
DNA Packaging Motor Assembly Intermediate of Bacteriophage ϕ29
19
DNA Packaging Motor Assembly Intermediate of Bacteriophage ϕ29 Jaya S. Koti 1 , Marc C. Morais 2 , Raj Rajagopal 1 , Barbara A. L. Owen 3,4 , Cynthia T. McMurray 3,4 and Dwight L. Anderson 1,5 ⁎ 1 Department of Diagnostic/ Biological Sciences, University of Minnesota, Minneapolis, MN 55455, USA 2 Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA 3 Department of Biochemistry and Molecular Biology, Mayo College of Medicine, Rochester, MN 55905, USA 4 Department of Medical Pharmacology and Experimental Therapeutics, Mayo College of Medicine, Rochester, MN 55905, USA 5 Department of Microbiology, University of Minnesota, Minneapolis, MN 55455, USA Received 1 March 2008; received in revised form 10 April 2008; accepted 11 April 2008 Available online 20 April 2008 Unraveling the structure and assembly of the DNA packaging ATPases of the tailed double-stranded DNA bacteriophages is integral to understanding the mechanism of DNA translocation. Here, the bacteriophage ϕ29 packaging ATPase gene product 16 (gp16) was overexpressed in soluble form in Bacillus subtilis (pSAC), purified to near homogeneity, and assembled to the ϕ29 precursor capsid (prohead) to produce a packaging motor inter- mediate that was fully active in in vitro DNA packaging. The formation of higher oligomers of the gp16 from monomers was concentration dependent and was characterized by analytical ultracentrifugation, gel filtration, and electron microscopy. The binding of multiple copies of gp16 to the prohead was dependent on the presence of an oligomer of 174- or 120-base prohead RNA (pRNA) fixed to the head–tail connector at the unique portal vertex of the prohead. The use of mutant pRNAs demonstrated that gp16 bound specifically to the A-helix of pRNA, and ribonuclease footprinting of gp16 on pRNA showed that gp16 protected the CC residues of the CCA bulge (residues 18–20) of the A-helix. The binding of gp16 to the prohead/pRNA to constitute the complete and active packaging motor was confirmed by cryo- electron microscopy three-dimensional reconstruction of the prohead/ pRNA/gp16 complex. The complex was capable of supercoiling DNA–gp3 as observed previously for gp16 alone; therefore, the binding of gp16 to the prohead, rather than first to DNA–gp3, represents an alternative packaging motor assembly pathway. © 2008 Elsevier Ltd. All rights reserved. Edited by J. Karn Keywords: DNA packaging; ATPase gp16; phage assembly; bacteriophage ϕ29; RNA binding Introduction The packaging of the genomes of the tailed double- stranded DNA (dsDNA) bacteriophages into pre- formed protein capsids (proheads) is a remarkable process in which the highly charged DNA is com- pacted to near crystalline density. 1,2 Molecular motors assembled at the unique portal vertex of the prohead drive packaging, converting energy ob- tained from ATP hydrolysis to power DNA translo- cation. The motor in bacteriophage ϕ29 is among the most powerful biological motors known, generating forces in the range of 60–100 pN. 3,4 The packaging *Corresponding author. Department of Diagnostic/ Biological Sciences, University of Minnesota, Minneapolis, MN 55455, USA. E-mail address: [email protected]. Present addresses: J. S. Koti, Instrument Systems Development Center, Beckman Coulter Inc., Chaska, MN 55318, USA; M. C. Morais, Department of Biochemistry and Molecular Biology, University of Texas Medical Branch, Galveston, TX 77555, USA. Abbreviations used: gp, gene product; pRNA, prohead RNA; dsDNA, double-stranded DNA; EM, electron microscopy; EMSA, electrophoretic mobility shift assay; HTH, helix–turn–helix; TCEP, Tris(2-carboxyethyl) phosphine. doi:10.1016/j.jmb.2008.04.034 J. Mol. Biol. (2008) 381, 1114–1132 Available online at www.sciencedirect.com 0022-2836/$ - see front matter © 2008 Elsevier Ltd. All rights reserved.
-
Upload
jaya-s-koti -
Category
Documents
-
view
213 -
download
0
Transcript of DNA Packaging Motor Assembly Intermediate of Bacteriophage ϕ29

doi:10.1016/j.jmb.2008.04.034 J. Mol. Biol. (2008) 381, 1114–1132
Available online at www.sciencedirect.com
DNA Packaging Motor Assembly Intermediate ofBacteriophage ϕ29
Jaya S. Koti1, Marc C. Morais2, Raj Rajagopal1, Barbara A. L. Owen3,4,Cynthia T. McMurray3,4 and Dwight L. Anderson1,5⁎
1Department of Diagnostic/Biological Sciences, Universityof Minnesota, Minneapolis,MN 55455, USA2Department of BiologicalSciences, Purdue University,West Lafayette, IN 47907, USA3Department of Biochemistryand Molecular Biology, MayoCollege of Medicine, Rochester,MN 55905, USA4Department of MedicalPharmacology and ExperimentalTherapeutics, Mayo Collegeof Medicine, Rochester,MN 55905, USA5Department of Microbiology,University of Minnesota,Minneapolis, MN 55455, USA
Received 1 March 2008;received in revised form10 April 2008;accepted 11 April 2008Available online20 April 2008
*Corresponding author. DepartmenBiological Sciences, University of MiMN 55455, USA. E-mail address: dlPresent addresses: J. S. Koti, Instr
Development Center, Beckman Cou55318, USA; M. C. Morais, Departmand Molecular Biology, University oBranch, Galveston, TX 77555, USA.Abbreviations used: gp, gene pro
RNA; dsDNA, double-stranded DNmicroscopy; EMSA, electrophoreticHTH, helix–turn–helix; TCEP, Tris(2phosphine.
0022-2836/$ - see front matter © 2008 E
Unraveling the structure and assembly of the DNA packaging ATPases ofthe tailed double-strandedDNAbacteriophages is integral to understandingthe mechanism of DNA translocation. Here, the bacteriophage ϕ29packaging ATPase gene product 16 (gp16) was overexpressed in solubleform inBacillus subtilis (pSAC), purified to near homogeneity, and assembledto the ϕ29 precursor capsid (prohead) to produce a packaging motor inter-mediate that was fully active in in vitro DNA packaging. The formation ofhigher oligomers of the gp16 from monomers was concentration dependentand was characterized by analytical ultracentrifugation, gel filtration, andelectron microscopy. The binding of multiple copies of gp16 to the proheadwas dependent on the presence of an oligomer of 174- or 120-base proheadRNA (pRNA) fixed to the head–tail connector at the unique portal vertex ofthe prohead. The use of mutant pRNAs demonstrated that gp16 boundspecifically to theA-helix of pRNA, and ribonuclease footprinting of gp16 onpRNA showed that gp16 protected the CC residues of the CCA bulge(residues 18–20) of theA-helix. The binding of gp16 to the prohead/pRNA toconstitute the complete and active packaging motor was confirmed by cryo-electron microscopy three-dimensional reconstruction of the prohead/pRNA/gp16 complex. The complex was capable of supercoiling DNA–gp3as observed previously for gp16 alone; therefore, the binding of gp16 to theprohead, rather than first to DNA–gp3, represents an alternative packagingmotor assembly pathway.
© 2008 Elsevier Ltd. All rights reserved.
Keywords: DNA packaging; ATPase gp16; phage assembly; bacteriophageϕ29; RNA binding
Edited by J. Karnt of Diagnostic/nnesota, Minneapolis,[email protected] Systemslter Inc., Chaska, MNent of Biochemistryf Texas Medical
duct; pRNA, proheadA; EM, electronmobility shift assay;-carboxyethyl)
lsevier Ltd. All rights reserve
Introduction
The packaging of the genomes of the tailed double-stranded DNA (dsDNA) bacteriophages into pre-formed protein capsids (proheads) is a remarkableprocess in which the highly charged DNA is com-pacted to near crystalline density.1,2 Molecularmotors assembled at the unique portal vertex of theprohead drive packaging, converting energy ob-tained from ATP hydrolysis to power DNA translo-cation. The motor in bacteriophage ϕ29 is among themost powerful biological motors known, generatingforces in the range of 60–100 pN.3,4 The packaging
d.

1115Viral Packaging Motor Intermediate
machines include a ring of ATPases (terminases), thehead–tail connector of the prohead, the prohead, andthe DNA substrate. These components show manybiochemical and structural similarities, suggesting acommon mechanism for DNA translocation.5–7 Ter-minases belong to a large family of ATPases involvedin DNA transactions, such as cell division, chromo-some segregation, recombination, strand separa-tion, and conjugation.8,9 Each terminase has largeand small subunits: The large subunit has proheadand ATP binding activities, potential magnesiumbinding motifs, and endonuclease activity.9–14 Theterminase small subunit has DNA binding andpackaging stimulation activities. Unraveling the struc-ture, assembly, and function of packaging ATPases isessential in gaining an understanding of the packagingmechanism.In the DNA packaging system of the Bacillus subtilis
bacteriophage ϕ29, gene product 16 (gp16) corre-sponds to the large terminase subunit, whereas gp3terminally bound to the 5′ DNA ends (DNA–gp3)represents the small subunit.15 In vitro and in vivoencapsidation of ϕ29 DNA–gp3 into proheads bothrequire gp16,16,17 a ~39,000 molecular weight proteinwith the Walker A and Walker B motifs needed forATP hydrolysis. Although ϕ29 is one of the bestcharacterized systems for study of the dsDNApackaging mechanism and the structure of thehead–tail connector is determined,18,19 limited struc-tural information is available for gp16 and itsassembly to constitute the packaging motor. Detailedphysical and biological characterization of gp16 hasbeen challenged by its insolubility after overexpres-sion andpurification. Expressionof gp16 inEscherichiacoli yielded inclusion bodies, and because the bulk ofthe proteinwas insoluble after renaturation, about 100copies of protein were needed to package each ϕ29DNA in the completely defined in vitro system.20
The following are unique features ofϕ29DNA–gp3packaging: (i) the DNA–gp3 packaging substrate issupercoiled by the concerted action of gp16 and gp3in the absence of the prohead, and the preferentialpackaging of this DNA/gp3/gp16 complex suggeststhat gp16 constitutes the packaging motor whilebound to DNA21; (ii) a 174-base ϕ29-encoded RNA(or its 120-base derivative) (prohead RNA, pRNA) isan essential packaging motor component22; (iii)pRNA binds to the connector of the prohead,23 andcryo-electron microscopy (EM) reconstructions showthat gp16 binds to the pRNA18; (iv) the ATPaseactivity of gp16 is pRNA dependent24 such that theATPase “subunits” of the packaging motor may begp16–pRNA heterodimers; and (v) in vitro packagingrivals in vivo packaging in efficiency,2,16,20 facilitatingstudy of the packaging mechanism.pRNA binds to the prohead in a specific, rapid,
irreversible, andmagnesium-dependent reaction.25–31Although pRNA and gp16 are essential to DNApackaging, their presence is transitory during phageassembly, as both components are displaced duringneck/tail assembly and are absent in the maturephage.32 Two functional regions of pRNA, a proheadbinding region and a region involved in DNA trans-
location, have been identified.33–36 Bases 22–84 of thepRNAwere identified to be the prohead binding do-main by ribonuclease footprinting, and this was con-firmed by mutant studies and competitive bindingexperiments.26,33,34 While the prohead binding do-main is well characterized, knowledge of the gp16binding domain has been rudimentary.A mechanistic understanding of the DNA packa-
ging process in ϕ29 and other dsDNA phages islacking in spite of extensive biochemical, structural,biophysical, and theoretical studies.2,14 A model forhow ϕ29 DNA is translocated into the capsid wasproposed in which ATP binding, hydrolysis, andrelease of products induce conformational changesin the ATPases that are directly involved in the trans-location of the DNA.37 DNA translocation is trig-gered or performed by the ring of ATPases, possiblyin concert with other motor components, such as thehead–tail connector, via a conformational changethat likely follows release of phosphate after ATPhydrolysis.37 Many of the packaging mechanismmodels have been based on rotation of the head–tailconnector.2,38 However, recent evidence suggestedthat the T4 connector does not rotate during DNAtranslocation,39 and a direct test by following theorientation of single fluorophores attached to theconnector provided evidence that the ϕ29 con-nector does not rotate.40 In phage SPP1, aminoacid loops in the connector channel that tightlyembrace DNA are proposed to form an undulating‘wave’ of conformational change as DNA is trans-located, which may coordinate ATP hydrolysiswhether force is applied by the ATPase itself or byATPase-induced conformational change in theconnector.41 On the basis of a crystal structure ofthe ATPase gp17 of phage T4 at 1.8 Å and thesimilarity of this structure to monomeric helicases, itwas proposed that DNA is translocated by the gp17itself, with each monomer in the ring of ATPasessequentially detecting the same DNA structuralcomponent after translocation of DNA by two basepairs.9
Here, overexpression of gp16 in its natural B.subtilis host is described. The protein has beenpurified to greater than 99% homogeneity to obtainsoluble and highly active protein in milligram quan-tities needed for structural and functional studies.The physical state of the protein in solution aswell asits pRNA and DNA binding properties are des-cribed. An alternative pathway of gp16 assembly tothe prohead prior to interaction with DNA–gp3 isdocumented. Multiple copies of gp16 interact withthe prohead-bound pRNA oligomer to constitute theDNA packaging motor, and the isolated prohead/pRNA/gp16 complex is active in DNA packagingwithout the aid of additional gp16. The pRNA–gp16interaction is ATP independent, and the gp16 bin-ding is shown to be A-helix specific by use of pRNAmutants and RNase footprinting. Cryo-EM re-construction demonstrates that the pRNA–gp16oligomers are both 5-fold symmetric and interfacedsuch that the motor subunits may be gp16–pRNAheterodimers.

Fig. 1. Purification and in vitro DNA–gp3 packagingactivity of the ATPase gp16. (a) SDS-PAGE of samples fromsteps in the purification of gp16 following expression in B.subtilis (pSACB-gp16). Lane 1, supernatant from the celllysate; lanes 2 and 3, P11 cation exchange column peakfractions; lanes 4–8, hydroxyapatite column peak fractions.(b) Packaging activity of purified gp16 determined by the invitro DNA–gp3 packaging assay (Materials and Methods).Lane 1, input DNA–gp3 added to the reaction; lanes 3–9,DNA that was packaged in the presence of 15, 10, 8, 6, 4, 3,and 2 copies (molecules) of gp16 per prohead, respectively;lanes 2 and 10, negative controls in which ATP and gp16were omitted, respectively, from the packaging reaction.
1116 Viral Packaging Motor Intermediate
Results
Production of soluble gp16, purification, andbiological activity
Production of gp16 in E. coli (pPLc2833) yields in-clusion bodies, and the bulk of the highly hydro-phobic protein is insoluble after treatment of isolatedinclusion bodies with guanidinium chloride, fol-lowed by urea,20 making detailed physical and bio-logical characterization of this DNA packagingATPase impossible. Gene 16 was expressed in B.subtilis to circumvent this, yielding soluble gp16 thathas long been the source of ATPase for packagingassays.21 However, the details of plasmid construc-tion and the purification of milligram amounts ofprotein to near homogeneity have not been pub-lished. Briefly, the EcoRI E/D segment of ϕ29 DNA–gp3 that includes gene 16,42 obtained by partial di-gestion, was cloned into the plasmid pUB18. Thesucrose inducible promoter SACB43 was inserted todrive expression, and the resulting plasmid pSACB-gp16 was transformed into B. subtilisWB30, an aspo-rogenic, protease-defective strain. After induction ofgene 16 expression with sucrose (Materials andMethods), about 70% of the gp16 was soluble. The~39,000 molecular weight gp16 was purified to nearhomogeneity by cation exchange followed by hydro-xyapatite chromatography (Fig. 1a). The pI of thepurified protein was estimated at 10.2 by isoelec-trofocusing, and its identity was confirmed byWestern blotting with polyclonal anti-gp16 serum.The activity of purified gp16 in in vitro DNA–gp3
packaging was determined by a standard DNaseprotection assay (Fig. 1b).44 Purified gp16was highlyactive, and multiple copies were required per pro-head for DNA–gp3 packaging (Fig. 1b). No DNA–gp3 packagingwas detected in the absence of ATP orgp16 (Fig. 1b, lanes 2 and 10). A 4-fold reduction ingp16 resulted in a 40-fold reduction in packaging.These results are consistent with the in vitro DNA–gp3 packaging in extracts, which shows a first-orderconcentration dependence for the prohead andDNA–gp3, while multiple copies of the packagingATPase gp16 are required.45
Monomeric/multimeric state of gp16
No large aggregate of the purified gp16 at a con-centration of 0.8 mg/ml was detected by dynamiclight scattering, and the hydrodynamic radius of 6–9 nm (data not shown) suggested that the proteinwassoluble and formed an oligomer. Sedimentation equi-librium centrifugation was performed to determinethe oligomeric state of gp16. The calculatedmonomermolecular weight of gp16 based on the amino acidsequence is ∼39,000. Sedimentation equilibrium cen-trifugation confirmed that gp16 self-associates, givinga molecular weight greater than the monomer and anapparent molecular mass that varied with the proteinconcentration (Table 1). The apparent molecular massof gp16 at various concentrations and speeds was
derived by fitting the data to a self-association modelusing NONLIN.46 Figure 2a shows the A280-versus-r2/2 plots for gp16 at three protein concentrations at11,000 rpm. Of the protein, 80%–85% was a trimer,and the rest was amonomer as determined by speciesanalysis using the sedimentation analysis softwareSEDPHAT.48 A trimer of gp16 would have a mole-cular weight of ∼117,000, and this is the most abun-dant species at 0.2 mg/ml or at 11,000 rpm (Table 1).However, fits of the data consistent with only mono-mer to trimer models were unsatisfactory, indicatingthat additional species may be present. Furthermore,the data at higher protein concentrations and lowercentrifugation speeds suggested that species largerthan the trimer were present. SEDPHAT48 was usedto determine the relative concentrations of additionalspecies. Figure 2b shows a plot of A280 versus theradial position at 0.4 mg/ml and that at 8000 rpm.The data were fit with four possible species: mono-mer, trimer, hexamer, and dodecamer. The best fit ofthe data yielded 22%, 68%, and 10% of monomer,trimer, and dodecamer, respectively. The relativelysmall and random distribution of the residuals atteststo the goodness of this fit. Indeed, other models tried,

Table 1. Apparent molecular mass (in kilodaltons) ofgp16 by sedimentation equilibrium
gp16 (mg/ml) 5000 rpm 8000 rpm 11000 rpm
0.2 100.5 104.7 96.50.4 157.7 128.7 107.50.8 156.2 126.8 104.4
Valueswere calculated from the fit of two cells at each concentrationusing NONLIN.46 Partial specific volume (υ) of 0.7375 ml/g,density (σ) of 1.02859 g/ml, and viscosity (η) of 0.0167 werecalculated using SEDNTERP.47 See Materials and Methods fordetails.
1117Viral Packaging Motor Intermediate
such as monomer, dimer, and tetramer, yielded poorfits to the data. A dissociation constant for theoligomerization could not be obtained because gp16was relatively difficult to maintain in a soluble state.At protein concentrations of 0.2–0.4 mg/ml, approxi-mately 40% of the total protein was irreversibly losteven at a speed as low as 5000 rpm. This lossincreased to 65% at 0.8 mg/ml, but additional losswas not seen at the higher centrifugation speeds of8000 and 11,000 rpm. Nonreducing PAGE was doneafter the centrifugation to look for degradation andoxidized cysteine-driven oligomerization, and noprotein degradation was detected.Secondary plots of the sedimentation equilibrium
primary data (data not shown) indicated that gp16was monomeric at protein concentrations below100 μg/ml. Gel filtration chromatography was per-formed at gp16 concentrations between 10 and100 μg/ml to confirm these findings. Buffer andtemperature conditions were exactly the same asthose used in the sedimentation equilibrium experi-ments. Figure 2c shows the elution profile of 100 μg/ml of gp16 on a Superdex 200 column (dashed line)with molecular weight markers (continuous line).gp16 eluted slightly ahead of a protein standardhaving a molecular weight of 44,000, consistent withgp16 being monomeric at concentrations less than100 μg/ml as suggested by the analytical ultracen-trifugation results.Transmission EM of the purified gp16 at 50 μg/ml
was done to determine the size and shape of gp16.Figure 2, panels (d) and (e), shows purified gp16 ne-gatively stained with uranyl acetate. gp16 appearedas rounded and porous oligomers with a diameter of9.5±1.5 nm, calculated from the area. Figure 2f showsa histogram of the multimer diameter measurements.
Fig. 2. Multimerization of the purified ATPase gp16. (a) Sedphosphate (pH 6.8), 400 mM sodium chloride, and 2 mM TCEP(r2/2) at 11,000 rpm are shown at three loading concentrationsself-associationmodel are shown in the upper panels. (b) Specieof the protein was trimer and that 15%–20%wasmonomer at thhighest speed of centrifugation, 11,000 rpm. Residuals to thiscentrations, analysis revealed predominantly trimers with vadecamer (see the text). (c) Gel filtration elution profiles of protstudies, plotted as protein concentration (A280) versus retentioprofile of gp16 at approximately 100 μg/ml at the peak. The costandards with molecular mass (in kilodaltons) indicated acromicrographs of gp16 negatively stainedwith 2% (w/v) uranyl ameasurement of the diameter of gp16 from electron micrograp
Monomers likely were not visualized because of theirsmall size, and it is not known as to what extentincrease in protein concentration during drying of thespecimen contributed to formation of the multimers.Variation of the pH level, salt conditions, stainingmethods, or the presence of ATP did not alter thestructure of gp16 by transmission EM.
Characterization of the gp16–pRNA interaction
Interaction between the ATPase gp16 and pRNAhas been documented by the stimulation of theATPase activity of gp16 by prohead-bound pRNA,which is needed continuously for ATP hydrolysis24;the presence of additional mass on the pRNA spokesof partially packaged particles in a cryo-EM three-dimensional reconstruction, interpreted as gp1618;the binding of pRNA to gp16 on nitrocellulose fil-ters49; theweak binding of pRNA to thioredoxinHis-tag gp16 fusion protein immobilized on a His-bindcolumn50; and recent delineation of the molecularboundaries of the components of the packagingmotor by cryo-EM reconstruction (Morais et al.,unpublished).One hundred twenty-base pRNA was incubated
with increasing concentrations of gp16, and theinteraction was assessed by electrophoretic mobilityshift assay (EMSA) in a native agarose gel (Fig. 3a) tocharacterize the gp16–pRNA interaction in solution.Mixed 1:1 (based on monomers), much of the pRNAdisappears from its fast migration position (comparelanes 1 and 2), suggesting that the pRNA and gp16interact to produce heterogeneous complexes that donot appear in the gel. Three copies of gp16 per pRNA(lane 3) were sufficient to shift the pRNA band in thegel, and as the copy number of gp16 per pRNAwasincreased, more of the pRNA/gp16 complex wasretained in the wells of the gel. gp16 bound to 174-base pRNA in the same manner (data not shown).The pRNA–gp16 interaction was independent ofATP. The complex was stable in the presence of100 mM ethylenediaminetetraacetic acid (EDTA) andovernight incubation at 4 °C but was disrupted by0.1% SDS. pRNA–gp16 (1:3) complexes were mixedand incubated with fiberless proheads (10:1), andfollowing isolation by electrophoresis on a nativeagarose gel, the particles were tested for pRNAbinding by staining with ethidium bromide and forgp16 binding following transfer to nitrocellulose and
imentation equilibrium analysis of gp16 in 50 mM sodium. Plots of protein concentration (A280) versus radial positionin the lower panel, and residuals of each set of data fit to as analysis of gp16 using SEDPHAT revealed that 80%–85%e lowest protein concentration, 0.2 mg/ml, analyzed at thefit are shown in the lower panel. At higher protein con-rying amounts of monomer, hexamer, and traces of do-eins in the same buffer used for sedimentation equilibriumn volume (milliliters). The dashed line shows the elutionntinuous line shows the elution profile of molecular weightss the top of the profile. (d) and (e) Transmission electroncetate (Materials andMethods). (f) Histogram showing thehs (n=88).

Fig. 2. Multimerization of the purified ATPase gp16. (a) Sedimentation equilibrium analysis of gp16 in 50 mM sodium phosphate(pH6.8), 400mMsodiumchloride,and2mMTCEP.Plotsofproteinconcentration (A280)versus radialposition (r
2/2)at11,000rpmareshownatthree loadingconcentrations inthe lowerpanel,andresidualsofeachsetofdatafit toaself-associationmodelareshownintheupperpanels.(b)Speciesanalysisofgp16usingSEDPHATrevealedthat80%–85%of theproteinwastrimerandthat15%–20%wasmonomerat the lowestproteinconcentration,0.2mg/ml,analyzedatthehighestspeedofcentrifugation,11,000rpm.Residualstothisfitareshowninthelowerpanel.At higher protein concentrations, analysis revealed predominantly trimers with varying amounts of monomer, hexamer, and traces ofdodecamer (see the text). (c)Gel filtration elutionprofiles of proteins in the samebuffer used for sedimentation equilibriumstudies, plotted asproteinconcentration(A280)versus retentionvolume(milliliters).Thedashedlineshowstheelutionprofileofgp16atapproximately100μg/mlat thepeak.Thecontinuous line showstheelutionprofileofmolecularweight standardswithmolecularmass (inkilodaltons) indicatedacrossthe top of the profile. (d) and (e) Transmission electronmicrographs of gp16 negatively stainedwith 2% (w/v) uranyl acetate (Materials andMethods). (f) Histogram showing themeasurement of the diameter of gp16 from electronmicrographs (n=88).
Fig. 2 (legend on previous page)
1118 Viral Packaging Motor Intermediate

Fig. 3. Binding of gp16 to pRNA, and the activity of thepRNA–gp16 complex in in vitro DNA–gp3 packaging. (a)EMSA of 120-base pRNA incubated with increasing con-centrations of gp16. Lane 1, 120-base pRNA alone; lanes 2–6, shift of pRNA in the presence of 1, 3, 5, 8, and 10 copies(molecules) of gp16 per pRNA molecule, respectively; lane7, empty; lane 8, 10 copies of gp16 alone. (b) 120-basepRNA/gp16 (1:3) was added in increasing amounts topRNA-free proheads and the mixtures tested for activity inthe in vitroDNA–gp3 packaging assay. Lane 1, input DNA–gp3 added to the reaction; lane 2, negative control withoutATP; lane 3, empty; lanes 4–8, packaged DNA in the pre-sence of pRNA:gp16 copies per prohead of 1:3, 2:6, 5:15,7:21, and 10:30, respectively.
1119Viral Packaging Motor Intermediate
development with anti-gp16 serum. Both pRNA andgp16 were found on the prohead, and excess pRNA/gp16 migrated as a complex (data not shown).pRNA can be removed from the ϕ29 prohead by
treatment with EDTA or with RNase A, resulting inloss of DNApackaging activity, and reconstitution ofthese pRNA-free proheads with purified pRNAresults in restoration of packaging activity in the
defined in vitro system.22,51 pRNA-free proheadswere reconstituted with 120-base pRNA/gp16 (1:3)complexes and the particles tested in the in vitroDNA–gp3 packaging system.44 The efficiency ofpackaging was dependent on the number of pRNA/gp16 complexes (mixed as 1 pRNA:3 gp16) perprohead (Fig. 3b). Proheads were twofold in excessover DNA–gp3, and all of the DNA–gp3 was pack-aged when five to seven copies of pRNA and its as-sociated gp16 were used per prohead in the in vitroDNA–gp3 packaging reaction. As pRNA is penta-meric in cryo-EM reconstruction30 (Fig. 7d), theresults suggested that nearly every pRNA moleculeadded was biologically active and that the gp16 mo-lecules were also highly active.
Isolation of a prohead/gp16 ATPase motorassembly intermediate
Prior evidence for prohead/gp16 interaction wasobtained by sucrose density gradient centrifugationand ELISA tests,15,49 but the role of pRNA in the in-teraction was not clear. Sucrose density gradient cen-trifugation was used to determine the role of pRNAin the prohead/gp16 interaction. Figure 4 shows thepredicted secondary structures of the various formsof pRNA tested for gp16 binding. Proheads with 174-base pRNA52 (see following paragraph) were incu-bated with 20 copies of gp16 per prohead, and thecomplexes were sedimented in a sucrose densitygradient (Fig. 5a). There was a clear shift in the po-sition of proheads incubated with gp16, and the fast-sedimenting particles were isolated and shown bySDS-PAGE to contain gp16 (Fig. 6a, lane 2). The co-pies of gp16 per prohead from densitometric analysiswere about half those of the head–tail connector,which is known to contain 12 copies of gp10.18 Thisprohead/174-base pRNA/gp16 interaction is Mg2+
dependent andATP independent. Proheads and gp16interacted at high ionic strength (100 mM NaCl), butlow ionic strength (10 mM) was needed to retain thebound gp16. The prohead/174-base pRNA/gp16complexes isolated from the sucrose gradient weretested for in vitro DNA–gp3 packaging activity uponaddition of DNA–gp3 and ATP (Fig. 6b). The isolatedprohead/174-base pRNA/ATPase intermediatepackaged DNA–gp3 without further addition ofgp16 (lane 5), and with the same efficiency asproheads with 174-base pRNA mixed with gp16 inthe in vitro reaction (lane 3), as the concentration ofproheads used in the reaction of lane 3 was approx-imately twofold higher than that represented in lane5. Figure 6b, lane 7, shows that packaging remainedabout the same upon addition ofmore gp16, and thusthe isolated prohead/174-base pRNA/gp16 complexis a true intermediate with an intact and active pack-aging motor. The intermediate is stable and active inpackaging after overnight storage at 4 °C. Also, justas EDTA treatment releases pRNA from the pro-head,22 EDTA releases pRNA–gp16 as a complexfrom the prohead (data not shown).One hundred seventy-four-base pRNA (with do-
mains I and II)52 is cleaved to a 120-base form (do-

Fig. 4. Secondary structure prediction of various forms of pRNA tested for gp16 binding. (a) 120-base pRNA. (b) 106-base pRNAwith a 14-base deletion at the 3′ end of the A-helix. (c) 71-base pRNAwith a deletion of the A-helix. (d) R7pRNAwith deletion of the CCA bulge (bases 18–20), indicated by an arrow.
1120 Viral Packaging Motor Intermediate
main I) by adventitious ribonucleases during pro-head isolation, and both forms are active in recons-tituting pRNA-free proheads for DNA–gp3 pack-aging.25 RNA-free proheads were reconstituted with120-base pRNA (Fig. 4a), purified by sucrose densitygradient centrifugation, incubated with gp16, andrun again on a sucrose gradient to determine the roleof the 54-base domain II in the prohead/gp16 inter-action. gp16 co-sediments with proheads containing120-base pRNA just as with proheads having 174-base pRNA, and the isolated prohead/120-basepRNA/gp16 complex is functional in DNA–gp3packaging (data not shown). Thus, domain II ofpRNA was confirmed to be dispensable for in vitropackaging as shown previously.25 Figure 6c, lane 2,shows the composition of the prohead/120-basepRNA/gp16 complexes on a native agarose gel. AWestern blot of the particle developedwith anti-gp16serum confirmed the presence of gp16 on these pro-heads (data not shown).
pRNA dependency of the prohead/gp16interaction and identification of the gp16 bindingsite on pRNA
The prohead/gp16 interaction is pRNA depen-dent, as pRNA-free proheads incubated with gp16did not show the characteristic faster sedimentationdemonstrated for the prohead/pRNA/gp16 parti-cles on a sucrose gradient (Fig. 5b), and gp16was not
found on the isolated particles by SDS-PAGE (datanot shown). Thus, stable binding of gp16 to proheadsrequired pRNA.Two functional domains of pRNA have been iden-
tified, a prohead binding domain and a segmentinvolved in DNA translocation.33–35 The proheadbinding domain consists of bases 22–84 and wasidentified by ribonuclease footprinting and competi-tive binding experiments, while the A-helix that con-tains bases 1–28 and 117–92 was predicted to interactwith gp16 (Fig. 4a).18 Mutant pRNAs with trunca-tions of the A-helix were employed to further definethe gp16 binding site on the A-helix (Fig. 4b–d). The71-base pRNA is an A-helix deletion mutant thatretains the prohead binding activity of 120-basepRNA but cannot support in vitro DNA–gp3 pack-aging.34 pRNA-free proheads reconstituted with 71-base pRNA and purified were incubated with gp16,and the mixture was sedimented in a sucrose densitygradient (Fig. 5c). The sedimentation profiles of par-ticles reconstituted with 71-base pRNA, without andwith gp16 incubation, were similar, and no gp16 wasdetected on particles by SDS-PAGE. These resultsconfirm that the prohead/gp16 interaction is me-diated by pRNA and show directly that the A-helix isthe site of gp16 binding.pRNA-free proheads were reconstituted with two
pRNA deletion mutants and studied for gp16 bin-ding by use of sucrose gradient centrifugation andSDS-PAGE of the isolated particles to further define

1121Viral Packaging Motor Intermediate
the sequences and elements of the pRNA A-helixneeded for gp16 binding. Mutants lacking the CCAbulge at positions 18–20 of pRNA (R7 mutant, Fig.4d) or the 14 residues at the 3′ end of theA-helix (106-base pRNA, Fig. 4b) have prohead binding compe-titor activity but no DNA–gp3 packaging activity.33
pRNA-free proheads reconstituted with thesemutant pRNAs were able to bind gp16, indicatingthat the CCA bulge or the 14 bases at the 3′ end of theA-helix were not required for the initial gp16–pRNAinteraction, although particles so constituted werenot active in DNA packaging (Table 2).
RNase footprinting of gp16 on pRNA
Ribonuclease footprinting analysis of 5′ end-labeled [32P]120-base pRNA was performed withthe RNases A, T1, and V1 to define the nucleotides ofpRNA protected by gp16. Proheads reconstitutedwith [32P]120-base pRNA were separated from un-bound labeled pRNA by centrifugation and used toproduce footprints. In addition, free [32P]120-basepRNAwas also complexedwith gp16 formapping ofthe gp16 interactive domain. Both free pRNA andprohead-bound pRNA complexed with gp16 wereprotected fromRNase A digestion at residues 8 to 19,5′ to 3′ (Fig. 7a and b). RNase T1 did not producecleavage outside the prohead binding domain andwas not useful for the current studies. Footprintsproduced with RNase V1 showed additional protec-tion at residues 5 and 31 (data not shown). The pre-sent results confirmed the ribonuclease footprint ofthe prohead on pRNA, which demonstrated protec-tion of residues 22–84 by the proheadwith RNase V1and enhanced cleavage of pRNA at residues 38–40(data not shown).26
Cryo-EM three-dimensional reconstruction ofthe prohead/pRNA/gp16 packaging intermediate
The electron-dense pRNA spokes that radiate fromthe head–tail connector in cryo-EM reconstructionswere postulated to be A-helices.18 A cryo-EM recon-struction of pRNA-free proheads reconstituted with71-base pRNA lacking theA-helix has confirmed thishypothesis (Morais et al., unpublished). Cryo-EMthree-dimensional reconstruction of the prohead/174-base pRNA/gp16 motor assembly intermediateshowed mass that bridged the pRNA spokes andwas interpreted as gp16 (Fig. 7d–g). The localizationof gp16 on the A-helix of pRNAwas consistent withthe sucrose density gradient sedimentation (Fig. 5)and ribonuclease footprinting (Fig. 7a and b) results.The presence of gp16 on the DNA-filled head was
also demonstrated by incubating proheads/pRNA
Fig. 5. The sedimentation rate of proheads/pRNAincreased in the presence of gp16 and was dependent onthe presence of the pRNA A-helix. (a) Sucrose density gra-dient centrifugation profiles of proheads/174-base pRNA(continuous line) and proheads/174-base pRNA incu-batedwith gp16 (dashed line). Sedimentation is from left toright. gp16 was bound to proheads containing 174-basepRNA in the faster-sedimenting peak as revealed by SDS-PAGE (Fig. 6a) and by in vitro DNA–gp3 packaging (Fig.6b). (b) Sucrose density gradient profile of pRNA-free pro-heads (continuous line) and pRNA-free proheads incu-bated with gp16 (dashed line), showing that gp16 did notalter the sedimentation rate of the pRNA-free proheads;gp16 was not found on these proheads by SDS-PAGE. (c)Sucrose density gradient profile of proheads/71-basepRNA (continuous line) and proheads/71-base pRNAincubated with gp16 (dashed line), showing that gp16 didnot alter the sedimentation rate of these proheads; gp16was not found on the particles. The gradients in (a–c) wererun at different times and under different conditions, andthe peak positions are not comparable.
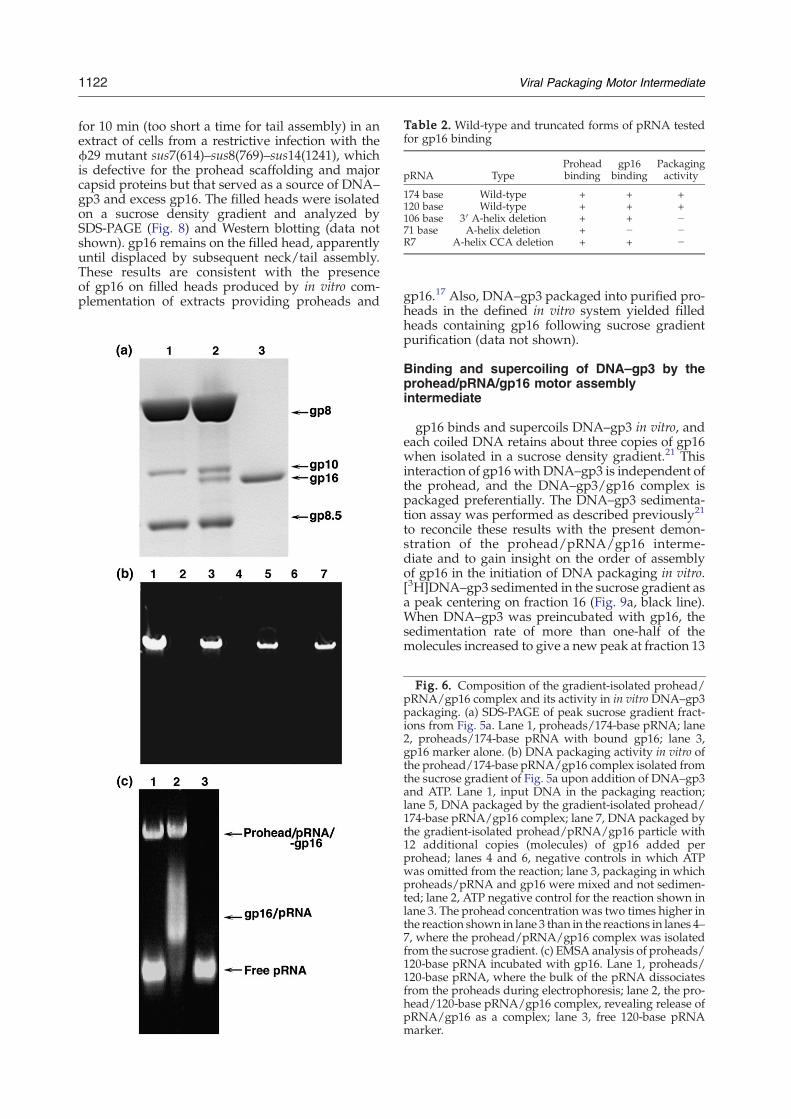
Table 2. Wild-type and truncated forms of pRNA testedfor gp16 binding
pRNA TypeProheadbinding
gp16binding
Packagingactivity
174 base Wild-type + + +120 base Wild-type + + +106 base 3′ A-helix deletion + + −71 base A-helix deletion + − −R7 A-helix CCA deletion + + −
1122 Viral Packaging Motor Intermediate
for 10 min (too short a time for tail assembly) in anextract of cells from a restrictive infection with theϕ29 mutant sus7(614)–sus8(769)–sus14(1241), whichis defective for the prohead scaffolding and majorcapsid proteins but that served as a source of DNA–gp3 and excess gp16. The filled heads were isolatedon a sucrose density gradient and analyzed bySDS-PAGE (Fig. 8) and Western blotting (data notshown). gp16 remains on the filled head, apparentlyuntil displaced by subsequent neck/tail assembly.These results are consistent with the presenceof gp16 on filled heads produced by in vitro com-plementation of extracts providing proheads and
gp16.17 Also, DNA–gp3 packaged into purified pro-heads in the defined in vitro system yielded filledheads containing gp16 following sucrose gradientpurification (data not shown).
Binding and supercoiling of DNA–gp3 by theprohead/pRNA/gp16 motor assemblyintermediate
gp16 binds and supercoils DNA–gp3 in vitro, andeach coiled DNA retains about three copies of gp16when isolated in a sucrose density gradient.21 Thisinteraction of gp16 with DNA–gp3 is independent ofthe prohead, and the DNA–gp3/gp16 complex ispackaged preferentially. The DNA–gp3 sedimenta-tion assay was performed as described previously21
to reconcile these results with the present demon-stration of the prohead/pRNA/gp16 interme-diate and to gain insight on the order of assemblyof gp16 in the initiation of DNA packaging in vitro.[3H]DNA–gp3 sedimented in the sucrose gradient asa peak centering on fraction 16 (Fig. 9a, black line).When DNA–gp3 was preincubated with gp16, thesedimentation rate of more than one-half of themolecules increased to give a new peak at fraction 13
Fig. 6. Composition of the gradient-isolated prohead/pRNA/gp16 complex and its activity in in vitro DNA–gp3packaging. (a) SDS-PAGE of peak sucrose gradient fract-ions from Fig. 5a. Lane 1, proheads/174-base pRNA; lane2, proheads/174-base pRNA with bound gp16; lane 3,gp16 marker alone. (b) DNA packaging activity in vitro ofthe prohead/174-base pRNA/gp16 complex isolated fromthe sucrose gradient of Fig. 5a upon addition of DNA–gp3and ATP. Lane 1, input DNA in the packaging reaction;lane 5, DNA packaged by the gradient-isolated prohead/174-base pRNA/gp16 complex; lane 7, DNA packaged bythe gradient-isolated prohead/pRNA/gp16 particle with12 additional copies (molecules) of gp16 added perprohead; lanes 4 and 6, negative controls in which ATPwas omitted from the reaction; lane 3, packaging in whichproheads/pRNA and gp16 were mixed and not sedimen-ted; lane 2, ATP negative control for the reaction shown inlane 3. The prohead concentration was two times higher inthe reaction shown in lane 3 than in the reactions in lanes 4–7, where the prohead/pRNA/gp16 complex was isolatedfrom the sucrose gradient. (c) EMSA analysis of proheads/120-base pRNA incubated with gp16. Lane 1, proheads/120-base pRNA, where the bulk of the pRNA dissociatesfrom the proheads during electrophoresis; lane 2, the pro-head/120-base pRNA/gp16 complex, revealing release ofpRNA/gp16 as a complex; lane 3, free 120-base pRNAmarker.
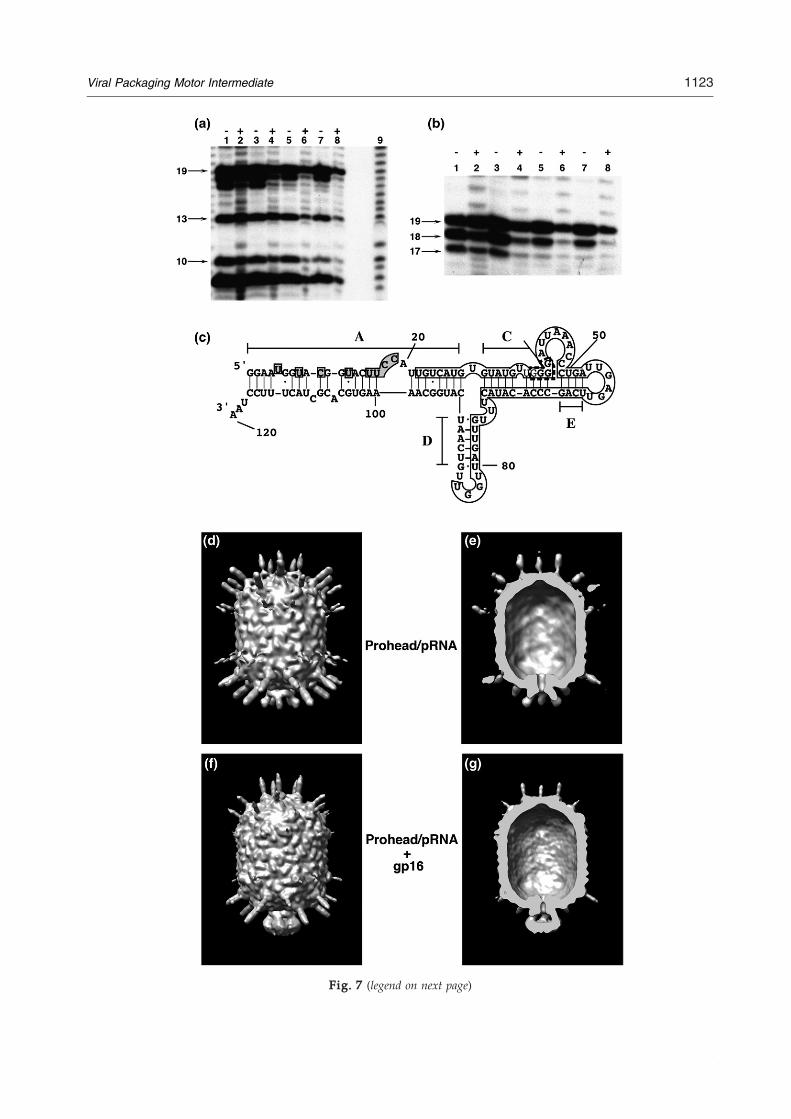
Fig. 7. Localizationofgp16onpRNA.(a–c)Ribonucleasefootprintingofgp16on120-basepRNA.[32P]120-basepRNAandprohead-bound[32P]120-basepRNAwereincubatedwithgp16,themixturesweretreatedwithRNaseA,andtheproductswereseparatedbydenaturinggelelectrophoresis(MaterialsandMethods).Analkalinehydrolysisladderwasgeneratedfrom5′end-labeledpRNA.(a)and(b)RNaseAdigestion.Lanes1–4,treatmentwith10−2μg/mlofRNaseA;lanes5–8,treatmentwith10−3μg/mlofRNaseA;lanes1and5,free[32P]120-basepRNA;lanes2and6,[32P]120-basepRNAwithgp16;lanes3and7,prohead-bound[32P]120-basepRNA;lanes4and8,prohead-bound[32P]120-basepRNAwithgp16.(c)gp16andproheadcompositefootprintonpRNA.Shadedregionsrepresentresiduesprotectedbygp16,theopenboxrepresentsresiduesprotectedbytheprohead,andregionsshownintheboxboundedbythediscontinuouslinerepresentenhancedcleavagesbyRNaseV1whenpRNAisboundtotheproheads.26(d)–(g)Cryo-EMthree-dimensionalreconstructionoftheprohead/174-basepRNA/gp16packagingintermediate.(d)proheadwith174-basepRNA.(e)Cross-sectionoftheprohead/pRNA,sameviewasin(d).(f)Prohead/174-basepRNA/gp16.(g)Cross-sectionoftheprohead/pRNA/gp16,sameviewasin(f).
Fig. 7 (legend on next page)
1123Viral Packaging Motor Intermediate

Fig. 8. The isolated DNA-filled head retains gp16. SDS-PAGE analysis of DNA-filled heads isolated in a sucrosedensity gradient. The gradient sample was prepared byincubating proheads/pRNA for 10 min with a head-defec-tive extract of cells from a restrictive infection with the ϕ29mutant sus7(614)–sus8(769)–sus14(1241), which served as asource of DNA–gp3 and excess gp16. Lane 1, filled headsconsisting of capsid (gp8), connector (gp10), gp16, andhead fibers (gp8.5); lane 2, gp16 marker; lane 3, proheadmarker.
1124 Viral Packaging Motor Intermediate
(Fig. 9a, pink line). AWestern dot blot of the sucrosegradient fractions, developed with anti-gp16 serum,confirmed the co-sedimentation of gp16 with DNA–gp3 (Fig. 9c) [Fig. 9b shows the position of gp16 alonein the gradient]. Addition of proheads/pRNA didnot alter the sedimentation state of DNA–gp3 (Fig.9d, green line). The prohead/pRNA/gp16 interme-diate (Fig. 9d, yellow line) produced a faster-sedi-menting DNA–gp3, just as gp16 did (Fig. 9d, pinkline). The presence of proheads and that of gp16 onthe fast-sedimenting DNA–gp3 were confirmed byWestern blotting with anti-gp10 and with anti-gp16sera, respectively. These results suggested that gp16,assembled as a component of the DNA packagingmotor onto proheads/pRNA, was able to bind andsupercoil the DNA–gp3 to produce an initiationcomplex for DNA packaging. While this interactionand formation of the higher-order complex are gp3dependent, EMSA showed that gp16 binds non-specifically to all HpaI fragments of proteinase K-treated DNA–gp3 (Fig. 9e).
Discussion
The ATPase gp16 is a central component in pack-aging of the ϕ29 genome, interacting with pRNAand DNA–gp3 to constitute the packaging motor atthe capsid portal vertex and then providing theenergy for DNA translocation. The ATPase (termi-
Fig. 7. Localization of gp16 on pRNA. (a–c) Ribonuclease foand prohead-bound [32P]120-base pRNA were incubated withproducts were separated by denaturing gel electrophoresis (Mgenerated from 5′ end-labeled pRNA. (a) and (b) RNase A diglanes 5–8, treatment with 10−3 μg/ml of RNase A; lanes 1 andpRNAwith gp16; lanes 3 and 7, prohead-bound [32P]120-basewith gp16. (c) gp16 and prohead composite footprint on pRNAopen box represents residues protected by the prohead, and rerepresent enhanced cleavages by RNase V1 when pRNA is boreconstruction of the prohead/174-base pRNA/gp16 packaginsection of the prohead/pRNA, same view as in (d). (f) ProheadpRNA/gp16, same view as in (f).
nase) large subunits of the dsDNA phages andherpesvirus tend to be insoluble,1,53 and productionof the highly hydrophobic gp16 in soluble form forstudy of structure and function has been proble-matic. Inclusion bodies of gp16 produced in E. coli(pPLc2833) could only be partially solubilized by theuse of guanidinium chloride followed by urea, andtherefore about 100 molecules were used to packageeach DNA–gp3 in vitro.20 Co-expression of gp16with GROESL in E. coli with the intent of producingsoluble protein resulted in an excess of GROESL,which had to be removed.49 The production of gp16with an N-terminal thioredoxin and His tag in E. coliresulted in an insoluble and inactive protein.54
Treatment with acetone or polyethylene glycol wasneeded to improve the solubility of the gp16–His–thioredoxin. However, cleaving away thioredoxincleaved gp16, making structural or functionalstudies of the native protein impossible. The presentpreparation of gp16 in a highly purified and solubleform in B. subtilis (pSAC) will facilitate studies ofgp16 structure and function.The number of copies of the large terminase
ATPase subunit required for optimal packagingactivity is not fully understood, although biochem-ical data suggest that six copies are needed in T311
and gp17 is predicted to form a ring surrounding theDNA on the procapsid in T4.55 All the known term-inase large subunits with ATPase activity exist pre-dominantly as monomers in solution.56–60 Self-association of terminase monomers into oligomershas been reported by sedimentation velocity inλ andby chemical cross-linking in T4 phage.13,14 gp16exists in solution predominantly in a monomericstate at a concentration of 0.1 mg/ml, with trimersand higher-order structures at concentrations of 0.2–0.8 mg/ml as seen by gel filtration, sedimentationequilibrium studies, and EM (Fig. 2). Early in vitropackaging studies demonstrated higher-order con-centration dependence for gp16, showing that multi-ple copies of gp16 were needed, while concentrationdependence for proheads and DNA–gp3 was firstorder.16 The present study of gp16 produced in B.subtilis (pSAC) shows that the quantity of gp16 thatwas found on the isolated, fully active prohead/pRNA/gp16 complex by SDS-PAGE was about halfthat of the dodecameric connector (Fig. 6a). Similarly,terminase large subunits of phages T3, T7, and P22bind the procapsid.61–63 In phage λ, packaging isinitiatedbydockingof theDNA/terminase (complex I)
otprinting of gp16 on 120-base pRNA. [32P]120-base pRNAgp16, the mixtures were treated with RNase A, and theaterials and Methods). An alkaline hydrolysis ladder wasestion. Lanes 1–4, treatment with 10−2 μg/ml of RNase A;5, free [32P]120-base pRNA; lanes 2 and 6, [32P]120-base
pRNA; lanes 4 and 8, prohead-bound [32P]120-base pRNA. Shaded regions represent residues protected by gp16, thegions shown in the box bounded by the discontinuous lineund to the proheads.26 (d)–(g) Cryo-EM three-dimensionalg intermediate. (d) proheadwith 174-base pRNA. (e) Cross-/174-base pRNA/gp16. (g) Cross-section of the prohead/

Fig. 9. Binding and supercoiling of DNA–gp3 by both gp16 and the prohead/pRNA/gp16 intermediate. (a) Thesedimentation rate of DNA–gp3 was increased by binding of gp16, previously shown to be due to gp3- and gp16-dependent supercoiling of DNA–gp3.21 The black line represents the gradient profile of DNA–gp3, and the pink linerepresents DNA–gp3 incubated with gp16. Sedimentation is from right to left. (b) Free gp16 (gradient not shown) wasfound primarily in the sucrose gradient fractions 23–20 in the dot blot developed with polyclonal gp16 antiserum. (c) Dotblot of fractions from the sucrose gradient of DNA–gp3 incubated with gp16 (a) showing co-sedimentation of gp16 withDNA–gp3, notably from fraction 16 to the bottom of the gradient. (d) gp16 on the prohead/pRNA supercoils DNA–gp3.Sucrose gradient profiles of DNA–gp3, black line; DNA–gp3+proheads, green line; DNA–gp3+gp16, pink line; DNA–gp3+gp16+proheads, sky blue line; DNA–gp3+gp16 complex incubated with proheads, blue line; prohead/gp16complex incubated with DNA–gp3, yellow line. All proheads contained 174-base pRNA. There was no alteration in thesedimentation rate of DNA–gp3 by proheads (green line). (e) Native agarose gel stained with ethidium bromide de-monstrating EMSA analysis of ϕ29 gp3-free DNAHpaI fragments (6784, 2549, 2341, 1781, 1777, 1714, 1608, and 731 bp) inthe presence of gp16. Lane 1, 0.5 μg of HpaI DNA fragments; lanes 2–6, the same DNA incubated with 6, 12, 24, 48, and 48copies of gp16 per DNA, respectively; lane 7, empty lane; lane 8, 48 copies of gp16 alone. gp16 binds to all of the HpaIfragments in a sequence-independent manner.
1125Viral Packaging Motor Intermediate
at the portal vertex, and amino acids of the largesubunit C-terminus are responsible for the capsidbinding.64,65
Amino acid sequence analysis indicates that the N-termini of many terminase large subunits have aconserved ATPase center.8 All known and predictedmembers of DNA packaging ATPase large subunitsbelong to the P-loop NTPase fold, which has con-served nucleotide binding (Walker A) and Mg2+
binding (Walker B)motifs.8,15 The ATPase domains ofT4 gp17 and ϕ29 gp16 appear to be similar. For ex-ample, theWalker Bmutant D118E/E119D in gp16 ofϕ29 is inactive in DNA packaging (data not shown),as is the mutant D255E/E256D in T4 gp17, whichbinds ATP but is unable to precisely orient the ATP
molecule.9 Also, the crystal structures of P4 of ϕ12and gp17 of T4 show a common ATPase domain.9,66
However, comparative genomic studies indicate thatgp16 of ϕ29 may be a diverging independent lineagefrom the HerA/FtsK ATPases.67,68 Sequence align-ment and mutation data suggest that the adeninebinding motif55 is not conserved among the gp16analogs and is different from that of T4 gp17 (data notshown). Massey et al.69 showed that FtsK ATPaseexists as a monomer in solution, and formation ofhexameric rings aroundDNAwas dependent only onthe presence of DNA and was independent of nuc-leotide or magnesium ions. The FtsK ATPase, likegp16, differs from terminases in lacking domain fu-sions with their nuclease partners.

1126 Viral Packaging Motor Intermediate
The ϕ29 DNA packaging motor differs primarilyfrom its counterparts among the dsDNA phages byhaving an essential RNA component, pRNA. To date,pRNA has been confirmed only in ϕ29 and itsrelatives,52 although Acidianus bottle-shaped virus,an archaeal phage, produces RNA with a predictedsecondary structure similar to pRNA.70 Binding ofpRNA to the head–tail connector requires the basicRKR residues at the gp10 N-terminus23 and is the laststep in morphogenesis of the mature prohead. gp16then binds to the pRNA oligomer to constitute theactive packaging motor, and this binding was strictlydependent on andmediated by the presence of pRNA(Figs. 5 and 6). Ribonuclease footprints of gp16 on theprohead/pRNA showed protection of residues of theA-helix, extending to residues 18 and 19, whichcorrespond to CC of the CCA bulge (Fig. 7). Thisresult was confirmed by demonstration that mutantpRNA lacking the A-helix did not bind gp16 (Fig. 5c)and by cryo-EM reconstruction of the prohead/gp16motor assembly intermediate, which showed thatgp16 was bound to the A-helix spokes of pRNA (Fig.7). The 120-base pRNA ΔCCA bulge mutant hasprohead binding competitor activity but noDNA–gp3packaging activity.33 Even though the CC residues inthe pRNA CCA bulge contribute to the gp16 binding,they were not required for the initial pRNA–gp16interaction (Fig. 4 and Table 2). This is similar to aprevious report that the CCA bulge is dispensable forgp16 binding.50 TheHis–gp16–thioredoxinwas foundto bind DNA nonspecifically and to bind the pRNA/prohead more strongly than the pRNA-free capsid,although none of these complexeswas tested for DNApackaging activity.50 The RNase footprinting of gp16on the prohead (Fig. 7) should help model the gp16–pRNA interaction. Endonuclease activity possessedby other phage large terminase subunits is not neededin ϕ29 as the DNA is unit length and not packagedfrom a DNA concatemer.In cryo-EM reconstruction, the pRNA and gp16
subunits have 5-fold symmetry and are intimatelyassociated (Fig. 7). This and the fact that gp16 is apRNA-dependent ATPase24 lend credence to the ideathat the ATPase subunits of the motor may be gp16/pRNA heterodimers. Once the gp16 has bound topRNA, the pRNA subunits have been shown bycryo-EM reconstruction to make contacts with thecapsid, which rationalizes the finding that the ring ofmotor subunits is 5-fold symmetric.30 Thus, pRNAultimately links the gp16 to the capsid, and whetherand how tightly pRNA remains associated with theconnector are unknown. The connector is shown bycryo-EM reconstruction to be essentially situated in abasket formed by pRNA.30 In conflict with the abovescenario, Ibarra et al.49 reported that, by the use ofELISA and immunoprecipitation experiments, theconnector N-terminus can bind gp16, as V8-treatedconnectors that lack the N-terminus did not interactwith gp16; pRNA–gp16 interaction was also reportedin this study. The gp16 used was in high excess inthese experiments (3137/prohead, 115/connector,and 3.3×104/RNA), and none of these complexeswas shown to be active in DNA packaging.
It is not known if the pRNA molecules retain theirnovel intermolecular base pairing interactions71,72 inthe active packaging motor, which might serve as ameans of communication among the ATPases in thecoordination of motor function. The pRNA subunitshave been hypothesized to function sequentially, asincorporation of one inactive subunit of a mutantpRNAhas been reported to be sufficient to stopDNApackaging.27,73 Tests of the sequential mode of actionof gp16 (and the proposed pRNA/gp16 heterodi-mers) are an aim of single-molecule packaging stu-dies in process.ϕ29 gp3, covalently bound at the DNA 5′ termini
(DNA–gp3), is needed for DNA packaging in bulkand has been viewed as a small terminase protein.10gp3 also greatly enhances the efficiency and selectiv-ity of DNA packaging in bulk in vitro.44 In phage P22,the small terminase subunit gp3 is shown to bindDNA in a sequence-independent manner and is aglobular decamer in solution with a central annulusthat can fit the DNA.60 In the phage λ small terminasesubunit gpNu1, the NMR structure shows a helix–turn–helix (HTH) motif for DNA binding,74 and theT4 gp16 small terminase also has a predicted HTHmotif, whereas gp3 of ϕ29 lacks an HTHmotif.75 Thegeneral strategy for genome packaging is thusretained in ϕ29 despite the apparent divergencefrom the conventional model with active participa-tion of pRNA in the packaging motor function.gp16 supercoils the DNA–gp3 packaging substrate
in a reaction that is dependent on gp3 covalentlybound at the DNA termini.21 This supercoiled DNA–gp3/gp16 complex is preferentially packaged intoproheads in vitro. Thus, the description here of astable and active prohead/pRNA/gp16 complex thatcan package DNA without additional gp16 (Fig. 6)represents an alternative pathway of gp16 assemblyand packaging initiation. Notably, however, this pro-head/pRNA/gp16 intermediate was also capable ofsupercoiling DNA–gp3, just as demonstrated earlierwith gp16 on free DNA–gp3 in the absence of pro-heads (Fig. 9).21 Figure 10 illustrates the alternativepathways for producing the prohead/pRNA/gp16/DNA–gp3 packaging initiation complex. Variousmixtures of the proheads/pRNA, gp16, and DNA–gp3, without or with ATP, or complexes stalled in thepackaging process with γS-ATP at different times,were prepared in BAC films,76 mounted on supportfilms, rotarymetal shadowed, and observed by trans-mission EM (data not shown) to directly observe theevents of initiation and progression of DNA–gp3packaging in vitro. DNA in the process of packagingappeared to be supercoiled and condensed, but theprovisional results have not yet provided a clearpicture of the structure of the initiation complex orthe progression of packaging. Proheads and DNA–gp3 were isolated at times ranging from 45 to 90 minfrom a lysate of a restrictive infection of B. subtiliswith the ϕ29 mutant sus14(1241), which providesdelayed lysis, in another attempt to determine thepathway and events in packaging initiation. gp16was found in multiple copies on both the DNA–gp3and the proheads (data not shown). The wrapping of

Fig. 10. Alternative pathways for producing the prohead/pRNA/gp16/DNA–gp3 packaging initiation complex. (a)DNA–gp3 forms a lariat by interaction of the terminal gp3 with DNA, independent of sequence, and gp16 binds to thelariat loop junction to effect supercoiling of the DNA in concert with gp3; this supercoiled DNA–gp3–gp16 complex ispreferentially packaged.21 (b) gp16 binds the prohead/pRNA to constitute a packaging intermediate that can be isolatedand is fully active without additional gp16. This prohead/pRNA/gp16 complex is hypothesized to bind and supercoilDNA–gp3 at the prohead portal vertex. Scaffolding protein exits as packaging proceeds upon hydrolysis of ATP, and thetail components are sequentially assembled on the DNA-filled head to yield ϕ29.
1127Viral Packaging Motor Intermediate
supercoiled DNA by the free ϕ29 head–tail connectorto remove negative supercoils77 is a provocativereminder that the initiation of packagingmay involveDNA–protein transactions within the motor thathave not been contemplated. Further experimenta-tion is needed to dissect the details of packaginginitiation.Isolation of the prohead/pRNA–gp16 motor as-
sembly intermediate has facilitated single-moleculestudies that allow DNA translocation to be measuredfrom initiation to completion, allowing study of thepreviously uncharacterized early steps of packagingand the demonstration of packaging forces N100pN.4,78 Prohead/pRNA/gp16 assembly intermedi-ates were attached to one bead, biotinylated DNAmolecules were tethered to streptavidin-coated mic-rospheres, and packaging was initiated by bringingthe beads into contact in the presence of ATP. In thesesingle-molecule studies, gp3 is not required, whereasDNAwithout gp3 is packaged 1 order of magnitudeless efficiently in bulk.44 The prohead/pRNA/gp16complex and DNA are rapidly brought into closephysical contact in the laser tweezers, possibly by-passing the need for gp3. It is also noteworthy thatuse of DNA–gp3 in the single-molecule studies re-sults in great variation in DNA tether length uponinitiation, indicating that the DNA–gp3 moleculeshave a higher-order structure.4
Materials and Methods
Purification of DNA–gp3
DNA–gp3 and [3H]DNA–gp3 were isolated fromlysates of B. subtilis RD2 (sup−) infected with the mutantsus4(369)–sus8(22), defective for late transcription andproduction of the major capsid protein, respectively, asdescribed previously.21 Proteinase K-treated DNA, furtherpurified in an isopycnic cesium chloride density gradient,
was used to generate an HpaI digest, which was phenolextracted and used for the EMSA determination of gp16binding.
Preparation of proheads
Proheads with 174-base pRNA were purified from alysate of B. subtilis SpoOA12 (sup−) cells infected with themutant sus16(300)–sus14(1241), which is defective for theDNA packaging ATPase gp16 as described previously.30
Production and purification of soluble gp16 fromB. subtilis
The purified E/D segment of ϕ29 DNA–gp3 containinggene 16 was obtained by partial digestion with EcoRI42and cloned into the plasmid pUB18, which encodeskanamycin resistance. The sucrose inducible promoterSACB43 was inserted before the gp16 gene, and theresulting pSACB-gp16 plasmid was moved into B. subtilisWB30 (asporogenic and protease negative) by protoplasttransformation.79 The maximum yield of the solubleprotein (10 mg/l) was obtained after induction of gene16 expression with 2% sucrose for 3 h at 37 °C. Theexpression of gp16 in B. subtilis did not result in inclusionbody formation as in E. coli,20 and about 70% of the proteinwas found in the soluble fraction (supernatant). Sucrose-induced cells were passed through a French press twice ina buffer containing 50 mM Tris–HCl, pH 7.8, 200 mMNaCl, 5% glycerol, and 2 mM Tris(2-carboxyethyl)phos-phine (TCEP). The lysate was clarified by centrifugation at10,000 rpm for 15 min at 4 °C in a SS34 rotor. The super-natant was subjected to P11 phosphocellulose (Whatman)cation exchange chromatography. The P11 column wasequilibrated with lysis buffer, washed with 8 bed volumesof 300 mM NaCl in 50 mM Tris–HCl, pH 7.8, 5% glycerol,and 2 mM TCEP, and gp16 was eluted with 350 mMNaClin the same buffer. The P11 eluate containing gp16 wasfurther purified on a hydroxyapatite column (Biorad),which was equilibrated with 300 mMNaCl in 50 mMTris–HCl, pH 7.8, 5% glycerol, and 2 mM TCEP. The columnwas washed with 3 bed volumes of 100 mM sodiumphosphate, pH 6.8, 5% glycerol, and 2 mMTCEP, followed

1128 Viral Packaging Motor Intermediate
by 2 bed volumes of 50 mM sodium phosphate, pH 6.8, inthe same buffer. The protein was eluted by a 10-bed-volume gradient of 100 to 900 mMNaCl in 50 mM sodiumphosphate, pH 6.8, 5% glycerol, and 2 mM TCEP. Frac-tions were analyzed by SDS-PAGE, and peak fractionscontaining pure gp16 were stored in aliquots at −70 °C.The identity of the protein was confirmed by a Westernblot developed with polyclonal anti-gp16 serum. Theaverage yield of the purified gp16 was 8 mg/l of culture.
In vitro DNA–gp3 packaging
Packaging in the defined in vitro system was performedas described previously.44 DNA–gp3 (1 μg) and proheadswere mixed in a ratio of 1:2 in a total volume of 20 μl in0.5× TMS buffer (25 mM Tris–HCl, pH 7.8, 5 mM MgCl2,and 50 mM NaCl) containing 0.5 mM ATP. Generally, 15copies (molecules) of gp16 per prohead were added, andthe mixture was incubated at ambient temperature for15 min. The gp16 copy number varied as indicated. DNaseI was added to 1 μg/ml to digest the unpackaged DNA,while the packaged DNAwas protected in the head. TheDNase was inhibited and the packaged DNA wasextracted from the filled heads by adding EDTA to25 mM and proteinase K to 500 μg/ml, and the efficiencyof packaging was quantified following electrophoresis ona 0.8% agarose gel run in TBE buffer (89 mM Tris, pH 8.3,89 mM boric acid, and 2.5 mM EDTA).
Sedimentation equilibrium
Sedimentation equilibrium experiments were per-formed with purified gp16 equilibrated in a buffer con-taining 50 mM sodium phosphate, pH 6.8, 400 mM NaCl,and 2 mM TCEP at 4 °C. Two samples at each con-centration of 0.2, 0.4, and 0.8 mg/ml were analyzed in anAn50Ti rotor in a Beckman Optima XL-I analytical cen-trifuge. Data were collected at 280 nm and at three rotorspeeds of 5000, 8000, and 11,000 rpm. Equilibrium wasreached when scans taken 4 h apart were superimposable.Each data curve was the result of at least three averagedscans. The partial specific volume (υ) for gp16 calculatedfrom the primary amino acid sequence using SEDNTERP47
was 0.7375 ml/g. Based on the primary amino acid se-quence, the calculated monomer molecular weight of gp16was 38,965. With the use of SEDNTERP, the calculateddensity (σ) of the buffer was 1.02859 g/ml and the viscosity(η) was 0.0167043.Data were initially fit to a self-association model using
NONLIN,46 which yielded molecular weights suggestiveof a trimer. Additional species that were included as fitswere not good for a simple monomer-to-trimer model. Thedata at lower centrifugation speeds and higher proteinconcentrations were fit using SEDPHAT.48 gp16 formshigher-molecular-weight species that are not reversibleand are lost to the bottom of the cell. The area of absorb-ance for each cell was determined at each centrifuge speedrelative to the initial absorbance at 3000 rpm to determinethe amount of loss. This amounted to loss of 40% of thegp16 at 0.2–0.4 mg/ml and that of 65% at 0.8 mg/ml. Theprotein remaining in solution was fit as described above.
Gel filtration
Gel filtration was performed using a Superdex 200 HR10/30 (Amersham Pharmacia Biotech) column at 4 °C in abuffer containing 50 mM sodium phosphate, pH 6.8,
400 mM NaCl, and 2 mM TCEP on an Akta FPLC sys-tem (Amersham Pharmacia Biotech). Molecular weightstandards were analyzed in the same buffer and con-tained thyroglobulin (669,000), human immunoglobulin G(158,000), ovalbumin (44,000), myoglobin (17,600), and vit-amin B12 (1355).
Transmission EM
Glow discharge-treated holey carbon films on 400-meshcopper grids were floated on samples of purified gp16(50 μg/ml in 50 mM Pipes, pH 6.8, and 400 mM NaCl),negatively stained with 2% (w/v) uranyl acetate, andimaged at a magnification of 57,000×. Micrographs weremade with a Philips EM301 electron microscope. Thediameter of gp16 particles from the electron micrographswas determined by the use of Image J (National Institutesof Health). Calibration was done from micrographs of acarbon-grating replica (Fullam, NY).
Preparation of pRNA
The 120- and 106-base pRNA forms were generated by invitro T7 transcription from pRT72 plasmid linearized withDdeI and ApaLI (Invitrogen), respectively, as describedpreviously.33 Alternatively, 120-base pRNA was pro-duced from in vitro transcription of the BamHI-linearizedpH120RNAH plasmid (Atz et al., unpublished results).The 71-base pRNA expression construct pH71RNAH wasgenerated by two rounds of PCR from the pRNA gene andby adding hammerhead ribozyme sequences to both endsof the template to generate uniform pRNA ends. Theplasmid was cloned into pUC19 and transformed intoE. coli DH5α. The purified plasmid was linearized withSmaI and transcribed in vitro to obtain the 71-base pRNA.R7 pRNAwas generated from B. subtilis 12A (pUM102) asdescribed previously.33 All forms of pRNA were purifiedby electrophoresis in denaturing urea acrylamide gels.80Individual pRNA bands were located by UV shadowingover polyethyleneimine UV254–cellulose plates with a 254-nm light source. pRNA bands were excised, and thepRNA eluted twice by diffusion into TEN buffer (10 mMTris–HCl, pH 8.0, 1 mM EDTA, and 100 mM NaCl) for 3 hat 4 °C.
EMSA
The 120-base pRNA was incubated with increasingconcentrations of gp16 in 50 mM Tris–HCl, pH 7.8, 10 mMMgCl2, and 100mMNaCl for 20 min at room temperature.Glycerol (5%), bromophenol blue (0.05%), and xylenecyanol (0.05%) were added to each reaction, the complexeswere resolved by electrophoresis in a 0.8% agarose gel inTB buffer (89 mM Tris–HCl, pH 8.3, and 89mM boric acid)for 3 h, and the gels were stained with ethidium bromide.EMSAwas also performed with HpaI-digested ϕ29 DNAfragments and gp16 in a similar manner.
Preparation of RNA-free proheads
Purified proheads were diluted 10-fold in TMS bufferand digested with 1 μg/ml of RNase A for 15 min at roomtemperature.44 These RNA-free proheads were purified ona 10%–40% sucrose density gradient in TMS buffer(50 mM Tris–HCl, pH 7.8, 10 mM MgCl2, and 100 mMNaCl) by centrifugation for 3 h at 35,000 rpm in an SW55

1129Viral Packaging Motor Intermediate
rotor. The prohead band was collected, and the particleswere pelleted in TMS buffer.
Reconstitution of pRNA-free proheads with pRNA
Ten copies of the pRNA produced from in vitro trans-cription were used per prohead, and the mixture wasincubated for 10 min at room temperature for reconstitu-tion. These reconstituted proheads were repurified on asucrose gradient to remove excess pRNA.
Isolation of the prohead/pRNA/gp16 ATPase motorassembly intermediate
Proheads with 174-base pRNA and gp16 were incubatedin TMS buffer without ATP at ambient temperature for20 min at a ratio of 20 gp16 molecules per prohead. Afterincubation, the reaction mixture was loaded on a 5%–20%sucrose gradient in TM buffer (50 mM Tris–HCl, pH 7.6,and 5 mMMgCl2) and centrifuged at 35,000 rpm for 30 minin an SW55 rotor at 4 °C. The peak band was collected witha syringe, and 20 μl was used for testing the in vitro DNA–gp3 packaging activity upon addition of DNA–gp3 andATP. A total of 100 μl of the fraction was precipitated with10% trichloroacetic acid and subjected to SDS-PAGE todetermine the composition of the prohead/pRNA/gp16intermediate. pRNA-free proheads and proheads reconsti-tuted with various forms of pRNA with A-helix deletions(71-base, 106-base and R7 pRNA, Fig. 4b–d) were used toscreen for binding of gp16 as described above.
RNase footprinting
The 120-base pRNA was 5′ end-labeled with Optiki-nase™ (USB Corp.) and [γ-32P]ATP after removal of the 5′terminal phosphatewith shrimp alkaline phosphatase. Theend-labeled pRNAwas further purified by electrophoresisin denaturing urea acrylamide gels. The pRNAbandswereeluted from the gel into TEN buffer and concentrated byisopropanol precipitation. pRNA obtained after precipi-tation was used for footprinting and reconstitution ofpRNA-free proheads for footprinting.A total of 140 μg of pRNA-free proheads was re-
constituted with 2.8 μg of 5′ end-labeled 120-base pRNA(6 pRNA/prohead; 7×105 CPM/μg of pRNA) in TMbuffer for 20 min at room temperature. Proheads wereseparated from the unbound pRNA by dilution into 5 mlof TM buffer and pelleting at 35,000 rpm for 5 h in theSW55 rotor at 4 °C as described previously.26
Prohead-bound [32P]pRNA, [32P]pRNA alone (2×104
CPM), and [32P]pRNA complexed with gp16 were digestedwith varying concentrations of RNase A (Ambion), RNaseT1, or RNase V1 (Ambion) in 10-μl reactions for 15 min atambient temperature. pRNAwas extracted with 200 μl of amixture containing one part 50 mM Tris–HCl, pH 7.6,10 mM EDTA, and 20 μg/ml of yeast tRNA (Ambion) andone part phenol. The aqueous phaseswere precipitatedwith0.6 volumes of isopropanol containing 20 μg/ml of gly-cogen. Pellets were resuspended in 4 M urea, 20% glycerol,0.05% xylene cyanol, and 0.05% bromophenol blue and runon a 10% denaturing 8.3 M urea polyacrylamide gel.
Cryo-EM
Proheads with 174-base pRNA and prohead/174-basepRNA/gp16 particles were flash-frozen on holey grids in
liquid ethane. Images were recorded at a magnification of39,000× with a CM200 FEGmicroscope, with electron doselevels of approximately 20 e−/Å2. All micrographs weredigitized at 4.24 Å pixel−1 using a Zeiss SCAI scanner.Individual particle images were boxed, floated, and
preprocessed to normalize mean intensities and variancesand to remove linear background gradients. Referenceprojections of a previously published prohead recons-truction30 were used to initially classify particles for three-dimensional reconstruction. The resulting model was usedto recalculate reference projections for better particleclassification. Several cycles of iterative particle classifica-tion and reconstruction were performed until convergencehad been reached. Structure factor phases and amplitudeswere modified as indicated by the parameters of thecontrast transfer function. All steps of the reconstructionprocess, including determination of the contrast transferfunction parameters, were performed with the programEMAN.81 Fivefold symmetry was assumed in the recon-struction. The number of particles incorporated in the finalreconstruction was 1705, and the final resolution of thereconstruction was 18.5 Å, as determined by the Fouriershell correlation method using a correlation coefficientof 0.5 between independent half data sets as the cutoffcriterion.
Sedimentation assay of DNA–gp16 andDNA–prohead/pRNA/gp16
[3H]DNA–gp3 (1 μg) was diluted in water and heatedfor 5 min at 37 °C. A 0.1 volume of 10× TM buffer and 12copies of gp16 per DNA were added to give a finalreaction volume of 100 μl. The mixture was incubated atambient temperature for 10 min and then centrifuged in a5%–20% linear sucrose density gradient containing TMbuffer (50 mM Tris–HCl, pH 7.6, and 5 mM MgCl2) in theSW55 rotor at 35,000 rpm for 2 h at 20 °C. Fractions werecollected, and the [3H]DNA–gp3 was quantified by liquidscintillation counting. Also, samples of the fractions weretreated with 5 μg/ml of DNase I at room temperaturefor 20 min, and dot blots of the samples were developedwith gp16 antiserum. As a control, gp16 alone was sedi-mented on a sucrose gradient and samples of the fractionswere used for dot blots and gp16 quantification. Prohead/pRNA/gp16 complexes (12 copies of gp16 per prohead)were prepared, incubated with [3H]DNA–gp3, and sedi-mented in a sucrose gradient to study their activityin binding and supercoiling DNA–gp3 as describedpreviously.21
Extract preparation
B. subtilis SpoOA12 (sup−) was infected with the mutantsus7(614)–sus8(769)–sus14(1241), which is defective for theprohead scaffold and capsid proteins and is a source ofgp16, and extracts of these infected cells were prepared asdescribed previously.16
Acknowledgements
This research was supported by the NationalInstitutes of Health through grants DE-003606 andGM-059604. M.C.M. was supported by the NationalScience Foundation through grant MCB-0443899. We

1130 Viral Packaging Motor Intermediate
thank Siddhartha Jena for dynamic light-scatteringanalysis, Steve Erickson and Rockney Atz for tech-nical assistance, and Daniel Cohen and CharlenePeterson for expertise with figures.
References
1. Catalano, C. E. (2005). Viral genome packagingmachines: an overview. In Viral Genome PackagingMachines: Genetics, Structure and Mechanism (Catalano,C. E., ed), pp. 1–4, Landes Bioscience/Eurekah.com,Georgetown, TX.
2. Jardine, P. J. & Anderson, D. L. (2006). DNA packagingin double-stranded DNA phages. In The Bacteriophages(Calendar, R., ed), pp. 49–65, Oxford University Press,New York, NY.
3. Smith, D. E., Tans, S. J., Smith, S. B., Grimes, S.,Anderson, D. L. & Bustamante, C. (2001). The bacterio-phage phi29 portal motor can package DNA against alarge internal force. Nature (London), 413, 748–752.
4. Rickgauer, J. P., Fuller, N., Grimes, S., Jardine, P. J.,Anderson, D. L. & Smith, D. E. (2008). Portal motorvelocity and internal force resisting viral DNA pack-aging in bacteriophage phi29. Biophys. J. 94, 159–167.
5. Murialdo, H. & Becker, A. (1978). Head morpho-genesis of complex double-stranded deoxyribonucleicacid bacteriophages. Microbiol. Rev. 42, 529–576.
6. Earnshaw, W. C. & Casjens, S. R. (1980). DNA pack-aging by the double-stranded DNA phages. Cell, 21,319–331.
7. Feiss, M. (1986). Terminase, a viral enzyme involved inthe recognition, cutting and packaging of bacterio-phage λ chromosomes. Trends Genet. 2, 100–104.
8. Mitchell, M. S., Matsuzaki, S., Imai, S. & Rao, V. B.(2002). Sequence analysis of bacteriophage T4 DNApackaging/terminase genes 16 and 17 reveals acommon ATPase center in the large subunit of viralterminases. Nucleic Acids Res. 30, 4009–4021.
9. Sun, S., Kondabagil, K., Gentz, P. M., Rossmann, M. G.& Rao, V. B. (2007). The structure of the ATPase thatpowers DNA packaging into bacteriophage T4 pro-capsids. Mol. Cell, 25, 943–949.
10. Guo, P., Peterson, C. & Anderson, D. L. (1987).Prohead and DNA–gp3-dependent ATPase activityof the DNA packaging protein gp16 of bacteriophageϕ29. J. Mol. Biol. 197, 229–236.
11. Fujisawa, H., Shibata, H. & Kato, H. (1991). Analysisof interactions among factors involved in the bacterio-phage T3 DNA packaging reaction in a defined in vitrosystem. Virology, 185, 788–794.
12. Kanamaru, S., Kondabagil, K., Rossmann, M. G. &Rao, V. B. (2004). The functional domains of bacterio-phage T4 terminase. J. Biol. Chem. 279, 40795–40801.
13. Kondabagil, K., Zhang, Z. & Rao, V. B. (2006). TheDNA translocating ATPase of bacteriophage T4packaging motor. J. Mol. Biol. 363, 786–799.
14. Feiss, M. & Catalano, C. E. (2005). Bacteriophagelambda terminase and the mechanism of viral DNApackaging. InViral Genome PackagingMachines: Genetics,Structure and Mechanism (Catalano, C. E., ed), pp. 5–39,Landes Bioscience/Eurekah.com, Georgetown, TX.
15. Guo, P., Peterson, C. & Anderson, D. L. (1987).Initiation events in in-vitro packaging of bacterio-phage ϕ29 DNA–gp3. J. Mol. Biol. 197, 219–228.
16. Bjornsti, M. A., Reilly, B. E. & Anderson, D. L. (1981).In vitro assembly of the Bacillus subtilis bacteriophageϕ29. Proc. Natl Acad. Sci. USA, 78, 5861–5865.
17. Bjornsti, M. A., Reilly, B. E. & Anderson, D. L. (1983).Morphogenesis of bacteriophage ϕ29 of Bacillussubtilis: oriented and quantized in vitro packaging ofDNA–gp3. J. Virol. 45, 383–396.
18. Simpson, A. A., Tao, Y., Leiman, P. G., Badasso, M. O.,He, Y., Jardine, P. J. et al. (2000). Structure of thebacteriophage phi29 DNA packaging motor. Nature,408, 745–750.
19. Gausch, A., Pous, J., Ibarra, B., Gomis-Ruth, F. X.,Valpuesta, J. M., Sousa, N. et al. (2002). Detailedarchitecture of a DNA translocating machine: thehigh-resolution structure of the bacteriophage ϕ29connector particle. J. Mol. Biol. 315, 663–676.
20. Guo, P., Grimes, S. & Anderson, D. L. (1986). Adefined system for in vitro packaging of DNA–gp3 ofthe Bacillus subtilis bacteriophage ϕ29. Proc. Natl Acad.Sci. USA, 83, 3505–3509.
21. Grimes, S. & Anderson, D. L. (1997). The bacterio-phage ϕ29 packaging proteins supercoil the DNAends. J. Mol. Biol. 266, 901–914.
22. Guo, P., Erickson, S. & Anderson, D. L. (1987). A smallviral RNA is required for in vitro packaging ofbacteriophage ϕ29 DNA. Science, 236, 690–694.
23. Atz, R., Ma, S., Gao, J., Anderson, D. L. & Grimes, S.(2007). Alanine scanning and FE-BABE probing of thebacteriophage ϕ29 prohead RNA–connector interac-tion. J. Mol. Biol. 369, 239–248.
24. Grimes, S. & Anderson, D. L. (1990). RNA dependenceof the bacteriophage ϕ29 DNA packaging ATPase.J. Mol. Biol. 215, 559–566.
25. Wichitwechkarn, J., Bailey, S., Bodley, J. W. &Anderson, D. L. (1989). Prohead RNA of bacterio-phage ϕ29: size, stoichiometry and biological activity.Nucleic Acids Res. 17, 3459–3468.
26. Reid, R. J., Bodley, J. W. & Anderson, D. L. (1994).Characterization of the prohead–pRNA interaction ofbacteriophage ϕ29. J. Biol. Chem. 269, 5157–5162.
27. Trottier, M. &Guo, P. (1997). Approaches to determinestoichiometry of viral assembly components. J. Virol.71, 487–494.
28. Ibarra, B., Caston, J. R., Llorca, O., Valle, M.,Valpuesta, J. M. & Carrascosa, J. L. (2000). Topologyof the components of the DNA packaging machineryin the phage ϕ29 prohead. J. Mol. Biol. 298, 807–815.
29. Morais, M. C., Tao, Y., Olson, N. H., Grimes, S.,Jardine, P. J., Anderson, D. L. et al. (2001). Cryoelec-tron-microscopy image reconstruction of symmetrymismatches in bacteriophage ϕ29. J. Struct. Biol. 135,38–46.
30. Morais, M. C., Choi, K. H., Koti, J. S., Chipman, P. R.,Anderson, D. L. & Rossmann, M. G. (2005). Conser-vation of capsid structure in tailed dsDNA bacterio-phages: the pseudoatomic structure of ϕ29. Mol. Cell,18, 149–159.
31. Shu, D., Zhang, H., Jin, J. & Guo, P. (2007). Counting ofsix pRNAs of phi29 DNA packaging motor withcustomized single-molecule dual-view system. EMBOJ. 26, 527–537.
32. Grimes, S., Jardine, P. J. & Anderson, D. L. (2002).Bacteriophage phi29 DNA packaging. Adv. Virus Res.58, 255–294.
33. Reid, R. J., Bodley, J. W. & Anderson, D. L. (1994).Identification of bacteriophage ϕ29 prohead RNA do-mains necessary for in vitro DNA–gp3 packaging.J. Biol. Chem. 269, 9084–9089.
34. Reid, R. J., Zhang, F., Benson, S. & Anderson, D. L.(1994). Probing the structure of bacteriophage ϕ29prohead RNA with specific mutations. J. Biol. Chem.269, 18656–18661.

1131Viral Packaging Motor Intermediate
35. Zhang, C., Lee, C. S. & Guo, P. (1994). The proximate5′ and 3′ ends of the 120-base viral RNA (pRNA) arecrucial for the packaging of bacteriophage phi29.Virology, 201, 77–85.
36. Zhang, C., Garver, K. & Guo, P. (1995). Inhibition ofphage phi29 assembly by antisense oligonucleotidestargeting viral pRNA essential for DNA packaging.Virology, 211, 568–676.
37. Chemla, Y. R., Aathavan, K., Michaelis, J., Grimes, S.,Jardine, P. J., Anderson, D. L. & Bustamante, C. (2005).Mechanism of force generation of a viral DNA pack-aging motor. Cell, 122, 683–692.
38. Hendrix, R. W. (1978). Symmetry mismatch and DNApackaging in large bacteriophages. Proc. Natl Acad. Sci.USA, 75, 4779–4783.
39. Baumann, R. G., Mullaney, J. & Black, L. W. (2006).Portal fusion protein constraints on function in DNApackaging of bacteriophage T4. Mol. Microbiol. 61,16–32.
40. Hugel, T., Michaelis, J., Hetherington, C. L., Jardine,P. J., Grimes, S., Walter, J. M. et al. (2007). Experimentaltest of connector rotation during DNA packaging intobacteriophage phi29 capsids. PLoS Biol. 5, e59.
41. Lebedev, A. E., Krause, M. H., Isidro, A. L., Vagin,A. A., Orlova, E. V., Turner, J. et al. (2007). Structuralframework for DNA translocation via the viral portalprotein. EMBO J. 26, 1984–1994.
42. Garvey, K. J., Saedi, M. S. & Ito, J. (1985). The completesequence of Bacillus phage ϕ29 gene 16: a proteinrequired for the genome encapsidation reaction. Gene,40, 311–316.
43. Li, M. & Wong, S. L. (1992). Cloning and character-ization of the GROESL operon from Bacillus subtilis.J. Bacteriol. 174, 3981–3992.
44. Grimes, S. & Anderson, D. L. (1989). In vitro packagingof bacteriophage ϕ29 DNA restriction fragments andthe role of the terminal protein gp3. J. Mol. Biol. 209,91–100.
45. Bjornsti, M. A., Reilly, B. E. & Anderson, D. L. (1984).Bacteriophageϕ29 proteins required for in vitroDNA–gp3 packaging. J. Virol. 50, 766–772.
46. Johnson, M., Correia, J. J., Yphantis, D. A. &Halvorson, H. (1981). Analysis of data from the ana-lytical ultracentrifuge by nonlinear least-squares tech-niques. Biophys. J. 36, 575–588.
47. Laue, T. M., Shah, B. D., Ridgeway, T. M. & Pelletier,S. L. (1992). In Analytical Ultracentrifugation in Biochem-istry and Polymer Science (Harding, S., Rowe, A. &Horton, J., eds), Royal Society of Chemistry, Cam-bridge, UK. (SEDNTERP: freeware from http://www.jphilo.mailway.com).
48. Vistica, J., Dam, J., Balbo, A., Yikilmaz, E., Mariuzza,R. A., Rouault, T. A. & Schuck, P. (2004). Sedimenta-tion equilibrium analysis of protein interactions withglobal implicit mass conservation constraints andsystematic noise decomposition. Anal. Biochem. 326,234–256. (SEDPHAT: freeware from http://www.analyticalultracentrifugation.com).
49. Ibarra, B., Valpuesta, J. & Carrascosa, J. L. (2001).Purification and functional characterization of p16,the ATPase of the bacteriophage ϕ29 packagingmachinery. Nucleic Acids Res. 29, 4264–4273.
50. Lee, T. & Guo, P. (2006). Interaction of gp16 withpRNA and DNA for genome packaging by the motorof bacterial virus phi29. J. Mol. Biol. 356, 589–599.
51. Guo, P., Bailey, S., Bodley, J. W. & Anderson, D. L.(1987). Characterization of the small RNA of thebacteriophage ϕ29 DNA packaging machine. NucleicAcids Res. 15, 7081–7090.
52. Bailey, S., Wichitwechkarn, J., Johnson, D., Reilly, B. E.,Anderson, D. L. & Bodley, J. W. (1990). Phylogeneticanalysis and secondary structure of the Bacillus subtilisbacteriophage RNA required for DNA packaging.J. Biol. Chem. 265, 22365–22370.
53. Przech, A. J., Yu, D. & Weller, S. K. (2003). Point muta-tions in exon I of the herpes simplex virus putativeterminase subunit, UL15, indicate that the most con-served residues are essential for cleavage and pack-aging. J. Virol. 77, 9613–9621.
54. Huang, L. P. & Guo, P. (2003). Use of PEG to acquirehighly soluble DNA-packaging enzyme gp16 ofbacterial virus phi29 for stoichiometry quantification.J. Virol. Methods, 109, 235–244.
55. Draper, B. & Rao, B. V. (2007). An ATP hydrolysissensor in the DNA packaging motor from bacterio-phage T4 suggests an inch-type translocation mechan-ism. J. Mol. Biol. 369, 79–94.
56. Gual, A., Camacho, A. G. & Alonso, C. J. (2000).Functional analysis of the terminase large subunit,G2P, of Bacillus subtilis bacteriophage SPP1. J. Biol.Chem. 275, 35311–35319.
57. Leffers, G. & Rao, V. B. (2000). Biochemical character-ization of an ATPase activity associated with the largepackaging subunit gp17 from bacteriophage T4.J. Biol. Chem. 275, 37127–37136.
58. Paris, W., Rubinchik, S., Yang, Y. & Gold, M. (1994).A new procedure for the purification of bacteriophageλ terminase enzyme and its subunits. Properties ofgene product A, the large subunit. J. Biol. Chem. 269,13564–13574.
59. Casjens, S. & Weigele, P. (2005). DNA packaging bybacteriophage P22. In Viral Genome PackagingMachines: Genetics, Structure and Mechanism (Catalano,C. E., ed), pp. 80–88, Landes Bioscience/Eurekah.com,Georgetown, TX.
60. Nemecek, D., Gilcrease, E. B., Kang, S., Prevelige, P. E.,Jr, Casjens, S. & Thomas, G. J., Jr (2007). Subunitconformations and assembly states of a DNA-translo-cating motor: the terminase of bacteriophage P22.J. Mol. Biol. 374, 817–836.
61. Serwer, P. (2007). Evolution and the complexity ofbacteriophages. Virol. J. 4, 30–39.
62. Morita, M., Tasaka, M. & Fujisawa, H. (1995). Struc-ture and functional domains of the large subunit of thebacteriophage T3 DNA packaging enzyme: impor-tance of the C-terminal region in prohead binding.J. Mol. Biol. 245, 635–644.
63. Eppler, K., Wyckoff, E., Goates, J., Parr, R. & Casjens,S. (1991). Nucleotide sequence of the bacteriophageP22 genes required for DNA packaging. Virology, 183,519–538.
64. Yeo, A. & Feiss, M. (1995). Specific interaction of ter-minase, the DNA packaging enzyme of bacteriophagelambda, with the portal protein of the prohead. J. Mol.Biol. 245, 141–150.
65. Sippy, J. & Feiss, M. (1992). Analysis of a mutationaffecting the specificity domain for the prohead bind-ing of the bacteriophage lambda terminase. J. Bacteriol.174, 850–856.
66. Kainov, D. E., Pirttimaa, M., Tuma, R., Butcher, S. J.,Thomas, G. J. J., Bamford, D. H. & Makeyev, E. V.(2003). RNA packaging device of double-strandedRNA bacteriophages, possibly as simple as hexamerof P4 protein. J. Biol. Chem. 278, 48084–48091.
67. Burroughs, A. M., Iyer, L. M. & Aravind, L. (2007).Comparative genomics and evolutionary trajectoriesof viral ATP dependent DNA-packaging systems.Genome Dyn. 3, 48–65.

1132 Viral Packaging Motor Intermediate
68. Iyer, L. M., Makarova, K. S., Koonin, E. V. & Aravind, L.(2004). Comparative genomics of the FtsK HerA super-family of pumping ATPases: implications for the originsof chromosome segregation, cell division and viralcapsid packaging. Nucleic Acids Res. 32, 5260–5279.
69. Massey, T. H., Mercogliano, C. P., Yates, J., Sherratt,D. J. & Lowe, J. (2006). Double-stranded DNAtranslocation: structure and mechanism of hexamericFtsK. Mol. Cell, 23, 457–469.
70. Peng, X., Basta, T., Haring, M., Garrett, R. A. &Prangishvili, D. (2007). Genome of Acidianus bottle-shaped virus and insights into the replication andpackaging mechanisms. Virology, 364, 237–243.
71. Zhang, F., Lemieux, S., Wu, X., Arnaud, S. D.,McMurray, C. T., Major, F. & Anderson, D. L. (1998).Function of hexameric RNA in packaging of bacter-iophage ϕ29 DNA in vitro. Mol. Cell, 2, 141–147.
72. Guo, P., Zhang, C., Chen, C., Garver, K. & Trottier, M.(1998). Inter-RNA interaction of phage ϕ29 pRNA toform a hexameric complex for viral DNA transporta-tion. Mol. Cell, 2, 149–155.
73. Chen, C. &Guo, P. (1997). Sequential action of six virus-encoded DNA-packaging RNAs during phage ϕ29genomic DNA translocation. J. Virol. 71, 3864–3871.
74. de Beer, T. D., Fang, J., Ortega, M., Yang, Q., Maes, L.,Duffy, C. et al. (2002). Insights into specific DNArecognition during the assembly of a viral genomepackaging machine. Mol. Cell, 9, 981–991.
75. Kamtekar, S., Berman, J. A., Wang, J., Lazaro, M. J.,Devega, M., Blanco, L. et al. (2006). The ϕ29 DNApolymerase: protein-primer structure suggests a modelfor the initiation to elongation transition. EMBO J. 25,1335–1343.
76. Vollenweider, H., Sogo, J. M. & Koller, T. (1975). Aroutine method for protein-free spreading of double-and single-stranded nucleic acid molecules. Proc. NatlAcad. Sci. USA, 72, 83–87.
77. Turnquist, S., Simon, M., Egelman, E. & Anderson, D.(1992). Supercoiled DNA wraps around the bacter-iophage ϕ29 head–tail connector. Proc. Natl Acad. Sci.USA, 89, 10479–10483.
78. Fuller, D. N., Rickgauer, J. P., Jardine, P. J., Grimes, S.,Anderson, D. L. & Smith, D. E. (2007). Ionic effects onviral DNA packaging and portal motor function inbacteriophage ϕ29. Proc. Natl Acad. Sci. USA, 104,11245–11250.
79. Chang, S. & Cohen, S. N. C. (1979). High frequencytransformation of Bacillus subtilis protoplasts byplasmid DNA. Mol. Gen. Genet. 168, 111–115.
80. Wichitwechkarn, J., Johnson, D. & Anderson, D. L.(1992). Mutant prohead RNAs in the in vitro packa-ging of bacteriophage ϕ29 DNA–gp3. J. Mol. Biol. 223,991–998.
81. Ludtke, S. J., Baldwin, P. R. & Chiu, W. (1999). EMAN:semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97.





![Bacteriophage [Compatibility Mode] (2)](https://static.fdocuments.net/doc/165x107/577cd7461a28ab9e789e8922/bacteriophage-compatibility-mode-2.jpg)












