cystic fibrosis STUDENT version
29
Cystic Fibrosis: Who Knew Mucus Could Cause So Much Trouble? Elva Angelique Van Devender, Ph.D., Pharm.D. PGY1 Pharmacy Practice Resident Providence Health and Services Portland, Oregon February 2012
-
Upload
elva-van-devender-phd-pharmd-bcps -
Category
Documents
-
view
72 -
download
1
Transcript of cystic fibrosis STUDENT version

Cystic Fibrosis:
Who Knew Mucus Could Cause So Much Trouble?
Elva Angelique Van Devender, Ph.D., Pharm.D.
PGY1 Pharmacy Practice Resident
Providence Health and Services
Portland, Oregon
February 2012

Learning Objectives By the end of this lecture, you should be able to….
1. Explain the etiology and pathophysiology of cystic fibrosis and its multi-organ
system involvement
2. Summarize the course/progression of the disease and the prognosis for
cystic fibrosis patients
3. Identify the clinical and diagnostic presentation of cystic fibrosis
4. Recommend appropriate pharmacologic and non-pharmacologic therapies to
manage cystic fibrosis
2

Famous People with (Suspected) Cystic Fibrosis
3

Epidemiology and Survival
The Cystic Fibrosis Foundation estimates about 30,000 children and adults in
the U.S. have the disease (70,000 worldwide).
• About 1,000 new cases each year.
• More than 70% of patients are diagnosed by age two
• More than 45% of the cystic fibrosis (CF) patient population is age 18 or
older
• 1 in 25 people are carriers
4
Median Survival of CF Patients
The predicted median age of
survival for a person with CF is
the late 30s.
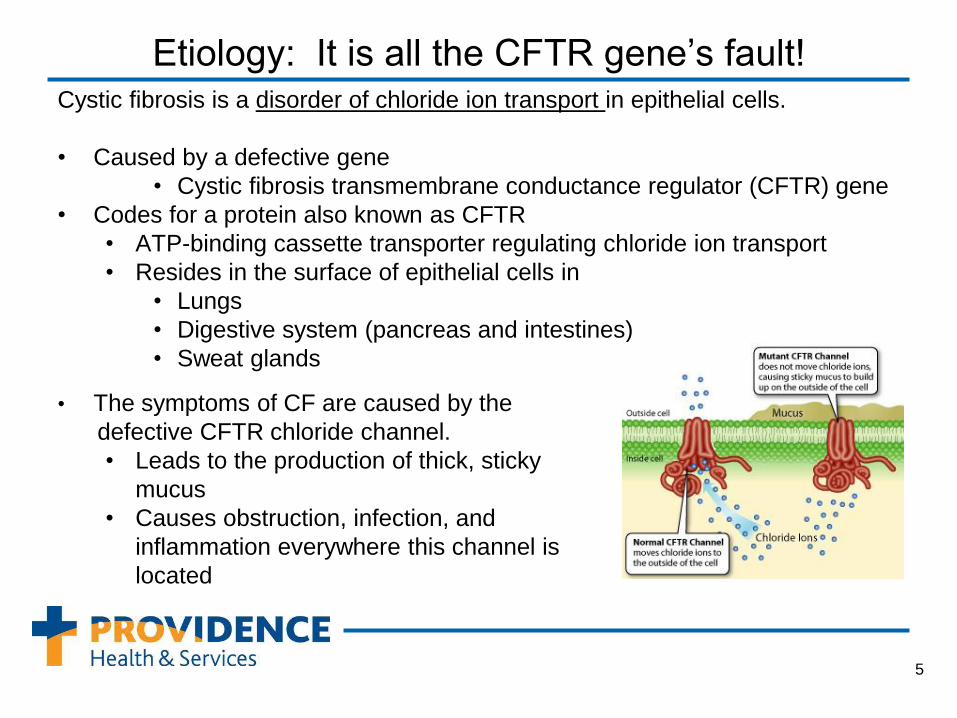
Etiology: It is all the CFTR gene’s fault! Cystic fibrosis is a disorder of chloride ion transport in epithelial cells.
• Caused by a defective gene
• Cystic fibrosis transmembrane conductance regulator (CFTR) gene
• Codes for a protein also known as CFTR
• ATP-binding cassette transporter regulating chloride ion transport
• Resides in the surface of epithelial cells in
• Lungs
• Digestive system (pancreas and intestines)
• Sweat glands
5
• The symptoms of CF are caused by the
defective CFTR chloride channel.
• Leads to the production of thick, sticky
mucus
• Causes obstruction, infection, and
inflammation everywhere this channel is
located

CFTR Mutation Determines Disease Severity Five functional classes of CF mutations have been described:
Class 1 mutations: Defective protein production with
premature termination of CFTR production; produce few or
no functioning CFTR chloride channels.
Class 2 mutations: (Most common affects~90% CF
patients); Defective trafficking of CFTR so that it does not
reach the apical surface membrane.
Class 3 mutations: Defective regulation of CFTR even
though it is able to reach the apical cell surface.
Class 4 mutations: CFTR reaches the apical surface but
chloride transport through the channel is defective.
Class 5 mutations: Reduced production of functional
CFTR. A small amount of functional CFTR may reach the
surface.
6

Mucus, Mucus, Mucus Everywhere!!! Cystic fibrosis is a multi-system disease!
Sinuses and Lungs
→ life-threatening lung infections
Pancreas
→ glucose intolerance (CF DM)
→ deficiencies of digestive enzymes leading
to↓digestion, ↓absorption, and ↓nutrition
Intestines
→ ↓nutrient absorption
→ problems with stool passage
Liver
→ cirrhosis
Vas deferens in men and the uterus in women
→ infertility
Sweat glands
→ salty perspiration
7

How Will Your Patients Present? Respiratory/Sinus
• Wheezing or shortness of breath
• Persistent coughing, with or without sputum
• Chronic sinusitis, sinus infections, and nasal polyps
• Recurrent or persistent lung infections with S. aureus or P. aeruginosa
Digestive/Nutrition
• Obstruction of the bowel at birth (meconium ileus)
• Frequent greasy, bulky stools, or difficulty in bowel movements.
• Poor growth/ difficulty with weight gain (failure to thrive)
• Malnutrition and vitamin deficiencies
Skin
• Tastes “salty”
• Clubbing of the fingers and toes
8

How is Cystic Fibrosis Diagnosed? Usually diagnosed in neonates (due to meconium ileus at
birth) or through newborn screening programs
• Diagnostic tests:
– Sweat test: gold standard
– Newborn screening
– Genetic testing
• What other tests might be ordered?
– WBC, glucose
– Liver function tests
– Pancreatic function tests
– Oropharyngeal culture for CF pathogens,
especially Pseudomonas
– Chest or abdominal radiograph
9
Chloride
Concentration
Result
< 40 mmol/L Normal
40-60 mmol/L Borderline or
indeterminate
> 60 mmol/L Abnormal

Pharmacokinetic Issues Patients with CF
• Have enhanced hepatic and renal drug clearance of medications due to
hypermetabolic state
– e.g. Septra, aminoglycosides, and vancomycin
• Have increased volumes of distribution for hydrophilic drugs due to
increased ratio of lean body mass to total body mass
– e.g. penicillins, cephalosporins, and aminoglycosides
• What does this mean for the patient?
– Potentially higher doses, more frequent dosing intervals
– Necessitates adequate monitoring of antibiotic levels to make sure
patient is getting optimal doses!
10

Overview: Mainstays of CF Therapy
• Medications for lung health maintenance
– Inhaled antibiotics
– Inhaled mucolytics
• Dornase alfa
• Hypertonic saline
– Oral anti-inflammatories
• Azithromycin
• High-dose ibuprofen
• Maintain adequate nutrition!
– High-calorie diet, including vitamin supplements
• Fat soluble vitamins: ADEK
– Supplements to boost immunity (antioxidants)
• Selenium, ascorbate and zinc
– Appropriate doses of pancreatic enzymes
11

Upper Respiratory Tract Complications Inflammation and infection of the upper airways and sinuses are extremely
common in people with CF.
• Upper airway disease is characterized by
– Runny nose/ post-nasal drip
– Nasal polyps
– Thick mucous secretions/ sinus obstruction
– Bacterial growth and infection!
• Chronic obstruction/ inflammation leads to
– Chronic sinusitis
– Exacerbation of lower respiratory symptoms
12

Lower Respiratory Tract Complications
• Pulmonary disease is characterized by
– Thick mucus secretions and impaired
clearance
– Obstruction of distal airways
– Bacterial colonization and infection!
• Chronic obstruction/ inflammation leads to
– Air trapping, atelectasis, bronchiectasis
– Scar tissue formation
– Chronic infection
– Respiratory failure!!!
• Decreased forced vital capacity (FVC)
• Decreased forced expiratory volume in 1
sec (FEV1)
13
Chronic lung disease is a hallmark of CF leading to death in 90% of patients.
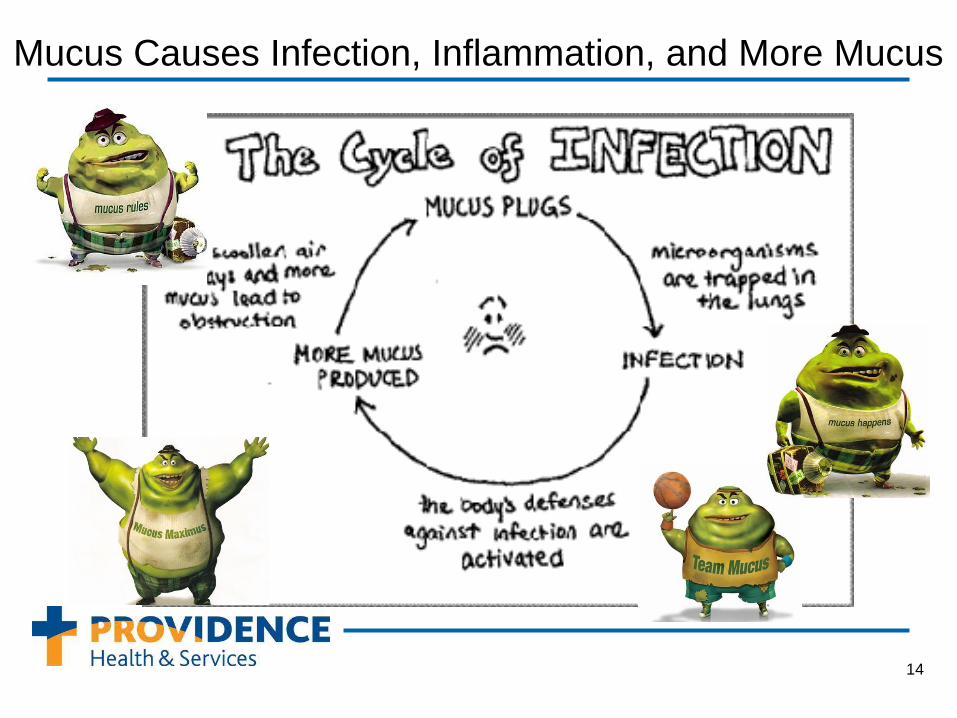
Mucus Causes Infection, Inflammation, and More Mucus
14

Cystic Fibrosis Respiratory Pathogens
Haemophilus influenzae, Staphylococcus aureus– mostly in children
Pseudomonas aeruginosa – most common and most important infection in adults
Burkholderia cepacia – rare; highly resistant
Stenotrophomonas maltophilia – difficult to eradicate
Aspergillus species – sputum often colonized
15

Treatment of Respiratory Complications • Antibiotics (Selection based on sputum cultures; double cover Gram- bugs)
– Oral antibiotics: mild exacerbations
• PCNs and Cephs: dicloxacillin, amox/clav, cephalexin
• Septra
• Fluoroquinolones: levofloxacin, moxifloxacin
• Macrolides: azithromycin, clarithromycin
– Inhaled antibiotics: acute exacerbations or chronic suppression
• Tobramycin
• Colistin
• Aztreonam
– IV antibiotics: moderate to severe exacerbations
• Aminoglycosides: tobramycin gentamicin
• PLUS beta-lactam/β-lactamase combo: piperacillin, ticarcillin
• OR 3rd or 4th gen ceph: ceftazidime, cefepime
• OR carbapenem: meropenem, imipenem
16

Respiratory Clearance Therapies • Mechanical devices
• Flutter valve devices with positive expiratory
pressure (PEP)
• High-frequency chest wall compression (HFCC)
devices
• Intrapulmonary percussive ventilation (IPV) devices
• Airway clearance methods
• Coughing
• Hand-held percussor
• Chest physical therapy
• Percussion and postural draining
• Active Cycle of Breathing Technique (ACBT)
• Autogenic or “self” drainage
17
HF
CC
devic
e
PE
P d
evic
e
IPV
devic
e

Other Inhaled Pharmacotherapy Agents Mucolytics
• Dornase alfa (Pulmozyme)
– MOA: cleaves DNA strands in the airway secretions to
help decrease mucus viscosity and improve lung function
– Dose: nebulized solution; 2.5 mg given 1-4x daily
• Hypertonic saline 7%
– MOA: improves the movement of salt in and out of cells,
allowing mucus to be more hydrated and cleared more
easily
– Dose: nebulized solution; 4 mL given 1-4x/daily
18
Bronchodilators
• Albuterol
− MOA: short-acting beta adrenergic agonist
− Dose: nebulized; 2.5 mg 2-4x daily
inhaler; 2 puffs 2-4x/daily

Treatment of Pulmonary Inflammation Anti-inflammatory agents
• MOA: ↓ inflammation in the lungs of people with cystic fibrosis, ↓chronic
damage to lung tissue
• High dose ibuprofren (oral)
– Dose: 15-30 mg/kg Q12H (most doses ~20mg/kg NTE1600mg/day)
• Cmax target: 50-100 mg/L (evaluate 1,2, and 3 hours after dose)
• Oral corticosteriods
– Short-term use only due to extensive side effect profile!
• Inhaled corticosteriods
– Consider when asthma, allergies, and/or upper respiratory symptoms
are present
• Azithromycin
– Dose: 250-500mg daily or MWF
19

Treatment of Nutritional Deficiencies Most CF patients have ↑caloric needs due to ↑energy
expenditure.
Recommendations
• High-calorie diet, including vitamin supplements when
needed
– Fat soluble vitamins: ADEK
• Products: AquADEKs, SourceCF, and Vitamax
– Calcium to promote bone health
– Iron to combat anemia
• Supplements to boost immunity (antioxidants)
– Selenium, ascorbate and zinc
• Appropriate doses of pancreatic enzymes
• Surgical placement of G-tube or J-tube if aggressive diet
and oral supplements fail
20
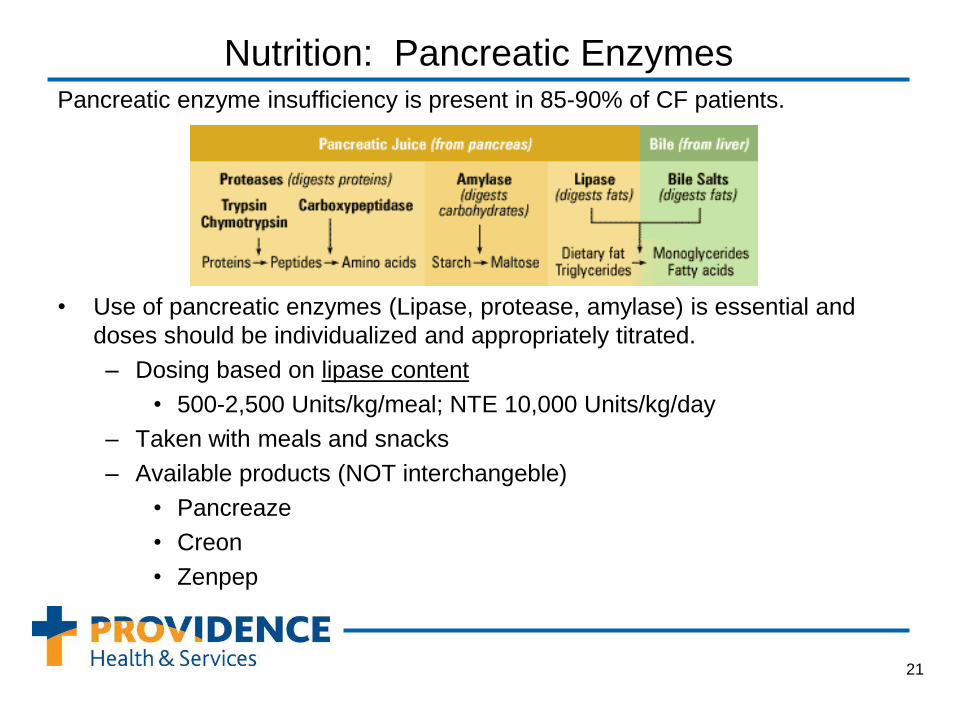
Nutrition: Pancreatic Enzymes Pancreatic enzyme insufficiency is present in 85-90% of CF patients.
• Use of pancreatic enzymes (Lipase, protease, amylase) is essential and
doses should be individualized and appropriately titrated.
– Dosing based on lipase content
• 500-2,500 Units/kg/meal; NTE 10,000 Units/kg/day
– Taken with meals and snacks
– Available products (NOT interchangeble)
• Pancreaze
• Creon
• Zenpep
21
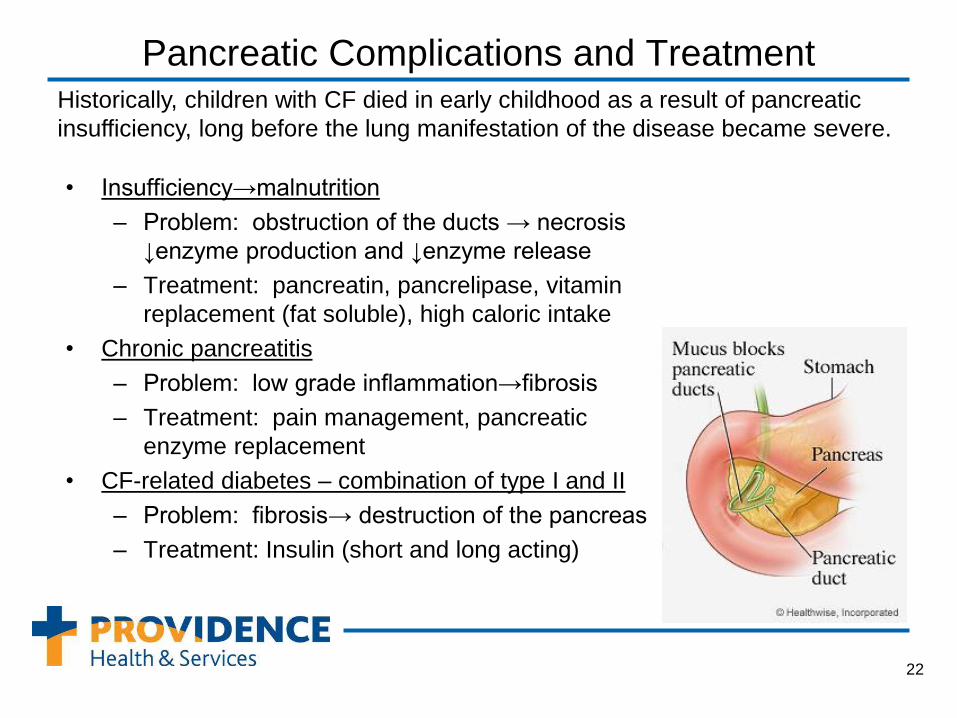
Pancreatic Complications and Treatment
• Insufficiency→malnutrition
– Problem: obstruction of the ducts → necrosis
↓enzyme production and ↓enzyme release
– Treatment: pancreatin, pancrelipase, vitamin
replacement (fat soluble), high caloric intake
• Chronic pancreatitis
– Problem: low grade inflammation→fibrosis
– Treatment: pain management, pancreatic
enzyme replacement
• CF-related diabetes – combination of type I and II
– Problem: fibrosis→ destruction of the pancreas
– Treatment: Insulin (short and long acting)
22
Historically, children with CF died in early childhood as a result of pancreatic
insufficiency, long before the lung manifestation of the disease became severe.

Intestinal Complications and Treatment • GERD
– Problem: Persistent coughing ↑abdominal pressure
↑lower esophageal sphincter (LES) pressure → acid
reflux
– Treatment: PPI, H2RA, sucralfate, surgery
• Meconium ileus (up to 20% of CF cases at birth)
– Problem: pancreatic insufficiency and Cl channel
dysfunction → thick fluids in intestine ↓ transit time of
stool in children
– Treatment: Irrigating products, surgery
• Distal interstitial obstructional syndrome (DIOS)
– Problem: Same as meconium ileus, found in older
children
– Treatment: laxatives (e.g. polyethylene glycol),
enemas, surgery
23

Hepatobiliary Complications and Treatment
• Gallstones
– Problem: gallbladder and cystic duct impacted by
thick mucosa
– Treatment: ursodiol (8-10mg/kg/day in 2-3 divided
doses)
• Focal biliary cirrhosis (multilobular cirrhosis)
– Problem: obstruction, inflammation → cirrhotic and
ductular changes
– Treatment: management of end-stage liver
disease, transplant, prevention, ursodiol (13-15
mg/kg/day in 2-4 divided doses)
• Liver Impairment
– Problem: toxic bile acid accumulation
– Treatment: ursodiol (15-20 mg/kg/day in 2 divided
doses)
24

Reproductive Complications and Treatment • Infertility
– Problem:
• men (95% are sterile)
– delayed puberty
– can’t release sperm
– lack vas deferens
– obstruction of epdidymis, vas deferens, and seminal vesicles
• women (20% infertile)
– delayed puberty
– abnormal cervical mucus
– amenorrhea
– Treatment:
• Males – in vitro fertilization
• Women – can usually conceive
25

Other Issues • Sweat glands
– Problem: ↑Na, ↑Cl (not reabsorbed)
– Treatment: stay hydrated, avoid overheating
• Musculoskeletal disorders
– Problem:
• ↑ Joint pain
• ↓ Bone density → osteopenia/osteoporosis
– Treatment:
• Analgesics
• Calcium, vitamin D, bisphosphonates
• Hematologic
– Problem: impaired erythropoietin regulation,
chronic inflammation and ↓ nutrition→ anemia
– Treatment: iron supplementation
26

Future Therapies • Gene Therapy (e.g. PLASmin)
• CFTR modulators (e.g. Ataluren, VX-809, Kalydeco)
• Surfactants (e.g. Bronchitol, Gilead GS9411)
• Anti-infectives (e.g. inhaled fluoroquinolones, fosfomycin/tobramycin)
• Anti-inflammatories (e.g. inhaled glutathione, sildenafil, DHA)
• Transplantation (e.g. inhaled cyclosporin)
• Nutrition (e.g. Liprotamase)
27

Ivacaftor (Kalydeco): New Drug for Cystic Fibrosis • Approval: The FDA approved ivacaftor (Kalydeco) for patients with CF
with a CFTR G551D mutation on January 31, 2012.
• MOA: CFTR potentiator: improves Cl transport through the channel
• SE: upper respiratory tract infection, headache, stomachache, rash,
diarrhea, and dizziness
• Claims to fame: New class of CF drugs; first treatment to target the
defective CFTR protein rather than only the symptoms of the disease
• Price tag: $294,000 for one year!!!
28
• Place in therapy: only in CF patients >6 years with a
CFTR G551D mutation!

References 1. Cystic Fibrosis Foundation. http://www.cff.org. Accessed January 30, 2012.
2. Johns Hopkins Cystic Fibrosis Center. http://www.hopkinscf.org . Accessed January 30, 2012.
3. Milavetz G. Cystic Fibrosis. In: Dipro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey LM, eds. Pharmacotherapy: A
Pathophysiologic Approach. 7th ed. New York: McGraw-Hill Companies, Inc., 2008: 535-546.
4. Kerr M. FDA Approves New Drug for a Form of Cystic Fibrosis. Medscape News [serial online]. January 31, 2012; Accessed
January 31, 2012. Available at http://www.medscape.com/viewarticle/757801.
5. Farrell PM, Rosenstein BJ, White TM, et al. Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults:
Cystic Fibrosis Foundation Consensus Report J Pediatr. 2008;153:S4-S14.
6. LeGrys VA, Yankaskas JR, Quittell LM, et. al. Diagnostic Sweat Testing: The Cystic Fibrosis Foundation Guidelines J Pediatr.
2007;151:85-9 .
7. Comeau AM, Accurso FJ, White TB, et. al. Guidelines for Implementationof Cystic Fibrosis Newborn Screening Programs:
Cystic Fibrosis Foundation Workshop Report Pediatrics. 2007;119:e495-e518.
8. Flume PA, Robinson, KA, O’Sullivan BP, et. al. Cystic Fibrosis Pulmonary Guidelines: Airway Clearance Therapies Respir
Care. 2009;54(4):522–537.
9. Flume PA, O’Sullivan BP, Robinson KA, et. al. Cystic Fibrosis Pulmonary Guidelines: Chronic Medications for Maintenance of
Lung Health Am J Respir Crit Care Med. 2007;176:957-969.
10. Flume PA, Mogayzel PJ, Robinson KA, et. al. Cystic Fibrosis Pulmonary Guidelines: Treatment of Pulmonary Exacerbations
Am. J. Respir Crit Care Med. 2009 180(9):802-8.
11. Stallings VA, Stark LJ, Robinson KA, et. al. Evidence-Based Practice Recommendations for Nutrition-Related Management of
Children and Adults with Cystic Fibrosis and Pancreatic Insufficiency: Results of a Systematic Review J Am Diet Assoc.
2008;108(5):832-839.
12. Borowitz D, Baker RD, Stallings V. Consensus Report on Nutrition for Pediatric Patients With Cystic Fibrosis J Pediatr
Gastroenterol Nutr. 2002 Sep;35(3):246-59.
13. Moran A, Brunzell C, Cohen RC, et. al. Clinical Care Guidelines for Cystic Fibrosis–Related Diabetes Diabetes Care. 2010;
33(12):2697-2708.
14. Aris RM, Merkel PA, Bachrach LK, et al. Consensus Statement: Guide to Bone Health and Disease in Cystic Fibrosis J Clin
Endocrinol Metab. 2005; 90(3):1888-96.
29