elektromos áram okozta baleseteknél a legelső teendő a sérült kiszabadításaaz áramkörből; utána a sérültet szellős helyre kell vinni, rajta mesterséges légzéstvégezni a normális légzés megindulásáig. Még a legkisebb mérvű hasonló jellegűbaleseteknél is feltétlenül orvosi felülvizsgálat szükséges!

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 351/366
SEBESÜLÉS — RAD IOAK TÍV FERTŐZÉS 353
75.4. Sebesülések
A laboratóriumi munka során, készülékek szerelésénél, üvegberendezések ex-plóziójánál vagy implóziójánál gyakori a nyílt sebbel járó sérülés. Ilyenkor azelsősegélynyújtó feladata az, hogy minél kisebbre csökkentse a fertőzési veszélyt. A sebet sem vízzel, sem más fertőtlenítő folyadékkal mosni nem szabad. Nyílt sebesetén az elsősegélynyújtás a következő:
A seb környékét, ha durván szennyezett, vizes vattával vagy tiszta vászonnalvízzel letisztítjuk. Ezt követően ugyancsak a seb környékét jódtinkturával megecseteljük, és sebkötöző pólyával vagy tiszta, fehér vászonnal bekötjük. A sebremagára soha sem teszünk vattát, sem semmiféle sebolajat; az elsősegélynyújtónem kezelőorvos!
Sérülés esetén nemegyszer szükség van arra, hogy az elsősegélynyújtó addig is,amíg az orvos a helyszínre érkezik, a bőr alatti kötőszövetből eredő hajszálér-vérzést vagy esetleg ütőeres vérzést csillapítsa. Kötőszövetes vérzésnél észrevesszük,hogy az általunk felhelyezett sebkötés aránylag gyorsan átvérzik. Ilyenkor ne vegyük le a kötést, hanem nyomókötést helyezzünk rá a következőképpen: gyermek-tenyérnyi nagyságú vattát vagy gézt kemény tömöttségűre összehajtogatunk, aztaz átvérzett kötés fölé helyezzük, majd egy újabb pólyával vagy kendővel szorosanátkötjük. Szoros nyomókötésre főleg ütőeres vérzésnél van szükség.- Ezt úgy ismer
jük fel, hogy a sebből ütemesen, mintegy lüktetve ömlik a vér. Ha a leszorításra
nem elég a nyomókötés, nagyobb erőt kell alkalmaznunk. Pl. ha a félkaron van azütőeres vérzés, akkor pólyával, vagy gumicsővel a seb felett szorítókötést alkalmazunk. Szükség esetén a szorító és a bőr közé fapálcát csúsztatunk, és a pálcafordításával húzzuk szorosabbra a szorítókötést. Ilyen szorítókötést maximum egyórán át szabad alkalmazni!
75.5 . Radioaktív sugárzás okozta fertőzések
Egészen más jellegű veszélyt jelentenek az emberi szervezetre a kémiai kuta
tásban egyre nagyobb jelentőségű radioaktív izotópok. Ezeknek azonban sem fel-használása, sem az óvórendszabályok részletei nem tartoznak e praktikum tárgykörébe. Alkalmazásuk biztonsága ugyanúgy, mint a robbanó anyagokkal valómunkáé, legegyszerűbben mikroméretben oldható meg.
Irodalom
Lazarev, N. V .: Mérgező hatású ipari anyagok. I. és H. kötet. Táncsics Könyvkiadó, Budapest, 1957.
Must, E., Ébert, A.: TJnfáUe bei chemischen Arbeiten. Rascher, Zürich, 1948.
Flörke, W.: Unfallverhütungim chemischen Unterricht. Quelle —Mayer, Heidelberg, 1951. Pieters, H. A. JCreyghton, J. W . : Safety in the Chemical Laboratory. Butterworth,
London, 1951.Schwartz, E.: Handbuch dér Feuer- und Explosionsgefahr. 4. Aufl. Jung, München, 1936.
Zimen, K. E .: Angewandte Radioaktivitát, Springer, Berlin, 1952.Munkavédelmi, balesetelhárítási és tűzbiztonsági útmutató. Összeállította az ELTE Szakszervezeti Bizottsága mellett működő munkavédelmi bizottság, Budapest, 1962.
2 3 Á lt . és szervetlen kémiai prakt.— 4 2 8 4 / 1 1 .

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 352/366
354 PERIÓDUSOS RENDSZER
V I I I
1
2
* §
2 H e
4 , 0 0 2 6
1 0 N e
'
2 0 , 1 8 3
1 0 A r
3 9 , 9 4 8 2
6 ' F e
2 7 C o
2 8 N i
5 5 , 8 4 7
5 8 , 9 3 3 2
5 8 , 7 1
3 6
K r
8 3 , 8 0 4
4 R u
4 5 R h
4
6 P d
1 0 1 , 0 7
1 0 2 , 9 0 5
1 0 6 , 4
5 4 , X e
1 3 1 , 3 0
5 8 C e
5 9 P r
6 0 N d
6 1 P m
6 2 S m
6 3 E u
6 4 G d
6 3 T b
6 6 D y
6 7 H
o
1 4 0 , 1 2
1 4 0 , 9 0 7
1 4 4 , 2 4
1 4 5
1 5 0 , 3 5
1 5 1 , 9 6
1
5 7 , 2 5
1 5 9 , 9 2 4
1 6 2 , 5 0
1 6 4
, 9 3 0
7 6 0 s
7 7 I r
7 8 P t
1 9 0 , 2
1 9 2 , 2 1
9 5 , 0 9
GPíCOC*OO(N
<N
9 0 T h
9 1
P a
9 2 U
9 3 N p
9 4 P u
9 5 A m
9 6 C m
9 7 B k
9 8 C í 9 9 E s
2 3 2 , 0 3 8
2 3
1
2 3 8 , 0 3
2 3 7
2 4 2
2 4
3
2 4 7
2 4 9
2 5 1
2
5 4
V I I
1
2
ffi o
Pí 00<*> \
** 000>«H 1 7 C l
3 5 , 4 5 3
2 5 M n
5 4
, 9 3 8 0
3 5 B r
7 9 , 9 0 9
4 3
T e 9 9
5 3
I
1 2 6 , 9 0
4 4 7
5
R e
1 8 6 , 2
8 5 A t
2 1 0
V I
1
2
« «K OPí Pí
8 0 1 5 , 9 9 9 4
1 6 S
3 2 , 0 6 4
2 4 C r
5 1 , 9 9 6
3 4 S e
7 8 , 9 6
4 2 M o
9 5 , 9 4
5 2 T e
1 2 7 , 6 0
7 4 W
1 8 3 , 8 5
8 4 P o
2 1 0
V
1
2
tű oPí Pí
7 N 1 4 , 0 0 6 7
1 5 P
3 0 , 9 7 3 8
2 3 V
5
0 , 9 4 2
3 3 A s
4 7 , 9 2 1 6
4
1 N b
9
2 , 9 0 6
5 1 S b
1 2 1 , 7 5
7 3 T a
1 8
0 , 9 4 8
8 3 B i
2 0 8 , 9 8 0
C4
í> ffi opH tí
6 C 1 2 , 0 1 1 1 5
1 4 S Í
2 8 , 0 8 6
2 2 T i
4 7 , 9 0
3 2 G e
7 2 , 5 9
4 0 Z r
9 1 , 2 2
5 0 S n
1 1 8 , 6 9
7 2 H f
1 7 8 , 4 9
8 2 P b
2 0 7 , 1 9
o
Z
I
I I I
n « rj O
Pí Pí
5 B 1 0 , 8 1 1
1 3 A 1
2 6 , 9 8 1 5
2 1
S e
4 4 , 9
5 6
3 1
G a
6 9 , 7 2
3 9
Y
8 8 , 9 0
5
4 9 I n
1 1 4 , 8 2
5 7 L
a
1 3 8 , 9 1
6 9 í m
7 0 Y b
7 1 L u
1 6 8 , 9 3 4
1 7 3 , 0 4
1 7 4 , 9
7
8 1 T I
2 0 4 , 3 7
o< b -
<NCT>oO
1 0 0
F m
1 0 1 M d
1 0 2 N o
1 0 3 L w
2 5
3
2 5 6
2 4 5
2 5 7
<N
l-H
•
a 2p í a
4 B e
9 , 0 1 2 2
1 2 M g
2 4 , 3 1 2
2 0 C a
4 0 , 0 8
,
3 0 Z n
6 5
, 3 7
3 8 S r
8 7 , 6 2
4 8 C d
1 1 2 , 4 0
1 5 6 B a
1 3 7 , 3 4
8 0 H g
2 0 0 , 5 9
8 8 R a
2 2 6
—<N
HH V O
Pí Pí
1 H 1 , 0 0 7 9 7
3 L i
6 , 9 3 9
1 1 N a
2 2 , 9 8 9 8
1 9 K
3 9 , 1 0 2
2 9 C u
6 3 , 5 4
3 7 R b
8 5 , 4 7
4 7 A g
1 0 7 . 8 7 0
5 5 C s
1 3 2 , 9 0 5
6 8 E r
1 6 7 , 2 6
7 9 A u
1 9 6 , 9 6 7
8 7 F r
2 2 3
_ OO /" --«N 00 OO OO CDIcD
Q. N-" w OO(A / T3 c* OO <D r-
Í3 M s o Ph o> Aí

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 353/366
összefoglaló irodalom a részletes részhez
Brauer, G .: Handbuch dér Práparativen Anorganiechen Chemie, Bd. I —II. Enke, Stutt
gart, 1962.K a w h h u k o s , H . r . : PyxoBOACTBO no H eopraHHqecKO My cHH Te3y. T o c x k mm s a s t , M c c K B a ,
1 9 5 3 . Dodd, R. E., Robinson, P. L .: Experimental inorganic chemistry. Elsevier Publ. Co.,
Amsterdam —Houston —London —New York, 1954.Inorganic Syntheses, Vol. I —VIII. MrGraw-Hill Book Co., New York —Toronto —London
1939-1965.Grubitsch, H .: Anorganisch-práparative Chemie. Springer, Wien, 1950.
Hecht, H . : Práparative Anorganische Chemie. Springer, Berlin —Göttingen —Heidelberg, 1951.
Vanino, L . : Handbuch dér Praparativen Chemie, I —II. Enke, Stuttgart, 1913 —1937. Jonassen, H. B. ,Weisberger, A.: Techniqúe of Inorganic Chemistry, Vol. I. Interscience
Publishers, New York, 1963.Goldschmidt, K . : Aluminothcrmio. Hirzel, Leipzig, 1925. van Arkel, A. E . : Reine Metalle. Springer, Berlin, 1939. A ngerer, É., Ebért., H .: Technishe Kunstgriffe bei physikalishen Untersuchungen. Vieweg,
Braunschweig, 1959.K y d p a , O ., r u TM a n , E . : E j ieKTpoj iHTHnecKoe no j iyqeHHe MeTajuiHMecKHX nopoiuKOB. H3Aa-
T e j i b c i B o A K ageM M H H a y K y K p a H H C K o n C C P , K n e B , 1 9 5 2 . Fűnk, H .: Darstellung dér Metalle im Laboratórium. Enke, Stuttgart, 1938. HaTancon, 3. M . : K o j u i o h a h u c MeTajiJibi. H3AaTenbCTBO AKageMHH H a y x yK paH H cKoi í C C P ,
K n e B , 1 9 5 9 . Mutter E .x Reuther H . : Elektrochemisches Praktikum. Th. Steinkopf Veri., Dresden —
Leipzig, 1950.
K A a y c , K - K . : M3ÖpaHHbie Tpygbi no x h m h h njiaTHH CBbix M cTajijiOB. H agaTejibCTEO A k s a c m h h H a y x C C C P , M o c K B a , 1 * 9 5 4 .
JJyKau iee, K . H . : Pe A xne M e ia j i j i b i m h x Hcno j ib30BaHHe b npoMHuuieHHocm. PteAaTenbCTBO
AnaAeM HH H a y x C C C P , M ocK B a , 1 9 5 6 .«2i u a m o e, B . A . : M oK raj ion ^H ue coeAHHeHHH. HsaaTejibCTBO AKaaeMHH Ha yK yxpaHHCK OÍi
C C P , K n e B , 1 9 5 8 .Jlee un , A . <p.t X h m h h öopa TO B. H3AaTenbCTB0 JlaTBHfócKOH AKaAeMHH H a y x JlaTBHHCKOH
C C P , P n r a , 1 9 5 3 . Heftm,ann,E.: Chromatography, Reinhold Publ. Corp. New York, 1963.Goehring, M .: Ergebnisse und Probleme dér Chemie dér Schwefelstickstoffverbindungen.
Akademie Verlag, Berlin, 1957.Grinbefg, A . A .: Bevezetés a komplex vegyületek kémiájába. Akadémiai Kiadó, Buda
pest, 1958. Bailar, J. G.\ The Chemistry of the Coordination Compounds. Reinhold Publ. Co., New
York, 1956. Hein, F .: Chemische Koordinationslehre. Hirzel Vorlag, Leipzig, 1950. Martell, A . E., Calvin, M . : Die Chemie dér Metallchelatverbindungen. Verlag Chemie,
Weinheim, 1958.T a m n a e e , H . B . : X h m h h peAKHx aneMeHTOB. HaAaTenbCTBO AxaAeMHH H a y x C C C P, M ocK Ba ,
1 9 5 4 .
2 3 *

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 354/366
356 Ö S S ZE F O G L A LÓ I B O D A L O M
TJer poe, A. J J.t Mu ponoe, B. 0., noHOMapewco, B . A., ^epnu ui ee, E . A.: Chhtc3 KpeMHHHop- raHHqecKHx MOHOMepoB. H3ÁaTejibCTB0 AKaAeiwHH HayK CCCP, MocKBa, 1961.
Andpuanos , K. A.: KpeMHMiíopraHHMecKHe coe;umeHHfl. rocyflapcTBeHHoe HaynHo-TexHH- MecKoe H3AaTejibCTB0, MocKBa, 1955.
Rochow, E . O .: Einführung in die Chemie dér Silikone. Verlag Chemie, Weinheim, 1952. Rochow, E . G., Hu r d , D . T., L ew i s, R. N . : The Chemistry of Organometallic Compounds, John Willey, New York, 1957.
K r au se, E . , Grosse, A . : Die Chemie dér metallorganischen Verbindungen. Berlin, 1937. Pasca l , P . : Nouveau traité de chimie minérale. Törne I —X X . Masson, Paris, 19 58 -. Qmél i n s Handbuch dér anorganiBchen Chemie. Verlag Chemie, Weinheim, 1926.TJllmans Encyklopadie dér technischen Chemie. Dritte Auflage, Úrban —Schwarzenberg,
München—Berlin, 1953 —.

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 355/366
Képletmutató
A Ba(H2POz)2•H 20 H 265
BaS20 8*2 H 20 II 275 Ag2[HgI4] I 312
BeCl2-4 H 20 n 232 AgMn04 I 301
BiBr3 II 212 A gN 02 I 302
BiH3 II 146 A1 II 146
Bil3 II 215 AlBr8 H 211
A1C18 n 221
Br2 II 138, 166
AlClIj H 224
A1I„ H 214, 216C
A120 8 I 255 c n 170
A1P II 281 CBr4 n 220
A12(S04)3*18 H 20 I 256 CCl2Br2 n 223
AljSes H 276 CI4 ÍI 221
Ar H 180 /CH3AlCl2/ 2 II 323
A b II 169 /(CH3)2AlCl/2 II 323
AsCl, I 221 (CH3)3A12C13 II 323
A sF8 II 222 CH3BC12 II 322
AsH, II 196 (CH3)3SiC6H 5 n 331
A bPS4 n 276 (CH3)3SiCeH 4CH8 II 333
CH3SiCl3 II 328
B (CH3)2SiCl2 II 328
(CH3)3SiCl H 328B I 231, II 173 (i-CaHjO^lCl n 326BBrs n 212, 220 (C2H 5)2SnCl2 II 339B(C6H s)3 H 320 (C2H 5)2Sn(OOCCH8)2 II 339BCla II 210, 216, 217 /(C2H 6)2SnO/n n 339BF8 II 222 (C2H e)20 -B F 3 n 320BF8.(02H 6)2O n 320 (CjHjOJaTiCl n 342
BN n 277 (C2H 6)3P b 0S02C6H 4CH8 II 341B(CH3COO)8 n 268 (CsHjCHgíaSnCla n 340B20 8«24 W 0 3-aq II 306 (CaH 3CH2)3SnCl H 340BaCS3 n 274 C6H 6HgCl n 317BaCl8-2 H 20 I 251 C6H 6HgOOCCH3 II 317BaH2 H 201 C6H 5SiCl3 II 336Ba3H 4(IOB)2 H 269 CflH 6Si(OC2H 6)3 II 332

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 356/366
358 KÉPLETMTTTATÓ
(CN)2 I I 234CO I 228C 0 2 I 227, II 129
COCl2 II 131Ca II 180
CaC2 II 281CaCl2-6 H 20 I 250
CaH2 II ,£01Ca3N , II 279Ca3P2 II 281CaS04-2 H jjO I 250
CaSi n 284CaSi2 II 284CaW 04 II 267Cd II 179
Cd(CH3COO)2 I 307Ce II 309Ce02 n 309Ce20 3 II 310Ce(S04)2 n 310Cla II 130, 138, 145, 166,a 0 2 n 245Co II 177[Co2(CO)8] II 301
[Co(NH3)4C 03]N 0 3‘ 1/2 H 20 I 287[CoíNHjJ^Cla I 287[Co(NH3)6]Cl3 I 288[Co (NH3)5C1]C12 I 288
[Co ((NH3)5H 20]C13 I 289
[Co (N H 3)6]{N 0 3)3I 291
Co (N H3)5N 0 2]C12 I 290
[Co(NH3)5ONO]Cl2 I 290
[Co (NH3)4(N 02)2]C1 I 291
[ C o í N H a J . Í N O a ) ^ ^ : , I 2 9 0
[Co(NH3)3(N 02)3] I 291Co2(S04)3*18 H 20 II 262[CoS(CO)5]n II 301
Cr I 268CrBr3 II 213C ra 2 n 228CrCl3 n 205[Cr(p5H 70 2)3] II 297
[Cr(H20 )6]Cl3 I 272, n 230
[Cr(HzO)4Cl2]Cl•2 H 20 I 271, n 231[Cr(NH3)5Cl]Cl2 I 272[Cr(NH3)a](N 03)3 I 272
Cr2Os I 268, II 254
Cr02Cl2 II 242
Cr2S3 II 273
Cs II 174, 185, 200
CsH n 200
CuH II 202
[Cu(C2H 40 2N)2] n 297 CuCl n 226
Cu C12« 2 H 20 I 297
Cu2[H g I4] I 298
Cu (N H 3)4]S 0 4.H 20 I 298
CuzO II 255
Cu (0H)2-CuC0 3.H20 I 298 CuS04 •5 H zO I 297
D
d 2 n 107
E
E u S 04 n 147
F
F2 II 145, 165 FaO II 244 Fe II 177
Fe3C II 282 FeCl2 n 227 FeCl3 II 204 [Fe(CO)s] n 299 [Fe(CO)4]H2 II 300 [Fe(CO)4I2] II 299 [Fe(NO)2(CO)2] II 302 Fe(N03)3*9 H 20 I 281
Fe3(P 04)2-8H 20 n 266 FeS 04-7 H 20 I 281
G
Ge II 178 GeH4 II 145
Gd n 149
HH a II 129, 165 H[BF4] II 292 H 3B 0 3 I 232

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 357/366
KÉPLETMUTATÓ 359
HBr II 139, 190
HC1 n 139, 190
HCN II 139 H 4C2N2S2 II 235
h [Co (CO)4] n 300
HF II 139, 189
H 4[Fe(CN)6] II 294
H l II 139, 192
H sI 0 6 1 183
H I0 3 I 181 H 258
H N 3 n 289
H 0 S 0 2C1 n 239 H 20 2 II 270
HOíCH^jSiOSiíCH^OH II 334
H 2[PtCl6] I 292
HPH 20 2 II 265
H 3P 0 3 II 264
H 3P 0 4 II 263
H 2S II 139, 193
H2St n 194
H 2Se H 195
H 2Te n 195
He II 130
H fH 108, 184
Hg(C3H 7)2 H 318
Hg(CH3COO)2 I 311
Hg2(CH3COO)2 I 309
Hg2(N 03)2-2 H 20 I 309
HgS II 273
Hg(SCN)2 I 312
H g2(SCN)2 I 312H gS 04 II 263
I
I2 II 167
IBr II 234
ICI II 233
IC13 n 233
120 4 II 246
120 5 I 180, II 246
K
K II 174, 185
KA1(S04)2*12 H 20 I 256
K[BF4] II 292
K 2BiS4 II 276
K B r0 3 II 258
KC103 I 239, n 256
KC104 I 178
K 3[Cr(C20 4)3]•3 H 20 I 273
K 3CrOg H 271
KC r(S04)2- 12H 20 I 269
KFeS2 H 276 K 3[Fe(CN)6] I 237
K 3[Fe(C20 4)3] .3 H 20 II 295
KH II 200
K^H^CN),] I 311
K 2 tHgI4] 131 1
KJHgíSCN),] I 312
K I0 4 n 259
K 2M n04 II 266
KN H 2 n 288
K N 0 3 I 243
2 K 20* S i02- 12 W 0 3-aq II 307
K[PbI3] •2 H 20 II 294
Kj[PbCl6] H 293
K 2S20 5 I 188
K 2S20 8 I 192, H 271
L
La II 308
LaCl3 H 217, 309
La2(S04)3 II 30ÖLi II 174, 179, 186
L í A 1H4 II 199
Li3As n 280
Li3Bi H 280
LiC6H 6 II 315
L iH II 198
Li3N H 277
LiNH2 II 288
Li3P II 280
Li3Sb II 280
Mg H 181
Mg(C104)2 H 128, 257
Mg3N 2 n 278
MgSCV 7 H 20 I 250
MgjjSi n 283
Mn H 173
MnS04-5H *0 I 250

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 358/366
360 K É P L E T M U T A T Ó
Mo II 173, 178
M o0 2 II 254 M o 0 3 II 250
Mo S jj H 274
N
N 2 n 129, 168N H 4BS0 8-4 H 20 I 234N H 4Fe(S04)2‘ 12 H 20 I 282
(NH4)2Fe(S04)2-6 H 20 I 281NH 2OH n 290
N H 3 II 130, 140, 196
(NH3OH)Cl II 289(NH30 H )3P 0 4 n 2905 (N H4)20 •12 Mo 0 3 •7 H 20 II 305
(NH4)2[PbCl6] n 293(NH ^P tCy n 295(NH4)2S20 8 II 271NH4V(S04)2-12H20 n 260(NH4)2V(S04)2'6 H20 n 261
(NH4)2Zn(S04)2-6 H 20 I 306N 2H 6S 04 I 202
N20 n 250NOC1 II 241Na II 174, 184Na3As II 280
N aB 02 •H 20 2•3 H 20 I 234Na3Bi II 280Na[(CBH 6)8GNB] n 322
Na2C 03 I 244Na3H 2I 0 6 n 258
N a I0 3 I 182
N a l04 n 268
NaCrS2 II 275Najj[Cu(OH)4] II 296Na2[Fe(CN)5N 0 ]- 2 H 20 II 303NaH II 200Na3M n04 x H 20 II 267NaN 3 II 288
NaNH2 II 286Na N 02 I 242
NaNO a I 242
Na20*4 M o03-aq II 3045N a20*12 M o03*aq II 304
Na20 2 n 270NajO-VjOj-aq II 3042N a20 -V 20 fi-aq II 304
3N a20*5 V 20 5*aq II 304 Na3P II 280
(NaP03)n n 266 Na4P20 7 n 266
Na2[Pb(OH)6] n 296
Na2S 03*7H20 I 240
Na2S2Ö3 I 241
Na3Sb n 280
Na3SbS4 •9 H 20 I 219
Na2[Sn(OH)6] II 296
NbClfi H 219
NbOCl3 n 243 Ni II 277
[Ni(CO)4] n 298
[Ni(NH3)6]Br2 I 287
N iS04-7 H 20 I 286
0
0 2 II 129, 145, 167
0 3 H 146
P
PBr3 n 213
PC13 n 207
PC15 n 208
PCl3Br2 II 238
PI3 II 214
P ^ n 215 (PNCl2)n n 291
P20 5 II 128
P0C13 n 236
P jsSs II 274 PSC13 n 238
Pb(C2H 5)4 II 341
Pb(CH3COO)4 II 267
PbCr04 I 263 PbCr04 PbO I 263
PbH 4 II 145 Pb(N3)2 II 289
Pb(N 03)2 I 263
P b 02 I 260
Pb30 4 I 260
Pb3(OH)2(CQ8)2 I 264
Pd n 187
Pt n 187

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 359/366
KÉ PLETMUTATÓ 361
R
ReCl* H 219Re207II 261Rb H 174, 184RbH H 200
s
S II 167SjClj I 184, n 210S02n 130, 140
S08n 247SOBr2n 237S0C12I 191, II 236S203II 247S02C12I 191S205C1ZII 240Sb II 169SbCl3H 225SbCl5I 221, II 206
SbH3n 146, 196Sb2(S04)3H 263Se II 167SeCl4n 210Se02n 247SeOCl2H 240Sin 173SiBr4II 213Si(CH3)4II 333Si(CH3COO)4n 268
SiCl4H 208, 216SinCl2n+2II 209SiCl jn 223SiH4II 197SiaH6n 197SiHF3n 8 0
Sil4II 214Si(OCaHfi)4n 327Si02-xH20n 268
Si02-12W03-aq n 306SnBr4II 211Sn(C2H5)4n 337Sn(C4Hg)4n 338SnCl4II 206SnH4H 145Snl4H 214SrH2II 201
TTaClsn 219
Ten 168Té02II 249Th n 184ThBr4n 216Ti II 175, 182TiB2II 286TiCII 282TiClan 80TiCl3II 226TiCI4II 215, 217Ti/H n 201Til4II 214TiNn 279Ti203II 253Ti(OC2H6)4n 342TiSi2II 284üUII 148UC14II 228U03II 251 V
V II 173 VC12II 228 VC13n 228 VC13-6H20 II 231 V203n 254 VOCl3II 243
wWII 178WBr6II 220WC16II 205W02II 254W03II 251ZZn(CH3COO)2I 306ZnC03•Zn(OH)2I 306ZnS04*7H20 I 305Zr II 108, 176, 184ZrBr4II 216[Zr(C5H702)4] n 297ZrCI4n 219Zrl4II 214V
YbS04n 147

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 360/366
Tárgymutató
A
.aceton 142acél 26acidokomplexek 292adagolófeltét 287adszorpció 106aktív szén 170 ~alacsony hőmérséklet előállítása 35
Alfatron vákuummérő 66
alkoholok 141alumínium146— -bromid 211
— klorid-dijodid 224—-foszfid 281—'jodid 214, 215—-klorid 221—-szelenid 276amalgámok 312 *v.ammónia 130, 140, 196ammónium- [hexakloro-platinát(IV)] 296— -[hexakloro-plumbát(lV)] 293— -molibdát 305
—-peroxi-diszulfát 271— 'Vanádium(II)-szulfát-hexahidrát 261 — vanádium-timsó 260. anódeffektus 148anódos oxidáció 147
Anhydron 128antimon 169---hidrogén 145, 196antimonidok 280antimon-pentaklorid 206—(OT)-szulfát 263— -triklorid 226
Apiezon-viasz 32—zsír 68aprítás 84argon 130arzén 169arzenidek 280. arzén-ortotiofoszfát 275—-trifluorid 222
autokláv 80automatikus adagoló 116— gáztovábbító 132—szűrő 86
A
áramlássebesség 132 áramütés 352
B
báriumamalgám 313 ~ bárium-ditionát-dihidrát 275
- -hidrid 201 —— -hipofoszfit-monohidrát 265 — perjodát 259 ~— -tiokarbonát 274 bepárlás 99
berillium(II)-klorid-tetrahidrát 232 bimetáll hőérzékelő 51 bizmut-hidrogén 146 bizmutidok 280 bizmut-tríbromid 212— -trijodid 215 Bodenstein-íele szelep 70 bombacső 81 ~bór 173 boridok 285 bór-nitrid 277— -triacetát 268
— -tribronajd 212, 220 — trifluorid 222
dietiléter 320— -triklorid 210, 215, 217 bórvolfrámsav 306 bróm 138, 166 Bunsen-csúcs 61

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 361/366
T Á R G Y M U T A T Ó 363
E
egykristály-előállítás 95elektromos kemencék 40elektroncserélő 109ellenállás-hőmérők 44—-kemencék 40elővákuum54Ergon-üveg 17ezüst-kloria-kitt 32extrakció 105extrakciós oszlop 91
C
Gartesius-féle manosztát 73centrifugálás 88cérium309—(Ill)-oxid 310—(IVJ-oxid 309-(IV)-szulfát 310cézium174, 185, 200—-hidrid 200Claisen-feMét 98
lombik 98cink-oxid —cink-klorid-kitt 32cirkónium108, 176, 184—-tetrabromid 216—-tetraklorid 219—jodid 214Cottrell—Möller-eljárás 118CVaig-készülék 90
Cs
csiszolatos illesztés 19
D
Dehydrit 128 Dekhotinsky-cement 32deszorpció 106desztillálás 96deutérium107 Dewar-edény 37, 52dibenzil-ón-klorid 340dicián 234dietiléter 141dietil-ón-diacetát 339--- -diklorid 339--- -oxid 339differenciális ionizáció 65diffúziós szivattyú 59difluor-monoxid 244difoszfor-tetrajodid 215[diglikokollát-réz{II)] 297dihidrogén-[tetrakarbonil-ferrát (—II)] 300di(izo-propil)-higany(II) 318d i( i z o -propoxi) -alumínium- kló rid 325dikén-diklorid .210—trioxid 247[dikloro-tetrakvo-króm(III)]-klorid-dihid-
rát 231dinátrium-[pentaciano-nitrozil-ferrát(III)]
303[dinitrozil-dikarbonil-ferrát(O)] 302disszugáz 130diszilícium-hexaklorid 209 Drechsel-féle mosópalack 118Durán-üveg 17
P
ferde furatú vákuumcsapok 67 fém-egykristály előállítása 95 fémorganikus vegyületek 314 fenil-higany(II)-acetát 317------ -klorid 317— lítium 315— -trietoxi-szilán- 332 — triklór-szilán 335 fluor 145, 165 Fluorex 21 folyadékhőmérő 43 folyadékszintjelző 88 folyadéktermosztátok 50 folyékony levegő 37 forrasztás 29 foszfidok 281 foszfor-nitrid-klorid 291 — oxid-klórid 236— pentaklorid 208— -pentaszulfid 274— -pentoxid 128— -triklorid 238— -tribromid 213 — trijodid 214 foszgén 131 foszforsav 263 foszforossav 264 frakcionáló feltét 97— szivattyú 59 frakcionált diffúzió 92— kristályosítás 93
G
Gaede-f é le f o r g ó n y e lv Q S o l a j s z i v a t t y ú 57 g a l l i u m 43 g a z o m é t e r 135gázadszorpció üveg- és fémfelületeken 73gázbüretta 136gázfejlesztés 115gázhőmérők 43gázkromatográfia 107gázszennyezések eltávolítása 121, 129gáztovábbító 131gáztüzelésű kemencék 39

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 362/366
364 TÁRGYMUTATÓ
gázok áramoltatása 131— ködtelenltáse 91, 118
—oldhatósága 134— portalanítása 91— tárolása 134—tisztítása, szárítása 117 Gerate-üveg 17 gérmánium 178— -hidrogén 145 getterezés 74 gőzsugárszivattyú 68 grafitbevonat 170 gumi 31gyapot 34
H
hafnium 108, 184 halogén elemek 165— -hidrogének 138, 189 hegesztés 29 heteropolisavak 308[hexakvo-króm(III)]-klorid 230 hélium 130 hidrogén 129, 165— -bromid 139, 190
-cianid 139— -fluorid 139, 189— -[hexaciano-ferrát(II)] 294— -jodid 139, 192— -klorid 139, 190 — peroxid 270— -poliszulfid 194— [tetrafluoro-borát] 292— -túlfeszültség 144 hidroxil-amin 290 —hidroxil-ammónium-foszfát 290 ~-------klorid 289 —hidroxokomplexek 295higany 43higanydesztilláció 101 higany(II)-szulfát 263— (Il)-szulfid 273 higanyos manométer 61 higanyzáras csapok 68 hipofoszforsav 265 " hőmérséklet jelző 46 hőmérsékletmérés 43 hőszabályozás 43 hőszigetelők 35 hűtőanyagok 35 hűtőfürdők 36 hfitőkeverékek 35
I
ioncsere 107ionizációs vákuummérŐk 65 ívfénykemencék 41
izo -pentán 44— polimolibdátok 306
— polisavak 304
J
jénai normál üveg 17 jód 167— -monobromid 234 — monoklorid 233— (ffi)-oxid-jódét 243— -pentoxid 246 —— triklorid 233
jódsav 258 **
E
kadmium 179 kalcium 180 kalciumamalgám 313 kalcium-diszilicid 284— -foszfid 281 — hidrid 201 - — karbid 281— -klorid 250
— nitrid 279 — szilicid 284 — volframát 267 Kapsenberg-féle szűrő 86 karbidok 281 karbonilok 298 kardszárú lombik 275 kationcserélő 109 kaucsukaszfalt 32 kálium 174, 185— -amid 288— -bizmut(IH)-Bzulfid 276
— [hexakloro-plumbát(IV)] 293 — hidrid 200 ~— bromát 268 -— -klorát 256 -— manganát(VI) 266— -perjodát 259 •" — peroxi-diszulfát 271— -sziliko-volframát 307— -[tetrafluoro-borát] 292— [tetraperoxo-kromát(V)] 271 — [trijodo-plumbit(II)] 294— -[trioxalato-ferrát(III)] 273
— -vas(IH)-szulfid 276 kelátkomplexek 297 keményforrasz 29 kerámiai anyagok 21 kén 167— dioxid 130, 140 — hidrogén 139, 193— -trioxid 247kétfokozatú rotációs szivattyú 67 kifagyasztás 67

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 363/366
TÁRGYMUTATÓ 365
kifagyasztócsapdák 67 Kinney-szivattyú 57
kioldás 105 kiolvasztás 104 kitt-tömítések 31 klór 130, 138, 145, 166 — dioxid 245 —
kloroform 140 kloro-kénsav 239 Knudsen-féle vákuummérő 63 kobalt 177—-karbonilok 301— szulfát-oktadekahidrát 262 komplex vegyöletek 292
kompresszióé vákuummérő 62 kondenzáció 104 kontakt hőmérő 51— manómé tér 73 kriohidrátok 35 kristálynövesztés 95 kristályosítás 93 kromatográfia 107 króm(II)-klorid 228 króm(III)-bromid 213— -klorid 205— -oxid 254“
— szulfid 273 kromil-klorid 242 kvarcüveg 17
L
lantán 308lantanidák 108, 146, 147, 149 lantán(in)-klorid 217, 309— -szulfát 309 Lávái-fuvóka 58
lágyforrasz 29
légtermoBztátok 50 lítium 174, 179, 186 — alumínium-hidrid 199 — amid 288— -antimonid 280— -arzenid 280 — bizmutid 280— -foszfid 280— -hidrid 198 — nitrid 277
M
M ocLeod-manométer 62 «*nagas hőmérséklet előállítása 39 magnézium 181— -nitrid 278— -perklorát 128, 257 —— -szilicid 283“ mangán 173 manosztát 73
metil-alumínium-kloridok 323— -bór-diklorid 322
— -klórszilánok 328 Meyer—Ronge-féXe oxigénmentesítő készülék 122
mérgezések 344 merülő hűtő 151 mikropreparatív módszerek 150 Moissan-kemence 42 molekuláris desztilláció 100— szűrők 110 molibdén 173, 178— -diszulfid 274—(IV)-oxid 254
— (VT)-oxid 250 mosópalack 118 mosótorony 120 Muencke-féle mosópalack 118 Muthmann-féle élektrolizáló edény 179 műanyag kittek 32műanyagok 30
N
nagyfrekvenciás indukciós kemence 42 nagynyomású manométerek 82
nátrium 174, 184— amalgám 312 nátrium-amid 286 —— antimonid 280— -arzenid 280 — azid 288 —— bizmutid 280 — foszfid 280— -[hexahidroxo-plumbát(IV)] 296 — [hexahidroxo-sztannát(IV)] 296 — hidrid 200— hipomanganát(V) 267
— metafoszfát 266 — metamolibdát 304 — metavanadát 304 — paramolibdát 304 — perjodát 258— -peroxid 270 — pirofoszfát 265— -pirovanadát 304— [tetrahidroxo-kuprát(II)] 296— -tiokromit 275 nemesfémek 28 nikkel 177
— -tetrakarbónil 298 nióbium 146— pentaklorid 219 — oxid-triklorid 243 nitridek 277 nitrogén 129, 168 — hidrogénsav 289 » nitroprusszid-nátrium 303 nitrozil-klorid 241 nitrozilok 302

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 364/366
366 TÁRGYMUTATÓ
íiormálcsiszolatok 19— méretei 19
Ny
nyitott differenciálmanométer 61 nyomásegységek átszámítása 64 nyomáspalack 80
0
180 feldúsítása vízgőzben 98 oldószerek 138
ólom-azid 289 — hidrogén 145 - ólom-oxid—glicerin kitt 32 ólom-tetraacetát 267 - olvadékelektrolízis 147 ón-hidrogén 145 ón(IV)-jodid 214— (IV)-klorid 206 optikai pirométer 48 ** ortohidrogén 107— kovasavas tetraetilészter 327 ózon 145oxigén 129, 145, 167 — túlfeszültség 144
0
önműködő hőmérséklet-szabályozók 50
P
palládiumazbeszt 187
— korom 188 parahidrogén 107 parafa 34 Peligot-cső 296 Penning-féle vákuummérő 66 [pentakarbonil-vas(O)] 299 pentavanadinsav nátriumsója 304 perforátor 89 perjódsav 260 picein 32P iram'-féle nyomásmérő 64 piridin 142
piroszulfuril-klorid 240 platina 25 platinaazbeszt 187 platinakorom 188 platinázó óidat 188 plexiüveg 30 polietilén 30 polimetakrilát 30 polikarbonátok 3C
polisztirol 30
poli(tetrafluor-etilén) 30 poli(vinil-klorid) 30
présszűrő 164 propán—bután-gáz 130 Prytz-féle manosztát 73 Pyrex-üveg- 17
B
Radiométer típusú nyomásmérő 63 Mamsay-zsír 68Rasotherm-üveg 17regulátorok 60reométer 133rénium(VII)-oxid 251rénium-pentaklorid 219réz-hidrogén 202 -réz(I)-klorid 226 -— oxid 255robbanás 347 Rossignol-szelep 313rotaméter 132rotációs légszivattyú 57Rotosil-kvarc 20rövidített manométer 61rubeán-hidrogénsav 235rubídium 174, 184— -hidrid 200
S
sárgaarzén 169 Seger-gúla 49 Soxhlet-ex. traktor 105 (Síoc^-féle szelep 71 stroncium-hidrid 201 Supremax-üveg 17
szárítóanyagok 126 szárítótorony 121 szelén 167— -dioxid 247
-hidrogén 195— tetraklorid 210 szelenil-klorid 240 szén 170— diklorid-dibromid 223— -dioxid 129 — diszulfid 142 szénhidrogének 140
szénsavhó 37 szén-tetrabromid 220—-tetrajodid 221 — tetraklodrid 140 szilícium-tetraacetát 268 — tetrabromid 213— tetraklorid 208, 215 szilikagél 268 szilikonok 30 szilikonzsírok 33

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 365/366
TÁRGYMUTATÓ 367
sziliko-volframsav 306 színtelen pecsétviasz 32
sziták szabványosítása 84 szívássebesség 64 szívógömb 152 szublimálás 102
trifenil-bór 320 trimetil-fenil-szilán 331
— p-tolil-szilán 333 trolitul 30 tűz 347tűzálló anyagok 21
T
Tammann-eljárás egykristály előállítására 96
tantál 146
— -pentaklorid 219 teflon 30tellúr 168— -dioxid 249-----hidrogén 195terelőlemezek 67 termisztor 44termisztoros áramlássebesség-mérés 133 termodiffúzió 92 termo elemek 47 térmokolor-festékek 48 termosztátok 49
teslázó 76[tetra-acetil-acetonát-cirkónium(IV)] 297 tetrabutil-ón 338 tetraetil-ólom 341— -ón 337tetraetoxi-szilán 327— -titán 342 tetrametil-szilán 333[tetrakarbonil-dijódid -vas(II)] 299 [tetrakarbonil-nikkel(O)] 298 thüringiai üveg 17 tionil-bromid 237
— -klorid 236 titán 176, 182 “" — -borid 285 — diklorid 80 — hidrogén 201— -karbid 282— -nitrid 279— -tetraiodid 214 “— -tetraklorid 215, 217— -triklorid 226— -trioxid 253 -— (IV)szilicid 284
toluolos regulátor 51 tórium 184— -tetrabromid 216 tömény kénsav 126 törőszelep 72 transzformációs pont 17 [tri-acetil-acetonát-króm(III)] 297 tribenzil-ón-klorid 340 trietil-ólom-p-toluol-szulfonát 341 trietoxi-titán-klorid 342
U
ultracentrifuga 88 ultraszűrők 86 urán 148— -(IV)-klorid 228— -(Vl)-oxid 251
Ü
ülepítés 88 üveg 17üvegalapú kitt 32üvegszűrő 119üvegek nyomásállósága 78
V
van Arkel —de Soer-eljárás 182 vanádium 173— (II)-klorid 228— (Ilí)-klorid 228-------hexahidrát 231— (Ill)-oxid 254— -oxid-triklorid 243 vas 177— -karbid 282— (Il)-klorid 227— (Ill)-klorid 204— (Il)-ortofoszfát-oktahidrát 266 vákuumcsapok 68 vákuumcsapzsír 68 vákuumdesztilláció 98 vákuumgyorsbepárló 99 vákuumindikálás 60 vákuummérés 60 vákuumszublimálás 103 vákuumtechnika 54 végvákuum 54
Vitreosil kvarc 21 víz 143vízdesztilláló 143 vízgőz-desztilláció 101 vízlégszivattyú 56 vízüveg kitt 31 volfrám 178—(Vl)-bromid 220’— (VI)-klorid 205— (Vl)-oxid 254— (IV)-oxid 251
X Wohl-féle csap 72 WUrtz-Teakció 333

7/24/2019 Dr. Lengyel Bela - Szervetlen Kemiai Praktikum 2
http://slidepdf.com/reader/full/dr-lengyel-bela-szervetlen-kemiai-praktikum-2 366/366
66/585. Franklin-nyomda Budapest, V il i., Szentkirályi a. 28.


















