
Development and Application of New Chiral β-Amino …160829/FULLTEXT01.pdf · development and...
43
Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 626 _____________________________ _____________________________ Development and Application of New Chiral β-Amino Alcohols in Synthesis and Catalysis Use of 2-Azanorboryl-3-Methanols as Common Intermediates in Synthesis and Catalysis BY PEDRO PINHO ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2001
-
Upload
hoangtuyen -
Category
Documents
-
view
217 -
download
0
Transcript of Development and Application of New Chiral β-Amino …160829/FULLTEXT01.pdf · development and...

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 626
_____________________________ _____________________________
Development and Application of New Chiral β-Amino Alcohols
in Synthesis and Catalysis
Use of 2-Azanorboryl-3-Methanols as Common Intermediates in Synthesis and Catalysis
BY
PEDRO PINHO
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2001

Dissertation for the Degree of Doctor of Philosophy in Organic Chemistry presented atUppsala University in 2001
ABSTRACT
Pinho, P. 2001. Development and Application of New Chiral β-Amino Alcohols in Synthesis andCatalysis. Use of 2-Azanorbornyl-3-Methanols as Common Intermediates in Synthesis and Catalysis.Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala Dissertations from the Facultyof Science and Technology 626. 43 pp. Uppsala. ISBN 91-554-5091-9.
The development and application of unnatural amino alcohols, prepared via hetero-Diels-Alderreactions, in synthesis and catalysis is described. The studies are concerned with the [i] scope of thehetero-Diels-Alder reaction and preparation of important intermediates in the synthesis of antiviralagents, [ii] application of amino alcohols in the ruthenium transfer hydrogenation of ketones, [iii] useof similar precursors in the in situ generation of oxazaborolidines for reduction of ketones, and [iv]development and application of new chiral auxiliaries for dialkylzinc additions to activated imines,respectively.
[i] The use of chiral exo-2-azanorbornyl-3-carboxylates in the preparation of enantiopure cyclopentyl-amines is described. At the same time the scope of the hetero-Diels-Alder reaction, used in theirpreparation, is extended by manipulations of the dienophiles.[ii] Application of 2-azanorbornyl-3-methanol as a very efficient ligand in the ruthenium-catalysedasymmetric transfer hydrogenation of aromatic ketones. This ligand (2 mol%) in combination with[RuCl2(p-cymene)]2 (0.25 mol%) gave rise to a very fast reaction (1.5 h) leading to the reducedproducts in excellent yields and enantioselectivities (up to 97% ee).[iii] Preparation of α-disubstituded 2-azanorbornyl-3-methanols, in situ generation of thecorresponding oxazaborolidines, and use of the latter in reduction of aromatic ketones. Concentration,solvent, and temperature effects on the reaction outcome are described.[iv] Development of two generations of chiral auxiliaries for the addition of dialkylzinc reagents to N-(diphenylphosphinoyl) imines. Studies using density functional computations allowed therationalisation of the reaction mechanism and the development of a second generation of ligands thatimproved the previously reported results. Up to 98% ee could be obtained with these new ligands.Solvent effects on the outcome of the reaction and extension of the work to a larger variety of N-(diphenylphosphinoyl) imines are described.
Key words: Asymmetric synthesis, hetero-Diels-Alder reactions, chiral cyclopentyl-amines, chiralligands and catalysts, amino alcohols, asymmetric reductions, ruthenium transfer hydrogenation,oxazaborolidines, asymmetric additions, dialkylzinc reagents.
Pedro Pinho, Department of Organic Chemistry, Institute of Chemistry, Uppsala University, Box 531,SE-751 21 Uppsala, Sweden. [email protected]
© Pedro Pinho 2001
ISSN 1104-232X
ISBN 91-554-5019-9
Printed in Sweden by Uppsala Universitet Tryck & Medier, Uppsala 2001

3
Watching fate as it flows down the path we have chose
-Trent Raznor

4
Papers included in the thesis
This thesis is based on the following papers and appendix, referred to in the text by theirRoman numerals I-VIII.
I. Diels-Alder Reaction of Heterocyclic Imine Dienophiles. Pinho, P.; Hedberg, C.;Roth, P.; Andersson, P. G. J. Org. Chem. 2000, 65, 2810-2812.
II. A novel synthesis of chiral cyclopentyl- and cyclohexyl-amines. Pinho, P.;Andersson, P. G. Chem. Commun. 1999, 597-598.
III. (1S, 3R, 4R)-2-Azanorbornylmethanol, an Efficient Ligand for Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of Ketones. Pinho, P.; Alonso, D.A.; Guijarro, D.; Temme, O.; Andersson, P. G. J. Org. Chem. 1998, 63, 2749-2751.
IV. (1S, 3R, 4R)-2-Azanorbornyl-3-methanol Oxazaborolidines in the AsymmetricReduction of Ketones. Pinho, P.; Guijarro, D.; Andersson, P. G. Tetrahedron 1998,54, 7897-7906.
V. Enantioselective Addition of Dialkylzinc Reagents to N-(Diphenylphosphinoyl)Imines Promoted by 2-Azanorbornylmethanols. Pinho, P.; Guijarro, D.;Andersson, P. G. J. Org. Chem. 1998, 63, 2530-2535.
VI. A Theoretical and Experimental Study of the Asymmetric Addition ofDialkylzinc to N-(Diphenylphosphinoyl)benzalimine. Pinho, P.; Brandt, P.;Hedberg, C.; Lawonn, K.; Andersson, P. G. Chem. Eur. J. 1999, 5, 1692-1699.
VII. Asymmetric Addition of Diethylzinc to N-(diphenylphosphinoyl) Imines. Pinho,P.; Andersson, P. G. Tetrahedron 2001, 57, 1615-1618.
VIII. Appendix: Supplementary Material. Pinho, P.
Reprints were made with permission from the publishers

5
Contents
Papers included in the thesis
List of abbreviations
1. Introduction 71.1 Towards enantiomerically pure or enriched compounds 81.2 Asymmetric synthesis – Ligands and metals 91.3 The use of simple β-amino alcohols as chiral ligands 11
2. Hetero-Diels-Alder reaction – Applications in synthesisand preparation of unnatural ββββ-amino alcohols 132.1 Introduction 132.2 Studies on the scope of the aza-Diels-Alder reaction – Towards nicotinic
acetylcholine receptors 142.3 Preparation of enantiomerically pure cyclopentyl- and cyclohexyl-amines 182.4 Access to unnatural β-amino alcohols 21
3. Ruthenium-catalysed asymmetric transfer hydrogenation of ketones 233.1 Introduction 233.2 The 2-azanorbornyl-3-methanol as a ligand for ruthenium 243.3.Reaction mechanism 26
4. Oxazaborolidines in the asymmetric reduction of ketones 294.1 Introduction 294.2 Reaction mechanism 294.3 Preparation of 2-azanorbornyl-3-methanol ligands and their application
in the form of the corresponding oxazaborolidines 30
5. Enantioselective addition of dialkylzinc reagentsto N-(diphenylphosphinoyl) imines 345.1 Introduction 345.2 The 2-azanorbornyl-3-methanols as chiral auxiliaries for the addition reaction 355.2.1 The first generation ligands – Synthesis and results obtained 355.2.2 The second generation ligands – Synthesis and results obtained 365.3 Reaction mechanism 39
Acknowledgements 42

6
List of abbreviations
Abs. Config. Absolute configurationBn Benzyln-Bu Butylt-Bu tert-ButylCat. CatalyticCBS Corey, Bakshi, ShibataConfig. ConfigurationCpH CyclopentadieneDIBAL-H Diisobutylaluminium hydrideee Enantiomeric excessequiv. EquivalentEt Ethylh Hour(s)HMB HexamethylbenzeneHPLC High Performance Liquid ChromatographyLAH Lithium aluminium hydrideM MetalMe Methylmin Minute(s)MS Molecular sieves1-Napht 1-NaphtylNMO N-methylmorpholine N-oxideNMR Nuclear Magnetic RessonancePh Phenyli-Pr iso-Propyln-Pr Propylrt Room temperatureStoich. StoichiometricTFA Triflouroacetic acidTHF TetrahydrofuranTIPSCl TriisopropylsilylchlorideTs p-ToluenesulphonylTS Transition StateX Halogen (Cl, Br, I)

7
“Life depends on chiral recognition,
because living systems interact with enantiomers
in decisively different manners.”
Noyori, R.1
1. Introduction
In 1849 Louis Pasteur resolved for the first time an enantiomeric pair by means of
mechanical separation of their differently shaped crystals. Since then chirality has been
recognised as of extreme importance, not only in chemistry and biology as academic subjects,
but also in life itself.
What then is chirality? A given molecule, or object in general is said to be chiral or
disymmetric if it does not possess any improper rotation axis Sn of any order n, where S1
corresponds to a symmetry plane (σ) and S2 to an inversion center (i).2 A consequence of this
definition is that chiral objects are not superimposable on their mirror images and are able to
rotate the plane of polarised light by the same angle, but in different directions, Figure 1.1.
NH
NH
COOH HOOC
(S)-Proline (R)-Proline
Mirrorplane
Figure 1.1 The two enantiomers (“mirror images”) of the amino acid proline
It is now widely accepted that Nature is chiral where amino acids, terpenes,
carbohydrates, and alkaloids are all natural occurring substances that are often enantiopure or
at least enantioenriched, i.e. one of the enantiomers predominates over the other. The
presence of chirality in Nature implies that usually only one enantiomer of a certain
compound is producing the correct response on a living organism. As a consequence
normally only one enantiomer of a given drug has the desired activity, hence, medicinal
1 In Asymmetric Catalysis in Organic Synthesis; John Wiley & Sons: New York, 1994.2 Klessinger, M.; Michl, J. Excited States and Photochemistry of Organic Molecules, Section 3.2; VCHPublishers, Inc.: New York, 1995.

8
chemistry has a very strong need for enantioselective processes in drug development.
However, this is not the only field where processes of this kind are being developed. Tastes
and smells may also be dependent on enantiomers, which raises the importance of chirality in
the food flavouring and perfumery industries. Agrochemicals may be easier or harder to
degrade depending on which enantiomer of the chemical substance is used. Due to the
growing concern about environmental aspects in modern society this branch of industry has
therefore an increasing need for enantioselective processes in the preparation of their
products.
These are only some of the reasons why synthetic organic chemistry has developed
enormously in the field of asymmetric synthesis during the last few decades.
1.1 Towards enantiomerically pure or enriched compounds
There are three basic means to perform the synthesis of enantiomerically pure or
enriched compounds.
a) Resolution; the oldest of all processes is based on the synthesis of the racemic
target molecule or intermediate in its synthetic sequence. The material is afterwards resolved
with the help of an enantiomerically pure compound. Resolution is an important and still
widely used process, but it suffers from a major drawback, i.e. the production of at least 50%
of unwanted material. This drawback can sometimes be overcome by recovering/recycling of
the unwanted enantiomer of the product, as for example in the dynamic kinetic resolution
approach.3 An example of classic resolution (max. 50% yield) is outlined in Scheme 1.1, in
this case the compound resolved is α-(1-naphthyl)ethylamine.4
NH2 COOH
OH
O H
O
O
O
H2O
(-)-DAG
NH2
(S)-(-)-α-(1-naphthyl)ethylamine> 99% ee
1)
2) 2M NaOH
Scheme 1.1 Resolution of α-(1-naphthy)ethylamine
3 For a review on dynamic kinetic resolution, see: Noyori, R.; Tokunaga, M.; Kitamura, N. Bull. Chem. Soc.Jpn. 1995, 68, 36.

9
b) “Chiral pool”; in this case the synthesis of the desired compound is based on a
commercially available and enantiomerically pure starting material. The “chiral pool”
approach strongly limits the possible synthetic strategies due to the still limited availability of
the appropriate starting materials. Besides this fact, usually only one of the enantiomers of the
starting material is naturally occurring further restricting the synthesis. Costs may also be a
problem since unnatural enantiomers, which are man made, are usually much more
expensive. An example of the “chiral pool” approach is depicted in Scheme 1.2 for the
synthesis of leukotriene A4.5
O OH
OHHO
HOsteps
from D-(-)-ribose
OCO2Me
(-)-Leukotriene A4
Scheme 1.2 Total synthesis of leukotriene A4 from D-(-)-ribose.
c) Asymmetric synthesis; involves the introduction of chirality by action of a chiral
reagent, auxiliary or catalyst, which is not incorporated in the final product. This process is
probably the choice, which provides the widest of possibilities. During the last few decades a
variety of asymmetric transformations have been developed. Due to its importance,
asymmetric synthesis and in particular asymmetric catalysis are treated in more detail in the
following sections.
1.2 Asymmetric synthesis – Ligands and metals
The chiral reagent and auxiliary methods6 require the use of at least one equivalent of
enantiopure material, usually the most expensive component of a synthetic sequence. For this
reason asymmetric synthesis is far more appreciated in the form of catalysis.
Still, there are important processes that involve the use of stoichiometric amounts of
enantiopure materials, such as hydroboration using the chiral (Icp)2BH reagent,7 Scheme 1.3.
4 For the resolution of this chiral building block, see: Leimgruber, W.; Mohacsi, E. Org. Synth. 1976, 55, 80.5 Marfat, A.; Corey, E. J. Advances in Prostaglandin, Thromboxane, and leukotriene Research; Pike, J. E. andMorton Jr., D. R. Eds.: Raven Press: New York 1985.6 For a review on recent chiral auxiliary applications, see: Regan, A. J. Chem. Soc., Perkin Trans. 1 1999, 357.7 (a) Brown. H. C.; Zweifel, G. Org. Synth. 1972, 52, 59; (b) Brown, H. C.; Jadhav, P. K.; Mandal, A. K.Tetrahedron 1981, 37, 3547.

10
3 NaBH44 BF3 OEt2
2 B2H6 3 NaBF4
8 4
BH
2
(Icp)2BH99.8% ee
94% ee
B
2
H HHO
95% ee
H2O2/NaOH
+
++ 4 Et2O
Scheme 1.3 Hydroboration of olefins using (Icp)2BH
Catalysis is a process by which a small amount of a foreign material, the catalyst,
increases the rate of a chemical transformation without itself being consumed. Metals are
known to be extremely efficient catalysts for a wide variety of organic transformations,
usually offering high selectivity under very mild reaction conditions.
During the last few decades the importance of metals in organic synthesis has seen a
tremendous growth and reactions catalysed by metals have become accepted as common
transformations.8 One early example of a metal catalysed transformation is the osmium
tetraoxide dihydroxylation of olefins,9 Scheme 1.4.
RR
RR
OH
OH
Cat. OsO4
Stoich. co-oxidant
racemic
Scheme 1.4 Catalytic dihydroxylation of olefins
If one then combines the chiral environment of an organic compound, able to co-
ordinate a metal, with the catalytic power of a metal itself, a chiral catalyst may be obtained
giving rise to a catalytic asymmetric process. Indeed, chiral metal complexes are among the
most powerful methods to achieve discrimination between functional groups and enantiotopic
faces of a pro-chiral substrate. Far are the days of the first reported example of a catalytic
asymmetric transformation using this type of complexes, Scheme 1.5.10
8 (a) For examples of metal catalysed reactions, see: Tonks, L.; Williams, J. M. J. J. Chem. Soc., Perkin Trans. 11998, 3637.9 For a review on catalytic non-asymmetric dihydoxylation, see: Schröder, M. Chem. Rev. 1980, 80, 187.10 Nozaki, H.; Moriuti, S.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1966, 5239.

11
Ph
CuN
O N
O
Ph
Ph
N2CHCOOEtcis/trans : 1/2.3
%ee for trans isomer = 6%
Ph CO2Et Ph
CO2Et
Scheme 1.5 The first reported example of a catalytic asymmetric process
It should be noted that the development of an asymmetric version of an existing
process is usually a difficult goal to achieve. The development may require many years of
research before the process becomes synthetically useful like the catalytic asymmetric
dihydroxylation of olefins11 shown in Scheme 1.6.
N
ONN
N
O
O
N
O
RR
RR
OH
OH
up to 99% ee
Cat.
Cat. OsO4
Stoich. NMO
N
Scheme 1.6 Catalytic asymmetric dihydroxylation of olefins
1.3 The use of simple ββββ-amino alcohols as chiral ligands
Due to their natural availability, it is not surprising that amino acids, or closely related
compounds, such as the corresponding amino alcohols are among the most common ligands
or ligand precursors for asymmetric catalysis.12 One successful example is the application of
the CBS catalyst derived from the amino acid (S)-(-)-proline. This catalyst was introduced by
11 (a) For a review on this process, see: Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994,94, 2483; (b) For a review regarding the application of this reaction in natural product synthesis, see: Cha, J. K.;Kim, N-S. Chem. Rev. 1995, 95, 1761.12 For a review on the application of β-amino alcohols in asymmetric transformations, see: Ager, D. J.; Prahash,I.; Schaad, D. R. Chem. Rev. 1996, 96, 835.

12
Corey and co-workers in the eighties and is used for the preparation of secondary alcohols
from pro-chiral ketones, Scheme 1.7.13
N BO
BH3
Ph
O
Ph
OHCat.
Stoich. BH3
99% yield, 97% ee,in 198713
PhPh
Scheme 1.7 Reduction of acetophenone using the CBS catalyst
Despite the progress made and the discovery of alternative processes this is still one
of the most efficient catalysts for this type of transformation. Many improved and simplified
reaction procedures have been developed in order to optimise the performance of this
powerful catalyst; acetophenone can nowadays be reduced to give the corresponding
secondary alcohol in more than 99% ee.13
13 The CBS catalyst was introduced in 1987 by: Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987,109, 5551. For further references and a more detailed discussion of the process, see Chapter 4 of this thesis.

13
2. Hetero-Diels-Alder reaction – Applications in synthesis and preparation
of unnatural ββββ-amino alcohols
2.1 Introduction
The Diels-Alder reaction has, since its discovery in 1928 by Otto Diels and Karl
Alder,14 been one of the cornerstones in synthetic organic chemistry. More recently, hetero-
Diels-Alder reactions and particularly very efficient catalytic and enantioselective versions of
this reaction have been developed.15
All the work presented in this thesis is based on chiral compounds containing the 2-
azanorbornyl skeleton, i.e. a product from a hetero-Diels-Alder reaction, and it is therefore
appropriate here to give special attention to this reaction.
S S
Re face
Major product
RR
exo endo
1a 1d
1c1b
δ
δ
Si face
N
H
CO2EtPh N
CO2Et
H
N
H
EtO2CN
CO2Et
HPh
O
O
O
O
O
NO
O
HN Ph
BF3
Ph1) TFA
CF3COO
exo endo
CpH
CpH CpH
CpH
H2N PhS
Ph
Ph
2) BF3 OEt2
Scheme 2.1 The aza-Diels-Alder reaction
14 Diels, O.; Alder, K. Liebigs Ann. Chem. 1928, 460, 98.15 For a review on catalytic asymmetric hetero-Diels-Alder reactions, see: (a) Jørgensen, K. A.; Johannsen, M.;Yao, S.; Audrain, H.; Thorhauge, J. Acc. Chem. Res. 1999, 32, 605; (b) Jørgensen, K. A. Angew. Chem. Int. Ed.2000, 39, 3558.

14
The dienophile for the aza-Diels-Alder reaction16 involved in the preparation of the 2-
azanorbornyl-3-methanols presented throughout this text is generated in situ from freshly
prepared methyl or ethyl glyoxylate and optically pure α-phenylethylamine,17 Scheme 2.1.
Unlike the amino acid derived β-amino alcohols, this inexpensive source of chirality is
available at the same price in both enantiomeric forms. This allows both enantiomers of the
ligands to be prepared via the same synthetic sequence, as shown above for (S)-α-
phenylethylamine. The selectivity in the cycloaddition between the formed imine and
cyclopentadiene was reported16b to be 96:2:2 (1a:1b:1c+1d). The major isomer can then
easily be obtained as a pure compound by means of flash chromatography. Moreover, it was
observed that if the methyl ester is produced the product could be crystallised from n-pentane
after the simple removal of polymeric material by filtration through silica.
2 . 2 Studies on the scope of the aza-Diels-Alder reaction – Towards nicotinic
acetylcholine receptors
Nitrogen containing bicyclic structures play an important role in the synthesis of
many natural products and ligands for asymmetric catalysis. A very efficient method for the
preparation of these structures is without any doubt the already mentioned aza-Diels-Alder
reaction.1 8 It is therefore of no surprise that there is growing interest in producing new
compounds by manipulations of the dienes and dienophiles used in the cycloaddition
reaction.
The observation that the scope of the reaction seemed to be limited to electron
deficient imines, derived from similar types of aldehydes16 or from very small aldehydes,19
led to new ideas.
16 The aza-Diels-Alder reaction used to prepare 1a, Scheme 2.1, has been previously reported by: (a) Stella, L.;Abraham, H.; Feneau-Dupont, J.; Tinant, B.; Declercq, J. P. Tetrahedron Lett. 1990, 18, 2603; (b) Abraham, H.;Stella, L. Tetrahedron 1992, 48, 9707. For other similar aza-Diels-Alder reactions, see for example: (c)Waldmann, H.; Braun, M. Liebigs Ann. Chem. 1991, 1045; (d) Bailey, P. D.; Wilson, R. D.; Brown, G. R. J.Chem. Soc., Perkin Trans. 1 1991, 1337; (e) Bailey, P. D.; Brown, G. R.; Korber, F.; Reed, A.; Wilson, R. D.Tetrahedron: Asymmetry 1991, 2, 1263; (f) Bailey, P. D.; Londesbrough, D. J.; Hancox, T. C.; Heffernan, J. D.;Holmes, A. B. J. Chem. Soc., Chem. Commun. 1994, 2543; (g) Bailey, P. D.; Millwood, P. A.; Smith, P. D.Chem. Commun. 1998, 633.17 For reviews on other applications of α-phenylethylamine, see: (a) Juaristi, E.; Escalante, J.; Léon-Romo, J. L.;Reyes, A. Tetrahedron: Asymmetry 1998, 9, 715; (b) Juaristi, E.; Escalante, J.; Léon-Romo, J. L.; Reyes, A.Tetrahedron: Asymmetry 1999, 10, 2441.18 Boger, D. L.; Weinreb, S. M. Hetero Diels-Alder Methodology in Organic Synthesis; Academic Press:London, 1987, Chapter 2.19 Larsen, S. D.; Grieco, P. A. J. Am. Chem. Soc. 1985, 107, 1768.

15
Isolation of the alkaloid epibatine20 (Figure 2.1) from the poisonous frogs,
epipedobates tricolor has led to a non-stop search for equally or even more powerful
analgesic analogues that might present themselves as being less toxic. During this research a
variety of compounds showing various properties have been prepared, especially compounds
containing the nitrogen in different locations of the norbornane framework attracted attention.
Me3NO
O
(-)-Epibatidine A(-)-NicotineAcetylcholine
HN
N Cl
N
H
N
N
N
Figure 2.1 Some biologically active compounds
Nicotine, epibatidine and some of their synthetic analogues are powerful
acetylcholine (one of the human transmitter substances) receptor agonists, i.e. analgesics.21
The major problem with compounds such as nicotine and epibatidine is their toxicity, which
makes dosage difficult and dangerous. More recently a patent report22 described the
preparation of the racemic form, followed by chiral HPLC separation, of compound A, which
has shown promising activity (Figure 2.1). It was therefore desirable to develop a general
method for the preparation of compounds having similar structures, i.e. bicyclic nicotine
analogues.
It was considered that the presence of a second nitrogen, if placed in conjugation with
the imine, might fulfil the role of an electron-withdrawing group under the usual acidic
reaction conditions of the aza-Diels-Alder reaction. This would open-up a general route for
isomers of epibatidine and/or nicotine and for new chiral ligands containing pyridine or a
general heterocyclic moiety (Paper I).
20 (a) Isolation of epibatidine: Spande, T. F.; Garraffo, H. M.; Edwards, M. W.; Yeh, H. J. C.; Pannell, L.; Daly,J. W. J. Am. Chem. Soc. 1992, 114, 3475; (b) For a review on the discovery of alkaloids in amphibian skin, see:Daly, J. W. J. Nat. Prod. 1998, 61, 162.21 For a review about drug research in this area, see: Holladay, M. W.; Dart, M. J.; Lynch, J. K. J. Med. Chem.1997, 40, 4169.22 Bencherif, M.; Caldwell, W. S.; Dull, G. M.; Lippielo, P. M. Pharmaceutical Compositions for the Treatmentsof Central Nervous System Disorders. U. S. Patent 5,583,140, 1996.

16
Indeed, when pyridine-2-carboxaldehyde (2a) was subsequently treated with (S)-(-)-
α-phenylethylamine and cyclopentadiene under acidic conditions a highly stereoselective
cycloaddition took place giving compound 3a in good yield, Table 2.1.
It was pleasing to confirm that the theory on the hetero-Diels-Alder reaction was
correct and that the general synthetic sequence outlined in Scheme 2.2 could be used with
heterocyclic imine dienophiles allowing the production, not only of potentially active
compounds, but also that of new chiral ligand precursors.
Reagents and conditions: (i) (S)-α-phenylethylamine, MS 4Å, CH2Cl2, rt; (ii) Acid(s), CpH, -78 °C to rt.Ar = Heteroaromatic system
i ii
2 3
NAr
Ph
Ar N PhAr O
Scheme 2.2 Aza-Diels-Alder reaction of heterocyclic imine dienophiles
The choice of acid also proved to be essential for the outcome of the reaction. The use
of Lewis acids only resulted in fast polymerisation of the aldehydes. A variety of protic acids
were screened where methane sulphonic acid and/or triflouroacetic acid were found to be the
most efficient. Furthermore, polymerisation could be reduced to a minimum by temperature
control. Performing the reactions at –78 °C afforded a crude product eventually containing no
polymers, but consisting in this case of simple diastereomeric mixtures that could be purified
by means of chromatography.
The need for conjugation of the second nitrogen with the imine system was also
confirmed. As expected aldehydes 2c and 2f both failed to react, even if the corresponding
imines were formed under the same conditions as for the remaining substrates. The results
obtained with the different dienophiles are summarised in Table 2.1.
As mentioned, these compounds could probably also find use in catalysis, since
pyridine is a very good ligand for a large variety of metals. Unfortunately all attempts to
deprotect compounds 3 failed, because no selectivity on these double bensylic-nitrogens
could be observed.

17
The (S)-α-phenylethylamine was also exchanged for p -methoxy- (S)-α-
phenylethylamine, but ammonium cerium (IV) nitrate cleavage did not afforded the desired
product in practical yields. According to the crude proton NMR analysis, the desired product
was one of the components in a complex crude mixture. Attempts to isolate this product, both
via acidic aqueous extraction, flash chromatography or preparative reversed phase HPLC
were unsuccessful.
Aldehydea Reactionconditions YieldbEntry
CH3SO3H / TFA 87:131
2
4
5
7
8
CH3SO3H / TFA 80:20
90:10CH3SO3H
CH3SO3H 90:10
CH3SO3H
CH3SO3H
75:25
90:10
3CH3SO3H / TFA or
CH3SO3H e
6CH3SO3H / TFA or
CH3SO3H e
Diastereoselectivityd
> 99 %
> 99 %
> 99 %
> 99 %
> 99 %
> 99 %
---
---
80
60
---
80
79
---
60
80
exo/endoselectivityc
aAll aldehydes were used as received from commercial sources; bRefers to the isolated yield over the two isomers; cNo endo isomer could be
observed; dDetermined by integration of the signals on the crude 1H-NMR; eThe imine of the corresponding aldehyde was formed but no
Diels-Alder reaction occurred.
2a
2b
2c
2d
2e
2f
2g
2h
Product
3a
3b
3d
3e
3g
3h
N
N
N
N
NH
N
NH
N
S
N
Ph
N
N
Ph
N
N
Ph
N
N
Ph
N
N
PhN
HN
N
PhN
S
O
NO
O
O
O
O
O
O
---
---
Table 2.1 Results of the cycloaddition reactions with heterocyclic imine dienophiles

18
Isoprene could also be used in the cycloaddition reaction, but this was a peculiar case.
Unlike cyclopentadiene the reaction was only effective using a Lewis acid (zinc etherate)23
and a non-conjugated imine, Scheme 2.3.
Reagents and conditions: (i) (S)-α-phenylethylamine, MS 4Å, CH2Cl2, rt; (ii) ZnCl2, isoprene, CH2Cl2/ether, rt.
i ii
2c 4
N NO N Ph N N
Ph
Scheme 2.3 The isoprene case
2.3 Preparation of enantiomerically pure cyclopentyl- and cyclohexyl-amines
A considerable variety of extremely important compounds in medicinal chemistry
contain multi-functionalised chiral cyclopentylamines. This structural unit is present in many
different antibiotics of the ribose mimic class. Amongst the more interesting are
amidomycin,24 aristeromycin25 and carbovir,26 Figure 2.2. All of these compounds have been
shown to have antiviral properties and carbovir is a promising antiviral agent used in the
treatment of AIDS.27
23 Pfrengle, W.; Kunz, H. J. Org. Chem. 1989, 54, 4263.24 (a) Sung, S-Y.; Frahm, A. W. Arch. Pharm. Pharm. Med. Chem., 1996, 329, 291; (b) Nakamura, S.;Karasawa, K.; Tanaka, N.; Yonehara, H.; Umezawa, H. J. Antibiot., Ser. A 1960, 392; (c) Nagata, H.; Taniguchi,T.; Ogasawara, K. Tetrahedron: Asymmetry 1997, 8, 2679.25 (a) Kusaka, T.; Yamamoto, H.; Shibata, M.; Muroi, M.; Kishi, T.; Mizuno, K. J. Antibiot. 1968, 255; (b)Arita, M.; Adachi, K.; Sawai, H.; Ohno, M. Nucleic Acids Research Symposium Series 1983, 12, 25.26 (a) White, E. L.; Parker, W. B.; Macy, L. J.; Shaddix, S. C.; McCaleb, G.; Secrist III, J. A.; Vince, R.;Shannon, W. M. Biochem. and Biophys. Research Commun. 1989, 161, 393; (b) Vince, R.; Hua, M.; Brownell,J.; Daluge, S.; Lee, F.; Shannon, W. M.; Lavelle, G. C.; Qualls, J.; Weislow, O. S.; Kiser, R.; Canonico, P. G.;Schultz, R. H.; Narayanan, V. L.; Mayo, J. G.; Shoemaker, R. H.; Boyd, M. R. Biochem. and Biophys. ResearchCommun. 1988, 156, 1046.27 Other related biologically active compounds: Neplanocin A: (a) Arita, M.; Adachi, K.; Sawai, H.; Ohno, M.Nucleic Acids Research Symposium Series 1983, 12, 25; (b) Lim, M-I.; Moyer, J. D.; Cysyk, R. L.; Marquez, V.E. J. Med. Chem. 1984, 27, 1536; Lim, M-I.; Marquez, V. E. Tetrahedron Lett. 1983, 24, 5559. Guaninederivatives: Reitz. A. B.; Goodman, M. G.; Pope, B. L.; Argentieri, D. C.; Bell, S. C.; Burr, L. E.; Chourmouzis,E.; Come, J.; Goodman, J. H.; Klaubert, D. H.; Maryanoff, B. E.; McDonnell, M. E.; Rampulla, M. S.; Schott,M. R.; Chen, R. J. Med. Chem. 1994, 37, 3561. Adenosin analogues: Shealy, Y. F.; Clayton, J. D. J. Am. Chem.Soc. 1966, 88, 3885. Tubercidin analogues: Montgomery, J. A.; Hewson, K. J. Med. Chem. 1967, 10, 665.Noraristeromycin: Siddiqi, S. M.; Oertel, F. P.; Chen, X.; Schneller, S. W. J. Chem. Soc., Chem. Commun.1993, 708. Adenosine deaminase inhibitors: Schaeffer, H. J.; Godse, D. D.; Liu, G. J. Pharm. Sci. 1964, 53,1510. γ-aminobutyric acid analogue: Milewska, M. J.; Polonski, T. Tetrahedron: Asymmetry 1994, 5, 359.Carboxylic sugars and nucleosides: (a) Ranganathan, S.; George, K. S. Tetrahedron 1997, 53, 3347; (b)Mulvihill, M. J.; Surman, M. D.; Miller, M. J. J. Org.Chem. 1998, 63, 4874.
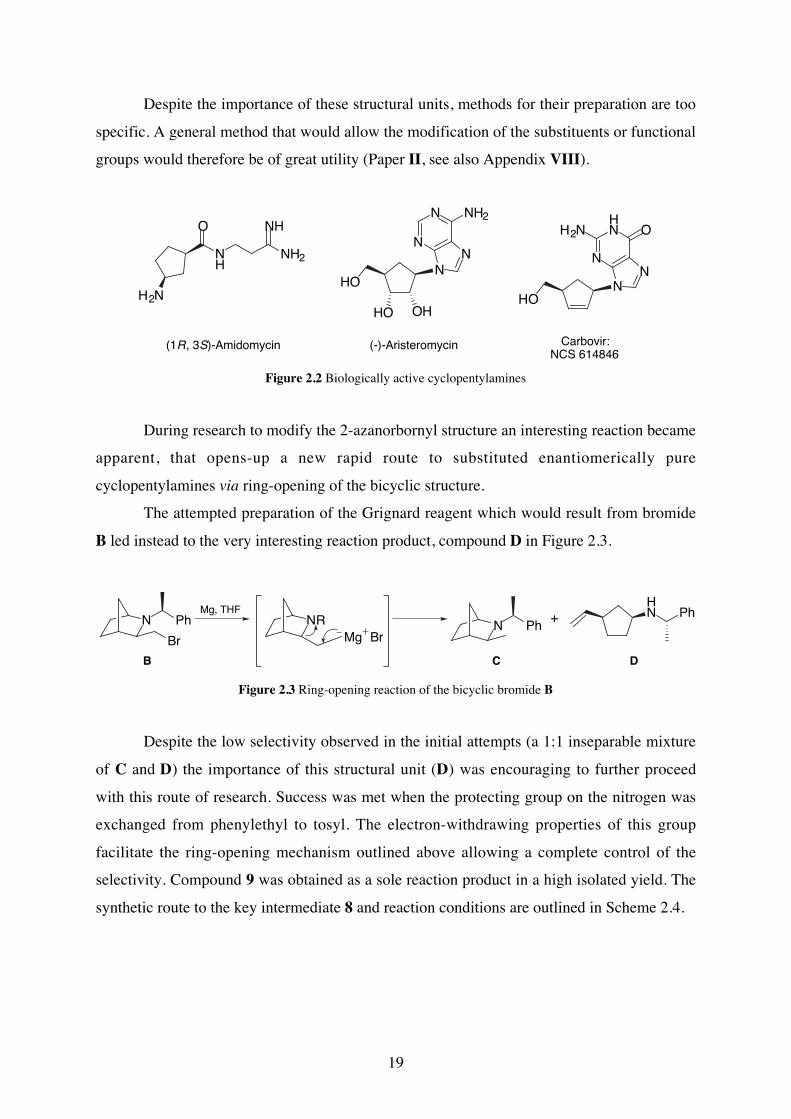
19
Despite the importance of these structural units, methods for their preparation are too
specific. A general method that would allow the modification of the substituents or functional
groups would therefore be of great utility (Paper II, see also Appendix VIII).
O
NH
NH2
NHN
N
NN
NH2
HO
HO OH
(1R, 3S)-Amidomycin (-)-Aristeromycin Carbovir:NCS 614846
H2N
HN
N
NN
O
HO
H2N
Figure 2.2 Biologically active cyclopentylamines
During research to modify the 2-azanorbornyl structure an interesting reaction became
apparent, that opens-up a new rapid route to substituted enantiomerically pure
cyclopentylamines via ring-opening of the bicyclic structure.
The attempted preparation of the Grignard reagent which would result from bromide
B led instead to the very interesting reaction product, compound D in Figure 2.3.
N N
HN
Ph
BrPh
Mg, THF+
B C D
PhNR
Mg Br
Figure 2.3 Ring-opening reaction of the bicyclic bromide B
Despite the low selectivity observed in the initial attempts (a 1:1 inseparable mixture
of C and D) the importance of this structural unit (D) was encouraging to further proceed
with this route of research. Success was met when the protecting group on the nitrogen was
exchanged from phenylethyl to tosyl. The electron-withdrawing properties of this group
facilitate the ring-opening mechanism outlined above allowing a complete control of the
selectivity. Compound 9 was obtained as a sole reaction product in a high isolated yield. The
synthetic route to the key intermediate 8 and reaction conditions are outlined in Scheme 2.4.

20
9
Reagents and conditions: (i) H2 (150 psi), 5% Pd-C, EtOH, rt, 48h, 98%; (ii) TsCl, Et3N, CH2Cl2, rt, overnight, 92%; (iii) LiAlH4, THF, rt, 2h, 95%; (iv) CBr4, Ph3P, CH2Cl2, rt, 24h, 60%; (v) Mg, BrCH2CH2Br,THF, reflux, 24h, 90%.
5 7
NHCO2Et
NTsOH
8
NTsBr
NHTs
N PhCO2Et
1a
i ii
6
NTsCO2Et
iii
ivv
Scheme 2.4 Ring-opening of the bicyclic bromide 8
Manipulation of the original aza-Diels-Alder adduct (1a) allows the possibility of
further functionalisation and as much as four functionalised chiral centers can be introduced
into the five carbons of cyclopentadiene. Dihydroxylation of 1a, followed by ketal protection
of the diol affords compound 11, which is converted into the analogue of 8 via the same
synthetic sequence. Compound 15 is then ring-opened to the multi-functionalised
cyclopentane 16 (Scheme 2.5).
Completely selective functionalisation of cyclopentadiene is then achieved using this
eight-step sequence.
Reagents and conditions: (i) OsO4, NMO/H2O, t-BuOH, 24h, 92%; (ii) (MeO)2C(CH3)2, TsOH, warm MeOH, 15 min, 87%; (iii) ammonium formate,10% Pd-C, EtOH, reflux, 1h, 99%; (iv) TsCl, Et3N, CH2Cl2 , rt, overnight, 90%; (v)LiAlH4, THF, rt, 2h, 92%; (vi) CBr4, Ph3P, CH2Cl2, rt, 24h, 59%; (vii) Mg, BrCH2CH2Br, THF, reflux, 32h, 89%.
NCO2Et
PhHO
HO NCO2Et
PhO
O
NTsO
OO O
NHTs
1a
NCO2Et
Ph i
10
ii
11
iii NHCO2Et
OO
12iv
OH
vi
14
NTsO
OBr
15
vii
16
NTsCO2Et
OO
v
13
Scheme 2.5 Multi-functionalisation of cyclopentadiene

21
This new methodology could also be extended to larger bicyclic structures as in the
case of the aza-bicyclo[2.2.2]octene (17), obtained using cyclohexa-1,3-diene in the hetero-
Diels-Alder reaction. The ring-opening of this compound shows that the reaction is not only a
consequence of the ring strain in the [2.2.1] system (Scheme 2.6).28
17 18
i -vii
Reagents and conditions: (i) OsO4, NMO/H2O, t-BuOH, 24h, 92%; (ii) (MeO)2C(CH3)2,TsOH, warm MeOH, 15 min, 87%; (iii) ammonium formate, 10% Pd-C, EtOH, reflux, 1h, 99%; (iv) TsCl, Et3N, CH2Cl2, rt, overnight, 91%; (v) LiAlH4, THF,rt, 2h, 94%; (vi) CBr4,Ph3P, CH2Cl2, rt, 24h, 62%; (vii) Mg, BrCH2CH2Br, THF, reflux, 32h, 85%.
NCO2Et
PhO
O
NHTs
Scheme 2.6 Ring-opening of the azabicyclo[2.2.2]octene 17
As mentioned before, this new methodology opens-up a practical route to multi-
functionalised chiral cyclopentyl- and cyclohexylamines, thus, allowing modification of
substituents or functional groups in a variety of antibiotics of the ribose mimic class.
2.4 Access to unnatural ββββ-amino alcohols
As mentioned in Chapter 1 (Section 1.3) β-amino alcohols derived from natural
occurring amino acids are widely used as ligands or ligand precursors in asymmetric
synthesis.12 Compounds 1a and 5 described above are direct intermediates in the synthesis of
unnatural amino alcohols, Figure 2.4. Besides this fact other nitrogen containing ligands, such
as amino-phosphines, amino-thiols or amino-oxazolines, which could also be prepared from
precursors like 5, are widely used in the field of catalysis.29
28 For interesting compounds containing this structure unity see for example: (a) Keck, G. E.; Fleming, S. A.Tetrahedron Lett. 1978, 48, 4763; (b) Hudlicky, T.; Olivo, H. F. Tetrahedron Lett. 1991, 32, 6077; (c) Chretien,F.; Ahmed, S. I.; Masion, A.; Chapleur, Y. Tetrahedron 1993, 49, 7463; (d) Grabowski, S.; Armbruster, J.;Prinzbach, H. Tetrahedron Lett. 1997, 38, 5485; (e) Noguchi, H.; Aoyama, T.; Shioiri, T. Tetrahedron Lett.1997, 38, 2883.29 For a recent review on nitrogen containing ligands, see: Fache, F.; Schultz, E.; Tommasino, M. L.; Lemair, M.Chem. Rev. 2000, 100, 2159.

22
The use of the referred type of amino alcohol ligands, 2-azanorbornyl-3-methanols,
will be described in the following chapters of this thesis.
N NR
NH NH
CO2Et
CO2Et
1a
5OH
OHR''
R'Ph
Figure 2.4 Preparation of unnatural β-amino alcohols from the aza-Diels-Alder adduct 1a

23
3. Ruthenium-catalysed asymmetric transfer hydrogenation of ketones
3.1 Introduction
Ruthenium-catalysed transfer hydrogenation30 from 2-propanol to ketones (Scheme
3.1) is one of the most attractive processes to prepare enantioenriched secondary alcohols.
Both from an industrial and economical point of view, 2-propanol is a very cheap hydrogen
source and the catalyst loadings typical for these experiments are low. In addition, it avoids
the use of explosive molecular hydrogen or reactive metal hydrides.
R R'
O
R R'
OHCat. "Ru", Cat. Chiral Ligand
Cat. Base, 2-Propanol
Scheme 3.1 Transfer hydrogenation reaction
Noyori and co-workers have reported one of the most successful examples in this
field with the introduction of the diamine ligand 19,31 Figure 3.1. Chiral phosphorous and
nitrogen ligands 20 to 2432 have also been used with variable levels of enantioselectivity
being obtained in the reduction of acetophenone.
NHTs
NH2
Ph
Ph
PhPNHHN
HO NHMe
Ph
OH
NH2
N
O
Ph2P
Fe
NHMe
NHMe
PhPh
19 20 21
242322
99% yield
98% ee 3195% yield91% ee 32a
96% yield20% ee 32d
70% yield91% ee32e
95% yield80% ee 32c
24% yield
94% ee 32b
Figure 3.1 Some ligands used in transfer hydrogenation
30 For reviews on the subject, see: (a) Gladiali, S.; Mestroni, G. Transition Metals for Organic Synthesis, Vol. 2,Chapter 1.3; Wiley-VCH: Toronto 1998; (b) Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97; (c)Palmer, M., J.; Wills, M. Tetrahedron: Asymmetry 1999, 10, 2045.31 Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996, 118, 2521 (resultobtained using formic acid-triethylamine mixture as hydrogen source).32 (a) Takehara, J.; Hashiguchi, S.; Fujii, A.; Inoue, S.; Ikariya, T.; Noyroi, R. Chem. Commun. 1996, 233; (b)Langer, T.; Helmchen, G. Tetrahedron Lett. 1996, 37, 1381; (c) Puntener, K.; Schwink, L.; Knochel, P.Tetrahedron Lett. 1996, 37, 8165; (d) Jiang, Y.; Jiang, Q.; Zhu, G.; Zhang, X. Tetrahedron Lett. 1997, 38, 6565;(e) Palmer, M.; Walsgrove, T.; Wills, M. J. Org. Chem. 1997, 62, 5226.

24
As mentioned before (Sections 1.3 and 2.4) the use of simple amino alcohols is
especially attractive, but in the case of the ruthenium-catalysed transfer hydrogenation of
ketones there are only a few successful applications of these kind of ligands, for example the
compounds 20 and 24 in Figure 3.1.
3.2 The 2-azanorbornyl-3-methanol as a ligand for ruthenium
Recently reported from this laboratory is the use of 2-azanorbornyl derivatives as
ligands in asymmetric catalysis.33 The results obtained prompted the application of a few of
these rigid proline analogues as ligands in the title transformation (Paper III).
The simplest of all 2-azanorbornyl-3-methanols (25) was prepared from ethyl-2-
azanorbornyl-3-carboxylate (5) in a single step as outlined in Scheme 3.2 (see Chapter 2 for
the preparation of 5).
i
Reagents and conditions: (i) LAH (2 equiv), THF, rt, 1h, 90%
5
NHCO2Et
25
NHOH
NH
OH
26, (S)-prolinol
Scheme 3.2 The simplest 2-azanorbornyl-3-methanol 25 and the (S)-prolinol analogue 26
Surprisingly, the use of (S)-prolinol (26) had never been reported in this reaction, so
comparison of this widely used ligand structure with the rigid and sterically more demanding
bicyclic analogue was a must. Moreover, it was also important to study the influence of α-
substitution, hence the α-dimethyl ligand (29) was also prepared, Scheme 3.3.
5 27 28 29
Reagents and conditions: (i) PhCH2Br, K2CO3, CH3CN, rt, 32h, 78%; (ii) MeMgBr, THF, rt, 2h, 84%; (iii) H2 (150psi), 5% Pd-C, EtOH, rt, 24h, 98%.
i ii iiiNHCO2Et
NCO2Et
Ph N
PhOH
NH
OH
Scheme 3.3 Synthesis of ligand 29
33 Södergren, M., J.; Andersson, P. G. Tetrahedron Lett. 1996, 37, 7577.

25
The different ligands were screened using acetophenone, a common model substrate
for this type of study, and ruthenium dichloride hexamethylbenzene dimer, [RuCl2(HMB)]2,
as the metal source.34 The results of this study are summarised in Table 3.1.
S
S
Entry Ligand Yield% %eea Configb
25
29
261
2
3
16
92
85
Time/h
6.5
5
16c
8
95
rac. - -
0.25 mol% [RuCl2(HMB)]2, 2 mol% Ligand
2.5 mol% i-PrOK, i-PrOH
aDetermined by HPLC analysis (ChiralCel OD-H; 5% i-PrOH in hexane; 0.5 mL/min); bDetermined from
the sign of rotation of the isolated product; cThe reaction was performed at 83 °C.
Ph
O
Ph
OH
NH
OH
NHOH
NH
OH
Table 3.1 The behaviour of the different ligands in the transfer hydrogenation of acetophenone
The use of (S)-prolinol (26) gave rise to an unselective reaction (Table 3.1, entry 1).
However, it was pleasing to observe that the rigid analogue 25 led to an excellent result
(Table 3.1, entry 2) after a reaction time of five hours. Unlike ligand 25, the sterically more
hindered α-dimethyl analogue 29 did not performed so well. No conversion into product was
observed at room temperature and only the racemic product was obtained at reflux
temperature (Table 3.1, entry 3).
As a natural consequence of these promising results it was decided to investigate a
variety of substrates, as well as a different metal source, ruthenium dichloride p-cymene
dimer, [RuCl2(p-cymene)]2.
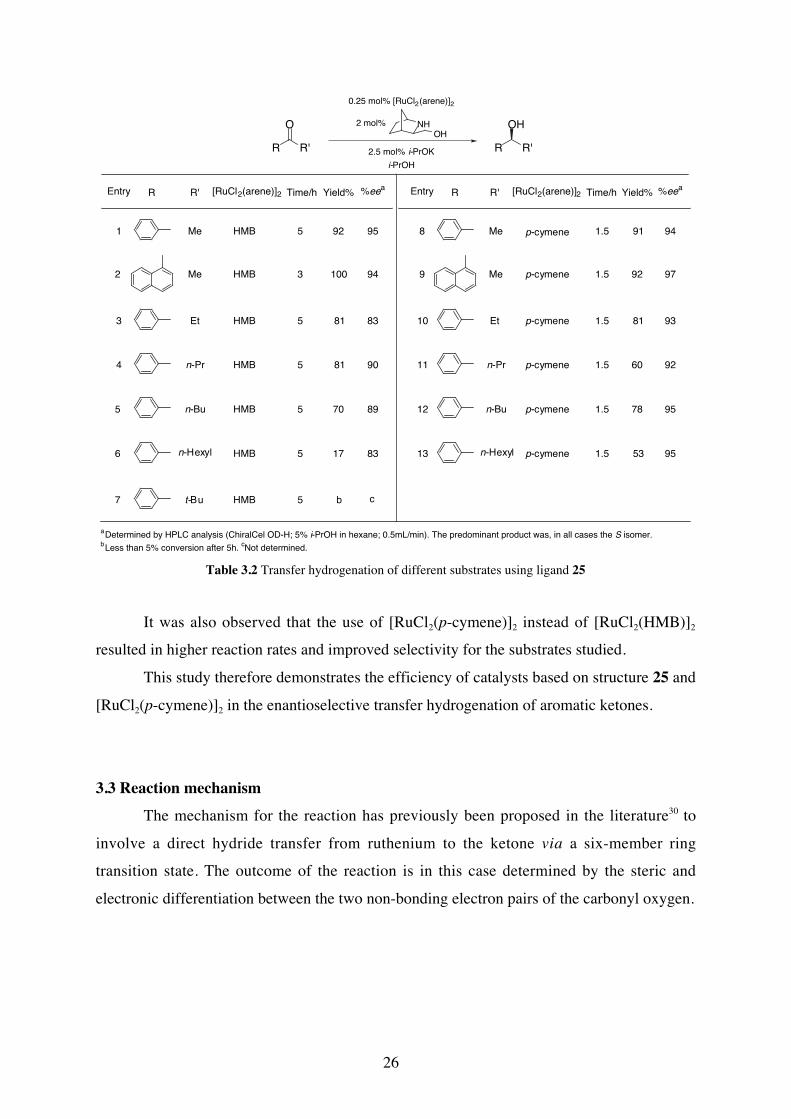
Indeed, this system turned out to be efficient for the reduction of other pro-chiral
ketones to the corresponding secondary alcohols, Table 3.2. High enantiomeric excesses were
obtained for most of the substrates, but in accordance with Noyori´s observations,30b ketones
with a bulky substituent reacted very slowly under the applied reaction conditions (Table 3.2,
entry 7).
34 For preparation of ruthenium complexes, see: (a) Bennet, M. A.; Smith, A. K. J. Chem. Soc., Dalton Trans.1974, 233; (b) Bennet, M. A.; Metheson, T. W.; Robertson, G. B.; Smith, A., K. Inorg. Chem. 1980, 19, 1014.
26
R R'
O
R R'
OH
Me HMB
Me HMB
Et HMB
n-Pr HMB
n-Bu HMB
n-Hexyl HMB
t-Bu HMB
Me p-cymene
Me p-cymene
Et p-cymene
n-Pr p-cymene
n-Bu p-cymene
n-Hexyl p-cymene
0.25 mol% [RuCl2(arene)]2
2.5 mol% i-PrOK
i-PrOH
Entry [RuCl2(arene)]2 Time/h Yield% %eeaR R'
1 5 92 95
2 3 100 94
3 5 81 83
4 5 81 90
5 5 70 89
6 5 17 83
7 5 b c
8 1.5 91 94
9 1.5 92 97
10 1.5 81 93
11 1.5 60 92
12 1.5 78 95
13 1.5 53 95
aDetermined by HPLC analysis (ChiralCel OD-H; 5% i-PrOH in hexane; 0.5mL/min). The predominant product was, in all cases the S isomer. bLess than 5% conversion after 5h. cNot determined.
Entry [RuCl2(arene)]2 Time/h Yield% %eeaR R'
NHOH
2 mol%
Table 3.2 Transfer hydrogenation of different substrates using ligand 25
It was also observed that the use of [RuCl2(p-cymene)]2 instead of [RuCl2(HMB)]2
resulted in higher reaction rates and improved selectivity for the substrates studied.
This study therefore demonstrates the efficiency of catalysts based on structure 25 and
[RuCl2(p-cymene)]2 in the enantioselective transfer hydrogenation of aromatic ketones.
3.3 Reaction mechanism
The mechanism for the reaction has previously been proposed in the literature30 to
involve a direct hydride transfer from ruthenium to the ketone via a six-member ring
transition state. The outcome of the reaction is in this case determined by the steric and
electronic differentiation between the two non-bonding electron pairs of the carbonyl oxygen.
27
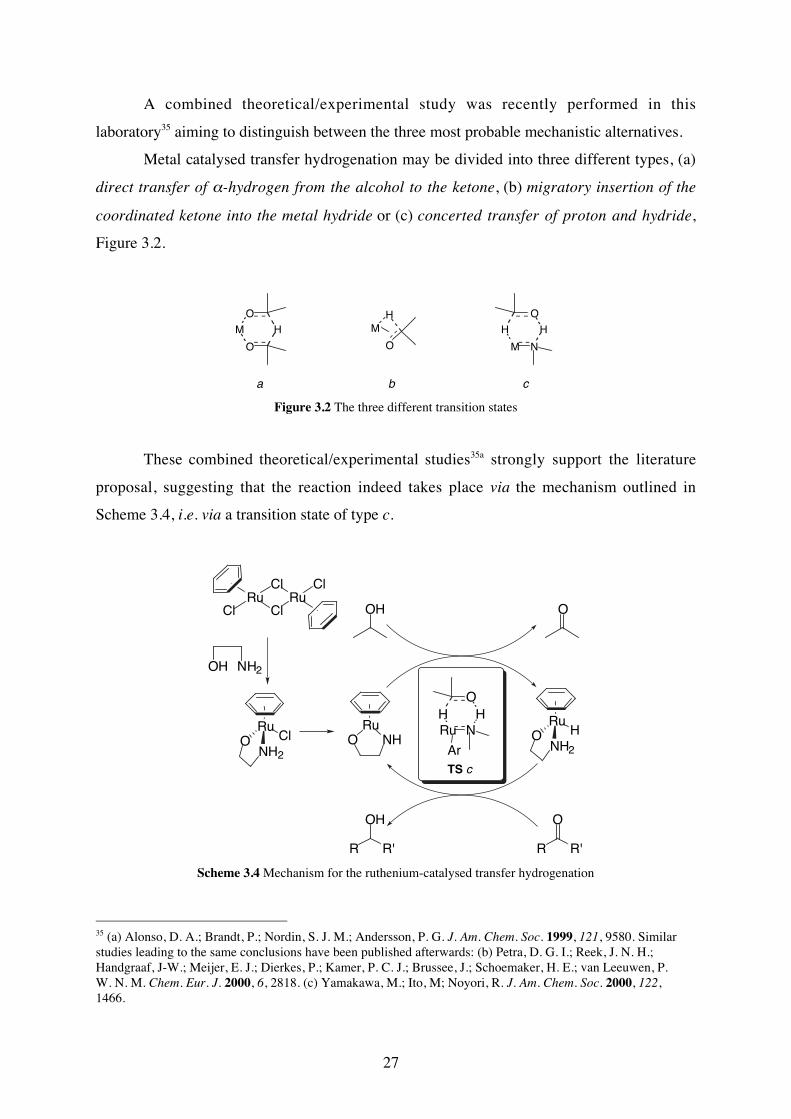
A combined theoretical/experimental study was recently performed in this
laboratory35 aiming to distinguish between the three most probable mechanistic alternatives.
Metal catalysed transfer hydrogenation may be divided into three different types, (a)
direct transfer of α-hydrogen from the alcohol to the ketone, (b) migratory insertion of the
coordinated ketone into the metal hydride or (c) concerted transfer of proton and hydride,
Figure 3.2.
O
M
O
H
O
H
M N
HH
M
O
a b c
Figure 3.2 The three different transition states
These combined theoretical/experimental studies35a strongly support the literature
proposal, suggesting that the reaction indeed takes place via the mechanism outlined in
Scheme 3.4, i.e. via a transition state of type c.
RuO NH
RuO
NH2
HRuO
NH2
Cl
O
R R'
O
OH
R R'
OH
HRu N
HO
ClRu
ClRu
Cl Cl
OH NH2
ArTS c
Scheme 3.4 Mechanism for the ruthenium-catalysed transfer hydrogenation
35 (a) Alonso, D. A.; Brandt, P.; Nordin, S. J. M.; Andersson, P. G. J. Am. Chem. Soc. 1999, 121, 9580. Similarstudies leading to the same conclusions have been published afterwards: (b) Petra, D. G. I.; Reek, J. N. H.;Handgraaf, J-W.; Meijer, E. J.; Dierkes, P.; Kamer, P. C. J.; Brussee, J.; Schoemaker, H. E.; van Leeuwen, P.W. N. M. Chem. Eur. J. 2000, 6, 2818. (c) Yamakawa, M.; Ito, M; Noyori, R. J. Am. Chem. Soc. 2000, 122,1466.

28
The mechanism is an example of metal-ligand bi-functional catalysis, i.e. the ligand is
not only providing the chiral environment for the metal, but also stabilising the transition
state through a hydrogen bond between the nitrogen and the carbonyl oxygen of the substrate.
As mentioned before the outcome of the reaction is determined by steric and electronic
differentiation between the two non-bonding electron pairs of the carbonyl oxygen. Although,
this is not the only reason for the high enantioselectivities obtained with ligand 25. The
rigidity of this ligand structure makes the face selectivity at the amine nitrogen complete
when the ligand co-ordinates to the metal. Hence, the generation of an enantiopure ruthenium
complex is achieved by the use of a rigid ligand (25) that strongly disfavours one specific
configuration of the co-ordinated amine.35a
It is important to notice that, as consequence of microscopic reversibility, the same
transition state is involved in the reverse process. This, if the oxidation/reduction potentials
will allow, can lead to racemisation of the secondary alcohol. However, this phenomena has
not been observed with our catalytic system, the enantiomeric excess remains constant at
different conversion levels and the reaction can reach completion without compromising the
enantiopurity of the product.

29
4. Oxazaborolidines in the asymmetric reduction of ketones
4.1 Introduction
An alternative route to the ruthenium-catalysed transfer hydrogenation in the
preparation of enantioenriched secondary alcohols (Chapter 3) is the use of chiral borane
complexes. This methodology already mentioned in Section 1.3, was first introduced by
Itsuno et al. in the early eighties.36 However, it was not until 1987 that the process became
attractive with the publication of Corey´s promising results on a catalytic version of this
reaction. The reaction as since undergone some major improvements37 and is now an
established process38 that finds application in the synthesis of many natural products.39
4.2 Reaction mechanism
In 1992 Corey and co-workers isolated and X-rayed crystals of the very air and
moisture sensitive (S)-2-(diphenylhydroxymethyl)-pyrrolidine40 borane complex.41 The
isolation of this oxazaborolidine borane complex (Scheme 4.1, 31) opened-up research
towards the understanding of the reaction mechanism leading to the proposal described in
Scheme 4.1.
The mechanism proposed by Corey and co-workers38b allows an explanation for the
absolute configuration of the product, the high enantiomeric excess, the rate enhancement of
the reaction, and the turnover of the catalyst. According to this proposal (S)-diphenyl prolinol
reacts first in an acid/base fashion (Brønsted sense) with the added borane forming species
30. Subsequent addition of a borane complex (Lewis acid) then leads to the formation of the
catalytic active species 31 via co-ordination to the lone pair of the nitrogen (Lewis base) of
the pyrrolidine moiety on the α face of 30.
36 (a) Hirao, A.; Itsuno, S.; Nakahama, S.; Yamazaki, N. J. Chem. Soc., Chem. Commun. 1981, 315; (b) Itsuno,S.; Hirao, A.; Nakahama, S.; Yamazaki, N. J. Chem. Soc., Perkin Trans. 1 1983, 1673.37 (a) Corey, E. J.; Bakshi, R. K.; Shibata, S.; Chen, C-P.; Singh, V. K. J. Am. Chem. Soc. 1987, 109, 7925; (b)Mathre, D. J.; Jones, T. K.; Xavier, L. C.; Blacklock, T. J.; Reamer, R. A.; Mohan, J. J.; Jones, E. T. T.;Hoogsteen, K.; Baum, M. W.; Grabowsky, E. J. J. J. Org. Chem. 1991, 56, 751; (c) Masui, M.; Shiori, T. Synlett1996, 49; (d) Masui, M.; Shiori, T. Synlett 1997, 273.38 For reviews on the process, see: (a) Wallbaum, S.; Martens, J. Tetrahedron: Asymmetry 1992, 12, 1475; (b)Corey, E. J.; Helal, C. J. Angew. Chem. Int. Ed. 1998, 37, 1986.39 Corey, E. J.; Cheng, X-M. The Logic of Chemical Synthesis; John Wiley & Sons: New York, 1995.40 For the preparation of diphenyl prolinol, see: Xavier, L. C.; Mohan, J. J.; Mathre, D. J. Thompson, A. S.;Carroll, J. D.; Corley, E. G.; Desmond, R. Org. Synth. 1996, 74, 50.41 Corey, E. J.; Azimioara, M.; Sarshar, S. Tetrahedron Lett. 1992, 33, 3429.

30
N BO
H
BH3
PhPh
Me
NHOH
H PhPh
NB
O
H PhPh
Me
MeB(OH)2
O
NB
O MePh
Ph H2BO
H
NB
O MePh
Ph BH2
OH
NB
O
H PhPh
H2BO
H B
HH
HBH3
HH2BO
HCl
OH
31
30
ab
BH3
Toluene, ∆-H2O
Scheme 4.1 Borane reduction mechanism
Complex 31 possesses an activated hydride donor (BH3 co-ordinated to nitrogen) as
well as a strong Lewis acid on the endocyclic boron atom. This last property allows the rapid
but selective co-ordination of the ketone, only via the less sterically hindered lone pair a, see
Scheme 4.1. The hydride transfer can then occur through a six-member transition state to
form the non-dissociated reduction product, which is released to regenerate the catalyst by
either of two possible pathways. Reaction of the alkoxide ligand attached to the endocyclic
boron atom with the adjacent boron atom regenerating 30 and the borinated product, or
addition of borane to form the depicted six-member borane bridged species that decomposes
into 31 and the borinated product. The borinated product is then hydrolysed to the secondary
alcohol upon quenching with hydrochloric acid.
4.3 Preparation of 2-azanorbornyl-3-methanols ligands and their application in the
form of the corresponding oxazaborolidines
As previously described in Chapter 3 of this thesis 2-azanorbornyl derivatives are
very efficient ligands in ruthenium-catalysed transfer hydrogenation. At the same time it had
been suggested38a that a rigid analogue of the CBS catalyst could further improve the
selectivity of this catalytic system. This can be explained in the same way as why Corey’s
catalyst (proline based) shows improved selectivity when compared to Itsuno’s catalyst

31
(valine based), Figure 4.1. Although a trans relation between the i-Pr group and the added
borane is preferred in Itsuno’s system, the cis relative conformation can not be completely
excluded. Due to the rigidity of Corey’s system, only the trans relationship is available. For
this reason higher enantioselectivities are obtained with the latter system.
NB
O MePh
PhH2B
OPhH
NH
BO
Ph
Ph
OPh
Me
HH2B
Corey's proline based catalyststrictly α co-ordination
Itsuno's valine based catalystpossibility for both α and β co-ordination
H
NB
O MePh
PhH2B
OPhH
2-azanorbornyl-3-methanol basedoxazaborolidine - increased rigidity
OPh
HB
NB
O
Me
HH
Ph
Ph
Figure 4.1 Comparison of different oxazaborolidine catalysts
With the objective of investigating different oxazaborolidines (Paper IV), a variety of
ligands based on the 2-azanorbornyl framework, i.e. increased rigidity in comparison to
diphenyl prolinol, were prepared, Scheme 4.2.
O
Me
32, R = 33, R =
34, R = 35, R =
36, R = 37, R =
38, R =
1a 5
32-3825, R = H29, R = CH3
i
ii iii 20-46%
N PhCO2Et
NHCO2Et
NHCR2OH
NHCR2OH
Cl
F3C
Reagents and conditions: (i) See Chapter 2; (ii) See Chapter 3; (iii) RMgBr, THF, rt, 1h
Scheme 4.2 Aza-norbornyl oxazaborolidine precursors
Using cyclohexa-1,3-diene in the aza-Diels-Alder reaction, an azabicyclo
[2.2.2]octene (17) was obtained (see Chapter 2) and converted to a less rigid analogue of 25,
ligand 39 in Scheme 4.3.

32
N PhCO2Et
NH
17 39
i
Reagents and conditions: (i) Same as for the transformation of 1a to 25
OH
Scheme 4.3 Preparation of ligand 39
All reductions were performed using a procedure37c,d where the catalytically active
oxazaborolidine is generated in situ using trimethyl borate, borane dimethyl sulphide
complex and the corresponding amino alcohol. The influence of different reaction parameters
on the enantiomeric excess was studied using acetophenone as the model substrate and amino
alcohol 32 as the oxazaborolidine precursor. During these studies it appeared that the
concentration of the solution had an influence on the outcome of the reaction and this was
therefore studied further. The results of this study are summarised in Table 4.1 and a clear
effect can be observed.
0.1 equiv 32, 0.12 equiv B(OMe)3
Entry Initial amino alcohol concentration/Ma % eeb
aInitial concentration means the concentration of 32 in THF before any other addition; bIsolated yields of the corresponding secondary
alcohol were in all cases >95%.
1equiv BH3Ph
O
Ph
OH
Me2S
12
34
0.050.1
0.20.9
8384
8787
Table 4.1 Ligand concentration effects
Since the reaction is known to be extremely solvent and temperature dependent, it was
decided to study these effects using a fixed ligand concentration at 0.2M. The results were in
complete agreement with those previously reported; i.e. the best conditions were found to be
tetrahydrofuran at room temperature42 (Table 4.2).
Entry Solvent Temperature / °C % eeTotal time / h
123456
THFCH2Cl2CH3CNToluene
THFTHF
rtrtrtrt
6 - 740
264282
87rac.23674979
Table 4.2 Solvents and temperatures
42 Stone, G. B. Tetrahedron: Asymmetry 1994, 5, 465.
33
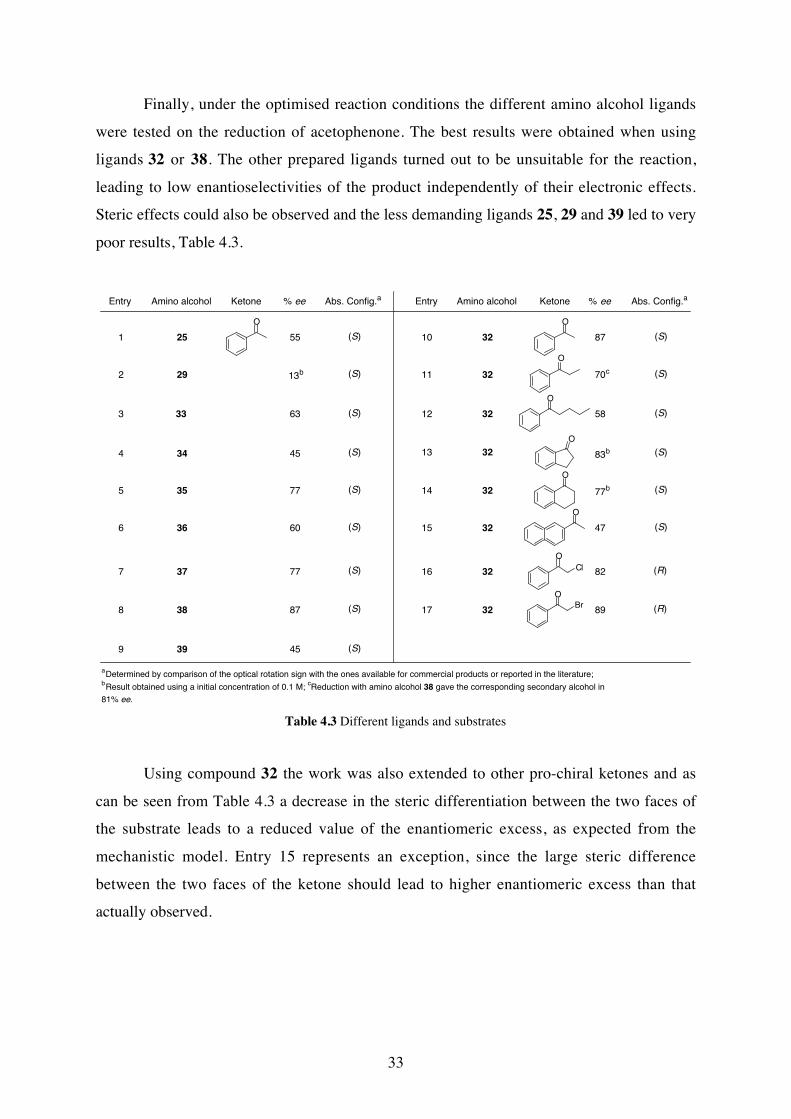
Finally, under the optimised reaction conditions the different amino alcohol ligands
were tested on the reduction of acetophenone. The best results were obtained when using
ligands 32 or 38. The other prepared ligands turned out to be unsuitable for the reaction,
leading to low enantioselectivities of the product independently of their electronic effects.
Steric effects could also be observed and the less demanding ligands 25, 29 and 39 led to very
poor results, Table 4.3.
aDetermined by comparison of the optical rotation sign with the ones available for commercial products or reported in the literature; bResult obtained using a initial concentration of 0.1 M; cReduction with amino alcohol 38 gave the corresponding secondary alcohol in
81% ee.
Entry Amino alcohol Ketone % ee Abs. Config.a
1 25 55 (S)
2 29 13b (S)
3 33 63 (S)
4 34 45 (S)
5 35 77 (S)
6 36 60 (S)
7 37 77 (S)
8 38 87 (S)
9 39 45 (S)
10 32 87 (S)
11 32 70c (S)
12 32 58 (S)
13 32 83b (S)
14 32 77b (S)
15 32 47 (S)
16 32 82 (R)
17 32 89 (R)
Entry Amino alcohol Ketone % ee Abs. Config.a
O O
O
O
O
O
O
OCl
OBr
Table 4.3 Different ligands and substrates
Using compound 32 the work was also extended to other pro-chiral ketones and as
can be seen from Table 4.3 a decrease in the steric differentiation between the two faces of
the substrate leads to a reduced value of the enantiomeric excess, as expected from the
mechanistic model. Entry 15 represents an exception, since the large steric difference
between the two faces of the ketone should lead to higher enantiomeric excess than that
actually observed.

34
5. Enantioselective addition of dialkylzinc reagents to N-(diphenyl
phosphinoyl) imines
5.1 Introduction
Chapters 3 and 4 of this thesis dealt with the asymmetric reduction of the carbonyl
functionality in ketones. Unlike these reactions43 the corresponding asymmetric reduction or
addition of organometallic reagents to imines, as a method for the preparation of
enantioenriched amines has not yet been subject of the same attention and only a few
successful examples can be found in the literature,44 Scheme 5.1.
HCO2H-Et3N
Et2Zn
99% yield95% ee45
90% yield91% ee46
1 equiv
nBuLi (2 equiv)
89% yield90% ee47
2.5 mol% [RuCl2(p-cymene)]2
5 mol%
1 equiv
N
O
O
Me
NH
O
OH Me
TsHN NH2
Ph Ph
Ph
N
O
Ph
HN
ONN
HH
HH
Ph NP
OPhPh Ph N
H
P
OPhPh
H
N OH
Me Ph
O
Scheme 5.1 Some successful examples of asymmetric transformations of imines
This laboratory has previously reported on the use of simple aziridino alcohols as
chiral ligands for the enantioselective addition of diethylzinc to N-(diphenylphosphinoyl)
43 For an extensive review on the addition of organozinc reagents to carbonyl compounds, see: Pu, L.; Yu, H-B.Chem. Rev. 2001, 101, 757.44 For an excellent review, see: Kobayashi, S.; Ishitani, H. Chem. Rev. 1999,99, 1069.45 Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996, 118, 4916. For areview of Noyori´s work on the field see reference 30b.46 (a) Denmark, S. E.; Nakajima, N.; Nicaise, O. J-C. J. Am. Chem. Soc. 1994, 116, 8797; (b) For a review, see:Denmark, S. E.; Nicaise, O. J-C. Chem. Commun. 1992, 999.47 Soai, K.; Hatanaka, T.; Miyazawa, T. J. Chem. Soc., Chem. Commun. 1992, 1097 and references cited therein.

35
imines,48 Scheme 5.2. At the same time, enantioselective addition of diethylzinc to aldehydes
had been successfully promoted using N-substituted 2-azanorbornyl-3-methanol and thiol as
ligands.49
Et2Zn63% yield94% ee
1 equiv
Ph NP
OPhPh Ph N
H
P
OPhPh
HNBn
MeOH
Scheme 5.2 Enantioselective addition promoted by simple aziridino alcohols
It was therefore decided to investigate the performance of these types of ligands in the
addition of dialkylzinc reagents to N-(diphenylphosphinoyl) imines (Papers V, VI and VII).
5.2 The 2-azanorbornyl-3-methanols as chiral auxiliaries for the addition reaction
5.2.1 The first generation ligands – Synthesis and results obtained
The first generation ligands were prepared from the ethyl-2-azanorbornyl-3-
carboxylate 5, using the synthetic sequence depicted in Scheme 5.3.
5
i
ii
iii
Reagents and conditions: (i) (R1)X, CH3CN, K2CO3; (ii) LAH, THF; (iii) (R2)MgBr, THF
27 (78%)40 (37%)
R1 = CH2PhR1 = Me
28 (84%)47 (20%)48 (47%)
R1 = CH2PhR1 = CH2PhR1 = CH2Ph
R2 = MeR2 = iPrR2 = Ph
NHCO2Et
NR1CO2Et
NR1OH
NR1C(R2)2OH
43 (74%)44 (76%)
R1 = CH2PhR1 = Me
45 (60%)46 (80%)
R1 = EtR1 = iPr
41 (80%)42 (82%)
R1 = EtR1 = iPr
Scheme 5.3 The first generation ligands
Compounds 28 and 43 to 48 were then used as chiral auxiliaries in the title
transformation and the results obtained are given in Table 5.1. As it can be seen from this
48 (a) Andersson, P. G.; Guijarro, D.; Tanner, D. Synlett 1996, 727; (b) Andersson, P. G.; Guijarro, D.; Tanner,D. J. Org. Chem. 1997, 62, 7364.49 (a) Nakano, H.; Kumagai, N.; Kabuto, C.; Matsuzaki, H.; Hongo, H. Tetrahedron: Asymmetry 1995, 6, 1233;(b) Nakano, H.; Kumagai, N.; Kabuto, C. Matsuzaki, H.; Hongo, H. Tetrahedron: Asymmetry 1997, 8, 1391; (c)Nakano, H.; Iwasa, K.; Hongo, H. Heterocycles 1991, 44, 435.

36
table structures 43 and 45 proved to be superior (entries 2 and 4) to all others and the
obtained enantioselectivities were in both cases above 90%.
S
49a, Ar = Ph49b, Ar = 1-naphthyl
50a, Ar = Ph; R = Et50a', Ar = Ph; R = Me50b, Ar = 1-naphthyl; R = Et
Chiral Ligand
R2Zn, toluene
Entry Ligand (equiv) R Yield% %ee
123456
PhPhPhPhPhPh
504359634638
759285918568
EtEtEtEtEtEt
44 (1)45 (1)46 (1)43 (1)
43 (0.25)43 (0.10)
a Isolated after flash chromatography (silica gel, pentane/acetone); bDetermined by HPLC analysis using a chiral column (ChiralCel OD-H)
Ar
Ar NPO
PhPh Ar N
H
PO
PhPh
R H
Entry Ligand (equiv) R Yield% %ee
7891011
1-NaphtPhPhPhPh
6532655233
9283884316
EtMeEtEtEt
43 (1)28 (1)28 (1)47 (1)48 (1)
Ar
Table 5.1 Results obtained with the first generation ligands
These promising results also prompted the use of substoichiometric amounts of the
chiral auxiliary. Unfortunately the selectivity in the reaction dropped as the amount of ligand
was decreased (entries 4, 5 and 6). Noteworthy is the fact that up to 90% of the auxiliary
could be recovered during purification and re-used without loss of asymmetric induction.
5.2.2 The second generation ligands – Synthesis and results obtained
The second generation ligands are compounds that contain a secondary alcohol
functionality. These were prepared via the 2-azanorbornyl-3-carboxaldehyde 53 (Scheme
5.4). The ligands from this generation were developed as a consequence of the theoretical
study of the reaction mechanism (Section 5.3), which suggested that secondary alcohols with
the correct absolute configuration would further improve the selectivity. Preparation of the
key intermediate (aldehyde 53) was straightforward, but a simple Grignard addition did not
led to the desired product in satisfactory yield and selectivity. This problem, however, was
overcome by the use of the corresponding organocerium reagent, prepared in situ by reaction
of anhydrous cerium (III) chloride and the organomagnesium species.50
50 (a) For a review on organocerium reagents in organic synthesis, see: Liu, H-J.; Shia, K-S.; Shang, X.; Zhu, B-Y.; Tetrahedron 1999, 55, 3803; For experimental procedure, see: (b) Imamoto, T.; Takiyama, N.; Nakamura,K.; Hatajima, T.; Kamiya, Y. J. Am. Chem. Soc. 1989, 111, 4392; (c) Imamoto, T.; Takeda, N. Org. Synth. 1998,76, 228.

37
1a 51 52 53
54, R = Ph (98%)55, R = Me (98%)56, R = iPr (78%)
57, R = Ph (96%)58, R = Me (93%)59, R = iPr (95%)
60, R = Ph (61%)61, R = Me (65%)62, R = iPr (64%)
i ii
iv
iii
vvi
Reagents and conditions: (i) H2 (1atm), Pd-C (10 wt%), EtOH, rt, 4h, 92%; (ii) LAH, THF, rt, 2h, 95%; (iii) Swern Oxidation, 92%;(iv) RMgX/CeCl3, THF, -78 °C, overnight; (v) H2 (300 psi), Pd(OH)2-C (20 wt%), EtOH, rt, 4days; (vi) PhCH2Br, K2CO3, CH3CN, rt, 32h.
N PhCO2Et
N PhCO2Et
N PhOH
N PhCHO
NR
PhOH
H
NHOH
H
RN
OHH
PhR
Scheme 5.4 The second generation ligands – Part I
Having isolated compounds 54, 55 and 56, it was intended to use the Mitsunobu
reaction51 to invert the absolute configuration at the secondary alcohol, since this would allow
verification of the computational results. However, the reaction did not take place, probably
due to steric hindrance of the substrate. Instead, another route to the desired diastereomers
had to be developed. If one diastereomer could be obtained via Grignard addition to the
aldehyde 53, the other one should be possible to prepare through hydride reduction of the
analogue ketone. This approach called for the development of a new aza-Diels-Alder reaction
involving imine dienophiles derived from keto-aldehydes instead of ester-aldehydes. This
approach proved to be effective and the synthetic route to these new ligands is described in
Scheme 5.5.
i ii iii
iv
v
63, R = Ph64, R = Me
65, R = Ph (42%)66, R = Me (31%)
67, R = Ph (95%)68, R = Me (95%)
69, R = Ph (71%)70, R = Me (61%)
71, R = Ph (96%), R = Me (not isolated)
72, R = Ph (65%)
Reagents and conditions: (i) (S)-α-phenylethylamine, CH2Cl2, 0 °C; TFA, BF3.Et2O, CpH, CH2Cl2, -78 °C, overnight; (ii) H2 (1 atm), Pd-C
(10 wt%), MeOH, K2CO3, rt, 4h; (iii) LAH, THF, -78 °C, overnight; (iv) H2 (300 psi), Pd(OH)2-C (20 wt%), EtOH, rt, 4 days; (v) PhCH2Br,
K2CO3, CH3CN, rt, 32 h.
R
O
ON Ph
CORN Ph
CORN Ph
H
ROH
NH
ROH
HN Ph
H
ROH
Scheme 5.5 The second generation ligands – Part II
51 Misunobu, O. Synthesis 1981, 1.

38
The use of sodium boron hydride in the reduction of compounds 67 and 68 led to very
low conversion to products 69 and 70 (20% conversion after two days). If DIBAL-H was
used a faster reaction was observed, although the selectivity remained lower than what was
desired (70:30). Finally, the use lithium aluminium hydride gave the right diastereomer in
good yield, > 60% over the pure major diastereomer, and selectivity, 80:20.
Compounds 60, 61, 62 and 72 were then tested in the addition reaction and indeed the
presence of an additional stereocenter turned out to be important. The right choice of the
absolute configuration at this new chiral center improved the enantioselectivity in the reaction
from 91% to 97% (Table 5.2), thus, turning this method into an attractive tool in the synthesis
of chiral amines from the corresponding N-(diphenylphosphinoyl) imines.
1 equiv Chiral Ligand
Et2Zn, toluene
Entry Ligand Yield%a %eeb
1234
70685952
97937971
60616272
aIsolated after flash chromatography (silica gel, pentane/acetone); bDetermined by HPLC analysis using a chiral column (ChiralCel OD-H)
Ph NPO
Ph
Ph Ph NH
PO
Ph
Ph
H
50a49a
Table 5.2 Results obtained with the second generation ligands
Due to the rather low number of publications dedicated to this transformation, solvent
studies are rare and only one example was found in the literature.52 It was therefore decided to
test this new and efficient chiral auxiliary performance in solvents other than toluene, using
the imine 49a as the model substrate. As it can be seen from the results of this study
summarised in Table 5.3, the non-aromatic solvents proved to be completely inadequate for
this transformation (entries 2 to 4), while different aromatic solvents led to product formation
in variable yields and selectivity.
12345678
toluenedichloromethane
diethylethertetrahydrofuran
benzeneethylbenzene
trifluorotolueneanisole
72------35385247
97------90909594
Entry Solvent Yield % ee %
9101112131415
p-methylanisolechlorobenzene
o-dichlorobenzenem-dichlorobenzene
o-chlorotoluenem-chlorotoluenep-chlorotoluene
43756059574669
92989694907795
Entry Solvent Yield % ee %
Table 5.3 The solvent effect
52 Soai, K.; Suzuki, T.; Shono, T. J. Chem. Soc., Chem. Commun. 1994, 317.

39
Although the difference in the results obtained using toluene or chlorobenzene as
solvents cannot be consider significant it was decided to try both solvents with the remaining
substrates.
Entry R Yield %
ee %
; Et2Zn (3 equiv)
toluene chlorobenzene
1 72 97 98
2 -- -- --
3 72 91 89
4 70 77 87
5 91 98 98
6 -- -- --
7 67 95 95
8 65 96 92
9 70 85 97
10 70 90 96
11 65 90 94
R NPO
Ph
Ph R NH
PO
Ph
Ph
H
Entry R Yield %
ee %
toluene chlorobenzene
O
N
O
O2N
Cl
Me
Br
NOH
H
PhPh
(1 equiv)
60
dry chlorobenzene or toluenert, 18h
Table 5.4 The addition of diethylzinc to different imines
From the results depicted in Table 5.4 it can be observed that some of the products
were obtained in considerably better enantiomeric excess when the reactions were performed
in chlorobenzene rather than toluene.
5.3 Reaction mechanism
The mechanism of the reaction was investigated in order to further improve the ligand
system, which in fact was achieved. As suggested by the theoretical calculations, ligand 60,
which gave the addition product in 97% ee, allowed an improvement on the previous result
obtained with ligand 43, 91% ee. This shows that secondary alcohols with the appropriate
stereochemistry are a way of improving the selectivity in the title transformation.
The addition of dialkylzinc reagents to aldehydes had recently been investigated by
quantum chemical methods53 and the transition state for this reaction was used as a starting
53 Yamakawa, M.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 6327.

40
point for the evaluation of five types of transition states for the corresponding addition to
imines, Figure 5.1.
NZn1
Et
PO(Ph)2
Et
Et
PhR
NZn1
Et
PO(Ph)2
EtEt
PhR
N Zn1
EtP
EtEt
PhR
O
PhPh
A
B D
NZn1
Et
N
Et
Et
OZn2
PhRC
P
O
PhPh
N Zn1
Et
OP
EtEt
OZn2R
N
Ph
Ph
E Ph
H
N
O
Zn2
NO
Zn2
NO
Zn2
Figure 5.1 The different transition states
According to the calculation results, transition state A can be excluded, since it is too
high in energy. Structures B and C optimised to structures A and D respectively. We had
previously invoked transition state D as a possible path for the reaction, but a fifth possibility
proved to be even more reliable. It is now believed that structure E is the actual transition
state for the reaction. The relative energies of the different transition states are listed in Table
5.5.
Entry TS type Product enantiomer Config. at Zn1 eq / axa HF/3-21GB3PW91/b
//HF/3-21G
123456789
ADDDEEEEE
eqeqaxeqeq
38.412.721.218.00.02.27.71.12.5
22.412.216.515.20.02.79.51.82.6
RRSRRSRRS
SSSRSSRRR
B3PW91/b
//B3PW91/c
11.7
0.0
1.4
aThis refers to the orientation of the aryl substituent of the imine in the TS of type E; bThe basis set used was 6-311+G* for Zn and 6-31G* for P, C,
N, O and H; c The basis set used was 6-311+G for Zn and 6-31G for P, C, N, O and H
Table 5.5 Transition states relative energies (in kcal/mol)
To rationalise the enantioselectivity for the addition reaction, sixteen different
transition states of type E were initially considered. Co-ordination of the nitrogen to Zn1
requires an R configuration at this position; this is due to the steric requirements of the ligand
in the (S)-Zn1 transition state, Figure 5.1. This places the –OH group and the co-ordinated

41
zinc center in a syn arrangement, thus, lowering the number of energetically viable transition
states to eight. The number of transition states was then further reduced by the
equatorial/axial selectivity concerning the orientation of the aryl substituent of the imine. The
equatorial configurations proved to be energetically favoured and the number of transition
states was now reduced to four. The lower transition states for the addition reaction are
depicted in Figure 5.2, these structures are the ones corresponding to entries 5 and 8 in Table
5.5.
1.981 2.191 2.374
1.370
2.1902.036
2.0272.212
1.984
Zn
P
O
N
C
1.985
2.033
2.018
2.1981.368
2.399
2.204
1.986
Figure 5.2 The two lowest transition states
The face selectivity of the imine then determines the enantioselectivity of the reaction,
and both calculations and experimental results pointed towards a preferential formation of the
S product. The difference between the lowest S and lowest R transition states arises from the
orientation of the four-member ring, Zn2-C-C-N, where an exo orientation is favoured, Figure
5.2. This last parameter was evaluated for ligands 43 and 60, showing that the latter should
lead to an improved enantioselectivity for the reaction, as seen by the energy differences in
Table 5.6. These theoretical results were confirmed experimentally and as mentioned in
Section 5.2 the level of enantioselectivity could be raised by up to 98%.
43604360
ChiralLigand
Productenantiomer
SSRR
B3PW91/a
//HF/3-21G
Out-of-plane angle
(Zn1-O-Zn2-(α-C))
Dihedral angle
∆(C-Zn2-N-C)
0.00.01.82.8
140°168°
-153°-155°
8°4°
-9°-12°
The comparison refers to the lowest S and the lowest R TS in table 5.5; aThe basis set used was 6-311G* for Zn and 6-31G*
for P, C, N, O and H
Table 5.6 Ligand substituent effects on the energies and selected geometrical parameters

42
Acknowledgements
I wish to express my gratitude to all the people of the department of Organic
Chemistry at Kemicum, especially Prof. Pher G. Andersson for accepting me as a PhD
student in his group, Assoc. Prof. Adolf Gogoll for all help with the different NMR
experiments, and the technical staff Lief, Tomas, Gunnar, Wik, and Eva for all the help in
solving the simple problems.
Thanks to all those with whom I had the pleasure to work with and learn from: Dr.
David Guijarro, Dr. Diego A. Alonso, Dr. Oliver Temme for all the nights out in Uppsala and
Münster, Dr. Peter Brandt, Dr. Klaus Lawonn, Mr. Christian Hedberg for the enthusiastic
discussions and some football games, and Ph. Lic. Peter Roth. Also acknowledged are all the
remaining past and present members of the PGA group with whom I did not had the pleasure
to work with or had less productive co-operation.
A very special thanks to all my other chemistry friends: Mr. Magnus Engqvist, ”That
which does not kill you makes you stronger”, Ms. Jenny Ekegren for the good fights in the
lab, Ph. Lic. Magnus Besev for your interest in chemistry, music and everything else, ”Ein
Stuhl in der Hoelle”, and Mr. Stefan Modin for all the jokes and help with artificial
intelligence?!
Thanks to all the ones that read the first raw versions of this thesis and made
constructive or destructive critics: Dr. Angelika Magnus, Mr. Christian Hedberg, Dr. Henrik
Ottosson, and Mr. Stefan Modin. Also acknowledge is Mr. Niclas Sandström for reading the
final version, for the good times in the office during my last months, and for helping me
playing with some calculations.
Thanks to my parents, sister, and family for all support in the past and care at present.

43
True and deep thanks to all my friends for making life worth living: In Portugal,
Victor Belo for visiting me in Sweden and for the good times with the rest of the Freaks –
keep going your own way!, Fred for the unforgettable good times in Porto’s night life – take
care and keep riding H.D., Carlos for all late night philosophic discussions, Rui another one
of the Freaks for all music, Júlio which was never a Freak for all good times and music,
Nando for the late film evenings, and all the ones I might have forgotten, but which will
always be remembered; In Sweden, Magnus Engqvist for the beers, parties, coffees, …, but
above everything for being who you are, Anna “space flower” Håkansson for all the good
times we spent and all dancing, Janne and Jordi, António Castillejo and Marta Castellote (the
spanish party friends), Arnaud Gayet, and Zina for sharing the same music taste. Without you
all life would have been impossible to enjoy! Sorry to the ones that I have not mentioned, you
are by no means less important to me.
Last, but most definitely not least a very special thanks to the person without whom I
would have never been able to do what I did, thank you Ida.















![Phosphine-Catalyzed Annulations of Azomethine Imines ...wag.caltech.edu/publications/sup/pdf/957.pdf · Pd-catalyzed [4 + 3] cycloadditions of azomethine imines with ... Chiral Phosphine-Catalyzed](https://static.fdocuments.net/doc/165x107/5af00cae7f8b9ad0618d9920/phosphine-catalyzed-annulations-of-azomethine-imines-wag-4-3-cycloadditions.jpg)


