
Directed evolution of artificial metalloenzymes for in...
17
29 SEPTEMBER 2016 | VOL 537 | NATURE | 661 LETTER doi:10.1038/nature19114 Directed evolution of artificial metalloenzymes for in vivo metathesis Markus Jeschek 1 , Raphael Reuter 2 , Tillmann Heinisch 2 , Christian Trindler 2 , Juliane Klehr 2 , Sven Panke 1 § & Thomas R. Ward 2 § The field of biocatalysis has advanced from harnessing natural enzymes to using directed evolution to obtain new biocatalysts with tailor-made functions 1 . Several tools have recently been developed to expand the natural enzymatic repertoire with abiotic reactions 2,3 . For example, artificial metalloenzymes, which combine the versatile reaction scope of transition metals with the beneficial catalytic features of enzymes, offer an attractive means to engineer new reactions. Three complementary strategies exist 3 : repurposing natural metalloenzymes for abiotic transformations 2,4 ; in silico metalloenzyme (re-)design 5–7 ; and incorporation of abiotic cofactors into proteins 8–11 . The third strategy offers the opportunity to design a wide variety of artificial metalloenzymes for non-natural reactions. However, many metal cofactors are inhibited by cellular components and therefore require purification of the scaffold protein 12–15 . This limits the throughput of genetic optimization schemes applied to artificial metalloenzymes and their applicability in vivo to expand natural metabolism. Here we report the compartmentalization and in vivo evolution of an artificial metalloenzyme for olefin metathesis, which represents an archetypal organometallic reaction 16–22 without equivalent in nature. Building on previous work 6 on an artificial metallohydrolase, we exploit the periplasm of Escherichia coli as a reaction compartment for the ‘metathase’ because it offers an auspicious environment for artificial metalloenzymes, mainly owing to low concentrations of inhibitors such as glutathione, which has recently been identified as a major inhibitor 15 . This strategy facilitated the assembly of a functional metathase in vivo and its directed evolution with substantially increased throughput compared to conventional approaches that rely on purified protein variants. The evolved metathase compares favourably with commercial catalysts, shows activity for different metathesis substrates and can be further evolved in different directions by adjusting the workflow. Our results represent the systematic implementation and evolution of an artificial metalloenzyme that catalyses an abiotic reaction in vivo, with potential applications in, for example, non-natural metabolism. In the past decade, the biotin–streptavidin technology has proven to be versatile for generating artificial metalloenzymes 12,23–26 . This ver- satility may be traced back to the non-covalent affinity of biotinylated organometallic cofactors for streptavidin (SAV), which ensures their quantitative anchoring in an evolvable protein scaffold. To customize this system for in vivo applications, we designed a strain for periplasmic expression of SAV (SAV peri ) in E. coli. The resulting strain revealed good secretion of functional SAV into the periplasm with no detectable contamination in the cytoplasm (Fig. 1b). Staining E. coli cells that harbour SAV peri with Atto-565-biotin yielded a persisting strong fluorescent signal as determined by flow cytometric analysis, whereas the dye-treated strain lacking SAV peri exhibited only low fluorescence (Fig. 1b). These experiments con- firm the biotin-binding activity of SAV peri and suggest that large hydrophobic biotinylated probes (weight-averaged molecular mass of Atto-565-biotin, M w (Atto-565-biotin) = 922 Da) are taken up into the periplasm. Next, we selected a biotinylated Hoveyda–Grubbs second-generation catalyst (biot-Ru hereafter) to complete the holoenzyme (Fig. 1c) 27 . Previous experiments using purified SAV revealed that, upon incorpo- ration within SAV, the biot-Ru–SAV complex catalyses the ring-closing metathesis (RCM) of the umbelliferone precursor 1 to afford umbellif- erone 2. Despite its poor performance as an RCM substrate in water 27 , we hypothesized that the formation of fluorescent umbelliferone 2, albeit in low yield, would offer an attractive means to monitor and evolve catalytic performance in vivo. Spiking an aqueous reaction mix- ture with glutathione (GSH; 10 equiv. versus 50 μM catalyst) led to complete inhibition of biot-Ru–SAV; no umbelliferone 2 was detected. By contrast, upon addition of glutathione disulfide (GSSG) instead of GSH (10 equiv. versus 50 μM catalyst), the metathesis activity of biot-Ru–SAV was maintained (Extended Data Table 1). This suggests that the periplasm may indeed offer a suitable environment to harbour the artificial metathase, because GSH is present in this compartment mainly in its oxidized GSSG form. To confirm the assembly of the artificial metalloenzyme biot-Ru– SAV within the periplasm, various E. coli strains were treated with the cofactor biot-Ru. Following incubation and washing, the ruthenium content was quantified by inductively coupled plasma optical emis- sion spectrometry (ICP-OES; Fig. 1c). Although some unspecifically bound ruthenium is present in cells either without SAV at all (SAV − ; approximately 26,000 atoms per cell) or with cytoplasmic SAV (SAV cyto ; approximately 31,000 atoms per cell), a threefold increase in ruthenium concentration was detected in the presence of SAV peri (approximately 80,000 atoms per cell), which suggests that biot-Ru transits through the outer membrane and accumulates in SAV peri . Next, we added the diolefin substrate 1 to E. coli cells bearing biot-Ru–SAV peri . RCM activity yielding umbelliferone 2 was moni- tored by fluorescence (Fig. 1d). In contrast to the strain containing only the apoenzyme SAV peri , metathesis activity could be unambiguously detected only for E. coli cells containing the artificial metalloenzyme biot-Ru–SAV peri (Fig. 1d, photograph). Most importantly, quantifica- tion of the fluorescent signal revealed detectable metathesis activity for the strain containing biot-Ru–SAV peri , whereas the free cofactor is largely inhibited in vivo in the absence of the SAV scaffold (Fig. 1d, bar chart). These experiments confirm: (i) that there is no E. coli-derived background RCM activity (that is, RCM is bio-orthogonal); (ii) that the bare metathesis cofactor biot-Ru is severely inhibited in E. coli; and (iii) that the periplasm is a propitious compartment to perform RCM with biot-Ru–SAV peri . To the best of our knowledge, these results represent the first report of the assembly of an artificial metalloenzyme in a whole-cell set-up and its subsequent use as a catalyst in an abiotic transformation in cellulo. By contrast, organometallic-mediated transformations performed so far in a cellular environment typically require multiple equivalents of the precious metal to achieve conversion 28 . 1 Department of Biosystems Science and Engineering, ETH Zurich, Basel CH-4058, Switzerland. 2 Department of Chemistry, University of Basel, Basel CH-4056, Switzerland. §These authors jointly supervised this work. © 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.
Transcript of Directed evolution of artificial metalloenzymes for in...

2 9 S E P T E M B E R 2 0 1 6 | V O L 5 3 7 | N A T U R E | 6 6 1
LETTERdoi:10.1038/nature19114
Directed evolution of artificial metalloenzymes for in vivo metathesisMarkus Jeschek1, Raphael Reuter2, Tillmann Heinisch2, Christian Trindler2, Juliane Klehr2, Sven Panke1§ & Thomas R. Ward2§
The field of biocatalysis has advanced from harnessing natural enzymes to using directed evolution to obtain new biocatalysts with tailor-made functions1. Several tools have recently been developed to expand the natural enzymatic repertoire with abiotic reactions2,3. For example, artificial metalloenzymes, which combine the versatile reaction scope of transition metals with the beneficial catalytic features of enzymes, offer an attractive means to engineer new reactions. Three complementary strategies exist3: repurposing natural metalloenzymes for abiotic transformations2,4; in silico metalloenzyme (re-)design5–7; and incorporation of abiotic cofactors into proteins8–11. The third strategy offers the opportunity to design a wide variety of artificial metalloenzymes for non-natural reactions. However, many metal cofactors are inhibited by cellular components and therefore require purification of the scaffold protein12–15. This limits the throughput of genetic optimization schemes applied to artificial metalloenzymes and their applicability in vivo to expand natural metabolism. Here we report the compartmentalization and in vivo evolution of an artificial metalloenzyme for olefin metathesis, which represents an archetypal organometallic reaction16–22 without equivalent in nature. Building on previous work6 on an artificial metallohydrolase, we exploit the periplasm of Escherichia coli as a reaction compartment for the ‘metathase’ because it offers an auspicious environment for artificial metalloenzymes, mainly owing to low concentrations of inhibitors such as glutathione, which has recently been identified as a major inhibitor15. This strategy facilitated the assembly of a functional metathase in vivo and its directed evolution with substantially increased throughput compared to conventional approaches that rely on purified protein variants. The evolved metathase compares favourably with commercial catalysts, shows activity for different metathesis substrates and can be further evolved in different directions by adjusting the workflow. Our results represent the systematic implementation and evolution of an artificial metalloenzyme that catalyses an abiotic reaction in vivo, with potential applications in, for example, non-natural metabolism.
In the past decade, the biotin–streptavidin technology has proven to be versatile for generating artificial metalloenzymes12,23–26. This ver-satility may be traced back to the non-covalent affinity of biotinylated organometallic cofactors for streptavidin (SAV), which ensures their quantitative anchoring in an evolvable protein scaffold. To customize this system for in vivo applications, we designed a strain for periplasmic expression of SAV (SAVperi) in E. coli. The resulting strain revealed good secretion of functional SAV into the periplasm with no detectable contamination in the cytoplasm (Fig. 1b).
Staining E. coli cells that harbour SAVperi with Atto-565-biotin yielded a persisting strong fluorescent signal as determined by flow cytometric analysis, whereas the dye-treated strain lacking SAVperi exhibited only low fluorescence (Fig. 1b). These experiments con-firm the biotin-binding activity of SAVperi and suggest that large hydrophobic biotinylated probes (weight-averaged molecular mass
of Atto-565-biotin, Mw(Atto-565-biotin) = 922 Da) are taken up into the periplasm.
Next, we selected a biotinylated Hoveyda–Grubbs second-generation catalyst (biot-Ru hereafter) to complete the holoenzyme (Fig. 1c)27. Previous experiments using purified SAV revealed that, upon incorpo-ration within SAV, the biot-Ru–SAV complex catalyses the ring-closing metathesis (RCM) of the umbelliferone precursor 1 to afford umbellif-erone 2. Despite its poor performance as an RCM substrate in water27, we hypothesized that the formation of fluorescent umbelliferone 2, albeit in low yield, would offer an attractive means to monitor and evolve catalytic performance in vivo. Spiking an aqueous reaction mix-ture with glutathione (GSH; 10 equiv. versus 50 μ M catalyst) led to complete inhibition of biot-Ru–SAV; no umbelliferone 2 was detected. By contrast, upon addition of glutathione disulfide (GSSG) instead of GSH (10 equiv. versus 50 μ M catalyst), the metathesis activity of biot-Ru–SAV was maintained (Extended Data Table 1). This suggests that the periplasm may indeed offer a suitable environment to harbour the artificial metathase, because GSH is present in this compartment mainly in its oxidized GSSG form.
To confirm the assembly of the artificial metalloenzyme biot-Ru–SAV within the periplasm, various E. coli strains were treated with the cofactor biot-Ru. Following incubation and washing, the ruthenium content was quantified by inductively coupled plasma optical emis-sion spectrometry (ICP-OES; Fig. 1c). Although some unspecifically bound ruthenium is present in cells either without SAV at all (SAV−; approximately 26,000 atoms per cell) or with cytoplasmic SAV (SAVcyto; approximately 31,000 atoms per cell), a threefold increase in ruthenium concentration was detected in the presence of SAVperi (approximately 80,000 atoms per cell), which suggests that biot-Ru transits through the outer membrane and accumulates in SAVperi.
Next, we added the diolefin substrate 1 to E. coli cells bearing biot-Ru–SAVperi. RCM activity yielding umbelliferone 2 was moni-tored by fluorescence (Fig. 1d). In contrast to the strain containing only the apoenzyme SAVperi, metathesis activity could be unambiguously detected only for E. coli cells containing the artificial metalloenzyme biot-Ru–SAVperi (Fig. 1d, photograph). Most importantly, quantifica-tion of the fluorescent signal revealed detectable metathesis activity for the strain containing biot-Ru–SAVperi, whereas the free cofactor is largely inhibited in vivo in the absence of the SAV scaffold (Fig. 1d, bar chart). These experiments confirm: (i) that there is no E. coli-derived background RCM activity (that is, RCM is bio-orthogonal); (ii) that the bare metathesis cofactor biot-Ru is severely inhibited in E. coli; and (iii) that the periplasm is a propitious compartment to perform RCM with biot-Ru–SAVperi.
To the best of our knowledge, these results represent the first report of the assembly of an artificial metalloenzyme in a whole-cell set-up and its subsequent use as a catalyst in an abiotic transformation in cellulo. By contrast, organometallic-mediated transformations performed so far in a cellular environment typically require multiple equivalents of the precious metal to achieve conversion28.
1Department of Biosystems Science and Engineering, ETH Zurich, Basel CH-4058, Switzerland. 2Department of Chemistry, University of Basel, Basel CH-4056, Switzerland.§ These authors jointly supervised this work.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

6 6 2 | N A T U R E | V O L 5 3 7 | 2 9 S E P T E M B E R 2 0 1 6
LETTERRESEARCH
After demonstrating the assembly and activity of the metathase in the periplasm, we sought to evolve biot-Ru–SAVperi towards higher activity. We thus established a streamlined workflow for the growth and expression of different SAVperi mutants in E. coli in a 96-well format (Fig. 2a). After the induction period, in which SAVperi is synthesized and exported to the periplasm, the cells are harvested by centrifuga-tion, resuspended in a buffer containing the cofactor and incubated for 30 min to allow for the assembly of the artificial metalloenzyme in the periplasm. The buffer containing excess cofactor is then removed by centrifugation, the cells are resuspended in the reaction buffer and RCM is initiated by the addition of substrate 1 and monitored in a microtiter plate reader. This procedure enables thousands of mutants to be screened in only a few days and represents a substantial increase in throughput compared to the conventional workflow that relies on purified mutants.
To improve the performance of the artificial metathase by directed evolution, we designed site saturation mutagenesis libraries of the twenty amino acid residues closest to the modelled position of ruthenium8 (Fig. 2b). Relying on degenerate NNK codons, analysis of 90 clones affords a > 94% probability of screening every possible amino acid residue at one position29. Fourteen of the twenty targeted positions revealed a beneficial impact on catalysis in the fluorescence assay and were further selected for iterative saturation mutagenesis30. After evaluation of more than 3,000 clones, this directed evo-lution protocol led to the identification of the quintuple mutant SAVperi(V47A/N49K/T114Q/A119G/K121R) (SAVmut) in complex with biot-Ru (biot-Ru–SAVmut), which displayed a cell-specific
activity 5.4 ± 1.2 (mean ± s.d.) times higher than that of biot-Ru–SAVperi (Fig. 2c).
Next, the kinetic behaviour of the free cofactor biot-Ru, biot-Ru–SAV and biot-Ru–SAVmut was determined in vitro using purified protein samples and the resulting fluorescence of umbelliferone 2. Even though the limited substrate solubility prevented reaching the maximum reaction velocity Vmax, all three systems clearly display Michaelis–Menten behaviour (Fig. 3a). As judged by the kinetic param-eters derived from the best fit of the experimental data to a Michaelis–Menten model, directed evolution led to an improvement of both Vmax and the substrate affinity KM. biot-Ru–SAVmut displays a (2.0 ± 0.6)-fold improvement in catalytic efficiency (kcat/KM, where kcat denotes the turnover frequency per cofactor molecule) over the wild-type metathase. To verify the improved catalytic behaviour, we tested the activity of biot-Ru–SAVmut on three independently produced and purified protein batches (Extended Data Fig. 1) and varied the ratio between cofactor and SAV binding sites to ensure full complexation (Extended Data Fig. 2). Both experiments confirmed the aforemen-tioned trend and the robustness of the proposed system.
The X-ray structure of biot-Ru–SAVmut (Protein Data Bank (PDB), 5F2B) reveals two conformations of the cofactor (I and II with 56% and 44% occupancy, respectively; Fig. 3b and Extended Data Figs 3, 4). Extensive steric clashes hamper the coexistence of two I conformers in adjacent cis-biotin binding sites. However, I–II and II–II confor-mations are possible. The evolved metathase reveals greater biotin- vestibule loop flexibility when compared to the biot-Ru–SAV structure (PDB, 5IRA), as highlighted by analysis of the corresponding B factors
Figure 1 | Artificial metalloenzymes for in vivo metathesis. a, Streptavidin (SAV, see Methods section for gene information) is secreted to the periplasm by fusion to an N-terminal signal peptide from the outer membrane protein A (OmpA). b, Top, the fusion to the OmpA signal peptide enables efficient secretion of SAV, which accumulates in the periplasmic protein fraction (‘peri’) without contamination in the cytoplasm (‘cyto’) (‘Vis’: exposure to visible light). Biotin binding is confirmed with a biotinylated fluorophore (‘UV’: exposure to UV light). Purified SAV (‘+ ’) was included as positive control. The masses between gels indicate the position of reference proteins of known mass. Bottom, flow cytometry analysis by in vivo staining with a biotinylated fluorescent dye. Cells expressing SAVperi (blue solid line) remain highly fluorescent after washing, whereas controls without SAV (SAV−; grey dashed line) and controls with SAVperi but without dye treatment (black dotted line) are non-fluorescent. Count is normalized by the maximum count; a.u.,
arbitrary units; horizontal axis is displayed in bi-exponential scale. c, Top, an artificial metathesis cofactor (biot-Ru) composed of a biotin anchor and a catalytic moiety is added to cells expressing SAVperi thus creating a metathase. Bottom, cofactor uptake as determined by ICP-OES quantification of ruthenium is more than twice as high for SAVperi than for SAV− or cytoplasmic SAV (SAVcyto). d, Top, the metathase converts the non-fluorescent substrate 1 into fluorescent umbelliferone 2 by a bio-orthogonal metathesis reaction. Bottom, only cells with SAVperi and biot-Ru develop a signal due to the freely diffusible product 2 in the supernatant (left two panels), which can be quantified against a low background for SAV− (right panel). Activity is given as the change in fluorescence Δ F in arbitrary units (a.u.) per optical density of the culture at 600 nm (OD600) and per minute. Bars in c and d show the mean of two (n = 2) and three (n = 3) independent replicate cultures, respectively, with error bars indicating 1 s.d.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

2 9 S E P T E M B E R 2 0 1 6 | V O L 5 3 7 | N A T U R E | 6 6 3
LETTER RESEARCH
Figure 2 | Directed evolution of an artificial metathase. a, A screening workflow (96-well) was developed that enables expression of SAV mutants and subsequent evaluation of their metathesis activity on the basis of a whole-cell fluorescent assay. To increase the throughput, the substrate surrogate 1 was selected, thus allowing kinetic monitoring in vivo. See Methods for a detailed description of the workflow. LB, lysogeny broth; O/N, overnight; ZYM5052mod, modified ZYM5052 medium; IPTG, isopropyl β -d-1-thiogalactopyranoside. b, The 20 closest residues to the catalytic ruthenium ion in SAV8 were randomized by saturation mutagenesis
(NNK codons), of which 14 revealed a positive impact (bold) on metathase activity. The different colours represent the four monomers of SAV. c, Iterative saturation mutagenesis30 on these 14 residues led to an optimized metathase SAVmut bearing five mutations. Data from independent replicate cultures (n = 8) are represented by boxes (median and first/third quartile) with whiskers (minimum/maximum of data). The horizontal dashed lines represent the mean cell-specific activity of each generation.
Figure 3 | Characterization of artificial metathases. a, Michaelis–Menten kinetics of free biot-Ru (grey circles), biot-Ru–SAV (blue squares) and biot-Ru–SAVmut (navy triangles) were performed, relying on umbelliferone 2 formation by metathesis. Symbols represent the mean of three (n = 3) kinetic in vitro experiments; error bars indicate 1 s.d. Dashed lines represent the best fit (R2 = 0.99, R2 = 0.98 and R2 = 0.97 for biot-Ru, biot-Ru–SAV and biot-Ru–SAVmut, respectively) to a Michaelis–Menten model with the maximum velocity (Vmax) and substrate affinity (KM) as summarized in the table together with the catalytic efficiency (kcat/KM; kcat is the turnover frequency per cofactor molecule). Bold figures in table highlight the ‘best’ value of each column. b, X-ray structure (PDB, 5F2B) of the evolved metathase biot-Ru–SAVmut in the two active site conformations I and II. biot-Ru and close-lying protein residues are depicted in a stick model. The flexibility of loop-7,81 is increased by hydrogen-bond swapping with mutation T114Q1 (red for green dashed line), entropy-raising mutation A119G1 and bulk-reducing V47A2. The superscripts refer to the respective SAV monomers (‘1’, blue monomer; ‘2’, tan monomer). The unresolved alkylidene ligand (magenta) in the crystal structure was modelled.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.
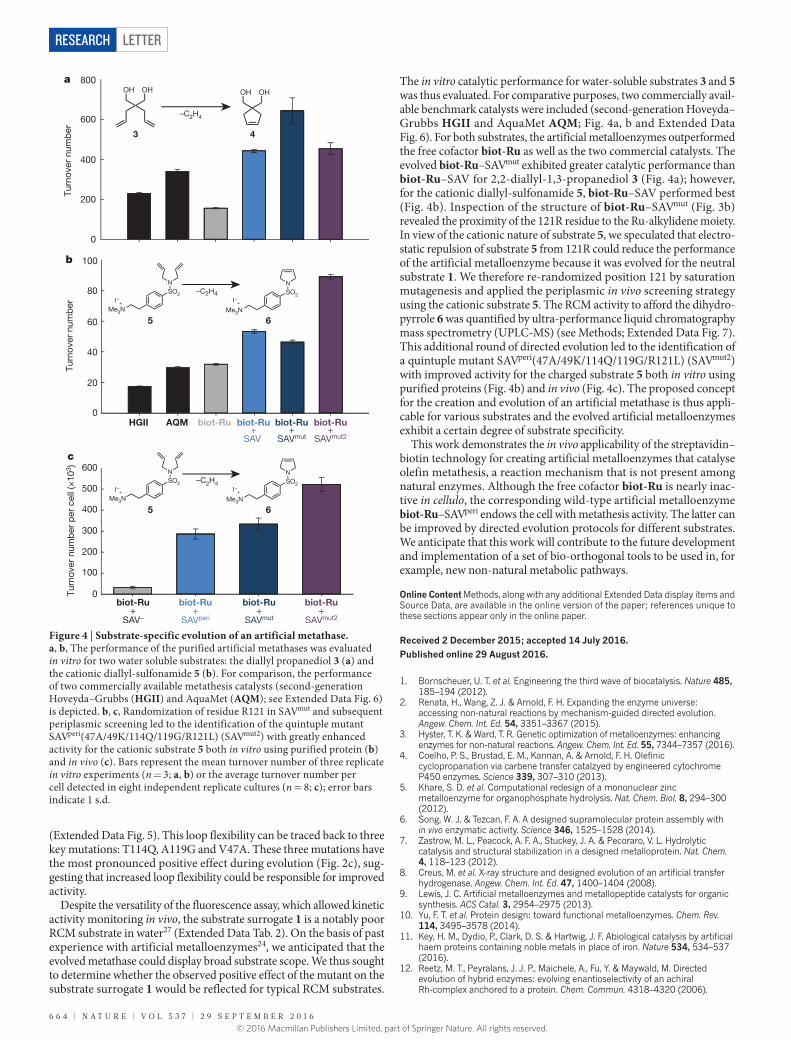
6 6 4 | N A T U R E | V O L 5 3 7 | 2 9 S E P T E M B E R 2 0 1 6
LETTERRESEARCH
(Extended Data Fig. 5). This loop flexibility can be traced back to three key mutations: T114Q, A119G and V47A. These three mutations have the most pronounced positive effect during evolution (Fig. 2c), sug-gesting that increased loop flexibility could be responsible for improved activity.
Despite the versatility of the fluorescence assay, which allowed kinetic activity monitoring in vivo, the substrate surrogate 1 is a notably poor RCM substrate in water27 (Extended Data Tab. 2). On the basis of past experience with artificial metalloenzymes24, we anticipated that the evolved metathase could display broad substrate scope. We thus sought to determine whether the observed positive effect of the mutant on the substrate surrogate 1 would be reflected for typical RCM substrates.
The in vitro catalytic performance for water-soluble substrates 3 and 5 was thus evaluated. For comparative purposes, two commercially avail-able benchmark catalysts were included (second-generation Hoveyda–Grubbs HGII and AquaMet AQM; Fig. 4a, b and Extended Data Fig. 6). For both substrates, the artificial metalloenzymes outperformed the free cofactor biot-Ru as well as the two commercial catalysts. The evolved biot-Ru–SAVmut exhibited greater catalytic performance than biot-Ru–SAV for 2,2-diallyl-1,3-propanediol 3 (Fig. 4a); however, for the cationic diallyl-sulfonamide 5, biot-Ru–SAV performed best (Fig. 4b). Inspection of the structure of biot-Ru–SAVmut (Fig. 3b) revealed the proximity of the 121R residue to the Ru-alkylidene moiety. In view of the cationic nature of substrate 5, we speculated that electro-static repulsion of substrate 5 from 121R could reduce the performance of the artificial metalloenzyme because it was evolved for the neutral substrate 1. We therefore re-randomized position 121 by saturation mutagenesis and applied the periplasmic in vivo screening strategy using the cationic substrate 5. The RCM activity to afford the dihydro-pyrrole 6 was quantified by ultra-performance liquid chromatography mass spectrometry (UPLC-MS) (see Methods; Extended Data Fig. 7). This additional round of directed evolution led to the identification of a quintuple mutant SAVperi(47A/49K/114Q/119G/R121L) (SAVmut2) with improved activity for the charged substrate 5 both in vitro using purified proteins (Fig. 4b) and in vivo (Fig. 4c). The proposed concept for the creation and evolution of an artificial metathase is thus appli-cable for various substrates and the evolved artificial metalloenzymes exhibit a certain degree of substrate specificity.
This work demonstrates the in vivo applicability of the streptavidin–biotin technology for creating artificial metalloenzymes that catalyse olefin metathesis, a reaction mechanism that is not present among natural enzymes. Although the free cofactor biot-Ru is nearly inac-tive in cellulo, the corresponding wild-type artificial metalloenzyme biot-Ru–SAVperi endows the cell with metathesis activity. The latter can be improved by directed evolution protocols for different substrates. We anticipate that this work will contribute to the future development and implementation of a set of bio-orthogonal tools to be used in, for example, new non-natural metabolic pathways.
Online Content Methods, along with any additional Extended Data display items and Source Data, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Received 2 December 2015; accepted 14 July 2016.
Published online 29 August 2016.
1. Bornscheuer, U. T. et al. Engineering the third wave of biocatalysis. Nature 485, 185–194 (2012).
2. Renata, H., Wang, Z. J. & Arnold, F. H. Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed. 54, 3351–3367 (2015).
3. Hyster, T. K. & Ward, T. R. Genetic optimization of metalloenzymes: enhancing enzymes for non-natural reactions. Angew. Chem. Int. Ed. 55, 7344–7357 (2016).
4. Coelho, P. S., Brustad, E. M., Kannan, A. & Arnold, F. H. Olefinic cyclopropanation via carbene transfer catalzyed by engineered cytochrome P450 enzymes. Science 339, 307–310 (2013).
5. Khare, S. D. et al. Computational redesign of a mononuclear zinc metalloenzyme for organophosphate hydrolysis. Nat. Chem. Biol. 8, 294–300 (2012).
6. Song, W. J. & Tezcan, F. A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014).
7. Zastrow, M. L., Peacock, A. F. A., Stuckey, J. A. & Pecoraro, V. L. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat. Chem. 4, 118–123 (2012).
8. Creus, M. et al. X-ray structure and designed evolution of an artificial transfer hydrogenase. Angew. Chem. Int. Ed. 47, 1400–1404 (2008).
9. Lewis, J. C. Artificial metalloenzymes and metallopeptide catalysts for organic synthesis. ACS Catal. 3, 2954–2975 (2013).
10. Yu, F. T. et al. Protein design: toward functional metalloenzymes. Chem. Rev. 114, 3495–3578 (2014).
11. Key, H. M., Dydio, P., Clark, D. S. & Hartwig, J. F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 534, 534–537 (2016).
12. Reetz, M. T., Peyralans, J. J. P., Maichele, A., Fu, Y. & Maywald, M. Directed evolution of hybrid enzymes: evolving enantioselectivity of an achiral Rh-complex anchored to a protein. Chem. Commun. 4318–4320 (2006).
Figure 4 | Substrate-specific evolution of an artificial metathase. a, b, The performance of the purified artificial metathases was evaluated in vitro for two water soluble substrates: the diallyl propanediol 3 (a) and the cationic diallyl-sulfonamide 5 (b). For comparison, the performance of two commercially available metathesis catalysts (second-generation Hoveyda–Grubbs (HGII) and AquaMet (AQM); see Extended Data Fig. 6) is depicted. b, c, Randomization of residue R121 in SAVmut and subsequent periplasmic screening led to the identification of the quintuple mutant SAVperi(47A/49K/114Q/119G/R121L) (SAVmut2) with greatly enhanced activity for the cationic substrate 5 both in vitro using purified protein (b) and in vivo (c). Bars represent the mean turnover number of three replicate in vitro experiments (n = 3; a, b) or the average turnover number per cell detected in eight independent replicate cultures (n = 8; c); error bars indicate 1 s.d.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

2 9 S E P T E M B E R 2 0 1 6 | V O L 5 3 7 | N A T U R E | 6 6 5
LETTER RESEARCH
13. Srivastava, P., Yang, H., Ellis-Guardiola, K. & Lewis, J. C. Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat. Commun. 6, 7789 (2015).
14. Sauer, D. F. et al. A highly active biohybrid catalyst for olefin metathesis in water: impact of a hydrophobic cavity in a β -barrel protein. ACS Catal. 5, 7519–7522 (2015).
15. Wilson, Y. M., Dürrenberger, M., Nogueira, E. S. & Ward, T. R. Neutralizing the detrimental effect of glutathione on precious metal catalysts. J. Am. Chem. Soc. 136, 8928–8932 (2014).
16. Grubbs, R. H. Olefin-metathesis catalysts for the preparation of molecules and materials (Nobel lecture). Angew. Chem. Int. Ed. 45, 3760–3765 (2006).
17. Schrock, R. R. Multiple metal–carbon bonds for catalytic metathesis reactions (Nobel lecture). Angew. Chem. Int. Ed. 45, 3748–3759 (2006).
18. Chauvin, Y. Olefin metathesis: the early days (Nobel lecture). Angew. Chem. Int. Ed. 45, 3740–3747 (2006).
19. Hoveyda, A. H. & Zhugralin, A. R. The remarkable metal-catalysed olefin metathesis reaction. Nature 450, 243–251 (2007).
20. Fürstner, A. Teaching metathesis “simple” stereochemistry. Science 341, 1229713 (2013).
21. Burtscher, D. & Grela, K. Aqueous olefin metathesis. Angew. Chem. Int. Ed. 48, 442–454 (2009).
22. Lin, Y. A., Chalker, J. M., Floyd, N., Bernardes, G. J. L. & Davis, B. G. Allyl sulfides are privileged substrates in aqueous cross-metathesis: application to site-selective protein modification. J. Am. Chem. Soc. 130, 9642–9643 (2008).
23. Wilson, M. E. & Whitesides, G. M. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J. Am. Chem. Soc. 100, 306–307 (1978).
24. Ward, T. R. Artificial metalloenzymes based on the biotin-avidin technology: enantioselective catalysis and beyond. Acc. Chem. Res. 44, 47–57 (2011).
25. Ilie, A. & Reetz, M. T. Directed evolution of artificial metalloenzymes. Isr. J. Chem. 55, 51–60 (2015).
26. Lo, C., Ringenberg, M. R., Gnandt, D., Wilson, Y. & Ward, T. R. Artificial metalloenzymes for olefin metathesis based on the biotin-(strept)avidin technology. Chem. Commun. 47, 12065–12067 (2011).
Supplementary Information is available in the online version of the paper.
Acknowledgements We thank P. Marlière for discussions. This work was supported by funding from the European Commission Seventh Framework Programme [289572-METACODE] and the Swiss National Science Foundation as part of the NCCR Molecular Systems Engineering. We thank M. Dessing and the single-cell facility (D-BSSE, ETH Zurich) for assistance with flow cytometry.
Author Contributions T.R.W. and S.P. conceived the project. M.J. developed the periplasmic screening platform and performed in vivo and directed evolution experiments. R.R. synthesized the cofactor and substrates. M.J. and R.R. performed the in vitro experiments. T.H. conducted the crystallography studies. C.T. performed ICP-OES and J.K. expressed and purified protein variants. T.R.W. and S.P. supervised the project. M.J., T.R.W. and S.P. wrote the manuscript.
Author Information The X-ray structures of the artificial metathases have been deposited in the Protein Data Bank (PDB) under accession numbers 5F2B and 5IRA. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to T.R.W. ([email protected]) or S.P. ([email protected]).
27. Kajetanowicz, A., Chatterjee, A., Reuter, R. & Ward, T. R. Biotinylated metathesis catalysts: synthesis and performance in ring closing metathesis. Catal. Lett. 144, 373–379 (2014).
28. Völker, T., Dempwolff, F., Graumann, P. L. & Meggers, E. Progress towards bioorthogonal catalysis with organometallic compounds. Angew. Chem. Int. Ed. 53, 10536–10540 (2014).
29. Reetz, M. T., Kahakeaw, D. & Lohmer, R. Addressing the numbers problem in directed evolution. ChemBioChem 9, 1797–1804 (2008).
30. Reetz, M. T. & Carballeira, J. D. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protocols 2, 891–903 (2007).
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTERRESEARCH
METHODSChemicals and reagents. If not stated otherwise, all chemicals and reagents were obtained from Sigma Aldrich. Restriction enzymes were obtained from New England Biolabs. The AquaMet catalyst was purchased from Apeiron Synthesis S.A.Cloning of SAV expression constructs. All plasmids used in this study are col-lated in Supplementary Table 1. To construct the periplasmic expression vector for SAV, the gene for T7-tagged SAV was amplified by polymerase chain reaction (PCR) from pET-11b-SAV31 using primers 1 and 2 (Supplementary Table 2) to add the 21 amino acid OmpA signal peptide (MKKTAIAIAVALAGFATVAQA) to the N terminus of SAV. The PCR product was digested with restriction enzymes NdeI and BamHI, gel purified and ligated into the target vector pET-30b(+ ) (Merck Millipore) pre-treated with the same enzymes. The resulting expression vector, designated pET-30b-SAVperi, carries the gene for the OmpA::SAV fusion protein under the control of a PT7 promoter. To construct a comparable cytoplasmic expres-sion construct, the gene for T7-tagged SAV from pET11b-SAV was PCR-amplified without adding additional amino acids using primers 3 and 2 (Supplementary Table 2) and subsequently cloned into pET-30b(+ ) by restriction digest (NdeI and BamHI) and ligation, resulting in plasmid pET-30b-SAVcyto.Construction of the periplasmic expression strain. All strains used in this study are summarized in Supplementary Table 3. A strain for periplasmic expression was constructed that combines the ease of library generation with the compatibility with the T7-expression system. Therefore, the gene of the T7 RNA polymerase was inte-grated into the chromosome of E. coli TOP10 (F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL(StrR) endA1 λ −, Thermo Fisher Scientific) using the λ DE3 Lysogenization Kit (Merck Millipore). The resulting lysogen was designated E. coli TOP10(DE3).Shake flask cultivation and expression of SAV. E. coli TOP10(DE3) containing the respective expression plasmid for SAV (pET-30b-SAVperi or pET-30b-SAVcyto) was cultivated in a Luria–Bertani (LB) medium (50 ml)32 supplemented with kan-amycin (50 mg l−1) in shake flasks (500 ml, 37 °C, 200 r.p.m.). An LB pre-culture was diluted 1:100 in fresh medium and incubation was performed until an optical density at 600 nm (OD600) of about 0.5 was reached. Subsequently, the cultivation temperature was lowered to 20 °C, and the expression of SAV was induced by addition of isopropyl-β -d-thiogalactopyranoside (IPTG, 50 μ M) and cells were harvested by centrifugation (6,000 r.c.f., 2 min) after four hours of induction. The cell pellet was further processed either for fractionation to analyse the cellular protein content or for flow cytometry.Fractioning of cellular protein and analysis. To separate the periplasmic and the cytoplasmic fraction of cellular proteins, the PeriPreps Periplasting Kit (Epicentre Technologies) was used. For in-gel staining of SAV, biotin-4-fluorescein (10 μ M) was added to the respective fractions before sodium dodecyl sulfate polyacryla-mide gel electrophoresis (SDS–PAGE) analysis. Owing to the binding to the bioti-nylated fluorescent dye, SAV can be visualized under ultraviolet light in the gel (before staining with Coomassie blue). Owing to its high stability, SAV remains tetrameric and fully functional even under the otherwise denaturing conditions of SDS–PAGE.Flow cytometry. To stain SAV in whole cells of E. coli, the pellet of a shake-flask expression culture was resuspended in phosphate buffer saline (PBS32) to a final OD600 = 1 before the addition of Atto-565-biotin (2 μ M, Atto-Tec GmbH). Cells were incubated on ice (30 min) and subsequently subjected to three wash cycles by centrifugation (6,000 r.c.f., 2 min) and resuspension of the pellet in PBS. The result-ing cell suspensions were analysed on a BD LSR Fortessa SORP (BD Biosciences) using a 561-nm laser for the excitation of Atto-565-biotin and a 610/20BP-600LP filter combination for analysis of the emitted fluorescence signal (peak area). The histograms displayed comprise data of 100,000 ungated events for each sample.Cultivation in 96-well plates. For expression of SAV (and mutants thereof) in the 96-deepwell format, LB medium (500 μ l) supplemented with kanamycin (50 mg l−1) was inoculated from a single colony and grown until stationary phase (37 °C, 300 r.p.m., 50-mm shaking amplitude). This pre-culture (aliquot of 30 μ l) was used to inoculate a main culture (1 ml) in a modified ZYM-5052 medium33 that lacked lactose as auto-induction agent, but contained kanamycin (50 mg l−1). Induction was performed by the addition of IPTG ([IPTG]final = 50 μ M) and continued for four hours before harvesting. Subsequently, a fraction of the cultures (100 μ l) was set aside for OD600 determination and the remaining suspension was subjected either to ICP-OES quantification or to metathesis with whole cells.Metathesis activity in the presence of GSH or GSSG. The activity of the artifi-cial metalloenzyme biot-Ru–SAV was quantified in the reaction buffer (100 mM sodium acetate, pH 4.0, 0.5 M MgCl2, 2.5% DMSO) both in the presence and absence of 10 equiv. of glutathione (GSH or GSSG; 500 μ M) relative to the amount of the cofactor biot-Ru (50 μ M). The reaction mixture (total volume of 200 μ l) was incubated shaking (16 h, 37 °C) and, after the reaction, 800 μ l of methanol contain-ing 2-phenylethanol (final concentration of 100 μ M) as an internal standard was added. After centrifugation (5 min, 21,000 r.c.f., 4 °C), 500 μ l of the supernatant
were diluted with 500 μ l of de-ionized water and the samples were subjected to UPLC analysis.
UPLC analysis was performed using a Waters H-Class Bio using a BEH C18 1.7 μ M column and a flow rate of 0.6 ml min−1 (eluent A, 0.1% formic acid in water; eluent B, 0.1% formic acid in acetonitrile; gradient at 0 min: 90% A, 10% B; at 0.5 min: 90% A, 10% B; at 2.5 min: 10% A, 90% B; at 3.5 min: 90% A, 10% B; at 4.5 min: 90% A, 10% B). The ultraviolet signal at 210 nm was used for quantifica-tion, and concentrations of the metathesis product umbelliferone 2 (retention time of 1.38 min) were determined on the basis of a standard curve with commercially available umbelliferone (Sigma Aldrich).ICP-OES quantification of cellular ruthenium content. To quantify the ruthenium content of cells, cultures from 96-deepwell plates were pelleted by centrifugation (3,220 r.c.f., 12 min, 4 °C) and resuspended in ice-cold Tris/HCl-buffer (1 ml, 50 mM, pH 7.4, 0.9% (w/v) NaCl) containing biot-Ru (10 μ M). After incubation on ice (30 min), the cells were spun down (1,260 r.c.f., 8 min, 4 °C), the supernatant- containing excess cofactor was discarded, and the pellet was washed by resuspen-sion in ice-cold Tris/HCl-buffer (1 ml), centrifugation and careful removal of the supernatant. Afterwards, the pellet was resuspended in de-ionized water (500 μ l). Twelve replicates of this suspension were combined and concentrated nitric acid was added (550 μ l, 65%) to fully digest the cellular material (48 h, 110 °C, pressure vials). The resulting clear solutions were diluted to a final volume of 10 ml with de-ionized water (containing 1 p.p.m. yttrium as internal standard) and subjected to ICP-OES quantification.
The obtained p.p.b.-values (1 p.p.b. = 1 μ g l−1) for ruthenium were trans-formed into a molar concentration and the average ruthenium atom count per cell was calculated from the OD600 of the cultures before decomposition assum-ing an OD600-to-cell-number correlation for E. coli in complex medium of 7.8 × 108 ml−1 OD600
−1, as described in ref. 34.Fluorescent metathesis assay with whole cells. The cellular metathesis activity was quantified using a fluorescent assay. For this purpose, cell cultures from 96-deepwell plate cultivations (see above) were pelleted by centrifugation (3,220 r.c.f., 12 min) and resuspended in ice-cold Tris/HCl-buffer (500 μ l, 50 mM, pH 7.4) containing biot-Ru (2.1 μ M). This buffer was supplemented with NaCl (0.9% (w/v)) to adjust to a physiological NaCl concentration. To allow cellular uptake of biot-Ru, the suspensions were incubated on ice for 30 min, spun down (1,260 r.c.f., 8 min) and the supernatant with its excess of cofactor was discarded. The pellets were resuspended in the reaction buffer (160 μ l, 100 mM sodium acetate, pH 4.0, 0.5 M MgCl2) and the reaction was initiated by addition of these cell suspensions (150 μ l) to the substrate solution (50 μ l reaction buffer containing 40 mM precursor 1 and 20% DMSO), leading to a final substrate concentration of 10 mM and 5% DMSO. As the metathesis product umbelliferone 2 is fluorescent, the reaction progress was monitored by fluorescence in a microtiter plate reader (excitation wavelength, 322 ± 4.5 nm; emission wavelength, 440 ± 10 nm; Infinite M1000 PRO, Tecan Group AG) at 37 °C and agitation (6-mm amplitude, orbital). Cell-specific metathesis activity is specified as the slope of the increasing fluorescence signal in the linear range of the reaction normalized by the OD600 of the respective culture.Site-saturation mutagenesis of SAV and screening. To generate diversity in the scaffold protein SAV, a focused, semi-rational strategy was pursued: the 20 amino acid residues closest to the ruthenium ion (see Supplementary Table 4) in a related artificial metalloenzyme structure8 were selected and individually randomized in SAVperi by site-saturation mutagenesis using NNK codons30. Degeneration was introduced by application of the Quikchange Site Directed Mutagenesis Protocol (Stratagene) using degenerate oligos (see Supplementary Table 4) and, after trans-formation, the libraries were checked for diversity by Sanger-sequencing of at least four individual clones before screening. To evaluate the performance of the SAVperi variants, the aforementioned fluorescent metathesis assay was carried out with at least 90 clones from the library in one 96-well plate (three replicates of strains producing ‘wild-type’ SAV (parent) and three replicates of strains carrying an empty vector control (pET-30b(+ ), lacking the gene for SAVperi). Evaluating 90 members of an NNK library ensures a > 94% likelihood of screening all 20 amino acid residues in the respective position29. To compensate for biological variance, promising clones were isolated and subjected to a replicate assay that was identical to the protocol described above, but using eight independent cultures per clone. After this first screening round, promising residues were ordered according to their potential impact on catalysis and mutations were combined using iterative saturation mutagenesis (ISM)30.Enzyme kinetics. To perform kinetic experiments on the artificial metathase, the initially used T7-tagged SAV as well as the quintuple mutant SAVmut that was isolated after ISM were cloned into a cytoplasmic expression vector (see Methods section ‘Cloning of SAV expression constructs’) and purified on an iminobiotin sepharose column as described elsewhere35. The biotin bind-ing capacity was determined using a fluorescent quenching assay36. Kinetic measurements were performed in reaction buffer (200 μ l total volume,
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTER RESEARCH
100 mM sodium acetate, pH 4.0, 0.5 M MgCl2, 11.5% (v/v) DMSO) containing biot-Ru (50 μ M) both in the presence and absence of purified SAV (100 μ M binding sites of either T7-tagged SAV or SAVmut) as well as the substrate 1 (variable concentrations, 0–5 mM) for fluorescent RCM. The reaction was mon-itored in a microtiter plate reader as described in Methods section ‘Fluorescent metathesis assay with whole cells’. The maximum velocity of the reaction was determined from the fluorescent signal curve by linear regression. To retrieve kinetic parameters, the reaction velocities were plotted over the respective substrate concentrations using the software GraphPad Prism (GraphPad Software, version 6.05) and applying the integrated ‘Michaelis–Menten’ least-squares fit with no constraints for the maximum velocity Vmax and substrate affinity KM.Protein crystallization and catalyst soaking. Crystals of SAV and variant SAVmut were obtained at 20 °C within two days by the sitting-drop vapour diffusion tech-nique mixing 1 μ l crystallization buffer (1.5 M ammonium sulfate, 0.1 M sodium acetate, pH 4.0) and 4 μ l protein solution (26 mg ml−1 lyophilized protein in water). The droplet was equilibrated against a reservoir solution of 100 μ l crystallization buffer. Subsequently, single crystals were soaked for two days at 20 °C in a soaking buffer, which was prepared by mixing 1 μ l of a 10 mM stock solution of complex biot-Ru (in 50% aqueous DMSO) and 9 μ l crystallization buffer. After the soak-ing, crystals were transferred for 30 s into a cryo-protectant solution consisting of 25% (v/v) glycerol in crystallization buffer. Next, crystals were shock-frozen in liquid nitrogen. Additional soaking of the above metathase crystals with substrate surrogate 1 did not lead to fluorescence. We therefore conclude that the multiple catalytic steps (for example, ligand displacement and cross-metathesis) required to ultimately liberate umbelliferone 2 cannot take place within a crystal.Data processing and structure refinement. X-ray diffraction data were collected at the Swiss Light Source beam line X06DA at a wavelength of 1 Å and processed with the software XDS37 and AIMLESS (CCP4 Suite)38. The struc-ture was solved by molecular replacement using the program PHASER (CCP4 Suite)38 and the structure 2QCB from the PDB as an input model with ligand and water molecules removed. For structure refinement REFMAC5 (CCP4 Suite)39 and PHENIX.REFINE40 were used. Ligand manipulation was carried out with the program REEL using the small-molecule crystal structure ABEJUM from the Cambridge Structural Database as an input model40. For water picking and elec-tron density and structure visualization, the software COOT41 was used. Figures were drawn with PyMOL (the PyMOL Molecular Graphics System, version 1.5.0.5, Schrödinger, LLC). Crystallographic details, processing and refinement statistics are given in Supplementary Table 5.Crystal structure of biot-Ru–SAVmut. There is one SAVmut monomer in the asym-metric unit from which a tetramer can be generated by application of two orthogonal crystallographic two-fold symmetry axes. The 12 N-terminal residues of the T7-tag and 25 residues at the C terminus are not resolved, probably owing to disorder.
Residual electron density in the biotin-binding site and the biotin vestibule as well as two strong anomalous dispersion density peaks in the biotin vestibule (Extended Data Fig. 3) suggested modelling of complex biot-Ru in two conformations I and II (56% and 44% occupancy, respectively). This projects the ruthenium atom in either one of the two densities, in close proximity to a crystallographic two-fold symmetry axis (Fig. 3b, Extended Data Figs 3, 4c, d). Only partial or no electron density was present for the mesityl linker and the terminal mesityl group, probably owing to high flexibility. In conformer I, the lengthy dimesitylimidazolidine (DMI)-Ru head group reaches into the neighbouring cis-related SAVmut monomer. The I conformer is stabilized mostly by hydrophobic interactions between the distal face of the DMI ligand and amino acid side chains within two neighbouring cis-related 7,8-loops (L1101, S1121, T114Q1, K121R1, L1241, S1122, W1202, K121R2, L1242; superscripts refer to monomers 1 or 2 of the tetrameric SAVmut; Extended Data Fig. 4c). Besides the DMI ligand, two chloride ions could be modelled binding to the ruthenium, but no density was found for the alkylidene, presumably owing to high flexibility and/or low occupancy. The orientation of the chlorides was very similar to that in a small-molecule crystal structure of the Grubbs–Hoyveda second- generation catalyst (ABEJUM from the Cambridge Structural Database), placing them nearly in trans position to each other. The ruthenium is largely solvent- exposed, which could facilitate substrate binding and product release. In Fig. 3b, the alkylidene was modelled binding to the ruthenium (magenta stick model) to highlight its orientation in the biotin vestibule. Conformer II is different from I by a rotation (about 60°) of the DMI-Ru moiety around an axis parallel to the cylinder axis of the SAVmut β -barrel (Fig. 3b, Extended Data Fig. 4d). This rotation places the hydrophobic distal side of the DMI-ligand in proximity to amino acid side chains within loop-5,61 (A86, H87), loop-7,81 (S112, T114Q) and loop-4,53 (D67, S69, A65) of a neighbouring trans-related SAV monomer. Atom N49K-Nε is located in close proximity to ruthenium (2.4 Å). No electron density was found to model chloride ions and the alkylidene ligand bound to the ruthenium. Because the cofactor is bound in close proximity to a two-fold crystal symmetry axis, forma-tion of a cis-symmetry-related neighbouring cofactor by application of the crystal
symmetry operation results in extensive steric clashes between the two cofactors in the orientation I. In contrast, coexistence of cofactor pairs I–I or II–II orientation is sterically accessible (Extended Data Fig. 3a, c, e).
Normalized B factors of residues within cofactor-flanking loop-7,8 are increased by up to about 0.5 when compared to related SAV structures that crystalized in the same space group and in a very similar unit cell (Extended Data Fig. 5b). This suggests increased loop-7,8 flexibility (Fig. 3b, Extended Data Fig. 4c, d). This flexibility is likely to be caused by three factors: (i) mutation T114Q cleaves a hydrogen-bond between threonine-Oγ H and T115-carbonyl oxygen (green dashed line in Fig. 3b and Extended Data Fig. 4a, b) and leads to a new hydrogen- bond between T114Q-glutamine-Nε and S112-Oγ H (red dashed line in Fig. 3b and Extended Data Fig. 4c, d); (ii) mutation A119G in the loop leads to increased entropy; and (iii) mutation V47A reduces steric hindrance between V47A in loop-3,42 and W120 in loop-7,81 (Fig. 3b and Extended Data Fig. 4c, d).Crystal structure of biot-Ru–SAV. The overall structure of complex biot-Ru–SAV is virtually identical to that of complex biot-Ru–SAVmut (root-mean-square deviation, r.m.s.d. = 0.25 Å). As in the mutant, two strong residual electron density peaks (Fo − Fc cofactor omit map: 12σ (conformer I) and 11σ (conformer II)) and two anomalous dispersion density peaks (9σ (conformer I) and 5σ (conformer II)) were located within the biotin vestibule of a SAV monomer at the interface between two symmetry-related SAV monomers (Extended Data Fig. 3b, d, f). The same two cofactor conformations I and II found in mutant SAVmut were modelled in the biotin binding vestibule of SAV with an occupancy of 50% for each conformer I and II (Extended Data Fig. 4a, b).
The side chain of residue L110 adopts two conformations with 50% occupancy each (Extended Data Fig. 4a, b). The close proximity of the terminal methyl group in L110 conformation A to the aromatic mesityl ring (distance between L110-Cδ H and mesitylcentroid of 4.4 Å) of cofactor conformation I suggests a stabilizing σ –π interactions (Extended Data Fig. 4a, red star). The L110 side chain conformation B can coexist only with cofactor conformation II (Extended Data Fig. 4b). This hypothesis is supported by the fact that the same L110 side chain conformation A is found in complex [(Cp* )Ir(Biot-p-L)Cl]-SAV-S112A (PDB, 3PK2), which has an aromatic ring located in the same position as the mesityl linker in structure biot-Ru–SAV, suggesting a similar σ –π interaction. In contrast, the side chain of L110 in apo-SAV (PDB, 2BC3) adopts conformation B. In complex biot-Ru–SAV, a water molecule is bound in proximity to L110 with 50% occupancy (Extended Data Fig. 4b). Steric clashes with L110 side chain in conformation A and the NHC ligand of biot-Ru conformer I suggest that the water is present only with L110 conformation B and biot-Ru conformer II. Additionally, the side chain of L124 adopts two conformations, each with 50% occupancy. Only conformation L124A does not undergo steric clashes with a methyl group of the bridging mesityl moiety of cofactor conformer I (Extended Data Fig. 4a, b). Together, the conformational side chain flexibility of residues L110 and L124 reflects the presence of the two cofactor conformations I and II.
In contrast to artificial metalloenzyme biot-Ru–SAVmut, the normalized B fac-tors of residues within the cofactor-flanking loop-7,8 in complex biot-Ru–SAV do not show increased values when compared to those in related crystal structures (Extended Data Fig. 5b). Indeed, a hydrogen-bond is formed between T114-Oγ H and T115-carbonyl oxygen in complex biot-Ru–SAV that could rigidify the loop (Extended Data Fig. 4a, b).In vitro metathesis assay for substrate 3 (2,2-diallyl-1,3-propanediol). The con-version for the product 4 was quantified by 1H-NMR. For this purpose, a deuter-ated reaction buffer was prepared from acetic acid-d4, dry MgCl2 and D2O with the same concentrations as for the reaction buffer used for substrate 1 (100 mM acetate, 0.5 M MgCl2). The pH was adjusted to 3.6 by addition of 1 M NaOD in D2O (with respect to pD = pH + 0.4).
For the reaction, 300 μ l of a substrate 3 stock solution (100 mM in deuterated reaction buffer) was mixed with 291 μ l of either a solution of SAV, SAVmut, or SAVmut2 (200 μM binding sites in deuterated reaction buffer) or plain deuterated reaction buffer (for samples without SAV or any of the SAV variants). Afterwards, 9 μ l of a biot-Ru (or HGII/AQM) stock solution (3.34 mM in DMSO-d6) was added to obtain a final concentration of 50 μ M and the reaction was performed for 16 h at 37 °C and 200 r.p.m. The mixture was analysed by 1H-NMR and the yield of the reaction product 4 was quantified by comparing integrals (I) of the product 4 peaks at 3.41 p.p.m. and 2.05 p.p.m. and the substrate 3 peaks at 3.33 p.p.m. and 1.91 p.p.m. using: yield = Iproduct/(Iproduct + Isubstrate).In vitro metathesis assay for substrate 5 (diallyl sulfonamide). To quantify the conversion of substrate 5, a 97 μ l aliquot of either SAV solution (200 μ M SAV bind-ing sites in reaction buffer) or plain reaction buffer (for samples without SAV variants) was mixed with 100 μ l of a stock solution of substrate 5 (20 mM in reac-tion buffer). Subsequently, 3 μ l of the respective catalyst/cofactor stock solution (3.34 mM in DMSO) was added to obtain a final concentration of 50 μ M. The reaction was performed for 16 h at 37 °C and 200 r.p.m.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTERRESEARCH
Then, an aqueous solution of benzyltriethylammonium chloride (100 μ l, 10 mM) was added as an internal standard and 700 μ l of methanol was added. The mixture was cleared by centrifugation and 250 μ l of the supernatant was mixed with 750 μ l of water for the final quantification of product 6 by UPLC-MS.
For the kinetic experiment, the reaction mixture was scaled up to a total volume of 1 ml and 50 μ l aliquots of this mixture were collected at different time points and immediately injected into 950 μ l of a quenching solution (0.5 mM potassium cyanoacetate, 0.25 mM benzyltriethyl-ammonium chloride (internal standard) in 50% aqueous methanol). After removal of precipitated protein by centrifugation, the supernatant was analysed by UPLC-MS.Metathesis assay with whole cells and diallyl sulfonamide 5. To quantify the cellular metathesis activity for substrate 5, a protocol analogous to that applied for the umbelliferone precursor 1 was applied. The substrate 5 (final concentration 10 mM) was added to whole cells and the samples were incubated at 37 °C and 300 r.p.m. for 16 h. To quantify the conversion for the non-fluorescent product 6, an extraction was performed: 800 μ l of methanol was added to each sample and an extraction was carried out (one hour with vigorous shaking, 800 r.p.m. at room temperature). The samples were cleared by centrifugation, the supernatant was diluted with water (factor four) and analysed by UPLC-MS using a calibration curve recorded for product 6.Sample size. No statistical methods were used to predetermine sample size.
31. Gallizia, A. et al. Production of a soluble and functional recombinant streptavidin in Escherichia coli. Protein Expr. Purif. 14, 192–196 (1998).
32. Sambrook, J. F. & Russell, D. W. (eds) Molecular Cloning: A Laboratory Manual 3rd edn (Cold Spring Harbor Laboratory, 2001).
33. Studier, F. W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005).
34. Volkmer, B. & Heinemann, M. Condition-dependent cell volume and concentration of Escherichia coli to facilitate data conversion for systems biology modeling. PLoS One 6, e23126 (2011).
35. Humbert, N., Schürmann, P., Zocchi, A., Neuhaus, J.-M. & Ward, T. R. in Avidin-Biotin Interactions: Methods and Applications (ed. McMahon, R. J.) 101–110 (Vol. 418 of Methods in Molecular Biology, Humana Press, 2008).
36. Kada, G., Falk, H. & Gruber, H. J. Accurate measurement of avidin and streptavidin in crude biofluids with a new, optimized biotin-fluorescein conjugate. Biochim. Biophys. Acta 1427, 33–43 (1999).
37. Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).38. Evans, P. R. An introduction to data reduction: space-group determination,
scaling and intensity statistics. Acta Crystallogr. D 67, 282–292 (2011).39. Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular
structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 (1997).
40. Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
41. Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTER RESEARCH
Extended Data Figure 1 | Batch-to-batch reproducibility of different protein preparations. The activity of the evolved artificial metathase biot-Ru–SAVmut was evaluated for three independently produced and purified protein batches and compared to two independent batches of non-mutated SAV (biot-Ru–SAV). Reactions were performed at 4 mM of substrate 1, 50 μ M of biot-Ru and 100 μ M of of SAV-variant binding sites, and the product 2 was quantified by fluorescence. The displayed activities represent the initial slopes of the fluorescence signal in the linear reaction phase relative to the activity of free biot-Ru, which was defined as 100%. Bar heights represent the average relative activity of four replicate in vitro experiments (n = 4), with 1 s.d. indicated as error bars.
Rel
ativ
e ac
tivity
(%)
0
100
200
300
400
batch II batch IIIbiot-Ru(:= 100%)
biot-Ru–SAVmut biot-Ru–SAV batch I batch II batch I
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTERRESEARCH
Extended Data Figure 2 | Influence of cofactor loading on the activity of the artificial metathase. The activity of the evolved artificial metathase biot-Ru–SAVmut was evaluated for different ratios between the cofactor biot-Ru and SAVmut binding sites (biot-Ru:SAVmut
b.s.). Reactions were performed at 4 mM of substrate 1, 5 μ M of biot-Ru and varying concentrations of SAVmut binding sites (5–50 μ M corresponding to ratios ranging from 1:1 to 1:10). The product 2 was quantified by fluorescence. The displayed activities represent the initial slopes of the fluorescence signal in the linear reaction phase. Bar heights represent the average relative activity of four replicate samples (n = 4), with 1 s.d. indicated as error bars.
Rel
ativ
e ac
tivity
(%)
0
100
20
60
40
80
120
1:2 1:3 1:4 1:5 1:101:1(:=100%)
biot-Ru:SAVmutb.s.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTER RESEARCH
a b
c d
e f
Extended Data Figure 3 | Conformers of the artificial metathases. a–f, Close-up views of the crystal structure of the complexes biot-Ru–SAVmut (a, c, e) and biot-Ru–SAV (b, d, f) displaying pairs of cis-symmetry-related biot-Ru cofactors (view perpendicular to a two-fold crystal symmetry axis,
displayed in red): conformers I–I (severe steric clash; a, b), conformers II–II (c, d) and conformers I–II (e, f). The protein was removed for displaying purposes. The cofactors are contoured with 2Fo − Fc electron density in blue at 1σ and anomalous dispersion density in red at 3.5σ.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.
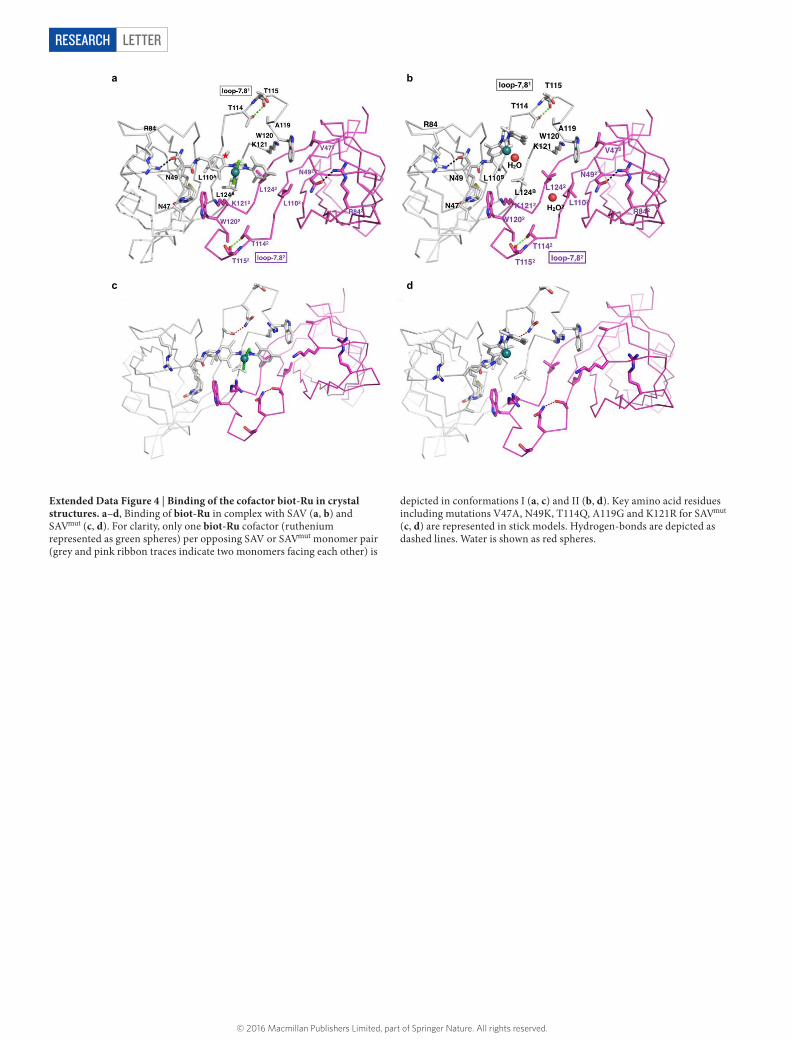
LETTERRESEARCH
a b
c d
Extended Data Figure 4 | Binding of the cofactor biot-Ru in crystal structures. a–d, Binding of biot-Ru in complex with SAV (a, b) and SAVmut (c, d). For clarity, only one biot-Ru cofactor (ruthenium represented as green spheres) per opposing SAV or SAVmut monomer pair (grey and pink ribbon traces indicate two monomers facing each other) is
depicted in conformations I (a, c) and II (b, d). Key amino acid residues including mutations V47A, N49K, T114Q, A119G and K121R for SAVmut (c, d) are represented in stick models. Hydrogen-bonds are depicted as dashed lines. Water is shown as red spheres.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTER RESEARCH
Extended Data Figure 5 | Flexibility of residues within loop-7,8. a, Comparison of mobility in loop-7,8 before (biot-Ru–SAV) and after (biot-Ru–SAVmut) directed evolution as highlighted by Cα atom B factors (colour scale). Mutations are indicated in two opposing monomers by asterisks: * 1/* 1′ , V47A; * 2/* 2′ , N49K; * 3/* 3′ , T114Q; * 4/* 4′ , A119G; and * 5/* 5′ , K121R. b, Normalized (BCα/Baverage) Cα atom B factors of residues
within loop-7,8 from different SAV structures that crystalize in the space group I4122 with similar unit cell dimensions. biot-Ru–SAVmut (dark blue) and biot-Ru–SAV (light blue) are compared to PDB reference structures (shades of grey): r1, 2QCB; r2, 3PK2; r3, 2WPU; r4, 4GJV; r5, 4OKA; r6, 2IZJ; and r7, 2IZB.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTERRESEARCH
RuCl
ClO
NN
RuCl
ClO
NN
N
NCl
a b
Extended Data Figure 6 | Structures of commercial metathesis catalysts used here. a, b, The second-generation Hoveyda–Grubbs (HGII) catalyst (a) was obtained from Sigma Aldrich (Buchs) and the Aquamet (AQM) catalyst (b) was purchased from Apeiron Synthesis S.A.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTER RESEARCH
Extended Data Figure 7 | Summary of the results obtained by UPLC-MS for the 96-well plate in vivo screen of the dallyl-sulfonamide 5. Saturation mutagenesis at position 121 (K121X) was performed on SAVmut(47A/49K/114Q/119G/121K). Heights of the navy and blue bars represent the average of three replicates (n = 3), with 1 s.d. indicated as error bars; purple bars represent the value of a single measurement (n = 1).
0
8‘000
10’000
6‘000
4‘000
2‘000
Pro
duct
6 c
once
ntra
tion
(nM
)
SAVperi (n=3)
SAVmut (n=3)
Saturation mutagenesis library (n=1)
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTERRESEARCH
Extended Data Table 1 | Influence of glutathione on the activity of biot-Ru–SAV
Both the reduced (GSH) and the oxidized (GSSG) form of glutathione were tested in the presence of 10 equiv. GSH or GSSG relative to the amount of the cofactor biot-Ru (50 μ M) in the presence of SAV (100 μ M free biotin binding sites). Reactions were performed in reaction buffer (100 mM sodium acetate, pH 4.0, 0.5 M MgCl2, 2.5% (v/v) DMSO) containing 10 mM of the precursor 1 at 37 °C for 16 h. Turnover numbers (TONs) were determined by UPLC quantification. Data are mean values of two independent assays (n = 2) with ± 1 s.d.
Redox conditions GSH GSSG TON
Buffer - - 1.7 ± 0.2
Buffer + GSH 10 equiv. - 0.0 ± 0.0
Buffer + GSSG - 10 equiv. 1.5 ± 0.1
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.

LETTER RESEARCH
Extended Data Table 2 | Turnover number of artificial metathases for the conversion of the umbelliferone precursor 1
Catalyst/ArM TON
biot-Ru 1.1 ± 0.1
biot-Ru–SAV 1.7 ± 0.2
biot-Ru–SAVmut 4.4 ± 0.2
2.5 ± 0.1biot-Ru–SAVmut2
Purified artificial metathases (ArMs; 100 μ M free biotin binding sites, 50 μ M biot-Ru) were tested and compared to the cofactor biot-Ru alone (50 μ M). Reactions were performed in reaction buffer (100 mM sodium acetate, pH 4.0, 0.5 M MgCl2, 2.5% (v/v) DMSO) containing 10 mM of the precursor 1 at 37 °C for 16 h. Turnover numbers (TONs) were determined by UPLC quantification. Data are mean values of three independent assays (n = 3) with ± 1 s.d.
© 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved.


















