
Expériences sur la décomposition et la recomposition de la ...

Année 2005
Thèse
Décomposition des matières organiques et stabilisation des métaux lourds dans les
sédiments de dragage
Présentée devant L’Institut National des Sciences Appliquées de Lyon
Pour obtenir
le grade de docteur
Ecole doctorale : CHIMIE LYON (Chimie, Procédé, Environnement) …………………………………………
Spécialité : Science et Technique du Déchet
par Souhila KRIBI
Soutenance le 23 septembre 2005 au laboratoire de Génie des Procédés
des Solides Divisés (LGPSD)
UMR CNRS 2392
Jury
NZIHOU Ange Professeur, EMAC, Albi Directeur SHARROCK Patrick Professeur, UPS, Toulouse Co-directeur MONTEL Jean-Marc Professeur, OMP-UPS Toulouse Rapporteur CARRERE Hélène HDR, INRA-LBE, Narbonne Rapporteur GOURDON Rémy Professeur, LAEPSI, INSA, Lyon Examinateur LECOMTE Didier Professeur, EMAC, Albi Examinateur DEPELSENAIRE Guy Ingénieur, SOLVAY HSE, Bruxelles Examinateur PIANTONE Patrice Ingénieur, BRGM, Orléans Invité

Table des matières Introduction générale 1 CHAPITRE I : Les sédiments de dragage 5
I.1. Problématique des sédiments de dragage ...........................................................................................6
I.1.1. Introduction........................................................................................................................................6 I.1.2. Le dragage et les quantités de sédiments draguées en France ..........................................................8
I.1.2.1. Les sédiments portuaires ..........................................................................................................8 I.1.2.2. Les sédiments continentaux......................................................................................................8
I.1.3. Réglementation régissant la gestion des sédiments .........................................................................11 I.1.3.1. Sédiments continentaux..........................................................................................................11
I.1.3.1.1. La gestion des sédiments continentaux..............................................................................11 I.1.3.2. Sédiments portuaires ..............................................................................................................16
I.1.3.2.1. La gestion des sédiments portuaires ..................................................................................16 I.1.3.3. Conclusions : ..........................................................................................................................18
I.1.4. Le devenir des sédiments dragués: ..................................................................................................19 I.1.4.1. Sédiments portuaires ..............................................................................................................19 I.1.4.2. Sédiments continentaux..........................................................................................................20
I.2. Procédés de traitement des sédiments...............................................................................................21
I.2.1. Introduction......................................................................................................................................21 I.2.2. Traitement physique et physico-chimique ........................................................................................22
I.2.2.1. Le cyclonage...........................................................................................................................23 I.2.2.2. L’attrition................................................................................................................................24 I.2.2.3. La gravimétrie ........................................................................................................................25 I.2.2.4. La séparation magnétique.......................................................................................................25 I.2.2.5. La flottation ............................................................................................................................26 I.2.2.6. L’électromigration..................................................................................................................27 I.2.2.7. Décontamination aux ultrasons ..............................................................................................27
I.2.3. Traitements biologiques...................................................................................................................28 I.2.3.1. Biolixiviation..........................................................................................................................28 I.2.3.2. Phytoextraction.......................................................................................................................29 I.2.3.3. Biodégradation .......................................................................................................................30
I.2.4. Traitements thermiques....................................................................................................................31 I.2.4.1. La désorption..........................................................................................................................31 I.2.4.2. Oxydation à l’air humide........................................................................................................31 I.2.4.3. La vitrification........................................................................................................................32 I.2.4.4. L’incinération .........................................................................................................................32
I.2.5. Traitements chimiques .....................................................................................................................33 I.2.5.1. Extraction par lixiviation........................................................................................................34 I.2.5.2. Extraction par complexation...................................................................................................34 I.2.5.3. Extraction par solvant.............................................................................................................35 I.2.5.4. Procédé d’échange cationique ................................................................................................36 I.2.5.5. Procédé d’immobilisation.......................................................................................................36
I.2.6. Les phosphates de calcium...............................................................................................................38 I.2.6.1. Les phosphates apatitiques .....................................................................................................39 I.2.6.2. L’utilisation des phosphates pour le traitement des déchets ...................................................42

I.2.6.3. Exemple d’utilisation des phosphates pour la stabilisation des métaux lourds dans les déchets : Procédé NOVOSOL ..................................................................................................................................49
I.3. Caractéristiques chimiques des sédiments........................................................................................52
I.3.1. Définition, origine et formation des sédiments ................................................................................52 I.3.2. Composition granulométrique des sédiments ..................................................................................52 I.3.3. Composition géochimique des sédiments.........................................................................................53
I.3.3.1. Les argiles...............................................................................................................................53 I.3.3.2. Les carbonates ........................................................................................................................54 I.3.3.3. Les sulfures.............................................................................................................................55 I.3.3.4. Les oxydes hydroxydes ..........................................................................................................55 I.3.3.5. La phase organique.................................................................................................................56
I.3.4. Polluants métalliques et organiques ................................................................................................60 I.3.4.1. Les métaux lourds...................................................................................................................60
I.3.5. Interactions entre les métaux lourds et les sédiments ......................................................................60 I.3.6. Paramètres influençant la répartition des contaminants :...............................................................61
I.3.6.1. Influence des paramètres physico-chimiques .........................................................................61 I.3.6.1.1. Le pH .................................................................................................................................61 I.3.6.1.2. Le potentiel d’oxydoréduction...........................................................................................62 I.3.6.1.3. La capacité d’échange cationique ......................................................................................63
I.3.7. Spéciation des métaux lourds dans les sédiments ............................................................................63 I.3.7.1. Extraction séquentielle ...........................................................................................................64
I.3.7.1.1. Extraction séquentielle suivant la méthode de Tessier ......................................................66
I.4. Conclusions..........................................................................................................................................69 CHAPITRE II : Caractérisation des sédiments 72
II.1. Introduction.........................................................................................................................................73
II.2. Préparation d’un échantillon de sédiment pour l’analyse...............................................................73
II.3. Caractérisation chimique ...................................................................................................................74
II.3.1. Détermination de l’humidité résiduelle............................................................................................74 II.3.2. Détermination du pH .......................................................................................................................75
II.3.2.1. Mode opératoire .....................................................................................................................75 II.3.3. Détermination de la conductivité électrique ....................................................................................76
II.3.3.1. Mode opératoire .....................................................................................................................76 II.3.4. Dosage des éléments chimiques .......................................................................................................77
II.3.4.1. Mode opératoire .....................................................................................................................78 II.3.5. Détermination du carbone total et du carbone organique...............................................................79
II.3.5.1. Mode opératoire .....................................................................................................................79 II.3.6. Détermination de la matière organique totale .................................................................................81
II.3.6.1. Mode opératoire .....................................................................................................................81 II.3.7. La diffraction des rayons X..............................................................................................................82 II.3.8. Détermination de la capacité d’échange cationique........................................................................84
II.3.8.1. Mode opératoire .....................................................................................................................84 II.3.9. Le pouvoir tampon ...........................................................................................................................85

II.3.9.1. Mode opératoire .....................................................................................................................85
II.4. Caractérisation physique ...................................................................................................................88
II.4.1. La densité .........................................................................................................................................88 II.4.2. La distribution granulométrique ......................................................................................................89
II.5. Conclusions..........................................................................................................................................90 CHAPITRE III : Traitement du sédiment de Vraimont 93
III.1. IntroductionEquation.........................................................................................................................94
III.2. Matériels et méthodes.........................................................................................................................94
II.2.1. Phosphatation et maturation............................................................................................................94 III.2.1.1. Le montage expérimentale de phosphatation..........................................................................94 III.2.1.2. Cinétique de phosphatation et de maturation..........................................................................97 III.2.1.3. Comportement rhéologique du sédiment................................................................................99
II.2.2. Séchage des sédiments .....................................................................................................................99 II.2.3. Calcination des sédiments..............................................................................................................102
III.2.3.1. Analyse thermogravimétrique (ATG)...................................................................................103 III.2.3.2. Mesure des propriétés physiques..........................................................................................103
III.3. Résultats et discussions.....................................................................................................................104
II.3.1. Phosphatation et maturation des sédiments...................................................................................104 III.3.1.1. La cinétique de phosphatation en réacteur tubulaire.............................................................104
III.3.1.1.1. Evolution du pH..........................................................................................................107 III.3.1.1.2. Evolution du phosphore ..............................................................................................107 III.3.1.1.3. Evolution du calcium ..................................................................................................110 III.3.1.1.4. Evolution d’autres éléments........................................................................................111 III.3.1.1.5. Discussion...................................................................................................................112
III.3.1.2. La cinétique de maturation ...................................................................................................118 III.3.1.2.1. Evolution du pH..........................................................................................................118 III.3.1.2.2. Evolution du phosphore ..............................................................................................119 III.3.1.2.3. Evolution du calcium ..................................................................................................120 III.3.1.2.4. Evolution des autres éléments.....................................................................................121 III.3.1.2.5. Discussion...................................................................................................................121
III.3.1.3. Synthèse ...............................................................................................................................123 II.3.2. Séchage des sédiments ...................................................................................................................124
III.3.2.1. Notion de séchage.................................................................................................................124 III.3.2.1.1. Définition du séchage .................................................................................................124 III.3.2.1.2. Humidité et taux d’humidité .......................................................................................124 III.3.2.1.3. Etat de siccité ..............................................................................................................125 III.3.2.1.4. Taux d’humidité à l’équilibre .....................................................................................125 III.3.2.1.5. Isotherme de sorption..................................................................................................125 III.3.2.1.6. Température de bulle humide .....................................................................................125 III.3.2.1.7. Mécanismes intervenant au cours du séchage.............................................................125
III.3.2.2. Résultats ...............................................................................................................................129 III.3.2.2.1. Cinétique de séchage du sédiment brut .......................................................................130 III.3.2.2.2. Influence de la vitesse de l’air et de la température ....................................................132 III.3.2.2.3. Cinétique de séchage des sédiments phosphatés.........................................................133 III.3.2.2.4. Influence de la température.........................................................................................135 III.3.2.2.5. Propriétés physiques des sédiments séchés.................................................................136
III.3.2.3. Discussion ............................................................................................................................138

II.3.3. Calcination des sédiments..............................................................................................................139 III.3.3.1. Dégradation des sédiments ...................................................................................................140 III.3.3.2. Propriétés physiques des sédiments calcinés ........................................................................142
III.3.3.2.1. La surface spécifique ..................................................................................................142 III.3.3.2.2. La densité....................................................................................................................143
III.4. Conclusions........................................................................................................................................144 CHAPITRE IV : Evaluation du procédé 148
IV.1. IntroductionEquation.......................................................................................................................149
IV.2. Méthodes et matériels.......................................................................................................................150
IV.2.1. Extraction séquentielle...................................................................................................................150 IV.2.2. Microsonde électronique................................................................................................................152 IV.2.3. Microscope électronique à balayage (MEB) .................................................................................153 IV.2.4. Test de lixiviation...........................................................................................................................153
IV.3. Résultats et discussions.....................................................................................................................154
IV.3.1. Extraction séquentielle...................................................................................................................154 IV.3.1.1. Distribution des éléments chimiques dans les sédiments bruts ............................................154 IV.3.1.2. Distribution des éléments chimiques dans les sédiments traités...........................................162
IV.3.2. Micro-cartographie des éléments ..................................................................................................185 IV.3.3. Microscope électronique à balayage .............................................................................................193 IV.3.4. Test de lixiviation...........................................................................................................................195
IV.4. Conclusions........................................................................................................................................197 Conclusions générales et perspectives 200Bibliographie 206Annexe A 217Liste des figures 219Liste des tableaux 220

Introduction générale
1
Introduction générale

Introduction générale
2
Introduction générale
Les sédiments d’origine diverses sont souvent très contaminés par l’activité humaine et
urbaine. Ils se déposent dans les milieux aquatiques et posent de nombreux problèmes pour
le devenir des écosystèmes en place à travers les risques de contamination de la chaîne
alimentaire mais aussi, lorsque pour des raisons hydrauliques et/ou des raisons de nuisance
extrêmes, des curages ou dragages deviennent nécessaires. Les volumes très abondant et la
probable toxicité des éléments prélevés sont autant de difficultés auxquelles les
gestionnaires sont appelés à répondre au mieux.
Depuis plusieurs décennies, des catastrophes écologiques liées au déversement dans
l’environnement accidentellement ou non de substances toxiques (métaux lourds ;
hydrocarbures aromatiques polycycliques (HAP), polychlorobiphényles (PCB)…) qui se
retrouvent finalement dans les écosystèmes aquatiques ont fort heureusement favorisé le
développement d’une prise de conscience collective. De nombreux travaux ont été
développés dans le but de mettre au point des traitements et des voies de valorisation de ces
sédiments tout en respectant les aspects environnementaux. Les efforts faits jusqu’à présent
restent néanmoins très faibles par rapport aux besoins. En effet, sur les 2.8Mm3 de sédiments
fluviaux dragués par an en France, uniquement 3% sont traités, quant aux sédiments marins,
sur les 50Mm3 dragués par an la grande majorité est restituée au milieu naturel par
immersion.
Dans une optique de traitement des sédiments de dragage et de leur valorisation dans le
domaine du génie civil. La société SOLVAY a développé le procédé NOVOSOL en
s’inspirant et en réadaptant le procédé REVASOL qu’elle a développé dans les années 96
pour le traitement des cendres volantes d’incinération des ordures ménagères. L’objectif du
procédé NOVOSOL est de traiter les sédiments de dragage fluviaux et marins par un
traitement chimique par phosphatation pour stabiliser la pollution inorganique (métaux
lourds) et un traitement thermique pour dégrader la pollution organique et renforcer la
stabilité des métaux lourds. Les résidus finaux obtenus ont des propriétés physiques
contrôlées et répondent aux exigences environnementales pour des valorisations matière.
Pour ce faire SOLVAY s’est associé avec des entreprises et des laboratoires choisis
suivant leur spécialité. Ce projet est notamment suivie par l’ADEME (Agence de
l’Environnement et de la Maîtrise de l’Energie) en tant que partenaire institutionnel et par
Bertin Technologie pour l’étude de la faisabilité technique et économique du procédé, ainsi
que pour la coordination du développement. Le BRGM (Bureau de Recherche Géologique et

Introduction générale
3
Minière) réalise l’étude minéralogique des sédiments de dragage à tous les stades du
procédé. Le laboratoire de l’INSA (Institut National des Sciences Appliqués) de Toulouse
ainsi que ECL (Ecole Centrale de Lille) effectuent des études au laboratoire pour déterminer
différents domaines d’application des sédiments de dragage traités dans les bâtiments et/ou
dans les travaux publics. L’ENTPE (Ecole Nationale des Travaux Publics de l’Etat) effectue
l’évaluation environnementale des sédiments traités et des produits valorisables. Le rôle de
l’EMAC (Ecole des Mines d’Albi-Carmaux) dans ce projet est d’approfondir l’étude
technique du procédé de traitement des sédiments de dragage.
Le travail de l’EMAC dans le cadre de cette thèse (contrat CIFRE) a été de bien
comprendre les trois phases du traitement des sédiments : la phase de traitement chimique
qui a pour but de stabiliser les métaux lourds sous forme de phosphates insolubles ; la phase
de séchage à l’air conduit à la réduction de l’humidité avant calcination et la phase de
calcination sous air qui a un double rôle la dégradation de la matière organique et de la
cristallisation des formes chimiques obtenues lors de la phosphatation dans le but de les
rendre plus stables.
Quatre grandes parties sont présentées dans ce document :
• Le chapitre I expose l’état actuel du dragage continental et marin en France et la gestion
de ces sédiments dragués. Une partie réglementaire est également abordée afin de montrer
les difficultés auxquelles sont affrontés les gestionnaires du dragage et les industriels pour
avoir un outil réglementaire adapté au sédiment et qui permettrai de les qualifier avant et
après traitement. Une présentation non exhaustive des différentes techniques de traitement
des sédiments a été faite ainsi que des exemples de traitement de différents types de déchets
utilisant le phosphate comme agent immobilisant des métaux lourds. Pour finir une
présentation de l’origine, de la formation ainsi que des principales phases chimiques des
sédiments a été abordée. L’interaction des polluants, principalement les polluants
métalliques, avec les sédiments de dragage, leur spéciation et l’influence des paramètres
physico-chimiques sur leur distribution a fait également l’objet de discussion dans cette
étude.
• Le chapitre II présente la caractérisation physico-chimiques des sédiments utilisés qui va
permettre une bonne compréhension et une analyse appropriée des résultats après traitement.
• Le chapitre III porte sur le traitement des sédiments de dragage par le procédé
NOVOSOL en utilisant des montages expérimentaux à l’échelle du laboratoire pour chaque
étape. Dans un premier temps nous présenterons la cinétique de phosphatation des sédiments
qui se fait dans un réacteur tubulaire afin d’essayer de comprendre les différents mécanismes

Introduction générale
4
mis en oeuvre lors de cette étape. En second lieu la cinétique de séchage des sédiments est
abordée afin de connaître l’influence des différents paramètres tels que la quantité d’acide, la
vitesse de l’air et de sa température sur les temps de séchage des sédiments. L’étude de
l’influence de la température de calcination sur la dégradation de la matière organique et sur
les propriétés physiques (densité et surface spécifique) des résidus solides obtenus fait
également l’objet d’une discussion dans ce chapitre..
• Le chapitre IV est consacré à l’évaluation de la capacité de stabilisation des métaux
lourds par le traitement effectué. Dans ce chapitre l’étude de la distribution des éléments
chimiques à travers l’utilisation de l’extraction séquentielle est réalisée. Cette méthode
permet de répartir les éléments chimiques en fractions plus ou moins disponibles en milieux
aqueux, acide, oxydant ou réducteur. Elle permet de classifier les éléments dans des
catégories métastables et stables. Les résultats obtenus pour le sédiment brut et après chaque
étape du traitement sont présentés et discutés en particulier pour les espèces majeurs telles
que Fe, Ca, Al et P, de même que les métaux lourds (As, Cd, Cr, CO, Cu, Pb, Zn). Une
étude minéralogique pour déterminer les associations minéralogiques des sédiments est la
caractérisation des phases qui se sont formées lors de la phosphatation a également été
réalisée. Pour répondre aux exigences réglementaires, des essais de lixiviation ont également
été menés pour évaluer la stabilisation des métaux lourds.
L’ensemble de ces travaux devrait constituer une base dans laquelle seront puisées les
données utiles pour l’application du procédé NOVOSOL aux différents sédiments.

Chapitre I : Les sédiments de dragage
5
CHAPITRE I
Les sédiments de dragage

Chapitre I : Les sédiments de dragage
6
I.1. Problématique des sédiments de dragage
I.1.1. Introduction
Le territoire français compte 525000 km de cours d’eau qui transportent chaque année, en
moyenne 6 millions de m3 de sédiments [Berteau 1993]. L’accumulation des sédiments, et
leurs pollutions éventuelles, au fond des canaux, cours d’eau, fossés et plan d’eau est
souvent accentuée par l’activité humaine. Le curage constitue alors une opération de
restauration, d’entretien voire d’assainissement indispensable pour prévenir les risques
d’inondation, pour rétablir le tirant d’eau pour la navigation mais aussi pour restaurer le
milieu naturel [AEAP 2000 (a)]. Le curage ne se pratique pas seulement au niveau des cours
d’eau et des canaux, il s’étend également aux structures portuaires. Ces dernières sont le
plus souvent établies dans des zones où la profondeur d'eau est relativement faible, telles que
les estuaires. Il est alors indispensable de réaliser des dragages pour permettre aux bateaux
d'accéder aux quais. Ces dragages sont effectués au moment de la construction du port, mais
également de façon périodique pour enlever les sédiments qui se sont accumulés dans les
chenaux et les darses (dragages d'entretien). C'est ainsi que les pays riverains de la mer du
Nord et de l'Atlantique draguent annuellement un peu plus de 110 millions de tonnes de
déblais [Alzieu 2003] ;
Lorsque la zone de dragage est à proximité de sites industriels polluants, les sédiments à
draguer peuvent s’avérer fortement contaminés. Parmi les substances chimiques émises dans
l’environnement, certaines s’accumulent dans les sédiments au fond des rivières et des lacs
ainsi que dans les estuaires et les fonds marins. Les effets écologiques et sanitaires de la
contamination des sédiments sont susceptibles de générer un impact environnemental et un
coût social réel. Ces effets sont liés à une diminution de la biodiversité avec des
conséquences indirectes sur les peuplements des poissons, à une perte de comestibilité des
poissons et des coquillages, mais également entraîner des restrictions des usages récréatifs et
augmenter significativement le coût de gestion des sédiments contaminés. Certains acteurs
de la gestion des sédiments continentaux notent déjà un ralentissement du rythme des
curages lié à la difficulté de gérer les sédiments pollués, et craignent à moyen terme une
aggravation des inondations et des problèmes de navigation [Clozel 2003].
Le devenir de ces produits dans ce cas est particulièrement complexe. Les solutions
traditionnelles de mise en immersion en milieu marin, pour les sédiments portuaires, et la
mise en dépôt ou la remise en suspension des sédiments continentaux sont inapplicables car
ces méthodes sont une voie de transfert des contaminants vers l’écosystème. Il convient

Chapitre I : Les sédiments de dragage
7
donc de prendre les dispositions de nature à en limiter les impacts sur l’écosystèmes. Ces
dispositions doivent être prises à deux niveaux : d’une manière préventive d’une part, en
limitant et en surveillant d’avantage les rejets anthropiques (industriels et urbains), et d’une
manière curative d’autre part en développant des méthodes de traitement et de valorisation
des matériaux de dragage à l’échelle industrielle. Ces méthodes doivent être
économiquement viables et en accord avec la réglementation environnementale du pays
concerné. C’est cette dernière voie qui suscite l’adhésion de la plupart des opérateurs des
secteurs concernés par la problématique des sédiments (pouvoir public et industriels).
Bien que seuls les sédiments fluviaux ont fait l’objet de cette thèse, dans ce chapitre nous
nous intéressons aux problématiques liées aux sédiments fluviaux (continentaux) et marins.
puisque les mêmes politiques de prévention et de traitement doivent être adaptées pour les
deux cas.
Une présentation des volumes de sédiments dragués ainsi que leur devenir en France sera
abordée. L’objectif étant de montrer la nécessité croissante de développer des procédés
industriels de traitement et de valorisation de ces déchets. Une partie réglementaire
concernant les deux types de sédiments en France est également présentée. Le but est de
montrer la difficulté liée au manque de réglementation spécifique à laquelle sont affrontés
les acteurs du dragage et les industriels impliqués dans le traitement de ces sédiments. C’est
ainsi par exemple qu’une étude sur le cas de la Belgique et l’Italie aurait pu être faite étant
donnée que les activités de Solvay dans ce domaine s’étendent également dans ces deux
pays là. Cependant nous nous sommes limités uniquement au cas de la France.
Ensuite, une présentation non exhaustive des différentes techniques de traitement
existantes a été faite. L’objectif est de situer le procédé NOVOSOL sur lequel nous
travaillons par rapport aux autres types de traitement. Dans cette même partie, des exemples
de traitement de différents types de déchets utilisant le même principe de traitement que
celui de NOVOSOL ont également été abordés.
Enfin, une présentation de l’origine, de la formation ainsi que des principales phases
chimiques existantes dans le sédiment a été arborée. L’interaction des polluants,
principalement les polluants métalliques, avec les sédiments de dragage, leur spéciation et
l’influence des paramètres physico-chimiques sur leur spéciation a fait également l’objet de
cette étude.

Chapitre I : Les sédiments de dragage
8
I.1.2. Le dragage et les quantités de sédiments draguées en France
I.1.2.1. Les sédiments portuaires
Le littoral français, avec environ 6500 km de côte d’une grande diversité, est devenu un
espace où se côtoient les activités marines et terrestres du commerce, de la pêche, des
cultures marines, de la plaisance et du tourisme en général.
Le dragage portuaire constitue une activité vitale pour l’exploitation des ports. On
distingue trois types de dragage : le dragage d’entretien, le dragage d’approfondissement et
le dragage d’aménagement de nouvelles aires portuaires. Ainsi, environ 50 millions de
mètres cubes de sédiments sont dragués en moyenne par an. Les volumes les plus importants
de matériaux déplacés le sont par les grands ports. Les trois principaux ports d’estuaire
(Rouen, Nantes-saint-Nazaire et Bordeaux) font état d’un volume moyen annuel d’environ
25 millions de mètres cubes, dont 6.5 millions de mètres cubes de sables, 9.3 millions de
mètres cubes de vase clapée dans des zones d’immersion et une quantité équivalente rejetée
en surverse. Les cinq grands ports maritimes, Dunkerque, Calais, Boulogne, Le Havre et la
Rochelle, draguent un volume moyen annuel de 6.2 millions de mètres cubes de sédiment.
C’est en Méditerranée que les volumes dragués sont les plus faibles, l’entretien du port de
Marseille, par exemple, nécessite le dragage d’environ 20000 mètres cubes par an. La part
importante que prennent ces grands ports (3/4 des volumes dragués) ne doit pas faire oublier
les dragages, moins conséquents en volume, des multiples autres enclaves portuaires, qui
peuvent poser des problèmes du fait de la nature et de la concentration des polluants
[Alzieu1999 ;2003].
I.1.2.2. Les sédiments continentaux
Pour les cours d'eau domaniaux et leurs dépendances faisant partie du domaine public, le
curage est à la charge de l'Etat au travers :
• Les Voies Navigables de France (VNF) pour les cours d'eau navigables ;
• Des ports autonomes fluviaux (exemple le Port Autonome de Paris),
• Des établissements publics comme la Compagnie Nationale du Rhône (CNR).
Néanmoins, sur les 525000 km de cours d'eau, canaux nationaux, environ 20000 seulement
font partie du domaine de l'Etat.
Pour la très grande majorité des cours d'eau, correspondant donc aux cours d'eau non
domaniaux, le propriétaire riverain est tenu à un curage régulier afin de lui redonner sa
largeur et sa profondeur naturelle. Le volume total serait donc plus élevé.
Chapitre I : Les sédiments de dragage
9
En 2002, une étude recommandée par le Ministère de l’Aménagement du Territoire et de
l’Environnement (MATE) [Agences de l’Eau 2002] sur l’historique des opérations de
curage d’entretien des sédiments continentaux en France entre 1990 et 2000 a été réalisée
dans le cadre du Comité Technique National sur la Gestion des Sédiments. Cette étude a
permis de montrer qu’actuellement, le coût des opérations de dragage au niveau national est
de 26 millions d’euros par an pour 2.8 millions de m3 de sédiments (Tableau I- 1) curés en
135 opérations.
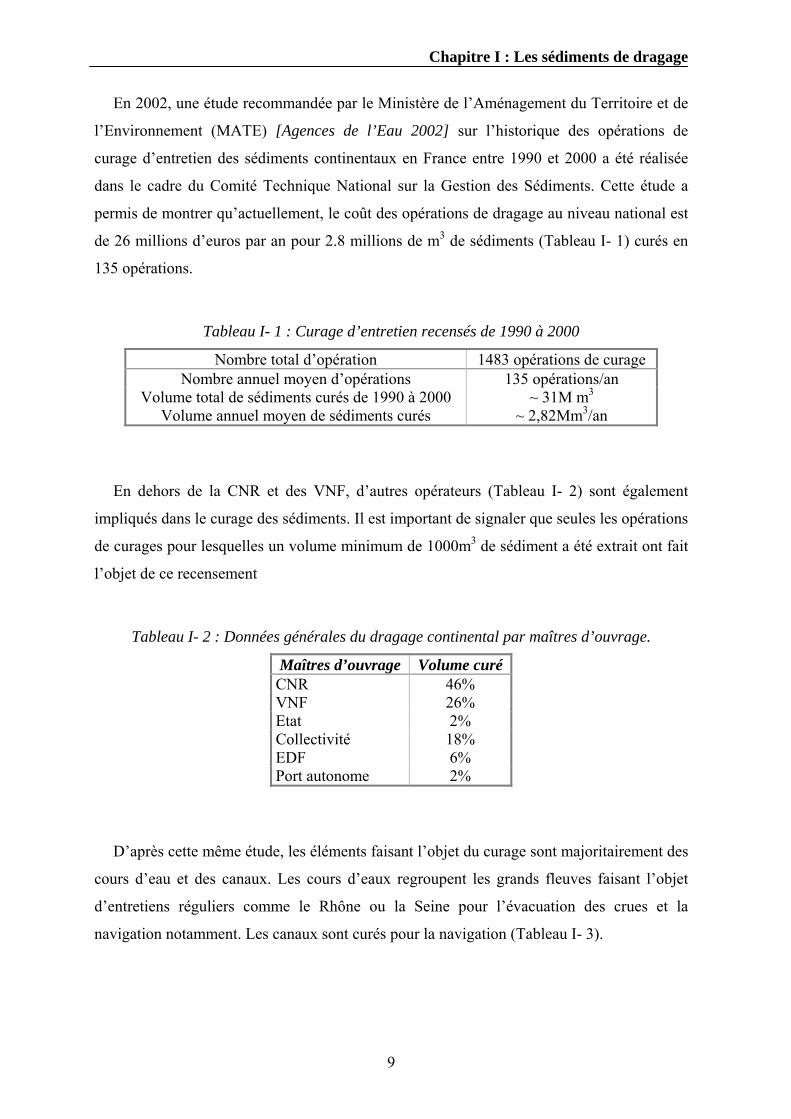
Tableau I- 1 : Curage d’entretien recensés de 1990 à 2000
Nombre total d’opération 1483 opérations de curage Nombre annuel moyen d’opérations 135 opérations/an
Volume total de sédiments curés de 1990 à 2000 ~ 31M m3
Volume annuel moyen de sédiments curés ~ 2,82Mm3/an
En dehors de la CNR et des VNF, d’autres opérateurs (Tableau I- 2) sont également
impliqués dans le curage des sédiments. Il est important de signaler que seules les opérations
de curages pour lesquelles un volume minimum de 1000m3 de sédiment a été extrait ont fait
l’objet de ce recensement
Tableau I- 2 : Données générales du dragage continental par maîtres d’ouvrage.
Maîtres d’ouvrage Volume curéCNR 46% VNF 26% Etat 2% Collectivité 18% EDF 6% Port autonome 2%
D’après cette même étude, les éléments faisant l’objet du curage sont majoritairement des
cours d’eau et des canaux. Les cours d’eaux regroupent les grands fleuves faisant l’objet
d’entretiens réguliers comme le Rhône ou la Seine pour l’évacuation des crues et la
navigation notamment. Les canaux sont curés pour la navigation (Tableau I- 3).

Chapitre I : Les sédiments de dragage
10
Tableau I- 3 : Données générales du dragage continental par type d’hydrosystème
Objet de curage Volume curéCours d’eau 70% Canal 8% Plan d’eau 13% Retenue d’eau 8% Zone portuaire fluviale 1%
Les motivations premières qui induisent la réalisation d’une opération de curage sont le
maintien de la navigation et la lutte contre les inondations (Tableau I- 4).
Tableau I- 4 : Principales motivations du curage
Motivation du curage Volume curéMaintien de la navigation 43% Lutte contre les inondations 32% Environnement 12% Exploitation 10% Tourisme 3%
L’étude a permis également de constater que les besoins en matière de curage pour les dix
ans à venir sont légèrement supérieurs de 8% au volume de sédiment curés entre 1990 et
2000.
A travers les chiffres donnés concernant les quantités de sédiment marins et continentaux
dragués, nous pouvons constater que les volumes produits par le dragage continental est
minime au regard du dragage marin. En effet, ils représentent respectivement 2,82 et 50Mm3
de sédiments dragués par an. Les besoins réguliers de dragage pour des raisons telles que
l’entretien, le maintien de la navigation ou la lutte contre les inondations laissent prévoir que
ces quantités risquent d’augmenter. Les prévisions les estiment d’ailleurs à la hausse. Des
solutions économiquement viables qui prennent en compte l’aspect environnemental et
réglementaire doivent donc être trouvées pour les gérer. Dans la partie qui suit nous allons
développer ce dernier point qui concerne la réglementation française qui régit la gestion des
sédiments.

Chapitre I : Les sédiments de dragage
11
I.1.3. Réglementation régissant la gestion des sédiments
La partie réglementaire qui concerne les sédiments continentaux à été en grande partie
tirée d’une note juridique du Ministère de l’Ecologie et du Développement Durable [MEDD
2002] sur la gestion des sédiments extraits des cours d’eau et de canaux.
I.1.3.1. Sédiments continentaux
I.1.3.1.1. La gestion des sédiments continentaux
L’analyse de la gestion des sédiments après enlèvement se heurte à la définition de la
notion de déchet et à la multiplicité des filières d’élimination de ces matériaux
La notion de déchet appliquée aux sédiments
La notion de déchet est définie à l’article L 541-1 [Legifrance] du code de
l’environnement. Cette définition indique qu’est considéré comme déchet tout résidu d'un
processus de production, de transformation ou d'utilisation, toute substance, matériau,
produit ou plus généralement tout bien meuble abandonné ou que son détenteur destine à
l'abandon.
Les sédiments sont des « sous-produits » d’une activité (le dragage) qui vise à rétablir un
libre écoulement et non pas à exploiter un matériau particulier en vue de son utilisation. Ils
ne sont pas la finalité même de l’opération de dragage et sont généralement destinés à
l’abandon. Pour ces raisons, les sédiments extraits des cours d’eau sont, dans de nombreux
cas, considérés comme des déchets.
Le statut de déchet n’interdit pas une utilisation future ou une vente à titre onéreux. Ainsi
certains laitiers de la sidérurgie sont des déchets même s’ils sont ensuite utilisés pour la
réalisation de routes.
Si le déchet subit un traitement pour le rendre directement utilisable, ou en extraire une
partie utilisable, les éléments utilisables peuvent être considérés comme ayant perdu le statut
de déchet.
Le décret n°2002-540 du 18 avril 2002 [Aida] qui reprend la nomenclature européenne
des déchets a classé les sédiments sous la rubrique 17 05 05* boues de dragage contenant
des substances dangereuses ou 17 05 06 boues de dragage autres que celles visées à la
rubrique 17 05 05*. L’astérisque qui suit le code indique qu’il s’agit alors d’un déchet
dangereux.

Chapitre I : Les sédiments de dragage
12
La réglementation communautaire comme nationale a également introduit la notion de
déchet dangereux. Pour de tels déchets, s’impose en sus une obligation de traçabilité.
Pour savoir si un déchet est dangereux et donc de quelle rubrique de la nomenclature
déchet il relève, il convient de déterminer s’il possède au moins un des 14 critères de
dangerosité définis à l’annexe I du décret (décret n°2002-540 du 18 avril 2002) qui vont de
H1 à H14. Certains critères ne sont pas adaptés à la gestion des sédiments (risque
explosif…). Très souvent et vraisemblablement dans le cas des sédiments, le critère H14
relatif à l’écotoxicité du déchet est déterminant pour savoir si un déchet est dangereux ou
non. Aucune méthode claire n’existe aujourd’hui pour déterminer si un déchet est
écotoxique ou non. Il y a actuellement une réflexion en cours sur des tests d’écotoxicité
utilisables pour caractériser le critère H14 mais les données actuellement disponibles ne
permettent pas d’envisager la fixation des seuils.
Au niveau français, la dangerosité des sédiments pourrait également être caractérisée à
partir des normes sols et boues utilisées dans le domaine des boues de station d’épuration
(décret n°97-1133 du 8 décembre 1997 et arrêté du 8 janvier 1998) [Aida]. Une décision a
été prise par le conseil de l’union européenne le 19 décembre 2002 établissant des critères et
procédures d’admission des déchets dans les décharges, conformément à l’article 16 et a
l’annexe II de la directive 1999/31/CE [Aida]. Elle a permis la définition de trois seuils
pour : les déchets inertes, les déchets non dangereux et les déchets dangereux (plus
d’informations seront données dans le prochain paragraphe).
En l’absence de valeurs guides, des acteurs de la gestion des curages/dragages comme par
exemple le port autonome de Rouen et les VNF ont été amenés à développer leur propres
valeurs guides [Clozel 2003] :
• Le port autonome de Rouen a proposé une grille de classification de la qualité des
produits de dragage continentaux dont il assure la gestion. Les sédiments sont classés selon
les indices de qualité. Une grille de catégorie de concentration est établie pour les métaux
lourds (Tableau I- 5), les PCB et les HAP. Cette classification n’a pas de statut
réglementaire officiel mais elle a été mise au point en collaboration avec la DRIRE locale.
Ces indices de qualité constituent essentiellement des indices de suivi de l’évolution de la
qualité chimique des sédiments sans définir spécifiquement de schéma de gestion.

Chapitre I : Les sédiments de dragage
13
Tableau I- 5 : Valeurs de référence du port autonome de Rouen
Catégorie As Cd Cr Cu Hg Pb Ni Zn 1 <10 <0.8 <50 <35 <0.3 <50 <25 <100 2 10-20 0.8-2.4 50-150 35-100 0.3-1 50-100 25-50 100-250 3 20-30 2.4-6 150-250 100-200 1-2 100-250 50-100 250-750 4 30-50 6-12 250-500 200-300 2-4 250-500 100-200 750-1500 5 50-100 12-20 500-1000 300-500 4-10 500-1000 200-500 1500-30006 >100 >20 >1000 >500 >10 >1000 >500 >3000
• Les VNF ont établi des consignes de caractérisation des sédiments continentaux à
draguer en terme qualitatif, quantitatif et devenir en découlant, visant à proposer à leur
services une procédure unifiée lors de la gestion des curages. Ces seuils, au nombre de deux,
permettent d’établir 3 catégories de sédiments :
1 Catégorie 1 : en dessous du seuil 1 : la valorisation des produits de dragage est
recommandée, elle peut être faite sur des terres agricoles alimentaires ; il n’existe aucune
restriction dans le devenir de ces produits
2 Catégorie 2 : aucune teneur des produits de dragage en l’un de ces éléments n’est
supérieure à la teneur de référence correspondante (seuil 2). Les conditions d’application de
la catégorie 2 permettent de valoriser ou de stocker les produits de dragage, seul le régalage
sur des terres agricoles est à proscrire.
3 Catégorie 3 : la teneur en un ou plusieurs éléments est supérieure à la teneur de
référence correspondante (seuil 2). La valorisation ou le stockage des produits nécessite la
mise en place d’une étude de faisabilité relative à la destination du produit de dragage.
Le seuil 1 correspond aux valeurs limites « sol » pour les métaux telles que définies dans
l’arrêté du 8 janvier 1998 (fixant les prescriptions techniques applicables aux épandages de
boues issues du traitement des eaux usées, sur les sols agricoles). Le seuil 2 a été défini
arbitrairement ; les valeurs qui composent sont comprises entre les valeurs limites « sol » et
« boues » de l’arrêté du 8 janvier 1998.
Il existe également des valeurs guides portant sur la qualité des sédiments dont la finalité
première est de préserver la qualité du milieu eau. Ce sont les données du SEQ (Système
d’Evaluation de la Qualité)
Les filières d’élimination des sédiments
Au regard des critères retenus, il semble que l’enlèvement des sédiments ne crée pas des
déchets dans tous les cas :
Chapitre I : Les sédiments de dragage
14
- Extraction de matériaux de carrière : dans le cas où l’entretien est soumis à la
réglementation sur les carrières (rubrique 2510 a de la nomenclature installations classées
[Aida]), les matériaux extraits sont considérés comme des matières brutes et non pas des
déchets. Toutefois, quand un traitement des matériaux est nécessaire, le résidu du traitement,
qui n’est pas valorisé en tant que matériau de carrière, est un déchet.
- Remise en suspension : les sédiments ne sont pas considérés comme des déchets. Les
dispositions sur les déchets seraient d’ailleurs inadaptées.
- Epandage et régalage : les sédiments sont des déchets car il font l’objet d’un abandon.
Toutefois, dans ce domaine il existe déjà une réglementation plutôt favorable à ceux qui
assurent l’entretien du cours d’eau.
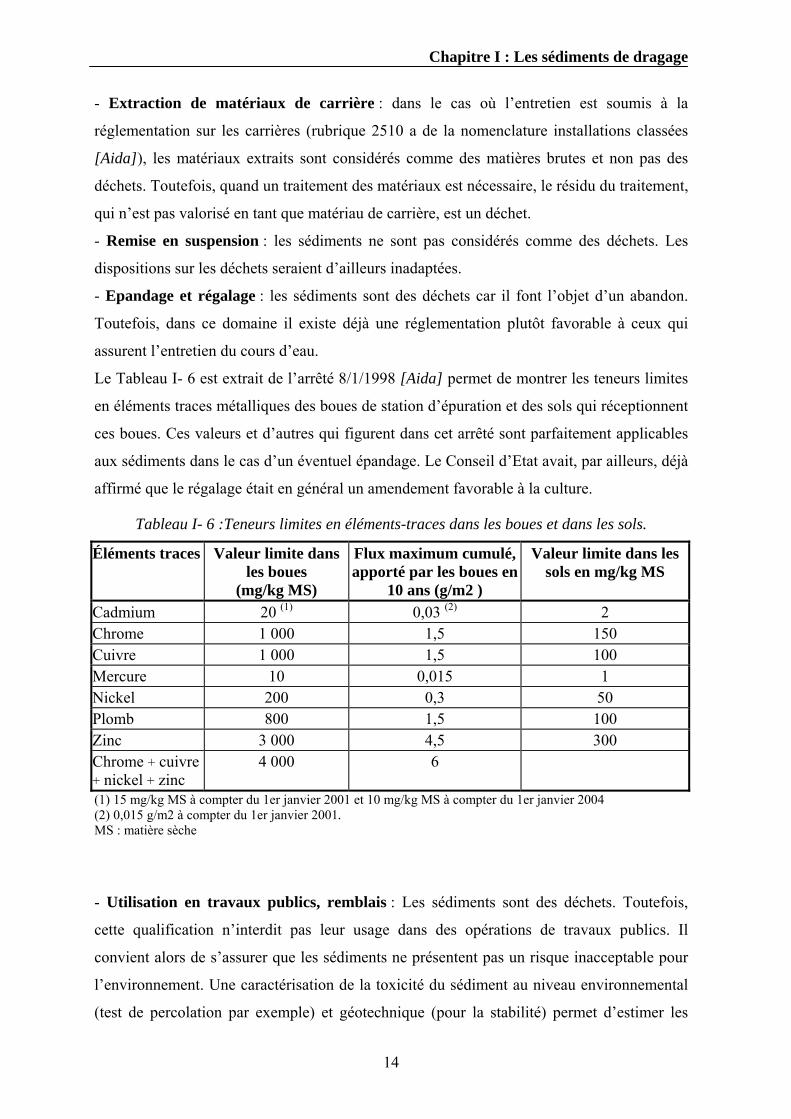
Le Tableau I- 6 est extrait de l’arrêté 8/1/1998 [Aida] permet de montrer les teneurs limites
en éléments traces métalliques des boues de station d’épuration et des sols qui réceptionnent
ces boues. Ces valeurs et d’autres qui figurent dans cet arrêté sont parfaitement applicables
aux sédiments dans le cas d’un éventuel épandage. Le Conseil d’Etat avait, par ailleurs, déjà
affirmé que le régalage était en général un amendement favorable à la culture.
Tableau I- 6 :Teneurs limites en éléments-traces dans les boues et dans les sols.
Éléments traces Valeur limite dans les boues
(mg/kg MS)
Flux maximum cumulé, apporté par les boues en
10 ans (g/m2 )
Valeur limite dans les sols en mg/kg MS
Cadmium 20 (1) 0,03 (2) 2 Chrome 1 000 1,5 150 Cuivre 1 000 1,5 100 Mercure 10 0,015 1 Nickel 200 0,3 50 Plomb 800 1,5 100 Zinc 3 000 4,5 300 Chrome + cuivre + nickel + zinc
4 000 6
(1) 15 mg/kg MS à compter du 1er janvier 2001 et 10 mg/kg MS à compter du 1er janvier 2004 (2) 0,015 g/m2 à compter du 1er janvier 2001. MS : matière sèche
- Utilisation en travaux publics, remblais : Les sédiments sont des déchets. Toutefois,
cette qualification n’interdit pas leur usage dans des opérations de travaux publics. Il
convient alors de s’assurer que les sédiments ne présentent pas un risque inacceptable pour
l’environnement. Une caractérisation de la toxicité du sédiment au niveau environnemental
(test de percolation par exemple) et géotechnique (pour la stabilité) permet d’estimer les

Chapitre I : Les sédiments de dragage
15
scénarios acceptables pour l’utilisation en travaux publics. L’inconvénient, est qu’il n’existe
pas de valeurs limites réglementaires pour la détermination de la toxicité.
- Dépôt ou stockage sur des parcelles, hors décharge (incluant le comblement d’ancienne
gravière ou d’ancienne carrière…) : les sédiments sont des déchets mais aucune rubrique
installation classée ne permet de classer ce stockage ou ce dépôt. Ces derniers peuvent
notamment être réglementés par le code de l’urbanisme [legifrance]
La directive 1999/31/CE du 26 avril 1999 concernant la mise en décharge des déchets
pourrait toutefois qualifier de tels dépôts en tant que décharge. Toutefois, certains
paragraphes de cette directive permettent de s’extraire du statut de décharge :
En conséquence peuvent être considérés comme exclus du champ de la directive :
• Les dépôts de boues de dragage non dangereuses sur les petites voies d’eau ;
• Les immersions de boues de dragage non dangereuses dans les eaux de surface dans le lit
des cours d’eau et leur soul-sol ;
• Les dépôts de boues de dragage inertes et non dangereux qui constitueraient des travaux
de remblai ou d’aménagement
- Mise en décharge : Une décision a été prise par le conseil de l’union européenne le 19
décembre 2002 [Aida] établissant des critères et procédures d’admission des déchets dans
les décharges. Trois types de décharges ont été définies pour : les déchets inertes, non
dangereux et dangereux. Les critères d’admission se basent en grande partie sur les tests de
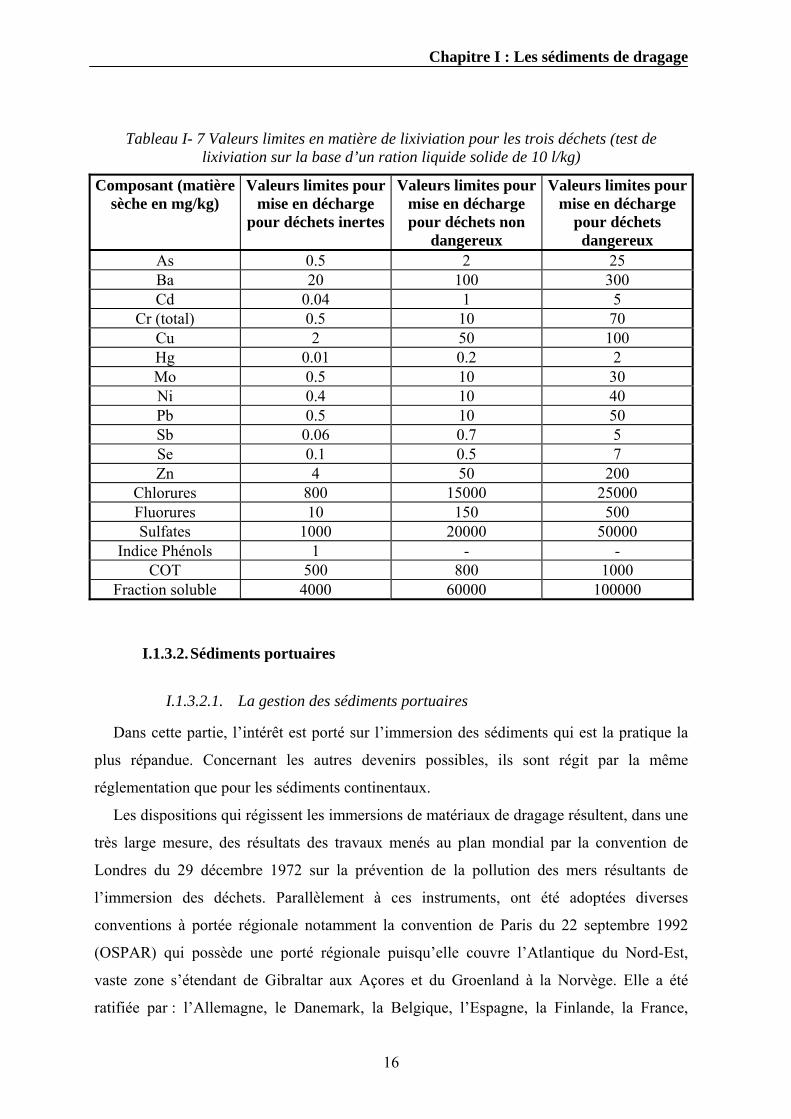
lixiviation. Le Tableau I- 7 regroupe les valeurs limites en matière de lixiviation pour les
trois types de déchets et uniquement pour les essais en batch avec un rapport liquide-solide
de 10 l/kg et une granulométrie inférieure à 4 mm (la norme NF EN 12457-2 (indice de
classement X30-402-2)) qui remplace la norme NF X 31-210
Chapitre I : Les sédiments de dragage
16
Tableau I- 7 Valeurs limites en matière de lixiviation pour les trois déchets (test de lixiviation sur la base d’un ration liquide solide de 10 l/kg)
Composant (matière sèche en mg/kg)
Valeurs limites pour mise en décharge
pour déchets inertes
Valeurs limites pour mise en décharge pour déchets non
dangereux
Valeurs limites pour mise en décharge
pour déchets dangereux
As 0.5 2 25 Ba 20 100 300 Cd 0.04 1 5
Cr (total) 0.5 10 70 Cu 2 50 100 Hg 0.01 0.2 2 Mo 0.5 10 30 Ni 0.4 10 40 Pb 0.5 10 50 Sb 0.06 0.7 5 Se 0.1 0.5 7 Zn 4 50 200
Chlorures 800 15000 25000 Fluorures 10 150 500 Sulfates 1000 20000 50000
Indice Phénols 1 - - COT 500 800 1000
Fraction soluble 4000 60000 100000
I.1.3.2. Sédiments portuaires
I.1.3.2.1. La gestion des sédiments portuaires
Dans cette partie, l’intérêt est porté sur l’immersion des sédiments qui est la pratique la
plus répandue. Concernant les autres devenirs possibles, ils sont régit par la même
réglementation que pour les sédiments continentaux.
Les dispositions qui régissent les immersions de matériaux de dragage résultent, dans une
très large mesure, des résultats des travaux menés au plan mondial par la convention de
Londres du 29 décembre 1972 sur la prévention de la pollution des mers résultants de
l’immersion des déchets. Parallèlement à ces instruments, ont été adoptées diverses
conventions à portée régionale notamment la convention de Paris du 22 septembre 1992
(OSPAR) qui possède une porté régionale puisqu’elle couvre l’Atlantique du Nord-Est,
vaste zone s’étendant de Gibraltar aux Açores et du Groenland à la Norvège. Elle a été
ratifiée par : l’Allemagne, le Danemark, la Belgique, l’Espagne, la Finlande, la France,
Chapitre I : Les sédiments de dragage
17
l’Irlande, l’Islande, la Norvège, les Pays-Bas, le Royaume-Uni, la suède, le Luxembourg et
la Suisse Elle concerne la protection du milieu marin et vise ainsi toutes les sources de
pollution.
Les stratégies de définition des valeurs guides sont largement laissées à l’appréciation des
autorités compétentes de chaque pays. En France elle sont le fruit d’un groupe de travail
intitulé Groupe d’Etude et d’Observation sur les Dragages et l’Environnement (GEODE),
qui a proposé des valeurs guides communément appelées « niveau Géode » pour les métaux
lourds et les PCB (Polychlorobiphényles) [Alzieu 1999]
La réglementation relative aux dragages et immersion a été révisée et complétée à partir
de juin 2000. Quatre textes traitent des conditions relatives à l’évaluation de la qualité des
sédiments et des procédures administratives à respecter pour la réalisation de travaux. Nous
ne citerons dans le cadre de ce travail que les textes qui concernent les niveaux de référence
pour l’immersion.
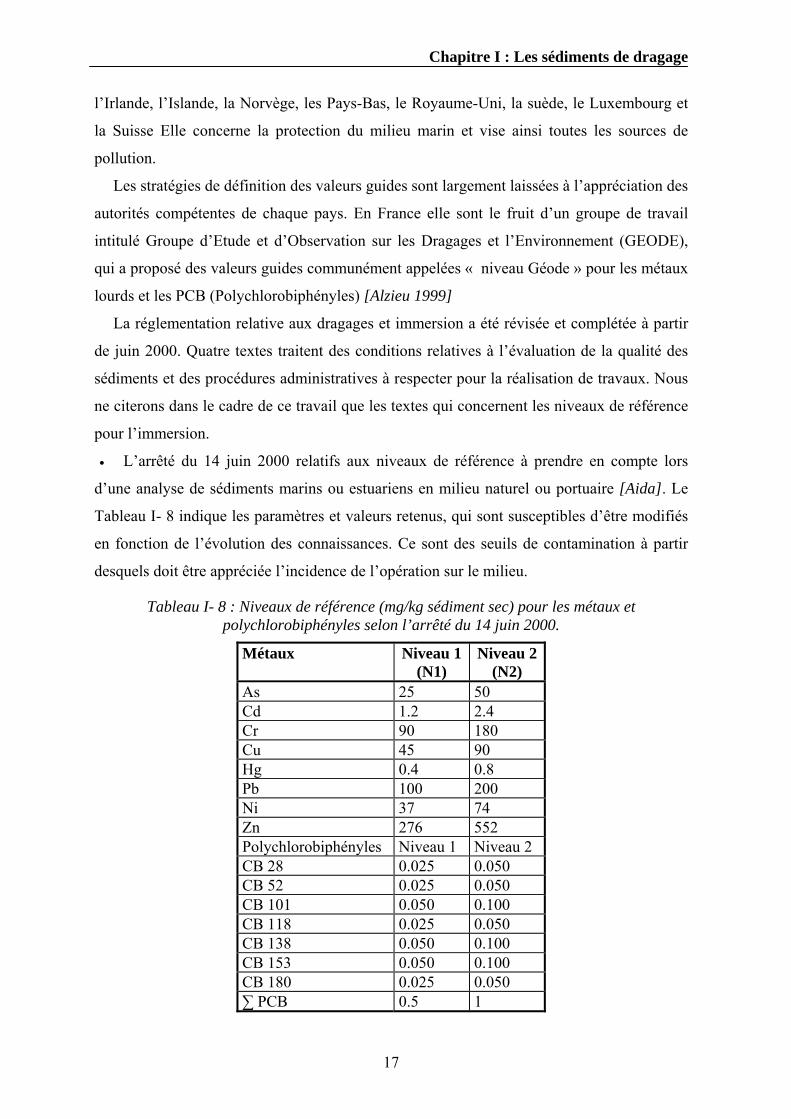
• L’arrêté du 14 juin 2000 relatifs aux niveaux de référence à prendre en compte lors
d’une analyse de sédiments marins ou estuariens en milieu naturel ou portuaire [Aida]. Le
Tableau I- 8 indique les paramètres et valeurs retenus, qui sont susceptibles d’être modifiés
en fonction de l’évolution des connaissances. Ce sont des seuils de contamination à partir
desquels doit être appréciée l’incidence de l’opération sur le milieu.
Tableau I- 8 : Niveaux de référence (mg/kg sédiment sec) pour les métaux et polychlorobiphényles selon l’arrêté du 14 juin 2000.
Métaux Niveau 1 (N1)
Niveau 2 (N2)
As 25 50 Cd 1.2 2.4 Cr 90 180 Cu 45 90 Hg 0.4 0.8 Pb 100 200 Ni 37 74 Zn 276 552 Polychlorobiphényles Niveau 1 Niveau 2 CB 28 0.025 0.050 CB 52 0.025 0.050 CB 101 0.050 0.100 CB 118 0.025 0.050 CB 138 0.050 0.100 CB 153 0.050 0.100 CB 180 0.025 0.050 ∑ PCB 0.5 1

Chapitre I : Les sédiments de dragage
18
• La circulaire relative aux conditions d’utilisation du référentiel de qualité des sédiments
marins ou estuariens en milieu naturel ou portuaire défini par arrêté interministériel du 14
juin 2000 [Aida], dont l’objet est d’expliciter auprès des services de l’Etat et de ces
établissements publics les conditions d’utilisation des niveaux de référence (Tableau I- 8).
Elle précise que : « Ces seuils constituent des points de repère permettant de mieux
apprécier l’incidence que peut avoir l’opération projetée, ainsi, au-dessous du niveau N1,
l’impact potentiel est en principe jugé d’emblée neutre ou négligeable, les teneurs étant
« normales » ou comparables au bruit de fond environnemental. Entre le niveau N1 et le
niveau N2, une investigation complémentaire peut s’avérer nécessaire en fonction du projet
considéré et du degré de dépassement du niveau N1. De façon générale, l’investigation
complémentaire doit être proportionnée à l’importance de l’opération envisagée. Au delà du
niveau N2, une investigation complémentaire est généralement nécessaire car des indices
notables laissent présager un impact potentiel négatif de l’opération. Il faut alors mener une
étude spécifique portant sur la sensibilité du milieu aux substances concernées, avec au
moins un test d’écotoxicité globale du sédiment, une évaluation de l’impact prévisible sur le
milieu »
Elle donne également des indications sur la stratégie d’échantillonnage et les paramètres à
prendre en compte [Alzieu 2003].
I.1.3.3. Conclusions :
On peut conclure au regard de toutes ces informations :
• La réglementation française aborde très peu, voire pas du tout, les problèmes essentiels
de la qualification (toxicité) des produits de curage et de leur devenir possible (mis à part les
seuils qui concernent les décharges et les niveaux de références Geode).
• En l’absence de valeurs guides réglementaires pour les sédiments, certains opérateurs
ont été amenés à développer leur propres grilles de valeurs. C’est par exemple le cas du port
autonome de Rouen et des voix navigables de France.
• La réglementation doit s’améliorer afin de pouvoir répondre à la question : quelle qualité
pour quel devenir et à quel risque ?
Cette partie à été longuement développée afin de mieux mettre en avant le vide juridique
auquel nous sommes affrontés. En effet la validation du procède NOVOSOL nécessite
l’utilisation d’un outil réglementaire, qui met en évidence son efficacité et surtout qui soit
approprié à l’usage final du produit valorisé (domaine du génie civil et dans d’autres filières
actuellement en étude). Ce manque d’outil approprié nous oblige à prendre comme méthode

Chapitre I : Les sédiments de dragage
19
de référence les valeurs de la décision 19 décembre 2002 établissant des critères et
procédures d’admission des déchets dans les décharges, soit l’application de la norme X30-
402-2, bien que le matériaux produit par le procédé NOVOSOL ne soit pas destiné à une
mise en décharge mais à la valorisation. L’idée est d’évaluer son degré de dangerosité avant
et après traitement.
I.1.4. Le devenir des sédiments dragués:
I.1.4.1. Sédiments portuaires
En dehors des indications fournies par l’IFREMER (Institut français de recherche pour
l'exploitation de la mer) et les valeurs données par la commission OSPAR concernant les
quantités de sédiment portuaires immergés, aucun organisme contacté ne possède des
chiffres exacts relatifs au devenir des sédiments portuaires en France hors immersion.
Selon les risques qu’ils présentent pour l’environnement, les matériaux dragués peuvent
être soit rejetés en mer par surverse au moment du dragage, soit immergé dans les zones
autorisées, soit déposés à terre en vue d’un stockage ou d’un traitement. Le dépôt à terre
constitue une solution alternative quand l’immersion présente des risques pour des zones
sensibles mais il nécessite d’importantes surfaces de stockage. Le stockage à terre des
matériaux sableux à des fins de génie civil est fréquemment utilisé. Ces matériaux sont
recherchés pour la construction, les remblais ou le rechargement des plages. Le recours au
stockage à terre pour les sédiments fins est très peu utilisé en France, vraisemblablement par
manque de sites appropriés [Alzieu 1999 ;2003].
De même, la mise en œuvre des techniques de traitement physico-chimiques, biologiques
ou thermiques est réservée, en raison de leur coût élevé, à de faibles volumes de sédiments
fortement pollués. Ceci explique que la majeure partie des sédiments dragués soit restituée
au milieu naturel [Alzieu 2003].
A titre indicatif, et afin de comparer les quantités immergées par la France avec le reste
des états membres de la convention OSPAR, les données des tonnages de matériaux dragués
fournies par ces états sont indiquées dans le Tableau I- 9. Ces résultats tendent à montrer que
plus de 80% de ces immersions sont effectuées par quatre pays : la Belgique, la France, le
Royaume-Uni et l’Allemagne. [OSPAR 2000 ; 2001].
L’élimination des matériaux de dragage en mer influe sur l’environnement, en raison des
contaminants qu’ils contiennent, ainsi que sur le plan physique. Selon les lignes directrices
OSPAR sur la gestion des matériaux de dragage, la minimisation du volume des matériaux
Chapitre I : Les sédiments de dragage
20
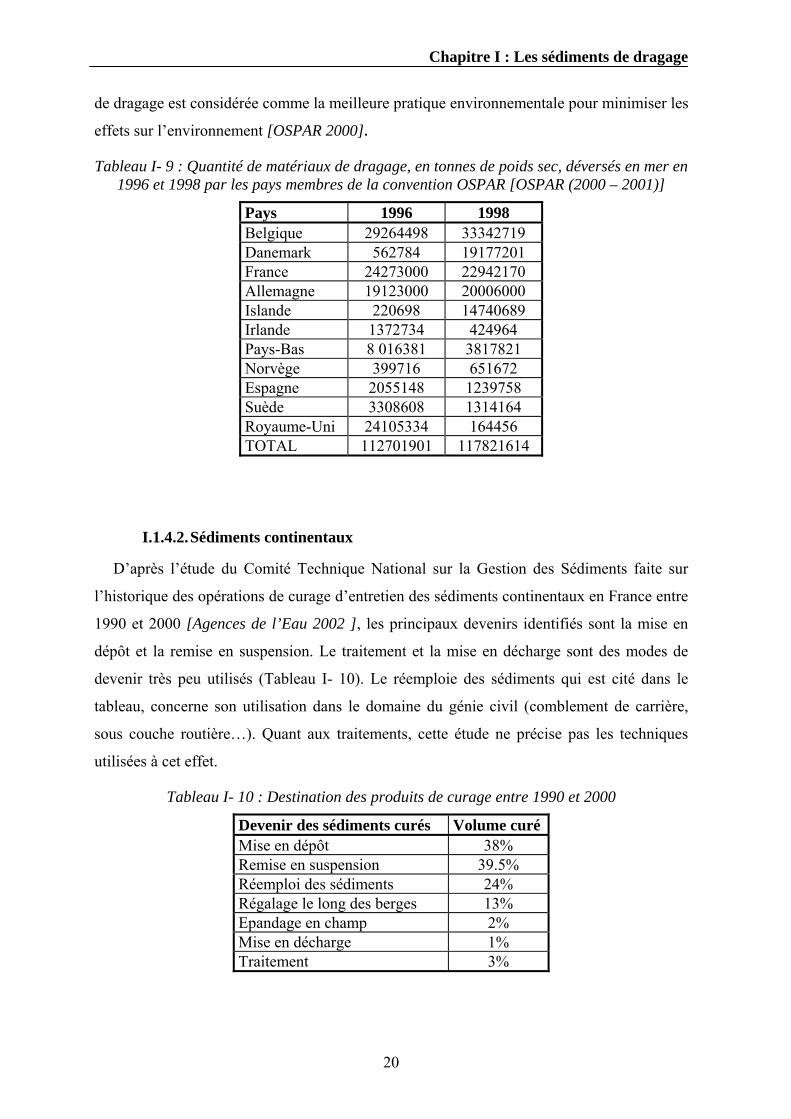
de dragage est considérée comme la meilleure pratique environnementale pour minimiser les
effets sur l’environnement [OSPAR 2000].
Tableau I- 9 : Quantité de matériaux de dragage, en tonnes de poids sec, déversés en mer en 1996 et 1998 par les pays membres de la convention OSPAR [OSPAR (2000 – 2001)]
Pays 1996 1998 Belgique 29264498 33342719 Danemark 562784 19177201 France 24273000 22942170 Allemagne 19123000 20006000 Islande 220698 14740689 Irlande 1372734 424964 Pays-Bas 8 016381 3817821 Norvège 399716 651672 Espagne 2055148 1239758 Suède 3308608 1314164 Royaume-Uni 24105334 164456 TOTAL 112701901 117821614
I.1.4.2. Sédiments continentaux
D’après l’étude du Comité Technique National sur la Gestion des Sédiments faite sur
l’historique des opérations de curage d’entretien des sédiments continentaux en France entre
1990 et 2000 [Agences de l’Eau 2002 ], les principaux devenirs identifiés sont la mise en
dépôt et la remise en suspension. Le traitement et la mise en décharge sont des modes de
devenir très peu utilisés (Tableau I- 10). Le réemploie des sédiments qui est cité dans le
tableau, concerne son utilisation dans le domaine du génie civil (comblement de carrière,
sous couche routière…). Quant aux traitements, cette étude ne précise pas les techniques
utilisées à cet effet.
Tableau I- 10 : Destination des produits de curage entre 1990 et 2000
Devenir des sédiments curés Volume curéMise en dépôt 38% Remise en suspension 39.5% Réemploi des sédiments 24% Régalage le long des berges 13% Epandage en champ 2% Mise en décharge 1% Traitement 3%

Chapitre I : Les sédiments de dragage
21
La totalité des valeurs annoncées excède les 100%. D’après les personnes qui se sont
chargées de cette étude, cette surestimation est due à une difficulté de gestion de la part des
acteurs du dragage. Néanmoins ces résultats nous permettent de dégager les grande
tendances des devenirs de ces sédiments. L’étude a permis également de conclure qu’aucune
modification très nette dans les types de devenir des sédiments curés n’est constatée entre
1990 et 2000. Il semble toutefois exister une tendance à l’augmentation de la mise en dépôt
et du régalage le long des berges ainsi qu’une diminution de l’épandage en champ.
La conclusion que nous pouvons tirées à travers les informations recueillies sur le devenir
des sédiments en France, est que très peu de sédiments sont traités. Uniquement 3% de
sédiments continentaux sont traités, et 24% réemployés. Concernant les sédiments marins,
nous n’avons pas réussi à avoir des informations exactes, toutefois, il semblerait que la
majorité des sédiments sont immergées.
Après avoir présenté les quantités de sédiment dragué en France, leur devenir et la
réglementation qui régit leur enlèvement et leur gestion. Nous présentons dans la partie
suivante, les différents procédés de traitement qui existent, dont celui sur lequel nous allons
travailler.
I.2. Procédés de traitement des sédiments
I.2.1. Introduction
Les premiers programmes de recherche sur les sédiments toxiques sont apparus à la fin
des années quatre-vingt aux Etats-Unis, au Canada, aux Pays-Bas et en Allemagne. Ces
programmes sont proches de ceux utilisés pour le traitement des sols. Les analogies entre ces
deux matériaux sont importantes, avec cependant, des particularités propres à chacun d’eux.
La problématique des sédiments est dans l’ensemble plus complexe. Plusieurs facteurs sont
à son origine :
Pour de nombreux sols contaminés, on est en mesure de déterminer l’origine de la
pollution (ancienne activité industrielle, déversement accidentel …).
La nature de la pollution est alors connue. Dans le cas des sédiments, la pollution
provient la plupart du temps de rejet d’origine variées (industriels, urbaines ou agricoles) qui
sont mélangés entre eux et véhiculés par l’eau et la matière en suspension. Tous ces
phénomènes qui favorisent la dispersion et la transformation chimique des polluants rendent
la caractérisation nettement plus difficile.

Chapitre I : Les sédiments de dragage
22
D’autre part, les caractéristiques physiques des produits sont différentes et influencent
beaucoup les procédés de traitement. Dans les deux cas, les polluants sont très liés aux
particules fines. Plusieurs études ont établi une relation entre la finesse des grains et la
concentration en métaux lourds et il s’avère que la relation entre les deux est
proportionnelle. Ainsi plus les particules sont fines, plus elles sont chargées en métaux
lourds [Cauwenberg 1998 ; Stevenson 1994]. Une simple séparation granulométrique peut
suffire à les récupérer. Pour un sol (distribution granulométrique étalée), la proportion de
matériaux grenus (dépollués) et potentiellement valorisable est nettement plus importante
que pour un sédiment, constitué d’une forte proportion de particules fines (argile,
colloïdes,…)
Plusieurs techniques de traitement ont été développées pour les deux types de matériaux.
Ces techniques peuvent être utilisées seules ou en combinaison. On classe ces techniques en
trois catégories:
• Procédés de traitement par concentration des polluants : les polluants sont isolés
de la matrice sédimentaire et le sédiment est décontaminé en totalité ou en partie ;
• Procédés de traitement par dégradation des polluants : ces procédés ne sont
applicables qu’à certaines contaminations organiques. Les polluants sont détruits
par voie chimique (oxydation) ou biologique (biodégradation).
• Procédés de traitement par neutralisation des polluants : le matériau contaminé est
alors soit stabilisé (mélange avec les liants hydrauliques, vitrification,…), soit
détruit (incinération).
Les techniques de traitement des sédiments contaminés sont multiples : certaines ont fait
la preuve de leur validité opérationnelle à l’échelles industrielle, d’autres en sont au stade du
laboratoire ou à l’échelle pilote. Les choix des techniques de traitement dépendent de la
nature physique des sédiments, des contaminants associés et de leurs niveaux de présence,
de l’importance des volumes à traiter ainsi que des valorisations possibles en fonction des
règlements locaux.
Dans ce qui suit, les techniques de traitement des sédiments vont être présentées suivant
la méthode de traitement : physique et physico-chimique, biologique, thermique et enfin
chimique.
I.2.2. Traitement physique et physico-chimique
Les polluants contenus dans un sédiment sont souvent présents dans des fractions
particulières de la matrice. Ainsi, selon les cas, les particules fines, lourdes ou légères,

Chapitre I : Les sédiments de dragage
23
peuvent présenter des teneurs en polluant très élevées, les travaux de caractérisation ont
montré que la quasi-totalité des polluants sont dans la fraction granulométrique inférieure à
60 µm. D’une façon générale, les procédés de traitement physique et physico-chimique
reposent sur ces spécificités, et reprennent techniquement des principes largement répandus
dans certains secteurs industriels (le traitement des minerais par exemple). L’objectif est
d’obtenir deux matériaux distincts :
• D’un coté, un produit correspondant à une part importante des solides du
sédiments, ne contenant plus qu’une infime proportion des contaminants présents au départ ;
• D’un autre coté, un résidu, de volume réduit, renfermant la quasi-totalité des
polluants. Le confinement ou l’inertage ne porte alors que sur cette fraction résiduelle, d’où
une réduction importante des coûts.
Les paramètres principaux qui peuvent influencer les performances de dépollution de ces
procédés sont :
• La nature et la concentration des polluants ;
• La granulometrie du sédiment ;
• La répartition chimique des contaminants en fonction de la granulométrie ;
• L’existence et l’amplitude de critères discriminants entre les porteurs de
polluants et autres constituants du sédiments : la densité, la susceptibilité magnétique,
l’hydrophobie, et la chimie de surface
Cela implique que, outre celle des phases porteuses de contaminants, la
connaissance/caractérisation des constituants principaux des sédiments est également une
information précieuse [Clozel 2002].
Cependant, l’utilisation de ces techniques peut conduire à la désagrégation des particules
élémentaires dans des proportions importantes. Cela entraîne une augmentation de la
fraction fine qui pénalise les bilans de décontamination. Les principales techniques sont les
suivantes.
I.2.2.1. Le cyclonage
Un cyclone est constitué d’un tube conique ouvert à ses extrémités. L’alimentation en
pulpe sous pression est effectuée en continue par une ouverture latérale située dans la partie
supérieure de l’appareil. Sous l’effet de la vitesse, un vortex se forme. Les particules les plus
grosses et les plus lourdes vont se coller à la paroi extérieure. Elle glissent vers le bas du
cyclone jusqu'à la sortie appelée souverse. Les particules les plus fines et les plus légères se
stabilisent au centre du vortex et sortent en débordement du cyclone par la surverse.

Chapitre I : Les sédiments de dragage
24
Cet outil permet d’effectuer des séparations granulométriques jusqu’à dix microns. Le
dimensionnement des orifices de sortie, la pression d’alimentation, la force centrifuge ainsi
que la dilution du sédiment d’entrée sont autant de paramètres qui permettent de faire varier
la coupure granulométrique. Son utilisation à grande échelle dans le traitement des
sédiments de dragage est assez courante. Il permet en effet de séparer les fractions argileuses
des fractions sableuses. Un tamisage grossier est cependant nécessaire afin d’éviter
l’obstruction de l’appareil tout en améliorant ses performances.
Dans certain cas le traitement par hydrocyclone est suffisant pour obtenir une fraction
sableuse propre. Cependant, il est parfois nécessaire de poursuivre le traitement par d’autres
procédés. L’hydrocyclone intervient alors comme pré-traitement, son coût est de 3 à 20 €
/tonnes de matière sèche, cette méthode est fiable à 90% pour les sédiments visqueux et
argileux [AEAP 2000 (b) ; Marot 1998]
I.2.2.2. L’attrition
Cette technique est utilisée en pré-traitement pour conditionner les matériaux à traiter.
Ainsi, lorsque la distribution granulométrique et chimique des polluants n’est pas
exclusivement sur la fraction fine, cela peut être dû à un enrobage des grosses particules
minérales par des fines polluées. C’est souvent le cas des matériaux argileux, et par
conséquent des sédiments. Des analyses particulières peuvent mettre en évidence ce type de
phénomène.
Dans ce cas, la séparation par cyclonage ne peut conduire à des résultats satisfaisants. Il
est alors nécessaire d’insérer en amont du circuit de traitement une étape supplémentaire de
nettoyage des surfaces. Le principe de l’attrition consiste à engendrer des forces de
frottement entre les grains au moyen d’une agitation vigoureuse. Pour cela, il existe des
cellules d’attrition qui sont équipées de pales inversées favorisant les contacts. Une attrition
efficace de sédiments contaminés conduit à la désagrégation des substances organiques
nocives d’une part, et au nettoyage des surfaces minérales sans concassage excessif des
composants minéraux cristallins d’autre part [AEAP 2000 (b) ; Marot 1998].
Actuellement, ce procédé est couramment employé dans les unités de lavage de
sédiments existant au Canada, aux Pays-Bas ou en Angleterre.
L’avantage de cette technique est la possibilité de l’employer à tout type de particules,
son inconvénient est la désagrégation des particules, son coût est inférieur à 10€/tonnes de
matière sèche et son efficacité est de 70%

Chapitre I : Les sédiments de dragage
25
I.2.2.3. La gravimétrie
Les polluants sont parfois fixés sur les fractions légères constituées de matière organique
et peuvent également être sur les fractions lourdes correspondant à des minéraux lourds ou à
des déchets particuliers. La séparation de ces constituants est alors intéressante pour
effectuer une opération de décontamination .
Les clés d’une séparation gravimétrique efficace sont à rechercher dans la connaissance
parfaite des paramètres minéralogiques du minerai à traiter (masse volumique des divers
constituants, répartition granulométrique des espèces minérales,…).
La séparation gravimétrique est un mode de concentration dans un fluide d’un matériau
mettant en œuvre la différence qui existe entre les masses volumiques des minéraux d’une
part et des gangues d’autre part. La concentration gravimétrique s’effectue dans un champ
de forces de masse, en général celui de la pesanteur, combiné à l’action d’autres forces telles
que la résistance offerte par le fluide (en général de l’eau pour la gravimétrie en voie
humide, plus rarement de l’air pour la gravimétrie en vois sèche) au mouvement des grains à
séparer ou les forces de frottement entre ces particules et une surface support fixe ou mobile.
Lorsque la pesanteur ne suffit pas, on a recours à une force centrifuge.
L’installation est constituée d’une cuve dans laquelle circule un flux d’eau de bas en eau
à un débit connu. L’alimentation en sédiment est effectuée en partie supérieure. Les fines
particules légères sont entraînées par le flux ascendant, tandis que les particules lourdes et de
taille plus importantes vont sédimenter dans l’appareil. Le diamètre des coupures est
contrôlé en faisant varier le débit d’eau. Les principaux procédés sont les spirales, les tables
à secousse ou les élutriateurs. La technique de séparation par spirale, table à secousses ne
semble pas avoir été essayées à grande échelle sur des sédiments. Le facteur limitant de cette
méthode semble être la granulométrie des particules, car pour qu’une concentration
gravimétrique puisse être obtenue, il faut qu’il existe une relation entre la masse volumique
d’un grain et sa teneur en contaminant [Alzieu 1999 ; Houot 1991].
I.2.2.4. La séparation magnétique
La séparation magnétique est un procédé qui utilise la force engendrée par un système
magnétique pour séparer les particules présentant des propriétés magnétiques différentes
[Gille1991], cela s’applique lorsque la pollution métallique du sédiment est due à la
présence de particules solides métalliques. Ainsi, pour les contaminants caractérisés par des
différences de comportement importantes entre les porteurs métalliques, le potentiel
d’application de ce procédé est limité. Des essais de séparation magnétique ont été faits aux

Chapitre I : Les sédiments de dragage
26
Canada et en Angleterre, ils ont montré sur des sols contaminés, que la fraction non
magnétique qui présente près de 70% pondéral de la fraction initiale ne contient plus que 4%
de Cr, 13% de Fe et 29 % de Zn. Le Cu et le Pb conduisent à des concentrations moins
favorables qui sont respectivement de 42 et 44%. Les affinités entre les métaux et l’oxyde de
fer sont bien connues, lorsque la caractérisation montre la présence de ce porteur métallique,
il devient intéressant de tester cet outil [Marot 1998].
Les limites de cette méthode est son application limitée par les caractéristiques du produit
de dragage et par la granulométrie qui doit être inférieur à 100 microns, elle est efficace à
80% et son coût est inférieur à 25 €/tonnes de matière sèche [AEAP 2000 (b)].
I.2.2.5. La flottation
La flottation est une technique de séparation des sols et sédiments qui à l’origine était
destinée aux traitements des minerais, mais qui a vu son extension à plusieurs champs
d’applications dans l’environnement tels que le traitement des eaux usées et la remédiation
des sols et sédiments [Cauwenberg 1998]. Cependant, compte tenu du grand nombre de
paramètres qu’il faut contrôler (pH, température, taille des particules concentration du
solide…) cette technique implique souvent des études préliminaires importantes [Vanthuyne
2003].
La flottation est basée sur les différences existant entre les propriétés superficielles des
solides dans une solution aqueuse et dans l’air. Pour ce faire, on disperse des bulles d’air
dans une suspension aqueuse de particules solides (sédiment) pour récupérer l’espèce
minérale à séparer, rendu préalablement hydrophobe par un ajout de collecteur (surfactant).
L’ensemble eau-bulles-particules hydrophobes est rassemblé sous forme d’une écume
surnageant stabilisée par un moussant.
La pratique de la flottation d’espèce minérale implique la compréhension [Blazy 2000] :
• des phénomènes électriques à l’interface minéral-eau ;
• de la modification des propriétés superficielles par le broyage ;
• de l’adsorption des surfactants sur les surfaces minérales ;
• de la nature des surfactants (collecteurs et moussants) et de l’action des
déprimants et des activants dont le rôle est d’inhiber ou de faciliter l’action des
collecteurs afin d’obtenir une séparation sélective.
L’utilisation de la flottation pour la matière organique semble satisfaisante [Marot 1998].
Cette méthode permet une réduction des volumes jusqu’à 85% et son coût est de 10 à 40 € /
tonnes de matière sèche [AEAP 2000 (b)].

Chapitre I : Les sédiments de dragage
27
I.2.2.6. L’électromigration
Le traitement par électromigration consiste à faire circuler un courant électrique entre
deux électrodes préalablement implantées dans le matériau à traiter. Cette différence de
potentiel génère une électrolyse qui favorise la formation d’une solution basique à la cathode
et d’une solution acide à l’anode. Cette dernière se charge progressivement des métaux
qu’elle dissout.
Les deux solutions sont ensuite récupérées et mélangées pour obtenir la neutralisation des
jus et la précipitation des métaux [Marot 1998].
Ce phénomène peut être vu comme un procédé à trois étapes [Leybros 1990] :
• électrodissolution du métal (oxydation) ;
• transport ionique (migration électrique, diffusion et /ou convection) ;
• électrodéposition (réduction de l’espèce métallique ionique sur les sites de
nucléation).
Les variables qui affectent la vitesse de migration d’un métal sont :
• La nature du métal et son état de surface ;
• La contrainte électrique
• L’espace entre les deux conducteurs ;
L’application de cette technique aux sédiments reste cependant assez délicate pour deux
raisons majeures. D’une part, le matériau doit être suffisamment perméable pour faciliter la
migration des polluants, la forte proportion de fines dans les sédiments est donc
préjudiciable. D’autre part, le passage en milieu acide est peu envisageable pour les
sédiments carbonatés.
I.2.2.7. Décontamination aux ultrasons
Les ultrasons sont souvent utilisés dans l’industrie pour une variété d’applications telles
que le décapage des surfaces métalliques, l’élimination des films d’oxyde, d’huile, de gras
ou autres contaminants sur les surfaces solides et également la dépollution des sols.
Les ultrasons sont des ondes d’une fréquence variant entre 16 kHz et 500 MHz qui
peuvent être transmises à travers l’eau, les gaz, et les suspensions de particules aqueuses.
Elles sont transmises sous forme d’énergie mécanique.
Une technique de décontamination des sédiments de dragage en couplant l’extraction par
ultrasons couplés à une pression sous vide a fait l’objet de recherche d’un laboratoire
américain. La technique se fait en trois étapes, la première consiste à appliquer les ultrasons
au sédiment pour traiter les grosses particules, une étape d’extraction sous vide permet

Chapitre I : Les sédiments de dragage
28
d’extraire les fines particules et l’eau contaminée contenant les polluants. La deuxième étape
consiste à appliquer les ultrasons aux particules fines récupérées, là aussi on récupère de
l’eau contaminée issue de l’extraction, sous vide. La troisième étape pour la
décontamination des eaux récupérées n’a pas été développée. Le contaminant étudié dans ce
cas est le chrome, en considérant que c’est un des métaux les plus dur à extraire et que si
l’efficacité de la technique est prouvée pour le Cr, elle le sera également pour les autres
métaux. Les résultats obtenus ont été satisfaisants. Il apparaît que cette technologie est
applicable de préférence aux sédiments à faible concentration d’argile, car l’extraction des
métaux à partir de cette dernière n’est pas très bonne [Meegoda 2001]. Cette méthode peut
également être appliquée à la pollution organique à condition d’utiliser un solvant qui
permet de faciliter leur extraction.
A travers la description de ces quelques techniques physiques et physico-chimiques, nous
pouvons conclure que mise à part l’électromigration et la décontamination par ultrason qui
présentent réellement des méthodes de dépollution, les autres techniques citées sont
d’avantage des pré-traitements. Ils permettent la séparation d’une partie du sédiment qui
comporte une concentration importante en polluants en se basant essentiellement sur les
propriétés physiques des sédiments. L’inconvénient de ces techniques est la nécessité de
traiter les volumes séparés dans lesquels la pollution est concentrée par d’autres méthodes de
dépollution. Néanmoins, l’avantage des pré-traitements est la réduction des volumes à
traiter. Cela permet la réduction des coûts de la dépollution proprement dite qui est souvent
onéreuse. Quant à l’électromigration et la décontamination par ultrason leur efficacité est
très limitée par la composition du sédiment et la spéciation des métaux lourds. Ils ne peuvent
donc pas être appliqués à tout les sédiments.
I.2.3. Traitements biologiques
I.2.3.1. Biolixiviation
La biolixiviation reprends le principe de la lixiviation chimique sauf que l’acidité du
milieu est générée par les microorganismes tels que le Thiobacillus ou les Paracoccus qui
réduisent les composés sulfurés sous conditions aérobiques et acides (pH=4) à températures
variant entre 15 et 55°C. La lixiviation peut être faite par voie indirecte, l’acidification des
composés sulfurés permet la libération d’acide sulfurique qui conduit par la suite à la
désorption des métaux lourds du sédiment par substitution de protons. La lixiviation directe

Chapitre I : Les sédiments de dragage
29
par contre permet la solubilisation du métal sulfuré par oxydation, cela conduit à la
formation de sulfate de métal [Mulligan 2001].
Cette méthode peut être statique ou dynamique. Dans le premier cas, le matériau à traiter
est mis en tas, et les solutions de lixiviation contenant les microorganismes sont introduites
par irrigation. Leur percolation favorise la dispersion des bactéries d’une part et la
récupération des métaux dissous d’autre part. Les jus de lixiviation sont collectés à la base
du tas dans des bassins pour être traités et recyclés. La biolixiviation dynamique se déroule
dans des réacteurs biologiques d’oxydation. Le choix des microorganismes dépend du pH,
de la source de carbone, de la vitesse d’agitation, de la température et du potentiel redox
[Marot 1998].
Au niveau des essais de laboratoire, les Thiobacillus ont permis l’extraction à partir de
sédiments contaminés de 70 à 75% de métaux lourds (à l’exception du Pb et de l'As)
[Mulligan 2001]
L’utilisation de la biolixiviation pour décontaminer des sédiments est relativement
séduisante. Cependant, elle n’est pas applicable dans tous les cas de figures. Dans ce cas
également. La nature des sédiments et la forme chimique des polluants sont des éléments
déterminants. Enfin, les temps de traitement ne paraissent pas un handicap trop important,
car ils sont compensées par des coûts assez faibles.
I.2.3.2. Phytoextraction
Certaines plantes (jacinthe d’eau, tournesol ou les herbes grasses) montrent une capacité
de rétention de métaux lourds très importante dans leurs organes (racines, les tiges et les
feuilles). Cette capacité d’accumulation a permis de développer cette méthode de
décontamination qui consiste tout simplement à les cultiver sur le site. Leur récolte permet
l’élimination d’une partie des polluants, les biomasses ainsi chargées en métaux lourds sont
séchées et incinérées. Les métaux sont alors récupérés dans les déchets ultimes de
l’incinération. L’efficacité de la méthode dépend de la nature du sédiment, des
contaminants, du degré de contamination et de l’épaisseur de la couche de matériau à traiter
[Alzieu 1999 ; Mulligan 2001]. Il dépend également de la température et de la salinité des
eaux de ruissellement dans lesquelles baignent le sédiment [Fritioff 2005]. Son coût est de
15 à 30 €/tonnes de matière sèche [AEAP 2000 (b)].
Il est important de signaler que cette méthode est une technique de phytoremediation.
Cette méthode peut être scindée sur la base du devenir des polluants et de leur localisation
dans les organes des plantes. Pour les sites contaminés avec les polluants inorganiques les

Chapitre I : Les sédiments de dragage
30
plantes peuvent stabiliser ou extraire ces polluants par trois mécanismes : phytostabilisation,
phytoextraction et rhizofiltration. Quand il s’agit de contaminants organiques comme le N,
le P et le K, les plantes peuvent extraire ou dégrader la pollution par trois mécanismes :
phytodégradation, rhizodégradation et phytovolatilisation [Mirck 2005].
Des obstacles s'opposent au développement de la phytoremédiation notamment le
transfert des contaminants dans la chaîne alimentaire par les animaux et les insectes, et le
temps requis pour l'absorption des contaminants d'un site.
I.2.3.3. Biodégradation
Le principe générale des traitements biologiques est d’exploiter certaines activités
microbiennes en les stimulant de manière contrôlée afin de réduire les nuisances potentielles
dues aux sédiments (odeur, risques sanitaires, caractère polluant au sens large du terme). De
ce fait les procédés biologiques sont en pratique généralement utilisés pour le traitement de
déchets essentiellement organiques présentant un caractère biodégradable [Gourdon 2001],
il favorise une minéralisation totale (H2O et CO2) de ces polluants. Cette technique nécessite
généralement peu d’énergie et peu de réactifs chimiques [Marot 1998]. Par contre elle
nécessite une régulation de la teneur en éléments nutritifs, en oxygène, du pH et de la
température afin d’assurer la croissance des bactéries [AEAP 2000 (b)]. La technique peut
être mise en œuvre dans des conditions statiques ou dynamiques, la première se fait en
épandage avec ajout de souches bactériennes appropriées (landfarming), il est possible
d’accélérer la biodégradation en utilisant des serres et la deuxième dans un bioréacteur ou en
bassin [Marot 1998].
Son efficacité pour les HAP peut atteindre les 99% en bioréacteur avec un coût de 60 à
100 €/tonnes de matière sèche, les coûts du traitement statique sont moins conséquents de 10
à 50 €/tonnes de matière sèche pour le landfarming simple et de 20 à 50 €/tonnes de matière
sèche pour le landfarming sous serre, l’efficacité de ces deux dernières méthodes est de 90%
[AEAP 2000 (b)].
En conclusions, on peut constater que dans les traitements biologiques deux types
d’espèces sont utilisées, les microorganismes et les plantes. Les microorganismes sont
utilisés pour traiter aussi bien les polluants organiques que inorganiques même si les
mécanismes mis en œuvre sont différents. Dans le premier cas, il s’agit d’une dégradation,
dans le deuxième, d’une lixiviation après solubilisation des polluants par les organismes. Le
premier cas est très souvent utilisé pour le traitement des boues de station d’épuration. Il est

Chapitre I : Les sédiments de dragage
31
efficace et ne présente pas d’inconvénients majeurs mise à part des temps de contacts
importants. Le second cas par contre, nécessite des temps de contact long, il dépend
fortement de la spéciation des métaux lourds et de la composition du sédiment.
Les plantes sont souvent utilisées pour le traitement des métaux lourds par extraction.
L’avantage de cette technique est de pouvoir l’appliquer in situ. Son inconvénient réside
dans le risque de contamination de la chaîne alimentaire à travers les animaux, la nécessité
de traiter les plantes par incinération, ce qui ne réduit pas forcement les coûts du traitement,
et la dépendance du traitement de la spéciation des métaux.
I.2.4. Traitements thermiques
I.2.4.1. La désorption
Cette méthode permet d’extraire les contaminants par évaporation ou désintégration.
L’évaporation est déterminée par le point d’évaporation du contaminant.
Les contaminants organiques se désintègrent (pyrolyse) en petite molécule entre 550 et
600°C [Rienks 1998]. Les composés organiques volatilisées sont récupérés par
condensation. Ainsi les pollutions sont concentrées sous une forme huileuse représentant un
volume réduit, facilement récupérable. La température et le temps de séjour sont les facteurs
déterminants de la décontamination. Son efficacité peut atteindre les 99% pour les PCB et
les HAP et son coût varie de 100 à 200 €/tonnes de matière sèche [AEAP 2000 (b)].
Dans le cas des pollutions métalliques, un traitement à une température de 800°C permet
d’évaporer les polluants tels que le mercure, l’arsenic et le cadmium. Certains métaux
restent dans les résidus solides et doivent être décontaminés autrement.
I.2.4.2. Oxydation à l’air humide
L’oxydation à l’air humide est un procédé qui permet d’accélérer la cinétique
d’oxydation chimique des composés organiques par addition d’oxygène. Les composés
organiques sont transformés en gaz carbonique et en eau. Cette méthode nécessite, pour une
bonne solubilité de l’oxygène dans l’eau, de travailler à pression importante de 20 à 100 bars
et température de 200-300°C [AEAP 2000 (b)]. Elle se fait en autoclave et il arrive que des
catalyseurs tel que le Fe+2 soient utilisés afin d’améliorer le rendement. Des résultats très
concluants ont été observés par un laboratoire au Pays-Bas, où la quasi-totalité des HAP
contenus dans le sédiment a été éliminés [Marot 1998 ; Rienks 1998]. Elle est également
très efficace pour le PCB, l’inconvénient de cette méthode est qu’elle ne permet pas la

Chapitre I : Les sédiments de dragage
32
décontamination des métaux lourds. Son coût est de 90 à 150 €/tonnes de matière sèche
[AEAP 2000 (b)].
I.2.4.3. La vitrification
Elle consiste à faire fondre les fractions argileuses des sédiments contaminés à une
température supérieure à 1250°C et à les refroidir rapidement : le refroidissement lent donne
un solide cristallin non lessivable. Elle permet la dégradation de la matière organique par
combustion et fixe les métaux non volatils ; le verre résultant du traitement occupe un faible
volume restreint par rapport à celui du sédiment de départ et peut être facilement valorisé.
Le coût élevé de l’énergie nécessaire à la fusion et l’émission de gaz renfermant des
substances toxiques en constituent les principaux inconvénients.
La vitrification peut également être obtenue directement sur un dépôt : la chaleur
nécessaire à la fusion est alors fournie par quatre électrodes de molybdène alimentées par un
courant triphasé à haute tension [Alzieu 1999].
Le coût de cette technique est de 90 à 130 €/tonnes de matière sèche pour une efficacité
qui peut atteindre les 99.99%. Ce procédé permet de donner des produits réutilisables
comme des matériaux de construction (briques, graviers …), des carreaux de verre, des
fibres de verre, de la laine de verre…[AEAP 2000 (b)].
I.2.4.4. L’incinération
Les matériaux subissent une oxydation à très haute température (800-1200°C) détruisant
les contaminants organiques et formant une structure cristalline qui fixe les métaux lourds.
Une partie des métaux lourds (Hg, Cd) est volatilisée au cours de l’opération [AEAP 2000
(b)], ce qui conduit dans le cas où la concentration en métaux lourds est très importante à un
transfert d’une partie de la pollution du sédiment vers les fumées. Cela nécessite donc un
traitement des fumées [Loannidis 2003]. Cette technique est très coûteuse en raison de la
consommation en énergie et du traitement des fumées. Son efficacité peut atteindre 99 %
pour les organiques et 100% pour les métaux lourds.
A partir de l’études de ces quatre techniques, nous pouvons conclure, qu’elles découlent
du même principe : un apport d’énergie thermique. Une température jusqu’à 600°C peut être
suffisante pour la dégradation de la matière organique. L’alternative est de chauffer à des
températures plus basses mais en augmentant la pression et en travaillant sous oxygène pour
améliorer le processus de dégradation.

Chapitre I : Les sédiments de dragage
33
Des températures supérieures à 600°C permettent la fixation des métaux lourds ou selon
les cas leur évaporation. Afin de bien stabiliser les métaux lourds, il est important de
chauffer à des températures plus importantes, voire jusqu’à 1250°C, pour s’assurer de la
stabilité des métaux dans la matrice solide, qui fond à ces températures. L’avantage de ces
techniques, contrairement à ce qu’on avait observé avec les méthodes physiques, est la
possibilité de décontaminer les deux types de pollution : organique et inorganique. Les
principaux inconvénients par contre sont :
• la nécessité de traiter les fumées émises essentiellement pour les sédiments ayant un fort
taux de polluants volatils. Pour palier à ce problème, il serait peut être nécessaire de rajouter
un agent qui permet de stabiliser les métaux susceptibles de se volatiliser
• la nécessité de la réduction du taux d’humidité des sédiments avant le traitement. Pour
cela, différentes techniques sont utilisées : la floculation-coagulation, le séchage et le
traitement mécanique (filtre presse, filtre bande, pressoir…). Cela ne fait qu’augmenter le
coût de la technique.
• le coût élevé du fait du coût de l’énergie. Ce coût peut être réduit par l’utilisation de
combustible peu onéreux tel que le charbon ou la coke de pétrole.
Après avoir présenté les techniques physiques et thermiques de traitement des sédiments,
nous allons présenter dans le paragraphe qui suit les techniques biologiques.
I.2.5. Traitements chimiques
Il repose sur la transformation chimique des polluants contenus dans le sédiment.
Différents traitements peuvent être utilisés :
• les polluants sont mis en solution puis récupérés (lixiviation, complexation) ;
• Les polluants sont piégés puis récupérés (extraction par solvant et échange d’ions) ;
• Les polluants sont stabilisés et laissés dans le sédiment, ce traitement permet de réduire
leur mobilités (ajout d’un agent chimique qui fixe les métaux lourds)
Compte tenu du type de procédés utilisés, les paramètres principaux qui ont une influence
sur les performances de dépollution sont :
• la granulométrie, et en particulier la proportion de fines ;
• la chimie globales du contaminant (majeurs et pollutions), y compris la teneur en
carbone organique (TOC)
• la minéralogie du sédiment et la spéciation des contaminants ;
• le pH et le pouvoir tampon ;

Chapitre I : Les sédiments de dragage
34
• la quantité d’argiles et de substances humiques, la capacité d’échange cationique (CEC)
[Clozel 2002].
I.2.5.1. Extraction par lixiviation
La lixiviation consiste à mettre en solution, sous forme ionique, le ou les métaux
recherchés. Le degré de solubilité des métaux est fonction de leur forme chimique [Rizet
2000]. La solution ionique est ensuite récupérée par filtration ou centrifugation puis, les
polluants qu’elle contient sont précipités.
En fonction du métal à extraire et du solide à traiter, différentes solutions de lixiviation
peuvent être employées, les plus courantes pour le traitement des contaminants sont les
acides (HCl, H2SO4 ou HNO3). Les bases (Na2CO3 ou NaOH) sont utilisées plutôt pour les
porteurs des contaminant tels que les argiles ou l’humus (substance humique), l’avantage de
la soude est son faible pouvoir de corrosion [Alzieu 1999 ; Clozel 2002].
Le développement de la lixiviation dans le traitement des sédiments paraît compromis,
d’une part, parce que certains contaminant inorganiques comme le cadmium [AEAP 2000
(b)], le mercure ou l’arsenic sont souvent présents sous des formes difficilement lixiviables,
de plus, le passage en milieu acide entraîne des conséquences néfastes pour la réutilisation
du matériau après traitement. D’autre part, la phase aqueuse contenant les polluants dissous
présente des toxicités importantes en raison de sa forte concentration en sels [Marot 1998].
Pour remédier a ces difficultés, les sédiments sont lavés à l’eau, ils sont alors propres et le
pH est maintenu. L’eau et l’acide récupérés, contenant des métaux et agents chélatants, sont
pompés et amenés dans un réservoir où l’on ajoute des billes de résine qui s’attachent à
l’extrémité des agents chélatants. Les billes sont extraites de la solution acide pour être
envoyées vers un second réservoir où le pH est neutre. Les billes sont ensuite récupérées par
filtration, les métaux par électrolyse et les agents chélatants sont recyclés. Cela entraîne des
surcoûts pouvant aller de 50 à 150 €/tonnes de matière sèche pour une efficacité de 70%
[AEAP 2000 (b)]. Enfin, cette technique demande des investissements technologiques
importants et nécessite des consommations de réactifs considérables lorsque les sédiments
sont riches en carbonates [Marot 1998].
I.2.5.2. Extraction par complexation
Ce procédé consiste à introduire dans la pulpe sédimentaire en agitation des agents
chimiques ayant de fortes propriétés complexantes vis-à-vis des polluants inorganiques.
L’avantage de cette méthode est la possibilité de recyclage et de réutilisation des agents

Chapitre I : Les sédiments de dragage
35
complexants. Parmi les milieux chimiques ayant un fort pouvoir complexant on peut citer :
milieu ammoniacal, les cyanures [Rizet 2000], l’EDTA ou encore l’acétate d’ammonium.
Ces deux derniers milieux sont les plus usuels. Le passage en solution de ces polluants
impose un traitement de la phase aqueuse.
Des essais au laboratoire sur des sédiments contaminés semblent donner des résultats
satisfaisants, la récupération métallique est comprise entre 70 et 90 %. Ces résultats
prometteurs ont conduit à mener une expérience pilote. Les résultats sont plus nuancés à
cause de la compétition entre les ions Ca et les ions métalliques qui semble ralentir le temps
d’extraction, cette technique paraît donc limitée [Marot 1998]. Le coût du traitement d’une
tonne de matière sèche peut varier de 50 à 130 € [AEAP 2000 (b)].
I.2.5.3. Extraction par solvant
L’extraction par solvant consiste à mettre en contact le sédiment et un solvant dans lequel
l’un des composants présents dans le sédiment est soluble. Ce composant est donc extrait et
se retrouve en solution dans le solvant, qui est ensuite séparé du sédiment. Ce procédé
permet de réaliser une purification du sédiment et une concentration du composant à
extraire. Il permet aussi d’extraire sélectivement le composé voulu, il suffit de choisir le
solvant adéquat.
La nature du solvant est variable, il peut s’agir d’eau, de vapeur d’eau, d’un solvant
organique ou inorganique, d’un gaz liquéfié ou d’un fluide supercritique. Le solvant est
choisi en fonction du soluté que l’on veut extraire et du diluant à purifier. La nature du
soluté est très variable. II peut s’agir de matière organique (hydrocarbures, huile, molécules
organiques complexe) ou minérale (sels, ion métalliques) [Bourgois 2000]. Pour la matière
organique, l’acétone représente le solvant le moins toxique, le plus miscible, le plus
disponible, le moins coûteux et le plus efficace avec une possibilité de recyclage [Rienks
1998].
La méthode au solvant organique a été utilisée par deux laboratoires aux Etats-Unis. Les
expériences du premier laboratoire ont permis d’extraire, à l’échelle du laboratoire à partir
de 1m3 de sédiment contaminé, jusqu’à 99,5% de PCB et entre 96 et 99% d’HAP, en
utilisant comme solvant du triéthylamine. Le deuxième laboratoire a obtenu des taux
d’extractions variant de 90 à 98% pour les PCB en utilisant du gaz liquéfié, il s’agit d’un
mélange de propane et butane.
Les résultats obtenus semblent être satisfaisants, mais l’application de cette méthode reste
limitée aux pollutions organiques, et n’a pas été testée à une échelle pilote. D’autre part

Chapitre I : Les sédiments de dragage
36
l’utilisation de solvants organiques nécessite une élimination complète après traitement
[Marot 1998].
Dans certains cas particuliers, l’extraction supercritique peut être utilisée pour extraire les
polluants organiques. L’extraction au fluide supercritique tel que le CO2 est une méthode qui
s’opère à haute pression et température, aux conditions supercritiques. Le CO2 se comporte
comme un composé organique non polaire. Pour optimiser l’extraction des composés
polaires tel que les HAP, il est donc nécessaire d’ajouter un agent entraîneur, qui peut être
un solvant organique qui augmente ainsi la solubilité des HAP dans le CO2 supercritique, et
augmente donc leur désorption des particules du sédiment. L’inconvénient de cette méthode
est la nécessité d’un séchage comme pré-traitement [Rienks 1998].
I.2.5.4. Procédé d’échange cationique
L’échange ionique est une technique de purification dans laquelle les ions dans une
solution sont éliminés par adsorption sur un matériau solide (résine) et remplacés par une
quantité équivalente d’un autre ion de même charge électrique émise par le solide. Lorsque
les ions échangés sont de charge positive, la résine est appelée cationique, et anionique dans
le cas contraire [Bourgois 2000]. Cette méthode est largement utilisée dans le traitement
d’effluents chargés en métaux lourds, son application pour les sédiments de dragage a été
testée au laboratoire. L’utilisation de polymère synthétique a été largement préférable aux
coagulants alumineux et ferriques. Cependant, pour être compétitive, la technique doit être
sélective à l’égard des métaux sans modification du pH naturel de la matrice. Ainsi, des
temps de contact importants sont indispensables et expliquent en partie le non-
développement de ce procédé [Marot 1998].
I.2.5.5. Procédé d’immobilisation
Le processus d’immobilisation conduit à la fixation des métaux lourds par addition d’un
agent d’immobilisation, qui se lie aux métaux lourds d’une manière irréversible diminuant
ainsi leur mobilité. Lorsque le réactif chimique conduit uniquement à stabiliser
chimiquement les polluants, on parle de stabilisation, par contre lorsque le traitement
conduit à la formation de masse solide ayant des propriétés mécaniques accrues, on parle de
solidification/stabilisation [Mulligan 2001 ; Rey F 2000, Jing 2004]. Les principaux réactifs
utilisés sont les liants hydrauliques tels que les ciments, la chaux, le carbonate de calcium,
les polymères. Grâce a leur importante capacité d’échange cationique, la zéolithe et l’argile
sont également des agents d’immobilisation utilisés dans les sols [Raicevic 2005 ; Zorpas

Chapitre I : Les sédiments de dragage
37
200 ; Castaldi 2005] et qui peuvent être utilisées pour la stabilisation des métaux lourds
dans les sédiments de dragage.
Parmi les agents d’immobilisation les plus efficaces couramment utilisés pour la
stabilisation des métaux lourds dans divers déchets (sols, effluents aqueux, cendre volantes,
sédiments …) on trouve les phosphates. L’ion orthophosphate des phosphates combiné avec
plus de 30 éléments peut former environ 300 minerais naturels, fréquemment trouvés dans la
nature (sol, sédiment…) [Eighmy 1998 ; Elliot 1994].
Les ciments sont les liants les plus utilisés à cause des propriétés mécaniques
remarquables qu’ils procurent [Rey F 2000]. Dans ce cas, la valorisation est envisageable.
Les substances humiques peuvent interférer fortement sur les processus d’hydratation et de
formation de la phase cimentaire, ils agissent comme retardateurs de prise, ces interférences
font baisser la résistance des matériaux à court et moyen terme. Pour éviter ces problèmes,
des traitements à la chaux, permettent l’oxydation des substances humiques, entraînant leur
solubilisation et minéralisation progressive.
L’inconvénient de la méthode de solidification/stabilisation est qu’elle conduit à une
augmentation du volume jusqu’à 30% [Mulligan 2001], par contre son avantage par rapport
au processus de stabilisation, est que son efficacité ne dépend pas de la spéciation des
métaux lourds.
A travers les exemples de technique chimique de traitement de sédiment qui ont été
présentées, nous pouvons conclure que ces méthodes ne s’appliquent qu’à une seule nature
de pollution, soit organique, soit inorganique. Nous pouvons également constater que mise à
part le procédé d’immobilisation, les autres procédés nécessitent des installations
technologiques importantes. Cela est dû à la nécessite de récupérer, à partir du sédiment, les
polluants piégés ou extraits ainsi que les réactifs qui ont été utilisés pour cet effet. Le
procédé d’immobilisation ne nécessite pas ces étapes car les métaux piégés restent dans le
sédiment. Cela conduit donc à la réduction des coûts du traitement.
Pour ce type de traitement, il est souvent nécessaire que le sédiment présente une
concentration en solide élevée (réduire les quantités d’agent stabilisant). Une étape de
réduction du taux de siccité est donc indispensable. L’efficacité de ces techniques dépend de
la composition des sédiments et de la forme sous laquelle se trouvent les métaux.
Dans les prochains paragraphes, une présentation succincte de la famille des phosphates
de calcium ainsi que leurs utilisations dans le traitement des déchets est faite. Ce sont les

Chapitre I : Les sédiments de dragage
38
matrices utilisées pour stabiliser les métaux lourds présents dans les sédiments de dragage
dans le cadre de cette thèse.
I.2.6. Les phosphates de calcium
Les phosphates de calcium font l’objet de nombreuses études. En effet, ces composés
présentent de nombreuses applications industrielles dans des domaines très différents
comme la fertilisation des sols, la catalyse hétérogène, la chromatographie, la pharmacie
(complément nutritionnel pour le traitement de l’ostéoporose), la médecine (revêtement des
prothèses, comblement osseux, …) et l’environnement (traitement des déchets).
Le système Ca(OH)2 – H3PO4 – H2O est constitué de plusieurs phases solides bien
caractérisées regroupées dans le Tableau I- 11 (le phosphate tetracalcique est indiqué dans
ce tableau bien qu’il n’existe qu’à haute température). Elles peuvent être sous forme
d’hydrate, d’hydroxyde ou d’anhydre et de structures cristallines très variées.
Tableau I- 11 : Différents phosphates de calcium
Phosphates de calcium
Abréviation Formule chimique
Ca/P
Phosphate monocalcique hydraté Phosphate monocalcique anhydre
MCP MCPA
Ca(H2PO4)2, H2O Ca(H2PO4)2
0.5 0.5
Phosphate dicalcique hydraté Phosphate dicalcique anhydre
DCPD DCPA
CaHPO4, 2H2O CaHPO4
1 1
Phosphate octocalcique OCP Ca8H2(PO4)6, 5H2O 1.33 Phosphate tricalcique α Phosphate tricalcique β
αTCP βTCP
α Ca3(PO4)2 β Ca3(PO4)2
1.5 1.5
Phosphate tricalcique apatitique Phosphate tricalcique amorphes
ACP
Ca9(HPO4)(PO4)5OH Ca9(PO4)6, nH2O
1.5 1.5
Hydroxyapatite calcique Ca-Hap Ca10(PO4)6(OH)2 1.67 Phosphate tétrécalcique TTCP Ca4(PO4)2O 2
Les neutralisations successives des différentes acidités de l’acide orthophosphorique
conduisent à des sels de calcium très peu soluble. Les constantes d’acidité de l’acide
phosphorique dans l’eau à 25°C sont les suivantes [Elmore 1965]
3 4 2 2 4 3 1 2.12H PO H O H PO H O pKa− ++ ↔ + = (1.1) 2
2 4 2 4 3 2 7.21H PO H O HPO H O pKa− − ++ ↔ + = (1.2)
3 34 2 4 3 3 12.7HPO H O PO H O pKa− − ++ ↔ + = (1.3)

Chapitre I : Les sédiments de dragage
39
La majorité des orthophosphates de calcium (Tableau I- 11) dérivent donc de la
neutralisation de la première, de la seconde ou de la troisième acidité de l’acide
phosphorique. Les phosphates de calcium sont fréquemment classés selon le rapport
atomique Calcium/Phosphore. Plus ce rapport augmente et plus la solubilité des phosphates
de calcium diminue en milieu aqueux. En effet, le produit de solubilité des phosphates
acides (contenant des protons) est beaucoup plus grand que celui des phosphates basiques
[Elliot 1994].
I.2.6.1. Les phosphates apatitiques
Le terme apatite désigne une famille de composés minéraux dont la formule chimique
peut s’écrire sous la forme générale suivante :
Me10(XO4)6Y2
dans laquelle Me représente généralement un cation bivalent (Me2+), XO4 un groupement
anionique trivalent (XO43-) et Y un anion monovalent (Y-).
La grande majorité des apatites cristallisent dans le système hexagonal et leur groupe
d’espace est P63/m. La structure cristallographique des apatites est représentée sur la Figure
I- 1 .Cette structure est constituée par un empilement quasi hexagonal compact de
groupements XO4, situés à la côte c/2. Cet empilement forme deux types de tunnels
parallèles à l’axe c dans lesquels se localisent les cations Me. Le premier type de tunnel,
d’environ 2,5 Å de diamètre, est situé entre trois groupements XO4 et son centre est un site
cationique Me appelé site I. Le second type de tunnel, plus large que le premier (son
diamètre est compris entre 3 et 4,5 Å) est localisé entre six groupements phosphate. C’est en
bordure de ces seconds tunnels que se situent six sites cationiques appelés sites II. Trois de
ces sites sont situés à la côte c/4 et les trois autres sont à la côte 3c/4 et forment
respectivement des triangles équilatéraux aux côtes c/4 et 3c/4. Le centre de ces tunnels est
occupé par les anions Y [Elliott 1994 ; Rey C 1984]. L’existence de ces tunnels confère aux
apatites des propriétés chimiques voisines de celles des zéolites. Les tunnels peuvent
contenir à la place des anions Y de petites molécules telles que O2 ou H2O.

Chapitre I : Les sédiments de dragage
40
Figure I- 1 : Projection de la structure apatitique selon l’axe c [Traina 1999].
La structure des apatites admet un grand nombre de substitutions qui peuvent être
partielles ou totales.
Ainsi les cations divalents (Me2+) peuvent être remplacés par des cations monovalents
Na+, K+, Li+, cations bivalents Ca2+, Sr2+, Pb2+, Zn2+, Mg2+, Mn2+, Co2+, Cd2+, cations
trivalents Al3+, La3+.
Le groupement anionique trivalent (XO43-) peut être remplacé par des PO4
3-, AsO43-, mais
aussi par des bivalents HPO42-, CO3
2-, SO42-, ou même par des monovalents et des
tétravalents MnO4-. SiO4
4-.
Les anions monovalents des tunnels (Y-) peuvent céder leurs place aux ions OH-, F-, Cl-,
Br- ou même aux ions bivalents O2-, CO32-, S2-
Une autre propriété remarquable des apatites est d’admettre des lacunes, qui n’existent
jamais en site (XO43-), mais peuvent se localiser en site cationique (Me2+) ou anionique (Y-).
La présence de lacune et de substitutions variées conduit à des modifications importantes
des équilibres de charges locales, des symétries locales et des distorsions sensibles du
réseau.
Les apatites naturelles les plus courantes sont les phosphates de calcium :

Chapitre I : Les sédiments de dragage
41
Ca10(PO4)6(OH, F, Cl)2, la forme apatitique la plus connue est la fluoroapatite de formule
idéale Ca10(PO4)6F2. Lorsque les ions fluor sont échangés par les ions OH-, on obtient
l’hydroxylapatite Ca10(PO4)6(OH)2 .
Du point de vue physico-chimique le Ca-Hap, est le composé le plus insoluble et le plus
basique du système Ca(OH)2 – H3PO4 – H2O (on ne tient pas compte du tétracalcique). C’est
pourquoi la plupart des phosphates de calcium de rapport Ca/P inférieur à 1.67 évoluent en
solution dans des conditions de température et de pH bien définie vers le Ca-HAP [Tung
1997].
Les différentes substitutions de l’apatite peuvent modifier le produit de solubilité du
minéral. Par exemple, les ions silicates et carbonates ont tendance à diminuer le solubilité
des apatites tandis que le fluor stabilise l’apatite (produit de solubilité de l’ordre de 10-120).
Du fait de la faible solubilité des minéraux de phosphates de type apatitique, et de leur
grande stabilité géochimique, sur un large domaine de pH, la stabilisation des métaux par
des phosphates est de plus en plus étudiée. Les métaux piégés dans la structure cristalline ne
peuvent être libérés que par la destruction du réseau cristallin [Haguenauer 1995].
Du point de vue thermique, l’apatite stoechiométrique peut contenir 2 à 3% d’eau, qui
peut être facilement éliminée par chauffage à 600°C. Néanmoins, une perte supplémentaire
d’eau au sein des tunnels du réseau cristallographique peut se produire à partie de 850°C
sous air par la réaction :
( ) ( ) ( ) ( ) ( )10 4 10 4 26 2 6 2 2 x xxCa PO OH Ca PO OH O xH O
−↔ Π + (1.4)
Cet équilibre indique qu’un chauffage sous vide en atmosphère sèche favorise
l’élimination d’eau et déplace l’équilibre à droite. Il y a donc une dégradation de Ca-Hap et
formation d’une apatite ne contenant pas de OH- appelée oxyapatite Ca10(PO4)6O. Le
chauffage en milieu humide déplace l’équilibre à gauche et stabilise le Ca-Hap.
A une température inférieure à 1000°C, une faible réhydratation se produit toujours pour
former un composé limite contenant des OH- : Ca10(PO4)6OH0.5(O)0.75.
De nombreux auteurs ont montré qu’au delà de 1200°C, une décomposition de Ca-Hap en
β-TCP et en phosphate tétracalcique peut se produire :
10 4 6 2 10 4 6 3 4 2 4 2 92
( ) ( ) ( ) 2 ( )Ca PO OH Ca PO O Ca PO Ca P O→ → + (1.5)

Chapitre I : Les sédiments de dragage
42
I.2.6.2. L’utilisation des phosphates pour le traitement des déchets
Divers auteurs se sont intéressés à l’utilisation de phosphates pour la stabilisation des
métaux lourds dans différents types de déchets, le Tableau I- 12 regroupe quelques études
menées dans ce sens. Ce tableau présente le type de déchet, la source de phosphate, les
métaux lourds visés ainsi que la méthode qui a été utilisée pour l’évaluation de la
stabilisation.
De cette analyse on peut constater que :
• l’utilisation du phosphate pour le traitement des sédiments comparé à d’autres
types de déchets reste très minime, néanmoins, la similitude qu’il y a entre les sols et les
sédiments fait que les résultats obtenues pour les sols peuvent être pris comme support de
discussion pour les sédiments.
• Les sources de phosphates sont très variées. Elles ont été choisies en fonction, de
leurs propriétés physiques, chimiques, de leur coût et de leur disponibilité. D’après
certains auteurs, [Basta 2004 ; Melamed 2003], les sources de phosphates hautement
solubles augmentent le potentiel de stabilisation du Pb comparé au phosphate naturel. Ces
auteurs précisent aussi qu’une source de phosphate hautement soluble mélangée avec du
phosphate naturel peut augmenter d’avantage son efficacité pour le traitement du Pb dans
les sols contaminés par exemple.
• Dans certains cas, ces phosphates ont été associées à d’autres composés tel que la
chaux, afin d’améliorer l’efficacité du traitement. Certains auteurs parlent de phosphates
solubles mais ne précisent pas forcement l’origine.
• Bien qu’elle ne figure pas sur le tableau, la quantité de phosphate employée est
variée, elle varie le plus souvent en fonction de la quantité des métaux à stabiliser.
Toutefois, il est important de prendre en compte les problèmes d’eutrophisation qui
peuvent être engendrés par une quantité trop importante de phosphates solubles pour le
traitement des sols [Melamed 2003].
• Les méthodes d’évaluation sont de nature chimique (test de lixiviation, étude
d’équilibre, l’extraction séquentielle…) ou spectroscopique (DRX, SEM…), le plus
souvent l’association des deux est requise pour mieux comprendre le processus mis en
œuvre.
• L’efficacité de chaque traitement dépend de différents paramètres : nature du
déchet, type de métal, sa spéciation, le pH du milieu, la présence d’autres métaux, de la
quantité et de la qualité de la source de phosphate, du temps de mise en contact…

Chapitre I : Les sédiments de dragage
43
• Ces mêmes paramètres influencent les mécanismes mis en jeu pour la stabilisation
des métaux. Parmi les mécanismes le plus souvent cités : l’adsorption en surface,
l’échange d’ions, la substitution et la dissolution-précipitation.
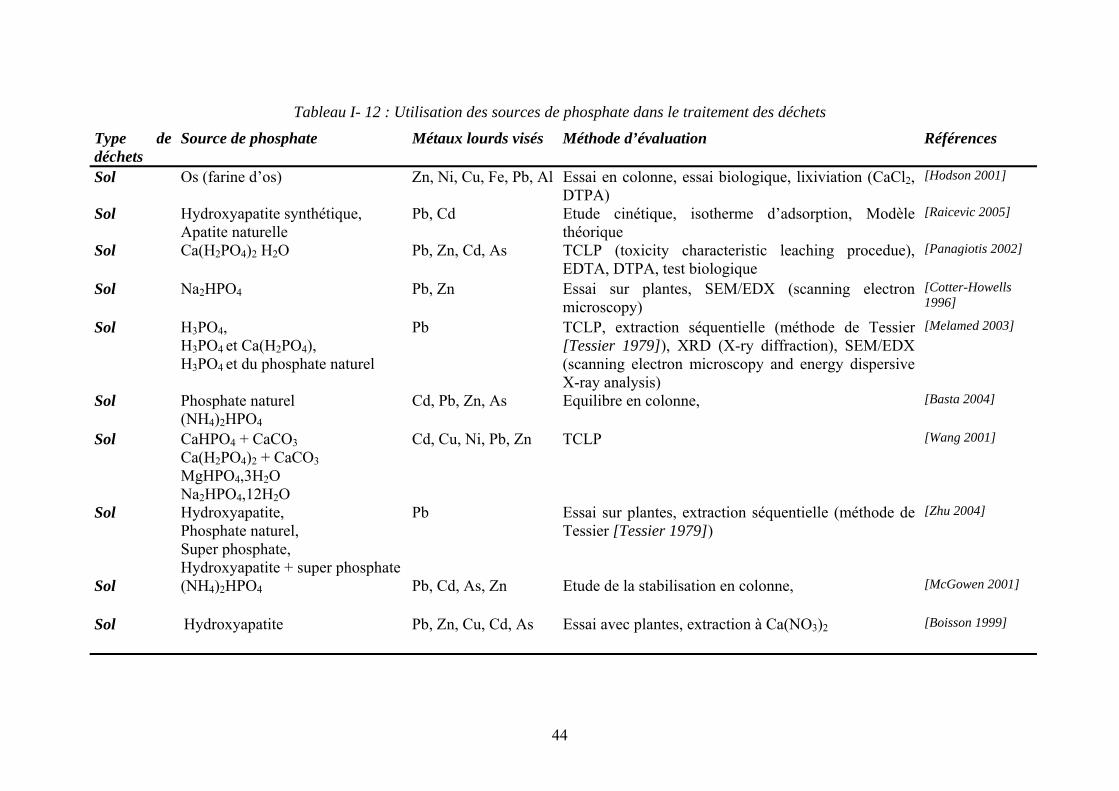
44
Tableau I- 12 : Utilisation des sources de phosphate dans le traitement des déchets
Type de déchets
Source de phosphate Métaux lourds visés Méthode d’évaluation Références
Sol Os (farine d’os) Zn, Ni, Cu, Fe, Pb, Al Essai en colonne, essai biologique, lixiviation (CaCl2, DTPA)
[Hodson 2001]
Sol Hydroxyapatite synthétique, Apatite naturelle
Pb, Cd Etude cinétique, isotherme d’adsorption, Modèle théorique
[Raicevic 2005]
Sol Ca(H2PO4)2 H2O
Pb, Zn, Cd, As TCLP (toxicity characteristic leaching procedue), EDTA, DTPA, test biologique
[Panagiotis 2002]
Sol Na2HPO4 Pb, Zn Essai sur plantes, SEM/EDX (scanning electron microscopy)
[Cotter-Howells 1996]
Sol H3PO4, H3PO4 et Ca(H2PO4), H3PO4 et du phosphate naturel
Pb TCLP, extraction séquentielle (méthode de Tessier [Tessier 1979]), XRD (X-ry diffraction), SEM/EDX (scanning electron microscopy and energy dispersive X-ray analysis)
[Melamed 2003]
Sol Phosphate naturel (NH4)2HPO4
Cd, Pb, Zn, As Equilibre en colonne, [Basta 2004]
Sol CaHPO4 + CaCO3 Ca(H2PO4)2 + CaCO3 MgHPO4,3H2O Na2HPO4,12H2O
Cd, Cu, Ni, Pb, Zn TCLP [Wang 2001]
Sol Hydroxyapatite, Phosphate naturel, Super phosphate, Hydroxyapatite + super phosphate
Pb Essai sur plantes, extraction séquentielle (méthode de Tessier [Tessier 1979])
[Zhu 2004]
Sol (NH4)2HPO4 Pb, Cd, As, Zn Etude de la stabilisation en colonne, [McGowen 2001]
Sol Hydroxyapatite Pb, Zn, Cu, Cd, As Essai avec plantes, extraction à Ca(NO3)2 [Boisson 1999]

45
Sol Phosphate naturel Pb Etude cinétique d’adsorption [Ma L. Q 1999]
Sol Hydroxyapatite synthétique Pb Etude de l’influence du pH sur la stabilisation, XRD, SEM
[Laperche 1996]
Sol Phosphate naturel Pb Extraction séquentielle (méthode de Tessier [Tessier 1979])
[Ma L. Q 1997]
Sol Triple superphosphate (réaction du phosphate naturel et H3PO4) + CaO Phosphate naturel + CaO H3PO4 + CaO
Pb Etude cinétique d’adsorption, physiologically Based Extraction (PBET), XRD
[Hettiarachchi 2001]
Sédiment Hydroxyapatite Hydroxyapatite + Illite Hydroxyapatite + Zéolithe
Cs-137, Sr-88, U Etude d’équilibre, testes de lixiviation (CaCl2, NH4Cl et TCLP), lixiviation en colonne,
[Seamam-Meehan mettre ref]
Sédiment Hydroxyapatite U, Ni, Al, Ba, Cd, Co, Pb
Etude d’équilibre, TCLP, extraction séquentielle (méthode de Miller [Miller 1986]), TEM (Transmission electron microscope)
[Seamam-Arey 2001]
Sol et solution aqueuse
Phosphate naturel Pb Etude cinétique, étude en colonne, XRD, SEM [Ma Q. Y 1995]
Solution aqueuse
Apatite synthetique Cd Etude cinétique, étude de l’influence du pH sur le satbilisation, micro-PIXE (micro-particle-induced X-ray émission)
[Kozai 2003]
Solution aqueuse
fluorapatite U(VI) Etude d’équilibre, étude cinétique, étude de l’influence du pH sur la réaction de stabilisation, PXRD (powder x-raydiffractometter), SEM/EDS (scanning electron microscopy équipé d’un energy dispersive X-ray analyzer), RBS (Rutherford bachscattering spectrometry)
[Ohnuki 2004]
Solution aqueuse
Hydroxyapatite Pb Etude cinétique d’adsorption, SEM, TEM, XRD [Mavropoulos 2004]

46
Solution aqueuse
β Ca3(PO4)2 CaHPO4, 2H2O Ca(H2PO4)2, H2O
Pb Etude cinétique, XRD, SEM [Sugiyama 2002]
Solution aqueuse
Apatite synthétique U, La, Ce, Sm, Gd, Tb, Er, Tm, Lu
Etude cinétique, XRD, SEM [Valsami-Jones 2000]
Solution aqueuse
Phosphate naturel Pb, Cu, Zn Isotherme d’adsorption, Influence du pH sur la désorption des métaux adsorbés, XRD, SEM/EDS.
[Cao 2004]
Solution aqueuse
Hydroxyapatite Cd, Zn, Co Etude d’équilibre, electrophoretic mobility [Gomez del rio 2004]
Solution aqueuse
Phosphate naturel activé (dans de l’acide nitrique puis lave et séché)
Pb Etude cinétique, étude d’équilibre [Mouflih 2005 ]
Solution aqueuse
Phosphate naturel calciné (900°C pendant 2 h lavé puis calcine à 900°C 0.5h )
Pb, Cu, Zn Etude cinétique, étude d’équilibre [Aklil 2004]
Solution aqueuse
Arête de poisson Pb, Cu, Cd, Ni, Zn Etude cinétique, étude d’équilibre [Admassu 1999]
Solution aqueuse
Hydroxyapatite Pb Stabilité en fonction du pH, SEM/EDS, XRD, [Arnich 2003]
Solution aqueuse
Hydroxyapatite Pb Etude cinétique, teste de lixiviation, XRD, SEM-WDX, XPS
[Bailliez 2003]
Solution aqueuse
CaHPO4, 2H2O, Ca3(PO4)2, 2H2O, Ca10(PO4)6(OH)2, Mg3(PO4)2 xH2O (x= 5.0; 8.7), MgNH4PO4 H2O
Pb, Cr, Fe, Zn, Ag, Cd, Cu, Co
Etude d’équilibre, stabilité en fonction du pH, [Shashkova1999]
Solution aqueuse
Hydroxyapatite synthétique Pb Atomic force microscopy (AFM), DRX, SEM/EDS, TEM
[Lower 1998]
Dust from the vitrification
Phosphate soluble (PO43-) Pb, Zn, Cu, Cd Neutron activation analysis (NAA), X-ray
fluorescence (XRF), X-ray photo-electron spectroscopy (XPS), XRD, Scanning-transmission
[Eighmy 1998]

47
of MSW conbustion residues
electron microscopy and X-ray microanalysis (STEM-XRM), magic angle spinning-nuclear magnetic resonance spectroscopy (MAS-NMR), teste de lixiviation en fonction du pH.
Scrubber residues from MSW combustion
Na2HPO4, NaH2PO4, H3PO4
Pb, Zn Teste de lixiviation allemand (DIN 38414-S4), lixiviation en fonction du pH
[Geysen 2004]
lead-loaded industrial solide waste
Hydroxyapatite Fluoroapatite
Pb Testes de lixiviation (DIN 38414-S4 et TCLP), SEM [Loannidis 2003]
Municipal solid waste combustion bottom ash
Phosphate soluble + Chaux Pb, Zn, Cu, Cd Lixiviation en fonction du pH, teste de lixiviation allemand (NEN 7341), XRPD, XPS,
[Crannell 2000]
Cendres volantes
H3PO4 + Chaux (avec calcinations à 900°C du mélange)
Cu Teste de lixiviation à l’EDTA, XRD [Nzihou 2002]
Cendre volante
Phosphate soluble + Chaux (grande surface spécifique)
Pb Teste de lixiviation JLT-13, lixiviation en fonction du pH
[Uchida 1996]
Mine tailings
H3PO4 + CaO Pb, Zn, Cu, Cd SEM, EDS, WDS, STEM-XRM, XRPD, MAS-NMR, XPS, lixiviation en fonction du pH.
[Eudsen Jr 2002]
Cendre volante
H3PO4 Cr, Pb, Zn, Cu, Cd Etude cinétique d’adsorption, XRD, analyse en microsonde, teste de lixiviation (norme AFNOR X31-210)
[Bournonville 2002]
Cendre volente
H3PO4 (calcination des cendres et l’acide entre 600 et 700°C)
Zn, Pb XRD, FTIR (Fourier Transform InfraRed spectroscopy), Micro-characterization, Microanalyse data processing, TCLP, AFNOR X31-210
[Piantone 2003]


Chapitre I : Les sédiments de dragage
49
I.2.6.3. Exemple d’utilisation des phosphates pour la stabilisation des métaux
lourds dans les déchets : Procédé NOVOSOL
En 1992, l’entreprise Solvay a développé le procédé REVASOL qui avait pour objectif la
stabilisation des métaux lourds dans les cendres volantes d’incinération d’ordures
ménagères. L’objectif était de limiter, dans sa globalité, la quantité de déchets ultimes à
stocker en CET de classe I, en valorisant l’essentiel. Ce procédé consiste en quatre étapes
successives [Bournonville 2002] :
• Le lavage des cendres volantes avec une eau alcaline de façon à éliminer les sels
solubles (chlorure et sulfate).
• La filtration afin de récupérer les sels solubles. Ces sels sont ensuite valorisés en
soudière
• La phosphatation se fait dans un réacteur batch par ajout d’acide phosphorique et
une quantité d’eau suffisante pour l’obtention d’un mélange réactionnel homogène par
malaxage. L’objectif de ce traitement est de former des phosphates calciques insolubles, de
grande stabilité chimique et thermique capable d’incorporer dans leur matrice cristalline des
métaux lourds. Les cendres sont ensuite séchées à une température de 300°C.
• La calcination sous atmosphère oxydante à des températures comprises entre 600
et 800°C. Cette étape permet de détruire les imbrûlés potentiels et la matière organique sans
favoriser la volatilisation des métaux lourds. Elle permet également une cristallisation, voire
un frittage des produits de la réaction de phosphatation.
Des essais de valorisation dans le domaine du génie civil des cendres volantes traitées ont
été réalisés notamment par la construction d’une route expérimentales contenant des cendres
volantes traités. Les résultats obtenus étant concluants, Solvay a développé le procédé
NOVOSOL ayant les mêmes objectifs que REVASOL mais pour d’autres types de déchet
tels que : les sols, les résidus de broyage automobiles et les sédiments de dragage. C’est sur
ce dernier déchet que le travail de cette thèse a été fait.
Les sédiments de dragage n’ayant pas les mêmes propriétés physiques, chimiques que les
cendres volantes, leurs comportements sont donc différents. Le procédé REVASOL a dû par
conséquent être adapté. Les principales modifications sont les suivantes :
• L’élimination des deux premières étapes de lavage et de filtration
• Un traitement à l’acide phosphorique du sédiment dans un mélange tubulaire sans
ajout d’eau étant donnée le taux d’humidité souvent élevé ses sédiments.

Chapitre I : Les sédiments de dragage
50
• Le séchage du sédiment phosphaté à l’air libre qui permet la réduction du taux
d’humidité et la maturation des sédiments.
• La calcination dans un four rotatif. Elle va permettre la dégradation de la matière
organique présente dans le sédiment et la stabilisation des phosphates métalliques
néoformés lors de la phosphatation.
La Figure I- 2 présente le procédé NOVOSOL avec ces deux unités, l’unité (A) de
phosphatation et de séchage et l’unité (B) de calcination. Ces deux unités sont munies d’un
dispositif de traitement des gaz. Pour l’unité (A), il s’agit des produits de la réaction de
l’acide phosphorique avec les carbonates et les sulfures présents dans les sédiments. Cette
réaction va conduire au dégagement du CO2 et du H2S (odeur très perceptible). En ce qui
concerne l’unité (B), le traitement des gaz se fait pour les produits de calcination de la
matière organique et en prévention d’une éventuelle évaporation des métaux lourds, entre
autre les volatils, qui n’ont pas été stabilisés.
A la sortie de l’unité (B) les résidus solides obtenus du fait de leur stabilité et
d’intéressantes propriétés d’usage sont prêts à être valorisés dans le domaine du génie civil.
En parallèle à nos travaux de compréhension des phénomènes mis en jeu, deux laboratoires
(Ecole Centrale de Lille et l’INSA Toulouse) travaillent pour identifier des filières de
valorisation viables en génie civil, notamment pour la fabrication de briques de construction,
de béton en collaborant avec Solvay et d’autres partenaires industriels dans ces différents
domaines.
En prenant en compte toutes les techniques de traitement des sédiments que nous avons
pu voir dans la partie 2 de ce chapitre. Nous pouvons dire que la combinaison de techniques
(chimique et thermique) comme dans le cas de NOVOSOL permet d’améliorer la
stabilisation des métaux lourds et confère également des propriétés bénéfiques pour une
valorisation. D’autre part, la combinaison des techniques va permettre de traiter les deux
types de pollution (organique et inorganique). Le traitement thermique va permettre de
stabiliser les métaux qui ne l’ont pas été lors du traitement chimique.

Chapitre I : Les sédiments de dragage
51
Figure I- 2 : Procédé NOVOSOL pour le traitement des sédiments.
Lors de la présentation des techniques de traitement des sédiments de dragage, nous
avons souvent relevé la nécessité de connaître la composition des sédiments à traiter ainsi
que la spéciation des polluants qu’ils contiennent. Nous allons donc présenter dans le
paragraphe qui suit les caractéristiques chimique des sédiments, la forme sous laquelle
peuvent se trouver les métaux lourds, ainsi que les paramètres qui peuvent influencer leur
spéciation. Nous présenterons par la suite une méthode de spéciation des métaux utilisés
dans le cadre de cette thèse.

Chapitre I : Les sédiments de dragage
52
I.3. Caractéristiques chimiques des sédiments
I.3.1. Définition, origine et formation des sédiments
Le nom vase est la définition générale d’une large famille de sédiments fins, argileux,
plus ou moins organiques, pouvant atteindre des teneurs en eau très supérieures à la limite
de liquidité. Les vases contiennent une phase minérale dont la granulométrie s’étale des
sables aux argiles et aux colloïdes, une phase organique et une phase liquide [Marot 1998].
Les particules solides qui constituent les sédiments peuvent être d’origine naturelle ou
anthropique [AEAP 2000 (a)] :
1 Origine naturelle : les particules peuvent être soit endogènes ou exogènes au cours
d’eau
• Les particules endogènes sont principalement constituées de matière organiques
essentiellement des organismes aquatiques appartenant aux règnes animal ou végétal
(plancton, plantes, algues,…)
• Les particules exogènes sont principalement des particules minérales provenant
d’une part de l’érosion éolienne des sols et d’autre part de l’érosion hydrique du bassin
versant et des phénomènes de ruissellement. Les particules exogènes peuvent également
être de nature organique, principalement des feuilles d’arbres transportées par le vent
dans le canal
2 Origine anthropique : les particules peuvent être de nature organique ou minérale
et proviennent des activités industrielles, urbaines et agricoles
I.3.2. Composition granulométrique des sédiments
Selon les régions géographiques, les sédiments peuvent avoir des granulométries très
différentes. On distingue les argiles de taille inférieure à 2 µm, les limons de 2 à 50 µm et
les sables de 50 µm à 2 mm. Les sédiments peuvent alors être typés selon la teneur
respective de ces trois composés en utilisant, par exemple, un système de coordonnées
triangulaires (Figure I- 3) [Bonnet 2000]

Chapitre I : Les sédiments de dragage
53
Figure I- 3 : Diagramme triangulaire de classification des sédiments en fonction de leur texture [Campy 1995]
Les pourcentages des différentes fractions dépendent de la région et de la nature du
milieu aquatique (rivières, estuaires, zones côtières,…). Dans tous les cas, ce sont les
fractions fines, et en particulier les argiles, qui sont responsables de la cohésion des
sédiments en raison de leurs propriétés électriques et de leurs structure en feuillets [AEAP
2000 (a)].
I.3.3. Composition géochimique des sédiments
I.3.3.1. Les argiles
En sédimentologie, les argiles regroupent l’ensemble des matériaux dont les particules
n’excèdent pas 2 mm de diamètre équivalent [Marot 1998].
En minéralogie, les argiles correspondent à des minéraux spécifiques que l’on ne
rencontre jamais en particule plus grosse. Ces minéraux sont des silicates d’aluminium
hydratés, ils appartiennent à la famille des phyllosilicates qui présentent une structure
cristalline en feuillet.
Dans les particules d’argiles, on distingue deux corps minéraux (la silice tétraédrique et
l’hydroxyde d’aluminium octaédrique) qui constituent les deux structures de base à partir
desquelles sont construites tout les argiles.
argile
limonsable
sablelimoneux argileux
sablelégèrement limoneux
limonlégèrement
sableux
sable légèrement
argileux
limonlégèrement
argileux
argile légèrement
sableux
argilelégèrement limoneux
100% SABLE 100% LIMON
100% ARGILE
75% argile
50% argile
25% argile
argile
limonsable
sablelimoneux argileux
sablelégèrement limoneux
limonlégèrement
sableux
sable légèrement
argileux
limonlégèrement
argileux
argile légèrement
sableux
argilelégèrement limoneux
argile
limonsable
sablelimoneux argileux
sablelégèrement limoneux
limonlégèrement
sableux
sable légèrement
argileux
limonlégèrement
argileux
argile légèrement
sableux
argilelégèrement limoneux
argile
limonsable
sablelimoneux argileux
sablelégèrement limoneux
limonlégèrement
sableux
sable légèrement
argileux
limonlégèrement
argileux
argile légèrement
sableux
argilelégèrement limoneux
100% SABLE 100% LIMON
100% ARGILE
75% argile
50% argile
25% argile

Chapitre I : Les sédiments de dragage
54
L’assemblage de ces feuillets entre eux forme différentes structures correspondant à
chacune des familles d’argile : la kaolinite, l’illite, la vermiculite, smectite et la chlorite
[Sposito 1984]. Compte tenu d’une granulométrie très fine des particules et d’une structure
en feuillet, ces minéraux présentent donc de très grandes surfaces spécifiques comprises
entre 5 et 800 m2/g [Marot 1998]. D’autre part, leurs feuillets élémentaires présentent une
charge électrique qui s’explique par deux phénomènes [Sposito 1984] :
Les substitutions isomorphiques dans la structure
Il s’agit de substitutions partielles d’ions trivalents (Al) par des ions tétravalents (Si),
dans les cavités tétraédriques, et de même entre les ions bivalents (ex : Mg) et trivalents (Al)
dans les cavités octaédriques.
Les dissociations des fonctions acides/ bases faibles
Elles sont dues à des groupements fonctionnels de type M-OH (essentiellement Si-OH
pour les argiles) qui se comportent comme des groupements acides/bases faibles.
Ces charges superficielles sont compensées par des cations (cations compensateurs) qui
assurent l’électroneutralité du feuillet. Les liaisons entre les feuillets sont plus ou moins
fortes selon les argiles et selon le degré d’hydratation de celles-ci. Elles sont dans une large
mesure à l’origine des propriétés particulières que possèdent ces minéraux
Les interactions argiles/contaminants
Leur faible taille leur confère une forte réactivité chimique et physique ainsi qu’une
importante surface de contact [Blanchard 2000]. Les charges négatives à la surface des
minéraux argileux sont neutralisées par des cations compensateurs. Ces derniers sont
relativement mobiles entre les feuillets, ils peuvent donc s’échanger avec les cations du
milieu pour former un équilibre stable. Ce phénomène explique en grande partie les affinités
qui existent entre les argiles et certains polluants organiques ou inorganiques (métaux
lourds) [Lin 1998 ; Laing 2002]. Outre le type d’argile, la capacité d’échange cationique
dépend également des dimensions du cations à échanger. Elle est d’autant plus importante
que le cation est petit.
I.3.3.2. Les carbonates
Les minerais de carbonate rencontrés le plus souvent dans les sols sont la calcite CaCO3,
la dolomite [CaMg(CO3)2], nahcolite (NaHCO3), trona [Na3H(CO3)2 H2O] et le soda
(Na2CO3 10H2O).
La co-précipitation du Ca avec le Mn, Fe(II), Co(II) ou le Cd par adsorption sur la calcite
n’est pas rare. Les métaux tels que le Zn et le Cu peuvent également co-précipiter avec la

Chapitre I : Les sédiments de dragage
55
calcite par inclusion comme l’hydroxycarbonate. Cela donne des minerais tels que :
hydrozincite Zn5(OH)6(CO3)2, malachite Cu2(OH)2CO3 ou azurite Cu3(OH)2(CO3)2 [Sposito
1984].
Les carbonates jouent un rôle important dans les sédiments, leur équilibre de dissolution
contrôle partiellement le pH et une teneur élevée en carbonates rend le sédiment alcalin,
favorisant ainsi l’ensemble des modes de fixation. De plus la surface des carbonates est le
siège de phénomène de sorption des ions métalliques : précipitation, adsorption
(accumulation de matière entre les phases solides et liquides, diffusion à l’intérieur de la
phase solide) [Blanchard 2000].
I.3.3.3. Les sulfures
Divers composés de soufre se constituent dans les sédiments. Les formes du soufre
inorganique incluent les sulfates (SO42-), le soufre élémentaire (S), les sulfures métalliques
(tel que le FeS) et la pyrite (FeS2).
Entre l’état d’oxydation du sulfate (VI) et le sulfure, différents états d’oxydation peuvent
exister tels que le bisulfure S22- (-I), polysulfure Sn2- (-II/n), disulfuroxyde S2O(g) (+I),
monoxyde de soufre SO(g) (+II), thiosulfate S2O32- (+II), dithionite S2O4
2-(+III), dioxyde de
soufre SO2(g) (+IV), sulfite SO32- (+IV) et trioxyde de soufre SO3(g) (+VI).
Quant au soufre organique, il se trouve généralement sous deux formes : soit dans le
groupe des esters, soit lié au carbone.
L’essentiel du soufre dans le sédiment est inorganique. Le soufre inorganique dans des
conditions anaérobiques (condition réductrice), se trouve sous forme de sulfure dans le
sédiment bien aéré et bien séché (condition oxydante), il s’oxyde et donne des sulfates (plus
mobile), l’oxydation des sulfures peut également donner lieu à des oxydes intermédiaires.
La réaction d’oxydation s’accompagne par une diminution du pH du milieu [Tack 1997]. La
plupart des sulfures métalliques sont fortement insolubles même dans des milieux très acides
[Tack 1998].
I.3.3.4. Les oxydes hydroxydes
Du fait de leur grande abondance dans la lithosphère et leur faible solubilité dans la
gamme de pH des sols, l’aluminum, le fer et la manganèse forment les plus importants
oxyde, oxyhydroxyde et hydroxyde dans le sol [Sposito 1984]. Les oxydes de fer et de
manganèse amorphes des sédiments sont les premiers intervenants dans les processus
d’adsorption. Ils ont une forte capacité de substitution isomorphique et ils lient les composés

Chapitre I : Les sédiments de dragage
56
organique et inorganique [Bonnet 2000]. Parmi les composés à base de fer les plus
communs : goethite (α-FeOOH), Ferrihydrite (5Fe2O3, 9H2O) et hématite (α-Fe2O3) ont une
adsorption préférentielle pour certains métaux. La Ferrihydrite adsorbe les métaux dans cet
ordre : Pb > Cu > Zn > Ni > Cd, l’hématite dans cet ordre : Pb > Cu > Zn > Ni et le geothite
dans cet ordre : Cu > Pb > Zn > Cd [Stephens 2001 (a)]
I.3.3.5. La phase organique
Tous les composés organiques naturels simples ou complexes sont susceptibles d’être
rencontrés dans les sédiments. Ils peuvent être à l’état libre ou en association plus ou moins
étroite avec les constituants minéraux. On distingue des constituants de nature bien définie,
hérités des plantes et des animaux du milieu ou biosynthétisés par la microflore, et des
constituants plus complexes, comme les substances humiques. Les substances humiques
sont des polymères naturels constitués principalement de carbone et d’oxygène, élaborés par
voie chimique et biochimique et défini par leur solubilité dans différents réactifs. Leur
décomposition est très lente (plusieurs centaines d’année) et elle combine de très fortes
propriétés tensioactives et de fortes propriétés complexantes. Leur rôle est de ce fait
prépondérant dans les interactions organo-minérales qui existent dans les sédiments.
On peut diviser les substances humiques SH en deux grands groupes aux propriétés
relativement indépendantes de leur origine et qui sont définis expérimentalement en fonction
de leur solubilité. :
Les acides fulviques (AF) : composés solubles en solution aqueuse pour une large
gamme de pH. Ils représentent souvent la fraction la plus importante des substances
humiques totales.
Les acides humiques (AH) : composés solubles uniquement à un pH supérieur à 2 et
parfois même totalement insolubles. Ils sont à l’origine de la couleur noire des sédiments.
L’humine est la fraction des SH insoluble pour toute condition de pH, donc difficilement
extractible et très mal connue [Sarret 1998].
Structure et composition de la substance humique
Les SH sont des polycondensats contenant de 50 à 70 % de composés phénoliques ou
benzène carboxyliques, et de 30 à 50 % de chaînes péptidiques et polysacchridiques. Il est
difficile de définir une formule chimique générique à cause de leur hétérogénéité. On préfère
généralement donner leur composition élémentaire. Comme on peut le voir sur le tableau 2,
les éléments majeurs dans les AH et AF sont le carbone France et l’oxygène. Le pourcentage
de carbone dans les AH varie généralement entre 53,8 à 58,7 % et celui de l’oxygène de

Chapitre I : Les sédiments de dragage
57
32,8 à 38,3%. L’AF à un pourcentage en carbone inférieur à celui des AH (40,7 – 50,6%)
mais un pourcentage en oxygène plus important (39,7 – 49,8%). Les pourcentages
d’hydrogène (H), d’azote (N) et de soufre (S) varient respectivement de 3,2 à 7, 0,8 à 4,3 et
de 0,1 à 3,6% [Stevenson 1994].
Eléments % acide humique % acide fulvique Carbone 53,8 – 58,7 40,7 – 50,6 Oxygène 32,8 – 38,3 39,7 – 49,8 hydrogène 3,2 – 6,2 3,8 – 7,0 Azote 0,8 – 4,3 0,9 – 3,3 soufre 0,1 – 1,5 0,1 – 3,6
Tableau I- 13 : Gamme de concentration élémentaire (%) pour les substances humiques [Stevenson 1994].
Les SH sont caractérisés par une forte densité des groupes fonctionnels, majoritairement
des groupes oxygénés : carboxyles, alcools, carbonyles (Tableau 3), mais aussi des groupes
azotés (amines) et soufrés (sulfate, acide sulfonique). La nature et la distribution de ces
groupes varient selon l’origine des SH. Les AF sont globalement plus riches en groupes
fonctionnels que les AH.
Les acides humiques possèdent une masse molaire de 10 000 à 100 000 g/moles environ,
alors que les acides fulviques varie de 500 à 5000 g/moles.
Groupement % acide humique % acide fulvique Acidité totale 5,6 – 7,7 6,4 – 14,2 COOH 1,5 – 5,7 6,1 – 11,2 OH phénolique 2,1 – 5,7 0,3 – 5,7 OH alcool 0,2 – 4,9 2,6 – 9,5 C=O quinconique 1,4 – 2,6 0,3 – 2,0 C=O cétonique 0,3 – 1,7 1,6 – 2,7 OCH3 0,3 – 0,8 0,3 – 1,2
Tableau I- 14 : Teneur des groupes fonctionnels principaux (m mol/g) pour les substances humiques [Stevenson 1994].
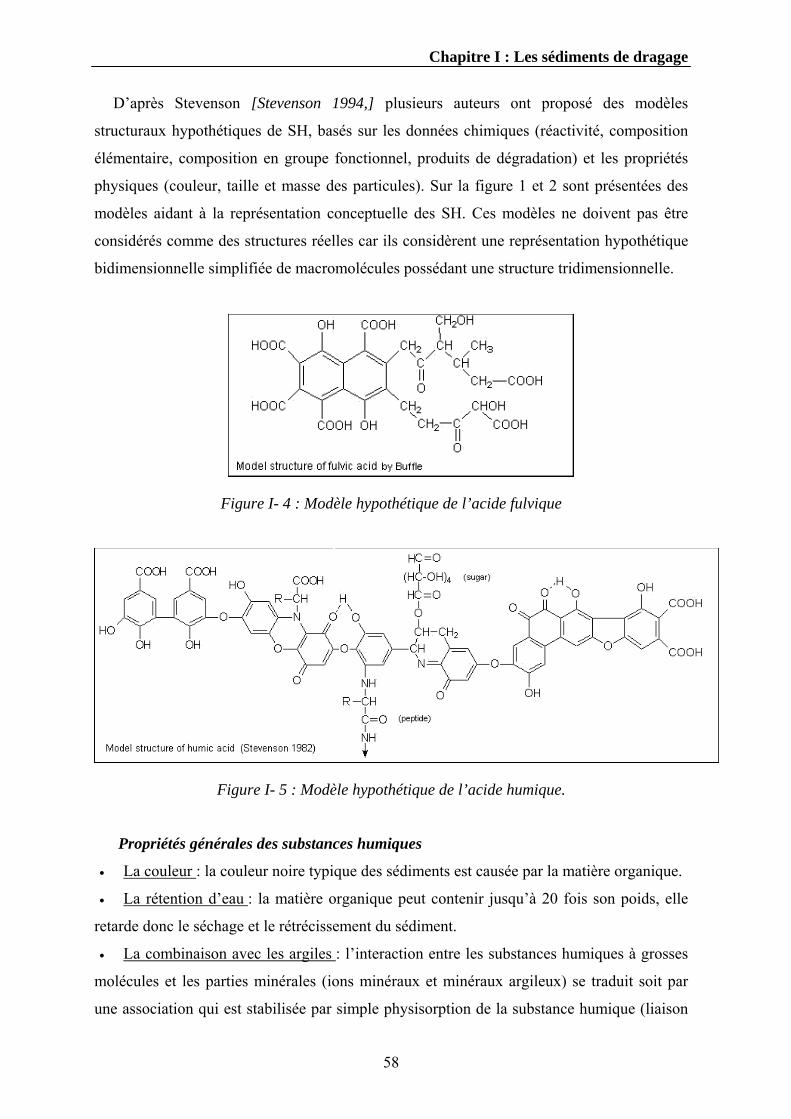
Chapitre I : Les sédiments de dragage
58
D’après Stevenson [Stevenson 1994,] plusieurs auteurs ont proposé des modèles
structuraux hypothétiques de SH, basés sur les données chimiques (réactivité, composition
élémentaire, composition en groupe fonctionnel, produits de dégradation) et les propriétés
physiques (couleur, taille et masse des particules). Sur la figure 1 et 2 sont présentées des
modèles aidant à la représentation conceptuelle des SH. Ces modèles ne doivent pas être
considérés comme des structures réelles car ils considèrent une représentation hypothétique
bidimensionnelle simplifiée de macromolécules possédant une structure tridimensionnelle.
Figure I- 4 : Modèle hypothétique de l’acide fulvique
Figure I- 5 : Modèle hypothétique de l’acide humique.
Propriétés générales des substances humiques
• La couleur : la couleur noire typique des sédiments est causée par la matière organique.
• La rétention d’eau : la matière organique peut contenir jusqu’à 20 fois son poids, elle
retarde donc le séchage et le rétrécissement du sédiment.
• La combinaison avec les argiles : l’interaction entre les substances humiques à grosses
molécules et les parties minérales (ions minéraux et minéraux argileux) se traduit soit par
une association qui est stabilisée par simple physisorption de la substance humique (liaison

Chapitre I : Les sédiments de dragage
59
hydrogène) soit par complexation mixte d’un même ion métallique entre le composé
organique et la particule minérale (argile). Cela favorise :
1. L’agglomération des particules entre elles, par combinaison des groupements
libres de la substance humique avec d’autres particules minérales.
2. La stabilité des composés formés entre l’ion métallique et le composé organique,
est suffisamment forte pour dissocier l’ion métallique de la particule minérale.
• Chélation : . Il existe une grande affinité entre la matière organique et les métaux lourds
[Coquery 1995 ; Lin 1998]. Staphens et al [Stephens 2001(b)] ont étudié le rapport entre la
concentration en métaux lourd, la matière organique et la couleur du sédiment, et trouvé une
relation bien claire entre les trois paramètres. Plus la couleur du sédiment est foncée, plus il
y a de matière organique, et plus il y a de métaux lourds, la matière organique forme alors
un complexe stable avec les cations divalents tels que : Cu2+, Mn2+, Zn 2+. Les acides à
petites molécules forment avec les cations métalliques des humates et fulvates plus ou moins
stables selon les conditions du milieu. Il semble que la très grande majorité de Pb (II) et Cu
(II) soit complexée par les acides fulviques. Coquery et Welbourn [Coquery 1995] ont
montré que l’affinité des substances humiques naturels vis à vis des cations métalliques suit
cet ordre : Hg > Cu > Pb > Zn > Cd> Fe. Par ailleurs Lin et Chen [Lin 1998] dans leur
études sur la relation entre les métaux lourds et la matière organique ont constaté que le Cr
s’adsorbait plus que le Cu, le Pb et le Zn. Adriano et al [Adriano 2001] ont montré que
l’ordre général de l’affinité complexation des métaux par la matière organique suit l’ordre :
Cu> Cd> Fe> Pb> Ni> Co> Mn> Zn. Ces quelques études nous montrent qu’il n’y a pas de
règle générale d’affinité des métaux par rapport à la matière organique. Cela dépend
fortement de la composition du sédiments, des métaux présents et des conditions physico-
chimiques…
• Solubilité dans l’eau : l’insolubilité de la matière organique est due à son association
avec l’argile. Les sels de cations divalents et trivalents associés à la matière organique sont
insolubles également, ainsi très peu de matière organique est perdue par lixiviation.
• Pouvoir tampon : ces fonctions COOH et OH lui confèrent un pouvoir tampon dans une
large gamme de pH.
• Capacité d’échange cationique (CEC) : l’acidité totale d’une fraction d’humus isolée
varie de 300 à 1400 cmole /kg, cela permet donc d’augmenter la CEC des sédiments de 20 à
70% [Stevenson 1994].

Chapitre I : Les sédiments de dragage
60
I.3.4. Polluants métalliques et organiques
Les polluants présents dans l’environnement peuvent être naturellement présents dans les
milieu ou être d’origine anthropique : métaux lourds (Cu, Pb, Cr…) issus des industries
minières et métallurgiques, fabrication de peinture et de colorant, combustion du charbon…
quant à la pollution organique (HAP, PCB, furanne, dioxyne…), elle est issue des industries
agroalimentaires et des rejets industriels, domestique et urbains. Ces substances ont un
caractère toxique affirmés. Dans le cadre de cette étude nous nous sommes intéressés plus
spécialement aux polluants métalliques.
I.3.4.1. Les métaux lourds
La dénomination « métaux lourds » représente selon les auteurs, un éventail d’éléments
parfois variable. Pendant longtemps, deux notions étaient le plus souvent prises en compte :
• une notion de densité : d > 5kg/dm3
• une notion de toxicité incluant parfois des éléments légers (Al, Be…).
La définition proposée au cours du congrès de Heidelberg de septembre 1983 inclut ces
deux notions en les associant dans une classification en trois groupes basée sur l’activité et
la toxicité biologique des éléments et sur leur chimie :
• classe A : affinité pour l’oxygène
• classe B ; affinité pour le soufre
• classe intermédiaire : entre les deux.
La toxicité étant croissante de la classe A à la classe B, et le terme « métaux lourds »
désignant les métaux et métalloïdes toxiques indépendamment de leur densité [Bicocchi
1998].
Certains de ces éléments, présents à l’état de trace, sont essentiels pour les organismes
vivants (Cu, Zn, Co, Fe, Mn, Ni, Cr, V, Mo, Se, Sn), mais l’augmentation de leur
concentration peut aboutir à des phénomènes de toxicité. D’autres éléments ne produisent
que des effets néfastes (Pb, Hg, Cd) [Bonnet 2000]. La toxicité des métaux lourds vis a vis
des organismes vivants dépend de leur nature, la concentration, le mode d’action, la
spéciation et de leur biodisponibilité [Alzieu 1999-Bonnet 2000].
I.3.5. Interactions entre les métaux lourds et les sédiments
Le sédiment est un milieu complexe qui possède de multiples possibilités d’association
avec les contaminants. En effet, les métaux lourds apportés dans les rivières s’adsorbent sur
les particules, le processus de sédimentation transfert rapidement les métaux lourds de l’eau

Chapitre I : Les sédiments de dragage
61
au sédiment. Selon les mécanismes d’accumulation des métaux lourds dans les sédiments,
on peut distinguer cinq fractions au propriétés différentes selon [Lin 1998 ; Tessier-1979 ;
Salomons 1980] : 1) échangeable, 2) lié aux carbonates, 3) lié aux oxyde de fer et de
manganèse, 4) lié à la matière organique et aux sulfures, et 5) les métaux résiduaires. Ainsi,
dans le milieu aquatique, les métaux lourds sont sujet à la précipitation, la complexation,
l’adsorption et la solubilisation, et cela dépend des propriétés physiques et chimiques du
milieu [Silva 2002]. Les métaux lourds dans les sédiments ne sont pas fixés d’une manière
permanente, le changement de pH et du potentiel redox, la teneur en matière organique
[Zoumis 2001 ; Gäbler 1997 ; Tack 1996], la teneur en argiles, la teneur en eau [Gäbler
1997] les sulfures [Zoumis 2001 ; Tack 1996] modifient leur spéciation et par conséquence,
leur mobilité. Les contaminants associés aux particules peuvent donc retourner dans la phase
aqueuse sous l’effet de processus physique (diffusion) ou chimique (désorption). Les
organismes vivants modifient également les caractéristiques physiques et chimiques du
milieu (biodégradation, bioturbation…), ils accumulent les contaminants à partir des phases
aqueuses et particulaires par adsorption [Bonnet 2000].
I.3.6. Paramètres influençant la répartition des contaminants :
Les facteurs qui contrôlent la répartition des métaux lourds dans les sédiments sont de
nature physico-chimique tels que le pH, le potentiel redox, la capacité d’échange cationique,
la salinité et la température. Ils dépendent fortement de constituants du sédiment : particules
inorganique, matière organique, sulfures, oxyde de fer et de manganèse et carbonates.
L’influence des constituants du sédiment ayant été abordés dans le paragraphe I.3.3, cette
partie ne porte que sur les paramètres physico-chimiques
I.3.6.1. Influence des paramètres physico-chimiques
I.3.6.1.1. Le pH
Dans les sédiments, le pH est contrôlé par les activités biologiques et dépend de la
capacité tampon du système : carbonates (Equilibre CO32-/HCO3
-…) [Bonnet 2000], les ions
échangeables, les argiles et les hydroxydes d’Aluminium [Tack 1996]. L’oxydation des
sulfures en sulfates entraîne une réduction du pH [Tack 1997], qui est proportionnelle au
pouvoir tampon [Peltola 2002]. La dénitrification (réduction des nitrates en azote) entraîne
également une diminution du pH [Bonnet 2000].

Chapitre I : Les sédiments de dragage
62
Les métaux et les métalloïdes sous forme d’oxyanions voient leurs concentration
augmenter dans les phases aqueuses quand le pH augmente [Bonnet 2000]. A l’inverse, la
solubilité des métaux lourds cationiques dans les solides augmente avec la diminution du pH
[Kedziorek 1996]. Cependant, il a été démontré que la présence de substances humiques
(substance complexante) pouvait changer significativement cette dernière tendance
[Kedziorek 1996], les métaux cationiques se comportent alors comme des anions [Bonnet
2000].
I.3.6.1.2. Le potentiel d’oxydoréduction
Le potentiel d’oxydoréduction ou potentiel redox (Eh en millivolts) mesure l’aptitude à
l’oxydation ou à la réduction d’une solution et permet de déterminer les possibilités de mise
en solution ou de précipitation de divers composés minéraux dans les sédiments. La
dégradation de la matière organique par les bactéries entraîne une diminution du potentiel
redox et une augmentation des conditions réductrices dans le sédiment [Bonnet 2000]. Le
dragage des sédiments et leur dépôt à terre (passage du milieu anaérobie au milieu aérobie
ou en d’autre terme du milieu réducteur en milieu oxydant), entraîne un changement du
potentiel redox qui conduit entre autre à l’oxydation des sulfures (1.6) et de la matière
organique entraînant ainsi une diminution du pH et la mobilisation des métaux lourds qui
ont été adsorbés ou complexés [Zoumis 2001 ; Law 1998].
3 22 2 2 47.5 2 4 2FeS O H O Fe SO H+ − ++ + → + + (1.6)
Une corrélation a été démontrée entre la lixiviabilité des métaux lourds et le rapport
sulfures/sulfates lors du séchage (oxydation), la relation entre les deux est inversement
proportionnelle. Le changement dans le rapport sulfures/sulfates du sédiment est une bonne
indication des conditions redox du sédiment, une diminution de ce rapport indique une
oxydation du milieu [Stephens 2001 (a)].
Différentes études concernant la géochimie des métaux lourds dans les sédiments dragués
et déposés sur terre, ont montré que les changement de paramètres tels que le pH et le redox
en même temps influencent la mobilité des métaux lourds. En effet, la solubilité du Cd, Cu,
Pb et Zn en fonction du pH augmente fortement dans l’environnement oxydant comparé à
un environnement réducteur [Tack 1996].
L’oxydoréduction du sédiment, ne change rien dans le comportement des carbonates de
calcium, si ce n’est la réaction avec les petites quantités d’acide qui peuvent se former lors
de l’oxydation du sédiment (oxydation des sulfures) [Tack 1996].

Chapitre I : Les sédiments de dragage
63
Dans le milieu réducteur, les oxydes de Fe et de Mn se réduisent et se dissolvent. Cela
s’accompagne par un départ des métaux lourds adsorbés, qui vont rapidement s’adsorber sur
d’autres composants du sédiment tels que la matière organique, l’argile et surtout les
sulfures [Tack 1998].
I.3.6.1.3. La capacité d’échange cationique
La réactivité des particules est d’autant plus importante que leur capacité d’échange
cationique (CEC) et leur surface spécifique sont importantes. La capacité d’échange
cationique de la fraction solide est fonction de l’importance et de la distribution des charges
dans sa structure, des cations adsorbés et de la nature des cations remplaçant. La surface
spécifique est inversement proportionnelle aux diamètres des particules [Bonnet 2000]. La
teneur en argiles et en matière organique procurent aux sédiments la capacité d’échange
cationique [Laing 2002].
I.3.7. Spéciation des métaux lourds dans les sédiments
Le terme spéciation fait référence aux formes spécifiques dans lesquelles un élément
chimique se trouve dans une matrice (niveaux d’oxydation, forme organométalliques…).
L’importance de la spéciation repose sur le fait que l’évaluation de la concentration totale
n’est pas suffisante pour déterminer l’impact environnemental d’un métal. En effet,
l’utilisation de la concentration totale comme critère d’évaluation des effets potentiels des
sédiments contaminés sous-entend que toute les formes d’un élément ont un même impact
sur l’environnement, ce qui n’est pas réellement le cas [Tessier 1979 ; Quevauviller 1997 ;
Tokalioglu 2000].
La spéciation des métaux lourds peut être étudiée par :
• La modélisation thermodynamique, mais qui soufre de manque de données [Singh
2005, Jain 2004].
• Les méthodes spectroscopiques : diffraction des rayons X, RMN, Microsonde,
spectrométrie infra-rouge, XPS, EXAFS…)
• Les méthodes chimiques (extraction séquentielle).
Les méthodes de caractérisation spectroscopique utilisées dans cette thèse seront détaillées
dans le chapitre 2 et 4. Nous détaillerons dans ce qui suit la procédure d’extraction
séquentielle.

Chapitre I : Les sédiments de dragage
64
I.3.7.1. Extraction séquentielle
L’extraction séquentielle a été utilisée afin d’identifier et d’évaluer la mobilité et la
disponibilité des métaux lourds dans l’environnement [Tessier 1979 ; Quevauviller 1997 ;
Tokalioglu 2000 ; Gómez Ariza 2000].
L’extraction séquentielle fournit plusieurs informations [Das 1995] :
• la procédure d’extraction utilisée est comparable à ce qui se passe dans la nature. Dans
l’environnement naturel, les sédiments sont sujets à des conditions similaires dues à : un
apport naturel ou anthropogénique d’électrolyte ou d’agent chélatant, des conditions
oxydantes ou réductrices, une diminution du pH (par le bais de pluies acide).
• c’est un outil essentiel dans la spéciation des éléments dans un échantillon naturel.
• l’extraction chimique peut être utilisée pour une estimation du potentiel de mobilisation
des métaux sous les conditions environnementales.
• la possibilité de vérifier le résultat, dans la mesure où la somme des fractions doit être
plus ou moins égale à 100%.
Différentes approches basées sur le fractionnement des métaux lourds contenus dans les
sédiments ont été décrites. Un des premiers schéma proposé est celui fait en 5 fractions
publié par Tessier et al [Tessier 1979]. Ce schéma est largement utilisé pour distinguer entre
les métaux échangeables, liés aux carbonates, liés aux oxydes de fer et de manganèse, liés
aux sulfures et à la matière organique et les métaux résiduels. Plusieurs auteurs ont modifié
ce schéma, en changeant le rapport solide/réactif, la concentration des réactifs, le temps
d’extraction, l’ordre des fractions. Das et al [Das 1995] ont fait un état de l’art de ces
différents schémas qui sont présentés dans le Tableau I- 15. Les auteurs de ces extractions
séquentielles ont utilisé différents réactifs afin de lixivier préférentiellement, étape par étape,
les métaux précipités, adsorbés, ou complexés. Les résultats obtenues dépendent du réactif
utilisé et des conditions opératoires [Gómez Ariza 2000]. Le choix du réactif est important
dans la mesure où un réactif comme l’électrolyte, acide faible ou un agent chélatant, libère le
métal du site de coordination, tandis qu’un acide fort ou un agent redox est capable de
libérer le métal et de décomposer à la même occasion la matrice solide [Gleyzes 2002].
Compte tenu de la diversité des procédures, The Community Bureau of Reference (BCR,
maintenant, Standard, Measurments and Testing Programme M&T) a lancé ces dernières
années un programme afin d’harmoniser le schéma de l’extraction séquentielle
[Quevauviller 1997]. Un schéma à 3 fractions a donc été développé : acido-soluble,
réductible, oxydable. L’objectif étant d’avoir une procédure de référence qui peut permettre
aux utilisateurs de comparer leurs résultats obtenus suivant la même procédure.

Chapitre I : Les sédiments de dragage
65
Tableau I- 15 : Méthodes d’extraction séquentielles communément utilisées [Das 1995] Auteurs Réactifs Fractions Gatehouse H2O
NH4Ac/Hac NH2OH HCl/Hac H2O2/HNO3 N2H4 HCl HclO4
Solubles à l’eau Echangeables Oxydes Sulfures et organiques Fe non lié aux silicates Résiduels
Tessier
MgCl2 NaAc/Hac NH2OH HCl/Hac H2O2/HNO3/NH4Ac HF/HclO4
Echangeables Carbonates Oxydes Sulfures et organiques Résiduels
Sposito KNO3 NaOH EDTA HNO3
Echangeables Eléments adsorbés Organiques Carbonates et sulfures
Miller et McFee
H2O KNO3 Na4P2O7 EDTA NH2OH HCl/HNO3 Na-citrate/NaHCO3/Na2S2O4 HNO3 HNO3/ H2O2
Solubles à l’eau Echangeables Organiques Carbonates, Fe (amorphe) Oxydes de Mn Oxydes de Fe cristallin Sulfures Résiduels
Psenner
HCO3-/S2O4
2- NaOH HCl NaOH chaud
Organiques et humiques (partielles) Humiques Carbonates, hydroxydes de Fe, sulfures (partielles) Kaolinite (partiellement) sulfures
Shuman et Hargrove
Mg(NO3)2 NaOCl NH2OH HCl/NH4Ac (NH4)2Ox Acide ascorbique/tampon d’oxalate HCl/HF/HNO3
Echangeables Organiques Oxydes de Mn Oxydes de Fe (amorphe) Oxydes de Fe (cristallin) Résiduels
Karsten et Förstner
NH4Ac NaAc/Hac NH2OH HCl/HNO3 Tampon d’oxalate H2O2/HNO3/NH4Ac HNO3
Echangeables Carbonates Oxydes de Mn Oxydes de Fe (amorphe) Sulfures et organiques Résiduels
Zeien et Brümmer
NH4NO3 NH4Ac NH2OH HCl/NH4Ac (NH4)2EDTA (NH4)2Ox Acide ascorbique/tampon d’oxalate HF/HclO4/HNO3
Echangeables (non adsorbés) Echangeables (adsorbés) Oxydes de Mn Organiques Oxydes de Fe (amorphe) Oxydes de Fe (cristallin) Résiduels
Hirner H2O NH4Ac C6H6/CH3OH C6H6/CH3OH/KOH HCl HF HF/HclO4/HNO3
Solubles à l’eau Echangeables Organique solubles (soluble au solvant) Organique solubles (acide humique et fulvique) Matrice minérale (soluble) Matrice minérale (très peu soluble) Organiques insolubles

Chapitre I : Les sédiments de dragage
66
I.3.7.1.1. Extraction séquentielle suivant la méthode de Tessier
Bien qu’il existe depuis 1997 la méthode du BCR, l’utilisation de la méthode d’extraction
séquentielle de Tessier a été choisie car c’est l’une des méthodes les plus utilisées [Melamed
2003 ; Zhu 2004 ; Ma L. Q 1997 ; Abd El Azim 2004 ; Gauthreaux 1998]. D’autre part, la
répartition de sédiment en 5 fractions au lieu de 3 comme la méthode BCR permet une
meilleure discrimination du sédiment et par conséquent une meilleure compréhension des
conséquences du traitement mis en œuvre.
Ainsi, la comparaison des résultats que nous avons obtenus dans le cadre de cette thèse
avec ceux de la littérature sur les sédiments de dragage sera plus simple.
Nos objectifs concernant l’usage de cette méthode consiste à une meilleur compréhension
de la distribution des métaux dans les sédiments après chaque étape du procédé, et ainsi,
évaluer l’efficacité de la phosphatation et de la calcination des métaux lourds étudiés. Ceci
en toute connaissance des problèmes de sélectivité, de réadsorption et de redistribution.
Tessier [Tessier 1979] dans son schéma d’extraction séquentielle a défini 5 fractions
différentes pour la spéciation métaux lourds dans les sols et sédiments :
• Fraction 1. Métaux échangeables
Ils correspond aux métaux lourds adsorbés sur la surface des argiles, des acides humiques
et des oxydes de fer et de manganèse et spécialement ceux liés par des interactions
électrostatiques faibles et ceux qui peuvent être libérés par échange d’ion. Les changements
de la composition ionique de l’eau affectent significativement le processus d’adsorption-
désorption.
Le réactif utilisé pour cette fraction doit permettre la libération des ions échangeables, sans
attaquer les carbonates (tel que le NH4Ac), et sans agir par la suite avec le calcium libéré de
la deuxième fraction (tel que le NaOAc). Suite à ces constats, l’utilisation du MgCl2 à pH 7
a été préconisée pour une durée d’une heure. Ce réactif combine les bonnes capacités
d’échange du Mg2+ et la faible capacité de complexation du Cl-. L’application du MgCl2
semble ne pas affecter les silicates, les sulfures et la matière organique. Les calculs
thermodynamiques ont également montrés qu’à pH 7, les oxydes de fer et de manganèse ne
sont pas significativement solubles. La très faible dégradation des carbonates peut être
réduite par la réduction du temps de contact sédiment/MgCl2.

Chapitre I : Les sédiments de dragage
67
• Fraction 2. Métaux liés aux carbonates :
Elle correspond aux métaux qui co-précipitent avec les carbonates. Les métaux de cette
fraction sont sensibles aux changement du pH et leur libération se fait après dissolution de
cette phase solide.
Pour l’obtention de cette fraction, l’utilisation du NaOAc/HOAc à pH 5 est recommandée.
Le temps de contact dépend de la concentration et du type de carbonates, de la taille des
grains et de la quantité d’échantillons. Tessier constate cependant, qu’au bout de 5h
l’extraction est achevée. Pour les sédiments ayant une forte concentration en carbonates, un
temps de contact plus important ainsi qu’un ajustement de pH sont nécessaires. Il est
important de préciser qu’à un pH plus faible, les oxydes de fer et de manganèse peuvent être
attaqués.
• Fraction 3. Métaux liés aux oxydes de fer et de manganèse :
Ces derniers sont à l’état de nodules ou de ciment entre les particules. Ces oxydes sont
d’excellents pièges pour les métaux lourds et sont thermodynamiquement instables dans les
conditions anoxiques (fraction réductible).
La meilleur méthode pour obtenir cette fraction est la combinaison de deux réactifs, le
premier pour réduire les oxydes de fer et de manganèse et le second pour maintenir en
solution les métaux qui ont été libérés suite à la réduction des oxydes. Tessier préconise
donc l’utilisation du couple NH2OH HCl/HOAc. Pour déterminer le temps nécessaire à
l’obtention de cette fraction, il a mesuré la concentration du fer libéré à différents temps de
contact et constate que l’extraction est complète au bout de 6h.
• Fraction 4. Métaux liés à la matière organique et aux sulfures :
Les métaux lourds peuvent être liés à différentes formes de matières organiques :
organismes vivants, détritus, enduit sur la particule minérale. Le pouvoir de complexation de
la matière organique naturelle (acides fulviques et humiques) sont bien identifiés. D’autre
part, les métaux peuvent être également sous forme de sulfure. Sous conditions oxydantes
dans l’eau naturelle, la matière organique peut être dégradée conduisant à une
immobilisation des métaux lourds solubles (fraction oxydable). Les sulfures peuvent
également être oxydés et transformés en sulfates plus solubles.
L’oxydation de la matière organique et des sulfures se fait généralement avec le peroxyde
d’hydrogène et un acide moyennement fort. L’inconvénient est que certaines formes
organiques ne sont pas parfaitement oxydées et en contre partie, l’utilisation d’acide fort

Chapitre I : Les sédiments de dragage
68
peut conduire à l’attaque des silicates. L’utilisation du couple H2O2/HNO3 à haute
température est un compromis qui permet une meilleure extraction. L’extraction est suivie
par l’utilisation du NH4Ac afin d’éviter que les métaux libérés précipitent à nouveau.
• Fractions 5. Métaux résiduels :
Une fois les 4 fractions extraites, il ne reste que les métaux piégés dans la matrice
cristalline d’aluminosilicate des minéraux primaires et secondaires. Dans les conditions
naturelles, ces métaux sont insolubles.
Afin de détruire les minéraux primaires et secondaire, le résidu de sulfure et la faible
quantité de matière organique qui n’ont pas été oxydés, le couple HF/HClO4 a été
recommandé pour une dissolution complète.
Ceci étant, les procédures d’extraction séquentielles sont toutes critiquables pour leur
manque de sélectivité et le problème de réadsorption et de redistribution [Gleyzes 2002 ;
Das 1995 ; Gómez-Ariza 1999].
Idéalement, un réactif est désigné pour dissoudre une phase minérale spécifique contenue
dans le sédiment, et en conséquence la solubilisation des métaux qui lui sont associés. Sauf
que plusieurs études théoriques et expérimentales ont prouvé le manque de sélectivité des
réactifs. Cela conduit le plus souvent à une surestimation de la quantité des métaux extraits
dans la phase en question et conduit à des erreurs d’interprétation des résultats. Ce genre
d’erreurs peut également se produire lorsque la phase à dissoudre est en concentration élevé
et que la quantité du réactif est insuffisante pour tout dissoudre. Dans ce cas, la
concentration des métaux liés à cette phase est sous-estimée, et la concentration des métaux
dans les autres fractions surestimée. Ce problème peut être résolu par l’addition d’un volume
plus important de réactif avec le risque de dissoudre d’autres phases. D’autre part, une
dissolution incomplète d’une phase peut entraîner un changement de pH conduisant à la
réadsorption des métaux ou leur redistribution sur d’autres. Cela peut conduire à des erreurs
d’interprétation.
Bien que cette méthode présente beaucoup d’inconvénient, elle permet d’apporter des
éclaircissements intéressants sur la répartition et le comportement des métaux lourds dans
les sédiments ainsi que l’évaluation de leur potentiel polluant. C’est une méthode largement
utilisée pour la spéciation des métaux lourds et pour l’évaluation de certains procédés de
traitement.

Chapitre I : Les sédiments de dragage
69
I.4. Conclusions
En France, on drague en moyenne 2.82Mm3/an de sédiments fluviaux et 50Mm3/an de
sédiments marins. Cela nécessitent des efforts considérables pour développer des techniques
de traitement et de valorisation afin de minimiser les méthodes traditionnelles d’élimination
(immersion en mer, mise en dépôt, remise en suspension) qui peuvent présenter des risques
certains de l’écosystème.
Le développement industriel de techniques de décontamination et de valorisation se
heurte aux lacunes de la réglementation dans ce domaine. Le vide juridique existant, ne
permet pas de répondre à une question essentielle: quelle qualité pour quel devenir et à quel
risque ? Des efforts doivent être faits par les institutions gouvernementales en collaboration
avec les industriels et les scientifiques afin de pouvoir y répondre. Cela peut se faire à
travers le développement de moyen de caractérisation de la toxicité des sédiments, la
définition de normes et de valeurs seuils pour chaque voie de valorisation possible. Dans le
cadre de cette thèse, nous sommes confrontés à ce problème. Il n’existe pas de normes ni de
seuils qui nous permettent de qualifier les sédiments, ni après ni avant le traitement.
Néanmoins, les chercheurs développent des techniques de traitement des sédiments. Elles
sont nombreuses et peuvent être physiques, chimiques, thermiques ou biologiques. Toutes
ces méthodes présentent des limites qui sont liées à la nature de la pollution, à la
composition du sédiment, au temps nécessaire pour le traitement ou à leur coût.
Le procédé NOVOSOL que nous allons utiliser pour les sédiments, combine une
technique chimique (traitement à l’acide phosphorique) et une technique thermique
(calcination) afin d’améliorer la qualité du traitement. Ce procédé va permettre de stabiliser
les métaux lourds et de dégrader la matière organique afin d’avoir un résidu principalement
minéral en composition ayant des propretés chimiques et physiques intéressantes pour des
valorisation dans divers secteurs.
La complexité des sédiments, la multitude de phases qu’ils peuvent contenir ainsi que les
différentes formes sous lesquelles peuvent se trouver les contaminants, impliquent une
bonne connaissance de leurs caractéristiques physico-chimiques. Celles-ci vont faciliter le
choix du traitement à appliquer et permettre de mieux prévoir le comportement des
sédiments lors du traitement, et finalement son optimisation. C’est pour ces raisons que le
chapitre II va porter sur la caractérisation des sédiments avec lesquels nous allons travailler.
Pour évaluer la spéciation des métaux et leur mobilité dans les sédiments, l’extraction
séquentielle est une méthode souvent utilisée. Son utilisation va nous permettre d’une part,

Chapitre I : Les sédiments de dragage
70
de connaître l’influence des traitements chimique et thermique sur le comportement des
métaux lourds, d’autre part elle va nous renseigner sur leur mobilité. L’extraction
séquentielle sera réalisée sur les sédiments bruts ainsi que sur les sédiments après chaque
étape de traitement.

Chapitre II : Caractérisation des sédiments de dragage
72
CHAPITRE II
Caractérisation des sédiments de dragage

Chapitre II : Caractérisation des sédiments de dragage
73
II.1. Introduction
L’étude expérimentale a été réalisée sur deux sédiments continentaux réels, La première
étape du travail réalisé est une caractérisation physico-chimique la plus large possible du
sédiment. L’objectif est d’arriver à une connaissance approfondie du milieu, (pH, teneur en
espèces métalliques, teneur en matière organique, granulométrie, densité, …), afin
d’identifier des grandeurs utiles pour le traitement par phosphatation. Une caractérisation
systématique permettra également une bonne compréhension et une analyse pertinente des
résultats.
II.2. Préparation d’un échantillon de sédiment pour l’analyse
Les deux sédiments réels sur lesquels nous avons travaillé sont d’origines différentes,
l’un provenant de la région de Lille (canal de la Marque) fournit à Solvay par les Voix
Navigable de France (VNF), l’autre de Vraimont (canal de Charleroi en Belgique) fournit à
Solvay par l’entreprise de dragage Ecoterre. Ces sédiments ont été choisis par Solvay car ils
représentent deux cas extrêmes en terme de pollution métallique.
Nous recevons ces sédiments tamisés à un diamètre < 5mm dans des bidons en
polyéthylène de 68l soigneusement fermés, afin d’éviter tout contact avec l’air. Ils sont
conservés dans une chambre froide à une température de 4°C afin de minimiser toute
activité bactérienne qui pourrait modifier leurs propriétés. Il est important de noter que les
caractérisations faites correspondent à l’état du sédiment à l’instant où l’analyse à été
réalisée. La caractérisation ne tient donc pas compte de toutes les modifications des
conditions environnementales telles que le changement de potentiel redox et de pH que
subissent les sédiments du moment où ils ont été dragués et tamisé, jusqu'à l’analyse au
laboratoire.
Pour la plupart des analyses physico-chimiques, les normes AFNOR sur la « Qualité des
sols » [AFNOR 1994] ont été utilisées. Ce choix a été motivé par les similitudes de
composition et de caractéristiques physico-chimiques entre les sols et les sédiments.
C’est ainsi que la norme NF X31-101 est utilisée afin d’avoir une fraction représentative
de l’échantillon brut pour les expériences. Cette norme consiste à sécher l’échantillon dans
une enceinte climatique à une température de 40°C jusqu’à ce que le masse soit constante.
Cette étape est suivie d’une réduction des mottes et tamisage de l’échantillon jusqu’à obtenir
un diamètre moyen ≤ 2mm. Ainsi, toutes les analyses physico-chimique seront menées avec
des échantillons préparés de cette façon.

Chapitre II : Caractérisation des sédiments de dragage
74
La teneur en humidité qui a été éliminée obtenu après lors du séchage à 40°C est notée
H40, il est exprimée en pourcentage par la relation suivante :
0 140
0
100m mHm
⎛ ⎞−= ×⎜ ⎟⎝ ⎠
(2.1)
Avec m0, la masse de l’échantillon (en gramme) avant séchage et m1, la masse de
l’échantillon après séchage (en gramme).
Le taux d’humidité à 40°C est estimé à 52.2% pour le sédiment de Vraimont (BV) et de
66.6% pour le sédiment de Lille (BL) (Tableau II- 1). Cette valeur dépend fortement des
conditions de dragage.
Tableau II- 1 : Humidité à 40°C des sédiments de Lille et de Vraimont.
Sédiment Humidité à 40°C (% massique)
Ecart type
BL 66.6 0.3 BV 52.2 0.3
II.3. Caractérisation chimique
II.3.1. Détermination de l’humidité résiduelle
Cette mesure permet de calculer la masse sèche utilisée lors des différentes expériences.
La méthode de détermination est définie par la norme NF X31-102. Le principe est le
séchage d’une masse d’échantillon dans une étuve à 103 ± 2°C jusqu’à ce que la masse soit
constante.
L’humidité résiduelle (HR) est calculée de la même manière que l’humidité à 40°C (H40).
Les résultats présentés dans le Tableau II- 2 montrent que le sédiment de Lille a une
humidité résiduelle deux fois plus importante que celle du sédiment de Vraimont. Il est
important de signaler que la perte de masse observé lors du séchage à 103°C ne correspond
pas uniquement à la perte d’eau, elle peut également correspondre à l’évaporation de la
matière organique volatile susceptible d’intervenir à partir de 60°C.
Ainsi l’humidité totale (HT) (somme de l’humidité à 40°C (H40) et de l’humidité
résiduelle (Hr)) des deux sédiments est estimée respectivement à 69.2% et 53.5% pour les
sédiments de Lille et de Vraimont. Cette mesure est importante pour l’étape de
phosphatation et de séchage.

Chapitre II : Caractérisation des sédiments de dragage
75
Tableau II- 2 : Humidité résiduelle des sédiments de Lille et de Vraimont.
Sédiment Humidité résiduelle
Ecart type
BL 2.6 0.1 BV 1.3 0.1
II.3.2. Détermination du pH
Le pH du sédiment est une donnée essentielle car l’existence d’une phase minérale, sa
spéciation et sa toxicité sont autant de paramètres liés au pH du milieu.
La mesure du pH se fait le plus fréquemment dans une suspension aqueuse, le rapport de
la masse du sédiment au volume d’eau varie suivant les méthodes ou suivant la texture du
milieu. Le ratio le plus souvent rencontré est de 1/2,5 [Guevara-Riba 2004 ; Cappuyns
2004] et 1/5 [Tack 1996 ; Vandecasteele 2002].
Les valeurs du pH obtenues par cette méthode de mesure sont considérées comme les
plus proches du pH du milieu considéré. Elles expriment l’acidité réelle et prennent en
compte les ions H3O+ libres dans la phase liquide.
Cette mesure est décrite par la norme NF X 31-103. Le principe de la méthode est la mise
en équilibre ionique d’une certaine masse de solide avec un volume donné d’eau
déminéralisée. Le ratio de la masse de sédiment au volume d’eau étant fixé à 1/2,5. La
mesure de la différence de potentiel existant entre une électrode de mesure et une électrode
de référence s’effectue dans la suspension aqueuse à l’équilibre.
II.3.2.1. Mode opératoire
Les échantillons (10 g) sont préparés pour analyse dans un bêcher, on y ajoute 25ml
d’eau distillée, on agite ensuite avec un agitateur magnétique pendant 60min dans une pièce
dont la température ambiante est de 20 ± 2°C. Cela permet de mettre en suspension la
totalité de l’échantillon et d’obtenir ainsi un équilibre entre la phase solide et la phase
liquide. La suspension est ensuite laissée au repos 2 heures à l’abri de l’air puis on mesure le
pH de la suspension. L’utilisation du sédiment brut lors de la phosphatation suppose une
mesure préalable de son pH. Pour cela, le pH des deux sédiments bruts à été également
mesuré (pH mètre METTLER TOLEDO MPC 227).

Chapitre II : Caractérisation des sédiments de dragage
76
Le Tableau II- 3 montre que le pH des deux sédiments bruts sont légèrement différents.
Le pH de BL est légèrement plus basique que celui de BV, cette basicité peut être attribuée à
la présence de carbonates. Notons une légère différence entre les pH des sédiments préparés
pour essai et les sédiments bruts, elle peut être attribuée à la différence du rapport
liquide/solide.
Tableau II- 3 : pH des sédiments de Lille et de Vraimont, bruts et séchés à 40°C
Sédiment pH Ecart type BL (brut) 7.4 0.1 BV (brut) 7.2 0.1
BL (à 40°C) 7.9 0.1 BV (à 40°C) 7.6 0.1
II.3.3. Détermination de la conductivité électrique
La conductivité électrique est une mesure qui donne une approximation de la
concentration des sels solubles présents dans l’échantillon.
La norme utilisée est la NF X 31-113 qui est basée sur l’extraction des sels d’un
échantillon, solubles dans l’eau, dans des conditions bien définies et dans un rapport
sédiment sec/eau égal à 1/5 (M/M). Deux électrodes en platine maintenues en parallèle dans
une colonne d’extrait aqueux de sédiment permettent de mesurer le courant conduit par les
ions présents.
II.3.3.1. Mode opératoire
On pèse 10g d’échantillon de sédiment préparé pour essai qu’on transvase dans un flacon
en polyéthylène, on y ajoute 50 ml d’eau distillée, on ferme le flacon et on le place dans
l’agitateur mécanique à mouvement horizontal, on agite pendant 30min. Après filtration, on
mesure la conductivité de la solution obtenue, la conductivité des sédiments bruts a été
également mesurée. L’analyse est faite dans une enceinte où la température a été contrôlée et
maintenue à 20 ± 1°C. Le conductimètre utilisé est un METTLER TOLEDO MPC 227.
Les mesures présentées dans le Tableau II- 4 révèlent un taux plus important de sels
solubles dans le sédiment de Lille que dans celui de Vraimont. Comme pour le pH, la
conductivité des sédiments préparés pour essai est différente de celle des sédiments bruts,
cette différence est ici encore probablement due à la différence de rapport solide/liquide.

Chapitre II : Caractérisation des sédiments de dragage
77
Tableau II- 4 : Conductivité des sédiments de Lille et de Vraimont bruts et séchés à 40°C
Sédiment Conductivité (mS)
Ecart type
BL (brut) 2 0.1 BV (brut) 1.6 0.1
BL (à 40°C) 0.7 0.03 BV(à 40°C) 0.4 0.01
II.3.4. Dosage des éléments chimiques
Le but de cette analyse est de mesurer la concentration globale des éléments considérés
comme polluants tels que le Pb, Zn, Co, Cd, As, Cr, Cu, ainsi que d’autres constituants de
l’échantillon tels que le Fe, P, Al. Ces données permettent de déterminer les quantités
initiales de ces éléments dans le sédiment.
La norme NF X 31-151 est utilisée pour décrire la mise en solution des éléments
métalliques traces, par attaque aux acides chlorhydrique et nitrique (communément dite
méthode de l’eau régale) ou par attaque à l’acide fluorhydrique. Ces méthodes conduisent à
l’obtention d’une solution destinée à des dosages par spectrométrie d’absorption ou
d’émission atomique.
La première méthode, celle à l’eau régale, à pour principe la mise en solution de
l’échantillon dans un mélange d’acide chlorhydrique et nitrique par ébullition sous
réfrigérant à reflux. L’inconvénient c’est qu’elle ne permet pas la mise en solution des
métaux liés à la structure silicatée. La méthode à l’acide fluorhydrique par contre, permet de
dissoudre les composés silicatés. Cette méthode nécessite une calcination préalable à 450°C
(afin de détruire la matière organique), suivie d’une mise en solution dans de l’acide
fluorhydrique concentré en présence d’acide perchlorique. Après élimination des acides
fluorhydrique et perchlorique par évaporation, le résidu est attaqué par les acides
chlorhydrique et nitrique. L’inconvénient de cette méthode est l’éventuelle perte d’éléments
sous forme de composés volatils tels que le Hg ou le plomb tétraéthyle lors de la calcination
ainsi que l’évaporation de la silice sous forme de SiF4. La méthode qui a été choisie pour
cette étude est celle de l’acide fluorhydrique.

Chapitre II : Caractérisation des sédiments de dragage
78
II.3.4.1. Mode opératoire
On pèse 0.25g d’échantillon préparé pour essai dans une capsule en quartz, on la place
dans un four (four Aubry) puis on calcine à 450°C pendant 3h, on retire la capsule et on
laisse refroidir.
Le résidu solide est ensuite transféré dans une capsule en PTFE, on ajoute 5ml d’acide
fluorhydrique (48%) et 1.5ml d’acide perchlorique (70%), on porte sur une plaque
chauffante et on laisse évaporer à sec.
Toujours sur la plaque, on ajoute 3,75ml d’acide chlorhydrique (38%) et 1.25ml d’acide
nitrique (65%), une fois la dissolution finie, on verse la solution dans une fiole jaugée de
100 ml, on complète au volume par de l’eau déminéralisée et on analyse en ICP-AES
(Spectromètre d’Emission Atomique Plasma à Couplage Inductif).
Le principe de l’analyse par ICP-AES est le suivant : l’échantillon liquide est prélevé,
nébulisé, puis transmis vers le plasma. Il subit là les différentes étapes de décomposition,
d’atomisation, d’ionisation ; les atomes et ions générés sont alors excités. L’intensité des
raies émises par ces espèces, lors de leur retour vers les niveaux d’énergie fondamentaux, est
ensuite mesurée. Le plasma est un gaz (argon) partiellement ionisé et macroscopiquement
neutre. La haute température (6000-10 000°C) générée permet d’éliminer les interférences
chimiques, et rend accessible à la fois les raies ioniques et atomiques. L’acquisition de tels
spectre très riches nécessite donc une haute résolution [Velasquez 2003].
L’appareil qui a été utilisé pour la mesure des concentrations des différents éléments est
un ULTIMA 2 de JOBIN YVON HORIBA. Les concentrations en mg/kg de matière sèche
des éléments analysés sont présentées dans le Tableau II- 5
Comme il a été mentionné dans le chapitre bibliographique, nous ne possédons pas de
valeurs limites réglementaires qui nous permettent de classer les deux sédiments comme
étant pollués ou non.
Nous pouvons cependant remarquer que le sédiment de Lille est plus pollué que le
sédiment de Vraimont. En effet mise à part les concentrations en As et en Co qui sont très
proches, le Cr, le Cu, le Pb ainsi que le Zn sont en moyenne 3.5 fois plus importants dans le
sédiment de Lille que dans le sédiment de Vraimont, la concentration de Cd par contre est 9
fois plus importante dans BL que dans le BV.
Toutefois l’analyse quantitative montre une certaine hétérogénéité des concentrations en
fonction de l’échantillon et de l’élément mesuré.
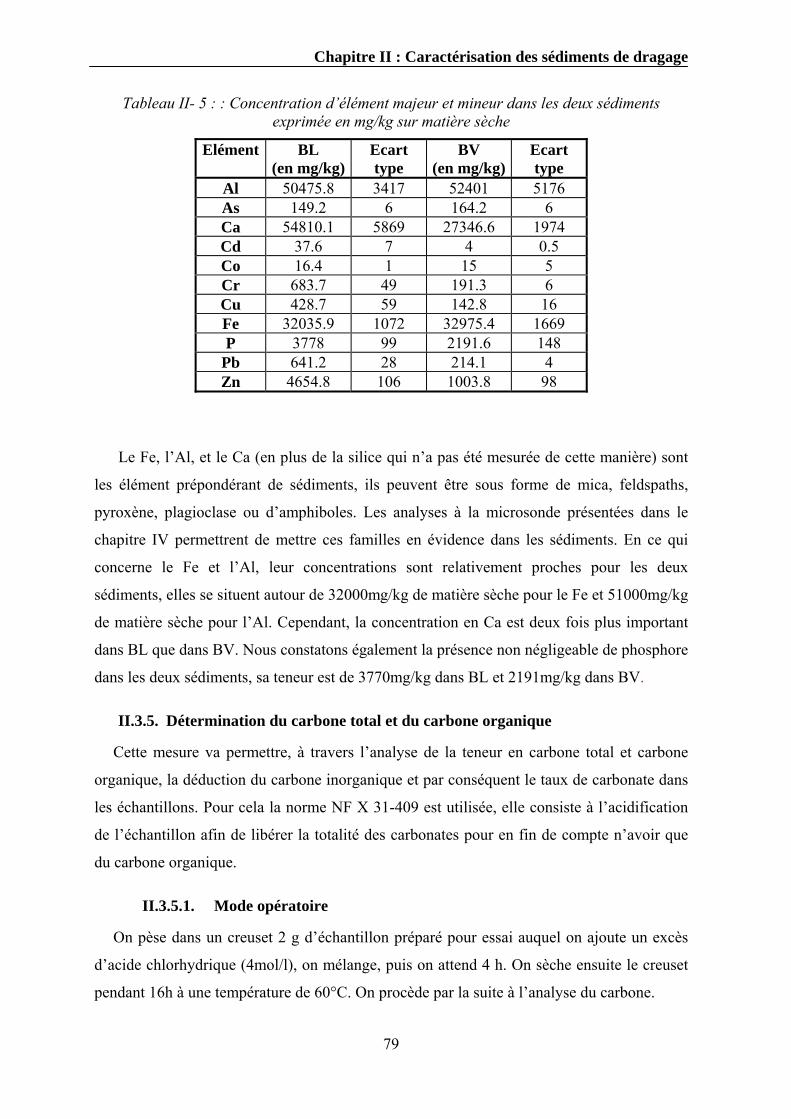
Chapitre II : Caractérisation des sédiments de dragage
79
Tableau II- 5 : : Concentration d’élément majeur et mineur dans les deux sédiments exprimée en mg/kg sur matière sèche
Elément BL (en mg/kg)
Ecart type
BV (en mg/kg)
Ecart type
Al 50475.8 3417 52401 5176 As 149.2 6 164.2 6 Ca 54810.1 5869 27346.6 1974 Cd 37.6 7 4 0.5 Co 16.4 1 15 5 Cr 683.7 49 191.3 6 Cu 428.7 59 142.8 16 Fe 32035.9 1072 32975.4 1669 P 3778 99 2191.6 148
Pb 641.2 28 214.1 4 Zn 4654.8 106 1003.8 98
Le Fe, l’Al, et le Ca (en plus de la silice qui n’a pas été mesurée de cette manière) sont
les élément prépondérant de sédiments, ils peuvent être sous forme de mica, feldspaths,
pyroxène, plagioclase ou d’amphiboles. Les analyses à la microsonde présentées dans le
chapitre IV permettrent de mettre ces familles en évidence dans les sédiments. En ce qui
concerne le Fe et l’Al, leur concentrations sont relativement proches pour les deux
sédiments, elles se situent autour de 32000mg/kg de matière sèche pour le Fe et 51000mg/kg
de matière sèche pour l’Al. Cependant, la concentration en Ca est deux fois plus important
dans BL que dans BV. Nous constatons également la présence non négligeable de phosphore
dans les deux sédiments, sa teneur est de 3770mg/kg dans BL et 2191mg/kg dans BV.
II.3.5. Détermination du carbone total et du carbone organique
Cette mesure va permettre, à travers l’analyse de la teneur en carbone total et carbone
organique, la déduction du carbone inorganique et par conséquent le taux de carbonate dans
les échantillons. Pour cela la norme NF X 31-409 est utilisée, elle consiste à l’acidification
de l’échantillon afin de libérer la totalité des carbonates pour en fin de compte n’avoir que
du carbone organique.
II.3.5.1. Mode opératoire
On pèse dans un creuset 2 g d’échantillon préparé pour essai auquel on ajoute un excès
d’acide chlorhydrique (4mol/l), on mélange, puis on attend 4 h. On sèche ensuite le creuset
pendant 16h à une température de 60°C. On procède par la suite à l’analyse du carbone.
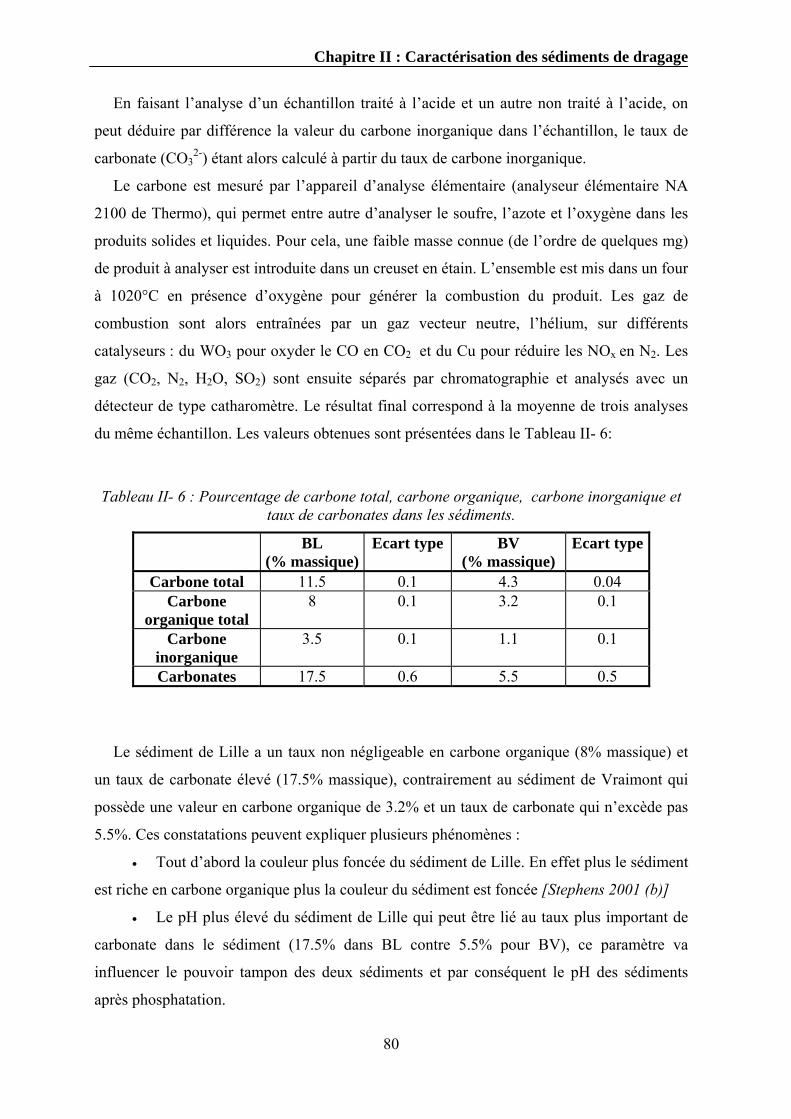
Chapitre II : Caractérisation des sédiments de dragage
80
En faisant l’analyse d’un échantillon traité à l’acide et un autre non traité à l’acide, on
peut déduire par différence la valeur du carbone inorganique dans l’échantillon, le taux de
carbonate (CO32-) étant alors calculé à partir du taux de carbone inorganique.
Le carbone est mesuré par l’appareil d’analyse élémentaire (analyseur élémentaire NA
2100 de Thermo), qui permet entre autre d’analyser le soufre, l’azote et l’oxygène dans les
produits solides et liquides. Pour cela, une faible masse connue (de l’ordre de quelques mg)
de produit à analyser est introduite dans un creuset en étain. L’ensemble est mis dans un four
à 1020°C en présence d’oxygène pour générer la combustion du produit. Les gaz de
combustion sont alors entraînées par un gaz vecteur neutre, l’hélium, sur différents
catalyseurs : du WO3 pour oxyder le CO en CO2 et du Cu pour réduire les NOx en N2. Les
gaz (CO2, N2, H2O, SO2) sont ensuite séparés par chromatographie et analysés avec un
détecteur de type catharomètre. Le résultat final correspond à la moyenne de trois analyses
du même échantillon. Les valeurs obtenues sont présentées dans le Tableau II- 6:
Tableau II- 6 : Pourcentage de carbone total, carbone organique, carbone inorganique et taux de carbonates dans les sédiments.
BL (% massique)
Ecart type BV (% massique)
Ecart type
Carbone total 11.5 0.1 4.3 0.04 Carbone
organique total 8 0.1 3.2 0.1
Carbone inorganique
3.5 0.1 1.1 0.1
Carbonates 17.5 0.6 5.5 0.5
Le sédiment de Lille a un taux non négligeable en carbone organique (8% massique) et
un taux de carbonate élevé (17.5% massique), contrairement au sédiment de Vraimont qui
possède une valeur en carbone organique de 3.2% et un taux de carbonate qui n’excède pas
5.5%. Ces constatations peuvent expliquer plusieurs phénomènes :
• Tout d’abord la couleur plus foncée du sédiment de Lille. En effet plus le sédiment
est riche en carbone organique plus la couleur du sédiment est foncée [Stephens 2001 (b)]
• Le pH plus élevé du sédiment de Lille qui peut être lié au taux plus important de
carbonate dans le sédiment (17.5% dans BL contre 5.5% pour BV), ce paramètre va
influencer le pouvoir tampon des deux sédiments et par conséquent le pH des sédiments
après phosphatation.

Chapitre II : Caractérisation des sédiments de dragage
81
• La concentration en métaux lourds plus importante dans BL. En effet, différents
auteurs [Coquery 1995 ; Lin 1998] ont relié la teneur en polluants métalliques à la
concentration en carbone organique, ils révèlent l’affinité entre les métaux lourds et le
carbone organique.
L’analyse du soufre dans les deux échantillons a été également faite, le pourcentage
massique de soufre dans le sédiment de Lille est de 1.2 ± 0.2 %.
Il est de 0.2 ± 0.02 % dans celui de Vraimont. Cette dernière concentration étant très voisine
du seuil de détection (0.2%) cette valeur reste incertaine.
II.3.6. Détermination de la matière organique totale
L’analyse élémentaire permet de donner le taux de carbone organique mais ne permet pas
de donner le pourcentage exact de matière organique dans les sédiments. Pour cela une
méthode est souvent utilisée pour le dosage de la matière organique dans les sédiments, elle
consiste à calciner l’échantillon à 450°C pendant 3h, la perte de masse observée est attribuée
à la matière organique et représente donc le pourcentage massique de matière organique
[Ausili 1998 ; Tack 1997 ; Guevara-Riba 2004].
II.3.6.1. Mode opératoire
On pèse 0.25 g d’échantillon préparé pour essai dans une capsule en quartz, on la place
dans un four (four Aubry) puis on calcine à 450°C pendant 3h, ensuite on retire la capsule et
on laisse refroidir. Le pourcentage de perte de masse engendrée par la calcination représente
le pourcentage en matière organique.
Les résultats obtenus sont présentés dans le Tableau II- 7.
Tableau II- 7 : Pourcentage massique de matière organique dans les sédiments
Sédiment % massique de matière
organique
Ecart type
BL 15.6 0.5
BV 6.7 0.2
Les pourcentages massiques de matière organique confirment bien les résultats de
l’analyse du carbone organique, en effet le sédiment de Lille renferme un taux de matière

Chapitre II : Caractérisation des sédiments de dragage
82
organique 2.3 fois plus important que celui de Vraimont. Le rapport matière organique sur
carbone est en moyenne de 2 pour les deux sédiments.
II.3.7. La diffraction des rayons X
La caractérisation aux rayons X a été utilisée pour identifier les phases cristallisées
contenues dans les deux sédiments. Les échantillons préparés pour essai sont fixés sur un
support cylindrique en PVC (Poly(chlorure de vinyl)) avec une surface la plus lisse possible.
Ils sont placés entre une source de rayon X et un détecteur. Les diffractogrammes sont
enregistrés entre 20° et 60° en 2θ avec un pas de comptage de 0.02° et un temps de
comptage par pas de 2 s. Les fentes avant et arrière sont respectivement de 1 mm et 1mm.
Les analyses ont été réalisées sur un diffractomètre D5000 de Siemens (30 mA, 40 kV) avec
une source de rayonnement du Cu Kα (1.543 Å), un filtre de nickel supprime la raie Kβ du
Cu.
Les informations obtenues à partir des diffractogrammes des rayons X des deux
sédiments (Figure II- 1) permettent la mise en évidence de trois phases cristallines
distinctes : la calcite, le quartz et le feldspath. On remarquera que l’intensité des pics des
différentes phases est plus importante pour le sédiment de Lille, ce qui traduit une plus forte
concentration de ces phases dans ce sédiment. Cette analyse nous permet de conclure qu’une
partie des carbonates présents dans les deux sédiments est de la calcite (CaCO3). On constate
que les métaux lourds ne sont pas détectables directement par le biais de cette méthode pour
deux raisons : d’une part, leur faible concentration dans les échantillons (inférieurs aux
seuils de détection) et d’autre part, leur présence sous des formes amorphes.
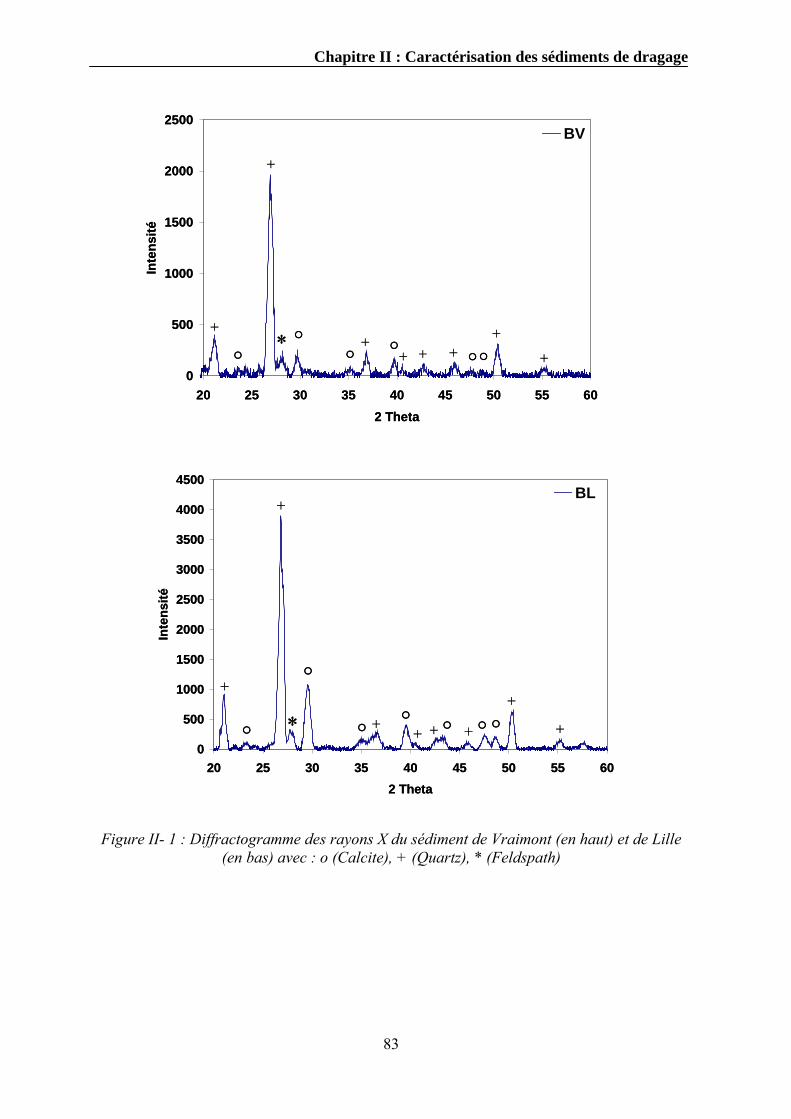
Chapitre II : Caractérisation des sédiments de dragage
83
Figure II- 1 : Diffractogramme des rayons X du sédiment de Vraimont (en haut) et de Lille (en bas) avec : о (Calcite), + (Quartz), * (Feldspath)
0
500
1000
1500
2000
2500
20 25 30 35 40 45 50 55 602 Theta
Inte
nsité
BV
+
+ ++
++
+
+
°°
° ° °°*
0
500
1000
1500
2000
2500
20 25 30 35 40 45 50 55 602 Theta
Inte
nsité
BV
+
+ ++
++
+
+
°°
° ° °°*
+
+ ++
++
+
+
°°
° ° °°*
0
500
1000
1500
2000
2500
3000
3500
4000
4500
20 25 30 35 40 45 50 55 602 Theta
Inte
nsité
BL
°*
+
+
++
+ +++
°
° °° °°0
500
1000
1500
2000
2500
3000
3500
4000
4500
20 25 30 35 40 45 50 55 602 Theta
Inte
nsité
BL
°*
+
+
++
+ +++
°
° °° °° °*
+
+
++
+ +++
°
° °° °°

Chapitre II : Caractérisation des sédiments de dragage
84
II.3.8. Détermination de la capacité d’échange cationique
La capacité d’échange représente la quantité maximale d’ions susceptible d’être échangée
sur un solide en présence d’un excès d’ions en solution. Cela correspond en fait à la charge
nécessaire pour atteindre l’électroneutralité du sédiment. Cette capacité d’échange peut être
soit cationique (C.E.C), soit anionique (C.E.A), selon que la surface du milieu est chargée
positivement ou négativement.
La C.E.C est intimement liée à la présence d’argile, de matière organique et par la même
occasion aux métaux lourds. En ce qui concerne les argiles, les sites d’échange d’un
sédiment donné sont généralement issus des substitutions isomorphiques dans le réseau
cristallin argileux et une liaison non compensée sur les bordures des feuillets d’argile.
La détermination de la C.E.C consiste à mesurer un paramètre caractérisant un état
d’équilibre du sédiment et un environnement expérimental donné. Les valeurs obtenues sont
fonction du milieu (cation saturant, pH …) ainsi que des conditions de réalisation
influençant le rendement des réactions d’échange.
La méthode la plus utilisée pour la mesure de la C.E.C est la norme X 31-13. Trois
méthodes sont proposées par cette norme. Celle que nous avons utilisée est la méthode au
chlorure de cobaltihexammine [Co(NH3)6Cl3] qui est réalisée dans les conditions suivantes :
• Echange entre les cations retenus par l’échantillon et les ions cobaltihexammine
d’une solution aqueuse ;
• Détermination de la capacité d’échange cationique par mesure de la
concentration dans le filtrat des ions cobalt libres, cette mesure s’effectue par ICP-AES
II.3.8.1. Mode opératoire
On ajoute à 5 g d’échantillon 50 ml d’une solution de chlorure de cobaltihexammine (pH
6.08), on mélange ensuite pendant 1 heure au moyen d’un agitateur culbuteur réglé à 40
tr/min placé dans un air ambiant à 20 ± 1°C, on filtre aussitôt et on dose le cobalt restant
dans le filtrat par absorption atomique.
En faisant la différence entre la valeur du cobalt ajouté et la valeur du cobalt restant en
solution on en deduit la CEC, elle est exprimée en meq.100g-1.
Nous constatons sur le Tableau II- 8 que le sédiment BL a une valeur de CEC plus
importante que celle du sédiment BV, qui pourrait être liée à la teneur plus importante
d’argile et à la matière organique. Cette mesure nous permet également une vérification des
mesures de la fraction échangeable lors des expériences d’extraction séquentielle

Chapitre II : Caractérisation des sédiments de dragage
85
Tableau II- 8 : Capacité d’échange cationique des sédiments de Lille et Vraimont exprimés en meq.100 g-1.
CEC (meq.100 g-1) Ecart typeBL 37.6 0.4 BV 29.4 0.2
II.3.9. Le pouvoir tampon
La présence de carbonate et d’argile dans le sédiment lui confère un pouvoir tampon non
négligeable [Åström 1998], ce pouvoir est proportionnel à la quantité de carbonates et
d’argile. Le pouvoir tampon d’un échantillon à un pH donné peut être déterminé par la
quantité d’acide ou de base qui peut être ajouté tout en gardant le pH de l’échantillon
constant [Gäbler 1997].
Le dosage acide est indispensable car il nous permet de connaître la valeur maximale
d’acide que nous pouvons ajouter sans que le pH du sédiment ne varie lors des expériences
de phosphatation. Ainsi, la mise en solution des polluants métalliques serait contrôlée.
Pour adapter cette méthode à la réaction de phosphatation que nous étudions, l’acide
phosphorique a été retenu pour cette mesure.
Actuellement, il n’existe pas de normes établies pour la mesure du pouvoir tampon du
sédiment, cependant il existe une méthode fréquemment utilisée pour les sols [Palaprat
2002] et qui peut s’appliquer aux sédiments, elle peut être conduite de deux façons
différentes :
• La première méthode : un échantillon de sédiment de 1 g est mis en contact avec
des volumes croissants d’acide de même concentration. Ainsi, nous avons une augmentation
de la quantité de protons apportés à force ionique contrôlée.
• La deuxième méthode : un échantillon de sédiment de 1 g est mis en contact avec
un même volume d’acide de concentration croissante. Dans ce cas la force ionique est
variable.
La première méthode est celle qui est la plus souvent utilisée.
II.3.9.1. Mode opératoire
22 échantillons contenant chacun 1 g de sédiment sont mis en contact, sous agitation,
avec 22 volumes croissants d’une solution d’acide phosphorique de concentration définie à
4.25 10-2 mol/l (entre 2 et 21 ml pour BL et entre 2 et 78 ml pour BV). Cette concentration

Chapitre II : Caractérisation des sédiments de dragage
86
est fixée lors d’essais préalables consistants à tester des concentrations croissantes d’acide
phosphorique. La concentration retenue correspond à celle où le pH de la suspension
commence à varier.
Le pH des 22 échantillons est mesuré par la suite au bout de dix jours de mise en contact
afin de s’assurer que l’équilibre est bien atteint. L’évolution de la quantité de H+ restant en
solution après dix jours de contact en fonction de la quantité d’acide ajouté est représentée
sur la Figure II- 2. A partir de là, il est possible de déterminer le nombre de mole de protons
nécessaire pour saturer les sites d’échanges de 1 g d’échantillon. Par conséquent, la quantité
d’acide qui fera baisser le pH est entraînera des mises en solution d’espèces.
L’évolution de la quantité d’H+ restant en solution après dix jours de contact par rapport à
la quantité apportée pour 1 g de sédiment BV et BL est représentée sur la Figure II- 2. On
constate qu’au départ, quelque soit la quantité d’H+ ajoutée, la concentration d’H+ restante
en solution est très faible et avoisine le zéro. A partir d’une certaine concentration d’H+
apportée, on constate que la concentration de H+ en solution augmente avec l’augmentation
de la quantité de protons apportés. Dans des conditions normales et pour des échantillons
purs, la pente de cette courbe (après 10 jours d’équilibre) doit être parallèle à celle de la
quantité d’H+ apportée, mais vu la composition très complexe du sédiment et la multitude de
réactions qui peuvent se produire, nous n’avons pas réussi à obtenir une telle droite. Par
contre nous avons essayé d’en tracer une en prenant le plus de points possibles. Ainsi, la
différence entre les deux parallèles nous permet de connaître le pouvoir tampon des deux
sédiments.
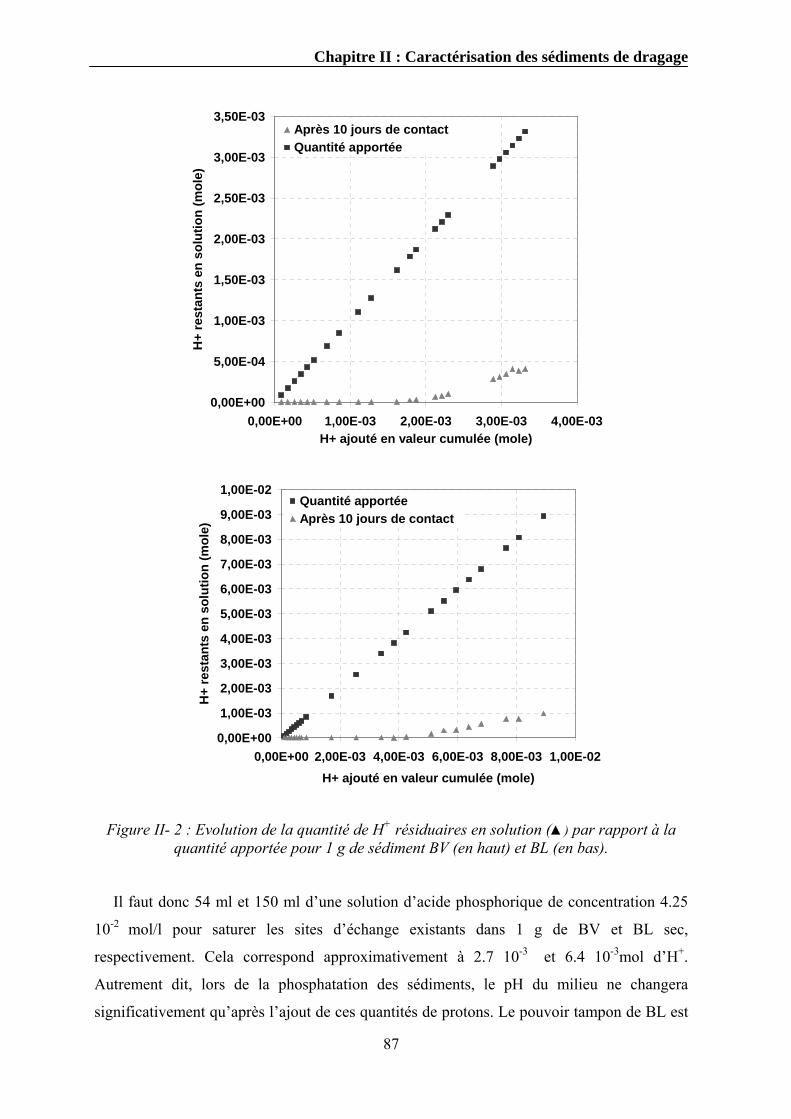
Chapitre II : Caractérisation des sédiments de dragage
87
Figure II- 2 : Evolution de la quantité de H+ résiduaires en solution (▲) par rapport à la quantité apportée pour 1 g de sédiment BV (en haut) et BL (en bas).
Il faut donc 54 ml et 150 ml d’une solution d’acide phosphorique de concentration 4.25
10-2 mol/l pour saturer les sites d’échange existants dans 1 g de BV et BL sec,
respectivement. Cela correspond approximativement à 2.7 10-3 et 6.4 10-3mol d’H+.
Autrement dit, lors de la phosphatation des sédiments, le pH du milieu ne changera
significativement qu’après l’ajout de ces quantités de protons. Le pouvoir tampon de BL est
0,00E+00
5,00E-04
1,00E-03
1,50E-03
2,00E-03
2,50E-03
3,00E-03
3,50E-03
0,00E+00 1,00E-03 2,00E-03 3,00E-03 4,00E-03H+ ajouté en valeur cumulée (mole)
H+
rest
ants
en
solu
tion
(mol
e)
Après 10 jours de contactQuantité apportée
0,00E+00
1,00E-03
2,00E-03
3,00E-03
4,00E-03
5,00E-03
6,00E-03
7,00E-03
8,00E-03
9,00E-03
1,00E-02
0,00E+00 2,00E-03 4,00E-03 6,00E-03 8,00E-03 1,00E-02H+ ajouté en valeur cumulée (mole)
H+
rest
ants
en
solu
tion
(mol
e)
Quantité apportéeAprès 10 jours de contact

Chapitre II : Caractérisation des sédiments de dragage
88
plus important que celui de BV, cela peut être en premier lieu lié à la plus grande quantité de
carbonates dans BL (17.5% pour BL contre 5.5% pour BV). En effet, en calculant les
quantités exactes de protons nécessaires pour la décarbonatation des carbonates des deux
sédiments, on constate qu’il faut 1.82 10-3 et 5.88 10-3mole d’H+ pour 1g de BV et BL sec
respectivement. Or ces valeurs sont inférieures à celles du pouvoir tampon, ce qui prouve
que d’autre espèces sont mises en jeu tels que les argiles et les oxydes de fer et de
manganèse. Néanmoins la contribution des carbonates reste prédominante.
II.4. Caractérisation physique
II.4.1. La densité
La densité sèche ou vraie est le rapport de la masse par le volume occupé par le solide,
l’identification de ce paramètre permet, par l’intermédiaire de la teneur en eau et du degré de
saturation, de connaître la porosité du matériau étudié. La mesure de la densité se fait par le
pycnomètre, dont le principe de mesure repose sur la détermination de la différence de
masse entre le volume du solide étudié et celui d’un fluide inerte dans lequel il est immergé.
Les deux pycnomètres les plus souvent utilisés pour les solides sont le pycnomètre à liquide,
appelé aussi pycnomètre à bouchon capillaire et le pycnomètre à gaz.
L’utilisation du pycnomètre à gaz semble être le plus approprié pour les sédiments
étudiés, à cause de la capacité des gaz à pénétrer les pores les plus fins. La densité des
échantillons bruts (BL et BV) a été mesurée par un pycnomètre à hélium Accupyc 1330 de
Micromeritics. Une masse connue d’échantillon (entre 5 et 10 g) est introduite dans une
cellule de 12 cm3 insérée dans le compartiment de mesure, l’analyse est répétée jusqu’à
l’obtention de cinq valeurs consécutives variant de moins de 0.1%.
Les résultats présentés sur le Tableau II- 9 montrent une différence de densité de 13%
entre les deux sédiments. Elle peut être liée à leur composition. Nous avons vu que BL
contient plus de minéraux cristallisés plus dense que les espèces amorphes. La différence
principale est la teneur en matière organique qui abaisse la densité de BL.
Tableau II- 9 : Densité des sédiments bruts de Lille et de Vraimont.
Sédiment Densité Ecart type
BL 1.23 0.005
BV 1.42 0.007

Chapitre II : Caractérisation des sédiments de dragage
89
II.4.2. La distribution granulométrique
L’analyse granulométrique permet de caractériser la distribution de taille des particules
d’un solide. Elle permet, également, d’identifier les différentes familles granulométriques
(sable, limon, argile) afin de les associer à une texture. Certaines caractéristiques d’un
sédiment qui peuvent avoir une influence sur la spéciation des polluants sont dépendantes de
la taille des grains qui le compose (réactivité, surface spécifique…).
Les méthodes de mesure de la taille des particules sont nombreuses et dépendent entre
autre de la distribution de taille des particules. Les sédiments avec lesquels nous travaillons
semblent avoir une large distribution granulométrique, pour cette raison, le granulométre à
diffraction laser semble être l’outil le plus approprié et le plus rapide. La gamme de mesure
est de 0,02 µm à 2 mm.
Les particules en suspension des sédiments bruts sont introduites (par voix sèche ou
humide) dans une cuve d’analyse grâce à une pompe de circulation. Dans la cuve, les
particules sont éclairées par un faisceau laser parallèle de 18 mm de diamètre avant d’être
projetés sur des photo-diodes de mesure suite à l’interaction entre le laser et les particules.
L’analyse est faite simultanément sur l’ensemble des particules circulant devant le faisceau
laser. La distribution est calculée soit en assimilant les particules à des sphères (distribution
en volume) soit en considérant les particules plates (distribution surfacique). Le problème de
ce type d’analyse effectuée sur l’ensemble de l’échantillon est la possibilité du masquage
des particules fines par les plus grosses.
La distribution granulométrique des échantillons bruts a été déterminée en voie humide
par un granulomètre laser Mastersizer 2000-Hydro 2000 de Malvern instruments. Elle est
souvent caractérisée par son diamètre moyen, noté d50, correspondant à une taille de
particules telle que 50% des particules aient une taille inférieure à d50. D’autre paramètres
tels que le d10 et le d90 sont parfois utilisés. Sur le Tableau II- 10, l’ensemble de ces
informations sont présentées. Le constat que nous pouvons faire est que les deux sédiments
ont une granulométrie très proche et ont une texture limon

Chapitre II : Caractérisation des sédiments de dragage
90
Tableau II- 10 : Distribution granulométrique des sédiments bruts de Lille et de Vraimont.
BL BV
Fraction de sable (en %) 5 7.1
Fraction de limon (en %) 70.1 72.7
Fraction d’argile (en %) 24.9 20.2
d10 (en µm) 3.3 2.7
d50 (en µm) 19.1 19.1
d90 (en µm) 148.4 94.9
II.5. Conclusions
La caractérisation physico-chimique des sédiments avant d’entamer un traitement de
stabilisation est une étape fondamentale. Cette étape permet de connaître la composition et
les caractéristiques propres du sédiment à traiter. A partir de ces donnés, des conditions de
traitement pertinentes peuvent être définies. Les données obtenues lors de la caractérisation
permettent en outre une meilleur interprétation des résultats expérimentaux et la
compréhension des phénomènes associés.
Dans le cadre de cette thèse, les sédiments de dragage vont subir un traitement chimique
qui vise à stabiliser les polluants inorganiques et un traitement thermique (calcination) visant
d’une part l’amélioration de la stabilisation des polluants inorganiques et d’autre part la
décomposition des matières organiques. Pour mieux comprendre les processus de
transformation qui seront observés lors de ces traitements, différentes caractéristiques
chimiques et physiques ont été déterminées puis récapitulées dans le Tableau II- 11.
Bien que les caractéristiques physiques des deux sédiments restent très proches, les
caractéristiques chimiques sont différentes. Les plus grandes différences observées le sont
au niveau de la concentration en polluants métalliques, de la teneur en carbonates et matière
organique. En effet, le sédiment de Lille est plus pollué que le sédiment de Vraimont, car il
contient un pourcentage massique en matière organique 2,3 fois plus importante que le
sédiment de Vraimont et un pourcentage de carbonates 3,2 fois plus important. Cela nous
inspire les conclusions suivantes:

Chapitre II : Caractérisation des sédiments de dragage
91
• La probabilité d’utilisation d’une quantité d’acide phosphorique plus importante pour
BL afin de stabiliser les métaux lourds. Cela dépendra de la spéciation des métaux lourds
dans les deux sédiments qui sera abordée au chapitre IV.
• La spéciation des métaux lourds est liée à la composition géochimique des sédiments
(argile, matière organique, carbonate…), la présence d’un taux de matière organique et
de carbonate plus important dans le BL laisse présager une différence non négligeable
dans leur spéciation et donc dans leur comportement chimique et thermique.
• Le pouvoir tampon plus important dans le sédiment de Lille que dans celui de
Vraimont indique qu’il va falloir plus d’acide pour mobiliser les métaux lourds dans le
sédiment de Lille lors de la phosphatation.
• La concentration plus importante de calcium dans le sédiment de Lille implique qu’il
y a une plus grande probabilité de former des phosphates de calcium précurseurs dans le
sédiment de Lille que dans celui de Vraimont. Ceci même si, à ce stade, nous ne savons
pas si le calcium est sous forme échangeable ou sous forme de calcite.
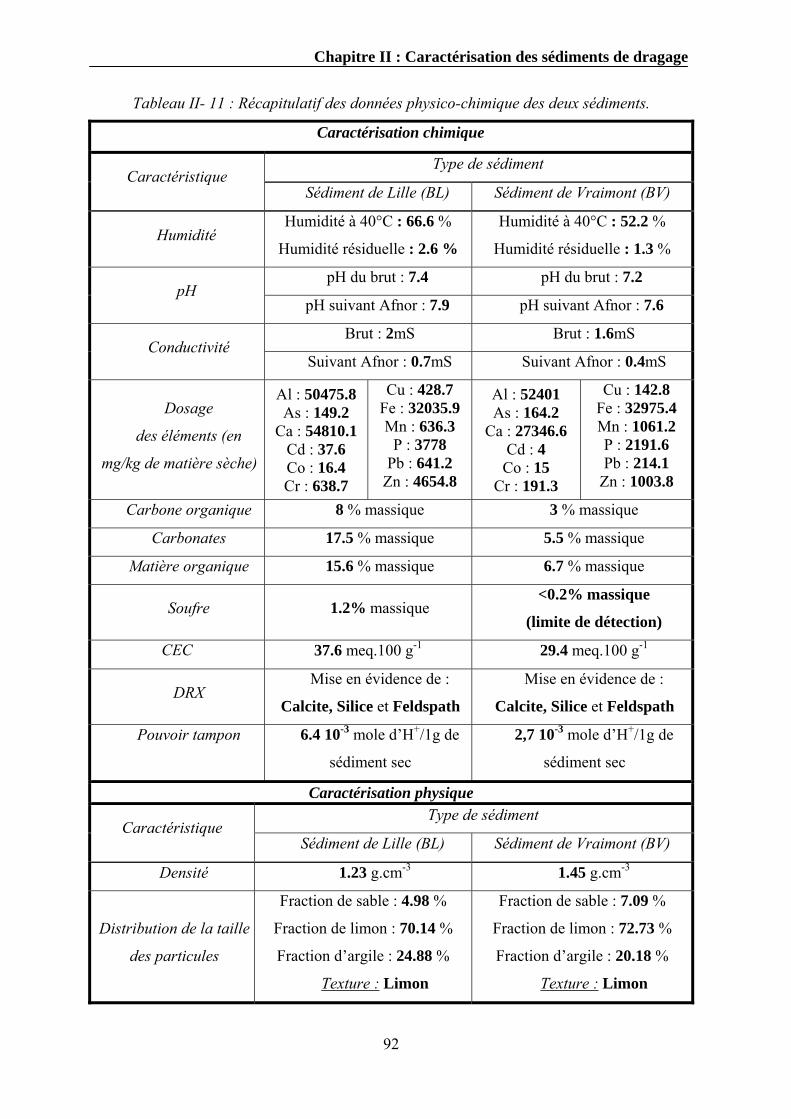
Chapitre II : Caractérisation des sédiments de dragage
92
Tableau II- 11 : Récapitulatif des données physico-chimique des deux sédiments.
Caractérisation chimique
Type de sédiment Caractéristique
Sédiment de Lille (BL) Sédiment de Vraimont (BV)
Humidité Humidité à 40°C : 66.6 %
Humidité résiduelle : 2.6 %
Humidité à 40°C : 52.2 %
Humidité résiduelle : 1.3 %
pH du brut : 7.4 pH du brut : 7.2 pH
pH suivant Afnor : 7.9 pH suivant Afnor : 7.6
Brut : 2mS Brut : 1.6mS Conductivité
Suivant Afnor : 0.7mS Suivant Afnor : 0.4mS
Dosage
des éléments (en
mg/kg de matière sèche)
Al : 50475.8 As : 149.2
Ca : 54810.1 Cd : 37.6 Co : 16.4 Cr : 638.7
Cu : 428.7 Fe : 32035.9 Mn : 636.3
P : 3778 Pb : 641.2
Zn : 4654.8
Al : 52401 As : 164.2
Ca : 27346.6 Cd : 4 Co : 15
Cr : 191.3
Cu : 142.8 Fe : 32975.4 Mn : 1061.2 P : 2191.6 Pb : 214.1
Zn : 1003.8
Carbone organique 8 % massique 3 % massique
Carbonates 17.5 % massique 5.5 % massique
Matière organique 15.6 % massique 6.7 % massique
Soufre 1.2% massique <0.2% massique
(limite de détection)
CEC 37.6 meq.100 g-1 29.4 meq.100 g-1
DRX Mise en évidence de :
Calcite, Silice et Feldspath
Mise en évidence de :
Calcite, Silice et Feldspath
Pouvoir tampon 6.4 10-3 mole d’H+/1g de
sédiment sec
2,7 10-3 mole d’H+/1g de
sédiment sec
Caractérisation physique Type de sédiment
Caractéristique Sédiment de Lille (BL) Sédiment de Vraimont (BV)
Densité 1.23 g.cm-3 1.45 g.cm-3
Distribution de la taille
des particules
Fraction de sable : 4.98 %
Fraction de limon : 70.14 %
Fraction d’argile : 24.88 %
Texture : Limon
Fraction de sable : 7.09 %
Fraction de limon : 72.73 %
Fraction d’argile : 20.18 %
Texture : Limon

Chapitre III : Traitement du sédiment de Vraimont
93
CHAPITRE III
Traitement des sédiments de Vraimont

Chapitre III : Traitement du sédiment de Vraimont
94
III.1. Introduction
Le procédé de stabilisation des métaux lourds et de dégradation de la matière organique
comporte trois étapes successives :
• le traitement chimique à l’acide phosphorique qui a pour objectif la stabilisation des
métaux lourds sous forme de phosphates métalliques insolubles.
• Le séchage convectif, à température, à vitesse de l’air et d’humidité relative ambiante
qui conduit à la diminution de la teneur en eau du sédiment. Lors de cette étape, la réaction
de phosphatation se poursuit, c’est la « maturation du sédiment ».
• l’étape de calcination, qui conduit à la dégradation de la matière organique (humus et
pollution organique) et à la cristallisation des formes chimiques obtenues lors de la
phosphatation. Les phosphates métalliques ainsi obtenus se caractérisent par une forte
insolubilisation.
Dans ce chapitre les trois étapes du procédé vont être étudiées, l’objectif étant de
comprendre les phénomènes qui ont lieu lors du traitement des sédiments avec l’acide
phosphorique. Cela se fera par la détermination de la cinétique de phosphatation et de
maturation du sédiment ainsi que le mécanisme qui sont mis en jeu. Dans la phase de
séchage convectif, c’est la cinétique de séchage qui sera étudiée avec l’objectif de connaître
les temps de séchage des sédiments ainsi que l’influence du traitement sur ces temps. Une
étude comparative de l’influence de la température et de la vitesse de l’air lors du séchage
sera également discutée. Quant à la phase de calcination, c’est l’influence de la température
sur la dégradation de la matière organique et sur les propriétés physiques du résidu final
obtenu qui sera étudiée.
III.2. Matériels et méthodes
II.2.1. Phosphatation et maturation
III.2.1.1. Le montage expérimentale de phosphatation
Compte tenu de l’hétérogénéité du sédiment et de la difficulté de mettre en œuvre un
dispositif permettant d’obtenir des mesures fiables, un réacteur a été construit pour la
phosphatation des sédiments. Pour cela, un réacteur de 1cm de diamètre muni d’une double
enveloppe thermostatée a été utilisé. L’objectif étant de maintenir la température du milieu à
20°C au cours des expérience.
Le réacteur est raccordée à deux pompes péristaltiques Masterflex L/S (Figure III- 1) qui
permettent de travailler avec des débits volumiques variant de 0.10 à 3400ml/min (soit 0.16

Chapitre III : Traitement du sédiment de Vraimont
95
10-3 à 56 10-3l/h). La première pompe péristaltique sert au transfert du sédiment brut stocké
dans un récipient approprié. L’alimentation se fait à travers un tuyau Masterflex Tygon LFL
06429 de diamètre de 9.7 mm (9.7 10-3m). Ce tuyau est relié au réacteur en verre grâce à un
T. L’autre coté du T est rattaché à la seconde pompe par un tuyau Masterflex C Flex 06424
d’un diamètre de 0.8 mm (0.8 10-3m) qui alimente le réacteur en acide phosphorique.
Afin de connaître la cinétique de phosphatation dans un réacteur tubulaire nous avons
décidé d’utiliser des réacteurs de taille croissante de façon à modifier le temps de contact.
Cette méthode d’étude a été préférée à celle qui aurait consisté pour un réacteur de taille
donnée à modifier les débits de pompage du sédiment et ainsi varier les temps. Ce choix a
été guidé par le dispositif expérimental.
L’étude de la cinétique de phosphatation n’a été réalisée que sur le sédiment de Vraimont.
Son taux d’humidité et sa consistance ne nous ont pas permis de le pomper tel quel. Pour
cela il a été dilué de sorte qu’on obtienne un taux d’humidité de 55%, qui le rendait ainsi
plus facilement pompable.
Un temps de contact maximal du sédiment dans le réacteur a été défini d’environ 20min,
ce choix a été inspiré par les résultats de Bournonville [Bournonville 2002]. En effet, elle a
démontré à travers les études cinétiques de la phosphatation des cendres volantes
d’incinération d’ordures ménagères que l’essentiel du phosphate injecté est consommé au
bout de 20 min. Même si les sédiments sont des matériaux sensiblement différents, ce travail
permettait d’avoir des repères cohérents.
Pour des raisons pratiques, le temps de contact maximal dans le réacteur a été défini à
22.5min (0.37h). Au tour de cette valeur et du diamètre du réacteur utilisé (1cm), la longueur
des réacteurs et les débits des deux pompes ont été calculés. De ce fait 10 réacteurs de
volume différents ont été utilisés, leur volumes et le temps de contact correspondant sont
indiqués sur le Tableau III- 1. Le temps de contact correspond au temps de contact théorique
calculé en prenant en compte le volume du réacteur et le débit utilisé.
Une détermination d’un temps de séjour n’a pas été faite car le milieu est très complexe.
Cette complexité est due à l’existence de trois phases dans le mélange réactionnel (liquide,
solide et gaz (dégagement gazeux lors de la phosphatation des sédiments)) qui rend donc la
détermination de temps de séjour des particules dans le réacteur très complexe.

Chapitre III : Traitement du sédiment de Vraimont
96
Tableau III- 1 : Volume des réacteurs de phosphatation et temps de contacts correspondant
Volume des réacteurs (x10-6 m3)
7.4 11.3 15.2 23.1 46.6 70.2 93.7 110.4 158.4 247.7
Temps de contact (min) 0.67 1.03 1.4 2.1 4.2 6.4 8.5 10 14.4 22.5
Le débit massique calculé pour la pompe d’alimentation en sédiment est de 11g/min (soit
0.18 10-3kg/h). Le débit massique de la pompe qui alimente le réacteur en acide
phosphorique (85% massique) est de 0.19, 0.32, et 0.44g/min, l’équivalent de 3.1 10-6, 5.3
10-6 et 7.3 10-6kg/h. Ces valeurs correspondent à un taux de phosphatation de 3, 5 et 7%
d’acide phosphorique sur matière sèche. Le choix de ces quantités d’acide s’est fait en
prenant en compte la somme des concentrations en mole des métaux lourds à stabiliser et du
Ca susceptible d’agir avec le phosphore ajouté.
Figure III- 1 : Dispositif expérimentale de phosphatation.

Chapitre III : Traitement du sédiment de Vraimont
97
III.2.1.2. Cinétique de phosphatation et de maturation.
Afin de déterminer la cinétique de phosphatation, un prélèvement est fait en sortie de
chaque réacteur. L’objectif est de déterminer la cinétique à travers l’analyse et le suivi de
l’évolution de la quantité de phosphore soluble, autrement dit, le phosphore qui n’a pas
réagit. L’analyse d’éléments tels que As, Al, Ca, Cd, Cr, Cu, Co, Pb, Zn, et Fe, a été
également faite afin de déterminer l’influence de la quantité d’acide sur leur comportement
durant la phosphatation et la maturation.
En sortie de chaque réacteur, un prélèvement est effectué sur le mélange réactionnelle
puis filtré. Le filtrat est ensuite analysé pour déterminer les concentrations du P résiduaire et
celle des éléments cités plus haut. Compte tenu de la difficulté de filtrer le mélange
réactionnel visqueux (55% d’humidité) sortant du réacteur. Un protocole opératoire a était
défini suite à une série d’essai.
Cette série d’essai a consisté à des expériences en batch dans des flacons en polyéthylène.
Nous avons rajouté à 7 échantillons d’une masse de 1g de sédiment phosphaté à 1% (sur
sec), et 7 autres d’une masse de 1g de sédiment phosphaté à 7% (sur sec), des quantités
d’eau déminéralisée croissante de tel sorte à avoir des rapports solide/liquide de 1/5, 1/10,
1/15, 1/20, 1/25, 1/30 et 1/35.
Ces taux de phosphatation choisit correspondent aux taux extrêmes de phosphatation
prévu en réacteur. Immédiatement après l’ajout de l’acide phosphorique au sédiment, le
mélange est agité pendant une minute avec un barreau magnétique. Au terme d’une minute,
l’eau déminéralisée est ajoutée, la suspension est ensuite agitée pendant une minute. Le
choix d’une minutes de mélange de la suspension sédiment/eau nous a paru être le bon
compromis pour éviter de modifier la cinétique et récupérer le phosphore qui n’a pas réagit
(libre). La suspension est ensuite filtrée sous-vide (filtre papier de 0.45µm), le filtrat est
récupéré et analysé. L’objectif étant d’analyser le P libre et voir l’influence du rapport
solide/liquide sur la quantité du P libre extrait de l’échantillon. Les résultats obtenus sont
représentés sur la Figure III- 2
Ces résultats montrent, qu’à partir d’un rapport solide liquide de 1/10, la concentration du
phosphore récupéré est pratiquement constate. En dessous de ce rapport, la solution sature et
nous n’arrivons donc pas à récupérer la totalité du phosphore qui n’a pas réagi. Au delà, la
totalité du P libre est récupéré. Nous avons opté donc pour la suite des expériences à
l’utilisation du rapport solide/liquide de 1/20 afin de récupérer tout le phosphore qui n’a pas
réagit.

Chapitre III : Traitement du sédiment de Vraimont
98
Finalement, le protocole choisit consiste à récupérer immédiatement en sortie de chaque
réacteur, 1g de sédiment, mélangé ensuite à 20ml d’eau déminéralisée. L’ensemble est agité
pendant 1 minute et filtré instantanément sous-vide (filtre papier de 0.45µm).
Nous analysons ensuite les éléments tels que le P, Ca, Cd, Co, Cr, Cu, As, Zn, Fe, Al, et
le Pb. Une mesure du pH est également effectuée. Ces analyses et ces mesures sont réalisées
afin de voir l’influence de l’acide phosphorique sur le pH des sédiments et sur le
comportement de ces éléments.
Figure III- 2 : Influence du rapport solide/liquide sur la concentration du P libre dans le sédiment phosphaté à 1 et 7%.
En ce qui concerne la maturation, il s’agit du comportement des composés chimiques du
sédiment à l’étape de séchage. Afin d’étudier leur évolution, les sédiments phosphatés sont
récupérés en sortie du réacteur dans des récipients en polyéthylène et séchés à l’air ambiant
(température du laboratoire entre 22 et 25°C). L’étude de la maturation commence à partir
du moment où le sédiment sort du réacteur. Pendant la première heure, des prélèvements
sont effectués toutes les 15min (1/4h). D’autres prélèvements sont effectués au bout de 2h,
6h et ensuite un prélèvement tous les jours jusqu’à 28 jours. La procédure pour l’extraction
des composés solubles notamment le P libre, se fait de la même manière que pour l’étude
cinétique de la phosphatation en réacteur.
0
200
400
600
800
1000
1200
1400
1600
1800
0 10 20 30 40Rapport solide/liquide (g/ml)
Phos
phor
e so
lubl
e (m
g/kg
)
1% d'acide 7% d'acide

Chapitre III : Traitement du sédiment de Vraimont
99
Les expériences sur l’étude de la cinétique de phosphatation en réacteur ainsi que la
cinétique de maturation, sont répétées trois fois afin de vérifier la reproductibilité des
données obtenues. L’analyses des composés chimiques cités précédemment se fait par ICP-
AES et la mesure du pH par un pH-mètre
III.2.1.3. Comportement rhéologique du sédiment
Comme nous l’avons souligné dans le paragraphe précédent, une des difficultés
rencontrées dans le pompage du sédiment est sa consistance. L’étude du comportement
rhéologique du sédiment dans différentes conditions représentatives des conditions d’étude
en réacteur (concentration d’acide, taux d’humidité) a été réalisée.
Le rhéomètre qui a été utilisé pour l’étude du comportement du sédiment est un
Rhéostress RS 150 de Rhéo (HAAKE). Il s’agit d’un rhéomètre rotatif à palier fluide, à
vitesse et contrainte imposées. Les spéciations du rhéomètre utilisé sont les suivantes : le
moment peut varier de 0.5.10-4 à 150 mNm et le gradient de cisaillement de 10-7 à 1920 s-1.
Les sédiments ont été étudiés avec une configuration de type rhéo-réacteur (43.4 mm de
diamètre, 119 mm de hauteur pour 175 ml de contenance) avec un double ruban hélicoïdal
pour améliorer le mélange [Cheng 1995, Bournonville 2002]. Pour permettre une meilleure
homogénéisation de la suspension, les mesures sont faites en fluage, c’est-à-dire que les
expériences ont été réalisées en plusieurs paliers, la mesure est effectuée après un temps de
mise à l’équilibre de chaque palier (30s). Toutes les expériences ont été réalisées à 21°C
pour des gradients de cisaillement variant entre 10-4 à 700 s-1.
Des expériences sur le sédiment brut ainsi que sur le sédiment phosphaté à 1, 3, 5 et 7 %
sur sec ont été réalisées dans le but d’étudier l’influence de la quantité d’acide sur la
viscosité du sédiment phosphaté. L'influence du taux d’humidité a également été étudiée.
Ainsi, des expériences ont été réalisées pour les sédiments de Vraimont pour des taux
d’humidité de 42.4, 53.5, 55, 57%. Pour cela, le sédiment de Vraimont brut à été dilué pour
obtenir des taux d’humidité de 55 et 57%. Par contre pour le sédiment à taux d’humidité de
42.4%, nous avons pris un échantillon du sédiment de Vraimont qui avait à la base cette
humidité. Nous n’avons donc pas eu à le diluer.
II.2.2. Séchage des sédiments
Le séchage est la deuxième étape du procédé NOVOSOL. Cette étape permet la
maturation des sédiments phosphatés ainsi que la réduction de leur taux d’humidité. La
réduction du taux d’humidité avant l’étape de calcination, permet la diminution du coût de
cette dernière étape.

Chapitre III : Traitement du sédiment de Vraimont
100
Le dispositif que nous utilisons pour nos expériences de séchage est une veine de séchage
(Figure III- 3). Celle-ci est composée d’une gaine en inox comportant une ouverture sur la
partie inférieure.
Le sédiment de dragage étudié est contenu dans une cuve qui affleure cette ouverture et
est séché par un flux d’air envoyé par la soufflerie située à l’entrée de la gaine. La cuve de 3
cm de hauteur et 8,5 cm de diamètre peut contenir environ 200g de sédiment (phosphaté ou
brut).
Nous avons réalisé les différentes expériences avec un air de ventilation ayant une
humidité relative, une température et une vitesse d’écoulement contrôlées.
Deux capteurs (d'humidité et de température), placés en amont de la cuve, nous
permettent de mesurer les données relatives à l’air de ventilation. Un troisième capteur,
positionné dans la couche supérieure du sédiment, permet de suivre l’évolution de la
température de celui-ci. L’acquisition de ces données est effectuée grâce à un ordinateur
relié aux capteurs. A noter qu’un étalonnage des deux thermocouples a été réalisé
Par ailleurs, l’échantillon est posé sur une balance, d’une précision de ± 0.1g, qui est
reliée à un second ordinateur. Nous avons pu enregistrer la perte de masse du sédiment au
cours du temps. Le montage expérimental est représenté sur la Figure III- 4.
Figure III- 3 : Veine de séchage.

Chapitre III : Traitement du sédiment de Vraimont
101
Les expériences menées ont été faites pour les sédiments de Vraimont brut et phosphaté à
3 et 5% sur sec. Les sédiments ont été séchés à trois températures différentes : 25, 35 et
45°C, à un taux d’humidité relative de l’air de 50% et pour des vitesses de l’air de 0.15, 0.5
et 1.5 m/s. Ces conditions de séchage à l’air ont été choisies car elle présentent des
conditions climatiques susceptibles d’être rencontrées dans différentes saisons. D’autre part,
il est en effet prévu dans le procédé industriel que les sédiments traités soit séchés à l’air
ambiant et pas dans un séchoir.
Ainsi, ces expériences vont nous permettre de réaliser l’étude de la cinétique de séchage
des sédiments à différentes conditions atmosphériques. Une étude de l’influence de la
vitesse de l’air, de la température ainsi que la quantité d’acide sur les temps de séchage est
également réalisée.
Figure III- 4 : Dispositif expérimental de séchage (a). Positionnement de l’échantillon en
cours d’expérience : vue latérale (b).
(a)
(b)

Chapitre III : Traitement du sédiment de Vraimont
102
II.2.3. Calcination des sédiments
La calcination en atmosphère oxydante des sédiments est la troisième étapes du procédé
NOVOSOL. Cette étape permet :
• La dégradation de la matière organique tels que les substances humiques et fulviques
ainsi que la pollution organique (HAP, PCB…) sous forme de CO2, H2O et SOx.
• La cristallisation des phases minérales présentes, notamment les phosphates crées lors de
l’étape de phosphatation et de maturation. Cela conduit à un renforcement de la stabilité des
métaux lourds.
• Une consolidation par frittage des sédiments traités.
Dans le procédé industriel, il est prévu que cette étape soit réalisée dans un four rotatif.
Au laboratoire par contre, la calcination des sédiments est faite dans un four classique type
four à lit fixé (four Aubry). Ce four de 2 litres est chauffé par des résistances en super
Kanthal. Un programmateur de température incorporé permet de monter jusqu’à 1650°C. De
plus un ordinateur permet de faire l’acquisition de la température d’un thermocouple placé
dans le four. Les calcinations ont été réalisées avec 5 à 10g d’échantillon disposés dans des
creusets cylindriques en alumine.
Le sédiment brut et les sédiments phosphatés à 1, 3, 5 et 7% sur sec puis séchés ont été
calcinés sous air. Une calcination de 3h à été effectuée pour des températures de 300, 500,
700, 900 et 1100°C, la vitesse de montée en température a été fixée à 10°C/min. L’objectif
est de connaître l’influence de la température de calcination sur :
• La dégradation du carbone total (carbone organique et inorganique), pour cela la mesure
du carbone total dans les résidus solides est faite avec l’appareil d’analyse élémentaire
(décrit au chapitre II, paragraphe II.3.5)
• Les propriétés physiques des résidus obtenus à travers la mesure de leur densité et de
leur surface spécifique. Les détails de ces mesures sont présentés dans le paragraphe
III.2.3.2
• Le comportement des métaux lourds, cette partie sera abordée dans le chapitre IV.
Afin de mieux comprendre les évolutions (variation de masse) observées lors de la
calcination, des analyses thermogravimétriques ont été réalisées. Les détails sont présentés
dans le paragraphe III.2.3.1 qui suit.

Chapitre III : Traitement du sédiment de Vraimont
103
III.2.3.1. Analyse thermogravimétrique (ATG)
L’étude thermogravimétrique (ATG) fournit pour un matériau la variation de masse au
cours du temps de chauffe, ce qui permet de recueillir des renseignements importants sur la
nature des transformations qui se produisent à des températures données. Le tracé des
courbes thermogravimétriques nécessite une balance de précision couplée à un système de
contrôle et de suivi de température. On peut alors concevoir d’enregistrer la courbe
cumulative ou intégrale, masse en fonction du temps.
L’appareil qui a été utilisé pour ces mesures est le STA 409 PC luxx de chez NETZSCH
monté en configuration TG seule qui nous permet de travailler sur des quantités de 200mg et
de monter à 1600°C avec des rampes en température inférieure à 30°C. Les creusets utilisés
sont en alumine.
Les ATG ont été menées sur le sédiments phosphatés à 7% sur sec puis séchés. Ainsi
200g d’échantillon finement broyés ont été calcinés sous air pendant 3h à 300, 500, 700, 900
et 1100°C avec une vitesse de montée en température de 10°C/min.
III.2.3.2. Mesure des propriétés physiques
Après les étapes de phosphatation, de séchage et de calcination, les propriétés physiques
des sédiments sont mesurés. Les propriétés mesurées sont la surface spécifique et la densité
car ce sont deux grandeurs importantes pour le choix des voies de valorisation.
La densité a été mesurée avec un pycnomètre à hélium comme pour le sédiment brut (voir
chapitre II, paragraphe 4.1).
La surface spécifique a été mesurée par l’analyseur BET (Brunauer, Emmet et Teller).
Environ 300mg d’échantillon sont introduits dans un tube qui est dégazé sous vide pendant
au moins 6h à 40°C. Après cette étape, l’échantillon est soumis à 5 pressions d’azote
différentes à la température de l’azote liquide (-196°C). Ainsi, la quantité de gaz adsorbée
sur l’échantillon est déterminée par la droite de l’isotherme d’adsorption, ce qui permet de
définir la surface spécifique. La mesure de la surface spécifique des échantillons est
déterminée sur un appareil Gemini de Micromeritics.
Ces deux mesures vont nous permettre de connaître l’influence de la quantité d’acide
ainsi que l’influence de la température de calcination sur les propriétés physiques des
sédiments.

Chapitre III : Traitement du sédiment de Vraimont
104
III.3. Résultats et discussions
II.3.1. Phosphatation et maturation des sédiments
III.3.1.1. La cinétique de phosphatation en réacteur tubulaire
L’étude de la cinétique de phosphatation du sédiment de Vraimont s’est faite comme il a
été précisé dans la partie « Matériels et méthodes » dans un réacteur tubulaire. Les taux de
phosphatation sont respectivement de 3, 5 et 7% d’acide phosphorique sur sec, ce qui est
équivalent à 137.8, 229.6 et 321.4mmoles de phosphore pour 1 kg d’un sédiment à 55%
d’humidité.
Lors de la phosphatation des sédiments de Vraimont, deux phénomènes sont observés
simultanément dès l’ajout de l’acide phosphorique avec le sédiment. Ces deux phénomènes
sont notamment un dégagement gazeux et un gonflement du sédiment (changement de la
structure apparente).
Concernant le dégagement gazeux, il se manifeste par une odeur très caractéristique de
sulfure d’hydrogène (H2S). L’intensité de cette odeur n’augmente pas spécialement avec
l’augmentation de la quantité d’acide. En effet, il est reconnu que la sensation olfactive
n’augmente pas forcement avec la concentration de H2S dans l’air.
Les gaz émis suite à la phosphatation contiennent également du CO2. En effet même si le
CO2, ne se manifeste pas par une odeur particulière. La présence 5.5% sur sec de carbonates
(CO32-), dans le sédiment de dragage, permet d’envisager un dégagement de CO2 lors du
contact sédiment-H3PO4.
La quantification des gaz dégagés (H2S et CO2) a été faite par l’analyseur élémentaire
(voir chapitre II, paragraphe 3.5). Le principe étant de mesurer à partir de l’échantillon
phosphaté et séché, la quantité de soufre et de carbone résiduaire. Connaissant la quantité de
carbone et de soufre totale dans l’échantillon brut, on détermine par différence, la quantité
de carbone et de soufre qui s’est dégagé lors de la phosphatation. La quantité de soufre total
dans le sédiment de Vraimont brut étant très proche de la limite de détection, il a été difficile
de suivre son évolution en fonction de la quantité d’acide injecté. Dans le cadre des travaux
de thèse qui se poursuivant dans notre laboratoire, l’identification et la détermination des
concentrations des gaz émis par d’autres moyens sera longuement abordée.
La Figure III- 5 montre l’évolution de la quantité de dioxyde de carbone exprimée en
pourcentage massique en fonction de la quantité d’acide injectée pour un sédiment à 55%
d’humidité. On peut constater à première vu que la relation entre les deux paramètres est
linéaire. En effet, plus la quantité d’acide ajoutée est importante, plus le taux de dioxyde de

Chapitre III : Traitement du sédiment de Vraimont
105
carbone émis est important. On constate que la totalité du carbone inorganique n’a pas été
transformé en CO2 même pour un taux d’acide phosphorique de 7% sur sec. En recalculant à
partir du carbone inorganique la quantité de dioxyde de carbone dégagée, on trouve 12.4g de
CO2/kg de sédiment humide pour un taux de phosphatation de 7% sur sec. Cela correspond à
281mmol de carbonate consommé ou de dioxyde de carbone dégagé.
Figure III- 5 : Influence de la quantité d’acide sur le taux de carbone inorganique (sous
dorme de CO2) dégagé.
Le dégagement gazeux s’accompagne d’une formation de bulles de gaz et par la
formation de mousse au sein de l’échantillon. Cela génère un gonflement du sédiment et par
conséquent à un changement significatif de sa texture
L’évolution de la texture et des propriétés du sédiment au moussage peut être évaluée par
une mesure rhéologique (viscosité). En effet, comme dans l’industrie agroalimentaire les
propriétés rhéologiques sont utilisées pour évaluer les propriétés mécaniques et texturales de
certains aliments (tendreté pour la viande, l’onctuosité ou le fermeté pour des fromages…)
[Scher 2003]. Cela peut bien s’appliquer à notre cas, ce type de mesure va nous permettre de
mieux expliquer l’effet du moussage sur les propriétés du sédiment. Cet intérêt se justifie par
le fait que la pompabilité et l’écoulement du sédiment ainsi que le temps de séchage sont
fortement influencés par le moussage.
Les résultats présentés sur la Figure III- 6 ont été réalisés pour différents taux de
phosphatation et d’humidité. Comme le montrent ces résultats, la viscosité des sédiments
bruts diminue avec l’augmentation du taux d’humidité. La viscosité varie de 523mPa.s pour
0
2
4
6
8
10
12
14
0 50 100 150 200 250 300 350Acide phosphorique (mmol/kg de sédiment humide)
Dio
xyde
de
carb
one
(g/k
g de
séd
imen
t hum
ide)
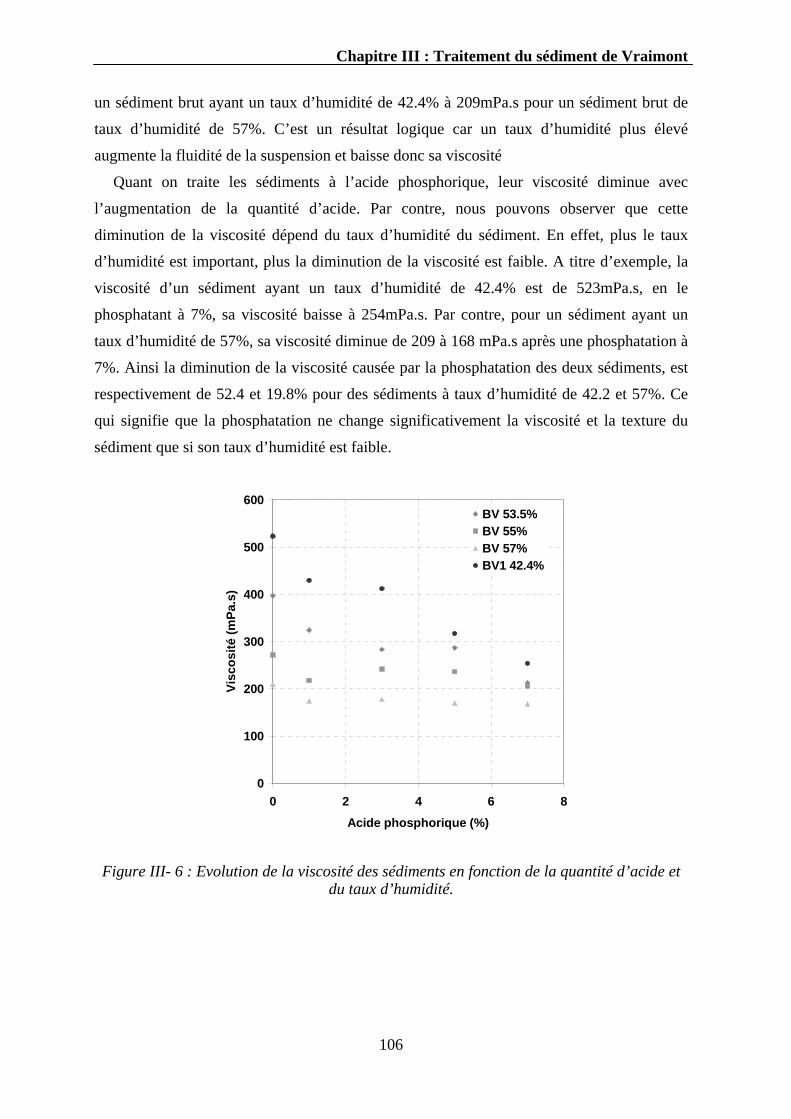
Chapitre III : Traitement du sédiment de Vraimont
106
un sédiment brut ayant un taux d’humidité de 42.4% à 209mPa.s pour un sédiment brut de
taux d’humidité de 57%. C’est un résultat logique car un taux d’humidité plus élevé
augmente la fluidité de la suspension et baisse donc sa viscosité
Quant on traite les sédiments à l’acide phosphorique, leur viscosité diminue avec
l’augmentation de la quantité d’acide. Par contre, nous pouvons observer que cette
diminution de la viscosité dépend du taux d’humidité du sédiment. En effet, plus le taux
d’humidité est important, plus la diminution de la viscosité est faible. A titre d’exemple, la
viscosité d’un sédiment ayant un taux d’humidité de 42.4% est de 523mPa.s, en le
phosphatant à 7%, sa viscosité baisse à 254mPa.s. Par contre, pour un sédiment ayant un
taux d’humidité de 57%, sa viscosité diminue de 209 à 168 mPa.s après une phosphatation à
7%. Ainsi la diminution de la viscosité causée par la phosphatation des deux sédiments, est
respectivement de 52.4 et 19.8% pour des sédiments à taux d’humidité de 42.2 et 57%. Ce
qui signifie que la phosphatation ne change significativement la viscosité et la texture du
sédiment que si son taux d’humidité est faible.
Figure III- 6 : Evolution de la viscosité des sédiments en fonction de la quantité d’acide et
du taux d’humidité.
0
100
200
300
400
500
600
0 2 4 6 8Acide phosphorique (%)
Visc
osité
(mPa
.s)
BV 53.5%BV 55%BV 57%BV1 42.4%
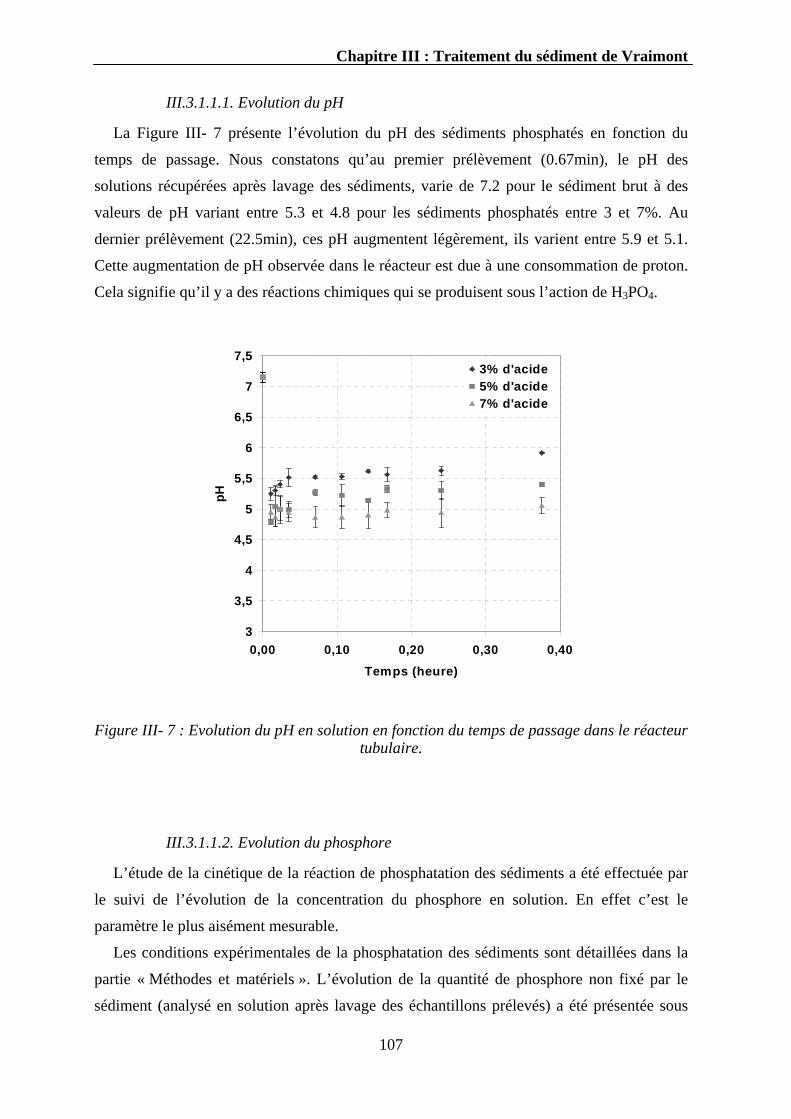
Chapitre III : Traitement du sédiment de Vraimont
107
III.3.1.1.1. Evolution du pH
La Figure III- 7 présente l’évolution du pH des sédiments phosphatés en fonction du
temps de passage. Nous constatons qu’au premier prélèvement (0.67min), le pH des
solutions récupérées après lavage des sédiments, varie de 7.2 pour le sédiment brut à des
valeurs de pH variant entre 5.3 et 4.8 pour les sédiments phosphatés entre 3 et 7%. Au
dernier prélèvement (22.5min), ces pH augmentent légèrement, ils varient entre 5.9 et 5.1.
Cette augmentation de pH observée dans le réacteur est due à une consommation de proton.
Cela signifie qu’il y a des réactions chimiques qui se produisent sous l’action de H3PO4.
Figure III- 7 : Evolution du pH en solution en fonction du temps de passage dans le réacteur tubulaire.
III.3.1.1.2. Evolution du phosphore
L’étude de la cinétique de la réaction de phosphatation des sédiments a été effectuée par
le suivi de l’évolution de la concentration du phosphore en solution. En effet c’est le
paramètre le plus aisément mesurable.
Les conditions expérimentales de la phosphatation des sédiments sont détaillées dans la
partie « Méthodes et matériels ». L’évolution de la quantité de phosphore non fixé par le
sédiment (analysé en solution après lavage des échantillons prélevés) a été présentée sous
3
3,5
4
4,5
5
5,5
6
6,5
7
7,5
0,00 0,10 0,20 0,30 0,40Temps (heure)
pH
3% d'acide 5% d'acide 7% d'acide
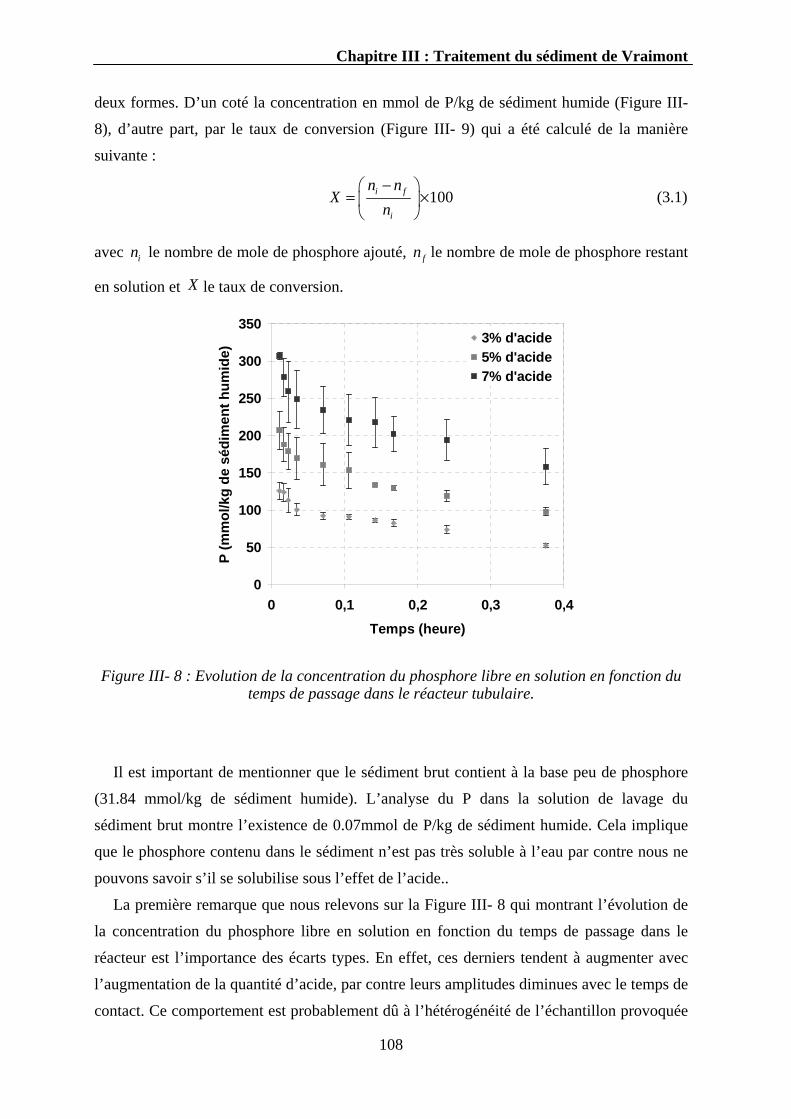
Chapitre III : Traitement du sédiment de Vraimont
108
deux formes. D’un coté la concentration en mmol de P/kg de sédiment humide (Figure III-
8), d’autre part, par le taux de conversion (Figure III- 9) qui a été calculé de la manière
suivante :
100i f
i
n nX
n−⎛ ⎞
= ×⎜ ⎟⎝ ⎠
(3.1)
avec in le nombre de mole de phosphore ajouté, fn le nombre de mole de phosphore restant
en solution et X le taux de conversion.
Figure III- 8 : Evolution de la concentration du phosphore libre en solution en fonction du
temps de passage dans le réacteur tubulaire.
Il est important de mentionner que le sédiment brut contient à la base peu de phosphore
(31.84 mmol/kg de sédiment humide). L’analyse du P dans la solution de lavage du
sédiment brut montre l’existence de 0.07mmol de P/kg de sédiment humide. Cela implique
que le phosphore contenu dans le sédiment n’est pas très soluble à l’eau par contre nous ne
pouvons savoir s’il se solubilise sous l’effet de l’acide..
La première remarque que nous relevons sur la Figure III- 8 qui montrant l’évolution de
la concentration du phosphore libre en solution en fonction du temps de passage dans le
réacteur est l’importance des écarts types. En effet, ces derniers tendent à augmenter avec
l’augmentation de la quantité d’acide, par contre leurs amplitudes diminues avec le temps de
contact. Ce comportement est probablement dû à l’hétérogénéité de l’échantillon provoquée
0
50
100
150
200
250
300
350
0 0,1 0,2 0,3 0,4Temps (heure)
P (m
mol
/kg
de s
édim
ent h
umid
e)
3% d'acide 5% d'acide 7% d'acide
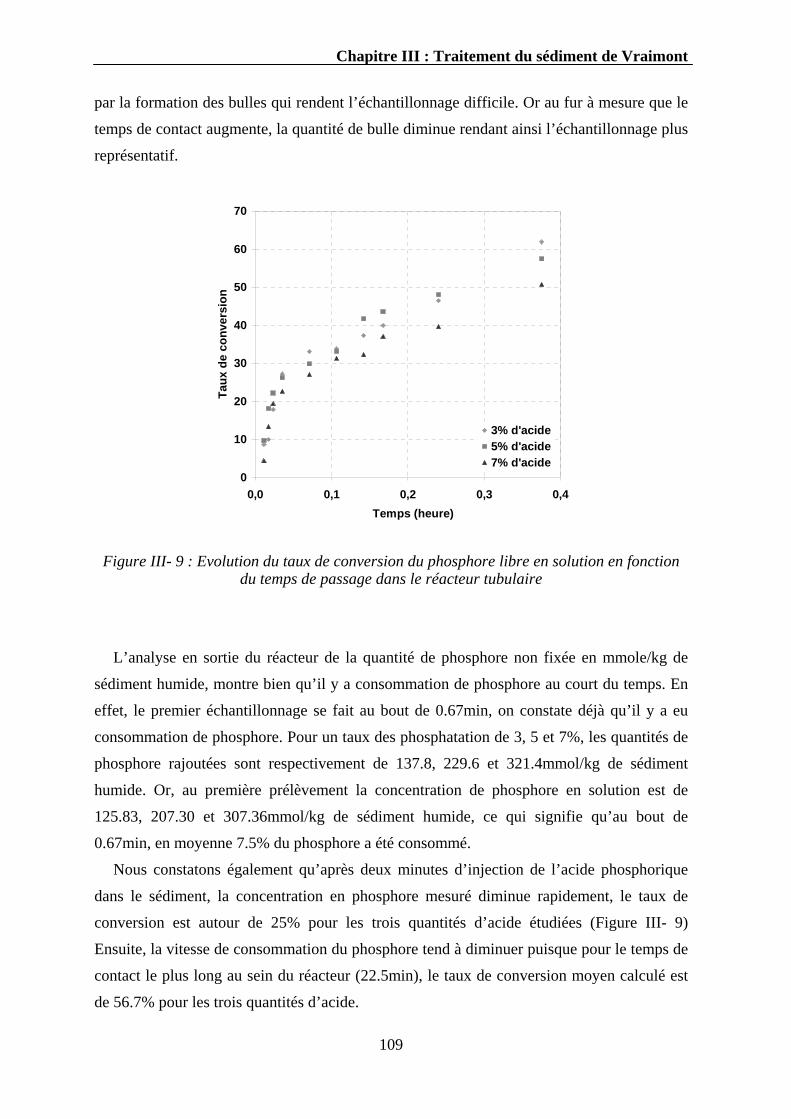
Chapitre III : Traitement du sédiment de Vraimont
109
par la formation des bulles qui rendent l’échantillonnage difficile. Or au fur à mesure que le
temps de contact augmente, la quantité de bulle diminue rendant ainsi l’échantillonnage plus
représentatif.
Figure III- 9 : Evolution du taux de conversion du phosphore libre en solution en fonction
du temps de passage dans le réacteur tubulaire
L’analyse en sortie du réacteur de la quantité de phosphore non fixée en mmole/kg de
sédiment humide, montre bien qu’il y a consommation de phosphore au court du temps. En
effet, le premier échantillonnage se fait au bout de 0.67min, on constate déjà qu’il y a eu
consommation de phosphore. Pour un taux des phosphatation de 3, 5 et 7%, les quantités de
phosphore rajoutées sont respectivement de 137.8, 229.6 et 321.4mmol/kg de sédiment
humide. Or, au première prélèvement la concentration de phosphore en solution est de
125.83, 207.30 et 307.36mmol/kg de sédiment humide, ce qui signifie qu’au bout de
0.67min, en moyenne 7.5% du phosphore a été consommé.
Nous constatons également qu’après deux minutes d’injection de l’acide phosphorique
dans le sédiment, la concentration en phosphore mesuré diminue rapidement, le taux de
conversion est autour de 25% pour les trois quantités d’acide étudiées (Figure III- 9)
Ensuite, la vitesse de consommation du phosphore tend à diminuer puisque pour le temps de
contact le plus long au sein du réacteur (22.5min), le taux de conversion moyen calculé est
de 56.7% pour les trois quantités d’acide.
0
10
20
30
40
50
60
70
0,0 0,1 0,2 0,3 0,4Temps (heure)
Taux
de
conv
ersi
on
3% d'acide5% d'acide7% d'acide

Chapitre III : Traitement du sédiment de Vraimont
110
Ces résultats montrent qu’en sortie du réacteur, une partie du P (56.7%) injecté a réagit
avec les constituants du sédiment puis précipite. L’évolution du P restant (qui n’a pas réagit)
sera discutée lors de l’étude du processus de maturation présentée plus tard.
III.3.1.1.3. Evolution du calcium
L’évolution du Ca en solution en fonction de la quantité d’acide ajouté a également été
suivie. Les résultats obtenus sont représentés sur la Figure III- 10. Nous pouvons remarqué
l’importance des écarts types et leur variation en fonction de la quantité d’acide et du temps
de contact. La raison de ces variations est identique que celle invoquée pour le P.
Figure III- 10 : Evolution de la consommation du calcium en solution en fonction du temps
de passage dans le réacteur tubulaire
Comme pour le P, l’analyse du calcium dans la solution de lavage du sédiment brut
montre la présence de 3.1mmole de Ca/kg de sédiment humide. Ce qui signifie que le
calcium présent dans le sédiment n’est pas très soluble à l’eau, et que la concentration
retrouvée en solution est assez faible pour modifier les résultats de l’étude cinétique.
Nous pouvons constater par contre la présence de Ca dès le premier prélèvement
(0.67min) dans les solutions de lavage des sédiments phosphatés. La concentration du Ca en
solution est proportionnelle à la quantité d’acide ajoutée et varie de 69.86 à
133.75mmoles/kg de sédiment humide phosphatés entre 3 et 7%. La présence du Ca en
0
20
40
60
80
100
120
140
160
0 0,1 0,2 0,3 0,4Temps (heure)
Ca
(mm
ol/k
g de
séd
imen
t hum
ide)
3% d'acide 5% d'acide 7% d'acide

Chapitre III : Traitement du sédiment de Vraimont
111
solution est probablement due à la solubilisation de carbonate de calcium par H3PO4. En
effet il est connu qu’à pH 5 les carbonates tel que la calcite peuvent se dissoudre. Le Ca qui
s’est solubilisé semble réagir puis précipiter par la suite au fur à mesure que le temps de
contact augmente. Son profil de consommation est identique à celui du P, sa cinétique est
rapide les deux premières minutes puis elle diminue. En sortie du réacteur, la concentration
de Ca restante varie de 27.61 à 79.33mmoles/kg de sédiment humide phosphaté entre 3 et
7%.
Nous pouvons penser que deux phénomènes se produisent sous l’action de l’acide, la
solubilisation du Ca suivie de sa précipitation. Le suivi de la consommation du Ca qui n’a
pas encore réagit se fera dans l’étape de maturation que nous allons aborder plus tard.
III.3.1.1.4. Evolution d’autres éléments
Les concentrations des autres éléments tels que Cd, Co, Cr, Cu, As, Zn, Fe, Al, et le Pb
dans les solutions provenant du sédiment traité à H3PO4 ont également été mesurées en
fonction du temps de passage dans le réacteur. Nous avons constaté qu’en dehors du Zn et
du Fe, aucun des autres éléments n’a été détecté dans le filtrat avec les moyens d’analyse
utilisés (ICP-AES).
Les mesures de la concentration du Zn et du Fe dans la solution de lavage du sédiment
brut montrent que leurs concentrations respective est 0.03 et 0.01 mmol/kg de sédiment
humide. Ces résultats montrent que la solubilité dans l’eau de ces deux éléments est faible,
néanmoins après l’ajout de l’acide phosphorique, les concentrations de ces éléments en
solution au premier prélèvement, sont plus importantes (Figure III- 11). Cela signifie que
l’acide phosphorique permet de solubiliser le Fe et le Zn présents dans le sédiment.
On remarque également sur la Figure III- 11 que concernant le Zn, les concentrations
mesurées dans les solutions du premier prélèvement au dernier prélèvement (22.5min) sont
faibles est varient en moyenne de 0.11 à 0.03, de 0.20 à 0.05 et de 0.37 à 0.08 mmol/kg de
sédiment humide respectivement pour les trois taux de phosphatation 3, 5 et 7%.
Le Fe par contre se trouve en concentration plus importante, sa concentration varie de
8.38 à 1.90, de 17.73 à 6.32, et de 31.5 à 9.38 mmol/kg de sédiment humide respectivement
pour les trois taux de phosphatation 3, 5 et 7% sur sec.
En conclusion, les résultats obtenues montrent que dans un premier lieu, ces deux
éléments se solubilisent sous l’effet de l’acide, cette solubilisation est proportionnelle à la
quantité d’acide ajoutée ensuite ils précipitent. Nous suivrons l’évolution de ces deux
éléments dans l’étape de maturation dans les paragraphes qui suivent.
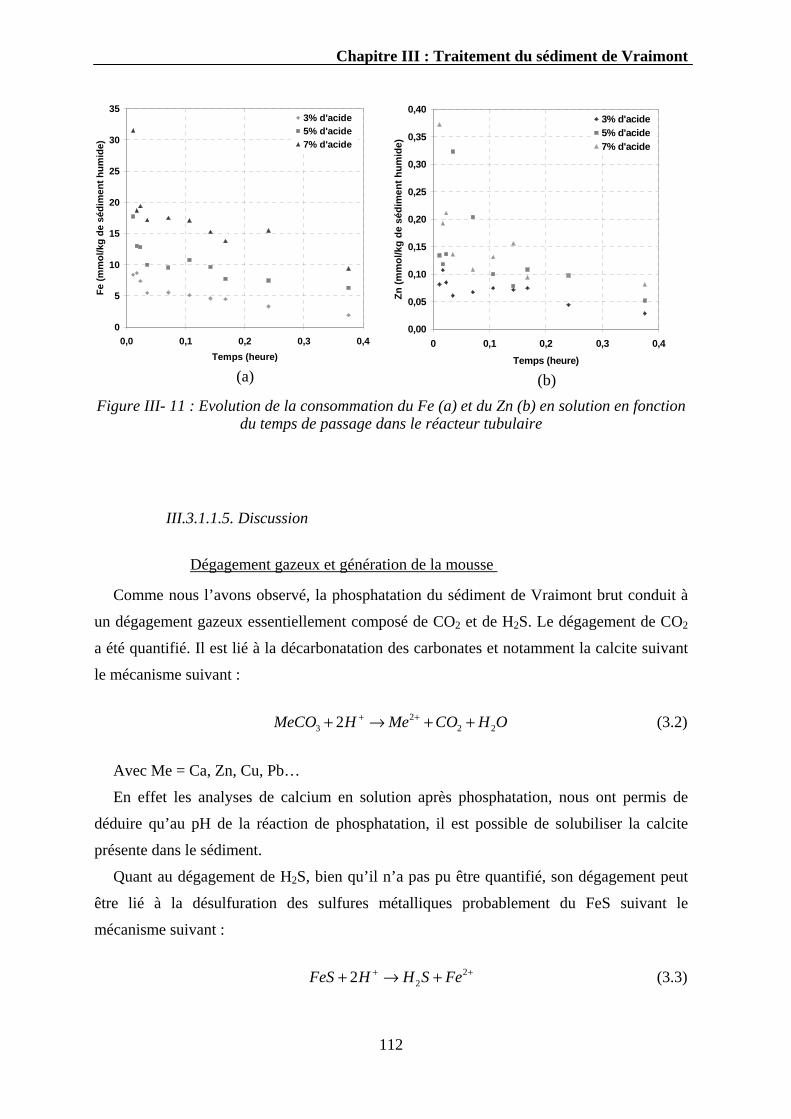
Chapitre III : Traitement du sédiment de Vraimont
112
(a) (b) Figure III- 11 : Evolution de la consommation du Fe (a) et du Zn (b) en solution en fonction
du temps de passage dans le réacteur tubulaire
III.3.1.1.5. Discussion
Dégagement gazeux et génération de la mousse
Comme nous l’avons observé, la phosphatation du sédiment de Vraimont brut conduit à
un dégagement gazeux essentiellement composé de CO2 et de H2S. Le dégagement de CO2
a été quantifié. Il est lié à la décarbonatation des carbonates et notamment la calcite suivant
le mécanisme suivant :
2
3 2 22MeCO H Me CO H O+ ++ → + + (3.2)
Avec Me = Ca, Zn, Cu, Pb…
En effet les analyses de calcium en solution après phosphatation, nous ont permis de
déduire qu’au pH de la réaction de phosphatation, il est possible de solubiliser la calcite
présente dans le sédiment.
Quant au dégagement de H2S, bien qu’il n’a pas pu être quantifié, son dégagement peut
être lié à la désulfuration des sulfures métalliques probablement du FeS suivant le
mécanisme suivant :
2
22FeS H H S Fe+ ++ → + (3.3)
0
5
10
15
20
25
30
35
0,0 0,1 0,2 0,3 0,4Temps (heure)
Fe (m
mol
/kg
de s
édim
ent h
umid
e)
3% d'acide 5% d'acide 7% d'acide
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0 0,1 0,2 0,3 0,4Temps (heure)
Zn (m
mol
/kg
de s
édim
ent h
umid
e)
3% d'acide 5% d'acide 7% d'acide

Chapitre III : Traitement du sédiment de Vraimont
113
En effet plusieurs auteurs [Gleyzes 2002 ; da Silva 2000] ont démontrés qu’à pH 5, le
FeS peut se dissoudre en provoquant le dégagement de H2S et la solubilisation du Fe. Ce
résultat est en adéquation avec les résultats que nous avons obtenues. En effet, nous avons
montré que l’ajout de l’acide phosphorique au sédiment permet entre autre la solubilisation
du Fe.
Le dégagement gazeux est accompagné d’une formation de bulles qui conduit à un
gonflement du sédiment. La compréhension du processus de création des bulles et de
gonflement du sédiment qui s’en suit n’a pas fait l’objet d’une étude détaillée dans cette
thèse. Une autre thèse en cours dans le laboratoire est consacrée à ces aspects. Par contre,
afin d’expliquer ces phénomènes nous nous sommes référés à des études similaires publiés
dans la littérature. En effet van Kessel et van Kesteren [van Kessel 2002] ont étudié ce genre
de phénomène dans le cas des sédiments stockés à l’air libre. Ils expliquent que lorsque la
matière organique se dégrade, en présence de bactéries et sous des conditions anaérobies,
cette dégradation s’accompagne d’un dégagement gazeux de méthane et de dioxyde de
carbone. Ce dégagement gazeux provoque la création de bulles et par conséquent le
gonflement des sédiments. Ils précisent aussi que le dégagement gazeux ne se fait pas
uniquement dans le cas des sédiments dragués et déposés, il se fait également dans le milieu
naturel des sédiments
Ce cas de figure n’est pas exactement le même que celui que nous observons lors de la
phosphatation des sédiments. En effet, le dégagement gazeux dans le cas de cette étude est
naturel quand à celui observé dans notre cas est dû à l’ajout de l’acide phosphorique.
Néanmoins, dans les deux cas cela se traduit par des dégagement gazeux qui gênèrent des
bulles et par conséquent le gonflement du sédiment.
Les auteurs de cette étude expliquent que quand le gaz émis transite dans la phase
gazeuse, le sédiment peut gonfler, et que le gonflement ou l’expansion du sédiment ne se fait
que lorsque le taux de production de gaz est supérieur au taux de gaz émis. En outre, un
gonflement significatif du sédiment ne se produit, que lorsque les gaz produits sont émis à
l’extérieur (phase gazeuse).
Ils précisent également que l’expansion ne se produit que si les molécules gazeuses qui se
dissolvent dans la phase aqueuse du sédiment transitent vers la phase gaz. Cela n’a lieu que
si la phase liquide est sursaturée en gaz (dans le cas de cette étude le CH4 et le CO2). En
effet, la solubilité des gaz dans une phase aqueuse est donnée par la loi d’Henry
i i ixρ κ= (3.4)

Chapitre III : Traitement du sédiment de Vraimont
114
où ρ est la pression de vapeur partiel du composé i, iκ est la constante d’Henry pour ce
composé et ix représente la fraction molaire de ce composé dans la phase aqueuse.
L’équation (3.4) montre que la pression de vapeur est proportionnelle à la concentration du
gaz dissout. Aussitôt que la pression de vapeur excède la pression du liquide, les bulles de
gaz sont formées. L’équation (3.4) implique également que la solubilité des gaz est
proportionnelle à la pression du liquide.
Quant l’eau interstitielle est sursaturée en gaz, la nucléation des bulles peut se faire, soit
de manière homogène soit de manière hétérogène. Pour que la nucléation homogène se
produise, il faut une grande sursaturation qui constitue la barrière d’énergie (énergie
d’activation) à surmonter pour générer les micro bulles. Cela est occasionné par la tension
de surface du liquide qui limite le processus de nucléation. L’excès de pression
p∆ nécessaire pour dépasser la tension de surface est donné par :
2
prσ
∆ =
où r est le rayon de la bulle et σ la tension de surface de liquide. A partir de cette équation,
il est clair que l’excès de pression (et la sursaturation) augmente avec la diminution de la
taille des bulles. La nucléation peut se produire aussi d’une manière hétérogène, les petites
dislocations sur lesquelles les cavités sont stables, sont toujours présentes sur la surface des
solides. C’est l’endroit idéal pour la nucléation des bulles, ces cavités initiales sous forme de
dislocations ont comme conséquence une forte diminution de la barrière d'énergie. La
nucléation hétérogène peut ainsi se produire à de faible sursaturation.
Après la nucléation, les bulles commencent à croître grâce à la diffusion du gaz dissout à
travers les bulles. Etant donnée que la diffusion est rapide et a lieu uniquement sur des
distances courtes, un grand nombre de bulles par kilogramme de sédiment sont formés. Le
nombre de bulles formées dépend de la quantité de gaz produit.
La grande différence de densité qui existe entre les bulles de gaz et le sédiment, conduit à
l’élévation des bulles et leur échappement. Cette élévation ne se fait que lorsque le rayon des
bulles atteint une valeur critique. Le départ de bulles de l’intérieur du sédiment vers
l’extérieur provoque la formation de canaux au sein du sédiment qui persistent pendant le
séchage.
Ce phénomène de création de bulle entraîne donc une modification de la texture du
sédiment en générant la formation de bulles. Cette évolution physique a des conséquences
sur la viscosité du sédiment notamment.
En mesurant la viscosité du sédiment en fonction de la quantité d’acide, nous avons
constaté une diminution de la viscosité en fonction de la quantité d’acide ajouté. Cette

Chapitre III : Traitement du sédiment de Vraimont
115
tendance diminue avec l’augmentation du taux d’humidité du sédiment. En effet, d’après les
explications données par van Kessel et van Kesteren, pour qu’il y est formation de bulle il
faut que la phase aqueuse du sédiment soit sursaturée en gaz. Or plus le taux d’humidité
dans le sédiment est important plus la concentration en gaz dissout dans la phase liquide doit
être importante pour atteindre la sursaturation nécessaire à la nucléation des bulles. C’est
entre autre pour cette raison que la viscosité des sédiments phosphatés à différents taux de
phosphatation n’est différente que lorsque le taux d’humidité est le plus faible. Cela
implique que la phase aqueuse se sursature plus facilement conduisant ainsi la formation des
bulles qui vont conduire à la diminution de la viscosité des sédiments.
L’apparition des bulles entraîne la formation de sillage dans le sédiment initialement
rhéofluidifiant. Ces sillages baissent le nombre de zone de contact entre « les films »
constituant le sédiment qui sont donc moins solidaires de l’ensemble et donc s’écoulent plus
facilement sous l’action d’une sollicitation quelconque. C’est ce qui explique la baisse de la
viscosité pour les sédiment à faible taux d’humidité.
Cinétique de phosphatation des sédiments en réacteur
L’étude de la cinétique de phosphatation des sédiments a permis de monter que lors de
l’ajout de l’acide phosphorique, deux réactions se produisent : la solubilisation du Ca, Zn et
Fe et la précipitation de ces mêmes éléments ainsi que celle du P.
L’ajout de l’acide phosphorique au sédiment provoque une diminution du pH des
sédiments (7.2 à 5.1 en moyenne pour les trois taux de phosphatation) qui conduit donc à la
solubilisation d’éléments tels que le Ca, le Fe et le Zn.
A ce pH, la solubilisation du Ca est probablement due à la dissolution de la calcite. En
effet nous avons montré dans le chapitre précédent l’existence de cette phase dans le
sédiment brut. La dissolution de cette phase s’accompagne également par le dégagement de
CO2 comme il a été précisé dans le paragraphe précédant. En comparant le nombre de mole
de CO2 dégagé (281mmol) au nombre de mole de Ca solubilisé (134mmol) pour une
phosphatation de 7%. Nous constatons que la calcite n’est pas le seul carbonate qui s’est
dissout car il y a plus de CO2 dégagé que de Ca qui s’est solubilisé. Le ZnCO3, peut être un
des carbonates dissout lors de la phosphatation. En effet, en présence de l’acide
phosphorique le Zn s’est également solubilisé. Sa concentration pour une phosphatation de
7% est de 0.37mmol/kg de sédiment humide. Cette concentration est faible et indique qu’il y
a sûrement d’autres types de carbonates (MnCO3 et MgCO3) qui se sont solubilisés mais que
nous n’avons pas analysés.

Chapitre III : Traitement du sédiment de Vraimont
116
Quant à la solubilisation du Fe, comme nous l’avons déjà expliqué, elle est probablement
due à la solubilisation de FeS qui s’accompagne d’un dégagement de H2S
Concernant les métaux tels que Cd, Co, Cr, Cu, As, Al, et le Pb, leurs absences des
solutions peut être expliques de différentes manières :
• la concentration des métaux dans les solutions est plus faible que le seuil de détection
de l’appareil de mesure (ICP-AES).
• la méthode qui a été choisie pour le mise en solution des métaux après phosphatation
n’est pas adéquate pour mettre en évidence leur évolution avec celle du phosphore.
• les métaux se solubilisent et réagissent rapidement, ce qui rend difficile le suivi de leur
évolution.
• le pH du mélange réactionnel n’est pas suffisamment faible pour mettre ces métaux en
solution. En effet, le pouvoir tampon étant de 1220mmol d’H+/kg de sédiment humide, une
phosphatation jusqu’à 7% d’acide, ne permet pas d’abaisser d’avantage le pH. Par
conséquent, cela ne permet pas la mise en solution de ces espèces. Il aurait peut être fallu
mettre plus d’acide pour permettre leur mise en solution.
La solubilisation de ces éléments se poursuit par une précipitation. Cette précipitation se
manifeste par la consommation du P injecté ainsi que par la consommation du Ca, du Fe et
du Zn solubilisé comme nous l’avons vu sur les courbes cinétiques (Figure III- 10 et Figure
III- 11). Lors de cette étape, le pH des sédiments augmente en moyenne de 5.1 à 5.5 en
sortie du réacteur (22.5min), ce qui se traduit par une consommation de protons.
Nous parlerons tout d’abord de la précipitation du Ca et du P car nous avons constaté que
leurs cinétiques sont identiques. En effet, nous avons tracé sur la Figure III- 12 la
concentration de P en solution en fonction de la concentration de Ca pour les trois taux de
phosphatation. Il est clair qu’il y a une bonne corrélation (en moyenne R2= 0.967) qui existe
entre les deux éléments et qui indique que ces deux éléments ont co-précipité. Pour savoir
sous quelle forme le P et le Ca ont précipité, il est importante de savoir en premier lieu sous
quelle forme le P peut se trouver en solution. Pour cela les différents équilibres de
dissociation de l’acide phosphorique dans l’eau à 25°C ainsi que leur constante d’acidité
sont présentés ci-dessous :
3 4 2 2 4 3 1 2.12H PO H O H PO H O pKa− ++ ↔ + = (3.5) 2
2 4 2 4 3 2 7.21H PO H O HPO H O pKa− − ++ ↔ + = (3.6)
2 34 2 4 3 3 12.7HPO H O PO H O pKa− − ++ ↔ + = (3.7)
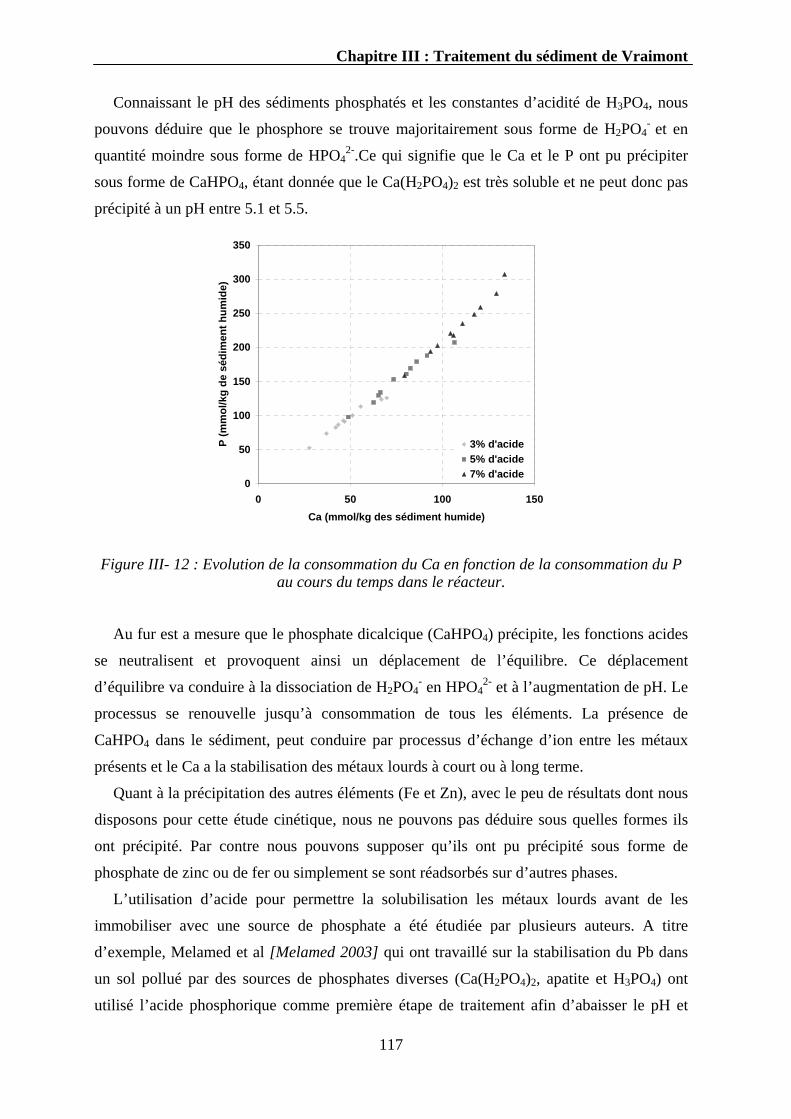
Chapitre III : Traitement du sédiment de Vraimont
117
Connaissant le pH des sédiments phosphatés et les constantes d’acidité de H3PO4, nous
pouvons déduire que le phosphore se trouve majoritairement sous forme de H2PO4- et en
quantité moindre sous forme de HPO42-.Ce qui signifie que le Ca et le P ont pu précipiter
sous forme de CaHPO4, étant donnée que le Ca(H2PO4)2 est très soluble et ne peut donc pas
précipité à un pH entre 5.1 et 5.5.
Figure III- 12 : Evolution de la consommation du Ca en fonction de la consommation du P
au cours du temps dans le réacteur.
Au fur est a mesure que le phosphate dicalcique (CaHPO4) précipite, les fonctions acides
se neutralisent et provoquent ainsi un déplacement de l’équilibre. Ce déplacement
d’équilibre va conduire à la dissociation de H2PO4- en HPO4
2- et à l’augmentation de pH. Le
processus se renouvelle jusqu’à consommation de tous les éléments. La présence de
CaHPO4 dans le sédiment, peut conduire par processus d’échange d’ion entre les métaux
présents et le Ca a la stabilisation des métaux lourds à court ou à long terme.
Quant à la précipitation des autres éléments (Fe et Zn), avec le peu de résultats dont nous
disposons pour cette étude cinétique, nous ne pouvons pas déduire sous quelles formes ils
ont précipité. Par contre nous pouvons supposer qu’ils ont pu précipité sous forme de
phosphate de zinc ou de fer ou simplement se sont réadsorbés sur d’autres phases.
L’utilisation d’acide pour permettre la solubilisation les métaux lourds avant de les
immobiliser avec une source de phosphate a été étudiée par plusieurs auteurs. A titre
d’exemple, Melamed et al [Melamed 2003] qui ont travaillé sur la stabilisation du Pb dans
un sol pollué par des sources de phosphates diverses (Ca(H2PO4)2, apatite et H3PO4) ont
utilisé l’acide phosphorique comme première étape de traitement afin d’abaisser le pH et
0
50
100
150
200
250
300
350
0 50 100 150Ca (mmol/kg des sédiment humide)
P (m
mol
/kg
de s
édim
ent h
umid
e)
3% d'acide 5% d'acide 7% d'acide

Chapitre III : Traitement du sédiment de Vraimont
118
permettre la dissolution du Pb méta stable. Au bout de quelques jours, ils ont introduit dans
le sol le Ca(H2PO4)2, l’apatite naturelle ou l’acide phosphorique afin de stabiliser le pb. En
effet, ils ont réussi a stabiliser plus de 60% du Pb à des pH entre 5.05 et 5.71.
Hettiarachchi et al [Hettiarachchi 2001] quant eux, préconisent l’utilisation de l’acide
acétique dans le sol pollué avant de le traiter avec une source de phosphate.
Dans le chapitre IV, nous essayerons d’expliquer le comportement des métaux lors de la
phosphatation et leur distribution dans le sédiment brut et dans les sédiment phosphatés.
En sortie du réacteur, nous avons constaté que la totalité des éléments (Zn, Fe, P, Ca)
n’avaient pas précipité, notamment le phosphore injecté. En effet, seulement 56.7% du
phosphore injecté a précipité. Pour cette raison nous avons continué à faire des prélèvements
pendant la phase de séchage et de maturation dans le but de suivre le comportement de ces
éléments.
III.3.1.2. La cinétique de maturation
Comme il a été expliqué dans le paragraphe III.2.1.2 (Matériels et méthodes), l’étude de
la cinétique de maturation vise à comprendre le comportement des constituants du sédiment
en sortie de réacteur et lors du séchage à l’air qui s’en suit. Les résultats discutés
précédemment ont montré que 56.7% du phosphore en sortie du réacteur a été consommé et
ceci pour les trois taux de phosphatation. Le temps maximal qui a été choisi pour cette étude
a été de 28 jours, ce qui ne signifie pas qu’il faut 28 jours pour sécher le sédiment, le temps
nécessaire pour cette étape sera abordé plus tard.
Il est important de rappeler que pour cette étude cinétique, nous avons utilisé le même
protocole opératoire que celui utilisé pour le lavage des sédiments lors de la phosphatation
en réacteur.
Ce qui est notable dans cette étape c’est l’évolution de la structure du sédiment en cours
de séchage. En effet, comme nous l’avons déjà dit, la phosphatation du sédiment se traduit
par la formation d’une mousse. Sous l’effet des bulles de gaz qui tendent à s’échapper du
sédiment, la mousse tombe et conduit à la formation de pores dans la structure du sédiment.
III.3.1.2.1. Evolution du pH
L’analyse de l’évolution du pH mesuré dans les solutions de lavage des sédiments en
fonction du temps de maturation est représentée sur la Figure III- 13, Cette figure comme
celles qui seront présentées plus tard regroupent les résultats obtenus dans le réacteur et en
phase de maturation.
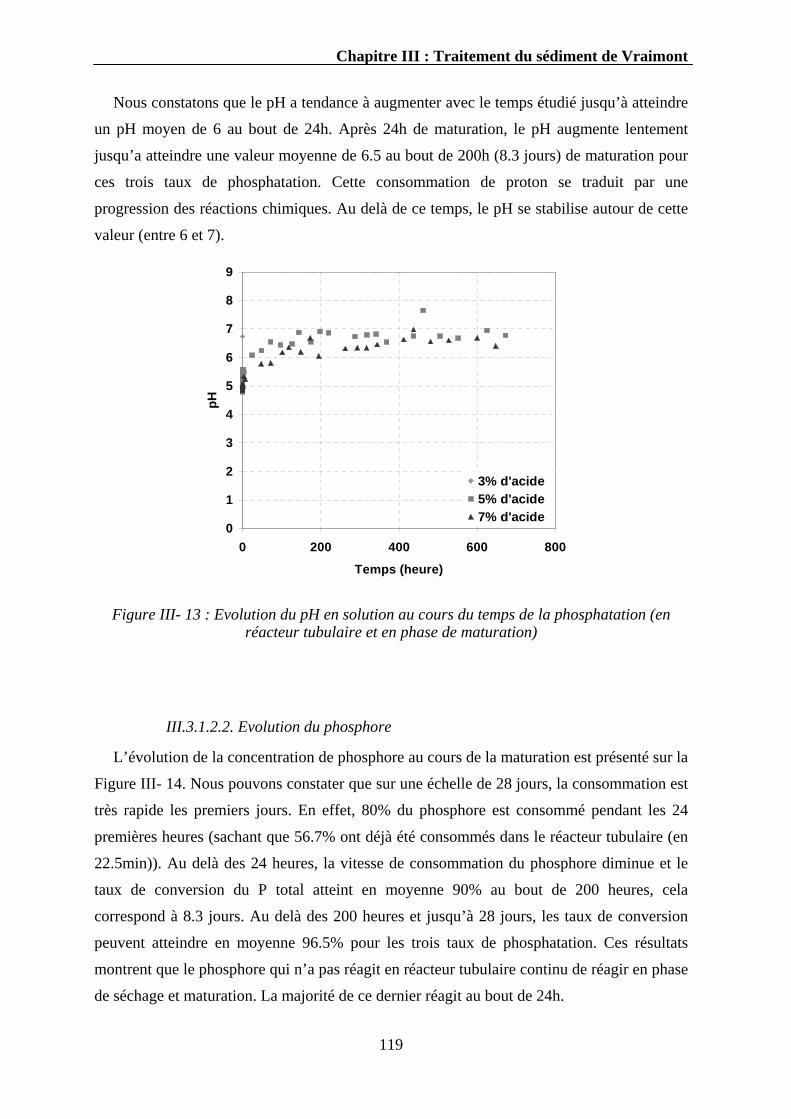
Chapitre III : Traitement du sédiment de Vraimont
119
Nous constatons que le pH a tendance à augmenter avec le temps étudié jusqu’à atteindre
un pH moyen de 6 au bout de 24h. Après 24h de maturation, le pH augmente lentement
jusqu’a atteindre une valeur moyenne de 6.5 au bout de 200h (8.3 jours) de maturation pour
ces trois taux de phosphatation. Cette consommation de proton se traduit par une
progression des réactions chimiques. Au delà de ce temps, le pH se stabilise autour de cette
valeur (entre 6 et 7).
Figure III- 13 : Evolution du pH en solution au cours du temps de la phosphatation (en
réacteur tubulaire et en phase de maturation)
III.3.1.2.2. Evolution du phosphore
L’évolution de la concentration de phosphore au cours de la maturation est présenté sur la
Figure III- 14. Nous pouvons constater que sur une échelle de 28 jours, la consommation est
très rapide les premiers jours. En effet, 80% du phosphore est consommé pendant les 24
premières heures (sachant que 56.7% ont déjà été consommés dans le réacteur tubulaire (en
22.5min)). Au delà des 24 heures, la vitesse de consommation du phosphore diminue et le
taux de conversion du P total atteint en moyenne 90% au bout de 200 heures, cela
correspond à 8.3 jours. Au delà des 200 heures et jusqu’à 28 jours, les taux de conversion
peuvent atteindre en moyenne 96.5% pour les trois taux de phosphatation. Ces résultats
montrent que le phosphore qui n’a pas réagit en réacteur tubulaire continu de réagir en phase
de séchage et maturation. La majorité de ce dernier réagit au bout de 24h.
0
1
2
3
4
5
6
7
8
9
0 200 400 600 800
Temps (heure)
pH
3% d'acide5% d'acide 7% d'acide
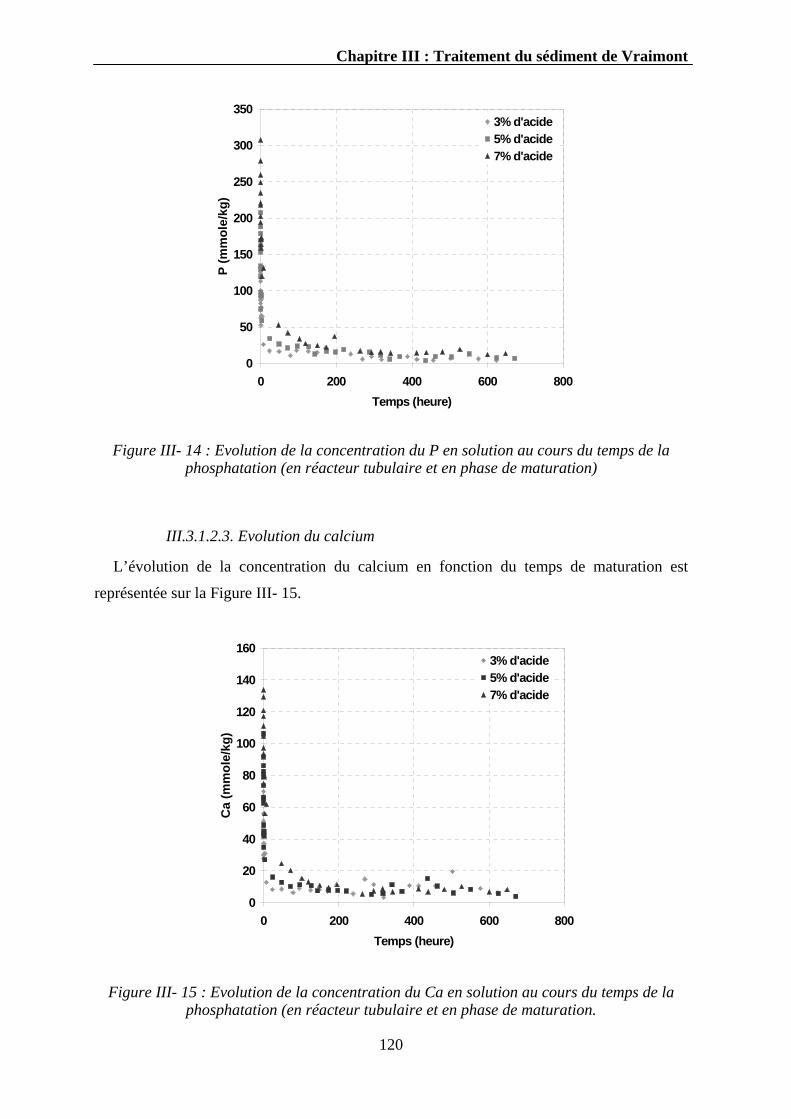
Chapitre III : Traitement du sédiment de Vraimont
120
Figure III- 14 : Evolution de la concentration du P en solution au cours du temps de la
phosphatation (en réacteur tubulaire et en phase de maturation)
III.3.1.2.3. Evolution du calcium
L’évolution de la concentration du calcium en fonction du temps de maturation est
représentée sur la Figure III- 15.
Figure III- 15 : Evolution de la concentration du Ca en solution au cours du temps de la phosphatation (en réacteur tubulaire et en phase de maturation.
0
50
100
150
200
250
300
350
0 200 400 600 800Temps (heure)
P (m
mol
e/kg
)
3% d'acide5% d'acide 7% d'acide
0
20
40
60
80
100
120
140
160
0 200 400 600 800Temps (heure)
Ca
(mm
ole/
kg)
3% d'acide5% d'acide7% d'acide

Chapitre III : Traitement du sédiment de Vraimont
121
Nous pouvons constater que cette évolution est identique à celle du P. Le Ca réagit
rapidement les premières 24h, puis son évolution décroît progressivement et au bout de 28
jours, il ne reste en solution qu’entre 5.70 et 8.1mmoles de Ca/kg de sédiment humide
phosphaté respectivement entre 3 et 7%. Là encore, nous pouvons constater que le Ca qui
n’a pas réagit en réacteur tubulaire, poursuit sa réaction lors de la phase de séchage et de
maturation.
III.3.1.2.4. Evolution des autres éléments
Comme pour la phosphatation en réacteur aucun des éléments chimiques tels que Cd, Co,
Cr, Cu, As, Al, et le Pb ne sont mesurés dans la solution en concentration détectable lors de
la maturation.
Concernant le Zn, les concentrations mesurées dans les solutions prélevées varient de
0.03 à 0.01, de 0.05 à 0.01 et de 0.08 à 0.01 mmol/kg de sédiment humide respectivement
pour les 3 taux de phosphatation 3, 5 et 7%. .
Le Fe par contre varie de 3.69 à 0.01, de 6.32 à 0.01, et de 13.51 à 0.01 mmol/kg de
sédiment humide respectivement pour les 3 taux de phosphatation 3, 5 et 7%.
Nous pouvons constater qu’au bout des 28 jours de maturation, les concentrations du Zn
et du Fe sont de 0.01 mmol/kg de sédiment humide quelque soit la quantité d’acide. Cela
implique que l’essentiel de ces éléments a précipité lors de la maturation.
III.3.1.2.5. Discussion
En sortie du réacteur, les sédiments vont être disposés dans des récipients en
polyéthylène, et laissés à l’air jusqu’à ce qu’ils sèchent, une fois que le taux d’humidité
minimal atteint, ils seront calcinés. Lors du séchage à des conditions ambiantes, la réaction
de phosphatation va se poursuivre c’est ce que nous avons appelé l’étape de maturation.
Il parait clair à partir de l’étude cinétique de maturation que les réactions de précipitation
des éléments (Ca, P, Fe, Zn) se poursuivent. Leurs cinétiques sont rapides les premières 24h,
puis elles ralentissent progressivement. En effet, dans le cas du P, le taux de conversion
atteint les 80% en moyenne au bout de 24h. Sa consommation ralentit par la suite, et son
taux de conversion n’atteint les 90% qu’au bout de 8.3 jours. Au bout de 28 jours, en
moyenne 96.5% du P est consommé.
La précipitation du P comme il a été indique lors de l’étude de la phosphatation en
réacteur, se fait sous forme de CaHPO4, il est d’autant plus vrai que la cinétique de
précipitation du Ca lors maturation est aussi identique à celle du P. Nous pouvons également
constater que le pH des solutions augmente au fur est à mesure que le P précipite. En effet,

Chapitre III : Traitement du sédiment de Vraimont
122
la cinétique de consommation des protons est identique a celle de consommation du P. Il
n’est pas exclu qu’à des pH entre 6 et 7, une faible concentration des ions PO43- soit présente
et favorise la formation d’hydroxyapatites (Ca10(PO4)6(OH)2 ou de calcium tricalcique
Ca3(PO4)2.
Les analyses en maturation du Zn et du Fe montrent également la même tendance, la
poursuite de la précipitation de ces deux éléments jusqu’à épuisement quasi total. Au bout
de 28 jours, il reste en solution seulement 0.01mmol de Zn et de Fe pour un kilogramme de
sédiment. A ce stade des résultats, nous ne savons pas sous quelle formes ces deux métaux
ont pu précipiter.
Certains auteurs qui travaillent sur le traitement des sols avec des sources de phosphate,
n’utilisent pas un réacteur pour la réaction. Ils injectent la source de phosphate au sol et
laisse réagir ou mûrir un certains temps pour permettre la stabilisation des métaux lourds.
A tire d’exemple, Melamed et al [Melamed 2003] utilisent (comme il a été cité plus haut)
différentes sources de phosphate pour la stabilisation du Pb dans un sol pollué. Leur
démarche consiste uniquement à injecter la source de phosphate dans le sol et laisser réagir
pendant 220 jours (après avoir ajouté de l’acide phosphorique pour solubiliser le Pb méta
stable). Ils ont démontré qu’à partir d’une source de phosphates tel Ca(H2PO4)2 ou le H3PO4
ils pouvaient stabiliser jusqu’à 60% du Pb sous forme de chloropyromorphite [Pb5(PO4)3Cl]
Pour stabiliser du Cd, du Cu, du Ni, du Pb et du Zn dans un sol pollué. Wang et al [Wang
2001] ont utilisé différentes sources de phosphate tels que Ca(H2PO4)2 combiné avec le
CaCO3, le CaHPO4 seul ou combiné avec le CaCO3, le Na2HPO4, et le MgHPO4. Ils ont
mélange pendant 10min le sol avec les différents sources de phosphate et ont laissé le tout
séché pendant 30 jours. Les résultats des tests de lixiviation TCLP (toxicity characteristic
leaching procedure) effectuées au termes des différents temps indiqués ont bien montrées
que les sels de phosphate permettent de stabiliser les métaux lourds extractibles, par contre
ils ne précisent pas sous quelles formes ils sont stabilisés.
Theodoratos et al [Theodoratos 2002] ont aussi utilisé le phosphate monobasique pour le
traitement d’un sol pollué par le Pb, Cd et As. Pour cela, ils ont simplement mélangé le sol
avec différents quantité de phosphate, et laissé agir deux mois. Au terme des deux mois, ils
ont appliqué entre autre la lixiviation par TCLP. Ils ont constaté que le phosphate
monobasique stabiliser efficacement le Pb et le Cd, leur valeurs sont en dessous des seuils
de toxicité TCLP. L’effet inverse se produisait par contre pour l’As due à la similitude qui
peut y avoir entre le phosphate et les ions arsenate (formation de Pb2(ASO4)Cl soluble).
Ces quelques exemples nous permettent de déduire deux informations importantes :

Chapitre III : Traitement du sédiment de Vraimont
123
• La maturation permet effectivement de stabiliser les métaux lourds. Il n’est peut être pas
nécessaire d’utiliser un réacteur pour effectuer la réaction de phosphatation. l’utilisation
d’un mélangeur (sediment-H3PO4) avant maturation serait peut être suffisant.
• Le phosphate dicalcique qui a précipité lors de la phosphatation, peut conduire à la
stabilisation des métaux lourds par des phénomène d’échange d’ions entre le Ca et les
métaux lourds.
Les métaux tels que le Cd, Co, Cr, Cu, As, Al, et le Pb n’ont pas été détectés en solution
lors de la maturation ceci bien qu’il soit connu que l’aération des sédiments conduit à
l’augmentation de la mobilité des métaux lourds. En effet le passage des conditions
anaérobies (réductrice) en conditions aérobies (oxydante) des sédiments conduit à
l’augmentation de la mobilité des métaux [Lee 1997 ; De Carvalho 1998 ; Alastuey 1999 ;
Tack 1999]. Parmi les raisons cités, l’oxydation des sulfure en sulfates qui peut conduire à la
solubilisation des métaux lourds qui leur sont associés [Tack 1997]. De la même sorte,
l’oxydation du la matière organique peut conduire à la libération des métaux lourds qui sont
complexés [Lau 1999]. Cependant il a été démontré dans certain cas que les métaux lourds
qui se solubilisent suite au changement de potentiel redox peuvent se réadsorber sur les
phases présentes dans les sédiments [Caille 2003]. Nous pouvons donc supposer, bien que le
potentiel redox du sédiment brut et des sédiments phosphatés durant la maturation n’a pas
été mesuré, que ces métaux ont pu se solubiliser puis se réadsorber sur d’autres phase ou
précipités par la suite sous forme de phosphates.
III.3.1.3. Synthèse
Le traitement chimique à l’acide phosphorique des sédiments de Vraimont à conduit à :
• La modification de la texture du sédiment. En effet l’acide solubilise les sulfures et les
carbonates présents dans le sédiment conduisant ainsi à un dégagement de H2S et de CO2.
Ces dégagements gazeux provoquent la formation de bulles qui conduisent à un gonflement
du sédiment et par conséquent à une modification de sa viscosité et de sa texture. Cette
modification dépend de la quantité d’acide ajouté et du taux d’humidité du sédiment. Pour
un même taux d’acide, plus le taux d’humidité est faible, plus le gonflement du sédiment est
important.
• La solubilisation des éléments tels que le Ca, le Zn et le Fe. En effet, pour un pH de
l’ordre de 5, il est probable que le CaCO3, le ZnCO3 ainsi que le FeS se serait dissouts. Part
contre, les autres éléments tels que Cd, Co, Cr, Cu, As, Al, et le Pb n’ont pas été en
concentration détectable en solution. Différentes raisons peuvent être à la base de ce résultat.
D’une part la méthode utilisée pour les mesurer n’est pas adéquate. D’autre part la forme

Chapitre III : Traitement du sédiment de Vraimont
124
chimique sous lesquelles ils se trouvent n’est pas soluble à des pH autour de 5, pour cela une
quantité plus importante d’acide aurait probablement conduit à leur solubilisation avant
stabilisation. Il est également possible que ces éléments se sont solubilisés et ont réagit
d’une manière très rapide
• La précipitation des éléments qui se sont solubilisés (Ca, Zn et Fe) ainsi que celle du P
injecté. En comparant les constantes d’acidité de l’acide phosphorique et en prenant en
compte le pH du milieu, il est clair que le Ca et le P ont co-précipité en formant un
phosphate dicalcique et probablement à des concentration faible de phosphate tricalcique et
d’hydoxyapatite lors de la maturation. Les phosphates de calcium précipités dans le
sédiment peuvent conduire à la stabilisation des métaux lourds par un mécanisme d’échange
d’ion. Quant au Zn et au Fe, nous pensons qu’ils ont pu précipiter sous forme de phosphates
de zinc et de fer ou se sont réadsorbés sous d’autre formes.
Cette étude a également permis de monter que la précipitation du P ainsi que des autres
éléments qui ont été solubilisés ne se termine pas à la sortie du réacteur tubulaire, elle se
poursuit pendant l’étape de séchage et de maturation. Un temps de maturation de 8.3 jours a
conduit à la consommation de 90% du phosphore injecté.
Une évaluation de la phosphatation sera présentée dans le chapitre IV afin de savoir si la
stabilisation des métaux lourds est effective.
II.3.2. Séchage des sédiments
Avant de présenter les résultats obtenus en séchage, des notions de séchage sont
présentées afin de mieux comprendre les phénomènes qui se passent lors de cette étape.
III.3.2.1. Notion de séchage
III.3.2.1.1. Définition du séchage
C’est l’opération unitaire ayant pour but d’éliminer par évaporation un liquide
imprégnant un solide ou un autre liquide. Le terme déshydratation a un sens plus restrictif :
il ne concerne que l’élimination de l’eau dans un solide ou dans un liquide [Charreau 1991].
III.3.2.1.2. Humidité et taux d’humidité
Ce terme désigne le liquide contenu dans le corps solide, liquide ou pâteux, et devant être
éliminé au cours du séchage. Le taux d’humidité est la masse de liquide contenue par unité
de masse de matière à sécher, Charreau et Cavaillé précisent que bien qu’il soit très souvent
fait référence à la matière humide, il est préférable d’exprimer le taux d’humidité par rapport

Chapitre III : Traitement du sédiment de Vraimont
125
à la matière anhydre (note ns). D’autres auteurs tel que Bonazzi et Bimbenet [Bonazzi 2003]
définissent cette grandeur par la teneur en eau (noté X), elle est exprimé dans les deux cas en
kg d’eau/kg de matière sèche.
III.3.2.1.3. Etat de siccité
Un corps anhydre est un corps dont le taux de d’humidité est nul. Un corps sec ou séché
correspond plus généralement au produit tel qu’il est obtenu à la sortie du sécheur. Dans ce
dernier cas, le taux d’humidité n’est pas forcément nul [Charreau 1991].
III.3.2.1.4. Taux d’humidité à l’équilibre
Un corps humide, placé dans une enceinte de volume important où l’humidité relative et
la température sont constantes, voit son taux d’humidité se stabiliser à une valeur dite
d’équilibre qui dépend de la nature de l’humidité et de celle du produit qui en est imprégné,
mais aussi de la pression partielle de la température [Charreau 1991].
III.3.2.1.5. Isotherme de sorption
C’est la courbe représentant la variation du taux d’humidité à l’équilibre en fonction de
l’humidité relative (HR). L’humidité relative est égale au rapport entre la pression partielle
du solvant et sa pression de vapeur saturante à la température considérée. Les courbes
obtenues en réhydratant le produit sont en général différentes de celles obtenues en le
déshydratant. Il se produit un phénomène d’hystérésis. En matière de séchage ne sont à
considérer que les isothermes obtenues en plaçant le produit dans des atmosphères dont
l’humidité relative est de plus en plus faible [Charreau 1991].
III.3.2.1.6. Température de bulle humide
La température de bulbe humide est la température indiquée par un thermomètre dont le
bulbe est recouvert d'une mince couche d'eau et placée dans un courant d’air
III.3.2.1.7. Mécanismes intervenant au cours du séchage
Mode de séchage
Deux mécanismes peuvent être mis en œuvre pour évaporer l’eau d’un produit :
l’ébullition ou l’entraînement. L’idée la plus simple consiste à porter le produit à la
température d’ébullition de l’eau, qui alors se vaporise. Mais pour obtenir une élimination
poussée de l’eau sans altération excessive de la qualité des produits, on préfère bien souvent
opérer à température plus basse en utilisant l’air comme gaz d’entraînement. Quel que soit le

Chapitre III : Traitement du sédiment de Vraimont
126
mode de séchage, c’est la pression de vapeur de l’eau dans le produit qui détermine les
échanges entre l’air et le produit [Bonazzi 2003]
Le séchage fait appel aux trois modes de transfert de chaleur : convection, conduction et
rayonnement. Dans le cadre de ce travail, le séchage des sédiments va être fait par
convection. Nous nous intéresserons donc qu’à ce type de transfert.
Séchage par convection
En séchage industriel, il s’agit probablement du mode de transfert d’énergie le plus
courant. Il consiste à mettre en contact un gaz (air) s’écoulant en régime généralement
turbulent autour du corps à sécher qui peut se présenter sous forme de particules, de
gouttelettes de film ou de plaque
Ce type de transfert obéit à l’équation suivante :
( )a sQ A T Tα= − (3.8)
avec Q (W) est la quantité de chaleur transférée par unité de temps. A (m2) est la surface
d’échange, Ta-Ts (K) est l’écart entre la température du gaz de séchage et la température
superficielle du produit à sécher et α (W. m2. K-1) est le coefficient d’échange par
convection . L’application au séchage de cette formule implique entre autre que la quantité
d’énergie transférée au produit à sécher est d’autant plus importante que la surface de
transfert est, elle même importante [Charreau 1991].
Différentes étapes du séchage d’un produit humide
Dans les paragraphes qui suivent, il ne sera fait mention que de l’eau, qui est un des
liquides le plus couramment évaporés. Les mécanismes qui vont être décrits d’applique
cependant aux autres solvants.
Obtention des courbes de séchage et interprétation
La courbe de séchage par entraînement d’un produit peut être obtenue en disposant celui-
ci en couche mince (1 a 3cm). On fait traverser celle-ci par un courant d’air chaud dont le
taux d’humidité et la température sont contrôlés et maintenus constants. Le taux d’humidité
du produit à intervalles de temps réguliers ou éventuellement en continu.
En portant le taux d’humidité du produit ou la teneur en eau en fonction du temps t, on
obtient une courbe de séchage dont la forme la plus complète (c’est à dire lorsque toutes les
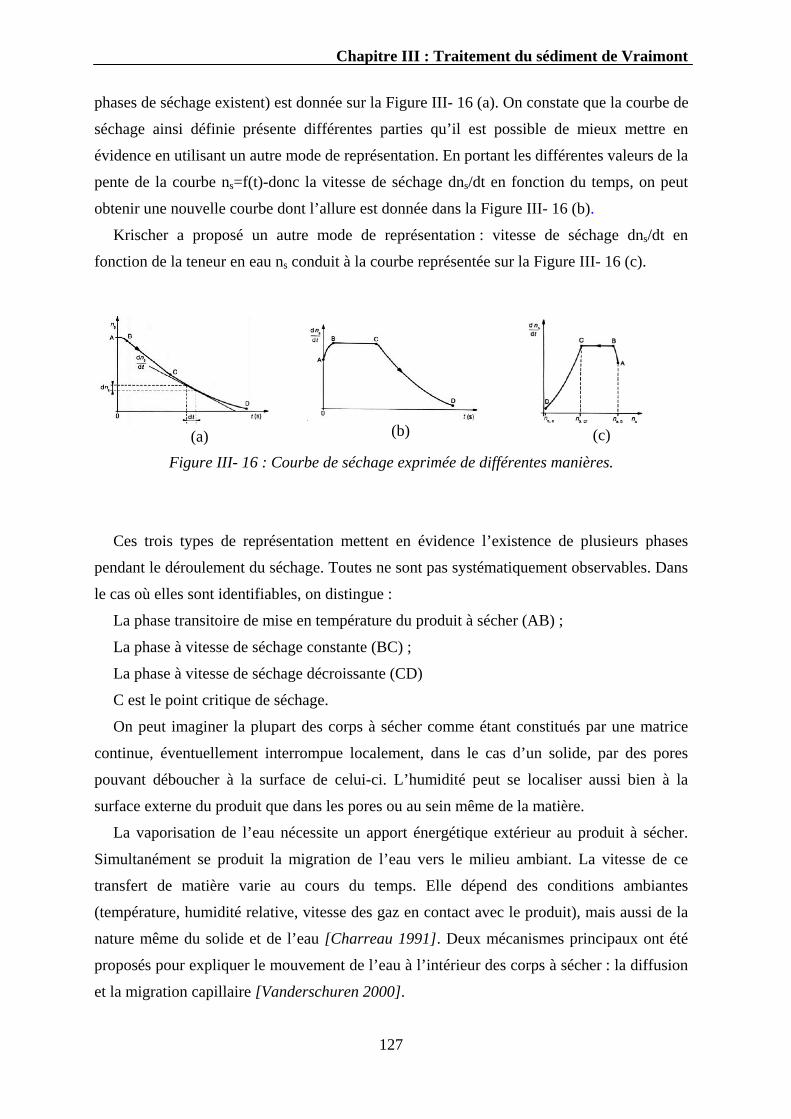
Chapitre III : Traitement du sédiment de Vraimont
127
phases de séchage existent) est donnée sur la Figure III- 16 (a). On constate que la courbe de
séchage ainsi définie présente différentes parties qu’il est possible de mieux mettre en
évidence en utilisant un autre mode de représentation. En portant les différentes valeurs de la
pente de la courbe ns=f(t)-donc la vitesse de séchage dns/dt en fonction du temps, on peut
obtenir une nouvelle courbe dont l’allure est donnée dans la Figure III- 16 (b).
Krischer a proposé un autre mode de représentation : vitesse de séchage dns/dt en
fonction de la teneur en eau ns conduit à la courbe représentée sur la Figure III- 16 (c).
(a) (b) (c)
Figure III- 16 : Courbe de séchage exprimée de différentes manières.
Ces trois types de représentation mettent en évidence l’existence de plusieurs phases
pendant le déroulement du séchage. Toutes ne sont pas systématiquement observables. Dans
le cas où elles sont identifiables, on distingue :
La phase transitoire de mise en température du produit à sécher (AB) ;
La phase à vitesse de séchage constante (BC) ;
La phase à vitesse de séchage décroissante (CD)
C est le point critique de séchage.
On peut imaginer la plupart des corps à sécher comme étant constitués par une matrice
continue, éventuellement interrompue localement, dans le cas d’un solide, par des pores
pouvant déboucher à la surface de celui-ci. L’humidité peut se localiser aussi bien à la
surface externe du produit que dans les pores ou au sein même de la matière.
La vaporisation de l’eau nécessite un apport énergétique extérieur au produit à sécher.
Simultanément se produit la migration de l’eau vers le milieu ambiant. La vitesse de ce
transfert de matière varie au cours du temps. Elle dépend des conditions ambiantes
(température, humidité relative, vitesse des gaz en contact avec le produit), mais aussi de la
nature même du solide et de l’eau [Charreau 1991]. Deux mécanismes principaux ont été
proposés pour expliquer le mouvement de l’eau à l’intérieur des corps à sécher : la diffusion
et la migration capillaire [Vanderschuren 2000].

Chapitre III : Traitement du sédiment de Vraimont
128
Phase transitoire de mise en température du produit à sécher
C’est souvent une période d’échauffement. La température du solide s’ajuste jusqu’au
moment où un état stationnaire est atteint. Cette période est de courte durée et généralement
négligeable [Vanderschuren 2000].
Phase à vitesse de séchage constante
Pendant toute la durée de cette phase, qui se produit jusqu’au point critique, la surface du
produit est saturée en eau non liée. Il s’y forme une couche limite de gaz où la pression de
vapeur d’eau est quasi égale à celle de l’eau pure dans les mêmes conditions de température
et de pression. Cette situation résulte soit de la présence de l’eau en quantité importante à la
surface du produit, soit d’une diffusion suffisamment rapide d’eau, du sein du produit vers
sa surface externe. Cette phase est comparable à l’évaporation d’un liquide. La température
du solide reste constante et égale à la température dite humide du fluide de séchage
(température du bulbe humide). La nature du solide n’intervient pas au cours de cette
période, par contre la forme du solide peut avoir une influence sur la vitesse de séchage (en
kg d’eau.s-1).
Différents paramètres influent sur la durée de la phase à vitesse de séchage constante.
Parmi les facteurs accélérant la vitesse, on peut citer :
• La diminution de la pression de vapeur de l’eau dans l’air, c’est à dire la diminution du
taux d’humidité de l’air de séchage ;
• L’augmentation des coefficients de transfert de la matière, représenté par l’évaporation
de l’eau, et de la chaleur thermique dirigés en sens inverse par l’augmentation de la vitesse
de l’air dans le sécheur ;
• L’accroissement de la surface spécifique : cela peut être obtenu en divisant le plus
possible les solides à traiter avec, cependant des limites : en effet plus les particules sont
petites et plus elles sont susceptibles d’être entraînées par le gaz de séchage ;
• L’élévation de la température du gaz de séchage.
La phase à vitesse constante n’est pas toujours identifiable, même pour des produits dont
l’humidité initiale est très importante (produits végétaux, produits carnés). L’explication
réside dans le fait que les parois cellulaires perturbent la migration rapide de l’humidité vers
la surface des produits.
De nombreux produits ont tendance à se rétracter au cours du séchage. Certains auteurs
ont établi des relations permettant de tenir compte de la variation de la surface [Charreau
1991].

Chapitre III : Traitement du sédiment de Vraimont
129
Phase à vitesse de séchage décroissante
Cette phase succède à la phase à vitesse constante quand cette dernière existe. La teneur
en eau à partir de laquelle la vitesse de séchage se met à décroître est la teneur en eau
critique Xcr qui n’est pas toujours identifiable et, quand elle peut être observée, dépend des
conditions de séchage.
On admet que pendant cette étape de séchage, le transfert de l’eau au sein du produit n’est
plus assez rapide pour saturer la pellicule de gaz entourant celui-ci. Il se crée dans les
produit poreux un front de vaporisation se dirigeant progressivement vers l’intérieur de la
matière à sécher. La vapeur d’eau ayant un chemin de plus en plus important à parcourir, sa
pression à la surface du produit diminue. La différence entre cette pression et la pression de
vapeur d’eau dans le milieu ambiant, c’est à dire le potentiel d’échange, va donc décroître
avec, comme conséquence, la diminution de la vitesse de séchage.
Dans les produits non poreux, la diffusion de l’eau au sein du produit est aussi à l’origine
de la diminution de la vitesse de séchage. De même, la diminution de le diffusivité de l’eau
au fur et à mesure que le produit sèche, ainsi que le croûtage éventuel du produit, peuvent
aussi expliquer la diminution de la vitesse de séchage [Charreau 1991].
La température du produit augmente alors à partir de ça surface. La teneur en eau du
produit diminue jusqu’à atteindre une valeur d’équilibre qui dépend des caractéristique de
l’air de séchage (température et humidité relative) ; neq se déduit de l’isotherme de
désorption (à cette température) [Bonazzi 2003].
III.3.2.2. Résultats
Le séchage est l’étape qui suit celle de la phosphatation, il permet la réduction du taux
d’humidité des sédiments avant la calcination.
Comme il a été indiqué dans le paragraphe II.2.2 (Matériels et méthode), le sédiment de
Vraimont brut ainsi que ceux phosphatés à 3 et a 5% sur sec ont subi un séchage convectif
dans une veine de séchage où les conditions de séchage ont été contrôlées et choisies
proches des conditions atmosphériques, c’est a dire à température de 25, 35 et 45°C, avec
des vitesses de l’air de 0.15, 0.5 et 1.5 m/s ce qui correspond à des brises d’air de 0.54, 1.8 et
5.4km/h. L’humidité relative de l’air a été fixée pour toutes les expériences à 50%, elle
correspond à l’humidité relative moyenne de l’atmosphère.
Ces expériences vont nous permettre de déterminer les cinétiques de séchage et
l’influence du traitement à l’acide sur cette cinétique de séchage. L’influence de la vitesse de
l’air ainsi que la température de séchage sera également abordée, le but étant d’évaluer
l’influence de ces deux paramètres sur la diminution des temps de séchage. Une réduction

Chapitre III : Traitement du sédiment de Vraimont
130
des temps de séchage permet le passage plus rapide à la troisième étape du procédé
(calcination).
III.3.2.2.1. Cinétique de séchage du sédiment brut
Afin de déterminer la cinétique de séchage du sédiment brut, nous avons séché le
sédiment à 45°C avec de l’air circulant à une vitesse de 1.5m/s avec un taux d’humidité
relative de 50%. La disposition de l’échantillon à sécher sur une balance, elle même reliée à
un système d’acquisition, nous permet de connaître en continu la perte de masse en fonction
du temps de séchage. A partir de ces données et de la connaissance de la teneur en eau dans
le sédiment brut nous pouvons déduire la teneur en eau dans l’échantillon au cours de
l’expérience de séchage. Cela permet ensuite de déterminer la cinétique de séchage en
représentant l’évolution de la teneur en eau en fonction du temps de séchage. Le sédiment
qui a été utilisé pour toutes les expériences est le sédiment de Vraimont qui a une teneur en
eau de 0.733 ± 0.017 kg d’eau/kg de matière sèche.
La Figure III- 17 représente l’évolution de la teneur en eau dans le sédiment brut en
fonction du temps. Cette figure montre bien que le séchage du sédiment de Vraimont brut se
fait en trois phases comme décrit dans les paragraphes précédents. Il s’agit de la mise en
température (qui n’est pas très claire sur cette figure mais qui est plus visible sur la Figure
III- 18 qui représente l’évolution de la température du produit), la phase de séchage à vitesse
constante appelée également phase stationnaire [Bonnazzi 2003] et la phase de
ralentissement qui se traduit par un changement de pente. Au regard des résultats présentés
sur cette courbe, nous pouvons déjà déduire que la teneur en eau critique se trouve entre
0.15 et 0.20 kg d’eau/kg de matière sèche.
Afin de bien délimiter la phase stationnaire de la phase de ralentissement et connaître
ainsi la teneur en eau critique, les évolutions de la température du produit ainsi que de celle
du bulbe humide en fonction du temps de séchage sont représentées sur la Figure III- 18.
La teneur en eau critique correspond au temps à partir duquel la température du produit
augmente et n’évolue plus de la même manière que celle du bulbe humide. Nous pouvons
constater sur ces deux courbes que la température du produit augmente à 36h de séchage, ce
qui correspond à une teneur en eau critique de à 0.183 kg d’eau/kg de matière sèche.
La courbe cinétique nous permet également de constater qu’il faut plus de 53.65h pour
sécher le sédiment de Vraimont brut au delà d’une teneur en eau de 0.066 kg d’eau/kg de
matière sèche dans des conditions dans lesquelles nous avons travaillé.

Chapitre III : Traitement du sédiment de Vraimont
131
Figure III- 17 : Courbe de séchage du sédiment brut (T = 45°C - HR=50% - v=1.5m/s).
Figure III- 18 : Evolution lors du séchage du sédiment brut de la température du bulbe humide et du produit (T = 45°C - HR=50% - v=1.5m/s)
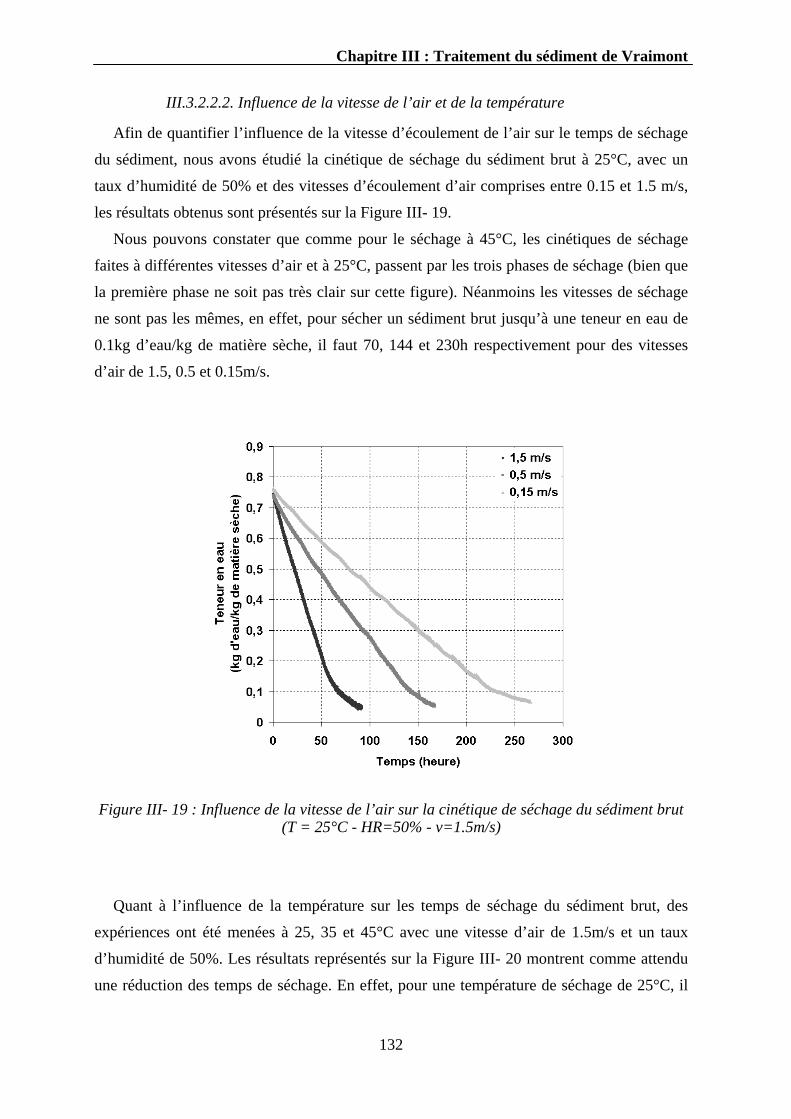
Chapitre III : Traitement du sédiment de Vraimont
132
III.3.2.2.2. Influence de la vitesse de l’air et de la température
Afin de quantifier l’influence de la vitesse d’écoulement de l’air sur le temps de séchage
du sédiment, nous avons étudié la cinétique de séchage du sédiment brut à 25°C, avec un
taux d’humidité de 50% et des vitesses d’écoulement d’air comprises entre 0.15 et 1.5 m/s,
les résultats obtenus sont présentés sur la Figure III- 19.
Nous pouvons constater que comme pour le séchage à 45°C, les cinétiques de séchage
faites à différentes vitesses d’air et à 25°C, passent par les trois phases de séchage (bien que
la première phase ne soit pas très clair sur cette figure). Néanmoins les vitesses de séchage
ne sont pas les mêmes, en effet, pour sécher un sédiment brut jusqu’à une teneur en eau de
0.1kg d’eau/kg de matière sèche, il faut 70, 144 et 230h respectivement pour des vitesses
d’air de 1.5, 0.5 et 0.15m/s.
Figure III- 19 : Influence de la vitesse de l’air sur la cinétique de séchage du sédiment brut
(T = 25°C - HR=50% - v=1.5m/s)
Quant à l’influence de la température sur les temps de séchage du sédiment brut, des
expériences ont été menées à 25, 35 et 45°C avec une vitesse d’air de 1.5m/s et un taux
d’humidité de 50%. Les résultats représentés sur la Figure III- 20 montrent comme attendu
une réduction des temps de séchage. En effet, pour une température de séchage de 25°C, il

Chapitre III : Traitement du sédiment de Vraimont
133
faut 70h pour sécher le sédiment jusqu’à une teneur en eau de 0.1kg d’eau/kg de matière
sèche, contre 54.2h pour une température de 35°C et 45.3h pour une température de 45°C.
A travers l’étude de l’influence de ces deux paramètres sur le temps de séchage du
sédiment, nous pouvons constater que pour sécher le sédiment jusqu’à une teneur en eau de
0.1kg d’eau/kg de matière sèche, il y un gain de temps de séchage de 160h ce qui correspond
à 6.66 jours pour une augmentation de vitesse d’air de 0.15 à 1.5m/s. Par contre pour une
augmentation de la température de séchage de 20°C (de 25 à 45°C), le gain de temps de
séchage est uniquement de 24.7h.
Figure III- 20 : Influence de la température de séchage sur les cinétique de séchage du
sédiment brut ( HR=50% - v=1.5m/s)
III.3.2.2.3. Cinétique de séchage des sédiments phosphatés
Afin d’étudier l’influence du traitement à l’acide phosphorique des sédiments sur le
temps de séchage, nous avons utilisé deux sédiments, l’un traité avec 3% d’acide sur sec et
l’autre à 5% sur sec. Le séchage s’est effectué à une température de 45°C, avec une vitesse
d’air de 1.5m/s et un taux d’humidité de 50%. Les cinétiques de séchage de ces deux
sédiments ainsi que celle du sédiment brut effectuées dans les mêmes conditions (présentées
plus haut) sont représentées sur la Figure III- 21. Nous avons également tracé pour les deux

Chapitre III : Traitement du sédiment de Vraimont
134
taux de phosphatation l’évolution de la température de bulbe humide et de la température du
produit en fonction des temps de séchage (Figure III- 22).
Deux remarques importantes peuvent être faites à partir des résultats obtenus :
• La phase de séchage à vitesse constante des sédiments est d’autant plus courte que la
quantité d’acide est importante, cette tendance est plus claire sur les courbes montrent
l’évolution de la température du produit et du bulbe humide. En effet, en comparant les
températures du produit du sédiment brut (présentées plus haut) à celle des sédiments
phosphatés à 3 et 5% sur sec, nous pouvons constater que la température du produit
augmente d’autant plus tôt que la quantité d’acide est importante. En reprenant le temps de
séchage à partir duquel la température augmente et en les superposant à la courbe cinétique,
nous en déduisons que la teneur en eau critique est de 0.183, 0.295 et 0.595kg d’eau/ kg de
matière sèche respectivement pour le sédiment brut, phosphaté à 3% et à 5%.
• Le temps de séchage global diminue avec l’augmentation de la quantité d’acide et ceci
bien que la phase stationnaire des sédiments phosphatés est plus courte que celle du
sédiment brut. En prenant comme repère la teneur en eau de 0.1kg d’eau/kg de matière
sèche, nous constatons, qu’il faut 45.3h pour sécher le sédiment brut, 38.7h pour le sédiment
phosphaté à 3% et 27.8h pour le sédiment phosphaté à 5%.
Figure III- 21 : Influence de la quantité d’acide sur la cinétique de séchage des sédiments
(T = 45°C - HR=50% - v=1.5m/s)
Chapitre III : Traitement du sédiment de Vraimont
135
(a) (b) Figure III- 22 : : Evolution lors du séchage de la température du bulbe humide et du produit
du sédiment phosphaté à 3% (a) et 5% (b) (T = 45°C - HR=50% - v=1.5m/s)
III.3.2.2.4. Influence de la température
Comme pour le sédiment brut, nous avons phosphaté un sédiment à 5% sur sec et nous
l’avons séché à 25, 35 et 45°C respectivement avec une vitesse de l’air de 1.5m/s et un taux
d’humidité de 50%. Les résultats sont présentés sur la Figure III- 23.
Nous pouvons relever comme attendu une diminution des temps de séchage avec
l’augmentation de la température. L’objectif premier étant de quantifier cette diminution,
nous notons donc un temps de séchage de 52h pour une température de séchage de 25°C à
41h pour 35°C et jusqu’à 27.8 pour une température de séchage de 45°C. La différence qui
peut y a voir pour sécher un sédiment phosphaté à 5% entre une température ambiante de
25°C et 45°C est de 24.2 h.

Chapitre III : Traitement du sédiment de Vraimont
136
Figure III- 23 :Influence de la température sur la cinétique de séchage des sédiments
phosphaté à 5% (HR=50% - v=1.5m/s)
III.3.2.2.5. Propriétés physiques des sédiments séchés
La connaissance des propriétés physiques des sédiments avant et après traitement va nous
permettre de connaître l’influence de la quantité d’acide sur le texture du sédiment, comme
nous l’avons vu dans l’étape de phosphatation, le traitement à l’acide phosphorique conduit
à un dégagement gazeux qui lui même conduit à la formation de bulles au sein du sédiment.
Le départ des bulles s’accompagne de la formation de canaux au sein du sédiment. Afin de
quantifier cet effet, la mesure de la surface spécifique a été effectuée sur le sédiment brut et
sur les sédiments phosphatés. Ces informations vont également pouvoir expliquer la
réduction des temps de séchage observée. Les résultats de cette mesure pour le sédiment brut
(0%) ainsi que pour les sédiments phosphatés à 1, 3, 5 et 7% sur sec après séchage sont
rapportés sur la Figure III- 24.
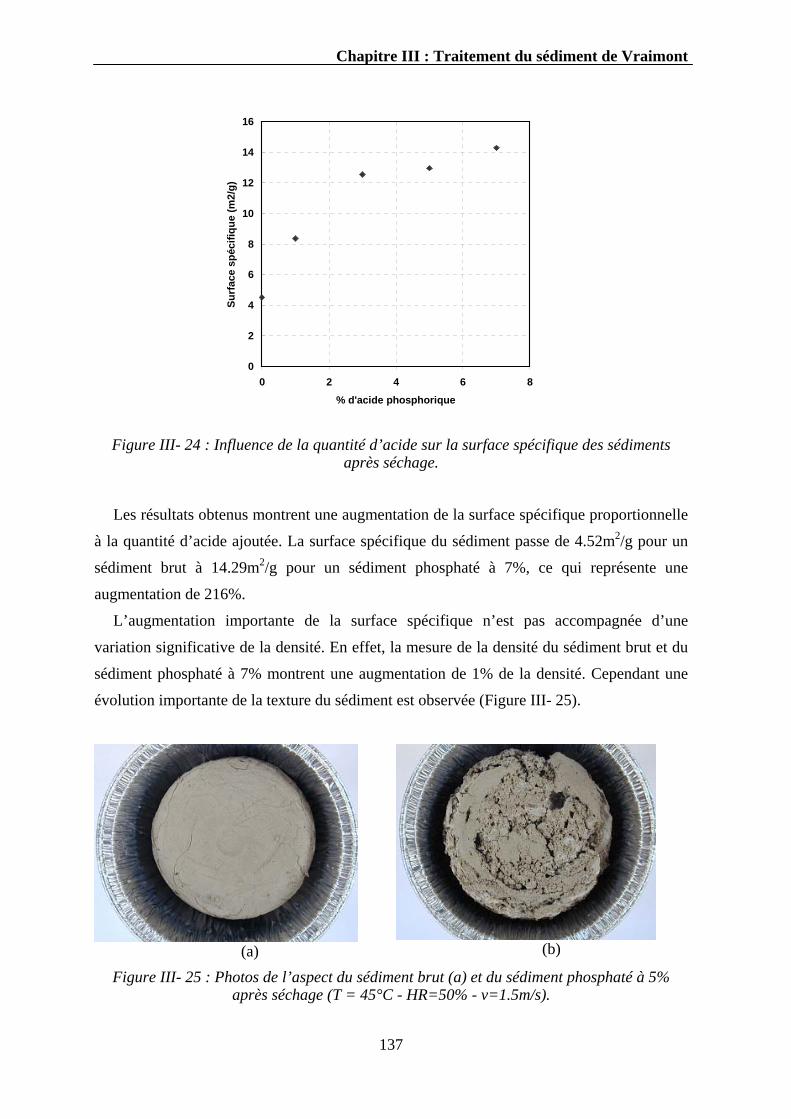
Chapitre III : Traitement du sédiment de Vraimont
137
Figure III- 24 : Influence de la quantité d’acide sur la surface spécifique des sédiments
après séchage.
Les résultats obtenus montrent une augmentation de la surface spécifique proportionnelle
à la quantité d’acide ajoutée. La surface spécifique du sédiment passe de 4.52m2/g pour un
sédiment brut à 14.29m2/g pour un sédiment phosphaté à 7%, ce qui représente une
augmentation de 216%.
L’augmentation importante de la surface spécifique n’est pas accompagnée d’une
variation significative de la densité. En effet, la mesure de la densité du sédiment brut et du
sédiment phosphaté à 7% montrent une augmentation de 1% de la densité. Cependant une
évolution importante de la texture du sédiment est observée (Figure III- 25).
(a) (b)
Figure III- 25 : Photos de l’aspect du sédiment brut (a) et du sédiment phosphaté à 5% après séchage (T = 45°C - HR=50% - v=1.5m/s).
0
2
4
6
8
10
12
14
16
0 2 4 6 8
% d'acide phosphorique
Surf
ace
spéc
ifiqu
e (m
2/g)

Chapitre III : Traitement du sédiment de Vraimont
138
III.3.2.3. Discussion
Les courbes de séchage du sédiment brut (Figure III- 17) et de celles des sédiments
phosphatés à 3 et à 5% (Figure III- 21) sont semblables à celles trouvées dans la littérature.
En effet, les sédiments passent par les trois phases de séchage :
• la phase de mise en température,
• la phase stationnaire, durant laquelle le produit est à température constante et la pression
partielle de la vapeur d’eau au niveau du produit est égale à la pression saturante. Le liquide
s’évapore directement à la surface du matériau. Cette phase est observable tant que la vitesse
de transport de l’eau liquide vers la surface du matériau reste supérieure à la vitesse
d’évaporation de l’eau. Dans ces conditions, ce sont les transferts externes qui gouvernent le
séchage. Il sera donc possible d’augmenter la vitesse de séchage en modifiant par exemple la
vitesse d’écoulement de l’air ou sa température.
• la phase de ralentissement, où la vitesse de transport de l’eau liquide vers la surface du
matériau devient inférieure à la vitesse d’évaporation de l’eau, la température du produit
augmente, la surface du matériaux commence à s’assécher et la vitesse de séchage diminue.
Dans cette phase, ce sont les transferts internes au produit qui gouvernent les échanges.
L’action sur les paramètres externes tels que la vitesse, la température ou l’humidité de l’air
ne permettent pas d’augmenter cette vitesse de séchage. Un des moyens qui pourrait
permettre l’augmentation des temps de séchage est de manipuler le sédiment phosphaté en le
retournant régulièrement. Cela permettrai d’une part de renouveler la surface et ensuite
favoriser le transfert d’humidité vers la surface.
Comme nous l’avons vu sur les courbes de séchage et confirmées par les courbes
montrant l’évolution de la température de bulbe humide et du produit en fonction du temps,
la phase de séchage stationnaire est d’autant plus courte que la quantité d’acide est
importante. D’autre part, l’ajout d’acide favorise une diminution du temps de séchage
pouvant atteindre jusqu’à un tiers du temps pour une phosphatation à 5%. Nous pensons que
ces deux observations sont fortement liées à l’augmentation de la surface spécifique que
nous avons observé lors de la mesure de ce paramètre, en effet une augmentation de la
surface spécifique de 4.52m2/s (sédiment brut) à 12.96m2/s (sédiment phosphaté à 5%) va
engendrer une augmentation du transfert thermique de l’extérieur de l’échantillon vers
l’intérieur favorisant ainsi l’augmentation rapide de la température du produit et par
conséquence la réduction de la phase stationnaire. D’autre part, l’augmentation de la surface
d’échange va conduire à la diminution des temps de séchage en favorisant le transfert de
matière de l’intérieur du produit vers l’extérieur.

Chapitre III : Traitement du sédiment de Vraimont
139
L’étude de l’influence de la température sur le comportement du sédiment brut et
phosphaté à 5% nous a permis de montrer qu’une augmentation de la température de 20°C,
conduit à une réduction des temps de séchage de 24.7h et 24.2h respectivement pour le
sédiment brut et pour le sédiment phosphaté. Cela implique que le temps de séchage n’est
pas fortement influencé par la quantité d’acide rajouté, ceci bien que la phase stationnaire
qui est sensible aux changements de température soit plus courte pour le sédiment
phosphaté.
Compte tenu de la similitude observée dans le comportement des sédiments bruts et
phosphatés face à une augmentation de la température. L’influence de la vitesse de l’air sur
les temps de séchage a également été étudiée. Cette étude n’a été entreprise que pour le
sédiment brut. Ainsi, une augmentation de la vitesse de l’air de 0.15 à 1.5m/s lors du
séchage du sédiment brut a montré une réduction maximum de temps de séchage de 160h.
Toutes les observations faites jusqu’à présent nous conduisent à dire que pour optimiser
le séchage des sédiments, il sera plus économique d’augmenter la vitesse de l’air que
d’augmenter la température. D’autre part, un retournement régulier des échantillons pourrait
également conduire à la réduction des temps de séchage.
Il est important de signaler que les expériences de séchage ont été menées sur 200g
d’échantillon et que le changement d’échelle à plusieurs tonnes de sédiment pourrait
modifier ces tendances. En effet, un accroissement de l’épaisseur de l’échantillon conduira
très certainement à la modification des transferts de chaleur et de matière. Dans ces
conditions, il serait souhaitable à grande échelle d’effectuer le séchage de couche
relativement minces de sédiment. Cela supposera des surfaces importantes de locaux ou un
processus d’alimentation discontinu des bacs de séchage des sédiments.
II.3.3. Calcination des sédiments
La phase de calcination est la dernière étape du procédé NOVOSOL, elle va d’abord
conduire à l’élimination de la matière organique par dégradation thermique. En effet, il est
reconnu qu’un traitement à des température varient de 550 et 600°C est suffisant pour
détruire jusqu’à 99% de la matière organique [Rienks 1998] contenu dans le sédiment.
Par matière organique nous sous-entendons d’aune part, la matière organique naturelle
tels que les substances humiques et fulviques qui peuvent réduire la résistance mécanique
des matériaux de construction à court ou a moyen terme [Rey F 2000]. Ce point est
important car les sédiments de dragage traité sont destinés à une valorisation dans le
domaine du génie civil.

Chapitre III : Traitement du sédiment de Vraimont
140
Dans un second lieu nous visons la matière organique polluante tels que l’hydrocarbures
aromatique polycyclique (HAP), polychlorobiphényle (PCB), furanne et dioxine...qui
peuvent présenter des risques pour la santé humaine et pour l’environnement [Belkessam
1998 ; Heemken 1997 ; Rossi 2004 ; de Pava 2005 ; Mandalakis 2003].
D’autre part, la calcination va permettre la cristallisation des phases présentes dans le
sédiment et notamment les phosphates métalliques qui ont pu être formés lors de l’étape de
phosphatation et de maturation. Cela conduira à l’augmentation de leur insolubilité [Nzihou
2002]. Les sédiments ainsi traités puis mis en forme, vont être valorisés dans le domaine du
génie civil. Pour cela la connaissance de leur propriétés physiques tels que la surface
spécifique et la densité est essentielle.
Afin de déterminer l’effet de la température de calcination sur la dégradation de la
matière (organique et inorganique), la surface spécifique et la densité du sédiment, la
calcination a été menée sous air dans un four à lit fixe pour les sédiments traités et non
traités. La vitesse de montée en température à été fixée à 10°C/min et la durée du palier de
calcination à 3h pour chaque température (300, 500, 700, 900 et 1100°C). Afin de suivre
l’évolution de la perte de masse due à la dégradation du sédiment, une ATG a été effectuée
sur le sédiment phosphaté à 7% puis séché. L’ATG a été effectuée dans les mêmes
conditions que celles de la calcination. Cette mesure va nous permettre également la
compréhension du changement de structure des sédiments en fonction des différentes
températures de calcination
L’évaporation éventuelle des métaux n’a pas fait l’objet d’étude au cours de cette thèse.
D’autres études menées dans notre laboratoire sur la calcination des sédiments phosphatés
s’intéressent particulièrement aux émissions gazeuses susceptibles de contenir des métaux
lourds. Par contre l’effet de la calcination sur le comportement des éléments chimiques
contenus dans le solide et leur mobilité sera présentée dans le chapitre IV consacré à
l’évaluation du procédé.
III.3.3.1. Dégradation des sédiments
Les résultats obtenues en ATG pour le sédiment phosphaté à 7% après séchage sont
présentés sur la Figure III- 26. Elle représente la perte de masse en fonction du temps.
Les expériences de l’ATG ayant commencées à une température de 20°C, pour une
vitesse de montée en température à 10°C/min, les paliers de température sont atteints après
28, 48, 68, 88 et 108min respectivement pour les expériences à 300, 500, 700, 900 et
1100°C. A ces temps de chauffe, nous pouvons constater que la perte de masse est

Chapitre III : Traitement du sédiment de Vraimont
141
respectivement de 6.52, 10, 12.17, 12.86 et 12.7%. Après 3h de maintien de la température,
la perte de masse est de 8.39, 11.36, 12.89, 13 et 13.03.
Nous constatons que la calcination de 3h à 500°C permet la dégradation de 11.36% de la
masse total du sédiment. Ce qui implique que si l’objectif est uniquement la dégradation de
matière organique (humus et pollution organique), 500°C est une température suffisante
étant donné que le pourcentage massique de matière organique dans le sédiment est de 6.7%.
A partir de 700°C, la perte de masse est maximale pour le sédiment phosphaté à 7%, elle est
en moyenne de 12.93%. Ce qui signifie qu’au delà de cette température il n’est plus
nécessaire de calciner étant donnée que toutes les phases susceptibles d’être dégradées le
sont déjà. De plus une durée de palier de 3h n’est pas indispensable car à partir de 700°C,
une perte de masse moyenne de 12.6% est observée dans la phase de montée en température.
Dans ces conditions, un palier d’une demie heure est largement suffisant.
Figure III- 26 : Courbe d’analyse thermogravimétrique du sédiment phosphaté à 7% à
différentes températures.
Après avoir calciné les sédiments dans le four à différentes températures de calcination,
la mesure du taux de carbone total contenu dans les sédiments a été effectuée par analyse
élémentaire. Les résultats obtenus sont présentés sur la Figure III- 27. Ces mesures montrent
qu’avant la calcination (à 20°C) l’écart entre le pourcentage de carbone total pour les
différents sédiments est important. Cette différence correspond au CO2 dégagé lors de la
phosphatation. Par contre plus la température de calcination augmente et plus cet écart
diminue.
86
88
90
92
94
96
98
100
102
0 50 100 150 200 250 300 350Temps (min)
Pert
e de
mas
se (%
)
300°C 500°C 700°C 900°C 1100°C
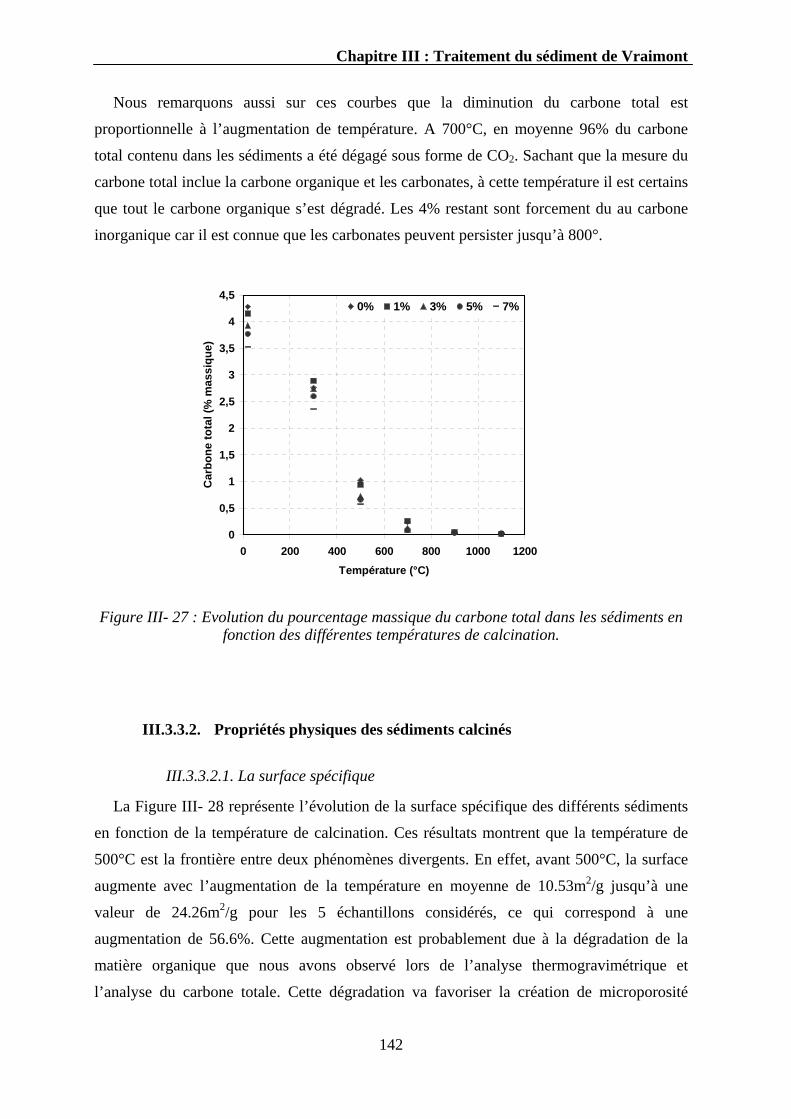
Chapitre III : Traitement du sédiment de Vraimont
142
Nous remarquons aussi sur ces courbes que la diminution du carbone total est
proportionnelle à l’augmentation de température. A 700°C, en moyenne 96% du carbone
total contenu dans les sédiments a été dégagé sous forme de CO2. Sachant que la mesure du
carbone total inclue la carbone organique et les carbonates, à cette température il est certains
que tout le carbone organique s’est dégradé. Les 4% restant sont forcement du au carbone
inorganique car il est connue que les carbonates peuvent persister jusqu’à 800°.
Figure III- 27 : Evolution du pourcentage massique du carbone total dans les sédiments en
fonction des différentes températures de calcination.
III.3.3.2. Propriétés physiques des sédiments calcinés
III.3.3.2.1. La surface spécifique
La Figure III- 28 représente l’évolution de la surface spécifique des différents sédiments
en fonction de la température de calcination. Ces résultats montrent que la température de
500°C est la frontière entre deux phénomènes divergents. En effet, avant 500°C, la surface
augmente avec l’augmentation de la température en moyenne de 10.53m2/g jusqu’à une
valeur de 24.26m2/g pour les 5 échantillons considérés, ce qui correspond à une
augmentation de 56.6%. Cette augmentation est probablement due à la dégradation de la
matière organique que nous avons observé lors de l’analyse thermogravimétrique et
l’analyse du carbone totale. Cette dégradation va favoriser la création de microporosité
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
0 200 400 600 800 1000 1200Température (°C)
Car
bone
tota
l (%
mas
siqu
e)
0% 1% 3% 5% 7%
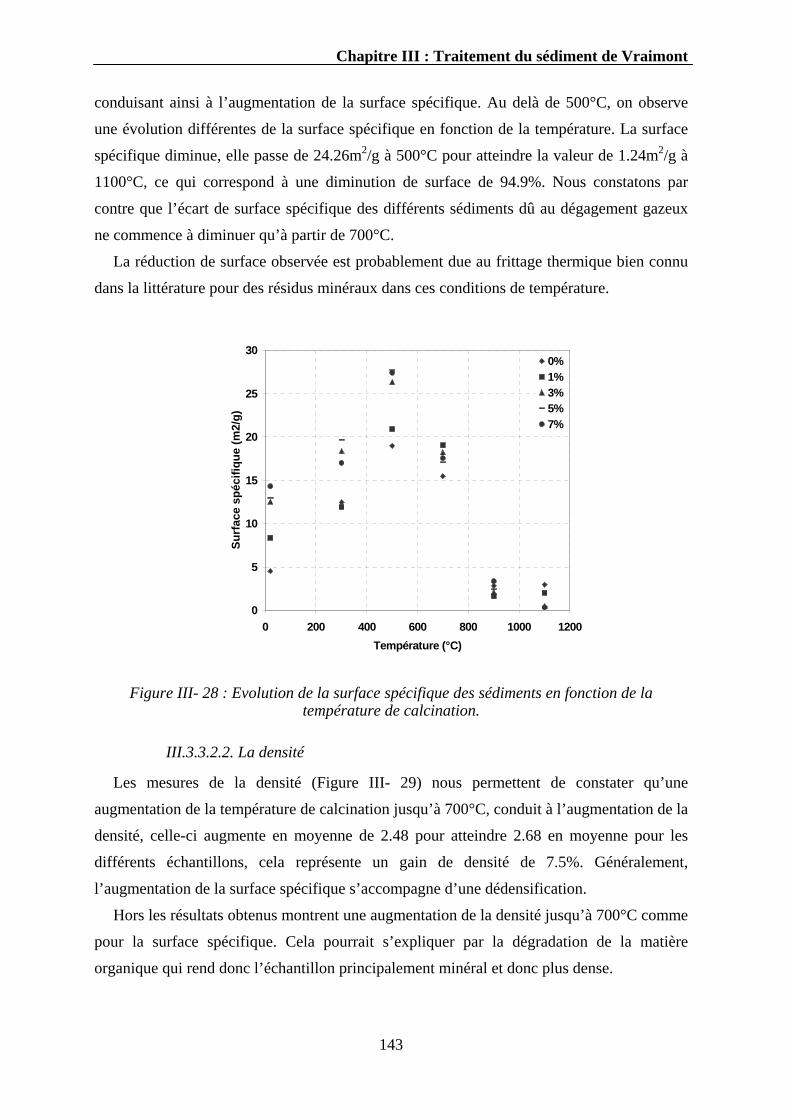
Chapitre III : Traitement du sédiment de Vraimont
143
conduisant ainsi à l’augmentation de la surface spécifique. Au delà de 500°C, on observe
une évolution différentes de la surface spécifique en fonction de la température. La surface
spécifique diminue, elle passe de 24.26m2/g à 500°C pour atteindre la valeur de 1.24m2/g à
1100°C, ce qui correspond à une diminution de surface de 94.9%. Nous constatons par
contre que l’écart de surface spécifique des différents sédiments dû au dégagement gazeux
ne commence à diminuer qu’à partir de 700°C.
La réduction de surface observée est probablement due au frittage thermique bien connu
dans la littérature pour des résidus minéraux dans ces conditions de température.
Figure III- 28 : Evolution de la surface spécifique des sédiments en fonction de la
température de calcination.
III.3.3.2.2. La densité
Les mesures de la densité (Figure III- 29) nous permettent de constater qu’une
augmentation de la température de calcination jusqu’à 700°C, conduit à l’augmentation de la
densité, celle-ci augmente en moyenne de 2.48 pour atteindre 2.68 en moyenne pour les
différents échantillons, cela représente un gain de densité de 7.5%. Généralement,
l’augmentation de la surface spécifique s’accompagne d’une dédensification.
Hors les résultats obtenus montrent une augmentation de la densité jusqu’à 700°C comme
pour la surface spécifique. Cela pourrait s’expliquer par la dégradation de la matière
organique qui rend donc l’échantillon principalement minéral et donc plus dense.
0
5
10
15
20
25
30
0 200 400 600 800 1000 1200Température (°C)
Surf
ace
spéc
ifiqu
e (m
2/g)
0%1%3%5%7%
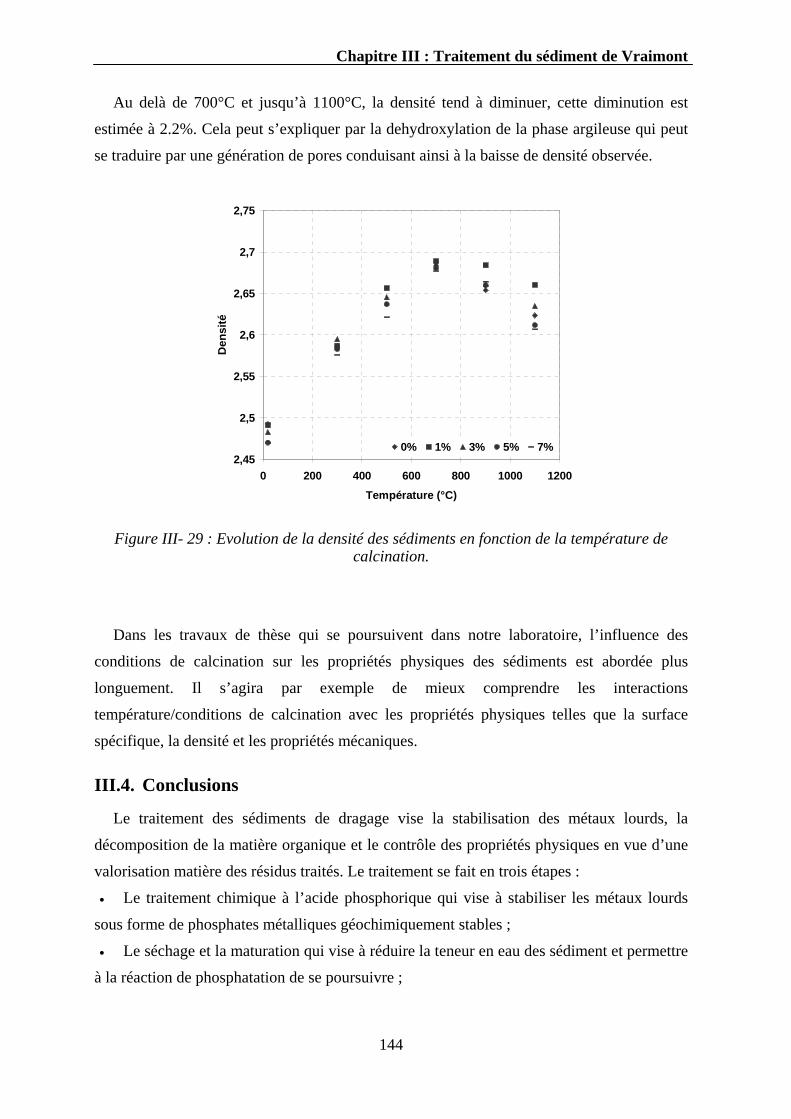
Chapitre III : Traitement du sédiment de Vraimont
144
Au delà de 700°C et jusqu’à 1100°C, la densité tend à diminuer, cette diminution est
estimée à 2.2%. Cela peut s’expliquer par la dehydroxylation de la phase argileuse qui peut
se traduire par une génération de pores conduisant ainsi à la baisse de densité observée.
Figure III- 29 : Evolution de la densité des sédiments en fonction de la température de
calcination.
Dans les travaux de thèse qui se poursuivent dans notre laboratoire, l’influence des
conditions de calcination sur les propriétés physiques des sédiments est abordée plus
longuement. Il s’agira par exemple de mieux comprendre les interactions
température/conditions de calcination avec les propriétés physiques telles que la surface
spécifique, la densité et les propriétés mécaniques.
III.4. Conclusions
Le traitement des sédiments de dragage vise la stabilisation des métaux lourds, la
décomposition de la matière organique et le contrôle des propriétés physiques en vue d’une
valorisation matière des résidus traités. Le traitement se fait en trois étapes :
• Le traitement chimique à l’acide phosphorique qui vise à stabiliser les métaux lourds
sous forme de phosphates métalliques géochimiquement stables ;
• Le séchage et la maturation qui vise à réduire la teneur en eau des sédiment et permettre
à la réaction de phosphatation de se poursuivre ;
2,45
2,5
2,55
2,6
2,65
2,7
2,75
0 200 400 600 800 1000 1200Température (°C)
Den
sité
0% 1% 3% 5% 7%

Chapitre III : Traitement du sédiment de Vraimont
145
• la calcination qui conduit à la dégradation de la matière organique et à la cristallisation
des formes chimiques obtenues lors de la phosphatation. Les phosphates métalliques ainsi
obtenus se caractérisent par une forte stabilité chimique et thermique.
Après l’étape de caractérisation physico-chimique, le sédiment de Vraimont a été choisi
pour le reste de l’étude. Néanmoins, dans le chapitre IV où nous présenterons l’évaluation
du procédé, les résultats obtenus avec le sédiment de Lille seront également présentés .
Concernant le traitement chimique du sédiment à l’acide, les réactions qui se produisent
ont lieu dans le réacteur tubulaire et se poursuivent pendant la phase de séchage. En effet les
cinétiques de consommation du phosphore dans le réacteur tubulaire, montrent bien que
dans nos conditions de travail, seulement 56.7% du P à réagit en sortie du réacteur, le reste
réagit dans la phase de séchage à l’air. Les résultats montrent qu’au bout de 8.3 jours de
maturation, 90% du phosphore a réagit et jusqu’à 98% au bout de 28 jours de contact. Ce qui
indique que la consommation du phosphore dans le réacteur est plutôt lente.
L’ajout de l’acide phosphorique dans le sédiment conduit à plusieurs transformations
physiques et chimiques.
La réaction de l’acide avec les carbonates et les sulfures contenus dans le sédiment
conduit à un dégagement gazeux de CO2 et H2S. Cela conduit à la génération de bulle de gaz
qui donnent un aspect mousseux aux sédiments. Comme nous l’avons expliqué au
paragraphe III.3.1.1.5, la formation des bulles est fortement liés aux taux d’humidité. En
effet pour un même taux d’acide, plus le taux d’humidité est important dans le sédiment
moins il y a de bulles et donc de gaz générés. Cela s’explique par le fait que le phénomène
de création des bulles est fortement lié à la sursaturation de l’eau interstitielle en ce gaz. Le
départ de ces bulles de gaz, conduit après séchage des sédiments à une augmentation de la
surface spécifique qui permet la réduction du temps de séchage des sédiments. Nous avons
montré que le temps de séchage peut-être réduit d’un tiers pour un sédiment de taux
d’humidité de 55% et phosphaté à 5% sur sec.
Une autre des conséquences de la réaction de l’acide phosphorique avec les carbonates
est la solubilisation du Ca qui va précipiter par la suite avec le P formant ainsi un phosphate
dicalcique. Lors de la phase de maturation et avec l’augmentation du pH, nous supposons
qu’une faible partie du phosphate dicalcique se transforme en phosphate plus stable tel que
l’hydroxyapatite ou le phosphate tricalcique. La présence dans le sédiment de l’une au
moins de ces formes de phosphate précurseurs de phosphates insoluble est susceptible de
stabiliser les métaux lourds mobilisables présents dans le sédiment à court ou à long terme.
L’influence de l’acide phosphorique sur le comportement des métaux tels que As, Al, Ca,
Cd, Cr, Cu, Co, Pb, Zn, et Fe a été étudiée. Mise à part le Fe et le Zn, aucun des autres

Chapitre III : Traitement du sédiment de Vraimont
146
éléments n’a été mis en évidence en concentration détectable en solution. Trois hypothèses
ont été faites :
• la non adaptation de la méthode utilisée pour la mesure de la mobilité des métaux ;
• leur réaction suffisamment rapide qui ne permet de les analyser facilement ;
• l’important pouvoir tampon du sédiment : la quantité de proton ajoutée lors de la
phosphatation n’a pas été suffisamment importante pour mobiliser une grande quantité de
métaux.
Ces trois hypothèses seront discutées dans le Chapitre IV dans lequel la spéciation des
métaux lourds et l’influence de l’acide sur leur mobilité seront abordées. Quant aux métaux
qui ont pu être étudiés, le Zn et le Fe, un mécanisme réactionnel basé sur deux processus
successives a été proposé : dissolution probable des carbonates de Zn et des sulfure de Fe
sous l’effet du traitement à l’acide suivie de leur précipitation probable sous forme de
phosphate de zinc ou de phosphate de fer ou leur réadsorption sous d’autres formes. Ces
processus feront l’objet de discussion plus détaillée au chapitre IV.
Le séchage qui s’en suit est fortement influencé par la quantité d’acide. Le résultat
évidant de cette influence est l’augmentation de la surface spécifique du sédiment qui
favorise le transfert de matière de l’intérieur de l’échantillon vers l’extérieur et vise versa. Il
n’est cependant pas envisageable ni nécessaire d’augmenter la quantité d’acide uniquement
pour ces raisons. La solution que nous proposons est de retourner régulièrement le sédiment
pour renouveler la surface et favoriser le transfert des espèces dans les deux sens et aussi
réduire les durées de séchage. Il est cependant important de tenir compte du temps
nécessaire à la maturation des sédiments. L’étude de l’influence de la vitesse de l’air et de la
température sur les temps de séchage nous a permis de constater qu’il serait
économiquement plus viable d’augmenter la vitesse de l’air pour réduire les temps de
séchage plutôt que d’augmenter sa température.
Pour la calcination, les résultats obtenus en analysant le carbone totale ainsi que les
analyses thermogravimetriques ont bien montrées qu’une calcination au delà de 700°C n’est
pas nécessaire car la totalité de la matière susceptible de se dégrader (matière organique et
carbonate) l’est dans ces conditions. Une durée d’une demie heure de chauffage à cette
température est suffisante pour décomposer les substances dégradables. Cependant si
l’objectif est uniquement de dégrader la matière organique, une température de 500°C est
suffisante pour faire face aux exigences environnementales concentrant la pollution
organique. La dégradation de la matière organique va conduire à la formation de
microporosité qui va favorise l’augmentation de la surface spécifique des sédiments d’une
part, et d’autre part à l’augmentation de la densité du sédiment (le sédiment étant composé

Chapitre III : Traitement du sédiment de Vraimont
147
principalement de phase minéral). Ces phénomènes sont observés jusqu’à une température
de calcination de 500°C. Au delà de cette température et jusqu’à 1100°C, la surface
spécifique du sédiment diminue fortement. Cela est probablement dû à une consolidation et
au frittage thermique de la phase minérale. La densité par contre tend à diminuer légèrement
probablement du fait de la dehydroxylation de la phase argileuse qui peut générer de la
porosité sur les particules de sédiments.

Chapitre IV : Evaluation du procédé
148
CHAPITRE IV
Evaluation du procédé

Chapitre IV : Evaluation du procédé
149
IV.1. IntroductionEquation Chapter (Next) Section 4
Après avoir effectué la caractérisation physico-chimique des sédiments et leur traitement,
nous procéderons à l’analyse de leur contenu en métaux lourds. Le traitement des sédiments
par l’acide phosphorique vise à neutraliser le caractère polluant des métaux lourds par la
formation de phosphates insolubles. En effet, le caractère polluant des métaux lourds dans
les sédiments est lié à leur biodisponibilité sous des formes assez solubles. L’évaluation du
procédé de stabilisation des métaux lourds par le traitement appliqué consiste à s’assurer que
les métaux lourds sont bien transformés en phosphates minéraux insolubles. Dans
l’évaluation globales du procédé, nous essayerons également de préciser l’effet de la
phosphatation et de la maturation ainsi que de la calcination sur la comportement des
métaux lourds.
Pour cela, nous avons utilisé l’extraction séquentielle. C’est une méthode qui permet de
décrire la mise en solution d’éléments en fonction de conditions prédéterminées plus ou
moins agressives. Cette méthode permet de répartir les éléments en fractions plus ou moins
disponibles en milieux aqueux, acide, oxydant et réducteur. Elle permet de classer les
éléments dans des catégories réputées métastables ou stables. L’extraction séquentielle
conduit à une spéciation par classification dans des catégories dont globalement on
comprend le devenir.
Nous procéderons donc à l’évaluation de l’impact sur les métaux lourds de la
phosphatation des sédiments en examinant l’effet de la concentration d’acide sur la
répartition des métaux lourds. Nous examinerons aussi l’effet de la température sur la
distribution des espèces. De plus, l’analyse minéralogique par microsonde permettra de
mettre en évidence les associations minéralogiques. Des observations aux MEB permettent
également de comprendre l’évolution de la microstructure sous l’effet du traitement. Enfin,
le test de lixiviation NF X30-402-2 est utilisé pour déterminer la nature inerte ou pas des
sédiments avant et après traitement.

Chapitre IV : Evaluation du procédé
150
IV.2. Méthodes et matériels
IV.2.1. Extraction séquentielle
Après avoir phosphaté, séché puis calciné les sédiments les sédiments de la manière qui a
été présentée dans le chapitre précédant. Nous leur avons appliqué la procédure d’extraction
séquentielle.
Pour effectuer l’extraction séquentielle, nous avons utilisé la méthode de Tessier et al
[Tessier 1979]. Elle est basée sur l’utilisation d’une série de réactifs plus ou moins sélectifs
choisi pour solubiliser successivement les différentes fractions minéralogiques. La
solubilisation des ces fractions, conduit à la libération des métaux qui leur sont liés par
différents mécanismes : l’échange d’ion, adsorption, co-précipitations, complexation….
Cinq fractions ont été définies et correspondent aux métaux : échangeables (F1), liés aux
carbonates (F3), liés aux oxydes de fer et de manganèse (F3), liés à la matière organique et
aux sulfures (F4) et résiduelles (F5).
Pour effectuer les expériences, on pèse 1g d’échantillon dans un tube à centrifugation en
polyéthylène de 40ml auquel on applique le protocole d’extraction séquentielle décrit dans
la Figure IV- 1. La cinquième fraction n’a pas été déterminée expérimentalement tel que
décrit dans le protocole. Elle a été calculée par différence entre la concentration totale des
éléments (après minéralisation complète (voir chapitre II, paragraphe II.3.4)), et la somme
des quatre fractions. Entre deux étapes d’extraction successives, le sédiment est centrifugé
pendant 15min à 8000rpm. Le surnageant est ensuite filtré (filtre papier 0.45µm), acidifié
avec quelques gouttes d’acide nitrique, puis complété à 20ml avec de l’eau déminéralisée.
Les filtrats sont conservés a 4°C dans des flacons en polyéthylène avant analyse. Le résidu
solide est utilisé pour l’étape qui suit après lavage à l’eau déminéralisée (10ml),
centrifugation et filtration.
La concentration des éléments As, Al, Ca, Cd, Co, Cr, Cu, Fe, P, Pb, et Zn dans les
filtrats obtenues après chaque étape est analysée en ICP-AES (Spectromètre d’Emission
Atomique Plasma à Couplage Inductif). Les expériences ont été répétées trois fois afin de
déterminer leur reproductibilité.

Chapitre IV : Evaluation du procédé
151
Figure IV- 1 : Protocole d’extraction séquentielle de Tessier et al.
L’extraction séquentielle a été appliquée aux sédiments de Vraimont (BV) et également
appliquée aux sédiments de Lille. Ces derniers ont fait l’objet du même traitement que le
Sédiment
1g
Résidu
Résidu
Résidu
Résidu
Fraction 5
Fraction 4
Fraction 3
Fraction 2
Fraction 1
8 ml (1M MgCl2. pH 7.0)1 h d’agitation à température ambiante
8 ml (1M NaOAc, pH 5.0)5 h d’agitation à température ambiante
20 ml (0.04 M NH2OH.HCl dans 25%v/v HOAc).6h à 96±3°C avecagitation occasionnelle
3ml (0.02M HNO3)+5ml (30% H2O2, pH 2), 2h à 85 ±2°C avec agitationoccasionnelle. 3 ml (30% H2O2, pH 2), 3h agitation occasionnelle à 85 ±2°C.A froid, 5 ml (3.2M NH4OAc dans 20%v/v HNO3, dilution à 20ml, agitation en continue 30 min
5:1 de HF : HCLO4Dissolution du résidu dans 12N HCl
Sédiment
1g
Résidu
Résidu
Résidu
Résidu
Fraction 5
Fraction 4
Fraction 3
Fraction 2
Fraction 1
8 ml (1M MgCl2. pH 7.0)1 h d’agitation à température ambiante
8 ml (1M NaOAc, pH 5.0)5 h d’agitation à température ambiante
20 ml (0.04 M NH2OH.HCl dans 25%v/v HOAc).6h à 96±3°C avecagitation occasionnelle
3ml (0.02M HNO3)+5ml (30% H2O2, pH 2), 2h à 85 ±2°C avec agitationoccasionnelle. 3 ml (30% H2O2, pH 2), 3h agitation occasionnelle à 85 ±2°C.A froid, 5 ml (3.2M NH4OAc dans 20%v/v HNO3, dilution à 20ml, agitation en continue 30 min
5:1 de HF : HCLO4Dissolution du résidu dans 12N HCl

Chapitre IV : Evaluation du procédé
152
sédiment de Vraimont. La différence est qu’au lieu de les phosphater à 3, 5 et 7% sur sec,
comme cela a été fait pour BV, ils ont été phosphaté jusqu’à 10% car ils présentent un
potentiel polluant plus important. Les sédiments suivants ont été utilisés :
• Le sédiment de Vraimont brut séché, nommé (BV0). Les sédiments de Vraimont
phosphatés à 3, 5 et 7% sur sec, puis séchés et maturés à l’air pendant 28 jours. Ils sont
nommés BV3, BV5 et BV7. Après leur calcination à 700°C pendant 3h, ils ont été
également étudiés. Ils sont nommés BV0C, BV3C, BV5C et BV7C
• Le sédiment de Lille brut séché, nommé (BL0). Les sédiments de Lille phosphatés à 3,
5 et 10% sur sec, puis séchés et maturés à l’air pendant 28 jours. Ils sont nommés BL3, BL5
et BL10. Après leur calcination à 700°C pendant 3h, ils ont été également étudiés. Ils sont
nommés BL0C, BL3C, BL5C et BL10C.
Le choix de l’étude d la température de calcination de 700°C a été dictée par le fait que
nous avons démontré que la calcination à 700°C permet la dégradation de la totalité des
éléments susceptibles de se dégrader.
Les résultats qui vont être présentés sont la moyenne de trois expériences menées pour
chaque sédiment, le pourcentage d’erreur dans ces mesures est entre 5 et 8%, cela peut être
dû d’une part à l’hétérogénéité des sédiments et d’autre part, à la perte de masse du résidu
observée lors de sa récupération du filtre.
IV.2.2. Microsonde électronique
Après avoir déterminé par voie chimique la distribution des éléments dans les sédiments
traités et non traités, nous procèderons à l’analyse par voie spectroscopique des associations
minéralogiques des éléments majeurs et mineurs contenus dans les sédiments par
l’utilisation de la microsonde. La microsonde électronique permet la visualisation
séquentielle des différents éléments d’une même zone géographique par cartographie.
L’échantillon sous forme de pastille est collé sur le porte échantillon avant d’être
métallisé à l’or pour les cartographies. Pour la quantification, l’échantillon est incorporé
dans une résine, l’araldite, avant polissage, et métallisé au carbone. Ensuite, il est inséré
dans la chambre d’analyse sous vide.
La microsonde électronique est de type SX 50 CAMECA travaillant en balayage de
longueur d’onde (WDS) avec une tension d’accélération de 15kV et une intensité
d’amplification de 40nA pour les cartographies et de 12nA pour la quantification. Elle est
équipée de quatre (ou cinq pour la quantification) spectromètres RX équipés des cristaux
monochromateurs disponibles en fonction des éléments à analyser.

Chapitre IV : Evaluation du procédé
153
L’analyse s’est faite uniquement sur le sédiment de Vraimont brut et sur le sédiment
phosphaté à 5% sur sec, séché puis calciné à 700°C.
IV.2.3. Microscope électronique à balayage (MEB)
Le microscope électronique à balayage permet de visualiser la microstructure des
sédiments sur des zones sélectionnées en SEM. Des analyses semi-quantitatives avec
détecteur EDS (Energy Dispersive Spectroscopy) ont également été effectuées dans le but
d’avoir une idée des constituants des produits. Les analyses ont été réalisées en mode
environnemental sur XL30 ESEM de PHILIPS sur des échantillons non métallisés.
L’analyse a été faite uniquement pour le sédiment de Vraimont phosphaté à 5% sur sec
puis calciné à 700 et 1100°C.
IV.2.4. Test de lixiviation
La lixiviation par définition est une méthode d’extraction liquide-solide d’un échantillon
solide par une solution aqueuse qui permet d’évaluer le largage des polluants vers le milieu
naturel. Comme il a été expliqué dans le chapitre I, le vide juridique auquel nous sommes
confrontés, ne nous permet pas d’avoir un outil réglementaire pour évaluer l’innocuité des
sédiments traités pour qu’ils soient appropriés à des usages dans le domaine du génie civil.
En d’autre terme savoir si le sédiment traité ne pose pas de problèmes environnementaux à
son usage dans le domaine du génie civil (briques pour la construction, sous couche
routière…). Ce manque d’outils appropriés nous oblige à prendre comme méthode de
référence les valeurs limites établissant des critères et procédures d’admission des déchets
dans les décharges (décision européenne du 19 décembre 2002). Ceci bien que le matériau
produit par le procédé NOVOSOL ne soit pas destiné à une mise en décharge pour déchets
inertes mais à la valorisation. Toutefois ce test nous permettra de savoir si le sédiment traité
est inerte, non dangereux ou dangereux. Pour cela nous appliquerons donc la norme de
lixiviation européenne NF EN 12457-2 (indice de classement français: X30-402-2) qui est
attribuée à cette décision.
Le test de lixiviation NF EN 12457-2 [NF EN 12457-2] consiste à mettre en contact 24 h
une masse l’échantillon (10g ont été utilisé) de taille inférieure à 4 mm et l’eau
déminéralisée à un rapport solide/liquide de 1:10 sous agitation rotative (10rpm). Au bout de
24 h la suspension est filtrée sur filtre à membrane de diamètre moyen de pore de 0,45 µm.
Le filtrat dit « lixiviat » est alors analysé par ICP-AES afin de déterminer la concentration

Chapitre IV : Evaluation du procédé
154
des métaux lourds : AS, Cd, Co, Cu, Pb, Zn. Le pH de ces lixiviats est mesuré avec un pH-
mètre
Cette norme à été appliquée aux sédiments de Vraimont traités et non traités. Les
concentrations de métaux lourds analysés dans les lixiviats sont ensuite comparés aux
valeurs limites pour mise en décharge pour déchets inertes qui sont présentées dans le
chapitre 1. Il est important de signaler que la liste des éléments chimiques à analyser pour
qu’un déchet soit admis dans une décharge est plus importante. Elle comporte en plus des
valeurs limites des éléments que nous avons cité plus haut, des valeurs limites pour d’autres
éléments tels que Ba, Hg, Mo, Ni, Sb, Se…auxquels nous nous sommes pas intéressés.
IV.3. Résultats et discussions
IV.3.1. Extraction séquentielle
IV.3.1.1. Distribution des éléments chimiques dans les sédiments bruts
Avant de montrer l’influence du traitement sur le distribution des éléments chimiques
dans les sédiments, la distribution de ces éléments dans les sédiments bruts (BV et BL) a été
déterminée et présentée sur la Figure IV- 2. La distribution en pourcentage massique de
éléments : As, Al, Ca, Cd, Co, Cr, Cu, Fe, P, Pb, et Zn dans le sédiment brut de Lille est
représentée sur la Figure IV- 2 (a). La même représentation a été faite sur la Figure IV- 2 (b)
pour le sédiment brut de Vraimont. Une autre représentation des concentrations de ces
éléments dans chaque fraction est proposée dans le Tableau IV- 1. Pour rappel, F1
représente les éléments échangeables. F2 représente les éléments liés aux carbonates. F3
représente les éléments liés aux oxydes de fer et de manganèse (appelée également fraction
réductible). F4 représente le éléments liés à la matière organique et aux sulfures (appelée
également fraction oxydable). F5 représente les éléments résiduels. Nous avons également
présenté dans ce même tableau, la somme des quatre fractions non résiduelles
(F1+F2+F3+F4). L’objectif étant d’évaluer la fraction de métaux lourds potentiellement
mobilisables. Il est reconnu que les quatre premières fractions sont plus mobilisables que la
fraction résiduelle [Ma L. Q 1999; Melamed 2003; Yuan 2004]. En effet, les métaux
adsorbés, échangés et liés aux carbonates sont généralement ceux apportés par l’activité
humaine. Ils sont faiblement liés aux sédiments et peuvent facilement s’équilibrer en phase
aqueuse et devenir plus biodisponibles [Salomons 1980, Pardo 1990]. Par contre les métaux
incorporés dans la phase minérale (F5) sont d’origine naturelle et sont donc très stables
[Salomons 1980 ; Abd El-Azim 2004]. Ils sont jugés comme étant chimiquement stables et
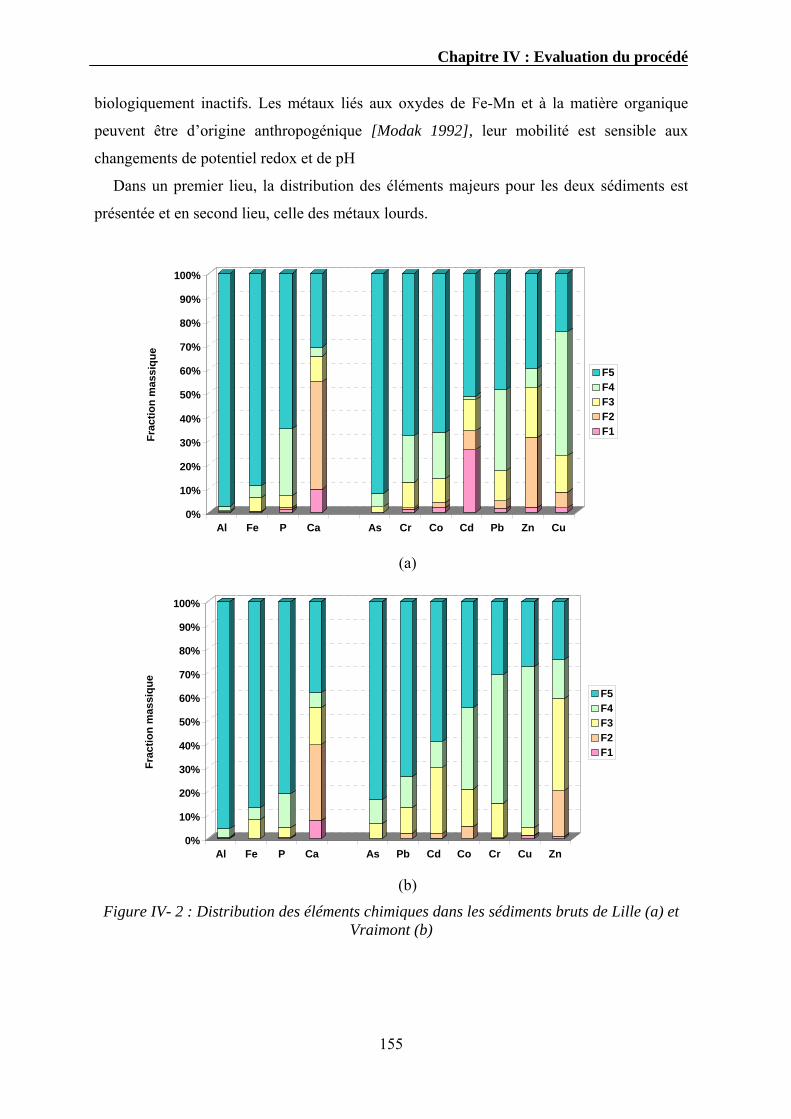
Chapitre IV : Evaluation du procédé
155
biologiquement inactifs. Les métaux liés aux oxydes de Fe-Mn et à la matière organique
peuvent être d’origine anthropogénique [Modak 1992], leur mobilité est sensible aux
changements de potentiel redox et de pH
Dans un premier lieu, la distribution des éléments majeurs pour les deux sédiments est
présentée et en second lieu, celle des métaux lourds.
(a)
(b)
Figure IV- 2 : Distribution des éléments chimiques dans les sédiments bruts de Lille (a) et Vraimont (b)
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Frac
tion
mas
s iq u
e
Al Fe P Ca As Cr Co Cd Pb Zn Cu
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Frac
tion
mas
siqu
e
Al Fe P Ca As Pb Cd Co Cr Cu Zn
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
156
Tableau IV- 1 : Concentration (mg/kg sur matière sèche) des différents éléments chimiques dans les différente fraction en du sédiment de Lille et Vraimont
IV.3.1.1.1. Eléments majeurs
Par éléments majeurs, nous sous entendons, le Fe, Al, Ca et le P présents naturellement
dans le sédiment. Nous présenterons la distribution de ces éléments un par un.
Le fer
A travers les résultats présentés sur la Figure IV- 2 et le Tableau IV- 1, nous pouvons
constater à première vu que la distribution du Fe est pratiquement identique pour les deux
sédiments. Le Fe se trouve majoritairement dans F5 dans BL et BV. En effet, sa fraction
massique est de 87 et 88.8% pour BL et BV respectivement. Dans les fractions F3 et F4, il
est plus faiblement présents (7.9% et 5% pour BL et 5.8% et 5.1% pour BV). Par contre il
est négligeable dans les deux premières fractions.
Contrairement à ce qu’on pouvait penser à priori, la fraction majoritaire est la fraction
résiduelle et non pas la fraction représentant les éléments liés aux oxydes de fer et de
manganèse (réductible). Ce résultat indique qu’un faible pourcentage de Fe se trouve sous
forme d’oxyde ou d’hydroxyde de Fe tel que la geothite, la ferrihydrite ou l’hématite. Par
contre la majorité du Fe dans les deux sédiments est très liée à la matrice cristalline des
silicates d’aluminium. Ce résultat est en adéquation avec les résultats trouvés par Yuan et al,
Usero et al et Singh et al [Yuan 2004 ; Usero 1998 ; Singh 2005] qui trouvent
respectivement 90, 89 et 78% du Fe sous forme résiduelle dans des sédiments. Quant à la
présence de Fe dans la fraction oxydable, elle peut être due à la complexation d’une partie
Al As Ca Cd Co Cr Cu Fe P Pb ZnF1 64,3 0,0 4202,2 0,0 0,0 1,2 5,3 0,0 7,9 0,0 39,3F2 8,8 0,0 17388,3 0,8 0,8 0,5 0,4 15,6 8,2 14,2 896,5F3 187,4 9,2 8684,1 10,4 2,6 100,0 14,1 2541,5 161,6 69,5 1812,4F4 1865,7 15,3 3464,4 4,1 5,6 370,5 290,8 1608,5 540,6 83,9 760,1F5 48349,6 124,7 21071,2 22,3 7,4 211,5 118,1 27870,3 3059,9 473,6 1146,6
F1+F2+F3+F4 2126,3 24,5 33738,9 15,3 9,0 472,2 310,6 4165,6 718,1 167,6 3508,2
F1 111,5 0,0 2633,6 1,0 0,3 2,2 2,4 72,8 24,9 3,3 20,0F2 11,9 0,0 12301,2 0,3 0,3 1,1 9,6 26,7 17,7 6,6 293,8F3 158,6 3,6 2915,4 0,5 1,5 20,7 21,6 1927,1 108,6 27,3 208,2F4 1009,7 9,5 1010,8 0,1 2,9 37,1 74,0 1665,8 612,7 72,6 81,2F5 51109,3 151,1 8485,6 2,1 10,0 130,2 35,2 29283,0 1427,7 104,4 400,6
F1+F2+F3+F4 1291,7 13,1 18861,0 1,9 5,0 61,1 107,6 3692,4 763,9 109,8 603,2
Sédiment de Lille
Sédiment de Vraimont

Chapitre IV : Evaluation du procédé
157
des oxydes et hydroxydes de Fe avec la substance humique [Fytianos 2004]. Elle peut
également être due à la présence de Fe sous forme de sulfure tel que le FeS2 (pyrite) ou FeS.
L’aluminium
Comme pour le Fe, l’Al se trouve majoritairement dans F5. Son pourcentage massique est
de 95.8% pour BL et 97.5 % pour BV. Quant aux autres fractions, elles sont négligeables,
mis à part la F4 contenant 3.7 et 1.9% d’Al pour BL et BV respectivement. Cela peut être
relié à la formation de complexes entre la matière organique et l’Al trivalent [da Silva
2002]. Ces résultats montrent que la grande majorité de l’Al se trouve sous forme de
silicates d’aluminium. Ces résultats sont en bon accord avec les résultats de Guevara-Riba et
al et ceux publiés par da Silva et al [Guevara-Riba 2004 ; da-Silva 2002]
Le calcium
La distribution du Ca est très différente de celle du Fe et de l’Al. Le Ca se reparti sur les
cinq fractions. Son ordre d’affinité aux différentes fractions est le suivant : F2 > F5 > F3>
F1 > F4 pour BL et F2 > F5 > F3 > F1 > F4 pour BV. Cela signifie que la majorité du Ca
dans les deux sédiments est liée aux carbonates et liée aux phases minérales primaires et
secondaires. Le Ca dans la F2 peut être sous forme de calcite (CaCO3) ou de dolomite
CaMg(CO3)2. Les résultats obtenus en diffraction des rayon X des deux sédiments (chapitre
II, paragraphe 3.7) ont permis de montrer la présence de calcite. Quant au Ca présent dans la
fraction résiduelle, il pourrait être sous forme de pyroxène ou d’amphibole.
Cependant, même si le pourcentage de Ca dans F2 est plus important que dans BV (45%)
que dans BV (31.7%). La concentration de Ca liée aux carbonates est plus importante dans
BL que dans BV (voir Tableau IV- 1), ce qui est en adéquation avec les résultats de DRX.
En effet les résultats de DRX ont également permis de montrer que la concentration en
calcite dans BL est plus importante que dans BV.
Le phosphore
Le phosphore se trouve majoritairement dans F5 dans les deux sédiments (81% pour BL
et 65.1% pour BV). Il est possible qu’il soit lié aux phases minérales primaires et
secondaires sous forme de phosphates métalliques ou de phosphates de calcium insolubles.
Nous le retrouvons également dans F4 (14.3% dans BL et 28% pour BV) et à des
concentration non négligeables dans F3 (4.3% dans BL et 5% dans BV). Par contre dans F1
et F2 le P est très faible concentration.

Chapitre IV : Evaluation du procédé
158
IV.3.1.1.2. Les métaux lourds
Après avoir présenté la distribution des éléments majeurs, nous présentons à présent la
distribution des métaux lourds toxiques (Figure IV- 2 (a) et (b)).
l’arsenic
L’arsenic est l’un des éléments toxiques les plus stables parmi ceux que nous avons
analysé. Nous pouvons constater qu’il se trouve dans la fraction résiduelle à 95.8% pour BL
et 97.5% pour BV. Ce qui signifie qu’il est inclus dans les minerais primaires et secondaires
et qu’il est donc très peu soluble. L’As se trouve également dans F3 et F4 à des proportions
faibles (respectivement 6.2 et 10.3% dans BL et 2.2 et 5.8% dans BV). Par contre il est
indétectable dans les fractions F1 et F2 pour les deux sédiments. La présence de l’As dans
F4 peut être due à l’accumulation des ions arsenates et arsenites à la surface de la matière
organique [Lombi 2000]
En faisant la somme des fractions non résiduelles, nous constatons que la concentration
de l’As potentiellement mobilisable est plus importante pour BL (24.5mg/kg) que pour BV
(13mg/kg).
Le chrome
Le comportement du Cr dans les deux sédiments est différent. Dans BL, il se trouve à
taux important dans F4 (54.2%) et à un taux plus faible F5 (30.9%). Dans BV par contre, le
Cr est majoritaire dans F5 (68.1%) et à un taux plus faible dans F4 (19.4%). Cela signifie
que le Cr dans BV est majoritairement incorporé dans la matrice de silicate d’alumine et
qu’il est donc stable. Ces résultats sont en accord avec les résultats trouvés par Singh et al et
Lee et al [Singh 2005 ; Lee 2005]. Par contre dans BV, un taux important de Cr est sous
forme oxydable. Cela a été observée également par Fytianos et Lourantou, Davidson el al
[Fytianos 2004 ; Davidson 1994]. Le Cr dans cette fraction est lié à la matière organique
peut aussi être sous forme d’oxyde de Cr(III) [Tokalioglu 2000]. Le Cr dans la fraction
oxydable n’est pas très stable, il est tributaire du changement de pH du milieu et à son
potentiel redox.
Nous pouvons noter cependant que la présence de Cr dans F1 et F2 est négligeable dans
les deux sédiments. En considérant les 4 fractions non résiduelles, nous pouvons noter que la
concentration de Cr potentiellement mobilisable dans BL (472.2 mg/kg) est beaucoup plus
importante que celle de BV (61.1mg/kg).

Chapitre IV : Evaluation du procédé
159
Le cobalt
L’étude de la distribution du cobalt dans BV et BL montre qu’il se trouve dans les 5
fractions. Cependant, le taux le plus important est retrouvé dans F5. En effet, dans BL il est
de 44.9% alors que dans BV il est plus important, son taux est de 66.7%. Nous pouvons
constater également que le Co a une bonne affinité pour la fraction oxydable. En effet, dans
BL sa fraction massique est de 34.4% alors que dans BV elle est de 19.3%. La somme des
concentrations massiques du Co dans les quatre fractions non résiduelles est de 5mg/kg pour
BV et 9mg/kg pour BL.
Le cadmium
Les résultats de l’extraction séquentielle montrent que l’affinité du Cd présent dans BL
pour les cinq fractions suit l’ordre suivant F5 > F3 > F4 > F2. Le Cd est indétectable dans
F1, toute fois il est présent majoritairement dans F5 (59.3%) et F3 (27.7%). Cela signifie que
la majorité du Cd est inclut dans les silicates d’alumine et sous forme réductible. Par contre
pour BV, l’ordre des affinités est différent : F5 > F1 > F3 > F2 > F4. Nous remarquons que
51.6% du Cd se trouve sous forme résiduaire, 26% sous forme échangeable et 8% lié aux
carbonates. La présence de Cd dans F1 a été expliquée par Modak et al [Modak 1992]. Ils
rapportent que le Cd possède une grande affinité pour les argiles à cause de son rayon
atomique qui est similaire à celui du K habituellement associé aux argiles. Cette hypothèse
s’applique pour BV mais ne s’applique pas pour BL, car le Cd est indétectable dans cette
fraction. En comparant la somme des fractions non résiduelles de BL et BV, nous constatons
qu’il y a 15.3mg/kg de Cd potentiellement mobilisable dans BL et 1.9mg/kg dans BV. Cela
implique que le Cd dans BV présente peu de risque comparé à BL.
Le plomb
Le Pb présent dans les deux sédiments est majoritair dans la fraction résiduaire et la
fraction oxydable. Pour BL, il se trouve à 73.9% dans F5 et 13.1% dans F4. Par contre, dans
BV, il se trouve à 48.8% dans F5 et 33.9% dans F4. L’affinité du Pb à la fraction résiduelle
et à la fraction oxydable a été démontrée par Caplat et al [Caplat 2005] ainsi que par Akcay
et al dans certains des sédiments avec lesquels ils ont travaillé [Akcay 2003]. Le Pb est donc
plus stable dans BL que dans BV, par contre en terme de concentration, 167.6mg/kg de Pb
sont potentiellement mobilisable dans BL contre 109.8 mg/kg dans BV.

Chapitre IV : Evaluation du procédé
160
Le zinc
Le Zn est réparti sur les différentes fractions de la manière suivante : F3 (38.9%) > F5
(24.7%) > F2 (19.3%) > F4 (16.3%) > F1 (0.8%) dans BL. Nous remarquons que le Zn est
bien réparti sur les quatre dernières fractions, sa présence dans la fraction échangeable est
négligeable. Quant à BV, la répartition du Zn est différente, son affinité pour les différentes
fractions est la suivante : F5 (39.9%) > F2 (29.3%) > F3 (20.7%) >F4 (8.1%) > F1 (2%). La
majeure partie du Zn dans BV est répartie dans F5, F2 et F3, sa présence sous forme
échangeable est négligeable.
Ces résultats montrent que le pourcentage de Zn stable dans BV est plus important que
celui de BL. Néanmoins, en additionnant les masses des fractions non résiduelles, nous
pouvons constater que la teneur en Zn potentiellement mobilisable est de 3058.2 mg/kg,
alors qu’elle n’est que de 603.2mg/kg dans BV.
Le cuivre
La distribution du Cu dans les deux sédiments révèle que ce dernier se trouve
majoritairement dans la fraction oxydable et en pourcentage moindre dans la fraction
résiduelle. Dans BL, son pourcentage massique est de 67.9% dans F4 et 27.6% dans F5.
Dans BV la fraction liée à F4 est moins importante que dans BL, elle est de 51.8%, quant à
la fraction résiduelle, elle est de 24.6%. Ces résultats sont en accord avec les résultats de
Stead-dexter et Ward [Stead-dexter 2004] et de Lee et al [Lee 2005] qui trouvent que le Cu
est fortement lié à la fraction oxydable. Pempkowiak et al [Pempkowiak 1999] expliquent
que le Cu de cette fraction est vraisemblablement lié aux substances humiques qui ont une
grande affinité pour ce métal.
On note également la présence de Cu sous forme de carbonate dans BV, alors que dans
BL, cette fraction est pratiquement inexistante. La somme des concentrations retrouvées
dans les fractions non résiduelles montrent que BL, avec 310.6mg/kg, a un potentiel polluant
supérieur à celui de BV qui ne présente que 107.6 mg/kg.
IV.3.1.1.3. Synthèse
Pour la détermination de la mobilité des métaux lourds et de leur impact sur
l’environnement, l’analyse de la totalité des métaux lourds présents dans les sédiments n’est
pas suffisante. Seule la forme chimique sous laquelle se trouve les métaux peut déterminer
leur mobilité et leur biodisponibilité et donc leur impact environnemental [Hickey 1984 ;
Wallman 1993]. L’extraction séquentielle représente donc l’outil qui nous permet de

Chapitre IV : Evaluation du procédé
161
répondre a ces questions à travers l’étude de la distribution des métaux lourds dans les
différentes fractions géochimiques du sédiment. Cette étude est faite ayant conscience des
limites de cette méthode, à savoir : le manque de sélectivité des réactifs ainsi que les
problèmes liés à la réadsorption et de redistribution des métaux aux cours de l’extraction.
L’application de cette méthode sur les deux sédiments sur lesquelles nous travaillons
permet de dégager les conclusions suivantes :
Les éléments majeurs se trouvent essentiellement dans la fraction résiduaire, excepté le
Ca qui se trouve sous forme de carbonates et inclus dans les silicates d’aluminium.
En considérant la proportion de métaux lourds contenus dans la fraction résiduaire, par
définition la plus stable, nous pouvons établir que la mobilité des métaux lourds dans BL
augmente dans l’ordre suivant As > Pb > Cd > Co > Cr > Cu > Zn. Pour BV, la tendance est
différente, la mobilité des métaux lourds augmente dans l’ordre As > Cr > Co > Cd > Pb >
Zn > Cu. Nous pouvons constater qu’il n’existe pas de règle générale qui définit l’ordre de
mobilité des métaux dans les sédiments. Plusieurs paramètres influencent cet ordre : la
composition du sédiment (argile, matière organique, carbonate…), la nature de la pollution,
source de la pollution (activité anthropogénique, ou naturelle), l’état d’oxydation du
sédiment, le pH du milieu, la taille des grains…
En comparant la somme des concentrations des métaux lourds des fractions non
résiduaires (F1+F2+F3+F4) pour chacun des sédiments, nous constatons que les métaux
lourds présents dans BL ont un potentiel de mobilisation plus important que ceux de BV. De
ce fait, la quantité de phosphore requise pour stabiliser un maximum de métaux dans BL
doit être plus importante que celle nécessaire pour BV. C’est la raison pour laquelle, BL a
été phosphaté jusqu’à 10% sur matière sèche alors que pour BV, le taux maximal d’acide
phosphorique appliqué a été de 7% sur sec.
L’affinité des différents métaux lourds pour les différentes fractions des sédiments
augmente dans l’ordre : F1 > F2 > F3 > F4 > F5, ceci pour les deux sédiments.
En faisant la somme des concentrations molaires des différents éléments qui se trouvent
dans la fraction échangeable et en les convertissant en meq.100g-1, nous constatons que ces
valeurs sont plus faible que la capacité d’échange cationique mesurée pour les deux
sédiments. Cela signifie qu’il existent d’autres éléments échangeables dans les deux
sédiments et que nous les avons pas mesurés.

Chapitre IV : Evaluation du procédé
162
IV.3.1.2. Distribution des éléments chimiques dans les sédiments traités
La distribution des métaux lourds dans les sédiments bruts permet une caractérisation du
potentiel polluant avant traitement. La distribution des métaux lourds dans les sédiments
phosphatés et séchés ainsi que pour ces mêmes sédiments après calcination à 700°C est
maintenant présentés. Le but est d’illustrer l’efficacité du traitement proposé et d’essayer de
comprendre l’influence de différents paramètres du traitement sur le métaux lourds contenus
dans les différentes fractions
Nous présenterons tout d’abord la distribution des éléments majeurs, puis celle des
métaux lourds dans les deux sédiments.
IV.3.1.2.1. Eléments majeurs
Le phosphore
Le phosphore est le premier élément que nous présentons. Le but est de connaître sa
distribution dans les sédiments afin de comprendre les phénomènes mis en oeuvre lors de la
phosphatation et de la maturation, ainsi que pendant la calcination des sédiments.
La Figure IV- 3 et Figure IV- 4 présentent la distribution du P (en mg/kg sur sec)
respectivement dans le sédiment de Lille et Vraimont traités et non traités. Il est important
de préciser que sur les figures qui représentent la distribution P et des autres éléments qui
seront représentés plus tard, les histogrammes de gauche ne représentent que les sédiments
phosphatés et séchés. Les histogrammes de droite représentent les sédiments phosphatés,
séchés et calcinés.
Contrairement aux autres éléments dont la distribution est présentée en pourcentage
massique, le P est représenté en mg/kg sur sec afin de mieux évaluer la répartition de ce
dernier sur les différentes fractions, d’autant plus que les quantités de P ajoutées sont
variables.
Avant calcination, nous pouvons constater que le P ajouté à BV et BL se répartit sur les 5
fractions, néanmoins, la fraction résiduaire reste la plus importante. En effet entre 58.2 et
69% du P ajouté à BV se trouve dans la fraction résiduelle pour les trois taux de
phosphatation. Dans BL par contre, entre 31.3 et 70.4% du P ajouté est présent dans F5.
Nous constatons que pour BL10, la taux de P dans F5 (31.3%) est faible, le P se répartit
d’avantage sur les autres fractions notamment F3 et F4. Cela peut être dû au faible pH du
milieu (causé par l’ajout de 10% de H3PO4 sur sec) qui ne permet pas la formation de sels de
phosphates comme il est observé pour les autres taux de phosphatation.
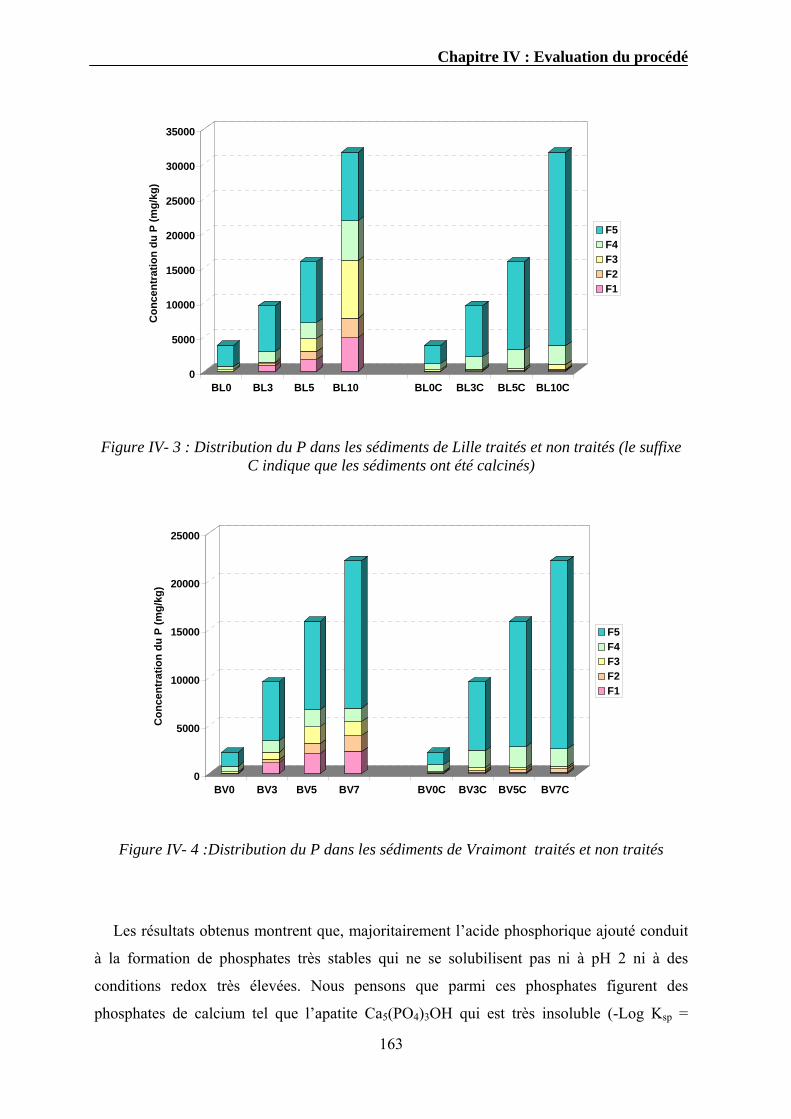
Chapitre IV : Evaluation du procédé
163
Figure IV- 3 : Distribution du P dans les sédiments de Lille traités et non traités (le suffixe C indique que les sédiments ont été calcinés)
Figure IV- 4 :Distribution du P dans les sédiments de Vraimont traités et non traités
Les résultats obtenus montrent que, majoritairement l’acide phosphorique ajouté conduit
à la formation de phosphates très stables qui ne se solubilisent pas ni à pH 2 ni à des
conditions redox très élevées. Nous pensons que parmi ces phosphates figurent des
phosphates de calcium tel que l’apatite Ca5(PO4)3OH qui est très insoluble (-Log Ksp =
0
5000
10000
15000
20000
25000
30000
35000
Con
cent
ratio
n du
P (m
g/kg
)
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
0
5000
10000
15000
20000
25000
Con
cent
ratio
n du
P (m
g/kg
)
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
164
38.15). D’autres types de phosphates peuvent se former lors de la phosphatation tels que
l’hydroxypyromorphite Pb5(PO4)3OH (-Log Ksp = 62.80), le Cu5(PO4)3OH (-Log Ksp =
51.62), Pb3(PO4)2 (-Log Ksp = 44.36), Cu3(PO4)2 (-Log Ksp = 36.85), vivianite
Fe3(PO4)2H2O. Nous pouvons également supposer que le P s’est introduit dans les matrices
silicatés formant des phosphosilicates stables.
L’existence du P dans les autres fractions non résiduelles peut être due à la formation de
phosphates de calcium et des phosphates métalliques plus solubles que ceux retrouvés dans
F5 tel que le phosphate dicalcique CaHPO4 (-Log Ksp = 19,09) ou même tricalcique (-Log
Ksp = 32.69). Cette hypothèse peut être confirmée par les résultats obtenus lors de l’étude
cinétique. En effet, nous avons démontré la corrélation qui existe entre la cinétique de
précipitation du Ca et celle du P et la possibilité de former un phosphate dicalcique. D’autre
hydrogenophosphates tels que le CuHPO4, PbHPO4, ZnHPO4…ont pu se formés lors de la
phosphatation et de la maturation
Après calcination, nous pouvons constater que la majorité du P qui a été injecté se trouve
sous forme résiduelle, et une faible partie sous forme oxydable. En effet, entre 74.7 et 88.5%
du P dans BV pour les trois taux de phosphatation est présent dans F5. Dans BL, cette
fraction présente entre 78.3 et 88.1% du P total ajouté. Cela implique qu’une partie du P qui
était présent sous des formes non résiduelles (comme ceux cités plus haut) s’est transformée
sous l’effet de la calcination en phosphates plus stables.
Ces résultats signifient d’une part que la phosphatation seule conduit à la formation de
phosphates stables, d’autre part que la calcination permet de transformer la majorité du P qui
était sous forme non résiduelle en forme plus stable. Tout indique que l’acide phosphorique
est bien réactif envers les composants tant majoritaires que minoritaires dans les sédiments.
Le calcium
La distribution de calcium en pourcentage massique pour les sédiments traités et non
traités pour BL et BV est représentée respectivement sur la Figure IV- 5 et Figure IV- 6.
Avant calcination, nous remarquons que pour BL, l’ajout d’acide (entre 3 et 10% sur sec)
provoque la diminution de la quantité de Ca lié à F2 (de 31.1 à 13.1%) et augmentation de
F1 (de 7.7 à 12.5), F3 (de 15.8 à 33.7) et F4 (de 6.3 et 11.2). Par contre nous constatons une
diminution légère de la fraction résiduelle.
Dans BV, la tendance générale est identique. L’ajout d’acide provoque la diminution du
Ca lié à F2 et augmentation des autres fractions, par contre contrairement à BL, la

Chapitre IV : Evaluation du procédé
165
concentration de Ca lié à F5 augmente légèrement avec l’augmentation de la quantité
d’acide .
Nous avons montré lors de la présentation de la distribution du Ca dans les deux
sédiments bruts que le Ca est présent sous forme de calcite dans F2. Cela implique que la
phosphatation permet la dissolution de la calcite et la redistribution du Ca sous d’autres
formes. Cela confirme les résultats obtenus lors de la phosphatation de BV. En effet, nous
avons constaté que sous l’effet de l’acide, le Ca se solubilise dans un premier temps, puis il
précipite par la suite. Nous avons démontré qu’il précipitait sous forme de phosphate
dicalcique et pouvait se transformer progressivement lors de la maturation vers la forme
thermodynamiquement la plus stable qu’est l’hydroxyapatite.
Après calcination, nous remarquons que dans BL, le Ca présent dans F5 augmente de
32.7% pour le sédiment brut à 67.3% pour le sédiment phosphaté à 10%. Cette augmentation
est proportionnelle à l’augmentation de la quantité d’acide. Dans tous les cas,
l’augmentation de la phase résiduaire s’accompagne par la réduction des autres fractions.
Pour BV, la tendance est la même, la fraction résiduaire augmente de 35.1% pour le
sédiment brut à 68.8% pour le sédiment phosphaté à 7% (BV7). Alors que les autres
fractions diminuent. Ceci implique que pendant la phosphatation, il y a formation de
phosphates de calcium métastables et qu’après calcination, ces phosphates se transforment
en phosphates plus stables tel que l’hydroxyapatite (-Log Ksp = 38.15)
Nous constatons cependant qu’après calcination de BV brut, le Ca lié à la fraction
échangeable augmente (de 9.6% BV0 à 33.9% pour BV0C). Néanmoins après
phosphatation, nous constatons la diminution de cette fraction avec l’augmentation de la
quantité d’acide. La même remarque peut être faite pour BL, sauf que l’augmentation de la
fraction échangeable (du sédiment brut) sous l’action de la calcination est beaucoup moins
importante. Cette augmentation de F1 peut être liée à la dégradation de la calcite sous l’effet
de la température et cela conduit à la formation de CaO beaucoup plus soluble. Cette
hypothèse est confirmée par la réduction de la fraction F2 lors de la calcination de BV0.
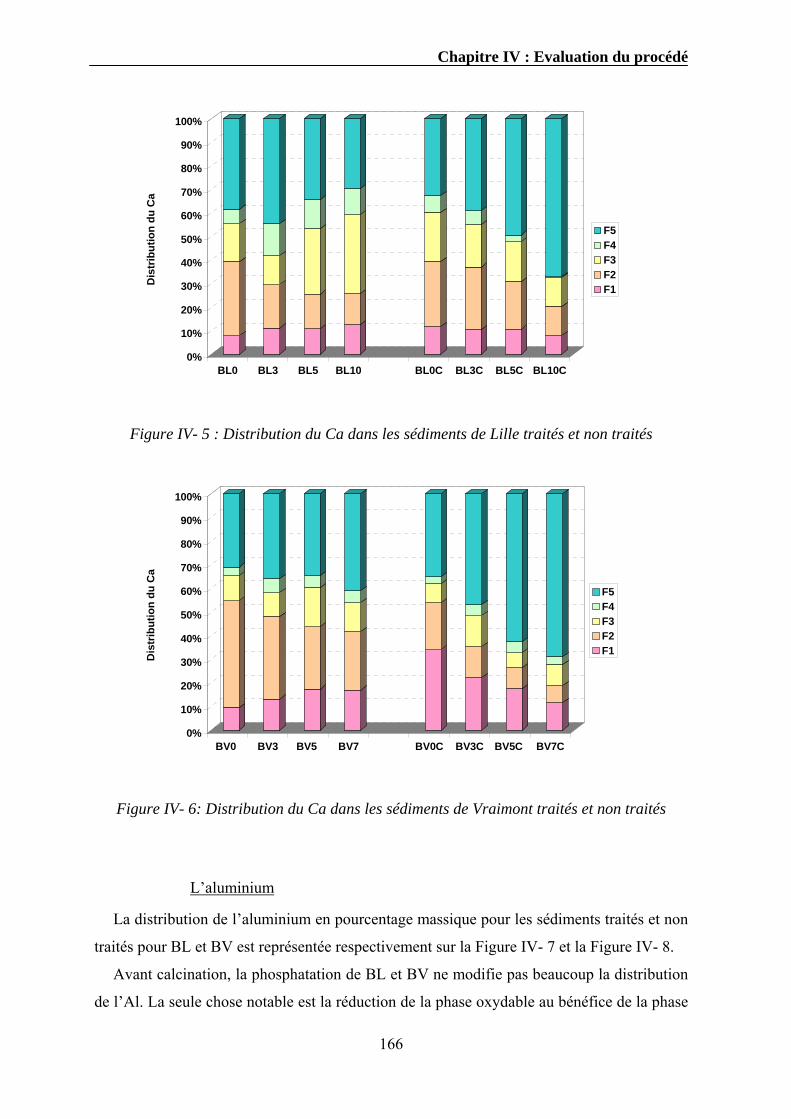
Chapitre IV : Evaluation du procédé
166
Figure IV- 5 : Distribution du Ca dans les sédiments de Lille traités et non traités
Figure IV- 6: Distribution du Ca dans les sédiments de Vraimont traités et non traités
L’aluminium
La distribution de l’aluminium en pourcentage massique pour les sédiments traités et non
traités pour BL et BV est représentée respectivement sur la Figure IV- 7 et la Figure IV- 8.
Avant calcination, la phosphatation de BL et BV ne modifie pas beaucoup la distribution
de l’Al. La seule chose notable est la réduction de la phase oxydable au bénéfice de la phase
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Ca
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Ca
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
167
résiduelle. Cela implique que l’acide phosphorique peut réagir avec l’aluminium pour le
rendre encore plus stable. Parmi les formes qui peuvent se former, on peut citer :
Al2(PO4)(OH)3, ZnAl6(PO4)4(OH)8 4H2O (Faustite) et les complexes de surfaces
La résultats de l’étude cinétique de phosphatation de BV ont permis de montrer que lors
de l’ajout de l’acide phosphorique, aucune solubilisation de l’Al n’a été enregistrée. Cela
implique que lors de la phosphatation et de la maturation, l’Al présent dans F4 a précipité
sous forme de phosphate d’Al insoluble.
Après calcination, nous constatons l’apparition d’une phase réductible qui était
inexistante dans BV0 et BL0. Cependant l’augmentation de la quantité d’acide permet sa
diminution. Cela signifie que la calcination conduit à la transformation d’une faible quantité
d’Al résiduelle en forme plus soluble, en occurrence de l’Al réductible. Par contre la
présence de P dans le sédiment permet sa stabilisation.
Figure IV- 7 : Distribution de l’Al dans les sédiments de Lille traités et non traités.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n de
l'A
l
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
168
Figure IV- 8 : Distribution de l’Al dans les sédiments de Vraimont traités et non traités
Le fer
La distribution du fer en pourcentage massique pour les sédiments traités et non traités de
BL et BV est présentée respectivement sur la Figure IV- 9 et la Figure IV- 10
Figure IV- 9 : Distribution de Fe dans les sédiments de Lille traités et non traités.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n de
l'A
l
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Fe
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
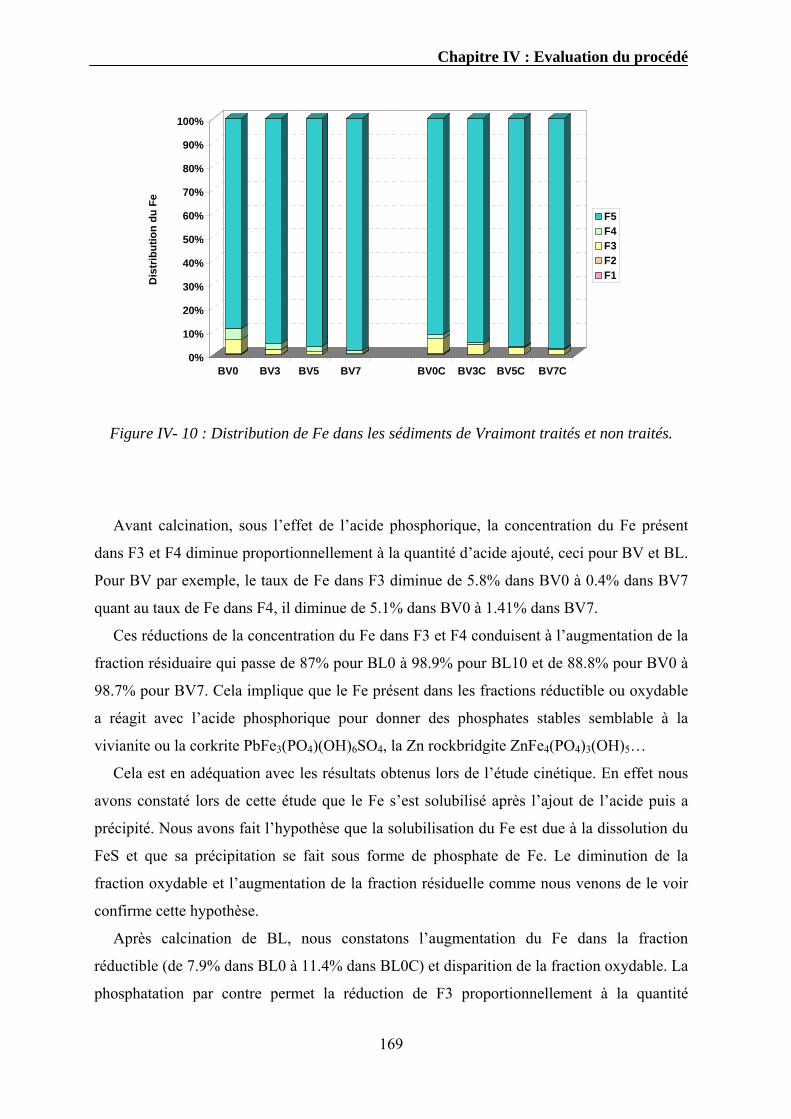
Chapitre IV : Evaluation du procédé
169
Figure IV- 10 : Distribution de Fe dans les sédiments de Vraimont traités et non traités.
Avant calcination, sous l’effet de l’acide phosphorique, la concentration du Fe présent
dans F3 et F4 diminue proportionnellement à la quantité d’acide ajouté, ceci pour BV et BL.
Pour BV par exemple, le taux de Fe dans F3 diminue de 5.8% dans BV0 à 0.4% dans BV7
quant au taux de Fe dans F4, il diminue de 5.1% dans BV0 à 1.41% dans BV7.
Ces réductions de la concentration du Fe dans F3 et F4 conduisent à l’augmentation de la
fraction résiduaire qui passe de 87% pour BL0 à 98.9% pour BL10 et de 88.8% pour BV0 à
98.7% pour BV7. Cela implique que le Fe présent dans les fractions réductible ou oxydable
a réagit avec l’acide phosphorique pour donner des phosphates stables semblable à la
vivianite ou la corkrite PbFe3(PO4)(OH)6SO4, la Zn rockbridgite ZnFe4(PO4)3(OH)5…
Cela est en adéquation avec les résultats obtenus lors de l’étude cinétique. En effet nous
avons constaté lors de cette étude que le Fe s’est solubilisé après l’ajout de l’acide puis a
précipité. Nous avons fait l’hypothèse que la solubilisation du Fe est due à la dissolution du
FeS et que sa précipitation se fait sous forme de phosphate de Fe. Le diminution de la
fraction oxydable et l’augmentation de la fraction résiduelle comme nous venons de le voir
confirme cette hypothèse.
Après calcination de BL, nous constatons l’augmentation du Fe dans la fraction
réductible (de 7.9% dans BL0 à 11.4% dans BL0C) et disparition de la fraction oxydable. La
phosphatation par contre permet la réduction de F3 proportionnellement à la quantité
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Fe
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
170
d’acide. Néanmoins, on constate que la calcination des sédiments phosphatés modifie de
réduction de 2% la phase résiduelle comparé à celle des sédiments uniquement phosphatés.
Lors de la calcination de BV, on constate que BV0C subit également une légère
augmentation de sa fraction réductible, par contre la fraction oxydable ne disparaît pas
entièrement. La phosphatation permet par contre la diminution de ces deux fractions (F3 et
F4) cela traduit la précipitation de phosphate de Fe insoluble. Au final la calcination des
sédiments phosphatés, contrairement à BL, n’a pas conduit à la modification de leur
fractions résiduelles.
IV.3.1.2.2. Les métaux lourds
Après avoir présenté la distribution des différents éléments majeurs avant et après
traitement, nous présenterons à présent le comportement des métaux lourds lors de la
phosphatation et de la calcination.
L’arsenic
La distribution de l’arsenic en pourcentage massique pour les sédiments traités et non
traités de BL et BV est présentée respectivement sur la Figure IV- 11 et la Figure IV- 12
Avant calcination, nous voyons que le phosphatation n’as pas d’effet particulier sur le
comportement de l’As dans les deux sédiments. Néanmoins l’ajout d’acide provoque une
légère augmentation de la fraction mobilisable de BV. Theodoratos et al [Theodoratos 2002]
ont observé ce type de comportement en traitant un sol avec du phosphate monobasique. En
effet ils ont remarqué une augmentation de l’arsenic lixiviable par TCLP (Toxicity
charactrestic Leaching procedure). Ils ont posé l’hypothèse que cela est dû à la similitude
chimique qui existe entre les ions phosphates et arsenates et qui peut conduire à une
substitution partielle des phosphates par des arsenates. Les analyses minéralogiques ont
révélé la présence de Pb2(AsO4)Cl solube.
La solubilisation de l’As sous l’effet de l’acide phosphorique dans BV aurait pu être
constatée lors de l’étude cinétique de phosphatation de BV. Or cela n’a pas été le cas, les
analyses de l’As en solution de lavage des sédiments phosphatés n’ont pas révélé sa
présence. Cela est probablement lié à la dilution étant donné que le sédiment prélevé est lavé
avec un rapport L/S de 20. Cela est d’autant plus probable que la mesure s’est faite pour un
sédiment humide et qu’ici, l’analyse est faite sur un sédiment sec.
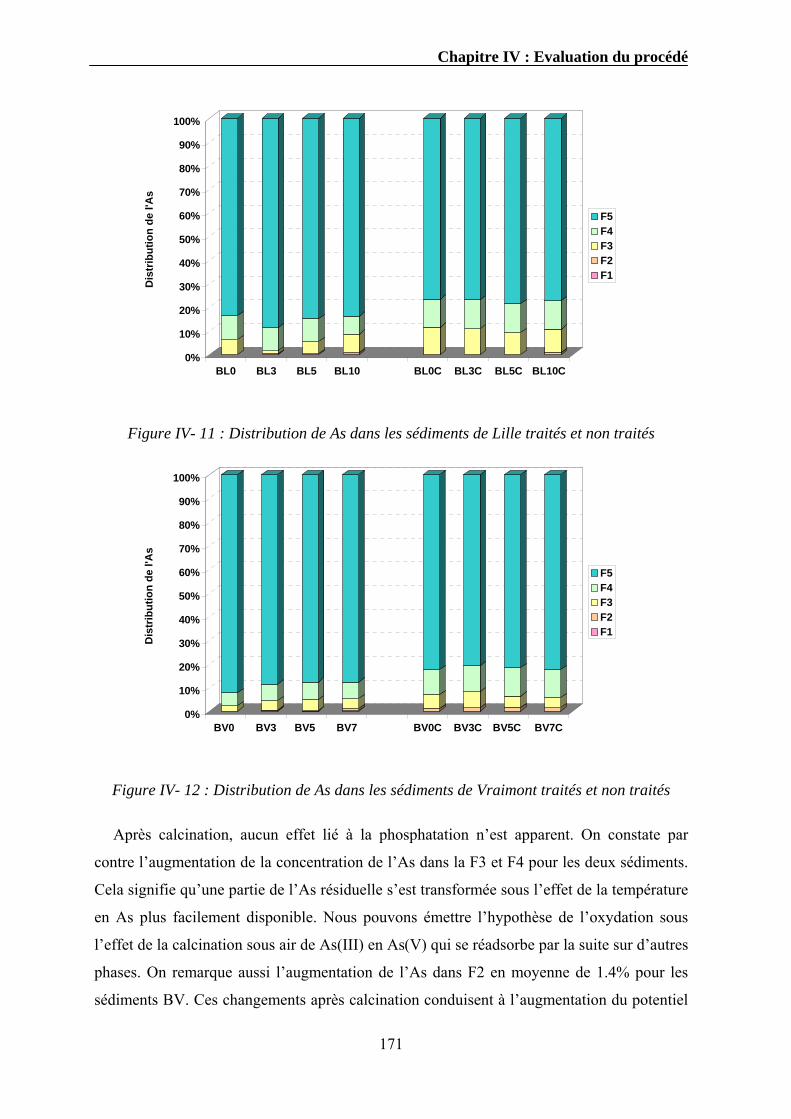
Chapitre IV : Evaluation du procédé
171
Figure IV- 11 : Distribution de As dans les sédiments de Lille traités et non traités
Figure IV- 12 : Distribution de As dans les sédiments de Vraimont traités et non traités
Après calcination, aucun effet lié à la phosphatation n’est apparent. On constate par
contre l’augmentation de la concentration de l’As dans la F3 et F4 pour les deux sédiments.
Cela signifie qu’une partie de l’As résiduelle s’est transformée sous l’effet de la température
en As plus facilement disponible. Nous pouvons émettre l’hypothèse de l’oxydation sous
l’effet de la calcination sous air de As(III) en As(V) qui se réadsorbe par la suite sur d’autres
phases. On remarque aussi l’augmentation de l’As dans F2 en moyenne de 1.4% pour les
sédiments BV. Ces changements après calcination conduisent à l’augmentation du potentiel
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n de
l'A
s
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n de
l'A
s
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
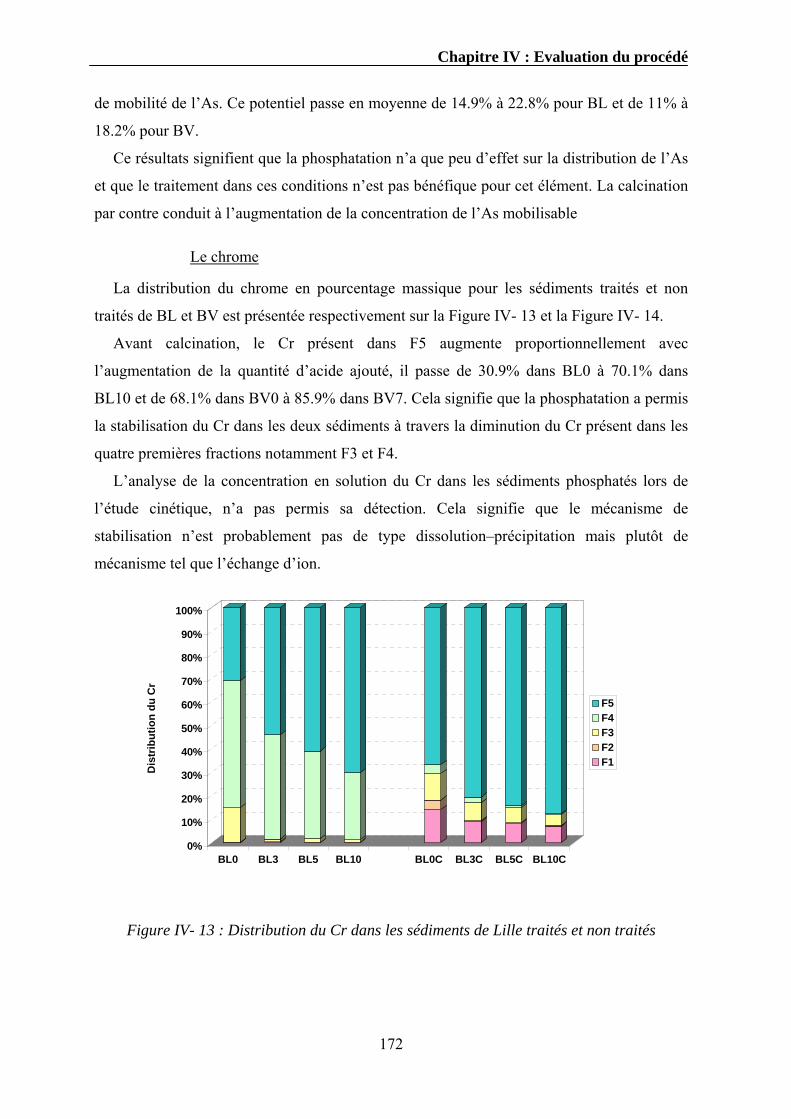
Chapitre IV : Evaluation du procédé
172
de mobilité de l’As. Ce potentiel passe en moyenne de 14.9% à 22.8% pour BL et de 11% à
18.2% pour BV.
Ce résultats signifient que la phosphatation n’a que peu d’effet sur la distribution de l’As
et que le traitement dans ces conditions n’est pas bénéfique pour cet élément. La calcination
par contre conduit à l’augmentation de la concentration de l’As mobilisable
Le chrome
La distribution du chrome en pourcentage massique pour les sédiments traités et non
traités de BL et BV est présentée respectivement sur la Figure IV- 13 et la Figure IV- 14.
Avant calcination, le Cr présent dans F5 augmente proportionnellement avec
l’augmentation de la quantité d’acide ajouté, il passe de 30.9% dans BL0 à 70.1% dans
BL10 et de 68.1% dans BV0 à 85.9% dans BV7. Cela signifie que la phosphatation a permis
la stabilisation du Cr dans les deux sédiments à travers la diminution du Cr présent dans les
quatre premières fractions notamment F3 et F4.
L’analyse de la concentration en solution du Cr dans les sédiments phosphatés lors de
l’étude cinétique, n’a pas permis sa détection. Cela signifie que le mécanisme de
stabilisation n’est probablement pas de type dissolution–précipitation mais plutôt de
mécanisme tel que l’échange d’ion.
Figure IV- 13 : Distribution du Cr dans les sédiments de Lille traités et non traités
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Cr
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
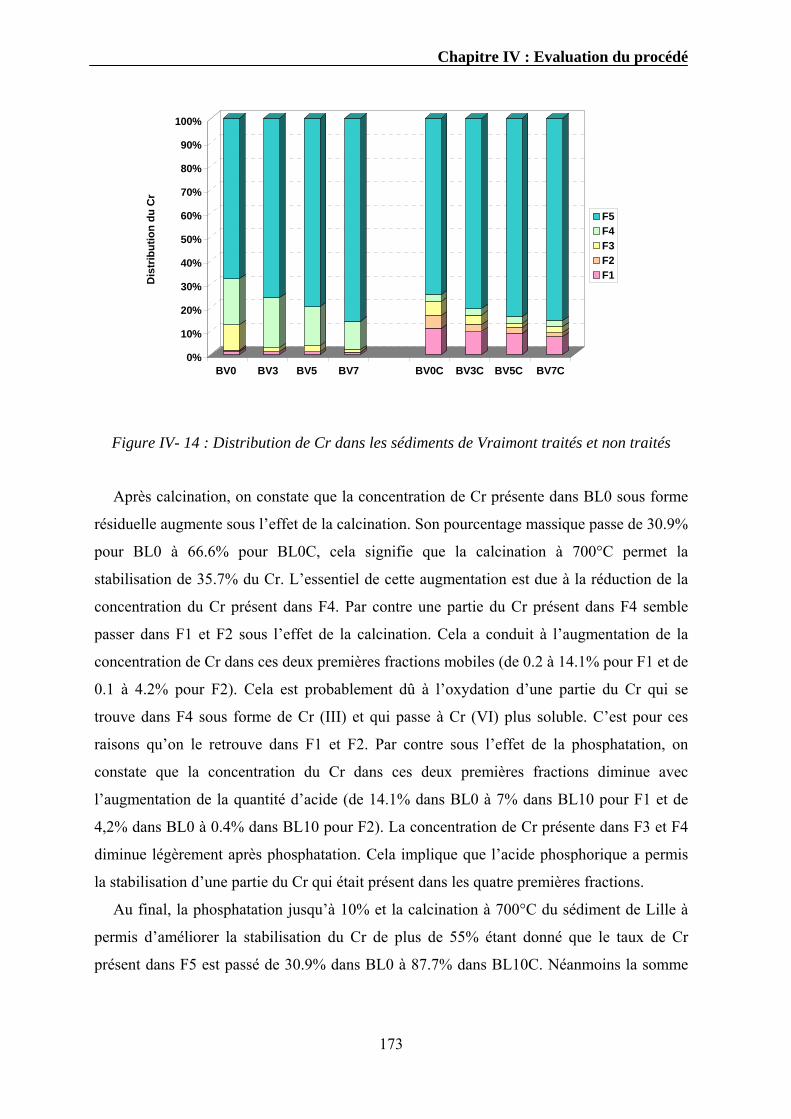
Chapitre IV : Evaluation du procédé
173
Figure IV- 14 : Distribution de Cr dans les sédiments de Vraimont traités et non traités
Après calcination, on constate que la concentration de Cr présente dans BL0 sous forme
résiduelle augmente sous l’effet de la calcination. Son pourcentage massique passe de 30.9%
pour BL0 à 66.6% pour BL0C, cela signifie que la calcination à 700°C permet la
stabilisation de 35.7% du Cr. L’essentiel de cette augmentation est due à la réduction de la
concentration du Cr présent dans F4. Par contre une partie du Cr présent dans F4 semble
passer dans F1 et F2 sous l’effet de la calcination. Cela a conduit à l’augmentation de la
concentration de Cr dans ces deux premières fractions mobiles (de 0.2 à 14.1% pour F1 et de
0.1 à 4.2% pour F2). Cela est probablement dû à l’oxydation d’une partie du Cr qui se
trouve dans F4 sous forme de Cr (III) et qui passe à Cr (VI) plus soluble. C’est pour ces
raisons qu’on le retrouve dans F1 et F2. Par contre sous l’effet de la phosphatation, on
constate que la concentration du Cr dans ces deux premières fractions diminue avec
l’augmentation de la quantité d’acide (de 14.1% dans BL0 à 7% dans BL10 pour F1 et de
4,2% dans BL0 à 0.4% dans BL10 pour F2). La concentration de Cr présente dans F3 et F4
diminue légèrement après phosphatation. Cela implique que l’acide phosphorique a permis
la stabilisation d’une partie du Cr qui était présent dans les quatre premières fractions.
Au final, la phosphatation jusqu’à 10% et la calcination à 700°C du sédiment de Lille à
permis d’améliorer la stabilisation du Cr de plus de 55% étant donné que le taux de Cr
présent dans F5 est passé de 30.9% dans BL0 à 87.7% dans BL10C. Néanmoins la somme
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Cr
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
174
des deux premières fractions les plus mobilisables à augmente de 0.3% (1.7 mg/kg sur sec)
dans BL0 à 7.5% dans BL10C (51.4mg/kg sur sec).
Les mêmes observations peuvent être faites pour BV, mais vu que la concentration de Cr
présente dans F4 dans ce cas est moins importante, la tendance après calcination est moins
évidente
Globalement, la phosphatation jusqu’à 7% et la calcination à 700°C du sédiment de Lille
a permis une augmentation du Cr stable étant donné que le taux de Cr présent dans F5 est
passé de 68.1% dans BV0 à 85.7% dans BV10C. Néanmoins la somme des deux premières
fractions plus facilement mobilisables a augmenté de 1.7% (3.3mg/kg sur sec) dans BV0 à
9.2% (17.6 mg/kg sur sec) dans BV10C.
Le cobalt
La distribution du cobalt en pourcentage massique pour les sédiments traités et non traités
de BL et BV est présentée respectivement sur la Figure IV- 15 et la Figure IV- 16.
Avant calcination, la phosphatation n’a pas d’effet particulier sur la stabilisation du Co
dans BL. En effet les fractions résiduelles sont pratiquement identiques pour les différents
sédiments, sauf pour BL10, où on constate une diminution de 7% du Co dans F5. On
constate également que le taux de Co présent dans F1 augment proportionnellement à la
quantité d’acide ajouté dans BL. En effet cette fraction, qui était inexistante dans BL0, a
atteint 13.5% dans BL10. Cela peut être lié à la solubilisation du Co présent dans F2 et F3
sous l’effet de l’acide, car le taux de Co lié à ces deux fractions a diminué.
Dans BV, on constate pratiquement le même comportement : pas d’effet de l’acide sur la
stabilisation des métaux, par contre une augmentation de la concentration de Co dans F1.
Ces résultats signifient que la phosphatation n’a aucun effet de stabilisation sur le Co,
mais qu’elle a tendance à augmenter la concentration du Co dans la fraction la plus
mobilisable (F1).
Bien que le Co dans BV existe dans les deux premières fractions, son absence dans les
solutions de lavage des différents échantillons lors de l’étude de cinétique de phosphatation
de BV est probablement due aux mêmes raisons de dilution citées précédemment.
Après calcination, on constate que le Co présent dans F2 dans BL0 à disparu dans BL0C,
et que la fraction résiduaire a augmenté de 10%. Quant aux sédiments phosphatés, leur
calcination a permis la disparition du Co de la fraction échangeable. On constate également
que l’augmentation de la quantité d’acide conduit à la réduction de la quantité de Co
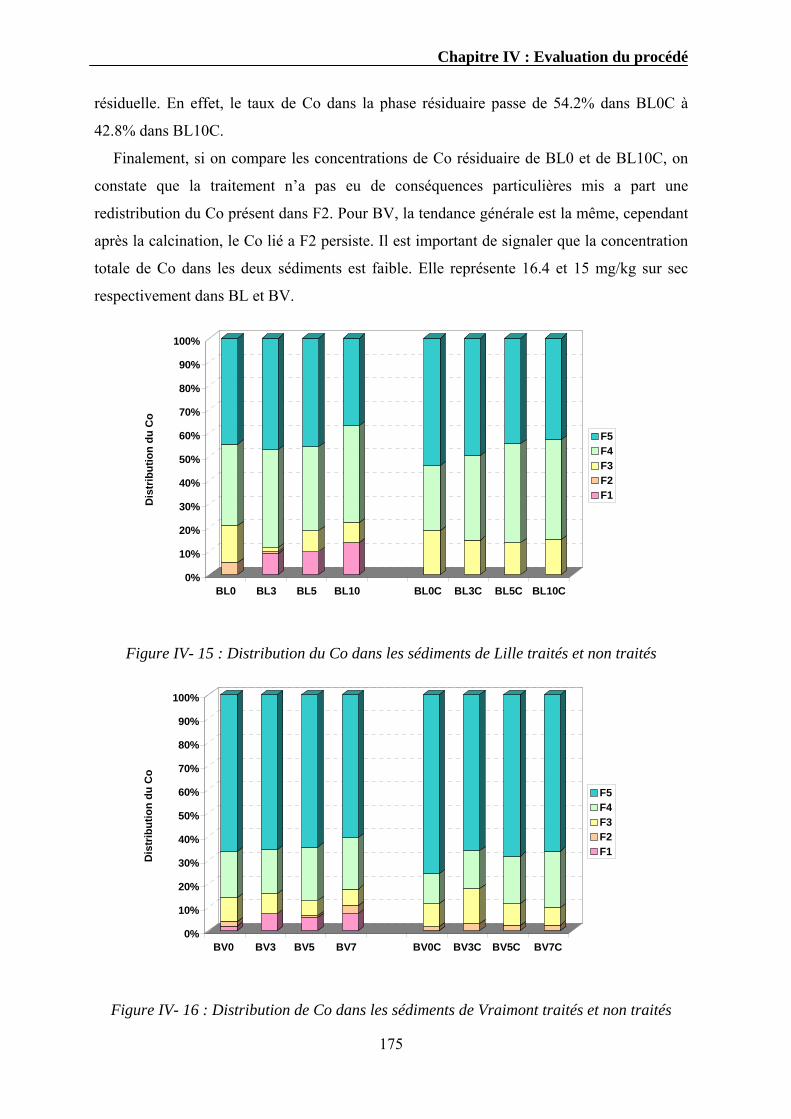
Chapitre IV : Evaluation du procédé
175
résiduelle. En effet, le taux de Co dans la phase résiduaire passe de 54.2% dans BL0C à
42.8% dans BL10C.
Finalement, si on compare les concentrations de Co résiduaire de BL0 et de BL10C, on
constate que la traitement n’a pas eu de conséquences particulières mis a part une
redistribution du Co présent dans F2. Pour BV, la tendance générale est la même, cependant
après la calcination, le Co lié a F2 persiste. Il est important de signaler que la concentration
totale de Co dans les deux sédiments est faible. Elle représente 16.4 et 15 mg/kg sur sec
respectivement dans BL et BV.
Figure IV- 15 : Distribution du Co dans les sédiments de Lille traités et non traités
Figure IV- 16 : Distribution de Co dans les sédiments de Vraimont traités et non traités
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Co
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Co
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
176
Le cadmium
La distribution du cadmium en pourcentage massique pour les sédiments traités et non
traités de BL et BV est présentée respectivement sur la Figure IV- 17 et la Figure IV- 18
Avant calcination, l’effet de la phosphatation sur le comportement du Cd n’a pas été
clairement mis en évidence. La concentration de Cd présente dans la fraction résiduelle est
pratiquement identique pour les différents sédiments, sauf pour le sédiment phosphaté à 3%
où l’on observe une augmentation de 10%, de cette fraction grâce sans doute à la faible
baisse du pH. On constate également l’apparition de Cd dans F1 suite à la phosphatation.
Elle peut être en partie attribuée à la libération du Cd présent lors de la décarbonatation de
F2.
Quant à BV, la tendance n’est pas très claire et la phosphatation ne semble pas avoir eu
un effet particulier sur le Cd. On constate néanmoins une légère diminution de la quantité de
Cd dans F1 avec l’augmentation de la quantité d’acide. Comme pour les autres métaux, le
Cd présent dans la fraction échangeable n’a pas pu être mesuré lors de l’étude cinétique dans
les solutions de lavage du sédiment brut et dans celle des sédiments phosphatés. Cela est
probablement dû aux mêmes raisons citées pour les autres métaux.
Après calcination, les tendances sont différentes pour les deux sédiments. Pour BL, la
concentration de Cd présente dans F5 augmente sous l’effet de la calcination. Elle passe de
59.3% pour BL0 à 76.6% pour BL0C, ceci est dû en grande partie à la réduction du Cd qui
se trouve dans F3. Cette tendance augmente avec l’augmentation de la quantité d’acide. En
effet pour une phosphatation de 10% la quantité de Cd présent dans la fraction résiduelle
augmente à 94.4%. Ces résultats signifient que la phosphatation jusqu’à 10% d’acide et une
calcination à 700°C permet l’amélioration de 35% la fraction du Cd stable. Cette
stabilisation de Cd après calcination, et pas avant, peut être due à la précipitation de
phosphate de Cd amorphe qui est ensuite stabilisé. Après calcination, ces phosphates se sont
transformés en espèce plus stables tels que Cd5(PO4)3OH (-Log ksp = 42.49) et Cd3(PO4)2 ,
(-Log ksp = 32.60).
Quant aux sédiments de Vraimont, après la calcination on constate que le Cd présent dans
la fraction résiduelle augmente. Le taux de Cd passe de 51.7% dans BV0 à 64.5% dans
BV0C. Par contre, après phosphatation cette fraction est variable. Elle va de 50.1% pour
BV5C à 69.5% pour BV7C.
Ces résultats montrent qu’il a été difficile de bien comprendre le comportement du Cd
dans BV suite au traitement. Cela est probablement dû a sa très faible concentration dans le
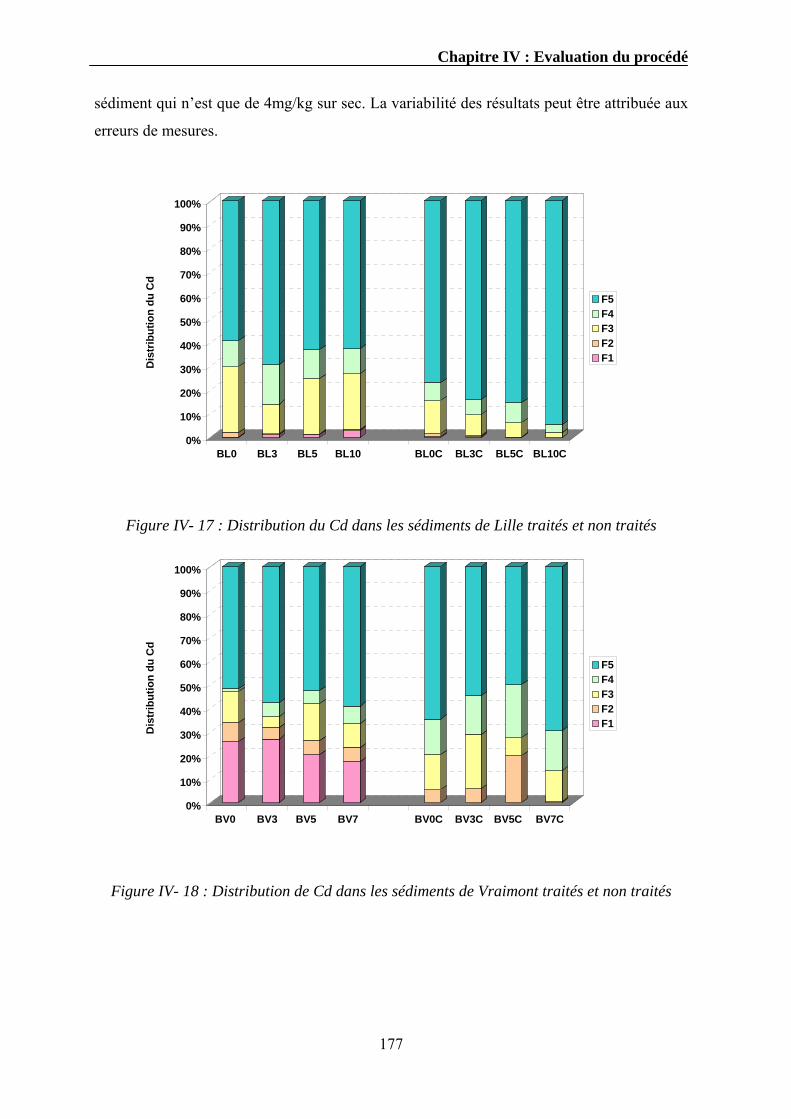
Chapitre IV : Evaluation du procédé
177
sédiment qui n’est que de 4mg/kg sur sec. La variabilité des résultats peut être attribuée aux
erreurs de mesures.
Figure IV- 17 : Distribution du Cd dans les sédiments de Lille traités et non traités
Figure IV- 18 : Distribution de Cd dans les sédiments de Vraimont traités et non traités
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Cd
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Cd
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1
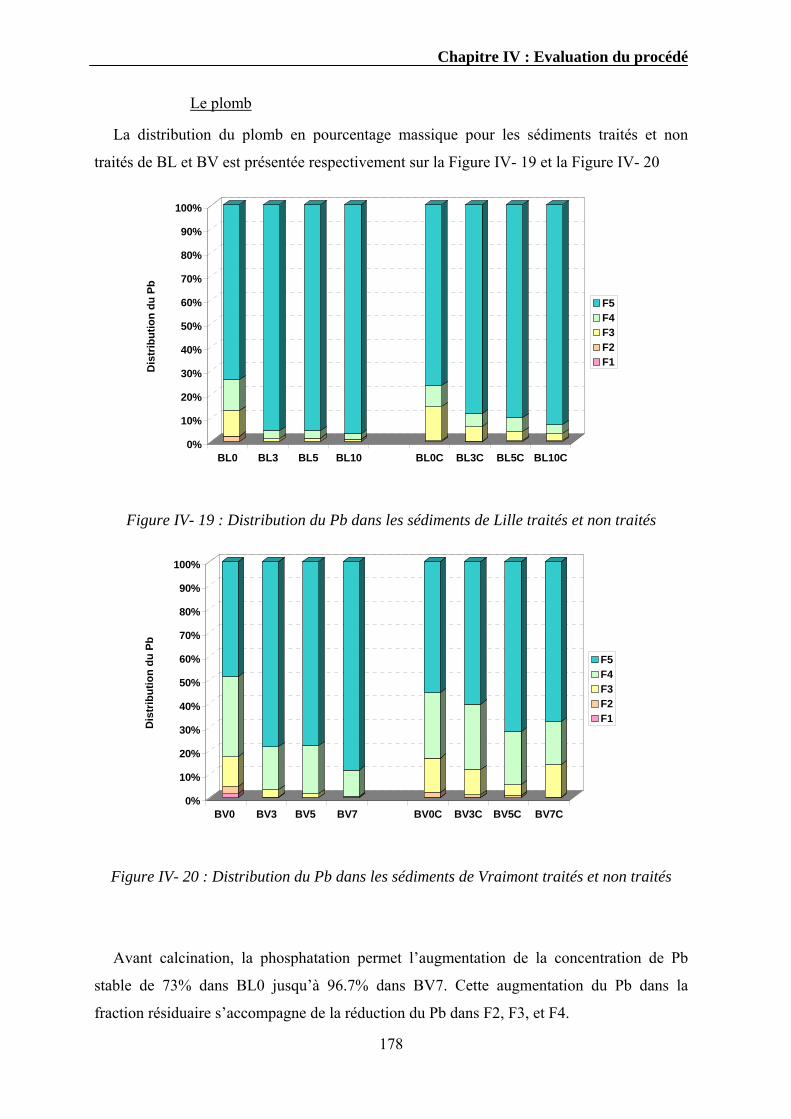
Chapitre IV : Evaluation du procédé
178
Le plomb
La distribution du plomb en pourcentage massique pour les sédiments traités et non
traités de BL et BV est présentée respectivement sur la Figure IV- 19 et la Figure IV- 20
Figure IV- 19 : Distribution du Pb dans les sédiments de Lille traités et non traités
Figure IV- 20 : Distribution du Pb dans les sédiments de Vraimont traités et non traités
Avant calcination, la phosphatation permet l’augmentation de la concentration de Pb
stable de 73% dans BL0 jusqu’à 96.7% dans BV7. Cette augmentation du Pb dans la
fraction résiduaire s’accompagne de la réduction du Pb dans F2, F3, et F4.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Pb
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Pb
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
179
Pour le sédiment de Vraimont, la même tendance est observée. Après phosphatation la
fraction résiduelle augmente de 48.8% pour BV0 à 88.3% pour BV7. L’absence du Pb des
solutions de lavage des sédiments lors de l’étude cinétique peut être due au fait que le
processus de stabilisation se fait par piégeage direct du Pb à la surface des phosphates
formés. L’hydroxypyromorphite (-Log Ksp= 62.80) est un des phosphates qui peut se former
en présence de ce metal. En effet plusieurs auteurs ont montré sa précipitation lors de la
stabilisation de Pb par des phosphates dans des sols pollués [Hettiarachchi 2001 ; Basta
2004 ; Cotter-Howwells 1995]. Ma et Rao ont [Ma L. Q 1999] ont montré lorsqu’ils ont
appliqué l’extraction séquentielle de Tessier à certains sols, que l’hydroxypyromorphite se
trouvait dans la fraction résiduaire, toujours très stable. D’autres phosphates de plomb
peuvent se former lors de la phosphatation, en particulier : Pb3(PO4)2 (-Log Ksp= 62.80),
Pb4O(PO4)2 (-Log Ksp= 36.86), et des solutions solides avec les phosphates de calcium.
Après calcination, la fraction résiduaire du sédiment brut (BL0C) est pratiquement
identique à celle du sédiment brut avant calcination (BL0). Après phosphatation par contre,
cette fraction diminue avec la quantité d’acide (de 76.6% pour BL0C à 93.7% pour BL10C).
Néanmoins, ces fractions résiduaires des sédiments phosphatés après calcination sont plus
faibles que celles des sédiments uniquement phosphatés (réduction moyenne de 5%).
Pour BV, la tendance est proche de celle de BL. La calcination du sédiment brut permet
une augmentation du Pb présent dans F5 de 48.8% dans BV0 à 55.5% pour BV0C. Après
phosphatation, cette fraction augmente avec la quantité d’acide. Par contre elle est moins
importante que celle des sédiments phosphatés et non calcinés. La calcination réduit le taux
de Pb présent dans F5 en moyenne de 15%. Sachant que les phosphates de plomb seuls sont
théoriquement stables, nos pouvons émettre l’hypothèse que la calcination contribue à une
redistribution de phosphate mixtes en faveurs d’autres éléments au détriment du plomb qui
reste tout de même bien stabilisé.
Le zinc
La distribution du zinc en pourcentage massique pour les sédiments traités et non traités
de BL et BV est présentée respectivement sur la Figure IV- 21 et la Figure IV- 22.
Avant calcination, l’ajout d’acide phosphorique à BL conduit à l’augmentation de la
concentration de Zn dans F1 et sa diminution dans F2. La concentration de Zn solubilisé
dans F2 solubilisé étant plus importante que celle récupérée en F1, nous pouvons déduire
que l’autre partie de F2 s’est redistribuée sous d’autres formes (F3 et F4). Les fractions
résiduaires des différents sédiments BL (brut et traités) étant pratiquement identiques sauf
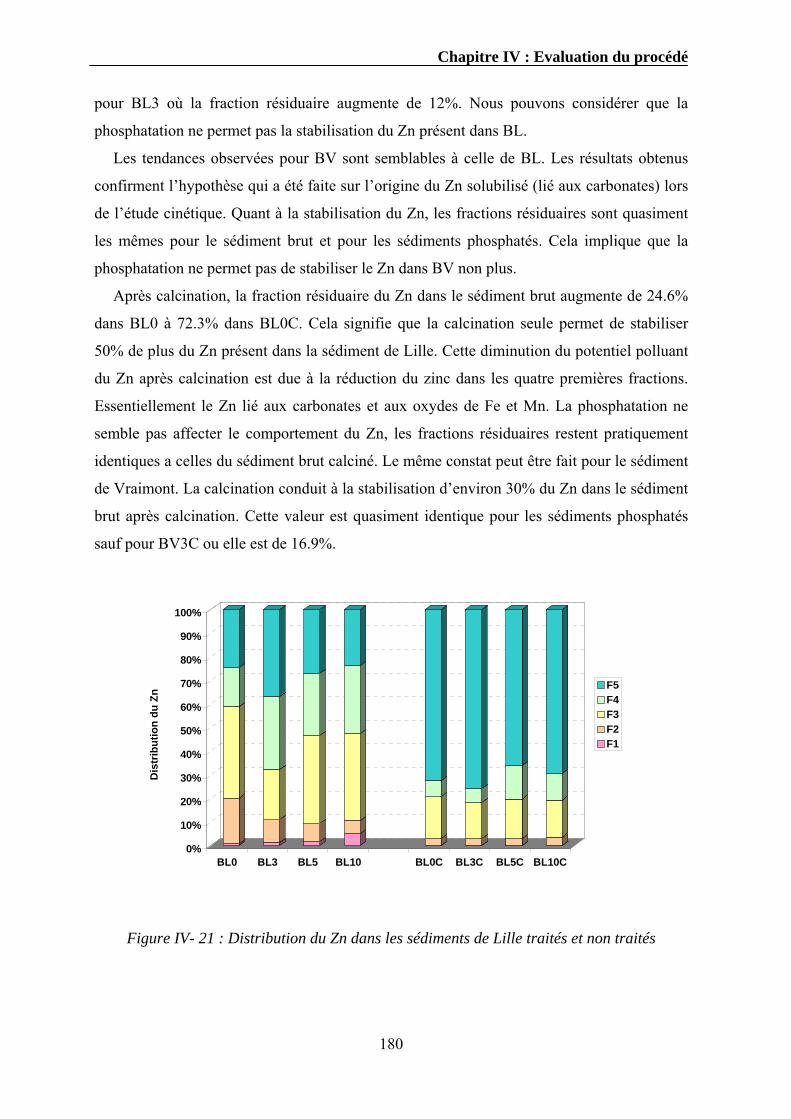
Chapitre IV : Evaluation du procédé
180
pour BL3 où la fraction résiduaire augmente de 12%. Nous pouvons considérer que la
phosphatation ne permet pas la stabilisation du Zn présent dans BL.
Les tendances observées pour BV sont semblables à celle de BL. Les résultats obtenus
confirment l’hypothèse qui a été faite sur l’origine du Zn solubilisé (lié aux carbonates) lors
de l’étude cinétique. Quant à la stabilisation du Zn, les fractions résiduaires sont quasiment
les mêmes pour le sédiment brut et pour les sédiments phosphatés. Cela implique que la
phosphatation ne permet pas de stabiliser le Zn dans BV non plus.
Après calcination, la fraction résiduaire du Zn dans le sédiment brut augmente de 24.6%
dans BL0 à 72.3% dans BL0C. Cela signifie que la calcination seule permet de stabiliser
50% de plus du Zn présent dans la sédiment de Lille. Cette diminution du potentiel polluant
du Zn après calcination est due à la réduction du zinc dans les quatre premières fractions.
Essentiellement le Zn lié aux carbonates et aux oxydes de Fe et Mn. La phosphatation ne
semble pas affecter le comportement du Zn, les fractions résiduaires restent pratiquement
identiques a celles du sédiment brut calciné. Le même constat peut être fait pour le sédiment
de Vraimont. La calcination conduit à la stabilisation d’environ 30% du Zn dans le sédiment
brut après calcination. Cette valeur est quasiment identique pour les sédiments phosphatés
sauf pour BV3C ou elle est de 16.9%.
Figure IV- 21 : Distribution du Zn dans les sédiments de Lille traités et non traités
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Zn
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
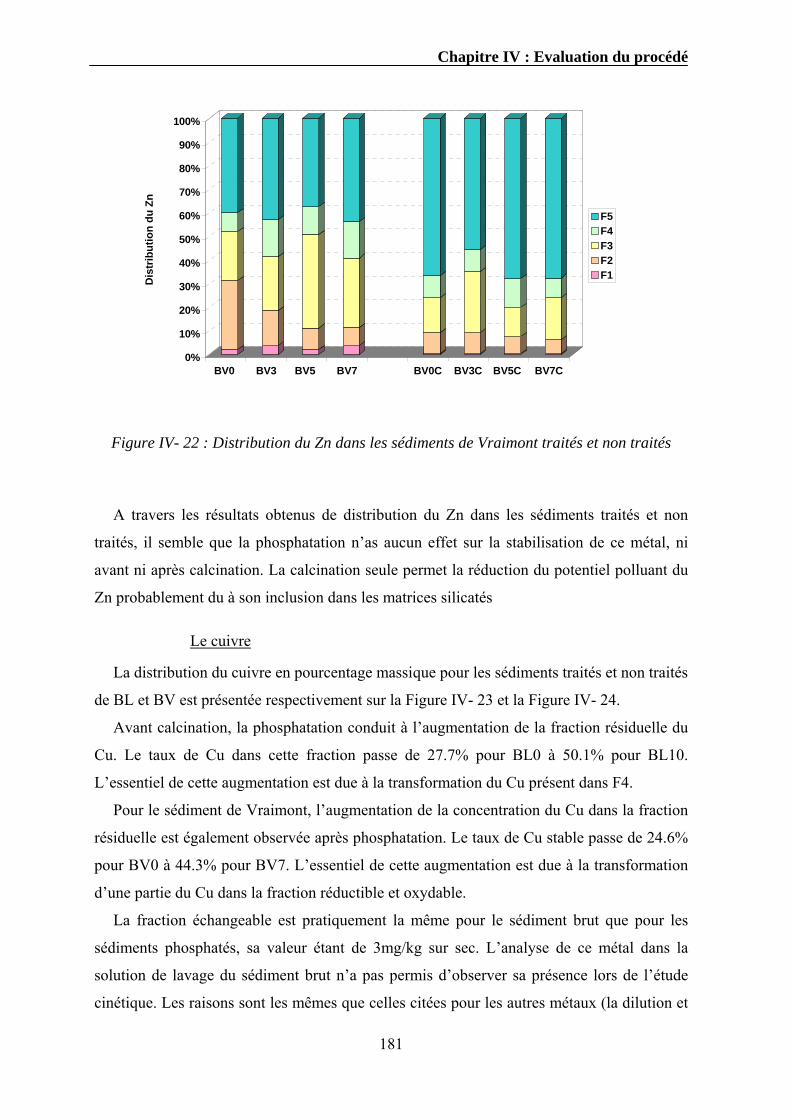
Chapitre IV : Evaluation du procédé
181
Figure IV- 22 : Distribution du Zn dans les sédiments de Vraimont traités et non traités
A travers les résultats obtenus de distribution du Zn dans les sédiments traités et non
traités, il semble que la phosphatation n’as aucun effet sur la stabilisation de ce métal, ni
avant ni après calcination. La calcination seule permet la réduction du potentiel polluant du
Zn probablement du à son inclusion dans les matrices silicatés
Le cuivre
La distribution du cuivre en pourcentage massique pour les sédiments traités et non traités
de BL et BV est présentée respectivement sur la Figure IV- 23 et la Figure IV- 24.
Avant calcination, la phosphatation conduit à l’augmentation de la fraction résiduelle du
Cu. Le taux de Cu dans cette fraction passe de 27.7% pour BL0 à 50.1% pour BL10.
L’essentiel de cette augmentation est due à la transformation du Cu présent dans F4.
Pour le sédiment de Vraimont, l’augmentation de la concentration du Cu dans la fraction
résiduelle est également observée après phosphatation. Le taux de Cu stable passe de 24.6%
pour BV0 à 44.3% pour BV7. L’essentiel de cette augmentation est due à la transformation
d’une partie du Cu dans la fraction réductible et oxydable.
La fraction échangeable est pratiquement la même pour le sédiment brut que pour les
sédiments phosphatés, sa valeur étant de 3mg/kg sur sec. L’analyse de ce métal dans la
solution de lavage du sédiment brut n’a pas permis d’observer sa présence lors de l’étude
cinétique. Les raisons sont les mêmes que celles citées pour les autres métaux (la dilution et
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Zn
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1
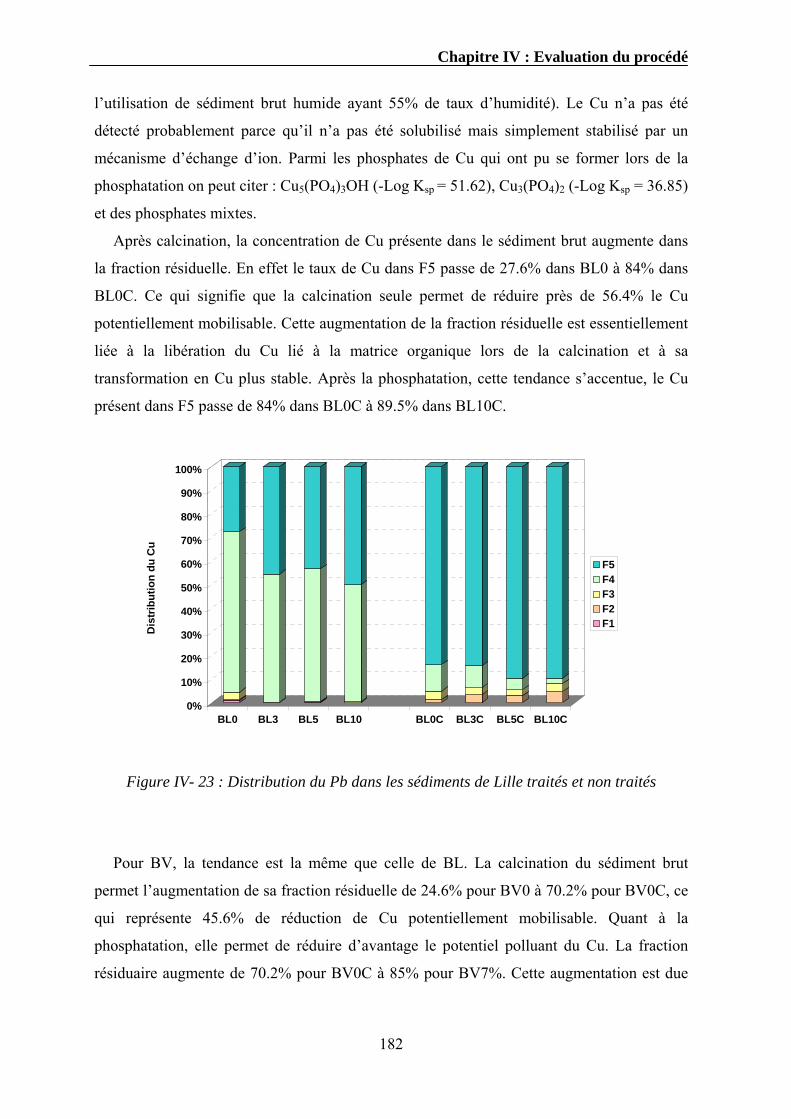
Chapitre IV : Evaluation du procédé
182
l’utilisation de sédiment brut humide ayant 55% de taux d’humidité). Le Cu n’a pas été
détecté probablement parce qu’il n’a pas été solubilisé mais simplement stabilisé par un
mécanisme d’échange d’ion. Parmi les phosphates de Cu qui ont pu se former lors de la
phosphatation on peut citer : Cu5(PO4)3OH (-Log Ksp = 51.62), Cu3(PO4)2 (-Log Ksp = 36.85)
et des phosphates mixtes.
Après calcination, la concentration de Cu présente dans le sédiment brut augmente dans
la fraction résiduelle. En effet le taux de Cu dans F5 passe de 27.6% dans BL0 à 84% dans
BL0C. Ce qui signifie que la calcination seule permet de réduire près de 56.4% le Cu
potentiellement mobilisable. Cette augmentation de la fraction résiduelle est essentiellement
liée à la libération du Cu lié à la matrice organique lors de la calcination et à sa
transformation en Cu plus stable. Après la phosphatation, cette tendance s’accentue, le Cu
présent dans F5 passe de 84% dans BL0C à 89.5% dans BL10C.
Figure IV- 23 : Distribution du Pb dans les sédiments de Lille traités et non traités
Pour BV, la tendance est la même que celle de BL. La calcination du sédiment brut
permet l’augmentation de sa fraction résiduelle de 24.6% pour BV0 à 70.2% pour BV0C, ce
qui représente 45.6% de réduction de Cu potentiellement mobilisable. Quant à la
phosphatation, elle permet de réduire d’avantage le potentiel polluant du Cu. La fraction
résiduaire augmente de 70.2% pour BV0C à 85% pour BV7%. Cette augmentation est due
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Cu
BL0 BL3 BL5 BL10 BL0C BL3C BL5C BL10C
F5F4F3F2F1
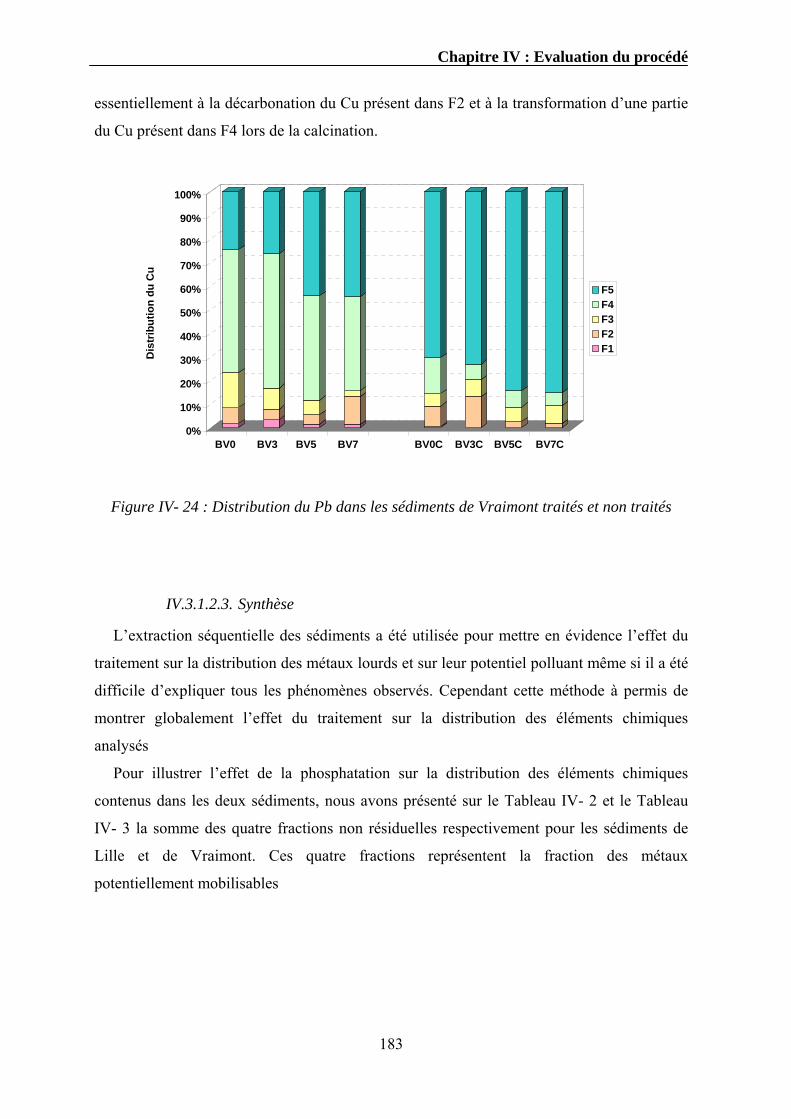
Chapitre IV : Evaluation du procédé
183
essentiellement à la décarbonation du Cu présent dans F2 et à la transformation d’une partie
du Cu présent dans F4 lors de la calcination.
Figure IV- 24 : Distribution du Pb dans les sédiments de Vraimont traités et non traités
IV.3.1.2.3. Synthèse
L’extraction séquentielle des sédiments a été utilisée pour mettre en évidence l’effet du
traitement sur la distribution des métaux lourds et sur leur potentiel polluant même si il a été
difficile d’expliquer tous les phénomènes observés. Cependant cette méthode à permis de
montrer globalement l’effet du traitement sur la distribution des éléments chimiques
analysés
Pour illustrer l’effet de la phosphatation sur la distribution des éléments chimiques
contenus dans les deux sédiments, nous avons présenté sur le Tableau IV- 2 et le Tableau
IV- 3 la somme des quatre fractions non résiduelles respectivement pour les sédiments de
Lille et de Vraimont. Ces quatre fractions représentent la fraction des métaux
potentiellement mobilisables
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Dis
trib
utio
n du
Cu
BV0 BV3 BV5 BV7 BV0C BV3C BV5C BV7C
F5F4F3F2F1

Chapitre IV : Evaluation du procédé
184
Tableau IV- 2 : Sommes des concentrations (mg/kg de matière sèche) des quatre fractions non résiduelles pour les sédiments de Lille traités et non traités
Tableau IV- 3 : Sommes des concentrations (mg/kg de matière sèche) quatre fractions non résiduelles pour les sédiments de Vraimont traités et non traités
Différentes remarques peuvent être faites des résultats obtenus par l’extraction
séquentielle :
le traitement à permis de réduire la concentration du Ca, Pb, Cu, Zn et Cr des fractions
non résiduelles au profit de la fraction résiduelle se qui peut être due à la formation de
phosphates de calcium et des phosphates métalliques stables. En effet, nous avons constaté
que l’essentiel du phosphore qui a été injecté se trouve également dans la fraction résiduelle.
Cependant, il paraît clair que dans le cas des ces éléments (Ca, Pb, Cu, Zn et Cr), leur
stabilisation est due aux effets combinés de la phosphatation et de la calcination. Par contre
la stabilisation du Zn est uniquement due à la calcination. Concernant le Cr, bien que la
calcination permet de réduire sa concentration dans les fractions non résiduelles, elle
présente l’inconvénient d’augmenter sa proportion dans la fraction la plus soluble (fraction
des métaux échangeables) se qui peut poser de réels problèmes environnementaux.
Néanmoins, l’ajout de l’acide phosphorique permet la réduction de la concentration de Cr
présent dans cette fraction.
Pour l’As, la phosphatation n’a aucun effet sur BL, par contre elle conduit à une légère
augmentation de sa fraction mobilisable dans BV. Quant à la calcination, elle augmente sa
Al As Ca Cd Co Cr Cu Fe P Pb ZnBL0 2126,3 24,5 33738,9 15,3 9,0 472,2 310,6 4165,6 718,1 167,6 3508,2BL3 811,1 17,0 30396,2 11,6 8,6 315,2 233,6 1005,7 2810,8 29,6 2946,7BL5 899,7 23,0 35976,0 13,9 8,9 267,5 243,6 804,5 6989,5 29,3 3393,5BL10 832,4 24,3 38503,0 14,1 10,3 204,7 214,0 347,9 21710,4 21,3 3550,9
BL0C 3346,0 35,0 36903,1 8,8 7,5 228,2 68,8 3715,8 1055,9 150,2 1289,0BL3C 1765,0 34,8 33287,1 5,9 8,3 133,2 67,5 2217,4 2053,2 75,7 1122,5BL5C 877,4 32,4 27677,4 5,6 9,1 109,1 45,1 1596,2 3150,0 64,6 1576,7BL10C 479,7 33,8 17921,2 2,1 9,4 84,4 45,1 1017,6 3753,4 44,7 1413,7
Al As Ca Cd Co Cr Cu Fe P Pb ZnBV0 1291,7 13,1 18861,0 1,9 5,0 61,1 107,6 3692,4 763,9 109,8 603,3BV3 1234,2 18,8 17552,0 1,7 5,2 46,0 105,5 1488,7 3416,3 46,2 573,0BV5 1028,5 20,1 17855,0 1,9 5,3 38,4 80,1 1115,8 6602,5 47,5 626,2BV7 861,3 20,2 16113,4 1,6 5,9 27,0 79,5 598,2 6794,5 25,2 565,8
BV0C 3545,3 28,7 17748,6 1,4 3,7 48,5 42,5 2753,2 906,0 95,2 333,6BV3C 2136,3 31,6 14490,2 1,8 5,1 36,8 38,5 1659,9 2399,7 85,0 445,9BV5C 1725,3 30,2 10221,0 2,0 4,8 30,8 22,7 1135,2 2749,8 60,0 321,8BV7C 1326,3 28,9 8528,0 1,2 5,1 27,5 21,4 837,7 2536,1 68,7 323,6

Chapitre IV : Evaluation du procédé
185
concentration dans les fractions non résiduelles. De la même façon que pour le Cr,
l’optimisation de la calcination en cours dans les travaux menés dans notre laboratoire
permettra probablement de mieux comprendre et expliquer les processus observés. Cela
permettra de mieux adapter le traitement aux attentes sur les produits finaux issus du
traitement
le comportement du Cd est différent dans les deux sédiments. Dans BV, aucun effet de
stabilisation n’est notable par contre dans BL, le traitement à conduit à le stabiliser
probablement sous forme de phosphate étant donnée que la diminution de la mobilité
augmente avec la quantité d’acide.
Le traitement des deux sédiments ne semble pas avoir d’effet particulier sur la
stabilisation du Co.
Le Fe et l’Al présents dans les fractions non résiduelles agissent avec le phosphore pour
former probablement des phosphates stables. Par contre la calcination conduit dans certains
cas à l’augmentation de ces éléments dans les fractions non résiduelles (fraction réductible et
oxydable).
Les résultats obtenus avec les deux sédiments à taux d’acide phosphorique de 5%
montrent que le sédiment de Vraimont comporte un risque de pollution inférieur à celui de
Lille. Cela d’une part parce que la concentration des métaux mobilisables pour BL est plus
importante que BV d’autre part parce que la distribution des métaux dans les deux sédiments
n’est pas identique et donc les processus qui s’y déroulent ne sont pas les mêmes. De plus la
composition des sédiments est différente dans la mesure où BL présente un pourcentage en
matière organique de soufre et en carbonates plus important que celui de BV.
Les mécanismes de dissolution-précipitation et d’échange d’ions ont été mis en évidence
comme étant les processus gouvernant le formation de phosphates métalliques utiles pour la
stabilisation des métaux lourds. ces résultats sont en adéquation avec ceux publiés dans la
littérature concernant le traitement des sols aux phosphates.
IV.3.2. Micro-cartographie des éléments
L’objectif de ces mesures est de mieux comprendre les résultats obtenus lors de
l’extraction séquentielle. Notamment dans l’identification des formes et des associations
chimiques des polluants dans les sédiments.
L’analyse microsonde a été effectuée sur le sédiment brut et le sédiment phosphaté à 5%
sur sec, séché puis calciné à 700°C. L’objectif étant d’une part la détermination des
associations minéralogiques des sédiments, d’autre part, l’évaluation de l’influence du

Chapitre IV : Evaluation du procédé
186
traitement sur le comportement des différentes phases et la détection de nouvelles phases
susceptibles d’être formées lors de la phosphatation.
Différents éléments chimiques contenus dans les sédiments de Vraimont ont été analysés
en microsonde au BRGM : Al, Si, Fe, Ca, P, S, Zn, Pb, Cu, Cr. Les micro-cartographies
obtenues permettent de définir la répartition de ces éléments dans le sédiment, ainsi, la
superposition de ces cartographies permet une mise en évidence des associations
minéralogiques. A partir des cartographies des éléments, des analyses quantitatives linéaires
de particules ont été effectuées. Les éléments quantifiés sont Al, Ca, Cu, Cr, Fe, K, Mg, Mn,
Na, Pb S, Si, Ti, Zn. Les résultats des analyses sont exprimés sous forme d’oxyde. A partir
de ces résultats, les formules structurales des différentes particules analysées peuvent être
calculées (Annexe A). L'objectif est de passer de la composition en % poids d'oxyde donnée
par la microsonde en fraction molaire. Le nombre de point d’analyse étant limité à 100
points, le choix des particules analysées s’est focalisé sur les associations de phosphates
ainsi que sur les associations silicoaluminieuses. Il est important de signaler qu’il ne s’agit
pas d’un échantillonnage rigoureux de toutes les phases minérales présentes dans les
sédiments.
Les faibles concentrations des métaux lourds (Zn, Pb, Cu, Cr) dans les sédiments
(<0.1%), n’a pas permis de les détecter et par conséquent de les micro-carographier. Ce qui
implique que leur associations minéralogiques n’ont pas pu être définies par cette méthode.
Les micro-cartographies de Al, Si, Fe, S, Ca et P contenus dans le sédiment brut ainsi
qu’une micro-cartographie globale représentant leur superposition sont représentées sur la
Figure IV- 25. Les cartographies des éléments montrent que les éléments majeurs sont bien
réparties dans l’échantillon, le soufre par contre l’est moins et ceci probablement du à sa
faible concentration.
Les résultats de l’analyse quantitative a permis la mise en évidence de phases
prédominantes tels que : silice, feldspath, calcite et mica. En effet les trois premières phases
ont déjà été identifiées par DRX (Chapitre II). D’autres phases moins importantes ont
également été détectées mais qui ne sont pas toutes présentées sur la légende de la
cartographie globale. Parmi ces phases ils existent : les oxydes de fer et d’aluminium,
pyroxènes, amphiboles, plagioclases et en plus faible fréquence les sulfates de calcium et les
phosphates de calcium.

Chapitre IV : Evaluation du procédé
187
Al Si
P Ca
Fe S
Superposition
Figure IV- 25 : Analyse en microsonde du Al, Si, P, Ca, Fe et S dans le sédiment de Vraimont. Image traitée en couleur arbitraire. Echelle des couleurs correspondant aux
concentrations relatives des éléments : rouge = maximum, violet = minimum et noir = inférieur au minimum. Superposition des différentes cartographie
Silice
Phosphate de calciumCalciteSulfate de calcium
Feldspath
Oxyde de fer
MicaOxyde d’aluminium
Silice
Phosphate de calciumCalciteSulfate de calcium
Feldspath
Oxyde de fer
MicaOxyde d’aluminium
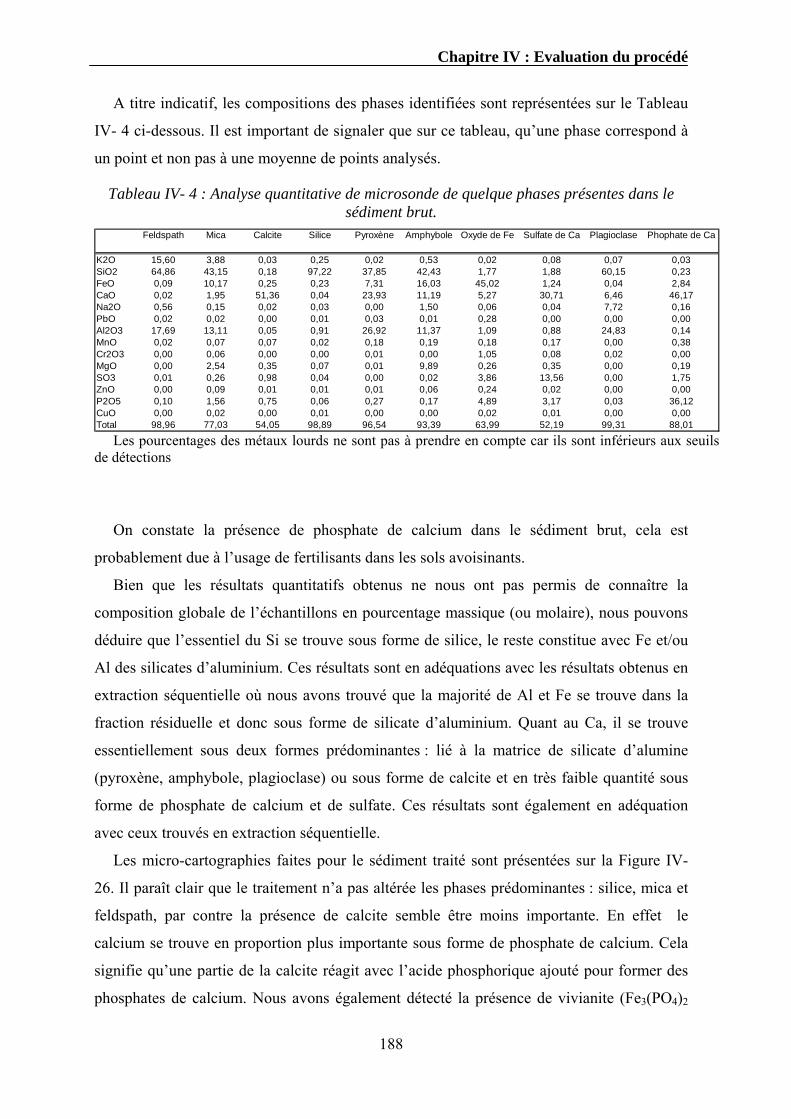
Chapitre IV : Evaluation du procédé
188
A titre indicatif, les compositions des phases identifiées sont représentées sur le Tableau
IV- 4 ci-dessous. Il est important de signaler que sur ce tableau, qu’une phase correspond à
un point et non pas à une moyenne de points analysés.
Tableau IV- 4 : Analyse quantitative de microsonde de quelque phases présentes dans le sédiment brut.
Les pourcentages des métaux lourds ne sont pas à prendre en compte car ils sont inférieurs aux seuils de détections
On constate la présence de phosphate de calcium dans le sédiment brut, cela est
probablement due à l’usage de fertilisants dans les sols avoisinants.
Bien que les résultats quantitatifs obtenus ne nous ont pas permis de connaître la
composition globale de l’échantillons en pourcentage massique (ou molaire), nous pouvons
déduire que l’essentiel du Si se trouve sous forme de silice, le reste constitue avec Fe et/ou
Al des silicates d’aluminium. Ces résultats sont en adéquations avec les résultats obtenus en
extraction séquentielle où nous avons trouvé que la majorité de Al et Fe se trouve dans la
fraction résiduelle et donc sous forme de silicate d’aluminium. Quant au Ca, il se trouve
essentiellement sous deux formes prédominantes : lié à la matrice de silicate d’alumine
(pyroxène, amphybole, plagioclase) ou sous forme de calcite et en très faible quantité sous
forme de phosphate de calcium et de sulfate. Ces résultats sont également en adéquation
avec ceux trouvés en extraction séquentielle.
Les micro-cartographies faites pour le sédiment traité sont présentées sur la Figure IV-
26. Il paraît clair que le traitement n’a pas altérée les phases prédominantes : silice, mica et
feldspath, par contre la présence de calcite semble être moins importante. En effet le
calcium se trouve en proportion plus importante sous forme de phosphate de calcium. Cela
signifie qu’une partie de la calcite réagit avec l’acide phosphorique ajouté pour former des
phosphates de calcium. Nous avons également détecté la présence de vivianite (Fe3(PO4)2
Feldspath Mica Calcite Silice Pyroxène Amphybole Oxyde de Fe Sulfate de Ca Plagioclase Phophate de Ca
K2O 15,60 3,88 0,03 0,25 0,02 0,53 0,02 0,08 0,07 0,03SiO2 64,86 43,15 0,18 97,22 37,85 42,43 1,77 1,88 60,15 0,23FeO 0,09 10,17 0,25 0,23 7,31 16,03 45,02 1,24 0,04 2,84CaO 0,02 1,95 51,36 0,04 23,93 11,19 5,27 30,71 6,46 46,17Na2O 0,56 0,15 0,02 0,03 0,00 1,50 0,06 0,04 7,72 0,16PbO 0,02 0,02 0,00 0,01 0,03 0,01 0,28 0,00 0,00 0,00Al2O3 17,69 13,11 0,05 0,91 26,92 11,37 1,09 0,88 24,83 0,14MnO 0,02 0,07 0,07 0,02 0,18 0,19 0,18 0,17 0,00 0,38Cr2O3 0,00 0,06 0,00 0,00 0,01 0,00 1,05 0,08 0,02 0,00MgO 0,00 2,54 0,35 0,07 0,01 9,89 0,26 0,35 0,00 0,19SO3 0,01 0,26 0,98 0,04 0,00 0,02 3,86 13,56 0,00 1,75ZnO 0,00 0,09 0,01 0,01 0,01 0,06 0,24 0,02 0,00 0,00P2O5 0,10 1,56 0,75 0,06 0,27 0,17 4,89 3,17 0,03 36,12CuO 0,00 0,02 0,00 0,01 0,00 0,00 0,02 0,01 0,00 0,00Total 98,96 77,03 54,05 98,89 96,54 93,39 63,99 52,19 99,31 88,01

Chapitre IV : Evaluation du procédé
189
H2O) signe que le Fe a réagit avec l’acide phosphorique. les oxydes de fer et d’aluminium,
les sulfates de calcium, les pyroxènes, les amphiboles et les plagioclases sont également
présents dans l’échantillon et ne semblent pas être altérées par le traitement.
Afin de déterminer le type de phosphate de calcium formé lors du traitement, un calcul de
la formule structural a été fait à partir de l’analyse quantitative faite sur un certain nombre
de particules (30). L'objectif du calcul des formules structurales est de passer de la
composition en % poids d'oxyde donnée par la microsonde en fraction molaire de la manière
décrite en annexe A. Ainsi, à partir des fractions molaires calculées, le rapport molaire Ca/P
peut être déterminé et par conséquence le type de phosphate formé peut être identifié. Les
résultats obtenus sont représentés sur le Tableau IV- 5.
Les résultats montrent la prédominance de phosphates de calcium de rapport Ca/P proche
de 1, ce qui correspond aux phosphates dicalcique (monétite). Ces résultats sont en
adéquation avec les résultats montrés précédemment. Nous constatons également la présence
en proportion moindre de phosphate de calcium de rapport Ca/P proche de 1.5. Ce
phosphate correspond aux phosphate tricalcique (Ca3(PO4)2) qui ont pu se former lors du
traitement.
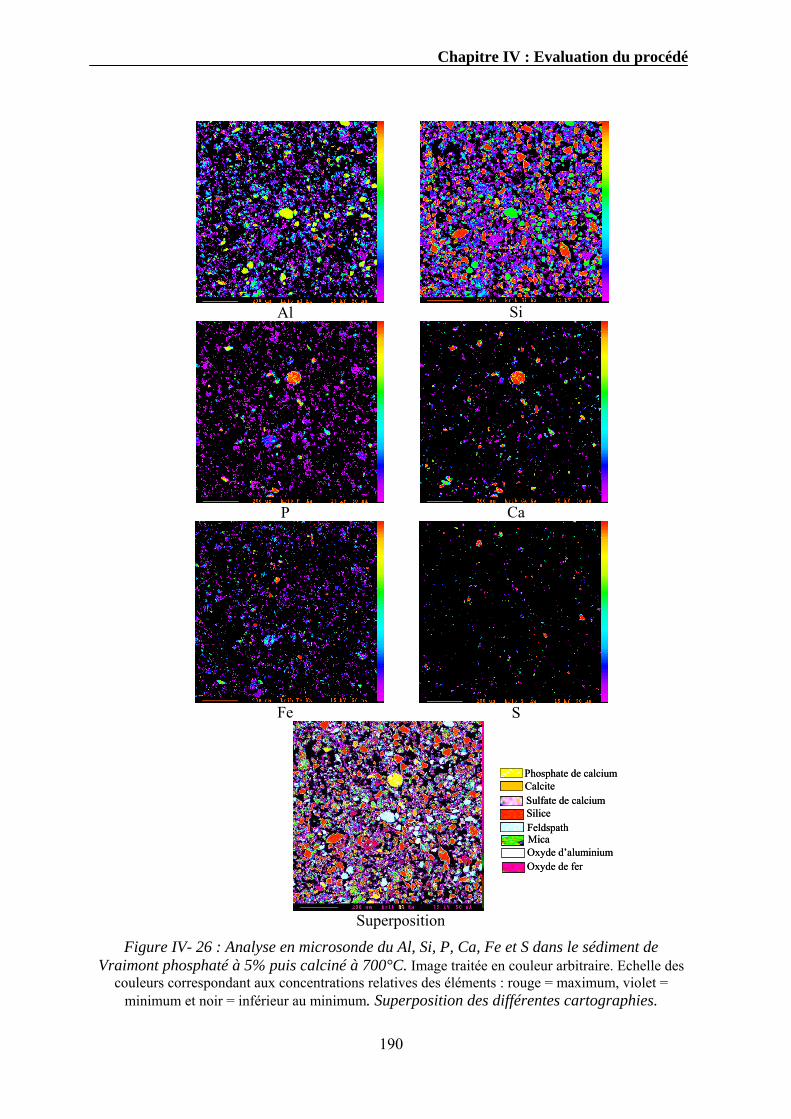
Chapitre IV : Evaluation du procédé
190
Al Si
P Ca
Fe S
Superposition
Figure IV- 26 : Analyse en microsonde du Al, Si, P, Ca, Fe et S dans le sédiment de Vraimont phosphaté à 5% puis calciné à 700°C. Image traitée en couleur arbitraire. Echelle des
couleurs correspondant aux concentrations relatives des éléments : rouge = maximum, violet = minimum et noir = inférieur au minimum. Superposition des différentes cartographies.
Silice
Phosphate de calciumCalciteSulfate de calcium
Feldspath
Oxyde de fer
MicaOxyde d’aluminium
Silice
Phosphate de calciumCalciteSulfate de calcium
Feldspath
Oxyde de fer
MicaOxyde d’aluminium

191
Tableau IV- 5 : Analyse quantitative des nouvelles phase de phosphate de calcium formées après traitements.
La formule structurale a été calculée sur la base de 25 oxygènes. Le points analysés ont été choisis aléatoirement
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30K2O 0,01 0,06 0,02 0,04 0,03 0,07 0,06 0,08 0,24 0,18 0,14 0,21 0,00 0,14 0,03 0,13 0,25 0,18 0,18 0,19 0,19 0,06 0,28 0,06 0,06 0,08 0,04 0,55 0,28 0,12SiO2 0,15 0,21 0,12 0,34 0,09 0,09 0,03 1,00 4,48 3,32 2,18 3,09 0,10 1,59 0,11 2,90 3,97 1,40 0,79 1,90 2,24 1,50 14,51 0,49 1,93 0,67 0,35 5,43 3,91 10,66FeO 0,03 0,00 0,17 0,08 1,10 0,99 1,14 0,45 1,27 1,24 1,14 1,05 0,26 0,56 0,20 0,77 1,22 0,50 0,61 1,56 0,88 0,64 1,36 0,55 0,54 0,63 0,43 1,25 1,02 0,64CaO 55,64 55,14 54,57 53,80 43,64 41,55 44,25 36,82 35,41 35,96 36,07 35,72 37,66 35,34 38,65 35,58 34,70 35,12 36,10 34,93 34,39 35,81 29,35 36,47 35,73 36,81 36,89 33,87 35,08 33,41Na2O 0,03 0,05 0,02 0,10 0,11 0,08 0,07 0,01 0,06 0,09 0,06 0,02 0,03 0,03 0,03 0,04 0,03 0,01 0,00 0,01 0,03 0,04 0,09 0,06 0,02 0,04 0,02 0,08 0,06 0,05PbO 0,00 0,00 0,02 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,02 0,00 0,00 0,00 0,05 0,00 0,00 0,00 0,00 0,00 0,00 0,00Al2O3 0,00 0,02 0,01 0,03 0,01 0,03 0,00 0,46 1,87 1,48 1,04 1,35 0,10 0,92 0,01 0,69 2,01 0,71 0,27 1,16 1,08 0,73 2,12 0,47 0,88 0,73 0,55 3,08 2,02 0,92MnO 0,01 0,02 0,07 0,03 0,18 0,26 0,17 0,00 0,01 0,06 0,11 0,00 0,00 0,07 0,00 0,00 0,12 0,00 0,10 0,09 0,06 0,04 0,11 0,00 0,07 0,09 0,13 0,14 0,00 0,03Cr2O3 0,00 0,00 0,02 0,01 0,00 0,00 0,00 0,00 0,03 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,02 0,01 0,01 0,00 0,00 0,00 0,00 0,00 0,01 0,00 0,00 0,00 0,00MgO 0,00 0,02 0,00 0,02 0,21 0,19 0,18 0,07 0,22 0,14 0,15 0,15 0,06 0,11 0,00 0,08 0,30 0,09 0,04 0,08 0,13 0,14 0,39 0,05 0,05 0,06 0,00 0,19 0,20 0,08SO3 0,00 0,09 0,01 0,14 0,45 0,32 0,23 1,06 0,47 0,50 0,54 0,49 0,77 0,49 0,18 0,49 0,36 0,53 0,55 0,47 0,85 0,67 0,38 0,56 0,48 0,43 0,42 0,46 0,38 0,28ZnO 0,00 0,00 0,00 0,00 0,00 0,04 0,00 0,03 0,00 0,00 0,00 0,00 0,00 0,09 0,00 0,00 0,05 0,00 0,00 0,06 0,00 0,00 0,04 0,00 0,03 0,00 0,00 0,09 0,03 0,00P2O5 44,87 44,48 44,37 44,09 38,91 38,15 41,18 48,61 46,77 48,35 48,52 48,15 50,86 48,11 52,72 48,64 47,45 48,04 49,53 48,15 47,50 49,87 40,95 51,04 50,10 51,76 52,14 48,47 50,45 49,31CuO 0,00 0,02 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,05 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,03 0,00 0,00 0,00 0,02 0,03 0,00 0,00Total 100,73 100,10 99,39 98,68 84,73 81,75 87,31 88,58 90,83 91,31 90,00 90,23 89,84 87,44 91,94 89,32 90,45 86,58 88,20 88,61 87,35 89,50 89,66 89,73 89,90 91,30 90,99 93,64 93,43 95,49K 0,00 0,01 0,00 0,01 0,01 0,02 0,01 0,02 0,05 0,04 0,03 0,04 0,00 0,03 0,01 0,03 0,05 0,04 0,04 0,04 0,04 0,01 0,06 0,01 0,01 0,02 0,01 0,11 0,06 0,02Si 0,02 0,03 0,02 0,06 0,02 0,02 0,01 0,17 0,74 0,54 0,36 0,51 0,02 0,27 0,02 0,48 0,65 0,24 0,13 0,32 0,38 0,25 2,36 0,08 0,32 0,11 0,06 0,86 0,62 1,62Fe 0,00 0,00 0,02 0,01 0,12 0,11 0,12 0,04 0,12 0,11 0,11 0,10 0,02 0,05 0,02 0,07 0,11 0,05 0,06 0,15 0,08 0,06 0,12 0,05 0,05 0,06 0,04 0,11 0,09 0,05Ca 9,62 9,59 9,56 9,47 8,87 8,72 8,68 6,66 6,24 6,31 6,43 6,33 6,70 6,46 6,73 6,35 6,13 6,48 6,55 6,32 6,27 6,37 5,11 6,48 6,32 6,43 6,46 5,74 5,95 5,44Na 0,01 0,02 0,01 0,03 0,04 0,03 0,03 0,00 0,02 0,03 0,02 0,01 0,01 0,01 0,01 0,01 0,01 0,00 0,00 0,00 0,01 0,01 0,03 0,02 0,01 0,01 0,01 0,03 0,02 0,02Pb 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00Al 0,00 0,00 0,00 0,00 0,00 0,01 0,00 0,09 0,36 0,29 0,20 0,26 0,02 0,19 0,00 0,14 0,39 0,14 0,05 0,23 0,22 0,14 0,41 0,09 0,17 0,14 0,11 0,57 0,38 0,16Mn 0,00 0,00 0,01 0,00 0,03 0,04 0,03 0,00 0,00 0,01 0,02 0,00 0,00 0,01 0,00 0,00 0,02 0,00 0,01 0,01 0,01 0,01 0,02 0,00 0,01 0,01 0,02 0,02 0,00 0,00Cr 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00Mg 0,00 0,01 0,00 0,00 0,06 0,05 0,05 0,02 0,05 0,03 0,04 0,04 0,02 0,03 0,00 0,02 0,07 0,02 0,01 0,02 0,03 0,04 0,09 0,01 0,01 0,01 0,00 0,05 0,05 0,02S 0,00 0,01 0,00 0,02 0,06 0,05 0,03 0,13 0,06 0,06 0,07 0,06 0,10 0,06 0,02 0,06 0,04 0,07 0,07 0,06 0,11 0,08 0,05 0,07 0,06 0,05 0,05 0,05 0,05 0,03Zn 0,00 0,00 0,00 0,00 0,00 0,01 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,01 0,00 0,00 0,01 0,00 0,00 0,01 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,01 0,00 0,00P 6,13 6,11 6,14 6,13 6,25 6,32 6,38 6,95 6,52 6,70 6,84 6,75 7,15 6,95 7,25 6,86 6,62 7,00 7,10 6,89 6,85 7,01 5,63 7,16 7,00 7,14 7,21 6,49 6,76 6,34Cu 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,01 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00 0,00Ca/P 1,57 1,57 1,56 1,54 1,42 1,38 1,36 0,96 0,96 0,94 0,94 0,94 0,94 0,93 0,93 0,93 0,93 0,93 0,92 0,92 0,92 0,91 0,91 0,90 0,90 0,90 0,90 0,88 0,88 0,86


Chapitre IV : Evaluation du procédé
193
IV.3.3. Microscope électronique à balayage
Dans le but de visualiser l’évolution de la structure des sédiments au cours du traitement,
des observations au Microscope électronique à balayage (MEB) ainsi que des analyses semi-
quantitatives (EDS) ont été effectuées. L’analyse a été faite sur le sédiment de Vraimont
phosphaté à 5% sur sec Figure IV- 27, phosphaté à 5% sur sec puis calciné à 700°C Figure
IV- 28 et phosphaté à 5% sur sec puis calciné à 1100°C Figure IV- 29.
Les observations et les analyses semi-quantitatives effectuées sur les différentes
échantillons ont permis de montrer la présence de phosphates de calcium sur la surface des
particule de silicate d’aluminium. Cela implique que lors de la phosphatation, le calcium et
le phosphore précipitent à la surface des silicates qui semblent présentés un support de
nucléation et de croissance des phosphates de calcium.
Néanmoins nous avons également observé la présence de particules de phosphate de
calcium qui ne recouvrent pas les surfaces des silicates, ainsi que certaines particules de
silicates qui semblent avoir de très faible quantité de phosphate de calcium sur leur surface
(figures non présenté)
Comme pour lea microsonde, les analyses semi-quantitatives faites par le MEB, ne nous
ont pas permis d’identifier des phase de phosphates métalliques susceptibles de s’être
formées lors de la phosphatation. Cela est due aux faibles concentrations des métaux dans la
sédiments. Les analyses présentées sur la Figure IV- 27 par contre montrent la présence de
Zn. Toutefois cette analyse ne permet pas de savoir sous quelle forme il se trouve. Nous
pouvons cependant faire l’hypothèse que les phosphates métallique qui ont pu se formé par
co-précipitation l’on fait sur les surface des silicates d’aluminium.
Comme il est perceptible sur la Figure IV- 27 (a) et (b), les phosphates de calcium qui ont
précipités sur les particules semblent êtres amorphes avant la calcination. Par contre,
l’augmentation de la température de calcination favorise sa cristallisation. En effet, à 700°C
(Figure IV- 28), cette cristallisation n’est pas très claire mais la structure semble différente
de celle du sédiment avant calcination. Par contre à 1100°C (Figure IV- 29) nous pouvons
constater un changement de la structure très net qui est probablement lié à la cristallisation
des phosphates de calcium.
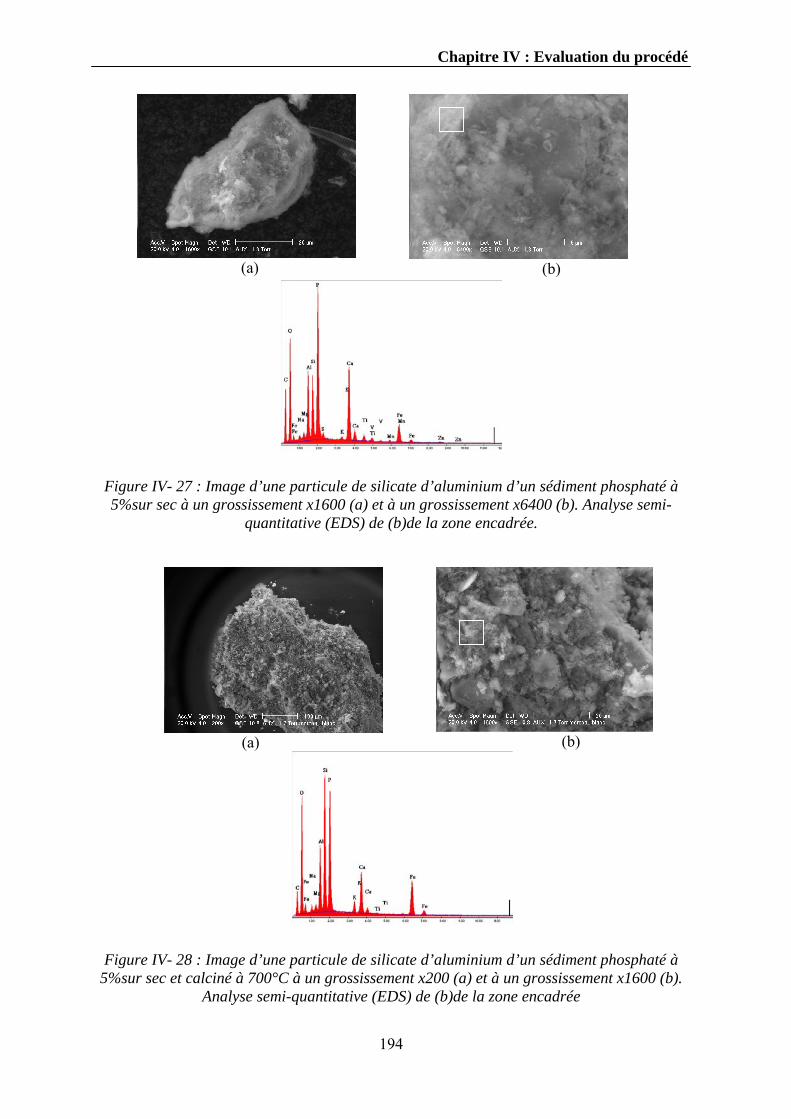
Chapitre IV : Evaluation du procédé
194
(a) (b)
Figure IV- 27 : Image d’une particule de silicate d’aluminium d’un sédiment phosphaté à 5%sur sec à un grossissement x1600 (a) et à un grossissement x6400 (b). Analyse semi-
quantitative (EDS) de (b)de la zone encadrée.
(a) (b)
Figure IV- 28 : Image d’une particule de silicate d’aluminium d’un sédiment phosphaté à 5%sur sec et calciné à 700°C à un grossissement x200 (a) et à un grossissement x1600 (b).
Analyse semi-quantitative (EDS) de (b)de la zone encadrée
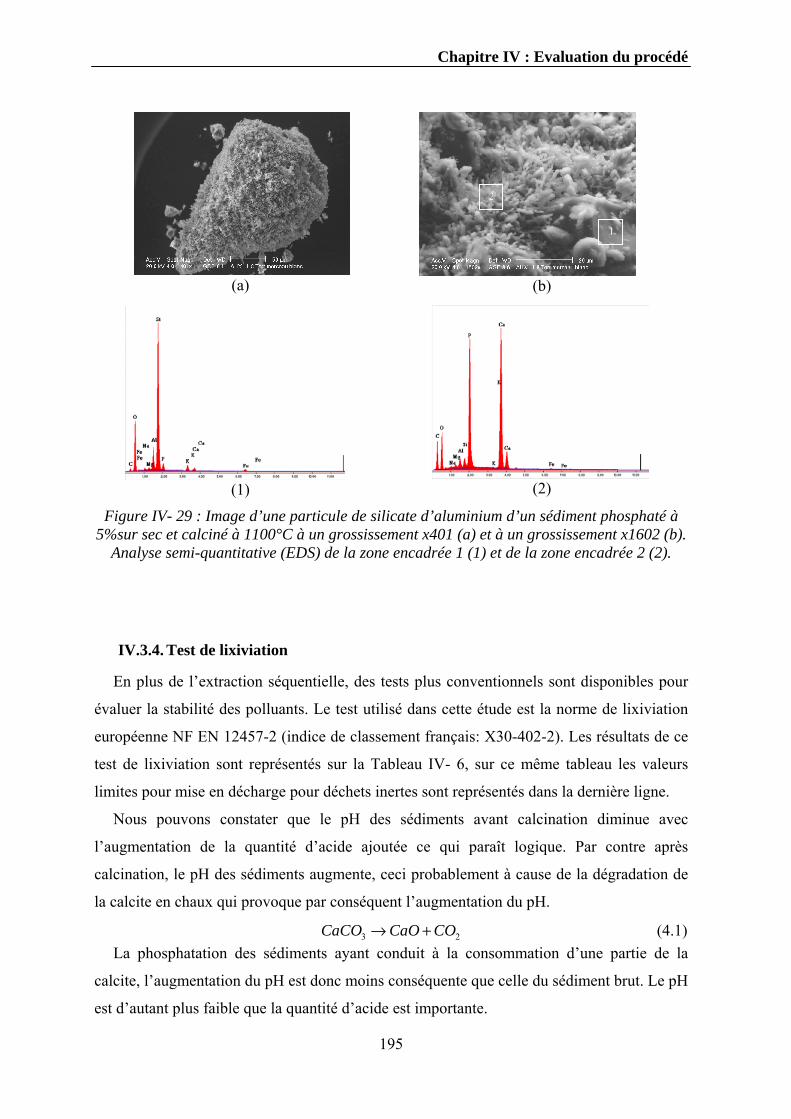
Chapitre IV : Evaluation du procédé
195
(a) (b)
(1) (2)
Figure IV- 29 : Image d’une particule de silicate d’aluminium d’un sédiment phosphaté à 5%sur sec et calciné à 1100°C à un grossissement x401 (a) et à un grossissement x1602 (b).
Analyse semi-quantitative (EDS) de la zone encadrée 1 (1) et de la zone encadrée 2 (2).
IV.3.4. Test de lixiviation
En plus de l’extraction séquentielle, des tests plus conventionnels sont disponibles pour
évaluer la stabilité des polluants. Le test utilisé dans cette étude est la norme de lixiviation
européenne NF EN 12457-2 (indice de classement français: X30-402-2). Les résultats de ce
test de lixiviation sont représentés sur la Tableau IV- 6, sur ce même tableau les valeurs
limites pour mise en décharge pour déchets inertes sont représentés dans la dernière ligne.
Nous pouvons constater que le pH des sédiments avant calcination diminue avec
l’augmentation de la quantité d’acide ajoutée ce qui paraît logique. Par contre après
calcination, le pH des sédiments augmente, ceci probablement à cause de la dégradation de
la calcite en chaux qui provoque par conséquent l’augmentation du pH.
3 2CaCO CaO CO→ + (4.1) La phosphatation des sédiments ayant conduit à la consommation d’une partie de la
calcite, l’augmentation du pH est donc moins conséquente que celle du sédiment brut. Le pH
est d’autant plus faible que la quantité d’acide est importante.
2
1

Chapitre IV : Evaluation du procédé
196
Concernant la concentration des métaux lourds dans le lixiviat du sédiment brut (BV0),
on constate que hormis le Cu et le Pb qui sont en concentration égale ou supérieure aux
valeurs limites, les concentrations des autres métaux sont en dessous de ces valeurs limites.
Cela signifie qu’en vu de cette norme et de ces seuils, le sédiment brut ne peut être considéré
comme inerte.
Après traitement et sauf pour le Cr, la concentration du Pb et du Cu deviennent
inférieures aux valeurs limites. En effet, la calcination du sédiment conduit à l’augmentation
de concentration de Cr lixiviable (10.4mg/kg de matière sèche pour BV0C), néanmoins
l’augmentation de la quantité d’acide conduit à la diminution de sa concentration (7mg/kg
de matière sèche pour BV7C). L’augmentation du Cr comme cela a était montré lors de
l’étude de l’extraction séquentielle, est probablement lié à l’oxydation du Cr(III) en Cr(VI)
plus soluble lors de la calcination.
Finalement, le traitement a permis réduire la concentration des métaux lourds lixiviables
à l’eau (Pb et Cu) par contre, il conduit à l’augmentation de la quantité de Cr lixivable. Cela
signifie que si on prend en compte uniquement les métaux lourds que nous avons étudiés, le
sédiment n’est pas considéré comme étant inerte. Des améliorations doivent être faites pour
permettre la réduction de la quantité de Cr lixiviable. L’amélioration peut se faire en
ajoutant la quantité d’acide phosphorique, toutefois cette solution n’est pas économiquement
viable et peut poser des problèmes liés à l’eutrophisation [Walpersdort 2004 ; Toor 2005,
de-Bashan 2004] qui peut être provoquée par des concentration en P lixivable importante.
La diminution de la température de calcination peut être aussi une solution pour diminuer la
concentration du Cr lixiviable. Des investigations dans ce sens sont en cours.
Ces résultats permettent de déduire aussi que cette méthode de lixiviation ne prend en
compte que les métaux lourds solubles dans l’eau pour déterminer le potentiel polluant des
sédiment. Or plusieurs études ont montrées que pour une lixiviation à l’eau (a pH neutre),
très peu de métaux lourds sont solubles [Carpentier 2002 ; Gäbler 1997]. Ce test de
lixiviation ne prend donc pas en compte les changements des conditions tels que le potentiel
redox et le pH qui peuvent influencer à long terme le relargage des métaux. L’étude de
l’extraction séquentielle a bien montré que le potentiel polluant des sédiments est plus
important que cela.
Chapitre IV : Evaluation du procédé
197
Tableau IV- 6 : Résultats de test de lixiviation NF X30-402-2 et valeurs limites pour mise en décharge pour déchets inertes exprimés en mg/kg de matière sèche.
IV.4. Conclusions
L’évaluation du traitement de stabilisation des métaux lourds a été faite par l’utilisation
de différentes méthodes chimiques et spectroscopiques.
Concernant la méthode chimique, l’extraction séquentielle de Tessier a été utilisée pour
évaluer la distribution des métaux lourds avant et après traitement. Cette méthode a été
utilisée en dépit des inconvénients liés au manque de sélectivité des réactifs, et des
problèmes liés à la réadsorption et la redistribution des éléments lors de l’extraction. Cette
méthode a cependant permis de fournir plusieurs informations sur les sédiments bruts et sur
les sédiments traités. Cette méthode a permis de montrer que :
Le sédiment brut de Lille présente un potentiel polluant plus important que celui de
Vraimont et que la mobilité des métaux lourds dans BL augmente dans l’ordre suivant As >
Pb > Cd > Co > Cr > Cu > Zn. Pour BV, la tendance est différente, la mobilité des métaux
lourds augmente dans l’ordre As > Cr > Co > Cd > Pb > Zn > Cu.
Les éléments majeurs se trouvent essentiellement dans la fraction résiduaire (la plus
stable), excepté le Ca qui se trouve essentiellement sous forme de carbonates et inclus dans
les silicates d’aluminium.
L’extraction séquentielle des deux sédiments phosphatés à différents taux d’acide et
calcinés à 700°C a permis de montrer que le traitement conduit à la réduction de la mobilité
du Pb, Ca, Cr, Cu et Zn ceci grâce aux effets combinés de l’acide phosphorique et de la
calcination sauf pour le Zn où seule la calcination à permis la réduction de sa mobilité. Cela
peut être due à son incorporation dans la matrice silicatée. Pour le Cr par contre bien qu’une
partie de sa fraction mobilisable soit stabilisée, on le retrouve suite à la calcination en
pH As Cd Co Cr Cu Pb ZnBV0 7,5 <0,003 <0,0002 <0,001 <0,001 2,0 2,2 2,6BV3 7,1 <0,003 <0,0002 <0,001 <0,001 <0,001 <0,003 0,7BV5 6,2 <0,003 <0,0002 <0,001 <0,001 <0,001 <0,003 0,8BV7 6,0 <0,003 <0,0002 <0,001 <0,001 <0,001 <0,003 1,5
BV0C 12,4 <0,003 <0,0002 <0,001 10,4 <0,001 <0,003 1,7BV3C 11,9 <0,003 <0,0002 <0,001 9,1 <0,001 <0,003 0,5BV5C 11,5 <0,003 <0,0002 <0,001 7,7 <0,001 <0,003 1,8BV7C 10,5 <0,003 <0,0002 <0,001 7,0 <0,001 <0,003 0,6Valeurs limites 0.5 0.04 0.5 2,0 0,5 4,0

Chapitre IV : Evaluation du procédé
198
concentration non négligeable dans la fraction échangeable plus mobile. Cela est
vraisemblablement dû à l’oxydation du Cr(III) en Cr(VI) plus mobile et plus toxique aussi.
Quant à la réduction de la mobilité du Pb, Ca, Cr et Cu, nous pensons qu’elle est
essentiellement due à la formation de phosphates métalliques et phosphate de Ca stables.
Cela concorde avec les résultats de la distribution du phosphore qui ont permis de montrer
que l’essentiel de ce constituant se trouve dans la fraction résiduaire.
Le Cd par contre réagit différemment dans les deux sédiments. Dans BV, aucun effet
notable du traitement n’est apparent, par contre, dans BL, sa mobilité diminue après la
calcination et avec l’augmentation de la quantité d’acide. Quant au Co le traitement ne
semble pas avoir d’effet particulier sur sa mobilité, néanmoins, son faible potentiel polluant
ne semble a priori pas poser de problème environnementaux majeurs. L’As réagit tout
autrement, la phosphatation n’a aucun effet sur le mobilité de l’As contenu dans BL par
contre elle conduit à une légère augmentation sa mobilité dans BV.
La calcination par contre conduit à une légère augmentation de la fraction mobilisable des
deux sédiments indépendamment de la phosphatation. Les travaux en cours dans notre
laboratoire portent spécifiquement sur une étude détaillée du processus de la calcination des
sédiments qui permettra d’améliorer la compréhension des processus de libération du Cr et
de l’Al. Cela devrait permettre de minimiser ces processus en maîtrisant mieux les condition
de calcination (température, vitesse de chauffe, atmosphère, etc…)
Finalement le traitement des sédiments de dragage nous a permis grâce à l’extraction
séquentielle d’observer l’influence du traitement sur la mobilité des métaux lourds et ceci
bien qu’il ne soit pas facile de déterminer les mécanismes qui ont été mis en jeu. Cependant
deux mécanismes sembles être à la base de la stabilisation des métaux : la dissolution des
différents formes chimiques lors de l’ajout de l’acide phosphorique ou même lors de la
maturation et leur précipitation par la suite sous forme de phosphate. En effet comme il a été
présenté dans le chapitre I et III, le séchage à l’air des sédiments peut conduire sous l’effet
de l’oxydation des sulfures en sulfates, ou sous l’effet de l’oxydation de la matière
organique à la libération des métaux lourds qui leur sont liés. En présence de phosphate, ces
éléments peuvent se réadsorber ou précipiter par la suite sous forme de phosphate. l’échange
d’ions est également un des mécanismes qui peut intervenir pour la stabilisation des métaux.
Il cependant nécessaire de rappeler que la calcination seule peut jouer un rôle important dans
le stabilisation des métaux lourds en permettant leur introduction dans la matrice silicaté.
En comparant la mobilité des métaux en prenant en compte la somme des fractions non
résiduelles, nous constatons que pour BV, la mobilité des métaux lourds après traitement

Chapitre IV : Evaluation du procédé
199
augmente dans l’ordre : Cd > Co > As > Pb ≈ Cu > Cr > Zn. Quant à BV, la mobilité des
métaux augmente dans l’ordre : Cd > Co > Cu > Cr ≈ As > Pb > Zn. L’extraction
séquentielle a également permis de montrer que dans des conditions de traitement
comparable (phosphaté à 5% et calciné à 700°C), le potentiel polluant de BV est inférieur à
celui de BL
Les fractions non résiduaires des éléments majeurs (Fe et Al) semblent également agir
avec le phosphore conduisant à l’augmentation de leur fractions résiduaires, cette
augmentation et probablement due à la formation de phosphates stables. Par contre la
calcination peut conduire dans certains cas à la diminution de cette fraction.
Les méthodes spectroscopiques n’ont été appliquée que pour les sédiments de Vraimont.
Nous avons utilisé la microsonde pour déterminer les associations minéralogiques et la
présence de nouveaux composés qui ont pu se former lors du traitement. Nous avons
également utilisé le microscope électronique à balayage pour essayer grâce à l’analyse semi
quantitative de détecter les nouvelles phases formées lors du traitement et l’évolution de la
structure au cours de traitement.
Compte tenu de la faible concentration en métaux lourds, il n’a pas été possible
d’analyser ces éléments dans les sédiments de Vraimont. Ce qui implique que seule
l’extraction séquentielle à pu nous donner des informations détaillées sur la distribution des
métaux lourds.
Cependant, la microsonde a permis grâce aux cartographies et aux analyses quantitatives
faites sur les éléments majeurs de montrer la présence prédominante dans le sédiment brut
de silice, mica et feldspath et calcite ainsi que dans une moindre mesure la présence
d’amphibole, pyroxène, plagioclase et d’oxyde de fer et d’aluminium. L’analyse du
sédiment de Vraimont phosphaté à 5% puis calciné à 700°C a montré que les phases majeurs
n’ont pas subit d’altération suite au traitement. Par contre nous avons pu montrer la
réduction de la quantité de calcite et la présence plus importante de phosphates dicalciques
et en quantité moindre le phosphate tricalcique. Ces résultats sont en adéquation avec les
résultats obtenus lors de l’étude cinétique et de l’extraction séquentielle. La présence de ces
phosphates de calcium est très intéressantes car ces phosphates ont tendance à évoluer vers
la forme thermodynamiquement la plus stable qui est l’hydroxyapatite. Nous avons
également pu montrer la présence de vivianite signe que le Fe à réagit avec le phosphore,
cela concorde avec les résultats de l’étude cinétique et l’extraction séquentielle.

200
Conclusions générales et perspectives

201
Conclusions générales et perspectives
Le traitement des sédiments de dragage par le procédé NOVOSOL comporte les trois
étapes suivantes :
• le traitement chimique à l’acide phosphorique en réacteur tubulaire ;
• le séchage convective à l’air ;
• la calcination sous air (en conditions oxydantes) des sédiments.
L’objectif de cette étude était la compréhension des trois phases du traitement à travers la
détermination de l’influence des paramètres expérimentaux sur :
• la stabilisation des métaux lourds ;
• les temps de séchage des sédiments ,
• la dégradation de la matière organique ;
• les propriétés physiques du résidu final.
Le but à terme de ce traitement est une valorisation matière du résidu solide final dans le
domaine du génie civil (briques de construction, béton, sous couche routière…)
La démarche adoptée pour cette étude consiste en une caractérisation physique chimique
des sédiments suivie d’un traitement des sédiments en faisant varier les différents paramètres
susceptibles d’influencer les trois étapes du traitement. Le traitement proprement dit est
suivi par une évaluation de la mobilité des espèces métalliques.
Deux sédiments fournis par la société Solvay ont été utilisés lors de cette étude. Un des
sédiments venait de Vraimont en Belgique et le deuxième de Lille. Les résultats obtenus
avec le sédiment de Vraimont lors du traitement chimique, du séchage, de la calcination de
même que sur l’évaluation de la stabilité des espèces chimiques après traitement ont été
présentés. Pour le sédiment de Lille, seuls les résultats obtenus lors de la caractérisation
physico-chimique et de l’évaluation de la mobilité des espèces après traitement ont été
présentés dans ce document.
La caractérisation physico-chimique s’est faite en se basant essentiellement sur les
normes AFNOR. Les différentes caractérisations physico-chimiques des deux sédiments ont
permis de mettre l’accent sur les principales différences entre les deux sédiments à savoir :
la concentration en métaux lourds, le pourcentage des carbonates, le taux la matière
organique et le pouvoir tampon.
Ainsi, des études cinétiques de phosphatation des sédiments de Vraimont ont été réalisées
dans un réacteur tubulaire à différents taux d’acide (entre 3 et 7%). Ces cinétiques ont
montré que dans les conditions définies, un temps de réaction de 22.5min dans le réacteur

202
est insuffisant pour la consommation et la précipitation totale du phosphore ajouté. En effet,
uniquement 56.7% du phosphore a été consommé, le reste du phosphore se consomme dans
l’étape de séchage à l’air que nous avons également appelée étape de séchage et de
maturation. L’étude de la cinétique de phosphatation en maturation a permis de montrer que
8.3 jours sont nécessaires pour la consommation de 90% du phosphore injecté.
Ces résultas impliquent que la phosphatation des sédiments n’est pas très rapide et de ce
fait, l’utilisation d’un réacteur tubulaire n’est peut être pas nécessaire, l’usage d’un
mélangeur (sédiment/H3PO4) pourrait être suffisant étant donné que la réaction de
stabilisation se poursuit lors du séchage
La phosphatation du sédiment de Vraimont a également permis de montrer que sous
l’action de l’acide phosphorique, un dégagement gazeux de CO2 et de H2S se produisait
conduisant à la formation de bulles au sein du sédiment. Cette formation de bulles génère un
changement de texture qui au fur à mesure du séchage se traduit par la formation de porosité
au sein du sédiment. Ce changement textural, qui a des conséquences importantes sur la
réduction du temps de séchage, est fortement lié au taux d’humidité et à la quantité d’acide
ajoutée. Pour un même taux d’acide, plus l’humidité est faible dans l’échantillon plus la
texture du sédiment final est poreuse.
Le dégagement gazeux de CO2 et de H2S suite à l’ajout d’acide est accompagné d’une
solubilisation du Ca, Zn et Fe. En effet l’acide phosphorique dissout une partie des
carbonates de calcium et de zinc ainsi que les sulfures de fer. Par contre nous n’avons pas
enregistré lors de l’étude cinétique en réacteur ou en maturation la présence en concentration
détectable d’autre éléments chimiques analysés tels que As, Cd, Cr, Cu, Co et Pb
Le Ca, Zn et Fe qui se sont solubilisés après l’ajout d’acide phosphorique précipitent par
la suite.
Concernant le Ca, sa cinétique étant identique à celle du P, il est possible que ces deux
éléments aient co-précipité pour former le phosphate dicalcique. La maturataion des
sédiments à conduit par la suite à la formation probable de phosphate tricalcique. La
présence de ces formes dans le sédiment peut conduire par des processus d’échange d’ions à
la stabilisation de métaux lourds comme il a été largement montré dans le littérature.
Quant au Zn et au Fe, la précipitation observés de ces deux éléments à travers l’étude
cinétique en réacteur et en maturation permet de faire deux hypothèses, la première est que
ces deux éléments ont précipité sous forme de phosphate ou bien ils se sont réadsorbés sur
d’autre phases.

203
Finalement l’étude cinétique à permis de montrer que la phosphatation des sédiments
peut se faire par deux mécanismes, la dissolution-précipitataion et l’échange d’ion entre Ca
et métaux lourds.
Après l’étude de cinétique et phosphatation et de maturation, le séchage convectif du
sédiment brut et des sédiments phosphatés a été réalisé. Il à permis de montrer que
l’augmentation de la surface d’échange due au dégagement gazeux produit par la
phosphatation peut conduire à des réductions des temps de séchage de l’ordre de un tiers
pour un sédiment phosphaté à 5% (sur sec). D’autre part, les résultats obtenus en faisant
varier la vitesse de l’air de séchage et sa température ont permis de montrer qu’il est
économiquement plus viable d’augmenter la vitesse de l’air pour réduire les temps de
séchage plutôt que d’augmenter sa température. D’autre part, la solution que nous proposons
pour réduire les temps de séchage est de retourner régulièrement le sédiment pour
renouveler la surface et favoriser le transfert de chaleur et de matière dans les deux sens.
Cependant, il est importante de prendre en compte le temps nécessaire à la maturation et
donc à la précipitation du P injecté.
Une fois que la teneur en eau dans le sédiment est minimale, la calcination du sédiment
peut se faire. La calcination des sédiments à différentes températures a permis de monter que
si l’objectif principal est uniquement la dégradation de la matière organique (humus et
pollution), une température de 500°C semble être suffisante. Par contre, si l’objectif est de
dégrader toute la matière susceptible de l’être, une calcination de 30 minutes à 700°C peut
être suffisante. La calcination des sédiments conduit à une modification de sa structure. En
effet, la calcination jusqu’à une température de 500°C qui conduit comme il a été dit en
partie à la dégradation de la matière organique favoris ainsi la formation de microporosité et
l’augmentation de la surface spécifique d’une part et à l’augmentation de la densité d’autre
part. Cette augmentation s’explique essentiellement par l’obtention d’un résidu
essentiellement constitué de phases minérales plus dense que les composés organique
décomposées. Ces résultats nous permettent de conclure d’une part que les propriétés
physiques des sédiments sont fortement liés à leur composition et aux conditions de
calcination utilisées. ces conditions (température, temps de calcination et vitesse de chauffe)
peuvent être modifiées afin d’améliorer les propriétés mécaniques du sédiment traité en
fonction de la voie de valorisation prévue.
Après avoir phosphaté, séché puis calciné les sédiments une étape d’évaluation du
procédé à été réalisée. Pour évaluer et déterminer la distribution des éléments chimiques
contenus dans le sédiment avant et après traitement l’extraction séquentielle a été utilisée

204
aussi bien pour le sédiment de Lille que pour celui de Vraimont. C’est une méthode qui
permet de décrire la mise en solution d’éléments en fonction de conditions prédéterminées
plus ou moins agressives. Cette méthode permet de répartir les éléments en fractions plus ou
moins disponibles en milieux aqueux, acide, oxydant et réducteur. Elle permet de classer les
éléments dans des catégories réputées métastables ou stables.
Les résultats obtenus montrent que l’effet combiné de la phosphatation et de la
calcination à 700°C permet de transformer l’essentiel du P ajouté en P très stable
probablement des phosphates métallique et du phosphate de calcium. Ces résultats signifient
que le P est très réactif vis à vis des éléments chimiques minéraux présents dans les
sédiment. Cela est d’autant plus vrai que l’analyse de la distribution du Pb, Cr, Cu, Zn Ca a
permis de montrer que sous l’effet du traitement appliqué, la fraction mobilisable des ces
éléments a diminué ce qui implique que leur stabilité augmenté probablement en formant
des phosphates métalliques ou des phosphates de calcium insolubles. L’hypothèse d’une
éventuelle incorporation de ces éléments dans la matrice silicaté n’est pas écartée.
Dans le cas du Cr, bien que la phosphatation et la calcination aient permis d’augmenter sa
fraction stable dans le sédiment sous l’effet de la calcination la fraction échangeable et donc
la plus mobile a augmenté. Cette augmentation peut être fortement liée à l’oxydation du
Cr(III) en Cr(VI) plus soluble qui peut nuire à la qualité du résidu final. La même
observation a été faite pour l’As, la différence est que le traitement n’a pas permis la
réduction de son potentiel polluant, mais en plus, la calcination a conduit à l’augmentation
de sa mobilité probablement sous l’effet de l’oxydation de l’As(III) en As(V). Cependant ,
une différence notable par rapport au Cr, c’est que la majeur partie de l’As qui s’est mobilisé
se trouve dans la fraction réductible et oxydable et une très faible quantité lié aux
carbonates. Ce qui n’a pas été le cas du Cr pour lequel il se retrouve après calcination dans
la fraction échangeable (la plus mobile).
La présence de nouveaux phosphates de calcium dans les sédiments de Vraimont a été
démontrée de deux manières, par le MEB et par la microsonde qui a révélé la présence de
phosphate dicalcique et en proportion moindre des phosphates tricalcique. Le MEB a
cependant montré que l’essentiel des phosphates de calcium précipités sont localisés à la
surface des silicates d’aluminium et que la calcination permet de les cristalliser. La présence
de phosphate de calcium dans les sédiments peut conduire à court ou à moyen terme à la
stabilisation des métaux qui sont encore sous forme soluble ou qui risquent de se solubiliser
avec le changement du potentiel redox ou du pH du milieu par exemple. Pour vérifier cela,
des études de suivi de la distribution des métaux lourds à long terme (voir une année) sur des

205
sédiment à différentes étapes du traitement doivent être faites. Ces deux méthodes ne nous
ont par contre pas permis par contre de détecter des phosphates métalliques et ceci du aux
faibles concentrations de ces éléments dans le sédiment de Vraimont.
L’évaluation du procédé par un test de lixiviation réglementaire (norme européenne NF
EN 12457-2 (indice de classement français: X30-402-2)) et la comparaison des résultats
obtenus avec les valeurs limites pour mise en décharge de déchets inertes, a permis de
montrer que seuls le Cu et le Pb contenus dans le sédiment de Vraimont brut sont en
concentrations supérieures ou égales aux valeurs limites. Après traitement (phosphatation et
calcination) des sédiments, leur concentrations été en dessous des valeurs limites ce qui
signifie que le traitement à permis de les stabiliser. Par contre, nous avons observé
l’augmentation de la concentration de Cr lixivible après la calcination, cela été prévisible vu
que la calcination a conduit à l’augmentation de sa fraction échangeable.
Des études doivent être faites pour améliorer la qualité du traitement et principalement
pour essayer de réduire les concentrations de Cr et d’As mobilisables qui augmentent après
calcination. L’évaluation plus détaillée des températures de calcination, des vitesses de
chauffe et l’atmosphère de calcination doivent être examinées. Autrement dit, travailler sous
atmosphère réductrice par exemple et non pas oxydante comme cela a été le cas pour ce
travail. Néanmoins il est important de signaler comme nous l’avons montré que l’efficacité
du traitement dépend de la composition des sédiments, de la spéciation des métaux et des
conditions du traitement (quantité d’acide, température de calcination…). Pour cela une
caractérisation détaillée préalable de chaque sédiment doit être faite pour ainsi mieux
adapter le traitement

206
Bibliographie Abd El Azim 2004
Abd El Azim H., El Moselhy Kh. M. Determination and partitioning of metals in sediments along the Suez Canal by sequential extraction. Journal of Marine Systems, 2004, vol.56, issue. 3-4, pp. 363-374
Admassu 1999 Admassu W., Breese T. Feasibility of using natural fishbone apatite as a substitute of hydroxyapatite in remediating aqueous heavy metals. Journal of Hazardous Materials, 1999, vol. B69, pp. 187-196.
Adriano 2004 Adriano D. C., Wenzel W. W., Vangronsveld J., Bolan N.S. Role of assisted natural remediation in environmental cleanup. Geoderma, 2004, vol. 122, pp. 121-142.
AFNOR 1994 Qualité des sols. Recueil des Normes Française. AFNOR, 1994, 250 p. Agence de l’Eau Artois Picardie 2000 (a)
Agence de l’Eau Artois Picardie. Enlèvement des sédiments – guide méthodologique. Evaluation détaillée des risques liés à la gestion des sédiments et aux opérations de curage [en ligne]. Picardie : Agence de l’Eau Artois Picardie, 2000, 146 p. Disponible sur : http://www.eau-artois-picardie.fr/IMG/pdf/etude_11.pdf (consulté le 5/09/2005)
Agence de l’Eau Artois Picardie 2000 (b)
Agence de l’Eau Artois Picardie. Méthode de gestion et de réutilisation des sédiments pollués [en ligne]. Picardie : Agence de l’Eau Artois Picardie, 2000, 126 p. Disponible sur : http://www.eau-artois-picardie.fr/IMG/pdf/gestionsediments.pdf (consulté le 5/09/2005)
Agences de l’eau 2002
Agences de l’eau. Historique national des opérations de curage et perspectives[en ligne]. Etude sur l’eau en france N°89.. Picardie : Agence de l’Eau Artois Picardie, 2002, 13 p. Disponible sur : http://www.eau.artois.picardie.fr (consulté le 5/09/2005)
Aida Tous les textes réglementaires organisés par thème et par rubrique de la nomenclatures [en ligne]. Disponible sur http ://aida.ineris.fr (consulté le 5/09/2005)
Akcay 2003 Akcay H., Oguz A., Karapire C. Study of heavy metal pollution speciation in Buyak Menderes and Gediz river sediments. Water Research, 2003, vol. 37, pp. 813-822.
Aklil 2004 Aklil A., Mouflih M., Sebti S. Removal of heavy metal ions from water by using calcined phosphate as a new adsorbent. Journal of Hazardous Materials, 2004, vol. A112, pp 183-190.
Alastuey 1999 Alastuey A., Garcia-Sánchez A., López F., Quero X. Evolution of pyrite mud weathering and mobility f heavy metals in the Guadiamar valley after the Aznalcóllar spill, south-west Spain. Sci Total Environ, 1999, vol. 242, pp. 41-55.
Alzieu 1999 C Alzieu., A Abarnou., P Bassoullet., B Boutier. Dragage et environnement marin : état des connaissances. Plouzane : Edition Ifremer, 1999, 223p
Alzieu 2001 C. Alzieu. Impact des dragages : programme de recherche IFREMER, Journées techniques et scientifiques du CETMEF, 2001, Paris
Alzieu 2003 C Alzieu., C Andral., P Bassoullet., B Boutier. Bioévaluation de la qualité environnemental des sédiments portuaires et des zones d’immersion. Plouzane : Edition Ifremer, 2003, 247p
Arnich 2003 Arnich N., Lanhers M-C., Laurensot F., Podor R. In vitro and in vivo studies of lead immobilization by synthetic hydroxyapatite. Environmental Pollution, 2003, vol. 124, pp. 139-149.
Åström 1998 Åström M. Partitioning of transition metals in oxidised and reduced zones

207
of sulphide-bearing fine-grained sediments. Applied geochemistry, 1998, vol. 13, n° 5, pp. 607-617.
Ausili 1998 Ausili A., Mecozzi M., Gabellini M., Ciuffa G. Physico chemical characteristics and multivariate analysis of contaminated harbour sediments. Wat. Sci. Tech, 1998, vol. 37, n°6-7, pp. 131-139.
Bailliez 2003 Bailliez S. Adsorption du plomb sur des hydroxyapatite et frittage thermique : processus cinétiques et transfert thermique. Thèse de doctorat. Lyon : INSA Lyon, 2003, 271p.
Basta 2004 Basta N. T., McGowen S. L. Evaluation of chemical immobilization treatment for reducing heavy metal transport in a smelter-contaminated soil. Environmental Pollution, 2004, vol. 127, pp. 73-82
Belkessam 1998 Belkessam L., Lecomte P., Milon V. Comparaison interlaboratoire de resultats d’analyse des hydrocarbures aromatiques polycycliques dans un sol. Déchets, 1998, vol.12 pp. 41
Berteau 1993 I. Berteau, S. Martin, A. Vassiliadis,. Le curage des cours d’eau et les éléments-traces toxiques. Le Courrier de l’environnement de l’INRA, 1993, vol.20, pp. 27-35
Bicocchi 1998 Bicocchi S. Les polluants et les techniques d’épuration des fumées. Paris : Tec & Doc- Lavoisier, 1998, 188p.
Blanchard Blanchard. C. Caractérisation de la mobilisation potentielle des polluants inorganiques dans les sols pollués, Thèse de doctorat. Lyon : INSA Lyon, 2000, 301p
Blazy 2000 Blazy P, Jdidi E. A. Flottation – Aspect pratique. Technique de l’ingénieur, 2000, vol. JP, J 3360.
Boisson 1999 Boisson J., Ruttens A., Mench M., Vangronsveld J. Evaluation of hydroxyapatite as a metal immobilizing soil additive for the remediation of polluted soil. Part 1 ; Influence of hydroxyapatite on metal echangeability in soil, plant growth and plant metal accumulation. Environmental Pollution, 1999, vol. 104, pp. 225-233.
Bonazzi 2003 Bonazzi C., Bimbenet J-J. Séchage des produits alimentaires - Principe. Technique de l’ingénieur, 2003, F3000, 14p
Bonnet 2000 Bonnet C. Développement de bioessais sur sédiments et applications de l’étude, en laboratoire, de la toxicité de sédiments dulçaquicoles contaminés. Thèse de doctorat. Metz : Université de Metz, 2000, 326 p.
Bourgois 2000 Bourgois J., Debry B., Laforeste V. Traitements chimiques et physico-chimiques des déchets. Technique de l’ingénieur, 2000, vol G, G 2070.
Bournonville 2002
Bournonville B. Stabilisation des métaux lourds dans les cendres volantes d’incinération – Comportement rhéologique, cinétique de phosphatation et évaluation du procédé. Thèse de doctorat. Perpignan : Université de Perpignan, 2002, 216 p.
Caille 2003 Caille N., Tiffreau C., Leyval C., Morel J. L. Solubility of metals in an anoxic sediment during prolonged aeration. The Science of the Total Environment, 2003, vol. 301, pp. 239-250.
Campy 1995 Campy M., Meybeck M. Les sediments lacustres. Limnologie générale. Paris : Masson, 1995, pp. 185-226.
Cao 2004 Cao X., Ma L. Q., Rhue D. R., Appel C. S. Mechanisms of lead, copper, and zinc retention by phosphate rock. Environmental Pollution, 2004, vol. 131, pp. 435-444.
Caplat 2005 Capalt C., Texier H., Barillier D., Lelievre C. Heavy metal mobility in harbour contaminated sediments :The case of Port-en-bessin. Marine

208
Pollution Bulletin, 2005, vol. 50, pp. 504-511. Cappuyns 2004 Cappuyns V., Swennen R., Verhulst J. Assessment of acid neutralizing
and potential mobilisation of trace metals from land-disposed dredged sediments. Science of the Total Environment, 2004, vol. 333, pp. 233-247.
Carpentier 2002 Carpentier S., Moilleron R., Beltran C., Hervé D. Quality of dredged material in the river Seine basin (France). II. Micropolluants. The Science of Total Environment, 2002, vol. 299, pp. 57-72.
Castaldi 2005 Castaldi P., Santona L., Melis P. Heavy metal immobilization by chemical amendments in a polluted soil and influence on white lupin growth. Chemosphere, 2005, vol. 60, Issue 3, pp. 365-371
Cauwenberg 1998
Cauwenberg F., Maes V. A. Flotation as a remediation technique for heavily polluted dredged material. 2. Characterisation of flotation fraction. The science of the total environment, 1998, vol. 209,pp 121-131.
CETMEF Charreau 1991 Charreau A., Cavaillé R. Séchage – Théorie et calculs. Technique de
l’ingénieur, 1991, J2480, 23p. Cheng 1995 Cheng J., Carreau P.J., Chhabra R. P. On the Effet of Wall and Bottom
Clearance on Mixing of Viscoelastic Fluids. AIChE symposium series, 1995, vol. 91, n° 305, pp. 115-122.
Clozel 2002 Clozel B., Battaglia F.,Conil P., Ignatiadis I. Traitabilité par méthodes physiques, chimiques et biologiques de sols contaminés. Rapport final. BRGM/RP-52065. Paris : BRGM, 2002, 356p.
Clozel 2003 B. Clozel-Leloup and Ph. Freyssinet. Valeurs guides intervenant dans la gestion des sédiments et méthodologie d’élaboration de ces valeur : synthèse bibliographique, Paris : BRGM, 2003
Coquery 1995 Coquery M., Welbourn P.M. The relationship between metal concentration and organic matter in sediments and metal concentration in aquatic macrophyte Eriocaulon Septangulare. Wat. Res, 1995, vol. 29, n°. 9, pp. 2094-2102.
Cotter-Howells 1996
Cotter-Howells J., Caporn S. Remediation of contaminated lead by formation of heavy metal phosphates. Applied Geochimistry, 1996, vol. 11, pp. 335-342
Crannell 2000 Crannell B. S., Eighmy T.T., Krzanowski J. E., Eudsen Jr J. D. Heavy metal stabilization in municipal solid waste combustion bottom ash using soluble phosphate. Waste Managment, 2000, vol. 20, pp. 135-148.
Das 1995 Das A. K., Chakraborty R., Cervera M.L., Guardia M. Metal speciation in solid matrices. Talanta, 1995, vol. 42, pp. 1007-1030.
da-Silva 2002 da-Silva I. S., Abate G., Lichtig J., Masini J. C. Heavy metal distribution in recent sediments of the Tietê-Pinheiros river system in Sâo Paulo state, Brazil. Applied Geochemistry, 2002, vol. 17, pp.105-116.
Davidson 1994 Davidson C. M., Thomas R. P., McVey S. E., Perala R. Evaluation of a sequential extraction procedure for the speciation of heavy metal in sediments. Analytica Chimica Acta, 1994, vol. 291. pp. 277-286;
De Carvalho 1998
De Carvalho P.S M., Zanardi E., Buratini S. V., Lamparelli M. C. Oxidizing effect on metal remobilization and Duphnia similis toxicity from a Brazilian reservoir sediment suspension. Water Res, 1998, vol. 32, pp. 193-199;
de Pava 2005 de Pava V. E., Battistel E. Polychlorinated dibenzo-dioxins and -furans detoxification of soil promoted by K-polyethylene glycol technology.

209
Chemosphere, 2005, vol.59, Issue 9, pp.1333-1342 de-Bashan 2004 de-Bashan L. E., Bashan Y. Recent advances in removing phosphorus
from wastewater and its future use as a fertilizer (1997-2003). Water Research, 2004, vol. 38, pp. 4222-4246.
Eighmy 1998 Eighmy T.T., Crannell B. S., Krzanowski J. E., Butler L. G. Characterization and phosphate stabilization of dusts from the vitrification of MSW combustion residues. Waste Management, 1998, vol. 18, pp.513-524.
Elliott 1994 Elliott J.C. in Studies in inorganic chemistry : Structure and chemistry of the apatites and other calcium orthophosphates. Amsterdam : Elsevier, 1994, 389 p. (Studies in inorganic chemistry, vol.18)
Elmore 1965 Elmore K.L., Hatfield J.D., Dunn R.L., Jones A.D. Dissociation of phosphoric acid solutions at 25°C. J.Phys, Chem, 1965, vol. 69, n°10, pp. 3520-3525.
Eudsen Jr 2002 Eudsen Jr J. D., Gallagher L., Eighmy T.T., Crannell B. S. Petrographic and spectroscopic characterization of phosphate-stabilization mine tailings from Leadville, Colorado. Waste Management, 2002, vol. 22, pp. 117-135.
Fritioff 2005 Frittiof A., Kautsky L., Greger M. Influence of temperature and salinity on heavy metal uptake by submersed plants. Environmental pollution, 2005, vol. 133, pp. 265-274.
Fytianos 2004 Fytianos K., Lourantou A. Spaciation of elements in sediment samples collected at lakes Volvi and Koronia, N. Grecce. Environment International, 2004, vol. 30, pp 11-17.
Gäbler 1997 Gäbler H. E. Mobility of heavy metal as a function of pH of samples from an overbank sediment profile contaminated by mining activies. Journal of Geochemical Exploration, 1997, vol. 58, pp. 185-194.
Gauthreaux 1998
Gauthreaux K., Noble C. O., Falgoust T., Beck M. J. Reliability and reproducibility of a sequential extraction procedure for trace metal determination in marsh sediments in southwest Louisiane. Microchemical Journal, 1998, vol. 60, pp. 175-183.
Geysen 2004 Geysen D., Imbrechts K., Vandecasteele C., Jaspers M. Immobilization of lead and zinc in scrubber residues from MSW combustion using soluble phosphate. Waste Management, 2004, vol. 24, pp.471-481.
Gille 1991 Gille G. Séparation magnétique. Technique de l’ingénieur, 1991, vol. JP, A 5220.
Gleyzes 2002 Gleyzes C., Tellier S., Astruc M. Fractionation studies of trace elements in contaminated soils and sediments : review of sequential extraction procedures. Trends in analytical chemistry, 2002, vol. 21, no 6+7, pp. 451-467.
Gomez del rio 2004
Gomez del rio J. A., Morando P. J., Cicerone D.S. Natural materials for treatment of industrial effluents : competitive study of the retention of cd, Zn and Co by calcite and hydroxyapatite. Journal of Environmental Management, 2004, vol. 71, pp. 169-177.
Gómez-Ariza 1999
Gómez-Ariza J. L., Giráldez I., Sánchez-Rodas D., Morales E. Metal readsorption and redistribution during the analytical fractionation of trace elements in oxic estuarine sediments. Analytica Chimica Acta, 1999, vol. 399, pp. 295-307.
Gómez-Ariza 2000
Gómez-Ariza J. L., Giráldez I., Sánchez-Rodas D., Morales E. Comparison of the feasibility of three extraction procedures for trace

210
metal partitioning in sediments from south-west Spain. The science of the Total Environment, 2000, vol. 246, pp. 271-283.
Gourdon 2001 Gourdon R. Traitement biologique des déchets. Technique de l’ingénieur, 2001, vol G, G2060.
Guevara-Riba 2004
Guevara-Riban A., Sahuquillo A., Rubio R., Rauret G. Assessment of mobility in dredged harbour sediments from Barcelona, Spain. Science of the Total Environment, 2004, vol. 321, pp. 241-255.
Haguenauer 1995
Huguenauer B., Nion S., Schreiner D., Albouy M. L’apatite des dents de requin fossiles de 60 millions d’années, garante du piégeage des métaux toxiques ou radioactifs ; In J.M. Cases and F. Thomas, editors, les procédés de solidification et de stabilisation des déchets, 28 Novembre-1 Décembre 1995, Nancy – France, pp 506-509
Heemken 1997 Heemken O.P., Theobald N., Wenclawiak B. W. Comparison of ASE and SFE with soxhlet, sonication, and methanolic saponification extraction for the determination of organic micropolluants in marine particulate matter. Ana. Chem, 1997, vol. 69, pp. 2180.
Hettiarachchi 2001
Hettiarachchi G. M., Pierzynski G. M., Ransom M. D. In situ stabilization of soil lead using phosphorus. Journal of Environmental Quality, 2001, vol. 30, pp. 1214-1221.
Hickey 1984 Hickey M. G., Kittrick J. A. Chemical partitioning of cadmium, copper, nickel and zinc in soils and sediments containing high levels of heavy metals. J Environ Qual, 1984, vol. 13, pp. 372-380
Hodson 2001 Hodson M. E., Valsami-Jones E., Cotter-Howells J. D., Dubbin W. E., Kemp A. J., Thornton I., Warren A. Effect of bone meal (calcium phosphate) amendements on metal release from contaminated soils-a leaching column study. Environmental pollution, 2001, vol. 112, pp. 233-243.
Houot 1991 Houot R., Joussemet R. Concentration gravimétrique. Technique de l’ingénieur, 1991, vol. JP,A 5190.
Jacobs 1999 Jacobs P. H., Förstner U. Concept of subaqueous capping of contaminated sediments with active barrier systems (ABS) using natural and modified zéolites. Wat. Res, 1999, vol. 33, n°9, p 2083-2087.
Jain 2004 Jain C.K. Metal fractionation study on bed sediments of River Yamuna. Water Research, 2004, vol. 38, pp. 569-578.
Jing 2004 Jing C., Meng X., Korfiatis G. P. Lead leachability in stabilized/solidified soil sample evaluated with different leaching tests. Journal of Hazardous Materials, 2004, vol. B114, pp. 101-110.
Kedziorek 1996 Kedziorek M. A. M., Bourg A. C. M. Acidification and solubilisation of heavy metals from single and dual-component model solids; Applied Geochemistry, 1996, vol. 11, pp. 299-304
Kozai 2003 Kozai N., Ohnuki T., Komarneni S., Kamiya T. Uptake of cadmium by synthetic mica and apatite : observation by micro-PIXE. Nuclear Instruments and methods in Physics Research, 2003, vol. 210, pp. 513-518.
Laing 2002 Laing G. D., Bogaert N., Tack F. M. G., Verloo M. G., Hendrickx F. Heavy metal contents (Cd, Cu, Zn) in spiders (Pirata piraticus) living in intertidial sediments of the river Scheldt estuary (Belgium) as affected by substrate characteristics. The science of the total environment, 2002, vol. 289, pp. 71-81.

211
Laing 2002 Laing G. D., Bogaert. N., Tack. F. M. G., Verloo. M. G., Hendrickx. F. Heavy metal contents (Cd, Cu, Zn) in spiders (Pirata piraticus) living in intertidial sediments of the river Scheldt estuary (Belgium) as affected by substrate characteristics. The Science of the Total Environment, 2002, vol. 289, pp. 71-81
Laperche 1996 Laperche V., Traina S. J., Gaddam P., Logan. T. Chemical and mineralogical characterizations of Pb in a contaminated soil : Reactions with synthetic apatite. Environ. Sci. Technol, 1996, vol. 30, pp. 3321-3326.
Lau 1999 Lau S. S. S., CHU L.M. Contaminant release from sediments in a coastal wetland. War. Res. 1999, vol. 33. n°4, pp. 909-918.
Law 1998 Law S. S. S., Chu L.M. Contaminant release from sediments in a coastal wetland. Wat. Res, 1998, vol. 33, n°. 4, pp. 909-918.
Lee 1997 Lee P. K., Touray J. C. Leaching of heavy metals (Zn, Cd, Pb) from contaminated sols and sediments in the vicinity of motorways: an experimental approach. Hydrogéologie, 1997, vol. 1, pp. 3-11.
Lee 2005 Lee P. K., Yu Y. H., Yun S. T., Mayer B. Metal contamination and solid phase partitioning of metals in urban roadside sediments. Chemosphere, 2005, vol. 60, Issue 5, pp. 672-689.
Legifrance Le service public de la diffusion du droit. Disponible sur : http://www.legifrance.gouv.fr
Leybros 1990 Leybros J., Frémeaux P. Extraction solide-liquide – 1. Aspects théoriques. Technique de l’ingénieur, 1990, vol. JP, J 2780.
Lin 1998 Lin J. G., Chen S. Y. The relationship between adsorption of heavy metal and organic matter in river sediments. Environment international, 1998, vol. 24, n° 3, pp. 345-352.
Loannidis 2003 Loannidis T. A., Zouboulis A. I. Detoxification of a highly toxic lead-loaded industrial solid waste by stabilization using apatite. Journal of Hazardous Materials, 2003, vol. B97, pp. 173-191.
Lombi 2000 Lombi E., Sletten R. S., Wenzel .W. W. Sequential extraction of arsenic from different size fractions of contaminated soils. Water. Air Soil Pollut, 2000. vol. 124, pp. 319-332.
Lower 1998 Lower S. K., Maurice P. A., Traina S. J. Simultaneous dissolution of hydroxylapatite and precipitation of hydroxypyromorphite: Direct evidence of homogeneous nucleation. Geochimica et cosmochimica Acta, 1998, vol. 62, n°10, pp. 1773-1780.
Ma L. Q 1999 Ma L. Q., Rao G. Aqueous Pb reduction in Pb-contaminated soils by Florida phosphate rocks. Water, Air, and Soil Pollution, 1999, vol.110, pp. 1-16.
Ma L.Q 1997 Ma L.Q., Rao G. N. Effects of phosphate rock on sequential chemical extraction of lead in contaminated soils. Journal of Environmental Quality, 1997, vol. 26, n° 3, pp. 788-794.
Ma Q. Y 1995 Ma Q. Y., Logan T. J., Traina S. J. Lead immobilization from aqueous solution and contaminated soils using phosphate rocks. Environ. Sci. Technol, 1995, vol. 29, pp. 1118-1128.
Mandalakis 2002
Mandalakis M., Tsapakis M., Tsoga A., Stephano E. G. Gas–particle concentrations and distribution of aliphatic hydrocarbons, PAHs, PCBs and PCDD/Fs in the atmosphere of Athens (Greece). Atmospheric Environment, 2002, vol. 36, Issue 25, pp. 4023-4035
Marot 1998 F. Marot. Caractérisation et traitement des sédiments de dragage

212
contenant des polluants métalliques. Paris : BRGM, 1998 Mavropoulos 2004
Mavropoulos E., Rocha N. C. C., Moreira J. C., Rossi A. M. Characterization of phase evolution during lead immobilization by synthetic hydroxyapatite. Materials Characterization, 2004, vol. 53, pp. 71-78.
McGowen 2001 McGowen S. L., Basta N. T., Brown G. O. Use of diammonium phoshate to reduce heavy metal solubility and transport in smelter-contaminated soil. Journal of Environmental Quality, 2001, vol. 30. pp. 493-500.
MEDD Ministère de l’écologie et du développement durable. Gestion des sédiments extraits des cours d’eau et des canaux. Direction de l’eau et direction de la prévention des pollutions et des risques. Note juridique, 2002, 7p (non publiée)
Meegoda 2001 Meegoda J. N., Perera. R. Ultrasound to decontaminate heavy metals in dredged sediments, Journal of hazardous materials. 2001, vol. 85, p 73-89.
Melamed 2003 Melamed R., Cao X., Chen M., Ma L. Q. Field assessment of lead immobilization in contaminated soil after phosphate application. The science of the Total Environment, 2003, vol. 305, pp. 117-127;
Miller 1986 Miller W. P., Martens D.C., Zelazny L.W. Effect of sequence in extraction of trace metals from soils. Soil Sci. Soc. Am. J, 1986, vol. 50, pp. 598-601.
Miquel 2001 Miquel G. Les effets des métaux lourds sur l’environnement et la santé. Office parlementaire d’évaluation des choix scientifiques et technologiques. Rapport 261. Paris : Assemblé Nationale, Sénat, 2001, 365p. Disponible sur http://senat.fr/rap/l00-261/l00-261.html (consulté le 5/09/2005)
Mirck 2005 Mirck J., Isebrands J.G., Verwijst T., Ledin S. Development of short-rotation willow coppice systems for environmental purposes in Sweden. Biomass and Bioenergy, 2005, vol. 28, pp. 219-228.
Modak 1992 Modak D.P., Singh K P., Chandra H., Ray P.K. Mobile and bound forms of trace metals in sediments of the lower Ganges. Water Res, 1992, vol. 26, n. 11, pp. 1541-1548.
Mouflih 2005 Mouflih M., Aklil A., Sebti S. Removal of lead from aqueous solutions by activated phosphate. Journal of Hazardous Materials, 2005, vol. B119, pp. 183-188.
Mulligan 2001 Mulligan C. N., Yong R. N., Gibbs B. F. An evaluation of technologies for the heavy metal remediation of dredged sediments. Journal of hazardous marerials, 2001, vol. 85, p 145-163.
Nada 1998 Nada Y. S, Biosolubilisation des métaux lourds à partir de sédiments pollués. Thèse de doctorat. Compiègne : Université de Technologie de Compiègne, 1998, 228p
NF EN 12457-2 AFNOR. Caractérisation des déchets - Lixiviation - Essai de conformité pour lixiviation des déchets fragmentés et des boues - Partie 2 : essai en bâchée unique avec un rapport liquide-solide de 10 l/kg et une granularité inférieure à 4 mm (sans ou avec réduction de la granularité). NF EN 12457-2. Paris : AFNOR, 2002, 29p.
Nzihou 2002 Nzihou A., Sharrock P. Calcium phosphate stabilization of fly ash with chloride extraction. Waste Management, 2002, vol. 22, pp. 235-239
Ohnuki 2004 Ohnuki T., Kozai N., Samadfam M., Yasuda R. The formation of autunite (Ca(UO2)2(PO4)2.nH2O) within the leached layer of dissolving apatite :

213
incorporation mechanism of uranium by apatite. Chemical Geology, 2004, vol. 211, pp. 1-14.
OSPAR 2000 Commission OSPAR 2000. Bilan de santé 2000 [en ligne]. Londres : Commission OSPAR, 2000, 115p. Disponible sur http://www.ospar.org/fr/html/welcome.html (consulté le 5/09/2005)
OSPAR 2001 OSPAR commision 2001. Dumping of wastes at sea in 1998[en ligne]. London : OSPAR commission, 2001, 13p. Disponible sur http ://www.ospar.org (consulté le 5/09/2005)
OSPAR 2003 Commission OSPAR 2003. Diversité biologique et matériaux de dragage : Evaluation générale de l’immersion des déchets en mer du milieu des années 1980 à 2001 dans la zone OSPAR[en ligne]. Londres : OSPAR, 2003. Disponible sur http://www.ospar.org (consulté le 5/09/2005)
Palaprat 2002 Palaprat S. Couplage géochimie/transport lors de la dépollution électrocinétique d’une terre polluée par des métaux lourds. Thèse de doctorat. Lorraine : Institut National Polytechnique de Lorraine, 2002, 233 p
Panagiotis 2002 Panagiotis T., Nymphodora P., Anthimos X. Evaluation of monobasic calcium phosphate for the immobilization of heavy metals in contaminated soils from Lavrion. Journal of Hazardous Materials, 2002, vol. B94, pp. 135-146.
Pardo 1990 Pardo R., Barrado E., Perez L., Vega M. Determination and association of heavy metals in sediments of the Pisucrga river. Water Res, 1990, vol. 24, n. 3, pp. 373-379;
Peltola 2002 Peltola P., Åström M. Concentrations and leachability of chemical elements in estuarine sulfure-rich sediments, W. Finland. The science of total environment, 2002, vol. 284, pp. 109-122.
Pempkowiak Pempkowiak J., Sikora A., Biernacka E. Speciation of heavy metals in marine sediment vs their bioaccumulation by mussels. Chemophere, 1999, vol. 39, pp. 313-321
Piantone 2003 Piantone P., Bodénan F., Derie R., Depelsenaire G. Monitoring the stabilization of municipal solid waste incineration fly ash by phosphatation : mineralogical and balance approach. Waste Management, 2003, vol. 23, pp. 225-243.
Quevauviller 1997
Quevauviller Ph., Rauret G., Lopez-Sanchez J.F., Rubio R. Certification of trace metal extractable contents in a sediment reference material (CRM 601) following a three-step sequential extraction procedure. The Science of the Total Environment, 1997, vol. 205, pp.223-234.
Raicevic 2005
Raicevic S., Kaludjerovic-Radoicic T., Zouboulis A. I. In situ stabilization of toxic metals in polluted soils using phosphates : theoretical prediction and experimental verification. Journal of Hazardous Materials, 2005, vol. B117, pp. 41-53.
Rapport 421 Rapport n°421- projet de loi autorisant la convention pour la protection du milieu marin de l’Atlantique du Nord-Est [en ligne]. Paris : Sénat, 1996/1997. Disponible sur http://www.senat.fr (consulté le 5/09/2005)
Rey C 1984 Rey C. Etude des relations entre apatites et composés moléculaires. Thèse d’état. Toulouse : I.N.P.T, 1984, 350 p.
Rey F 2000 Rey F., Levacher D., Quénec’h J. L. Solidification /stabilisation des vases à base de chaux et additifs, concepts comportementaux et lixiviation. Déchet – Sciences et techniques, 2000, vol. 18, pp. 27-32.

214
Rienks 1998 Rienks J. Comparison of results for chemical and treatement of contaminated dredged sediments. Wat. Sci. Tech, 1998, vol. 37, n°6-7, p 355-362.
Rizet 2000 Rizet L., Charpentier P. E. Métallurgie extractive – hydrométallurgie. Technique de l’ingénieur, 2000, vol. ME, M 2235.
Rossi 2004 Rossi L., de Alencastro L., Kupper T., Tarradellas J. Urban stormwater contamination by polychlorinated biphenyls (PCBs) and its importance for urban water systems in Switzerland. Science of The Total Environment, 2004, vol. 322, Issues 1-3, pp.179-189
Ryba 2002 Ryba S. A., Burgess R. M. Effects of sample preparation on the measurement of organic carbon, hydrogen, nitrogen, sulphur, and oxygen concentrations in marine sediments. Chemosphere, 2002, vol. 48, pp. 139-147
Salomons 1980 Salomons W., Forstner U., Trace metal analysis on polluted sediments : part II : Evaluation of environmental impact. Environ. Technol, 1980, lett. 1, pp. 506-517.
Salomons 1980 Salomons W., Förstner U. Trace metal analysis on polluted sediments Prt II : evaluation of environmental impact. Environ. Technol, 1980, lett. 1, pp. 506-517.
Sarret 1998 Sarret G. Biogéochimie structurale du zinc et du plomb par spectroscopie EXAFS : Interaction avec des acides humiques, des parois cellulaires de champignon, et de lichens. Thèse de doctorat. Grenoble 1 : Université Joseph Fourier, 1998, 189 p.
Scher Scher J. Rhéologie, texture et texturation des produits alimentaires. Technique de l’ingénieur, 2003, F3300, 11p
Seamam-Arey 2001
Seamam J. C., Arey J. S. Bertsch P. M. Immobilization of nickel and other metals in contaminated sediments by hydroxyapatite addition. Journal of Environmental Quality, 2001, vol. 30, pp. 460-469.
Shashkova 1999 Shashkova I., Rat’ko A.I., Kitikova N. Removal of heavy metal ions from aqueous solutions by alkaline-earth metal phosphates. Colloids And Surface, 1999, vol. 160, pp. 207-215.
Silva 2002 Silva I. S. D., Abate G., Lichtig J., Masini J. C. Heavy metal distribution in recent sediments of the Tietê-Pinheiros river system in São Paulo state, Brazil. Applied geochemistry, 2002, vol. 17, pp. 105-116
Singh 2005 Singh K. P., Mohan D., Singh V. K., Malik A. Studies on distribution and fractionation of heavy metals in Gomti river sediments – a tributary on the Ganges, India. Journal of Hydrology, 2005, pp. 1-14.
Sposito 1984 Sposito G. The chemistry of soils. New Work. Oxford : Oxford University press., 1984, 277p.
Stead-dexter 2004
Stead-dexter K., Ward N. I. Mobility of heavy metals within freshwater sediments affected by motorway stormwater. Science of the Total Environment, 2004, vol. 334-335, pp. 271-277.
Stephens 2001 (a)
Stephens S. R., Alloway B. J., Parker A., Carter J. E., Hodson M. E. Change in the leachability of metal from dredged canal sediments during drying and oxidation. Environmental pollution, 2001,vol. 114, pp 407-413.
Stephens 2001 (b)
Stephens S.R., Alloway B. J., Carter J. E., Parker A. Towards the characterisation of heavy metals in dredged canal sediments and an appreciation of availability : two example from the UK. Environmental pollution, 2001, vol. 113, p 395-401.

215
Stevenson 1994 Stevenson F. J. Humus Chemistry, genesis, composition, reactions. Second ed. New York : J. Wiley & Sons., 1994. 496 p.
Sugiyama 2002 Sugiyama S., Ichii T., Hayashi H., Tomida T. Lead immobilisation by non-apatite-type calcium phosphates in aqueous solutions. Inorganic Chemistry Communications, 2002, vol. 5, pp. 156-158.
Tack 1996 Tack F. M., Callewaert O. W. J. J., Verloo M.G. Metal solubility as a function of pH in a contaminated dredged sediment affected by oxidation. Environmental Pollution, 1996, vol. 91, n°. 2, pp. 199-208
Tack 1997 Tack F. M., Lapauw F., Veloo M. G. Determination and fractionation of sulphur in a contaminated dredged sediment. Talante, 1997, vol. 44, pp. 2185-2192.
Tack 1998 Tack F. M., Singh S.P., Veloo M. G. Heavy metal concentrations in consecutive saturation extract of dredged sediment derived surface soils. Environmental Pollution, 1998, vol. 103, pp. 109-115.
Tack 1999 Tack F. G. M., Singh S. P., Verloo M. G. Leaching behaviour of Cd, Cu, Pb and Zn in surface soil derived from dredged sediments. Environ Pollut, 1999, vol. 106, pp. 107-114 ;
Tessier 1979 Tessier A., Campbell P.G. C., Bisson M. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem, 1979, vol. 51, pp. 844-851
Theodoratos 2002
Theodoratos P., Papassiopi N., Xenidis A. Evaluation of monobasic calcium phosphate for the immobilisation of heavy metals in contaminated soils from Laviron. Journal of hazardous materials, 2002, vol. B94, pp. 135-146.
Tokalioglu 2000 Tokalioglu S., Kartal S., Elci L. Detemination of heavy metals and speciation in lake sediments by flame absorption spectrometry after a four-stage sequential extraction procedure. Analytical Chimica Acte, 2000, vol. 413, pp. 33-40.
Toor 2005 Toor G. P., Sims J. T., Dou Z. Reducing phosphorus in dairy diets improves farm nutrient balances and increases the risk of nonpoint pollution of surface and ground waters. Agriculture Ecosystems & Environment, 2005, vol. 105, pp; 401-411.
Traina 1999 Traina S. J., Laperche V. Contaminant bioavailability in soils, sediments, and aquatic environments. Proc. Natl. Acad. Sci. USA, 1999, vol. 96, pp. 3365-3371.
Tung 1997 Tung M.S. Calcium phosphates in biological and industrial systems- In Zahid Amjed. Calcium phosphates : structure, composition, solubility and stability. Boston : Kluwer academic publishers, 1997, 515 p
Uchida 1996 Uchida T., Itoh I., Harada K. Immobilization of heavy metals contained in incinerator fly ash by application of soluble phosphate – treatment and disposal cost reduction by combined use of “high specific surface area lime”. Waste Management, 1996, vol. 16, pp. 475-481.
Usero 1998 Usero J., Gamero M., Morillo J., Gracia I. Comparative study of three sequential extraction procedures of metals in marins sediments. Environment International, 1998, vol. 24, pp 478-496.
Valsami-Jones Valsami-Jones E., Fields M., Ragnarsdottir K. V. Apatite reactivity in the presence of rare earth elements (REE) and uranium. Journal of conference abstract, 2000, vol. 5, pp. 1030.
van Kessel 2002 van Kessel T., van Kesteren W. G.M. Gas production and transport in artificial sludge depots. Waste Management, 2002, vol 22, pp 19-28.

216
Vandecasteele 2002
Vandecasteele B., De Vos B., Tack F. M. G. Heavy metal contents in surface soils along the Upper Scheldt river (Belgium) affected by historical upland disposal of dreged materials. The Science of the Total Environment, 2002, vol. 290, pp.1-14.
Vandecasteele 2002
Vandecasteele B., De Vos B., Tack F. M. G. Heavy metal contents in surface soils along the Upper Scheldt river (Belgium) affected by historical upland disposal of dreged materials. The science of the total environment, 2002, vol. 290, p 1-14
Vanderschuren 2000
Vanderschuren J. Séchage. Les techniques de l’industrie minérale, 2000, n° 8, 128p.
Vanthuyne 2003 Vanthuyne M., Maes A., Cauwenberg P. The use of flotation techniques in the remediation of heavy metal contaminated sediments and soils : an overview of controlling factors. Minerals Engineering,2003, vol. 16, pp. 1131-1141.
Velasquez Velasquez S. Application ICP-AES : présentation de l’ULTIMA 2 et des performances analytiques typiques. Rapport interne de démonstration. Longjumeaux: Jobin Yvon Horiba, 2003, 10 p
VNF 2002 Voies Navigables de France. Historique national des opérations de curage et perspectives. Béthune : VNF, 2002
Wallman 1993 Wallman W., Kersten M., Gruber J. Forstner U. Artifacts in the determination of trace metal binding forms in anoxic sediments by sequential extractions. Int Environ Anal Chem, 1993, vol. 51, pp. 187-200.
Walpersdort 2004
Walpertsdort E., Neumann T., Stüben D. Efficiency of natural calcite precipitation compared to lake marl allocation used for water quality improvement in an eutrophic lake. Applied Geochemistry, 2004, vol. 19, pp. 1687-1698.
Wang 2001 Wang Y. M., Chen T.C., Yeh K. J., Shue M. F. Stabilization of an elevated heavy metal contaminated site. Journal of hazardous Materials, 2001, vol. B88, pp. 63-74.
Yuan 2004 Yuan C-G., Shi J-B., He B., Liu J-F. Speciation of heavy metals in mrine sediments from east China Sea by ICP-S with sequential extraction. Environment International, 2004, vol. 30, pp 769-783.
Zhu 2004 Zhu Y. G., Chen S. B., Yang J. C. Effects of soil amendments on lead uptake by two vegetable crops from lead-contaminated soil from Anhui, China. Environment International, 2004, vol. 30, pp. 351-356.
Zorpas 2000 Zorpas A. A., Constantinides T., Vlyssides A. G., Haralambous I. Heavy metal uptake by natural zeolite and metals partitioning in sewage sludge compost. Bioresource Technology, 2000, vol. 72, Issue 2, pp 113-119
Zoumis 2001 Zoumis T., Schmidt A., Grigorova L., Calmano W. Contamination in sediments : remobilisation and demobilisation. The Science of the Total Environment, 2001, vol. 266, pp. 195-202.

217
Annexe A
Calcul des formules structurales : principe
Exemple de l'olivine L'objectif du calcul des formules structurales est de passer de la composition en %poids
d'oxyde donnée par la microsonde en fraction molaire, en respectant la formule de
l'architecture du minéral. Par exemple, on va calculer la formule d'une olivine. Les olivines
forment une solution solide entre la fayalite (pôle ferrifère) et la forstérite (pôle magnésien).
Soit une olivine dans la composition est donnée par la colonne [1] :
OLIVINE [1] Masse Mol. [2]
Prop. mol. [3]
Cations/mol[4]
Nb cations [5]
Ox/mol[6]
Nb oxygènes [7]
Nb/cations 4
[8] SiO2 34,96 60,09 0,58 1 0,58 2 1,164 0,99 FeO 36,77 71,85 0,51 1 0,51 1 0,512 0,87 MgO 27,04 40,3 0,67 1 0,67 1 0,671 1,14 MnO 0,52 70,94 0,01 1 0,01 1 0,007 0,01 Total 99,29 2,354
Colonne [2] : masse molaire moléculaire
Colonne [3] : proportions moléculaires. Résultat de la division de la composition par la
masse molaire de l'oxyde
Colonne [4] : nombre de cations dans une mole d'oxyde
Colonne [5] : nombre de cations à partir de la composition. Multiplier [3] par [4]
Colonne [6] : nombre d'oxygène dans chaque oxyde. Comptabiliser le nombre d'oxygènes
dans chaque oxyde
Colonne [7] : multiplier la proportion moléculaire par [3] le nombre d'oxygène [6]. Faire
le total
Colonne [8] : diviser le résultat de l'étape [5] par le total de l'étape [7] et multiplier par le
nombre d'oxygène du modèle d'architecture (attention : les oxygènes liés à l'eau ne sont pas
comptabilisés). Dans le cas de l'olivine, c'est 4.
La formule structurale est :
[Si0,99O4](Fe0,87Mg1,14Mn0,0) Ce même principe a été appliqué pour la détermination de la formule structurale des
phosphates de calcium en prenant en compte 25 d’oxygène.

218
Liste des figures Figure I- 1 : Projection de la structure apatitique selon l’axe c [Traina 1999].................................................40 Figure I- 2 : Procédé NOVOSOL pour le traitement des sédiments. ..................................................................51 Figure I- 3 : Diagramme triangulaire de classification des sédiments en fonction de leur texture [Campy 1995]............................................................................................................................................................................53 Figure I- 4 : Modèle hypothétique de l’acide fulvique........................................................................................58 Figure I- 5 : Modèle hypothétique de l’acide humique.......................................................................................58 Figure II- 1 : Diffractogramme des rayons X du sédiment de Vraimont (en haut) et de Lille (en bas) avec : о (Calcite), + (Quartz), * (Feldspath) ...................................................................................................................83 Figure II- 2 : Evolution de la quantité de H+ résiduaires en solution (▲) par rapport à la quantité apportée pour 1 g de sédiment BV (en haut) et BL (en bas). ......................................................................................................87 Figure III- 1 : Dispositif expérimentale de phosphatation..................................................................................96 Figure III- 2 : Influence du rapport solide/liquide sur la concentration du P libre dans le sédiment phosphaté à 1 et 7%. ..................................................................................................................................................................98 Figure III- 3 : Veine de séchage. ......................................................................................................................100 Figure III- 4 : Dispositif expérimental de séchage (a). Positionnement de l’échantillon en cours d’expérience : vue latérale (b). .................................................................................................................................................101 Figure III- 5 : Influence de la quantité d’acide sur le taux de carbone inorganique (sous dorme de CO2) dégagé...........................................................................................................................................................................105 Figure III- 6 : Evolution de la viscosité des sédiments en fonction de la quantité d’acide et du taux d’humidité...........................................................................................................................................................................106 Figure III- 7 : Evolution du pH en solution en fonction du temps de passage dans le réacteur tubulaire. ......107 Figure III- 8 : Evolution de la concentration du phosphore libre en solution en fonction du temps de passage dans le réacteur tubulaire. ................................................................................................................................108 Figure III- 9 : Evolution du taux de conversion du phosphore libre en solution en fonction du temps de passage dans le réacteur tubulaire .................................................................................................................................109 Figure III- 10 : Evolution de la consommation du calcium en solution en fonction du temps de passage dans le réacteur tubulaire .............................................................................................................................................110 Figure III- 11 : Evolution de la consommation du Fe (a) et du Zn (b) en solution en fonction du temps de passage dans le réacteur tubulaire ...................................................................................................................112 Figure III- 12 : Evolution de la consommation du Ca en fonction de la consommation du P au cours du temps dans le réacteur.................................................................................................................................................117 Figure III- 13 : Evolution du pH en solution au cours du temps de la phosphatation (en réacteur tubulaire et en phase de maturation) ........................................................................................................................................119 Figure III- 14 : Evolution de la concentration du P en solution au cours du temps de la phosphatation (en réacteur tubulaire et en phase de maturation)..................................................................................................120 Figure III- 15 : Evolution de la concentration du Ca en solution au cours du temps de la phosphatation (en réacteur tubulaire et en phase de maturation. ..................................................................................................120 Figure III- 16 : Courbe de séchage exprimée de différentes manières. ............................................................127 Figure III- 17 : Courbe de séchage du sédiment brut (T = 45°C - HR=50% - v=1.5m/s)................................131 Figure III- 18 : Evolution lors du séchage du sédiment brut de la température du bulbe humide et du produit (T = 45°C - HR=50% - v=1.5m/s) ........................................................................................................................131 Figure III- 19 : Influence de la vitesse de l’air sur la cinétique de séchage du sédiment brut (T = 25°C - HR=50% - v=1.5m/s)........................................................................................................................................132 Figure III- 20 : Influence de la température de séchage sur les cinétique de séchage du sédiment brut ( HR=50% - v=1.5m/s)........................................................................................................................................................133 Figure III- 21 : Influence de la quantité d’acide sur la cinétique de séchage des sédiments (T = 45°C - HR=50% - v=1.5m/s)........................................................................................................................................................134 Figure III- 22 : : Evolution lors du séchage de la température du bulbe humide et du produit du sédiment phosphaté à 3% (a) et 5% (b) (T = 45°C - HR=50% - v=1.5m/s) ....................................................................135 Figure III- 23 :Influence de la température sur la cinétique de séchage des sédiments phosphaté à 5% (HR=50% - v=1.5m/s)........................................................................................................................................................136 Figure III- 24 : Influence de la quantité d’acide sur la surface spécifique des sédiments après séchage. .......137 Figure III- 25 : Photos de l’aspect du sédiment brut (a) et du sédiment phosphaté à 5% après séchage (T = 45°C - HR=50% - v=1.5m/s). ....................................................................................................................................137 Figure III- 26 : Courbe d’analyse thermogravimétrique du sédiment phosphaté à 7% à différentes températures...........................................................................................................................................................................141
219
Figure III- 27 : Evolution du pourcentage massique du carbone total dans les sédiments en fonction des différentes températures de calcination. ...........................................................................................................142 Figure III- 28 : Evolution de la surface spécifique des sédiments en fonction de la température de calcination...........................................................................................................................................................................143 Figure III- 29 : Evolution de la densité des sédiments en fonction de la température de calcination. .............144 Figure IV- 1 : Protocole d’extraction séquentielle de Tessier et al. .................................................................151 Figure IV- 2 : Distribution des éléments chimiques dans les sédiments bruts de Lille (a) et Vraimont (b)......155 Figure IV- 3 : Distribution du P dans les sédiments de Lille traités et non traités (le suffixe C indique que les sédiments ont été calcinés)................................................................................................................................163 Figure IV- 4 :Distribution du P dans les sédiments de Vraimont traités et non traités ...................................163 Figure IV- 5 : Distribution du Ca dans les sédiments de Lille traités et non traités.........................................166 Figure IV- 6: Distribution du Ca dans les sédiments de Vraimont traités et non traités ..................................166 Figure IV- 7 : Distribution de l’Al dans les sédiments de Lille traités et non traités........................................167 Figure IV- 8 : Distribution de l’Al dans les sédiments de Vraimont traités et non traités................................168 Figure IV- 9 : Distribution de Fe dans les sédiments de Lille traités et non traités. ........................................168 Figure IV- 10 : Distribution de Fe dans les sédiments de Vraimont traités et non traités................................169 Figure IV- 11 : Distribution de As dans les sédiments de Lille traités et non traités........................................171 Figure IV- 12 : Distribution de As dans les sédiments de Vraimont traités et non traités................................171 Figure IV- 13 : Distribution du Cr dans les sédiments de Lille traités et non traités .......................................172 Figure IV- 14 : Distribution de Cr dans les sédiments de Vraimont traités et non traités................................173 Figure IV- 15 : Distribution du Co dans les sédiments de Lille traités et non traités.......................................175 Figure IV- 16 : Distribution de Co dans les sédiments de Vraimont traités et non traités ...............................175 Figure IV- 17 : Distribution du Cd dans les sédiments de Lille traités et non traités.......................................177 Figure IV- 18 : Distribution de Cd dans les sédiments de Vraimont traités et non traités ...............................177 Figure IV- 19 : Distribution du Pb dans les sédiments de Lille traités et non traités .......................................178 Figure IV- 20 : Distribution du Pb dans les sédiments de Vraimont traités et non traités ...............................178 Figure IV- 21 : Distribution du Zn dans les sédiments de Lille traités et non traités .......................................180 Figure IV- 22 : Distribution du Zn dans les sédiments de Vraimont traités et non traités ...............................181 Figure IV- 23 : Distribution du Pb dans les sédiments de Lille traités et non traités .......................................182 Figure IV- 24 : Distribution du Pb dans les sédiments de Vraimont traités et non traités ...............................183 Figure IV- 25 : Analyse en microsonde du Al, Si, P, Ca, Fe et S dans le sédiment de Vraimont. Image traitée en couleur arbitraire. Echelle des couleurs correspondant aux concentrations relatives des éléments : rouge = maximum, violet = minimum et noir = inférieur au minimum. Superposition des différentes cartographie......................................................................................................................................................187 Figure IV- 26 : Analyse en microsonde du Al, Si, P, Ca, Fe et S dans le sédiment de Vraimont phosphaté à 5% puis calciné à 700°C. Image traitée en couleur arbitraire. Echelle des couleurs correspondant aux concentrations relatives des éléments : rouge = maximum, violet = minimum et noir = inférieur au minimum. Superposition des différentes cartographies...................................................................................190 Figure IV- 27 : Image d’une particule de silicate d’aluminium d’un sédiment phosphaté à 5%sur sec à un grossissement x1600 (a) et à un grossissement x6400 (b). Analyse semi-quantitative (EDS) de (b)de la zone encadrée............................................................................................................................................................194 Figure IV- 28 : Image d’une particule de silicate d’aluminium d’un sédiment phosphaté à 5%sur sec et calciné à 700°C à un grossissement x200 (a) et à un grossissement x1600 (b). Analyse semi-quantitative (EDS) de (b)de la zone encadrée....................................................................................................................................................194 Figure IV- 29 : Image d’une particule de silicate d’aluminium d’un sédiment phosphaté à 5%sur sec et calciné à 1100°C à un grossissement x401 (a) et à un grossissement x1602 (b). Analyse semi-quantitative (EDS) de la zone encadrée 1 (1) et de la zone encadrée 2 (2). .............................................................................................195
220
Liste des tableaux Tableau I- 1 : Curage d’entretien recensés de 1990 à 2000.................................................................................9 Tableau I- 2 : Données générales du dragage continental par maîtres d’ouvrage. .............................................9 Tableau I- 3 : Données générales du dragage continental par type d’hydrosystème .........................................10 Tableau I- 4 : Principales motivations du curage...............................................................................................10 Tableau I- 5 : Valeurs de référence du port autonome de Rouen .......................................................................13 Tableau I- 6 :Teneurs limites en éléments-traces dans les boues et dans les sols. .............................................14 Tableau I- 7 Valeurs limites en matière de lixiviation pour les trois déchets (test de lixiviation sur la base d’un ration liquide solide de 10 l/kg) ..........................................................................................................................16 Tableau I- 8 : Niveaux de référence (mg/kg sédiment sec) pour les métaux et polychlorobiphényles selon l’arrêté du 14 juin 2000. ..................................................................................................................................................17 Tableau I- 9 : Quantité de matériaux de dragage, en tonnes de poids sec, déversés en mer en 1996 et 1998 par les pays membres de la convention OSPAR [OSPAR (2000 – 2001)] ................................................................20 Tableau I- 10 : Destination des produits de curage entre 1990 et 2000.............................................................20 Tableau I- 11 : Différents phosphates de calcium ..............................................................................................38 Tableau I- 12 : Utilisation des sources de phosphate dans le traitement des déchets ........................................44 Tableau I- 13 : Gamme de concentration élémentaire (%) pour les substances humiques [Stevenson 1994]....57 Tableau I- 14 : Teneur des groupes fonctionnels principaux (m mol/g) pour les substances humiques [Stevenson 1994]. ..................................................................................................................................................................57 Tableau I- 15 : Méthodes d’extraction séquentielles communément utilisées [Das 1995].................................65 Tableau II- 1 : Humidité à 40°C des sédiments de Lille et de Vraimont.............................................................74 Tableau II- 2 : Humidité résiduelle des sédiments de Lille et de Vraimont. .......................................................75 Tableau II- 3 : pH des sédiments de Lille et de Vraimont, bruts et séchés à 40°C .............................................76 Tableau II- 4 : Conductivité des sédiments de Lille et de Vraimont bruts et séchés à 40°C...............................77 Tableau II- 5 : : Concentration d’élément majeur et mineur dans les deux sédiments exprimée en mg/kg sur matière sèche ......................................................................................................................................................79 Tableau II- 6 : Pourcentage de carbone total, carbone organique, carbone inorganique et taux de carbonates dans les sédiments...............................................................................................................................................80 Tableau II- 7 : Pourcentage massique de matière organique dans les sédiments ..............................................81 Tableau II- 8 : Capacité d’échange cationique des sédiments de Lille et Vraimont exprimés en meq.100 g-1. ..85 Tableau II- 9 : Densité des sédiments bruts de Lille et de Vraimont. .................................................................88 Tableau II- 10 : Distribution granulométrique des sédiments bruts de Lille et de Vraimont. ............................90 Tableau II- 11 : Récapitulatif des données physico-chimique des deux sédiments. ............................................92 Tableau III- 1 : Volume des réacteurs de phosphatation et temps de contacts correspondant...........................96 Tableau IV- 1 : Concentration (mg/kg sur matière sèche) des différents éléments chimiques dans les différente fraction en du sédiment de Lille et Vraimont ....................................................................................................156 Tableau IV- 2 : Sommes des concentrations (mg/kg de matière sèche) des quatre fractions non résiduelles pour les sédiments de Lille traités et non traités .......................................................................................................184 Tableau IV- 3 : Sommes des concentrations (mg/kg de matière sèche) quatre fractions non résiduelles pour les sédiments de Vraimont traités et non traités .....................................................................................................184 Tableau IV- 4 : Analyse quantitative de microsonde de quelque phases présentes dans le sédiment brut. ......188 Tableau IV- 5 : Analyse quantitative des nouvelles phase de phosphate de calcium formées après traitements.191 Tableau IV- 6 : Résultats de test de lixiviation NF X30-402-2 et valeurs limites pour mise en décharge pour déchets inertes exprimés en mg/kg de matière sèche. .......................................................................................197


















