
Development and application of a PCR diagnostic assay for the accurate and rapid identification of...
5
Development and application of a PCR diagnostic assay for the accurate and rapid identification of the nematophagous fungus Duddingtonia flagrans Paula KELLY a,b , Barbara GOOD a, *, Richard FITZPATRICK a , J. P. HANRAHAN a , Theo D. T. DE WAAL b a Teagasc, Animal Production Research Centre, Mellows Campus, Athenry, Co. Galway, Ireland b University College Dublin, School of Agriculture Food Science and Veterinary Medicine, Belfield, Dublin 4, Ireland article info Article history: Received 29 June 2007 Received in revised form 13 December 2007 Accepted 14 February 2008 Corresponding Editor: Judith K. Pell Keywords: Biocontrol Nematodes Nematogenous fungi Soil fungi Species-specific primers abstract The nematophagous fungus, Duddingtonia flagrans is a potential biocontrol agent against nematode parasites of ruminants. Improved methods for the rapid and accurate detection of D. flagrans would aid the evaluation of this fungus as a biocontrol agent and its suitability for environmental release. To date, detection and identification of D. flagrans is reliant on morphological methods, which can be laborious, time-consuming, and error prone. In this study, a PCR assay using species-specific primers located in the ITS regions was developed for the rapid and accurate identification of D. flagrans. The PCR assay was specific to five different isolates of D. flagrans and was capable of detecting a minimum concentration of 100 chlamydospores per gram of soil. In contrast to cultured-based detection and iden- tification methods, this assay is amenable to high throughput screening of environmental samples. The assay detected D. flagrans in faecal, leaf litter, and soil samples collected from 80 % of the Irish farms tested indicating that the fungus is abundant in Ireland. ª 2008 The British Mycological Society. Published by Elsevier Ltd. All rights reserved. Introduction Parasitic gastrointestinal nematodes impose a significant pro- duction penalty in pasture-based livestock systems world- wide. Since the 1960s, parasite control has been enhanced with the availability of broad-spectrum anthelmintics. How- ever, anthelmintic resistance has now emerged as a worldwide phenomenon, and therefore, there is a need to develop alter- native strategies to control parasitic nematodes. The use of nematophagous fungi is now emerging as an alternative nem- atode control method. In particular, Duddingtonia flagrans (Ascomycota: Ascomycetes: Orbiliaceae) has shown promising results in controlling the main ruminant endoparasites (Chandrawathani et al. 2004; Fontenot et al. 2003; Larsen et al. 1998; Terrill et al. 2004; Waller et al. 2004). D. flagrans traps and digests the nematode larvae using three-dimensional ad- hesive nets. Several studies have shown the beneficial effects of feeding chlamydospores of D. flagrans to animals on the subsequent level of nematode challenge on pasture and ulti- mately on host worm burden (Faedo et al. 1998; Larsen 1999; Mendoza de Gives et al. 1998; Waller et al. 1994). It was first de- scribed by Duddington (1950) as Trichothecium flagrans and was later renamed as D. flagrans (Cooke 1969). If D. flagrans is to be used as a biocontrol agent in a country, it is important to establish that it is indigenous to that country before being released into the environment. D. flagrans has * Corresponding author. Tel.: þ353 91 845 848; fax: þ353 91 845 847. E-mail address: [email protected] journal homepage: www.elsevier.com/locate/mycres mycological research 112 (2008) 1026–1030 0953-7562/$ – see front matter ª 2008 The British Mycological Society. Published by Elsevier Ltd. All rights reserved. doi:10.1016/j.mycres.2008.02.005
-
Upload
paula-kelly -
Category
Documents
-
view
212 -
download
1
Transcript of Development and application of a PCR diagnostic assay for the accurate and rapid identification of...

m y c o l o g i c a l r e s e a r c h 1 1 2 ( 2 0 0 8 ) 1 0 2 6 – 1 0 3 0
j ourna l homepage : www.e lsev ier . com/ loca te /mycres
Development and application of a PCR diagnostic assayfor the accurate and rapid identification of thenematophagous fungus Duddingtonia flagrans
Paula KELLYa,b, Barbara GOODa,*, Richard FITZPATRICKa, J. P. HANRAHANa,Theo D. T. DE WAALb
aTeagasc, Animal Production Research Centre, Mellows Campus, Athenry, Co. Galway, IrelandbUniversity College Dublin, School of Agriculture Food Science and Veterinary Medicine, Belfield, Dublin 4, Ireland
a r t i c l e i n f o
Article history:
Received 29 June 2007
Received in revised form
13 December 2007
Accepted 14 February 2008
Corresponding Editor: Judith K. Pell
Keywords:
Biocontrol
Nematodes
Nematogenous fungi
Soil fungi
Species-specific primers
* Corresponding author. Tel.: þ353 91 845 8E-mail address: [email protected]
0953-7562/$ – see front matter ª 2008 The Bdoi:10.1016/j.mycres.2008.02.005
a b s t r a c t
The nematophagous fungus, Duddingtonia flagrans is a potential biocontrol agent against
nematode parasites of ruminants. Improved methods for the rapid and accurate detection
of D. flagrans would aid the evaluation of this fungus as a biocontrol agent and its suitability
for environmental release. To date, detection and identification of D. flagrans is reliant on
morphological methods, which can be laborious, time-consuming, and error prone. In this
study, a PCR assay using species-specific primers located in the ITS regions was developed
for the rapid and accurate identification of D. flagrans. The PCR assay was specific to five
different isolates of D. flagrans and was capable of detecting a minimum concentration
of 100 chlamydospores per gram of soil. In contrast to cultured-based detection and iden-
tification methods, this assay is amenable to high throughput screening of environmental
samples. The assay detected D. flagrans in faecal, leaf litter, and soil samples collected from
80 % of the Irish farms tested indicating that the fungus is abundant in Ireland.
ª 2008 The British Mycological Society. Published by Elsevier Ltd. All rights reserved.
Introduction (Chandrawathani et al. 2004; Fontenot et al. 2003; Larsen et al.
Parasitic gastrointestinal nematodes impose a significant pro-
duction penalty in pasture-based livestock systems world-
wide. Since the 1960s, parasite control has been enhanced
with the availability of broad-spectrum anthelmintics. How-
ever, anthelmintic resistance has now emerged as a worldwide
phenomenon, and therefore, there is a need to develop alter-
native strategies to control parasitic nematodes. The use of
nematophagous fungi is now emerging as an alternative nem-
atode control method. In particular, Duddingtonia flagrans
(Ascomycota: Ascomycetes: Orbiliaceae) has shown promising
results in controlling the main ruminant endoparasites
48; fax: þ353 91 845 847.
ritish Mycological Society
1998; Terrill et al. 2004; Waller et al. 2004). D. flagrans traps
and digests the nematode larvae using three-dimensional ad-
hesive nets. Several studies have shown the beneficial effects
of feeding chlamydospores of D. flagrans to animals on the
subsequent level of nematode challenge on pasture and ulti-
mately on host worm burden (Faedo et al. 1998; Larsen 1999;
Mendoza de Gives et al. 1998; Waller et al. 1994). It was first de-
scribed by Duddington (1950) as Trichothecium flagrans and was
later renamed as D. flagrans (Cooke 1969).
If D. flagrans is to be used as a biocontrol agent in a country,
it is important to establish that it is indigenous to that country
before being released into the environment. D. flagrans has
. Published by Elsevier Ltd. All rights reserved.

Table 1 – Fungal species, source material, country oforigin and Centraalbureau voor Schimmelcultures (CBS)accession number
Species Source material Countryof origin
CBSref. no.
Arthrobotrys oligospora – USA 111.37
Drechmeria coniospora Nematodes Sweden 615.82
Duddingtonia flagrans Solidago canadensis,
dead stem
USA 343.94
D. flagrans Leaf litter UK 143.83
D. flagrans Solidago canadensis,
dead stem
Germany 342.94
D. flagrans – – 561.92
D. flagransa Greenhouse soil,
Camellia house
Germany 583.91
Fusarium coeruleum Nematode egg Germany 618.76
F. incarnatum Medicago sativa, seed Denmark 621.87
F. oxysporum Cucumis melo
(Cucurbitaceae)
Israel 420.90
Haptocillium balanoides – – 250.82
H. sphaerosporum Soil in potato field Canada 589.92
Harposporium helicoides Nematodes Canada 944.70
H. lilliputanum Nematode in soil Japan 111353
Paecilomyces lilacinus Meloidogyne, egg
mass
Philippines 431.87
Pochonia chlamydosporia
var. cantenulata
Heterodera
avenae, egg
Sweden 250.83A
Pyricularia higginsii Cyperus rotundus Israel 665.79
Pochonia chlamydosporia
(syn. Verticillium
chlamydosporium)
Egg of slug (Mollusca) Brazil 101244
V. coccospora Lymantria dispar, egg USA 691.86
V. sphaerosporum Ditylenchus triformis
on compost
Germany 522.80
– No information available.
a Isolate used in testing the sensitivity of the PCR assay.
Development and application of a PCR diagnostic assay 1027
been detected in Denmark (Larsen et al. 1992), France (Chartier
& Pors 2003), Germany (CBS 342.94, isolated by A. Rubner), UK
(CBS 143.83, isolated by R. C. Cooke), South Africa (Durand et al.
2005), Australia (Larsen et al. 1994), USA (CBS 343.94, isolated
by A. Rubner), Mexico (Mendoza de Gives et al. 1998), Malaysia
(Chandrawathani et al. 2002), New Zealand (Skipp et al. 2002),
and Iran (Ghahfarokhi et al. 2004). However, it has not been
detected in Ireland (Gray 1983). In addition, for regulatory pur-
poses, it is important to have methods that permit the reliable
and sensitive monitoring of biocontrol agents after release
into the environment (Avis et al. 2001).
Routine detection and identification of nematophagous
fungi typically involves nematode baiting, repeated incuba-
tions, and re-isolations in order to obtain a pure culture, fol-
lowed by microscopic examination of hyphae and spores.
Discrimination between closely related species requires addi-
tional detailed microscopic examination of particular phases
of the life cycle. Morphological identification is, therefore, la-
borious, can take several weeks, requires specialised training
in mycology, and is prone to contamination and error, which
delays definitive identification. Therefore, a simple and reli-
able method for the detection and identification of D. flagrans
would be highly desirable in the evaluation of this fungus as
a potential biocontrol agent.
Molecular approaches are increasingly being used to de-
velop PCR-based species-specific diagnostic assays for a broad
range of organisms (Atkins & Clark 2004). Molecular tools have
been employed to identify and monitor a number of fungi,
namely, Pochonia chlamydosporia var. catenulata (Atkins et al.
2003a), Plectophaerella cucumerina (Atkins et al. 2003b), Paecilo-
myces lilacinus (Atkins et al. 2005), Pochonia chlamydosporia
(syn. Verticillium chlamydosporium) (Hirsch et al. 2000), and Fusa-
rium sp. (Abd-Elsalam et al. 2003). These molecular approaches
are rapid, sensitive, easy to interpret, and provide a useful
alternative to culture-based detection and identification
methods. The ITS regions are commonly used as target re-
gions to design species-specific primers for PCR amplification.
The ITS regions, located between the 18S and 28S ribosomal
sequences, are hypervariable DNA regions, which make
them ideal target sequences for the generation of species-
specific primers. There are also multiple copies of the ITS re-
gion per genome, which increases the template number for
the PCR assay, thereby improving assay sensitivity of the diag-
nostic assay.
This study describes the development and application of
a PCR diagnostic assay for the rapid detection and identifica-
tion of D. flagrans in the environment.
Materials and methods
Fungal cultures
The 15 fungal species and five Duddingtonia flagrans isolates
used in these study were obtained from the Centraalbureau
voor Schimmelcultures (CBS), Utrecht, The Netherlands and
are listed in Table 1. All fungi were grown on oatmeal agar
with the exception of Cantenaria anguillulae and Hirsutella rhois-
siliensis, which were grown on archimycetes and malt extract
agar, respectively (CBS 2007: www.cbs.knaw.nl).
Field samples
To determine whether Duddingtonia flagrans was present in
Ireland, faecal (fresh) (n¼ 5), leaf litter (n¼ 2), and soil
(n¼ 5) samples were collected at random from permanent
sheep pastures on each of ten Irish farms, between 5–6
September, 2006. The criterion for inclusion of the farm in
the survey was that it was a well-established sheep enter-
prise. The three habitats were chosen based on previous ev-
idence, which reported the detection of D. flagrans in these
habitats, with the greatest occurrence in fresh faecal sam-
ples (Chandrawathani et al. 2002; Larsen et al. 1994; Skipp
et al. 2002) followed by decaying vegetation or leaf-litter
(Durand et al. 2005; Skipp et al. 2002). Samples were kept
in air-tight plastic bags at 4 �C before processing. Samples
collected from each farm were pooled and mixed by type
prior to processing.
DNA extraction from fungi
Genomic DNA was isolated from pure cultures using mechan-
ical disruption and enzymatic digestion. Briefly, approximately
500 mg frozen (liquid nitrogen) fungal mycelium was

1028 P. Kelly et al.
homogenized (Heidolph Diax 900, Sigma, UK) for 30 s in 3 ml
phosphate buffer saline (PBS, Sigma, UK, pH 7.0) containing
3 mg of lysing enzyme from Trichoderma harzianum (Sigma,
UK) and incubated at 37 �C for 3 h. This was followed by 1 h in-
cubation at 65 �C in 3 ml extraction buffer [50 mM Tris–HCl (pH
8), 100 mM EDTA, 100 mM NaCl2, 1 % sodium dodecyl sulphate
(SDS)] containing 100 mg proteinase K (Sigma, UK). RNA was
digested by the addition of 100 mg RNase (QIAGEN, UK) and
the sample incubated for a further 30 min at 37 �C. Genomic
DNA was then extracted with an equal volume of phenol and
chloroform and precipitated using 0.6 volumes of isopropanol.
The precipitated DNA was washed with 70 % ethanol, resus-
pended in 50 ml of nuclease-free water and stored at �20 �C.
The quantity and quality of the isolated DNA was evaluated
by absorbance at 260 and 280 nm.
DNA extraction from field samples
Total DNA was extracted from 5 g samples using the Ultra-
Clean� Mega Soil Kit (Mo Bio Laboratories, CA , USA) according
to manufacturer’s instructions. In order to remove humic acid
and other PCR inhibitors, the recovered DNA was then cleaned
using the PowerClean� DNA Clean-up Kit (Mo Bio Laborato-
ries, CA, USA) and subjected to PCR analysis as described
below. The identity of the PCR product was confirmed by
sequencing (Macrogen) Universal fungal primers, as described
by Turenne et al. 1999, were used as a positive control.
Design of selective primers and optimizationof PCR conditions
The previously published Duddingtonia flagrans ITS1–5.8S–ITS2
DNA sequence (GenBank accession no. U51961) was aligned
using ClustalW (EMBL-EBI) with homologous DNA sequences
from five closely related species, namely, Arthrobotrys
oligospora (accession no. AY773462), Pyricularia higginsii
(accession no. AB274438), Fusarium incarnatum (accession no.
EF158029), Pochonia chlamydosporia (accession no. AM412780),
and Paecilomyces lilacinus (accession no. AY213668). From this
alignment, D. flagrans species specific primers were designed;
forward primer: DFITS1F (50-CTT CGT CCG GTC TTG ACC G-30)
and reverse primer: DFITS2R (50-TTC GGG TTC AAA ACC AGC
GTT-30). The optimum PCR reaction consisted of 100 ng geno-
mic DNA, 1� PCR buffer (Promega, WI), 0.2 mM dNTPs, 4 mM
MgCl2, 100 ng of each primer and 2.5 U of Taq DNA polymerase
(Promega, WI) in a final reaction volume of 50 ml. Optimised
PCR amplification conditions consisted of 34 cycles of dena-
turation at 95 �C for 30 s, annealing at 66.6 �C for 30 s, and ex-
tension at 72 �C for 30 s. PCR products were separated on 1.5 %
ethidium bromide-stained agarose gels and visualised using
a Molecular Imager FX (Bio Rad Laboratories, CA, USA). The
identity of the PCR product was confirmed by sequencing
(Macrogen). The primers and PCR reaction conditions used
for universal fungal amplification were as previously de-
scribed (Turenne et al. 1999).
PCR assay specificity
The specificity of the Duddingtonia flagrans PCR assay using
primers DFITS1 and DFITS2 was tested on 15 different fungal
species and five D. flagrans isolates with different geographical
origins (Table 1).
PCR assay sensitivity
The sensitivity of the PCR assay was determined in two ways.
In the first approach, a 5 g soil sample taken from a recrea-
tional site (lawn) was spiked with 106 Duddingtonia flagrans
chlamydospores and subjected to DNA extraction as described
above (Table 1). The extracted DNA was diluted to provide the
equivalent of 120, 50, ten, five, and one chlamydospores and
then subjected to PCR analysis. In the second approach, DNA
was extracted from 5 g soil samples that had been spiked
with either 5� 105, 5� 104, 5� 103, 5� 102, 50 or five chla-
mydospores. Control samples consisted of soil from the
same source with no chlamydospores added. The extracted
DNA was subjected to PCR and the presence or absence of
the D. flagrans specific 299 bp PCR product determined by aga-
rose gel electrophoresis.
Statistical analysis
Fisher’s exact test (SAS, 2000) was used to compare the fre-
quency of Duddingtonia flagrans between the three habitats
examined.
Results
PCR assay specificity
The primers, DFITS1 and DFITS2, were highly specific when
tested against 15 different fungal species (Fig 1A). A single
PCR product of the expected size (299 bp) was exclusively am-
plified from Duddingtonia flagrans demonstrating the specific-
ity of the PCR assay. The control PCR reaction using
universal fungal primers was positive for all fungal isolates
(Fig 1B). The PCR assay was positive when tested on five differ-
ent D. flagrans isolates (data not shown).
PCR assay sensitivity
The first approach which examined a serial dilution of DNA
extracted from a specific number of chlamydospores demon-
strated that Duddingtonia flagrans could be detected in a soil
sample containing ten chlamydospores (data not shown).
The second approach using DNA from a range of spiked
samples demonstrated that a minimum concentration of 100
chlamydospores per gram of soil was necessary for the reli-
able amplification and detection of D. flagrans (Fig 2). No PCR
product was detected in the negative control (no chlamydo-
spores added).
PCR detection in environmental samples
The PCR assay detected Duddingtonia flagrans in material from
eight out of the ten Irish farms surveyed. The fungus was
found in faecal, leaf litter, and soil samples, from seven, five,
and three farms respectively (Table 2). There was no
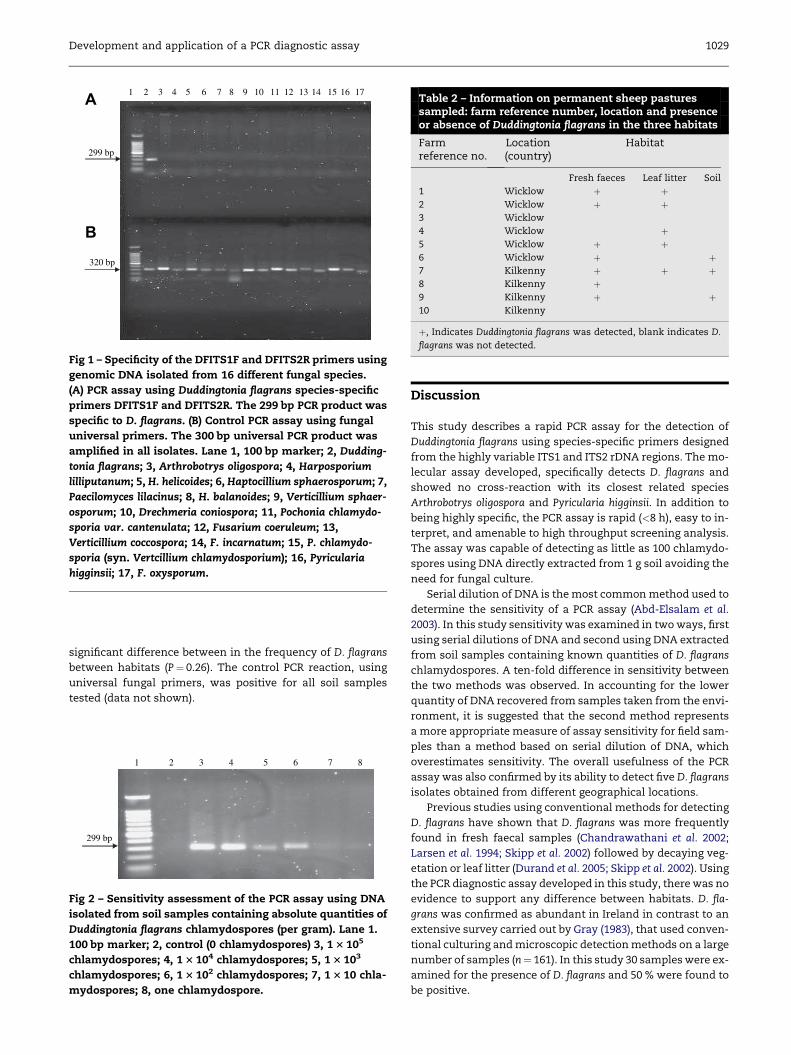
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17A
B
299 bp
320 bp
Fig 1 – Specificity of the DFITS1F and DFITS2R primers using
genomic DNA isolated from 16 different fungal species.
(A) PCR assay using Duddingtonia flagrans species-specific
primers DFITS1F and DFITS2R. The 299 bp PCR product was
specific to D. flagrans. (B) Control PCR assay using fungal
universal primers. The 300 bp universal PCR product was
amplified in all isolates. Lane 1, 100 bp marker; 2, Dudding-
tonia flagrans; 3, Arthrobotrys oligospora; 4, Harposporium
lilliputanum; 5, H. helicoides; 6, Haptocillium sphaerosporum; 7,
Paecilomyces lilacinus; 8, H. balanoides; 9, Verticillium sphaer-
osporum; 10, Drechmeria coniospora; 11, Pochonia chlamydo-
sporia var. cantenulata; 12, Fusarium coeruleum; 13,
Verticillium coccospora; 14, F. incarnatum; 15, P. chlamydo-
sporia (syn. Vertcillium chlamydosporium); 16, Pyricularia
higginsii; 17, F. oxysporum.
Table 2 – Information on permanent sheep pasturessampled: farm reference number, location and presenceor absence of Duddingtonia flagrans in the three habitats
Farmreference no.
Location(country)
Habitat
Fresh faeces Leaf litter Soil
1 Wicklow þ þ2 Wicklow þ þ3 Wicklow
4 Wicklow þ5 Wicklow þ þ6 Wicklow þ þ7 Kilkenny þ þ þ8 Kilkenny þ9 Kilkenny þ þ10 Kilkenny
þ, Indicates Duddingtonia flagrans was detected, blank indicates D.
flagrans was not detected.
Development and application of a PCR diagnostic assay 1029
significant difference between in the frequency of D. flagrans
between habitats (P¼ 0.26). The control PCR reaction, using
universal fungal primers, was positive for all soil samples
tested (data not shown).
1 2 3 4 5 6 7 8
299 bp
Fig 2 – Sensitivity assessment of the PCR assay using DNA
isolated from soil samples containing absolute quantities of
Duddingtonia flagrans chlamydospores (per gram). Lane 1.
100 bp marker; 2, control (0 chlamydospores) 3, 1 3 105
chlamydospores; 4, 1 3 104 chlamydospores; 5, 1 3 103
chlamydospores; 6, 1 3 102 chlamydospores; 7, 1 3 10 chla-
mydospores; 8, one chlamydospore.
Discussion
This study describes a rapid PCR assay for the detection of
Duddingtonia flagrans using species-specific primers designed
from the highly variable ITS1 and ITS2 rDNA regions. The mo-
lecular assay developed, specifically detects D. flagrans and
showed no cross-reaction with its closest related species
Arthrobotrys oligospora and Pyricularia higginsii. In addition to
being highly specific, the PCR assay is rapid (<8 h), easy to in-
terpret, and amenable to high throughput screening analysis.
The assay was capable of detecting as little as 100 chlamydo-
spores using DNA directly extracted from 1 g soil avoiding the
need for fungal culture.
Serial dilution of DNA is the most common method used to
determine the sensitivity of a PCR assay (Abd-Elsalam et al.
2003). In this study sensitivity was examined in two ways, first
using serial dilutions of DNA and second using DNA extracted
from soil samples containing known quantities of D. flagrans
chlamydospores. A ten-fold difference in sensitivity between
the two methods was observed. In accounting for the lower
quantity of DNA recovered from samples taken from the envi-
ronment, it is suggested that the second method represents
a more appropriate measure of assay sensitivity for field sam-
ples than a method based on serial dilution of DNA, which
overestimates sensitivity. The overall usefulness of the PCR
assay was also confirmed by its ability to detect five D. flagrans
isolates obtained from different geographical locations.
Previous studies using conventional methods for detecting
D. flagrans have shown that D. flagrans was more frequently
found in fresh faecal samples (Chandrawathani et al. 2002;
Larsen et al. 1994; Skipp et al. 2002) followed by decaying veg-
etation or leaf litter (Durand et al. 2005; Skipp et al. 2002). Using
the PCR diagnostic assay developed in this study, there was no
evidence to support any difference between habitats. D. fla-
grans was confirmed as abundant in Ireland in contrast to an
extensive survey carried out by Gray (1983), that used conven-
tional culturing and microscopic detection methods on a large
number of samples (n¼ 161). In this study 30 samples were ex-
amined for the presence of D. flagrans and 50 % were found to
be positive.

1030 P. Kelly et al.
r e f e r e n c e s
Abd-Elsalam KA, Aly IN, Abdel-Satar MA, Khalil MS, Verreet JA, 2003.PCR identificationofFusariumgenus based onnuclearribosomal-DNA sequence data. African Journal of Biotechnology 2: 82–85.
Atkins S, Clark IM, 2004. Fungal molecular diagnostics: a minireview. Journal of Applied Genetics 45: 3–15.
Atkins S, Hidalgo-Diaz L, Clark IM, Norton CO, Montes de Oca N,Gray PA, Kerry B, 2003a. Approaches for monitoring the releaseof Pochonia chlamydosporia var. catenulata, a biocontrol agent ofroot-knot nematodes. Mycological Research 107: 206–212.
Atkins SD, Clark IM, Pande S, Hirsch PR, Kerry BR, 2005. The use ofreal-time PCR and species-specific primers for the identifica-tion and monitoring of Paecilomyces lilacinus. FEMS MicrobiologyEcology 51: 257–264.
Atkins SD, Clark IM, Sosnowska D, Hirsch PR, Kerry BR, 2003b.Detection and quantification of Plectophaerella cucumerina,a potential biological control agent of potato cyst nematodes,by using conventional PCR, real-time PCR, selective media andbaiting. Applied and Environmental Microbiology 69: 4788–4793.
Avis TJ, Hamelin RC, Belamger RR, 2001. Approaches to molecularcharacterization of fungal biocontrol agents: some casestudies. Canadian Journal of Plant Pathology 23: 8–12.
Chandrawathani P, Jamnah O, Adran M, Waller PJ, Larsen M,Gillespie AT, 2004. Field study on the biological control ofnematode parasites of sheep in the tropics, using microfungusDuddingtonia flagrans. Veterinary Parasitology 120: 177–187.
Chandrawathani P, Jamnah O, Waller PJ, Hoglund J, Larsen M,Zahari WM, 2002. Nematophagous fungi as a biological controlagent for nematode parasites of small ruminants in Malaysia:a special emphasis on Duddingtonia flagrans. VeterinaryParasitology 33: 685–696.
Chartier C, Pors I, 2003. Effects of nematophagous fungus,Duddingtonia flagrans, on larval development of goat parasiticnematodes: plot study. Veterinary Research 34: 221–230.
Cooke RC, 1969. Two nematode-trapping hyphomycetes,Duddingtonia flagrans gen. et comb. nov. and Monacrosporiummutabilis sp. nov. Transactions of the British Mycological Society53: 315–318.
Duddington CL, 1950. A new predacious species of Trichothecium.Transactions of the British Mycological Society 32: 284–287.
Durand DT, Boshoff HM, Micheal LM, Krecek RC, 2005. Survey ofnematophagous fungi in South Africa. Onderstepoort Journal ofVeterinary Research 72: 185–187.
Faedo M, Barnes EH, Dobson RJ, Waller PJ, 1998. The potential ofnematophagous to control the free-living stages of nematodeparasites of sheep: pasture plot study with Duddingtoniaflagrans. Veterinary Parasitology 76: 129–135.
Fontenot ME, Miller JE, Pena MT, Larsen M, Gillespie A, 2003.Efficiency of feeding Duddingtonia flagrans chlamydospores tograzing ewes on reducing availability of parasitic nematodeslarvae on pasture. Veterinary Parasitology 118: 203–213.
Ghahfarokhi SM, Abyaneh MR, Bahadori SR, Eslami A, Zare A,Ebrahimi M, 2004. Screening of soil and sheep samples forpredacious fungi: isolation and characterisation of the
nematode-trapping fungus Arthrobotrys oligospora. Iranian Bio-medical Journal 8: 135–142.
Gray NF, 1983. Ecology of nematophagous fungi: distribution andhabitat. Association of Applied Biologists 102: 501–509.
Hirsch PR, Mauchline TM, Mendum TA, Kerry B, 2000. Detectionof nematophagous fungus Verticillium chlamydosporium innematode-infested plant roots using PCR. Mycological Research104: 435–439.
Larsen M, 1999. Biological control of helminths. InternationalJournal of Parasitology 29: 139–146.
Larsen M, Faedo M, Waller PJ, 1994. The potential of nematoph-agous fungi to control the free-living stages of nematodeparasites of sheep: survey for the presence of fungi in freshfaeces of grazing livestock in Australia. Veterinary Parasitology53: 275–281.
Larsen M, Faedo M, Waller PJ, Hennessy DR, 1998. The potential ofnematophagous fungi to control the free-living stages ofnematode parasites of sheep: studies with Duddingtoniaflagrans. Veterinary Parasitology 76: 121–128.
Larsen M, Wolstrup J, Henriksen SA, Gronvold J, Nansen P, 1992. Invivo passage through calves of nematophagous fungi selectedfor biocontrol of parasitic nematodes. Journal of Helminthology66: 137–141.
Mendoza de Gives P, Crespo JF, Rodriguez DH, Prats VV,Hernandez EL, Fernandez GEO, 1998. Biological control ofHaemonchus contortus infective larvae in ovine faeces byadministering an oral suspension of Duddingtonia flagranschlamydospores to sheep. Journal of Helminthology 72:343–347.
SAS Institute, Inc., 2000. SAS/STAT User’s Guide, Version 8. SASInstitute, Inc., Cary, North Carolina, USA.
Skipp RA, Yeates GW, Chen LY, Glare TR, 2002. Occurrence,morphological characteristics and ribotyping of New Zealandisolates of Duddingtonia flagrans, a candidate for biocontrol ofanimal parasitic nematodes. New Zealand Journal of AgriculturalResearch 45: 187–196.
Terrill TH, Larsen M, Samples O, Husted S, Miller JE, Kaplan RM,Gelaye S, 2004. Capability of nematode-trapping fungus Dud-dingtonia flagrans to reduce infective larvae of gastrointestinalnematodes in goat faeces in the south-eastern United States:dose titration and dose time interval studies. VeterinaryParasitology 120: 285–296.
Turenne CY, Sanche SE, Hoban DJ, Karlowsky JA, Kabani AJ,1999. Rapid identification of fungi by using the ITS2genetic region and an automated fluorescent cappilaryelectrophoresis system. Journal of Clinical Microbiology 37:1846–1851.
Waller PJ, Larsen M, Faedo M, Hennessy DR, 1994. The potential ofnematopaghous fungi to control the free-living stages ofnemtode parasites of sheep: in vitro and in vivo studies.Veterinary Parasitology 51: 289–299.
Waller PJ, Schwan O, Ljungstorm B-L, Rydzik A, Yeates GW,2004. Evaluation of biological control of sheep parasitesusing Duddingtonia flagrans under commercial farmingconditions on the island of Gotland, Sweden. VeterinaryParasitology 126: 299–315.







![Basidiomycetes as Potential Biocontrol Agents against ... · entomophagous and nematophagous specialized fungi (YANG& al. [9]). These organisms have the advantage of being effective](https://static.fdocuments.net/doc/165x107/5ed69c0d843ed9152066b50e/basidiomycetes-as-potential-biocontrol-agents-against-entomophagous-and-nematophagous.jpg)










