
DISCUSSION ADDENDUM for: 2 … · 34 Org. Synth. 2012, 89, ... report some of the more recent...
10
34 Org. Synth. 2012, 89, 34-43 Published on the Web 8/24/2011 © 2012 Organic Syntheses, Inc. DISCUSSION ADDENDUM for: 2-Trimethylsilylethanesulfonyl Chloride (SES-Cl) Me 3 Si NaHSO 3 PhCO 3 t-Bu aq. MeOH 50 °C Me 3 Si SO 3 Na SOCl 2 cat. DMF 0 °C to rt Me 3 Si SO 2 Cl Prepared by Joshua R. Sacher and Steven M. Weinreb.* 1 Original article: Weinreb, S. M.; Chase, C. E; Wipf, P.; Venkatraman, S. Org. Synth. 1997, 75, 161. Sulfonamides are among the most stable amine protecting groups that are able to tolerate a broad range of reaction conditions. This stability, however, can sometimes be problematic for protecting group removal, often requiring harsh conditions. The 2-(trimethylsilyl)ethanesulfonyl (SES) group provides a sulfonamide that combines the compatibility with many synthetic transformations with relatively benign removal conditions. The chemistry of reactions involving the SES group has previously been reviewed; 2 herein, we report some of the more recent examples of its use in methodology and total synthesis. The SES group can be installed on a primary or secondary amine using 2-(trimethylsilyl)ethanesulfonyl chloride (SES-Cl) and a base, typically a tertiary amine or sodium hydride. 3 An alternate procedure using silver cyanide has been used to improve yields in difficult protections. 4 Generally, cesium fluoride in DMF or tetrabutylammonium fluoride (TBAF) in acetonitrile at elevated temperature is sufficient to remove the SES group, 3 producing TMS-F, ethylene, sulfur dioxide, and the free amine. Additionally, the SES group can be cleaved by HF, 5 tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF), 6 or refluxing 6 N HCl. 7 In certain special cases, judicious selection of the deprotection strategy can lead to different products, as demonstrated in the formation of either pyrrole 2 or pyrroline 3 from the corresponding SES- protected pyrroline 1 (Scheme 1). 8 DOI:10.15227/orgsyn.089.0034
Transcript of DISCUSSION ADDENDUM for: 2 … · 34 Org. Synth. 2012, 89, ... report some of the more recent...

34 Org. Synth. 2012, 89, 34-43 Published on the Web 8/24/2011
© 2012 Organic Syntheses, Inc.
DISCUSSION ADDENDUM for:
2-Trimethylsilylethanesulfonyl Chloride (SES-Cl)
Me3Si
NaHSO3PhCO3t-Bu
aq. MeOH50 °C
Me3SiSO3Na
SOCl2cat. DMF
0 °C to rtMe3Si
SO2Cl
Prepared by Joshua R. Sacher and Steven M. Weinreb.*1
Original article: Weinreb, S. M.; Chase, C. E; Wipf, P.; Venkatraman, S.
Org. Synth. 1997, 75, 161.
Sulfonamides are among the most stable amine protecting groups that
are able to tolerate a broad range of reaction conditions. This stability,
however, can sometimes be problematic for protecting group removal, often
requiring harsh conditions. The 2-(trimethylsilyl)ethanesulfonyl (SES) group
provides a sulfonamide that combines the compatibility with many synthetic
transformations with relatively benign removal conditions. The chemistry of
reactions involving the SES group has previously been reviewed;2
herein, we
report some of the more recent examples of its use in methodology and total
synthesis.
The SES group can be installed on a primary or secondary amine
using 2-(trimethylsilyl)ethanesulfonyl chloride (SES-Cl) and a base,
typically a tertiary amine or sodium hydride.3 An alternate procedure using
silver cyanide has been used to improve yields in difficult protections.4
Generally, cesium fluoride in DMF or tetrabutylammonium fluoride (TBAF)
in acetonitrile at elevated temperature is sufficient to remove the SES
group,3 producing TMS-F, ethylene, sulfur dioxide, and the free amine.
Additionally, the SES group can be cleaved by HF,5
tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF),6 or
refluxing 6 N HCl.7 In certain special cases, judicious selection of the
deprotection strategy can lead to different products, as demonstrated in the
formation of either pyrrole 2 or pyrroline 3 from the corresponding SES-
protected pyrroline 1 (Scheme 1).8
DOI:10.15227/orgsyn.089.0034

Org. Synth. 2012, 89, 34-43 35
NSES
F- or base
MeO2C
RHN
MeO2C
R HF, 0 °CH2N
MeO2C
R
F
12 3
Scheme 1
In addition to its use in introducing a protecting group, SES-Cl can
also effectively acidify an amine nitrogen and simultaneously provide a
leaving group, as demonstrated in a synthesis of L-azetidine-2-carboxylic
acid (9), a naturally occurring non-proteogenic amino acid.9 N-Sulfonylation
of amino alcohol 4 with SES-Cl, followed by treatment of the resulting bis-
SES compound 5 with base, gave the orthogonally-protected cyclic amino
acid 6 (Scheme 2). The SES and t-butyl ester protecting groups were
removed selectively with TBAF or TFA, to yield amine 7 and acid 8,
respectively, while global deprotection to form amino acid 9 was
accomplished with HF.
HO NH2
CO2tBu SES-Cl (2 eq)
Et3N, DMF, 0 °C66%
SESO NH
CO2tBu
SESCs2CO3, MeCN
rt, 12 h, 97%or
20 min, μW, 99%
N
CO2tBu
SES
TBAF
75%
TFA
99%
HF0 °C
97%
NH
CO2tBu
N
CO2H
SES
NH
CO2H
L-azetidine-2-carboxylic acid (9)
4 5 6
6
7 8
Scheme 2
Although the traditional manner of SES group installation involves
protection of an existing amine, it has become increasingly common to use
related derivatives to introduce a SES-protected nitrogen into a molecule.
One of the most common SES derivatives, 2-
(trimethylsilyl)ethanesulfonamide (SES-NH2), is easily prepared from the
chloride by reaction with anhydrous ammonia10
or ammonium hydroxide.11

36 Org. Synth. 2012, 89, 33-43
SES-NH2 has been shown to be effective in palladium-catalyzed
sulfonamidations involving electron deficient aryl and heteroaryl halides
(Scheme 3).12
Both aryl bromides 10 and chlorides 11 give good to excellent
yields of the corresponding aryl sulfonamides 12 and 13, and the reaction
tolerates a wide range of functionality.
XR
Br
H2N SESPd(OAc)2, Xantphos
Cs2CO3, dioxane100–120 °C
67–98%
XR
HN
SES
X
X
R
Cl
X
X
R
HN
SES
X = C, NR = H, COR, CO2R, CHO, CN, F, NO2, CF3
10
11
12
13
Scheme 3
SES-NH2 has also been N-alkylated in high yield with benzyl alcohol
(14) under ruthenium13
and copper14
catalysis to give benzyl sulfonamide 15
(Scheme 4). When benzylic acetate 16 was used in place of the alcohol, the
sulfonamide alkylation to form 17 has been shown to take place rapidly at
ambient temperature.15
OH
orCu(OAc)2, K2CO3air, 150 °C, 12 h
94%
NH
SES+
Ru/Fe3O4, K2CO3150 °C, 12 h
95%SES NH2
SES NH2 OAc
MeO
NH
MeO
SESCu(OTf)2, t-BuOOAc
DCE, 25 °C, 6 h78%
+
14 15
16 17
Scheme 4
SES-Sulfonamides bearing a terminal alkene moiety can undergo a
cyclization in the presence of a metal catalyst or oxidant (Scheme 5). For
example, utilizing a chiral copper catalyst, Chemler and coworkers

Org. Synth. 2012, 89, 34-43 37
performed an enantioselective carboamination of 18, which they proposed
occurs via a single-electron mechanism involving radical 19 to form
hexahydro-1H-benz[f]indole 20.16
In contrast to the 5-exo cyclization observed with metal catalysts,
Michael and coworkers demonstrated that oxidative cyclization of 21 with
hypervalent iodine gave solely the 6-endo product 24.17
A plausible
rationalization for this transformation involves opening of an oxidatively-
formed aziridinium intermediate 22 with trifluoroacetate resulting in the
observed endo selectivity. Hydrolysis of the trifluoroacetate 23 gave the
SES-protected hydroxypiperidine 24. In this system, the SES protecting
group gave slightly better yields than the corresponding tosyl, or 2- and 4-
nosylsulfonamides.
NH
SESPhI(OTFA)2, TFA
CH2Cl2, rt, 24 h94%
NSES
OR
NH
SES
PhPh
Cu(OTf)2, (R,R)-Ph-Box
K2CO3, MnO2PhCF3, 120 °C, 24 h
93%, dr >20:1, 83% ee
NSES
Ph
NSES
Ph
NSES
23 R = TFA24 R = H
18 19 20
21 22
Scheme 5
Iodosulfonamidation methodology using SES-NH2 and a cationic
iodine complex developed by Danishefsky18
was recently adopted by
Jurczak and Chaladaj in a formal synthesis of galantinic acid (25) (Scheme
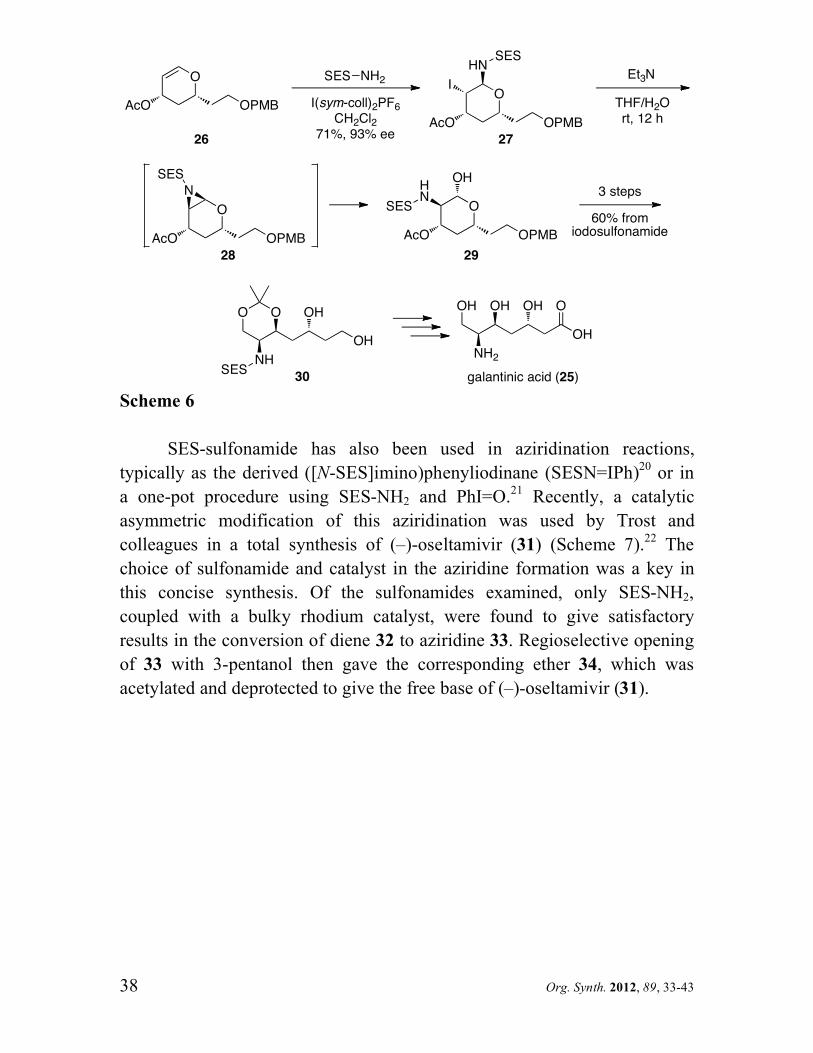
6).19
Activation of the enol ether 26 with I(sym-coll)2PF6, followed by
opening of the iodonium intermediate by SES-NH2, gave the
iodosulfonamide 27 with good diastereoselectivity. Under basic aqueous
conditions, sulfonamidoalcohol 29 was produced by hydrolysis of the SES-
aziridine intermediate 28. Reductive opening of the pyran ring, protection of
the resulting diol as the acetonide, and hydrogenolysis of the PMB group
gave advanced intermediate 30 that could easily be converted to galantinic
acid (25).
38 Org. Synth. 2012, 89, 33-43
O
AcO OPMB I(sym-coll)2PF6CH2Cl2
71%, 93% ee
SES NH2
O
AcO OPMB
I
HNSES
Et3N
THF/H2Ort, 12 h
O
AcO OPMB
NSES
O
AcO OPMB
HN
SES
OH3 steps
O O
OHNH
SES
OH
60% from iodosulfonamide
OH OH
OHNH2
OH O
galantinic acid (25)
26 27
28 29
30
Scheme 6
SES-sulfonamide has also been used in aziridination reactions,
typically as the derived ([N-SES]imino)phenyliodinane (SESN=IPh)20
or in
a one-pot procedure using SES-NH2 and PhI=O.21
Recently, a catalytic
asymmetric modification of this aziridination was used by Trost and
colleagues in a total synthesis of (–)-oseltamivir (31) (Scheme 7).22
The
choice of sulfonamide and catalyst in the aziridine formation was a key in
this concise synthesis. Of the sulfonamides examined, only SES-NH2,
coupled with a bulky rhodium catalyst, were found to give satisfactory
results in the conversion of diene 32 to aziridine 33. Regioselective opening
of 33 with 3-pentanol then gave the corresponding ether 34, which was
acetylated and deprotected to give the free base of (–)-oseltamivir (31).

Org. Synth. 2012, 89, 34-43 39
NPhth CO2Et
H2N SESRh2(esp)2 (2 mol%)
PhI(O2CCMe3)2MgO, PhCl, 0 °C to rt
86%NPhth CO2Et
NSES
BF3•OEt2
3-pentanol75 °C65%
NPhth CO2Et
HN
O
SES
1) Ac2O DMAP, Py μW, 150 °C, 1h, 84%
2) TBAF, THF, rt, 95%
3) H2NNH2 EtOH, 68 °C, 100% H2N CO2Et
AcHNO
(–)-oseltamivir (31)
32 33
34
Scheme 7
Recently, Komatsu and coworkers developed a high yielding, metal-
free sulfonamide aziridination method using SES-NH2 and t-butyl
hypoiodite to convert styrene (35) into SES-aziridine 36 (Scheme 8).23
In a
screening of sulfonamides, SES-NH2 was found to be superior to 2-
nitrophenyl-, n-butyl-, and p-toluenesulfonamides. The authors note that the
SES group is particularly useful in aziridine chemistry because it can be
removed without intervention of undesirable side reactions.
H2N SES
t-BuOCl, NaI
MeCN, rt, 3 h97%
NSES
35 36
Scheme 8
Due to the electron-withdrawing nature of the sulfonamide moiety,
SES aziridines are useful substrates for ring opening reactions. For example,
in a synthesis of (+)-preussin (37), SES-aziridine 39 was opened by the
lithium anion of allyl sulfone 38 to give SES-sulfonamide 40.24
Isomerization of the alkene to intermediate 41 by treatment with TBAF at
elevated temperature promoted a stereoselective 5-endo-trig cyclization that
took place with concomitant SES removal. The resulting pyrrolidine 42 was
then converted to the natural product 37 in 3 steps.

40 Org. Synth. 2012, 89, 33-43
n-C9H19
NSES
Ph
PhO2S n-BuLiTHF/TMEDA, -78 °C
95%
n-C9H19
HNSES
PhO2S
Ph
TBAF
n-C9H19
HNSES
PhO2S
PhNH
n-C9H19
PhO2S
Ph
THF, 65 °C78%
N
Ph
HO
(+)-preussin (37)Me
3 steps
38 39 40
41 42
Scheme 9
2-(Trimethylsilyl)ethanesulfonyl azide (SES-N3), which is easily
prepared by reaction of SES-Cl with NaN3 in acetone,25a
has also been used
in metal-catalyzed aziridination reactions. For instance, Katsuki and
coworkers used SES-N3 to effect an asymmetric transformation of alkenes
43 to aziridines 44 using a chiral ruthenium-salen catalyst (Scheme 10).25
Interestingly, SES-N3 showed higher enantioselectivity than the
corresponding 2- or 4-nosylazides, while maintaining a similar yield.
RRu(salen)CO (1-5%)
R = Ph, 4-BrPh, C6H13, CO2Bn, CON(OMe)Bn, PhC C
28-100%, 77-99% ee RN
SESN3 SES
43 44
Scheme 10
SES-N3 has also been shown to participate in alkyne-azide 1,3-dipolar
cycloaddition reactions (Scheme 11). Thus, in the presence of a catalytic
amount of CuI and 2,6-lutidine, the reaction of the azide with
phenylacetylene (45) proceeds smoothly to give the 4-phenyl 1,2,3-triazole
46.26
By changing the base to triethylamine and adding an alcohol to the
reaction mixture, the corresponding N-protected imidate 48 can be isolated.27
If water is used, the acylated sulfonamide 49 is obtained in high yield.28
The

Org. Synth. 2012, 89, 34-43 41
reactions involving alcohol and water most likely proceed through addition
to a transient ketenimine intermediate 47.
SES N3
CuI (10 mol %)CHCl3
2,6-lutidine NN
N
Ph
SES
Ph25 °C, 12 h
84%
PhOBn
NSES
H2O, Et3NPh
HN
OSES
BnOH, Et3N
0 °C, 12 h84%
25 °C, 12 h97%
45
46
48
49
NSESX
Ph
47X = H or Cu
Scheme 11
N-Acyl-SES-sulfonimides can also be obtained from carboxylic acids
and SES-N3 via a one-pot, three-step sequence (Scheme 12).29
For example,
activation of acids 50 with t-butyl chloroformate, followed by treatment with
lithio trimethylsilyl thiolate, and methanolysis of the resulting silylated
compound gives the thioacid, which is immediately combined with SES-N3
to give the N-acylated sulfonimides 51. These sulfonimides can be N-
alkylated and the SES group can be selectively removed under mild
conditions.
BocHN
OR
OH
O1) t-butyl chloroformate, 2,6-lutidine, THF, 0 °C, 30 min
2) TMS-SLi, THF, 0 °C3) SES N3
BocHN
OR
NH
OSES
51a: R = H, 94%51b: R = Bn, 96%
50a: R = H50b: R = Bn
Scheme 12
1. Department of Chemistry, The Pennsylvania State University,
University Park, PA, 16802. [email protected]
2. For recent reviews, see: a) Ribière, P.; Declerck, V.; Martinez, J.;
Lamaty, F. Chem. Rev. 2006, 106, 2249; b) Chambert, S.; Désiré, J.;
Décout, J.-L. Synthesis 2002, 2319.
3. Weinreb, S. M.; Demko, D. M.; Lessen, T. A. Tetrahedron Lett. 1986,
27, 2099.

42 Org. Synth. 2012, 89, 33-43
4. Hale, K. J.; Domostoj, M. M.; Tocher, D. A.; Irving, E.; Scheinmann, F.
Org. Lett. 2003, 5, 2927.
5. Boger, D. L.; Ledeboer, M. W.; Kume, M.; Searcey, M.; Jin, Q. J. Am.
Chem. Soc. 1999, 121, 11375.
6. Dauban, P.; Dodd, R. H. J. Org. Chem. 1999, 64, 5304.
7. Decicco, C. P.; Grover, P. Synlett 1997, 529.
8. Declerck, V.; Allouchi, H.; Martinez, J.; Lamaty, F. J. Org. Chem.
2007, 72, 1518.
9. Bouazaoui, M.; Martinez, J.; Cavelier, F. Eur. J. Org. Chem. 2009,
2729.
10. Stien, D.; Anderson, G. T.; Chase, C. E.; Koh, Y.-H., Weinreb, S. M. J.
Am. Chem. Soc. 1999, 121, 9574.
11. Benohoud, M.; Leman, L.; Cardoso, S. H.; Retailleau, P.; Dauban, P.;
Thierry, J.; Dodd, R. H. J. Org. Chem. 2009, 74, 5331.
12. Anjanappa, P.; Mullick, D.; Selvakumar, K.; Sivakumar, M.
Tetrahedron Lett. 2008, 49, 4585.
13. Shi, F.; Tse, M. K.; Zhou, S.; Pohl, M.-M.; Radnik, J.; Hübner, S.;
Jähnisch, K.; Brückner, A.; Beller, M. J. Am. Chem. Soc. 2009, 131,
1775.
14. Shi, F.; Tse, M. K.; Cui, X.; Gördes, D.; Michalik, D.; Thurow, K.;
Deng, Y.; Beller, M. Angew. Chem. Int. Ed. 2009, 48, 5912.
15. Powell, D. A.; Pelletier, G. Tetrahedron Lett. 2008, 49, 2495.
16. Miao, L.; Haque, I.; Manzoni, M. R.; Tham, W. S.; Chemler, S. R. Org.
Lett. 2010, 12, 4739.
17. Lovick, H. M.; Michael, F. E. J. Am. Chem. Soc. 2010, 132, 1294
18. Griffith, D. A.; Danishefsky, S. J. J. Am. Chem. Soc. 1990, 112, 5811
19. Chaladaj, W.; Jurczak, J. Eur. J. Org. Chem. 2011, 1223.
20. Dauban, P.; Dodd, R. H. J. Org. Chem. 1999, 64, 5304.
21. Dauban, P.; Sanière, L.; Tarrade, A.; Dodd, R. H. J. Am. Chem. Soc.
2001, 123, 7707.
22. a) Trost, B. M.; Zhang, T. Angew. Chem. Int. Ed. 2008, 47, 3759; b)
Trost, B. M.; Zhang, T. Chem. Eur. J. 2011, 17, 3630.
23. Minakata, S.; Morino, Y.; Oderaotoshi, Y.; Komatsu, M. Chem.
Commun. 2006, 3337.
24. Caldwell, J. J.; Craig, D.; East, S. P. ARKIVOC 2007, 7, 67.
25. a) Kawabata, H.; Omura, K.; Uchida, T.; Katsuki, T. Chem. Asian. J.
2007, 2, 248; b) Kawabata, H.; Omura, K.; Katsuki, T. Tetrahedron
Lett. 2006, 47, 1571.

Org. Synth. 2012, 89, 34-43 43
26. Yoo, E. J.; Ahlquist, M.; Kim, S. H.; Bae, I.; Fokin, V. V.; Sharpless,
K. B.; Chang, S. Angew. Chem. Int. Ed. 2007, 46, 1730.
27. Yoo, E. J.; Bae, I.; Cho, S. H.; Han, H.; Chang, S. Org. Lett. 2006, 8,
1347.
28. Cho, S. H.; Yoo, E. J.; Bae, I.; Chang, S. J. Am. Chem. Soc. 2005, 127,
16046.
29. Barlett, K. N.; Kolakowski, R. V.; Katukojvala, S.; Williams, L. J. Org.
Lett. 2006, 8, 823.
Steven M. Weinreb was born in Brooklyn, and grew up in Rochester, NY. He did his undergraduate work at Cornell University, and received a doctorate from the University of Rochester with Marshall Gates in 1967. He then held NIH Postdoctoral Fellowships at Columbia University during 1966-67 with Gilbert Stork and at MIT during 1967-70 with George Büchi. He began his independent scientific career at Fordham University in New York City as an Assistant Professor of Chemistry in 1970. In 1978, he joined the faculty at Penn State and was named Russell and Mildred Marker Professor of Natural Products Chemistry in 1987. Throughout his career he has been involved in research on the total synthesis of natural products, heterocyclic chemistry and the development of new synthetic methods
Joshua Sacher was born in Wilmington, Delaware in 1984. He obtained his B.S. degree in biochemistry from the University of Delaware in 2005. While at Delaware, he did undergraduate research with Douglass Taber and had an internship at Cephalon, Inc. He then joined the Weinreb group at Penn State University, where he is currently pursuing his Ph.D degree.


















