
Department of Neural and Behavioral Sciences
154
The Pennsylvania State University The Graduate School Department of Neural and Behavioral Sciences FUNCTIONAL INTERACTIONS BETWEEN OPIOIDS AND A CANNABINOID RECEPTOR 2 AGONIST IN INFLAMMATORY PAIN. A Dissertation in Neuroscience by Matthew B. Yuill 2018 Matthew B. Yuill Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2018
Transcript of Department of Neural and Behavioral Sciences

The Pennsylvania State University
The Graduate School
Department of Neural and Behavioral Sciences
FUNCTIONAL INTERACTIONS BETWEEN OPIOIDS AND A CANNABINOID
RECEPTOR 2 AGONIST IN INFLAMMATORY PAIN.
A Dissertation in
Neuroscience by
Matthew B. Yuill
2018 Matthew B. Yuill
Submitted in Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
May 2018

The dissertation of Matthew B. Yuill was reviewed and approved* by the following:
Daniel Morgan Professor of Neuroscience Dissertation Adviser Chair of Committee
Patricia S. Grigson Professor of Neural and Behavioral Sciences
Robert Levenson Distinguished Professor of Pharmacology
John Ellis Professor of Psychiatry and Pharmacology
Jossee Guindon Assistant Professor of Pharmacology Neuroscience Texas Tech University Health Sciences Center Special Member Colin J. Barnstable Chair of Department of Neural and Behavioral Sciences
*Signatures are on file in the Graduate School

iii
ABSTRACT
The goal of this study was to test the hypothesis that the Cannabinoid 2
Receptor (CB2R) functionally interacts with the opioid system to modulate
inflammatory pain. Additionally, we tested mechanisms mediating tolerance to
multiple opioids, and to the prototypical cannabinoid Δ9-THC. CB2R agonists
produce low levels of side effects and no tolerance relative to other opioid and
cannabinoid agonists, making them an attractive pharmacotherapeutic target.
This study assessed the anti-nociceptive effects of a selective CB2R agonist
(JWH-133) in pathological pain using mice subjected to inflammatory pain using
the formalin test. Furthermore, we examined several ways in which JWH-133
may interact with the activity of opioids in this model.
JWH-133 produces dose-dependent anti-nociception during both the
acute pain and inflammatory pain phases of the formalin test. This was observed
in both male and female mice. However, a maximally efficacious dose of JWH-
133 (1 mg/kg) was not associated with somatic withdrawal symptoms, motor
impairment, or hypothermia. The efficacy of JWH-133 was blocked by application
of a CB2R selective antagonist (SR144528).
After eleven once-daily injections of 1 mg/kg JWH-133, no tolerance was
observed in the formalin test. Conversely, wild-type mice become tolerant to Δ9-
THC, morphine, and fentanyl within eleven days. Cross-tolerance for the anti-
nociceptive effects of JWH-133 and morphine were assessed to gain insight into
physiologically relevant CB2R and Mu opioid receptor (MOR) interaction. Mice

iv
made tolerant to the effects of morphine exhibited a lower JWH-133 response in
both phases of the formalin test compared to vehicle treated morphine-naïve
animals. However, repeated daily JWH-133 administration did not cause cross-
tolerance for morphine. Similar results were found for cross-tolerance between
JWH-133 and fentanyl, suggesting opioid and CB2R cross-tolerance is
unidirectional in this model. However, preliminary data suggests co-
administration of JWH-133 with morphine modestly attenuates morphine
tolerance in the formalin model. Furthermore, isobolographic analysis revealed
that co-administration of a fixed-ratio combination of JWH-133 and morphine has
an additive effect on anti-nociception in the formalin test. Overall these findings
show that CB2R may functionally interact with MOR to modulate anti-nociception
and tolerance in inflammatory pain, which suggests clinical utility.

v
TABLE OF CONTENTS
List of Figures .......................................................................................................... vii
List of Tables ........................................................................................................... viii
List of Common Abbreviations ................................................................................. ix
Chapter 1 - Introduction and literature review................................................................... 1
Pain .................................................................................................................................. 1 Anatomical pain circuitry....................................................................................... 1 Pathological pain ................................................................................................... 3
The endogenous opioid system .................................................................................. 4 Overview ................................................................................................................. 4 Mu Opioid Receptor and pain .............................................................................. 5 Clinical use of opioid drugs .................................................................................. 6
The endogenous cannabinoid system ....................................................................... 7 Overview ................................................................................................................. 7 Cannabinoid Receptor 1 signaling and localization ......................................... 9 Cannabinoid Receptor 2 signaling and localization ......................................... 10 Cannabinoids in pain............................................................................................. 12 Cannabinoid Receptor 2 in pain .......................................................................... 14 Clinical use of Cannabinoids ............................................................................... 16 Interactions between the cannabinoid system and opioid system ................. 18 Cannabinoid Receptor 2 and opioid interactions. ............................................. 20
Chapter 2 - General methods .............................................................................................. 22
Subjects .......................................................................................................................... 22 Drugs ............................................................................................................................... 22 Procedures ..................................................................................................................... 23
Tail-flick and hotplate anti-nociception ............................................................... 23 Formalin test ........................................................................................................... 24
Physical side-effects ..................................................................................................... 25 Body temperature .................................................................................................. 25 Rotarod test ............................................................................................................ 26 Precipitated Withdrawal ........................................................................................ 26
Data analysis .................................................................................................................. 27 Isobolographic analysis ........................................................................................ 28
Chapter 3 ................................................................................................................................ 30
Tolerance to the anti-nociceptive and hypothermic effects of morphine are mediated by multiple isoforms of c-Jun N-terminal Kinase ............................. 30
Chapter 4 ................................................................................................................................ 46

vi
Anti-nociceptive interactions between opioids and a cannabinoid receptor 2 agonist in inflammatory pain. ............................................................................... 46
Unpublished Experiment 1: Efficacy of JWH-133 in hotplate and tail-flick assays ..................................................................................................................... 88 Rationale ................................................................................................................. 88 Procedure ............................................................................................................... 89 Results .................................................................................................................... 89 Discussion .............................................................................................................. 91
Unpublished Experiment 2: JWH-133 and fentanyl cross-tolerance. .................... 92 Rationale ................................................................................................................. 92 Procedure ............................................................................................................... 92 Results .................................................................................................................... 93 Discussion .............................................................................................................. 95
Chapter 5 – Mechanisms of Cannabinoid Tolerance through the CB1 Receptor ....... 98
Results .................................................................................................................... 99
Chapter 6 - General Discussion and Conclusion ............................................................. 102
Future directions .................................................................................................... 109 Conclusion .............................................................................................................. 110
Appendix ................................................................................................................................. 112
Supplementary Data and Figures ............................................................................... 112
Works Cited ............................................................................................................................ 114

vii
LIST OF FIGURES
Figure 1. Cannabinoid Pain Circuitry. ................................................................................ 14
Figure 2. Tolerance to morphine in the tail-flick test. ....................................................... 39
Figure 3. Tolerance to morphine in the hotplate test. ...................................................... 40
Figure 4. Tolerance to morphine-induced hypothermia. ................................................. 42
Figure 5. Anti-nociceptive efficacy of JWH-133. ............................................................... 61
Figure 6. Comparison of morphine and JWH-133 in the formalin test. ......................... 62
Figure 7. JWH-133 acts through the CB2 Receptor. ....................................................... 63
Figure 8. Lack of JWH-113 adverse effects. ..................................................................... 68
Figure 9. Lack of observed tolerance to JWH-133. ......................................................... 70
Figure 10. Cross-tolerance between JWH-133 and morphine. ...................................... 73
Figure 11. JWH-133 co-administration modestly protects against morphine tolerance. ........................................................................................................................ 76
Figure 12. JNK signaling is partially responsible for morphine-induced cross-tolerance to JWH-133 ................................................................................................... 78
Figure 13. Non-linear isobolographic analysis of 1:10 fixed ratio JWH-133 and morphine in the formalin test. ...................................................................................... 80
Figure 14. JWH-133 dose responses in acute thermal pain .......................................... 90
Figure 15. JWH-133 and fentanyl cross tolerance. .......................................................... 94
Figure 16. Tolerance to the anti-nociceptive effects of fentanyl is not blocked by SP6. ................................................................................................................................. 97
Figure 17. Δ9-THC tolerance in the formalin test. ............................................................ 100
Figure 18. Δ9-THC and SP6 in the formalin test. .............................................................. 101
Figure 19. Morphine with a fixed-dose of JWH-133 in the formalin test. ...................... 112

viii
LIST OF TABLES
Table 1. Opioid Receptors ................................................................................................... 5
Table 2. CB2R-agonist induced anti-nociception in various pain models. ................... 16
Table 3. ED50 Values in Formalin. ...................................................................................... 113
Table 4. Paw edema following formalin. ............................................................................ 113

ix
List of Common Abbreviations
GPCR G-protein coupled receptor
2-AG 2-arachidonylglycerol
CB1R Cannabinoid Receptor 1
CB2R Cannabinoid Receptor 2
CNS central nervous system
eCB endocannabinoid
GRK G protein-coupled receptor kinase
JNK c-Jun N-terminal kinase
MAPK mitogen-activated protein kinase
MOR mu-opioid receptor
KOR Kappa opioid receptor
DOR Delta opioid receptor
PAG Periaqueductal gray
VTA Ventral tegmental area
CPP Conditioned Place Preference
SR2 SR-144,528
SP6 SP600125
i.p. intraperitoneal
%MPE Percent of Maximum possible effect
AUC Area under the curve
SEM Standard error of the mean

x
CNS Central Nervous System
RVM rostral ventral medulla
NGF Nerve Growth Factor
cAMP cyclic adenosine monophosphate
AEA N-arachidonoylethanolamide
2-AG 2-arachidonylglycerol
MAPK mitogen-activated protein kinase
PKA protein kinase A
PEA palmitoylethanolamide
GRK G-protein coupled receptor kinase
JNK c-Jun N-terminal Kinase
CPS Composite Pain Score

1
Chapter 1 - Introduction and literature review
Pain
Pain can be broadly characterized as an unpleasant sensation resulting
from intense or damaging stimuli [Basbaum 2009]. The propagation of pain is
initiated with the activation of physiological receptors, called nociceptors.
Normally, this painful sensation results from specific activation of the nociceptors
by mechanical, thermal, or chemical stimulus and is short-lived. However,
chronic or persistent pain in the absence of injury is a serious clinical challenge.
Pathologically, the modality of pain encompasses more than a simple physical
sensation. Pain can better be defined as a combination of sensory, cognitive, and
emotional aspects associated with real or potential injuries. Knowing that pain
represents a complex sensory modality accompanied by affective, motivational
and cognitive aspects highlights some of the challenges related to its treatment.
Anatomical pain circuitry
Pain is initiated primarily by stimulation of nociceptors, specialized sensory
receptors widely distributed throughout the periphery. There are multiple classes
of nociceptors which detect specific stimulus modalities, such as thermal,
mechanical, and chemical stimuli. Activated nociceptors then signal through
peripheral afferent fibers which terminate in the spinal cord. These first-order

2
afferent fibers are categorized according to structure, diameter, and conduction
velocity. C-type fibers are unmyelinated, under 2 μm in diameter and have a
conduction velocity of 0.5–2.0 m/s [Beasou 1969; Almeida 2004]. Aσ fibers are
lightly myelinated, are 2-6 μm and have a conduction velocity of 12–30
m/s[Burgess 1967]. The myelinated Aβ fibers have a diameter of more than 10
μm and a velocity of 30–100 m/s [Perl 1968].
After information from nociceptors reaches the dorsal horn of the spinal
cord, pain is signaled by the release of glutamate from primary afferent fibers,
generating excitatory post-synaptic currents (EPSCs) to second order dorsal
horn neurons. Second order neurons transmit signals among multiple ascending
pathways to different areas of the central nervous system (CNS) depending on
the type of information [Wall 1967; Fields 2004]. The spinothalamic tract
transmits information to the somatosensory cortex via the thalamus, providing
information about the location and intensity of the painful stimulus [Halliday
1972]. Other projection neurons along the spinomesencephalic tract engage the
cingulate and insular cortices via the parabrachial nucleus and amygdala,
contributing to the affective component of pain [Bernard 1994]. In addition to pain
signals ascending from the spine, signals engage neurons of the Rostral Ventral
Medulla (RVM) and Periaqueductal Gray (PAG) to engage descending feedback
tracts to regulate the output from the spinal cord and provide endogenous pain
control [Wang 1990].

3
Pathological pain
Pathological pain states can result from direct nerve injury (neuropathic
pain), or from persistent stimulation of nociceptors due to injury and
inflammation. In addition to activation of nociceptors, inflammatory processes
also sensitize nociceptors to generate pain a response to milder stimuli
(hyperalgesia). Peripheral sensitization results from the numerous chemical
changes that accompany inflammation. Bradykinins, prostaglandins, and Nerve
Growth Factor (NGF) are among the inflammatory molecules released which
can directly bind to and sensitize nociceptors [Raja 1984; Coderre TJ 1997]. In
animal models of inflammation, primary afferent fibers are made more sensitive,
and normally silent mechanoreceptors are activated due to release of
inflammatory mediators (prostaglandins, histamine, and others) from mast cells
[Di Rosa 1971; Friedman 1990; Leon 1994].
Chronic pain is one of the most pervasive clinical challenges facing
medicine today, afflicting over an estimated 100 million people in the U.S. alone
[Gaskin 2012]. As life expectancy and survivability of conditions like cancer and
HIV increase, the prevalence of chronic pain is expected to steadily increase.
Chronic pain is comorbid for serious conditions such as depression, anxiety, and
suicidal ideation [Braden 2008]. As a result, chronic pain costs the U.S. an
estimated $635 billion, annually [Gaskin and Richard 2012]. Currently available
agents (antidepressants, anticonvulsants, opioids and nonsteroidal anti-
inflammatory drugs) are either unable to completely mitigate the symptoms or, as

4
will be described below, carry significant adverse effects in doing so [Lynch
2015]. There is a critical need for new treatments.
Opioids are extensively used in treatment of both acute and chronic pain
[CDC 2011]. While drugs in the opioid class have remarkable anti-nociceptive
efficacy, there are severe adverse consequences of prolonged use. High doses
of opioid drugs result in a rapid development of tolerance, and carry a high
potential for physical dependence and use disorder. This has generated great
interest in investigating the mechanisms of opioid drug action, tolerance, and
dependence.
The endogenous opioid system
Overview
The endogenous opioid system contains three putative receptor subtypes
(mu, delta, and kappa) and three major groups of endogenous opioid peptides
(endorphins, enkephalins, and dynorphins) [Corbett 2006]. Opioid receptors
belong to the superfamily of seven transmembrane receptors and produce their
cellular effects via coupling with Gi/Go GTP-binding proteins [Ueda 1988; Wong
1988] [Waldhoer 2004]. The primary pathway involves stimulation of inwardly
rectifying potassium conductance, inhibition of adenylyl cyclase and cyclic
adenosine monophosphate. Opioid receptor activation also inactivates voltage
gated calcium channels. The net of these signals is the reduction of
5
neurotransmitter release. Activation of the opioid receptor subtypes produces
similar cellular responses, so the differences in physiological effects result
primarily from different anatomical distribution of the receptor subtypes, and
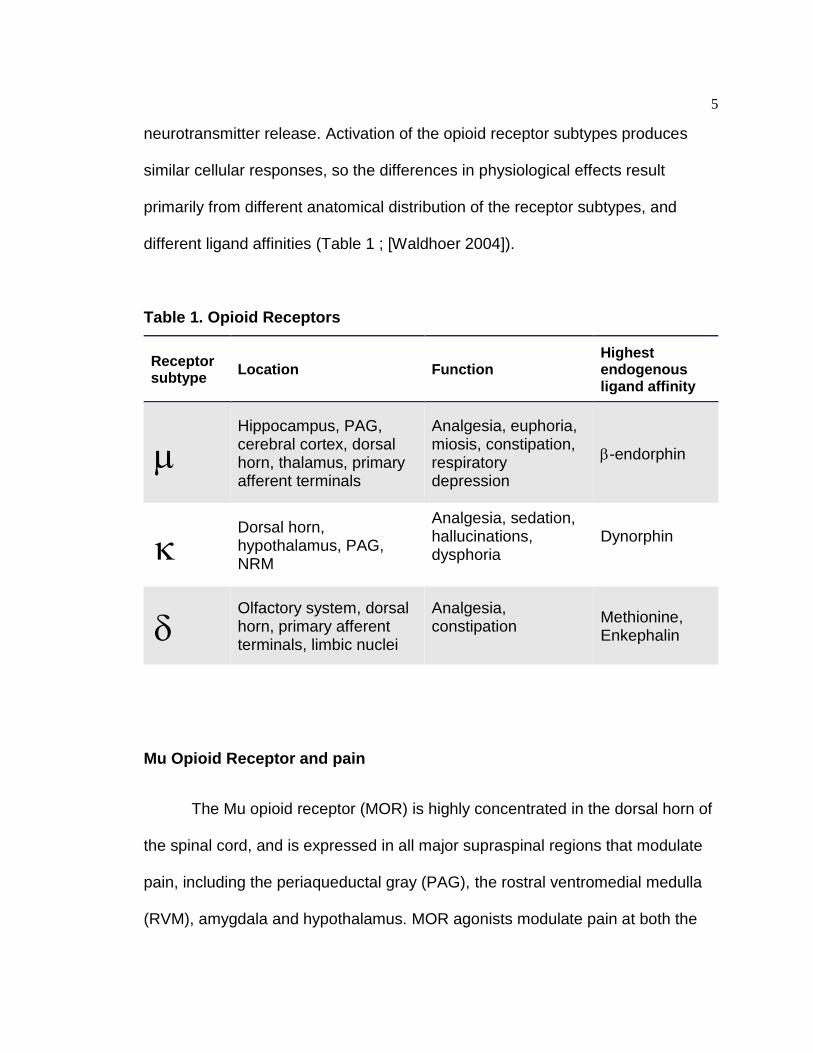
different ligand affinities (Table 1 ; [Waldhoer 2004]).
Table 1. Opioid Receptors
Receptor subtype Location Function
Highest endogenous ligand affinity
Hippocampus, PAG, cerebral cortex, dorsal horn, thalamus, primary afferent terminals
Analgesia, euphoria, miosis, constipation, respiratory depression
-endorphin
Dorsal horn, hypothalamus, PAG, NRM
Analgesia, sedation, hallucinations, dysphoria
Dynorphin
Olfactory system, dorsal horn, primary afferent terminals, limbic nuclei
Analgesia, constipation
Methionine, Enkephalin
Mu Opioid Receptor and pain
The Mu opioid receptor (MOR) is highly concentrated in the dorsal horn of
the spinal cord, and is expressed in all major supraspinal regions that modulate
pain, including the periaqueductal gray (PAG), the rostral ventromedial medulla
(RVM), amygdala and hypothalamus. MOR agonists modulate pain at both the

6
spinal and supraspinal level. In the spinal cord, Opioids regulate nociceptive
transmission both post-synaptically by hyperpolarization of dorsal horn neurons
(potassium) and by binding to presynaptic sensory neurons to inhibit
neurotransmitter release.[Wall 1967; Corbett 2006]. Among supraspinal pain
circuitry, MOR is located pre-synaptically on GABAergic interneurons. Thus,
activation of MOR leads to disinhibition, and increased descending inhibition of
pain pathways. This was first demonstrated in the RVM of rats, which have
distinct neuronal populations that are directly inhibited by opioid agonists [Pan
1990]. Similarly, analgesia resulting from direct electrical stimulation of the PAG
is blocked by naloxone [Akil 1976].
Clinical use of opioid drugs
The majority of opioid drugs in clinical use specifically target MOR as this
produces the strongest analgesic effect, in addition to significant side effects;
euphoria, constipation, respiratory depression and others. [Yaksh 1985].
Activation of MOR also causes euphoria, which plays a role in the high addictive
liability of opiates [Koob 2006a; Savage 2009]. Opioid dependence is
compounded by the development of tolerance, which can cause an escalation of
dose and development of dependence [Williams 2013]. The rate of prescription
opioid overdose has more than tripled since the 1990s, and is still on the rise
[CDC 2011]. Due to the diminishing efficacy of repeated opioid use, and the
resulting prevalence and severity of opioid drug dependence, there is great

7
interest in alternatives. As such, some attempts have been made to target other
opioid receptors.
Kappa opioid receptors (KOR) are activated by endogenous dynorphins.
Systemic KOR agonists also produce robust analgesia [Kolesnikov 1996].
However, activation of KOR negatively modulates mood and is aversive
[Wadenberg 2003]. The negative effects of Kappa agonists have limited their
clinical utility [Williams 2013].
Delta opioid receptors (DOR) are activated by endogenous enkephalins.
Generally, activation of DOR produces minimal analgesia. However, DOR
appear to be up-regulated in rodent models of chronic pain, where they may
become slightly effective. [Holdridge 2007; Kabli 2007].
The endogenous cannabinoid system
Overview
Increasing interest in the potential therapeutic value of cannabis has given
rise to a growing number of states legalizing cannabis for medical use. The
increasing public interest is accompanied by recent studies demonstrating that
states allowing use of medical cannabis show lower than predicated rates of
opiate overdose [Bachhuber 2014; Hayes 2014].
The endocannabinoid (eCB) system is a neuromodulatory system
comprised of two receptors, (CB1R and CB2R) ligands for those receptors, and

8
enzymes involved in the synthesis and degradation of those components. The.
The eCB system is of increasing interest medically, as it has been shown to play
a role in a wide variety of neuronal and immunological processes including:
analgesia, memory, neurogenesis, appetite, metabolism, stress/anxiety,
thermoregulation sleep and immune cell function [Howlett 2004].
The drugs targeting the eCB system can be divided in to several primary
categories; exogenous compounds from the cannabis plant (phytocannabinoids),
synthetic cannabinoids, and endogenous cannabinoids (endocannabinoids). The
major psychoactive component of marijuana, D9-tetrahydrocannabinol (Δ9-THC),
was isolated more than 50 years ago [Gaoni 1964]. The Major endocannabinoids
were not identified until decades later. The two major endocannabinoids,
anandamide (N-arachidonoylethanolamide: AEA, [Devane 1992]) and 2-
arachidonylglycerol (2-AG, [Mechoulam 1995]) are the most extensively studied.
Despite interest in cannabinoid drugs, our understanding of how they function
remains under investigation. In particular, the receptors upon which these
cannabinoids act are of significant interest.
Both CB1R and CB2R are 7-transmembrane GPCRs that couple primarily
to the pertussis (PTX) toxin sensitive Gi/Go subfamily of proteins to modulate a
variety of similar signaling pathways [Howlett 1986; Howlett 2004]. Human CB1R
and CB2R share 44% amino acid sequence identity throughout the total protein,
and differ in terms of localization and levels of expression in the body [Munro
1993]. This warrants that they are examined individually.

9
Cannabinoid Receptor 1 signaling and localization
CB1Rs are located primarily on presynaptic terminals both of
glutamatergic and GABAergic neurons in the CNS [Katona 1999]. Activation of
CB1R is occurs in response to increased neuronal activity, and leads to the
inhibition of neurotransmitter release. Activation of synaptic CB1R by
endogenous and exogenous agonists triggers a canonical G-protein pathway and
inhibits neurotransmitter release directly, via inhibitory coupling to voltage-
dependent calcium channels [Howlett 1989; Mackie 1992];[Sullivan 1999], or
through activation of potassium channels, which shortens action potential
duration and lessens the amount of neurotransmitter released per action
potential. CB1R can also act through secondary messenger systems, and
activate intracellular mediators such as mitogen-activated protein kinase (MAPK;
[Korzh 2008]).
CB1R is the most abundantly expressed GPCR in the CNS, and exerts a
wide variety of effects according to location. [Howlett 2004; Gong 2006]. The
high density of CB1R in hippocampus impacts memory [Herkenham 1991a].
Control over motor function results from the high occurrence of CB1R in the
basal ganglia [Herkenham 1991b], and loss of CB1R is associated with
Parkinson’s Disease [Sañudo-Peña 1998]. The presence of CB1R in cerebellum
further explains the motor effects of cannabinoids [Matsuda 1990]. Metabolism
and food intake are impacted by expression of hypothalamic CB1R, which
interacts with neuropeptides controlling energetic homeostasis and lipogenesis

10
[Cota 2003]. CB1R also impacts reward and motivational circuits, and is present
both on glutamatergic and GABAergic neurons in the Ventral Tegmental Area
(VTA) and Nucleus accumbens (NAc). This is further supported by behavior of
the genes encoding cannabinoid receptors. Polymorphisms of the CNR1 gene
encoding CB1R are correlated with increased dependence of multiple drugs of
abuse [Lopez-Moreno 2012]. Moreover, administration of morphine, cocaine, or
ethanol increases CB1R mRNA expression in limbic and striatal regions
[Gonzalez S 2002].
Cannabinoid Receptor 2 signaling and localization
CB2R is usually coupled to a pertussis toxin-sensitive Gi/Go protein that
triggers the same canonical signaling pathway as CB1R; with inhibition of
adenylyl cyclase activity leading to reduced cAMP levels and lower activation of
protein kinase A (PKA). CB2R also initiates secondary signaling pathways
through protein kinase B and β-arrestin. Unlike CB1R, CB2R appears to poorly
modulate calcium channels or inwardly rectifying potassium channels [Felder
1995; McAllister 1999]. However, because CB2R is principally located on
immune cells, the end results differ significantly.
The CB2 receptor was first cloned and discovered in 1993 and was
reported to be expressed in macrophages and to a lesser extent in the spleen
[Munro 1993]. The expression profile of the CB2 receptor is currently well
established. The ubiquitous presence of the CB2R in immune cells was initially

11
reported in 1995, and has since been extensively characterized [Galiegue 1995].
CB2R expression in the immune cells follows: B-cells > natural killer cells >>
monocytes > neutrophil cells > T8 cells > T4 cells. [Derocq 2000; Carlisle 2002].
As evidenced by CB2R’s ubiquity, it plays a significant role in immune
modulation. CB2R deficient mice lose all immune modulation in response to
cannabinoids [Buckley 2000]. Moreover, both the mRNA and CB2R protein levels
in immune cells correlate with the level of cellular activity. Many believe that
CB2R is activated in response to immune conditions to direct cell activity towards
appropriate response [Carayon 1998]. Furthermore, the ability of the
endocannabinoid 2-AG to induce immune cell migration is blocked by a CB2R-
selective antagonist [Jorda 2002; Tanikawa 2007].
Despite being initially described as an immune cell cannabinoid receptor,
CB2R has been identified in numerous other peripheral cell types. CB2R has
been found in pulmonary endothelial cells [Zoratti 2003], and can also be found
in bone (in osteocytes, osteoblasts and osteoclasts) where it controls bone
formation [Ofek 2006]. The gastrointestinal system also contains CB2R [Storr
2002; Duncan 2008].
The role of the CB2R “peripheral cannabinoid receptor” within the central
nervous system has been largely overlooked due to the belief that it was not
expressed in the CNS [Munro 1993; Atwood 2010]. However, recent discoveries
have suggested that expression of CB2R in the CNS is highly inducible under
pathological conditions. Increase in the expression of CB2 receptors in microglia
and astrocytes occurs in animal models of pain [Beltramo 2006], inflammation,

12
chronic constriction injury. [Brownjohn 2012; JC 2102], ischemia-induced hypoxia
[Ashton 2007], Alzheimer’s disease , and multiple sclerosis [Yiangou 2006].
CB2R activation has been shown to modulate multiple aspects of
neuroinflammation, including the attenuation of pro-inflammatory factors in
microglia and astrocytes [Stella 2004; Maresz 2005]. Moreover, it has been
shown that a selective CB2 agonist, JWH-015 modulates glial marker expression
[Ehrhart 2005].
Far more controversial is the assertions some have made that CB2R is
also expressed neuronally in several brain regions, including the cerebellum
[Ashton 2006], brainstem [Van Sickle 2005], PAG, thalamus, striatum, cortex,
amygdala and hippocampus [Gong 2006]. The exact nature of expression and
distribution of CB2R in the CNS is still a matter of some controversy due to
questions about antibody specificity [Baek 2013], choice of PCR probes, lack of
controls [Marchalant 2014], and species differences [Liu 2009].
Cannabinoids in pain
Systemic administration of nonselective cannabinoid agonists produces
Anti-nociception in animal models of acute and tonic pain [Pertwee 2001]
For example, the nonselective cannabinoid agonist CP55940 is anti-nociceptive
in the tail-flick test [Pugh 1997] and WIN55212-2 inhibits inflammation-induced
nociceptive behavior [Martin 1999b].

13
In order to determine where cannabinoids may produce their anti-
nociceptive effects, the first experiments involved stereotaxic administration of
cannabinoid agonists into specific regions along rat pain circuits. The regions
(see Figure 1) that produced anti-nociception included the Periaqueductal Gray
(PAG; [Lichtman 1996] ), the Rostral Ventromedial Medulla (RVM; [Martin 1998]
), and the lateral posterior nuclei of the thalamus [Martin 1999a]. It should also be
noted that this anti-nociceptive effect is ablated by administration of the CB1R-
selective antagonist, SR141716A [Lichtman 1997]. However, in preclinical rodent
models, repeated administration of cannabinoids can cause tolerance to effects
such as anti-nociception, hypothermia, and catalepsy [Nguyen 2012].
Additionally, the development of dependence is observed to Δ9-THC [Tsou
1995].

14
Figure 1. Cannabinoid Pain Circuitry.
Regions in rat pain circuitry in which injection of a cannabinoid agonist resulted in thermal anti-nocieption. Triangles indicate injections that caused the response. (PAG: Periaqueductal gray), (RVM: Rostral Ventromedial Medulla), (DH: Dorsal Horn), (+: excitatory, -: inhibitory).
Cannabinoid Receptor 2 in pain
Traditionally, the anti-nociceptive effects of Δ9-THC were thought to be a
result exclusively of CB1R activation. However, there has been increased
interest in finding alternatives to CB1R, due to adverse effects. CB2R agonists
are known to generate fewer of the psychotomimetic and sedative effects seen

15
from CB1R agonists [Fernandez-Ruiz 2009], while potentially providing
peripheral anti-nociception [Malan TP Jr 2001]. The anti-nociceptive potential of
CB2 receptor agonists was first indicated by studies using the endogenous fatty
acid derivative palmitoylethanolamide (PEA). PEA was demonstrated to have
marked anti-inflammatory, as well as anti-nociceptive effects when administered
in vivo. [Facci 1995; Calignano 1998] Although PEA is not an agonist at CB1 or
CB2 receptors, its anti-nociceptive effects were blocked by the CB2 receptor
selective antagonist, SR144528, which implicates an indirect role of CB2
receptors in the effects of PEA, or the involvement of a CB2-like receptor.
Following this, it was also observed that, mice lacking CB1R still
demonstrate Δ9-THC -induced anti-nociception in acute thermal pain tests
[Zimmer 1999]. Furthermore, using three different assays of nociception, it was
demonstrated that CB2R knockout mice have a lower anti-nociceptive response
to cannabinoids relative to wild-type mice [Ibrahim 2006]. While this alone does
not prove CB2R activity, it is intriguing. This information, in addition to the
development of a number of CB2R selective agonists has encouraged numerous
labs to investigate (see Table 2). While studies such as these show promise
individually, there remains significant discord in the field as a whole. Results
often cannot be replicated across species, different agonists, or pain modalities.
Furthermore, there are no putative mechanistic explanations for the anti-
nociceptive effects in many cases.

16
Table 2. CB2R-agonist induced anti-nociception in various pain models.
CB2R agonist Pain Model Result Reference
HU308 Formalin 2nd phase anti-nociception
[Hanus L 1999]
HU308 Post-operative pain Anti-allodynia [LaBuda 2005]
JWH133 Sciatic nerve ligation Anti-allodynia [Yamamoto 2008]
A796260 Post-operative pain Anti-allodynia [Yao 2008]
A796260 Chronic constriction injury
Anti-allodynia Yao 2008]
GW842166X Chronic constriction injury
Anti-allodynia [Clayton 2004]
AM-1241 Formalin inflammation Anti-nociception [Quartilho 2003]
AM-1241 λ-carrageenan inflammation
Anti-nociception [Nackley 2003]
AM-1241 Thermal Paw withdrawal
Anti-nociception [Malan TP Jr 2001]
GW405833 Complete Freund’s adjuvant inflammation
Anti-nociception [Whiteside 2005]
A-836339 Complete Freund’s adjuvant inflammation
Anti-nociception [Nackley 2003]
Clinical use of Cannabinoids
Preparations of the hemp plant (Cannabis sativa) have been used for the
treatment of pain for more than 4,000 years. However, there has been a
reluctance to use cannabis-based products over the past century [Jhaveri 2007].
However renewed interest in their development began in the 1960s when the
most psychoactive component of cannabis extract, Δ9-tetrahydrocannabinol (Δ9-
THC), was isolated and partially synthesized [Gaoni 1964].

17
The increased academic interest has been complemented by the growing
number of states legalizing the Cannabis sativa plant for both medical and
recreational use. For example, the drug Sativex (50% Δ9-THC and 50%
cannabidiol), has been approved in Canada and several other countries for the
treatment of neuropathic pain and cancer pain [Pacher 2013]. Δ9-
tetrahydrocannabinol (Δ9-THC;dronabinol; marinol) and its synthetic analogue,
Nabilone (Cesamet), were FDA approved over 25 years ago as medicines for
suppressing nausea and vomiting produced by chemotherapy. Subsequently, the
use of dronabinol was approved as an appetite stimulant for example in AIDS
[Pertwee 2012]. However, the FDA has not approved and cannabinoid drug for
treatment of pain. While showing potential, current attempts to target the eCB
system for pharmacotherapy have yielded inconsistent and often unexpected
results, usually resulting from the intensity of psychotropic side effects [Tanda
2003; Pacher 2006; Pertwee 2012; Pacher and Kunos 2013]. A recent
systematic review and meta-analysis of cannabinoids for medical use found that
among 28 clinical trials, cannabis was associated with greater pain reduction but
also significant side effects relative to placebo [Whiting 2015]. This highlights the
current need to more fully explore the nature of cannabinoid drugs and how they
may interact with current clinical approaches to develop more viable approaches.
In the case of pain management, this means opioids.

18
Interactions between the cannabinoid system and opioid system
It has been established that cannabinoids and endocannabinoids enhance
the anti-nociceptive effects of opiates, in addition to promoting peripheral anti-
nociception by themselves [Malan TP Jr 2001; Cichewicz 2004]. This is
correlated with studies reporting a similar distribution of CB1R and MOR in areas
that are involved in pain modulation, including the periaqueductal gray and the
dorsal horn of the spinal cord [Vigano 2005; Svizenska 2008]. Not only are the
two receptors expressed in similar brain areas, but they are co-expressed in
individual neurons in rat striatum and dorsal horn [Canals 2008].
In addition, opioid and cannabinoid receptors also share similar signal
transduction properties. Both are GPCRs that couple to Gi, and when activated
under similar circumstances will initiate similar signaling responses. For example,
both receptor types are generally found on presynaptic terminals, and will cause
inhibition of neurotransmitter release through the same mechanisms [Vigano
2005]: (1) By blocking cAMP production; (2) via activation of MAP kinases
through other second messenger systems; and (3) by inhibition of
neurotransmitter release via inhibition of calcium channels and activation of
potassium channels. As a result of these similarities, it is unsurprising that
activation of opioid or cannabinoid receptors can produce similar behavioral
effects, including anti-nociception, hypothermia, sedation, hypotension, inhibition
of intestinal motility, and motor depression.

19
Like opioids, Cannabinoid 1 receptor agonists and dual cannabinoid
agonists (including Δ9-THC) are also associated with the development of
tolerance [Tanda and Goldberg 2003; Pacher and Kunos 2013]. Moreover, opioid
and cannabinoid tolerance appear to be related. Chronic exposure to opioid
agonists induced tolerance to the anti-nociceptive effect of Δ9-THC [Bloom 1978;
Hine 1985]. Similarly, chronic Δ9-THC induced tolerance to the anti-nociceptive
effect of opioids [Smith 1994; Welch 1997].
While the exact mechanisms of opioid and cannabinoid tolerance are still
under investigation, there are definite similarities. There are multiple mechanisms
that may act to mediate opioid tolerance [Savage 2009; Williams 2013]. One
process termed desensitization occurs with the loss of MOR-effector coupling
following opioid administration, and appears to be mediated through
phosphorylation of the receptor by G-protein coupled receptor kinases and/or
second messenger regulated protein kinases. Several previous studies have
demonstrated the loss of MOR-effector coupling following agonist treatment
[Bailey 2009; Williams 2013]. This phenomenon appears to result from
phosphorylation of MORs at 3 specific C-terminal residues. [Doll 2011]. These
phosphorylation events are thought to be the result of G protein-coupled receptor
kinase (GRK) and PKC activity, and cause β-arrestin 2 recruitment [Koch 2008].
β-arrestin 2 recruitment causes both the uncoupling of MOR from its associated
G proteins and endocytosis of the receptor [Zhang 1998; Williams 2013].
Desensitization of CB1R shares similar features. Upon agonist stimulation, CB1R
recruits G-protein coupled receptor kinase 3 (GRK3; [Hsieh 1999; Jin 1999] or G-

20
protein coupled receptor kinase 2 (GRK2; [Kouznetsova 2002; Rubino 2006]) to
phosphorylate serine 426 and/or 430 in the CB1R C terminal tail.
However, this sequence of events occurs in an agonist-specific manner,
with certain ligands not causing phosphorylation sites. Some studies suggest that
internalization of MOR does not occur efficiently in response to morphine [Zhang
1998]. Several studies have demonstrated that GRK is responsible for tolerance
to fentanyl (which is strongly internalizing [Imai 2006]) but not to morphine
[Terman 2004; Melief 2010]. Previous work has demonstrated that tolerance to
the anti-nociceptive and anti-allodynic effects of morphine require JNK signaling
[Melief EJ 2010; Hervera 2012; Marcus 2015]. The agonist-selective mechanisms
of opioid tolerance, as well as their interaction with other GPCR systems is of
significant interest for this project.
Cannabinoid Receptor 2 and opioid interactions.
While there is a great deal of work with CB1R and opioids, there is much
less information on how the CB2R receptor might interact with the opioid system
in a clinically significant manner. CB2R receptor has several advantages over
CB1R as a target for the treatment of pathological pain including fewer adverse
side effects, strong anti-inflammatory properties, and a low potential of tolerance
and dependence for CB2R-directed agonists. These advantages make CB2R a
better option for many types of chronic pathological pain including inflammatory
and neuropathic pain CB2R-MOR interactions have also been suggested by

21
studies indicating that the two receptors influence the signaling of one-another
[Cichewicz 2004; Vigano 2005; Paldyova 2008; Merighi 2012]. CB2R agonists
result in decreases to MOR mRNA expression and G-protein activation by MOR
agonists in forebrain of both wild-type and CB1-Knock-out mice [Paldyova 2008].
Additionally, rates of CB2R mRNA expression are altered by drug use.
Administration of heroin or cocaine increases CB2R mRNA levels in the CNS.
[Onaivi 2008; Paldyova 2008].
Overall the current literature suggests CB2R agonists may be useful for
pain management, and some of the potential efficacy may be due to interaction
with the opioid system. This suggests CB2R activation may be a useful way to
enhance concurrent opiate anti-nociception, particularly in cases of inflammatory
pain, while also minimizing tolerance and potential side effects associated with
CB1R activation. If CB2R agonists are to be used clinically, it is essential to know
how their use impacts, and is impacted by opiate tolerance. This information will
provide context for the circumstances under which CB2R agonists may prove
beneficial, as well as limitations to their use.

22
Chapter 2 - General methods
Subjects
Experiments were carried out with wild-type C57BL6/J mice obtained from
Jackson Laboratories (Bar Harbor, Maine). All mice were group-housed, and kept
on a standard 12:12h light-dark cycle with ad libitum access to standard rodent
chow (Teklad 18% Protein Diet, Harlan Teklad, Indianapolis, IN) and water. Mice
were tested between 8-14 weeks of age, and each animal was only exposed to a
single drug or drug combination. All animal care and procedures conformed to
the guidelines of the National Institutes of Health on the Care and Use of
Animals, and were approved by the Institutional Animal Care and Use Committee
of the Penn State University College of Medicine.
Drugs
Opioid agonists morphine sulfate and fentanyl were obtained from the
National Institute on Drug Abuse Drug Supply (Bethesda, MD). JWH-133 (a CB2
receptor agonist with a Ki of 3.4 nm and approximately 200 fold selectivity for
CB2R over CB1R [Huffman 1999]), SR-144,528 (SR2, CB2R antagonist with a
Ki = 0.6 nM and over 700 fold selectivity for CB2R over CB1R [Rinaldi-Carmona
1998] ), naloxone (MOR antagonist), and rimonabant (SR1, CB1R antagonist)
were obtained from Cayman Chemical (Ann Arbor, MI.) JNK inhibitor SP600125
(SP6) was obtained from Sigma-Aldrich (St. Louis, MO [Bennett 2001].)

23
Drugs were prepared daily through dissolution either in isotonic 0.9%
saline, or saline with 5% Cremaphor and 5% ethanol, and administered via
intraperitoneal (i.p.) injection in a volume of 10 mL/kg body weight. When
animals were given multiple i.p. injections in one day, the second injection was
administered on the opposite side of the body cavity. When testing the effects of
co-administered agonists (JWH-133 and morphine), each drug was injected at
the same time. When testing for agonist selectivity, antagonists were
administered via i.p. injection 30 min prior to agonist treatment.
Procedures
Tail-flick and hotplate anti-nociception
Tail-flick anti-nociception was assessed with a Columbus Instruments TF-
1 analgesia meter (Columbus, OH). The apparatus was calibrated to elicit an
average tail-flick latency of 3-4 s in wild-type mice. A cutoff time of 10 seconds
was used to prevent tissue damage. Mice were restrained, allowing their tail to
be exposed to the radiant heat source. The latency until reflexive tail withdrawal
from the heat source was recorded.
Hotplate anti-nociception was measured with a Columbus Instruments
hotplate set to 55o C (Columbus, OH). A 30s cutoff was used to avoid paw
damage. The latency between an animal being placed on the hotplate and
withdrawal from the heated surface (jumping, shaking or licking of paws) was

24
recorded. Both the hotplate and tail-flick responses were used to calculate the
percentage of maximal possible effect (%MPE). This value was calculated using
the formula below as described previously [Morgan 2014; Marcus 2015].
(%MPE) =(post drug latency)– (pre drug latency)
(cutoff time − pre drug latency)× 100
Formalin test
The formalin test is an extensively used model of acute inflammatory pain
[Tjolsen 1992]. This method utilizes injection of formalin into an animal’s paw to
elicit a biphasic pattern of pain behavior, with a phase of acute pain followed by a
phase of inflammatory pain. The early (acute) phase is generated by the activation
of C and Aδ fibers as a result of needle penetration into the hind paw. The late
phase involves an inflammatory reaction due to intraplantar formalin in the paw
[Tjolsen 1992], Mice were subjected to the formalin test to assess basal differences
in inflammatory pain response and the anti-nociceptive effect of morphine and
JWH-133 on this type of pathological pain. Prior to testing, mice were acclimated
for 20 min in a Plexiglas (5”x5”x5”) observation chamber placed on a transparent
elevated platform. A mirror angled at 45° was placed below the platform to allow
for constant observation of the animal’s paws. Following acclimation, mice were
administered 10μL of a 2.5% formalin solution into the plantar surface of a single
hind paw using a 28 ½ gauge needle (Becton Dickinson, Franklin Lakes, NJ).

25
Immediately after the formalin injection, mice were returned to the Plexiglas
observation unit and nociceptive behavior was continuously measured in 12 five-
min intervals for a total testing time of 60 min. During each five-min time bin, the
duration spent performing pain-response behaviors was recorded. The nociceptive
behaviors were separated into three categories: (0) the injected paw has little
weight placed on it; (1) the injected paw is raised off of the ground; (2) the injected
paw is licked, shaken, or bitten. The amount of time spent in each category was
quantified and weighted with the composite pain score-weighted scores technique
(CPS-WST0,1,2), resulting in a Composite Pain Score (CPS) for each five-min
interval between 0 (no pain behaviors) to 2 (maximal pain behavior; [G. Stennis
Watsona 1997]). The Area Under the Curve (AUC, CPS x time (min)) was
calculated using the trapezoidal rule for the acute phase (0-15 min; phase I) and
the inflammatory phase (15-60 min; phase 2). To assess the anti-nociceptive
effects of drugs, mice were injected (i.p.) 60 min prior to the formalin injection.
Physical side-effects
Body temperature
Body temperature was measured using a mouse rectal thermometer
probe (Physitemp, Clifton, NJ). Temperature was measured immediately prior to,
and 60 min following drug administration. Hypothermia was reported as a percent

26
change in body temperature between pre-drug and 60 min post-drug
measurements (°C), as described by the formula:
(%∆BT) =(post − drug temperature)– (pre − drug temperature)
pre − drug temperature× 100
Rotarod test
Motor impairment was measured using a Med Associates ENV-577-M
Rotarod apparatus (Fairfax, VA). Animals were trained by undergoing two
consecutive days of six 300 second training trials. Mice were placed on a rotating
drum (3 cm in diameter), which accelerated at a constant rate from 4 to 40 rpm
over a 5 minute period. The time spent walking on top of the rod until the mouse
either fell off the rod, or slipped and held onto the rod to ride completely around
was recorded. Impairment was determined through comparison of performance
prior to, and 60 minutes following drug administration.
Precipitated Withdrawal
Physical dependence was induced using a series of 20 injections that
were given twice-daily for 10 days (5 mg/kg morphine, i.p.; 1 mg/kg JWH-133,
i.p.). Following 10 days of daily drug administration, withdrawal was precipitated
using an i.p. injection of vehicle, 10 mg/kg naloxone (to counter morphine), or 10
mg/kg SR2 (to counter JWH-133) 30 min after the final drug injection on the 11th

27
day. Somatic withdrawal symptoms (paw tremors, body tremors, diarrhea, and
jumps) were video recorded for 60 min after injection of naloxone, SR2, or
vehicle. Withdrawal symptoms were scored in alternating 5 min time intervals (5–
10, 15–20, 25–30, 35–40, 45–50, and 55–60 min, as described previously
[Morgan 2014]).
Data analysis
Values for anti-nociception, hypothermia, motor coordination, and
precipitated withdrawal were expressed as the mean ± standard error of the
mean (SEM). Data was analyzed using either one-way or two-way ANOVA,
followed by Bonferroni or Dunnett post-hoc testing as appropriate. Additional
analyses were performed using SPSS statistical software (SPSS Incorporated,
Chicago, IL). P<0.05 was considered significant.
Dose response curves and related values (ED50) were calculated using
the curve fitting functions in GraphPad PRISM. Experimental data (mean+SEM of
individual dose points) were fitted to a sigmoidal curve with variable slope,
according to:
Y=Bottom + (Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)).

28
Isobolographic analysis
This analysis was performed to determine whether the combined anti-
nociceptive effects of morphine and JWH-133 were sub-additive, additive, or
synergistic (super-additive).
Full dose response curves were generated in the formalin test (as
described above) for JWH-133, morphine, and then a combination that was co-
administered in a fixed 1:10 dose ratio (see [Grabovsky 2004; Tallarida 2010;
Kazantzis 2016] for detailed explanation and formulas.) ED50 values for this
combination were determined and compared to a theoretically calculated ED50
value [Tallarida 2002]. This theoretical value was determined using the dose-
response curves of JWH-133 and morphine, alone, to generate a predicted
additive curve using the formula below [Tallarida and Raffa 2010; Kazantzis
2016].
𝐸(𝑎, 𝑏) = 𝐸𝐵
(𝑏 + 𝑏𝑒𝑞(𝑎) )𝑝
(𝑏 + 𝑏𝑒𝑞(𝑎) )𝑝 + 𝐶𝑏𝑝
Where the effect (E) of specific doses of two drugs (a,b) in combination is
estimated using the dose of drug b (beq(a)) that gives and equivalent response to
a specific dose of drug a (a), the ED50 of drug b (Cb), and the Hill slope of drug b
(p). If the experimentally determined ED50 of the combination is significantly lower
than the predicted value (according to a t-test), the combination is deemed
synergistic. If the two ED50 values are equal, the combination has only an

29
additive effect. The variance for the theoretical ED50 value is calculated by
combination of the variances of both JWH-133 and morphine according to the
formula below [Miranda 2014].
Var ED50 (combination) = (0.5)2Var ED50 Morphine + (0.5)2Var ED50 JWH-133
Isobolograms also allow visual comparison of experimental and theoretical
values. The ED50 of the first drug is plotted on the abscissa and the ED50 of the
second drug is plotted on the ordinate. A straight line is drawn connecting the two
values and is termed the line of additivity. ED50s of drug combinations falling
below this line demonstrate synergism, while values on the line demonstrate
additivity.

30
Chapter 3
Tolerance to the anti-nociceptive and hypothermic effects of morphine are mediated by multiple isoforms of c-Jun N-terminal Kinase
Matthew B Yuill1,2,3,†, Michael L Zee1,†, David Marcus1, Daniel J Morgan1,2,3*
1Department of Anesthesiology, Penn State University College of Medicine,
Hershey, PA 17033; 2Department of Pharmacology, Penn State University College
of Medicine, Hershey, PA 17033; 3Department of Neural and Behavioral Sciences,
Penn State University College of Medicine, Hershey PA 17033;
† These authors contributed equally
* To whom correspondence should be sent: [email protected]
Acknowledgements
This work has been supported by NIH grants DA036385 (DJM), DA037355
(DJM), and is also funded, in part, under a grant from the Pennsylvania
Department of Health using Tobacco CURE Funds (DJM).
Conflicts of interest
There are no conflicts of interest

31
Abstract
The abuse and overdose of opioid drugs is a growing public health
problem, globally. While progress has been made towards understanding the
mechanisms governing tolerance to opioids, the exact cellular machinery
involved remains unclear. However, there is growing evidence to suggest that c-
Jun N-terminal Kinases (JNKs) play a major role in mu opioid receptor regulation
and morphine tolerance. In this study, we aimed to determine the potential role
of different isoforms of JNK in tolerance to the anti-nociceptive and hypothermic
effects of morphine. We used the hotplate and tail-flick tests for thermal pain to
measure tolerance to the anti-nociceptive effects of once daily sub-cutaneous
injections with 10 mg/kg morphine. Body temperature was also measured to
determine tolerance to the hypothermic effects of morphine. Tolerance to
morphine was assessed in wild-type mice and compared to single knockout (KO)
mice lacking each of the three c-jun N-terminal kinase (JNK) isoforms (JNK 1,
JNK2, or JNK3). We found that loss of each individual JNK isoform causes
impairment in tolerance for the anti-nociceptive and hypothermic effects of daily
morphine. However, disruption of JNK2 seems to have the most profound effect
on morphine tolerance. These results demonstrate a clear role for c-jun N-
terminal kinase (JNK) signaling pathways in morphine tolerance. This
complements previous studies suggesting that the JNK2 isoform is required for
morphine tolerance, but presents additional novel data suggesting that additional
JNK isoforms also contribute to this process.

32
Keywords: tolerance, morphine, JNK, anti-nociception, opioids, mu opioid
receptor, desensitization, GPCR
Introduction
Opioid drugs, such as morphine, fentanyl and oxycodone, remain a
preferred and commonly prescribed class of drug for pain management [CDC
2011]. While they demonstrate remarkable efficacy for treating acute pain, there
are several limitations to their use. Opioid drugs are associated with rapid
development of tolerance and also high abuse potential. Despite this, they
remain the default approach to treatment of many chronic pain conditions. As a
result, abuse and overdose of opioids are the fastest growing issues for narcotic
drugs in the US. This is punctuated by a tripling in the rate of prescription opioid
overdose in just two decades [CDC 2011].
The opioid system is comprised of multiple opioid receptors, each with a
unique distribution and function. The anti-nociceptive effects of many opioid
drugs are mediated through the mu-opioid receptor (MOR, [Waldhoer 2004]);
making this receptor one of the most extensively studied G protein-coupled
receptors (GPCRs,[Corbett 2006]). MOR is expressed in numerous regions of
the central nervous system, among them: the dorsal horn of the spinal cord, the
periaqueductal gray, and the cortex. As a result, it is a crucial receptor in the
modulation of pain circuitry at both the supra-spinal and spinal level. Activation of
MOR also causes euphoria, which plays a role in the high addictive liability of
opiates [Koob 2006a; Savage 2009]. Opioid dependence is compounded by the

33
development of tolerance, which can cause an escalation of dose and a
progression to dependence [Williams 2013]. Due to the diminishing efficacy of
repeated opioid use, and the resulting prevalence and severity of opioid drug
dependence, understanding the mechanisms behind tolerance to these drugs is
of significant interest.
There are multiple mechanisms that may act to mediate opioid tolerance
[Savage 2009; Williams 2013]. One process termed desensitization occurs with
the loss of MOR-effector coupling following opioid administration, and appears to
be mediated through phosphorylation of the receptor and recruitment of β-
arrestin proteins [Koch and Hollt 2008]. Several previous studies have
demonstrated the loss of MOR-effector coupling resulting from the use of
agonists [Bailey 2009; Williams 2013]. This phenomena appears to result from
phosphorylation of MORs at C-terminal threonine 370 and/or serine 375 [Doll
2011]. These phosphorylation events are thought to be the result of G-protein
coupled receptor kinase (GRK)2 and/or GRK3 activity, and cause β-arrestin 2
recruitment [Koch and Hollt 2008]. β -arrestin 2 causes both the uncoupling of
MOR from its associated G proteins, and also results in endocytosis [Zhang
1998; Williams 2013]. This sequence of events occurs in an agonist-specific
manner, and some studies suggest that internalization of MOR does not occur
efficiently in response to morphine [Zhang 1998].
Several studies have demonstrated that GRK is responsible for fentanyl
but not morphine tolerance [Terman 2004; Melief 2010]. Recent work has shown
that tolerance to morphine is attenuated through the use of the JNK inhibitor,

34
SP600125 [Chen 2008; Guo 2009; Hervera 2012; Marcus 2015]. SP600125 is
an anthrapyrazolone capable of inhibiting JNK1, JNK2, and JNK3 with high
affinity [Zhuang 2006]. It has been demonstrated that this compound prevents
phosphorylation of JNK in the spinal cord, resulting in attenuation of tolerance to
the anti-nociceptive and anti-allodynic effects of morphine [Guo 2009; Hervera
2012]. However, use of this JNK inhibitor is non-selective for the different JNK
isoforms and does not allow determination of which one(s) are responsible for
morphine tolerance. It has been suggested by recent studies that JNK2 is
required for tolerance to the anti-nociceptive effects of morphine [Kuhar 2015].
However, no studies thus far have characterized morphine tolerance in JNK1,
JNK2, and JNK3 Knock-Out (KO) mice.
Therefore, this novel work examined tolerance to the anti-nociceptive
effects of morphine in JNK1, JNK2, and JNK3 KO mice. We tested the
hypothesis that tolerance to morphine would be disrupted in JNK 2 KO mice, and
found that, while this was indeed the case, JNK1, (and to a lesser extent) JNK3
also contribute to morphine tolerance. This finding represents a novel and
significant addition to recent work demonstrating the role of JNK signaling in
morphine tolerance and MOR regulation.

35
Methods
Subjects
Experiments were carried out with wild-type C57BL6/J mice obtained from
Jackson Laboratories (Bar Harbor, Maine), and three strains of JNK mutant mice.
Mice lacking either JNK1, JNK2, or JNK3 were generously provided by Dr.
Charles Chavkin at the University of Washington School of Medicine. The
generation of JNK1 KO [Dong 1998], JNK2 KO [Yang 1998], and JNK3 KO mice
[Yang 1997] has been described previously. All mice were kept on a standard
12:12h light-dark cycle with ad libitum access to standard mouse chow and
water. All animal care and procedures conformed to the guidelines of the
National Institutes of Health on the Care and Use of Animals, and were approved
by the Institutional Animal Care and Use Committee of the Penn State University
College of Medicine.
Drugs
Morphine sulfate was obtained from the National Institute on Drug Abuse
Drug Supply (Bethesda, MD). Morphine was dissolved in isotonic 0.9% saline
and administered sub-cutaneously in an injection volume of 10 mL/kg body
weight.

36
Tail-flick and hotplate anti-nociception
Tail-flick anti-nociception was assessed with a Columbus Instruments TF-
1 analgesia meter (Columbus, OH). The apparatus was calibrated to elicit an
average tail-flick latency of 3-4 s in wild-type mice. A cutoff time of 10 seconds
was used to prevent tissue damage. Mice were restrained, allowing their tail to
be exposed to the radiant heat source. The latency until reflexive tail withdrawal
from the heat source was recorded.
Hotplate anti-nociception was measured with a Columbus Instruments
hotplate set to 55o C (Columbus, OH). A 30s cutoff was used to avoid paw
damage. The latency between an animal being placed on the hotplate and
withdrawal from the heated surface (jumping, shaking of paws) was recorded.
Both the hotplate and tail-flick responses were used to calculate the percentage
of maximal possible effect (%MPE). This value was calculated using the formula
%MPE = (post-drug latency – pre-drug latency)/(cutoff time- pre-drug latency) x
100. These procedures and calculations have been described previously
[Morgan 2014; Marcus 2015].
Measurement of body temperature
Body temperature was measured using a mouse rectal thermometer
probe (Physitemp, Clifton, NJ). Hypothermia was reported as a % change in
body temperature between pre-drug and post-drug measurements, as
demonstrated by the formula:

37
(%∆BT)=[(pre-morphine temperature)–(post-morphine temperature)]/[pre-
morphine temperature] x 100.
Procedures
Anti-nociception and hypothermia were measured in groups of mice [wild-
type (n=17), JNK1 KO (n=15), JNK2 KO (n=14), and JNK3 KO (n=20)] receiving
daily sub-cutaneous (s.c.) injections of morphine (10 mg/kg x 10 days). Mice
were tested for body temperature, tail-flick and hotplate latency immediately prior
to, and one hour after morphine injection on each day.
Data analysis
Anti-nociception and hypothermia values were expressed as mean ±
SEM. Values were analyzed using two-way mixed factorial ANOVA (genotype x
day) followed by Bonferroni post-hoc testing. Differences in baseline tail-flick and
hotplate latencies and basal body temperatures were analyze by one-way
ANOVA. Analyses of the initial responses to the first injection of morphine were
also analyzed by one-way ANOVA. Analyses were performed using PRISM6
statistical software (Graphpad, La Jolla, CA). P<0.05 was considered significant.

38
Results
Tolerance to the anti-nociceptive effects of 10 mg/kg morphine
Baseline tail-flick latencies were different between WT (3.35±0.09 sec)
and JNK 1 KO (4.39±0.28 sec; p<0.001), JNK 2 KO (4.04±0.20 sec; p<0.001),
and JNK 3 KO (4.34±0.16 sec; p<0.0001) mice. However, the response to the
first injection of morphine was not different (F3,70 = 1.164, P=0.33) between WT
(90.9±4.6% MPE), JNK 1 KO (90.0±3.4% MPE), JNK 2 KO (98.7±1.2% MPE),
and JNK 3 KO (94.4±2.4% MPE) mice.
Wild-type mice rapidly developed tolerance to the anti-nociceptive effects
of daily s.c. morphine (10 mg/kg) injections in the tail-flick test (Figure 2).
Tolerance to the anti-nociceptive effect of 10 mg/kg morphine, in the tail-flick test,
developed in a time-dependent manner (F9,637 = 12.77, P<0.0001) that was also
dependent on genotype (F3,637 = 105.6, P<0.0001). There was also a significant
day x genotype interaction effect (F27,637 = 3.26, P<0.0001). Bonferroni post-hoc
tests show that tolerance to morphine, in the tail-flick test, was different between
wild-type mice and JNK 1 KO (p<0.0001), JNK 2 KO (p<0.0001), and JNK 3 KO
mice (p<0.0001). However, post-hoc testing also indicated that JNK2 KO mice
also developed less tolerance than either JNK1 KO (p<0.0001) or JNK3 KO
(p<0.001) mice. There was no difference in morphine tolerance between JNK1
KO and JNK3 KO mice.
Baseline hotplate latencies were also different between WT (7.88 ± 0.57
sec.), JNK 1 KO (11.48±0.77 sec; p<0.001), JNK 2 KO (6.84±0.57 sec; p<0.001),
and JNK 3 KO (5.96±0.26 sec; p<0.0001) mice. However, responses to the first

39
injection of morphine were not different (F3,64 = 1.747, P=0.17) between WT
(67.7±6.1% MPE), JNK 1 KO (83.9±5.7% MPE), JNK 2 KO (67.1±8.9% MPE),
and JNK 3 KO (65.3±2.4% MPE) mice.
Figure 2. Tolerance to morphine in the tail-flick test.
Tolerance to the anti-nociceptive effects of morphine in the tail-flick test is altered in mutant mice lacking JNK 1, JNK 2, or JNK 3. Wild-type (WT; black squares and line), JNK1 KO (red circles and line), JNK2 KO (orange circles and line), and JNK3 KO (magenta circles and line) mice were injected (s.c.) with 10 mg/kg morphine once daily for ten days. All three knockout mouse lines showed impaired tolerance to the anti-nociceptive effects of morphine, in the tail-flick test, relative to wild-type animals (p<0.0001). Data are expressed as mean ±SEM (n=15-20 per group).
Similar results were observed for tolerance to the anti-nociceptive effects
of 10 mg/kg morphine, in the hotplate test (Figure 3). However, all mutant mouse
groups showed delayed onset of tolerance to the anti-nociceptive effects of
morphine, relative to wild-type mice. Two-way ANOVA analysis reveals main

40
effects of genotype (F3,577 = 39.39, P<0.0001) and time (F9,577 = 32.2, P<0.0001).
However, no significant day x interaction effect was detected (F27,577 = 1.28,
P=0.155).
Figure 3. Tolerance to morphine in the hotplate test.
Tolerance to morphine-induced anti-nociception, in the hotplate test, is altered in mutant mice lacking JNK 1, JNK 2, or JNK 3. Wild-type (WT; black squares and line), JNK1 KO (red circles and line), JNK2 KO (orange circles and line), and JNK3 KO (magenta circles and line) mice were injected (s.c.) with 10 mg/kg morphine once daily for ten days. All three knockout mouse lines showed impaired tolerance to the anti-nociceptive effects of morphine, in the hotplate test, relative to wild-type animals (p<0.0001). Data are expressed as mean ±SEM (n=15-20 per group). Tolerance to morphine-induced hypothermia
Basal body temperature was also different between WT (38.6 ± 0.1 °C),
and JNK 1 KO (37.7±0.1 °C; p<0.001), JNK 2 KO (36.8±0.1 °C; p<0.0001), and
JNK 3 KO (37.1±0.1 °C; p<0.0001) mice. There were also differences in basal

41
body temperature between JNK 1 and JNK 2 KO mice (p<0.01) as well as JNK 1
KO and JNK 3 KO mice (p<0.5). There were also genotype differences in the
hypothermic response to morphine (F3,68 = 6.537, P=0.0006), with Bonferroni
post-tests revealing that the main effect of genotype was due to differences
between WT (-4.2 ± 0.7 % change) and JNK 3 KO (-1.6 ± 0.5 % change) mice
(p<0.1) and between JNK 1 (-4.6 ± 0.4 % change) and JNK 3 KO mice (p<0.01).
There were no differences between JNK 2 KO mice (-2.5 ± 0.7 % change) and
the other genotypes.
Rapid tolerance developed to the hypothermic effects of once daily 10
mg/kg morphine in wild-type mice (Figure 4). Tolerance to the hypothermic effect
of 10 mg/kg morphine developed in a time-dependent manner (F9,607 = 18.44,
P<0.0001) that also depended on genotype (F3,607 = 31.24, P<0.0001). There
was also a significant day x genotype interaction effect (F27,607 = 3.33, P<0.0001).
Bonferroni post-hoc tests show that tolerance to morphine was different between
wild-type mice and JNK 1 KO (p<0.001), JNK 2 KO (p<0.0001), and JNK 3 KO
mice (p<0.05). However, post-hoc testing also indicated that JNK 2 KO mice
also developed less tolerance to morphine hypothermia than JNK3 KO (p<0.05)
mice. There was also a difference in tolerance to the hypothermic effects of
morphine between JNK1 KO and JNK 3 KO mice (p<0.0001). However, there
was no difference in morphine tolerance between JNK1 KO and JNK2 KO mice.

42
Figure 4. Tolerance to morphine-induced hypothermia.
Tolerance to morphine-induced hypothermia is altered in mutant mice lacking JNK 1, JNK 2, or JNK 3. Wild-type (WT; black squares and line), JNK1 KO (red circles and line), JNK2 KO (orange circles and line), and JNK3 KO (magenta circles and line) mice were injected (s.c.) with 10 mg/kg morphine once daily for ten days. All three knockout mouse lines showed impaired tolerance to the hypothermic effect of morphine relative to wild-type animals (p<0.0001). Data are expressed as mean ±SEM (n=15-20 per group).

43
Discussion
The primary finding of this study is that all three isoforms of JNK (JNK1,
JNK2, and JNK3) are involved in tolerance to the anti-nociceptive and
hypothermic effects of morphine. Although deletion of JNK2 had the greatest
impact on morphine tolerance, the loss of JNK1 or JNK 3 in KO mice also
attenuated morphine tolerance. Our results suggest that JNK3 plays the least
prominent role in morphine tolerance. Our results are novel in the finding that all
three forms of JNK contribute to morphine tolerance, and they are consistent with
previous work demonstrating that JNK2 can mediate tolerance to morphine
[Melief 2010; Kuhar 2015].
Interestingly, disruption of JNK2 appears to have a greater impact on
tolerance than the other isoforms in the tail-flick but not the hotplate test. This
may be due to the mediation of tail-flick response primarily by spinal circuitry,
versus the mediation of hotplate response by both spinal and supraspinal
circuitry [Langerman 1995]. It should also be noted that hotplate responses are
not reflexive, and thus the possibility of a learning effect among the mice altered
their responses. While the JNK3 KO mice showed a significant diminishment in
hypothermic tolerance relative to wild-type mice, it is important to note that JNK 3
KO mice exhibit profound differences in the hypothermic response to the first
morphine injection. Further investigation is warranted to determine the cause for
reduced morphine-induced hypothermia in JNK 3 KO mice.
Morphine appears to be different from many other opioids, as it does not
produce MOR desensitization through the common GRK/β-arrestin pathway

44
[Terman 2004; Melief 2010]. As such, the question of how JNK is mechanistically
involved in morphine tolerance is of significant interest. Application of morphine
results in an increase in spinal JNK phosphorylation through a protein kinase C
(PKC)-dependent process [Melief 2010]. Furthermore, this effect was abolished
in JNK2 KO, but not JNK1 KO or JNK3 KO mice [Kuhar 2015]. Both of these
studies also demonstrated that JNK2 KO mice have a reduction in acute
morphine tolerance. Despite these results, it should not be concluded that
morphine tolerance is strictly the result of JNK2 phosphorylation by PKC. While
other opioids such as fentanyl also cause JNK2 phosphorylation, their tolerance
is largely JNK-independent [Kuhar 2015; Marcus 2015]. Moreover, while spinal
MOR-desensitization by morphine requires JNK2, the desensitization of MOR in
the locus ceruleus appears to be JNK-independent [Levitt 2012].
Our finding that all three JNK isoforms appear to impact morphine
tolerance further suggests that morphine tolerance is not always mediated by
JNK2 alone. This raises the question of how the other two JNK isoforms might be
involved. While all three JNK isoforms are expressed in parts of the CNS, they
have distinct localization patterns and levels of expression in vivo [Bogoyevitch
2006]. It is an intriguing possibility that development of tolerance to distinct
effects of morphine are mediated to varying degrees by each JNK isoform.
Continued investigation of JNK pathways are thus essential to integrate the
currently disparate findings in the literature to develop a comprehensive
understanding of MOR desensitization and tolerance.

45
Conclusions
Our findings suggest a possible role for multiple isoforms of JNK on the
development of tolerance to morphine. We found a diminishment of tolerance to
the anti-nociceptive and hypothermic effects of morphine not only in JNK 2 KO
mice, but also JNK 1 KO and JNK 3 KO mice. This may be a significant factor in
the developing picture regarding the mechanisms behind morphine tolerance.
Determining the nature through which these signaling pathways act on MOR will
be an important task if JNKs become a therapeutic target in pain management
and opioid abuse.

46
Chapter 4
Anti-nociceptive interactions between opioids and a cannabinoid receptor
2 agonist in inflammatory pain.
Matthew B Yuill1,2,3, David E Hale1, Josée Guindon4,†,* and Daniel J Morgan1,2,3,†,*
Abstract
The cannabinoid 1 receptor (CB1R) and cannabinoid 2 receptor (CB2R) can both
be targeted in the treatment of pain, yet they have some important differences.
CB1R is expressed at high levels in the central nervous system (CNS), whereas
CB2R is found predominantly, although not exclusively, outside the CNS. The
objective of this study was to investigate potential interactions between CB2R
and the mu opioid receptor (MOR) in pathological pain. The low level of adverse
side effects and lack of tolerance for CB2R agonists are attractive
pharmacotherapeutic traits. This study assessed the anti-nociceptive effects of a
selective CB2R agonist (JWH-133) in pathological pain using mice subjected to
inflammatory pain using the formalin test. Furthermore, we examined several
ways in which JWH-133 may interact with morphine. JWH-133 produces dose-
dependent anti-nociception during both the acute and inflammatory phases of the
formalin test. This was observed in both male and female mice. However, a
maximally efficacious dose of JWH-133 (1 mg/kg) was not associated with

47
somatic withdrawal symptoms, motor impairment, or hypothermia. After eleven
once-daily injections of 1 mg/ JWH-133, no tolerance was observed in the
formalin test. Cross-tolerance for the anti-nociceptive effects of JWH-133 and
morphine were assessed to gain insight into physiologically relevant CB2R and
MOR interaction. Mice made tolerant to the effects of morphine exhibited a lower
JWH-133 response in both phases of the formalin test compared to vehicle
treated morphine-naïve animals. However, repeated daily JWH-133
administration did not cause cross-tolerance for morphine, suggesting opioid and
CB2R cross-tolerance is unidirectional. However, preliminary data suggests co-
administration of JWH-133 with morphine modestly attenuates morphine
tolerance. Isobolographic analysis revealed that co-administration of JWH-133
and morphine has an additive effect on anti-nociception in the formalin test.
Overall these findings show that CB2R may functionally interact with MOR to
modulate anti-nociception in the formalin test.
Keywords:
CB2R, morphine, pain, tolerance, formalin, JWH-133, opioid, cannabinoid
agonist
1Department of Anesthesiology and Perioperative Medicine, Penn State University College of Medicine,
Hershey, PA
2Department of Pharmacology, Penn State University College of Medicine, Hershey, PA
3Department of Neural and Behavioral Sciences, Penn State University College of Medicine, Hershey PA

48
4Department of Pharmacology and Neuroscience, Texas Tech University Health Sciences Center, Lubbock,
TX
† These authors contributed equally
Corresponding authors:
Daniel J Morgan. Department of Anesthesiology and Perioperative Medicine, Penn State University College
of Medicine, Hershey, PA
Email: [email protected]
Josée Guindon. Department of Pharmacology and Neuroscience, Texas Tech University Health Sciences
Center, Lubbock, TX
Email: [email protected]

49
Introduction
Pain is one of the most widespread and costly clinical challenges facing
medicine today, afflicting an estimated 120 million Americans and costing $600
billion annually in medical expenses, loss of work productivity, and long-term
insurance disability [Gaskin and Richard 2012; Nahin 2015]. Long term pain is
also characterized by the high occurrence of comorbid side effects such as
depression, anxiety, and suicidal ideation [Braden and Sullivan 2008]. Opioid
drugs, which exert analgesic activity through MOR, are the current gold standard
for the treatment of acute and long term chronic pain [CDC 2011]. While opioid
drugs have remarkable anti-nociceptive efficacy for certain types of pain, there
are severe adverse consequences that can occur with prolonged use. For
example, chronic use of opioids causes tolerance and a high potential risk for
physical dependence and abuse [Koob 2006b; Savage 2009; Williams 2013].
The rate of overdose from prescription opioids has more than tripled since the
early 1990s, and is still on the rise [CDC 2011]. More than 33,000 deaths in the
United States were attributed to opioid overdose in 2015 [CDC 2017].
Consequently, there is a current unmet medical need to find possible alternatives
or adjuvants to opioids for the treatment of chronic pain. Therefore, an interest in
understanding the potential interactions of the opioid system with other pathways
involved in alleviating pain is crucial.
The evidence is mounting in terms of the use of cannabinoids for the
treatment of unalleviated pain. They constitute a new class of agents that can be
added to the pharmaceutical toolbox for the management of chronic pain

50
[Eisenberg 2014; Lynch and Ware 2015]. In human clinical trials and case
studies, drugs targeting the endocannabinoid (eCB) system have shown promise
for treatment of numerous pathologies, including chronic and acute pain [Pacher
2006]. For example, the drug Sativex (50% Δ9-THC and 50% cannabidiol), has
been approved in Canada and several other countries for the treatment of
neuropathic pain and cancer pain [Pacher and Kunos 2013]. While CB1R
agonists and dual cannabinoid receptor agonists (including Δ9-THC) demonstrate
potent analgesic effects in rodent models and in human use, they also have
several disadvantages. Use of these drugs is associated with the development of
tolerance, psychotomimetic effects, and numerous other physical side effects
[Tanda and Goldberg 2003; Pacher and Kunos 2013].
However, selective activation of CB2R in rats and mice does not produce
these psychotropic adverse side effects [Guindon 2008; Kinsey 2011; Deng
2015b], making CB2R an attractive target for the treatment of pain and other
pathologies. Indeed, CB2R agonists have been shown to alleviate acute,
inflammatory, and chronic pain causing them to garner increased attention as a
potential alternative to the use of opioids for treatment of pain [Guindon and
Hohmann 2008].
In recent years, mounting evidence of the importance for CB2R in
pathological pain has increased interest as demonstrated by the synthesis of a
variety of CB2R-selective cannabinoid agonists [Atwood 2010]. CB2R agonists
have been shown to have efficacy in multiple models of pathological pain in
preclinical rodent models including post-operative pain [Romero-Sandoval 2007],

51
inflammatory pain [Guindon and Hohmann 2008], chemotherapeutic pain [Deng
2015b], and cancer-induced pain [Curto-Reyes 2010]. However, the mechanisms
through which the anti-nociceptive and analgesic effects of CB2R agonists are
mediated are not completely well characterized.
The CB2R agonists have anti-inflammatory properties, and many pain
studies have suggested a mechanism of action through the regulation of
inflammation (including neuroinflammation, see [Ehrhart 2005; Benito 2008]).
CB2R activation attenuates the release of pro-inflammatory agents (Tumor
Necrosis Factor-α, Nitric Oxide) and increases release of anti-inflammatory
agents (Interleukin 10) from microglia and astrocytes [Ehrhart 2005; Romero-
Sandoval 2009; Correa 2010]. The release of these pro-inflammatory factors is
stimulated by activation of MOR on microglia, and this inflammatory response is
thought to facilitate opioid tolerance [Watkins 2009; Merighi 2012]. As such, the
use of CB2R agonists could both relieve pain and help mitigate the
consequences of opioid treatment. This possibility is supported more generally
by previous work both in animals and humans showing inhaled cannabis and Δ9-
THC enhance the pain relief of opioids [Malan TP Jr 2001; Cichewicz 2004].
However, the interaction between opioids and CB2R selective agonists must be
more extensively investigated.
Therefore, the objectives of this study were to investigate the efficacy of a
CB2R agonist on inflammatory pain and to examine potential interactions
between CB2R and MOR in this pathological pain model. First, we determined
the anti-nociceptive efficacy of a CB2R agonist (JWH-133) in an inflammatory

52
pain model. Second, we evaluated potential side effects and tolerance to JWH-
133. Co-administration of JWH-133 and morphine was also done to assess
potential functional interactions between CB2R and MOR agonists. Together, the
results of this study suggest that co-administration of a CB2R agonist with an
opioid could result in opioid-sparing effects in the treatment of inflammatory pain,
thus minimizing adverse effects and potentially protecting against the
development of opioid tolerance.
Materials and Methods
Subjects
Experiments were carried out with wild-type C57BL6/J mice obtained from
Jackson Laboratories (Bar Harbor, Maine). Mice used were male unless
specifically noted otherwise, and were tested between 8-14 weeks of age. All
mice were group-housed, and were kept on a standard 12:12h light-dark cycle
with ad libitum access to standard mouse chow and water. All animal care and
procedures conformed to the guidelines of the National Institutes of Health on the
Care and Use of Animals, and were approved by the Institutional Animal Care
and Use Committee of the Penn State University College of Medicine.
Drugs
Morphine sulfate was obtained from the National Institute on Drug Abuse
Drug Supply (Bethesda, MD). JWH-133 (CB2R agonist), SR144528 (SR2, CB2R

53
antagonist), naloxone (MOR antagonist), and Rimonabant (SR1, CB1R
antagonist) were obtained from Cayman Chemical (Ann Arbor, MI.). SP600125
(SP6), an inhibitor of c-Jun N-terminal kinases (JNK), was obtained from Sigma-
Aldrich (St. Louis, MO [Bennett 2001].) Drugs were dissolved in isotonic 0.9%
saline (90 %) with Cremaphor (5 %) and ethanol (5 %), and administered via
intraperitoneal (i.p.) injection in a volume of 10 mL/kg body weight.
General Experimental Protocols
JWH-133 dose–response curves. To assess the anti-nociceptive efficacy of JWH-
133, groups of age- and sex-matched drug-naïve mice (n=4-7 per dose) were
given log-scale doses of JWH-133 (0.01, 0.03, 0.1, 0.3, 1, 3, 10 mg/kg) via i.p.
injection. Anti-nociception was measured using the formalin test 60 min after JWH-
133 injection. A similar dose-response analysis was performed for morphine (0.01-
10 mg/kg, i.p.). The data from these experiments were fitted to standard sigmoidal
curves with variable slope to determine the ED50 values and maximal doses for
both drugs. For subsequent experiments the minimum dose of JWH-133 (1 mg/kg)
required to produce a maximal anti-nociceptive response in the formalin test was
used. To determine which receptor was responsible for mediating the anti-
nociceptive effect of 1 mg/kg JWH-133 in the formalin test, it was co-administered
with CB2R antagonist SR2 (10 mg/kg), CB1R antagonist SR1 (10 mg/kg) or MOR
antagonist naloxone (10 mg/kg).

54
To assess tolerance, mice were injected (i.p.) once-daily with either 10
mg/kg morphine or 1 mg/kg JWH-133 for up to eleven consecutive days. This
duration of repeated dosing is sufficient to cause complete tolerance to the anti-
nociceptive effects of morphine. The mice were assessed in the formalin model 60
min following drug administration on the final day. To assess cross-tolerance
between morphine and JWH-133, some groups were given a challenge dose of
the alternate drug (i.e., mice given 10 days of morphine were given a JWH-133
challenge and vice versa) on the eleventh day. Some groups were co-
administered 10 mg/kg morphine and 1 mg/kg JWH-133 for ten days and then
tested for anti-nociception using morphine alone on day 11 to assess the potential
protective effects of JWH-133 co-administration on morphine tolerance.
Since previous published work from our laboratory has demonstrated that
JNK signaling is required for morphine tolerance, we examined whether JNK
signaling was required for morphine-induced cross-tolerance to JWH-133
[Marcus 2015; Yuill 2016]. This experiment was done by examining the efficacy
of JWH-133 (1 mg/kg) anti-nociception in the formalin model using mice that
received either five days of repeated morphine alone (10 mg/kg), mice that
received daily pre-treatment with SP6 (3 mg/kg) prior to morphine (10 mg/kg) and
mice receiving daily vehicle injections. Pre-treatment with SP6 was given 60 min
before injection of morphine.

55
Formalin Test
The formalin test is an extensively used model of inflammatory pain [Tjolsen
1992]. This method elicits a biphasic pattern of pain behavior, with a phase of acute
pain followed by a phase of inflammatory pain. The early (acute) phase is
generated by the activation of C and Aδ fibers as a result of needle penetration
into the hind paw. The late phase involves an inflammatory reaction due to
presence of formalin in peripheral tissue [Tjolsen 1992], the development of central
sensitization and additionally the activation of primary afferent nociceptors [Puig
1996; Coderre TJ 1997]. Mice were subjected to the formalin test to assess basal
differences in inflammatory pain response and the anti-nociceptive effect of
morphine and JWH-133 on pathological pain. Prior to testing, mice were
acclimated for 20 min in a Plexiglas (5”x5”x5”) observation chamber placed on a
transparent elevated platform. A mirror angled at 45° was placed below the
platform to allow for constant observation of the animal’s paws. Following
acclimation, mice were administered 10μL of a 2.5% formalin solution into the
plantar surface of a single hind paw using a 28 ½ gauge needle (Becton Dickinson,
Franklin Lakes, NJ). Immediately after the formalin injection, mice were returned
to the Plexiglas observation unit and nociceptive behavior was continuously
measured in 12 five-min intervals for a total testing time of 60 min. During each
five-min time bin, the duration spent performing pain-response behaviors was
recorded. The nociceptive behaviors were separated into three categories: (0) the
injected paw has little weight placed on it; (1) the injected paw is raised off of the
ground; (2) the injected paw is licked, shaken, or bitten. The amount of time spent

56
in each category was quantified and weighted with the composite pain score-
weighted scores technique (CPS-WST0,1,2), resulting in a Composite Pain Score
(CPS) for each five-min interval between 0 (no pain behaviors) to 2 (maximal pain
behavior; [G. Stennis Watsona 1997]). The Area Under the Curve (AUC, CPS x
time(min)) was calculated using the trapezoidal rule for the acute phase (0-15 min;
phase I) and the inflammatory phase (15-60 min; phase 2). To assess the anti-
nociceptive effects of drugs, mice were injected (i.p.) 60 min prior to the formalin
injection.
Measurement of body temperature
Body temperature was measured using a mouse rectal thermometer probe
(Physitemp, Clifton, NJ). Mice were measured immediately prior to, and 60 min
following drug administration. Hypothermia was reported as a percent change in
body temperature between pre-drug and 60 min post-drug measurements, as
described by the formula:
(%∆BT) =(post − drug temperature)– (pre − drug temperature)
pre − drug temperature× 100
Rotarod test
Motor impairment was measured using a Med Associates ENV-577-M
rotarod apparatus (St. Albans, VT). Animals were trained by undergoing six

57
training trials per day over two consecutive days. The maximum cut-off for
training trails was limited to 300s. Mice were placed on a rotating rod (3 cm in
diameter), which accelerated at a constant rate from 4 to 40 rpm over the 5 min
testing period. The time spent walking on top of the rod until the mouse either fell
off the rod, or slipped and held onto the rod to ride completely around was
recorded. Motor impairment was determined by calculating the change in
performance between the pre-test and post-test given 60 min after JWH-133,
morphine or vehicle injection.
Precipitated withdrawal
Physical dependence was induced using a series of 20 injections that
were given twice-daily for 10 days (5 mg/kg morphine, i.p.; 1 mg/kg JWH-133,
i.p.). Following 10 days of daily drug administration, withdrawal was precipitated
using an i.p. injection of vehicle, 10 mg/kg naloxone (to counter morphine), or 10
mg/kg SR2 (to counter JWH-133) 30 min after the final drug injection on the 11th
day. Somatic withdrawal symptoms (paw tremors, body tremors, diarrhea, and
jumps) were video recorded for 60 min after injection of naloxone, SR2, or
vehicle. Withdrawal symptoms were scored in alternating 5 min time intervals (5–
10, 15–20, 25–30, 35–40, 45–50, and 55–60 min, as described previously
[Morgan 2014]).

58
Co-administration
When animals were given multiple i.p. injections, the second injection was
administered on the opposing side of the body cavity. When testing the effects of
co-administered agonists (JWH-133 and morphine), each drug was injected at
the same time. When testing for agonist selectivity, antagonists were
administered via i.p. injection 30 min prior to agonist treatment.
Isobolographic analysis
This analysis was performed to determine whether the combined anti-
nociceptive effects of morphine and JWH-133 were sub-additive, additive, or
synergistic (super-additive). Full dose response curves were generated in the
formalin test (as described above) for JWH-133, morphine, and then a
combination that was co-administered in a fixed 1:10 dose ratio (see [Grabovsky
and Tallarida 2004; Tallarida and Raffa 2010; Kazantzis 2016] for detailed
explanation and formulas.) ED50 values for this combination were determined and
compared to a theoretically calculated ED50 value [Tallarida 2002]. This
theoretical value was determined using the dose-response curves of JWH-133
and morphine, alone, to generate a predicted additive curve using the formula
below [Tallarida and Raffa 2010; Kazantzis 2016].

59
𝐸(𝑎, 𝑏) = 𝐸𝐵
(𝑏 + 𝑏𝑒𝑞(𝑎) )𝑝
(𝑏 + 𝑏𝑒𝑞(𝑎) )𝑝 + 𝐶𝑏𝑝
Where the effect (E) of specific doses of two drugs (a,b) in combination is
estimated using the dose of drug b (beq(a)) that gives and equivalent response to
a specific dose of drug a (a), the ED50 of drug b (Cb), and the Hill slope of drug b
(p). If the experimentally determined ED50 of the combination is significantly lower
than the predicted value (according to a t-test), the combination is deemed
synergistic. If the two ED50 values are equal, the combination has only an
additive effect.
Data analysis
Values for anti-nociception, hypothermia, motor coordination, and
precipitated withdrawal were expressed as the mean ± standard error of the
mean (SEM). Data was analyzed using either one-way (Figure 3, Figure 4(c),
Figure 6(d) or two-way ANOVA (all other figures), followed by Bonferroni or
Dunnett post-hoc testing as appropriate. Analyses were performed using SPSS
statistical software (SPSS Incorporated, Chicago, IL). P<0.05 was considered
significant.

60
Results
Anti-nociceptive effect of JWH-133 in the formalin test
The anti-nociceptive effect of JWH-133 was assessed by performing a
dose-response analysis in the formalin test using male (Figure 1(a), N=32) and
female (Figure 1(b), N=28) mice. In males, JWH-133 diminished both acute (0-15
min post-formalin, ED50=0.23 mg/kg, [F(7,72)=9.72, p<0.0001]) and inflammatory
pain (15-60 min post-formalin, ED50=0.23 mg/kg, [F(7,216) = 8.42, p<0.00001]) in a
dose-dependent manner (Figure 5(a)). There was also a significant dose-
dependent effect in the levels of both acute pain (ED50=0.24 mg/kg, [F(7,60)= 4.62,
p=0.0004]) and inflammatory pain (ED50= 0.20 mg/kg, [F(7,180)=3.69, p=0.0014]) in
female mice (Figure 5(b)). Both male and female mice displayed maximal anti-
nociceptive effects with 1 mg/kg JWH-133.
Comparison of the anti-nociceptive effect of JWH-133 and morphine in the
formalin test
The anti-nociceptive effect of a maximal dose of JWH-133 (1 mg/kg) was
compared to a maximal dose of morphine in groups of male mice (10 mg/kg,
Figure 6, N=4 per group). Analysis of the AUC of pain behavior revealed that
both morphine and JWH-133 reduced pain behavior relative to the vehicle group
in the acute (F(2,9)= 32.28, P < 0.0001, Figure 6(a)) and inflammatory phases
(F(2,9)= 132.47, P < 0.0001, Figure 6(b)) of the formalin test. Moreover, morphine
produced a greater anti-nociceptive effect than JWH-133 in both phases (P <
0.037 acute; P < 0.0001 inflammatory).

61
Figure 5. Anti-nociceptive efficacy of JWH-133.
Wild-type male (a) and female (b) mice (N=4-7 per dose) were tested using the formalin model. Testing was conducted 60 min after i.p. injection of JWH-133 (0.01, 0.03, 0.1, 0.3, 1, 3, 10 mg/kg). The area under the curve (AUC) represents the pain behavior obtained from a composite pain score. Treatment with JWH-133 reduced both acute (F(6,35)= 16.02, p<0.0001) and inflammatory (F(6,35)= 21.53, p<0.0001) pain in a dose-dependent fashion, with a maximal effect occurring at a dose of 1 mg/kg. The calculated ED50 values for acute and
inflammatory phases were 0.23 mg/kg and 0.23 mg/kg for males. For females the ED50 values were 0.24 mg/kg and 0.20 mg/kg.

62
Figure 6. Comparison of morphine and JWH-133 in the formalin test.
Mice (n=4–6 per dose) were tested in the formalin model 60 min after i.p. administration of maximal doses of either JWH-133 (1 mg/kg) or morphine (10 mg/kg). The area under the curve (AUC) represents the pain behavior obtained from the composite pain score. Both morphine and JWH-133 reduced pain behavior relative to the vehicle group in the acute (a) and inflammatory (b) phases. Morphine produced a greater anti-nociceptive effect than JWH-133 in both phases. *p<0.0001 for JWH-133 and morphine versus vehicle group (ANOVA with Bonferroni post hoc) and *+p<0.037 for morphine versus JWH-133 group (ANOVA with Bonferroni post hoc). ANOVA: analysis of variance.

63
Figure 7. JWH-133 acts through the CB2 Receptor.
The efficacy of JWH-133 (1 mg/kg, i.p.) in the formalin test was examined in the presence of multiple antagonists administered 30 minutes prior to JWH-133 treatment. (n=4-5 per treatment) The effect of JWH-133 was blocked by CB2R antagonist SR2 (10 mg/kg, i.p.), in both acute (p<0.05) and inflammatory pain (p<0.001) According to a one-way ANOVA with Bonferonni post-hoc. JWH-133 was not impacted by CB1R antagonist rimonabant (SR1, 10 mg/kg) nor by MOR antagonist naloxone (10 mg/kg).

64
JWH-133 acts through the CB2 Receptor
To establish that JWH-133 was selective for the CB2R, it was co-
administered with antagonists for CB1R (SR1, 10 mg/kg) or CB2R (SR2, 10
mg/kg). The impact of these antagonists on the anti-nociceptive effects of JWH-
133 was then measured using male mice in the formalin test (Figure 7, N=4-5 per
group). We found that JWH-133 alleviates formalin-induced pain behaviors in
both the acute (Figure 7(a), F(5,18)= 9.21, P < 0.0001) and inflammatory (Figure
5(b), F5,18= 18.69, P < 0.0001) phases. The anti-nociceptive effect of JWH-133
was blocked by SR2 (P < 0.006, acute; P < 0.0001, inflammatory), but not by
SR1 (P = 1.00, acute; P = 0.83 inflammatory). Indeed, the effect of JWH-133 is
not mediated through CB1R since its combination with a CB1R antagonist (SR1)
produced anti-nociceptive effects when compared to SR1 alone for the acute (P
< 0.012) and inflammatory (P < 0.03) phases. There were no difference in pain
values between JWH-133 alone or JWH-133 combined with SR1 (P = 1.00 acute
and P = 0.833 inflammatory).
JWH-133 does not act through mu-opioid receptor
JWH-133 suppressed formalin-induced pain scores in both the acute (F(3,12)
= 10.39, P < 0.001) and inflammatory (F(3,12) = 58.09, P < 0.0001) phases. This
anti-nociceptive effect is not blocked by the MOR antagonist naloxone (p = 0.688,
acute; P = 0.751, inflammatory). Formalin-induced pain behavior was similar in
groups receiving JWH-133 alone or co-administered naloxone (p = 0.69, acute; P
= 0.75, inflammatory (Figure 7(c) and (d)).

65
Evaluation of side effects
Male mice were injected daily with JWH-133 (1 mg/kg) or morphine (10
mg/kg) for seven days to examine the possibility of either acute or cumulative
negative side effects such as hypothermia. JWH-133 had no impact on body
temperature (F(1,27)=2.78, p=0.11), and the effects of JWH-133 on body
temperature were not significantly different from vehicle after one (p=0.530), four
(p=0.7359), or seven (p=0.8917) days of repeated administration according to
Dunnett’s post-hoc testing (Figure 8(a)). By contrast, morphine does have a
significant impact on hypothermia (F(1,27)=9.88, p<0.004), and was significantly
different from vehicle on day one (P<0.0001) but not on days 4 (p=0.1334) or 7
(p=0.1673, Figure 8(a)).
Motor impairment was measured using a rotarod apparatus because this
is a common side effect for both opiates and many cannabinoids. Similarly to the
hypothermia test, JWH-133 treatment did not produce motor impairment
(F(1,27)=0.17, p=0.68), and did not differ from vehicle on days 1 (p=0.999), 4
(p=0.7774), or 7 (p=0.9998, Figure 8(b)). Comparatively, morphine treatment had
a significant effect on motor impairment (F(1,27)=21.32, p<0.0001) and was
significantly different from vehicle on day 1 (p=0.0001), but not on days 4
(p=0.2120), or 7 (p=0.8429, Figure 8(b)).

66
Precipitated somatic withdrawal
Following ten days of twice daily injections of either JWH-133 or morphine,
antagonists were given to determine whether somatic withdrawal symptoms
could be precipitated. Precipitation of physical withdrawal from JWH-133 using
the CB2R antagonist SR2 did not result in any detectable somatic withdrawal
events following repeated JWH-133. Similarly, SR2 did not elicit somatic
withdrawal symptoms in mice treated with daily saline (Figure 8(c), N=4 per
group). However, treatment of morphine-tolerant mice with naloxone elicited an
increase in paw tremors (p<0.001), body tremors (p<0.001), and jumping
behavior (p<0.001) compared to vehicle treated mice.

67

68
Lack of observed tolerance to 11 once-daily injections of JWH-133
In order to investigate the development of tolerance, male mice (N= 3-5
per group) were assessed using the formalin model following 1, 6, or 11 days of
JWH-133 (1 mg/kg; Figure 9(a)) or morphine (10 mg/kg; Figure 9(b)) treatment.
Analysis of the AUC of pain behavior revealed that JWH-133 reduced pain
behavior relative to the vehicle group in the acute (F(3,15) = 15.01, P < 0.0001)
and inflammatory phases (F(3,15) = 41.21, P < 0.0001) of the formalin test after 1,
6 or 11 days of repeated administration ( P <0.0002 acute and P < 0.0001
inflammatory). The magnitude of JWH-133’s anti-nociceptive efficacy did not
diminish following 1, 6 or 11 days of continuous administration (P = 1.000). In the
acute phase, morphine reduced pain behavior in comparison to the vehicle group
(F(3,15) = 47.05, P < 0.0001) after 1 and 6 days of repeated administration (P <
0.0001), but not after 11 days (P = 0.149). In the inflammatory phase, morphine
Figure 8. Lack of JWH-113 adverse effects.
Mice (n=5-7 per group) were chronically injected with a maximal dose of JWH-133 (1 mg/kg), or morphine (10 mg/kg) for seven days to look for either acute or cumulative physical side effects. The hypothermic effects of JWH-133 were not significantly different from vehicle according to a two-way ANOVA with Dunnett’s post-hoc test (A). The same trends were observed when measuring rota-rod motor impairment of JWH-133 (B). Comparatively, morphine injected mice showed significant hypothermic effects on day 1(p=0.0001), but not 4(p=0.1334), or 7(p=0.1673). Morphine also showed significant motor impairment on day 1(p=0.0001), but not 4(p=0.2120), or 7(p=0.8429, Fig 3B). Following ten days of chronic drug administration, antagonists and frequency of withdrawal behaviors were recorded for60 minutes (C). While SR2 administration following chronic JWH-133 did not result in withdrawal behavior, morphine-treated mice challenged with naloxone showed significant amounts of paw tremors (p<0.001). Body tremor (p<0.001), and jumping (p<0.001) relative to vehicle.

69
reduced pain behavior relative to the vehicle group (F(3,15) = 55.03, P < 0.0001)
after 1, 6 or 11 days of repeated administration (P <0.031). However, this anti-
nociceptive effect of morphine was different in a time-dependent manner since
greater anti-nociceptive effect is observed at day 1 relative to day 6 (P < 0.001)
and from day 6 relative to day 11 (P < 0.039).

70
Figure 9. Lack of observed tolerance to JWH-133.
Mice (n=3-5 per group) were measured in the formalin test following 1, 6, or 11 days of JWH-133 (1 mg/kg, a) or morphine (10 mg/kg, b) administration. Data are expressed as mean ± S.E.M. * P < 0.0001 for JWH-133 or morphine vs vehicle group (ANOVA with Bonferroni post-hoc); + P < 0.001 for morphine day 1 vs morphine day 6 group (ANOVA with Bonferroni post-hoc); # P < 0.039 for morphine day 6 vs morphine day 11 group (ANOVA with Bonferroni post-hoc).

71
JWH-133 and morphine cross-tolerance
Male mice (N= 5-8 per group) were injected for ten days with either vehicle
or JWH-133 (1 mg/kg), and then given a challenge dose of morphine (10 mg/kg)
on day 11 to assess cross-tolerance in the formalin test (Figure 10(a)). The
repeated (10 days) administration of vehicle or JWH-133 and a challenge dose (on
day 11) of morphine (F(2,13) = 325.04, p < 0.0001) suppressed CPS relative to
control group in a time-dependent manner ( F(22,143) = 11.46, p < 0.0001, Figure
10(a)). This suppression was observed from 5 to 15 min (acute phase; F(2,13)
=22.24 (5), 5.86 (10), 4.57 (15); p < 0.0001 for all time points) and from 20 to 50
min (inflammatory phase 2; F(2,13) = 25.92 (20), 37.56 (25), 71.57 (30), 71.67 (35),
57.10 (40), 12.30 (45) and 4.37 (50); p < 0.035 for all time points) post-formalin
injection compared to the control group.
Analysis of the AUC of pain behavior revealed that both the repeated
vehicle group and the repeated JWH-133 group showed anti-nociceptive efficacy
of morphine challenge relative to the vehicle-only control group in both phase 1
(F(2,13) = 63.58, p < 0.0001; Figure 6(b)) and phase 2 (F(2,13) = 356.04, p < 0.0001;
Figure 10(c)) of the formalin test.
Male mice were also injected for ten days with either vehicle or morphine
and then given a challenge dose of the JWH-133 drug on day 11 to assess cross-
tolerance in the opposing direction. Only the repeated (10 days) administration of
vehicle and a challenge dose (on day 11) of JWH-133 (F(2,14) = 17.61, p < 0.0001)
suppressed CPS relative to control group in a time-dependent manner (F(22,154) =
6.30, p < 0.0001, Figure 10(d)). This suppression was observed at 5 (acute phase;

72
F(2,14) =5.65; p < 0.001) min and from 30 to 50 min (inflammatory phase 2; F(2,14) =
5.65 (30), 24.97 (35), 24.93 (40), 13.84 (45); p < 0.016 for all time points) post-
formalin injection compared to the control or repeated morphine + JWH-133
challenge groups. There was no difference in formalin-induced pain values
between the control and repeated morphine + JWH-133 challenge groups from 5
to 60 min (P > 0.064 for all time points) except at 20 (P < 0.001) and 45 (P < 0.004)
min.
In these mice, analysis of the AUC of pain behavior revealed that only the
repeated vehicle with JWH-133 challenge group produced anti-nociception relative
to the control or repeated morphine + JWH-133 challenge groups in both Phase 1
(F(2,14) = 22.39, p < 0.0001; Figure 10(e)) and Phase 2 (F(2,14) = 6.57, p < 0.01;
Figure 10(f)) of the formalin test. Similar formalin-induced pain values are observed
in the control and repeated morphine + JWH-133 challenge groups in both phases
(P=1.00 acute and inflammatory).
In groups of female mice (N= 4-7), analysis of the AUC of pain behavior
revealed that only the repeated vehicle with JWH-133 challenge group produced
anti-nociception relative to the control or repeated morphine + JWH-133 challenge
groups in both Phase 1 (F(2,8) = 13.72, p < 0.003; Figure 10(g)) and Phase 2 (F(2,8)
= 9.71, p < 0.01; Figure 10(h)) of the formalin test. Similar formalin-induced pain
values were observed in the control and repeated morphine + JWH-133 challenge
groups in both phases (P = 0.164 acute; 1.00 inflammatory).

73
Figure 10. Cross-tolerance between JWH-133 and morphine.
The formalin model of inflammatory pain was utilized to examine cross-tolerance for the anti-nociceptive effects of a challenge dose of morphine (10 mg/kg, i.p.) after 10 days of chronic JWH-133 (1 mg/kg i.p.) administration (A), and cross-tolerance for the anti-nociceptive effects of JWH-133 after 10 days of chronic morphine administration (B, n=5-8 per group). A two-way ANOVA failed to reveal an effect of chronic JWH-133 administration on morphine anti-nociceptive efficacy in either acute pain (p=0.8583) or inflammatory pain (p=0.2992). However, chronic injection of morphine significantly reduced the anti-nociceptive efficacy of JWH-133 both in acute [F(2,20)=16.63, p<0.0001] and inflammatory pain [F(2,20=28.13, p<0.001].

74
JWH-133 co-administration modestly attenuates morphine tolerance
Male mice (N=4 per group) were co-administered 10 mg/kg morphine and
1 mg/kg JWH-133, given morphine alone, or given vehicle for up to ten days. All
groups were then tested for morphine-induced anti-nociception using a challenge
dose morphine alone on (days 2, 6, 11) to assess potential protective effects of
JWH-133 on morphine tolerance.
On day 2, analysis of the AUC of pain behavior revealed that
vehicle+morphine challenge, morphine+morphine challenge or combination
(morphine+JWH-133)+morphine challenge groups showed lower pain values
relative to the control group for both phases (F(3,12) = 60.26 acute and 360.55
inflammatory, p < 0.0001, Figure 11). In the inflammatory phase, the vehicle +
morphine challenge group shows lower pain values relative to morphine +
morphine challenge group (P < 0.004).
On day 6, the AUC of pain behavior also revealed that vehicle+morphine
challenge, morphine+morphine challenge or combination (morphine+JWH-
133)+morphine challenge groups showed lower pain values relative to the control
group for both phases (F(3,12) = 117.69 acute and 165.98 inflammatory, p <
0.0001). In the acute and inflammatory phases, the vehicle +morphine challenge
or the combination+morphine challenge groups showed lower pain values
relative to morphine+morphine challenge group (P < 0.002, acute and P < 0.047,
inflammatory). These results show the protective effect of JWH-133 co-
administration with repeated administration of morphine on both phases of the

75
formalin test. In the inflammatory phase only, the vehicle+morphine challenge
group showed the lowest pain values (P < 0.004).
On day 11, the AUC of pain behavior revealed that vehicle+morphine
challenge and combination (morphine+JWH-133)+morphine challenge groups
showed lower pain values relative to the control group for both phases (F(3,13) =
74.44 acute and 179.36 inflammatory, p < 0.0001). The control group showed
similar values to the morphine + morphine challenge group for both phases (P =
1.00 acute and P = 0.05 inflammatory). In the acute and inflammatory phases,
the vehicle+morphine challenge or the combination + morphine challenge groups
show lower pain values relative to morphine + morphine challenge group (P <
0.019, acute and P < 0.037, inflammatory). In both phases, the vehicle+morphine
challenge group shows the lowest pain values (P < 0.0001 acute and
inflammatory phases).

76
Figure 11. JWH-133 co-administration modestly protects against morphine
tolerance.
The efficacy of morphine (10 mg/kg) phase I (a) and phase 2 (b) anti-nociception in the formalin model compared between mice receiving daily morphine alone (10 mg/kg), and mice receiving daily morphine (10 mg/kg) and daily JWH-133 (1 mg/kg). Data are expressed as mean ± S.E.M. (n = 4 per group). * P < 0.004 vs vehicle group (ANOVA with Bonferroni post-hoc); + P < 0.047 vs morphine + morphine challenge group (ANOVA with Bonferroni post-hoc); # P < 0.003 vs combination + morphine challenge group (ANOVA with Bonferroni post-hoc).

77
JNK signaling is partially responsible for morphine-induced cross-tolerance to
JWH-133
The efficacy of a challenge dose of JWH-133 (1 mg/kg) in the formalin
model was compared between groups of male mice that received either five days
of morphine alone (10 mg/kg), five days of the JNK inhibitor SP600125 (3
mg/kg) and morphine (10 mg/kg), or five days of vehicle (Figure 12, N=4 per
group). Pre-treatment with SP600125 prior to morphine caused a significant
increase in JWH-133 efficacy (F(2,9)=91.86 acute, and F(2,9)=55.37 inflammatory,
p<0.0001 for both phases). Therefore, repeated co-administration of SP600125
(3 mg/kg) with morphine appears to reduce observed cross-tolerance to JWH-
133. In both phases of the formalin test, the vehicle and JWH-133 challenge
group shows the lowest pain values (P < 0.0001 acute and inflammatory
phases).
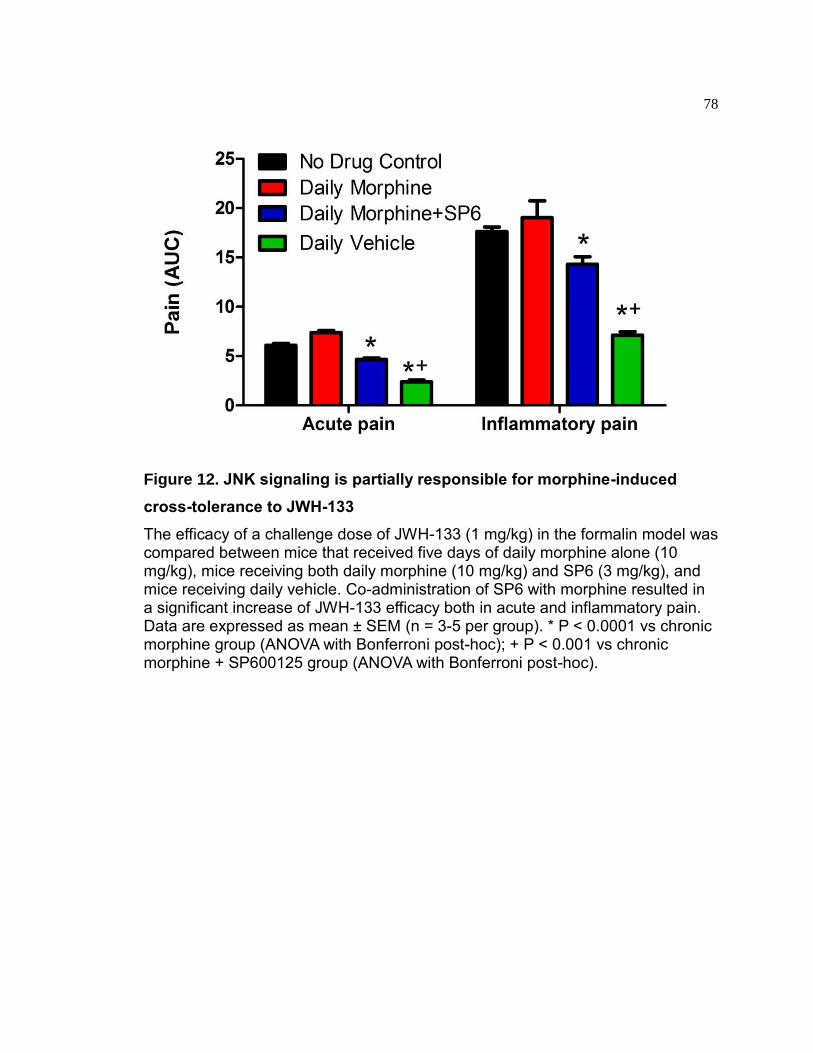
78
Figure 12. JNK signaling is partially responsible for morphine-induced
cross-tolerance to JWH-133
The efficacy of a challenge dose of JWH-133 (1 mg/kg) in the formalin model was compared between mice that received five days of daily morphine alone (10 mg/kg), mice receiving both daily morphine (10 mg/kg) and SP6 (3 mg/kg), and mice receiving daily vehicle. Co-administration of SP6 with morphine resulted in a significant increase of JWH-133 efficacy both in acute and inflammatory pain. Data are expressed as mean ± SEM (n = 3-5 per group). * P < 0.0001 vs chronic morphine group (ANOVA with Bonferroni post-hoc); + P < 0.001 vs chronic morphine + SP600125 group (ANOVA with Bonferroni post-hoc).

79
Isobolographic analysis
To examine the potential synergy between JWH-133 and morphine,
isobolographic analysis was used to compare the theoretical and experimental
dose-response curves for a 1:10 fixed ratio dose combination of JWH-133 and
morphine in the formalin test (Figure 13(a) and (b)). In this analysis, there is a
substantial difference in the maximal efficacies of JWH-133 and morphine.
Therefore, a non-linear isobolographic analysis [Tallarida and Raffa 2010] was
used. In phase I of the formalin test, the theoretical ED50 of 1:10 JWH-
133/morphine combination (0.78 mg/kg) was found to overlap with the
experimentally determined value (0.72 mg/kg, Figure 13(c)). This indicates that
the 1:10 fixed ratio combination is likely to be additive, but not synergistic. In
phase II of the formalin test, the experimentally determined ED50 (0.62 mg/kg)
appears to be lower than the predicted ED50 (0.93 mg/kg, Figure 13(d)).
However, the 95% confidence intervals of the two values are overlapping. Thus
our results demonstrate that the combined anti-nociceptive effects of JWH-133
and morphine are additive, but likely not synergistic.
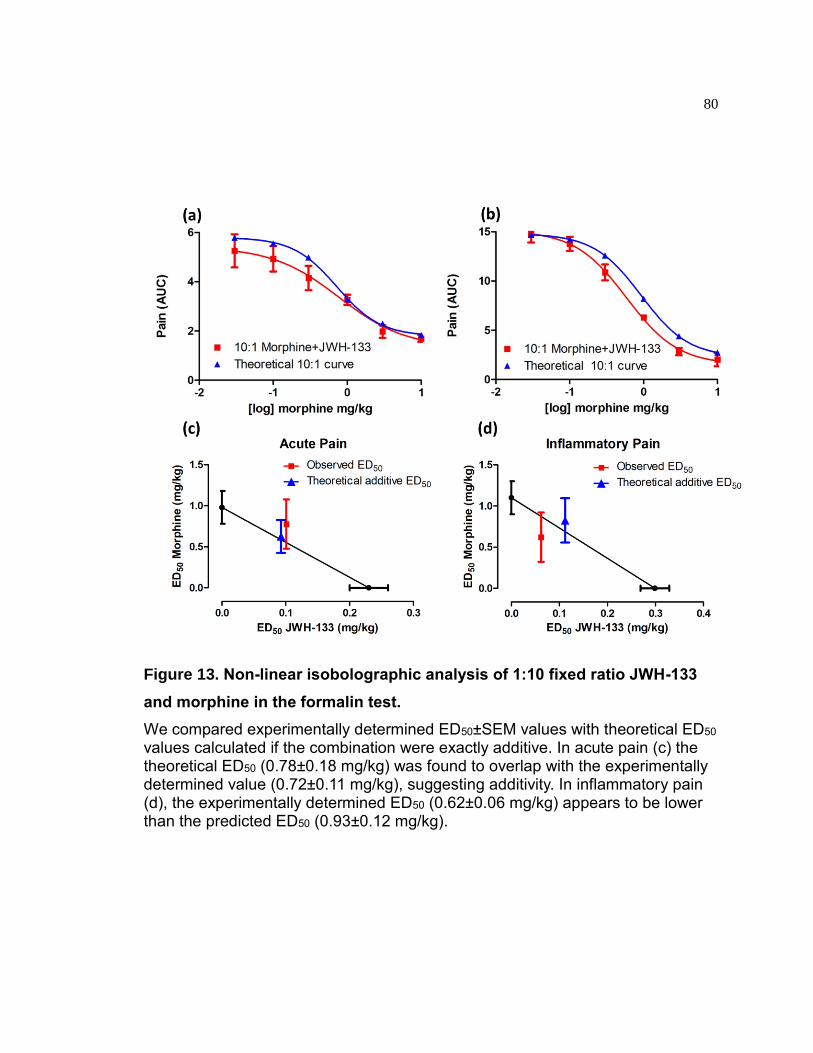
80
Figure 13. Non-linear isobolographic analysis of 1:10 fixed ratio JWH-133
and morphine in the formalin test.
We compared experimentally determined ED50±SEM values with theoretical ED50 values calculated if the combination were exactly additive. In acute pain (c) the theoretical ED50 (0.78±0.18 mg/kg) was found to overlap with the experimentally determined value (0.72±0.11 mg/kg), suggesting additivity. In inflammatory pain (d), the experimentally determined ED50 (0.62±0.06 mg/kg) appears to be lower than the predicted ED50 (0.93±0.12 mg/kg).

81
Discussion
The primary goal of this study was to test the hypothesis that CB2R
functionally interacts with the opioid system to modulate inflammatory pain. First,
we examined the effects of CB2R activation alone and found that JWH-133
causes dose-dependent anti-nociception in both the acute and inflammatory
phases of the formalin test (Figure 4). Using the CB2R-seletive antagonist SR2,
we demonstrated that the anti-nociceptive effects of JWH-133 occur through
activation of CB2R (Figure 6). The efficacy of JWH-133 is consistent with
previous work demonstrating that CB2R agonists can produce robust anti-
nociception in other inflammatory and neuropathic pain models [Gutierrez 2011;
Hsieh 2011; Brownjohn and Ashton 2012]. For example, systemic administration
of the CB2R agonist GW405833 to mice and rats was able to reverse
hyperalgesia in a model of inflammatory pain involving complete Freund’s
adjuvant (CFA, [Whiteside 2005]). In addition, two other CB2R agonists A-
836339 and AM-1241 were anti-nociceptive in a rat CFA model when
administered spinally (intrathecally) or via direct intraplantar injection to the
inflamed paw [Hsieh 2011]. Previous studies report that AM-1710 was able to
completely reverse paclitaxel-induced neuropathic pain in mice [Deng 2015b].
Interestingly, continuous administration of AM-1710 before and after paclitaxel
treatment can prevent the development of neuropathic pain for several weeks
[Rahn 2014].
Additionally, we sought to examine potential sex differences in JWH-133
anti-nociception between males and females using the formalin test (Fig 5(a),

82
5(b)). Females displayed higher levels of pain behavior at every JWH-133 dose.
However this difference appears to be due to higher basal levels of pain in
female mice rather than reduced sensitivity to the anti-nociceptive effects of
JWH-133. The finding that females generally exhibit more pain behaviors is
consistent with animal and human studies of pain [Cepeda 2003; Fillingim 2009;
Bergeson 2016; Blanton 2016; Henderson-Redmond 2016]. Our study did not
suggest that female mice were more sensitive to JWH-133, as the calculated
ED50 values and maximally efficacious doses did not differ between male and
female mice. However, there is evidence for sex differences in the response to
cannabinoids. Previous studies have shown that female rats are more sensitive
to Δ9-THC, and develop tolerance to it more quickly [Wakley 2014]. It is possible
that this discrepancy is due to the fact that many previously reported sex
differences in cannabinoid response involved compounds that also activate
CB1R. It is also possible that previously reported sex differences in rats are
partially species-dependent.
We also sought to assess potential negative side effects of JWH-133. Our
results closely aligned with previous studies that failed to demonstrate adverse
effects of other CB2R agonists (AM-1241, AM-1710 and HU-308) in rodents
[Hanus L 1999; Kinsey 2011; Deng 2015b]. We did not observe negative side
effects typically associated with agonists for the CB1R and/or opioid receptors
which includes; hypothermia, motor incoordination, or antagonist-induced
somatic withdrawal symptoms (tremors, jumping, diarrhea) for JWH-133 (Figure
8). However, it is important to recognize that this list of potential adverse effects

83
is not exclusive, and more research studies are needed to assess the impact of
these adverse side effects and possibly sex-specific differences and their impact
on pain pathways and alleviation of pain. In particular, the short duration of
repeated JWH-133 administration and limited number of observed somatic
withdrawal symptoms here are not sufficient to fully assess the possibility that
subtle somatic symptoms might occur. Furthermore, since we only examined
possible somatic symptoms, it is possible that precipitated JWH-133 withdrawal
could cause affective and behavioral withdrawal symptoms such as anxiety,
depression, and anhedonia. Lastly, sex-differences could also be found.
Regardless of the pain model used, multiple studies observe a lack of
tolerance to repeated administration of CB2R agonists. For example, daily
systemic administration of AM-1710 attenuated chemotherapy-evoked
neuropathic pain for up to 8 days with no evidence of tolerance [11]. Similarly,
the CB2R agonist, JWH015 (intrathecal, once daily), reversed surgery-induced
allodynia for up to 5 days without tolerance [Romero-Sandoval 2008]. In this
study we report a lack of tolerance to the anti-nociceptive effects of JWH-133
after 11 consecutive days of administration (Figure 9). However, it is possible
that tolerance to JWH-133 may occur with longer periods of chronic dosing
and/or the affective component of pain (anxiety, depression, and anhedonia)
could be dysregulated. Previous studies evaluating the absence of tolerance of
CB2 agonists failed to evaluate the affective component of pain.
To fully understand why CB2R agonists do not cause obvious tolerance, it
is important to increase our understanding of the mechanisms through which

84
they produce anti-nociception and possible role of changes in the affective
component of pain and/or sex-differences may shed light into this lack of
tolerance to sensory stimulation. First, the anti-nociceptive effect of JWH-133
might be caused by the ability of CB2R activation to suppress inflammation at the
site of injury [Ehrhart 2005; Benito 2008]. For example, paclitaxel-induced
neuropathic pain in rats and mice can be prevented if the CB2R agonist, MDA7 is
co-administered during treatment with paclitaxel [Naguib 2012]. Rats receiving
MDA7 did not display the expected increase of markers for microglial and
astrocyte activation in the spinal cord that are associated with inflammatory
response during paclitaxel treatment. One possible explanation for this finding is
that CB2R activation blocked the normal inflammatory response to paclitaxel,
thus preventing inflammation, nerve damage, and pain.
Beyond mediating inflammation, a second proposed mechanism for this
crosstalk between cannabinoid and the opioid systems is agonist-stimulated
release of endogenous ligands. We have directly examined the possibility that
JWH-133-mediated anti-nociception might be mediated through endogenous
opioid release. Our results indicate that anti-nociceptive efficacy of JWH-133 was
not prevented by antagonism of MOR with naloxone (Figure 6). This is consistent
with previous studies using CB2R agonist GW405833 to reduce CFA-induced
inflammatory allodynia in the presence of naltrexone, another MOR antagonist
[Whiteside 2005]. These findings suggest that certain CB2R agonists (including
JWH-133) can attenuate inflammatory pain through a MOR-independent
mechanism. Differences in the ability of CB2R agonists to stimulate endogenous

85
opioid release might be due to signaling bias. For example, JWH-133 appears to
be strongly biased towards G protein dependent mechanisms and biased against
signaling through β-arrestin recruitment [Atwood 2012; Dhopeshwarkar 2016;
Soethoudt 2017]. However, there are other mechanisms of action that could
explain these results including morphine-stimulated endocannabinoid release
and/or morphine-stimulated down-regulation of CB2R.
Despite the lack of tolerance to the anti-nociceptive effects of JWH-133,
we observe morphine-induced cross-tolerance for JWH-133 following repeated
daily injections of morphine (Figure 9). However, this effect is unidirectional since
repeated daily injections of JWH-133 do not cause cross-tolerance to a challenge
dose of morphine. Previous work has demonstrated that tolerance to the anti-
nociceptive and anti-allodynic effects of morphine require JNK signaling [Melief
EJ 2010; Hervera 2012; Marcus 2015]. Therefore, examined the possibility that
morphine-induced cross-tolerance to JWH-133 was also mediated through a JNK
signaling mechanism. We found that this cross-tolerance to JWH-133 is only
partially attenuated by SP600125, a broad-spectrum inhibitor of all three JNK
isoforms (Figure 11). However, there are other mechanisms of action that could
explain these results including morphine-stimulated endocannabinoid release
and/or morphine-stimulated down-regulation of CB2R. Thus, the specific
mechanism of action for opioid-induced cross-tolerance to JWH-133 is not
understood and requires additional study.
Interestingly, our results suggest that JWH-133 co-administration may be
protective against the development of morphine tolerance (Figure 10). This

86
finding is in agreement with a previous study of cancer pain that showed a delay
in morphine tolerance when morphine was co-administered with a sub-analgesic
dose of AM-1241 [Zhang 2016]. While intriguing, the magnitude of the JWH-133
effect on morphine tolerance is quite modest in our study.
Co-administration of CB2R agonists with opioids may also result in
increased anti-nociceptive efficacy due to drug synergism. One potential benefit
of a synergistic interaction between JWH-133 is that it could allow effective anti-
nociception with considerably lower morphine doses. Such an opioid-sparing
effect for JWH-133 could help minimize the adverse effects of opioids. Human
studies have shown increased pain relief in patients when inhaled cannabis use
is combined with opioids [Abrams 2011]. Therefore, we performed isobolographic
analysis of morphine and JWH-133 in combination to test for potential synergy
(Figure 12). We were able to demonstrate an additive effect of this combination,
but were not able to demonstrate a greater-than-additive (synergistic) effect.
However, only one fixed ratio (10:1) drug combination was used. Nonetheless,
even an additive effect of CB2R agonists could be beneficial due to the lack of
adverse effects [Stone 2014].
Conclusion
Our study demonstrates unidirectional morphine-induced cross-tolerance
to JWH-133. This finding raises the possibility that CB2R may functionally
interact with the opioid system to modulate inflammatory pain. Interestingly, we
also find that co-administration of JWH-133 with morphine produces an additive

87
anti-nociceptive effect in the formalin test while also potentially protecting against
morphine tolerance. Taken together, these findings highlight the potential
therapeutic applications for different CB2R ligands in pathological pain states
without tolerance or adverse effects associated with currently available
treatments. In particular, further investigation into the use of CB2R agonist as
adjuvant treatments to opioid therapy should be investigated.

88
Unpublished Experiment 1: Efficacy of JWH-133 in hotplate and tail-flick assays
Rationale
Used together, the hotplate and tail flick assays serve to measure acute
thermal nociceptive thresholds in subjects tested. While both utilize heat as a
noxious stimuli, tail flick elicits a more reflexive response at the level of the spinal
cord and hotplate involves more cognitive and emotional components [Espejo
1993]. These assays are beneficial as preliminary measurements of a drug’s
anti-nociceptive efficacy for several reasons. These assays can be performed
quickly and without any injury or lasting effects on the subjects, and are simple to
perform. However, acute pain as a result of noxious stimuli is entirely different
from clinically relevant chronic and pathological pain modalities. Furthermore, the
possibility of a learning effect often makes interpretation of long-term testing in
these assays difficult. They could still be valuable in the case of JWH-133 as the
literature surrounding this agonist in pain models is so limited.
There is considerable clinical evidence that opiate analgesics differentially
affect responses to pain produced by brief noxious stimulation and those
associated with persistent pain states [Fields 2004]. Acute pain is usually highly
sensitive to opiate therapy yet high doses of opiates are required to relieve
inflammatory and neuropathic pain states because they are longer term and
involve sensitization [Basbaum 2009]. Similar results are observed with CB1R-
selective agonists [Liu 2006; da Fonseca Pacheco 2008]. However, CB2R may

89
offer a more effective target in pathological pain relative to acute pain due to its
role in inflammatory modulation. Therefore, comparison of JWH-133 effect in
thermal and inflammatory models may be informative.
Procedure
The anti-nociceptive effect of JWH-133 was assessed in male mice by
performing dose-response analyses in the hotplate and tail flick tests as
described previously. Animals (n=6 per dose) were given log-scale doses of the
CB2R agonist JWH-133 ( 0.01, 0.1, 0.3, 1, 3 mg/kg) via i.p. injection. Post-drug
measurements were taken 60 min following JWH-133 administration in both
assays. Values are reported as the percentage of the maximal possible effect
(%MPE).
Results
In the hotplate test, JWH-133 increased %MPE in a dose-dependent
fashion [F(4,29)=7.1, p=0.0006] until a maximum value of 20±4 %MPE was
reached (Figure 14). Similarly, JWH-133 had a dose-dependent effect in the tail
flick test [F(4,29)=4.9, (Figure 14)} up to a maximum of 15±4 %MPE.
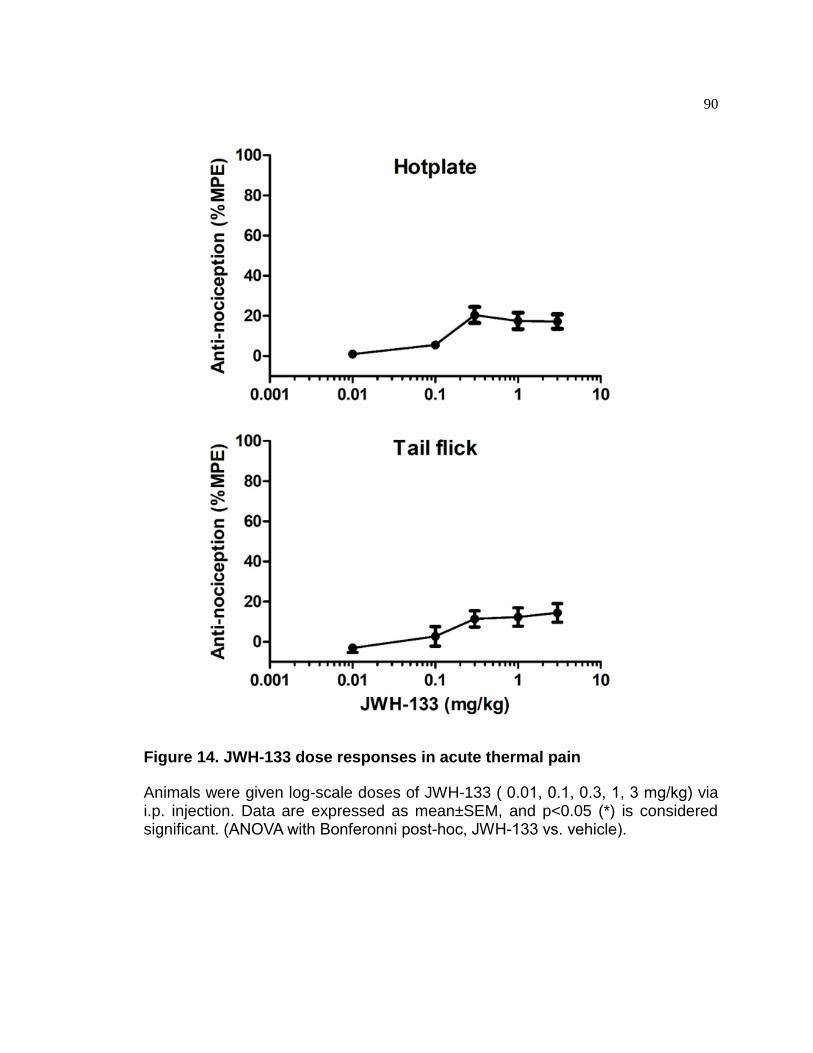
90
Figure 14. JWH-133 dose responses in acute thermal pain
Animals were given log-scale doses of JWH-133 ( 0.01, 0.1, 0.3, 1, 3 mg/kg) via i.p. injection. Data are expressed as mean±SEM, and p<0.05 (*) is considered significant. (ANOVA with Bonferonni post-hoc, JWH-133 vs. vehicle).

91
Discussion
While JWH-133 demonstrated anti-nociceptive efficacy in both models of
acute pain, the results and possible analyses are limited. Even at maximal doses,
the highest effect was between 15-20% MPE. Comparatively, opioids such as
morphine and fentanyl can demonstrate 100% MPE (Figures 2, 3, 15).
Moreover, the shift from almost no effect to maximal effect occurred over only 1
order of magnitude increase in dose. The low effect for JWH-133 suggests that
using these assays to measure subtle changes or interactions with other
compounds would not be worthwhile.

92
Unpublished Experiment 2: JWH-133 and fentanyl cross-tolerance.
Rationale
Tolerance to fentanyl, another mu opioid receptor agonist, is not affected
by pre-treatment with SP600125 (figure 16, [Marcus 2015]). Interestingly,
tolerance to fentanyl is absent in G protein-coupled receptor kinase (GRK) 3 KO
mice suggesting that tolerance for this agonist is mediated exclusively through a
classic mechanism involving GRK 3 phosphorylation and β-arrestin2 recruitment
[Melief 2010]. Because cross-tolerance with morphine appeared to be partially
dependent upon JNK signaling, we wanted to examine interaction with an opiate
that desensitizes through non-JNK mechanisms. Therefore, we examined
whether daily fentanyl injections could cause cross-tolerance to JWH-133.
Procedure
Following the procedures for morphine tolerance, male mice were injected
(i.p.) once-daily with either 0.3 mg/kg fentanyl or 1 mg/kg JWH-133 for up to eleven
consecutive days. The mice were assessed in the formalin model 60 min following
drug administration on the final day. To assess cross-tolerance between fentanyl
and JWH-133, some groups were given a challenge dose of the alternate drug
(i.e., mice given 10 days of fentanyl were given a JWH-133 challenge and vice
versa) on the eleventh day. Some groups were co-administered 0.3 mg/kg
fentanyl and 1 mg/kg JWH-133 for ten days and then tested for anti-nociception

93
using morphine alone on day 11 to assess the potential protective effects of JWH-
133 co-administration on morphine tolerance.
Results
We found that fentanyl also causes cross-tolerance to JWH-133, and that
it is similar in magnitude to what we observe for morphine. In these mice,
analysis of the AUC of pain behavior revealed that only the repeated vehicle with
JWH-133 challenge group produced anti-nociception relative to the control or
repeated morphine + JWH-133 challenge groups in both Phase 1 (F(2,14) = 22.39,
p < 0.0001; Figure 15) and Phase 2 (F(2,14) = 6.57, p < 0.01; Figure 15) of the
formalin test.

94
Figure 15. JWH-133 and fentanyl cross tolerance.
The formalin model of inflammatory pain was utilized to examine cross-tolerance between fentanyl and JWH-133. The AUCs for acute and inflammatory pain are shown for male mice that received a challenge dose of JWH-133 after 10 days of repeated fentanyl administration Data are expressed as mean ± S.E.M. (n = 5-8 per group). *** P < 0.0001 vs control group (ANOVA with Bonferroni post-hoc); ** P < 0.01 vs control (ANOVA with Bonferroni post-hoc).

95
Discussion
While chronic morphine administration diminishes CB2R agonist efficacy,
the reverse effect is not observed (Figure 9). Similar trends were observed with
JWH-133 and fentanyl (Figure 15). The results of these experiments suggest that
there is unidirectional cross-tolerance between CB2R agonists and opioids. Our
lab and others have demonstrated that the mechanisms governing tolerance to
morphine and fentanyl occur through different cellular mechanisms [Melief EJ
2010; Hervera 2012; Marcus 2015]. Furthermore, we found that the cross
tolerance of JWH-133 due to chronic morphine was partially attenuated by the
addition of the JNK inhibitor SP6 (Fig 11.) This agrees with previous findings
showing that JNKs mediate morphine tolerance, and also suggests that they are
also involved in the mechanism causing this tolerance to cross over to JWH-133
(Figure 16 below, [Marcus 2015]). Thus the specific mechanism of action for
opioid-induced cross-tolerance to JWH-133 is not understood and requires
additional study.

96

97
Figure 16. Tolerance to the anti-nociceptive effects of fentanyl is not
blocked by SP6.
JNK inhibition attenuates chronic tolerance to the hypothermic but not the anti-nociceptive effects of repeated treatment with fentanyl (0.3 mg/kg). Wild-type mice were treated with vehicle (black line with triangles), 3 mg/kg SP600125 (dashed black line with diamonds), or 10 mg/kg SP600125 (black line with diamonds) 60 min prior to administration of fentanyl (0.3 mg/kg) for ten consecutive days. Tail-flick anti-nociception (a), hotplate nociception (b), and body temperature (c) were measured 60 min later. Treatment with only SP600125 (3 mg/kg) attenuated tolerance to the hypothermic effects of chronically administered 0.3 mg/kg fentanyl alone. Data are expressed as mean ± S.E.M. (n = 10–24 per group). *p < 0.0001 for SP600125 (3 mg/kg) vs. vehicle or SP600125 (10 mg/kg) groups (ANOVA, Bonferroni post hoc).

98
Chapter 5 – Mechanisms of Cannabinoid Tolerance through the CB1 Receptor
Δ9-THC produces potent anti-nociceptive effects but, like opiates, is
subject to the onset of tolerance [Morgan 2014]. As discussed, there is also
overlap in the mechanisms of CB1R and opioid desensitization. The objective of
this study was to examine potential mechanisms of tolerance from daily
administration of Δ9-THC using a model of pathological pain. As with MOR,
CB1R desensitization could involve GRK or JNK phosphorylation. S426A/S430A
mutant mice express two serine to alanine point mutations on the CB1R sites
phosphorylated by GRK, and exhibit enhanced response to Δ9-THC [Morgan
2014]. Therefore, we used the formalin test to compare the development of Δ9-
THC tolerance in these animals and in wild-type mice. Additionally, The impact of
c-Jun N-terminal kinase (JNK) inhibitor SP600125 (SP6) on the development of
Δ9-THC tolerance was also assessed in wild-type mice, as previous research has
suggested it is significant in the development of tolerance to morphine [Marcus
2015].

99
Results
Δ9-THC Tolerance in S426A/S430A mice
Male wild-type C57BL/6 mice, or S426A/S430A mutant mice were
subjected to the formalin test after receiving daily injections of Δ9-THC (6 mg/kg)
ranging from zero to twelve days (Figure 17). Wild-type mice exhibited complete
tolerance to the anti-nociceptive effects of Δ9-THC in the formalin test after eight
days of daily Δ9-THC. These results are consistent with the findings of studies
assessing Δ9-THC tolerance in other models of pain, which often show onset of
tolerance by three days of chronic administration. Interestingly, we find that Wild-
type mice show development of tolerance in inflammatory pain, however,
S426A/S430A mice showed reduced tolerance to Δ9-THC specifically for
inflammatory pain.
SP6 impairs Δ9-THC tolerance in the formalin test
In a second experiment, groups of wild-type mice were also tested for up
to twelve days receiving SP6 (3 mg/kg), Δ9-THC (6 mg/kg), or SP6 and Δ9-THC
(Figure 18). Anti-nociception was then measured in the formalin test to determine
the role of JNK signaling in the acquisition of tolerance. Administration of SP6
alone provides effective anti-nociception in both pain types with no observed
tolerance within 12 days of chronic administration. Moreover, co-administration of
SP6 with Δ9-THC reduces the tolerance developed to Δ9-THC in both acute (A),
and inflammatory (B) pain.

100
Figure 17. Δ9-THC tolerance in the formalin test.
Wild-type (n=34, black) and S426A/S430A mutant (n=27, red) mice were given chronic daily doses of Δ9-THC (6 mg/kg, i.p). The anti-nociceptive efficacy of Δ9-THC was then assessed using the formalin model, which allows measurement of both acute (A) and inflammatory (B) pain.

101
Figure 18. Δ9-THC and SP6 in the formalin test.
Male wild-type mice (n=23) were given chronic daily injection of either vehicle (black), SP600125 (SP6) alone (3 mg/kg, red), Δ9-THC (6 mg/kg, blue), or SP6 in combination with Δ9-THC (6 mg/kg, white). The formalin model was used to assess acute (A) and inflammatory (B) pain.

102
Chapter 6 - General Discussion and Conclusion
The primary goal of this study was to test the hypothesis that CB2R
functionally interacts with the opioid system to modulate inflammatory pain.
Additionally, we tested mechanisms mediating tolerance to multiple opioids, and
to the prototypical cannabinoid Δ9-THC.
First, we examined the effects of CB2R activation alone both in acute and
inflammatory pain. In the case of our study, we found mild anti-nociceptive effect
in acute thermal pain (Figure 4). Moreover, we found that JWH-133 causes
considerable dose-dependent anti-nociception in both the acute and
inflammatory phases of the formalin test (Figures 5&6). As discussed, there are
considerable discrepancies among the literature investigating the potential
mechanisms and location of action for the anti-nociceptive effects of CB2R
agonists [Malan TP Jr 2001; Cichewicz 2004]. In the case of our study, the first
possibility is inhibition of inflammation at the site of formalin injection. The
efficacy of JWH-133 in inflammatory pain compared to the limited efficacy in the
tail-flick and hotplate tests (Figure 4) suggests that the anti-nociceptive effect of
JWH-133 is related to reduction of inflammation [Ehrhart 2005; Benito 2008].
This is further supported by the literature. For example, paclitaxel-induced
neuropathic pain in rats and mice can be prevented if the CB2R agonist, MDA7 is
co-administered during treatment with the chemotherapeutic agent paclitaxel
[Naguib 2012]. Rats receiving MDA7 did not display the expected increase of
markers for microglial and astrocyte activation in the spinal cord that are

103
associated with inflammatory response during paclitaxel treatment. One possible
explanation for this finding is that CB2R activation blocked the normal
inflammatory response to paclitaxel, thus preventing inflammation, nerve
damage, and pain. Rats subjected to the spared nerve injury procedure were
found to self-administer AM-1241 for its anti-allodynic effects [Gutierrez 2011].
The dual CB1/CB2 agonist CP55,940 was found to dose-dependently reduce
paclitaxel-induced allodynia in CB1 KO mice [Deng 2015a]. Another CB2R-
selective agonist, AM-1710, was found to completely reverse paclitaxel-induced
neuropathic pain in mice [Deng 2015b]. Neuropathic allodynia can not only be
reduced, but also reversed by a CB2R agonist (MDA7) through reduction of
microglial activity and cytokine release [Naguib 2012]. Similarly, CB2R agonist O-
3223 reduces LPS-induced paw edema [Kinsey 2011]. Conversely, we did not
see a difference in formalin-induced paw edema between animals given JWH-
133 or vehicle as measured by microcaliper (appendix, Table 4), which agrees
with literature showing that JWH-133 also does not diminish λ-carrageenan-
induced inflammation [Elmes 2004]. However, this does not rule out mediation of
inflammation as a possibility, as paw edema is a very gross measure of
inflammation. More quantitative measurements, such as cytokine levels in tissue
of treated animals would be a more informative test to address mechanism.
A second potential mechanism is mediation of spinal inflammatory
response and sensitization. CB2R agonists could activate CB2R on mast or other
immune cells in the spine, thereby decreasing the inflammation-evoked release

104
of sensitizing molecules such as NGF, cytokines, or histamine. This could
decrease the sensitivity of primary afferent neurons and inhibit pain response.
It has been shown that Microglial CBR2 activation inhibits pERK in spinal cord,
which results in a reduction of TNF expression and microglial motility. Overall,
this would result in a reduction in pro-algesic factors that would create
hyperalgesia or allodynia in response to pathology.
An alternative explanation for CB2 receptor-mediated attenuation
of acute nociceptive responses is the putative presence of CB2R on primary
afferent fibers. Currently, there is conflicting evidence for the expression of CB2R
receptors on dorsal root ganglia [Hohmann 1999; Ross 2001]. However, it has
been demonstrated that intraplantar injection of JWH-133 directly inhibited
mechanically evoked responses of rat dorsal horn neurons [Elmes 2004].
Some studies suggest that CB2R activation triggers endogenous opioid
release in the spinal cord [Ibrahim 2005; Curto-Reyes 2010]. In our case, the
anti-nociceptive efficacy of JWH-133 was not prevented by antagonism of MOR
with naloxone (Figure 7). Similarly, the CB2R agonist GW405833 reduces CFA-
induced inflammatory allodynia even with co-administration of naltrexone,
another MOR antagonist [Whiteside 2005]. These findings suggest that certain
CB2R agonists (including JWH-133) can attenuate inflammatory pain through a
MOR-independent mechanism. Similarly, the inability of a CB1R antagonist to
block the effect of JWH-133 in our study reinforces that the mechanism occurs
directly through CB2R.

105
Differences between these studies may be attributable to signaling biases
of specific ligands. In recent years, mounting evidence of the importance for
CB2R in pathological pain has increased interest as demonstrated by the
synthesis of a variety of CB2R-selective cannabinoid agonists [Atwood 2010].
JWH-133 appears to be strongly biased towards G protein dependent
mechanisms and biased against signaling through β-arrestin recruitment;
meaning that this agonist also very weakly desensitizes and internalizes CB2R
[Dhopeshwarkar and Mackie 2016]. JWH-133 is among the most CB2R-selective
cannabinoids, and recent analysis found that it has no activity at any off-target
proteins [Soethoudt 2017].
In addition to mechanism, there is also the question of location of effect.
Numerous studies have suggested ability of CB2R agonists to reduce allodynia
and hyperalgesia is spinally mediated, and that intrathecal administration is
effective [Romero-Sandoval 2008; Yamamoto 2008; Curto-Reyes 2010; Deng
2015b]. Conversely, some studies report CB2R agonists show efficacy from
systemic but not intrathecal administration [Brownjohn and Ashton 2012]. As
such, caution should be taken to assume the same mechanism of action across
different CB2R ligands or pain modalities. This further suggests that more
localized administration of JWH-133 should be performed in the future.
Additionally, we sought to examine potential sex differences in JWH-133
anti-nociception between males and females using the formalin test. The finding
that females generally exhibit more pain behaviors is consistent with animal and
human studies of pain [Cepeda and Carr 2003; Fillingim 2009; Bergeson 2016;

106
Blanton 2016; Henderson-Redmond 2016]. Our study did not suggest that female
mice were more sensitive to JWH-133, as the calculated ED50 values and
maximally efficacious doses did not differ between male and female mice.
However, there is evidence for sex differences in the response to cannabinoids.
Previous studies have shown that female rats are more sensitive to Δ9-THC, and
develop tolerance to it more quickly [Wakley 2014]. Female rats also exhibit more
severe adverse effects (sedation, catalepsy) and precipitated withdrawal from Δ9-
THC [Marusich 2014]. In human studies, the same trends (faster onset of
tolerance, more severe adverse effects) are reported for female cannabis
smokers (for review, see [Craft 2013]). It is frequently suggested that this is due
to a higher percentage of adipose tissue in females. Cannabinoids are highly
lipophilic and stored in adipose cells.
Regardless of the pain model used, multiple studies have observed a lack
of tolerance to repeated administration of CB2R agonists. For example, daily
systemic administration of AM-1710 attenuated chemotherapy-evoked
neuropathic pain for up to 8 days with no evidence of tolerance [Deng 2015b].
Similarly, the CB2R agonist, JWH015 (intrathecal, once daily), reversed surgery-
induced allodynia for up to 5 days without tolerance [Romero-Sandoval 2008]. In
this study we report a lack of tolerance to the anti-nociceptive effects of JWH-133
after 11 consecutive days of administration (Figure 9). This duration of dosing in
the formalin model was sufficient to generate complete tolerance to morphine,
fentanyl, and Δ9-THC. However, it is possible that tolerance to JWH-133 may
occur with longer periods of chronic dosing and/or the affective component of

107
pain (anxiety, depression, and anhedonia) could be dysregulated. Previous
studies evaluating the absence of tolerance of CB2R agonists failed to evaluate
the affective component of pain.
Previous work has demonstrated that tolerance to the anti-nociceptive and
anti-allodynic effects of morphine require JNK signaling [Melief EJ 2010; Hervera
2012; Marcus 2015]. Recent work has also shown that tolerance to morphine is
attenuated through the use of the JNK inhibitor SP600125 [Chen 2008; Guo
2009; Hervera 2012; Marcus 2015]. SP600125 is an anthrapyrazolone capable of
inhibiting JNK1, JNK2, and JNK3 with high affinity [Zhuang 2006]. It has been
demonstrated that systemic administration of SP600125 prevents
phosphorylation of JNK in the spinal cord, resulting in attenuation of tolerance to
the anti-nociceptive and anti-allodynic effects of morphine [Guo 2009; Hervera
2012]. We have also previously demonstrated that multiple JNK isoforms
mediate morphine tolerance [Yuill 2016]. We also demonstrated that inhibition of
JNK attenuates the onset of tolerance to Δ9-THC. Therefore, we wanted to
examine the possibility that morphine-induced cross-tolerance to JWH-133 was
also mediated through a JNK signaling mechanism. We found that this cross-
tolerance to JWH-133 is only partially attenuated by SP600125, a broad-
spectrum inhibitor of all three JNK isoforms (Figure 12). Moreover, there was
cross-tolerance between JWH-133 and fentanyl, despite fentanyl tolerance being
JNK-independent (Figure 15). However, there are other mechanisms of action
that could explain these results including morphine-stimulated endocannabinoid
release and/or morphine-stimulated down-regulation of CB2R.

108
Interestingly, our results suggest that JWH-133 co-administration may be
protective against the development of morphine tolerance (Figure 11). This
finding is in agreement with a previous study of cancer pain that showed a delay
in morphine tolerance when morphine was co-administered with a sub-analgesic
dose of AM-1241 [Zhang 2016]. While intriguing, the magnitude of the JWH-133
effect on morphine tolerance is quite modest in our study. Furthermore, our
results do not suggest any specific mechanisms to explain the effect. However,
one possibility is that CB2R activation might suppress the release of microglial
cytokines involved in the development of opioid tolerance [Watkins 2009]. JWH-
133 blocks JNK phosphorylation on microglia [Correa 2010]. In vitro models
suggest that opioid mediated pro-inflammatory cytokine release is attenuated
through concurrent CB2R activation [Merighi 2012]. This protective effect of
CB2R against opioid tolerance raises the possibility that they might be useful as
an adjuvant to traditional opiate treatment. It should also be noted that
Cannabioids do impact cytochrome-mediated metabolism, which could alter the
pharmacokinetic profile of opioids [Abrams 2011].
Co-administration of CB2R agonists with opioids may also result in
increased anti-nociceptive efficacy due to drug synergism. One potential benefit
of a synergistic interaction between JWH-133 is that it could allow effective anti-
nociception with considerably lower morphine doses. Therefore, we performed
isobolographic analysis of morphine and JWH-133 in combination to test for
potential synergy (Figure 13). We were able to demonstrate an additive effect of
this combination, but were not able to demonstrate a greater-than-additive

109
(synergistic) effect. However, only one fixed ratio (10:1) drug combination was
used. Nonetheless, even an additive effect of CB2R agonists could be beneficial
due to the lack of adverse effects [Stone 2014].
Future directions
While both inflammatory pain and neuropathy are types of pathological
pain, literature suggests that CB2R may impact the two differently [Zhang 2003].
Rats subjected to the spared nerve injury procedure were found to self-
administer AM-1241 for its anti-allodynic effects [Gutierrez 2011]. The dual
CB1R/CB2R agonist CP55,940 was found to dose-dependently reduce
paclitaxel-induced allodynia in CB1RKO mice [Deng 2015a]. Another CB2R-
selective agonist, AM-1710, was found to completely reverse paclitaxel-induced
neuropathic pain in mice [Deng 2015b]. Interestingly, continuous administration
of AM-1710 before and after paclitaxel treatment can prevent the development of
neuropathic pain for several weeks [Rahn 2014]. Due to the marked differences
in physiology between neuropathic and inflammatory pain our results from the
formalin test should be replicated in models of neuropathy.
From a scientific perspective, it is also important to investigate the
mechanisms governing CB2R/MOR interactions. Measuring in vivo changes to
CB2R expression and activity may explain any observed cross-tolerance
between CB2R and MOR agonists. Tissue from mice given chronic morphine
could be analyzed to measure changes in CB2R mRNA and protein expression,

110
as well as CB2R binding and G-protein coupling. Previous studies have
demonstrated that changes in CB2R expression during pathological conditions
are measurable in the dorsal root ganglia (DRG [Svizenska 2013]). As CB2R
functions by coupling to Gi/o protein [Izzo 2001], measurement of G protein
activity using agonist-stimulated [35S]GTPγS binding assays would provide a
direct measurement of CB2R desensitization. This technique has been used to
measure CB2R coupling and desensitization previously [Bouaboula 1999; Marini
2013].
Conclusion
The results of these experiments suggest that there is unidirectional cross-
tolerance between CB2R agonists and multiple different opioids. While chronic
morphine administration diminishes CB2R agonist efficacy, the reverse effect is
not observed. Overall these findings suggest that CB2R may functionally interact
with the opioid system to modulate anti-nociception in the formalin test in
response to inflammatory pain. This information will provide context for the
circumstances under which CB2R agonists may prove beneficial clinically, as
well as limitations to their use. Diminished efficacy of CB2R agonists due to
opiate tolerance demonstrates the importance of applying them proactively as an
adjuvant to opiates rather than as a reactionary measure after opiates have been
administered. Furthermore, co-administration of a CB2R agonist with morphine

111
suggests both anti-nociceptive synergy and protection against tolerance. Further
investigation of this combination in treatment of pathological pain is warranted.
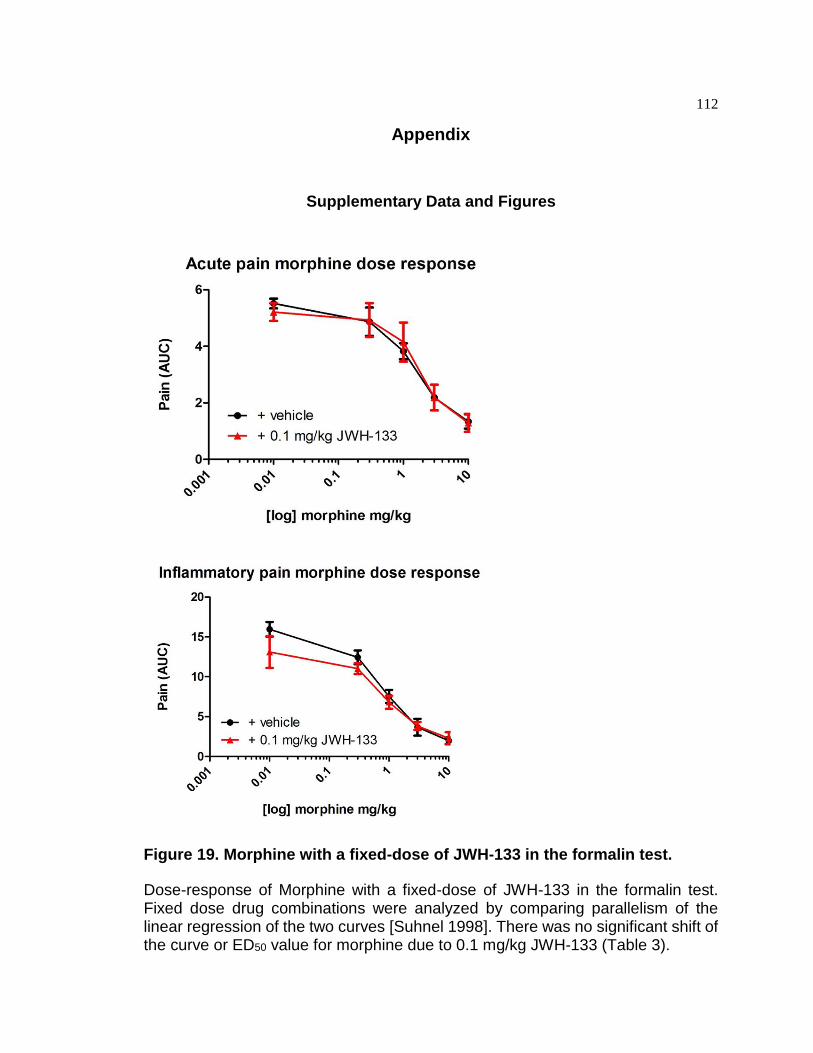
112
Appendix
Supplementary Data and Figures
Figure 19. Morphine with a fixed-dose of JWH-133 in the formalin test.
Dose-response of Morphine with a fixed-dose of JWH-133 in the formalin test. Fixed dose drug combinations were analyzed by comparing parallelism of the linear regression of the two curves [Suhnel 1998]. There was no significant shift of the curve or ED50 value for morphine due to 0.1 mg/kg JWH-133 (Table 3).

113
Table 3. ED50 Values in Formalin.
Drug Combination Phase I ED50 ( mg/kg + 95% CI)
Phase II ED50 ( mg/kg + 95% CI)
Morphine Alone 0.98 (0.89 to 1.08)
0.79 (0.54 to 0.98)
JWH-133 Alone 0.23 (0.89 to 1.08)
0.25 (0.89 to 1.08)
Morphine + 0.1 mg/kg fixed dose JWH-133
1.45 (1.20 to 1.70)
1.77 (1.51 to 2.04)
Experimental 1:10 JWH-133 and morphine fixed ratio
0.78 (0.61 to 0.90)
0.62 (0.54 to 0.72)
Theoretical 1:10 JWH-133 and morphine fixed ratio
0.72 (0.54 to 0.98)
0.93 (0.80 to 1.05)
Table 4. Paw edema following formalin.
Treatment Paw edema (% increase in thickness)
JWH-133 (1 mg/kg) 39.9 ± 6.6
JWH-133 (10 mg/kg) 36.1 ± 5.4
Morphine (10 mg/kg) 42.2 ± 5.8
Vehicle 38.9 ± 3.8
Paw edema (Table 4) was compared in mice (n=4-5 per group) receiving vehicle, JWH-133 (1 mg/kg), and morphine (10 mg/kg) to see if JWH-133 measurably reduced formalin inflammation. Thickness of the base of the right hind paw was measured immediately before and following testing using a digital micrometer.

114
Works Cited
Abrams, D. I., P. Couey, S. B. Shade, M. E. Kelly and N. L. Benowitz (2011).
"Cannabinoid-opioid interaction in chronic pain." Clin Pharmacol Ther
90(6): 844-851.
Akil, H., Mayer, DJ, Liebeskind, JC. (1976). "Antagonism of stimulation-produced
analgesia by naloxone, a narcotic antagonist." Science 5(191): 961-962.
Almeida, T. F., S. Roizenblatt and S. Tufik (2004). "Afferent pain pathways: a
neuroanatomical review." Brain Res 1000(1-2): 40-56.
Ashton, J. C., Friberg D, Darlington CL, Smith PF (2006). "Expression of the
cannabinoid CB2 receptor in the rat cerebellum: an immunohistochemical
study." Neurosci Lett(396): 113-116.
Ashton, J. C., R. M. Rahman, S. M. Nair, B. A. Sutherland, M. Glass and I.
Appleton (2007). "Cerebral hypoxia-ischemia and middle cerebral artery
occlusion induce expression of the cannabinoid CB2 receptor in the brain."
Neurosci Lett 412(2): 114-117.
Atwood, B. K., & Mackie, K. (2010). "CB2: a cannabinoid receptor with an identity
crisis." Br J Pharmacol 160: 467-479.
Atwood, B. K., J. Wager-Miller, C. Haskins, A. Straiker and K. Mackie (2012).
"Functional selectivity in CB(2) cannabinoid receptor signaling and
regulation: implications for the therapeutic potential of CB(2) ligands." Mol
Pharmacol 81(2): 250-263.

115
Bachhuber, M. A., B. Saloner, C. O. Cunningham and C. L. Barry (2014).
"Medical cannabis laws and opioid analgesic overdose mortality in the
United States, 1999-2010." JAMA Intern Med 174(10): 1668-1673.
Baek, J. H., C. L. Darlington, P. F. Smith and J. C. Ashton (2013). "Antibody
testing for brain immunohistochemistry: brain immunolabeling for the
cannabinoid CB(2) receptor." J Neurosci Methods 216(2): 87-95.
Bailey, C., J. Llorente, B. Gabra, F. Smith, W. Dewey, E. Kelly and G. Henderson
(2009). "Role of protein kinase C and mu-opioid receptor (MOPr)
desensitization in tolerance to morphine in rat locus coeruleus neurons."
Eur J Neurosci 29(2): 307-318.
Basbaum, A. I., D. M. Bautista, G. Scherrer and D. Julius (2009). "Cellular and
molecular mechanisms of pain." Cell 139(2): 267-284.
Beasou, P. (1969). "Response of cutaneous sensory units with unmyelinated
fibers to noxious stimuli." J Neurophysiol 32: 1025-1043.
Beltramo, M., N. Bernardini, R. Bertorelli, M. Campanella, E. Nicolussi, S.
Fredduzzi and A. Reggiani (2006). "CB2 receptor-mediated
antihyperalgesia: possible direct involvement of neural mechanisms." Eur
J Neurosci 23(6): 1530-1538.
Benito, C., R. M. Tolon, M. R. Pazos, E. Nunez, A. I. Castillo and J. Romero
(2008). "Cannabinoid CB2 receptors in human brain inflammation." Br J
Pharmacol 153(2): 277-285.
Bennett, B. L., D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J.
C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, S. S. Bhagwat, A. M.

116
Manning and D. W. Anderson (2001). "SP600125, an anthrapyrazolone
inhibitor of Jun N-terminal kinase." Proc Natl Acad Sci U S A 98(24):
13681-13686.
Bergeson, S. E., H. Blanton, J. M. Martinez, D. C. Curtis, C. Sherfey, B.
Seegmiller, P. C. Marquardt, J. A. Groot, C. L. Allison, C. Bezboruah and
J. Guindon (2016). "Binge Ethanol Consumption Increases Inflammatory
Pain Responses and Mechanical and Cold Sensitivity: Tigecycline
Treatment Efficacy Shows Sex Differences." Alcohol Clin Exp Res 40(12):
2506-2515.
Bernard, J., Huang, G., Besson, J. (1994). "The parabrachial area:
electrophysiological evidence for an involvement in visceral nociceptive
processes." J Neurophysiol 71: 1646-1660.
Blanton, H. e. a. (2016). "Gender-specific pain responses in the formalin test
using a synthetic tetracycline compound." J Pain 17(4): 50-51.
Bloom, A. S. and W. L. Dewey (1978). "A comparison of some pharmacological
actions of morphine and delta9-tetrahydrocannabinol in the mouse."
Psychopharmacology (Berl) 57(3): 243-248.
Bogoyevitch, M. and B. Kobe (2006). "Uses for JNK: the many and varied
substrates of the c-Jun N-terminal kinases." Microbiol Mol Biol Rev 70(4):
1061-1095.
Bouaboula, M., Desnoyer, N., Carayon, P., Combes, T., and Casellas, P. (1999).
"Gi Protein Modulation Induced by a Selective Inverse Agonist for the
Peripheral Cannabinoid Receptor CB2: Implication for
Intracellular Signalization Cross-Regulation." Mol Pharmacol 55: 473-480.

117
Braden, J. B. and M. D. Sullivan (2008). "Suicidal thoughts and behavior among
adults with self-reported pain conditions in the national comorbidity survey
replication." J Pain 9(12): 1106-1115.
Brownjohn, P. W. and J. C. Ashton (2012). "Spinal cannabinoid CB2 receptors as
a target for neuropathic pain: an investigation using chronic constriction
injury." Neuroscience 203: 180-193.
Buckley, N. E., McCoy,K.L., Mezey,E., Bonner,T., Zimmer,A., Felder,C.C.,
Glass,M., and Zimmer,A. (2000). "Immunomodulation by cannabinoids is
absent in mice deficient for the cannabinoid CB(2) receptor." Eur J
Pharmacol(396): 141-149.
Burgess, P. R. a. P., E.R. (1967). "Myeiinated afferent fibres responding
specifically to noxious stimulation on the skin." J. Physiol. (Lond.)(190):
541-562. .
Calignano, A., La Rana G, Giuffrida A, Piomelli D (1998). "Control of pain
initiation by endogenous cannabinoids." Nature(394): 277-281.
Canals, M. and G. Milligan (2008). "Constitutive activity of the cannabinoid CB1
receptor regulates the function of co-expressed Mu opioid receptors." J
Biol Chem 283(17): 11424-11434.
Carayon, P., J. Marchand, D. Dussossoy, J. M. Derocq, O. Jbilo, A. Bord, M.
Bouaboula, S. Galiegue, P. Mondiere, G. Penarier, G. L. Fur, T. Defrance
and P. Casellas (1998). "Modulation and functional involvement of CB2
peripheral cannabinoid receptors during B-cell differentiation." Blood
92(10): 3605-3615.

118
Carlisle, S. J., F. Marciano-Cabral, A. Staab, C. Ludwick, G.A. Cabral (2002).
"Differential expression of the CB cannabinoid receptor by 2 rodent
macrophages and macrophage-like cells in relation to cell activation." Int
Immunopharm(2): 69-82.
CDC (2011). "Vital signs: Overdoses of prescription opioid pain relievers –United
States, 1999-2008." Morbidity and Mortality Weekly Report 60 1487-1492.
CDC (2017). Wide-ranging online data for epidemiologic research (WONDER).
Atlanta, GA: CDC,, National Center for Health Statistics.
Cepeda, M. S. and D. B. Carr (2003). "Women Experience More Pain and
Require More Morphine Than Men to Achieve a Similar Degree of
Analgesia." Anesthesia & Analgesia: 1464-1468.
Chen, Y., C. Geis and C. Sommer (2008). "Activation of TRPV1 contributes to
morphine tolerance: involvement of the mitogen-activated protein kinase
signaling pathway." J Neurosci 28(22): 5836-5845.
Cichewicz, D. L. (2004). "Synergistic interactions between cannabinoid and
opioid analgesics." Life Sci 74(11): 1317-1324.
Clayton, N. M. W., A.W.; Collins, S.D.; Giblin, G.M.; Mitchell, B.L.; Goldsmith, P.;
Chessell, I.P.; O'Shaughnessy, C.; Bountra, C. (2004). "Anti-
hypersensitive and anti-inflammatory activity of the potent and selective
CB2 agonist GW842166X." Proc Brit Pharm Soc.
Coderre TJ, K. J. (1997). "Peripheral and central hyperexcitability: differential
signs and symptoms in persistent pain." Behav Brain Sci 20: 404-419.

119
Corbett, A., G. Henderson, A. McKnight and S. Paterson (2006). "75 years of
opioid research: the exciting but vain quest for the Holy Grail." Br J
Pharmacol 147 Suppl 1: S153-162.
Correa, F., M. Hernangomez, L. Mestre, F. Loria, A. Spagnolo, F. Docagne, V. Di
Marzo and C. Guaza (2010). "Anandamide enhances IL-10 production in
activated microglia by targeting CB(2) receptors: roles of ERK1/2, JNK,
and NF-kappaB." Glia 58(2): 135-147.
Cota, D., Marsicano G, Tschop M, et al (2003). "The endogenous cannabinoid
system affects energy balance via central orexigenic drive and peripheral
lipogenesis." J Clin Invest(112): 423-431.
Craft, R. M., J. A. Marusich and J. L. Wiley (2013). "Sex differences in
cannabinoid pharmacology: a reflection of differences in the
endocannabinoid system?" Life Sci 92(8-9): 476-481.
Curto-Reyes, V., Llames, S., Hidalgo, A., Menendez, L., and Baamonde, A.
(2010). "Spinal and peripheral analgesic effects of the CB2 cannabinoid
receptor agonist AM1241 in two models of bone cancer-induced pain." Br
J Pharmacol 160: 561-573.
da Fonseca Pacheco, D., A. Klein, A. de Castro Perez, C. M. da Fonseca
Pacheco, J. N. de Francischi and I. D. Duarte (2008). "The mu-opioid
receptor agonist morphine, but not agonists at delta- or kappa-opioid
receptors, induces peripheral antinociception mediated by cannabinoid
receptors." Br J Pharmacol 154(5): 1143-1149.

120
Deng, L., B. L. Cornett, K. Mackie and A. G. Hohmann (2015a). "CB1 Knockout
Mice Unveil Sustained CB2-Mediated Antiallodynic Effects of the Mixed
CB1/CB2 Agonist CP55,940 in a Mouse Model of Paclitaxel-Induced
Neuropathic Pain." Mol Pharmacol 88(1): 64-74.
Deng, L., J. Guindon, B. L. Cornett, A. Makriyannis, K. Mackie and A. G.
Hohmann (2015b). "Chronic cannabinoid receptor 2 activation reverses
paclitaxel neuropathy without tolerance or cannabinoid receptor 1-
dependent withdrawal." Biol Psychiatry 77(5): 475-487.
Derocq, J. M., Jbilo,O., Bouaboula,M., Segui,M., Clere,C., and Casellas,P.
(2000). "Genomic and functional changes induced by the activation of the
peripheral cannabinoid receptor CB2 in the promyelocytic cells HL-60.
Possible involvement of the CB2 receptor in cell differentiation." J Biol
Chem(275): 15621-15628.
Devane, W. A., Hanus, L., Breuer, A., Pertwee, R.G., Stevenson, L.A., Griffin, G.,
Gibson, D., Mandelbaum ,A., Etinger, A., and Mechoulam, R. (1992).
"Isolation and structure of a brain constituent that binds to the cannabinoid
receptor." Science(258): 1946-1949.
Dhopeshwarkar, A. and K. Mackie (2016). "Functional Selectivity of CB2
Cannabinoid Receptor Ligands at a Canonical and Noncanonical
Pathway." J Pharmacol Exp Ther 358(2): 342-351.
Di Rosa, M., Giroud JP, Willoughby DA. (1971). "Studies on the mediators of the
acute inflammatory response induced in rats in different sites by
carrageenan and turpentine." J Pathol 104: 15-29.

121
Doll, C., J. Konietzko, F. Poll, T. Koch, V. Hollt and S. Schulz (2011). "Agonist-
selective patterns of m-opioid receptor phosphorylation revealed by
phosphosite-specific antibodies." British Journal of Pharmacology(164):
298–307.
Dong, C., D. Yang, M. Wysk, A. Whitmarsh, R. Davis and R. Flavell (1998).
"Defective T Cell Differentiation in the Absence of JNK1." Science 282:
2092-2095.
Duncan, M., A. Mouihate, K. Mackie, C. M. Keenan, N. E. Buckley, J. S. Davison,
K. D. Patel, Q. J. Pittman and K. A. Sharkey (2008). "Cannabinoid CB2
receptors in the enteric nervous system modulate gastrointestinal
contractility in lipopolysaccharide-treated rats." Am J Physiol Gastrointest
Liver Physiol 295(1): G78-g87.
Ehrhart, J., D. Obregon, T. Mori, H. Hou, N. Sun, Y. Bai, T. Klein, F. Fernandez,
J. Tan and R. D. Shytle (2005). "Stimulation of cannabinoid receptor 2
(CB2) suppresses microglial activation." J Neuroinflammation 2: 29.
Eisenberg, E., M. Ogintz and S. Almog (2014). "The pharmacokinetics, efficacy,
safety, and ease of use of a novel portable metered-dose cannabis inhaler
in patients with chronic neuropathic pain: a phase 1a study." J Pain Palliat
Care Pharmacother 28(3): 216-225.
Elmes, S. J., M. D. Jhaveri, D. Smart, D. A. Kendall and V. Chapman (2004).
"Cannabinoid CB2 receptor activation inhibits mechanically evoked
responses of wide dynamic range dorsal horn neurons in naive rats and in
rat models of inflammatory and neuropathic pain." Eur J Neurosci 20(9):
2311-2320.

122
Espejo, E. M., D. (1993). "Structure of the rat's behaviour in the hot plate test."
Behavioural Brain Research 56: 171-176.
Facci, L., Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A (1995). "Mast
cells express a peripheral cannabinoid receptor with differential sensitivity
to anandamide and palmitoylethanolamide." Proc Natl Acad Sci U S A(92):
3376-3380.
Felder, C. C., K. E. Joyce, E. M. Briley, J. Mansouri, K. Mackie, O. Blond, Y. Lai,
A. L. Ma and R. L. Mitchell (1995). "Comparison of the pharmacology and
signal transduction of the human cannabinoid CB1 and CB2 receptors."
Molecular Pharmacology 48(3): 443-450.
Fernandez-Ruiz, J. (2009). "The endocannabinoid system as a target for the
treatment of motor dysfunction." Br J Pharmacol 156(7): 1029-1040.
Fields, H. (2004). "State-dependent opioid control of pain." Nat Rev Neurosci
5(7): 565-575.
Fillingim, R. B., C. D. King, M. C. Ribeiro-Dasilva, B. Rahim-Williams and J. L.
Riley, 3rd (2009). "Sex, gender, and pain: a review of recent clinical and
experimental findings." J Pain 10(5): 447-485.
Friedman, H., S. Specter, T. Klein, C. Newton, M. Rivenbark, D. Rowlands and
D. T. Walz (1990). "Auranofin-induced suppression of autoimmune
antibody production and inflammation in genetically autoimmune-prone
mice." Inflammation 14(4): 463-470.
G. Stennis Watsona, K. J. S., b, Terence J. Coderrec (1997). "Optimal scoring
strategies and weights for the formalin test in rats." Pain 70: 53-58.

123
Galiegue, S., Mary,S., Marchand,J., Dussossoy,D., Carriere,D., Carayon,P.,
Bouaboula,M., Shire,D., Le Fur,G., and Casellas,P. (1995). "Expression of
central and peripheral cannabinoid receptors in human immune tissues
and leukocyte subpopulations." Eur J Biochem(232): 54-61.
Gaoni, Y., Mechoulam, R. (1964). "Isolation, structure and partial synthesis of the
active constituent of hashish." J Am Chem Soc 86: 1646-1647.
Gaskin, D. J. and P. Richard (2012). "The economic costs of pain in the United
States." J Pain 13(8): 715-724.
Gong, J. P., E. S. Onaivi, H. Ishiguro, Q. R. Liu, P. A. Tagliaferro, A. Brusco and
G. R. Uhl (2006). "Cannabinoid CB2 receptors: immunohistochemical
localization in rat brain." Brain Res 1071(1): 10-23.
Gonzalez S, F.-R. J., Sparpaglione V, Parolaro D, Ramos JA. (2002). "Chronic
exposure to morphine, cocaine or ethanol in rats produced different effects
in brain cannabinoid CB(1) receptor binding and mRNA levels." Drug
Alcohol Depend 66(1): 77-84.
Grabovsky, Y. and R. J. Tallarida (2004). "Isobolographic analysis for
combinations of a full and partial agonist: curved isoboles." J Pharmacol
Exp Ther 310(3): 981-986.
Guindon, J. and A. G. Hohmann (2008). "Cannabinoid CB2 receptors: a
therapeutic target for the treatment of inflammatory and neuropathic pain."
Br J Pharmacol 153(2): 319-334.

124
Guo, R. X., M. Zhang, W. Liu, C. M. Zhao, Y. Cui, C. H. Wang, J. Q. Feng and P.
X. Chen (2009). "NMDA receptors are involved in upstream of the spinal
JNK activation in morphine antinociceptive tolerance." Neurosci Lett
467(2): 95-99.
Gutierrez, T., J. D. Crystal, A. M. Zvonok, A. Makriyannis and A. G. Hohmann
(2011). "Self-medication of a cannabinoid CB2 agonist in an animal model
of neuropathic pain." Pain 152(9): 1976-1987.
Halliday, A., Logue, V. (1972). "Painful sensations evoked by electrical
stimulation in the thalamus." Neurophysiology: 221-230.
Hanus L, B. A., Tchilibon S, Shiloah S, Goldenberg D, Horowitz M et al. (1999).
"HU-308: A specific agonist for CB2, a peripheral cannabinoid receptor."
Proc Natl Acad Sci U S A 96: 14228-14233.
Hayes, M. J. and M. S. Brown (2014). "Legalization of medical marijuana and
incidence of opioid mortality." JAMA Intern Med 174(10): 1673-1674.
Henderson-Redmond, A. N., M. B. Yuill, T. E. Lowe, A. M. Kline, M. L. Zee, J.
Guindon and D. J. Morgan (2016). "Morphine-induced antinociception and
reward in "humanized" mice expressing the mu opioid receptor A118G
polymorphism." Brain Res Bull 123: 5-12.
Herkenham, M., Lynn, A.B., Johnson, M.R., Melvin, L.S., de Costa, B.R., Rice,
K.C. (1991a). "Characterization and localization of cannabinoid receptors
in rat brain: a quantitative in vitro autoradiographic study." J Neurosci 11:
563-583.

125
Herkenham, M., Lynn, AB, de Costa, BR, Richfield, EK. (1991b). "Neuronal
localization of cannabinoid receptors in basal ganglia of the rat." Brain Res
Bull(547): 267-274.
Hervera, A., S. Leanez and O. Pol (2012). "The inhibition of the nitric oxide-
cGMP-PKG-JNK signaling pathway avoids the development of tolerance
to the local antiallodynic effects produced by morphine during neuropathic
pain." Eur J Pharmacol 685(1-3): 42-51.
Hine, B. (1985). "Morphine and delta 9-tetrahydrocannabinol: two-way cross
tolerance for antinociceptive and heart-rate responses in the rat."
Psychopharmacology (Berl) 87(1): 34-38.
Hohmann, A. G. and M. Herkenham (1999). "Cannabinoid receptors undergo
axonal flow in sensory nerves." Neuroscience 92(4): 1171-1175.
Holdridge, S. V. and C. M. Cahill (2007). "Spinal administration of a delta opioid
receptor agonist attenuates hyperalgesia and allodynia in a rat model of
neuropathic pain." Eur J Pain 11(6): 685-693.
Howlett, A. C., C. S. Breivogel, S. R. Childers, S. A. Deadwyler, R. E. Hampson
and L. J. Porrino (2004). "Cannabinoid physiology and pharmacology: 30
years of progress." Neuropharmacology 47 Suppl 1: 345-358.
Howlett, A. C., J. M. Qualy and L. L. Khachatrian (1986). "Involvement of Gi in
the inhibition of adenylate cyclase by cannabimimetic drugs." Mol
Pharmacol 29(3): 307-313.

126
Howlett, A. C., D. K. Scott and G. H. Wilken (1989). "Regulation of adenylate
cyclase by cannabinoid drugs. Insights based on thermodynamic studies."
Biochem Pharmacol 38(19): 3297-3304.
Hsieh, C., S. Brown, C. Derleth and K. Mackie (1999). "Internalization and
recycling of the CB1 cannabinoid receptor." J Neurochem 73(2): 493-501.
Hsieh, G. C., M. Pai, P. Chandran, B. A. Hooker, C. Z. Zhu, A. K. Salyers, E. J.
Wensink, C. Zhan, W. A. Carroll, M. J. Dart, B. B. Yao, P. Honore and M.
D. Meyer (2011). "Central and peripheral sites of action for CB(2) receptor
mediated analgesic activity in chronic inflammatory and neuropathic pain
models in rats." Br J Pharmacol 162(2): 428-440.
Huffman , J. W. e. a. (1999). "3-(10,10-Dimethylbutyl)-1-deoxy-D8-THC and
Related Compounds: Synthesis of Selective Ligands for the CB2
Receptor." Bioorganic & Medicinal Chemistry 7: 2905-2914.
Ibrahim, M., Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW (2006). "CB2
cannabinoid receptor mediation of antinociception." pain 122: 36-42.
Ibrahim, M. M., F. Porreca, J. Lai, P. J. Albrecht, F. L. Rice, A. Khodorova, G.
Davar, A. Makriyannis, T. W. Vanderah, H. P. Mata and T. P. Malan, Jr.
(2005). "CB2 cannabinoid receptor activation produces antinociception by
stimulating peripheral release of endogenous opioids." Proc Natl Acad Sci
U S A 102(8): 3093-3098.
Imai, S., M. Narita, S. Hashimoto, A. Nakamura, K. Miyoshi, H. Nozaki, N.
Hareyama, T. Takagi, M. Suzuki, M. Narita and T. Suzuki (2006).
"Differences in tolerance to anti-hyperalgesic effects between chronic

127
treatment with morphine and fentanyl under a state of pain." Nihon Shinkei
Seishin Yakurigaku Zasshi 26(5-6): 183-192.
Izzo, A., Mascolo, N., and Capasso, F. (2001). "The gastrointestinal
pharmacology of cannabinoids." Curr Opin Pharmacol 1: 597-603.
JC, A. (2102). "The use of knockout mice to test the specificity of
antibodies for cannabinoid receptors." Hippocampus 22: 643-644.
Jhaveri, M. D., D. R. Sagar, S. J. Elmes, D. A. Kendall and V. Chapman (2007).
"Cannabinoid CB2 receptor-mediated anti-nociception in models of acute
and chronic pain." Mol Neurobiol 36(1): 26-35.
Jin, W., S. Brown, J. P. Roche, C. Hsieh, J. P. Celver, A. Kovoor, C. Chavkin and
K. Mackie (1999). "Distinct domains of the CB1 cannabinoid receptor
mediate desensitization and internalization." J Neurosci 19(10): 3773-
3780.
Jorda, M. A., S. E. Verbakel, P. J. Valk, Y. V. Vankan-Berkhoudt, M. Maccarrone,
A. Finazzi-Agro, B. Lowenberg and R. Delwel (2002). "Hematopoietic cells
expressing the peripheral cannabinoid receptor migrate in response to the
endocannabinoid 2-arachidonoylglycerol." Blood 99(8): 2786-2793.
Kabli, N. and C. M. Cahill (2007). "Anti-allodynic effects of peripheral delta opioid
receptors in neuropathic pain." Pain 127(1-2): 84-93.
Katona, I., Sperlagh B, Sik A, Käfalvi A, Vizi ES, Mackie K (1999).
"Presynaptically located CB1 cannabinoid receptors regulate GABA
release from axon terminals of specific hippocampal interneurons." J
Neurosci(19): 4544-4558.

128
Kazantzis, N. P., S. L. Casey, P. W. Seow, V. A. Mitchell and C. W. Vaughan
(2016). "Opioid and cannabinoid synergy in a mouse neuropathic pain
model." Br J Pharmacol 173(16): 2521-2531.
Kinsey, S. G., A. Mahadevan, B. Zhao, H. Sun, P. S. Naidu, R. K. Razdan, D. E.
Selley, M. Imad Damaj and A. H. Lichtman (2011). "The CB2 cannabinoid
receptor-selective agonist O-3223 reduces pain and inflammation without
apparent cannabinoid behavioral effects." Neuropharmacology 60(2-3):
244-251.
Koch, T. and V. Hollt (2008). "Role of receptor internalization in opioid tolerance
and dependence." Pharmacol Ther 117(2): 199-206.
Kolesnikov, Y., S. Jain, R. Wilson and G. W. Pasternak (1996). "Peripheral kappa
1-opioid receptor-mediated analgesia in mice." Eur J Pharmacol 310(2-3):
141-143.
Koob, G. (2006a). "Drug addiction: The yin and yang of hedonic homeostasis."
Neuron 16: 893-896.
Koob, G. (2006b). "The neurobiology of addiction: a neuroadaptational view
relevantfor diagnosis." Addiction 101: 23-30.
Korzh, A., O. Keren, M. Gafni, H. Bar-Josef and Y. Sarne (2008). "Modulation of
extracellular signal-regulated kinase (ERK) by opioid and cannabinoid
receptors that are expressed in the same cell." Brain Res 1189: 23-32.
Kouznetsova, M., B. Kelley, M. Shen and S. A. Thayer (2002). "Desensitization of
cannabinoid-mediated presynaptic inhibition of neurotransmission

129
between rat hippocampal neurons in culture." Mol Pharmacol 61(3): 477-
485.
Kuhar, J. R., A. Bedini, E. J. Melief, Y. C. Chiu, H. N. Striegel and C. Chavkin
(2015). "Mu opioid receptor stimulation activates c-Jun N-terminal kinase 2
by distinct arrestin-dependent and independent mechanisms." Cell Signal
27(9): 1799-1806.
LaBuda, C. J. K., M.; Little, P.J. (2005). "Cannabinoid CB2 receptor agonist
activity in the hindpaw incision model of postoperative pain." Eur J
Pharmacol 527: 172-174.
Langerman, L., M. I. Zakowski, B. Piskoun and G. J. Grant (1995). "Hot plate
versus tail flick: evaluation of acute tolerance to continuous morphine
infusion in the rat model." J Pharmacol Toxicol Methods. 34: 23-27.
Leon, A., A. Buriani, R. Dal Toso, M. Fabris, S. Romanello, L. Aloe and R. Levi-
Montalcini (1994). "Mast cells synthesize, store, and release nerve growth
factor." Proc Natl Acad Sci U S A 91(9): 3739-3743.
Levitt, E. S. and J. T. Williams (2012). "Morphine desensitization and cellular
tolerance are distinguished in rat locus ceruleus neurons." Mol Pharmacol
82(5): 983-992.
Lichtman, A. H., Cook, S.A., Martin, B.R (1996). "Investigation of brain sites
mediating cannabinoid-induced antinociception in rats: evidence
supporting periaqueductal gray involvement." Pharmacol Biochem
Behav(57): 7-12.

130
Lichtman, A. H., Martin, B.R., (1997). "The selective cannabinoid antagonist SR
141716A blocks cannabinoid-induced antinociception in rats." Pharmacol
Biochem Behav(57): 7-12.
Liu, C. and J. M. Walker (2006). "Effects of a cannabinoid agonist on spinal
nociceptive neurons in a rodent model of neuropathic pain." J
Neurophysiol 96(6): 2984-2994.
Liu, Q. R., C. H. Pan, A. Hishimoto, C. Y. Li, Z. X. Xi, A. Llorente-Berzal, M. P.
Viveros, H. Ishiguro, T. Arinami, E. S. Onaivi and G. R. Uhl (2009).
"Species differences in cannabinoid receptor 2 (CNR2 gene): identification
of novel human and rodent CB2 isoforms, differential tissue expression
and regulation by cannabinoid receptor ligands." Genes Brain Behav 8(5):
519-530.
Lopez-Moreno, J. A., V. Echeverry-Alzate and K. M. Buhler (2012). "The genetic
basis of the endocannabinoid system and drug addiction in humans." J
Psychopharmacol 26(1): 133-143.
Lynch, M. E. and M. A. Ware (2015). "Cannabinoids for the Treatment of Chronic
Non-Cancer Pain: An Updated Systematic Review of Randomized
Controlled Trials." J Neuroimmune Pharmacol 10(2): 293-301.
Mackie, K. a. H., B. (1992). "Cannabinoids inhibit N-type calcium channels in
neuroblastoma-glioma cells." Proc Natl Acad Sci U S A(89): 3825-3829.
Malan TP Jr, I. M., Deng H, Liu Q, Mata HP, Vanderah T, Porreca F, Makriyannis
A. (2001). "CB2 cannabinoid receptor-mediated peripheral
antinociception." Pain 93(3): 239-245.

131
Marchalant, Y., P. W. Brownjohn, A. Bonnet, T. Kleffmann and J. C. Ashton
(2014). "Validating Antibodies to the Cannabinoid CB2 Receptor: Antibody
Sensitivity Is Not Evidence of Antibody Specificity." J Histochem
Cytochem 62(6): 395-404.
Marcus, D. J., M. Zee, A. Hughes, M. B. Yuill, A. G. Hohmann, K. Mackie, J.
Guindon and D. J. Morgan (2015). "Tolerance to the antinociceptive
effects of chronic morphine requires c-Jun N-terminal kinase." Mol Pain
11: 34.
Maresz, K., E. J. Carrier, E. D. Ponomarev, C. J. Hillard and B. N. Dittel (2005).
"Modulation of the cannabinoid CB2 receptor in microglial cells in
response to inflammatory stimuli." J Neurochem 95(2): 437-445.
Marini, P., M. G. Cascio, A. King, R. G. Pertwee and R. A. Ross (2013).
"Characterization of cannabinoid receptor ligands in tissues natively
expressing cannabinoid CB2 receptors." Br J Pharmacol 169(4): 887-899.
Martin, W. J., Coffin, P.O., Attias, E., Balinsky, M., Tsou, K (1999a).
"Cannabinoid receptormediated inhibition of the rat tail-flick reflex after
microinjection into the rostral ventromedial medulla." Brain Res(822): 237-
242.
Martin, W. J., C. M. Loo and A. I. Basbaum (1999b). "Spinal cannabinoids are
anti-allodynic in rats with persistent inflammation." Pain 82(2): 199-205.
Martin, W. J., Tsou, K., Walker, J.M., (1998). "Cannabinoid receptormediated
inhibition of the rat tail-flick reflex after microinjection into the rostral
ventromedial medulla." Neurosci Lett(242): 33-36.

132
Marusich, J. A., T. W. Lefever, K. R. Antonazzo, R. M. Craft and J. L. Wiley
(2014). "Evaluation of sex differences in cannabinoid dependence." Drug
Alcohol Depend 137: 20-28.
Matsuda, L. A., Lolait, S.J., Brownstein, M.J., Young, A.C., Bonner, T.I.. (1990).
"Structure of a cannabinoid receptor and functional expression of the
cloned cDNA." Nature 346: 561-564.
McAllister, S. D., Griffin G, Satin LS, Abood ME (1999). "Cannabinoid receptors
can activate and inhibit G protein-coupled inwardly rectifying potassium
channels in a xenopus oocyte expression system." J Pharmacol Exp
Ther(291): 618-626.
Mechoulam, R., Ben Shabat,S., Hanus,L., Ligumsky,M., Kaminski,N.E.,
Schatz,A.R., Gopher,A., Almog,S., Martin,B.R., Compton,D.R.. and
(1995). " Identification of an endogenous 2-monoglyceride, present in
canine gut, that binds to cannabinoid receptors." Biochem. Pharmacol(50):
83-90.
Melief, E. J., M. Miyatake, M. R. Bruchas. and C. Chavkin (2010). "Ligand-
directed c-Jun N-terminal kinase activation disrupts opioid receptor
signaling." PNAS 107: 11608-11613.
Melief EJ, M. M., Bruchas MR, Chavkin C (2010). "Ligand Directed c Jun N
Terminal Kinase Activation Disrupts Opioid Receptor Signaling." Proc Natl
Acad Sci USA 107(25): 11608-11613.
Merighi, S., S. Gessi, K. Varani, D. Fazzi, P. Mirandola and P. A. Borea (2012).
"Cannabinoid CB(2) receptor attenuates morphine-induced inflammatory

133
responses in activated microglial cells." Br J Pharmacol 166(8): 2371-
2385.
Miranda, H. F., Noriega, V., Zanetta, P., Prieto, J.C., Aranda, N., Sierralta, F.
(2014). "Isobolographic analysis of the opioid-opioid interactions in a tonic
and a phasic mouse model of induced nociceptive pain." J Biomed Sci
21(62).
Morgan, D. J., B. J. Davis, C. S. Kearn, D. Marcus, A. J. Cook, J. Wager-Miller,
A. Straiker, M. H. Myoga, J. Karduck, E. Leishman, L. J. Sim-Selley, T. A.
Czyzyk, H. B. Bradshaw, D. E. Selley and K. Mackie (2014). "Mutation of
putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R)
confers resistance to cannabinoid tolerance and hypersensitivity to
cannabinoids in mice." J Neurosci 34(15): 5152-5163.
Munro, S., Kerrie, L., Abu-Shaar, M., and Abu-Shaar T. (1993). "Molecular
characterization of a peripheral receptor for cannabinoids." Nature 365:
61-65.
Nackley, A. G., A. Makriyannis and A. G. Hohmann (2003). "Selective activation
of cannabinoid CB2 receptors suppresses spinal fos protein expression
and pain behavior in a rat model of inflammation." Neuroscience 119(3):
747-757.
Naguib, M., J. J. Xu, P. Diaz, D. L. Brown, D. Cogdell, B. Bie, J. Hu, S. Craig and
W. N. Hittelman (2012). "Prevention of paclitaxel-induced neuropathy
through activation of the central cannabinoid type 2 receptor system."
Anesth Analg 114(5): 1104-1120.

134
Nahin, R. L. (2015). "Estimates of pain prevalence and severity in adults: United
States, 2012." J Pain 16(8): 769-780.
Nguyen, P. T., C. L. Schmid, K. M. Raehal, D. E. Selley, L. M. Bohn and L. J.
Sim-Selley (2012). "beta-arrestin2 regulates cannabinoid CB1 receptor
signaling and adaptation in a central nervous system region-dependent
manner." Biol Psychiatry 71(8): 714-724.
Ofek, O., Karsak,M., Leclerc,N., Fogel,M., Frenkel,B., Wright,K., Tam,J., Attar-
Namdar,M., Kram,V., Shohami,E., Mechoulam,R., Zimmer,A., and Bab,I.
(2006). "Peripheral cannabinoid receptor, CB2, regulates bone mass."
Proc Natl Acad Sci U S A(103): 696-701.
Onaivi, E. S., H. Ishiguro, J. P. Gong, S. Patel, P. A. Meozzi, L. Myers, A.
Perchuk, Z. Mora, P. A. Tagliaferro, E. Gardner, A. Brusco, B. E.
Akinshola, Q. R. Liu, S. S. Chirwa, B. Hope, J. Lujilde, T. Inada, S.
Iwasaki, D. Macharia, L. Teasenfitz, T. Arinami and G. R. Uhl (2008).
"Functional expression of brain neuronal CB2 cannabinoid receptors are
involved in the effects of drugs of abuse and in depression." Ann N Y Acad
Sci 1139: 434-449.
Pacher, P., S. Batkai and G. Kunos (2006). "The endocannabinoid system as an
emerging target of pharmacotherapy." Pharmacol Rev 58(3): 389-462.
Pacher, P. and G. Kunos (2013). "Modulating the endocannabinoid system in
human health and disease--successes and failures." FEBS J 280(9):
1918-1943.
Paldyova, E., E. Bereczki, M. Santha, T. Wenger, A. Borsodi and S. Benyhe
(2008). "Noladin ether, a putative endocannabinoid, inhibits mu-opioid

135
receptor activation via CB2 cannabinoid receptors." Neurochem Int 52(1-
2): 321-328.
Pan, Z. Z., Williams, J. T. & Osborne, P. B. (1990). "Opioid actions on single
nucleus raphe magnus neurons from rat and guinea pig in vitro." J.
Physiol. (Lond.) 427: 519-532.
Perl, E. (1968). "Myelinated afferent fibres innervating the primate skin and their
response to noxious stimuli." J Physiol (Lond): 593-615.
Pertwee, R. G. (2001). "Cannabinoid receptors and pain." Progress in
Neurobiology 63: 569-611.
Pertwee, R. G. (2012). "Targeting the endocannabinoid system with cannabinoid
receptor agonists: pharmacological strategies and therapeutic
possibilities." Philos Trans R Soc Lond B Biol Sci 367(1607): 3353-3363.
Pugh, G., Jr., D. J. Mason, Jr., V. Combs and S. P. Welch (1997). "Involvement
of dynorphin B in the antinociceptive effects of the cannabinoid CP55,940
in the spinal cord." J Pharmacol Exp Ther 281(2): 730-737.
Puig, S., Sorkin, L. (1996). "Formalin-evoked activity in identified primary afferent
fibers: systemic lidocaine suppresses phase-2 activity." Pain 64(2): 345-
355.
Quartilho, A., Mata HP, Ibrahim MM, Vanderah TW, Porreca F, Makriyannis A,
Malan TP Jr (2003). "Inhibition of inflammatory hyperalgesia by activation
of peripheral CB2 cannabinoid receptors." Anesthesiology(99): 955-960.

136
Rahn, E. J., L. Deng, G. A. Thakur, K. Vemuri, A. M. Zvonok, Y. Y. Lai, A.
Makriyannis and A. G. Hohmann (2014). "Prophylactic cannabinoid
administration blocks the development of paclitaxel-induced neuropathic
nociception during analgesic treatment and following cessation of drug
delivery." Mol Pain 10: 27.
Raja, S. N., Campbell, J.N., Meyer, R.A. (1984). "Evidence for different
mechanisms of primary and secondary hyperalgesia following heat injury
to the glabrous skin." Brain 107: 1179-1188.
Rinaldi-Carmona, M. E. a. (1998). "SR 144528, the first potent and selective
antagonist of the CB2 cannabinoid receptor." J Pharmacol Exp Ther 284:
644-650.
Romero-Sandoval, A., Eisenach, J. (2007). "Spinal Cannabinoid Receptor Type 2
Activation Reduces Hypersensitivity and Spinal Cord Glial Activation after
Paw Incision." Anesthesiology 106: 787-794.
Romero-Sandoval, A., N. Nutile-McMenemy and J. A. DeLeo (2008). "Spinal
microglial and perivascular cell cannabinoid receptor type 2 activation
reduces behavioral hypersensitivity without tolerance after peripheral
nerve injury." Anesthesiology 108(4): 722-734.
Romero-Sandoval, E. A., R. Horvath, R. P. Landry and J. A. DeLeo (2009).
"Cannabinoid receptor type 2 activation induces a microglial anti-
inflammatory phenotype and reduces migration via MKP induction and
ERK dephosphorylation." Mol Pain 5: 25.
Ross, R. A., A. A. Coutts, S. M. McFarlane, S. Anavi-Goffer, A. J. Irving, R. G.
Pertwee, D. J. MacEwan and R. H. Scott (2001). "Actions of cannabinoid

137
receptor ligands on rat cultured sensory neurones: implications for
antinociception." Neuropharmacology 40(2): 221-232.
Rubino, T., D. Vigano, F. Premoli, C. Castiglioni, S. Bianchessi, R. Zippel and D.
Parolaro (2006). "Changes in the expression of G protein-coupled receptor
kinases and beta-arrestins in mouse brain during cannabinoid tolerance: a
role for RAS-ERK cascade." Mol Neurobiol 33(3): 199-213.
Sañudo-Peña, M., Patrick SL, Khen S, Patrick RL, Tsou K,Walker JM (1998).
"Cannabinoid effects in basal ganglia in a rat model of Parkinson's
disease." Neurosci Lett(248): 171-174.
Savage, S. R. (2009). "Management of Opioid Medications in Patients With
Chronic Pain and Risk of Substance Misuse." Current Psychiatry Reports
11: 377-384.
Smith, P. B., S. P. Welch and B. R. Martin (1994). "Interactions between delta 9-
tetrahydrocannabinol and kappa opioids in mice." J Pharmacol Exp Ther
268(3): 1381-1387.
Soethoudt, M., U. Grether, J. Fingerle, T. W. Grim, F. Fezza, L. de Petrocellis, C.
Ullmer, B. Rothenhausler, C. Perret, N. van Gils, D. Finlay, C. MacDonald,
A. Chicca, M. D. Gens, J. Stuart, H. de Vries, N. Mastrangelo, L. Xia, G.
Alachouzos, M. P. Baggelaar, A. Martella, E. D. Mock, H. Deng, L. H.
Heitman, M. Connor, V. Di Marzo, J. Gertsch, A. H. Lichtman, M.
Maccarrone, P. Pacher, M. Glass and M. van der Stelt (2017).
"Cannabinoid CB2 receptor ligand profiling reveals biased signalling and
off-target activity." Nat Commun 8: 13958.
Stella, N. (2004). "Cannabinoid signaling in glial cells." Glia 48(4): 267-277.

138
Stone, L., Geman, J., Kitto, KF, Fairbanks, CA, Wilcox, GL (2014). "Morphine
and Clonidine Combination Therapy Improves Therapeutic Window in
Mice: Synergy in Antinociceptive but Not in Sedative or Cardiovascular
Effects." PLoS One 9(10).
Storr, M., E. Gaffal, D. Saur, V. Schusdziarra and H. D. Allescher (2002). "Effect
of cannabinoids on neural transmission in rat gastric fundus." Can J
Physiol Pharmacol 80(1): 67-76.
Suhnel, J. (1998). "Parallel Dose–Response Curves in Combination
Experiments." Bulletin of Mathematical Biology 60: 197-213.
Sullivan, J. M. (1999). "Mechanisms of cannabinoid-receptor-mediated inhibition
of synaptic transmission in cultured hippocampal pyramidal neurons." J
Neurophysiol 82(3): 1286-1294.
Svizenska, I., P. Dubovy and A. Sulcova (2008). "Cannabinoid receptors 1 and 2
(CB1 and CB2), their distribution, ligands and functional involvement in
nervous system structures--a short review." Pharmacol Biochem Behav
90(4): 501-511.
Svizenska, I. H., V. Brazda, I. Klusakova and P. Dubovy (2013). "Bilateral
changes of cannabinoid receptor type 2 protein and mRNA in the dorsal
root ganglia of a rat neuropathic pain model." J Histochem Cytochem
61(7): 529-547.
Tallarida, R. J. (2002). "The interaction index: a measure of drug synergism."
pain 98: 163-168.

139
Tallarida, R. J. and R. B. Raffa (2010). "The application of drug dose equivalence
in the quantitative analysis of receptor occupation and drug combinations."
Pharmacol Ther 127(2): 165-174.
Tanda, G. and S. R. Goldberg (2003). "Cannabinoids: reward, dependence, and
underlying neurochemical mechanisms--a review of recent preclinical
data." Psychopharmacology (Berl) 169(2): 115-134.
Tanikawa, T., K. Kurohane and Y. Imai (2007). "Induction of preferential
chemotaxis of unstimulated B-lymphocytes by 2-arachidonoylglycerol in
immunized mice." Microbiol Immunol 51(10): 1013-1019.
Terman, G. W., W. Jin, Y. P. Cheong, J. Lowe, M. G. Caron, R. J. Lefkowitz and
C. Chavkin (2004). "G-protein receptor kinase 3 (GRK3) influences opioid
analgesic tolerance but not opioid withdrawal." Br J Pharmacol 141(1): 55-
64.
Tjolsen, A., Berge, O., Hunskaar, S., Rosland, J., and Hole, K. (1992). "The
formalin test: an evaluation of the method." Pain 51: 5-17.
Tsou, K., S. L. Patrick and J. M. Walker (1995). "Physical withdrawal in rats
tolerant to delta 9-tetrahydrocannabinol precipitated by a cannabinoid
receptor antagonist." Eur J Pharmacol 280(3): R13-15.
Ueda, H., Harada, H., Nozaki, M., Katada, T., Ui, M., Satoh, M., and Takagi, H.
(1988). "Reconstitution of rat brain mu opioid receptors with purified
guanine nucleotide-binding regulatory proteins, Gi and Go." Proc Natl
Acad Sci U S A(85).

140
Van Sickle, M. D., M. Duncan, P. J. Kingsley, A. Mouihate, P. Urbani, K. Mackie,
N. Stella, A. Makriyannis, D. Piomelli, J. S. Davison, L. J. Marnett, V. Di
Marzo, Q. J. Pittman, K. D. Patel and K. A. Sharkey (2005). "Identification
and functional characterization of brainstem cannabinoid CB2 receptors."
Science 310(5746): 329-332.
Vigano, D., T. Rubino and D. Parolaro (2005). "Molecular and cellular basis of
cannabinoid and opioid interactions." Pharmacol Biochem Behav 81(2):
360-368.
Wadenberg, M. L. (2003). "A review of the properties of spiradoline: a potent and
selective kappa-opioid receptor agonist." CNS Drug Rev 9(2): 187-198.
Wakley, A. A., J. L. Wiley and R. M. Craft (2014). "Sex differences in
antinociceptive tolerance to delta-9-tetrahydrocannabinol in the rat." Drug
Alcohol Depend 143: 22-28.
Waldhoer, M., S. E. Bartlett and J. L. Whistler (2004). "Opioid receptors." Annu
Rev Biochem 73: 953-990.
Wall, P. D. (1967). "The laminar organisation of dorsal horn and effects of
descending impulses." J. Physiol. (Lond.) 188: 403-423.
Wang, Q., Mao L., Shi, Y., Han, J. (1990). "Lumbar intrathecal administration of
naloxone antagonizes analgesia produced by electrical stimulation of the
hypothalamic arcuate nucleus in pentobarbital-anesthetized rats."
Neuropharmacology 29: 1123-1129.

141
Watkins, L. R., M. R. Hutchinson, K. C. Rice and S. F. Maier (2009). "The "toll" of
opioid-induced glial activation: improving the clinical efficacy of opioids by
targeting glia." Trends Pharmacol Sci 30(11): 581-591.
Welch, S. P. (1997). "Characterization of anandamide-induced tolerance:
comparison to delta 9-THC-induced interactions with dynorphinergic
systems." Drug Alcohol Depend 45(1-2): 39-45.
Whiteside, G. T., S. L. Gottshall, J. M. Boulet, S. M. Chaffer, J. E. Harrison, M. S.
Pearson, P. I. Turchin, L. Mark, A. E. Garrison and K. J. Valenzano
(2005). "A role for cannabinoid receptors, but not endogenous opioids, in
the antinociceptive activity of the CB2-selective agonist, GW405833." Eur
J Pharmacol 528(1-3): 65-72.
Whiting, P. F., R. F. Wolff, S. Deshpande, M. Di Nisio, S. Duffy, A. V. Hernandez,
J. C. Keurentjes, S. Lang, K. Misso, S. Ryder, S. Schmidlkofer, M.
Westwood and J. Kleijnen (2015). "Cannabinoids for Medical Use: A
Systematic Review and Meta-analysis." Jama 313(24): 2456-2473.
Williams, J. T., S. L. Ingram, G. Henderson, C. Chavkin, M. von Zastrow, S.
Schulz, T. Koch, C. J. Evans and M. J. Christie (2013). "Regulation of mu-
opioid receptors: desensitization, phosphorylation, internalization, and
tolerance." Pharmacol Rev 65(1): 223-254.
Wong, Y. H., C. D. Demoliou-Mason and E. A. Barnard (1988). "ADP-ribosylation
with pertussis toxin modulates the GTP-sensitive opioid ligand binding in
digitonin-soluble extracts of rat brain membranes." J Neurochem 51(1):
114-121.

142
Yaksh, T. L. N., R. (1985). "The physiology and pharmacology of spinal opiates."
Ann Rev Phamacol Toxicol 25(433-462).
Yamamoto, W., T. Mikami and H. Iwamura (2008). "Involvement of central
cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse
model of neuropathic pain." Eur J Pharmacol 583(1): 56-61.
Yang, D., D. Conze, A. Whitmarsh, T. Barrett, R. Davis, M. Rincon and R. Flavell
(1998). "Differentiation of CD4+ T Cells to Th1 Cells Requires MAP
Kinase JNK 2." Immunity 9: 575-585.
Yang, D., C. Kuan, A. Whitmarsh, M. Rincon, T. Zheng, R. Davis, P. Rakic and
R. Flavell (1997). "Absence of excitotoxicity induced apopotosis in the
hippocampus of mice lacking the Jnk3 gene." Nature 389.
Yao, B. B. H. G., Frost JM, Fan Y, Garrison TR, Daza AV, Grayson GK, Zhu CZ,
Pai M, Chandran P, Salyers AK, Wensink EJ, Honore P, Sullivan JP, Dart
MJ, Meyer MD (2008). "In vitro and in vivo characterization of A-796260: a
selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in
rodent pain models." Br J Pharmacol 153: 390-401.
Yiangou, Y., Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati
RR, Anand P (2006). "COX-2, CB2 and P2X7-immunoreactivities are
increased in activated microglial cells/macrophages of multiple sclerosis
and amyotrophic lateral sclerosis spinal cord." BMC Neurol(6): 12.
Yuill, M. B., M. L. Zee, D. Marcus and D. J. Morgan (2016). "Tolerance to the
antinociceptive and hypothermic effects of morphine is mediated by
multiple isoforms of c-Jun N-terminal kinase." Neuroreport 27(6): 392-396.

143
Zhang, J., Ferguson, SSG, Barak, LS, Bodduluri, SR, Laporte, SA, Law, PY,
Caron, MG (1998). "Role for G protein-coupled receptor kinase in agonist-
specific regulation of m-opioid receptor responsiveness." Neurobiology 95:
7`157-7162.
Zhang, J., Hoffert, C., Huy, K., Groblewski, T., Ahmad, S., O'Donnell, D. (2003).
"Induction of CB2 receptor expression in the rat spinal cord of neuropathic
but not inflammatory chronic pain models." Eur J Neurosci 17: 2750-2754.
Zhang, M., K. Wang, M. Ma, S. Tian, N. Wei and G. Wang (2016). "Low-Dose
Cannabinoid Type 2 Receptor Agonist Attenuates Tolerance to Repeated
Morphine Administration via Regulating mu-Opioid Receptor Expression in
Walker 256 Tumor-Bearing Rats." Anesth Analg 122(4): 1031-1037.
Zhuang, Z. Y., Y. R. Wen, D. R. Zhang, T. Borsello, C. Bonny, G. R. Strichartz, I.
Decosterd and R. R. Ji (2006). "A peptide c-Jun N-terminal kinase (JNK)
inhibitor blocks mechanical allodynia after spinal nerve ligation: respective
roles of JNK activation in primary sensory neurons and spinal astrocytes
for neuropathic pain development and maintenance." J Neurosci 26(13):
3551-3560.
Zimmer, A., Zimmer, A.M., Hohmann, A.G., Herkenham, M., Bonner, T.I. (1999).
"Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1
receptor knockout mice." Proc Natl Acad Sci U S A 96: 5780-5785.
Zoratti, C., Kipmen-Korgun,D., Osibow,K., Malli,R., and Graier,W.F. (2003).
"Anandamide initiates Ca(2+) signaling via CB2 receptor linked to
phospholipase C in calf pulmonary endothelial cells." Br J
Pharmacol(140): 1351-1362.

144
VITA
Education
Franklin and Marshall College B.A. (Neuroscience) (2007-2011)
Penn State University Ph.D. (Neuroscience, expected) (2011-2017)
Publications Yuill, MB., Hale, DE., Guindon J, Morgan DJ. (2017). “Anti-nociceptive interactions between opioids and a cannabinoid receptor 2 agonist in inflammatory pain.” Molecular Pain.
Yuill, MB., Marcus, DJ., Zee, ML., Morgan DJ (2016). “Tolerance to the anti-nociceptive and hypothermic effects of morphine are mediated by multiple isoforms of c-Jun N-terminal Kinase.” Neuroreport. Marcus DJ, Zee M, Hughes, A., Yuill, MB., Hohmann AG, Mackie K, Guindon J, Morgan DJ (2015). Tolerance to the antinociceptive effects of chronic morphine requires c-Jun N-terminal kinase. Molecular Pain. Henderson-Redmond, AN., Yuill, MB., Lowe, TE., Kline, AM., Zee, ML., Guindon, J., Morgan, DJ., (2015) “Morphine-induced antinociception and reward in "humanized" mice expressing the mu opioid receptor A118G polymorphism” Brain Res. Bul. Nealon, C., Davis, BJ., Henderson Redmond, A., Yuill, MB., Muller, J., Haskins, CP., Marcus, DJ., Czyzyk, TA., Mackie, K., Guindon, J., Morgan, DJ.“c-Jun N terminal kinase signaling pathways mediate cannabinoid tolerance in an agonist-specific manner.” (In preparation).
Abstracts Yuill, MB., Morgan, DJ., and Guindon, J. (2016). Impact of a CB2R Cannabinoid Agonist On Inflammatory Pain And Morphine Tolerance. International Cannabinoid Research Society Annual Meeting. Bukovina, Poland. Yuill, MB., Morgan, DJ., and Guindon, J. (2015). Mu opioid receptor and cannabinoid Receptor 2 in inflammatory pain. International Cannabinoid Research Society Annual Meeting. Wolfville, Nova Scotia. Yuill, MB., Morgan, DJ., and Guindon, J. (2015). Tolerance to the antinociceptive effects of Δ9-THC in the formalin model of inflammatory pain. Society for Neuroscience Annual Meeting. Chicago, Illinois. Yuill, MB., Morgan, DJ. Levenson, R., and Guindon, J. (2015). Cross-tolerance between opioid receptor and cannabinoid receptor 2 agonists in inflammatory pain. Gill Symposium annual meeting. Bloomington, Indiana.

















